 Fernando Alvarez
Fernando Alvarez Zhiyang Liu
Zhiyang Liu Alexandre Bay
Alexandre Bay Ciriaco A. Piccirillo
Ciriaco A. Piccirillo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 28 March 2024
Sec. T Cell Biology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1331846
This article is part of the Research TopicRegulatory T cells in Immune-mediated diseasesView all 14 articles
Foxp3+ TREG cells have been at the focus of intense investigation for their recognized roles in preventing autoimmunity, facilitating tissue recuperation following injury, and orchestrating a tolerance to innocuous non-self-antigens. To perform these critical tasks, TREG cells undergo deep epigenetic, transcriptional, and post-transcriptional changes that allow them to adapt to conditions found in tissues both at steady-state and during inflammation. The path leading TREG cells to express these tissue-specialized phenotypes begins during thymic development, and is further driven by epigenetic and transcriptional modifications following TCR engagement and polarizing signals in the periphery. However, this process is highly regulated and requires TREG cells to adopt strategies to avoid losing their regulatory program altogether. Here, we review the origins of tissue-resident TREG cells, from their thymic and peripheral development to the transcriptional regulators involved in their tissue residency program. In addition, we discuss the distinct signalling pathways that engage the inflammatory adaptation of tissue-resident TREG cells, and how they relate to their ability to recognize tissue and pathogen-derived danger signals.
The immune system is capable of both effectively eliminating internal and external dangers and preventing exacerbated immune-mediated tissue pathology. These biological properties, coined disease resistance and disease tolerance, respectively, are complementary and require a controlled balance between pro-inflammatory and regulatory immune responses (1). This is particularly the case in mammalian hosts, where adaptive immunity allows antigen specificity to sustain long-lasting effector and memory responses that can become a potential threat to the function and homeostasis of an affected tissue long after the elimination of the danger. Amongst the mechanisms capable of controlling inflammation-generated pathology, a lymphocyte of thymic origin, a suppressor or regulatory T cell (TREG), first described in the late 1960s (2), was shown to be particularly adept at immune suppression. These CD4+ T cells express the Forkhead-Box P3 (Foxp3), a lineage-defining transcription factor that governs a large part of their transcriptional program through the repression of pro-inflammatory genes (e.g. Il2, Ifng) and the activation of genes essential for their suppressive functions (e.g. Il2ra (CD25), Ctla4, Lag3, Entpd1 (CD39), Nt5a (CD73), Il10, Tgfb1, Gzmb) (3, 4). In addition, some key signature genes are prominently expressed by these cells when compared to conventional T cells, including Ikzf2 (Helios), Tnfrsf18 (GITR), Nrp1 (Neuropilin 1), and Itgae (CD103) (5). In their capacity, TREG cells occupy a central position in the immune response, and are required to ensure tolerance to self-antigens (6, 7), innocuous allergens (8, 9), and commensal microflora (10), promote tissue function and regeneration (11), and prevent and control immunopathology (12).
In a mature immune system, TREG cells isolated from tissues encompass a pool of antigen-experienced CD45RA−CD69+CD45RO+ cells that differ in developmental origin, possess unique functions, and display distinct stages of activation (13). A prominent population of TREG cells found in all organs are tissue-resident TREG (TR-TREG) cells that differ from effector memory TREG cells (emTREG) in that they display higher levels of the alpha E integrin (CD103) (14), lose CCR7 expression, and lose the ability to re-circulate to lymphoid organs (15). Despite the lack of a consensus on the markers to distinguish TR-TREG and emTREG cells in tissues, recent studies have been able to capture the high degree of transcriptional and post-transcriptional modifications that “precursor” TR-TREG cells acquire to localize to non-lymphoid organs, survive, and adjust their specialized functions in situ amidst unfavorable inflammatory, osmotic, or metabolic conditions (16). This program involves the expression of a set of core genes that are typically upregulated, albeit at different levels, by TR-TREG isolated from distinct organs, including the expression of the IL-33 receptor ST2 (17), RORα (18, 19), Icos (20, 21) and Gata3 (22–24). Amongst these differentially expressed proteins, ST2 was recently proposed to distinguish TR-TREG from emTREG (17). Moreover, while there is evidence TR-TREG cells seed non-lymphoid organs, such as the lungs, as early as 8 days of life (25, 26), other TR-TREG cells, like visceral adipose tissue TREG (VAT-TREG), accumulate progressively with age (27), suggesting a highly dynamic and developmental path that is largely organ-specific. Critically, there is novel evidence on the developmental trajectory that lead TR-TREG cells to fully establish in the tissue. For example, recent evidence highlights how the TCR repertoire is a central determinant of TR-TREG localisation (16, 28).
Currently, much remains to be understood regarding the origin of TR-TREG cells. Can TR-TREG cells be generated from emTREG cells after the resolution of inflammatory events (29), or do they constitute stable and distinct populations of TREG cells? Seeing that TREG cells found in tissues can originate from the thymic selection process (thymic-derived; tTREG) or be generated from the induction of Foxp3 in naïve CD4+ T cells in the periphery (peripherally-induced; pTREG), can both subsets be considered TR-TREG cells? Thus, a better understanding of the origin, function, and fate of TR-TREG cells is required before we can harness their therapeutic potential.
In this review, we describe the steps required for the generation of TR-TREG, starting from thymic selection and spanning to TCR engagement in the periphery, the switch to distinct metabolic strategies, and the modulation of Foxp3 expression that enables the adoption of key epigenetic and transcriptional changes, which, in turn, lead to the expression of a program that is highly adapted to the target tissue (Figure 1). These processes involve signaling pathways that can, when in excess, hinder, either temporarily or permanently, the stability of their core transcriptional program, revealing mechanisms by which local inflammation guides the timing and potency of immune suppression. Finally, we attempt to guide the reader through the unique signaling events that can lead tissue-resident TREG cells to control type 1, type 2, and type 3-driven inflammation.
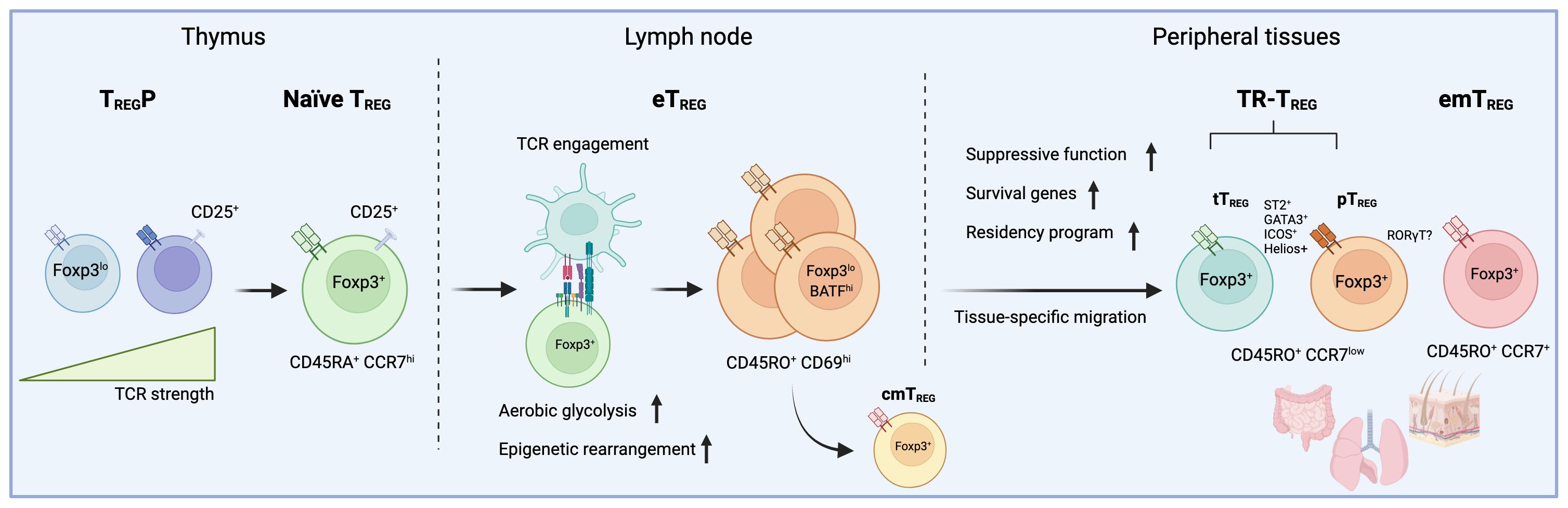
Figure 1 The developmental trajectory of tissue-resident TREG cells involves a series of events starting from thymic selection to peripheral TCR engagement. In this figure, the trajectory of peripheral regulatory T (TREG) cells is depicted, as currently defined by recent multi-omics approaches conducted in various lymphoid and non-lymphoid tissues. During thymic selection, precursor regulatory T cells (TREGP) expressing self-reactive T-cell receptors (TCR) give rise to a pool of naive CD45RA+CCR7hi regulatory T cells (TREG). Once in circulation, these TREG cells encounter their specific antigen, triggering an activation cascade that results in a metabolic shift and chromatin remodeling. Subsequently, CD45RO+CD69hi effector regulatory T cells (eTREG) can either stay in lymph nodes as central memory (cmTREG) or migrate to tissues, where they become tissue-resident (TR-TREG) or effector memory regulatory T cells (emTREG). While thymic-derived TR-TREG cells comprise a large portion of TREG cells in tissues, TREG located in the gut, for example, include peripherally-induced regulatory T cells (pTREG). The absence of clear markers poses a challenge in distinguishing between these two populations in situ. In addition, while TR-TREG cells isolated from various tissues typically display a conserved phenotype marked by the expression of ICOS, ST2, Helios, and GATA3, a significant portion of TREG cells in the gut exhibit a distinctive RORγT-driven phenotype. Interestingly, there is cumulating evidence that TREG cells lacking Helios expression may be more driven to express RORγT, suggesting a possible segregation between TR-TREG cells derived from the thymus or induced in the periphery.
Commitment of the TREG cell lineage can occur at various stages of the T cell life cycle. During their development in the thymus, immature thymocytes are selected for the establishment of a functional TCR repertoire. Subsequently, self-reactive thymocytes are either clonally deleted or diverged into a regulatory cell fate as part of a process known as central tolerance. Despite this, a very small fraction of thymocytes escape central tolerance stochastically and must be kept in check by self-reactive thymic-derived TREG cells (tTREG), making them critical mediators of peripheral tolerance. Importantly, the events giving rise to tTREG cells require optimal TCR signals and a unique combination of cytokines. However, the peptide pool to which thymocytes are exposed to during this selection process does not ensure complete tolerance towards innocuous non-self-antigens such as commensal bacterial peptides or allergens.
This type of peripheral tolerance often requires the in situ induction of peripheral TREG cells (pTREG) that possess unique non-self TCR repertoires (30–32) and confer them with non-redundant roles in maintaining homeostatic conditions at barrier sites like the lung and colon. In adoptive transfer models, pTREG cells are capable of suppressing local inflammation in both the colon and the lungs (32–34), but are less efficient at suppressing systemic inflammation (31). Indeed, the distinct transcriptional profiles of tTREG and pTREG cells indicate they favour different suppressive mechanisms that vary in effectiveness in a context-dependent manner (31). Yet, despite these potential differences, attempts at identifying markers in pTREG cells that are distinct from tTREG have so far failed (35, 36), rendering them mostly undistinguishable at barrier sites. While Helios and Neuropilin 1 (Nrp1) are highly expressed by tTREG cells (30, 37), neither Helios (36) nor Nrp1 (38), were found to be exclusively expressed by these cells. Thus, despite their distinct origin, TCR repertoire, and functions, pTREG cells cannot be distinguished from the pool of tTREG cells in mucosal tissues, and further investigation into features that demarcate each subset is warranted.
Thymic-derived TREG cells undergo the same early core processes of thymic selection as conventional CD4+ T cells (39, 40). Namely, newly seeded thymocytes undergo V(D)J recombination in the thymic cortex to generate productive TCR chains capable of self-MHC recognition. Upon successful TCR signaling, committed thymocytes migrate into the thymic medulla where they encounter medullary thymic epithelial cells (mTECs) that express promiscuous transcription factors AIRE and Fezf2, allowing them to transcribe and present tissue-restricted antigens (TRAs) to developing thymocytes (41, 42). Here, thymocytes that are strongly reactive toward TRAs and other self-antigens are deleted, while weaker stimulation and the presence of certain cytokines such as TGF-β and IL-2 can skew their fate toward TREG cell differentiation (43–47).
Optimal TCR signaling is the predominant factor driving TREG cell lineage commitment in the thymus. TCR:peptide-MHC interaction triggers a series of phosphorylation events resulting in downstream activation of NFAT, AP-1, and NF-κB family transcription factors (48, 49). Together, these events lead to different T cell lineage specification in the thymus, as well as T cell survival, expansion, and effector function in the periphery. Expression level of the orphan nuclear receptor Nur77 (Nr4a1) has been directly linked to TCR signaling strength, and its expression level is elevated in TREG cells compared to conventional T cells in a TCR-dependent manner (50). Unsurprisingly, since co-stimulatory molecules such as CD28 profoundly augment TCR signaling strength via NF-κB activation, they were found to play an essential role in tTREG cell differentiation (46, 51–53). Foxp3 transcription is intricately regulated by transcription factor complexes binding at its promoter and four conserved noncoding sequences (CNS), termed CNS0 to CNS3. Upon TCR stimulation, downstream activation of the NF-κB pathway results in the recruitment of c-Rel to the Foxp3 locus at CNS3, which acts as a Foxp3 transcriptional enhancer that is responsive to TCR signaling alone (54, 55). By dissecting each CNS region through targeted mutations, Zheng and colleagues demonstrated that CNS3 is the region that acts as a pioneer element to the generation of tTREG cells, while CNS1, a region known to bind TGF-β-induced SMAD factors, and CNS2, a region targeted by CREB and STAT5 signals, were not essential to the induction of Foxp3 in tTREG precursors (55), which still require cytokine signaling to become mature and functional tTREG cells (43–47).
Cytokines, particularly common γ (γc) cytokines, are critical for TREG cell development. IL-2 is known to be essential for commitment to the TREG cell lineage (56, 57), as well as its maintenance (58). IL-2 signaling mediates STAT5 binding to the distal enhancer CNS0 as well as the promoter of Foxp3 (56, 59), and sustains the constitutive expression of Foxp3 through CNS2 binding (57, 59). Not only does STAT5 directly facilitate Foxp3 transcription, Foxp3 also binds to the IL-2 receptor alpha chain (IL2Rα) as a transcriptional activator (60). Completion of this feedforward loop via paracrine IL-2 signaling is obligatory for TREG cell development and homeostasis. Other STAT5-activating γc cytokines have also been linked to TREG cell development, albeit mostly as a compensatory mechanism for impaired IL-2 signaling (45). In addition, TGF-β has also been implicated in tTreg development. While either of its downstream transcription factors, SMAD2 or SMAD3, can directly regulate Foxp3 transcription (61, 62), deletion of the SMAD binding site in the Foxp3 locus predominantly affects the induction of pTREG, but not tTREG cells (62, 63). Yet, deletion of the TGF-β receptor TβRI during thymocyte development results in severely reduced TREG cell numbers and defective TREG cell function (64). Nonetheless, a recent study might reconcile the paradoxical discoveries. SMAD3/4 can trigger a PKA-dependent signaling cascade that causes the cessation of TCR signaling (65). Thus, the role of TGF-β in tTREG differentiation could most likely be attributed to its effects on TCR signaling rather than direct transcriptional regulation of Foxp3.
In recent years, accumulating evidence shows that the nature of TCR signaling during thymic selection influences TREG cell response to signals long after thymus egress. Notably, TCR engagement during thymic selection is a critical step in the establishment of a CpG hypomethylation pattern that characterises the epigenetic background of tTREG cells (66). Numerous studies have identified two distinct tTREG precursor (TREGP) populations thought to develop into CD25+Foxp3+ tTREG cells (47, 67–69). The more common CD25- Foxp3low and less abundant CD25+ Foxp3- TREGP cells were shown to have distinct TCR repertoires with affinity to auto-antigens (67). In the thymus, the two TREGP populations display different cytokine and TCR-signaling requirements (47). Importantly, CD25+ TREGP-derived TREG cells are specifically capable of suppressing experimental autoimmune encephalitis (EAE), whereas Foxp3low TREGP-derived TREG cells cannot (67), suggesting a functional bias within the TREG population. For example, murine TREG cells from the colonic lamina propria that express the same TCRα/β sequence have related transcriptional programs (70), illustrating the close relationship between TCR and the transcriptional fate of antigen-experienced memory TREG cells.
Interestingly, while the relationship between TCR specificity and the establishment of TR-TREG cells is not entirely understood, there are experimental examples that suggest the TCR repertoire generated during thymic selection is critical to the destination of both tTREG and naïve T cells. For example, TREG cells transgenic for a VAT-TREG-derived TCRα/TCRβ gene arrangement will preferentially migrate to adipose tissue and differentiate into VAT TREG cells (28). Yet, while these observations suggest TR-TREG cells possess a largely self-specific TCR repertoire, earlier work in viral infection mouse models demonstrated that antigen-experienced TREG cells with predominantly non-self TCR repertoires are generated during tissue injury and activate during re-infection (13, 71), suggesting they also contribute to the TR-TREG pool. In addition, in transgenic mice possessing a fixed TCR-β sequence isolated from a Foxp3+RORγT+ colonic TREG cell, T cells upregulate Foxp3 in the mesenteric lymph node prior to expressing RORγT in the colon (72). As such, both self and non-self-reactive TCR repertoires are key drivers in the generation of TR-TREG cells.
While the strength of TCR signaling acts as the predominant driving force for tTREG cell differentiation, cytokines play a more influential role in the periphery both in maintaining tTREG homeostasis and generating pTREG cells. The signals that lead to the generation of pTREG cells involve chronic suboptimal TCR signaling (73–75) and cytokines such as TGF-β and IL-2 to generate Foxp3-expressing TREG cells in vitro (76, 77) and in tissues (78–80). In addition, TGF-β has been shown to strongly promote Foxp3 induction through its downstream transcription factors (SMAD2 or SMAD3) that target CNS1 (61). Consequently, a deletion of CNS1 predominantly affects the induction of pTREG, but not tTREG cells (63). Lastly, pTREG cell induction via TGF-β can be further augmented by DC-derived retinoic acid in the lamina propria as well as short chain fatty acid metabolites of commensal bacteria (81, 82), ensuring the establishment of tolerance at mucosal surfaces. While these examples of signals that promote pTREG induction are part of a complex signaling system that merits its own review, they share the common outcome of facilitating Foxp3 expression in tissue-resident T cells, and further the importance of this transcription factor in forming the regulatory program of tissue-resident CCR7lowCD69+CD45RO+ TREG cells.
The factors that regulate the differentiation of TR-TREG remain to be fully understood. Miragaia and colleagues demonstrated through single-cell RNA-seq analysis of lymphoid and non-lymphoid (colon and skin) TREG cells that these tissue-specific adaptations originate from events happening in their respective draining lymph node (19). By tracing TCR clonotypes from draining lymph nodes to their respective tissue, the authors were able to establish a pseudo-space relationship detailing the series of events that drive the generation of specialized TREG cells. They were able to establish that TREG cells are activated, switch to a glycolytic metabolism, and cycle rapidly prior to acquiring genes involved in migration to the tissue (19), revealing conserved stages involved in the generation of TR-TREG cells. As such, this seminal work provided confirmation that progressive transcriptional changes guide the generation of eTREG cells that become TR-TREG cells and highlighted how, despite tissue-specific differences, these cells share a series of epigenetic modifications that allow them to migrate, survive, and function at specific non-lymphoid sites.
The engagement of the TCR of naïve TREG cells is an important prerequisite for the development of tissue-specialized TREG cells (83, 84), as it promotes a signaling cascade that elicits the expression of key regulatory genes leading to the suppressive activity of TREG cells (85). Additionally, TCR engagement can induce epigenetic and transcriptional changes in TREG cells, some of which are directly influenced by Foxp3, while others act independently (66). People affected by a loss-of-function mutations in STIM1 or ORAI1, proteins involved in store-operated calcium entry (SOCE), encounter a loss of peripheral tolerance despite some cases displaying normal TREG numbers in circulation (86, 87). Similarly, impairing the normal Ca2+ influx during TCR engagement by deleting proteins that form the Ca2+ release-activated Ca2+ (CRAC) channels (STIM1 and STIM2) in mice specifically prevents the differentiation of activated TREG cells into follicular and tissue-resident memory TREG cells and generates a cascade of inflammation leading to multiorgan autoimmunity (88).
Another critical factor involved in the differentiation and clonal expansion of activated TREG cells is the adoption of aerobic glycolysis. This was notably demonstrated in the skin, as aerobic glycolysis by activated TREG cells is required prior to their migration (89). This may, at first glance, seem counter-intuitive, as there is ample evidence that mature TREG cells adopt fatty-acid oxidation (FAO) as a critical metabolic strategy to survive and suppress immune responses in tissues (90). Yet, while less efficient than oxidative phosphorylation (OXPHOS), adopting aerobic glycolysis is a critical step that occurs during T cell activation by rapidly providing the needed energy for expansion and migration, all-the-while maintaining fatty acid and amino acid reserves for cell division and protein synthesis (91). This is further evidenced by the fact that the mammalian target of rapamycin 1 (mTORC1) which is required for aerobic glycolysis, is not necessary for the thymic or peripheral development of TREG cells, but essential to the function and activation of TREG cells (92). Indeed, to avoid losing their suppressive program, TREG cells balance the intensity of the mTORC1 and mTORC2 pathways (93), a process that is critical during their differentiation. Importantly, however, increasing glycolytic metabolism in TREG cells temporarily deprives them of their suppressive capacity (90, 94), providing further evidence that the differentiation and clonal expansion of TREG cells is contained within a short window of time. As such, the maturational process leading TREG to become eTREG cells requires both TCR engagement and a shift in their metabolic strategy (Figure 1).
The Foxp3-driven transcriptome of TREG cells is comprised of a TREG-specific gene signature and a gene set associated with an activation program which is shared with conventional T cells (95). A lymphoproliferative pathology had been previously observed in “Scurfy” mice where the X-linked Foxp3 gene encountered a frame-shift mutation that completely disrupts the transcription of Foxp3 (96), confirming the key role of Foxp3 in establishing the suppressive program of TREG cells. Point mutations in Foxp3 that interfere with its function are the cause of a frequently fatal pediatric hereditary syndrome called immune dysregulation, polyendocrinopathy, enteropathy X-linked (IPEX) syndrome (97), featuring early onset diabetes, severe diarrhea, and eczema, which highly reflects the pathology of ‘Scurfy” mice. Restoring Foxp3 transcription in mice whose TREG cells were genetically engineered to block Foxp3 expression rescues them from severe autoimmunity as it effectively reinstates their suppressive function (12). However, while Foxp3 is essential for the establishment of TREG cells, it does not determine, by itself, the entire epigenetic and transcriptional identity of mature TREG cells (5, 98, 99). Rather, Foxp3 ensures that inflammatory and non-inflammatory signals encountered in the periphery do not destabilise the core suppressive program of TREG cells (98, 100).
Evidence for the unique roles of Foxp3 in non-lymphoid tissues comes from the observation that functional single nucleotide polymorphisms (SNPs) in the human Foxp3 gene do not generate a homogeneous pathology (97), with multiple accounts of IPEX-related mutations having distinct functional consequences on TREG cells (101). By transposing human-isolated Foxp3 mutations in conserved murine Foxp3 motifs, Leon and colleagues confirmed that spontaneous multiorgan autoimmunity is largely attributed to mutations in the DNA-binding motifs, while mutations outside these motifs, notably in the N-terminal regions, lead to organ-specific dysregulation of TREG cell function (101). In particular, a K199del mutation situated in the zing-finger (ZF) domain or mutations R51Q or C168Y in the N-terminal regions are prone to generating symptoms of enteropathy and skin disorders, while a R337Q mutation in the DNA-binding Fork-head domain can, in addition to these symptoms, lead to the development of diabetes mellitus (101). In addition, a murine model mimicking an A384 mutation in Foxp3 was shown to specifically impair TREG cell function in the periphery, directly impairing the ability of Foxp3 to recognize target genes and altering BATF expression (102), a key transcription factor required for TR-TREG generation (103). As such, the ability of Foxp3 to interact with multiple partners is required to preserve the functional integrity of TREG cells in peripheral tissues.
Although there are elements that suggest protein-protein interactions are critical to this process, we are currently limited in our understanding of how the different molecular complexes that partner with the N-terminal region of Foxp3 (104, 105), such as Tip60, Hdac7, Hdac9, Gata3, c-Rel, Foxp3, Runx1 or Eos, influence the specialization of TREG cells. This is imparted by the fact that it is particularly difficult to dissociate their functions during the early events leading to the differentiation of these cells and the events that happen later in the tissues. One such example is the interaction of Foxp3 with the chromatin remodeling transcription factors TCF1 (encoded by Tcf7) and lymphoid enhancer binding factor 1 (Lef1) of the high-mobility group (HMG) family. In mice, the combined knock-out of both Tcf7 and Lef1 (Foxp3CRETcf7fl/flLef1fl/fl) does not perturb lymphoid TREG cells but hinders the capacity of colonic TREG cells to suppress DSS-mediated colitis (106). Mechanistically, the molecular complexes TCF1 and Lef1 form with Foxp3 allow TREG cells to control inflammation by repressing genes associated with excessive cycling and cytotoxic function (GzmB, Prf1, Ifng) and promoting genes associated to a TREG suppressive program (106). Bulk RNAseq of murine mesenteric TREG cells deficient in TCF1 (Foxp3CRETcf7fl/fl) show enhanced expression of core genes (including Il2ra, Foxp3, Tgfb1 and Lef1), and a concomitant increase in both pro-inflammatory genes (including Il6ra, Ifngr2, Stat3) and genes involved in TCR activity compared to TREG cells from control mice (107). These data suggest that TCF1 helps maintain a core TREG program and suppress the expression of pro-inflammatory genes during TCR engagement. Similarly, Lef1 is part of an activated TREG program (108), and in vitro gain-of-function experiments reveal it reinforces the expression of Foxp3 target genes (108). As such, these observations indicate that when Foxp3 is abundantly expressed, it interacts with both TFs to suppress pro-inflammatory gene expression and reinforce its own transcriptional profile (109). Yet, both murine and human activated (CD45RO+) TREG cells display lower Tcf7 and Lef1 expression than conventional T cells (TCONV) (110) as Foxp3 directly suppresses Tcf7 transcription and protein production, and reduces chromatin accessibility in regions targeted by TCF1 (95). As such, the highly-regulated chromatin-remodelling effect of TCF1 and Lef1 on TREG cells are likely required for their further differentiation and effector function. Furthermore, pseudo-time analysis from single cell RNA-seq data of lymphoid and non-lymphoid activated TREG cells reveals Tcf7 and Lef1 to be particularly expressed by lymphoid TREG cells prior to their tissue migration (19), reinforcing the notion that TCF1 and Lef1 are involved during the early specialization events of TREG cells. For example, a TREG-specific depletion of Lef1 abolishes the generation of follicular TREG (TFR) (107), suggesting Lef1 promotes the generation of these cells in a process similar to what is observed in follicular helper T cells (TFH) (111). In addition, when compared to murine activated TCF1- TREG cells, TCF1+ TREG cells display higher mRNA expression of transcription factors associated to helper T cells, including Gata3, Tbx21 and Rorc (107). Collectively, these examples highlight how changes in chromatin accessibility in TREG cells happen mostly after TCR engagement in the lymph node. Nonetheless, Lef1 and TCF1 are but a part of a wide network of known Foxp3-binding partners (104) whose role in defining the specialisation of TREG cells remain ill-defined.
To effectively reach the tissue, TREG must undergo a series of epigenetic and transcriptional changes that ensure chromatin accessibility in key genes (112). Interestingly, direct comparison between human and murine TREG cells reveal evolutionarily conserved epigenetic mechanisms involved in defining a TREG cell program (110). Histone methylation is an important component in the processes that govern DNA accessibility and, ultimately, a TREG cell transcriptional signature. Importantly, while TREG cells undergo a series of chromatin remodeling events, they actively maintain CpG motif demethylation within the intronic enhancer CNS2 of the Foxp3 locus (55, 113, 114). Maintaining an open chromatin structure in the CNS2 allows for the robust transcription of Foxp3 by multi-molecular complexes including Foxp3 itself, NFAT, c-Rel, STAT5, Runx1-CBFβ, CREB/ATFx and Ets1 (114–118). Incidentally, a loss of any of these transcription factors or the methylation of CNS2 impairs the transcription of Foxp3 and, ultimately, the suppressive function of TREG cells in the periphery (114–119), confirming that Foxp3 is critical for the stability of the transcriptional program of tissue-localised TREG cells.
Tagmentation-based whole-genome bisulfide sequencing of lymph node and tissue-isolated murine TREG reveals these cells undergo multiple rounds of DNA alterations before adopting a tissue-residency program, with up to 4000 genes involved in either gain or loss of methylation (120). The processes that govern the establishment of a TREG program by histone modifications have been elegantly reviewed by Joudi and colleagues (121). Globally, a delicate balance between DNA methyltransferases (DNMTs), ten-eleven translocation dioxygenases (TET), histone acetyltransferases (HATs), and histone deacetylases (HDAC) govern the stability of the TREG cell transcriptional program (119, 121), but can be directly influenced by polarizing signals provided during TCR engagement.
Methylation of cytosines located in CpG-rich regions are largely governed by Dnmt1, Dnmt3a and Dnmt3b (122, 123). Interestingly, the conditional deletion of Dnmt1, but not Dnmt3a, in murine TREG cells causes a loss of peripheral tolerance by 3 to 4 weeks of life, yet the cells maintain their expression of Foxp3 (124). However, these TREG cells display enhanced expression of pro-inflammatory cytokines (IFNγ, IL-6, IL-12, IL-17, IL-22), chemokine receptors (CCR1, CXCR6), and transcription factors (Runx2, Stat3), highlighting the role of Dnmt1 as a non-redundant epigenetic silencer (124). During the S phase, Dnmt1 acts in partnership with the epigenetic regulator ubiquitin-like with plant homeodomain and RING finger domains 1 (Uhrf1) to govern the suppression of these gene loci (125, 126), making both Dnmt1 and Uhrf1 important therapeutical targets for the control of TREG stability and function. Yet, because of the necessity of TREG cells to acquire a set of genes associated with pro-inflammatory T cells, it remains to be understood how both regulators act during TREG cell generation. For example, pharmacological inhibition of PI3K through its PIP4K-associated kinase results in a specific decrease in Uhrf1 in human TREG cells but not TCONV cells (127), suggesting that the strength of TCR signaling plays a role in the way TREG cells govern DNA accessibility of pro-inflammatory genes. In addition, signaling by TGF-β leads to the phosphorylation and subsequent sequestration of Uhrf1 outside the nucleus (128), possibly preventing its partnering with Dnmt1.
On the other hand, the modulation of histone acetylation and deacetylation on the epigenetic adaptation of TREG cells remains ill-defined. Foxp3+ TREG cells have been found to express histone acetyltransferases (HAT), including p300, Tip60 and CBP, as well as most members of the histone deacetylase family (HDAC) (129). Pan-HDAC inhibitors, for example, promote the acetylation of Foxp3 and the suppressive functions of TREG cells (130), confirming the importance of regulating histone acetylation to maintain a TREG transcriptional program. Interestingly, HATs and HDACs are clearly involved in the helper differentiation of TCONV cells (131), and further investigation is required to understand how they govern the differentiation of TREG cells.
During these early differentiating steps, some transcriptional regulators are found to be particularly critical for the generation of TR-TREG over other emTREG subsets. At its core, the acquisition of a tissue residency program of TR-TREG cells is closely matched to the expression of basic leucine zipper ATF-like transcription factor (BATF) and its downstream targets (16). Delacher and colleagues identified a BATF-dependent transcriptional program that drives, notably, the expression of the IL-33 receptor ST2 (120), a receptor specifically found in TR-TREG (17). A TREG-specific BATF deficiency in mice (Foxp3CREBatffl/fl; BATF-/-), results in a multiorgan autoimmune disease with death initiating at 6 weeks of age (103). BATF-/- TREG cells fail to accumulate in the lungs, colon, liver, and spleen, and display reduced chromatin accessibility to genes involved in TREG survival in tissue, including Gata3, Irf4, Ikzf4, Ets1 and Icos (103). In addition, Foxp3CREBatffl/fl mice generate exTREG cells that lose TREG-associated genes (Ctla4, Tgfb1, Foxp3) and adopt inflammatory genes (Rorc, Il6ra, Stat3) (103). Specifically, ATAC-seq of murine BATFWT and BATF-/- TREG cells reveals BATF acts as a chromatin regulator, facilitating the expression of TR-TREG-associated genes, including Ctla4, Icos, Gata3, and Irf4, and preserving the demethylated state of the CNS2 region of Foxp3 (103), positioning BATF as the epigenetic guardian of TREG cells as they undergo their differentiation into specialized memory TREG cells.
Another transcription factor (TF) observed to be highly expressed by TREG cells following TCR engagement is the interferon regulatory factor 4 (Irf4) (132). Foxp3 can directly promote the transcription of Irf4 (133) and the BATF-JUN complex (134). In turn, Irf4 collaborates with BATF to further promote TREG activation, proliferation, and transcriptional differentiation (135). Ding and colleagues demonstrated that upon TCR engagement, TREG cells express the SUMO-conjugating enzyme UBC9 to specifically stabilise Irf4 function (136). While not affecting thymic development of murine TREG cells, a TREG-specific deletion of UBC9 causes an early and fatal inflammatory disorder at 3 weeks of age (136), mimicking the dynamics observed in scurfy mice (96). These TREG cells show defects in TCR activation, migration, and peripheral accumulation (136). However, we do not observe such dramatic outcomes when knocking out Irf4 in murine TREG cells, suggesting other factors may compensate for the loss of Irf4. Mice harboring a conditional knock-down (Foxp3CRE Irf4fl/fl) develop spontaneous dermatitis, blepharitis, and lymphadenopathy disease by 5-6 weeks, and die by 3-4 months from a mostly TH2-mediated autoimmune disease (133). Co-immunoprecipitation of Irf4 and Foxp3 shows that both TF interact to, facilitate the transcription of genes such as Icos, Il1rl1, Maf and Ccr8 (133). In addition, Irf4 allows TREG cells to exert their suppressive functions. For example, a knock-out or a disruption of Irf4 expression in murine or human TREG cells, impacts the expression of key suppressive genes, including Il10 (137). Moreover, while there is evidence Irf4 is an important contributor during the early transcriptional events involved in the specialisation of activated TREG cells, this TF is also readily detected in some populations of memory TREG cells in the tissue, suggesting its expression is maintained long after TCR engagement. Finally, BATF and Irf4 are particularly upregulated in relation to the strength of the TCR signal (138, 139), and, together, directly suppress Foxp3 transcription in TREG cells induced in vitro (139). Collectively, these observations imply that BATF and Irf4 hinder Foxp3 transcription during the early events that define eTREG formation (Figure 2).
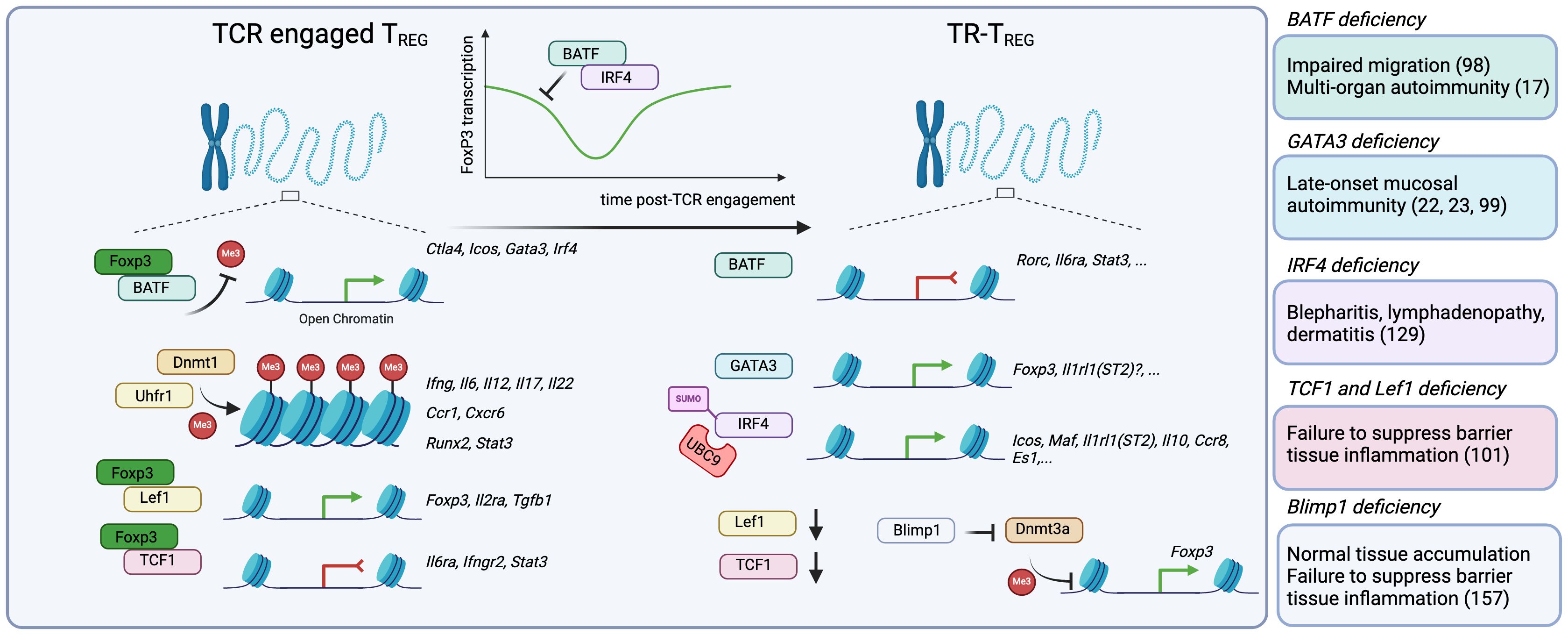
Figure 2 The acquisition of a tissue-resident program requires a series of epigenetic and transcriptional changes that involve modulation of Foxp3 expression or activity. After thymic egress into the periphery, TREG cells are TCR-activated by self or non-self-antigens, and undergo a series of epigenetic and transcriptional changes that guide their maturation into TR-TREG cells. While not entirely understood, this process seems to happen in a step-wise manner. First, TCR-engaged TREG cells upregulate key transcriptional programs in part driven by the transcription factor BATF, which, in conjunction with Foxp3, promotes the accessibility of Foxp3 and expression of BATF-driven genes including Ctla4, Icos, Gata3, Irf4. Key to the stability of their epigenetic landscape, TREG cells require Dnmt1 and its partner Uhrf1 to promote the methylation of CpG-rich regions and control the accessibility to inflammatory genes, including Ifng, Il6, Il12, Il17a, Il22, Ccr1, Cxcr6, Runx2 and Stat3. Finally, Foxp3 partners with Lef1 to promote the expression of genes involved in its core program, including Foxp3, Il2ra and Tgfb1, and also with TCF1 to suppress the expression of genes associated with inflammation like Il6ra, Ifngr2 and Stat3. Importantly, BATF and IRF4 can, in turn, suppress Foxp3 expression, a process that, while not fully understood, may enable the temporal accessibility of genes normally repressed by FoxP3. Once in the tissue, BATF enables the continued suppression of genes like Rorc (RORγT), Il6ra and Stat3. GATA3 promotes the transcription of Foxp3, but may be further involved in the expression of other GATA3-associated genes, like Il1rl1(ST2). IRF4 is also required for the expression of core TR-TREG genes, including Icos, Il1rl1 and Il10. Moreover, there is evidence that Lef1 and Tcf7 (TCF1) mRNA expression are significantly decreased in TR-TREG cells, suggesting they are no longer required. Finally, BLIMP-1 expression is increased, and can actively inhibit the action of Dnmt3a, promoting the accessibility of key genes in TREG cells such as Foxp3. Consistently, murine models with Foxp3-conditional deletion of BATF, GATA3, IRF4, TCF1 and BLIMP-1 reveal how critical these regulators are for the function of TR-TREG cells.
As discussed above, the pool of TREG cells residing in tissues is highly dependent on the organ and is composed in adults of both TR-TREG and emTREG cells whose fate remains ill-defined. Moreover, while the establishment of a peripheral TREG population in mucosal tissues happens in a relatively short amount of time after birth, this is not the case for VAT TREG cells that follow a more gradual accumulation (27), complexifying our understanding of the events that govern TR-TREG accumulation. Notably, fate-mapping systems (Foxp3eGFPCreERT2x ROSA26STOP-eYFP) in neonate mice reveal that TREG cells seed non-lymphoid organs like the lungs and liver in the first 8 days of life, persisting for up to 12 weeks with little renewal (25). Critically, exposure to an inflammatory event prior to day 8, but not after, significantly reduces TCR diversity of liver and lung TR-TREG and causes long-lasting alterations to their transcriptional program (25), revealing how critical the neonatal period is to the establishment of tissue homeostasis. Here, the establishment of TR-TREG cells is heavily dependent on the acquisition of a core of transcriptional factors. Single-cell RNA-seq (19), bulk RNA-seq (17), microarray and ATAC-seq (16, 112) analysis of TREG from visceral adipose tissue (VAT), lung, skin or colon reveal the epigenetic and transcriptional landscape of these cells is primarily determined by the organ, with only a small set of core genes shared between them. In various non-lymphoid tissues, TR-TREG cells express a shared a set of core genes, including Il1rl1 (ST2), Gata3, Tnfrsf4, Rora, Il10 and Gzmb (16, 19). On the other hand, there is a significant difference in gene expression between the transcriptional signature and DNA methylation profile of colonic and skin-isolated TREG, including increased Dgat2, a gene involved in lipid synthesis (16, 19), in skin TR-TREG cells, revealing these cells acquire tissue-specific abilities that allow them to persist in these microenvironments.
Following TCR engagement and clonal expansion, the development of TR-TREG involves the adoption of migratory properties through the acquisition and loss of chemokine receptors and other adhesion molecules. Indeed, as they undergo deep transcriptional changes and rapid clonal expansion, they also begin to express chemokine receptors that lead them to egress from the lymph node and migrate to a selected tissue. As with other T cells, activated TREG cells downregulate the surface expression of the L-selectin CD62L and upregulate the expression of the glycoprotein type I CD44 (132). Similarly, TREG cells from human tumors (140) and skin (141), as well as murine TREG cells isolated from multiple non-lymphoid organs (15), display low levels of CCR7, preventing their recirculation in lymphoid organs (142). However, the combination of chemokine receptors TR-TREG cells possess is specific to the type of tissue these cells travelled to. In adult mice, RNA sequencing of two distinct populations of TREG cells isolated from barrier tissues reveals that CCR7- TREG possess an organ-specific chemokine receptor signature, regardless of their expression of the IL-33 receptor ST2 (17), suggesting that the migration of all TR-TREG cells is determined by a shared group of chemokine receptors. This combination of chemokine receptors can also be appreciated in the seminal work by Miragaia and colleagues, as they observed skin-localised TREG cells preferentially expressed Ccr6, while colonic TREG cells displayed higher levels of Ccr1 and Ccr5; yet, both subsets showed similar levels of Ccr4, Ccr8 and Ccxr4 (19). Unfortunately, we have yet to determine which combination of chemokine receptors is part of their migratory program and which are locally upregulated to provide further movement inside the tissues.
Interestingly, while these experiments highlight the transcriptional diversity of TR-TREG, so did they help identify a core identity that govern their residency program (19). Some members of this list include transcriptional regulators that have been clearly associated to tissue residency in other T cell subsets, like tissue-resident TRM CD8+ cells (143, 144), including Runx3 and Blimp1 (145). In addition, murine and human TR-TREG also possess unique key markers including transcription factors Ikzf2, Gata3, and Rora.
An important transcription factor associated with TR-TREG cells is Helios. While the majority of TREG cells in circulation readily express Helios, siRNA-mediated silencing of Helios expression in human and murine TREG cells does not impede their survival and suppressive capacity in vitro (146, 147). On the other hand, the conditional deletion of Helios in murine TREG cells (Foxp3CRE Ikzf2fl/fl) leads to the development of a progressive, rather than a scurfy-like, lymphoproliferative disease in adult mice (147), revealing it is not required for the development of TREG cells, but rather for the preservation of TREG cell fitness at barrier tissues. Importantly, Helios potentiates the suppressive function of TREG by directly interacting with Foxp3 and promoting histone deacetylation (148), providing further evidence Helios plays a supportive role to the program provided by Foxp3.
However, not all lymphoid and tissue-resident TREG cells express Helios. Originally thought to be solely expressed by tTREG cells (30), it is now well-appreciated that Helios expression in both murine and human Helios- TREG cells is inducible (31, 149) in vivo and in vitro, respectively. Some of the key features that differentiate splenic Helios+ from Helios- TREG is the little overlap they share between their respective TCR repertoire, and the expression of genes involved in the differentiation of specialized TH17 cells, including Rorc, Il6ra and Il23r (31), suggesting a division of labor between two TREG subsets that may have long-reaching consequences in the tissue adaptation of TR-TREG cells. For example, Cruz-Morales et al. showed that colonic Helios+Gata3+ TREG differ greatly from Helios- RORγT+ TREG cells by their requirement of CD28, but not MHC-II, to proliferate locally (20), providing a potential point of distinction between colonic Helios+ TR-TREG and RORγT+ emTREG. Nonetheless, further investigation into the role of Helios in the differentiation and maintenance of TR-TREG cells is required.
Gata3 is the transcription factor 3 of the Gata-binding family that comprises six known members. In T cells, it has been shown to govern T cell development, proliferation and maintenance (150) and is particularly important to promote the transcriptional signature of helper type 2 T cells (TH2) (151). Skin, gastro-intestinal, visceral adipose tissue, and pulmonary TR-TREG cell were all shown to express Gata3 (22, 152), albeit with different intensities. This observation could be explained by the different states of activity of these TREG cells, as Gata3 expression is significantly increased in both murine and human TREG cells upon TCR engagement (22). Interestingly, the signaling pathway that leads TREG to express this TF does not require IL-4 – a cytokine associated with Gata3 expression in conventional T cells (153) – and depends largely on exogenous IL-2 (22). Deletion of Gata3 in murine TREG cells does not lead to the development of spontaneous autoimmunity before 6 months of age (22), after which the mice develop intestinal pathology and dermatitis (104). This is because Gata3-deprived TR-TREG are not hindered in their development, but rather fail to respond to an inflammatory threat, displaying decreased tissue migration, proliferation, transcriptional stability, and suppressive capacity (22, 23, 104).
While not necessary for the maintenance of peripheral tolerance, Gata3 contributes to the functional adaptation of TR-TREG cells. Gata3 recognizes the CNS2 region of Foxp3 (23), promoting Foxp3 activity and stabilising the transcriptional program of TREG cells to avoid their conversion to pro-inflammatory T cells under stress (22). In addition, Gata3 partners with Foxp3 to form a complex that contributes to the regulation of a wide array of TREG-associated genes (104). Gastro-intestinal, skin, pulmonary, and VAT TR-TREG cells express the IL-33 receptor ST2 (17, 24, 154), a known target of Gata3 in T cells (155). Unfortunately, while Gata3 is known to remodel the Il10 locus in CD4+ T cells (156), the link between Gata3 and IL-10 has yet to be established in TR-TREG cells. As such, there are many indicators that Gata3 is an important contributor to the tissue adaptation of TREG cells, and future investigation into the epigenetic, transcriptional, and post-transcriptional impact of this TF is warranted.
Another gene that is consistently found in RNA-seq data from TR-TREG cells is Rora. This gene codes for the retinoic acid receptor-related orphan receptor alpha (RORα), a transcription factor which has been found to be expressed in differentiated T cells, including TH1, TH2 and TH17 (157) cells. Unfortunately, we know very little on the role of RORα in TR-TREG. In T cells, Rora is expressed upon TCR activation, and is closely associated with the expression of their lineage defining TH1, TH2 or TH17 signature (158). Similarly, RORα plays a supporting role in the transcriptional signature of TR-TREG cells. For example, a Foxp3 conditional deletion of RORα does not alter the accumulation of skin localised TR-TREG cells but enables the evasion of immune responses during skin treatment with MC903, a chemical inducer of atopic dermatitis (18). Thus, as with Gata3, RORα is not required during the transcriptional transformation of tissue-migrating eTREG cells, but rather for their function once in the tissue.
The B lymphocyte-induced maturation protein-1 (Blimp 1) is a transcriptional regulator that is particularly expressed by TREG cells located in secondary lymphoid organs or non-lymphoid tissues (159). A conditional knock out of Prdm1 (Blimp-1) in murine TREG (Foxp3Cre Prdm1fl/fl) generates an increase in the accumulation of TREG cells, accompanied by small increase in TCONV cell abundance that is insufficient to induce autoimmunity (159), confirming Blimp-1 is not essential to the generation, migration or even function of eTREG. Rather, Blimp-1 prevents the methylation of multiple genes, including CNS2 in the Foxp3 locus, by inhibiting the action of the methyltransferase Dmnt3a downstream of IL-6 (160). In doing so, Blimp-1 prevents the full conversion of colonic TREG to non-suppressive RORγT+ eTREG cells (161), suggesting that the role of Blimp-1 is to preserve the transcriptional program of TR-TREG cells.
TR-TREG cells have shown a remarkable capacity to communicate with their immediate environment, adopting cytokine receptors, sensing molecular changes in its environment, and providing direct cell-to-cell contact with immune and non-immune cells (162). TR-TREG achieve this by adopting unique phenotypic characteristics, such as the ability to sense local danger signals and compete in microenvironments with limited IL-2 availability, allowing them to maintain their identity in non-lymphoid organs.
IL-33 is a cytokine of the IL-1 family of alarmins constitutively expressed by endothelial and epithelial cells (163) and by activated macrophages and dendritic cells (164). The IL-33 receptor ST2 is transcriptionally upregulated and detected on the surface of TR-TREG (17, 120). This is consistent with the fact that the expression of Il1rl1 (ST2) is closely associated to the expression of BATF and is part of the transcriptional signature elicited after DNA methylation in TR-TREG cells (16, 120). However, not all tissue isolated TREG express ST2 in mice at the steady state, nor do skin, lung, gut, or VAT-isolated TREG cells express ST2 with the same intensity (17). As such, while suggested as a marker of TR-TREG cells (17), there is currently no clear evidence that ST2 expression is exclusive to TR-TREG cells, and further investigation into this receptor is warranted. Moreover, the importance of ST2 in the differentiation and function of TR-TREG cells remains ill-defined. For example, while IL-33 can directly promote the homeostatic expansion of TREG cells (24, 165), a Foxp3-specific conditional knock-down of ST2 (Foxp3CREIl1rl1fl/fl) does not impair TREG accumulation in the lungs (166). Rather, IL-33 orchestrates TREG-mediated suppression of local γδ T (166), TH1, and TH17 cells during tissue injury (24, 167). To complicate things, it is unclear if these mechanisms depend entirely on the expression of ST2 by TREG cells (168). Indeed, innate immune cells can readily respond to IL-33 and provide proliferative signals to promote TR-TREG expansion and survival (169). As such, rather than providing a survival signal, ST2 may act as a sensing mechanism for local TR-TREG to rapidly reactivate and produce suppressing cytokines.
While not exclusive to TR-TREG, the inducible co-stimulator Icos plays a crucial role in both TR-TREG and emTREG cells to maintain their identity and survival within non-lymphoid organs (21). In mice, a Foxp3 conditional knock out of Icos (Foxp3YFP-CRE Icosfl/fl) does not generate autoimmunity, but rather prevents tissue-localised TREG cells from suppressing oxalone-induced dermatitis (170), suggesting Icos is particularly required for TREG cells to control tissue injury. Specifically, Icos coordinates with mTORC1 signaling to support TREG proliferation and the expression of suppressive molecules (171), and is particularly critical for TR-TREG and emTREG cells to persist in the absence of IL-2 signaling by providing anti-apoptotic signals (15). Together, Icos and CD28 act as potent activators of the PI3K/Akt signaling pathway that triggers the phosphorylation of the transcription factor Foxo1 (171, 172). In turn, this sequesters Foxo1 in the cytoplasm and leads to down-regulation of genes like Klf2 and Ccr7 (173). In the absence of IL-2, TREG cells become susceptible to apoptosis, highlighting the critical role of sustained Icos-IcosL signaling in their survival as they migrate to the tissue (15). On the other hand, abrogating the PI3K-activating capacity of Icos by removing a YMFM motif in its cytoplasmic tail increases VAT TR-TREG accumulation and function (174), suggesting that Icos may have tissue-specific roles for TREG cells. Thus, while there is abundant evidence that Icos promotes the activation and survival of TR-TREG cells, tissue-specific differences are likely at play and must be considered when investigating TR-TREG cell sub-populations.
Genes involved in fatty acid β-oxidation (FAO) can be readily detected in antigen-experienced TREG cells isolated from non-lymphoid tissues, including in visceral adipose tissue (VAT), the skin, the colon, and the lungs, suggesting TR-TREG default to FAO in non-inflamed tissues (19, 120). However, these transcriptional approaches have not formally demonstrated that TR-TREG cells require FAO to persist in all tissues. Most of the current evidence comes from VAT-isolated TR-TREG, which express the peroxisome proliferator-activated receptor gamma (PPARγ), a ligand-activated transcription factor. Functionally, PPARγ provides a complex signal to engage FAO in VAT TREG cells (175), providing them with a competitive advantage over TCONV cells to survive, accumulate, and function (176). This crucial metabolic strategy enables VAT TREG cells to catabolize long-chain fatty acids (LCFAs) from the environment, turning to FAO to sustain their demand for energy (177, 178). While this process is shared between TREG and TCONV cells, TREG cells utilise fatty acids differently as they do not build endogenous fatty acids from acetyl-CoA, but rely on the acquisition of exogenous fatty acids to meet their metabolic needs (179). Concomitantly, efficient lipid storage by VAT TR-TREG cells is essential to protect them against lipo-toxicity and to provide the metabolic precursors needed for energy generation. These include scavenger proteins such as CD36 and enzymes involved in triglyceride production, such as DGAT1 and DGAT2. Skin and VAT-isolated PPARγ+ TREG cells readily express CD36, providing them with the ability to capture and secure LCFAs (175, 180). DGAT are a family of enzymes involved in triglyceride production and lipid droplet (LD) formation that are preferentially expressed in activated TREG cells (181). Foxp3 itself is a strong repressor of Glut1 (182), the glucose transporter, and favors the expression of FAO genes (178). Yet, this mechanism acts in a feed-back loop, with DGAT1 promoting Foxp3 expression by diminishing protein kinase C (PKC) activity downstream of the TCR (181, 183). Interestingly, by tracing the tissue distribution of splenic TREG cells with shared TCR sequences, Li et al. demonstrated that PPARγ-expressing eTREG cells localise to other non-lymphoid sites, including the skin and the liver (184), providing new translational evidence that FAO proteins are expressed by other TR-TREG cells. Nonetheless, while these observations highlight the importance of FAO for VAT TR-TREG cells to sustain their bioenergetic demands, it remains to be determined if this metabolic strategy is required to sustain other TR-TREG cells.
One of the most recent and exciting discoveries has been the observation that activated eTREG can further specialize to adopt TH1, TH2, TH17, and even TFH-like features. Importantly, they can express master transcription factors that are part of a transcriptional program typically expressed by helper T cells, including T-bet (TH1), RORγT(TH17), Gata3(TH2), and BCL6 (TFH). The differentiation, migration, and tissue accumulation of functionally-specialized TREG cells in tissues is a dynamic process that can occur in microbiota-rich barrier sites (10) or during tissue injury (185, 186). Indeed, contrary to the core genes necessary for the generation and maintenance of TR-TREG cells, the role of these “master” transcription factors is not associated with a residency program; rather, these TFs promote a set of specialized functions that allow TREG cells to suppress or orchestrate local immune responses (Figure 3). For example, single-cell analysis performed at distinct times during an Influenza A infection in mice portrays how Gata3+ TREG cells are progressively replaced by antigen-specific T-bet+CXCR3+ TREG cells in the course of disease, suggesting that, contrary to the permanent presence of TR-TREG, TH1-specialized TREG cells are generated concurrently with the antiviral TH1 response and follow the pattern of accumulation of these cells (185, 187).
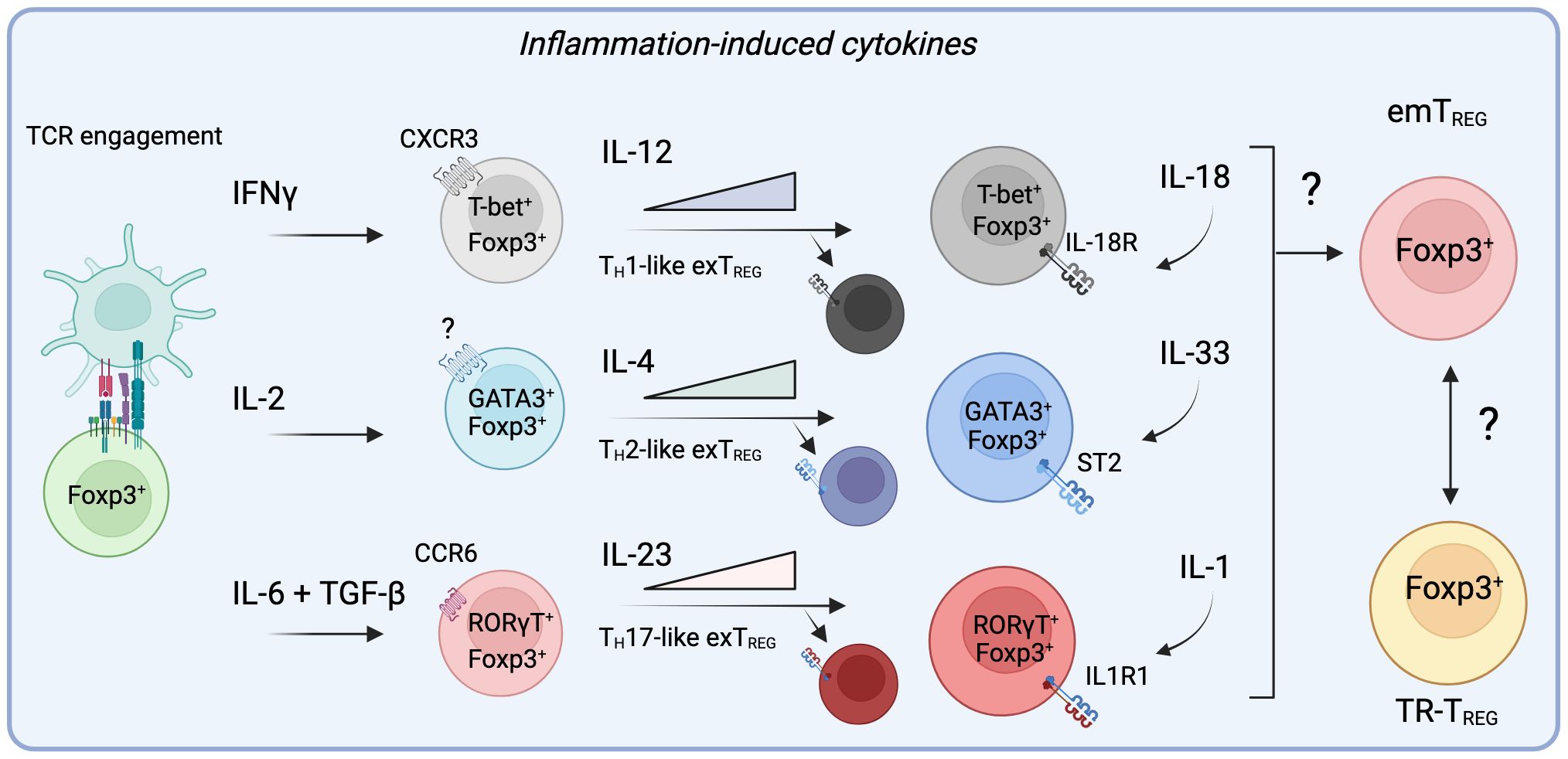
Figure 3 Specific inflammatory signals alter the trajectory of TREG cells in non-lymphoid sites by engaging specialized programs prior and during their migration to inflamed tissues. During active inflammation, the presence of cytokines such as IFNγ, IL-2, IL-6, and TGF-β can divert the differentiation of TREG cells to adopt helper-like phenotypes, allowing them to migrate to specific sites of inflammation alongside conventional T cells. Importantly, by acquiring these master transcription factors, effector TREG cells (eTREG) become responsive to signals provided by IL-12, IL-4 or IL-23. While these cytokines further promote the transcriptional program engaged by these specialized TREG cells, they can ultimately diminish their suppressive functions and allow them them to contribute to inflammation as exTREG cells. Importantly, it remains to be determined if the resulting population of emTREG cells in the tissue after inflammation acquire a residency program that lead them to form part of the TR-TREG cell population.
Interestingly, some of these specialized TREG cells (RORγT+ TREG) are present at the steady-state in mucosal tissues such as the colon, blurring attempts at defining what constitutes the bona fide TR-TREG phenotype in these tissues. Indeed, key events leading to the generation of specialized TREG cells include the requirement for TCR signaling and aerobic glycolysis to facilitate clonal expansion and differentiation (188). Moreover, Irf4 (27) is a necessary stepping-stone for the differentiation of specialized TREG cells (9, 28, 29). A typical example of these specialized TREG cells is observed in the colon, where resident TREG cells displaying two distinct TCR repertoires can be segregated based on their transcriptional program. Indeed, both RORγT+ TREG and Gata3+ TREG are readily detected in the colon; however, absence of a local microflora only hinders the specific generation of RORγT+ TREG (189, 190) since their TCR repertoire is largely biased towards bacterial antigens (72, 191, 192). Since specific signals are required for TREG cells to acquire these programs, it is possible to dissect the required pathways that lead TREG cells to acquire these specialized programs.
Some of the better described signals that promote the generation of specialized TREG cells include cytokines that drive the phosphorylation and nuclear translocation of STAT and SMAD proteins (193). In turn, these signals promote the expression of genes that define T cell fate, including the acquisition of master transcription factors T-bet, Gata3, or RORγT. What is particularly interesting, however, is that the pathways that lead TREG to adopt these TFs can also undermine their Foxp3-dependent transcriptional program, either through the loss of Foxp3 expression, the expression of pro-inflammatory genes, or the engagement of apoptosis. As such, at the time when activated TREG cells undergo important epigenetic and transcriptional changes, certain inflammatory signals can promote the loss of Foxp3 expression and their conversion into inflammatory “exTREG” cells. Several key transcription factors have been described to be involved in this inflammatory adaptation process of TREG cells.
T-bet is a T-box transcription factor expressed in a wide variety of immune cells, and mostly recognized for its role in defining the transcriptional landscape of TH1 cells (194). Using a unique murine model that enables the tracking of murine T-bet-expressing TREG (Foxp3Thy1.1 Tbx21tdTomato-T2A-CreERT2 R26YFP-fl-stop-fl), Levine and colleagues showed that the conditional deletion of T-bet in Foxp3+ TREG cells does not lead to autoimmunity in adult mice, although it does generate a mild increase in TH1 activity (195), suggesting T-bet has little to no impact on the way TREG preserve tissue function at the steady-state. Notably, T-bet is a critical regulator for the expression of CXCR3 (196), a chemokine receptor that orchestrates eTREG migration to sites of TH1-driven inflammation (196, 197). Highlighting the role of TCR engagement, T-bet+ eTREG cells that progressively accumulate in the lungs of mice infected with acute Influenza A infection recognize viral proteins (185, 198). Thus, as with TH1 cell polarization, the generation of T-bet+ eTREG occurs progressively during inflammation and is closely associated to the clonal expansion of antigen-specific CD4+ TH1 cells.
The signals that promote the generation of TH1 cells include IFNγ (STAT1) and IL-12 (STAT4). Interestingly, an IFNγ-STAT1 signal drives the initial expression of T-bet during TCR engagement, while a subsequent IL-12-STAT4 signal is required for their definitive differentiation (199, 200). This initial T-bet expression can, in turn, promote the expression of the IL-12 receptor (IL-12Rβ2) (201–203). However, contrary to TH1 cells, eTREG cells seem to depend exclusively on the presence of IFNγ for the acquisition of T-bet (196, 204). By activating murine CD4+Foxp3+ cells in vitro, Koch and colleagues demonstrated that TREG cells acquire T-bet expression and its associated target, CXCR3, only if they possess the receptor IFNγR1 (205), suggesting that IFNγ-producing TH1 cells are responsible for the polarization of TH1-like eTREG cells.
The control of IL-12 signalling by TREG cells is critical, as excessive pSTAT4 can lead TREG cells to lose Foxp3 expression (206) by, notably, limiting chromatin accessibility of STAT5 to the Foxp3 locus (207). Yet, STAT4 is a major regulator of Ifng in CD4+ T cells (208), and both human and murine TREG exposed to IL-12 produce low levels of IFNγ (187, 205, 206, 209–212), revealing excessive IL-12 can still be perceived by TH1-like eTREG cells. However, contrary to STAT1, STAT4 signaling is associated with less suppressive TREG cells and can even lead to the complete loss of Foxp3 expression (187, 205, 206, 209–211), suggesting T-bet+ eTREG are in a constant struggle to avoid the loss of genes involved in their suppressive functions. In this regard, T-bet+ TREG cells possess mechanisms to avoid overt STAT4 signaling. For example, IFNγ-induced T-bet+ eTREG cells suppress IL-12Rβ2 surface expression, preventing excessive phosphorylation of STAT4 and further TH1-like commitment (205). Moreover, non-labelled proteomics on circulating human TREG cell populations revealed that, compared to memory or naïve TREG, eTREG maintain low cytosolic levels of STAT4 (213).
There is growing evidence for the role of IL-18 on the function of tissue-resident T-bet+ eTREG cells. While the origin of IL-18R1+ eTREG cell remain to be fully understood, TH1 polarizing conditions, and particularly IL-12, allow TREG cells to adopt the expression of both T-bet and IL-18R1 (187), suggesting that, like for Tconv cells, eTREG require a STAT4-dependent chromatin remodeling to express IL-18R1 (214, 215). In vitro, IL-18 promotes the expansion and suppressive capacity of IL-12-generated T-bet+ TREG cells (187), suggesting this signal can counter the destabilising effects of IL-12. In vivo, T-bet+ eTREG cells express IL-18R1 when they accumulate in the lungs during an Influenza A infection (187). Here, IL-18 enhances the production of amphiregulin in local TREG cells, facilitating tissue restoration after pulmonary Influenza A infection (216). In addition, a Foxp3 conditional knock-out of Il18r1 (Foxp3ERT2-CRE Il18r1fl/fl) allowed us to demonstrate that IL-18 is specifically required for eTREG cells to suppress IL-17A responses in the lungs after an Influenza A infection (187). Similarly, IL-18R1 deficiency in TREG cells fails to control the onset of a T cell-mediated colitis (217) as well as inflammation in an experimental model of ovalbumin-induced asthma (218), confirming IL-18 is an important contributor to eTREG function. However, these observations do not necessarily mean that IL-18R1 expression is restricted to T-bet+ TREG, as we have observed RORγT expression among a subset of IL-18R1+ TREG cells (187) and IL-18R1 expression has been described in TH17 cells (217). Collectively, these observations illustrate how the TH1 adaptation of eTREG cells allows for the suppression of tissue inflammation.
The transcription factor Gata3, which is an important component of the transcriptional program of TR-TREG, is best described for its role in driving TH2 cell differentiation (219). In both human and murine CD4+ T cells, Gata3 promotes TH2-associated genes, allowing for the expression of genes associated to their function, such as IL-4, IL-5, and IL-13 (151, 219). There are numerous accounts of tissue-homing TREG cells expressing high levels of Gata3 during acute TH2-driven immunity, such as what is observed during asthma (220) or helminth infections (221, 222).
The signals driving GATA-3 expression in TREG cells are not fully understood. Two signals have been described to be sufficient to induce Gata3 expression during TH2 differentiation, namely an IL2/STAT5-dependent and an IL-4/STAT6-dependent signal (223–225). In homeostatic conditions, IL-2 (STAT5) is sufficient to promote the expression of Gata3 during TCR engagement (22). However, in TH2-driven responses, TREG cells require IL-4R to acquire GATA-3 expression and their TH2-like characteristics (226). This distinction between STAT5 and STAT6-dependent induction of Gata3 may pave the way towards understanding how TH2-like eTREG cells differ from TR-TREG cells. For example, mice with a Foxp3-specific conditional knock-down of Il4ra (Foxp3CRE Il4rafl/fl) fail to prevent exacerbated asthma-like symptoms when challenged with house-dust-mite (HDM) (226) and helminth-driven inflammation, despite the presence of TREG cells in situ (221).
While IL-4 can favor TREG cell-mediated functions, sustained IL-4 can also force TREG cells to lose Foxp3 expression and their suppressive capacity both in vitro (227) and in vivo (221, 222, 227). STAT6 can promote the activity of the histone deacetylase HDAC9, which decreases chromatin accessibility to the Foxp3 locus (228). To prevent this, eTREG cells require strategies to avoid excessive IL-4 signaling. First, by maintaining high levels of CD25 expression, eTREG cells remain sensitive to IL-2, whose STAT5 signal competes with STAT6 activity (229). Second, tissue-localised TREG cells prevent further commitment into the TH2 lineage by producing the E3 ubiquitin ligase Itch (230, 231). Finally, murine in vitro-induced TREG cells exposed to IL-4 express higher levels of the JAK/STAT inhibitor SOCS2 to prevent further STAT6 phosphorylation and the expression of pro-inflammatory cytokines (232). Thus, while it remains to be fully confirmed in tissue-resident TREG cells, there is cumulating evidence that IL-4 is important for the commitment of Gata3+ eTREG cells, and responsible for their transcriptional destabilisation and conversion into TH2-like ex-TREG cells.
Finally, IL-33, which contributes to the proliferation of TR-TREG cells (165), can also govern the function of Gata3+ eTREG cells during inflammation. In this regard, IL-33-responding activated TREG cells where shown to produce high amounts of IL-10 and TGF-β (233), playing a key role in maintaining intestinal homeostasis (24). Similarly, ST2+ TREG cells promote the suppression of anti-tumor immune responses (234–236). However, IL-33 can also drive the production of the TH2 associated cytokines IL-5 and IL-13 in pulmonary eTREG cells (233, 237, 238) and interfere with their capacity to supress TH2 responses (238). Thus, the role of IL-33 on Gata3+ TREG cells is specific to the inflammatory context and may depend on whether it targets TR-TREG cells or eTREG cells accompanying TH2 responses.
While complex and not entirely defined, the signaling events that lead TREG cells to adopt a TH17-like phenotype include some of the same polarizing JAK-STAT and SMAD signals that are required for the generation of TH17 cells. Indeed, the promoter functions of both Stat3 (239) and RORγT (240) are required to establish a TH17 cell transcriptional program (241), and TREG cells have been shown to share part of this transcriptional program through the acquisition of these TFs (239). In the gut, RORγT+ TREG cells play an essential part in maintaining gut homeostasis and contribute to maintain local homeostasis by, notably, suppressing TH17-driven responses (242). Transcriptionally, RORγT+ TREG cells from the mouse colon at steady-state express higher levels of Il23r, Il1r1, Maf, Irf4, and Ikzf3 than their RORγT- counterparts (191), revealing they possess a unique landscape that encompasses some key TH17-associated genes. Moreover, RORγT is required for IL-10 production by colonic TREG cells and prevention of T cell-mediated colitis (191). Similarly, RORγT is required for TREG cells to control TH17-mediated autoimmune arthritis and EAE (192, 243), suggesting that RORγT expression allows emTREG cells to target and suppress TH17-driven responses. However, the role of RORγT and its impact on the transcriptional landscape of emTREG cells remains ill-defined and is likely driven by the inflammatory microenvironments these cells are exposed to.
While many cytokines can promote the nuclear translocation of Stat3 in TH17 cells, the simultaneous signals provided by TGF-β (SMAD2/3) and IL-6 (Stat3) are sufficient, in vitro, to induce RORγT expression in TREG cells (162, 192). Interestingly, a delicate balance is achieved between the signal provided by TGF-β and IL-6. For example, TGF-β and IL-6 synergistically promote the proteasome-dependent degradation pathway of Foxp3 (244), contributing to a partial loss of Foxp3 function. Interestingly, once colonic RORγT+ TREG cells are generated, they display a significantly stable phenotype, with maintained demethylation of TREG-specific genes like Foxp3, Ikzf2, Ctla4, Gitr and Ikzf4 (Eos) (191). In fact, these cells possess intrinsic mechanisms to avoid their full conversion towards TH17 cells. As with IL-12 and TH1 cells, subsequent signals provided by IL-23(Stat3) can further destabilise the transcriptional program of RORγT+ TREG cells and even engage an apoptotic cascade in these cells (245). Indeed, Il23r is amongst the genes upregulated by Stat3 and RORγT (246), making RORγT+ eTREG particularly sensitive to IL-23 (245). In a recent report, Jacobse and colleagues demonstrated IL-23R expression is restricted to RORγT+ TREG under homeostatic conditions in the colon, and colonic TREG cells maintain a competitive advantage over WT TREG cells to survive in these conditions (245). Concomitantly, the authors demonstrate that TREG cells isolated from the lamina propria of patients with active IBD express high levels of Il23r and pro-apoptotic genes (126), suggesting an evolutionary conserved mechanism that orchestrates RORγT+ eTREG survival and function.
In addition to IL-23, IL-1β was found to promote the differentiation of human CD4+CD25highCD127low Foxp3+ TREG cells into IL-17-producing cells (247, 248), suggesting IL-1 may promote a pro-inflammatory phenotype in TREG cells. However, the role of IL-1 in RORγT+ eTREG cells remains ill-defined. Through a T-cell mediated colitis model in mice, we demonstrated that a knock-out of IL-1R1 in TREG cells favors an accumulation of Gata3+ TREG cells over RORγT+ TREG cells in the colon, as IL-1 directly promotes RORγT+ TREG expansion (167). Despite this effect, a lack of IL-1 signaling in TREG cells results in more abundant accumulation in the colon compared to WT TREG (167), suggesting IL-1 is a negative signal for the maintenance of colonic TREG cells. Interestingly, there are specific situations where this effect is beneficial. For example, IL-1R1-/- mice infected with Cryptococcus neoformans are particularly sensitive to the infection, as they cannot mount an effective T cell response (249). In their lungs, these mice lack RORγT+ TREG cells and have increased ST2+ TREG cells in the lungs compared to WT mice (167), suggesting sustained immunosuppression. To counter this, activated TREG cells express high levels of the decoy receptor IL-1R2, which allows them to neutralize IL-1 signalling (250–252).
In this review, we aimed to detail some of the major elements that govern the trajectory of a precursor TREGP cell to a highly specialized TR-TREG cell. It is particularly interesting that the trajectory of a TREG cells is, in most regards, highly like that of the conventional T cell as it undergoes further polarization prior to reaching peripheral tissues. Importantly, the epigenetic malleability of TREG cells is central to their ability to perform outside of the thymus, as these transformations allow them to sense tissue-derived signals that, in turn, modulate their suppressive functions. However, while we have accumulated a lot of information in recent years, much remains to be understood on how these tissue and inflammation-specific adaptations govern the function of TR-TREG cells. For example, the notion that TREG cells can adopt a specific differentiation path and revert to their previous state, labelled “plasticity” (253, 254), remains to be proven experimentally.
Finally, recent reviews have addressed how Foxp3 gene editing, IL-2 therapy, and the use of TREG cells as cellular therapies represent key strategies to engage human TREG cells (255). However, most of our current knowledge on TR-TREG cells has not been specifically exploited by TREG-targeting therapeutical approaches. There is, however, some evidence these strategies may facilitate the expression of a tissue residency program. For example, the development of muteins or low-dose therapies (256) aimed at promoting IL-2 signaling in TREG cells can promote the expression of genes associated with TR-TREG cell function, such as Il1rl1(ST2), as well as migratory and other tissue resident genes (257). Thus, it is of interest to understand how TREG targeting strategies can influence both the developmental trajectory and the function of tissue resident TREG cells. In addition, understanding the migratory cues that enable TR-TREG cells to recognize specific tissues can have long reaching therapeutical benefits. Chimeric antigen receptor (CAR) TREG cells have been proposed as a new avenue to circumvent the constraints of low TREG cells numbers and the unknown TCR repertoire of TREG in autoimmune or graft-versus-host (GvHD) diseases (258). However, this approach is still very novel, and, in the absence of additional modifications, is expected to suffer from the same limitations of CAR-T cells (258, 259), including failing to adopt metabolic strategies to survive, preventing exhaustion, and maintaining their function in tissues. Thus, it is by establishing a solid understanding of the entire pathway leading TREG cells to adapt to non-lymphoid organs that we provide the basis for the development of better TREG cell-based therapies.
FA: Writing – original draft, Writing – review & editing. ZL: Writing – original draft, Writing – review & editing. AB: Writing – original draft, Writing – review & editing. CP: Writing – original draft, Writing – review & editing, Conceptualization, Funding acquisition, Project administration, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The funds for this study came from the Canadian Institutes of Health Research (CIHR) operating grants (MOP67211).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
eTREG, effector TREG; emTREG, effector memory TREG; TR-TREG, tissue-resident TREG; TH, T helper cell; Foxp3, Forkhead box P3; Irf4, Interferon regulatory factor 4; BATF, Basic leucine zipper transcription factor; ST2, Suppressor of tumorigenicity 2, IL-33 receptor; TCR, T cell receptor; RORα, Retinoic acid–related orphan receptor α; Gata3, GATA-binding protein 3; T-bet, T-box transcription factor TBX21; RORγt, RAR-related orphan receptor gamma; VAT, Visceral adipose tissue; CNS, Conserved non-coding regions of the Foxp3 locus; STAT, signal transducer and activator of transcription; mTOR, mammalian target of rapamycin; TGF-β, Transforming growth factor beta; TCF1, T cell factor 1; Icos, Inducible T cell costimulator, CD278; OXPHOS, oxidative phosphorylation; Dnmt, DNA methyltransferases.
1. McCarville JL, Ayres JS. Disease tolerance: concept and mechanisms. Curr Opin Immunol. (2018) 50:88–93. doi: 10.1016/j.coi.2017.12.003
2. Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. (1970) 18:723–37.
3. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
4. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
5. Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. (2007) 27:786–800. doi: 10.1016/j.immuni.2007.09.010
6. Rajendeeran A, Tenbrock K. Regulatory T cell function in autoimmune disease. J Transl Autoimmun. (2021) 4:100130. doi: 10.1016/j.jtauto.2021.100130
7. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
8. Satitsuksanoa P, Jansen K, Globinska A, van de Veen W, Akdis M. Regulatory immune mechanisms in tolerance to food allergy. Front Immunol. (2018) 9:2939. doi: 10.3389/fimmu.2018.02939
9. Bacher P, Heinrich F, Stervbo U, Nienen M, Vahldieck M, Iwert C, et al. Regulatory T Cell Specificity Directs Tolerance versus Allergy against Aeroantigens in Humans. Cell. (2016) 167:1067–78 e16. doi: 10.1016/j.cell.2016.09.050
10. Russler-Germain EV, Rengarajan S, Hsieh CS. Antigen-specific regulatory T-cell responses to intestinal microbiota. Mucosal Immunol. (2017) 10:1375–86. doi: 10.1038/mi.2017.65
11. Astarita JL, Dominguez CX, Tan C, Guillen J, Pauli ML, Labastida R, et al. Treg specialization and functions beyond immune suppression. Clin Exp Immunol. (2023) 211:176–83. doi: 10.1093/cei/uxac123
12. Hu W, Wang ZM, Feng Y, Schizas M, Hoyos BE, van der Veeken J, et al. Regulatory T cells function in established systemic inflammation and reverse fatal autoimmunity. Nat Immunol. (2021) 22:1163–74. doi: 10.1038/s41590-021-01001-4
13. Khantakova JN, Bulygin AS, Sennikov SV. The regulatory-T-cell memory phenotype: What we know. Cells. (2022) 11(10):1687. doi: 10.3390/cells11101687
14. Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. (2004) 199:303–13. doi: 10.1084/jem.20031562
15. Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med. (2014) 211:121–36. doi: 10.1084/jem.20131142
16. Delacher M, Imbusch CD, Hotz-Wagenblatt A, Mallm JP, Bauer K, Simon M, et al. Precursors for nonlymphoid-tissue treg cells reside in secondary lymphoid organs and are programmed by the transcription factor BATF. Immunity. (2020) 52:295–312.e11. doi: 10.1016/j.immuni.2019.12.002
17. Spath S, Roan F, Presnell SR, Hollbacher B, Ziegler SF. Profiling of Tregs across tissues reveals plasticity in ST2 expression and hierarchies in tissue-specific phenotypes. iScience. (2022) 25:104998. doi: 10.1016/j.isci.2022.104998
18. Malhotra N, Leyva-Castillo JM, Jadhav U, Barreiro O, Kam C, O'Neill NK, et al. RORalpha-expressing T regulatory cells restrain allergic skin inflammation. Sci Immunol. (2018) 3(21):eaao6923. doi: 10.1126/sciimmunol.aao6923
19. Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, et al. Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity. (2019) 50:493–504.e7. doi: 10.1016/j.immuni.2019.01.001
20. Cruz-Morales E, Hart AP, Fossett GM, Laufer TM. Helios(+) and RORgammat(+) Treg populations are differentially regulated by MHCII, CD28, and Icos to shape the intestinal Treg pool. Mucosal Immunol. (2023) 16:264–74. doi: 10.1016/j.mucimm.2023.02.007
21. McGee MC, Zhang T, Magazine N, Islam R, Carossino M, Huang W. PD-1 and Icos counter-regulate tissue resident regulatory T cell development and IL-10 production during flu. Front Immunol. (2022) 13:984476. doi: 10.3389/fimmu.2022.984476
22. Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, et al. Gata3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J Clin Invest. (2011) 121:4503–15. doi: 10.1172/JCI57456
23. Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity. (2011) 35:337–48. doi: 10.1016/j.immuni.2011.08.012
24. Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. (2014) 513:564–8. doi: 10.1038/nature13577
25. Yang J, Zou M, Chu X, Floess S, Li Y, Delacher M, et al. Inflammatory perturbations in early life long-lastingly shape the transcriptome and TCR repertoire of the first wave of regulatory T cells. Front Immunol. (2022) 13:991671. doi: 10.3389/fimmu.2022.991671
26. Han Y, Ouyang X, Chen Y, Lai S, Sun H, Wu N, et al. scRNA-seq profiling of neonatal and adult thymus-derived CD4+ T cells by a T cell origin-time tracing model. J Mol Cell Biol. (2023) 14(12):mjac072. doi: 10.1093/jmcb/mjac072
27. Cipolletta D, Cohen P, Spiegelman BM, Benoist C, Mathis D. Appearance and disappearance of the mRNA signature characteristic of Treg cells in visceral adipose tissue: age, diet, and PPARgamma effects. Proc Natl Acad Sci U S A. (2015) 112:482–7. doi: 10.1073/pnas.1423486112
28. Li C, DiSpirito JR, Zemmour D, Spallanzani RG, Kuswanto W, Benoist C, et al. TCR transgenic mice reveal stepwise, multi-site acquisition of the distinctive fat-treg phenotype. Cell. (2018) 174:285–99.e12. doi: 10.1016/j.cell.2018.05.004
29. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol. (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
30. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. (2010) 184:3433–41. doi: 10.4049/jimmunol.0904028
31. Thornton AM, Lu J, Korty PE, Kim YC, Martens C, Sun PD, et al. Helios(+) and Helios(-) Treg subpopulations are phenotypically and functionally distinct and express dissimilar TCR repertoires. Eur J Immunol. (2019) 49:398–412. doi: 10.1002/eji.201847935
32. Haribhai D, Lin W, Edwards B, Ziegelbauer J, Salzman NH, Carlson MR, et al. A central role for induced regulatory T cells in tolerance induction in experimental colitis. J Immunol. (2009) 182:3461–8. doi: 10.4049/jimmunol.0802535
33. Curotto de Lafaille MA, Kutchukhidze N, Shen S, Ding Y, Yee H, Lafaille JJ. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity. (2008) 29:114–26. doi: 10.1016/j.immuni.2008.05.010
34. Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature. (2012) 482:395–9. doi: 10.1038/nature10772
35. Szurek E, Cebula A, Wojciech L, Pietrzak M, Rempala G, Kisielow P, et al. Differences in expression level of helios and neuropilin-1 do not distinguish thymus-derived from extrathymically-induced CD4+Foxp3+ Regulatory T cells. PloS One. (2015) 10:e0141161. doi: 10.1371/journal.pone.0141161
36. Elkord E. Helios should not be cited as a marker of human thymus-derived tregs. Commentary: Helios(+) and helios(-) cells coexist within the natural Foxp3(+) T regulatory cell subset in humans. Front Immunol. (2016) 7:276. doi: 10.3389/fimmu.2016.00276
37. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. (2012) 209:1723–42, S1. doi: 10.1084/jem.20120914
38. Chen W, Huang W, Xue Y, Chen Y, Qian W, Ma J, et al. Neuropilin-1 identifies a new subpopulation of TGF-beta-induced Foxp3(+) regulatory T cells with potent suppressive function and enhanced stability during inflammation. Front Immunol. (2022) 13:900139. doi: 10.3389/fimmu.2022.900139
39. Xing Y, Hogquist KA. T-cell tolerance: central and peripheral. Cold Spring Harb Perspect Biol. (2012) 4(6):a006957. doi: 10.1101/cshperspect.a006957
40. Owen DL, Sjaastad LE, Farrar MA. Regulatory T cell development in the thymus. J Immunol. (2019) 203:2031–41. doi: 10.4049/jimmunol.1900662
41. Takaba H, Morishita Y, Tomofuji Y, Danks L, Nitta T, Komatsu N, et al. Fezf2 orchestrates a thymic program of self-antigen expression for immune tolerance. Cell. (2015) 163:975–87. doi: 10.1016/j.cell.2015.10.013
42. Peterson P, Org T, Rebane A. Transcriptional regulation by AIRE: molecular mechanisms of central tolerance. Nat Rev Immunol. (2008) 8:948–57. doi: 10.1038/nri2450
43. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. (2001) 2:301–6. doi: 10.1038/86302
44. Ouyang W, Beckett O, Ma Q, Li MO. Transforming growth factor-beta signaling curbs thymic negative selection promoting regulatory T cell development. Immunity. (2010) 32:642–53. doi: 10.1016/j.immuni.2010.04.012
45. Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA. IL-2, -7, and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol. (2008) 181:3285–90. doi: 10.4049/jimmunol.181.5.3285
46. Watanabe N, Wang Y-H, Lee HK, Ito T, Wang Y-H, Cao W, et al. Hassall's corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature. (2005) 436:1181–5. doi: 10.1038/nature03886
47. Tai X, Indart A, Rojano M, Guo J, Apenes N, Kadakia T, et al. How autoreactive thymocytes differentiate into regulatory versus effector CD4+ T cells after avoiding clonal deletion. Nat Immunol. (2023) 24:637–51. doi: 10.1038/s41590-023-01469-2
48. Bhattacharyya ND, Feng CG. Regulation of T helper cell fate by TCR signal strength. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.00624
49. Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signalling. Nat Rev Immunol. (2018) 18:485–97. doi: 10.1038/s41577-018-0020-8
50. Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med. (2011) 208:1279–89. doi: 10.1084/jem.20110308
51. Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol. (2005) 6:152–62. doi: 10.1038/ni1160
52. Hanabuchi S, Ito T, Park W-R, Watanabe N, Shaw JL, Roman E, et al. Thymic stromal lymphopoietin-activated plasmacytoid dendritic cells induce the generation of Foxp3+ Regulatory T cells in human thymus. J Immunol. (2010) 184:2999–3007. doi: 10.4049/jimmunol.0804106
53. Dong M, Chang J, Lebel M, Gervais N, Fournier M, Mallet Gauthier È, et al. The Icos-IcosL pathway tunes thymic selection. Immunol Cell Biol. (2022) 100(3):205–17. doi: 10.1111/imcb.12520
54. Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity. (2009) 31:921–31. doi: 10.1016/j.immuni.2009.09.022
55. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. (2010) 463:808–12. doi: 10.1038/nature08750
56. Dikiy S, Li J, Bai L, Jiang M, Janke L, Zong X, et al. A distal Foxp3 enhancer enables interleukin-2 dependent thymic Treg cell lineage commitment for robust immune tolerance. Immunity. (2021) 54:931–46.e11. doi: 10.1016/j.immuni.2021.03.020
57. Ogawa C, Tone Y, Tsuda M, Peter C, Waldmann H, Tone M. TGF-β-mediated Foxp3 gene expression is cooperatively regulated by Stat5, Creb, and AP-1 through CNS2. J Immunol. (2014) 192:475–83. doi: 10.4049/jimmunol.1301892
58. Permanyer M, Bošnjak B, Glage S, Friedrichsen M, Floess S, Huehn J, et al. Efficient IL-2R signaling differentially affects the stability, function, and composition of the regulatory T-cell pool. Cell Mol Immunol. (2021) 18:398–414. doi: 10.1038/s41423-020-00599-z
59. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. (2007) 178:280–90. doi: 10.4049/jimmunol.178.1.280
60. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. Foxp3 controls regulatory T cell function through cooperation with NFAT. Cell. (2006) 126:375–87. doi: 10.1016/j.cell.2006.05.042
61. Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, et al. Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J Immunol. (2010) 185:842–55. doi: 10.4049/jimmunol.0904100
62. Schlenner SM, Weigmann B, Ruan Q, Chen Y, von Boehmer H. Smad3 binding to the Foxp3 enhancer is dispensable for the development of regulatory T cells with the exception of the gut. J Exp Med. (2012) 209:1529–35. doi: 10.1084/jem.20112646
63. Holohan DR, Van Gool F, Bluestone JA. Thymically-derived Foxp3+ regulatory T cells are the primary regulators of type 1 diabetes in the non-obese diabetic mouse model. PloS One. (2019) 14:e0217728. doi: 10.1371/journal.pone.0217728
64. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. (2008) 9:632–40. doi: 10.1038/ni.1607
65. Cattley RT, Lee M, Boggess WC, Hawse WF. Transforming growth factor β (TGF-β) receptor signaling regulates kinase networks and phosphatidylinositol metabolism during T-cell activation. J Biol Chem. (2020) 295:8236–51. doi: 10.1074/jbc.RA120.012572
66. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. (2012) 37:785–99. doi: 10.1016/j.immuni.2012.09.010
67. Owen DL, Mahmud SA, Sjaastad LE, Williams JB, Spanier JA, Simeonov DR, et al. Thymic regulatory T cells arise via two distinct developmental programs. Nat Immunol. (2019) 20:195–205. doi: 10.1038/s41590-018-0289-6
68. Park J-E, Botting RA, Domínguez Conde C, Popescu D-M, Lavaert M, Kunz DJ, et al. A cell atlas of human thymic development defines T cell repertoire formation. Science. (2020) 367:eaay3224. doi: 10.1126/science.aay3224
69. Herppich S, Toker A, Pietzsch B, Kitagawa Y, Ohkura N, Miyao T, et al. Dynamic imprinting of the treg cell-specific epigenetic signature in developing thymic regulatory T cells. Front Immunol. (2019) 10:2382. doi: 10.3389/fimmu.2019.02382
70. Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol. (2018) 19:291–301. doi: 10.1038/s41590-018-0051-0
71. Sanchez AM, Zhu J, Huang X, Yang Y. The development and function of memory regulatory T cells after acute viral infections. J Immunol. (2012) 189:2805–14. doi: 10.4049/jimmunol.1200645
72. Solomon BD, Hsieh CS. Antigen-specific development of mucosal Foxp3+RORgammat+ T cells from regulatory T cell precursors. J Immunol. (2016) 197:3512–9. doi: 10.4049/jimmunol.1601217
73. Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. (2004) 199:1401–8. doi: 10.1084/jem.20040249
74. Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. (2005) 6:1219–27. doi: 10.1038/ni1265
75. Thorstenson KM, Khoruts A. Generation of anergic and potentially immunoregulatory CD25+CD4 T cells in vivo after induction of peripheral tolerance with intravenous or oral antigen. J Immunol (Baltimore Md 1950). (2001) 167:188–95. doi: 10.4049/jimmunol.167.1.188
76. Wan YY, Flavell RA. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci U S A. (2005) 102:5126–31. doi: 10.1073/pnas.0501701102
77. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
78. Wan YY, Flavell RA. 'Yin-Yang' functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol Rev. (2007) 220:199–213. doi: 10.1111/j.1600-065X.2007.00565.x
79. Wang J, Zhao X, Wan YY. Intricacies of TGF-beta signaling in Treg and Th17 cell biology. Cell Mol Immunol. (2023) 20:1002–22. doi: 10.1038/s41423-023-01036-7
80. Tran DQ. TGF-beta: the sword, the wand, and the shield of Foxp3(+) regulatory T cells. J Mol Cell Biol. (2012) 4:29–37. doi: 10.1093/jmcb/mjr033
81. Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. (2007) 204:1775–85. doi: 10.1084/jem.20070602
82. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. (2013) 504:451–5. doi: 10.1038/nature12726
83. Hoeppli RE, MacDonald KG, Levings MK, Cook L. How antigen specificity directs regulatory T-cell function: self, foreign and engineered specificity. HLA. (2016) 88:3–13. doi: 10.1111/tan.12822
84. Attias M, Al-Aubodah T, Piccirillo CA. Mechanisms of human Foxp3(+) T(reg) cell development and function in health and disease. Clin Exp Immunol. (2019) 197:36–51. doi: 10.1111/cei.13290
85. Schmidt AM, Lu W, Sindhava VJ, Huang Y, Burkhardt JK, Yang E, et al. Regulatory T cells require TCR signaling for their suppressive function. J Immunol. (2015) 194:4362–70. doi: 10.4049/jimmunol.1402384
86. Fuchs S, Rensing-Ehl A, Speckmann C, Bengsch B, Schmitt-Graeff A, Bondzio I, et al. Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J Immunol. (2012) 188:1523–33. doi: 10.4049/jimmunol.1102507
87. Lacruz RS, Feske S. Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci. (2015) 1356:45–79. doi: 10.1111/nyas.12938
88. Vaeth M, Wang YH, Eckstein M, Yang J, Silverman GJ, Lacruz RS, et al. Tissue resident and follicular Treg cell differentiation is regulated by CRAC channels. Nat Commun. (2019) 10:1183. doi: 10.1038/s41467-019-08959-8
89. Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D, et al. Regulatory T cell migration is dependent on glucokinase-mediated glycolysis. Immunity. (2017) 47:875–89.e10. doi: 10.1016/j.immuni.2017.10.017
90. Kempkes RWM, Joosten I, Koenen H, He X. Metabolic pathways involved in regulatory T cell functionality. Front Immunol. (2019) 10:2839. doi: 10.3389/fimmu.2019.02839
91. van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev. (2012) 249:27–42. doi: 10.1111/j.1600-065X.2012.01150.x
92. Sun IH, Oh MH, Zhao L, Patel CH, Arwood ML, Xu W, et al. mTOR complex 1 signaling regulates the generation and function of central and effector Foxp3(+) regulatory T cells. J Immunol. (2018) 201:481–92. doi: 10.4049/jimmunol.1701477
93. Zeng H, Chi H. mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr Opin Immunol. (2017) 46:103–11. doi: 10.1016/j.coi.2017.04.005
94. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-like receptor signaling balance T(reg) cell anabolic metabolism for suppression. Nat Immunol. (2016) 17:1459–66. doi: 10.1038/ni.3577
95. van der Veeken J, Glasner A, Zhong Y, Hu W, Wang ZM, Bou-Puerto R, et al. The transcription factor Foxp3 shapes regulatory T cell identity by tuning the activity of trans-acting intermediaries. Immunity. (2020) 53:971–84.e5. doi: 10.1016/j.immuni.2020.10.010
96. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. (2001) 27:68–73. doi: 10.1038/83784
97. van der Vliet HJ, Nieuwenhuis EE. IPEX as a result of mutations in Foxp3. Clin Dev Immunol. (2007) 2007:89017. doi: 10.1155/2007/89017
98. Lam AJ, Lin DTS, Gillies JK, Uday P, Pesenacker AM, Kobor MS, et al. Optimized CRISPR-mediated gene knockin reveals Foxp3-independent maintenance of human Treg identity. Cell Rep. (2021) 36:109494. doi: 10.1016/j.celrep.2021.109494
99. Sugimoto N, Oida T, Hirota K, Nakamura K, Nomura T, Uchiyama T, et al. Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int Immunol. (2006) 18:1197–209. doi: 10.1093/intimm/dxl060
100. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. (2007) 445:771–5. doi: 10.1038/nature05543
101. Leon J, Chowdhary K, Zhang W, Ramirez RN, Andre I, Hur S, et al. Mutations from patients with IPEX ported to mice reveal different patterns of Foxp3 and Treg dysfunction. Cell Rep. (2023) 42:113018. doi: 10.1016/j.celrep.2023.113018
102. Hayatsu N, Miyao T, Tachibana M, Murakami R, Kimura A, Kato T, et al. Analyses of a mutant Foxp3 allele reveal BATF as a critical transcription factor in the differentiation and accumulation of tissue regulatory T cells. Immunity. (2017) 47:268–83.e9. doi: 10.1016/j.immuni.2017.07.008
103. Khatun A, Wu X, Qi F, Gai K, Kharel A, Kudek MR, et al. BATF is required for treg homeostasis and stability to prevent autoimmune pathology. Adv Sci (Weinh). (2023) 10(28):e2206692. doi: 10.1002/advs.202206692
104. Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol. (2012) 13:1010–9. doi: 10.1038/ni.2402
105. Ono M. Control of regulatory T-cell differentiation and function by T-cell receptor signalling and Foxp3 transcription factor complexes. Immunology. (2020) 160:24–37. doi: 10.1111/imm.13178
106. Xing S, Gai K, Li X, Shao P, Zeng Z, Zhao X, et al. Tcf1 and Lef1 are required for the immunosuppressive function of regulatory T cells. J Exp Med. (2019) 216:847–66. doi: 10.1084/jem.20182010
107. Yang BH, Wang K, Wan S, Liang Y, Yuan X, Dong Y, et al. TCF1 and Lef1 control treg competitive survival and tfr development to prevent autoimmune diseases. Cell Rep. (2019) 27:3629–45.e6. doi: 10.1016/j.celrep.2019.05.061
108. Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS, et al. A multiply redundant genetic switch 'locks in' the transcriptional signature of regulatory T cells. Nat Immunol. (2012) 13:972–80. doi: 10.1038/ni.2420
109. Osman A, Yan B, Li Y, Pavelko KD, Quandt J, Saadalla A, et al. TCF-1 controls T(reg) cell functions that regulate inflammation, CD8(+) T cell cytotoxicity and severity of colon cancer. Nat Immunol. (2021) 22:1152–62. doi: 10.1038/s41590-021-00987-1
110. Arvey A, van der Veeken J, Plitas G, Rich SS, Concannon P, Rudensky AY. Genetic and epigenetic variation in the lineage specification of regulatory T cells. Elife. (2015) 4:e07571. doi: 10.7554/eLife.07571
111. Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F, et al. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol. (2015) 16:980–90. doi: 10.1038/ni.3226
112. DiSpirito JR, Zemmour D, Ramanan D, Cho J, Zilionis R, Klein AM, et al. Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci Immunol. (2018) 3(27):eaat5861. doi: 10.1126/sciimmunol.aat5861
113. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. (2008) 38:1654–63. doi: 10.1002/eji.200838105
114. Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced Foxp3 gene expression: a role for DNA methylation. J Exp Med. (2007) 204:1543–51. doi: 10.1084/jem.20070109
115. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell. (2014) 158:749–63. doi: 10.1016/j.cell.2014.07.031
116. Li X, Liang Y, LeBlanc M, Benner C, Zheng Y. Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell. (2014) 158:734–48. doi: 10.1016/j.cell.2014.07.030
117. Polansky JK, Schreiber L, Thelemann C, Ludwig L, Kruger M, Baumgrass R, et al. Methylation matters: binding of Ets-1 to the demethylated Foxp3 gene contributes to the stabilization of Foxp3 expression in regulatory T cells. J Mol Med (Berl). (2010) 88:1029–40. doi: 10.1007/s00109-010-0642-1
118. Kitoh A, Ono M, Naoe Y, Ohkura N, Yamaguchi T, Yaguchi H, et al. Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of Foxp3+ regulatory T cells. Immunity. (2009) 31:609–20. doi: 10.1016/j.immuni.2009.09.003
119. Someya K, Nakatsukasa H, Ito M, Kondo T, Tateda KI, Akanuma T, et al. Improvement of Foxp3 stability through CNS2 demethylation by TET enzyme induction and activation. Int Immunol. (2017) 29:365–75. doi: 10.1093/intimm/dxx049
120. Delacher M, Imbusch CD, Weichenhan D, Breiling A, Hotz-Wagenblatt A, Trager U, et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat Immunol. (2017) 18:1160–72. doi: 10.1038/ni.3799
121. Joudi AM, Reyes Flores CP, Singer BD. Epigenetic control of regulatory T cell stability and function: Implications for translation. Front Immunol. (2022) 13:861607. doi: 10.3389/fimmu.2022.861607
122. Lan J, Hua S, He X, Zhang Y. DNA methyltransferases and methyl-binding proteins of mammals. Acta Biochim Biophys Sin (Shanghai). (2010) 42:243–52. doi: 10.1093/abbs/gmq015
123. He XJ, Chen T, Zhu JK. Regulation and function of DNA methylation in plants and animals. Cell Res. (2011) 21:442–65. doi: 10.1038/cr.2011.23
124. Wang L, Liu Y, Beier UH, Han R, Bhatti TR, Akimova T, et al. Foxp3+ T-regulatory cells require DNA methyltransferase 1 expression to prevent development of lethal autoimmunity. Blood. (2013) 121:3631–9. doi: 10.1182/blood-2012-08-451765
125. Helmin KA, Morales-Nebreda L, Torres Acosta MA, Anekalla KR, Chen SY, Abdala-Valencia H, et al. Maintenance DNA methylation is essential for regulatory T cell development and stability of suppressive function. J Clin Invest. (2020) 130:6571–87. doi: 10.1172/JCI137712
126. Obata Y, Furusawa Y, Endo TA, Sharif J, Takahashi D, Atarashi K, et al. The epigenetic regulator Uhrf1 facilitates the proliferation and maturation of colonic regulatory T cells. Nat Immunol. (2014) 15:571–9. doi: 10.1038/ni.2886
127. Poli A, Abdul-Hamid S, Zaurito AE, Campagnoli F, Bevilacqua V, Sheth B, et al. PIP4Ks impact on PI3K, Foxp3, and UHRF1 signaling and modulate human regulatory T cell proliferation and immunosuppressive activity. Proc Natl Acad Sci U.S.A. (2021) 118(31):e2010053118. doi: 10.1073/pnas.2010053118
128. Sun X, Cui Y, Feng H, Liu H, Liu X. TGF-beta signaling controls Foxp3 methylation and T reg cell differentiation by modulating Uhrf1 activity. J Exp Med. (2019) 216:2819–37. doi: 10.1084/jem.20190550
129. Wang L, Beier UH, Akimova T, Dahiya S, Han R, Samanta A, et al. Histone/protein deacetylase inhibitor therapy for enhancement of Foxp3+ T-regulatory cell function posttransplantation. Am J Transplant. (2018) 18:1596–603. doi: 10.1111/ajt.14749
130. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. (2007) 13:1299–307. doi: 10.1038/nm1652
131. Pieniawska M, Izykowska K. Role of histone deacetylases in T-cell development and function. Int J Mol Sci. (2022) 23(14):7828. doi: 10.3390/ijms23147828
132. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. (2014) 15:1070–8. doi: 10.1038/ni.3004
133. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor Irf4 to control T(H)2 responses. Nature. (2009) 458:351–6. doi: 10.1038/nature07674
134. Li P, Spolski R, Liao W, Wang L, Murphy TL, Murphy KM, et al. BATF-JUN is critical for Irf4-mediated transcription in T cells. Nature. (2012) 490:543–6. doi: 10.1038/nature11530
135. Alvisi G, Brummelman J, Puccio S, Mazza EM, Tomada EP, Losurdo A, et al. Irf4 instructs effector Treg differentiation and immune suppression in human cancer. J Clin Invest. (2020) 130:3137–50. doi: 10.1172/JCI130426
136. Ding X, Wang A, Ma X, Demarque M, Jin W, Xin H, et al. Protein SUMOylation is required for regulatory T cell expansion and function. Cell Rep. (2016) 16:1055–66. doi: 10.1016/j.celrep.2016.06.056
137. Tang M, Cheng L, Li F, Wu B, Chen P, Zhan Y, et al. Transcription factor Irf4 dysfunction affects the immunosuppressive function of treg cells in patients with primary immune thrombocytopenia. BioMed Res Int. (2019) 2019:1050285. doi: 10.1155/2019/1050285
138. Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, et al. The transcription factor Irf4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol. (2013) 14:1155–65. doi: 10.1038/ni.2710
139. Arnold PR, Wen M, Zhang L, Ying Y, Xiao X, Chu X, et al. Suppression of Foxp3 expression by the AP-1 family transcription factor BATF3 requires partnering with Irf4. Front Immunol. (2022) 13:966364. doi: 10.3389/fimmu.2022.966364
140. Tosello V, Odunsi K, Souleimanian NE, Lele S, Shrikant P, Old LJ, et al. Differential expression of CCR7 defines two distinct subsets of human memory CD4+CD25+ Tregs. Clin Immunol. (2008) 126:291–302. doi: 10.1016/j.clim.2007.11.008
141. Sanchez Rodriguez R, Pauli ML, Neuhaus IM, Yu SS, Arron ST, Harris HW, et al. Memory regulatory T cells reside in human skin. J Clin Invest. (2014) 124:1027–36. doi: 10.1172/JCI72932
142. Menning A, Hopken UE, Siegmund K, Lipp M, Hamann A, Huehn J. Distinctive role of CCR7 in migration and functional activity of naive- and effector/memory-like Treg subsets. Eur J Immunol. (2007) 37:1575–83. doi: 10.1002/eji.200737201
143. Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. (2016) 352:459–63. doi: 10.1126/science.aad2035
144. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
145. Li L, Patsoukis N, Petkova V, Boussiotis VA. Runx1 and Runx3 are involved in the generation and function of highly suppressive IL-17-producing T regulatory cells. PloS One. (2012) 7:e45115. doi: 10.1371/journal.pone.0045115
146. Lam AJ, Uday P, Gillies JK, Levings MK. Helios is a marker, not a driver, of human Treg stability. Eur J Immunol. (2022) 52:75–84. doi: 10.1002/eji.202149318
147. Sebastian M, Lopez-Ocasio M, Metidji A, Rieder SA, Shevach EM, Thornton AM. Helios controls a limited subset of regulatory T cell functions. J Immunol. (2016) 196:144–55. doi: 10.4049/jimmunol.1501704
148. Baine I, Basu S, Ames R, Sellers RS, Macian F. Helios induces epigenetic silencing of IL2 gene expression in regulatory T cells. J Immunol. (2013) 190:1008–16. doi: 10.4049/jimmunol.1200792
149. Gottschalk RA, Corse E, Allison JP. Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J Immunol. (2012) 188:976–80. doi: 10.4049/jimmunol.1102964
150. Wang Y, Misumi I, Gu AD, Curtis TA, Su L, Whitmire JK, et al. GATA-3 controls the maintenance and proliferation of T cells downstream of TCR and cytokine signaling. Nat Immunol. (2013) 14:714–22. doi: 10.1038/ni.2623
151. Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. (1997) 89:587–96. doi: 10.1016/S0092-8674(00)80240-8
152. Xu K, Yang WY, Nanayakkara GK, Shao Y, Yang F, Hu W, et al. Gata3, HDAC6, and BCL6 regulate Foxp3+ Treg plasticity and determine treg conversion into either novel antigen-presenting cell-like treg or th1-treg. Front Immunol. (2018) 9:45. doi: 10.3389/fimmu.2018.00045
153. Seki N, Miyazaki M, Suzuki W, Hayashi K, Arima K, Myburgh E, et al. IL-4-induced GATA-3 expression is a time-restricted instruction switch for Th2 cell differentiation. J Immunol. (2004) 172:6158–66. doi: 10.4049/jimmunol.172.10.6158
154. Ding Z, Cai T, Tang J, Sun H, Qi X, Zhang Y, et al. Setd2 supports Gata3(+)ST2(+) thymic-derived Treg cells and suppresses intestinal inflammation. Nat Commun. (2022) 13:7468. doi: 10.1038/s41467-022-35250-0
155. Hayakawa M, Yanagisawa K, Aoki S, Hayakawa H, Takezako N, Tominaga S. T-helper type 2 cell-specific expression of the ST2 gene is regulated by transcription factor GATA-3. Biochim Biophys Acta. (2005) 1728:53–64. doi: 10.1016/j.bbaexp.2005.01.012
156. Shoemaker J, Saraiva M, O'Garra A. GATA-3 directly remodels the IL-10 locus independently of IL-4 in CD4+ T cells. J Immunol. (2006) 176:3470–9. doi: 10.4049/jimmunol.176.6.3470
157. Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. (2008) 28:29–39. doi: 10.1016/j.immuni.2007.11.016
158. Haim-Vilmovsky L, Henriksson J, Walker JA, Miao Z, Natan E, Kar G, et al. Mapping Rora expression in resting and activated CD4+ T cells. PloS One. (2021) 16:e0251233. doi: 10.1371/journal.pone.0251233
159. Bankoti R, Ogawa C, Nguyen T, Emadi L, Couse M, Salehi S, et al. Differential regulation of Effector and Regulatory T cell function by Blimp1. Sci Rep. (2017) 7:12078. doi: 10.1038/s41598-017-12171-3
160. Garg G, Muschaweckh A, Moreno H, Vasanthakumar A, Floess S, Lepennetier G, et al. Blimp1 prevents methylation of Foxp3 and loss of regulatory T cell identity at sites of inflammation. Cell Rep. (2019) 26:1854–68.e5. doi: 10.1016/j.celrep.2019.01.070
161. Ogawa C, Bankoti R, Nguyen T, Hassanzadeh-Kiabi N, Nadeau S, Porritt RA, et al. Blimp-1 functions as a molecular switch to prevent inflammatory activity in Foxp3(+)RORgammat(+) regulatory T cells. Cell Rep. (2018) 25:19–28.e5. doi: 10.1016/j.celrep.2018.09.016
162. Alvarez F, Al-Aubodah TA, Yang YH, Piccirillo CA. Mechanisms of T(REG) cell adaptation to inflammation. J Leukoc Biol. (2020) 108:559–71. doi: 10.1002/JLB.1MR0120-196R
163. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel 'alarmin'? PloS One. (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
164. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
165. Matta BM, Turnquist HR. Expansion of regulatory T cells in vitro and in vivo by IL-33. Methods Mol Biol. (2016) 1371:29–41. doi: 10.1007/978-1-4939-3139-2_3
166. Faustino LD, Griffith JW, Rahimi RA, Nepal K, Hamilos DL, Cho JL, et al. Interleukin-33 activates regulatory T cells to suppress innate gammadelta T cell responses in the lung. Nat Immunol. (2020) 21:1371–83. doi: 10.1038/s41590-020-0785-3
167. Alvarez F, Istomine R, Shourian M, Pavey N, Al-Aubodah TA, Qureshi S, et al. The alarmins IL-1 and IL-33 differentially regulate the functional specialisation of Foxp3(+) regulatory T cells during mucosal inflammation. Mucosal Immunol. (2019) 12:746–60. doi: 10.1038/s41385-019-0153-5
168. Hemmers S, Schizas M, Rudensky AY. T reg cell-intrinsic requirements for ST2 signaling in health and neuroinflammation. J Exp Med. (2021) 218(2):e20201234. doi: 10.1084/jem.20201234
169. Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, et al. IL-33 is an unconventional Alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol. (2014) 193:4010–20. doi: 10.4049/jimmunol.1400481
170. Chang J, Bouchard A, Bouklouch Y, Panneton V, Li J, Diamantopoulos N, et al. Icos-deficient regulatory T cells can prevent spontaneous autoimmunity but are impaired in controlling acute inflammation. J Immunol. (2022) 209:301–9. doi: 10.4049/jimmunol.2100897
171. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. (2013) 499:485–90. doi: 10.1038/nature12297
172. Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature. (2012) 491:554–9. doi: 10.1038/nature11581
173. Ouyang W, Li MO. Foxo: in command of T lymphocyte homeostasis and tolerance. Trends Immunol. (2011) 32:26–33. doi: 10.1016/j.it.2010.10.005
174. Mittelsteadt KL, Hayes ET, Campbell DJ. Icos signaling limits regulatory T cell accumulation and function in visceral adipose tissue. J Exp Med. (2021) 218(6):e20201142. doi: 10.1084/jem.20201142
175. Miao Y, Zhang C, Yang L, Zeng X, Hu Y, Xue X, et al. The activation of PPARgamma enhances Treg responses through up-regulating CD36/CPT1-mediated fatty acid oxidation and subsequent N-glycan branching of TbetaRII/IL-2Ralpha. Cell Commun Signal. (2022) 20:48. doi: 10.1186/s12964-022-00849-9
176. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. (2012) 486:549–53. doi: 10.1038/nature11132
177. Sanderlin EJ, Leffler NR, Lertpiriyapong K, Cai Q, Hong H, Bakthavatchalu V, et al. GPR4 deficiency alleviates intestinal inflammation in a mouse model of acute experimental colitis. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:569–84. doi: 10.1016/j.bbadis.2016.12.005
178. Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, et al. Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight. (2017) 2:e89160. doi: 10.1172/jci.insight.89160
179. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
180. Sivasami P, Elkins C, Diaz-Saldana PP, Goss K, Peng A, Hamersky MT, et al. Obesity-induced dysregulation of skin-resident PPARgamma(+) Treg cells promotes IL-17A-mediated psoriatic inflammation. Immunity. (2023) 56:1844–61.e6. doi: 10.1016/j.immuni.2023.06.021
181. Howie D, Ten Bokum A, Cobbold SP, Yu Z, Kessler BM, Waldmann H. A novel role for triglyceride metabolism in Foxp3 expression. Front Immunol. (2019) 10:1860. doi: 10.3389/fimmu.2019.01860
182. Basu S, Hubbard B, Shevach EM. Foxp3-mediated inhibition of Akt inhibits Glut1 (glucose transporter 1) expression in human T regulatory cells. J Leukoc Biol. (2015) 97:279–83. doi: 10.1189/jlb.2AB0514-273RR
183. Graham KL, Werner BJ, Moyer KM, Patton AK, Krois CR, Yoo HS, et al. DGAT1 inhibits retinol-dependent regulatory T cell formation and mediates autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. (2019) 116:3126–35. doi: 10.1073/pnas.1817669116
184. Li C, Munoz-Rojas AR, Wang G, Mann AO, Benoist C, Mathis D. PPARgamma marks splenic precursors of multiple nonlymphoid-tissue Treg compartments. Proc Natl Acad Sci U S A. (2021) 118(13):e2025197118. doi: 10.1073/pnas.2025197118
185. Elfaki Y, Yang J, Boehme J, Schultz K, Bruder D, Falk CS, et al. Tbx21 and Foxp3 are epigenetically stabilized in T-bet(+) tregs that transiently accumulate in influenza A virus-infected lungs. Int J Mol Sci. (2021) 22(14):7522. doi: 10.3390/ijms22147522
186. Sjaastad LE, Owen DL, Joo S, Knutson TP, O’Connor CH, McCluskey B, et al. Influenza infection recruits distinct waves of regulatory T cells to the lung that limit lung resident IgA+ B cells. bioRxiv. (2022) 2022:2022.09.19.508325. doi: 10.1101/2022.09.19.508325
187. Alvarez F, Istomine R, Da Silva Lira Filho A, Al-Aubodah TA, Huang D, Okde R, et al. IL-18 is required for the T(H)1-adaptation of T(REG) cells and the selective suppression of T(H)17 responses in acute and chronic infections. Mucosal Immunol. (2023) 16:462–75. doi: 10.1016/j.mucimm.2023.05.004
188. Procaccini C, De Rosa V, Galgani M, Abanni L, Cali G, Porcellini A, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. (2010) 33:929–41. doi: 10.1016/j.immuni.2010.11.024
189. Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science. (2015) 349:993–7. doi: 10.1126/science.aaa9420
190. Rankin LC, Kaiser KA, de Los Santos-Alexis K, Park H, Uhlemann AC, Gray DHD, et al. Dietary tryptophan deficiency promotes gut RORgammat(+) Treg cells at the expense of Gata3(+) Treg cells and alters commensal microbiota metabolism. Cell Rep. (2023) 42:112135. doi: 10.1016/j.celrep.2023.112135
191. Yang BH, Hagemann S, Mamareli P, Lauer U, Hoffmann U, Beckstette M, et al. Foxp3(+) T cells expressing RORgammat represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol. (2016) 9:444–57. doi: 10.1038/mi.2015.74
192. Kim BS, Lu H, Ichiyama K, Chen X, Zhang YB, Mistry NA, et al. Generation of RORgammat(+) antigen-specific T regulatory 17 cells from Foxp3(+) precursors in autoimmunity. Cell Rep. (2017) 21:195–207. doi: 10.1016/j.celrep.2017.09.021
193. Seif F, Khoshmirsafa M, Aazami H, Mohsenzadegan M, Sedighi G, Bahar M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal. (2017) 15:23. doi: 10.1186/s12964-017-0177-y
194. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. (2000) 100:655–69. doi: 10.1016/S0092-8674(00)80702-3
195. Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature. (2017) 546:421–5. doi: 10.1038/nature22360
196. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. (2009) 10:595–602. doi: 10.1038/ni.1731
197. Paust HJ, Riedel JH, Krebs CF, Turner JE, Brix SR, Krohn S, et al. CXCR3+ Regulatory T cells control TH1 responses in crescentic GN. J Am Soc Nephrol. (2016) 27:1933–42. doi: 10.1681/ASN.2015020203
198. Bedoya F, Cheng GS, Leibow A, Zakhary N, Weissler K, Garcia V, et al. Viral antigen induces differentiation of Foxp3+ natural regulatory T cells in influenza virus-infected mice. J Immunol. (2013) 190:6115–25. doi: 10.4049/jimmunol.1203302
199. Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, et al. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity. (2008) 29:679–90. doi: 10.1016/j.immuni.2008.08.017
200. Wei L, Vahedi G, Sun HW, Watford WT, Takatori H, Ramos HL, et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity. (2010) 32:840–51. doi: 10.1016/j.immuni.2010.06.003
201. Murphy KM, Ouyang W, Szabo SJ, Jacobson NG, Guler ML, Gorham JD, et al. T helper differentiation proceeds through Stat1-dependent, Stat4-dependent and Stat4-independent phases. Curr Top Microbiol Immunol. (1999) 238:13–26. doi: 10.1007/978-3-662-09709-0_2
202. Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, et al. STATs shape the active enhancer landscape of T cell populations. Cell. (2012) 151:981–93. doi: 10.1016/j.cell.2012.09.044
203. Schulz EG, Mariani L, Radbruch A, Hofer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity. (2009) 30:673–83. doi: 10.1016/j.immuni.2009.03.013
204. Di Giovangiulio M, Rizzo A, Franze E, Caprioli F, Facciotti F, Onali S, et al. Tbet expression in regulatory T cells is required to initiate th1-mediated colitis. Front Immunol. (2019) 10:2158. doi: 10.3389/fimmu.2019.02158
205. Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor beta2. Immunity. (2012) 37:501–10. doi: 10.1016/j.immuni.2012.05.031
206. Zhao J, Zhao J, Perlman S. Differential effects of IL-12 on Tregs and non-Treg T cells: roles of IFN-gamma, IL-2 and IL-2R. PloS One. (2012) 7:e46241. doi: 10.1371/journal.pone.0046241
207. O'Malley JT, Sehra S, Thieu VT, Yu Q, Chang HC, Stritesky GL, et al. Signal transducer and activator of transcription 4 limits the development of adaptive regulatory T cells. Immunology. (2009) 127:587–95. doi: 10.1111/j.1365-2567.2008.03037.x
208. O'Shea JJ, Lahesmaa R, Vahedi G, Laurence A, Kanno Y. Genomic views of STAT function in CD4+ T helper cell differentiation. Nat Rev Immunol. (2011) 11:239–50. doi: 10.1038/nri2958
209. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol. (2011) 186:3918–26. doi: 10.4049/jimmunol.1003099
210. Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med. (2011) 17:673–5. doi: 10.1038/nm.2389
211. Kitz A, Dominguez-Villar M. Molecular mechanisms underlying Th1-like Treg generation and function. Cell Mol Life Sci. (2017) 74:4059–75. doi: 10.1007/s00018-017-2569-y
212. Koenecke C, Lee CW, Thamm K, Fohse L, Schafferus M, Mittrucker HW, et al. IFN-gamma production by allogeneic Foxp3+ regulatory T cells is essential for preventing experimental graft-versus-host disease. J Immunol. (2012) 189:2890–6. doi: 10.4049/jimmunol.1200413
213. Cuadrado E, van den Biggelaar M, de Kivit S, Chen YY, Slot M, Doubal I, et al. Proteomic analyses of human regulatory T cells reveal adaptations in signaling pathways that protect cellular identity. Immunity. (2018) 48:1046–59.e6. doi: 10.1016/j.immuni.2018.04.008
214. Yu Q, Chang HC, Ahyi AN, Kaplan MH. Transcription factor-dependent chromatin remodeling of il18r1 during Th1 and Th2 differentiation. J Immunol. (2008) 181:3346–52. doi: 10.4049/jimmunol.181.5.3346
215. Yu Q, Thieu VT, Kaplan MH. Stat4 limits DNA methyltransferase recruitment and DNA methylation of the IL-18Ralpha gene during Th1 differentiation. EMBO J. (2007) 26:2052–60. doi: 10.1038/sj.emboj.7601653
216. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell. (2015) 162:1078–89. doi: 10.1016/j.cell.2015.08.021
217. Harrison OJ, Srinivasan N, Pott J, Schiering C, Krausgruber T, Ilott NE, et al. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3(+) Treg cell function in the intestine. Mucosal Immunol. (2015) 8:1226–36. doi: 10.1038/mi.2015.13
218. Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, et al. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest. (2012) 122:1082–96. doi: 10.1172/JCI61029
219. Pai SY, Truitt ML, Ho IC. GATA-3 deficiency abrogates the development and maintenance of T helper type 2 cells. Proc Natl Acad Sci U S A. (2004) 101:1993–8. doi: 10.1073/pnas.0308697100
220. Chen T, Hou X, Ni Y, Du W, Han H, Yu Y, et al. The imbalance of Foxp3/Gata3 in regulatory T cells from the peripheral blood of asthmatic patients. J Immunol Res. (2018) 2018:3096183. doi: 10.1155/2018/3096183
221. Abdel Aziz N, Nono JK, Mpotje T, Brombacher F. The Foxp3+ regulatory T-cell population requires IL-4Ralpha signaling to control inflammation during helminth infections. PloS Biol. (2018) 16:e2005850. doi: 10.1371/journal.pbio.2005850
222. Pelly VS, Coomes SM, Kannan Y, Gialitakis M, Entwistle LJ, Perez-Lloret J, et al. Interleukin 4 promotes the development of ex-Foxp3 Th2 cells during immunity to intestinal helminths. J Exp Med. (2017) 214:1809–26. doi: 10.1084/jem.20161104
223. Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity. (2003) 19:739–48. doi: 10.1016/S1074-7613(03)00292-9
224. Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. (1996) 4:313–9. doi: 10.1016/S1074-7613(00)80439-2
225. Zhu J. Transcriptional regulation of Th2 cell differentiation. Immunol Cell Biol. (2010) 88:244–9. doi: 10.1038/icb.2009.114
226. Khumalo J, Kirstein F, Hadebe S, Brombacher F. IL-4Ralpha signaling in CD4+CD25+Foxp3+ T regulatory cells restrains airway inflammation via limiting local tissue IL-33. JCI Insight. (2020) 5(20):e136206. doi: 10.1172/jci.insight.136206
227. Pace L, Pioli C, Doria G. IL-4 modulation of CD4+CD25+ T regulatory cell-mediated suppression. J Immunol. (2005) 174:7645–53. doi: 10.4049/jimmunol.174.12.7645
228. Cui J, Xu H, Yu J, Li Y, Chen Z, Zou Y, et al. IL-4 inhibits regulatory T cells differentiation by HDAC9-mediated epigenetic regulation. Cell Death Dis. (2021) 12:501. doi: 10.1038/s41419-021-03769-7
229. Zhou JY, Alvarez CA, Cobb BA. Integration of IL-2 and IL-4 signals coordinates divergent regulatory T cell responses and drives therapeutic efficacy. Elife. (2021) 10:e57417. doi: 10.7554/eLife.57417
230. Jin HS, Park Y, Elly C, Liu YC. Itch expression by Treg cells controls Th2 inflammatory responses. J Clin Invest. (2013) 123:4923–34. doi: 10.1172/JCI69355
231. Chen W. Regulatory T cells use "Itch" to control asthma. J Clin Invest. (2013) 123:4576–8. doi: 10.1172/JCI72477
232. Knosp CA, Schiering C, Spence S, Carroll HP, Nel HJ, Osbourn M, et al. Regulation of Foxp3+ inducible regulatory T cell stability by SOCS2. J Immunol. (2013) 190:3235–45. doi: 10.4049/jimmunol.1201396
233. Siede J, Frohlich A, Datsi A, Hegazy AN, Varga DV, Holecska V, et al. IL-33 receptor-expressing regulatory T cells are highly activated, th2 biased and suppress CD4 T cell proliferation through IL-10 and TGFbeta release. PloS One. (2016) 11:e0161507. doi: 10.1371/journal.pone.0161507
234. Wen YH, Lin HQ, Li H, Zhao Y, Lui VWY, Chen L, et al. Stromal interleukin-33 promotes regulatory T cell-mediated immunosuppression in head and neck squamous cell carcinoma and correlates with poor prognosis. Cancer Immunol Immunother. (2019) 68:221–32. doi: 10.1007/s00262-018-2265-2
235. Ameri AH, Moradi Tuchayi S, Zaalberg A, Park JH, Ngo KH, Li T, et al. IL-33/regulatory T cell axis triggers the development of a tumor-promoting immune environment in chronic inflammation. Proc Natl Acad Sci United States America. (2019) 116:2646–51. doi: 10.1073/pnas.1815016116
236. Pastille E, Wasmer MH, Adamczyk A, Vu VP, Mager LF, Phuong NNT, et al. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol. (2019) 12:990–1003. doi: 10.1038/s41385-019-0176-y
237. Liu Q, Dwyer GK, Zhao Y, Li H, Mathews LR, Chakka AB, et al. IL-33-mediated IL-13 secretion by ST2+ Tregs controls inflammation after lung injury. JCI Insight. (2019) 4(6):e123919. doi: 10.1172/jci.insight.123919
238. Chen CC, Kobayashi T, Iijima K, Hsu FC, Kita H. IL-33 dysregulates regulatory T cells and impairs established immunologic tolerance in the lungs. J Allergy Clin Immunol. (2017) 140:1351–63 e7. doi: 10.1016/j.jaci.2017.01.015
239. Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. Stat3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. (2007) 282:9358–63. doi: 10.1074/jbc.C600321200
240. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
241. Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood. (2013) 121:2402–14. doi: 10.1182/blood-2012-09-378653
242. Yang BH, Hagemann S, Mamareli P, Lauer U, Hoffmann U, Beckstette M, et al. Foxp3(+) T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol. (2016) 9:444–57. doi: 10.1038/mi.2015.74
243. Furuyama K, Kondo Y, Shimizu M, Yokosawa M, Segawa S, Iizuka A, et al. RORgammat+Foxp3+ regulatory T cells in the regulation of autoimmune arthritis. Clin Exp Immunol. (2022) 207:176–87. doi: 10.1093/cei/uxab007
244. Gao Z, Gao Y, Li Z, Chen Z, Lu D, Tsun A, et al. Synergy between IL-6 and TGF-beta signaling promotes Foxp3 degradation. Int J Clin Exp Pathol. (2012) 5:626–33. doi: 10.1016/j.joca.2019.04.014
245. Jacobse J, Brown RE, Li J, Pilat JM, Pham L, Short SP, et al. Interleukin-23 receptor signaling impairs the stability and function of colonic regulatory T cells. Cell Rep. (2023) 42:112128. doi: 10.1016/j.celrep.2023.112128
246. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. (2007) 8:967–74. doi: 10.1038/ni1488
247. Li L, Kim J, Boussiotis VA. IL-1beta-mediated signals preferentially drive conversion of regulatory T cells but not conventional T cells into IL-17-producing cells. J Immunol. (2010) 185:4148–53. doi: 10.4049/jimmunol.1001536
248. Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. (2008) 112:2340–52. doi: 10.1182/blood-2008-01-133967
249. Shourian M, Ralph B, Angers I, Sheppard DC, Qureshi ST. Contribution of IL-1RI Signaling to Protection against Cryptococcus neoformans 52D in a Mouse Model of Infection. Front Immunol. (2017) 8:1987. doi: 10.3389/fimmu.2017.01987
250. Nikolouli E, Elfaki Y, Herppich S, Schelmbauer C, Delacher M, Falk C, et al. Recirculating IL-1R2(+) Tregs fine-tune intrathymic Treg development under inflammatory conditions. Cell Mol Immunol. (2021) 18:182–93. doi: 10.1038/s41423-019-0352-8
251. Mercer F, Kozhaya L, Unutmaz D. Expression and function of TNF and IL-1 receptors on human regulatory T cells. PloS One. (2010) 5:e8639. doi: 10.1371/journal.pone.0008639
252. Tran DQ, Andersson J, Hardwick D, Bebris L, Illei GG, Shevach EM. Selective expression of latency-associated peptide (LAP) and IL-1 receptor type I/II (CD121a/CD121b) on activated human Foxp3+ regulatory T cells allows for their purification from expansion cultures. Blood. (2009) 113:5125–33. doi: 10.1182/blood-2009-01-199950
253. Liston A, Piccirillo CA. Developmental plasticity of murine and human Foxp3(+) regulatory T cells. Adv Immunol. (2013) 119:85–106. doi: 10.1016/B978-0-12-407707-2.00003-5
254. Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol. (2013) 13:461–7. doi: 10.1038/nri3464
255. Georgiev P, Benamar M, Han S, Haigis MC, Sharpe AH, Chatila TA. Regulatory T cells in dominant immunologic tolerance. J Allergy Clin Immunol. (2024) 153:28–41. doi: 10.1016/j.jaci.2023.09.025
256. Harris F, Berdugo YA, Tree T. IL-2-based approaches to Treg enhancement. Clin Exp Immunol. (2023) 211:149–63. doi: 10.1093/cei/uxac105
257. Ma S, So M, Ghelani A, Srivas R, Sahoo A, Hall R, et al. Attenuated IL-2 muteins leverage the TCR signal to enhance regulatory T cell homeostasis and response in vivo. Front Immunol. (2023) 14:1257652. doi: 10.3389/fimmu.2023.1257652
258. Arjomandnejad M, Kopec AL, Keeler AM. CAR-T regulatory (CAR-treg) cells: Engineering and applications. Biomedicines. (2022) 10(2):287. doi: 10.3390/biomedicines10020287
Keywords: Foxp3 + eTREG cells, transcriptional adaptation, tissue residency, polarization, inflammation, TREG development, mucosal immunity
Citation: Alvarez F, Liu Z, Bay A and Piccirillo CA (2024) Deciphering the developmental trajectory of tissue-resident Foxp3+ regulatory T cells. Front. Immunol. 15:1331846. doi: 10.3389/fimmu.2024.1331846
Received: 01 November 2023; Accepted: 14 February 2024;
Published: 28 March 2024.
Edited by:
Nirupama Darshan Verma, University of New South Wales, AustraliaReviewed by:
Jochen Huehn, Helmholtz Association of German Research Centers (HZ), GermanyCopyright © 2024 Alvarez, Liu, Bay and Piccirillo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ciriaco A. Piccirillo, Y2lyby5waWNjaXJpbGxvQG1jZ2lsbC5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.