 Anastasios Karamanakos1,2
Anastasios Karamanakos1,2 Olga Vougiouka3
Olga Vougiouka3 Evdoxia Sapountzi4
Evdoxia Sapountzi4 Aliki I. Venetsanopoulou5
Aliki I. Venetsanopoulou5 Maria G. Tektonidou1
Maria G. Tektonidou1 Anastasios E. Germenis6
Anastasios E. Germenis6 Petros P. Sfikakis1
Petros P. Sfikakis1 Katerina Laskari1*
Katerina Laskari1*- 1Joint Academic Rheumatology Program, First Department of Propaedeutic and Internal Medicine, National and Kapodistrian University of Athens, Athens, Greece
- 2Department of Rheumatology, Evangelismos General Hospital, Athens, Greece
- 3Second Department of Pediatrics, National and Kapodistrian University School of Medicine, “P. A. Kyriakou” Children’s Hospital, Athens, Greece
- 4Second Department of Pediatrics, School of Medicine, Faculty of Health Sciences, Aristotle University of Thessaloniki, American Hellenic Educational Progressive Association (AHEPA) University General Hospital, Thessaloniki, Greece
- 5Rheumatology Clinic, Department of Internal Medicine, Medical School, University of Ioannina, Ioannina, Greece
- 6Department of Immunology and Histocompatibility, School of Medicine, University of Thessaly, Larissa, Greece
Objective: To assess the impact conferred by NOD2 variants on the clinical spectrum of patients with systemic autoinflammatory diseases (SAIDs) in Greece.
Methods: Consecutive patients (n=167) with confirmed SAIDs who underwent screening by next generation sequencing (NGS) targeting 26 SAID-associated genes, and carried at least one NOD2 gene variant, were retrospectively studied. The demographic, clinical and laboratory parameters were recorded.
Results: In total, 24 rare NOD2 variants in 23/167 patients (14%) were detected. Notably, 18 patients had at least one co-existing variant in 13 genes other than NOD2. Nine patients had juvenile- and 14 adult-onset disease. All patients presented with symptoms potentially induced by the NOD2 variants. In particular, the candidate clinical diagnosis was Yao syndrome (YAOS) in 12 patients (7% of the whole SAID cohort). The clinical spectrum of patients with YAOS (mean episode duration 8 days) was fever (n=12/12), articular symptoms (n=8), gastrointestinal symptoms (n=7; abdominal pain/bloating in 7; diarrhea in 4; oral ulcers in 3), serositis (n=7), and rash (n=5), while the inflammatory markers were elevated in all but one patient. Most of these patients showed a poor response to nonsteroidal anti-inflammatory drugs (n=7/9), colchicine (n=6/8) and/or anti-TNF treatment (n=3/4), while a complete response was observed in 6/10 patients receiving steroids and 3/5 on anti-IL1 treatment. Another 8 patients were diagnosed with either FMF (n=6) or PFAPA syndrome (n=2) presenting with prominent diarrhea (n=7), oral ulcers (n=2), periorbital swelling and sicca-like symptoms (n=1), or maculopapular rash (n=1). One patient had a clinically undefined SAID, albeit characterized by oral ulcers and diarrhea. Finally, one patient presented with chronic relapsing urticaria with periorbital edema and inflammatory markers, and another one had a Crohn-like syndrome with good response to anti-IL-1 but refractory to anti-TNF treatment.
Conclusion: NOD2 variants were detected in 1 out of 7 SAID patients and seem to have an impact on disease phenotype and treatment response. Further studies should validate combined molecular and clinical data to better understand these distinct nosological entities.
1 Introduction
Systemic autoinflammatory diseases (SAIDs) are a group of genetically heterogenous disorders presenting as sterile, episodic and unprovoked inflammatory attacks driven by the innate immune system. Characteristically, SAIDs lack the high autoantibody titers and autoreactive T cells characterizing autoimmune disorders (1, 2). Historically they are classified as monogenic autoinflammatory disorders (mAIDs), inherited in a mendelian pattern, and polygenic, in which no straightforward pattern of inheritance is observed (3). Interestingly, a new concept in genomic medicine termed genetically transitional disease (GTD) seems to better define Yao syndrome (YAOS) (4), a NOD2-associated AID (NAID), linked to the nucleotide-binding oligomerization domain containing 2 (NOD2) gene, encoding a cytosolic NOD-like receptor (NLR) and innate immune sensor (Online Mendelian Inheritance in Man [OMIM] 617321) (5). According to this novel nomenclature, GTDs are conditions where mutations are necessary, but not sufficient alone to cause disease (4). YAOS represents a new disease entity with still expanding phenotypic and genotypic spectrum (6). It is linked to specific NOD2 sequence variants, which are distinct from those described in Blau syndrome and early onset sarcoidosis, representing the familial and sporadic forms of the same pediatric noncaseating granulomatous SAID, respectively (7, 8), but also in multifactorial Crohn’s disease (9); on the other hand, there is also evidence supporting the presence of shared variants among these nosological entities. At the same time, NOD2 variants have been shown to influence the phenotype of well-defined mAIDs such as Familial Mediterranean Fever (FMF) (10) or other SAIDs (11–28). Due to insufficient awareness, overlapping features and/or lack of extensive genetic screening, NAIDs are often underrecognized or described as a syndrome of undifferentiated recurrent fever (29). Nevertheless, in the era of next generation sequencing (NGS), the ability to screen multiple genes simultaneously has provided significant impact on diagnosing SAIDs and guiding therapy (30). Indeed, recent studies highlight the high frequency of NOD2 variants and diagnosis of YAOS among patients with SAIDs (31). In this respect, we retrospectively studied patients with SAIDs who underwent genetic screening by NGS and carried at least one NOD2 gene variant, in order to describe their genotypic and phenotypic characteristics, and provide evidence on the role of NOD2 variants on SAID phenotypes in a Greek patient cohort.
2 Patients and methods
2.1 Study population
Consecutive patients (n=167) with SAIDs who underwent genetic screening by NGS targeting 26 SAID-associated genes (2015–2023) and carried at least one NOD2 gene variant (n=23), were retrospectively studied. Medical information on demographic characteristics, clinical symptoms, laboratory parameters, and treatment details was retrieved from medical charts and a personal interview. A standard questionnaire was completed for each patient. The same physician in the Rheumatology Unit of the First Department of Propaedeutic Internal Medicine at the University of Athens (center of excellence for rare rheumatic diseases) confirmed disease diagnosis in all patients. The following classification/diagnostic criteria for autoinflammatory syndromes were recorded: Eurofever/Printo classification criteria for mAIDs and PFAPA (1), Yamaguchi diagnostic criteria for Still’s disease (32), YAOS diagnostic criteria (33). Good response to treatment was defined as complete resolution of clinical and biological disease-related manifestations, whereas partial response as any improvement without full recovery. The study was approved by the Ethics Committee of the National and Kapodistrian University of Athens. Informed consent was obtained from each patient or her/his legal guardians for participation in the study according to the Declaration of Helsinki. All data will be made available on request.
2.2 Genetic screening
Genetic screening was performed as previously described (34) at the Department of Immunology & Histocompatibility, University of Thessaly Medical School, in order to identify variants in the coding regions of 26 genes associated with SAIDs (Supplementary Table 1).
2.3 Variant classification
Variants with a worldwide frequency of >1% (1000 Genomes Global Minor Allele Frequency, ExAC) and polymorphisms (UCSC Common SNPs) for which no disease associations are reported in the public ClinVar disease database (https://www.ncbi.nlm.nih.gov/clinvar/), as well as synonymous single-nucleotide variants (SNVs) were excluded from further analysis. To assess the pathogenicity of variants, the ClinVar and INFEVERS databases (https://infevers.umai-montpellier.fr/web/) as well as Varsome, an aggregator and impact analysis tool for human genetic variation (https://varsome.com/about/general/varsome-citations/), were used in addition to literature search. We classified genetic variants as likely benign, benign, VUS (variant of uncertain significance), likely pathogenic or pathogenic for a certain disease, according to the ACMG guidelines and Eurofever criteria (1, 35).
2.4 Disease classification
After considering both clinical findings and genetic results, each patient got a final diagnosis. The genetic results supported a final diagnosis if the patient had clinical symptoms compatible with this diagnosis. A definite disease diagnosis was set if clinical signs and symptoms were compatible with a single SAID, especially in the presence of a confirmatory or a non-confirmatory genotype (35). The disease diagnosis was considered undefined when a particular diagnosis could not be made. If the clinical and genetic characteristics were compatible with more than one SAIDs, the diagnosis of an overlap or mixed syndrome has been suggested. According to the diagnostic criteria for YAOS (33), identification of certain NOD2 variants, including rare ones, was required for this diagnosis.
3 Results
At least one NOD2 variant was detected in 14% of SAID patients (23 out of 167) (Figure 1A). In particular, 15% of 158 patients with positive genetic testing and 20% of 114 patients with variants possibly influencing/causing disease, carried NOD2 variants. Among these 23 patients, 17 were adults (74%) and 6 children; 14 patients (61%) had adult-onset and 9 juvenile-onset disease. Thirteen of them (57%) were females. All were of Greek origin except for three patients of Turkish, Armenian or Arabic descent. Overall, variants were found in 14 out of 26 SAID-associated genes screened (NOD2 included). In total, 24 rare NOD2 (Figures 2A, B) and 36 variants in the other genes were detected; 18 out of 23 patients had at least one variant located in 13 genes other than NOD2. Among these 13 genes the most commonly affected was MEFV (16 variants in 12 patients), followed by TNFRSF11A (3 variants in 2 patients) and LPIN2 (3 variants in 3 patients) (Figure 3). A detailed report of patients’ genotype is provided in Supplementary Table 2.
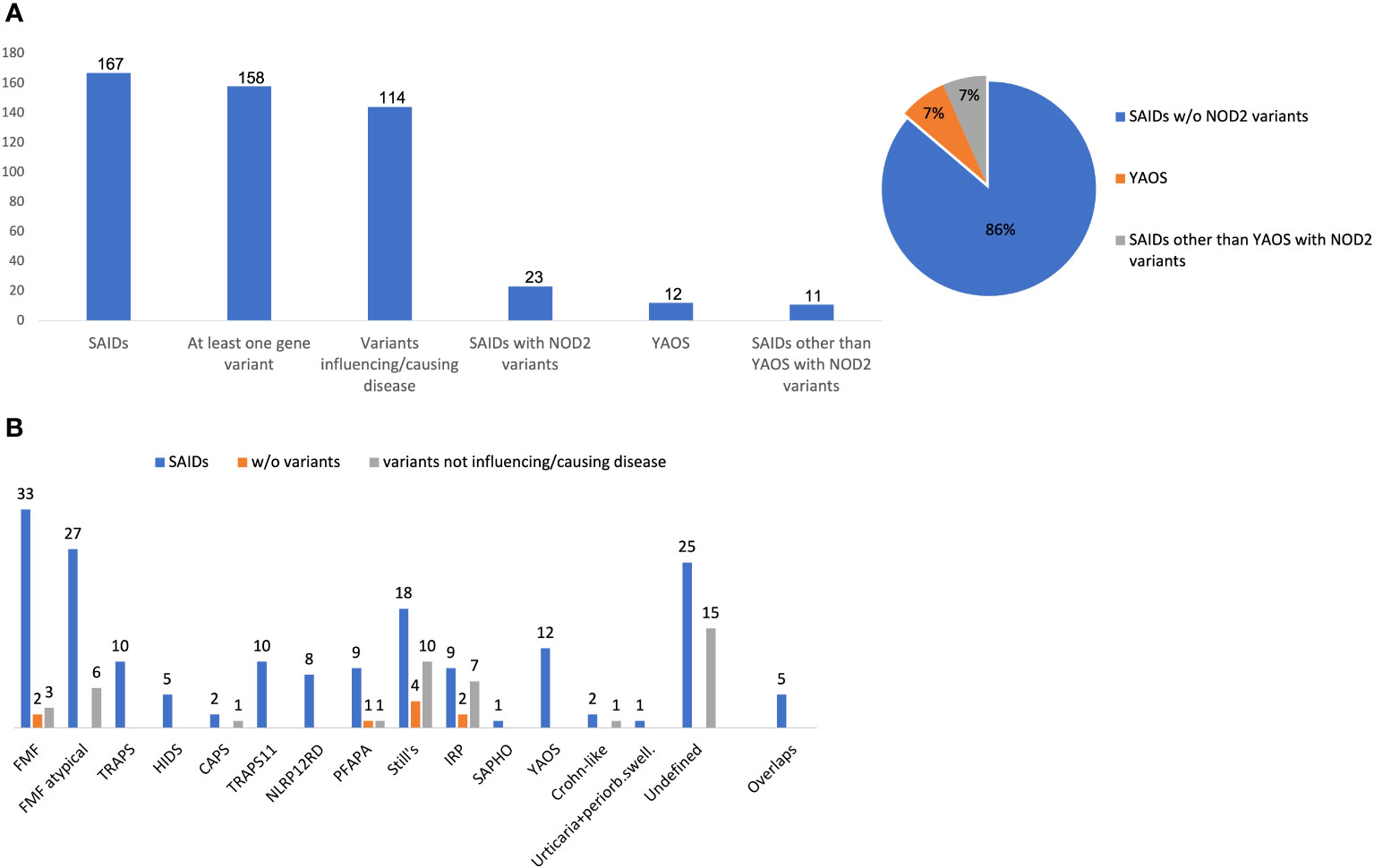
Figure 1 (A) Frequency of NOD2 variants, Yao syndrome or other diagnoses among 167 patients with systemic autoinflammatory diseases. (B) Disease diagnosis (blue bar), patients without variants (orange bar) or carrying only variants not influencing/causing disease (grey bar) in each disease. The y-axis indicates the absolute number of patients. Variants not influencing/causing disease are defined as non-contributory genotypes in MEFV, TNFRSF1A, MVK, NLRP3, NLRP12 (Eurofever criteria) (1), and/or variants in other genes not related to phenotype.Overlap diagnoses in 5 patients include the following: FMF/TRAPS11, n= 3; TRAPS/TRAPS11, n=1; HIDS/NLRP12RD, n=1. w/o, without; SAID, systemic autoinflammatory disease; YAOS, Yao syndrome; FMF, Familial Mediterranean Fever; PFAPA, Periodic Fever, Aphthous Stomatitis, Pharyngitis, Adenitis; NLRP12 RD, NLRP12 related disease; CAPS, cryopyrin-associated periodic syndrome; HIDS, Hyperimmunoglobulinemia D with periodic fever syndrome; TRAPS, Tumor necrosis factor receptor-associated periodic syndrome; TRAPS11, TNFRSF11A associated hereditary fever disease; IRP, idiopathic relapsing pericarditis; SAPHO, Synovitis, Acne, Pustulosis, Hyperostosis, and Osteitis; periorb. swell, periorbital swelling.
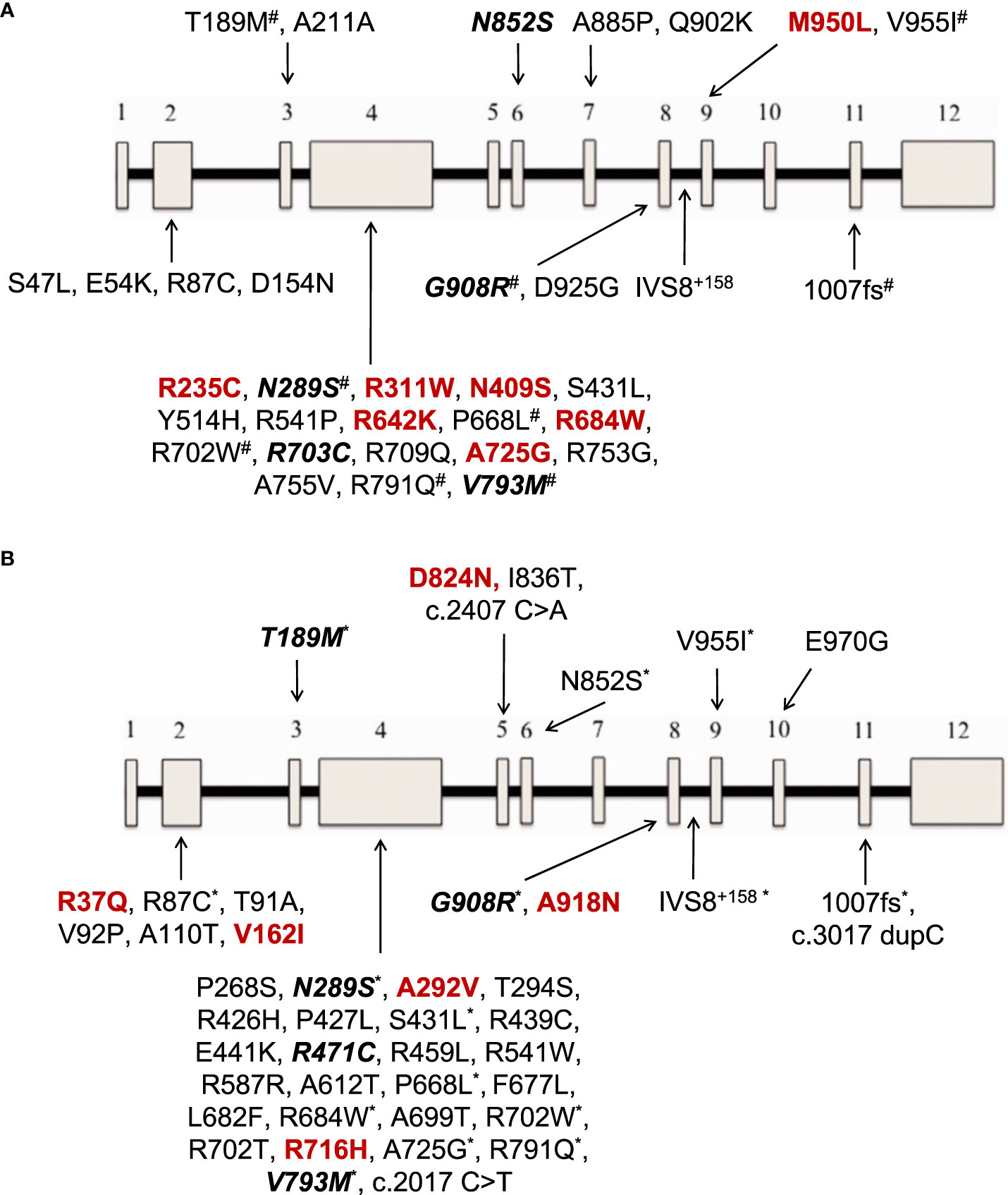
Figure 2 (A) NOD2 gene variants reported in 12 patients with Yao syndrome in the present study and/or in literature. (B) NOD2 gene variants reported in patients with diagnoses other than Yao syndrome in 11 patients in the present study and/or in literature. In colored font are depicted variations found only in our series, in bold italics font those found both in our cohort and in literature, and the rest (regular font) are variants reported only in literature. The 12 exons of the gene are depicted as boxes, and the black line connecting the exons represents the intronic gene regions. #variants also found in patients with diagnoses other than Yao syndrome *variants also found in patients with Yao syndrome.
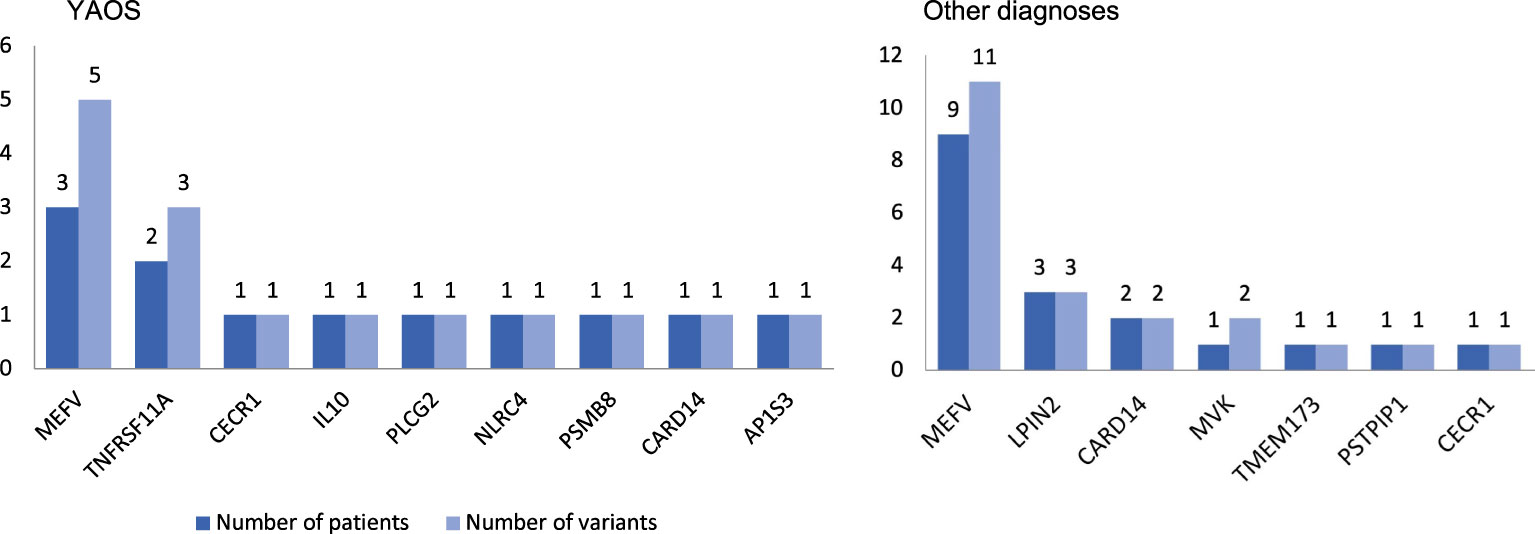
Figure 3 Coexisting variants in genes other than NOD2 in 8 out of 12 patients with Yao syndrome (YAOS) (67%) and 10 out of 11 patients with other diagnoses (91%). The y-axis indicates the absolute number of patients (dark blue bars)/variants (light blue bars).
3.1 Characteristics of patients fulfilling criteria for YAOS
Twelve out of 23 patients carrying NOD2 variants (52%) received the diagnosis of YAOS (7% of the whole cohort). YAOS was the most frequent diagnosis among patients with NOD2 variants and the fourth in order of frequency SAID in our whole cohort following FMF, unclassified disease and Still’s disease (Figures 1A, B). The detailed patient characteristics are shown in Table 1; Supplementary Table 2. The slight majority of patients (58%) was male, and all of them were adults at disease onset (mean age 36 years), with a mean disease duration of 6.3 ± 6.5 years at the time of assessment. The most common symptom was high grade fever, which was present in all patients and had an intermediate duration of 6.5 ± 4.7 days. Most of the patients had almost monthly episodes (the mean number of attacks per year was 13). Polyarthritis/arthralgia was the second common symptom (67%), while approximately 2/3 had gastrointestinal involvement, mainly with abdominal pain, and 1/4 had oral ulcers. One patient was found to have terminal ileitis in colonoscopy but without Crohn’s disease findings in biopsy. Serositis was present in 58% of patients, myalgias in 33%, and almost 1/4 had a maculopapular or urticarial rash. One patient suffered from sicca symptoms, and one presented with lower leg swelling. All but one patient had increased inflammatory markers during episodes. With regard to genetic analysis, all patients were single heterozygous except for one compound heterozygous NOD2 genotype (Supplementary Table 2). Eight patients had 15 variants in genes other than NOD2 (Figure 3); most frequently MEFV (in total 3 pathogenic and 2 VUS variants in 3 patients) and TNFRSF11A (3 variants in 2 patients), but also NLRC4, CECR1, IL10, PLCG2, AP1S3, PSMB8 and CARD14.
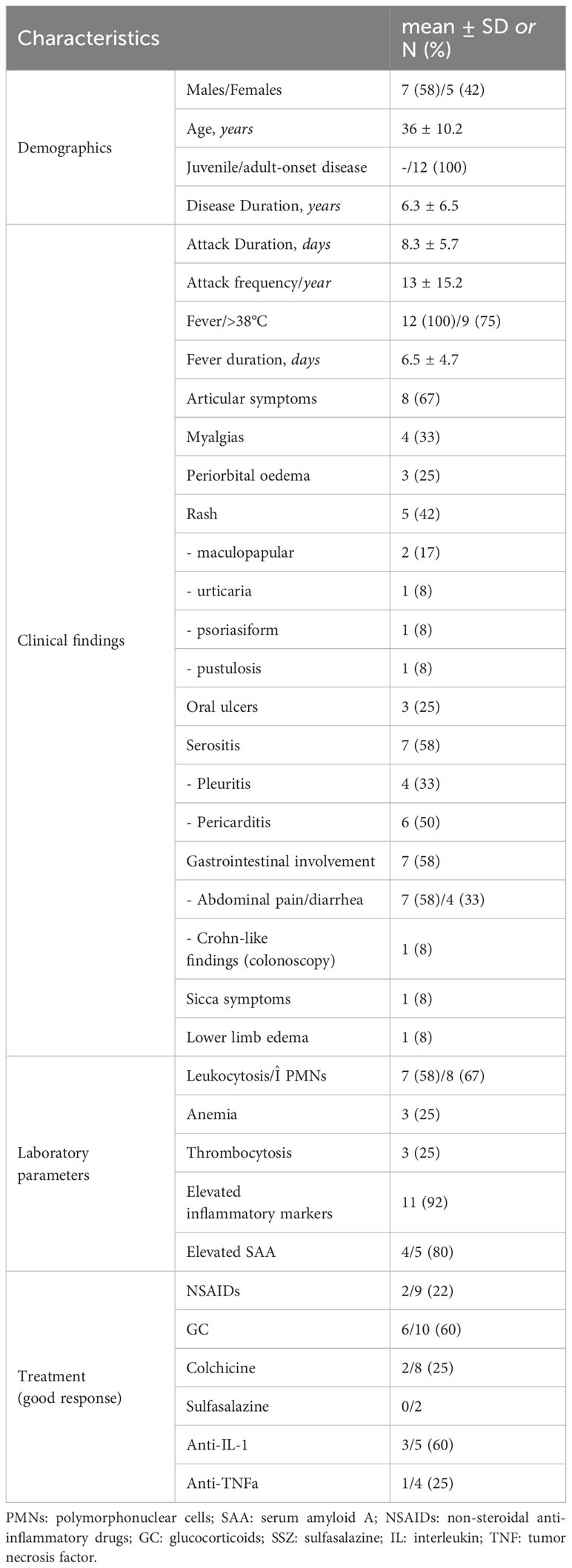
Table 1 Characteristics of 12 patients with Yao syndrome in the present study.
The co-existing variants in genes other than NOD2 might have an impact on disease phenotype in 4 patients with YAOS with features of mAIDs [TNFRSF11A-associated hereditary fever disease - TRAPS11 (36–38), n=2 and/or FMF, n=3] (Supplementary Table 2). In particular, patient no. 10 in Supplementary Table 2, had a pathogenic variant in the MEFV gene and a rare variant in TNFRSF11A. The patient presented with recurrent periodic episodes of high fever lasting 72 hours, accompanied by serositis, periorbital edema, and maculopapular rash, with good response to steroids. Another patient (no. 11 in Supplementary Table 2) had two rare variants in the TNFRSF11A gene as well as a variant in PSMB8. Clinical features included fever episodes lasting up to 10 days, pericarditis, abdominal pain, splenomegaly, partial response to non-steroidal anti-inflammatory drugs (NSAIDs) and colchicine, albeit good response to steroids. Finally, two other patients with pathogenic MEFV variants (patients no. 1 and 2 in Supplementary Table 2), the first showed a good response to colchicine, while the second one had one-day fever episodes.
Overall, patients with YAOS showed a poor response to NSAIDs (n=7/9), colchicine (n=6/8) and/or anti-TNFa treatment (n=3/4), while more than half of the patients receiving steroids (n=6/10) and most of the patients receiving anti–IL-1 agents (n=3/5) had a complete resolution of symptoms and normalization of laboratory parameters. Lastly, two patients received sulfasalazine and one of them showed a partial improvement of his gastrointestinal symptoms.
3.2 Characteristics of patients with diagnoses other than YAOS
In total, 11 patients with SAIDs and diagnoses other than YAOS carried NOD2 variants (48% of all patients carrying NOD2 variants and 7% of the whole cohort) (Figures 1A, 4). In particular, we found a frequency of 7% among patients with positive genetic testing, while 10% among patients with variants possibly influencing/causing disease. The characteristics of non-YAOS patients are summarized in Table 2 and shown in detail in Supplementary Table 2. Most of the patients with NOD2 gene variants and a disease diagnosis other than YAOS were females (n=8), they had juvenile-onset disease (n=9) and a mean age of 8 years at first symptom. All patients were heterozygous for NOD2 variants (Figure 2B) and all but one, had at least one variant in 7 other SAID-associated genes. Among patients with other gene variants, all but one had at least one variant in the MEFV gene (11 variants in 9 patients), whereas fewer patients in LPIN2 (3 variants in 3 patients), CARD14 (2 variants in 2 patients), MVK (2 variants in 1 patient), TMEM173, PSTPIP1, and CECR1 (Figure 3).
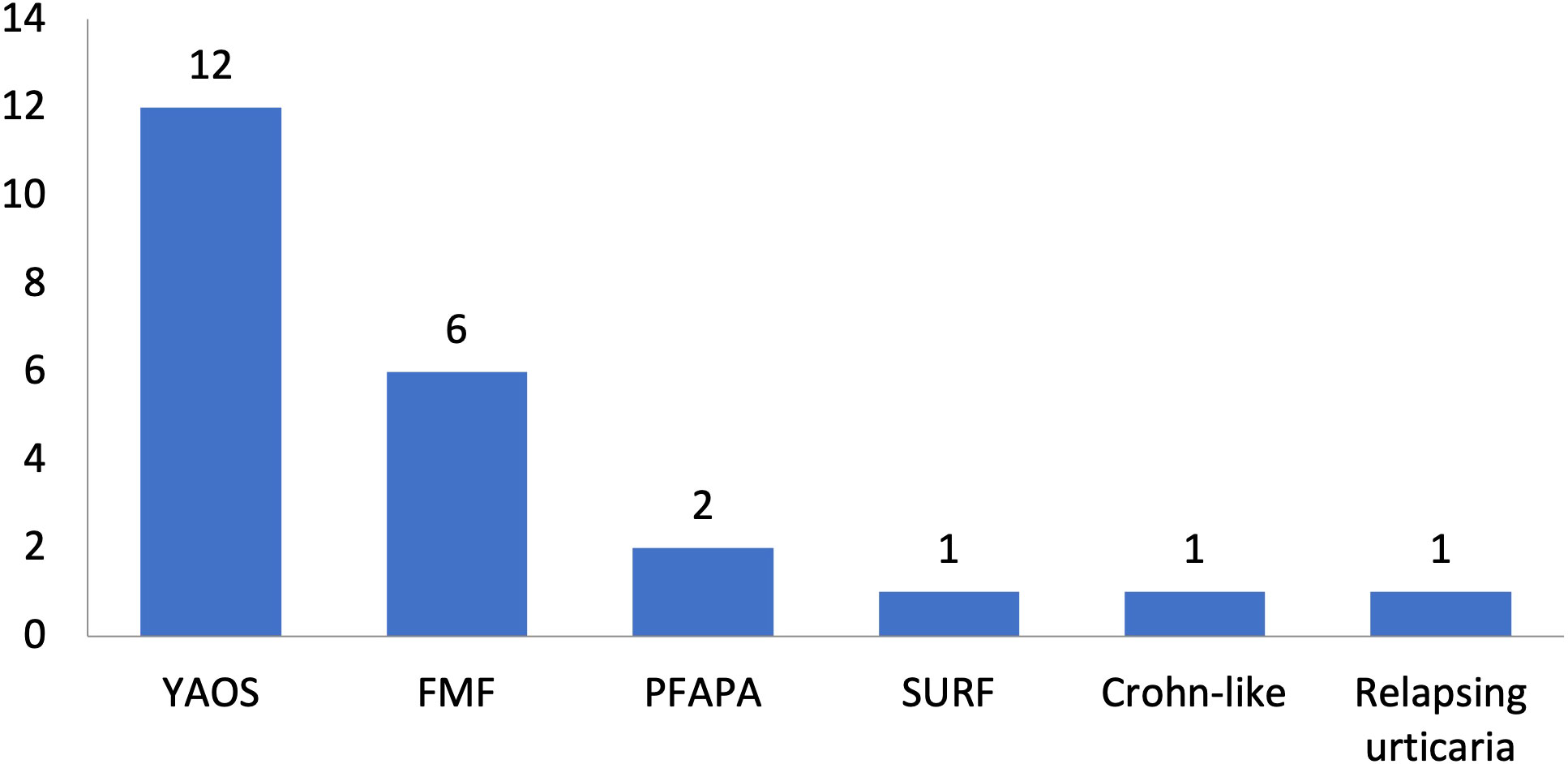
Figure 4 Disease diagnoses among 23 patients with systemic autoinflammatory diseases and rare NOD2 variants. Twelve patients had Yao syndrome (52%) and 11 other diagnoses (48%). YAOS, Yao syndrome; FMF, Familial Mediterranean Fever; PFAPA, Periodic Fever, Aphthous Stomatitis, Pharyngitis, Adenitis; TRAPS11, TNFRSF11A associated hereditary fever disease; SURF, syndrome of undifferentiated recurrent fever.

Table 2 Characteristics of 11 patients with NOD2 variants and diagnoses other than Yao syndrome in the present study.
All patients presented with symptoms potentially induced by variants in the NOD2 gene. In particular, among 6 patients receiving the diagnosis of FMF, 5 presented with prominent diarrhea, of which one had also oral ulcers and showed a poor response to colchicine, while one developed periorbital swelling and sicca-like symptoms later during disease course; the sixth patient, being compound heterozygous for two pathogenic MEFV variations, exhibited an extensive maculopapular rash (Supplementary Table 2). Another two patients fulfilled the criteria for PFAPA syndrome, albeit, they exhibited prominent diarrhea (n=2) and oral ulcers (n=1). One patient was diagnosed with an undifferentiated SAID (syndrome of undifferentiated recurrent fever – SURF); she presented with fever, myalgias, urticarial rash, prominent recurrent oral ulcers and diarrhea. Finally, one patient carrying the G908R NOD2 variant, presented with a clinical syndrome resembling Crohn’s disease, albeit he was refractory to anti-TNFa treatment and showed good response to anti-IL-1 agents, and another one had relapsing urticaria refractory to steroids, periorbital swelling, and increased inflammatory markers.
4 Discussion
NOD2 variants seem to be common when screening patients with SAIDs. Indeed, since the first mention of the NOD2 gene on chromosome 16q12 and its protein in 2001 (39), up to 187 NOD2 variants have been reported in Infevers database (https://infevers.umai-montpellier.fr/web/search.php?n=6). In our Greek cohort with SAIDs, 14% of patients carried NOD2 variants; 15% of cases with positive genetic testing and 20% of those with variants possibly influencing/causing disease. More than half of these patients fulfilled the YAOS disease criteria (7% of the whole cohort). In agreement with our study, in a Chinese population with monogenic or polygenic SAIDs, Hua et al. detected NOD2 variants in 10 out of 68 patients (15%); one third of them suffered from YAOS (4% of the whole cohort) (24), whereas Qin et al. reported 6 out of 80 (8%) patients with NOD2 variants, all were finally diagnosed with YAOS (40). In an aforementioned study including 143 patients with clinical phenotypes suspicious for NOD2-associated disease, almost half of them (47%) carried NOD2 variants. Among them, four-fifths received the diagnosis of YAOS (38% of the whole cohort) (41).
YAOS, first described in 2011 in 7 patients in the United States (3), seems to be more common than originally thought, especially when compared to other SAIDs (31), with an estimated prevalence of 1–10/100,000 (41). Indeed, YAOS was the fourth in order of frequency SAID in our whole cohort following FMF, unclassified disease and Still’s disease, and the most frequent diagnosis among patients with NOD2 variants (52%), as also previously described (20). According to the largest studies (20, 33, 41, 42), YAOS is principally sporadic and is predominantly reported in white adults with a female-to-male ratio of 2:1; the disease occurs at any age, however, it is most common between age 20 and 50 (6), and recurrent episodes last from days to several weeks alternating with asymptomatic periods of weeks, months or years (5). The inflammatory nature of YAOS is supported by studies showing spontaneous IL-6 production by monocytes derived from the patient peripheral blood, as well as aberrant NOD2 transcriptional levels and its signal NF-κB pathway; nevertheless, plasma levels of pro-inflammatory cytokines such as TNFa, IL-1β, IFNγ, IL-6 and IL-17 are not elevated (43). Others support the role of Th17 cells as well as latency-associated peptide-positive T cells, and not Th1/Th2 cells, in disease pathogenesis (44). Interestingly, infections and/or interferon release have been described as possible triggers, and vasoactive intestinal polypeptide as a possible NOD2 pathway activator (45, 46).
To date, fewer than 100 patients have been described worldwide (Supplementary Table 3), suggesting that YAOS is underdiagnosed. Patients present with fever (60-80%), non-destructive polyarthritis/polyarthralgia (≥80%), skin disease (90%), gastrointestinal symptoms (65-90%) with no convincing endoscopic or histologic evidence of inflammatory bowel disease, serositis (10-40%), lower unilateral/bilateral extremity swelling (30-60%), sicca-like symptoms (50-60%), oral ulcers (25-50%) and eyelid swelling (up to 50%) (47–49). Rare manifestations such as ocular myositis have been also described (50, 51). Interestingly, phenotypical differences are observed among different ethnic groups, such as the lack of skin disease in patients of Asian descent (40, 52).
In our Greek cohort of 12 patients, we observed an almost equal gender distribution. In accordance to literature, all but one patient, were diagnosed in adulthood (mean age 36 years), and disease episodes lasted (mean) 8 days. We observed a higher frequency of fever, usually high grade, present in all our patients, and of serositis (58%), whereas a lower frequency of periorbital edema (25%), maculopapular rash (17%), sicca-like symptoms (8%), and lower limb swelling (8%). Interestingly, a recent study comparing 35 patients with YAOS to 28 patients with NOD2 variants but other diagnoses, demonstrated more frequent lower leg edema and less headache in the former group (20).
With regard to YAOS treatment, glucocorticoids and sulfasalazine seem to be effective treatment options (33), however, up to half of the patients suffer major relapses despite therapy (33) (Supplementary Table 4). In these cases, anti-TNFa therapy seems to provide only modest or no response, while promising efficacy has been observed with the IL-1 inhibitor canakinumab (53, 54). Indeed, in a retrospective study of 7 refractory patients, all showed a complete clinical response with canakinumab starting from day 7 post treatment and reaching peak response on day 14 (53). Other drugs, such as IL-6 inhibitors, may be an option in refractory patients, nevertheless, more data are awaited to confirm their efficacy (43). In our study, despite previous favorable experience (33, 55), sulfasalazine did not completely control symptoms in both YAOS patients treated with the drug. Nevertheless, the small number of patients makes it impossible to reach a firm conclusion on the effectiveness of sulfasalazine in our patient cohort. On the other hand, in consistency with literature, more than half of our patients receiving glucocorticoids and/or IL-1 inhibitors (anakinra or canakinumab) showed a good response to treatment, in contrast to NSAIDs, colchicine and anti-TNFa agents.
Among patients with SAIDs and diagnoses other than YAOS (Supplementary Table 5) the frequency of NOD2 variants varies between 3-20% (13, 16, 21, 23, 24) – 7% in our study. In particular, Demir et al. (14) observed a frequency of 6% among patients with positive genetic testing, whereas Karacan et al. (26) noted 7% among patients with at least one pathogenic variant. In accordance with these studies, we also found a frequency of 7% among patients with positive genetic testing, while 10% among patients with variants possibly influencing/causing disease. With regard to disease diagnosis, NOD2 variants have been described among patients with mAIDs, e.g. FMF (4-10%) (10,16,18,20,24,26,27, present study), CAPS (21, 23), HIDS (21), patients with TNFRSF1A variants (15%) (12), NLRP12-related disease (13%) (21, 22, 24), VEXAS syndrome (19), as well as among patients with polygenic SAIDs such as Still’s disease (11), PFAPA (22%) (present study), Schnitzler syndrome (17), and also unclassified SAIDs (4-11% - 12% of patients with positive genetic testing) (13–15,21,23,25,26, present study), Crohn-like disease (present study) and relapsing urticaria (23, present study). In our study, FMF was the most frequent disease among patients with concurrent NOD2 variants and a diagnosis other than YAOS; in particular, 22% of atypical FMF cases and 10% of the whole FMF cohort carried NOD2 variants.
Interestingly, NOD2 variants alter the phenotype of patients with the above monogenic or polygenic diagnoses, which implies a strong influence of NOD2 in phenotypic expression beside other genes. For example, more severe disease has been observed in FMF patients carrying NOD2 variants (10); at the same time, atypical disease features and manifestations possibly attributed to NOD2 variants include resistance to colchicine (10,15,18,21, present study), pericarditis (21), abdominal pain (14, 16, 22) even between fever episodes (21), oral ulcers (15,22, present study) diarrhea (14, present study), Crohn’s like disease (22, present study) resistant to anti-TNFa but responsive to anti-IL-1 treatment (present study), conjunctivitis (14), erythematous rash (15, present study), and periorbital edema (present study). Interestingly, the patient with Crohn-like syndrome (Supplementary Table 2, patient no. 23) carried the NOD2 G908R variant. G908R has been characterized a common coding risk variant for Crohn’s disease, nevertheless, it has been also reported in YAOS (9, 56, 57, present study). Finally, it is worth mentioning that some reports argue for the presence of intermediate entities between inflammatory bowel disease or Blau syndrome and NAIDs in patients carrying NOD2 variants other than those usually observed in the former diseases (58–60).
With regard to NOD2 genotype, most patients in literature, in accord with our results, carry a single gene variant in heterozygous state, that probably acts as a low penetrance variant in a gain-of-function fashion (9). In YAOS, the vast majority of cases (85%), derived from one center in the United States, carry the variant IVS8 + 158 residing in intron 8 splicing region of NOD2 in heterozygosity; a quarter of patients also carry the variant R702W or 1007fs, residing in exon 4 and region encoding the leucin-rich repeat (31, 33, 41–43, 56). Nevertheless, several other rare variants have been previously reported. In the present study, even though our screening method did not allow examining intronic regions, we were able to detect variants already described by others, in addition to new ones (Figure 2A). The same was the case for patients with diagnoses other than YAOS carrying already known, but also newly detected variants (Figure 2B). Interestingly, patients with YAOS and those with other diagnoses shared common variants. This observation points towards the hypothesis that carriage of specific NOD2 variants may confer susceptibility to disease by modifying or amplifying inflammation rather than triggers a specific disease (17). Indeed, the meaning of gene dosage or synergistic gene effect is of growing interest (20, 34, 44). In this context, phenotypes are mainly determined by the high penetrance variant, whereas a synergistic effect between high-low penetrance variants may create phenotypic and therapeutic variability (4).
In the current study, 18 out of 23 patients with NOD2 variants had at least one variant located in 13 genes other than NOD2 (67% of patients with YAOS vs. 91% other diagnoses). In a recent study (20), 44 patients were found to have coexisting -mostly digenic- variants in NOD2 and another gene vs. 19 patients with variants in NOD2 only (47% of patients with YAOS vs. 100% other diagnoses). Except for being more frequently diagnosed with YAOS, patients with NOD2 variants solely, experienced more frequently chest pain. On the other hand, in agreement with the synergistic gene effect hypothesis, those carrying variants in different genes presented more often with other diagnoses or mixed clinical features. Indeed, in the present study, YAOS patients with TNFRSF11A and/or MEFV variants displayed features also compatible with TRAPS11 syndrome (36–38) and/or FMF.
In line with the literature, in the present study, the most frequently co-existing mutated gene was MEFV, both among patients with YAOS and those with other diagnoses. Indeed, variants in MEFV and NOD2 have been also previously speculated to be synergistic (2, 18). Based on the evidence that NOD2 contributes to disease expression in conjunction with other gene variants, Nomani et al. (20) suggested a new term, mixed NLR-associated Autoinflammatory Disease (NLR-AID), to describe mixed presentations heavily influenced by NOD2 variants. In our cohort, we encountered a patient with SURF (patient no. 20 in Supplementary Table 2) with symptoms possibly attributed to NOD2 variants, carrying a single low penetrance variant in MEFV, and representing, per se, a case of mixed NLR-AID. This new nosological entity remains a challenge to be further addressed in future studies.
Limitations of the present study include the following: Our screening method did not allow examining intronic gene regions that are of special interest to YAOS. Whole NOD2 gene sequencing should be included in future studies. Nevertheless, we provided evidence on the presence of not previously described rare NOD2 variants. Our cohort consists of a small number of patients, however, in contrast to studies with common diseases, SAIDs are exceedingly rare. Besides, due to the retrospective design of the study we might have missed information regarding clinical or therapeutic features. On the other hand, the mono-centric approach offers the benefit of enhanced standardization of the study population. Finally, one should always consider discrepancies between studies attributed to the different clinical SAID phenotypes tested, the screening methods used, number of genes tested, overlapping features between SAIDs, as well as intrinsic divergence among different populations.
To conclude, this is the first study focusing on SAIDs with NOD2 variants, including YAOS, in a Greek patient cohort. It emphasizes the high frequency of NOD2 variants and their impact on disease phenotype as well as the potential additive role of other coexisting gene variants. Additional studies, using whole exome or genome sequencing, to reveal the exact role of underlying gene variants in disease expression, gene-to-gene interactions that result in overlap syndromes, the type of inheritance explaining variability in penetrance and expressivity of disease, and, finally, to better describe the expanding phenotypic spectrum of NOD2-associated disease, are warranted.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Ethics Committee of the National and Kapodistrian University of Athens. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
AK: Data curation, Writing – original draft, Writing – review & editing. OV: Data curation, Writing – review & editing. ES: Data curation, Writing – review & editing. AV: Data curation, Writing – review & editing. MT: Writing – review & editing. AG: Writing – review & editing. PS: Writing – review & editing. KL: Conceptualization, Data curation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1342668/full#supplementary-material
References
1. Gattorno M, Hofer M, Federici S, Vanoni F, Bovis F, Aksentijevich I, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis (2019) 78(8):1025–32. doi: 10.1136/annrheumdis-2019-215048
2. Yao Q. Systemic autoinflammatory disease and genetic testing. Rheumatol Immunol Res (2021) 2(4):209–11. doi: 10.2478/rir-2021-0028
3. Yao Q, Zhou L, Cusumano P, Bose N, Piliang M, Jayakar B, et al. A new category of autoinflammatory disease associated with NOD2 gene mutations. Arthritis Res Ther (2011) 13(5). doi: 10.1186/ar3462
4. Yao Q, Gorevic P, Shen B, Gibson G. Genetically transitional disease: a new concept in genomic medicine. Trends Genet (2023) 39(2):98–108. doi: 10.1016/j.tig.2022.11.002
5. Yao Q, Li E, Shen B. Autoinflammatory disease with focus on NOD2-associated disease in the era of genomic medicine. Autoimmunity (2019) 52(2):48–56. doi: 10.1080/08916934.2019.1613382
6. Yao Q, Kontzias A. Expansion of phenotypic and genotypic spectrum in yao syndrome: A case series. J Clin Rheumatol (2022) 28(1):E156–60. doi: 10.1097/RHU.0000000000001655
7. Chamaillard M, Philpott D, Girardin SE, Zouali H, Lesage S, Chareyre F, et al. Gene-environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proc Natl Acad Sci U S A. (2003) 100(6):3455–60. doi: 10.1073/pnas.0530276100
8. Caso F, Costa L, Rigante D, Vitale A, Cimaz R, Lucherini OM, et al. Caveats and truths in genetic, clinical, autoimmune and autoinflammatory issues in Blau syndrome and early onset sarcoidosis. Autoimmun Rev (2014) 13(12):1220–9. doi: 10.1016/j.autrev.2014.08.010
9. Ashton JJ, Seaby EG, Beattie RM, Ennis S. NOD2 in crohn’s disease-unfinished business. J Crohns Colitis. (2023) 17(3):450–8. doi: 10.1093/ecco-jcc/jjac124
10. Berkun Y, Karban A, Padeh S, Pras E, Shinar Y, Lidar M, et al. NOD2/CARD15 gene mutations in patients with familial Mediterranean fever. Semin Arthritis Rheumatol (2012) 42(1):84–8. doi: 10.1016/j.semarthrit.2011.12.002
11. Garcia-Melchor E, Grados D, González-Roca E, Arostegui JI, Yague J, Narváez FJ, et al. CIAS1 and NOD2 genes in adult-onset Still’s disease. J Rheumatol (2014) 41(7):1566–7. doi: 10.3899/jrheum.131563
12. Ueda N, Ida H, Washio M, Miyahara H, Tokunaga S, Tanaka F, et al. Clinical and genetic features of patients with TNFRSF1A variants in Japan: findings of a nationwide survey. Arthritis Rheumatol (Hoboken NJ). (2016) 68(11):2760–71. doi: 10.1002/art.39793
13. Sözeri B, Demir F, Sönmez HE, Karadağ ŞG, Demirkol YK, Doğan ÖA, et al. Comparison of the clinical diagnostic criteria and the results of the next-generation sequence gene panel in patients with monogenic systemic autoinflammatory diseases. Clin Rheumatol (2020) 40(6):2327–37. doi: 10.1007/s10067-020-05492-8
14. Demir F, Doğan ÖA, Demirkol YK, Tekkuş KE, Canbek S, Karadağ ŞG, et al. Genetic panel screening in patients with clinically unclassified systemic autoinflammatory diseases. Clin Rheumatol (2020) 39(12):3733–45. doi: 10.1007/s10067-020-05108-1
15. Papa R, Rusmini M, Volpi S, Caorsi R, Picco P, Grossi A, et al. Next generation sequencing panel in undifferentiated autoinflammatory diseases identifies patients with colchicine-responder recurrent fevers. Rheumatol (United Kingdom). (2020) 59(2):344–60. doi: 10.1093/rheumatology/kez270
16. Hidaka Y, Fujimoto K, Matsuo N, Koga T, Kaieda S, Yamasaki S, et al. Clinical phenotypes and genetic analyses for diagnosis of systemic autoinflammatory diseases in adult patients with unexplained fever. Mod Rheumatol (2021) 31(3):704–9. doi: 10.1080/14397595.2020.1784542
17. Navetta-Modrov B, Yao Q. Macroglobulinemia and autoinflammatory disease. Rheumatol Immunol Res (2021) 2(4):227–32. doi: 10.2478/rir-2021-0031
18. Lee J, Bizzocchi L, Jain R, Tagoe CE. An autoinflammatory syndrome with compound heterozygous MEFV and NOD2/ CARD15 gene mutations successfully treated with tocilizumab. Rheumatol Adv Pract (2022) 6(2). doi: 10.1093/rap/rkac035
19. Rivera EG, Patnaik A, Salvemini J, Jain S, Lee K, Lozeau D, et al. SARS-CoV-2/COVID-19 and its relationship with NOD2 and ubiquitination. Clin Immunol (2022) 238. doi: 10.1016/j.clim.2022.109027
20. Nomani H, Deng Z, Navetta-Modrov B, Yang J, Yun M, Aroniadis O, et al. Implications of combined NOD2 and other gene mutations in autoinflammatory diseases. Front Immunol (2023) 14:1265404. doi: 10.3389/fimmu.2023.1265404
21. Rusmini M, Federici S, Caroli F, Grossi A, Baldi M, Obici L, et al. Next-generation sequencing and its initial applications for molecular diagnosis of systemic auto-inflammatory diseases. Ann Rheum Dis (2016) 75(8):1550–7. doi: 10.1136/annrheumdis-2015-207701
22. Kostik MM, Suspitsin EN, Guseva MN, Levina AS, Kazantseva AY, Sokolenko AP, et al. Multigene sequencing reveals heterogeneity of NLRP12-related autoinflammatory disorders. Rheumatol Int (2018) 38(5):887–93. doi: 10.1007/s00296-018-4002-8
23. Hoang TK, Albert DA. Novel presentations of periodic fever syndromes: Discrepancies between genetic and clinical diagnoses. Eur J Rheumatol (2019) 6(1):12–8. doi: 10.5152/eurjrheum.2018.18023
24. Hua Y, Wu D, Shen M, Yu K, Zhang W, Zeng X. Phenotypes and genotypes of Chinese adult patients with systemic autoinflammatory diseases. Semin Arthritis Rheumatol (2019) 49(3):446–52. doi: 10.1016/j.semarthrit.2019.05.002
25. Ter Haar NM, Eijkelboom C, Cantarini L, Papa R, Brogan PA, Kone-Paut I, et al. Clinical characteristics and genetic analyses of 187 patients with Undefined autoinflammatory diseases. Ann Rheum Dis (2019) 78(10):1405–11. doi: 10.1136/annrheumdis-2018-214472
26. Karacan İ, Balamir A, Uğurlu S, Aydın AK, Everest E, Zor S, et al. Diagnostic utility of a targeted next-generation sequencing gene panel in the clinical suspicion of systemic autoinflammatory diseases: a multi-center study. Rheumatol Int (2019) 39(5):911–9. doi: 10.1007/s00296-019-04252-5
27. Fujimoto K, Hidaka Y, Koga T, Kaieda S, Yamasaki S, Nakashima M, et al. Clinical and genetic analysis of 22 Japanese patients with familial Mediterranean fever: An examination of MEFV and 10 other genes related to autoinflammatory syndromes. Intern Med (2020) 59(11):1373–8. doi: 10.2169/internalmedicine.3778-19
28. Bozgeyik E, Mercan R, Arslan A, Tozkir H. Next-generation screening of a panel of genes associated with periodic fever syndromes in patients with Familial Mediterranean Fever and their clinical characteristics. Genomics (2020) 112(4):2755–62. doi: 10.1016/j.ygeno.2020.03.012
29. Broderick L, Hoffman HM. Pediatric recurrent fever and autoinflammation from the perspective of an allergist/immunologist. J Allergy Clin Immunol (2020) 146(5):960–966.e2. doi: 10.1016/j.jaci.2020.09.019
30. Papa R, Penco F, Volpi S, Sutera D, Caorsi R, Gattorno M. Syndrome of undifferentiated recurrent fever (SURF): an emerging group of autoinflammatory recurrent fevers. J Clin Med (2021) 10(9). doi: 10.3390/jcm10091963
31. Yao Q, Lacbawan F, Li J. Adult autoinflammatory disease frequency and our diagnostic experience in an adult autoinflammatory clinic. Semin Arthritis Rheumatol (2016) 45(5):633–7. doi: 10.1016/j.semarthrit.2015.10.012
32. Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, Kashiwagi H, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol (1992) 19(3):424–30.
33. Yao Q, Shen B. A systematic analysis of treatment and outcomes of NOD2-associated autoinflammatory disease. . Am J Med (2017) 130(3):365.e13–365.e18. doi: 10.1016/j.amjmed.2016.09.028
34. Karamanakos A, Tektonidou M, Vougiouka O, Gerodimos C, Katsiari C, Pikazis D, et al. Autoinflammatory syndromes with coexisting variants in Mediterranean FeVer and other genes: Utility of multiple gene screening and the possible impact of gene dosage. Semin Arthritis Rheum (2022) 56. doi: 10.1016/j.semarthrit.2022.152055
35. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
36. Jéru I, Cochet E, Duquesnoy P, Hentgen V, Copin B, Mitjavila-Garcia MT, et al. Brief Report: Involvement of TNFRSF11A molecular defects in autoinflammatory disorders. Arthritis Rheumatol (Hoboken NJ). (2014) 66(9):2621–7. doi: 10.1002/art.38727
37. Martorana D, Bonatti F, Mozzoni P, Vaglio A, Percesepe A. Monogenic autoinflammatory diseases with mendelian inheritance: genes, mutations, and genotype/phenotype correlations. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00344
38. De Jesus AA, Goldbach-Mansky R. Newly recognized Mendelian disorders with rheumatic manifestations. Curr Opin Rheumatol (2015) 27(5):511–9. doi: 10.1097/BOR.0000000000000207
39. Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Núñez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem (2001) 276(7):4812–8. doi: 10.1074/jbc.M008072200
40. Qin W, Wu D, Luo Y, Zhao M, Wang Y, Shi X, et al. Neurological manifestations of autoinflammatory diseases in Chinese adult patients. Semin Arthritis Rheumatol (2020) 50(6):1500–6. doi: 10.1016/j.semarthrit.2019.12.003
41. Yao Q, Shen M, Mcdonald C, Lacbawan F, Moran R, Shen B. NOD2-associated autoinflammatory disease: a large cohort study. Rheumatol (Oxford). (2015) 54(10):1904–12. doi: 10.1093/rheumatology/kev207
42. Navetta-Modrov B, Nomani H, Yun M, Yang J, Salvemini J, Aroniadis O, et al. A novel nucleotide-binding oligomerization domain 2 genetic marker for Yao syndrome. J Am Acad Dermatol (2023) 89(1):166–8. doi: 10.1016/j.jaad.2023.02.029
43. McDonald C, Shen M, Johnson EE, Kabi A, Yao Q. Alterations in nucleotide-binding oligomerization domain-2 expression, pathway activation, and cytokine production in Yao syndrome. Autoimmunity (2018) 51(2):53–61. doi: 10.1080/08916934.2018.1442442
44. Yao Q, Myles J, Shen B, Mcdonald C. NOD2-associated autoinflammatory disease: an exploratory study of its pathogenesis. Rheumatol (Oxford). (2014) 53(5):958–60. doi: 10.1093/rheumatology/ket384
45. Trueb B, Zhuang L, Keller I, Köckritz LV, Kuchen S, Dufour JF, et al. Coincidence of NOD2-associated autoinflammatory disease (Yao syndrome) and HCV infection with fatal consequences: interaction between genes and environment. J Clin Rheumatol (2021) 27(8S):S592–4. doi: 10.1097/RHU.0000000000000963
46. Navetta-Modrov B, Ghebrehiwet B, Yao Q. Yao syndrome: A potential role and association of vasoactive intestinal peptide with NOD2. Rheumatol Immunol Res (2021) 2(1):57–9. doi: 10.2478/rir-2021-0005
47. Yao Q, Su LC, Tomecki KJ, Zhou L, Jayakar B, Shen B. Dermatitis as a characteristic phenotype of a new autoinflammatory disease associated with NOD2 mutations. J Am Acad Dermatol (2013) 68(4):624–31. doi: 10.1016/j.jaad.2012.09.025
48. Yao Q, Schils J. Distal lower extremity swelling as a prominent phenotype of NOD2-associated autoinflammatory disease. Rheumatol (Oxford). (2013) 52(11):2095–7. doi: 10.1093/rheumatology/ket143
49. Yzeiraj E, Kumar A, Kontzias A, Klein AL. Pericardial enhancement using multimodality imaging in a rare auto-inflammatory disorder. Int J Cardiol (2016) 220:654–5. doi: 10.1016/j.ijcard.2016.06.257
50. Yao Q, Ruggieri P, Lowder C. Ocular myositis occurring with NOD2-associated autoinflammatory disease. J Rheumatol (2013) 40(10):1768–9. doi: 10.3899/jrheum.130332
51. Esse I, Kincaid C, Horton L, Arnold JD, Mesinkovska NA. Yao syndrome: Cyclical folliculitis, fevers, and abdominal pain. JAAD Case Rep (2023) 35:71–3. doi: 10.1016/j.jdcr.2023.01.039
52. Yang X, Wu D, Li J, Shen M, Zhang W. A Chinese case series of Yao syndrome and literature review. Clin Rheumatol (2018) 37(12):3449–54. doi: 10.1007/s10067-018-4274-0
53. Yao Q. Effectiveness of canakinumab for the treatment of patients with Yao syndrome. J Am Acad Dermatol (2023) 88(3):653–4. doi: 10.1016/j.jaad.2019.09.020
54. Brailsford CJ, Khamdan F, Elston DM. Treatment of refractory Yao syndrome with canakinumab. JAAD Case Rep (2022) 29:37–40. doi: 10.1016/j.jdcr.2022.08.035
55. Estephan M, Yao Q, Springer J. Case of NOD2-associated autoinflammatory disease successfully treated with sulfasalazine. J Clin Rheumatol (2017) 23(1):58–9. doi: 10.1097/RHU.0000000000000468
56. Yao Q. AB1069 Consideration of Yao syndrome as a differential diagnosis for hereditary periodic fever syndromes. Ann Rheum Dis (2020) 79(Suppl 1):1823–4. doi: 10.1136/annrheumdis-2020-eular.1449
57. Yao Q, Shen M, Gorevic P. NOD2 versus MEFV: differential diagnosis of yao syndrome and familial mediterranean fever. Rheumatol Immunol Res (2021) 2(4):233–9. doi: 10.2478/rir-2021-0032
58. Dziedzic M, Marjańska A, Babol-Pokora K, Urbańczyk A, Grześk E, Młynarski W, et al. Co-existence of Blau syndrome and NAID? Diagnostic challenges associated with presence of multiple pathogenic variants in NOD2 gene: a case report. (2017) 15(1). doi: 10.1186/s12969-017-0188-7
59. Seril DN, Yao Q, Shen B. Auto-inflammatory diseases in ileal pouch patients with NOD2/CARD15 mutations. Gastroenterol Rep (2016) 4(1):73. doi: 10.1093/gastro/gou069
Keywords: NOD2, systemic autoinflammatory disease, Yao syndrome, coexisting gene variants, NGS
Citation: Karamanakos A, Vougiouka O, Sapountzi E, Venetsanopoulou AI, Tektonidou MG, Germenis AE, Sfikakis PP and Laskari K (2024) The expanding clinical spectrum of autoinflammatory diseases with NOD2 variants: a case series and literature review. Front. Immunol. 15:1342668. doi: 10.3389/fimmu.2024.1342668
Received: 22 November 2023; Accepted: 08 January 2024;
Published: 29 January 2024.
Edited by:
Qingping Yao, Stony Brook University, United StatesReviewed by:
Dorota Monika Rowczenio, University College London, United KingdomMilos Jesenak, Comenius University, Slovakia
Min Shen, Peking Union Medical College Hospital (CAMS), China
Copyright © 2024 Karamanakos, Vougiouka, Sapountzi, Venetsanopoulou, Tektonidou, Germenis, Sfikakis and Laskari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katerina Laskari, a2F0ZXJpbmFfbGFza2FyaUB5YWhvby5ncg==