 Wonyoung Jo
Wonyoung Jo Taejoon Won
Taejoon Won Abdel Daoud
Abdel Daoud Daniela Čiháková
Daniela Čiháková
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 05 February 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1340373
This article is part of the Research TopicBiomarkers of Immunotoxicity from Immune Checkpoint InhibitorsView all 13 articles
Immune checkpoint inhibitors (ICIs) are specialized monoclonal antibodies (mAbs) that target immune checkpoints and their ligands, counteracting cancer cell-induced T-cell suppression. Approved ICIs like cytotoxic T-lymphocyte antigen-4 (CTLA-4), programmed death-1 (PD-1), its ligand PD-L1, and lymphocyte activation gene-3 (LAG-3) have improved cancer patient outcomes by enhancing anti-tumor responses. However, some patients are unresponsive, and others experience immune-related adverse events (irAEs), affecting organs like the lung, liver, intestine, skin and now the cardiovascular system. These cardiac irAEs include conditions like myocarditis, atherosclerosis, pericarditis, arrhythmias, and cardiomyopathy. Ongoing clinical trials investigate promising alternative co-inhibitory receptor targets, including T cell immunoglobulin and mucin domain-containing protein 3 (Tim-3) and T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT). This review delves into the mechanisms of approved ICIs (CTLA-4, PD-1, PD-L1, and LAG-3) and upcoming options like Tim-3 and TIGIT. It explores the use of ICIs in cancer treatment, supported by both preclinical and clinical data. Additionally, it examines the mechanisms behind cardiac toxic irAEs, focusing on ICI-associated myocarditis and atherosclerosis. These insights are vital as ICIs continue to revolutionize cancer therapy, offering hope to patients, while also necessitating careful monitoring and management of potential side effects, including emerging cardiac complications.
Remarkable advances have been made in recent years in cancer treatment (1). Immunotherapy represents a category of cancer treatment that harnesses the immune system’s elements to combat tumor cells (2). Techniques like adoptive cell transfer (ACT) and immune checkpoint inhibitors (ICIs) fall under this approach, offering promising methods in the fight against cancer (3). This innovative approach, either administered alone or in combination with conventional treatments like radiotherapy and chemotherapy, has become a prevailing standard for numerous cancers, exhibiting considerable success (4). A particularly promising strategy to trigger therapeutic anti-tumor immunity involves obstructing immune checkpoints (5).
The balance between co-stimulatory and inhibitory signals governs the ultimate amplitude and quality of the T-cell response, triggered by antigen recognition via the T-cell receptor (TCR) (5). The immune system relies on inhibitory pathways, known as immune checkpoints, to maintain self-tolerance and regulate the duration and strength of the immune response in peripheral tissues, thus minimizing collateral tissue damage (5). Unfortunately, tumor cells exploit these inhibitory molecules by expressing immune checkpoint proteins as a means to induce immune resistance and T-cell exhaustion (3). Given that ligand-receptor interactions initiate many immune checkpoints, these can be readily blocked by antibodies or modulated through recombinant forms of ligands and receptors (5). The treatment involving ICIs, particularly targeting cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), programmed death-1 (PD-1), programmed death ligand-1 (PD-L1), and lymphocyte activation gene-3 (LAG-3), has been proven effective in activating anti-tumor T-cell activity and dynamically regulating the anti-tumor immune response (6).
However, despite the promising outcomes observed with immunotherapy in certain cancers, not all patients exhibit a favorable response to ICIs, and the overall response rate (ORR) is influenced by tumor type and specific drugs, ranging from 10.9% for single-agent ipilimumab (monoclonal antibody (mAbs) targeting CTLA-4, anti-CTLA-4 mAb) in previously treated melanoma to 69% for pembrolizumab (anti-PD-1 mAb) in relapsed/refractory classic Hodgkin’s lymphoma (7–9). Additionally, some patients may develop resistance to these treatments over time (10). Furthermore, a new set of side effects termed irAEs, immune-related adverse events, have been associated with ICI therapy (11). These irAEs manifest as autoimmune conditions affecting various organs throughout the body following ICI administration, and they have distinct characteristics compared to non-ICI therapy-related autoimmune diseases (11). Among the reported cases of irAEs in cancer patients treated with ICI, cardiotoxicity has emerged as a concerning issue in recent years (12). Although immune-related cardiotoxicity is less frequent than other irAEs, it is often fatal (13). Studies analyzing safety databases have identified potential ICI-associated cardiac toxicities, including myocarditis, cardiomyopathy, conduction defects (heart block), vasculitis, atrial and ventricular arrhythmias, pericarditis/pericardial effusion, venous thrombosis, acute coronary syndrome, tachycardia, hypotension, and cardiac dysfunction (13–23). Significant focus of ICI cardiotoxicity research is centered on myocarditis, a rare cardiac irAE with notable mortality (24, 25). Recently attention has also expanded towards ICI-associated atherosclerosis, given the ICI role in accelerating atherosclerotic cardiovascular disease with substantial implications for long-term vascular toxicity (26). In this review, ICI-myocarditis and atherosclerosis are chosen as the primary focus, emphasizing the clinical significance of these two major cardiotoxic irAEs. This strategic selection underscores the critical need for a comprehensive exploration and management of cardiovascular irAEs associated with ICI therapy.
In an effort to increase the percentage of responsive cancers to these therapies, researchers are exploring novel pathways and molecules to enhance the response and effectiveness of ICI therapy (27). Among the promising options are ICIs targeting T-cell immunoglobulin and mucin-domain-containing-3 (Tim-3) and T-cell immunoglobulin and ITIM domain (TIGIT), which show potential for treating solid tumors and are currently under active investigation in clinical trials (6). This review aims to provide a comprehensive overview of the mechanisms and cardiotoxicity associated with Food and Drug Administration (FDA)-approved ICIs. Additionally, we focus on next-generation ICIs, Tim-3 and TIGIT inhibitors, exploring their influence on T-cell function, role in cancer treatment, and potential for cardiac irAEs.
The mAbs that inhibit immune checkpoints have displayed remarkable efficacy in clinical trials and have made significant advancements in oncology (7, 28–32). While targeted therapies often result in short-lived clinical responses due to the development of drug resistance within a few months, immune checkpoint blockade therapies have demonstrated durable clinical responses (7, 32, 33). Some patients have experienced prolonged periods without cancer progression, spanning several years (7, 32, 33). These ICIs, including ipilimumab (anti-CTLA-4 mAb), pembrolizumab, nivolumab, cemiplimab (anti-PD-1 mAb), as well as atezolizumab, avelumab, and durvalumab (anti-PD-L1 mAb), have become standard in clinical practice (7, 33–37). They have demonstrated remarkable effectiveness against diverse types of cancer that was previously unparalleled (7, 33–37). In recent developments, two agents directed at LAG-3 (eftilagimod alpha and relatlimab, anti-LAG-3) have received approval for use in combination with anti-PD-1 to treat advanced solid tumors due to their crucial role in modulating the immune system (38).
CTLA-4, also referred to as CD152, represents one of the initial negative modulators that have been targeted in clinical settings (39). It is predominantly present in conventional T cells following activation and Foxp3+ regulatory T lymphocytes (Treg) cells, and its main function is to regulate the intensity of early T cell activation (40–43). The use of ipilimumab (anti-CTLA-4 mAb) either alone or in combination with nivolumab (anti-PD-1 mAb) is currently approved for the treatment of melanoma, colorectal cancer (CRC), hepatocellular carcinoma (HCC), non-small cell lung cancer (NSCLC), and renal cell carcinoma (RCC) (44).
CTLA-4 functions by counteracting CD28, a co-stimulatory receptor on T cells (45–47), and shares ligands [CD80 (B7.1) and CD86 (B7.2)] with CD28 (48–52). While the exact mechanisms of CTLA-4 action are debated (5), one theory proposes that its higher affinity for CD80 and CD86 dampens T cell activation by outcompeting CD28 for binding (53–58). Upon binding to CD80 and CD86 on antigen-presenting cells, CTLA-4 triggers inhibitory reactions via protein phosphatases SHP2 and PP2A, resulting in immune suppression (59), including the blockade of T lymphocyte response, reduced T lymphocyte proliferation, increased Treg activity, decreased cytokine secretion, and overall immunosuppression (60–62). Furthermore, it has been shown that ipilimumab (anti-CTLA-4 mAb) selectively depletes CTLA-4+ FOXP3+ Treg cells via antibody-dependent cell-mediated (ADCC) cytotoxicity (40).
PD-1 and PD-L1 inhibitors function by blocking inhibitory signals, thereby supporting the eradication of tumor cells through sustained activation of T lymphocytes (63, 64). PD-1 is expressed on various immune cells, including monocytes, T cells, B cells, dendritic cells (DCs), and tumor-infiltrating lymphocytes (TILs) (65, 66). On the other hand, PD-L1 is typically found on APCs and tumor cells (66). In human cancers, PD-1 has been predominantly detected in a wide range of malignancies, such as melanoma, lung cancer, RCC, head and neck cancer, bladder cancer, ovarian cancer, and gastrointestinal cancer (67).
In contrast to CTLA-4, which primarily regulates T-cell proliferation in lymph nodes during early immune responses, PD-1 plays a crucial role in limiting T cell activity in peripheral tissues during inflammatory responses and contributes to autoimmune control (68–74). PD-1 plays a significant role as an immune resistance mechanism within the tumor microenvironment (TME) (75–77). PD-1 expression counteracts positive signaling events initiated by TCR and CD28 interactions, leading to the inhibition of transcription factors vital for T cell activation, proliferation, effector functions, and survival (39). This includes suppression of activator protein 1 (AP-1), nuclear factor of activated T cells (NFAT), nuclear factor-κB (NF-κB), and anti-apoptotic proteins like Bcl-2 and Bcl-xL, impairing T-cell survival (78). Ligand activation of PD-1 triggers a negative feedback pathway, reducing cytokine production (59). Similarly, to CTLA-4, PD-1 is highly expressed on Treg cells, potentially enhancing their proliferation in the presence of its ligand (79).
Identified more than two decades ago as a CD4 homologue (80), LAG-3’s role as an immune checkpoint was clarified in 2005 when it was discovered to enhance Treg cell function (81, 82). Beyond its influence on Treg cells, LAG-3 independently inhibits CD8+ effector T cells (83). Its sole known ligand is MHC class II molecules, upregulated on certain epithelial cancers in response to interferon-γ (IFN-γ), also found on tumor-infiltrating macrophages and DCs (5). While the precise function of the LAG-3-MHC class II interaction in inhibiting T cell responses is not fully understood, LAG-3 antibodies that do not block this interaction have been observed to enhance T cell proliferation and improve effector cell functions in vitro and in vivo (5).
The specific molecular mechanisms underlying LAG-3 signaling and its interaction with other immune checkpoints are largely uncertain (84). However, LAG-3 has shown remarkable synergistic effects with PD-1, inhibiting immune responses in various scenarios (85, 86). Particularly, the combination therapy of relatlimab (anti-LAG-3 mAb; BMS-986016) with nivolumab (anti-PD-1 mAb) has demonstrated impressive clinical effectiveness in melanoma patients unresponsive to anti-PD-1/PD-L1 therapy (87). Moreover, LAG-3 expression was significantly higher in various cancers, including kidney renal clear cell carcinoma (KIRC), pancreatic adenocarcinoma (PAAD), skin cutaneous melanoma (SKCM), testicular germ cell tumors (TGCT), lymphoid neoplasm diffuse large B‐cell lymphoma (DLBC), and head and neck squamous cell carcinoma (HNSC), compared to their corresponding normal tissues (88). This suggests that targeting LAG-3 could have a substantial antitumor impact in these cancer types (88).
Immunotherapy with ICIs offers numerous benefits compared to traditional cancer treatments (27). Clinical studies focusing on melanoma, RCC, squamous cell carcinoma, and NSCLC have shown remarkable increases in patient survival with both CTLA-4 and PD-1 checkpoint inhibitors compared to conventional chemotherapy (89). Particularly, ICIs have demonstrated the potential for durable responses and even the possibility of cure in metastatic diseases (27). Another advantage of ICI treatment is the availability of biomarkers to predict the response to therapy (27). These biomarkers help identify patients who are likely to benefit the most from immunotherapy, making it a more personalized and targeted approach (90).
As the inhibitory roles of CTLA-4 and PD-1 in immune responses, including antitumor responses, are distinct, their effectiveness in cancer treatment depends significantly on the cancer type and tumor size (27, 44). Notably, in melanoma, while CTLA-4 blockade with ipilimumab was the first treatment to extend overall survival in patients with advanced melanoma in a randomized setting, anti-PD-1 mAb treatment (nivolumab) demonstrated higher efficacy in patients with smaller tumors (7, 91–94). A phase 3 clinical trial directly comparing the two ICIs found that patients treated with nivolumab (anti-PD-1 mAb) had a better response rate (44%) and longer progression-free survival (6.9 months) than those treated with ipilimumab (anti-CTLA-4 mAb) (response rate of 19% and progression-free survival of 2.8 months) (95).
The combination of CTLA-4 and PD-1 blockers has been proposed to have a synergistic effect on activating anti-tumor immune responses, leading to increased response rates in patients (96). Numerous clinical studies have been conducted to assess the safety and efficacy of this combination in various cancer subtypes (97). Recent data indicates that the dual inhibition of PD-1 and CTLA-4 may enhance the activity observed with single-agent therapy by promoting the recruitment of peripheral T-cells and reducing resident Tregs (96). Clinical results have shown that when PD-1/PD-L1 inhibitors and CTLA-4 inhibitors are administered individually, the response rates are typically around 20-25% at best (95). However, when these inhibitors are combined, the response rate can reach up to 60%, with an associated survival benefit of 11.5 months (95). This significant increase in response rates and median survival times has been observed in melanoma and RCC, leading to the approval of the ipilimumab (anti-CTLA-4 mAb) and nivolumab (anti-PD-1 mAb) combination for the treatment of these cancers (98, 99).
Apart from CTLA-4 and PD-1/PD-L1, LAG-3 has emerged as a promising target for tumor immunotherapy (100). Currently, more than 80 clinical trials are ongoing worldwide to assess drug candidates targeting LAG-3 (100). However, the efficacy of relatlimab (anti-LAG-3 mAb) alone is limited, and it is typically utilized in combination with other checkpoint inhibitors like ipilimumab (anti-CTLA-4 mAb) or nivolumab (anti-PD-1 mAb) to achieve synergistic enhancement of efficacy (101). Especially noteworthy is the combination of relatlimab (anti-LAG-3) and nivolumab (anti-PD-1), which has shown promising preliminary efficacy in melanoma patients previously unresponsive to anti-PD-1/PD-L1 therapy (NCT01968109) (86, 87). As a result of these findings, the FDA approved relatlimab (anti-LAG-3 mAb) in combination with nivolumab (anti-PD-1 mAb) in March 2022, making it the first mAb to be approved for the treatment of unresectable or metastatic melanoma (100).
In clinical settings, ICIs, despite their proven efficacy, are associated with numerous irAEs affecting various organ systems (102). Approximately 60% to 80% of individuals undergoing ICI treatment experience irAEs, with 13% to 23% encountering severe grade 3 to 4 adverse events as per Common Terminology Criteria for Adverse Events (102). Common mild side effects include symptoms like diarrhea, fatigue, itching, rash, nausea, and reduced appetite (89). Serious adverse responses encompass severe diarrhea, colitis, elevated alanine aminotransferase levels, pneumonitis-related inflammation, and interstitial nephritis (95, 103, 104). Patients may also experience exacerbation of existing autoimmune conditions or the emergence of new ones, such as type 1 diabetes mellitus (105–108). Severe adverse effects may necessitate treatment discontinuation, although subsequent positive responses could still occur (109). Particularly, specific treatment-related autoimmune reactions, like rashes and vitiligo, have shown a connection with improved disease prognosis, suggesting a potential convergence between autoimmune and anti-tumor immune responses (110).
Under normal physiological circumstances, PD-1 and CTLA-4 prevent autoimmunity and curb immune activation to safeguard against unwanted inflammation (89). Using therapeutic antibodies to inhibit these receptors for cancer treatment is linked to adverse effects similar to autoimmune responses (89). Clinical studies contrasting various ICIs observed a higher incidence of side effects in individuals undergoing anti-CTLA-4 treatment (27.3%) compared to those receiving anti-PD-1 treatment (16.7%) (95). Animal studies corroborate this, with CTLA-4-deficient mice experiencing spontaneous severe myocarditis and pancreatitis, while PD-1-deficient mice develop lupus-like proliferative arthritis and glomerulonephritis on C57BL/6 background and myocarditis on BALB/c background (71, 72, 111). Thus, the nature of inflammation and adverse reactions induced in these knockout mice depends on their specific genetic background (72, 111–113).
Combined CTLA-4 and PD-1 blockade has increased anti-cancer efficacy, but it may also result in heightened toxicity (44). In melanoma or relapsed small cell lung cancer patients, combined blockade led to more severe grade 3-4 adverse events (54% to 55%) compared to solely anti-CTLA-4 (24% to 27%) or anti-PD-1 (15% to 16%) treatment (114–116). Similarly, despite the therapeutic enhancement achieved through combined PD-1 and LAG-3 targeting, there is a potential for increased toxicity. A study in melanoma patients showed that relatlimab (anti-LAG-3 mAb) plus nivolumab (anti-PD-1 mAb) led to a higher occurrence of adverse events (81.1% vs. 69.9%) and grade 3–4 irAEs (18.9% vs. 9.7%) compared to nivolumab (anti-PD-1 mAb) alone, with a notable rise in hepatitis cases (3.9% vs. 1.1%) (117). Overall, 14.6% of patients receiving the combinatory treatment discontinued it due to irAEs, compared to 6.7% in the nivolumab (anti-PD-1 mAb)-only group (117).
ICIs, by intensifying T cell activation, can enhance anti-tumor immune reactions but may also perturb peripheral T cell tolerance, potentially resulting in hyperinflammatory responses and autoimmune conditions (118). irAEs are associated with the infiltration of various inflammatory immune cells into affected organs, often showing a significant increase in CD8+ lymphocytes and a markedly elevated CD8+/CD4+ ratio (119, 120).
The majority of irAEs are manageable during their early stages; however, approximately 10–17% of these instances result in fatal outcomes (24, 25). While the incidence of cardiovascular adverse events caused by ICIs is relatively low, the associated mortality rate is alarmingly high (111). The spotlight on cardiac irAEs began in 2016, with Johnson et al. documenting two instances of fatal myocarditis occurring after ICI treatment (121, 122). Subsequently, the spectrum of cardiovascular irAEs has expanded to include conditions such as myocardial infarction, atrioventricular (AV) block, various forms of arrhythmias (including supraventricular and ventricular), sudden cardiac death, Tako-Tsubo cardiomyopathy, non-inflammatory cardiomyopathy, pericarditis, pericardial effusion, ischemic stroke, venous thromboembolism and accelerated atherosclerosis (123).
In a retrospective analysis encompassing 424 patients who underwent treatment involving at least one ICI, it was found that 62 individuals (14.6%) received diagnoses of new cardiovascular disorders subsequent to the commencement of ICI therapy (124). Within this group, 5.6% experienced the development of heart failure during ICI monotherapy (124). This rate escalated to 6.1% when a sequential administration of two ICIs was implemented (124). A similar pattern emerged from a recent meta-analysis, involving a patient cohort of 13,646 individuals who were administered anti-CTLA-4, anti-PD-1, and/or anti-PD-L1 therapies (125). In the subset of patients receiving ICI as a sole therapeutic approach, the incidence of cardiovascular adverse events stood at 3.1% (125). Remarkably, this incidence nearly doubled to 5.8% among patients who received dual immunotherapy (125). The introduction of chemotherapy alongside ICI did not substantially alter the incidence rate, which remained at 3.7% (125). This data aligns with findings from clinical trials, suggesting that CTLA-4 inhibition is prone to causing immune-related cardiotoxicity, inclusive of conditions like pericarditis and myocarditis (126, 127). Likewise, instances of myocarditis have been recorded following the administration of anti-PD-1 drugs such as nivolumab or pembrolizumab (128, 129). An independent meta-analysis conducted by Dolladille et al., involving 32,518 patients, reinforced the association between ICI usage and an elevated risk of myocarditis, pericardial diseases, heart failure, dyslipidemia, myocardial infarction, and cerebral arterial ischemia (130). Significantly, ICI-treated patients, heart failure was more frequently observed adverse events. The calculated “number needed to harm”, indicating the number of individuals who must be exposed to a specific risk factor or treatment for one person to experience a particular adverse event, was found to be 462 for myocarditis and only 260 for heart failure (130).
Myocarditis, characterized by inflammation of the heart muscle, has seen a remarkable increase in its association with ICIs in recent years (15, 131, 132). Several preclinical studies have demonstrated a correlation between ICI treatment and myocarditis, as illustrated in Table 1 (133–138). Notably, the initial trials of ICIs did not include proactive screenings for myocarditis, which raises concerns that some cases may have gone undetected (139). It was reported that approximately 0.04% to 1.14% of cases involving ICI treatments are linked to myocarditis (18, 121). Myocarditis stands out as the most frequently reported cardiac irAE, primarily due to its significant morbidity rates (13, 16, 140). In contrast to other irAEs, it carries a significantly elevated mortality rate ranging from 25% to 50% (15, 16, 18, 122).
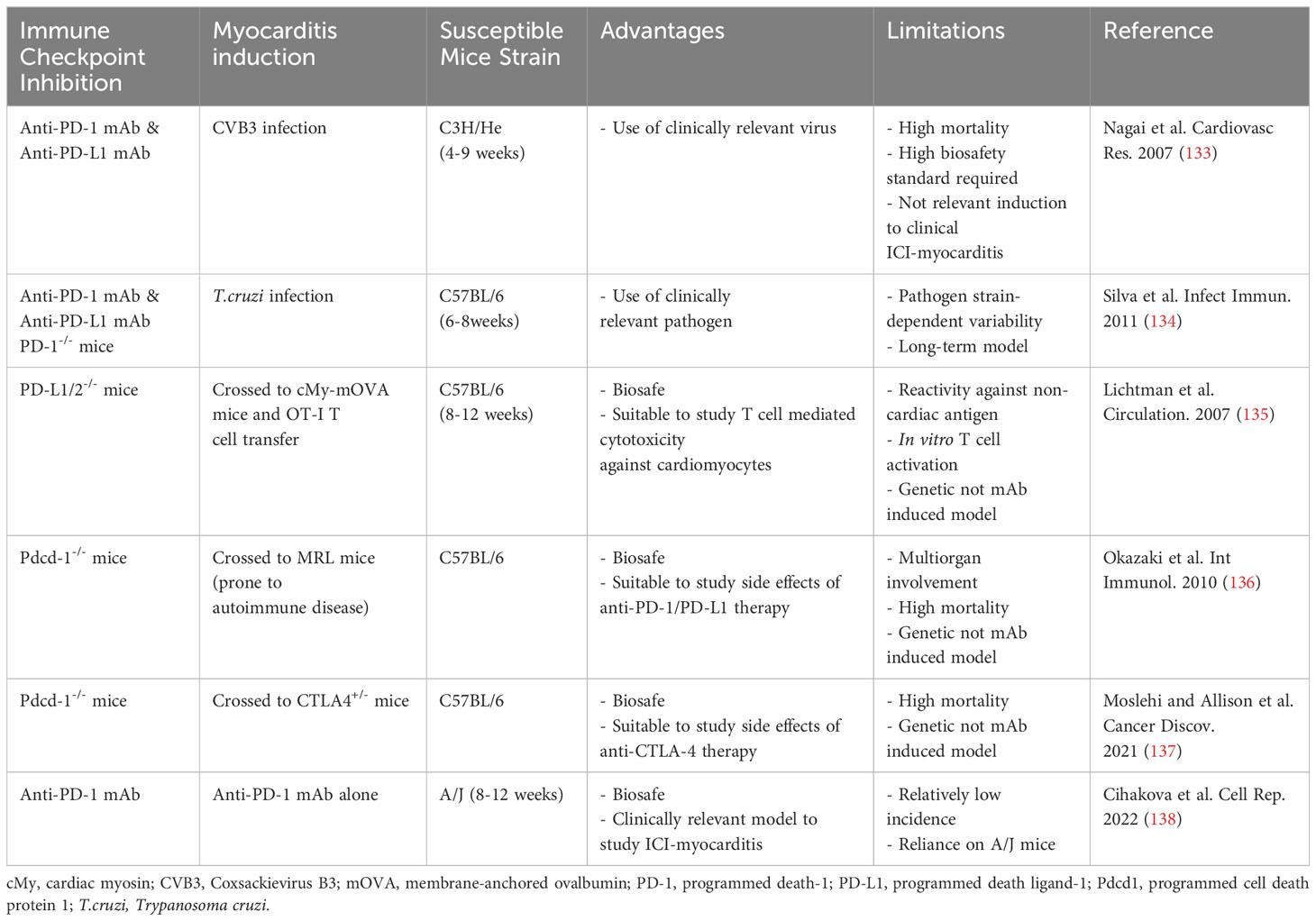
Table 1 Preclinical models for immune checkpoint inhibitor associated myocarditis studies.
A risk factor for myocarditis associated with ICIs is the utilization of combined ICI therapy (121). For instance, the combined usage of nivolumab (anti-PD-1 mAb) and ipilimumab (anti-CTLA-4 mAb) leads to a 4.74-fold increase in the risk of myocarditis when contrasted with the utilization of nivolumab (anti-PD-1 mAb) as a standalone therapy (122). Similarly, recent trials involving combined nivolumab (anti-PD-1 mAb) and relatlimab (anti-LAG-3 mAb) therapy revealed slightly elevated instances of myocarditis in comparison to the usage of single nivolumab (anti-PD-1 mAb) therapy (1.7% versus 0.6%, respectively) (117). Furthermore, instances of myocarditis stemming from combination therapies tend to have higher severity and higher rate of fatality (15). One study revealed that the mortality rate associated with the combination of anti-PD-1/PD-L1 and anti-CTLA-4 therapies was 67%, in contrast to the 36% fatality rate observed in anti-PD-1/PD-L1 monotherapy (15). However, it is important to note that due to the study’s limited sample size of only 59 patients, the reported fatality rate may not accurately reflect the true extent of the risk.
Patients who develop myocarditis may present with a diverse range of symptoms, including chest pain, elevated cardiac troponin levels, and abnormalities detected through electrocardiograms (ECGs), echocardiograms, or cardiac magnetic resonance imaging (127, 129, 141–143). However, it’s important to note that clinical presentations can vary among cases (127, 129, 141–143). Similar to viral myocarditis, ICI-related myocarditis has been characterized by the infiltration of T cells into the myocardium (122). On a histological level, the infiltrating cells observed in cases of ICI-associated myocarditis are primarily composed of T cells and macrophages (122, 127, 143–145). Among these infiltrating T cells, CD8+ T cells tend to predominate over CD4+ T cells in instances of ICI-associated myocarditis (122, 127, 143–145). Researchers have postulated that T cells play a crucial role in the development of myocarditis associated with ICIs, given the predominant expression of PD-1 and CTLA-4 in T cells (138).
In the first case report of ICI-associated myocarditis, the activation of shared T cell clones targeting both myocardium and tumor has been suggested as the mechanism of its onset and progress (122). Recently, our preclinical study revealed that autoreactive T cells specific for cardiac myosin heavy chain, a well-known heart autoantigen, lead to the development of ICI-myocarditis in mice (138). This finding is supported by an independent study reported by other researchers (146). In their study, the experimental and clinical findings involving T cells responsive to cardiac myosin heavy chain in patients imply a clear immunological relevance of cardiac myosin heavy chain expression levels in the human ventricle concerning myocarditis associated with ICIs (146). Notably, in those two studies, the development of ICI-associated myocarditis in mice did not require tumor cells, indicating that the activation of autoreactive T cells targeting the heart by an ICI treatment is sufficient to cause ICI-associated myocarditis. Furthermore, the findings from independent studies, which highlight the involvement of cardiac myosin heavy chain-reactive T cells in the development of ICI myocarditis, emphasize the critical function of immune checkpoint molecules, such as PD-1/PD-L1 and CTLA-4, in maintaining cardiac autoimmunity under normal conditions (Figure 1) (147).
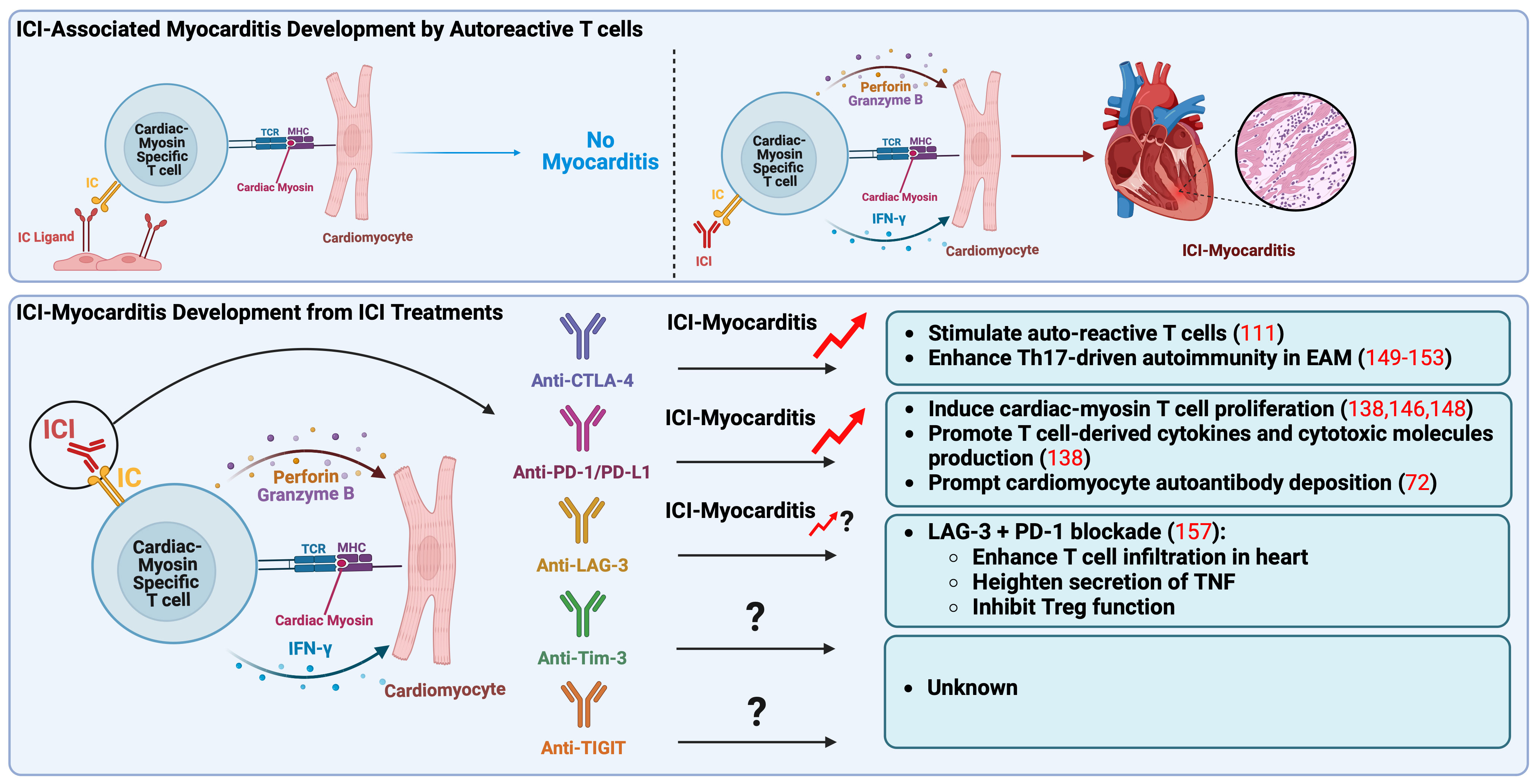
Figure 1 Pathogenic mechanisms of immune checkpoint inhibitors associated myocarditis. Autoreactive T cells, which recognize cardiac myosin heavy chain, main autoantigen in the heart, play a crucial role in the onset of ICI-myocarditis. These autoreactive T cells may be present in the heart in a naïve state due to impaired thymic selection, expressing elevated levels of IC as a peripheral tolerance mechanism to prevent their activation. In a mouse model, ICI treatment seems to directly activate these autoreactive T cells by obstructing the inhibitory IC pathway, specifically targeting the heart. Recent reports have indicated that anti-CTLA-4 and anti-PD-1/PD-L1 treatments result in the clonal expansion of T cells specific to cardiac myosin, observed in peripheral blood mononuclear cells of patients with ICI-associated myocarditis, indicating their pathogenic role in clinical scenarios. Anti-CTLA-4 is associated with the promotion of Th17-mediated autoimmunity in EAM, while anti-PD-1/PD-L1 is linked to excessive production of T cell derived cytokines and cytotoxic molecules like IFN-γ, perforin, and granzyme B, ultimately contributing to myocarditis development. Notably, anti-LAG-3 has only been associated with ICI-myocarditis when combined with anti-PD-1. CTLA-4, cytotoxic T-lymphocyte antigen-4; EAM, experimental autoimmune myocarditis; IC, immune checkpoint; ICI, immune checkpoint inhibitor; IFN-γ, interferon-γ; LAG-3, lymphocyte activation gene-3; MHC, major histocompatibility complex; PD-1, programmed death-1; PD-L1, programmed death ligand-1, TCR, T-cell receptor; Th17, IL-17-producing T cell (type 17 helper T cell); TIGIT, T cell immunoreceptor with immunoglobulin and ITIM domain; Tim-3, T cell immunoglobulin and mucin domain-containing protein 3; TNF, tumor necrosis factor; Treg, regulatory T lymphocytes.
It is reported that autoreactive T cells specific for cardiac myosin can be present in the mouse heart during a naïve state due to the impairment of thymic selection and that they express a high level of PD-1 to avoid autoimmune reactions (138, 148). In our mouse model, anti-PD-1 mAb treatment seems to directly activate autoreactive T cells targeting the heart by blocking the inhibitory PD-1/PD-L1 pathway, leading to the excessive production of effector cytokines and cytotoxic molecules such as IFN-γ, perforin, and granzyme B followed by myocardial damage and myocarditis development (138). The clonal expansion of T cells specific for cardiac myosin was observed also in the peripheral blood mononuclear cells of the patients with ICI-associated myocarditis, suggesting their pathogenic role in clinical settings (146).
Unlike the anti-PD-1 mAb treatment, it is unclear whether the anti-CTLA-4 mAb regimen mediates ICI-associated myocarditis development through the activation of autoreactive T cells targeting cardiac myosin heavy chain. However, it is reported that CTLA-4-deficient mice spontaneously develop lethal myocarditis at an early age, indicating the activation of autoreactive pathogenic T cells targeting the heart by the blockade of CTLA-4 (111). Additionally, in our mouse model for ICI-associated myocarditis, cardiac myosin-specific T cells exhibited an elevated level of CTLA-4 expression compared to bystander T cells (138). Thus, CTLA-4 inhibition using mAbs may cause fulminant myocarditis by activating autoreactive T cells targeting cardiac myosin in clinical settings as well as PD-1 inhibition.
IL-17-producing T cells, also known as Th17 cells, play a pivotal role in the pathophysiology of different types of myocarditis in both clinical and preclinical settings, as evidenced by various studies (149–152). These cells are initiators of inflammation and contribute to the progression from acute myocarditis to dilated cardiomyopathy (149–152). Notably, disrupting the interaction between CTLA-4 and B7 molecules has been shown to enhance the differentiation of Th17 cells, both in laboratory settings and in live animals, subsequently promoting Th17-mediated autoimmunity in murine models (153). Furthermore, multiple studies have indicated CTLA-4’s role in maintaining peripheral heart tolerance (42, 111, 151–155). Collectively, these findings suggest that blocking CTLA-4 pathways has the potential to contribute to the development of myocarditis and worsen the severity of the disease.
Since LAG-3 has recently gained FDA approval, there is a limited number of reported cases concerning ICI-related myocarditis involving this factor. In animal experiments focusing on LAG-3, it has been previously documented that the knockout of LAG-3 in mice did not result in the onset of myocarditis (156). However, when both LAG-3 and PD-1 were knocked out, a lethal form of myocarditis emerged, characterized by T-cell infiltration, heightened secretion of tumor necrosis factor (TNF), and persistent inhibitory function of Tregs (157).
The management of ICI-associated myocarditis has predominantly relied on the administration of glucocorticoids, encompassing both oral prednisone and intravenous methylprednisolone (18, 158). According to a limited dataset, approximately 86% of patients treated with glucocorticoids exhibited improved outcomes, particularly when high-dose glucocorticoids were employed (18). Therefore, it is advisable to initiate high-dose steroid therapy at the outset, followed by a gradual tapering regimen, contingent upon clinical progress and ongoing troponin level monitoring (159). If there is an insufficient clinical or biomarker response to steroids, alternative immune modulators may be contemplated (159).
Case reports and small case series have documented successful treatment of ICI-related myocarditis with various agents, including intravenous immunoglobulin (160), mycophenolate (161), infliximab (anti-TNF mAb) (162), anti–thymocyte globulin (145), plasmapheresis (162), alemtuzumab (anti-CD52 mAb) (163), and abatacept (CTLA-4 agonistic mAb) (164). In particular, the use of abatacept either by itself or in combination with Ruxolitinib (mAb targeting IFN-γ/JAK2/STAT1 signaling pathway) has led to a notable decrease in mortality among patients suffering from ICI-associated myocarditis and myositis (165). The effectiveness of these agents in ICI-related myocarditis remains uncertain and is generally reserved for patients who do not respond adequately to glucocorticoids (159).
In addition to immunosuppressive therapy, conventional systematic therapy is essential (159). For acute decompensated heart failure, it is advisable to administer intravenous diuretics, inotropes, and mechanical circulatory support, as outlined in the American College of Cardiology/American Heart Association heart failure guidelines (166).
Due to the substantial mortality risk associated with ICI-related myocarditis, the prevailing consensus suggests discontinuing ICI therapy even in cases of mild toxicity when there is suspicion of this condition (159). Furthermore, it remains uncertain whether ICI therapy can be safely resumed following the successful treatment of ICI-related myocarditis (159). A case report indicates that resuming ICI therapy with a different agent after myocarditis developed on nivolumab resulted in worsened heart failure hospitalization with pembrolizumab (167). Consequently, it is not advisable to consider reattempting ICI therapy in patients with a history of ICI-related myocarditis (159).
Atherosclerosis represents the predominant pathological process underlying cardiovascular diseases (168). This ailment pertains to the development of atherosclerotic plaques within large- and medium-sized arteries (169). These plaques include necrotic cores, calcified regions, accumulated modified lipids, inflamed smooth muscle cells, endothelial cells, leukocytes, and foam cells (169). The intricate composition of these plaques underscores the complexity of atherosclerosis, involving numerous components of the vascular, metabolic, and immune systems (168).
Emerging data propose that ICIs might expedite the progression of atherosclerosis, potentially resulting in an elevation of atherosclerosis-related cardiovascular events like acute myocardial infarction, ischemic stroke, and peripheral arterial disease (21, 170–175). Specifically, substantial foundational cellular and animal studies indicate that the immune checkpoint proteins such as CTLA-4, PD-1, PD-L1, and LAG-3 serve as crucial negative regulators of atherosclerosis (176–178). Consequently, their inhibition could potentially accelerate atherosclerosis by intensifying effector T cell responses, constraining the function of Treg cells, and infiltrating the vascular endothelium (177–181). Moreover, a growing body of clinical evidence reinforces these preclinical discoveries by demonstrating that ICI therapy contributes to the hastened advancement of atherosclerotic plaque, consequently amplifying the susceptibility to atherosclerotic cardiovascular ailments (182–184).
In a comprehensive study, Bar et al. performed a retrospective analysis to examine the occurrence of acute vascular complications in a group of 1,215 cancer patients receiving ICI therapy (185). 1% of these patients underwent myocardial infarction or ischemic stroke within 6 months of initiating ICI treatment (185). Furthermore, a recently issued systematic review of 17 studies, comprising a total of 10,106 participants, examined the frequency of arterial thrombotic events, specifically focusing on stroke and myocardial infarction, following the administration of ICI therapy (186). They found an incidence rate of arterial thrombotic events among ICI-treated patients at 1.1% (186). Particularly, this risk appeared consistent regardless of whether patients were subjected to single or combined ICI treatment strategies (186, 187).
There have been no direct mechanistic studies in humans to evaluate accelerated atherosclerosis in the setting of ICI therapy (26). However, extrapolating data from mouse models for ICI-associated atherosclerosis and ICI-myocarditis provides insight into several plausible pathways for immune checkpoint blockade to cause plaque progression (26).
One potential mechanism is that the revitalization of T cells by ICI may lead to excess inflammation, resulting in pro-atherogenic effects. The interaction between immune checkpoints expressed on T cells and their ligands on DCs within the microenvironment plays a crucial role in T-cell inactivation and the maintenance of an immunosuppressive microenvironment within plaques (Figure 2) (188). However, ICIs can disrupt this process by activating T cells, triggering the production of proinflammatory cytokines such as TNF and IFN-γ (188). These cytokines, in turn, induce various detrimental effects, including smooth muscle cell proliferation, collagen deposition, and macrophage activation (188). These changes contribute to increased phagocytosis of low-density lipoprotein (LDL) in macrophages, leading to their transformation into foam cells (173, 177). Ultimately, these structural alterations in the plaque lead to the formation of a necrotic core, rendering the plaque more unstable (188).
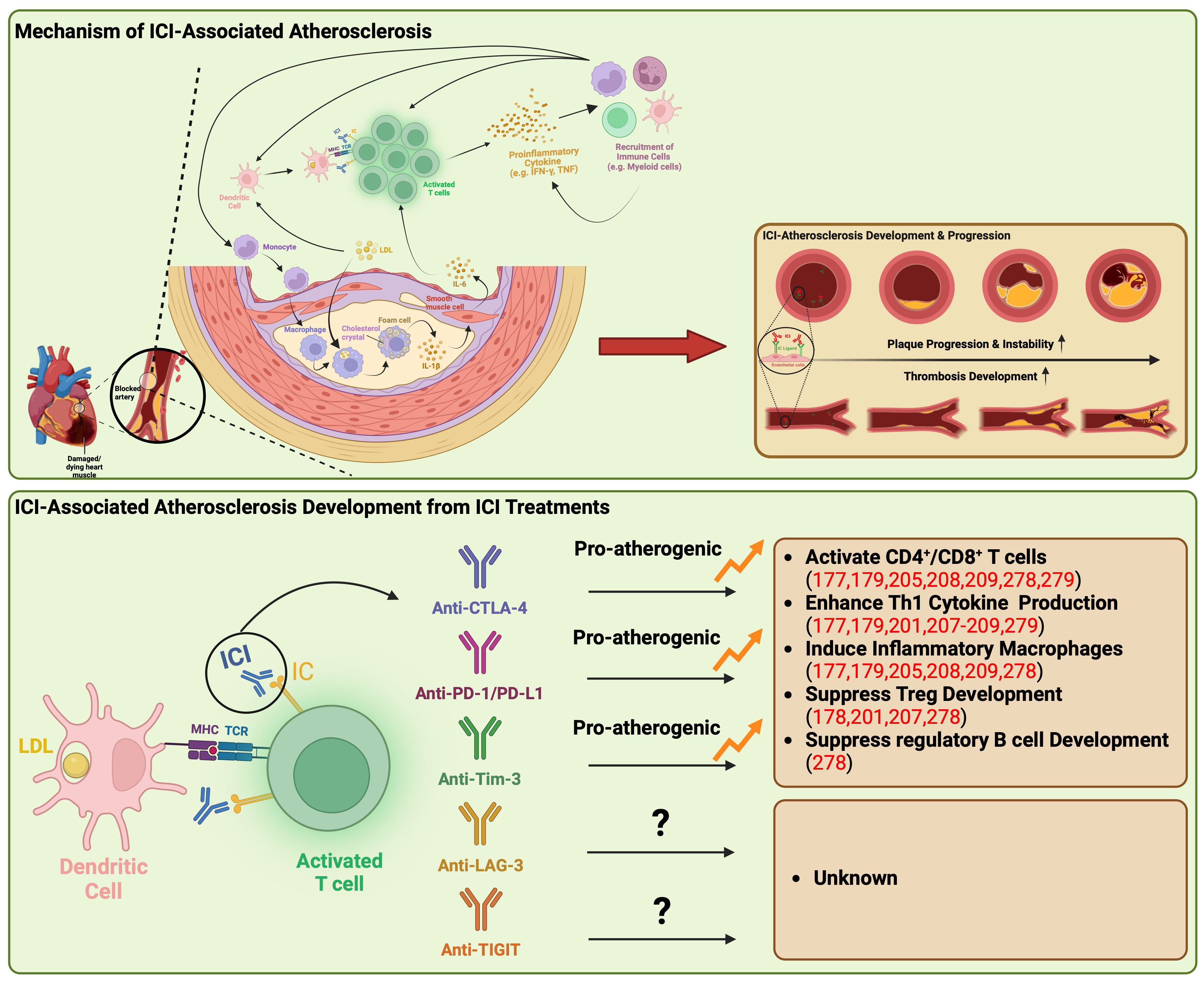
Figure 2 Underlying mechanisms of atherosclerosis linked to immune checkpoint inhibitors. ICI-associated atherosclerosis encompasses an intricate process of plaque formation within arterial walls, commencing with the retention of LDL-cholesterol that triggers inflammation and recruit monocytes into artery walls. These monocytes evolve into cholesterol-laden macrophages, eventually transforming into foam cells, creating a central necrotic area within the plaque. Persistent lipoprotein uptake and macrophage proliferation contribute to plaque enlargement, attracting immune cells, particularly T cells, to the plaque’s periphery. Th1 cells release IFN-γ and TNF, promoting macrophage activation and destabilizing the plaque, potentially leading to heart attacks or strokes. ICIs disrupt this process by activating T cells, producing proinflammatory cytokines like TNF and IFN-γ. These cytokines induce detrimental effects such as immune cell recruitment, smooth muscle cell proliferation, collagen deposition, and macrophage activation, triggering further proinflammatory cytokine release syndrome. This results in increased LDL phagocytosis and foam cell formation, ultimately causing structural changes within the plaque, including the formation of a necrotic core, rendering the plaque more unstable. Anti-CTLA-4, PD-1/PD-L1, and Tim-3 have been demonstrated to be pro-atherogenic, fostering the proliferation of inflammatory immune cells while suppressing the development of Treg and regulatory B cells. CTLA-4, cytotoxic T-lymphocyte antigen-4; IC, immune checkpoint; ICI, immune checkpoint inhibitor; IFN-γ, interferon-γ; IL, interleukin; LAG-3, lymphocyte activation gene-3; LDL, low-density lipoprotein; MHC, major histocompatibility complex; PD-1, programmed death-1; PD-L1, programmed death ligand-1, TCR, T-cell receptor; Th1, type 1 helper T cell; TIGIT, T cell immunoreceptor with immunoglobulin and ITIM domain; Tim-3, T cell immunoglobulin and mucin domain-containing protein 3; TNF, tumor necrosis factor; Treg, regulatory T lymphocytes.
Another proposed mechanism for atherosclerosis associated with ICI involves the activation of T cells that recognize autoantigens specific to atherosclerosis (26). This recognition is analogous to the phenomenon observed in ICI myocarditis, where cardiac antigen-specific T cells contribute. Recent single-cell RNA analysis of human coronary atherosclerotic plaques has revealed clonal expansion of antigen-experienced T cells (189). This finding underscores a potential pathway through which self-reactive epitopes may expedite atherosclerosis by interacting with smooth muscle cells and macrophages within the plaque microenvironment (189).
The interaction between CTLA-4 and CD80/CD86 binding appears to have immunoregulatory effects that suggest a protective role in atherosclerosis (190). Poels et al. conducted an assessment of the impact of antibody-mediated CTLA-4 inhibition on the progression of atherosclerosis (191). Their findings indicated a two-fold increase in the size of atherosclerotic lesions in mice treated with CTLA-4 inhibiting antibodies (191). This increase was predominantly attributed to a shift towards an activated T-cell profile, with limited alterations observed in the macrophage inflammatory profile (191). Furthermore, the inhibition of CTLA-4 was associated with the progression of plaques towards more advanced phenotypes (191). These advanced phenotypes were characterized by reduced collagen content, augmented intimal thickening, and larger necrotic core areas (191). Consistent with the observations from studies involving antibody-mediated CTLA-4 inhibition, or mice overexpressing CTLA-4 exhibited decreased intimal thickening, with a remarkable reduction of 58.5% (181, 191, 192). These mice also displayed reduced numbers of CD4 T-cells, diminished proliferation activity, and a decrease in proinflammatory cytokine production (181, 191, 192).
PD-1’s ability to suppress Th1 cytokine production and facilitate Treg cell development points towards a plausible role in safeguarding against atherosclerosis (193). This protective effect of PD-1/PD-L1 activity becomes evident in knockout mice that manifest enlarged plaques characterized by heightened T-cell and macrophage populations, elevated levels of TNF, intensified T-cell activation by APCs, and amplified cytotoxicity exhibited by CD8+ T cells (177, 179). These combined factors contribute to escalated inflammation and the formation of plaques (177, 179). Additionally, the interaction of PD-1 with PD-L1 leads to the inhibition of cytokine production in differentiated Treg cells, effectively targeting Th1 cell cytokines such as IFN-γ and TNF (194). Since IFN-γ plays a pivotal role in atherogenesis by stimulating T-cell and macrophage recruitment, driving cytokine secretion, and enhancing antigen presentation by endothelial cells, the PD-1/PD-L1 binding serves to curtail both plaque size and the inflammatory response of T cells (195, 196). Similarly, in the context of humans, multiple studies have reported reduced expression of PD-1 or its ligands in individuals with coronary artery disease (CAD) and acute coronary syndrome (180, 197). This observation further substantiates the protective role of PD-1 in atherogenesis and the progression towards advanced plaque phenotypes (180, 197). Notably, within human atherosclerotic plaques, T cells exhibit elevated PD-1 expression, indicative of T-cell exhaustion in the setting of chronic inflammation (178). This complex interplay of PD-1-expressing T cells coexisting with activated counterparts within human plaques raises concerns that the inhibition of PD-1 could potentially exacerbate atherosclerosis (190).
Instances of atherosclerosis or CAD associated with anti-LAG-3 therapy haven’t been documented in clinical trials. Nevertheless, independent findings have shown that the presence of LAG-3 on exhausted T cells present within atherosclerotic plaques (198, 199). Additionally, an observational study conducted on the Multiethnic Study of Atherosclerosis (MESA) cohort revealed that individuals with CAD displayed elevated levels of LAG-3 (199). This study further established LAG-3 as a noteworthy predictor of CAD risk (199).
The treatment for ICI-associated atherosclerosis is currently in the exploratory phase (26). Statins and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors have proven to be safe, effective, and well-established treatments for atherosclerosis (26). However, further investigation is needed to fully characterize the risks and benefits of their application in the context of ICIs and cancer within this complex patient population (26).
Recent studies have highlighted the effectiveness of statin treatment in significantly slowing the annual progression rate of plaque volume among atherosclerosis patients (26, 183). Notably, concurrent statin use has shown promise in enhancing ICI activity, leading to increased cytotoxic CD8 T cell function and reduced pro-inflammatory cytokines (200–202). However, it is unclear if concurrent statin and ICI therapy could further elevate the low risk of statin-induced myopathy (203).
PCSK9 inhibitors, a novel class of monoclonal antibodies used in atherosclerosis treatment, operate by reducing the deviation of LDL receptors and increasing LDL-cholesterol clearance in the bloodstream (204). Alirocumab and evolocumab are two FDA-approved PCSK9 inhibitors (204). Similar to studies on statins, recent findings reveal that PCSK9 inhibitors can enhance ICI therapeutic efficacy (205–207). Studies have shown that combining PCSK9 with ICIs improves intra-tumoral cytotoxic T cell infiltration, antigen presentation, and the expression of co-inhibitory checkpoint molecules (205–207).
Despite nonspecific immunomodulatory agents, such as steroids, being associated with a lower annual rate of plaque progression, they are not recommended for preventing or suppressing ICI-induced plaque development due to their side effects (183) and the linkage of chronic glucocorticoid therapy to adverse cardiovascular outcomes (208–212).
While targeted therapies addressing the underlying immune-mediated mechanisms are still in development, the optimal management of ICI atherosclerosis should prioritize recognizing immunotherapy as a significant risk factor for atherosclerotic cardiovascular disease (26). This involves early risk stratification and optimization, emphasizing a multidisciplinary approach involving both oncologists and cardiologists to determine appropriate screening and medical management strategies (26).
To address the challenges posed by ICIs, researchers are exploring alternative approaches involving different immune checkpoints within the TME (6). Novel pathways and molecules are being studied with the aim of enhancing the effectiveness and broader application of immune checkpoint inhibition therapy (27). Among these options, Tim-3, also referred to as hepatitis A virus cellular receptor 2 (HAVCR2), and TIGIT, known by various names including Washington University cell adhesion molecule (WUCAM), V-set and transmembrane domain containing protein 3 (Vstm3), and V-Set and immunoglobulin domain containing protein 9 (VSIG9), have emerged as viable and promising candidates for the treatment of solid tumors (6). Ongoing clinical trials are actively investigating their therapeutic potential as immune checkpoints (6, 213). Employing antibodies to target these receptors, either independently or in conjunction with other ICIs such as anti-PD-1, has demonstrated the ability to enhance the immune response against tumors in animal models (214–227). These findings highlight the potential of Tim-3 and TIGIT as promising therapeutic targets to address the limitations observed in previous ICI treatments (6).
Tim-3, formerly known as HAVCR2, belongs to the Tim gene family, which includes Tim-1 and Tim-4 (228). Tim-3 has four ligands: soluble ligands Galectin-9 (LGALS9) and high-mobility group protein B1 (HMGB1), and surface-bound ligands carcinoembryonic antigen cell adhesion molecule 1 (CEACAM-1) and phosphatidylserine (PtdSer) (229–233). Unlike other immune checkpoints, Tim-3 is exclusively upregulated by CD4+ and CD8+ cells producing IFN-γ (214, 215). Initially identified on CTLs and Th1 cells, Tim-3 inhibits type 1 immune responses, suppressing cytokine generation, including TNF and INF-γ (228, 234, 235) and is expressed in various tumor cells and immune cells including Th17, Tregs, TILs, natural killer (NK) cells, macrophages, and DCs (228, 236–243).
Understanding Tim-3 signaling pathways is incomplete, with its interactions with intracellular partners for its inhibitory function largely unexplored (234). Research suggests both inhibitory and stimulatory roles for Tim-3 (234). Tim-3 lacks conventional inhibitory signaling motifs and structural elements for recruiting inhibitory phosphatases in its cytoplasmic tail (244). It has five conserved tyrosine residues, with phosphorylation of Tyr256 and Tyr263 (Tyr265 and Tyr272 in humans) being crucial (234). In the current model, upon T-cell activation, Tim-3 migrates to lipid rafts and interacts with BAT3 and LCK (245, 246). In the absence of ligand binding, BAT3 remains bound to Tim-3’s tail, recruiting an active form of LCK, promoting T-cell proliferation and survival (246, 247). However, ligand engagement displaces BAT3, allowing recruitment of tyrosine phosphatases (CD45 and CD148), inactivating LCK, downregulating TCR signaling, suppressing T-cell proliferation and survival (247). Galectin-9 and CEACAM-1 induce phosphorylation of Tyr256 and Tyr263 by ITK, leading to BAT3 release, allowing Tim-3 to exert its inhibitory function (232, 248). BAT3-mediated regulation of Tim-3 signaling is associated with inhibitory actions towards T cells, and its potential application in other cell types like DCs is under investigation (247).
TIGIT, previously known as Vstm3, VSIG9, or WUCAM, is characterized by a protein structure with an extracellular IgV domain and an intracellular domain containing both a canonical ITIM and an immunoglobulin tyrosine tail (ITT) motif (249, 250). Its expression is specifically limited to lymphocytes, prominently observed in subsets such as NK cells, effector and regulatory CD4+ T cells, follicular helper CD4+ T cells, and effector CD8+ T cells (217, 250–254). Notably, TIGIT is found in various cancers, including melanoma, NSCLC, colon cancer, HCC, gastric cancer, glioblastoma, and hematological malignancies (218–220, 255–263). Moreover, TIGIT is significantly overexpressed by Tregs in the peripheral blood mononuclear cells of both healthy individuals and cancer patients, with further upregulation within TME (255, 264).
TIGIT exhibits binding affinity for two members of the nectin family, CD155 (poliovirus receptor, Necl-5), and CD112 (poliovirus receptor related-2, Nectin-2) (249). Its affinity for CD155 (Kd=1-3 nM) surpasses its affinity for CD112 (Kd remains unmeasurable) (249). CD112 has a stronger affinity for CD112R (PVRIG) than for TIGIT, primarily suppressing T cells through binding to CD112R rather than TIGIT (265–267). Consequently, TIGIT’s modulation of T cell and NK cell functions mainly occurs through its interaction with CD155 (251).
Functionally, TIGIT has the ability to hinder CD8+ T cell proliferation and activation by directly influencing TCR expression, leading to the downregulation of the TCR-α chain and other components forming the TCR complex (268). Additionally, TIGIT can curtail TCR-induced p-ERK signaling within CD8+ T cells, thereby suppressing CD8+ T cell priming, differentiation, and cytotoxic activity (269). Similar to CTLA-4’s inhibition of CD28’s co-stimulatory interaction with shared ligands CD80 and CD86, TIGIT exerts its effects indirectly (251). It competes for ligand binding with CD226 (DNAM-1), consequently diminishing T-cell co-stimulation through CD226 (253). Furthermore, TIGIT can obstruct co-stimulatory signaling via CD226 by impeding CD226’s homo-dimerization (217). Ultimately, TIGIT indirectly suppresses T cells by modulating the functions of cells expressing its ligand CD155 (251).
Extensive evidence from studies utilizing preclinical cancer models and in vitro cultures using patient samples has provided substantial support for considering Tim-3 inhibition as a promising avenue for enhancing antitumor immunity (214, 216, 220, 221). The advancement of Tim-3 as an immunotherapeutic target has been strengthened by results similar to those seen with PD-1 inhibitors (221). Experimental evidence showcases that obstructing Tim-3 expression leads to enhanced T cell proliferation and increased cytokine production, thereby stimulating immune activation (215). Furthermore, it has been documented that heightened Tim-3 levels are linked to the emergence of resistance against PD-1 blockade, evident in both lung cancer patient samples and lung cancer models, as well as samples taken from patients with head and neck cancer (222, 223). This intriguing observation implies that Tim-3 might serve as a viable alternative for patients exhibiting resistance to PD-1 blocking antibodies (222). Moreover, in preclinical settings, the combination treatment targeting Tim-3 and PD-1 has exhibited a synergistic effect, reinvigorating T cell function and augmenting the overall anti-tumor immune response (187, 195). Consequently, the concurrent blockade of PD-1 and Tim-3 emerges as a promising and feasible therapeutic strategy (214, 216, 221, 222, 224, 270–274). Currently, there are seven mAbs targeting Tim-3 and one bispecific antibody targeting both PD-1 and Tim-3 (RO7121661) that are undergoing clinical trials (247).
The outcome of single TIGIT blockade has exhibited limited or moderate antitumor effectiveness in experimental tumor models and in enhancing the in vitro functionality of human tumor-infiltrating CD8+ T cells (217, 218, 220, 225, 275). Nonetheless, it’s noteworthy that the interaction between CD155 and TIGIT plays a role in conferring resistance to PD-1 blockade within the context of cancer (226, 276). Studies have indicated that in vitro PD-1 blockade leads to an increase in TIGIT expression on NY-ESO-1-reactive CD8+ T cells extracted from melanoma patients (219). Additionally, TIGIT emerges as the most prominently upregulated immune checkpoint on CD8+ TILs following anti-PD-1 treatment in a PD-1 non-responsive HCC mouse model (275). This observation suggests that inhibiting TIGIT might hold promise as a strategy to enhance the efficacy of PD-1 blockade therapy, particularly against tumors that have developed resistance to PD-1 inhibitors (275). Furthermore, preclinical studies have both demonstrated and mechanistically elucidated the synergy between TIGIT blockade and PD-1 blockade in augmenting the antitumor CD8+ T cell response (226, 227, 275). These findings underscore the potential of combining PD-1 and TIGIT blockade as a promising approach to overcome resistance observed with single PD-1/PD-L1 blockade (226, 227, 275). In the context of mice carrying HCC and displaying resistance to PD-1 blockade, the dual inhibition of TIGIT and PD-1 has been shown to expand the population of effector memory CD8+ T cells and elevate the ratio of CTLs to Tregs within tumors (275). This, in turn, resulted in the suppression of tumor growth and extended survival (275, 277). Moreover, in an MC38 colon tumor mouse model, animals treated with dual anti-TIGIT/PD-1 antibodies exhibited heightened secretion of IFN-γ by CD4+ TILs in comparison to those treated solely with anti-PD-1 therapy (277). Presently, an array of nine human anti-TIGIT mAbs are undergoing evaluation in a total of 43 Phase 1/2/3 clinical trials (251). These trials include monotherapy usage, but more frequently involve combinations with anti-PD-1/PD-L1 antibodies or chemotherapies (251).
As investigations into the safety of Tim-3 blockade continue, there is a limited number of reported cases regarding cardiac toxic adverse events associated with anti-Tim-3 treatment. Nevertheless, a recent report has proposed that Tim-3 may function as a negative regulator or atherosclerosis (278). This particular study has demonstrated that anti-Tim-3 treatment enhances the development of atherosclerotic lesions, leading to an increased presence of monocytes, macrophages, and CD4+ T cells, while simultaneously reducing Tregs and regulatory B cells (278). Furthermore, an independent study has observed that co-blockade of PD-1 and Tim-3 results in a decrease in the production of anti-atherogenic cytokines by PD1+ Tim-3+ CD8+ T cells and an increase in the production of TNF and IFN-γ, both of which contribute to the development and progression of atherosclerosis (279).
While clinical trials have not reported any occurrences of cardiac toxic irAEs linked to anti-TIGIT therapy so far, ongoing investigations are actively exploring the potential emergence of cardiac toxic irAEs resulting from TIGIT blockade treatment. This exploration is particularly focused on conditions characterized by elevated TIGIT expression, especially in cardiovascular diseases. The heightened expression of TIGIT ligands raises the question of whether it signifies a potential risk of cardiotoxic irAEs or suggests a protective function of TIGIT against cardiovascular diseases. This underscores the need for further research to elucidate the potential association between anti-TIGIT treatment and cardiac toxic irAEs.
In cancer treatment, inhibitory targeting of CTLA-4 and PD-1/PD-L1 has evolved into a fundamental approach (27). Despite the proven viability and effectiveness of these ICIs, significant obstacles persist within the field of cancer immunotherapy (6). These hurdles include a restricted response rate and the occurrence of irAEs (6). Notably, among these challenges, the impact on the heart is particularly concerning, with instances of severe cardiac toxicity and a growing number of cases linking CAD to ICIs (91, 218). These challenges underscore the pressing requirement for innovative tactics to mitigate the adverse effects associated with therapy. In response to these challenges, there has been a gradual diversification in the array of co-inhibitory receptor pathways, extending beyond the established CTLA-4 and PD-1 pathways (280). Particularly noteworthy is the incorporation of LAG-3, Tim-3, and, more recently, TIGIT, which has significantly enriched the existing repertoire of approaches aimed at addressing these obstacles (280).
While the occurrence of cardiovascular adverse events related to ICIs is relatively infrequent, their associated mortality rate is notably high (281). Recently, several cardiovascular irAEs have come to light, encompassing conditions such as myocarditis, atherosclerosis, pericarditis, arrhythmias, and cardiomyopathy (123). Among these, myocarditis has exhibited a significant increase in its association with ICIs in recent years and is particularly concerning due to its significantly elevated mortality rate ranging from 25 to 50% (13, 15, 16, 18, 122, 131, 132, 140). Our recent preclinical study has illuminated the crucial role of ICIs like PD-1/PD-L1 and CTLA-4 in maintaining cardiac autoimmunity under normal conditions, emphasizing their importance in this context (138). Furthermore, emerging data suggest that ICIs may expedite the progression of atherosclerosis, potentially leading to an increased risk of atherosclerosis-related cardiovascular events such as acute myocardial infarction, ischemic stroke, and peripheral arterial disease (173–176). Notably, the blockade of the CTLA-4 and PD-1/PD-L1 pathways is significantly associated with the occurrence and progression of both myocarditis and atherosclerosis (42, 138, 146, 148, 151–155, 178–181, 191–193, 195–198, 282). While combining ICIs had significantly improved therapeutic effectiveness, it also carries a considerably higher risk of cardiac irAEs, resulting in increased incidence and mortality rates compared to single-agent therapy (5, 15, 117, 121, 122).
While ongoing clinical trials are actively evaluating the therapeutic efficacy and safety of next-generation ICIs like LAG-3, Tim-3, and TIGIT, Anderson and colleagues have put forth a hierarchical model illustrating the role of co-inhibitory receptors (280). This model proposes that LAG-3, Tim-3, or TIGIT could potentially serve as safer alternatives to existing ICIs based on their roles in regulating self-tolerance and autoimmune toxicity, with CTLA-4 and PD-1 being the primary regulators of self-tolerance and LAG-3, Tim-3, and TIGIT forming a second tier of co-inhibitory molecules with distinct roles in immune response regulation. Furthermore, the latest generation of ICIs offers the potential for targeted regulation of immune responses within specific tissue environments, leveraging the expression of their corresponding ligands to uphold tissue tolerance and prevent immune-related damage (280). This concept underscores the proposed tissue-specific immunoregulatory roles of LAG-3, Tim-3, and TIGIT.
However, it is important to note that increased levels of these ligands must not be exclusively construed as a conclusive sign that blocking these ICIs or their ligands would inherently lead to significant therapeutic advantages. Heightened ligand expression might stem from an inflammatory response triggered by the disease, or it could play a role in a defensive mechanism against the disease. Consequently, pursuing this strategy might potentially trigger serious irAEs that carry the potential for life-threatening outcomes. Moreover, as previously mentioned, the majority of clinical trials have underscored that the use of standalone therapy involving the new generation ICI does not exhibit notable therapeutic effectiveness when contrasted with the outcomes of anti-CTLA-4 or PD-1 treatments—except when combined with anti-CTLA-4 or PD-1 agents (101, 214, 216, 221, 222, 224, 226, 227, 270, 271, 273–275). As a result, further investigation is necessary to unveil the precise therapeutic efficacy and safety profile of these next-generation ICIs. This pursuit could potentially establish them as secure alternatives for ICI-based cancer treatments.
WJ: Conceptualization, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. TW: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. AD: Formal analysis, Investigation, Resources, Visualization, Writing – review & editing. DČ: Conceptualization, Project administration, Resources, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the American Heart Association (AHA) 20TPA35490421 and 23EIA1040103 to DČ, the National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (NHLBI) R01HL118183 and R01HL136586 to DČ, the Global Autoimmune Institute, and the Matthew Poyner MVP Memorial Myocarditis Research Fund to DČ.
Figures 1 and 2 were originally created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Smith SM, Wachter K, Burris Iii HA, Schilsky RL, George DJ, Peterson DE, et al. Clinical cancer advances 2021: ASCO’s report on progress against cancer. J Clin Oncol (2021) 39(10):1165–84. doi: 10.1200/JCO.20
2. Shiravand Y, Khodadadi F, Kashani SMA, Hosseini-Fard SR, Hosseini S, Sadeghirad H, et al. Immune checkpoint inhibitors in cancer therapy. Curr Oncol (2022) 29:3044–60. doi: 10.3390/CURRONCOL29050247
3. Sadeghi Rad H, Monkman J, Warkiani ME, Ladwa R, O’Byrne K, Rezaei N, et al. Understanding the tumor microenvironment for effective immunotherapy. Med Res Rev (2021) 41:1474–98. doi: 10.1002/MED.21765
4. Barbari C, Fontaine T, Parajuli P, Lamichhane N, Jakubski S, Lamichhane P, et al. Immunotherapies and combination strategies for immuno-oncology. Int J Mol Sci (2020) 21:1–28. doi: 10.3390/IJMS21145009
5. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/NRC3239
6. Lee JB, Ha SJ, Kim HR. Clinical insights into novel immune checkpoint inhibitors. Front Pharmacol (2021) 12:681320. doi: 10.3389/FPHAR.2021.681320
7. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi: 10.1056/NEJMOA1003466
8. Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open (2019) 2:e192535. doi: 10.1001/JAMANETWORKOPEN.2019.2535
9. Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic hodgkin lymphoma. J Clin Oncol (2017) 35:2125–32. doi: 10.1200/JCO.2016.72.1316
10. Zhang L, Reynolds KL, Lyon AR, Palaskas N, Neilan TG. The evolving immunotherapy landscape and the epidemiology, diagnosis, and management of cardiotoxicity: JACC: cardioOncology primer. JACC CardioOncol (2021) 3:35–47. doi: 10.1016/J.JACCAO.2020.11.012
11. Conroy M, Naidoo J. Immune-related adverse events and the balancing act of immunotherapy. Nat Commun (2022) 13:1–4. doi: 10.1038/s41467-022-27960-2
12. Tajiri K, Ieda M. Cardiac complications in immune checkpoint inhibition therapy. Front Cardiovasc Med (2019) 6:3. doi: 10.3389/FCVM.2019.00003
13. Li C, Bhatti SA, Ying J. Immune checkpoint inhibitors—Associated cardiotoxicity. Cancers (Basel) (2022) 14:1145. doi: 10.3390/CANCERS14051145/S1
14. Shalata W, Abu-salman A, Steckbeck R, Jacob BM, Massalha I, Yakobson A. Cardiac toxicity associated with immune checkpoint inhibitors: A systematic review. Cancers (Basel) (2021) 13:5218. doi: 10.3390/CANCERS13205218
15. Moslehi JJ, Salem JE, Sosman JA, Lebrun-Vignes B, Johnson DB. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet (2018) 391:933. doi: 10.1016/S0140-6736(18)30533-6
16. Salem JE, Manouchehri A, Moey M, Lebrun-Vignes B, Bastarache L, Pariente A, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective, pharmacovigilance study. Lancet Oncol (2018) 19:1579–89. doi: 10.1016/S1470-2045(18)30608-9
17. Zhang L, Zlotoff DA, Awadalla M, Mahmood SS, Nohria A, Hassan MZO, et al. Major adverse cardiovascular events and the timing and dose of corticosteroids in immune checkpoint inhibitor-associated myocarditis. Circulation (2020) 141:2031–4. doi: 10.1161/CIRCULATIONAHA.119.044703
18. Mahmood SS, Fradley MG, Cohen JV, Nohria A, Reynolds KL, Heinzerling LM, et al. Myocarditis in patients treated with immune checkpoint inhibitors. J Am Coll Cardiol (2018) 71:1755–64. doi: 10.1016/J.JACC.2018.02.037
19. Gong J, Drobni ZD, Zafar A, Quinaglia T, Hartmann S, Gilman HK, et al. Pericardial disease in patients treated with immune checkpoint inhibitors. J Immunother Cancer (2021) 9. doi: 10.1136/JITC-2021-002771
20. Gong J, Drobni ZD, Alvi RM, Murphy SP, Sullivan RJ, Hartmann SE, et al. Immune checkpoint inhibitors for cancer and venous thromboembolic events. Eur J Cancer (2021) 158:99–110. doi: 10.1016/J.EJCA.2021.09.010
21. Suero-Abreu GA, Zanni MV, Neilan TG. Atherosclerosis with immune checkpoint inhibitor therapy: evidence, diagnosis, and management: JACC: cardioOncology state-of-the-art review. JACC CardioOncol (2022) 4:598–615. doi: 10.1016/J.JACCAO.2022.11.011
22. Ganatra S, Neilan TG. Immune checkpoint inhibitor-associated myocarditis. Oncologist (2018) 23:879–86. doi: 10.1634/THEONCOLOGIST.2018-0130
23. Ganatra S, Parikh R, Neilan TG. Cardiotoxicity of immune therapy. Cardiol Clin (2019) 37:385–97. doi: 10.1016/J.CCL.2019.07.008
24. Wu S, Bai H, Zhang L, He J, Luo X, Wang S, et al. Cardiovascular adverse events induced by immune checkpoint inhibitors: A real world study from 2018 to 2022. Front Cardiovasc Med (2022) 9:969942/BIBTEX. doi: 10.3389/FCVM.2022.969942/BIBTEX
25. Wang DY, Salem JE, Cohen JV, Chandra S, Menzer C, Ye F, et al. Fatal toxic effects associated with immune checkpoint inhibitors: A systematic review and meta-analysis. JAMA Oncol (2018) 4:1721–8. doi: 10.1001/JAMAONCOL.2018.3923
26. Chan A, Torelli S, Cheng E, Batchelder R, Waliany S, Neal J, et al. Immunotherapy-associated atherosclerosis: A comprehensive review of recent findings and implications for future research. Curr Treat Options Cardiovasc Med (2023) 25:1–21. doi: 10.1007/S11936-023-01024-0/TABLES/1
27. Marin-Acevedo JA, Kimbrough EMO, Lou Y. Next generation of immune checkpoint inhibitors and beyond. J Hematol Oncol (2021) 14:1–29. doi: 10.1186/S13045-021-01056-8
28. Lee JY, Lee HT, Shin W, Chae J, Choi J, Kim SH, et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat Commun (2016) 7:13354. doi: 10.1038/NCOMMS13354
29. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest (2015) 125:3384–91. doi: 10.1172/JCI80011
30. Gunturi A, McDermott DF. Nivolumab for the treatment of cancer. Expert Opin Investig Drugs (2015) 24:253–60. doi: 10.1517/13543784.2015.991819
31. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature (2014) 515:558–62. doi: 10.1038/NATURE13904
32. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med (2015) 372:2018–28. doi: 10.1056/NEJMOA1501824
33. Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med (2013) 369:134–44. doi: 10.1056/NEJMOA1305133
34. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med (2015) 372:320–30. doi: 10.1056/NEJMOA1412082
35. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med (2015) 373:123–35. doi: 10.1056/NEJMOA1504627
36. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med (2015) 373:1627–39. doi: 10.1056/NEJMOA1507643
37. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med (2015) 373:1803–13. doi: 10.1056/NEJMOA1510665
38. Ibrahim R, Saleh K, Chahine C, Khoury R, Khalife N, Le Cesne A. LAG-3 inhibitors: novel immune checkpoint inhibitors changing the landscape of immunotherapy. Biomedicines (2023) 11:1878. doi: 10.3390/BIOMEDICINES11071878
39. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2017) 18:153–67. doi: 10.1038/nri.2017.108
40. Ha D, Tanaka A, Kibayashi T, Tanemura A, Sugiyama D, Wing JB, et al. Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc Natl Acad Sci U.S.A. (2019) 116:609–18. doi: 10.1073/PNAS.1812186116
41. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity (2009) 30:636–45. doi: 10.1016/J.IMMUNI.2009.04.010
42. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:271–5. doi: 10.1126/SCIENCE.1160062
43. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Sci (1979) (2011) 332:600–3. doi: 10.1126/SCIENCE.1202947
44. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
45. Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell (1992) 71:1065–8. doi: 10.1016/S0092-8674(05)80055-8
46. Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol (1996) 14:233–58. doi: 10.1146/ANNUREV.IMMUNOL.14.1.233
47. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev (2009) 229:12–26. doi: 10.1111/J.1600-065X.2009.00770.X
48. Hathcock KS, Laszlo G, Dickler HB, Bradshaw J, Linsley P, Hodes RJ. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science (1993) 262:905–7. doi: 10.1126/SCIENCE.7694361
49. Freeman GJ, Gribben JG, Boussiotis VA, Ng JW, Restivo VA, Lombard LA, et al. Cloning of B7-2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science (1993) 262:909–11. doi: 10.1126/SCIENCE.7694363
50. Azuma M, Ito D, Yagita H, Okumura K, Phillips JH, Lanier LL, et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature (1993) 366:76–9. doi: 10.1038/366076A0
51. Linsley PS, Clark EA, Ledbetter JA. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc Natl Acad Sci U.S.A. (1990) 87:5031–5. doi: 10.1073/PNAS.87.13.5031
52. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med (1991) 174:561–9. doi: 10.1084/JEM.174.3.561
53. Linsley PS, Greene JAL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity (1994) 1:793–801. doi: 10.1016/S1074-7613(94)80021-9
54. Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, et al. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc Natl Acad Sci U.S.A. (2002) 99:11790–5. doi: 10.1073/PNAS.162359999
55. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR stop signal by CTLA-4. Science (2006) 313:1972–5. doi: 10.1126/SCIENCE.1131078
56. Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity (2002) 16:23–35. doi: 10.1016/S1074-7613(01)00259-X
57. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
58. Schneider H, Mandelbrot DA, Greenwald RJ, Ng F, Lechler R, Sharpe AH, et al. Cutting edge: CTLA-4 (CD152) differentially regulates mitogen-activated protein kinases (extracellular signal-regulated kinase and c-Jun N-terminal kinase) in CD4+ T cells from receptor/ligand-deficient mice. J Immunol (2002) 169:3475–9. doi: 10.4049/JIMMUNOL.169.7.3475
59. Wojtukiewicz MZ, Rek MM, Karpowicz K, Górska M, Polityńska B, Wojtukiewicz AM, et al. Inhibitors of immune checkpoints-PD-1, PD-L1, CTLA-4-new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev (2021) 40:949–82. doi: 10.1007/S10555-021-09976-0
60. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discovery (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
61. Zhao Y, Yang W, Huang Y, Cui R, Li X, Li B. Evolving roles for targeting CTLA-4 in cancer immunotherapy. Cell Physiol Biochem (2018) 47:721–34. doi: 10.1159/000490025
62. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood (2018) 131:58–67. doi: 10.1182/BLOOD-2017-06-741033
63. Koh SA. An update on immunotherapy with PD-1 and PD-L1 blockade. Yeungnam Univ J Med (2021) 38:308. doi: 10.12701/YUJM.2021.01312
64. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U.S.A. (2002) 99:12293–7. doi: 10.1073/PNAS.192461099
65. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol (2017) 8:561. doi: 10.3389/FPHAR.2017.00561
66. Sun Z, Fourcade J, Pagliano O, Chauvin JM, Sander C, Kirkwood JM, et al. IL10 and PD-1 cooperate to limit the activity of tumor-specific CD8+ T cells. Cancer Res (2015) 75:1635–44. doi: 10.1158/0008-5472.CAN-14-3016
67. Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther (2016) 9:5023–39. doi: 10.2147/OTT.S105862
68. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11:3887–95. doi: 10.1002/J.1460-2075.1992.TB05481.X
69. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192:1027–34. doi: 10.1084/JEM.192.7.1027
70. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med (2006) 203:883–95. doi: 10.1084/JEM.20051776
71. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science (2001) 291:319–22. doi: 10.1126/SCIENCE.291.5502.319
72. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11:141–51. doi: 10.1016/S1074-7613(00)80089-8
73. Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol (2007) 19:813–24. doi: 10.1093/INTIMM/DXM057
74. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/ANNUREV.IMMUNOL.26.021607.090331
75. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi: 10.1038/NM730
76. Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res (2004) 64:1140–5. doi: 10.1158/0008-5472.CAN-03-3259
77. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med (2012) 4:127ra37. doi: 10.1126/SCITRANSLMED.3003689
78. Wang C-Y, Guttridge DC, Mayo MW, Albert S. Baldwin jr. NF-κB induces expression of the bcl-2 homologue A1/bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol Cell Biol (1999) 19:5923. doi: 10.1128/MCB.19.9.5923
79. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206:3015–29. doi: 10.1084/JEM.20090847
80. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med (1990) 171:1393–405. doi: 10.1084/JEM.171.5.1393
81. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21:503–13. doi: 10.1016/J.IMMUNI.2004.08.010
82. Goldberg MV, Drake CG. LAG-3 in cancer immunotherapy. Curr Top Microbiol Immunol (2011) 344:269–78. doi: 10.1007/82_2010_114
83. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest (2007) 117:3383–92. doi: 10.1172/JCI31184
84. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012) 72:917–27. doi: 10.1158/0008-5472.CAN-11-1620
85. Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y, et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer (2018) 9:176–89. doi: 10.18632/GENESANDCANCER.180
86. Ascierto PA, Melero I, Bhatia S, Bono P, Sanborn RE, Lipson EJ, et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. J Clin Oncol (2017) 35:9520–0. doi: 10.1200/JCO.2017.35.15_SUPPL.9520
87. Ascierto PA, Bono P, Bhatia S, Melero I, Nyakas MS, Svane I-M, et al. Efficacy of BMS-986016, a monoclonal antibody that targets lymphocyte activation gene-3 (LAG-3), in combination with nivolumab in pts with melanoma who progressed during prior anti–PD-1/PD-L1 therapy (mel prior IO) in all-comer and biomarker-enriched populations. Ann Oncol (2017) 28:v611–2. doi: 10.1093/annonc/mdx440.011
88. Wang M, Du Q, Jin J, Wei Y, Lu Y, Li Q. LAG3 and its emerging role in cancer immunotherapy. Clin Transl Med (2021) 11. doi: 10.1002/CTM2.365
89. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol (2018) 8:86. doi: 10.3389/FONC.2018.00086
90. Sankar K, Ye JC, Li Z, Zheng L, Song W, Hu-Lieskovan S. The role of biomarkers in personalized immunotherapy. biomark Res (2022) 10:1–13. doi: 10.1186/S40364-022-00378-0
91. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364:2517–26. doi: 10.1056/NEJMOA1104621
92. SChadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol (2015) 33:1889–94. doi: 10.1200/JCO.2014.56.2736
93. Farolfi A, Ridolfi L, Guidoboni M, Nicoletti SVL, Piciucchi S, Valmorri L, et al. Ipilimumab in advanced melanoma: reports of long-lasting responses. Melanoma Res (2012) 22:263–70. doi: 10.1097/CMR.0B013E328353E65C
94. Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA (2016) 315:1600–9. doi: 10.1001/JAMA.2016.4059
95. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New Engl J Med (2015) 373:23–34. doi: 10.1056/NEJMOA1504030/SUPPL_FILE/NEJMOA1504030_DISCLOSURES.PDF
96. Lim DWT, Kao HF, Suteja L, Li CH, Quah HS, Tan DSW, et al. Clinical efficacy and biomarker analysis of dual PD-1/CTLA-4 blockade in recurrent/metastatic EBV-associated nasopharyngeal carcinoma. Nat Commun (2023) 14:2781. doi: 10.1038/S41467-023-38407-7
97. Lingel H, Brunner-Weinzierl MC. CTLA-4 (CD152): A versatile receptor for immune-based therapy. Semin Immunol (2019) 42:101298. doi: 10.1016/J.SMIM.2019.101298
98. Liu J, Chen Z, Li Y, Zhao W, Wu JB, Zhang Z. PD-1/PD-L1 checkpoint inhibitors in tumor immunotherapy. Front Pharmacol (2021) 12:731798. doi: 10.3389/FPHAR.2021.731798
99. Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res (2019) 38:1–12. doi: 10.1186/S13046-019-1259-Z
100. Huo JL, Wang YT, Fu WJ, Lu N, Liu ZS. The promising immune checkpoint LAG-3 in cancer immunotherapy: from basic research to clinical application. Front Immunol (2022) 13:956090. doi: 10.3389/FIMMU.2022.956090
101. Yu X, Huang X, Chen X, Liu J, Wu C, Pu Q, et al. Characterization of a novel anti-human lymphocyte activation gene 3 (LAG-3) antibody for cancer immunotherapy. MAbs (2019) 11:1139–48. doi: 10.1080/19420862.2019.1629239
102. Ball S, Ghosh RK, Wongsaengsak S, Bandyopadhyay D, Ghosh GC, Aronow WS, et al. Cardiovascular toxicities of immune checkpoint inhibitors: JACC review topic of the week. J Am Coll Cardiol (2019) 74:1714–27. doi: 10.1016/J.JACC.2019.07.079
103. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372:2521–32. doi: 10.1056/NEJMOA1503093
104. Abdel-Rahman O, Fouad M. A network meta-analysis of the risk of immune-related renal toxicity in cancer patients treated with immune checkpoint inhibitors. Immunotherapy (2016) 8:665–74. doi: 10.2217/IMT-2015-0020
105. Nonomura Y, Otsuka A, Ohtsuka M, Yamamoto T, Dummer R, Kabashima K. ADAMTSL5 is upregulated in melanoma tissues in patients with idiopathic psoriasis vulgaris induced by nivolumab. J Eur Acad Dermatol Venereol (2017) 31:e100–1. doi: 10.1111/JDV.13818
106. Nonomura Y, Otsuka A, Nakashima C, Seidel JA, Kitoh A, Dainichi T, et al. Peripheral blood Th9 cells are a possible pharmacodynamic biomarker of nivolumab treatment efficacy in metastatic melanoma patients. Oncoimmunology (2016) 5:e1248327. doi: 10.1080/2162402X.2016.1248327
107. Kato Y, Otsuka A, Miyachi Y, Kabashima K. Exacerbation of psoriasis vulgaris during nivolumab for oral mucosal melanoma. J Eur Acad Dermatol Venereol (2016) 30:e89–91. doi: 10.1111/JDV.13336
108. Chae YK, Chiec L, Mohindra N, Gentzler R, Patel J, Giles F. A case of pembrolizumab-induced type-1 diabetes mellitus and discussion of immune checkpoint inhibitor-induced type 1 diabetes. Cancer Immunol Immunother (2017) 66:25–32. doi: 10.1007/S00262-016-1913-7
109. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med (2016) 8:328rv4. doi: 10.1126/SCITRANSLMED.AAD7118
110. Freeman-Keller M, Kim Y, Cronin H, Richards A, Gibney G, Weber JS. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res (2016) 22:886–94. doi: 10.1158/1078-0432.CCR-15-1136
111. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
112. Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol (1985) 37:269–390. doi: 10.1016/S0065-2776(08)60342-9
113. Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol (1991) 9:243–69. doi: 10.1146/ANNUREV.IY.09.040191.001331
114. Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol (2015) 16:375–84. doi: 10.1016/S1470-2045(15)70076-8
115. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med (2015) 372:2006–17. doi: 10.1056/NEJMOA1414428
116. Antonia SJ, Bendell JC, Taylor MH, Calvo E, Jaeger D, De Braud FG, et al. Phase I/II study of nivolumab with or without ipilimumab for treatment of recurrent small cell lung cancer (SCLC): CA209-032. J Clin Oncol (2015) 33:7503–3. doi: 10.1200/JCO.2015.33.15_SUPPL.7503
117. Tawbi HA, SChadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutiérrez E, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med (2022) 386:24–34. doi: 10.1056/NEJMOA2109970
118. Lee DJ, Lee HJ, Farmer JR, Reynolds KL. Mechanisms driving immune-related adverse events in cancer patients treated with immune checkpoint inhibitors. Curr Cardiol Rep (2021) 23:1–12. doi: 10.1007/S11886-021-01530-2
119. Takahashi Y, Nagaya T, Iwaya Y, Okamura T, Hirayama A, Iwaya M, et al. CD8+ Lymphocyte infiltration is a specific feature of colitis induced by immune checkpoint inhibitors. Dig Dis Sci (2023) 68:451–9. doi: 10.1007/S10620-022-07598-2
120. Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol (2019) 16:563–80. doi: 10.1038/S41571-019-0218-0
121. Wang D, Bauersachs J, Berliner D. Immune checkpoint inhibitor associated myocarditis and cardiomyopathy: A translational review. Biol (Basel) (2023) 12:472. doi: 10.3390/BIOLOGY12030472
122. Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med (2016) 375:1749–55. doi: 10.1056/NEJMOA1609214
123. D’Souza M, Nielsen D, Svane IM, Iversen K, Rasmussen PV, Madelaire C, et al. The risk of cardiac events in patients receiving immune checkpoint inhibitors: a nationwide Danish study. Eur Heart J (2021) 42:1621–31. doi: 10.1093/EURHEARTJ/EHAA884
124. Waheed N, Fradley MG, DeRemer DL, Mahmoud A, Shah CP, Langaee TY, et al. Newly diagnosed cardiovascular disease in patients treated with immune checkpoint inhibitors: a retrospective analysis of patients at an academic tertiary care center. Cardiooncology (2021) 7:1–7. doi: 10.1186/S40959-021-00097-9
125. Rubio-Infante N, Ramírez-Flores YA, Castillo EC, Lozano O, García-Rivas G, Torre-Amione G. Cardiotoxicity associated with immune checkpoint inhibitor therapy: a meta-analysis. Eur J Heart Fail (2021) 23:1739–47. doi: 10.1002/EJHF.2289
126. Geisler BP, Raad RA, Esaian D, Sharon E, Schwartz DR. Apical ballooning and cardiomyopathy in a melanoma patient treated with ipilimumab: a case of takotsubo-like syndrome. J Immunother Cancer (2015) 3:1–4. doi: 10.1186/S40425-015-0048-2
127. Heinzerling L, Ott PA, Hodi FS, Husain AN, Tajmir-Riahi A, Tawbi H, et al. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J Immunother Cancer (2016) 4:1–11. doi: 10.1186/S40425-016-0152-Y
128. Honton B, Despas F, Dumonteil N, Rouvellat C, Roussel M, Carrie D, et al. Bortezomib and heart failure: case-report and review of the French Pharmacovigilance database. Fundam Clin Pharmacol (2014) 28:349–52. doi: 10.1111/FCP.12039
129. Läubli H, Balmelli C, Bossard M, Pfister O, Glatz K, Zippelius A. Acute heart failure due to autoimmune myocarditis under pembrolizumab treatment for metastatic melanoma. J Immunother Cancer (2015) 3:11. doi: 10.1186/S40425-015-0057-1
130. Dolladille C, Akroun J, Morice PM, Dompmartin A, Ezine E, Sassier M, et al. Cardiovascular immunotoxicities associated with immune checkpoint inhibitors: a safety meta-analysis. Eur Heart J (2021) 42:4964–77. doi: 10.1093/EURHEARTJ/EHAB618
131. Lampejo T, Durkin SM, Bhatt N, Guttmann O. Acute myocarditis: aetiology, diagnosis and management. Clin Med (2021) 21:e505. doi: 10.7861/CLINMED.2021-0121
132. Peretto G, Sala S, Rizzo S, De Luca G, Campochiaro C, Sartorelli S, et al. Arrhythmias in myocarditis: State of the art. Heart Rhythm (2019) 16:793–801. doi: 10.1016/J.HRTHM.2018.11.024
133. Seko Y, Yagita H, Okumura K, Azuma M, Nagai R. Roles of programmed death-1 (PD-1)/PD-1 ligands pathway in the development of murine acute myocarditis caused by coxsackievirus B3. Cardiovasc Res (2007) 75:158–67. doi: 10.1016/J.CARDIORES.2007.03.012
134. Gutierrez FRS, Mariano FS, Oliveira CJF, Pavanelli WR, Guedes PMM, Silva GK, et al. Regulation of trypanosoma cruzi-induced myocarditis by programmed death cell receptor 1. Infect Immun (2011) 79:1873. doi: 10.1128/IAI.01047-10
135. Grabie N, Gotsman I, DaCosta R, Pang H, Stavrakis G, Butte MJ, et al. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell–mediated injury in the heart. Circulation (2007) 116:2062–71. doi: 10.1161/CIRCULATIONAHA.107.709360
136. Wang J, Okazaki IM, Yoshida T, Chikuma S, Kato Y, Nakaki F, et al. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int Immunol (2010) 22:443–52. doi: 10.1093/INTIMM/DXQ026
137. Wei SC, Meijers WC, Axelrod ML, Anang NAAS, Screever EM, Wescott EC, et al. A genetic mouse model recapitulates immune checkpoint inhibitor-associated myocarditis and supports a mechanism-based therapeutic intervention. Cancer Discovery (2021) 11:614. doi: 10.1158/2159-8290.CD-20-0856
138. Won T, Kalinoski HM, Wood MK, Hughes DM, Jaime CM, Delgado P, et al. Cardiac myosin-specific autoimmune T cells contribute to immune-checkpoint-inhibitor-associated myocarditis. Cell Rep (2022) 41. doi: 10.1016/J.CELREP.2022.111611
139. Groarke JD, Cheng S, Moslehi J. Cancer-drug discovery and cardiovascular surveillance. N Engl J Med (2013) 369:1779–81. doi: 10.1056/NEJMP1313140
140. Escudier M, Cautela J, Malissen N, Ancedy Y, Orabona M, Pinto J, et al. Clinical features, management, and outcomes of immune checkpoint inhibitor-related cardiotoxicity. Circulation (2017) 136:2085–7. doi: 10.1161/CIRCULATIONAHA.117.030571
141. Waliany S, Lee D, Witteles RM, Neal JW, Nguyen P, Davis MM, et al. Immune checkpoint inhibitor cardiotoxicity: understanding basic mechanisms and clinical characteristics and finding a cure. Annu Rev Pharmacol Toxicol (2021) 61:113–34. doi: 10.1146/ANNUREV-PHARMTOX-010919-023451
142. Lehmann LH, Cautela J, Palaskas N, Baik AH, Meijers WC, Allenbach Y, et al. Clinical strategy for the diagnosis and treatment of immune checkpoint inhibitor-associated myocarditis: A narrative review. JAMA Cardiol (2021) 6:1329–37. doi: 10.1001/JAMACARDIO.2021.2241
143. Tadokoro T, Keshino E, Makiyama A, Sasaguri T, Ohshima K, Katano H, et al. Acute lymphocytic myocarditis with anti-PD-1 antibody nivolumab. Circ Heart Fail (2016) 9:e003514. doi: 10.1161/CIRCHEARTFAILURE.116.003514
144. Koelzer VH, Rothschild SI, Zihler D, Wicki A, Willi B, Willi N, et al. Systemic inflammation in a melanoma patient treated with immune checkpoint inhibitors-an autopsy study. J Immunother Cancer (2016) 4:1–8. doi: 10.1186/S40425-016-0117-1
145. Tay RY, Blackley E, McLean C, Moore M, Bergin P, Gill S, et al. Successful use of equine anti-thymocyte globulin (ATGAM) for fulminant myocarditis secondary to nivolumab therapy. Br J Cancer (2017) 117:921–4. doi: 10.1038/BJC.2017.253
146. Axelrod ML, Meijers WC, Screever EM, Qin J, Carroll MG, Sun X, et al. T cells specific for α-myosin drive immunotherapy-related myocarditis. Nature (2022) 611:818–26. doi: 10.1038/S41586-022-05432-3
147. Ramos G, Frantz S, Hofmann U. Myosin-specific T cells in myocardial diseases revisited. Circulation (2023) 148:4–6. doi: 10.1161/CIRCULATIONAHA.123.064453
148. Lv HJ, Havari E, Pinto S, Gottumukkala RVSRK, Cornivelli L, Raddassi K, et al. Impaired thymic tolerance to α-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest (2011) 121:1561–73. doi: 10.1172/JCI44583
149. Hou X, Chen G, Bracamonte-Baran W, Choi HS, Diny NL, Sung J, et al. The cardiac microenvironment instructs divergent monocyte fates and functions in myocarditis. Cell Rep (2019) 28:172–189.e7. doi: 10.1016/J.CELREP.2019.06.007
150. Wu L, Ong SF, Talor MV, Barin JG, Baldeviano GC, Kass DA, et al. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J Exp Med (2014) 211:1449–64. doi: 10.1084/JEM.20132126
151. Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res (2010) 106:1646–55. doi: 10.1161/CIRCRESAHA.109.213157
152. Myers JM, Cooper LT, Kem DC, Stavrakis S, Kosanke SD, Shevach EM, et al. Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight (2016) 1. doi: 10.1172/JCI.INSIGHT.85851
153. Ying H, Yang L, Qiao G, Li Z, Zhang L, Yin F, et al. Cutting edge: CTLA-4–B7 interaction suppresses th17 cell differentiation. J Immunol (2010) 185:1375. doi: 10.4049/JIMMUNOL.0903369
154. Blanco-Domínguez R, Sánchez-Díaz R, de la Fuente H, Jiménez-Borreguero LJ, Matesanz-Marín A, Relaño M, et al. A novel circulating noncoding small RNA for the detection of acute myocarditis. N Engl J Med (2021) 384:2014–27. doi: 10.1056/NEJMOA2003608
155. Hua X, Hu G, Hu Q, Chang Y, Hu Y, Gao L, et al. Single-cell RNA sequencing to dissect the immunological network of autoimmune myocarditis. Circulation (2020) 142:384–400. doi: 10.1161/CIRCULATIONAHA.119.043545
156. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res (2015) 3:412–23. doi: 10.1158/2326-6066.CIR-14-0150
157. Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med (2003) 9:1477–83. doi: 10.1038/NM955
158. Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: american society of clinical oncology clinical practice guideline. J Clin Oncol (2018) 36:1714. doi: 10.1200/JCO.2017.77.6385
159. Palaskas N, Lopez-Mattei J, Durand JB, Iliescu C, Deswal A. Immune checkpoint inhibitor myocarditis: pathophysiological characteristics, diagnosis, and treatment. J Am Heart Association: Cardiovasc Cerebrovascular Dis (2020) 9:e013757. doi: 10.1161/JAHA.119.013757
160. Norwood TG, Westbrook BC, Johnson DB, Litovsky SH, Terry NL, McKee SB, et al. Smoldering myocarditis following immune checkpoint blockade. J Immunother Cancer (2017) 5:1–6. doi: 10.1186/S40425-017-0296-4
161. Arangalage D, Delyon J, Lermuzeaux M, Ekpe K, Ederhy S, Pages C, et al. Survival after fulminant myocarditis induced by immune-checkpoint inhibitors. Ann Intern Med (2017) 167:683–4. doi: 10.7326/L17-0396
162. Frigeri M, Meyer P, Banfi C, Giraud R, Hachulla AL, Spoerl D, et al. Immune checkpoint inhibitor-associated myocarditis: A new challenge for cardiologists. Can J Cardiol (2018) 34:92.e1–3. doi: 10.1016/J.CJCA.2017.09.025
163. Esfahani K, Buhlaiga N, Thébault P, Lapointe R, Johnson NA, Miller WH. Alemtuzumab for immune-related myocarditis due to PD-1 therapy. N Engl J Med (2019) 380:2375–6. doi: 10.1056/NEJMC1903064
164. Salem J-E, Allenbach Y, Vozy A, Brechot N, Johnson DB, Moslehi JJ, et al. Abatacept for severe immune checkpoint inhibitor-associated myocarditis. N Engl J Med (2019) 380:2377–9. doi: 10.1056/NEJMC1901677
165. Salem JE, Bretagne M, Abbar B, Leonard-Louis S, Ederhy S, Redheuil A, et al. Abatacept/ruxolitinib and screening for concomitant respiratory muscle failure to mitigate fatality of immune-checkpoint inhibitor myocarditis. Cancer Discovery (2023) 13:1100–15. doi: 10.1158/2159-8290.CD-22-1180/716628/AM/ABATACEPT-RUXOLITINIB-AND-SCREENING-FOR
166. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation (2013) 128:e147–e239. doi: 10.1161/CIR.0B013E31829E8776
167. Tajmir-Riahi A, Bergmann T, Schmid M, Agaimy A, Schuler G, Heinzerling L. Life-threatening autoimmune cardiomyopathy reproducibly induced in a patient by checkpoint inhibitor therapy. J Immunother (2018) 41:35–8. doi: 10.1097/CJI.0000000000000190
168. Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol (2009) 27:165–97. doi: 10.1146/ANNUREV.IMMUNOL.021908.132620
169. Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol (2008) 8:802–15. doi: 10.1038/NRI2415
170. Seijkens TTP, Lutgens E. Cardiovascular oncology: exploring the effects of targeted cancer therapies on atherosclerosis. Curr Opin Lipidol (2018) 29:381–8. doi: 10.1097/MOL.0000000000000538
171. Poels K, Neppelenbroek SIM, Kersten MJ, Antoni ML, Lutgens E, Seijkens TTP. Immune checkpoint inhibitor treatment and atherosclerotic cardiovascular disease: an emerging clinical problem. J Immunother Cancer (2021) 9. doi: 10.1136/JITC-2021-002916
172. Lutgens E, Seijkens TTP. Cancer patients receiving immune checkpoint inhibitor therapy are at an increased risk for atherosclerotic cardiovascular disease. J Immunother Cancer (2020) 8. doi: 10.1136/JITC-2019-000300
173. Johnson DB, Sullivan RJ, Ott PA, Carlino MS, Khushalani NI, Ye F, et al. Ipilimumab therapy in patients with advanced melanoma and preexisting autoimmune disorders. JAMA Oncol (2016) 2:234–40. doi: 10.1001/JAMAONCOL.2015.4368
174. Menzies AM, Johnson DB, Ramanujam S, Atkinson VG, Wong ANM, Park JJ, et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann Oncol (2017) 28:368–76. doi: 10.1093/ANNONC/MDW443
175. Tison A, Quéré G, Misery L, Funck-Brentano E, Danlos FX, Routier E, et al. Safety and efficacy of immune checkpoint inhibitors in patients with cancer and preexisting autoimmune disease: A nationwide, multicenter cohort study. Arthritis Rheumatol (2019) 71:2100–11. doi: 10.1002/ART.41068
176. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol (2020) 5:eaay1863. doi: 10.1126/SCIIMMUNOL.AAY1863
177. Gotsman I, Grabie N, Dacosta R, Sukhova G, Sharpe A, Lichtman AH. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J Clin Invest (2007) 117:2974–82. doi: 10.1172/JCI31344
178. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir E ad D, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med (2019) 25:1576. doi: 10.1038/S41591-019-0590-4
179. Bu DX, Tarrio M, Maganto-Garcia E, Stavrakis G, Tajima G, Lederer J, et al. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler Thromb Vasc Biol (2011) 31:1100–7. doi: 10.1161/ATVBAHA.111.224709
180. Lee J, Zhuang Y, Wei X, Shang F, Wang J, Zhang Y, et al. Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J Mol Cell Cardiol (2009) 46:169–76. doi: 10.1016/J.YJMCC.2008.10.028
181. Matsumoto T, Sasaki N, Yamashita T, Emoto T, Kasahara K, Mizoguchi T, et al. Overexpression of cytotoxic T-lymphocyte-associated antigen-4 prevents atherosclerosis in mice. Arterioscler Thromb Vasc Biol (2016) 36:1141–51. doi: 10.1161/ATVBAHA.115.306848
182. Amiri-Kordestani L, Moslehi J, Cheng J, Tang S, Schroeder R, Sridhara R, et al. Cardiovascular adverse events in immune checkpoint inhibitor clinical trials: A U.S. Food and Drug Administration pooled analysis. J Clin Oncol (2018) 36:3009–9. doi: 10.1200/JCO.2018.36.15_SUPPL.3009
183. Drobni ZD, Alvi RM, Taron J, Zafar A, Murphy SP, Rambarat PK, et al. Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation (2020) 142:2299–311. doi: 10.1161/CIRCULATIONAHA.120.049981
184. Wang Y, Zhou S, Yang F, Qi X, Wang X, Guan X, et al. Treatment-related adverse events of PD-1 and PD-L1 inhibitors in clinical trials: A systematic review and meta-analysis. JAMA Oncol (2019) 5:1008–19. doi: 10.1001/JAMAONCOL.2019.0393
185. Bar J, Markel G, Gottfried T, Percik R, Leibowitz-Amit R, Berger R, et al. Acute vascular events as a possibly related adverse event of immunotherapy: a single-institute retrospective study. Eur J Cancer (2019) 120:122–31. doi: 10.1016/J.EJCA.2019.06.021
186. Solinas C, Saba L, Sganzerla P, Petrelli F. Venous and arterial thromboembolic events with immune checkpoint inhibitors: A systematic review. Thromb Res (2020) 196:444–53. doi: 10.1016/J.THROMRES.2020.09.038
187. Razak NBA, Jones G, Bhandari M, Berndt MC, Metharom P. Cancer-associated thrombosis: an overview of mechanisms, risk factors, and treatment. Cancers (Basel) (2018) 10:380. doi: 10.3390/CANCERS10100380
188. Inno A, Chiampan A, Lanzoni L, Verzè M, Molon G, Gori S. Immune checkpoint inhibitors and atherosclerotic vascular events in cancer patients. Front Cardiovasc Med (2021) 8:652186. doi: 10.3389/FCVM.2021.652186
189. Chowdhury RR, D’addabbo J, Huang X, Veizades S, Sasagawa K, Louis DM, et al. Human coronary plaque T cells are clonal and cross-react to virus and self. Circ Res (2022) 130:1510–30. doi: 10.1161/CIRCRESAHA.121.320090
190. Vuong JT, Stein-Merlob AF, Nayeri A, Sallam T, Neilan TG, Yang EH. Immune checkpoint therapies and atherosclerosis: mechanisms and clinical implications: JACC state-of-the-art review. J Am Coll Cardiol (2022) 79:577–93. doi: 10.1016/J.JACC.2021.11.048
191. Poels K, van Leent MMT, Reiche ME, Kusters PJH, Huveneers S, de Winther MPJ, et al. Antibody-mediated inhibition of CTLA4 aggravates atherosclerotic plaque inflammation and progression in hyperlipidemic mice. Cells (2020) 9:1987. doi: 10.3390/CELLS9091987
192. Ewing MM, Karper JC, Abdul S, De Jong RCM, Peters HAB, De Vries MR, et al. T-cell co-stimulation by CD28-CD80/86 and its negative regulator CTLA-4 strongly influence accelerated atherosclerosis development. Int J Cardiol (2013) 168:1965–74. doi: 10.1016/J.IJCARD.2012.12.085
193. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol (2001) 2:261–8. doi: 10.1038/85330
194. Frostegård J, Ulfgren AK, Nyberg P, Hedin U, Swedenborg J, Andersson U, et al. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis (1999) 145:33–43. doi: 10.1016/S0021-9150(99)00011-8
195. Amento EP, Ehsani N, Palmer H, Libby P. Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb (1991) 11:1223–30. doi: 10.1161/01.ATV.11.5.1223
196. Gotsman I, Lichtman AH. Targeting interferon-gamma to treat atherosclerosis. Circ Res (2007) 101:333–4. doi: 10.1161/CIRCRESAHA.107.155838
197. Li SH, Chen WJ, Yan M, Shu YW, Liao YH. Expression of coinhibitory PD-L1 on CD4+CD25+FOXP3+ regulatory T cells is elevated in patients with acute coronary syndrome. Coron Artery Dis (2015) 26:598–603. doi: 10.1097/MCA.0000000000000282
198. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15:486–99. doi: 10.1038/NRI3862
199. Golden D, Kolmakova A, Sura S, Vella AT, Manichaikul A, Wang XQ, et al. Lymphocyte activation gene 3 and coronary artery disease. JCI Insight (2016) 1:88628. doi: 10.1172/JCI.INSIGHT.88628
200. Cantini L, Pecci F, Hurkmans DP, Belderbos RA, Lanese A, Copparoni C, et al. High-intensity statins are associated with improved clinical activity of PD-1 inhibitors in Malignant pleural mesothelioma and advanced non-small cell lung cancer patients. Eur J Cancer (2021) 144:41–8. doi: 10.1016/J.EJCA.2020.10.031
202. Xia Y, Xie Y, Yu Z, Xiao H, Jiang G, Zhou X, et al. The mevalonate pathway is a druggable target for vaccine adjuvant discovery. Cell (2018) 175:1059–1073.e21. doi: 10.1016/J.CELL.2018.08.070
203. Drobni ZD, Murphy SP, Alvi RM, Lee C, Gong J, Mosarla RC, et al. Association between incidental statin use and skeletal myopathies in patients treated with immune checkpoint inhibitors. Immunother Adv (2021) 1:ltab014. doi: 10.1093/IMMADV/LTAB014
204. Hajar R. PCSK 9 inhibitors: A short history and a new era of lipid-lowering therapy. Heart Views (2019) 20:74. doi: 10.4103/HEARTVIEWS.HEARTVIEWS_59_19
205. Liu X, Bao X, Hu M, Chang H, Jiao M, Cheng J, et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature (2020) 588:693–8. doi: 10.1038/S41586-020-2911-7
206. Wang R, Liu H, He P, An D, Guo X, Zhang X, et al. Inhibition of PCSK9 enhances the antitumor effect of PD-1 inhibitor in colorectal cancer by promoting the infiltration of CD8+ T cells and the exclusion of Treg cells. Front Immunol (2022) 13:947756. doi: 10.3389/FIMMU.2022.947756
207. Almeida CR, Ferreira BH, Duarte IF. Targeting PCSK9: a promising adjuvant strategy in cancer immunotherapy. Signal Transduct Target Ther (2021) 6:111. doi: 10.1038/S41392-021-00530-6
208. De Leo M, Pivonello R, Auriemma RS, Cozzolino A, Vitale P, Simeoli C, et al. Cardiovascular disease in Cushing’s syndrome: heart versus vasculature. Neuroendocrinology (2010) 92 Suppl 1:50–4. doi: 10.1159/000318566
209. Tune JD, Goodwill AG, Sassoon DJ, Mather KJ. Cardiovascular consequences of metabolic syndrome. Transl Res (2017) 183:57–70. doi: 10.1016/J.TRSL.2017.01.001
210. Walker BR. Glucocorticoids and cardiovascular disease. Eur J Endocrinol (2007) 157:545–59. doi: 10.1530/EJE-07-0455
211. Pujades-Rodriguez M, Morgan AW, Cubbon RM, Wu J. Dose-dependent oral glucocorticoid cardiovascular risks in people with immune-mediated inflammatory diseases: A population-based cohort study. PloS Med (2020) 17:e1003432. doi: 10.1371/JOURNAL.PMED.1003432
212. Wei L, MacDonald TM, Walker BR. Taking glucocorticoids by prescription is associated with subsequent cardiovascular disease. Ann Intern Med (2004) 141:764–70. doi: 10.7326/0003-4819-141-10-200411160-00007
213. Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer (2020) 8. doi: 10.1136/JITC-2020-000957
214. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207:2187–94. doi: 10.1084/JEM.20100643
215. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PloS One (2012) 7:e30676. doi: 10.1371/JOURNAL.PONE.0030676
216. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207:2175–86. doi: 10.1084/JEM.20100637
217. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell (2014) 26:923–37. doi: 10.1016/J.CCELL.2014.10.018
218. Ge Z, Zhou G, Campos Carrascosa L, Gausvik E, Boor PPC, Noordam L, et al. TIGIT and PD1 Co-blockade Restores ex vivo Functions of Human Tumor-Infiltrating CD8+ T Cells in Hepatocellular Carcinoma. Cell Mol Gastroenterol Hepatol (2021) 12:443. doi: 10.1016/J.JCMGH.2021.03.003
219. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J Clin Invest (2015) 125:2046–58. doi: 10.1172/JCI80445
220. Ostroumov D, Duong S, Wingerath J, Woller N, Manns MP, Timrott K, et al. Transcriptome profiling identifies TIGIT as a marker of T-cell exhaustion in liver cancer. Hepatology (2021) 73:1399–418. doi: 10.1002/HEP.31466
221. Ngiow SF, Von Scheidt B, Akiba H, Yagita H, Teng MWL, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res (2011) 71:3540–51. doi: 10.1158/0008-5472.CAN-11-0096
222. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/NCOMMS10501
223. Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology (2016) 6:e1261779. doi: 10.1080/2162402X.2016.1261779
224. Cai C, Xu YF, Wu ZJ, Dong Q, Li MY, Olson JC, et al. Tim-3 expression represents dysfunctional tumor infiltrating T cells in renal cell carcinoma. World J Urol (2016) 34:561–7. doi: 10.1007/S00345-015-1656-7
225. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology (2018) 7:e1466769. doi: 10.1080/2162402X.2018.1466769
226. Li XY, Das I, Lepletier A, Addala V, Bald T, Stannard K, et al. CD155 loss enhances tumor suppression via combined host and tumor-intrinsic mechanisms. J Clin Invest (2022) 132:2613–25. doi: 10.1172/JCI159825
227. Braun M, Aguilera AR, Sundarrajan A, Corvino D, Stannard K, Krumeich S, et al. CD155 on tumor cells drives resistance to immunotherapy by inducing the degradation of the activating receptor CD226 in CD8+ T cells. Immunity (2020) 53:805–823.e15. doi: 10.1016/J.IMMUNI.2020.09.010
228. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415:536–41. doi: 10.1038/415536A
229. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6:1245–52. doi: 10.1038/NI1271
230. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood (2009) 113:3821–30. doi: 10.1182/BLOOD-2008-10-185884
231. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol (2012) 13:832–42. doi: 10.1038/NI.2376
232. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2015) 517:386–90. doi: 10.1038/NATURE13848
233. Kang CW, Dutta A, Chang LY, Mahalingam J, Lin YC, Chiang JM, et al. Apoptosis of tumor infiltrating effector TIM-3+CD8+ T cells in colon cancer. Sci Rep (2015) 5:15659. doi: 10.1038/SREP15659
234. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev (2017) 276:97–111. doi: 10.1111/IMR.12520
235. Tian T, Li Z. Targeting tim-3 in cancer with resistance to PD-1/PD-L1 blockade. Front Oncol (2021) 11:731175. doi: 10.3389/FONC.2021.731175
236. Huang X, Bai X, Cao Y, Wu J, Huang M, Tang D, et al. Lymphoma endothelium preferentially expresses Tim-3 and facilitates the progression of lymphoma by mediating immune evasion. J Exp Med (2010) 207:505–20. doi: 10.1084/JEM.20090397
237. Jan M, Chao MP, Cha AC, Alizadeh AA, Gentlese AJ, Weissmana IL, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci U.S.A. (2011) 108:5009–14. doi: 10.1073/PNAS.1100551108
238. Anderson AC. Tim-3, a negative regulator of anti-tumor immunity. Curr Opin Immunol (2012) 24:213–6. doi: 10.1016/J.COI.2011.12.005
239. Datar I, Sanmamed MF, Wang J, Henick BS, Choi J, Badri T, et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin Cancer Res (2019) 25:4663–73. doi: 10.1158/1078-0432.CCR-18-4142
240. So EC, Khaladj-Ghom A, Ji Y, Amin J, Song Y, Burch E, et al. NK cell expression of Tim-3: First impressions matter. Immunobiology (2019) 224:362–70. doi: 10.1016/J.IMBIO.2019.03.001
241. Yong YK, Saeidi A, Tan HY, Rosmawati M, Enström PF, Al Batran R, et al. Hyper-expression of PD-1 is associated with the levels of exhausted and dysfunctional phenotypes of circulating CD161++TCR iVα7.2+ Mucosal-associated invariant T cells in chronic hepatitis B virus infection. Front Immunol (2018) 9:472/BIBTEX. doi: 10.3389/FIMMU.2018.00472/BIBTEX
242. Gleason MK, Lenvik TR, McCullar V, Felices M, O’Brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood (2012) 119:3064–72. doi: 10.1182/BLOOD-2011-06-360321
243. Li Y, Zhang J, Zhang D, Hong X, Tao Y, Wang S, et al. Tim-3 signaling in peripheral NK cells promotes maternal-fetal immune tolerance and alleviates pregnancy loss. Sci Signal (2017) 10:eaah4323. doi: 10.1126/SCISIGNAL.AAH4323/SUPPL_FILE/AAH4323_SM.PDF
244. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol (2020) 20:173–85. doi: 10.1038/S41577-019-0224-6
245. Clayton KL, Haaland MS, Douglas-Vail MB, Mujib S, Chew GM, Ndhlovu LC, et al. T cell Ig and mucin domain-containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J Immunol (2014) 192:782–91. doi: 10.4049/JIMMUNOL.1302663
246. Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat Med (2012) 18:1394–400. doi: 10.1038/NM.2871
247. Acharya N, Acharya N, Sabatos-Peyton C, Anderson AC, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer (2020) 8. doi: 10.1136/JITC-2020-000911
248. van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem Biophys Res Commun (2006) 351:571–6. doi: 10.1016/J.BBRC.2006.10.079
249. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10:48–57. doi: 10.1038/NI.1674
250. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol (2011) 41:902–15. doi: 10.1002/EJI.201041136
251. Ge Z, Peppelenbosch MP, Sprengers D, Kwekkeboom J. TIGIT, the next step towards successful combination immune checkpoint therapy in cancer. Front Immunol (2021) 12:699895. doi: 10.3389/FIMMU.2021.699895
252. Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG, et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol (2009) 39:695–703. doi: 10.1002/EJI.200839116
253. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol (2012) 188:3869–75. doi: 10.4049/JIMMUNOL.1103627
254. Stengel KF, Harden-Bowles K, Yu X, Rouge L, Yin J, Comps-Agrar L, et al. Structure of TIGIT immunoreceptor bound to poliovirus receptor reveals a cell-cell adhesion and signaling mechanism that requires cis-trans receptor clustering. Proc Natl Acad Sci U.S.A. (2012) 109:5399–404. doi: 10.1073/PNAS.1120606109
255. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40:569–81. doi: 10.1016/J.IMMUNI.2014.02.012
256. Lozano E, Mena MP, Díaz T, Martin-Antonio B, Leon S, Rodríguez-Lobato LG, et al. Nectin-2 expression on Malignant plasma cells is associated with better response to TIGIT blockade in multiple myeloma. Clin Cancer Res (2020) 26:4688–98. doi: 10.1158/1078-0432.CCR-19-3673
257. Inozume T, Yaguchi T, Furuta J, Harada K, Kawakami Y, Shimada S. Melanoma cells control antimelanoma CTL responses via interaction between TIGIT and CD155 in the effector phase. J Invest Dermatol (2016) 136:255–63. doi: 10.1038/JID.2015.404
258. Hu F, Wang W, Fang C, Bai C. TIGIT presents earlier expression dynamic than PD-1 in activated CD8+ T cells and is upregulated in non-small cell lung cancer patients. Exp Cell Res (2020) 396:112260. doi: 10.1016/J.YEXCR.2020.112260
259. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med (2018) 24:994–1004. doi: 10.1038/S41591-018-0057-Z
260. Xu D, Zhao E, Zhu C, Zhao W, Wang C, Zhang Z, et al. TIGIT and PD-1 may serve as potential prognostic biomarkers for gastric cancer. Immunobiology (2020) 225:112260. doi: 10.1016/J.IMBIO.2020.151915
261. Lucca LE, Lerner BA, Park C, DeBartolo D, Harnett B, Kumar VP, et al. Differential expression of the T-cell inhibitor TIGIT in glioblastoma and MS. Neurology(R) neuroimmunol Neuroinflamm (2020) 7. doi: 10.1212/NXI.0000000000000712
262. Yang ZZ, Kim HJ, Wu H, Jalali S, Tang X, Krull JE, et al. TIGIT expression is associated with T-cell suppression and exhaustion and predicts clinical outcome and anti-PD-1 response in follicular lymphoma. Clin Cancer Res (2020) 26:5217–31. doi: 10.1158/1078-0432.CCR-20-0558
263. Kong Y, Zhu L, Schell TD, Zhang J, Claxton DF, Ehmann WC, et al. T-cell immunoglobulin and ITIM domain (TIGIT) associates with CD8+ T-cell exhaustion and poor clinical outcome in AML patients. Clin Cancer Res (2016) 22:3057–66. doi: 10.1158/1078-0432.CCR-15-2626
264. Fourcade J, Sun Z, Chauvin JM, Ka M, Davar D, Pagliano O, et al. CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight (2018) 3. doi: 10.1172/JCI.INSIGHT.121157
265. Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT, et al. Identification of CD112R as a novel checkpoint for human T cells. J Exp Med (2016) 213:167–76. doi: 10.1084/JEM.20150785
266. Murter B, Pan X, Ophir E, Alteber Z, Azulay M, Sen R, et al. Mouse PVRIG has CD8+ T cell-specific coinhibitory functions and dampens antitumor immunity. Cancer Immunol Res (2019) 7:244–56. doi: 10.1158/2326-6066.CIR-18-0460
267. Whelan S, Ophir E, Kotturi MF, Levy O, Ganguly S, Leung L, et al. PVRIG and PVRL2 are induced in cancer and inhibit CD8+ T-cell function. Cancer Immunol Res (2019) 7:257–68. doi: 10.1158/2326-6066.CIR-18-0442
268. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol (2011) 186:1338–42. doi: 10.4049/JIMMUNOL.1003081
269. Josefsson SE, Huse K, Kolstad A, Beiske K, Pende D, Steen CB, et al. T cells expressing checkpoint receptor TIGIT are enriched in follicular lymphoma tumors and characterized by reversible suppression of T-cell receptor signaling. Clin Cancer Res (2018) 24:870–81. doi: 10.1158/1078-0432.CCR-17-2337
270. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood (2011) 117:4501–10. doi: 10.1182/BLOOD-2010-10-310425
271. Fourcade J, Sun Z, Pagliano O, Chauvin JM, Sander C, Janjic B, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8+ T cells induced by melanoma vaccines. Cancer Res (2014) 74:1045–55. doi: 10.1158/0008-5472.CAN-13-2908
272. Cheng L, Ruan Z. Tim-3 and Tim-4 as the potential targets for antitumor therapy. Hum Vaccin Immunother (2015) 11:2458–62. doi: 10.1080/21645515.2015.1056953
273. Liu J, Zhang S, Hu Y, Yang Z, Li J, Liu X, et al. Targeting PD-1 and tim-3 pathways to reverse CD8 T-cell exhaustion and enhance ex vivo T-cell responses to autologous dendritic/tumor vaccines. J Immunother (2016) 39:171–80. doi: 10.1097/CJI.0000000000000122
274. Lu X, Yang L, Yao D, Wu X, Li J, Liu X, et al. Tumor antigen-specific CD8+ T cells are negatively regulated by PD-1 and Tim-3 in human gastric cancer. Cell Immunol (2017) 313:43–51. doi: 10.1016/J.CELLIMM.2017.01.001
275. Chiu DKC, Yuen VWH, Cheu JWS, Wei LL, Ting V, Fehlings M, et al. Hepatocellular carcinoma cells up-regulate PVRL1, stabilizing PVR and inhibiting the cytotoxic T-cell response via TIGIT to mediate tumor resistance to PD1 inhibitors in mice. Gastroenterology (2020) 159:609–23. doi: 10.1053/J.GASTRO.2020.03.074
276. Lepletier A, Madore J, O’Donnell JS, Johnston RL, Li XY, McDonald E, et al. Tumor CD155 expression is associated with resistance to anti-PD1 immunotherapy in metastatic melanoma. Clin Cancer Res (2020) 26:3671–81. doi: 10.1158/1078-0432.CCR-19-3925
277. Dixon KO, Schorer M, Nevin J, Etminan Y, Amoozgar Z, Kondo T, et al. Functional anti-TIGIT antibodies regulate development of autoimmunity and antitumor immunity. J Immunol (2018) 200:3000–7. doi: 10.4049/JIMMUNOL.1700407
278. Foks AC, Ran IA, Wasserman L, Frodermann V, Ter Borg MND, De Jager SCA, et al. T-cell immunoglobulin and mucin domain 3 acts as a negative regulator of atherosclerosis. Arterioscler Thromb Vasc Biol (2013) 33:2558–65. doi: 10.1161/ATVBAHA.113.301879
279. Qiu MK, Wang SC, Dai YX, Wang SQ, Ou JM, Quan ZW. PD-1 and tim-3 pathways regulate CD8+ T cells function in atherosclerosis. PloS One (2015) 10:e0128523. doi: 10.1371/JOURNAL.PONE.0128523
280. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989. doi: 10.1016/J.IMMUNI.2016.05.001
281. Moey MYY, Tomdio AN, McCallen JD, Vaughan LM, O’Brien K, Naqash AR, et al. Characterization of immune checkpoint inhibitor-related cardiotoxicity in lung cancer patients from a rural setting. JACC CardioOncol (2020) 2:491–502. doi: 10.1016/J.JACCAO.2020.07.005
Keywords: immune checkpoint inhibitors, myocarditis, atherosclerosis, CTLA-4, PD-1, LAG-3, TIM-3, TIGIT
Citation: Jo W, Won T, Daoud A and Čiháková D (2024) Immune checkpoint inhibitors associated cardiovascular immune-related adverse events. Front. Immunol. 15:1340373. doi: 10.3389/fimmu.2024.1340373
Received: 17 November 2023; Accepted: 19 January 2024;
Published: 05 February 2024.
Edited by:
Michelle Krogsgaard, New York University, United StatesReviewed by:
Iryna Voloshyna, New York University, United StatesCopyright © 2024 Jo, Won, Daoud and Čiháková. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Čiháková, Y2loYWtvdmFAamhtaS5lZHU=
†These authors have contributed equally to this work and share first authorship
‡ORCID: Daniela Čiháková, orcid.org/0000-0002-8713-2860
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.