 Guoping Zheng1
Guoping Zheng1 Guanguan Qiu
Guanguan Qiu Qiang Shu
Qiang Shu Jianguo Xu
Jianguo Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 05 February 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1332440
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the third human coronavirus to cause acute respiratory distress syndrome (ARDS) and contains four structural proteins: spike, envelope, membrane, and nucleocapsid. An increasing number of studies have demonstrated that all four structural proteins of SARS-CoV-2 are capable of causing lung injury, even without the presence of intact virus. Therefore, the topic of SARS-CoV-2 structural protein-evoked lung injury warrants more attention. In the current article, we first synopsize the structural features of SARS-CoV-2 structural proteins. Second, we discuss the mechanisms for structural protein-induced inflammatory responses in vitro. Finally, we list the findings that indicate structural proteins themselves are toxic and sufficient to induce lung injury in vivo. Recognizing mechanisms of lung injury triggered by SARS-CoV-2 structural proteins may facilitate the development of targeted modalities in treating COVID-19.
The genome of SARS-CoV-2 contains a single-stranded positive RNA approximately 30 kb in length (1). It encodes 4 structural proteins [spike (S), envelope (E), membrane (M), and nucleocapsid (N)], 9 accessory proteins (open reading frames 3a, 3b, 6, 7a, 7b, 8, 9b, 9c, and 10), and 16 non-structural proteins (NSP 1-16) (1). All four structural proteins are required to complete an infectious event, which includes entering host cells, replication of the viral genome, packaging, assembly, trafficking, and release of virus particles (2). The S protein mediates viral attachment to the target cells via association with angiotensin-converting enzyme 2 (ACE2). S protein is then cleaved by proteases to cause fusion of viral and cellular membranes. Following cellular entry, the viral RNA is replicated, translated, and packaged, resulting in exocytosis of virions and activation of host immune response (3).
COVID-19 initially presents with upper and lower respiratory tract manifestations and later progresses to systemic diseases such as gastrointestinal diseases, myocardial inflammation, and acute respiratory distress syndrome (ARDS), which results from severe acute lung injury (4). A global literature survey at the early phase of global pandemic showed that the fatality ratio of COVID-19-associated ARDS (CARDS) was almost 50%. Moreover, the prevalence of CARDS in non-survivors was as high as 90% (5). In the early stage of CARDS development, alveolar macrophages are activated following viral infection and death of alveolar epithelial cells. Activated alveolar macrophages generate inflammatory cytokines and chemokines that recruit immune cells, including but not limited to T cells, monocytes, and neutrophils, to the alveolar space. In the meantime, the recruited cells augment the production of proinflammatory mediators and culminate in a cytokine storm (6). Concurrently, infection of SARS-CoV-2 elicits direct as well as indirect activation of complement pathways (7). Cytokine storm and complement activation disrupt the epithelial-endothelial barrier and drive endothelialitis, causing elevated permeability as well as accumulation of protein-rich fluid in alveolar and interstitial spaces. Lung pathology and chest computed tomography show evidence of pneumonitis (8). Endothelialitis enhances procoagulant activity and represses fibrinolytic activity, resulting in COVID-19-associated coagulopathy (9). About 80% of patients with viral pneumonitis improve with no specific interventions. Patients with old age, hypertension, diabetes, and obesity are at increased risk of exacerbation and development of CARDS 7-10 days after onset of symptoms (3, 10).
The pathogenesis of CARDS is quite complex and remains to be further explored. S protein of SARS-CoV-1 was reported to aggravate acid-stimulated lung injury via binding with ACE2. In addition, the injury was mitigated by an inhibitor for angiotensin II receptor type 1 (11). Intranasal delivery of recombinant SARS-CoV-1 N protein triggered progressive pulmonary edema in mice (12). Many reports have found that structural proteins of SARS-CoV-2 are sufficient to cause acute lung injury independent of viral infection (13, 14). Our group documented that SARS-CoV-2 N protein prompted acute lung injury in mice via binding with receptor for advanced glycation endproducts (RAGE) and activation of nuclear factor kappa B (NF-ĸB) pathway (15, 16). This article discusses the structural characteristics of SARS-CoV-2 structural proteins, structural protein-provoked proinflammatory responses in vitro, and structural protein-evoked lung injury.
S protein mediates cell recognition, membrane fusion, and entry of the SARS-CoV-2 virus. Variants of SARS-CoV-2 with S protein mutations such as Alpha, Beta, Gamma, Delta, and Omicron augment infectivity and immune escape (17). S protein is comprised of two subunits, S1 and S2. The S1 subunit is composed of a N-terminal domain (NTD) and a long C-terminal receptor-binding domain (RBD), which binds with ACE2 in the membrane of host cells (18). The S2 subunit carries a fusion peptide for viral entry, two heptad repeats, a transmembrane domain, and a C-terminal tail (19). ACE2 is highly expressed in type I and type II alveolar epithelial cells. In contrast, ACE2 has low expression in airway epithelial cells, endothelial cells, and macrophages (20, 21). Single-cell transcriptomic analysis revealed that SARS-CoV-2 infected type I and type II alveolar epithelial cells, basal cells, club cells, and alveolar macrophages (22, 23). After the binding of S1 subunit with cell surface ACE2, the S2 subunit is subjected to proteolytic digestion by transmembrane serine protease 2 (TMPRSS2), resulting in virus-plasma membrane fusion (24). SARS-CoV-2 virion can also gain entry to host cells via the endosomal pathway, in which S2 subunit is cleaved by cathepsin L (24). Upon cell entry, the viral genome is replicated and transcribed by RNA-dependent RNA polymerase (RdRp), which is composed of catalytic nsp12 subunit and nsp7-nsp8 cofactors in a replication–transcription complex (RTC) (25). The RTC also includes nsp13 helicase for RNA unwinding, nsp10/nsp14 exonuclease for proofreading to enhance replication fidelity, and nsp10/nsp14/nsp16 methyltransferase for RNA capping (26). The viral replication culminates in an exorbitant inflammatory response, which is accompanied by systemic cytokine storm and complement activation, as well as excessive activation of macrophages in the lung. The cytokine storm and complement activation cause endothelial dysfunction and elevated vascular permeability. Activated macrophages in the lung also release excessive proinflammatory chemokines, resulting in infiltration of neutrophils and monocyte-derived macrophages (27).
E protein is the smallest SARS-CoV-2 structural protein and is indispensable in viral assembly, release, and pathogenesis. E protein is highly conserved during evolution, as evidenced by 96% similarity between SARS-CoV-1 and SARS-CoV-2 (28). It contains three domains: a negatively charged N-terminus, an uncharged transmembrane domain, and a C-terminus containing diverse motifs for posttranslational modification (28). A small percentage of E protein is integrated into the virions, while the bulk of the protein is localized at Golgi and endoplasmic reticulum–Golgi intermediate compartment (ERGIC), participating in viral assembly and release (29). A recombinant SARS-CoV-1 virus without the E protein gene replicated at a slower rate and caused milder lung inflammation compared with the recombinant wild-type virus in a hamster model (30). A SARS-CoV-2 variant with a 12 base pair deletion at the E protein gene showed higher S protein levels in viral culture, indicating the deletion may enhance viral replication (31). E protein possesses two unique structural features. First, it can form cation selective channels named viroporins via homo-oligomerization, in which asparagine 15 (N15) and valine 25 (V25) are essential for the function (32). SARS-CoV-2 E protein elevated the pH value in ERGIC and lysosomes via the viroporin activity (33). A SARS-CoV-1 virus with E protein mutation for viroporin activity showed reduced edema and production of proinflammatory cytokines in mouse lung (34). Second, E protein contains a C-terminal motif (DLLV) which mediates the binding with PDZ proteins to disrupt epithelial barrier and promote viral spread (35).
M protein is also essential in the assembly and release of SARS-CoV-2 (36). It is comprised of a short NTD, three transmembrane domains, and a C terminal domain (CTD) situated in the interior of virion (37). The assembly of SARS-CoV-2 virion takes place inside of ERGIC. M protein encompasses a trans-Golgi network localization signal and is transported to the ERGIC a little earlier than S and E, indicating its function in originating the assembly of SARS-CoV-2 (38, 39). M protein itself binds weakly with N protein, however the interaction is strongly elevated with the co-presence of the N protein and RNA (40). It associates with E protein on the membrane of virus like particles and mediates virion release. The binding between M and E is enhanced by ubiquitinating M at position K15 (41). M protein as well as E protein induce the intracellular retention of S protein and ensure that SARS-CoV-2 viral particles are assembled (36).
SARS-CoV-2 N protein packages the viral genomic RNA to form helical ribonucleoprotein complex encompassed within viral capsid (42). In addition, N protein enhances viral RNA transcription and replication via liquid–liquid phase separation (43). N protein contains a NTD, a linker region with abundance in serine and arginine residues, and a CTD. NTD forms a right‐handed fist shape with a core of β‐sheet as well as a β‐hairpin region, while CTD exhibits as tightly interlocked homodimer with a rectangular slab shape (44). There is a positively charged RNA binding groove on the surface of NTD (45). Both NTD and CTD participate in binding with the RNA genome, while phosphorylation of the linker region impacts RNA binding (46). The linker region is indispensable for anchoring the ribonucleoprotein to the viral membrane (45). There is a phosphorylation-dependent association between N protein at the linker region and human 14-3-3 family proteins, which may regulate nucleocytoplasmic shuttling of N protein (47). Binding between N protein and ubiquitin-like domain 1 of NSP3 of SARS-CoV-2 induces ribonucleoprotein dissociation (48). N protein also triggers humoral and cellular immune response, suggesting the potential benefits of future COVID-19 vaccine formation containing N component (49, 50).
Both ACE2 shedding and downregulation of ACE2 play a role in S protein-induced inflammatory response. ACE2 is a transmembrane protein and can be cleaved into soluble but enzymatically active ACE2 through a process called shedding. Binding of S protein to ACE2 induced ectodomain shedding of ACE2 by tumor necrosis factor-alpha convertase (ADAM17) for SARS-CoV-1 (51) and membrane-type 1 matrix metalloproteinase for SARS-CoV-2 (52). Soluble ACE2 interacted with S protein to promote receptor-mediated endocytosis of SARS-CoV-2 (53). Inhibition of the generation of soluble ACE2 reduced the production of TNF-α in vitro (54). Soluble ACE2 occurred early in COVID-19 patients and was reported as a predictor of disease severity (55). In terms of ACE2 downregulation, Gao et al. reported that S protein downregulated ACE2 via enhancing the degradation mRNA of ACE2 (56). ACE2 countered the effect of angiotensin II (Ang II) by converting Ang II into Ang 1-7. Reduced ACE2 enhanced the activation of Ang II/Ang II type 1 receptor pathway, leading to proinflammation response and vasoconstriction (57). Higher levels of Ang II were detected in COVID-19 patients (56). In contrast, Lu et al. found that S protein downregulated ACE2 expression via clathrin and AP-2-mediated endocytosis. S protein-primed cells presented with a gene expression pattern of activated cytokine signaling (58). Additionally, downregulation of ACE2 induced by S protein was responsible for endothelial dysfunction, resulting in oxidative stress and inflammation (59).
Several studies have demonstrated that S protein has proinflammatory activity in vitro. Villacampa et al. reported that S protein triggered the activation of NF-κB and NLR family pyrin domain containing 3 (NLRP3) inflammasome in endothelial and immune cells (60). NLRP3 inflammasome activated caspase-1, which enhanced the generation of proinflammatory and proapoptotic IL-1β and IL-18 (61). Petruk et al. discovered that S protein bound lipopolysaccharide (LPS) with high affinity and induced NF-κB activation and cytokine responses in several cell types (62). Khan et al. found that S protein was recognized by Toll-like receptor 2 (TLR2), which dimerized with TLR1 or TLR6, to trigger the activation of NF-κB and induce proinflammatory cytokines such as IL-6, TNF-α, and IL-1β (63). Umar et al. revealed that production of S protein-induced proinflammatory cytokines was blocked by TLR2 or TLR7 knockdown in macrophages, indicating the involvement of both TLR2 and TLR7 in S protein signaling (64). However, Zhao et al. showed that S protein bound and activated TLR4 (65). Patra et al. demonstrated that S protein promoted Ang II type 1 receptor signaling, which activated MAPK/NF-κB pathway and induced IL-6 release in epithelial cells (66). Barhoumi et al. uncovered that S protein promoted M1 macrophage polarization, leading to apoptosis, production of reactive oxygen species, and elevated proinflammatory cytokines. These effects were partially blocked by an ACE inhibitor (67). Li et al. showed that S protein promoted autophagy via PI3K/AKT/mTOR pathway in ACE2 expressing cells and enhanced inflammation and apoptotic responses (68). In addition, Olajide et al. reported that recombinant S1 protein stimulated the release of proinflammatory cytokines from peripheral blood mononuclear cells through activation of NF-κB, p38 MAPK, and NLRP3 inflammasome (69).
Ion-channeling viroporins formed by E protein promoted the activity of NLRP3 inflammasome, which controlled activation and release of IL-1β and IL-18 (70). E protein interacted with PDZ domain 2 of human zona occludens-1 and caused damages to tight junction and epithelial barrier, contributing to virus spread and accumulation of water in the lungs (71). Equilibrium and kinetic analysis indicated that E protein bound with the tight junction-associated PALS1 with high affinity, resulting in epithelial barrier disruption and amplified tissue remodeling (72). E protein induced the dysfunction of the blood-brain barrier and triggered inflammatory response in a blood-brain barrier model (73). In macrophages primed with LPS and stimulated with an analogue of viral double-stranded RNA (poly I:C), E protein elevated NLRP3 inflammasome activation (74). E protein was recognized by TLR2 to trigger the release of inflammatory mediators including TNF-α and IFN-γ (75). Additionally, intracisternal injection of E protein prompted depression-like symptoms and dysosmia via TLR2-dependent neuroinflammation (76). Furthermore, E protein was documented to trigger the production of high-mobility group box 1 (HMGB1), which elicited proinflammatory response via TGF-β1/SMAD2/3 pathway, resulting in renal fibrosis (77).
Interferons (IFNs) type I (IFN-α and IFN-β) and type III (IFN-λ) are cytokines with inherent antiviral activity, which impairs viral replication in infected cells (78). Galani et al. reported that reduced production of type I and III IFNs, as evidenced by enhanced proinflammatory responses, were present in peripheral blood mononuclear cells in a group of severe COVID-19 patients (79). M protein was found to modulate type I IFN generation via binding with TANK-binding kinase 1 (TBK1) and enhancing its degradation via ubiquitination. The reduced TBK1 blocked the activation of interferon regulation factor 3 (IRF3), resulting in diminished production of type I IFN (80). Lei et al. documented that SARS-CoV-2 infection triggered overt but delayed IFN-β production, while M protein inhibited virus-induced IFN-β promoter activation (81). Zheng et al. revealed that M protein functioned to reduce the release of IFN-β and IFN-λ via interacting with proteins in RIG-I/MDA-5 signaling, which recognized cytosolic double-stranded viral RNA and mediated the generation of IFNs. M protein bound with RIG-I, MAVS, and TBK1 and subsequently reduced the binding between MAVS and TBK1, resulting in diminished phosphorylation of IRF3 (82). Ren et al. found that M protein induced cell apoptosis via binding with phosphoinositide-dependent protein kinase-1 (PDK1) and blocking the activation of PKB/Akt pathway, while N protein served as a scaffold for the function of M protein (83).
Chen et al. discovered that N protein bound with SMAD3, which suppressed the expression of cystic fibrosis transmembrane conductance regulator (CFTR), leading to increased intracellular Cl− concentration in airway epithelial cells. Subsequently, serum/glucocorticoid regulated kinase 1 sensed the elevated Cl− concentration and triggered an inflammatory response (84). Another study showed that SARS-CoV-2 N protein had the most dramatic effect in stimulating antiviral cytokines and proinflammatory chemokines in comparison to the other six N proteins from coronaviruses. N protein promoted endocytosis of nucleic acids, which was enhanced by RANTES and lactate. Moreover, wild-type SARS-CoV-2 N protein prompted more prominent endocytosis of nucleic acid compared with Omicron counterpart (85). Lopez-Munoz et al. discovered that N protein bound to heparan sulfate in cell surface with high affinity. N protein had high affinity to 11 chemokines and impaired chemokine functions, which may facilitate viral replication and transmission (86). Karwaciak et al. found that N protein induced production of IL-6 from human monocytes and macrophages (87), while the effect was blocked by chlorpromazine via impairing MEK/ERK signaling (88). Qian et al. revealed that N protein elevated the expression of proinflammatory TNF-α, IL-1β, and MCP-1 in addition to ICAM-1 and VCAM-1 in endothelial cells. Endothelial cells were activated by the N protein via TLR2/NF-κB and TLR2/MAPK signal pathways (89). Wu et al. showed that viral RNA triggered liquid-liquid phase separation of N protein. As a result, N protein associated with and activated the TAK1 and IKK enzyme complex, which promoted NF-κB activation and inflammatory response (90).
N protein has also been reported to participate in the suppression of innate immune response. Savellini et al. uncovered that N protein bound with TRIM25, an E3 ubiquitin ligase enzyme, and blocked TRIM25-facilitated RIG-I activation and IFN-β production (91). Zheng et al. documented that N protein subdued expression of ISG56, CXCL10, IFN-β, and IFN-λ, induced by poly (I:C). N protein bound with Ras GTPase-activating protein-binding protein 1 (G3BP1) to reduce the formation of antiviral stress granule and block the activation of RIG-1 by double-stranded RNA (92). Another group identified that CTD of N protein was crucial in the liquid-liquid phase separation of N protein. This separation blocked ubiquitination and aggregation of mitochondrial antiviral signaling protein and inhibited innate immunity (93).
In transgenic mice overexpressing human ACE2, intratracheal instillation of recombinant S1 subunit of S protein elevated cell infiltration and protein concentration in the bronchoalveolar lavage (BAL) at 72 h after exposure. It also upregulated inflammatory cytokines in BAL/serum and induced histological characteristics of acute lung injury. Mechanistically, S protein activated the NF-κB and STAT3 pathways in the lungs (94) (Table 1) (Figure 1). The same group also reported that S1 subunit of S protein exacerbated lung injury in human ACE2 transgenic mice on an alcohol diet in comparison with mice on a control diet. Concurrently, the S1 subunit activated NF-κB, STAT3, and NLRP3 (95). Cao et al. found that lentivirus expressing S protein targeted type II alveolar cells and M1 macrophages and induced acute lung inflammation in mice at 24 h. Lentiviral S protein also elevated proinflammatory cytokines in the lungs as well as in the RAW264.7 macrophage cell line (96). Using NF-κB reporter mice, Puthia et al. demonstrated that co-administration of S protein and LPS via aerosol synergistically increased NF-κB induction compared with LPS alone. Co-administration of S protein and LPS significantly elevated infiltration of macrophages and neutrophils as well as proinflammatory cytokines in the BAL at 24 h compared with LPS alone. Mice treated with S protein and LPS also had a higher lung injury score in histological analysis. The coadministration model mimicked lung injury observed in COVID-19 (97). In Syrian hamsters, Lei et al. showed that intratracheal administration of pseudovirus expressing S protein induced lung damage with thickened alveolar septa and elevated infiltration of mononuclear cells at day 5. There were decreased levels of phospho-AMP-activated protein kinase, phosphor-ACE2, and ACE2 in the damaged lungs. The alterations in the protein expression were recapitulated in pulmonary arterial endothelial cells infected with pseudovirus expressing S protein (59). Zhang et al. discovered that intraperitoneal injection of recombinant RBD of S protein aggravated LPS-induced acute lung injury in mice at day 3. RBD of S protein bound with ACE2 and downregulated its expression, resulting in an elevation in Ang II. Ang II activated its receptor and downstream NF-κB-NOX1/2 signaling pathway, leading to oxidative stress and redox imbalance as well as proinflammatory response in the lung. In addition, recombinant ACE2 blocked lung injury induced by RBD of S protein (98). In transgenic C57BL/6 mice expressing human ACE2, Liang et al. revealed that intratracheal administration of recombinant RBD of S or S1 protein for 10 days elevated IL-18 mRNA expression in the blood. The treatment augmented infiltration of neutrophils in the lung and lung injury scores. S protein administration also elevated expression of NLRP3-dependent IL-18 in the lung, while IκBα levels were decreased. In addition, S protein increased IL-18 expression via reducing mitophagy and enhancing mitochondrial reactive oxygenation species in vitro and in vivo (99). Elevated IL-18 levels have been correlated with disease severity and clinical outcomes of COVID-19 patients (99, 102). Satta et al. revealed that intravenous injection of lentivirus expressing S protein increased proinflammatory cytokines in the BAL and macrophages in the lung, which were abolished by liposome-human ACE2 (100). In BALB/c mice expressing human ACE2, Gu et al. found that co-administration of recombinant extracellular domain of S protein and poly (I:C) aggregated lung injury in histology compared with poly (I:C) alone at 24 h. The co-administration also increased neutrophil infiltration and proinflammatory cytokines in the BAL, while S protein or poly (I:C) alone lacked the effect (101). These findings contradicted some other reports cited in this review and indicate that S protein itself does not directly induce significant lung injury but requires coadministration of a pathogen-associated molecular pattern (PAMP). Furthermore, S protein was able to induce lung injury in an ACE2-independent manner. Biering et al. found that S protein triggered endothelial hyperpermeability in cells that do not express ACE2 in vitro. Intranasal administration of recombinant S protein triggered vascular leak in the lungs of mice that do not express human ACE2. In vitro studies revealed that glycosaminoglycans, integrins, and the TGF-β signaling pathways were all essential for S-mediated barrier dysfunction (13).
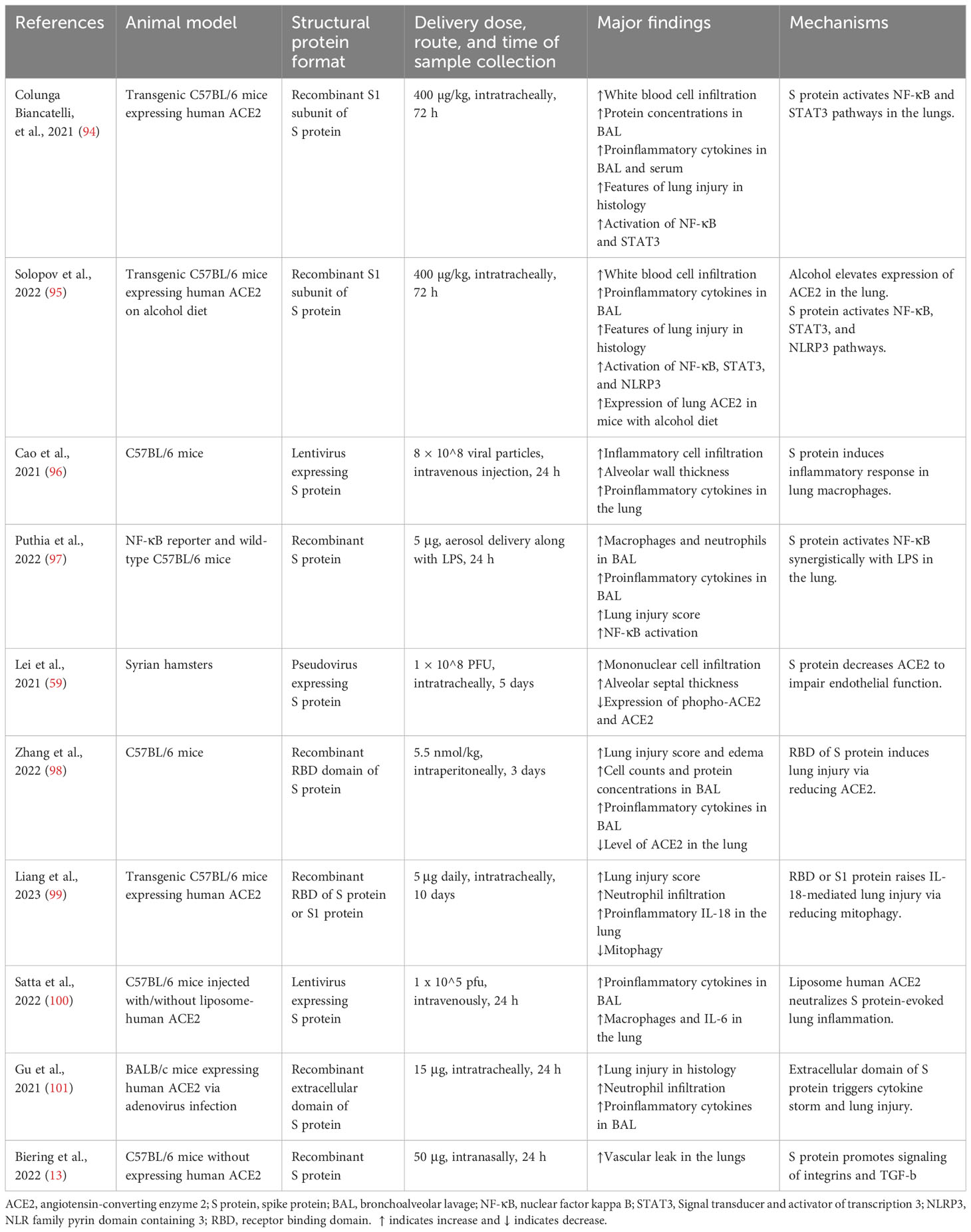
Table 1 Studies demonstrating the effects of SARS-CoV-2 structural proteins on acute lung injury in animal models.
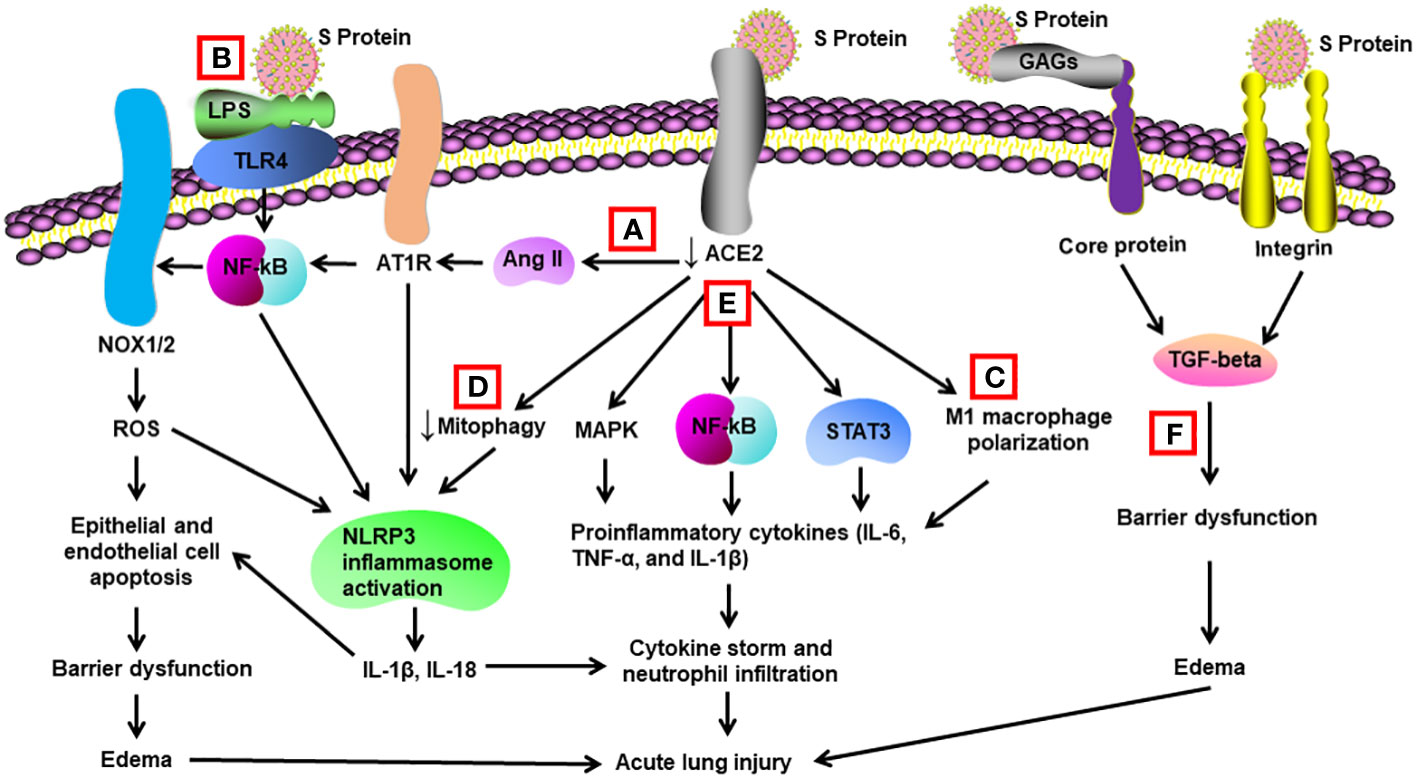
Figure 1 Schematic diagram of mechanisms for S protein-induced lung injury. (A) S protein binds with ACE2, leading to ACE2 shedding and downregulation. Reduced ACE2 promotes the activation of Ang II/angiotensin II type 1 receptor (AT1R)/NF-κB/NOX1/2 pathway, leading to generation of reactive oxygen species (ROS). ROS enhances apoptosis of alveolar epithelial cells and endothelial cells, resulting in barrier dysfunction and edema. Activated AT1R also activates NLR family pyrin domain containing 3 (NLRP3) inflammasome in alveolar macrophages and triggers cell apoptosis and cytokine storm. (B) S protein activates Toll-like receptors such as TLR4 through binding of lipopolysaccharide (LPS) and enhances NF-κB activity, resulting in activation of NLRP3 inflammasome and cytokine storm. (C) S protein promotes M1 polarization of alveolar macrophages through binding to ACE2, which enhances the proinflammatory response. (D) S protein reduces mitophagy through binding to ACE2, resulting in activation of NLRP3 inflammasome and cell apoptosis. (E) S protein binds to ACE2 and triggers activation of STAT3, MAP kinases (MAPK), and NF-κB via other unidentified mechanisms, leading to cytokine storm and neutrophil infiltration. (F) S protein triggers vascular leak and edema independent of ACE2 binding. S protein binds with glycosaminoglycans and integrins, which leads to the activation of TGF-β signaling pathway and barrier dysfunction.
S protein also causes injuries in other systems. Nasal inoculation of adenovirus vector expression S1 protein caused olfactory bulb damage and brain inflammation in mice via elevating calcium and decreasing intracerebral acetylcholine production (103). In mice with collagen-induced arthritis, Lee et al. showed that injection of a plasmid encoding S protein exacerbated arthritis via inducing inflammation, autoantibody, and thrombosis (104). Liang et al. documented that intratracheal delivery of S1 and RBD of S protein prompted cardiac dysfunction and elevated expression of IL-18 and NLRP3 in the heart. IL-18 inhibition alleviated S protein-induced cardiac dysfunction (99). Robles et al. discovered that S protein bound with integrin α5β1 in endothelial cells via RGD motif in the RBD domain. The binding activated NF-κB in endothelial cells, resulting in elevated expression of adhesion molecules (VCAM1 and ICAM1) and proinflammatory cytokines, as well as the hyperpermeability of the endothelial cells in vitro and in vivo (105). Another study observed that S protein elevated VEGF levels in enterocytes via Ras‐Raf‐MEK‐ERK pathway, enhancing vascular hyperpermeability and inflammation. S protein-induced intestinal inflammation was alleviated by both ERK and VEGF inhibitors in vivo (106).
Intratracheal administration of recombinant E protein induced the accumulation of inflammatory cells and cell death in the lungs of wild-type (WT) mice at 24 h, which was not present in the TLR2–/– mice. E protein also elevated proinflammatory cytokines in the BAL of WT mice. Blockage of TLR2 reduced SARS-CoV-2 virus-induced mortality and elevation in proinflammatory cytokines in mice (75) (Table 2) (Figure 2). Another group found that E protein formed pH-sensitive cation channels in an environment of lipid bilayer. Intravenous administration of recombinant E protein produced the hallmarks of acute lung injury with infiltration of inflammatory cells, pulmonary hemorrhage and edema, and interstitial hyperemia in mice at 72 h. There was also an upregulation of proinflammatory cytokines in the serum. In SARS-CoV-2-infected transgenic mice, administration of inhibitors for the channels decreased the viral load, extent of injury, and proinflammatory cytokines in the lungs (107).
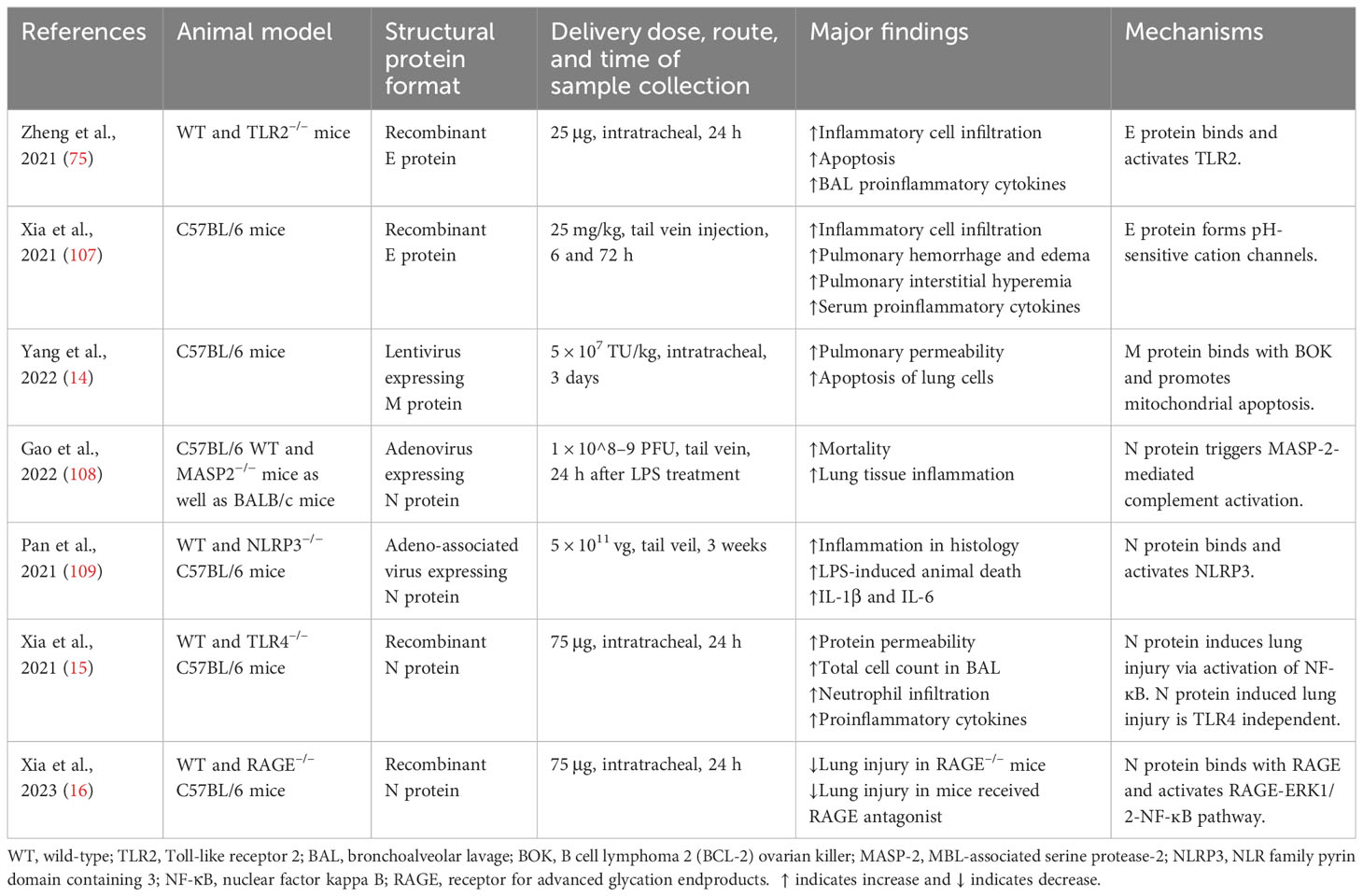
Table 2 Studies demonstrating the effects of SARS-CoV-2 structural proteins on acute lung injury in animal models.
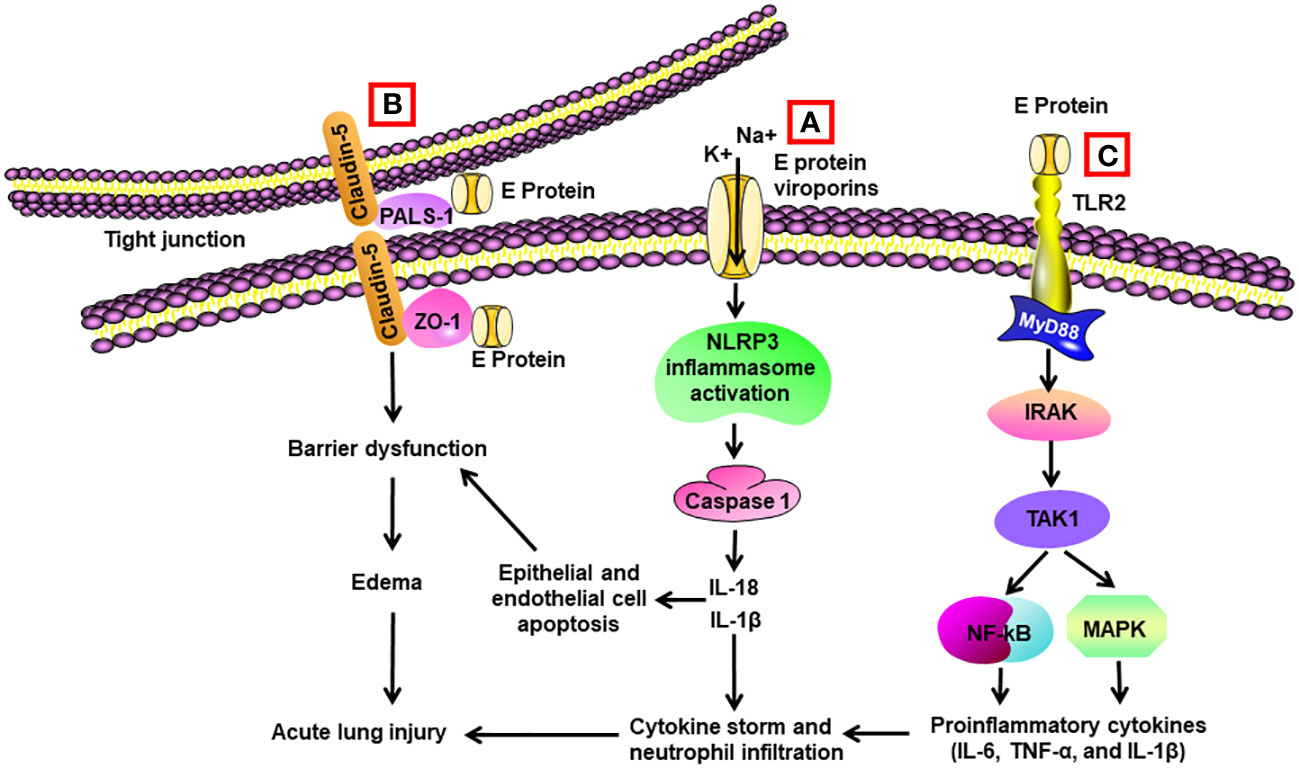
Figure 2 Schematic diagram of mechanisms for E protein-induced lung injury. (A) E protein forms ion-channeling viroporins for cations and activates NLRP3 inflammasome in alveolar macrophages, leading to caspase 1 activation and elevated levels of Il-1β and IL-18. IL-1β and IL-18 are the sources of cytokine storm. They also trigger apoptosis of alveolar epithelial cells and endothelial cells, resulting in barrier dysfunction and edema. (B) E protein binds with proteins associated with tight junctions such as zona occludens-1 (ZO-1) and PALS-1, leading to barrier dysfunction in epithelial and endothelial cells. (C) E protein binds with TLR2 and activates NF-κB and MAPK through activation of the IRAK/TAK1 pathway, resulting in cytokine storm.
Yang et al. reported that M protein bound to BH2 region of B cell lymphoma 2 (BCL-2) ovarian killer (BOK), blocked the ubiquitination of BOK, elevated BOK levels, and promoted mitochondrial apoptosis in vitro. Lentiviral expression of M protein elevated pulmonary permeability and prompted apoptosis of lung cells in vivo. Knockdown of BOK ameliorated alveolar-capillary permeability and pulmonary edema induced by M protein (14) (Table 2) (Figure 3).
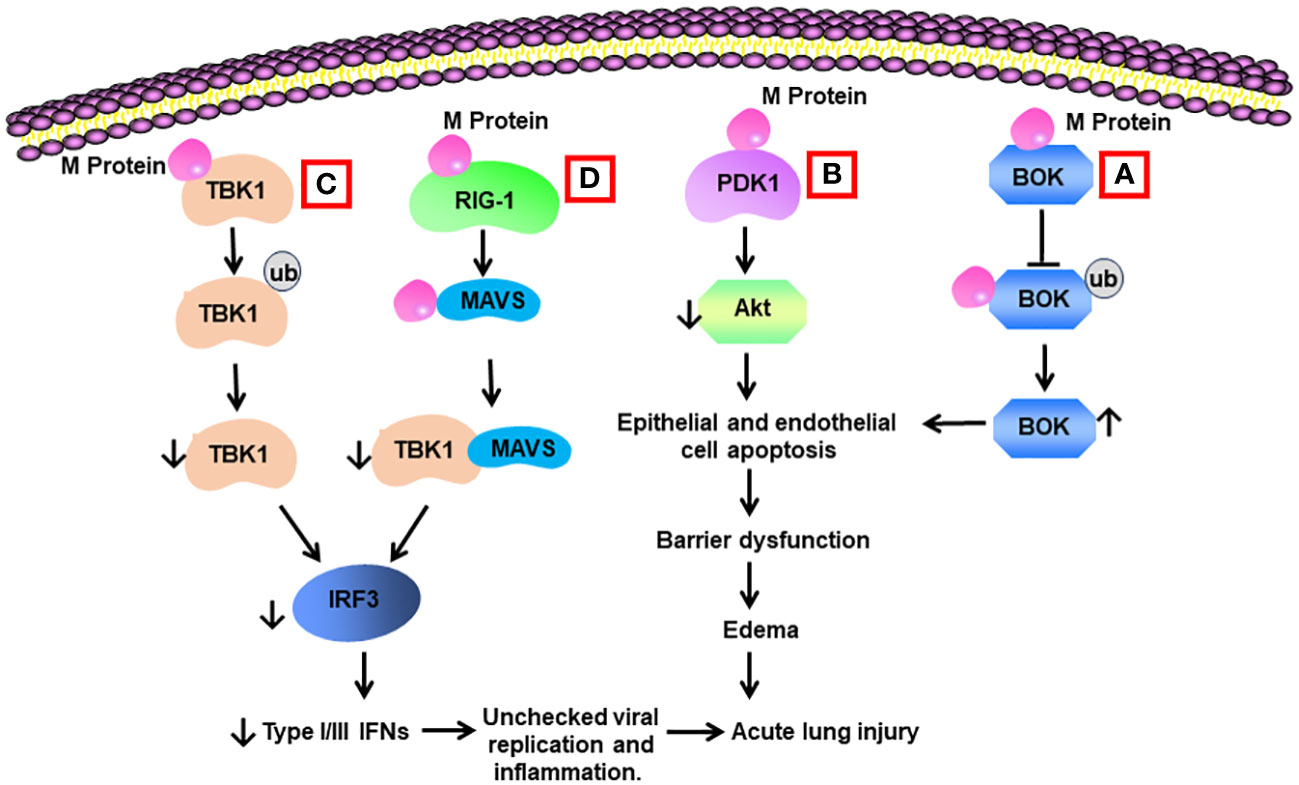
Figure 3 Schematic diagram of mechanisms for M protein-induced lung injury. (A) M protein binds with B cell lymphoma 2 (BCL-2) ovarian killer (BOK). The association inhibits the ubiquitination of BOK and increases BOK levels. BOK induces apoptosis of alveolar epithelial cells and endothelial cells, resulting in barrier dysfunction and edema. (B) M protein binds with phosphoinositide-dependent protein kinase-1 (PDK1) and downregulates the activity of AKT, leading to cell apoptosis. (C) M protein binds with TANK-binding kinase 1 (TBK1) and promotes its degradation via ubiquitination. Reduced levels of TBK1 cause decreased activity of interferon regulation factor 3 (IRF3), leading to low production of type I and type III IFNs. Low type I and type III IFNs contribute to unchecked viral replication and inflammation. (D) M protein binds with RIG-1, MAVS, and TBK1 and decreases the phosphorylation of IRF3 via blocking the association between MAVS and TBK1.
Gao et al. revealed that N protein bound and activated MBL-associated serine protease-2 (MASP-2), leading to activation of complements 3 and 5b-9. Adenovirus expressing N protein aggravated LPS-induced mortality and lung tissue inflammation in mice. The impact of N protein in vivo was blocked by an inhibitor of MASP-2 and antibodies for N protein and MASP-2 (108) (Table 2) (Figure 4). Pan et al. discovered that N protein bound with NLRP3 to enhance the assembly of NLRP3 inflammasome. Adeno-associated virus expressing N protein induced lung inflammation in histology, aggravated LPS-induced animal death, and elevated the expression of IL-1β and IL-6 in serum as well as in the lung. N protein-evoked lung injury was hindered by inhibitors for caspase-1 and NLRP3 (109). Our group showed that administration of recombinant N protein to C57BL/6 mice prompted acute lung injury, as reflected by increased protein permeability, proinflammatory cytokines, and infiltration of neutrophils in the BAL. N protein also induced M1 macrophage polarization of alveolar macrophages and phosphorylation of NF-ĸB p65 (15). Our group recently revealed that N protein is a ligand for RAGE. N protein triggered proinflammatory response via RAGE-ERK1/2-NF-ĸB pathway. In mice, RAGE knockout and inhibition partially alleviated N-protein-evoked lung injury (16). Wick et al. demonstrated that N protein levels of plasma samples harvested within 72 h of hospital admission were strongly associated with RAGE and correlated with ICU admission as well as mechanical ventilation at 28 days (110). Furthermore, Matthay et al. found that high levels of N protein and RAGE at admission were significantly correlated with the development of severe COVID-19 (111).
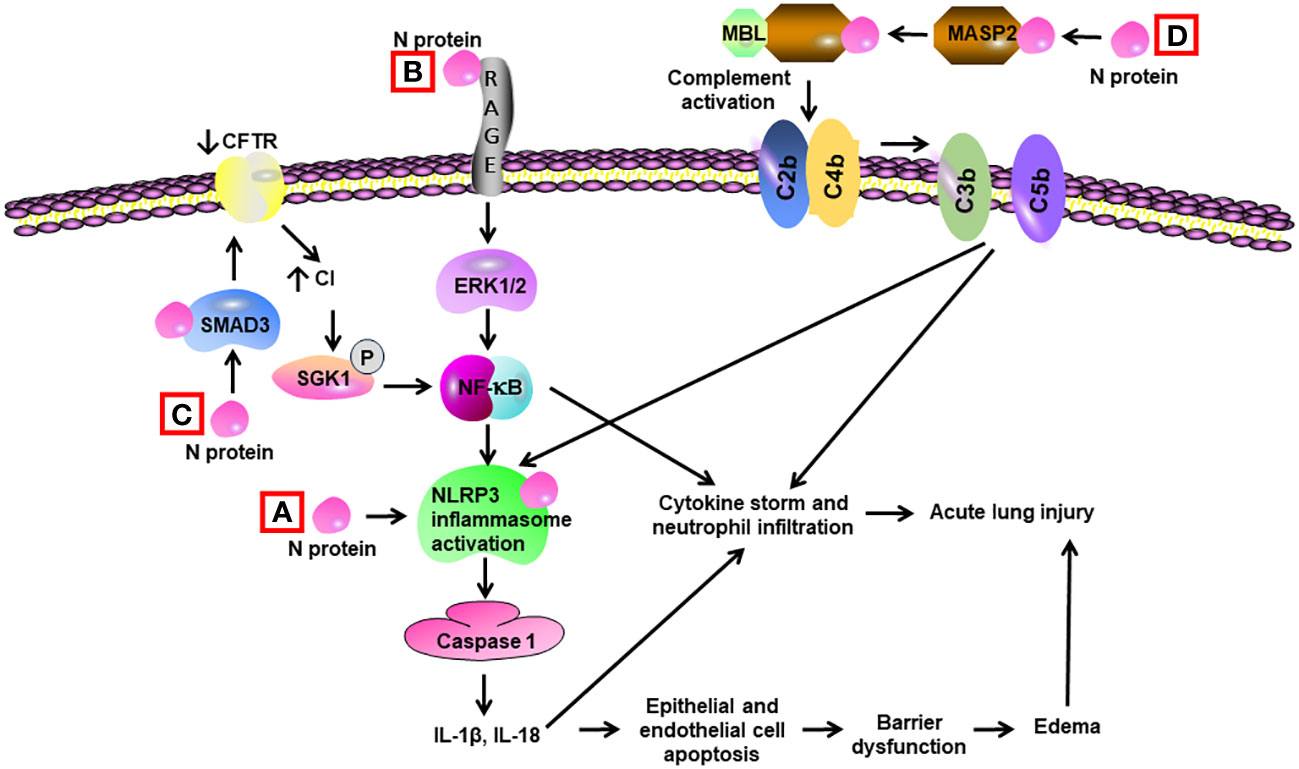
Figure 4 Schematic diagram of mechanisms for N protein-induced lung injury. (A) N protein binds and activates NLRP3 inflammasome in alveolar macrophages, leading to activation of caspase 1 and elevated levels of Il-1β and IL-18. These cytokines induce a cytokine storm and neutrophil infiltration. They also trigger apoptosis of alveolar epithelial cells and endothelial cells, resulting in barrier dysfunction and edema. (B) N protein binds with RAGE and subsequently activates ERK1/2 in alveolar macrophages. ERK1/2 triggers the activation of NF-κB, leading to cytokine storm. (C) N protein binds with SMAD3 and subsequently decreases the expression of cystic fibrosis transmembrane conductance regulator (CFTR), resulting in elevated intracellular Cl− concentration in airway epithelial cells. An increase in concentrations of Cl− causes phosphorylation of serum/glucocorticoid-regulated kinase 1 (SGK1) and activation of NF-κB. (D) N protein binds and activates the MBL-associated serine protease-2 (MASP-2), leading to activation of complement cascade via cleavage of C2 and C4 into C3/C5 convertases (C4bC2b). Complement activation leads to cytokine storm and activation of NLRP3 inflammasome.
Up to the present, there have been three zoonotic coronaviruses (SARS-CoV-1, MERS-CoV, and SARS-CoV-2) that cause human ARDS. With the ever-increasing intrusion of natural habits, it is foreseeable that novel coronavirus diseases will emerge and spread via the respiratory system. All four structural proteins of SARS-CoV-2 are essential in assembly and release of the virion. The N protein binds to the genomic RNA of SARS-CoV-2, while S is indispensable in viral attachment and entry to target cells. Existing findings have demonstrated that all four structural proteins of SARS-CoV-2 are able to trigger lung injury independent of viral infection. Much work remains to be performed to decipher the molecular mechanisms of structural protein-evoked lung injury and the implications for the injury in humans. Antibody cocktail of structural proteins may represent a new therapeutic tool for treating COVID-19.
GZ: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. GQ: Writing – original draft, Writing – review & editing. HQ: Writing – original draft, Writing – review & editing. QS: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. JX: Conceptualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Natural Science Foundation of Zhejiang Province (Grant No. LGF22H150010), the Health Commission of Zhejiang Province (Grant No. 2024KY491), the National Natural Science Foundation of China (Grant No. 82070074, 82272191, an 82370080), the Fundamental Research Funds for the Central Universities (Grant No. 226202200060), the Health Science Commission of Shaoxing (Grant No. 2023SKY 100 and 2023SKY 103).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
SARS-CoV-2, Severe acute respiratory syndrome coronavirus 2; ARDS, acute respiratory distress syndrome; CARDS, COVID-19-associated ARDS; S, spike; E, envelope; M, membrane; N, nucleocapsid; NSP, non-structural protein; ACE2, angiotensin converting enzyme 2; RAGE, receptor for advanced glycation endproducts; NF-ĸB, nuclear factor kappa B; RBD, receptor-binding domain; TMPRSS2, transmembrane protease serine 2; ERGIC, endoplasmic reticulum–Golgi intermediate compartment; NTD, N-terminal domain; CTD, C terminal domain; LPS, lipopolysaccharide; TLR2, Toll-like receptor 2; NLRP3, NLR family pyrin domain containing 3; TBK-1, TANK-binding kinase 1; BAL, bronchoalveolar lavage; BOK, B cell lymphoma 2 (BCL-2) ovarian killer; MASP-2, MBL-associated serine protease-2.
1. Ren LL, Wang YM, Wu ZQ, Xiang ZC, Guo L, Xu T, et al. Identification of a novel coronavirus causing severe pneumonia in human: a descriptive study. Chin Med J (Engl) (2020) 133:1015–24. doi: 10.1097/CM9.0000000000000722
2. Rahman MS, Hoque MN, Islam MR, Islam I, Mishu ID, Rahaman MM, et al. Mutational insights into the envelope protein of SARS-CoV-2. Gene Rep (2021) 22:100997. doi: 10.1016/j.genrep.2020.100997
3. Matheson NJ, Lehner PJ. How does SARS-CoV-2 cause COVID-19? Science (2020) 369:510–1. doi: 10.1126/science.abc6156
4. Nalbandian A, Sehgal K, Gupta A, Madhavan MV, McGroder C, Stevens JS, et al. Post-acute COVID-19 syndrome. Nat Med (2021) 27:601–15. doi: 10.1038/s41591-021-01283-z
5. Tzotzos SJ, Fischer B, Fischer H, Zeitlinger M. Incidence of ARDS and outcomes in hospitalized patients with COVID-19: a global literature survey. Crit Care (2020) 24:516. doi: 10.1186/s13054-020-03240-7
6. Morris G, Bortolasci CC, Puri BK, Olive L, Marx W, O'Neil A, et al. The pathophysiology of SARS-CoV-2: A suggested model and therapeutic approach. Life Sci (2020) 258:118166. doi: 10.1016/j.lfs.2020.118166
7. Afzali B, Noris M, Lambrecht BN, Kemper C. The state of complement in COVID-19. Nat Rev Immunol (2022) 22:77–84. doi: 10.1038/s41577-021-00665-1
8. Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, Zerbi P, et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis (2020) 20:1135–40. doi: 10.1016/S1473-3099(20)30434-5
9. Conway EM, Mackman N, Warren RQ, Wolberg AS, Mosnier LO, Campbell RA, et al. Understanding COVID-19-associated coagulopathy. Nat Rev Immunol (2022) 22:639–49. doi: 10.1038/s41577-022-00762-9
10. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese center for disease control and prevention. JAMA (2020) 323:1239–42. doi: 10.1001/jama.2020.2648
11. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med (2005) 11:875–9. doi: 10.1038/nm1267
12. Zhu YG, Qu JM. Differential characteristics of the early stage of lung inflammation induced by SARS-CoV Nucleocapsid protein related to age in the mouse. Inflammation Res (2009) 58:312–20. doi: 10.1007/s00011-009-8062-9
13. Biering SB, Gomes de Sousa FT, Tjang LV, Pahmeier F, Zhu C, Ruan R, et al. SARS-CoV-2 Spike triggers barrier dysfunction and vascular leak via integrins and TGF-beta signaling. Nat Commun (2022) 13:7630. doi: 10.1038/s41467-022-34910-5
14. Yang Y, Wu Y, Meng X, Wang Z, Younis M, Liu Y, et al. SARS-CoV-2 membrane protein causes the mitochondrial apoptosis and pulmonary edema via targeting BOK. Cell Death Differ (2022) 29:1395–408. doi: 10.1038/s41418-022-00928-x
15. Xia J, Tang W, Wang J, Lai D, Xu Q, Huang R, et al. SARS-CoV-2 N protein induces acute lung injury in mice via NF-kB activation. Front Immunol (2021) 12:791753. doi: 10.3389/fimmu.2021.791753
16. Xia J, Wang J, Ying L, Huang R, Zhang K, Zhang R, et al. RAGE is a receptor for SARS-CoV-2 N protein and mediates N protein-induced acute lung injury. Am J Respir Cell Mol Biol (2023) 69:508–20. doi: 10.1165/rcmb.2022-0351OC
17. Willett BJ, Grove J, MacLean OA, Wilkie C, De Lorenzo G, Furnon W, et al. SARS-CoV-2 Omicron is an immune escape variant with an altered cell entry pathway. Nat Microbiol (2022) 7:1161–79. doi: 10.1038/s41564-022-01143-7
18. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol (2020) 94. doi: 10.1128/JVI.00127-20
19. Seyran M, Takayama K, Uversky VN, Lundstrom K, Palu G, Sherchan SP, et al. The structural basis of accelerated host cell entry by SARS-CoV-2dagger. FEBS J (2021) 288:5010–20. doi: 10.1111/febs.15651
20. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol (2004) 203:631–7. doi: 10.1002/path.1570
21. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. Am J Respir Crit Care Med (2020) 202:756–9. doi: 10.1164/rccm.202001-0179LE
22. Grant RA, Morales-Nebreda L, Markov NS, Swaminathan S, Querrey M, Guzman ER, et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature (2021) 590:635–41. doi: 10.1038/s41586-020-03148-w
23. Liu J, Li Y, Liu Q, Yao Q, Wang X, Zhang H, et al. SARS-CoV-2 cell tropism and multiorgan infection. Cell Discovery (2021) 7:17. doi: 10.1038/s41421-021-00249-2
24. Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol (2022) 23:3–20. doi: 10.1038/s41580-021-00418-x
25. Hillen HS, Kokic G, Farnung L, Dienemann C, Tegunov D, Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature (2020) 584:154–6. doi: 10.1038/s41586-020-2368-8
26. Malone B, Urakova N, Snijder EJ, Campbell EA. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat Rev Mol Cell Biol (2022) 23:21–39. doi: 10.1038/s41580-021-00432-z
27. Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol (2020) 20:355–62. doi: 10.1038/s41577-020-0331-4
28. Santos-Mendoza T. The envelope (E) protein of SARS-coV-2 as a pharmacological target. Viruses (2023) 15. doi: 10.3390/v15041000
29. Schoeman D, Fielding BC. Coronavirus envelope protein: current knowledge. Virol J (2019) 16:69. doi: 10.1186/s12985-019-1182-0
30. DeDiego ML, Alvarez E, Almazan F, Rejas MT, Lamirande E, Roberts A, et al. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated. Vitro vivo J Virol (2007) 81:1701–13. doi: 10.1128/JVI.01467-06
31. Sun YS, Xu F, An Q, Chen C, Yang ZN, Lu HJ, et al. A SARS-CoV-2 variant with the 12-bp deletion at E gene. Emerg Microbes Infect (2020) 9:2361–7. doi: 10.1080/22221751.2020.1837017
32. Zhou S, Lv P, Li M, Chen Z, Xin H, Reilly S, et al. SARS-CoV-2 E protein: Pathogenesis and potential therapeutic development. BioMed Pharmacother (2023) 159:114242. doi: 10.1016/j.biopha.2023.114242
33. Wang WA, Carreras-Sureda A, Demaurex N. SARS-CoV-2 infection alkalinizes the ERGIC and lysosomes through the viroporin activity of the viral envelope protein. J Cell Sci (2023) 136. doi: 10.1242/jcs.260685
34. Nieto-Torres JL, DeDiego ML, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PloS Pathog (2014) 10:e1004077. doi: 10.1371/journal.ppat.1004077
35. Chai J, Cai Y, Pang C, Wang L, McSweeney S, Shanklin J, et al. Structural basis for SARS-CoV-2 envelope protein recognition of human cell junction protein PALS1. Nat Commun (2021) 12:3433. doi: 10.1038/s41467-021-23533-x
36. Boson B, Legros V, Zhou B, Siret E, Mathieu C, Cosset FL, et al. The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J Biol Chem (2021) 296:100111. doi: 10.1074/jbc.RA120.016175
37. Marques-Pereira C, Pires MN, Gouveia RP, Pereira NN, Caniceiro AB, Rosario-Ferreira N, et al. SARS-coV-2 membrane protein: from genomic data to structural new insights. Int J Mol Sci (2022) 23. doi: 10.3390/ijms23062986
38. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol (2015) 1282:1–23. doi: 10.1007/978-1-4939-2438-7_1
39. Scherer KM, Mascheroni L, Carnell GW, Wunderlich LCS, Makarchuk S, Brockhoff M, et al. SARS-CoV-2 nucleocapsid protein adheres to replication organelles before viral assembly at the Golgi/ERGIC and lysosome-mediated egress. Sci Adv (2022) 8:eabl4895. doi: 10.1126/sciadv.abl4895
40. Zhang Z, Nomura N, Muramoto Y, Ekimoto T, Uemura T, Liu K, et al. Structure of SARS-CoV-2 membrane protein essential for virus assembly. Nat Commun (2022) 13:4399. doi: 10.1038/s41467-022-32019-3
41. Yuan Z, Hu B, Xiao H, Tan X, Li Y, Tang K, et al. The E3 ubiquitin ligase RNF5 facilitates SARS-coV-2 membrane protein-mediated virion release. mBio (2021) 13:e0316821. doi: 10.1101/2021.02.28.433287
42. Wu W, Cheng Y, Zhou H, Sun C, Zhang S. The SARS-CoV-2 nucleocapsid protein: its role in the viral life cycle, structure and functions, and use as a potential target in the development of vaccines and diagnostics. Virol J (2023) 20:6. doi: 10.1186/s12985-023-01968-6
43. Savastano A, Ibanez de Opakua A, Rankovic M, Zweckstetter M. Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat Commun (2020) 11:6041. doi: 10.1038/s41467-020-19843-1
44. Peng Y, Du N, Lei Y, Dorje S, Qi J, Luo T, et al. Structures of the SARS-CoV-2 nucleocapsid and their perspectives for drug design. EMBO J (2020) 39:e105938. doi: 10.15252/embj.2020105938
45. Dinesh DC, Chalupska D, Silhan J, Koutna E, Nencka R, Veverka V, et al. Structural basis of RNA recognition by the SARS-CoV-2 nucleocapsid phosphoprotein. PloS Pathog (2020) 16:e1009100. doi: 10.1371/journal.ppat.1009100
46. Wu C, Qavi AJ, Hachim A, Kavian N, Cole AR, Moyle AB, et al. Characterization of SARS-CoV-2 nucleocapsid protein reveals multiple functional consequences of the C-terminal domain. iScience (2021) 24:102681. doi: 10.1016/j.isci.2021.102681
47. Tugaeva KV, Hawkins D, Smith JLR, Bayfield OW, Ker DS, Sysoev AA, et al. The mechanism of SARS-coV-2 nucleocapsid protein recognition by the human 14-3-3 proteins. J Mol Biol (2021) 433:166875. doi: 10.1016/j.jmb.2021.166875
48. Ni X, Han Y, Zhou R, Zhou Y, Lei J. Structural insights into ribonucleoprotein dissociation by nucleocapsid protein interacting with non-structural protein 3 in SARS-CoV-2. Commun Biol (2023) 6:193. doi: 10.1038/s42003-023-04570-2
49. Smits VAJ, Hernandez-Carralero E, Paz-Cabrera MC, Cabrera E, Hernandez-Reyes Y, Hernandez-Fernaud JR, et al. The Nucleocapsid protein triggers the main humoral immune response in COVID-19 patients. Biochem Biophys Res Commun (2021) 543:45–9. doi: 10.1016/j.bbrc.2021.01.073
50. Huang R, Ying L, Wang J, Xia J, Zhang Y, Mao H, et al. Non-spike and spike-specific memory T cell responses after the third dose of inactivated COVID-19 vaccine. Front Immunol (2023) 14:1139620. doi: 10.3389/fimmu.2023.1139620
51. Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem (2005) 280:30113–9. doi: 10.1074/jbc.M505111200
52. Guo X, Cao J, Cai JP, Wu J, Huang J, Asthana P, et al. Control of SARS-CoV-2 infection by MT1-MMP-mediated shedding of ACE2. Nat Commun (2022) 13:7907. doi: 10.1038/s41467-022-35590-x
53. Yeung ML, Teng JLL, Jia L, Zhang C, Huang C, Cai JP, et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell (2021) 184:2212–2228 e2212. doi: 10.1016/j.cell.2021.02.053
54. Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, et al. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci U.S.A. (2008) 105:7809–14. doi: 10.1073/pnas.0711241105
55. Mariappan V, Ranganadin P, Shanmugam L, Rao SR, Balakrishna Pillai A. Early shedding of membrane-bounded ACE2 could be an indicator for disease severity in SARS-CoV-2. Biochimie (2022) 201:139–47. doi: 10.1016/j.biochi.2022.06.005
56. Gao X, Zhang S, Gou J, Wen Y, Fan L, Zhou J, et al. Spike-mediated ACE2 down-regulation was involved in the pathogenesis of SARS-CoV-2 infection. J Infect (2022) 85:418–27. doi: 10.1016/j.jinf.2022.06.030
57. Iwasaki M, Saito J, Zhao H, Sakamoto A, Hirota K, Ma D. Inflammation triggered by SARS-coV-2 and ACE2 augment drives multiple organ failure of severe COVID-19: molecular mechanisms and implications. Inflammation (2021) 44:13–34. doi: 10.1007/s10753-020-01337-3
58. Lu Y, Zhu Q, Fox DM, Gao C, Stanley SA, Luo K. SARS-CoV-2 down-regulates ACE2 through lysosomal degradation. Mol Biol Cell (2022) 33:ar147. doi: 10.1091/mbc.E22-02-0045
59. Lei Y, Zhang J, Schiavon CR, He M, Chen L, Shen H, et al. SARS-coV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ Res (2021) 128:1323–6. doi: 10.1161/CIRCRESAHA.121.318902
60. Villacampa A, Alfaro E, Morales C, Diaz-Garcia E, Lopez-Fernandez C, Bartha JL, et al. SARS-CoV-2 S protein activates NLRP3 inflammasome and deregulates coagulation factors in endothelial and immune cells. Cell Commun Signal (2024) 22:38. doi: 10.1186/s12964-023-01397-6
61. Paik S, Kim JK, Silwal P, Sasakawa C, Jo EK. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol (2021) 18:1141–60. doi: 10.1038/s41423-021-00670-3
62. Petruk G, Puthia M, Petrlova J, Samsudin F, Stromdahl AC, Cerps S, et al. SARS-CoV-2 spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J Mol Cell Biol (2020) 12:916–32. doi: 10.1093/jmcb/mjaa067
63. Khan S, Shafiei MS, Longoria C, Schoggins JW, Savani RC, Zaki H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-kappaB pathway. Elife (2021) 10. doi: 10.7554/eLife.68563
64. Umar S, Palasiewicz K, Meyer A, Kumar P, Prabhakar BS, Volin MV, et al. Inhibition of IRAK4 dysregulates SARS-CoV-2 spike protein-induced macrophage inflammatory and glycolytic reprogramming. Cell Mol Life Sci (2022) 79:301. doi: 10.1007/s00018-022-04329-8
65. Zhao Y, Kuang M, Li J, Zhu L, Jia Z, Guo X, et al. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res (2021) 31:818–20. doi: 10.1038/s41422-021-00495-9
66. Patra T, Meyer K, Geerling L, Isbell TS, Hoft DF, Brien J, et al. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PloS Pathog (2020) 16:e1009128. doi: 10.1371/journal.ppat.1009128
67. Barhoumi T, Alghanem B, Shaibah H, Mansour FA, Alamri HS, Akiel MA, et al. SARS-coV-2 coronavirus spike protein-induced apoptosis, inflammatory, and oxidative stress responses in THP-1-like-macrophages: potential role of angiotensin-converting enzyme inhibitor (Perindopril). Front Immunol (2021) 12:728896. doi: 10.3389/fimmu.2021.728896
68. Li F, Li J, Wang PH, Yang N, Huang J, Ou J, et al. SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ROS-suppressed PI3K/AKT/mTOR signaling. Biochim Biophys Acta Mol Basis Dis (2021) 1867:166260. doi: 10.1016/j.bbadis.2021.166260
69. Olajide OA, Iwuanyanwu VU, Lepiarz-Raba I, Al-Hindawi AA. Induction of exaggerated cytokine production in human peripheral blood mononuclear cells by a recombinant SARS-coV-2 spike glycoprotein S1 and its inhibition by dexamethasone. Inflammation (2021) 44:1865–77. doi: 10.1007/s10753-021-01464-5
70. Farag NS, Breitinger U, Breitinger HG, El Azizi MA. Viroporins and inflammasomes: A key to understand virus-induced inflammation. Int J Biochem Cell Biol (2020) 122:105738. doi: 10.1016/j.biocel.2020.105738
71. Shepley-McTaggart A, Sagum CA, Oliva I, Rybakovsky E, DiGuilio K, Liang J, et al. SARS-CoV-2 Envelope (E) protein interacts with PDZ-domain-2 of host tight junction protein ZO1. PloS One (2021) 16:e0251955. doi: 10.1371/journal.pone.0251955
72. Toto A, Ma S, Malagrino F, Visconti L, Pagano L, Stromgaard K, et al. Comparing the binding properties of peptides mimicking the Envelope protein of SARS-CoV and SARS-CoV-2 to the PDZ domain of the tight junction-associated PALS1 protein. Protein Sci (2020) 29:2038–42. doi: 10.1002/pro.3936
73. Ju J, Su Y, Zhou Y, Wei H, Xu Q. The SARS-CoV-2 envelope protein disrupts barrier function in an in vitro human blood-brain barrier model. Front Cell Neurosci (2022) 16:897564. doi: 10.3389/fncel.2022.897564
74. Yalcinkaya M, Liu W, Islam MN, Kotini AG, Gusarova GA, Fidler TP, et al. Modulation of the NLRP3 inflammasome by Sars-CoV-2 Envelope protein. Sci Rep (2021) 11:24432. doi: 10.1038/s41598-021-04133-7
75. Zheng M, Karki R, Williams EP, Yang D, Fitzpatrick E, Vogel P, et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol (2021) 22:829–38. doi: 10.1038/s41590-021-00937-x
76. Su W, Ju J, Gu M, Wang X, Liu S, Yu J, et al. SARS-CoV-2 envelope protein triggers depression-like behaviors and dysosmia via TLR2-mediated neuroinflammation in mice. J Neuroinflamm (2023) 20:110. doi: 10.1186/s12974-023-02786-x
77. Zhou S, Yu Z, Chen Z, Ning F, Hu X, Wu T, et al. Olmesartan alleviates SARS-CoV-2 envelope protein induced renal fibrosis by regulating HMGB1 release and autophagic degradation of TGF-beta1. Front Pharmacol (2023) 14:1187818. doi: 10.3389/fphar.2023.1187818
78. Makris S, Paulsen M, Johansson C. Type I interferons as regulators of lung inflammation. Front Immunol (2017) 8:259. doi: 10.3389/fimmu.2017.00259
79. Galani IE, Rovina N, Lampropoulou V, Triantafyllia V, Manioudaki M, Pavlos E, et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat Immunol (2021) 22:32–40. doi: 10.1038/s41590-020-00840-x
80. Sui L, Zhao Y, Wang W, Wu P, Wang Z, Yu Y, et al. SARS-coV-2 membrane protein inhibits type I interferon production through ubiquitin-mediated degradation of TBK1. Front Immunol (2021) 12:662989. doi: 10.3389/fimmu.2021.662989
81. Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun (2020) 11:3810. doi: 10.1038/s41467-020-17665-9
82. Zheng Y, Zhuang MW, Han L, Zhang J, Nan ML, Zhan P, et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct Target Ther (2020) 5:299. doi: 10.1038/s41392-020-00438-7
83. Ren Y, Wang A, Fang Y, Shu T, Wu D, Wang C, et al. SARS-coV-2 membrane glycoprotein M triggers apoptosis with the assistance of nucleocapsid protein N in cells. Front Cell Infect Microbiol (2021) 11:706252. doi: 10.3389/fcimb.2021.706252
84. Chen L, Guan WJ, Qiu ZE, Xu JB, Bai X, Hou XC, et al. SARS-CoV-2 nucleocapsid protein triggers hyperinflammation via protein-protein interaction-mediated intracellular Cl(-) accumulation in respiratory epithelium. Signal Transduct Target Ther (2022) 7:255. doi: 10.1038/s41392-022-01048-1
85. Wu JL, Kuan II, Guo JY, Hsu WC, Tang WC, Chan HJ, et al. SARS-CoV-2 N protein mediates intercellular nucleic acid dispersion, a feature reduced in Omicron. iScience (2023) 26:105995. doi: 10.1016/j.isci.2023.105995
86. Lopez-Munoz AD, Kosik I, Holly J, Yewdell JW. Cell surface SARS-CoV-2 nucleocapsid protein modulates innate and adaptive immunity. Sci Adv (2022) 8:eabp9770. doi: 10.1126/sciadv.abp9770
87. Karwaciak I, Salkowska A, Karas K, Dastych J, Ratajewski M. Nucleocapsid and spike proteins of the coronavirus SARS-coV-2 induce IL6 in monocytes and macrophages-potential implications for cytokine storm syndrome. Vaccines (Basel) (2021) 9. doi: 10.3390/vaccines9010054
88. Karwaciak I, Karas K, Salkowska A, Pastwinska J, Ratajewski M. Chlorpromazine, a clinically approved drug, inhibits SARS-coV-2 nucleocapsid-mediated induction of IL-6 in human monocytes. Molecules (2022) 27. doi: 10.3390/molecules27123651
89. Qian Y, Lei T, Patel PS, Lee CH, Monaghan-Nichols P, Xin HB, et al. Direct activation of endothelial cells by SARS-coV-2 nucleocapsid protein is blocked by simvastatin. J Virol (2021) 95:e0139621. doi: 10.1128/JVI.01396-21
90. Wu Y, Ma L, Cai S, Zhuang Z, Zhao Z, Jin S, et al. RNA-induced liquid phase separation of SARS-CoV-2 nucleocapsid protein facilitates NF-kappaB hyper-activation and inflammation. Signal Transduct Target Ther (2021) 6:167. doi: 10.1038/s41392-021-00575-7
91. Gori Savellini G, Anichini G, Gandolfo C, Cusi MG. SARS-coV-2 N protein targets TRIM25-mediated RIG-I activation to suppress innate immunity. Viruses (2021) 13. doi: 10.3390/v13081439
92. Zheng Y, Deng J, Han L, Zhuang MW, Xu Y, Zhang J, et al. SARS-CoV-2 NSP5 and N protein counteract the RIG-I signaling pathway by suppressing the formation of stress granules. Signal Transduct Target Ther (2022) 7:22. doi: 10.1038/s41392-022-00878-3
93. Wang S, Dai T, Qin Z, Pan T, Chu F, Lou L, et al. Targeting liquid-liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat Cell Biol (2021) 23:718–32. doi: 10.1038/s41556-021-00710-0
94. Colunga Biancatelli RML, Solopov PA, Sharlow ER, Lazo JS, Marik PE, Catravas JD. The SARS-CoV-2 spike protein subunit S1 induces COVID-19-like acute lung injury in Kappa18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells. Am J Physiol Lung Cell Mol Physiol (2021) 321:L477–84. doi: 10.1152/ajplung.00223.2021
95. Solopov PA, Colunga Biancatelli RML, Catravas JD. Alcohol increases lung angiotensin-converting enzyme 2 expression and exacerbates severe acute respiratory syndrome coronavirus 2 spike protein subunit 1-induced acute lung injury in K18-hACE2 transgenic mice. Am J Pathol (2022) 192:990–1000. doi: 10.1016/j.ajpath.2022.03.012
96. Cao X, Tian Y, Nguyen V, Zhang Y, Gao C, Yin R, et al. Spike protein of SARS-CoV-2 activates macrophages and contributes to induction of acute lung inflammation in male mice. FASEB J (2021) 35:e21801. doi: 10.1096/fj.202002742RR
97. Puthia M, Tanner L, Petruk G, Schmidtchen A. Experimental model of pulmonary inflammation induced by SARS-coV-2 spike protein and endotoxin. ACS Pharmacol Transl Sci (2022) 5:141–8. doi: 10.1021/acsptsci.1c00219
98. Zhang L, Zhang Y, Qin X, Jiang X, Zhang J, Mao L, et al. Recombinant ACE2 protein protects against acute lung injury induced by SARS-CoV-2 spike RBD protein. Crit Care (2022) 26:171. doi: 10.1186/s13054-022-04034-9
99. Liang S, Bao C, Yang Z, Liu S, Sun Y, Cao W, et al. SARS-CoV-2 spike protein induces IL-18-mediated cardiopulmonary inflammation via reduced mitophagy. Signal Transduct Target Ther (2023) 8:108. doi: 10.1038/s41392-023-01368-w
100. Satta S, Meng Z, Hernandez R, Cavallero S, Zhou T, Hsiai TK, et al. An engineered nano-liposome-human ACE2 decoy neutralizes SARS-CoV-2 Spike protein-induced inflammation in both murine and human macrophages. Theranostics (2022) 12:2639–57. doi: 10.7150/thno.66831
101. Gu T, Zhao S, Jin G, Song M, Zhi Y, Zhao R, et al. Cytokine signature induced by SARS-CoV-2 spike protein in a mouse model. Front Immunol (2020) 11:621441. doi: 10.3389/fimmu.2020.621441
102. Satis H, Ozger HS, Aysert Yildiz P, Hizel K, Gulbahar O, Erbas G, et al. Prognostic value of interleukin-18 and its association with other inflammatory markers and disease severity in COVID-19. Cytokine (2021) 137:155302. doi: 10.1016/j.cyto.2020.155302
103. Oka N, Shimada K, Ishii A, Kobayashi N, Kondo K. SARS-CoV-2 S1 protein causes brain inflammation by reducing intracerebral acetylcholine production. iScience (2023) 26:106954. doi: 10.1016/j.isci.2023.106954
104. Lee AR, Woo JS, Lee SY, Lee YS, Jung J, Lee CR, et al. SARS-CoV-2 spike protein promotes inflammatory cytokine activation and aggravates rheumatoid arthritis. Cell Commun Signal (2023) 21:44. doi: 10.1186/s12964-023-01044-0
105. Robles JP, Zamora M, Adan-Castro E, Siqueiros-Marquez L. Martinez de la Escalera G, Clapp C: The spike protein of SARS-CoV-2 induces endothelial inflammation through integrin alpha5beta1 and NF-kappaB signaling. J Biol Chem (2022) 298:101695. doi: 10.1016/j.jbc.2022.101695
106. Zeng FM, Li YW, Deng ZH, He JZ, Li W, Wang L, et al. SARS-CoV-2 spike spurs intestinal inflammation via VEGF production in enterocytes. EMBO Mol Med (2022) 14:e14844. doi: 10.15252/emmm.202114844
107. Xia B, Shen X, He Y, Pan X, Liu FL, Wang Y, et al. SARS-CoV-2 envelope protein causes acute respiratory distress syndrome (ARDS)-like pathological damages and constitutes an antiviral target. Cell Res (2021) 31:847–60. doi: 10.1038/s41422-021-00519-4
108. Gao T, Zhu L, Liu H, Zhang X, Wang T, Fu Y, et al. Highly pathogenic coronavirus N protein aggravates inflammation by MASP-2-mediated lectin complement pathway overactivation. Signal Transduct Target Ther (2022) 7:318. doi: 10.1038/s41392-022-01133-5
109. Pan P, Shen M, Yu Z, Ge W, Chen K, Tian M, et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun (2021) 12:4664. doi: 10.1038/s41467-021-25015-6
110. Wick KD, Leligdowicz A, Willmore A, Carrillo SA, Ghale R, Jauregui A, et al. Plasma SARS-CoV-2 nucleocapsid antigen levels are associated with progression to severe disease in hospitalized COVID-19. Crit Care (2022) 26:278. doi: 10.1186/s13054-022-04153-3
111. Matthay ZA, Fields AT, Wick KD, Jones C, Lane HC, Herrera K, et al. Association of SARS-CoV-2 nucleocapsid viral antigen and the receptor for advanced glycation end products with development of severe disease in patients presenting to the emergency department with COVID-19. Front Immunol (2023) 14:1130821. doi: 10.3389/fimmu.2023.1130821
Keywords: SARS-CoV-2, COVID-19, acute respiratory distress syndrome (ARDS), structural proteins, acute lung injury
Citation: Zheng G, Qiu G, Qian H, Shu Q and Xu J (2024) Multifaceted role of SARS-CoV-2 structural proteins in lung injury. Front. Immunol. 15:1332440. doi: 10.3389/fimmu.2024.1332440
Received: 02 November 2023; Accepted: 22 January 2024;
Published: 05 February 2024.
Edited by:
Zhiyong Li, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Zhanbo Zhu, Heilongjiang Bayi Agricultural University, ChinaCopyright © 2024 Zheng, Qiu, Qian, Shu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Shu, c2h1cWlhbmdAemp1LmVkdS5jbg==; Jianguo Xu, anh1NUB5YWhvby5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.