
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 13 March 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1333170
This article is part of the Research TopicImmunity and Inflammatory Responses in Kidney Disease: from Mechanisms to Novel Therapeutic StrategiesView all 17 articles
 Xiao-min Hao1†
Xiao-min Hao1† Yu Liu1†
Yu Liu1† Dilizhawaer Hailaiti2
Dilizhawaer Hailaiti2 Yu Gong3
Yu Gong3 Xu-dong Zhang4
Xu-dong Zhang4 Bing-nan Yue3
Bing-nan Yue3 Ji-peng Liu3Xiao-li Wu3
Ji-peng Liu3Xiao-li Wu3 Ke-zhen Yang5*Jun Wang1*Qing-guo Liu3*
Ke-zhen Yang5*Jun Wang1*Qing-guo Liu3*Hypertensive nephropathy (HTN) is the second leading cause of end-stage renal disease (ESRD) and a chronic inflammatory disease. Persistent hypertension leads to lesions of intrarenal arterioles and arterioles, luminal stenosis, secondary ischemic renal parenchymal damage, and glomerulosclerosis, tubular atrophy, and interstitial fibrosis. Studying the pathogenesis of hypertensive nephropathy is a prerequisite for diagnosis and treatment. The main cause of HTN is poor long-term blood pressure control, but kidney damage is often accompanied by the occurrence of immune inflammation. Some studies have found that the activation of innate immunity, inflammation and acquired immunity is closely related to the pathogenesis of HTN, which can cause damage and dysfunction of target organs. There are more articles on the mechanism of diabetic nephropathy, while there are fewer studies related to immunity in hypertensive nephropathy. This article reviews the mechanisms by which several different immune cells and inflammatory cytokines regulate blood pressure and renal damage in HTN. It mainly focuses on immune cells, cytokines, and chemokines and inhibitors. However, further comprehensive and large-scale studies are needed to determine the role of these markers and provide effective protocols for clinical intervention and treatment.
Hypertension is a major public health concern (1), affecting approximately 30% of adults. It is a major contributor to cardiovascular disease morbidity and mortality (2) and is strongly associated with chronic kidney disease(CKD) (3–7). The global prevalence of hypertension-induced chronic CKD exceeds 23.6 million,is characterized by a high degree of hypertension, and severely affects the patient’s quality of life (8, 9). As hypertension progresses, it eventually leads to hypertensive nephropathy(HTN) with irreversible glomerular injury, glomerulosclerosis, tubular atrophy, and interstitial fibrosis (10, 11), which is the second leading cause of end-stage renal disease (12). According to previous studies, the treatment of HTN is relatively limited to controlling blood pressure and regulating lifestyle habits (13, 14). Once the patient develops end-stage renal disease(ESRD), blood pressure control program should be implemented. Notably, dialysis and kidney transplantation are the only available approaches (15), and the treatment of HTN and ESRD is limited. In addition, the incidence of HTN and ESRD is expected to increase in the coming decades (16), placing a heavy burden on healthcare resources.
HTN is first diagnosed based on hypertension and CKD; however, secondary hypertension must be excluded. Hypertension affects the renal vasculature, glomeruli, and tubular interstitium. In general, renal immune cell aggregation promotes an immune-inflammatory response, which in turn disrupts renal blood pressure regulation. Although a large number of studies have been published on HTN in the past, the pathogenesis of hypertension and HTN remains unclear, despite the prevalence of hypertension and associated CKD. Hypertension is closely associated with a continuously activated immune system, which can cause target organ damage and dysfunction, eventually leading to the development of complications such as HTN (17) (Figure 1). The first step in innate immune activation that causes hypertension is the pathogen recognition receptor (PRR), which senses a pathogen-associated molecular pattern (PAMP) or damage-associated molecular pattern (DAMP) from stressed or injured tissues and is thus activated to initiate host defense and inflammation (18). Determining the mechanisms by which different immune cells and their subtypes regulate inflammation could help identify new strategies for treating this disease and preventing its progression.
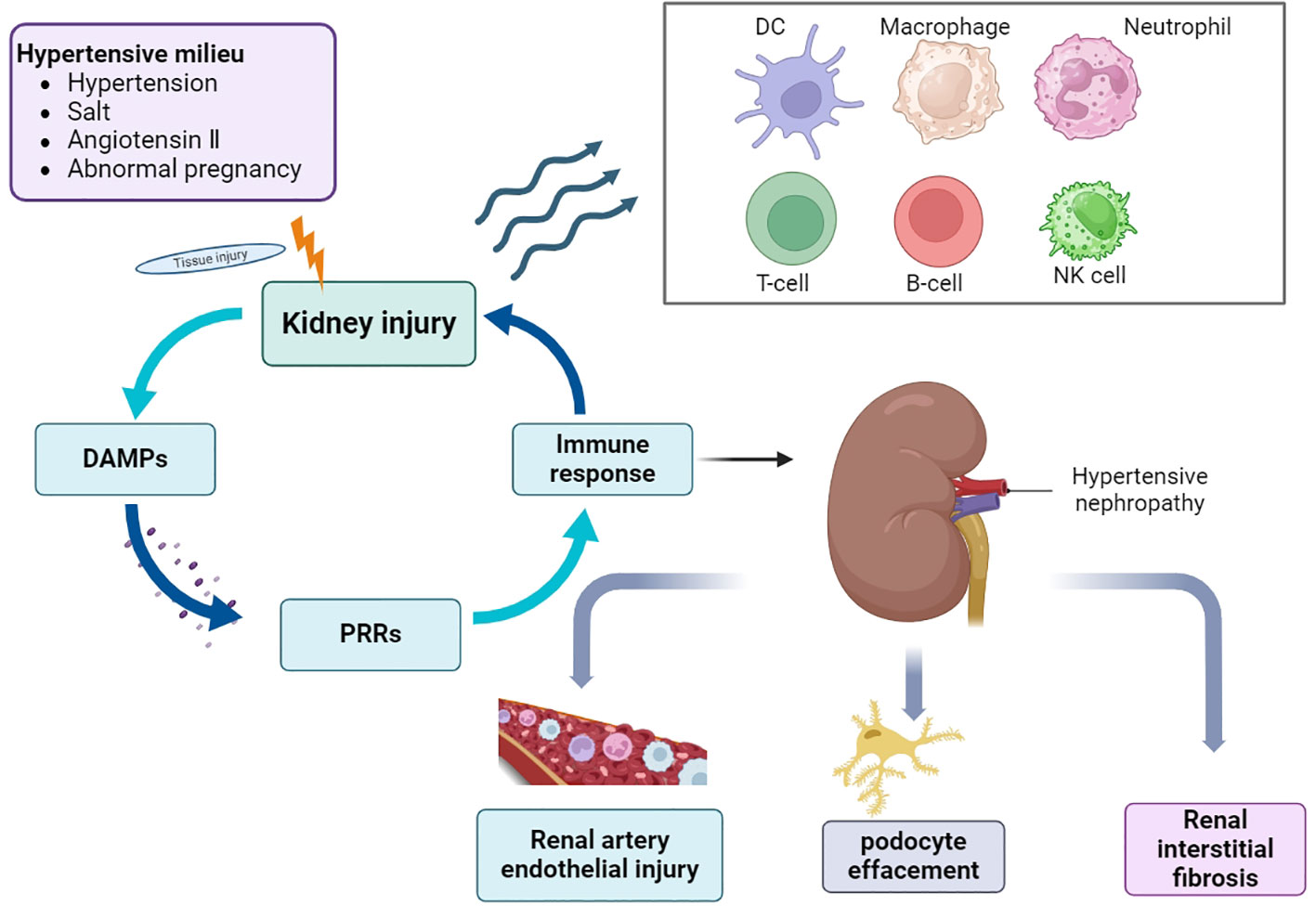
Figure 1 Overview of the pathogenesis of HTN. In the environment of hypertension, hypertension, high salt, angiotensin II and abnormal pregnancy activate a variety of signaling cascades, which promote the recruitment and activation of immune cells, cause the occurrence and development of inflammation, and eventually lead to a series of pathological changes of HTN. DAMPs, damage-associated molecular patterns; PRRs, pattern recognition receptors; DC, dendritic cells; NK cells, natural killer cells.
Renal injury is a common complication of essential hypertension and is most commonly observed 5–10 years after the onset of hypertension (19). It is also a major cause of kidney disease deterioration. Blood pressure affects the course of kidney disease. A study evaluating ambulatory blood pressure in African Americans found that occult hypertension may be associated with the development of chronic kidney disease (20). Further, insidious hypertension may be associated with the onset of CKD (20). This suggests that regulating blood pressure reduces the risk of hypertension and is also a key factor in reducing the cardiovascular burden of kidney disease (21).
The pathogenesis of CKD is complex, encompassing vascular, glomerular, and tubulointerstitial injuries, and is influenced by environmental and genetic factors, including the severity and duration of hypertension. Remodeling small input arteries is a hallmark of HTN in humans.Hypertension leads to injury and subsequent apoptosis of podocytes, which form the final filtration barrier of the glomerulus, ultimately leading to glomerulosclerosis (22–25).
The progression of hypertension leads to changes in small arteries, resulting in intraglomerular hemodynamics changes. A clinical study found that hypertension combined with renal small artery hyalinosis was closely associated with proteinuria and that the accumulation of hyalinized material in the small renal arteries was more likely to lead to hypertensive kidney damage (26). Renal sympathetic nerve activity is usually increased in pathophysiological conditions such as hypertension and chronic and ESRD. Numerous studies (27) have confirmed that elevated sympathetic nerve levels in patients with hypertension lead to deterioration of renal function and abnormal cardiovascular homeostasis due to activation of the renin–angiotensin system (RAS) and renal vascular injury (28, 29). Angiotensin II (Ang II) is most closely related to the RAS (30). Once hypertension occurs, the RAS becomes hyperactivated in vivo, potentially leading to glomerulosclerosis and interstitial fibrosis (31). Overproduction of Ang II in the kidney directly constricts vascular endothelial cells (ECs), causing changes in diastolic and contractile substances that are involved in hypertension and kidney injury. In hypertensive rats, the miRNA-mediated mTOR signaling pathway may play a role in Ang II-induced renal artery endothelial cell injury by driving glycolysis and activating autophagy (32).
Glomerular injury is a prominent feature of hypertensive nephropathy (33). Any change primarily involving small blood vessels eventually translates into glomerular injury (24). In some clinical studies, patients with a history of hypertension showed a significant reduction in glomerular number (34) and an increase in glomerular volume compared with patients with normal blood pressure (35). Podocytes are epithelial cells that form a glomerular barrier to prevent protein loss (36–39). Podocyte injury is involved in RAS activation and is an essential factor in the progression of HTN and diabetic nephropathy (40). Ang II is a central active molecule of the RAS, plays a role in regulating hypertension-induced inflammation in organ damage (41, 42), and can directly contribute to podocyte injury (43, 44). Podocyte injury was positively correlated with mean arterial pressure, suggesting that higher blood pressure was more likely to cause glomerular injury (45). Hypertensive disease causes podocyte damage and apoptosis, leading to glomerulosclerosis (46). As a pro-apoptotic protein, septin4 is an important marker of organ damage. Recent studies have shown that septin4 promotes hypertensive kidney injury by activating the acetylation of K174 (K174Q). Septin4–K174R is one of the renoprotective factors that ameliorates Ang II-induced hypertensive kidney injury (47). In addition, an increase in blood pressure leads to arterial stiffness, which eventually affects the arteries and causes glomerular lesions (48).
Vascular endothelial growth factor (VEGF), produced by podocytes, is a significant regulator of vascular biology and plays a key role in glomerular development and function (49). Further, it has been reported in various types of glomerulonephritis, including diabetic nephropathy, which is usually associated with increased VEGF-A (50).
Sustained hypertension causes renal tubular cell damage and tubulointerstitial fibrosis (TIF). In patients with hypertension, numerous scattered inflammatory cells may trigger TIF (51). Renal interstitial injury is a dynamic and heterogeneous structure involving a variety of molecules, such as collagen, glycosaminoglycans, and glycoproteins. Notably, type I, III, V, VI, VII, and XV collagen are all expressed in the kidney under physiological conditions. The expression of type III procollagen and type V collagen is significantly increased in spontaneously hypertensive rats(SHR) kidneys, suggesting that these collagens play a role in hypertension-induced renal fibrosis (52). Further, complement C3 expression was significantly increased in the renal mesenchymal tissues of SHR, inducing the synthesis and overgrowth of mesenchymal cells (53). Similar results were confirmed in a clinical study in which enhanced interstitial complement C3 expression in patients with HTN was significantly associated with interstitial macrophage density, serum creatinine levels, interstitial fibrosis, glomerulosclerosis, and arteriopathy scores (54). The results of another clinical study revealed (55) that mast cells were mainly present in the renal tubulointerstitium and increased in number in human HTN, suggesting the involvement of renal mast cells in the progression of HTN.
The complex role of immune system in hypertension was proposed as early as the 1960s (56). Hypertension is a chronic inflammatory disease, and hypertensive stimuli lead to the accumulation of pro-inflammatory cells, such as macrophages and T lymphocytes, which affect the development and progression of hypertension (57). In renal diseases, inflammation, immune cell aggregation, and cell death form a vicious cycle of fibrous matrix deposition that destroys renal tissue structure and ultimately leads to renal dysfunction (58). Immune cells can maintain renal homeostasis and attenuate renal damage. In addition, immunosuppressive agents have been used to treat kidney disease (59). The cells of the innate immune system in renal immunity include dendritic cells (DCs), macrophages/monocytes, natural killer (NK) cells, T cells, B cells, and neutrophils. HTN is an inflammatory disease caused by metabolic disorders in nature (60), which can be manifested as an increase in lymphocyte count (61), thereby increasing the risk of HTN.
Considerable overlap has been reported between DCs and macrophages regarding cell origin morphology, function, and cell surface antigen expression (62). As innate immune cells, DCs and macrophages can act as sentinels and messengers and are closely related to individual development and the triggering of inflammatory responses. Simultaneously, they complement each other in their surveillance functions (63). DCs focus on antigen presentation, whereas macrophages play a role in clearing dead cells and repairing damaged tissues. These cells induce and regulate the inflammatory response and protect the kidney from infection (64).
DCs form a dendritic network that spans the barrier surface and entire organs, including the kidney, performing important sentinel and messenger functions (65). DCs are the most potent antigen-presenting cells (APCs) in the body, located in the interstitial space of the renal tubules, with immature DCs located closer to the vasculature (66). CD103+ cells mainly activate cytotoxic T cells (67), whereas CD11b+ cells activate CD4+ T cells and produce inflammatory chemokines (68). Notably, DCs are the effectors of renal inflammation, as depletion of CD11c+ cells ameliorates antibody-induced inflammation in the glomerular basement membrane (69). Renal DCs mediate the transfer of T cells to the kidney in an antigen-dependent manner during bacterial infection, suggesting that renal DCs not only monitor systemic infections but are also crucial in localized infections and autoimmunity (70). Similar findings were observed in Ang II-induced hypertension. Renal mid-renal DCs also present antigens to T cells under appropriate instructions (71). In hypertensive mice (72), activated DCs improved sensitivity to hypertension; however, only responsive T lymphocytes can trigger activation. In another mouse experiment (73), mice lacking CD70 did not aggregate T (EM) T-cells and did not develop hypertension to the point of attenuated kidney injury under high-salt conditions or a second Ang II challenge. In addition, typical FLT3L-dependent DCs promote T-cell activation in the kidney, leading to oxidative stress, fluid retention, and elevated blood pressure (74).
Macrophages participate in the innate immune response, immune surveillance, and the maintenance of renal homeostasis (75, 76). Macrophages have various functions in different microenvironments (77), responding differently depending on the nature and duration of renal injury (78–80). Macrophage phenotypes are influenced by the microenvironment, ranging from pro-inflammatory (termed classical M1-type activation) to anti-inflammatory or pro-catabolic (termed selective M2-type activation) (81, 82). The main functions are maintenance of homeostasis and phagocytosis to remove various foreign bodies (83, 84). M1 macrophages favor the clearance of infections and promote immune responses (85). M2 macrophages have a wide range of functions that inhibit inflammation and promote tissue repair (86). In the kidneys, immune activation significantly increases the number of macrophages, leading to the infiltration of monocyte-derived macrophages involved in inflammation in the stenotic kidney and the promotion of fibrosis, especially during the chronic phase of kidney disease (87–89). As previously mentioned (54), inflammation with complement C3 activation and macrophage infiltration and their interaction may play an important role in the pathogenesis of human HTN. There are also data confirming (90) that increased macrophage infiltration is closely correlated with increased YM1/Chi3l3 expression, suggesting that YM1/Chi3l3 may be a biomarker for HTN. Recent evidence shows (91) that VEGFC inhibits inflammasome activation by promoting high salt-induced autophagy in macrophages to ameliorate hypertensive kidney injury.
Neutrophils are the first line of defense in the innate immune system and help maintain blood vessel tone (92). A study found that neutrophils in the aorta increased after 7 days of Ang II-induced hypertension (93). An increase in neutrophil count can aggravate the risk of chronic renal failure. However, direct evidence to confirm the role of neutrophils in HTN remains lacking.
T-lymphocytes are immune cells with the greatest phenotypic and functional diversity. Growing evidence suggests that T cells are involved in the development and progression of HTN. Further, the FASL/FAS (CD95L-CD95) pathway is activated in the kidneys of hypertensive rats, and cytotoxic T lymphocytes (CTLs) kill target cells through this apoptotic pathway. Activation of the FAS/FASL pathway may also be a mechanism underlying renal tubular epithelial cell loss and mesangial fibrosis, which are prominent renal characteristics of hypertensive kidneys (94). Earlier studies (95) also found lymphocytic infiltration in the renal interstitial space in patients with hypertensive renal injury. Some studies have reported (96) that lymphocytes can reduce sodium excretion during Ang II-dependent hypertension by regulating NOS3 and COX-2 expression in the kidneys. Notably, the main source of interferon (IFN)-γ and TNF are CTLs, and mice lacking any of these cytokines showed attenuated hypertension and renal injury in the presence of hypertensive stimuli (97, 98), which aligns with the pathogenic role of CTLs. CTLs play a critical role in hypertension. However, only the CD8+ T-cell population is antigenically activated in the kidneys of hypertensive mice, which may be the main site of immune activation in hypertension (99). This study also found that Cd8 mice had an approximately 50% reduction in the hypertensive response to Ang II or deoxycorticosterone acetate (DOCA) and salt treatment. CD8+ mice had fewer kidney leukocytes than wild-type mice, thinner small renal arteries, and less endothelial dysfunction (99). In a recent study, during DOCA- and salt-induced hypertension, CD8+ T cells acted directly on renal distal tubules to maintain renal retention (100). Therefore, CTLs may influence the blood pressure by enhancing renal reabsorption of sodium and water.
Activation of the immune system contributes to the pathogenesis of hypertension and the progression of chronic kidney disease. An imbalance in the helper T cell subpopulation is associated with Ang II-induced hypertensive kidney injury (101). T regulatory (Treg) cells are key regulators that mediate immunity and are involved in the activation and proliferation of auto T cells, which play an essential role in autoimmune diseases, immune homeostasis, graft tolerance, and immune regulation of tumors (102). In addition, Tregs exert an inhibitory effect on inflammation by secreting the cytotoxic factors, perforin, and granzyme.
The discovery of Tregs has led to a better understanding of CKD and HTN pathogenesis. Notably, the number of Treg cells in the serum of patients with CKD is significantly decreased, and the percentage of Th17 cells, serum IL-17 levels, and ratio of Th17/Treg cells are increased with the progression of CKD (103). Further studies found that transferring Tregs from healthy mice to mice treated for kidney injury attenuated the condition (104). Moreover, macrophages work synergistically with Tregs to attenuate kidney injury by enhancing chemokine expression (105). Tregs also reduce kidney injury by inhibiting macrophage activation and reducing pro-inflammatory cytokines (106). Clinical and basic data have shown that the Th17 pathway has specific effects on hypertensive kidney injury, ultimately leading to kidney injury (107–109). The Th17 pathway has been shown to affect hypertension (110) specifically.
A study found that NK cells play an important role in Ang II-induced vascular dysfunction (111). A laboratory study confirmed that the long-term infusion of IL-17 into pregnant rats results in NK cell activation and proliferation (112). In another study (113), CD1d-dependent NKT cell activation was protective against Ang II-induced cardiac remodeling. However, the specific mechanisms underlying hypertension and concomitant renal disease require further investigation.
B cells play a role in hypertension by producing antibodies. For example, the B cell/IgG pair is closely associated with Ang II-induced hypertension and vascular remodeling in mice (114). It has been reported (4, 114) that B-cell activation and IgG production are closely associated with the pathogenesis of hypertension and end-organ damage. Targeting B cells may be able to ameliorate renal lipid peroxidative damage in patients with hyperhomocysteinemia (HHcy) and HTN (115).
Inflammation is a natural and necessary response of the body to stimuli. It is responsible for migrating immune system cells to the stimulus target after a series of steps facilitated and coordinated by cytokines, chemokines, and acute-phase proteins (58). Activation of the immune system is involved in the pathogenesis of hypertension and contributes to CKD progression (116). The accumulation of immune cells in the kidneys promotes a chronic inflammatory response that disrupts blood pressure regulation (117). Inflammation is a key factor in the pathogenesis of hypertension and cardiovascular diseases. Some studies have found that inflammation, immune cell infiltration, and alterations in chemokines and cytokines characterize hypertensive kidney damage (118, 119).
Cytokines are small proteins involved in the development and activity of the immune system, have a wide range of biological activities, and help coordinate the body’s response to infection (120, 121). Hypertension is associated with the aggregation of T cells and monocytes/macrophages in the blood vessels and kidneys, which produce potent cytokines that affect vascular and renal function (122). Innate immune cells express several receptors that recognize microorganisms, induce rapid defense, and delay cellular responses. Toll-like receptors on the cell surface and endosomal membranes monitor extracellular microbes and activate multiple host defense signaling pathways (123). The NF-κB and interferon regulatory factor (IRF) pathways downstream of these receptors induce the expression of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6, among others (Table 1).
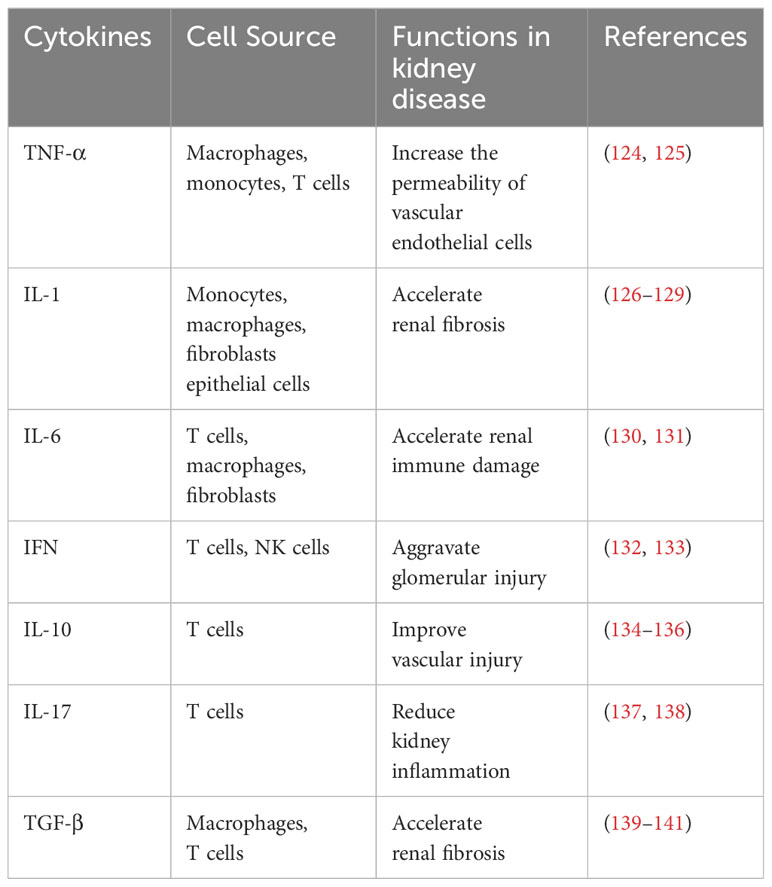
Table 1 Cytokines involved in HTN pathogenesis.
TNF-α, a typical macrophage factor appearing earliest during the inflammatory response, is another cytokine released by immune cells (142). TNF-α activates neutrophils and lymphocytes, increases the permeability of vascular endothelial cells, regulates tissue metabolism, and promotes cytokine synthesis and release (143, 144). When inflammatory lesions occur in the kidney, mononuclear macrophages are infiltrated and can express and secrete large amounts of TNF-α, promoting the secretion of IL-6 by the tethered cells. Many experimental studies have found that TNF-α contributes to the proliferation of tethered cells and stroma and the widening of tethered zones, which promotes the onset and development of inflammation. For example, TNF deficiency in mice attenuates the Ang II-induced hypertensive response and renal injury (97, 116, 124). TNF is toxic to glomerular epithelial cells (125, 145), regulates blood pressure, and ameliorates the extent of renal injury (146–149). Inflammatory factors such as TNF-α, IL-1β and MCP-1 were significantly expressed in the kidney of SHR (150). In humans, the effect of TNF on blood pressure is more complex. For example, in patients with heart failure, TNF antagonism does not improve blood pressure or symptoms (151, 152).
In contrast, in animal models of CKD, TNF elevates renal and systemic FGF23 levels, which may be involved in CKD (153). Similarly, in an animal model of diabetic nephropathy, it was found that macrophage-produced TNF-α plays a role in diabetic kidney injury, mainly reducing albuminuria, plasma creatinine, histopathological changes, renal macrophage recruitment, and plasma inflammatory cytokine levels (154). In a mouse study (116), TNF in the kidney elevated blood pressure by reducing NO production. In other experiments, TNF-α was downregulated through Cyp2c23 and upregulated through sEH (148), activation of NF-κB-mediated pathways (147), and increased renal cortical NF-kappa B activity (149) that are involved in renal damage caused by multiple causes of hypertension. In rats (146), the direct infusion of TNF inhibitors into the renal interstitium attenuated salt-induced blood pressure elevation and renal damage. Another study confirmed (126) that during prolonged administration of Ang II and hypertension on a high-salt diet, TNF receptor 2 may mediate the inflammatory response to renal injury without activating TNF receptor 1. Therefore, TNF-α is an essential mediator of hypertension and kidney injury.
IL-1 acts as an inflammatory cytokine with two active isoforms, IL-1α and IL-1β, which are recognized by IL-1 receptor. NLRP3 is an inflammatory vesicle that promotes the maturation and secretion of IL-1β and IL-18 (127–129, 155). IL-1β can act on pericytes to accelerate renal fibrosis (156) and activate helper T cell 17 (TH17) cells in the kidneys (157). Activation of NLRP3 inflammatory vesicles causes elevated blood pressure and/or kidney injury (158–161), exacerbating cardiovascular risk (128). The guanidinylation of apolipoprotein C3 (ApoC3) activates NLRP3 inflammatory vesicles in human monocytes (162). Guanidinylated ApoC3 (gApoC3) accumulates in the kidneys and plasma of patients with CKD to promote inflammation. gApoC3 promotes renal fibrosis and impedes vascular regeneration (163). Mechanistically, NLRP3 directly promotes the epithelial-to-mesenchymal transition of renal tubular epithelial cells, independent of inflammatory vesicles, by inducing phosphorylation and activity of SMAD3 (164). In SHR, BCL6 attenuates renal inflammation by negatively regulating NLRP3 transcription (165). In a recent study (91), VEGFC was shown to inhibit the activation of NLRP3 inflammatory vesicles by promoting autophagy to ameliorate sensitized hypertension and nephritis, which may be a potential therapeutic target for HTN. We found that the infusion of exogenous IL-1 promotes urinary sodium excretion (166–168). Further, IL-1 is involved in developing pulmonary or systemic hypertension through several other mechanisms (169, 170). Several studies have also reported the use of IL-1 inhibitors (171). For example, the IL-1 receptor antagonist anakinra attenuates hypertension and renal fibrosis but does not affect inflammation and leukocyte infiltration in saline-induced hypertensive mice (1K/DOCA/salt) (172). Therefore, investigating the renal-specific mechanisms of IL-1 in hypertension is crucial.
IL-6, known for its pro-inflammatory effects, is one of the most studied cytokines in renal disease and plays a role in systemic inflammation (121, 130). IL-6 has been reported to be involved in hypertension-induced kidney injury (131, 173). IL-6 induces B-cell differentiation and antibody production as a pro-inflammatory response, causes T-cell proliferation and differentiation, and regulates the immune response. IL-6 promotes the expression of cell adhesion molecules by inducing cytokine production of acute-phase response proteins, which leads to the aggregation of inflammatory cells. The expression of IL-6 is directly proportional to the number of glomeruli in glomerular disease. In addition, it stimulates the production of platelets by tethered cells activating factors and other inflammatory mediators, exacerbating renal immune damage. Notably, the risk of cardiovascular death, myocardial infarction, and stroke is associated with decreased glomerular filtration rate (eGFR) and elevated IL-6 levels (174). IL-6 levels in CKD are involved in atherosclerosis (175, 176). Notably, the results of these studies are mutually contradictory. Other studies also reported conflicting findings, such as that IL-6 levels at baseline or changes during the observation period do not show a consistent and significant correlation with renal function in older adults (177). Recently, drug studies on IL-6 and IL-6 receptors have also gained momentum (121, 132, 178–180).
IFN is an inflammatory cytokine produced by T lymphocytes and macrophages that enhances sodium transport by activating intrarenal Ang II production to stimulate transport proteins (133) indirectly. Therefore, IFN deficiency attenuates the response to Ang II-induced chronic hypertension (98) and exacerbates glomerular injury (181). IRF is a multifunctional transcription factor, and IRF-4 deficiency improves renal function, albuminuria, and attenuates renal injury and fibrosis but does not affect blood pressure (182). IRF-4 is a multifunctional transcription factor.
Cytokines in the kidneys act indirectly by inducing other inflammatory mediators and their direct effects on tissue cells. NF-κ B is a critical transcription factor in regulating the inflammatory response and the expression of many pro-inflammatory genes (183). The complex hypertensive environment (i.e., hypertension, oxidative stress, glomerular leakage of free fatty acids, and activated RAS) can exacerbate renal inflammation and the progression of HTN by activating the NF-κB signaling system (184, 185). During glomerular disease progression, NF-κB activation triggers an inflammatory cascade that produces inflammatory factors, such as IL-10 and MCP-1.Studies have found (186) that TGF-β/SMAD and NF-κB signaling pathways play an important role in HTN, and SMAD7, as a downstream inhibitor of these two pathways, can inhibit proteinuria and serum creatinine, and improve glomerular filtration rate, thereby alleviating the condition of HTN. In a recent study (187), ubiquitin-specific protease 25 (USP25) inhibited TGF-β-induced SMAD4 ubiquitination, which reduced the expression of renal fibrosis and renal injury related genes and alleviated renal injury caused by HTN. It has also been confirmed (134) that exercise training may prevent and alleviate renal abnormalities in hypertensive patients by down-regulating TGF-β signaling. Renal fibrosis is an important pathological feature of hypertensive renal injury, which is related to the up-regulation of pro-fibrotic factor TGF-β1 (150).
IL-10 is strongly associated with inflammation in the kidney (135, 136, 188). For example, IL-10 ameliorates hypertension-induced renal and vascular injury (189), and genetic polymorphisms in IL-10 are also protective against diabetic nephropathy (139). MCP-1 promotes chemotaxis of monocytes or macrophages to reach damaged renal tissues, resulting in sustained cytokine activation and localized protease disruption of the renal basement membrane. MCP-1 increases the production of IL-8 and TGF-β1 by affecting the inflammatory response of diseased renal tissues. TGF-β, a cytokine that can regulate cellular proliferation, plays a role in the process of membranous nephropathy, diabetic nephropathy, and renal fibrosis (140, 141, 190).
Chemokine ligand 16 (CXCL16) plays a role in Ang II-induced renal injury and fibrosis by regulating macrophage and T cell infiltration and fibroblast aggregation (191). During RAS-activated hypertension, the chemokine CCL5 attenuates renal injury and fibrosis by reducing the number of infiltrating macrophages in the kidneys (137).
IL-17 is also involved in Ang II-induced hypertension and vascular function (138). In a mouse study (192), IL-17A and IL-17RA antibodies (but not IL-17F) reduced blood pressure and attenuated renal and vascular inflammation. Notably, in other experimental models of kidney injury, IL-17 deficiency exacerbated disease progression (193–195). In a recent transcriptomics study (196), genes upregulated in the renal tubules of patients with HTN were associated with IFN-γ, NF-κB, IL-12, and Wnt signaling pathways, all of which are involved in the inflammatory response, providing a broader perspective on the pathogenesis and treatment of human HTN. Although the number of immune cells is small, the released cytokines play key roles in renal changes involved in fibrosis, glomerular injury, and sodium transport (58).
The signaling pathways between oxidative stress and HTN are complex in the pathophysiology of HTN (197, 198). In the kidney, ROS exists in arterioles, glomerular and tubular cells and podocytes, etc. Vasoactive substances and metabolic factors stimulate the production of cellular inflammation and ROS, inducing NADPH oxidase or mitochondrial production (199). The endoplasmic reticulum (ER) is a key player in the redox pathophysiology of the cardiovascular system that maintains protein homeostasis. ER stress (ERS) is typically characterized by impaired protein and lipid synthesis and disturbed intracellular calcium levels (200–202). Excessive ERS damages glomerular mesangial cells, podocytes, and tubular epithelial cells (203–205). However, ERS activity may impair renal function and worsen renal damage during CKD development. Oxidative stress affects T-cell activation and function (206). Septin4 is a non-histone protein that promotes apoptosis, is regulated by SIRT2, and is a marker of organ damage (207–210). SIRT2 deficiency increases oxidative stress and alters mitochondrial morphology (211). Notably, SIRT2–septin4 axis prevents hypertensive kidney injury by attenuating apoptosis and oxidative stress. SIRT2 attenuated oxidative stress in podocytes through deacetylation of septin4-K174, ameliorating hypertension-induced kidney injury in mice. Therefore, septin4-K174R (mimicking deacetylation via SIRT2) may be a new target for alleviating renal injury in hypertension (47), which may help design precise therapeutic regimens and develop targeted drugs in the future. Some studies have demonstrated that SIRT3 is involved in hypertensive kidney injury by inhibiting the epithelial–mesenchymal transition (EMT) and ameliorating hypertension-induced kidney injury in mice (212, 213). Another study in rats (214) suggested that cagliflozin regulates renal EMT and oxidative stress through the SIRT3 pathway, thereby inhibiting high-salt diet-induced hypertensive renal fibrosis.
Modern medical research has found that many glomerular diseases are immune-mediated inflammatory lesions. The involvement of various inflammatory mediators, such as inflammatory cytokines and complement factors based on immune response, leads to glomerular injury and the development of corresponding clinical symptoms (215). The immune system plays an important role in developing hypertension and related endpoints, such as the associated end-organ damage. Sodium provides a strong stimulus for the development of hypertension. High concentrations of extracellular natriuretic ions have been reported to directly activate the inflammatory response in APCs, leading to hypertension, T-cell activation, and ultimately vascular and renal injury (216). APCs respond to hypertension-stimulated inflammatory processes, and DCs, macrophages, and B cells are the classical APCs of the immune system, which coordinate the immune response by activating T cells through antigen-MHT receptor complexes and expressing cytokines, among others (217). Large amounts of sodium-activated DCs produce IL-1β and promote the production of IL-17A and IFN-γ by T cells, which in turn lead to mediated hypertension and end-organ dysfunction, indicating that the mechanistic link between salt, inflammation, and hypertension includes increased oxidative stress and IsoLG production in DCs (218). Macrophages and T cells play important roles in developing HTN-induced kidney disease (219). Pro-inflammatory cytokines, tumor necrosis factor-α, IL-6, and IL-1β have been implicated in the pathogenesis of Ang II-induced target organ damage (220). For example, as mentioned previously, the deletion of CXCL16 affects the expression of pro-inflammatory cytokines in the kidney. TGF-β1, a key pro-fibrotic cytokine, causes renal damage and tubulointerstitial fibrosis. One study showed that CXCL16 deficiency caused a significant reduction in Ang II-induced TGF-β1 gene expression, which may be an alternative mechanism for developing renal fibrosis.
GPR97, a member of the G protein-coupled receptor subfamily, modulates the inflammatory response (221, 222). GPR97 exacerbates AKI by regulating Sema3A. GPR97 was expressed in tubular epithelial cells in mice with AKI (223). Recent animal experiments have also found (224) that GPR97 causes renal injury and tubulointerstitial fibrosis (TIF) in deoxycorticosterone acetate (DOCA)/salt-induced hypertensive mice by regulating TGF-β signaling. Therefore, GPR97 leads to TIF in patients with hypertension, indicating that it may be a novel therapeutic target for delaying or attenuating renal injury in hypertension.
HTN, as a cross-discipline, presents difficulties and challenges in diagnosis and intervention. Patients with HTN should monitor and manage their blood pressure and set appropriate control goals in their daily lives, make assessments before treatment, choose appropriate antihypertensive medications to actively prevent complications (225–229). Moreover, lifestyle modification is important to improve the progression and prognosis of HTN. In addition to genetic and environmental interactions, appropriate lifestyle changes, such as weight loss, healthy diet, reduction of dietary sodium, increased physical activity, and cessation of smoking and excessive alcohol consumption, should be implemented according to the recommendations of the hypertension guidelines (230). However, these non-pharmacological interventions alone are insufficient for HTN treatment. For patients with HTN, salt restriction should be the priority as patients with advanced renal disease who excrete a low sodium load exhibit significant salt and water retention (227, 231). Simultaneously, attention should be paid to the intake of adequate amounts of vegetables and fruits that can protect kidneys from renal injury (232).
In conclusion, inflammation mediates the end-organ damage associated with hypertension. Although reducing blood pressure is crucial, preventing the localized inflammation accompanying this disease, resulting in damage to the vital target organs, should be prioritized (122). In recent years, an increasing number of studies have been published on the pathogenesis of HTN; however, the specific mechanisms are still not fully understood. This review was conducted based on previous studies on the role of immune cells, inflammatory cytokines, and related components in HTN. We analyzed the results at the molecular level to reveal some of the relevant mechanisms to better understand the development of HTN. Future research needs to fill the existing gaps and add more evidence and therapeutic strategies for the prevention and treatment of HTN and avoiding hypertension-related complications.
Hypertension and kidney disease are strongly co-related, and this link should be used effectively to prevent hypertension or treat it at an early stage. Although hypertension is very common today, complications due to hypertension are not encouraging, and many problems remain unresolved. In future studies, antihypertensive drugs, immunosuppressive drugs, and life management must be considered to identify more effective therapeutic targets for HTN.
Although this review focused on inflammatory markers, it is insufficient because the relationship between immunity and inflammation is complex. As the next step, the mechanism of HTN must be explored to alleviate or solve renal injury caused by hypertension.
XH: Writing – original draft. YL: Writing – review & editing. DH: Conceptualization, Writing – review & editing. YG: Writing – review & editing. XZ: Writing – review & editing. BY: Methodology, Writing – review & editing. JL: Writing – review & editing. XW: Writing – review & editing. KY: Writing – review & editing. JW: Supervision, Writing – review & editing. QL: Resources, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by the National Natural Science Foundation of China (grant no. 82074553).
The authors thank the patients for consenting to the publication of this report.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Visseren FLJ, Mach F, Smulders YM, Carballo D, Koskinas KC, Bäck M, et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J. (2021) 42:3227–337. doi: 10.1093/eurheartj/ehab484
2. Poulter NR, Prabhakaran D, Caulfield M. Hypertension. Lancet (London England). (2015) 386:801–12. doi: 10.1016/S0140-6736(14)61468-9
3. NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. Lancet (London England). (2021) 398:957–80. doi: 10.1016/S0140-6736(21)01330-1
4. Drummond GR, Vinh A, Guzik TJ, Sobey CG. Immune mechanisms of hypertension. Nat Rev Immunol. (2019) 19:517–32. doi: 10.1038/s41577-019-0160-5
5. Pan X, Shao Y, Wu F, Wang Y, Xiong R, Zheng J, et al. FGF21 prevents angiotensin II-induced hypertension and vascular dysfunction by activation of ACE2/angiotensin-(1-7) axis in mice. Cell Metab. (2018) 27:1323–37.e5. doi: 10.1016/j.cmet.2018.04.002
6. Brouwers S, Sudano I, Kokubo Y, Sulaica EM. Arterial hypertension. Lancet (London England). (2021) 398:249–61. doi: 10.1016/S0140-6736(21)00221-X
7. Carey RM, Moran AE, Whelton PK. Treatment of hypertension: A review. JAMA. (2022) 328:1849–61. doi: 10.1001/jama.2022.19590
8. Kakitapalli Y, Ampolu J, Madasu SD, Sai Kumar MLS. Detailed review of chronic kidney disease. Kidney Dis (Basel Switzerland). (2020) 6:85–91. doi: 10.1159/000504622
9. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet (London England). (2018) 392:1789–858. doi: 10.1016/S0140-6736(18)32279-7
10. Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol. (2006) 17:17–25. doi: 10.1681/ASN.2005070757
11. Coresh J, Byrd-Holt D, Astor BC, Briggs JP, Eggers PW, Lacher DA, et al. Chronic kidney disease awareness, prevalence, and trends among U.S. adults, 1999 to 2000. J Am Soc Nephrol. (2005) 16:180–8. doi: 10.1681/ASN.2004070539
12. Charles C, Ferris AH. Chronic kidney disease. Primary Care. (2020) 47:585–95. doi: 10.1016/j.pop.2020.08.001
13. Cai A, Calhoun DA. Resistant hypertension: an update of experimental and clinical findings. Hypertension (Dallas Tex: 1979). (2017) 70:5–9. doi: 10.1161/HYPERTENSIONAHA.117.08929
14. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines (Accessed 23rd October 2023).
15. Hart PD, Bakris GL. Hypertensive nephropathy: prevention and treatment recommendations. Expert Opin Pharmacother. (2010) 11:2675–86. doi: 10.1517/14656566.2010.485612
16. Johansen KL, Chertow GM, Gilbertson DT, Ishani A, Israni A, Ku E, et al. US renal data system 2022 annual data report: epidemiology of kidney disease in the United States. Am J Kidney Dis. (2023) 81:A8–A11. doi: 10.1053/j.ajkd.2022.12.001
17. Lopez Gelston CA, Mitchell BM. Recent advances in immunity and hypertension. Am J Hypertens. (2017) 30:643–52. doi: 10.1093/ajh/hpx011
18. Bomfim GF, Rodrigues FL, Carneiro FS. Are the innate and adaptive immune systems setting hypertension on fire? Pharmacol Res. (2017) 117:377–93. doi: 10.1016/j.phrs.2017.01.010
19. Lin L, Ren J, Wang C, Mei M, Zheng L, Yang J. A set of urinary peptides can predict early renal damage in primary hypertension. J Hypertens. (2023) 41:1653–60. doi: 10.1097/HJH.0000000000003539
20. Mwasongwe S, Min YI, Booth JN, Katz R, Sims M, Correa A, et al. Masked hypertension and kidney function decline: the Jackson Heart Study. J Hypertens. (2018) 36:1524–32. doi: 10.1097/HJH.0000000000001727
21. Ameer OZ. Hypertension in chronic kidney disease: What lies behind the scene. Front Pharmacol. (2022) 13:949260. doi: 10.3389/fphar.2022.949260
22. Kretzler M, Koeppen-Hagemann I, Kriz W. Podocyte damage is a critical step in the development of glomerulosclerosis in the uninephrectomised-desoxycorticosterone hypertensive rat. Virchows Archiv: Int J Pathol. (1994) 425:181–93. doi: 10.1007/BF00230355
23. Wei SY, Wang YX, Zhang QF, Zhao SL, Diao TT, Li JS, et al. Multiple mechanisms are involved in salt-sensitive hypertension-induced renal injury and interstitial fibrosis. Sci Rep. (2017) 7:45952. doi: 10.1038/srep45952
24. Nagase M, Shibata S, Yoshida S, Nagase T, Gotoda T, Fujita T. Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension (Dallas Tex: 1979). (2006) 47:1084–93. doi: 10.1161/01.HYP.0000222003.28517.99
25. Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. (2006) 69:2131–47. doi: 10.1038/sj.ki.5000410
26. Zamami R, Kohagura K, Miyagi T, Kinjyo T, Shiota K, Ohya Y. Modification of the impact of hypertension on proteinuria by renal arteriolar hyalinosis in nonnephrotic chronic kidney disease. J Hypertens. (2016) 34:2274–9. doi: 10.1097/HJH.0000000000001091
27. Sata Y, Head GA, Denton K, May CN, Schlaich MP. Role of the sympathetic nervous system and its modulation in renal hypertension. Front Med. (2018) 5:82. doi: 10.3389/fmed.2018.00082
28. Campese VM, Mitra N, Sandee D. Hypertension in renal parenchymal disease: why is it so resistant to treatment? Kidney Int. (2006) 69:967–73. doi: 10.1038/sj.ki.5000177
29. DiBona GF, Kopp UC. Neural control of renal function. Physiol Rev. (1997) 77:75–197. doi: 10.1152/physrev.1997.77.1.75
30. Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. (2007) 59:251–87. doi: 10.1124/pr.59.3.3
31. Kashiwagi M, Shinozaki M, Hirakata H, Tamaki K, Hirano T, Tokumoto M, et al. Locally activated renin-angiotensin system associated with TGF-beta1 as a major factor for renal injury induced by chronic inhibition of nitric oxide synthase in rats. J Am Soc Nephrol: JASN. (2000) 11:616–24. doi: 10.1681/ASN.V114616
32. Liu Y, Jiang Y, Li W, Han C, Qi Z. MicroRNA and mRNA analysis of angiotensin II-induced renal artery endothelial cell dysfunction. Exp Ther Med. (2020) 19:3723–37. doi: 10.3892/etm.2020.8613
33. Durvasula RV, Shankland SJ. The renin-angiotensin system in glomerular podocytes: mediator of glomerulosclerosis and link to hypertensive nephropathy. Curr Hypertens Rep. (2006) 8:132–8. doi: 10.1007/s11906-006-0009-8
34. Keller G, Zimmer G, Mall G, Ritz E, Amann K. Nephron number in patients with primary hypertension. N Engl J Med. (2003) 348:101–8. doi: 10.1056/NEJMoa020549
35. Hill GS, Heudes D, Jacquot C, Gauthier E, Bariéty J. Morphometric evidence for impairment of renal autoregulation in advanced essential hypertension. Kidney Int. (2006) 69:823–31. doi: 10.1038/sj.ki.5000163
36. Zhou D, Wang Y, Gui Y, Fu H, Zhou S, Wang Y, et al. Non-canonical Wnt/calcium signaling is protective against podocyte injury and glomerulosclerosis. Kidney Int. (2022) 102:96–107. doi: 10.1016/j.kint.2022.02.029
37. Nagata M. Podocyte injury and its consequences. Kidney Int. (2016) 89:1221–30. doi: 10.1016/j.kint.2016.01.012
38. Sun H, Li H, Yan J, Wang X, Xu M, Wang M, et al. Loss of CLDN5 in podocytes deregulates WIF1 to activate WNT signaling and contributes to kidney disease. Nat Commun. (2022) 13:1600. doi: 10.1038/s41467-022-29277-6
39. Seccia TM, Caroccia B, Calò LA. Hypertensive nephropathy. Moving from classic to emerging pathogenetic mechanisms. J Hypertens. (2017) 35:205–12. doi: 10.1097/HJH.0000000000001170
40. Fu EL, Evans M, Clase CM, Tomlinson LA, van Diepen M, Dekker FW, et al. Stopping renin-angiotensin system inhibitors in patients with advanced CKD and risk of adverse outcomes: A nationwide study. J Am Soc Nephrol: JASN. (2021) 32:424–35. doi: 10.1681/ASN.2020050682
41. Angiotensin II as an inflammatory mediator: evolving concepts in the role of the renin angiotensin system in the failing heart. Available online at: https://pubmed.ncbi.nlm.nih.gov/12085982/ (Accessed 26th October 2023).
42. Ruiz-Ortega M, Rupérez M, Esteban V, Rodríguez-Vita J, Sánchez-López E, Carvajal G, et al. Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol Dialysis Transplant. (2006) 21:16–20. doi: 10.1093/ndt/gfi265
43. Ma Y, Yang Q, Zhong Z, Liang W, Zhang L, Yang Y, et al. Role of c-Abl and nephrin in podocyte cytoskeletal remodeling induced by angiotensin II. Cell Death Dis. (2018) 9:185. doi: 10.1038/s41419-017-0225-y
44. Yang Q, Hu J, Yang Y, Chen Z, Feng J, Zhu Z, et al. Sirt6 deficiency aggravates angiotensin II-induced cholesterol accumulation and injury in podocytes. Theranostics. (2020) 10:7465–79. doi: 10.7150/thno.45003
45. Naik AS, Le D, Aqeel J, Wang SQ, Chowdhury M, Walters LM, et al. Podocyte stress and detachment measured in urine are related to mean arterial pressure in healthy humans. Kidney Int. (2020) 98:699–707. doi: 10.1016/j.kint.2020.03.038
46. Yu D, Petermann A, Kunter U, Rong S, Shankland SJ, Floege J. Urinary podocyte loss is a more specific marker of ongoing glomerular damage than proteinuria. J Am Soc Nephrol. (2005) 16:1733–41. doi: 10.1681/ASN.2005020159
47. Zhang Y, Zhang N, Zou Y, Song C, Cao K, Wu B, et al. Deacetylation of septin4 by SIRT2 (Silent mating type information regulation 2 homolog-2) mitigates damaging of hypertensive nephropathy. Circ Res. (2023) 132:601–24. doi: 10.1161/CIRCRESAHA.122.321591
48. Hill GS. Hypertensive nephrosclerosis. Curr Opin Nephrol Hypertens. (2008) 17:266–70. doi: 10.1097/MNH.0b013e3282f88a1f
49. Eremina V, Quaggin SE. The role of VEGF-A in glomerular development and function. Curr Opin Nephrol Hypertens. (2004) 13:9–15. doi: 10.1097/00041552-200401000-00002
50. Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. (2005) 54:1626–34. doi: 10.2337/diabetes.54.6.1626
51. Costantino VV, Gil Lorenzo AF, Bocanegra V, Vallés PG. Molecular mechanisms of hypertensive nephropathy: renoprotective effect of losartan through hsp70. Cells. (2021) 10:3146. doi: 10.3390/cells10113146
52. Stanchev S, Stamenov N, Kirkov V, Dzhambazova E, Nikolov D, Paloff A. Differential collagen expression in kidney and heart during hypertension. Bratislava Med J. (2020) 121:73–8. doi: 10.4149/BLL_2020_011
53. Chen L, Fukuda N, Matsumoto T, Abe M. Role of complement 3 in the pathogenesis of hypertension. Hypertens Res. (2020) 43:255–62. doi: 10.1038/s41440-019-0371-y
54. Cui J, Wan J, You D, Zou Z, Chen Y, Li Z, et al. Interstitial complement C3 activation and macrophage infiltration in patients with hypertensive nephropathy. Clin Nephrol. (2017) 88:328–37. doi: 10.5414/CN109154
55. Welker P, Krämer S, Groneberg DA, Neumayer HH, Bachmann S, Amann K, et al. Increased mast cell number in human hypertensive nephropathy. Am J Physiol Renal Physiol. (2008) 295:F1103–1109. doi: 10.1152/ajprenal.00374.2007
56. White FN, Grollman A. AUTOIMMUNE FACTORS ASSOCIATED WITH INFARCTION OF THE KIDNEY. Nephron. (1964) 1:93–102. doi: 10.1159/000179322
57. Crowley SD, Jeffs AD. Targeting cytokine signaling in salt-sensitive hypertension. Am J Physiol Renal Physiol. (2016) 311:F1153–8. doi: 10.1152/ajprenal.00273.2016
58. Petreski T, Piko N, Ekart R, Hojs R, Bevc S. Review on inflammation markers in chronic kidney disease. Biomedicines. (2021) 9:182. doi: 10.3390/biomedicines9020182
59. Zhang L, Wang F, Wang L, Wang W, Liu B, Liu J, et al. Prevalence of chronic kidney disease in China: a cross-sectional survey. Lancet (London England). (2012) 379:815–22. doi: 10.1016/S0140-6736(12)60033-6
60. Venkatraghavan L, Tan TP, Mehta J, Arekapudi A, Govindarajulu A, Siu E. Neutrophil Lymphocyte Ratio as a predictor of systemic inflammation - A cross-sectional study in a pre-admission setting. F1000Research. (2015) 4:123. doi: 10.12688/f1000research.6474.1
61. Pang L, Ding Z, Chai H, Shuang W. The causal relationship between immune cells and different kidney diseases: A Mendelian randomization study. Open Med. (2023) 18:20230877. doi: 10.1515/med-2023-0877
62. Rogers NM, Ferenbach DA, Isenberg JS, Thomson AW, Hughes J. Dendritic cells and macrophages in the kidney: a spectrum of good and evil. Nat Rev Nephrol. (2014) 10:625–43. doi: 10.1038/nrneph.2014.170
63. Higaki A, Mogi M. Dendritic cells as potential initiators of immune-mediated hypertensive disorders. Hypertens Res. (2022) 45:527–9. doi: 10.1038/s41440-021-00830-y
64. Brähler S, Zinselmeyer BH, Raju S, Nitschke M, Suleiman H, Saunders BT, et al. Opposing roles of dendritic cell subsets in experimental GN. J Am Soc Nephrol. (2018) 29:138–54. doi: 10.1681/ASN.2017030270
65. Jiao J, Dragomir AC, Kocabayoglu P, Rahman AH, Chow A, Hashimoto D, et al. Central role of conventional dendritic cells in regulation of bone marrow release and survival of neutrophils. J Immunol (Baltimore Md.: 1950). (2014) 192:3374–82. doi: 10.4049/jimmunol.1300237
66. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Sci (New York N.Y.). (2008) 322:1097–100. doi: 10.1126/science.1164206
67. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets. vivo Sci (New York N.Y.). (2007) 315:107–11. doi: 10.1126/science.1136080
68. Hochheiser K, Engel DR, Hammerich L, Heymann F, Knolle PA, Panzer U, et al. Kidney dendritic cells become pathogenic during crescentic glomerulonephritis with proteinuria. J Am Soc Nephrol. (2011) 22:306–16. doi: 10.1681/ASN.2010050548
69. Yatim KM, Gosto M, Humar R, Williams AL, Oberbarnscheidt MH. Renal dendritic cells sample blood-borne antigen and guide T-cell migration to the kidney by means of intravascular processes. Kidney Int. (2016) 90:818–27. doi: 10.1016/j.kint.2016.05.030
70. Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, et al. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. (2010) 122:2529–37. doi: 10.1161/CIRCULATIONAHA.109.930446
71. Kirabo A, Fontana V, de Faria APC, Loperena R, Galindo CL, Wu J, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. (2014) 124:4642–56. doi: 10.1172/JCI74084
72. Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, et al. CD70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. (2016) 118:1233–43. doi: 10.1161/CIRCRESAHA.115.308111
73. Lu X, Rudemiller NP, Privratsky JR, Ren J, Wen Y, Griffiths R, et al. Classical dendritic cells mediate hypertension by promoting renal oxidative stress and fluid retention. Hypertension (Dallas Tex: 1979). (2020) 75:131–8. doi: 10.1161/HYPERTENSIONAHA.119.13667
74. Viehmann SF, Böhner AMC, Kurts C, Brähler S. The multifaceted role of the renal mononuclear phagocyte system. Cell Immunol. (2018) 330:97–104. doi: 10.1016/j.cellimm.2018.04.009
75. Rodriguez-Iturbe B, Pons H, Johnson RJ. Role of the immune system in hypertension. Physiol Rev. (2017) 97:1127–64. doi: 10.1152/physrev.00031.2016
76. Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. (2017) 18:234. doi: 10.1186/s13059-017-1362-4
77. Wise AF, Williams TM, Kiewiet MBG, Payne NL, Siatskas C, Samuel CS, et al. Human mesenchymal stem cells alter macrophage phenotype and promote regeneration via homing to the kidney following ischemia-reperfusion injury. Am J Physiol Renal Physiol. (2014) 306:F1222–1235. doi: 10.1152/ajprenal.00675.2013
78. Lech M, Gröbmayr R, Ryu M, Lorenz G, Hartter I, Mulay SR, et al. Macrophage phenotype controls long-term AKI outcomes–kidney regeneration versus atrophy. J Am Soc Nephrol. (2014) 25:292–304. doi: 10.1681/ASN.2013020152
79. Ma R, Jiang W, Li Z, Sun Y, Wei Z. Intrarenal macrophage infiltration induced by T cells is associated with podocyte injury in lupus nephritis patients. Lupus. (2016) 25:1577–86. doi: 10.1177/0961203316646861
80. Dai X, Mao C, Lan X, Chen H, Li M, Bai J, et al. Acute Penicillium marneffei infection stimulates host M1/M2a macrophages polarization in BALB/C mice. BMC Microbiol. (2017) 17:177. doi: 10.1186/s12866-017-1086-3
81. Tang PMK, Nikolic-Paterson DJ, Lan HY. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol. (2019) 15:144–58. doi: 10.1038/s41581-019-0110-2
82. Das A, Sinha M, Datta S, Abas M, Chaffee S, Sen CK, et al. Monocyte and macrophage plasticity in tissue repair and regeneration. Am J Pathol. (2015) 185:2596–606. doi: 10.1016/j.ajpath.2015.06.001
83. Rosenberger CM, Finlay BB. Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat Rev Mol Cell Biol. (2003) 4:385–96. doi: 10.1038/nrm1104
84. Gordon S, Plüddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol. (2017) 15:53. doi: 10.1186/s12915-017-0392-4
85. Huen SC, Cantley LG. Macrophage-mediated injury and repair after ischemic kidney injury. Pediatr Nephrol (Berlin Germany). (2015) 30:199–209. doi: 10.1007/s00467-013-2726-y
86. Saad A, Herrmann SMS, Crane J, Glockner JF, McKusick MA, Misra S, et al. Stent revascularization restores cortical blood flow and reverses tissue hypoxia in atherosclerotic renal artery stenosis but fails to reverse inflammatory pathways or glomerular filtration rate. Circ Cardiovasc Interv. (2013) 6:428–35. doi: 10.1161/CIRCINTERVENTIONS.113.000219
87. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. (2017) 214:2387–404. doi: 10.1084/jem.20162152
88. Gloviczki ML, Keddis MT, Garovic VD, Friedman H, Herrmann S, McKusick MA, et al. TGF expression and macrophage accumulation in atherosclerotic renal artery stenosis. Clin J Am Soc Nephrol. (2013) 8:546–53. doi: 10.2215/CJN.06460612
89. Cavalcante PAM, Alenina N, Budu A, Freitas-Lima LC, Alves-Silva T, Agudelo JSH, et al. Nephropathy in hypertensive animals is linked to M2 macrophages and increased expression of the YM1/chi3l3 protein. Mediators Inflamm. (2019) 2019:9086758. doi: 10.1155/2019/9086758
90. Wu Q, Meng W, Zhu B, Chen X, Fu J, Zhao C, et al. VEGFC ameliorates salt-sensitive hypertension and hypertensive nephropathy by inhibiting NLRP3 inflammasome via activating VEGFR3-AMPK dependent autophagy pathway. Cell Mol Life Sci. (2023) 80:327. doi: 10.1007/s00018-023-04978-3
91. Morton J, Coles B, Wright K, Gallimore A, Morrow JD, Terry ES, et al. Circulating neutrophils maintain physiological blood pressure by suppressing bacteria and IFNgamma-dependent iNOS expression in the vasculature of healthy mice. Blood. (2008) 111:5187–94. doi: 10.1182/blood-2007-10-117283
92. Lu X, Crowley SD. Actions of immune cells in the hypertensive kidney. Curr Opin Nephrol Hypertens. (2020) 29:515–22. doi: 10.1097/MNH.0000000000000635
93. Sanders PW, Wang PX. Activation of the Fas/Fas ligand pathway in hypertensive renal disease in Dahl/Rapp rats. BMC nephrol. (2002) 3:1. doi: 10.1186/1471-2369-3-1
94. Sommers SC, Relman AS, Smithwick RH. Histologic studies of kidney biopsy specimens from patients with hypertension. Am J Pathol. (1958) 34:685–715.
95. Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regulatory Integr Comp Physiol. (2010) 298:R1089–1097. doi: 10.1152/ajpregu.00373.2009
96. Sriramula S, Haque M, Majid DSA, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension (Dallas Tex: 1979). (2008) 51:1345–51. doi: 10.1161/HYPERTENSIONAHA.107.102152
97. Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR, et al. Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end-organ inflammation. J Clin Invest. (2015) 125:1189–202. doi: 10.1172/JCI76327
98. Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, et al. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension (Dallas Tex: 1979). (2014) 64:1108–15. doi: 10.1161/HYPERTENSIONAHA.114.04147
99. Liu Y, Rafferty TM, Rhee SW, Webber JS, Song L, Ko B, et al. CD8+ T cells stimulate Na-Cl co-transporter NCC in distal convoluted tubules leading to salt-sensitive hypertension. Nat Commun. (2017) 8:14037. doi: 10.1038/ncomms14037
100. Shao J, Nangaku M, Miyata T, Inagi R, Yamada K, Kurokawa K, et al. Imbalance of T-cell subsets in angiotensin II-infused hypertensive rats with kidney injury. Hypertension (Dallas Tex: 1979). (2003) 42:31–8. doi: 10.1161/01.HYP.0000075082.06183.4E
101. Scheinecker C, Göschl L, Bonelli M. Treg cells in health and autoimmune diseases: New insights from single cell analysis. J Autoimmunity. (2020) 110:102376. doi: 10.1016/j.jaut.2019.102376
102. Zhu X, Li S, Zhang Q, Zhu D, Xu Y, Zhang P, et al. Correlation of increased Th17/Treg cell ratio with endoplasmic reticulum stress in chronic kidney disease. Medicine. (2018) 97:e10748. doi: 10.1097/MD.0000000000010748
103. Lu J, Zhang J, Chen M, Chen C, Li Z, Liao P. Regulatory T cells as a novel candidate for cell-based therapy in kidney disease. Front Physiol. (2020) 11:621. doi: 10.3389/fphys.2020.00621
104. Zhang N, Schröppel B, Lal G, Jakubzick C, Mao X, Chen D, et al. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity. (2009) 30:458–69. doi: 10.1016/j.immuni.2008.12.022
105. Mahajan D, Wang Y, Qin X, Wang Y, Zheng G, Wang YM, et al. CD4+CD25+ regulatory T cells protect against injury in an innate murine model of chronic kidney disease. J Am Soc Nephrol. (2006) 17:2731–41. doi: 10.1681/ASN.2005080842
106. Higaki A, Mahmoud AUM, Paradis P, Schiffrin EL. Role of interleukin-23/interleukin-17 axis in T-cell-mediated actions in hypertension. Cardiovasc Res. (2021) 117:1274–83. doi: 10.1093/cvr/cvaa257
107. Wang Z, Wang J, Yang P, Song X, Li Y. Elevated Th17 cell proportion, related cytokines and mRNA expression level in patients with hypertension-mediated organ damage: a case control study. BMC Cardiovasc Disord. (2022) 22:257. doi: 10.1186/s12872-022-02698-3
108. Taylor LE, Gillis EE, Musall JB, Baban B, Sullivan JC. High-fat diet-induced hypertension is associated with a proinflammatory T cell profile in male and female Dahl salt-sensitive rats. Am J Physiol Heart Circ Physiol. (2018) 315:H1713–23. doi: 10.1152/ajpheart.00389.2018
109. Mehrotra P, Patel JB, Ivancic CM, Collett JA, Basile DP. Th-17 cell activation in response to high salt following acute kidney injury is associated with progressive fibrosis and attenuated by AT-1R antagonism. Kidney Int. (2015) 88:776–84. doi: 10.1038/ki.2015.200
110. Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M, et al. Angiotensin II-induced vascular dysfunction depends on interferon-γ-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler Thromb Vasc Biol. (2013) 33:1313–9. doi: 10.1161/ATVBAHA.113.301437
111. Travis OK, White D, Pierce WA, Ge Y, Stubbs CY, Spradley FT, et al. Chronic infusion of interleukin-17 promotes hypertension, activation of cytolytic natural killer cells, and vascular dysfunction in pregnant rats. Physiol Rep. (2019) 7:e14038. doi: 10.14814/phy2.14038
112. Wang HX, Li WJ, Hou CL, Lai S, Zhang YL, Tian C, et al. CD1d-dependent natural killer T cells attenuate angiotensin II-induced cardiac remodelling via IL-10 signalling in mice. Cardiovasc Res. (2019) 115:83–93. doi: 10.1093/cvr/cvy164
113. Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, et al. Obligatory role for B cells in the development of angiotensin II-dependent hypertension. Hypertension (Dallas Tex: 1979). (2015) 66:1023–33. doi: 10.1161/HYPERTENSIONAHA.115.05779
114. Du X, Ma X, Tan Y, Shao F, Li C, Zhao Y, et al. B cell-derived anti-beta 2 glycoprotein I antibody mediates hyperhomocysteinemia-aggravated hypertensive glomerular lesions by triggering ferroptosis. Signal Transduct Target Ther. (2023) 8:103. doi: 10.1038/s41392-023-01313-x
115. Zhang J, Patel MB, Griffiths R, Mao A, Song Ys, Karlovich NS, et al. Tumor necrosis factor-α produced in the kidney contributes to angiotensin II-dependent hypertension. Hypertension (Dallas Tex: 1979). (2014) 64:1275–81. doi: 10.1161/HYPERTENSIONAHA.114.03863
116. Mattson DL. Immune mechanisms of salt-sensitive hypertension and renal end-organ damage. Nat Rev Nephrol. (2019) 15:290–300. doi: 10.1038/s41581-019-0121-z
117. Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, et al. Stimulation of lymphocyte responses by angiotensin II promotes kidney injury in hypertension. Am J Physiol Renal Physiol. (2008) 295:F515–524. doi: 10.1152/ajprenal.00527.2007
118. Ren J, Crowley SD. Role of T-cell activation in salt-sensitive hypertension. Am J Physiol Heart Circulatory Physiol. (2019) 316:H1345–53. doi: 10.1152/ajpheart.00096.2019
119. Yazdi AS, Ghoreschi K. The interleukin-1 family. Adv Exp Med Biol. (2016) 941:21–9. doi: 10.1007/978-94-024-0921-5_2
120. Rose-John S. Interleukin-6 family cytokines. Cold Spring Harbor Perspect Biol. (2018) 10:a028415. doi: 10.1101/cshperspect.a028415
121. McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. (2015) 116:1022–33. doi: 10.1161/CIRCRESAHA.116.303697
122. Wenzel UO, Ehmke H, Bode M. Immune mechanisms in arterial hypertension. Recent advances Cell Tissue Res. (2021) 385:393–404. doi: 10.1007/s00441-020-03409-0
123. Majid DSA. Tumor necrosis factor-α and kidney function: experimental findings in mice. Adv Exp Med Biol. (2011) 691:471–80. doi: 10.1007/978-1-4419-6612-4_48
124. Bertani T, Abbate M, Zoja C, Corna D, Perico N, Ghezzi P, et al. Tumor necrosis factor induces glomerular damage in the rabbit. Am J Pathol. (1989) 134:419–30.
125. Gómez-Chiarri M, Ortíz A, Lerma JL, López-Armada MJ, Mampaso F, González E, et al. Involvement of tumor necrosis factor and platelet-activating factor in the pathogenesis of experimental nephrosis in rats. Lab Invest. (1994) 70:449–59.
126. Xiao H, Lu M, Lin TY, Chen Z, Chen G, Wang WC, et al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. (2013) 128:632–42. doi: 10.1161/CIRCULATIONAHA.113.002714
127. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. (2010) 464:1357–61. doi: 10.1038/nature08938
128. Folco EJ, Sukhova GK, Quillard T, Libby P. Moderate hypoxia potentiates interleukin-1β production in activated human macrophages. Circ Res. (2014) 115:875–83. doi: 10.1161/CIRCRESAHA.115.304437
129. Rajamäki K, Lappalainen J, Oörni K, Välimäki E, Matikainen S, Kovanen PT, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PloS One. (2010) 5:e11765. doi: 10.1371/journal.pone.0011765
130. Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C, et al. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension (Dallas Tex: 1979). (2012) 59:136–44. doi: 10.1161/HYPERTENSIONAHA.111.173328
131. Hashmat S, Rudemiller N, Lund H, Abais-Battad JM, Van Why S, Mattson DL. Interleukin-6 inhibition attenuates hypertension and associated renal damage in Dahl salt-sensitive rats. Am J Physiol Renal Physiol. (2016) 311(3):F555–61. doi: 10.1152/ajprenal.00594.2015
132. Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, et al. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-γ-/- and interleukin-17A-/- mice. Hypertension (Dallas Tex: 1979). (2015) 65:569–76. doi: 10.1161/HYPERTENSIONAHA.114.04975
133. Markó L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, et al. Interferon-γ signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension (Dallas Tex: 1979). (2012) 60:1430–6. doi: 10.1161/HYPERTENSIONAHA.112.199265
134. Lv W, Booz GW, Wang Y, Fan F, Roman RJ. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur J Pharmacol. (2018) 820:65–76. doi: 10.1016/j.ejphar.2017.12.016
135. Steen EH, Wang X, Balaji S, Butte MJ, Bollyky PL, Keswani SG. The role of the anti-inflammatory cytokine interleukin-10 in tissue fibrosis. Adv Wound Care. (2020) 9:184–98. doi: 10.1089/wound.2019.1032
136. Sziksz E, Pap D, Lippai R, Béres NJ, Fekete A, Szabó AJ, et al. Fibrosis related inflammatory mediators: role of the IL-10 cytokine family. Mediators Inflamm. (2015) 2015:764641. doi: 10.1155/2015/764641
137. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension (Dallas Tex: 1979). (2010) 55:500–7. doi: 10.1161/HYPERTENSIONAHA.109.145094
138. Saleh MA, Norlander AE, Madhur MS. Inhibition of Interleukin 17-A but not Interleukin-17F Signaling Lowers Blood Pressure and Reduces End-organ Inflammation in Angiotensin II-induced Hypertension. JACC. Basic to Trans science. (2016) 1:606–16. doi: 10.1016/j.jacbts.2016.07.009
139. Mou X, Zhou DY, Zhou DY, Ma JR, Liu YH, Chen HP, et al. Serum TGF-β1 as a biomarker for type 2 diabetic nephropathy: A meta-analysis of randomized controlled trials. PloS One. (2016) 11:e0149513. doi: 10.1371/journal.pone.0149513
140. Isaka Y. Targeting TGF-β Signaling in kidney fibrosis. Int J Mol Sci. (2018) 19:2532. doi: 10.3390/ijms19092532
141. Sutariya B, Jhonsa D, Saraf MN. TGF-β: the connecting link between nephropathy and fibrosis. Immunopharmacol Immunotoxicol. (2016) 38:39–49. doi: 10.3109/08923973.2015.1127382
142. Wallach D. The tumor necrosis factor family: family conventions and private idiosyncrasies. Cold Spring Harbor Perspect Biol. (2018) 10:a028431. doi: 10.1101/cshperspect.a028431
143. Vanamee ÉS, Faustman DL. Structural principles of tumor necrosis factor superfamily signaling. Sci Signaling. (2018) 11:eaao4910. doi: 10.1126/scisignal.aao4910
144. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. (2007) 204:2449–60. doi: 10.1084/jem.20070657
145. Huang B, Cheng Y, Usa K, Liu Y, Baker MA, Mattson DL, et al. Renal tumor necrosis factor α Contributes to hypertension in Dahl salt-sensitive rats. Sci Rep. (2016) 6:21960. doi: 10.1038/srep21960
146. Venegas-Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C, Racusen LC, et al. Tumor necrosis factor-alpha antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension (Dallas Tex: 1979). (2010) 56:643–9. doi: 10.1161/HYPERTENSIONAHA.110.157685
147. Elmarakby AA, Quigley JE, Pollock DM, Imig JD. Tumor necrosis factor alpha blockade increases renal Cyp2c23 expression and slows the progression of renal damage in salt-sensitive hypertension. Hypertension (Dallas Tex: 1979). (2006) 47:557–62. doi: 10.1161/01.HYP.0000198545.01860.90
148. Elmarakby AA, Quigley JE, Imig JD, Pollock JS, Pollock DM. TNF-alpha inhibition reduces renal injury in DOCA-salt hypertensive rats. Am J Physiol Regulatory Integr Comp Physiol. (2008) 294:R76–83. doi: 10.1152/ajpregu.00466.2007
149. Xu SB, Xu B, Ma ZH, Huang MQ, Gao ZS, Ni JL. Peptide 17 alleviates early hypertensive renal injury by regulating the Hippo/YAP signalling pathway. Nephrol (Carlton Vic.). (2022) 27:712–23. doi: 10.1111/nep.14066
150. Mann DL, McMurray JJV, Packer M, Swedberg K, Borer JS, Colucci WS, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation. (2004) 109:1594–602. doi: 10.1161/01.CIR.0000124490.27666.B2
151. Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT, Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. (2003) 107:3133–40. doi: 10.1161/01.CIR.0000077913.60364.D2
152. Egli-Spichtig D, Imenez Silva PH, Glaudemans B, Gehring N, Bettoni C, Zhang MYH, et al. Tumor necrosis factor stimulates fibroblast growth factor 23 levels in chronic kidney disease and non-renal inflammation. Kidney Int. (2019) 96:890–905. doi: 10.1016/j.kint.2019.04.009
153. Awad AS, You H, Gao T, Cooper TK, Nedospasov SA, Vacher J, et al. Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int. (2015) 88:722–33. doi: 10.1038/ki.2015.162
154. Singh P, Bahrami L, Castillo A, Majid DSA. TNF-α type 2 receptor mediates renal inflammatory response to chronic angiotensin II administration with high salt intake in mice. Am J Physiol Renal Physiol. (2013) 304:F991–999. doi: 10.1152/ajprenal.00525.2012
155. Leaf IA, Nakagawa S, Johnson BG, Cha JJ, Mittelsteadt K, Guckian KM, et al. Pericyte MyD88 and IRAK4 control inflammatory and fibrotic responses to tissue injury. J Clin Invest. (2017) 127:321–34. doi: 10.1172/JCI87532
156. Pindjakova J, Hanley SA, Duffy MM, Sutton CE, Weidhofer GA, Miller MN, et al. Interleukin-1 accounts for intrarenal Th17 cell activation during ureteral obstruction. Kidney Int. (2012) 81:379–90. doi: 10.1038/ki.2011.348
157. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
158. Wen Y, Liu Y, Tang T, Lv L, Liu H, Ma K, et al. NLRP3 inflammasome activation is involved in Ang II-induced kidney damage via mitochondrial dysfunction. Oncotarget. (2016) 7:54290–302. doi: 10.18632/oncotarget.11091
159. Krishnan SM, Dowling JK, Ling YH, Diep H, Chan CT, Ferens D, et al. Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt-induced hypertension in mice. Br J Pharmacol. (2016) 173:752–65. doi: 10.1111/bph.13230
160. Shirasuna K, Karasawa T, Usui F, Kobayashi M, Komada T, Kimura H, et al. NLRP3 deficiency improves angiotensin II-induced hypertension but not fetal growth restriction during pregnancy. Endocrinology. (2015) 156:4281–92. doi: 10.1210/en.2015-1408
161. Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol. (2020) 21:30–41. doi: 10.1038/s41590-019-0548-1
162. Schunk SJ, Hermann J, Sarakpi T, Triem S, Lellig M, Hahm E, et al. Guanidinylated apolipoprotein C3 (ApoC3) associates with kidney and vascular injury. J Am Soc Nephrol: JASN. (2021) 32:3146–60. doi: 10.1681/ASN.2021040503
163. Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, et al. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol (Baltimore Md.: 1950). (2013) 190:1239–49. doi: 10.4049/jimmunol.1201959
164. Chen D, Xiong XQ, Zang YH, Tong Y, Zhou B, Chen Q, et al. BCL6 attenuates renal inflammation via negative regulation of NLRP3 transcription. Cell Death Dis. (2017) 8:e3156. doi: 10.1038/cddis.2017.567
165. Schreiner GF, Kohan DE. Regulation of renal transport processes and hemodynamics by macrophages and lymphocytes. Am J Physiol. (1990) 258:F761–767. doi: 10.1152/ajprenal.1990.258.4.F761
166. Kohan DE, Merli CA, Simon EE. Micropuncture localization of the natriuretic effect of interleukin 1. Am J Physiol. (1989) 256:F810–813. doi: 10.1152/ajprenal.1989.256.5.F810
167. Takahashi H, Nishimura M, Sakamoto M, Ikegaki I, Nakanishi T, Yoshimura M. Effects of interleukin-1 beta on blood pressure, sympathetic nerve activity, and pituitary endocrine functions in anesthetized rats. Am J Hypertens. (1992) 5:224–9. doi: 10.1093/ajh/5.4.224
168. Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, et al. Brain microglial cytokines in neurogenic hypertension. Hypertension (Dallas Tex: 1979). (2010) 56:297–303. doi: 10.1161/HYPERTENSIONAHA.110.150409
169. Voelkel NF, Tuder RM, Bridges J, Arend WP. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am J Respir Cell Mol Biol. (1994) 11:664–75. doi: 10.1165/ajrcmb.11.6.7946395
170. Nowak KL, Chonchol M, Ikizler TA, Farmer-Bailey H, Salas N, Chaudhry R, et al. IL-1 inhibition and vascular function in CKD. J Am Soc Nephrol: JASN. (2017) 28:971–80. doi: 10.1681/ASN.2016040453
171. Ling YH, Krishnan SM, Chan CT, Diep H, Ferens D, Chin-Dusting J, et al. Anakinra reduces blood pressure and renal fibrosis in one kidney/DOCA/salt-induced hypertension. Pharmacol Res. (2017) 116:77–86. doi: 10.1016/j.phrs.2016.12.015
172. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Et Biophys Acta. (2011) 1813:878–88. doi: 10.1016/j.bbamcr.2011.01.034
173. Batra G, Ghukasyan Lakic T, Lindbäck J, Held C, White HD, Stewart RAH, et al. Interleukin 6 and cardiovascular outcomes in patients with chronic kidney disease and chronic coronary syndrome. JAMA Cardiol. (2021) 6:1440–5. doi: 10.1001/jamacardio.2021.3079
174. Hassan MO, Duarte R, Dickens C, Dix-Peek T, Naidoo S, Vachiat A, et al. Interleukin-6 gene polymorhisms and interleukin-6 levels are associated with atherosclerosis in CKD patients. Clin Nephrol. (2020) 93:82–6. doi: 10.5414/CNP92S114
175. Desjardins MP, Sidibé A, Fortier C, Mac-Way F, Marquis K, De Serres S, et al. Association of interleukin-6 with aortic stiffness in end-stage renal disease. J Am Soc Hypertension: JASH. (2018) 12:5–13. doi: 10.1016/j.jash.2017.09.013
176. Salimi S, Shardell MD, Seliger SL, Bandinelli S, Guralnik JM, Ferrucci L. Inflammation and trajectory of renal function in community-dwelling older adults. J Am Geriatrics Society. (2018) 66:804–11. doi: 10.1111/jgs.15268
177. Chen W, Yuan H, Cao W, Wang T, Chen W, Yu H, et al. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics. (2019) 9:3980–91. doi: 10.7150/thno.32352
178. Rovin BH, van Vollenhoven RF, Aranow C, Wagner C, Gordon R, Zhuang Y, et al. A multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of treatment with sirukumab (CNTO 136) in patients with active lupus nephritis. Arthritis Rheumatol (Hoboken N.J.). (2016) 68:2174–83. doi: 10.1002/art.39722
179. Pergola PE, Devalaraja M, Fishbane S, Chonchol M, Mathur VS, Smith MT, et al. Ziltivekimab for treatment of anemia of inflammation in patients on hemodialysis: results from a phase 1/2 multicenter, randomized, double-blind, placebo-controlled trial. J Am Soc Nephrol: JASN. (2021) 32:211–22. doi: 10.1681/ASN.2020050595
180. Fukuda M, Sawa N, Hoshino J, Ohashi K, Motoaki M, Ubara Y. Tocilizumab preserves renal function in rheumatoid arthritis with AA amyloidosis and end-stage kidney disease: Two case reports. Clin Nephrol. (2021) 95:54–61. doi: 10.5414/CN109971
181. Gao Y, Liu B, Guo X, Nie J, Zou H, Wen S, et al. Interferon regulatory factor 4 deletion protects against kidney inflammation and fibrosis in deoxycorticosterone acetate/salt hypertension. J Hypertens. (2023) 41:794–810. doi: 10.1097/HJH.0000000000003401
182. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harbor Perspect Biol. (2009) 1:a001651. doi: 10.1101/cshperspect.a001651
183. Hirohama D, Fujita T. Evaluation of the pathophysiological mechanisms of salt-sensitive hypertension. Hypertens Res. (2019) 42:1848–57. doi: 10.1038/s41440-019-0332-5
184. Yan Y, Zhou XE, Xu HE, Melcher K. Structure and physiological regulation of AMPK. Int J Mol Sci. (2018) 19:3534. doi: 10.3390/ijms19113534
185. Liu GX, Li YQ, Huang XR, Wei LH, Zhang Y, Feng M, et al. Smad7 inhibits AngII-mediated hypertensive nephropathy in a mouse model of hypertension. Clin Sci (London England: 1979). (2014) 127:195–208. doi: 10.1042/CS20130706
186. Zhao Y, Chen X, Lin Y, Li Z, Su X, Fan S, et al. USP25 inhibits renal fibrosis by regulating TGFβ-SMAD signaling pathway in Ang II-induced hypertensive mice. Biochim Et Biophys Acta Mol Basis Dis. (2023) 1869:166713. doi: 10.1016/j.bbadis.2023.166713
187. Huang CC, Lin YY, Yang AL, Kuo TW, Kuo CH, Lee SD. Anti-renal fibrotic effect of exercise training in hypertension. Int J Mol Sci. (2018) 19:613. doi: 10.3390/ijms19020613
188. Wen Y, Crowley SD. Renal effects of cytokines in hypertension. Curr Opin Nephrol Hypertens. (2018) 27:70–6. doi: 10.1097/MNH.0000000000000385
189. Naing C, Htet NH, Basavaraj AK, Nalliah S. An association between IL-10 promoter polymorphisms and diabetic nephropathy: a meta-analysis of case-control studies. J Diabetes Metab Disord. (2018) 17:333–43. doi: 10.1007/s40200-018-0349-3
190. Xia Y, Entman ML, Wang Y. Critical role of CXCL16 in hypertensive kidney injury and fibrosis. Hypertension (Dallas Tex: 1979). (2013) 62:1129–37. doi: 10.1161/HYPERTENSIONAHA.113.01837
191. Rudemiller NP, Patel MB, Zhang JD, Jeffs AD, Karlovich NS, Griffiths R, et al. C-C motif chemokine 5 attenuates angiotensin II-dependent kidney injury by limiting renal macrophage infiltration. Am J Pathol. (2016) 186:2846–56. doi: 10.1016/j.ajpath.2016.07.015
192. Mohamed R, Jayakumar C, Chen F, Fulton D, Stepp D, Gansevoort RT, et al. Low-dose IL-17 therapy prevents and reverses diabetic nephropathy, metabolic syndrome, and associated organ fibrosis. J Am Soc Nephrol: JASN. (2016) 27:745–65. doi: 10.1681/ASN.2014111136
193. Hamour S, Gan PY, Pepper R, Florez Barros F, Wang HH, O’Sullivan K, et al. Local IL-17 production exerts a protective role in murine experimental glomerulonephritis. PLoS One. (2015) 10:e0136238. doi: 10.1371/journal.pone.0136238
194. Krebs CF, Lange S, Niemann G, Rosendahl A, Lehners A, Meyer-Schwesinger C, et al. Deficiency of the interleukin 17/23 axis accelerates renal injury in mice with deoxycorticosterone acetate+angiotensin ii-induced hypertension. Hypertension (Dallas Tex: 1979). (2014) 63:565–71. doi: 10.1161/HYPERTENSIONAHA.113.02620
195. Tang R, Lin W, Shen C, Hu X, Yu L, Meng T, et al. Single-cell transcriptomics uncover hub genes and cell-cell crosstalk in patients with hypertensive nephropathy. Int Immunopharmacol. (2023) 125:111104. doi: 10.1016/j.intimp.2023.111104
196. Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension (Dallas Tex: 1979). (2017) 70:660–7. doi: 10.1161/HYPERTENSIONAHA.117.07802
197. Case AJ, Tian J, Zimmerman MC. Increased mitochondrial superoxide in the brain, but not periphery, sensitizes mice to angiotensin II-mediated hypertension. Redox Biol. (2017) 11:82–90. doi: 10.1016/j.redox.2016.11.011
198. Mennuni S, Rubattu S, Pierelli G, Tocci G, Fofi C, Volpe M. Hypertension and kidneys: unraveling complex molecular mechanisms underlying hypertensive renal damage. J Hum Hypertens. (2014) 28:74–9. doi: 10.1038/jhh.2013.55
199. Rainbolt TK, Saunders JM, Wiseman RL. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol metabolism: TEM. (2014) 25:528–37. doi: 10.1016/j.tem.2014.06.007
200. Young CN. Endoplasmic reticulum stress in the pathogenesis of hypertension. Exp Physiol. (2017) 102:869–84. doi: 10.1113/EP086274
201. Zito E. ERO1: A protein disulfide oxidase and H2O2 producer. Free Radical Biol Med. (2015) 83:299–304. doi: 10.1016/j.freeradbiomed.2015.01.011
202. Inagi R. Endoplasmic reticulum stress in the kidney as a novel mediator of kidney injury. Nephron. Exp Nephrol. (2009) 112:e1–9. doi: 10.1159/000210573
203. Chiang CK, Hsu SP, Wu CT, Huang JW, Cheng HT, Chang YW, et al. Endoplasmic reticulum stress implicated in the development of renal fibrosis. Mol Med (Cambridge Mass.). (2011) 17:1295–305. doi: 10.2119/molmed.2011.00131
204. He F, Chen S, Wang H, Shao N, Tian X, Jiang H, et al. Regulation of CD2-associated protein influences podocyte endoplasmic reticulum stress-mediated apoptosis induced by albumin overload. Gene. (2011) 484:18–25. doi: 10.1016/j.gene.2011.05.025
205. Larbi A, Kempf J, Pawelec G. Oxidative stress modulation and T cell activation. Exp Gerontol. (2007) 42:852–8. doi: 10.1016/j.exger.2007.05.004
206. Edison N, Curtz Y, Paland N, Mamriev D, Chorubczyk N, Haviv-Reingewertz T, et al. Degradation of Bcl-2 by XIAP and ARTS promotes apoptosis. Cell Rep. (2017) 21:442–54. doi: 10.1016/j.celrep.2017.09.052
207. Larisch S, Yi Y, Lotan R, Kerner H, Eimerl S, Tony Parks W, et al. A novel mitochondrial septin-like protein, ARTS, mediates apoptosis dependent on its P-loop motif. Nat Cell Biol. (2000) 2:915–21. doi: 10.1038/35046566
208. Gottfried Y, Rotem A, Lotan R, Steller H, Larisch S. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J. (2004) 23:1627–35. doi: 10.1038/sj.emboj.7600155
209. Edison N, Zuri D, Maniv I, Bornstein B, Lev T, Gottfried Y, et al. The IAP-antagonist ARTS initiates caspase activation upstream of cytochrome C and SMAC/Diablo. Cell Death Differ. (2012) 19:356–68. doi: 10.1038/cdd.2011.112
210. Liu G, Park SH, Imbesi M, Nathan WJ, Zou X, Zhu Y, et al. Loss of NAD-dependent protein deacetylase sirtuin-2 alters mitochondrial protein acetylation and dysregulates mitophagy. Antioxidants Redox Signaling. (2017) 26:849–63. doi: 10.1089/ars.2016.6662
211. He P, Li Z, Yue Z, Gao H, Feng G, Wang P, et al. SIRT3 prevents angiotensin II-induced renal tubular epithelial-mesenchymal transition by ameliorating oxidative stress and mitochondrial dysfunction. Mol Cell Endocrinol. (2018) 460:1–13. doi: 10.1016/j.mce.2017.04.027
212. Lin JR, Zheng YJ, Zhang ZB, Shen WL, Li XD, Wei T, et al. Suppression of endothelial-to-mesenchymal transition by SIRT (Sirtuin) 3 alleviated the development of hypertensive renal injury. Hypertension (Dallas Tex: 1979). (2018) 72:350–60. doi: 10.1161/HYPERTENSIONAHA.118.10482
213. Wang Z, Zhai J, Zhang T, He L, Ma S, Zuo Q, et al. Canagliflozin ameliorates epithelial-mesenchymal transition in high-salt diet-induced hypertensive renal injury through restoration of sirtuin 3 expression and the reduction of oxidative stress. Biochem Biophys Res Commun. (2023) 653:53–61. doi: 10.1016/j.bbrc.2023.01.084
214. Hallan SI, Øvrehus MA, Bjørneklett R, Aasarød KI, Fogo AB, Ix JH. Hypertensive nephrosclerosis: wider kidney biopsy indications may be needed to improve diagnostics. J Internal Med. (2021) 289:69–83. doi: 10.1111/joim.13146
215. Ertuglu LA, Kirabo A. Dendritic cell epithelial sodium channel in inflammation, salt-sensitive hypertension, and kidney damage. Kidney360. (2022) 3:1620–9. doi: 10.34067/KID.0001272022
216. Kambayashi T, Laufer TM. Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell? Nature Reviews. Immunology. (2014) 14:719–30. doi: 10.1038/nri3754
217. Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, et al. Dendritic cell Amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. (2017) 21:1009–20. doi: 10.1016/j.celrep.2017.10.002
218. Luft FC, Dechend R, Müller DN. Immune mechanisms in angiotensin II-induced target-organ damage. Ann Med. (2012) 44:S49–54. doi: 10.3109/07853890.2011.653396
219. Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. (2007) 292:C82–97. doi: 10.1152/ajpcell.00287.2006
220. Zugasti O, Bose N, Squiban B, Belougne J, Kurz CL, Schroeder FC, et al. Activation of a G protein-coupled receptor by its endogenous ligand triggers the innate immune response of Caenorhabditis elegans. Nat Immunol. (2014) 15:833–8. doi: 10.1038/ni.2957
221. Causton B, Ramadas RA, Cho JL, Jones K, Pardo-Saganta A, Rajagopal J, et al. CARMA3 is critical for the initiation of allergic airway inflammation. J Immunol (Baltimore Md.: 1950). (2015) 195:683–94. doi: 10.4049/jimmunol.1402983
222. Fang W, Wang Z, Li Q, Wang X, Zhang Y, Sun Y, et al. Gpr97 exacerbates AKI by mediating sema3A signaling. J Am Soc Nephrol: JASN. (2018) 29:1475–89. doi: 10.1681/ASN.2017080932
223. Wu JC, Wang XJ, Zhu JH, Huang XY, Liu M, Qiao Z, et al. GPR97 deficiency ameliorates renal interstitial fibrosis in mouse hypertensive nephropathy. Acta Pharmacologica Sinica. (2023) 44:1206–16. doi: 10.1038/s41401-022-01041-y
224. Fu EL, Clase CM, Evans M, Lindholm B, Rotmans JI, Dekker FW, et al. Comparative effectiveness of renin-angiotensin system inhibitors and calcium channel blockers in individuals with advanced CKD: A nationwide observational cohort study. Am J Kidney Dis. (2021) 77:719–729.e1. doi: 10.1053/j.ajkd.2020.10.006
225. Unger T, Borghi C, Charchar F, Khan NA, Poulter NR, Prabhakaran D, et al. 2020 International Society of Hypertension global hypertension practice guidelines. J Hypertens. (2020) 38:982–1004. doi: 10.1097/HJH.0000000000002453
226. Rovin BH, Adler SG, Barratt J, Bridoux F, Burdge KA, Chan TM, et al. KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney Int. (2021) 100:S1–S276. doi: 10.1016/j.kint.2021.05.021
227. Chen TK, Knicely DH, Grams ME. Chronic kidney disease diagnosis and management: A review. JAMA. (2019) 322:1294–304. doi: 10.1001/jama.2019.14745
228. Yang X, Zhou B, Zhou L, Cui L, Zeng J, Wang S, et al. Development and validation of prediction models for hypertensive nephropathy, the PANDORA study. Front Cardiovasc Med. (2022) 9:794768. doi: 10.3389/fcvm.2022.794768
229. Kokubo Y, Padmanabhan S, Iwashima Y, Yamagishi K, Goto A. Gene and environmental interactions according to the components of lifestyle modifications in hypertension guidelines. Environ Health Prev Med. (2019) 24:19. doi: 10.1186/s12199-019-0771-2
230. Sun N, Mu J, Li Y, Working Committee of Salt evaluation, Blood Pressure Management, Chinese Medical Association Hypertension Professional Committee, Hypertension Group, Chinese Society of Cardiology. An expert recommendation on salt intake and blood pressure management in Chinese patients with hypertension: A statement of the Chinese Medical Association Hypertension Professional Committee. J Clin Hypertension (Greenwich Conn.). (2019) 21:446–50. doi: 10.1111/jch.13501
231. Goraya N, Simoni J, Jo C, Wesson DE. Dietary acid reduction with fruits and vegetables or bicarbonate attenuates kidney injury in patients with a moderately reduced glomerular filtration rate due to hypertensive nephropathy. Kidney Int. (2012) 81:86–93. doi: 10.1038/ki.2011.313
Keywords: hypertensive nephropathy, hypertension, immunity, inflammation, cytokines
Citation: Hao X-m, Liu Y, Hailaiti D, Gong Y, Zhang X-d, Yue B-n, Liu J-p, Wu X-l, Yang K-z, Wang J and Liu Q-g (2024) Mechanisms of inflammation modulation by different immune cells in hypertensive nephropathy. Front. Immunol. 15:1333170. doi: 10.3389/fimmu.2024.1333170
Received: 04 November 2023; Accepted: 15 February 2024;
Published: 13 March 2024.
Edited by:
Xu-jie Zhou, Peking University, ChinaReviewed by:
InKyeom Kim, Kyungpook National University, Republic of KoreaCopyright © 2024 Hao, Liu, Hailaiti, Gong, Zhang, Yue, Liu, Wu, Yang, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ke-zhen Yang, eWt6MTk5NkB6anUuZWR1LmNu; Jun Wang, d2FuZ2p1bmVlQHllYWgubmV0; Qing-guo Liu, bGl1cWluZ2d1bzg4OEB2aXAuc2luYS5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.