 Zhaoyang Yin
Zhaoyang Yin Bowen Wan2†
Bowen Wan2† Jian Yin
Jian Yin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 23 January 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1330678
The damage to the central nervous system and dysfunction of the body caused by spinal cord injury (SCI) are extremely severe. The pathological process of SCI is accompanied by inflammation and injury to nerve cells. Current evidence suggests that oxidative stress, resulting from an increase in the production of reactive oxygen species (ROS) and an imbalance in its clearance, plays a significant role in the secondary damage during SCI. The transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) is a crucial regulatory molecule for cellular redox. This review summarizes recent advancements in the regulation of ROS-Nrf2 signaling and focuses on the interaction between ROS and the regulation of different modes of neuronal cell death after SCI, such as apoptosis, autophagy, pyroptosis, and ferroptosis. Furthermore, we highlight the pathways through which materials science, including exosomes, hydrogels, and nanomaterials, can alleviate SCI by modulating ROS production and clearance. This review provides valuable insights and directions for reducing neuronal cell death and alleviating SCI through the regulation of ROS and oxidative stress.
Spinal cord injury (SCI) is a prevalent form of central nervous system injury that leads to motor, sensory, and autonomic dysfunction. In the United States alone, the annual incidence of spinal cord injuries ranges from 12,000 to 20,000 (1). According to a study conducted by the Global Burden of Disease in 2016, there are approximately 93,500 new cases of SCI worldwide each year, with a total of 27 million existing cases (2). The pathophysiological process of SCI primarily consists of two stages: primary injury and secondary injury. Primary injury involves the physical forces that occur during the initial traumatic event, such as compression, tearing, shearing, and stretching (3). Secondary injury refers to the continuation of primary injury and is characterized by vasogenic edema, as mentioned above, mitochondrial dysfunction, and excessive production of reactive oxygen species (ROS) (4, 5). The overproduction of ROS plays a crucial role in the secondary injury process, further exacerbating the inflammatory response and various regulatory cell death (RCD) pathways (6). Due to the complexity of SCI’s pathophysiology, there is currently no satisfactory clinical intervention method that can significantly improve neurological function. Effectively mitigating the extensive cell death caused by secondary injury is the key to preserving the neurological function of the damaged spinal cord.
The current understanding of secondary injury mechanisms in SCI primarily involves oxidative stress and nitrosative stress (7). Oxidative and nitrosative stress are thought to be an imbalance between the presence of high levels of ROS and reactive nitrogen species (RNS), and antioxidant defense mechanisms (8). Oxidative stress response can be considered a double-edged sword. In the early stages, low levels of oxidative stress can provide some protection to damaged tissues and cells (9, 10), However, sustained high levels of oxidative stress can worsen inflammation and cell death (11, 12). During cellular metabolic activities, free radicals are generated and can be counteracted by endogenous antioxidants, maintaining redox balance. The primary molecule involved in oxidative stress is the superoxide O2·- radical, which is produced when electrons in O2 molecules are reduced (13). Interestingly, this radical can function both as an oxidizing agent and a reducing agent. ROS and RNS, which are highly reactive free radicals, can cause damage to cell membranes, proteins, and DNA, triggering inflammatory cascade events (14). Oxygen free radicals are particularly harmful to polyunsaturated fatty acids, leading to lipid peroxidation (15). There is increasing evidence suggesting that oxidative stress, induced by excessive ROS, plays a critical role in the secondary injury of spinal cord injury (SCI) (9, 16, 17). ROS include superoxide anion radical (O2-), hydrogen peroxide (H2O2), hydroxyl radical (·OH), and peroxynitrite (18). These ROS are produced through enzymatic and non-enzymatic pathways during normal metabolic processes (19). While natural ROS scavenging enzymes like superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) have limited stability, availability, and high cost, current research focuses on developing exogenous ROS scavengers. Additionally, researchers are continuously updating material systems to address challenges related to antioxidant loading and delivery. This article provides a comprehensive review of the role of reactive ROS in apoptosis, autophagy, pyroptosis, and ferroptosis during SCI. It also explores the potential crosstalk mechanisms involved, aiming to offer new insights and directions for mitigating nerve damage through the elimination of excess ROS.
Signal regulation between ROS and apoptosis is a highly significant area of research in life sciences. In addition to H2O2, oxidized low-density lipoprotein and peroxynitrite free radicals have been found to induce various types of apoptosis (20). Antioxidants play a role in suppressing signaling cascades that lead to apoptosis (21). Some studies have shown that depletion of reduced glutathione is associated with the initiation of apoptosis (22, 23). Recently, there has been extensive research on the control of SCI through microRNA (miRNA) modulation. MiRNAs are non-coding small RNA molecules composed of 20-24 nucleotides, and they actively participate in the regulation of oxidative stress (24, 25). Two independent studies have demonstrated similar modes of action, where H2O2 and LPS inhibit the expression of MiR-495 and MiR−122−5p in neural cells respectively, resulting in reduced ROS production and cell apoptosis. Mechanistically, MiR-495 downregulates the expression of STAT3, while MiR-122-5p represses CPEB1, targeting the PI3K/AKT signaling pathway (26, 27). Another study on traditional Chinese medicine suggests that calycosin promotes the expression level of p-AKT by up-regulating heat shock protein 90 (HSP90), while inhibiting the expression levels of p-ASK1 (pThr845) and p-p38, thereby alleviating ROS, oxidative stress, and apoptosis in neurons (28). In contrast to what was mentioned above (26), the role of AKT phosphorylation in promoting or inhibiting ROS generation and apoptosis is still controversial. This discrepancy could potentially be attributed to differences in phosphorylation sites.
The reduction of mitochondrial membrane potential (MMP), intracellular calcium overload, opening of mitochondrial permeability transition pore (mPTP), and generation of reactive oxygen species (ROS) during spinal cord ischemia-reperfusion injury (SCIRI) lead to mitochondrial dysfunction and apoptosis of neural cells (29, 30). In the SCIRI or oxygen glucose deprivation/reperfusion (OGD/R) model, polydatatin promotes the nuclear import of nuclear factor erythroid 2-related factor 2 (Nrf2) to bind to antioxidant response elements (ARE) by accelerating the dissociation of Kelch-like ECH-associated protein 1 (Keap1)/Nrf2 complex. This enhances the antioxidant activity of spinal motor neurons (SMNs), which is beneficial for increasing MMP, clearing ROS, and restoring mitochondrial function. By reversing damaged mitochondria, polydatin inhibits the apoptosis of SMNs through the classic Bcl-2/cytochrome c pathway (29). Ischemia-reperfusion-induced changes in cellular metabolism lead to inflammation-related epigenetic modifications, post-transcriptional regulation, and post-translational modifications of proteins (31–33). Phosphoglycerate kinase 1 (PGK1) is a key enzyme involved in ATP generation during glycolysis in cellular metabolism and is linked to oxidative stress through the Keap1-Nrf2 pathway (34–36). Dimerization of Keap1 inhibits Nrf2 ubiquitination, promoting Nrf2 nuclear import and antioxidant effects (37–39). A recent study suggested that G-protein-coupled receptor (GPCR) kinase 2-interacting protein-1 (Git1), a GTPase-activating protein, regulates the phosphorylation of PGK1 at S203 in 293T cells under OGD/R conditions, thus affecting Keap1-Nrf2 signaling and oxidative stress (40). Git1 in neurons enhances Nrf2 signaling by promoting Keap1 dimerization to reduce reperfusion-induced ROS levels, providing a potential target for SCIRI treatment.
Peng Zou et al. (41) observed a significant upregulation of the expression of excision repair cross-complementing group 6 (ERCC6, also known as CSB) in SCI mice and LPS-induced microglia. ERCC6 is a DNA-binding protein with ATPase activity that inhibits apoptosis and contributes to tumor proliferation (42, 43). Silencing ERCC6 not only hindered the activation of astrocytes and microglia, thereby reducing the inflammatory response, but also prevented microglial apoptosis and alleviated neuronal damage. Mechanistically, ERCC6 silencing reduced cellular oxidative stress by decreasing the expression of 4-HNE, Nrf2, and Keap1, as well as reducing excessive ROS accumulation in the spinal cord after SCI (41). Imatinib, a tyrosine kinase inhibitor, promoted the expression of HO-1 and SOD by activating Nrf2, thus providing resistance against oxidative stress and apoptosis (44). Additionally, imatinib, an anti-tumor drug used for chronic lymphocytic leukemia, can protect the blood-brain barrier and alleviate inflammation (45). The V-raf-1 murine leukemia viral oncogene homolog 1 (Raf-1)/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (Erk) pathway, a classic MAPK pathway, plays a role in cell proliferation, differentiation, inflammatory response, and apoptosis. Activation of Raf kinase leads to the activation of downstream MEK 1/2, enabling ERK1/2 to translocate from the cytoplasm to the nucleus and exert biological effects (46–49). Research by Haocong Zhang et al. (50) demonstrated that palmitoylethanolamide (PEA), an agonist of peroxisome proliferator-activated receptor α (PPARα), activates Nrf2 through phosphorylation of the Raf-1/MEK/ERK pathway, thereby inhibiting oxidative stress, inflammatory response, apoptosis, and promoting motor function recovery in SCI rats (Figure 1) (Table 1).
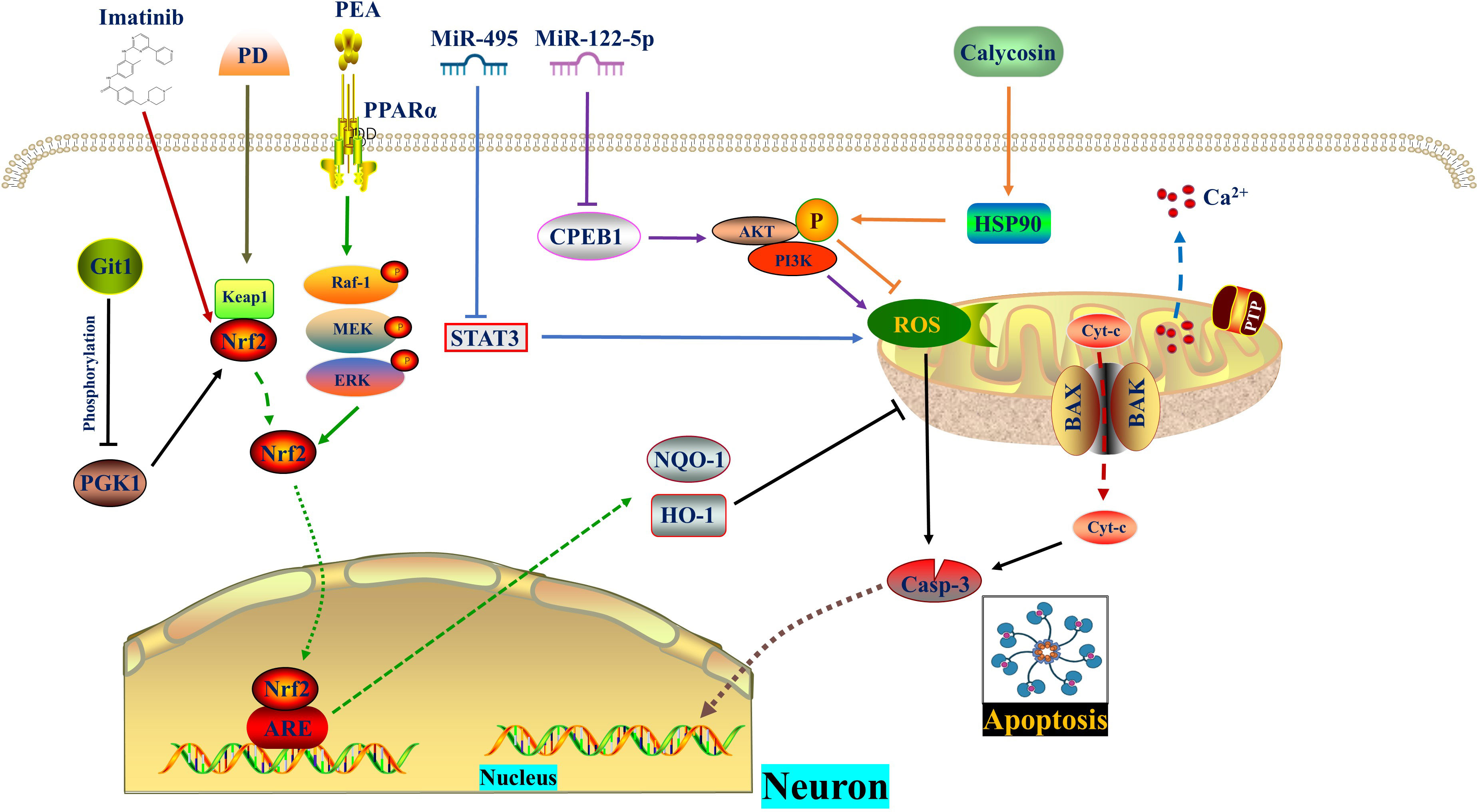
Figure 1 ROS in apoptosis. MiR-495 and MiR−122−5p reduce ROS level and apoptosis by inhibiting STAT3 and CPEB1, respectively. Calycosin promotes AKT phosphorylation by increasing HSP90 expression, thereby diminishing ROS generation and apoptosis. PD promotes the dissociation of Nrf2 from Keap1. Subsequently, Nrf2 enters the nucleus and binds to AREs, thereby enhancing cellular oxidation resistance and reversing mitochondrial damage as well as apoptosis. Git1 regulates PGK1 phosphorylation to counteract ROS through the Keap1/Nrf2 axis. Imatinib relieves oxidative stress and apoptosis by activating Nrf2. The PPARα agonist PEA activates Nrf2 through phosphorylation of the Raf‐1/MEK/ERK pathway, thereby inhibiting subsequent oxidative stress and apoptosis. CPEB1, Cytoplasmic polyadenylation element binding protein 1; ERK, Extracellular signal-regulated kinase; Git1, G-protein-coupled receptor (GPCR) kinase 2-interacting protein-1; HSP90, Heat shock protein 90; IR, Ischemia reperfusion; MEK, Mitogen-activated protein kinase; PD, Polydatin; PEA, Palmitoylethanolamide, a peroxisome proliferator-activated receptor alpha (PPARα) agonist; PGK1, Phosphoglycerate kinase 1; PPARα, Peroxisome proliferator-activated receptor α; Raf-1, V-raf-1 murine leukemia viral oncogene homolog 1; ROS, Reactive oxygen species.
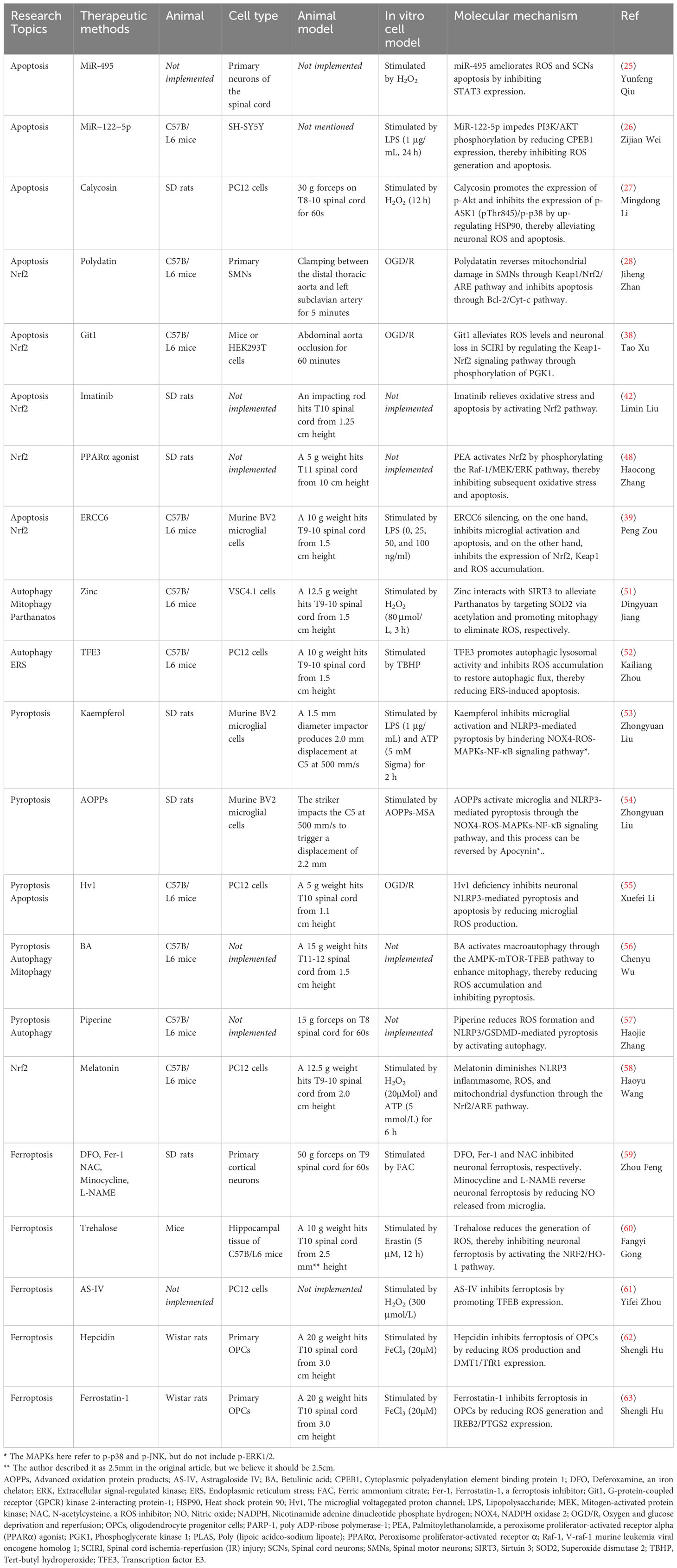
Table 1 Experimental study on ROS as a therapeutic target in SCI - molecular mechanism.
Autophagy is a unique phenomenon in eukaryotic cells that involves the degradation of damaged proteins and organelles for recycling through the lysosomal pathway. It is important to note that autophagy has both positive and negative effects (64, 65). There are three categories of autophagy: macroautophagy, chaperone-mediated autophagy, and microautophagy. When we refer to autophagy, we usually mean macroautophagy (66). The occurrence of autophagy is regulated by a series of autophagy protein families called ATG proteins, which are structurally and functionally conserved (67). Autophagy is induced in the body by factors such as deprivation of amino acids, oxygen, and sugar, low insulin levels, and reduced ATP levels. In the case of SCI, secondary pathological changes like hypoxia, hemorrhage, and edema lead to mitochondrial dysfunction and increased activity of ROS-generating enzymes, resulting in the production of large amounts of ROS (68). ROS then oxidizes unsaturated fatty acids in the lysosomal membrane, causing proteolytic enzyme extravasation and disruption of lysosomal function, which in turn blocks autophagic flux in SCI (69, 70). Impaired autophagy has been shown to inhibit ubiquitination, weaken mitochondrial function, and accumulate ROS, thereby disrupting cellular homeostasis (66). On the other hand, excessive autophagy can lead to autophagic death of spinal cord nerve cells (71, 72). Therefore, it is crucial to strictly control the degree of autophagy to maintain optimal levels (73). Mitochondria, which are not only essential for cellular energy metabolism and mitochondrial homeostasis, but also the main organelles that generate ROS after SCI, play a significant role. Interestingly, ROS can worsen mitochondrial damage and DNA oxidation, creating a vicious cycle that ultimately leads to cell death (56, 74, 75). Mitophagy, a selective autophagic degradation process specifically targeting damaged mitochondria, helps maintain mitochondrial homeostasis and cell survival by eliminating these damaged organelles (76, 77).
In 2007, Ted Dawson and Valina Dawson from Johns Hopkins University School of Medicine discovered a new form of programmed brain cell death called Parthanatos, also known as poly ADP-ribose polymerase-1 (PARP-1) dependent cell death. Parthanatos is a type of programmed cell death that occurs due to DNA damage and the activation of PARP-1 (78). After spinal cord injury (SCI), there is an increase in oxidative stress which leads to the production of large amounts of ROS. These ROS molecules play a crucial role in activating Parthanatos (79, 80). Zinc is an essential nutrient that binds with various enzymes and transcription factors (81, 82). The neuroprotective effect of Zinc has been extensively studied (51, 83). Sirtuin 3 (SIRT3), a major mitochondrial acetyl-lysine deacetylase, is responsible for regulating mitochondrial homeostasis and ROS production (52). Recent findings suggest that Zinc acts on SIRT3 to prevent SCI by directly eliminating ROS through acetylation, targeting SOD2 to inhibit Parthanatos. Additionally, Zinc promotes mitophagy, indirectly eliminating ROS and alleviating Parthanatos. In summary, Zinc mediates anti-oxidative stress through SIRT3, reducing the expression of PARP-1 (84). This discovery reinforces the understanding of how Zinc protects against SCI by scavenging ROS.
As mentioned above, the fusion of autophagosomes and lysosomes plays a crucial role in autophagy. In a study by Kailiang Zhou et al. (54) it was discovered that the excessive accumulation of ROS caused lysosomal dysfunction and blocked autophagic flux in SCI. This inhibition of autophagy then led to endoplasmic reticulum stress-dependent neuronal apoptosis. The researchers further confirmed these findings in neurons from ATG5-/+ mice. They also found that the transcription factor E3, which is partially regulated by the AMPK-mTOR and AMPK-SKP2-CARM1 signaling pathways, promoted autophagy-lysosomal activity and inhibited ROS accumulation. This restoration of autophagic flux after SCI helped alleviate neuronal damage. Interestingly, under SCI stimulation, AMPK phosphorylation in the cytoplasm inhibited the level of m-TOR and the nuclear translocation of TFE3. However, p-AMPK in the nucleus increased the levels of CARM1, which binds to TFEB to induce TFEB transcription (Figure 2) (Table 1).
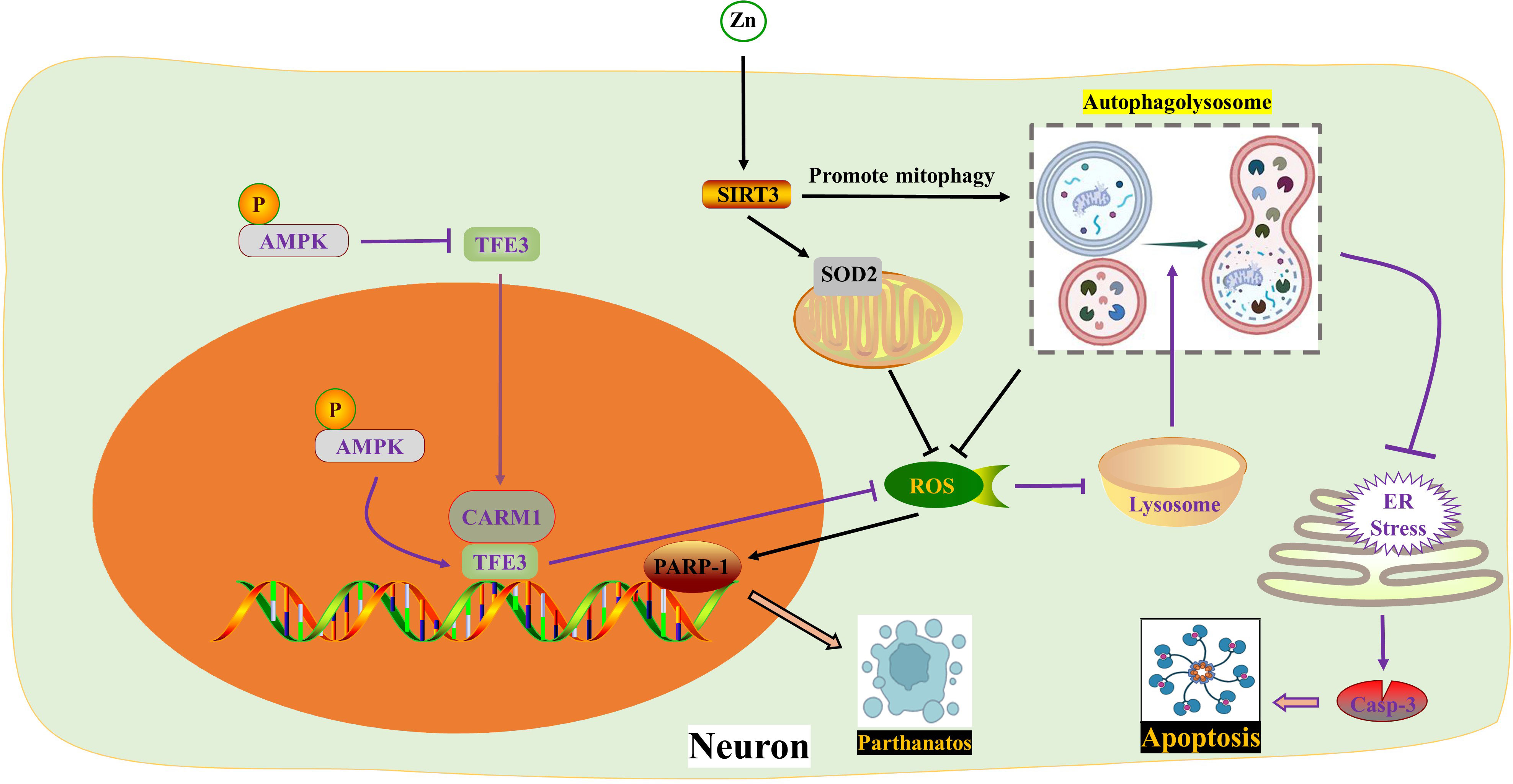
Figure 2 ROS in autophagy. Zinc interacts with SIRT3 to inhibit PARP-1-mediated Parthanatos by targeting mitochondrial SOD2 and promoting mitophagy to eliminate ROS respectively. TFE3 reduces ROS formation and enhances lysosomal activity to restore autophagy, thereby impeding endoplasmic reticulum stress and apoptosis. The activity of TFE3 is regulated by p-AMPK in both the cytoplasm and nucleus. AMPK, Adenosine 5’-monophosphate (AMP)-activated protein kinase; ER, Endoplasmic reticulum; PARP-1, poly ADP-ribose polymerase-1; ROS, Reactive oxygen species; SIRT3, Sirtuin 3; SOD2, Superoxide dismutase 2; TFE3, Transcription factor E3; Zn, zinc.
Autophagy is a complex self-degradation process that involves multiple steps. In current literature, the assessment of autophagy levels often relies on the detection of autophagosomes or the expression of autophagy proteins. However, these measures may not provide a comprehensive evaluation of the smoothness of autophagic flow. It is important to note that a decrease in the degradation rate of autophagy substrates, caused by reduced lysosomal function due to endoplasmic reticulum stress, can weaken the overall autophagy process. This weakening can lead to an accumulation of autophagosomes and increased expression of autophagy proteins (85, 86). Electron microscopy is considered the gold standard for observing autophagy as it can capture both the induction of autophagy and the consumption of autophagosomes. Autophagy generally plays a protective role in cells, but disruption of the autophagy mechanism or excessive autophagy flux can result in cell death, known as autophagy-dependent cell death or autophagy-mediated cell death (80, 87, 88). Restoring lysosomal function and blocking autophagy flux can protect cells from apoptosis (89). Interestingly, blocking autophagic flux may also contribute to cell death or apoptosis (90). In oncology research, candesartan has been found to induce cell death in trail-resistant lung cancer cells by blocking autophagy flux (91). Taken together, regulating autophagic flux to protect body cells from damage or promote tumor death is a challenging task.
The hallmark event of pyroptosis is the formation of pores in the cytoplasmic membrane by the Gasdermin (GSDM) protein family, primarily GSDMD and GSDME, which are cleaved by caspases. When exposed to external stimuli, inflammasomes recognize pathogen-related molecular patterns (PAMPs) or host-derived danger signal molecules (DAMPs) to activate caspase-1 (53, 92). In the classic pyroptosis pathway, inflammasomes such as NLRP1, NLRP2, NLRP3, NLRC4, and AIM2 dissociate and activate caspase-1. Activated caspase-1 has two main functions: cleaving GSDMD into GSDMD-NT to form membrane pores, and promoting the maturation of pro-IL-1β/18 into IL-1β/18, which is released from the GSDMD-NT pores, thereby amplifying the inflammatory response. In the non-classical pyroptosis pathway, external bacteria or viruses activate caspase-11 (in mice) or caspase-4/5 (in humans) through lipopolysaccharide (LPS) to cleave GSDMD and induce pyroptosis (93, 94). The GSDM family includes GSDM-A/B/C/D/E and DFNB59. However, DFNB59 does not form membrane pores through the previously mentioned pathways. The NLRP3 inflammasome, which consists of NLRP3, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and caspase-1, is the most extensively studied. Studies have shown that NLRP3 is activated and plays a crucial role in spinal cord injury (SCI) in various cell types, including neurons, microglia, astrocytes, and pericytes (94). Oxidative stress and the production of reactive oxygen species (ROS) are critical in the activation of the NLRP3 inflammasome and the initiation of pyroptosis. Currently, the main mechanisms of NLRP3 inflammasome activation include ROS formation, lysosomal rupture, and ion channel gating. Among these mechanisms, NADPH oxidase (NOX) is considered the most important trigger, mediating ROS generation (95, 96).
MAPKs, such as ERK1/2, JNK, and p38, are a group of highly conserved serine/threonine protein kinases that play a role in various processes including stress, inflammation, proliferation, differentiation, survival, apoptosis, and more. Previous studies have shown that MAPKs and ROS mutually affect each other’s activities (97). ROS can enhance the inflammatory response by activating multiple pro-inflammatory signaling pathways, such as MAPK, NF-κB, and JAK-STAT 9-12 (98). Additionally, the hyperphosphorylation of MAPKs amplifies subsequent inflammatory responses by activating NF-κB (99). Recent studies by Zhongyuan Liu and colleagues have confirmed that both LPS+ATP (55) and AOPPs (Advanced oxidation protein products) (92) activate BV2 cells through the ROS-dependent MAPKs-NF-κB signaling pathway, leading to NLRP3 inflammasome-mediated pyroptosis. Specifically, this process activates p-p38 and p-JNK MAPKs, but not p-ERK1/2. The natural polyphenol kaempferol and the selective NADPH oxidase inhibitor apocynin both inhibit the translocation of NF-κB p65 subunit into the nucleus and alleviate neuroinflammation by inhibiting the aforementioned pathways. In the upstream signaling pathway, the NADPH oxidase family is the primary source of ROS in BV2 cells (100). NOX2 (NADPH oxidase 2) and NOX4 are mainly expressed in spinal cord microglia. Interestingly, the authors found that kaempferol and AOPPs only regulate the expression of NOX4.
The voltage-gated proton channel (Hv1), a unique member of the superfamily of voltage-gated cation channels, facilitates the production of ROS (101, 102). Ion channels, including Hv1, play a crucial role in the physiological functions of neurons and glial cells in the nervous system (57, 103). Hv1 is a selective ion channel for H+ and plays a key role in regulating nicotinamide adenine dinucleotide phosphate NOX activity and pH in phagocytes (104). Hv1-mediated ROS production contributes to brain damage after cerebral ischemia (105). In microglia, Hv1 accelerates ROS generation by inducing NOX (59). A recent study revealed that Hv1 deficiency reduced microglial ROS production during SCI, leading to inhibition of neuronal apoptosis and NLRP3-mediated pyroptosis. Hv1−/− mice showed improved axonal regeneration and motor function recovery after SCI (106). Interestingly, this study found that the peak of neuronal apoptosis occurred earlier than neuronal pyroptosis, while the duration of pyroptosis was longer than that of apoptosis. However, the researchers did not provide further insights into the crosstalk between pyroptosis and apoptosis, which warrants further investigation.
The role of autophagy after SCI is a topic of debate among scholars. Many believe that moderate activation of autophagy helps nerve cells adapt and survive (58). Recent studies have shown that increased autophagy can reduce inflammation and pyroptosis (107, 108). In the context of alleviating SCI, betulinic acid (BA) and piperine have been found to activate autophagy, leading to a decrease in ROS formation and NLRP3/GSDMD-mediated pyroptosis (75, 109). Specifically, BA activates macroautophagy through the AMPK-mTOR-TFEB pathway, which in turn reduces ROS accumulation by enhancing mitophagy, thereby inhibiting pyroptosis (75). The interaction between autophagy and pyroptosis is complex, with ROS and mitochondrial homeostasis playing a crucial role in their communication.
The Nrf2/ARE pathway is a major target for neuroprotection due to its anti-inflammatory and antioxidant effects (110, 111). Melatonin, a hormone primarily synthesized in the pineal gland that delays aging, resists oxidation, and improves sleep, is considered a positive regulator of Nrf2 (112–114). A study on SCI confirmed that Melatonin inhibited the NLRP3 inflammasome by activating Nrf2/ARE signaling. This resulted in the downregulation of inflammatory factors and reduced levels of reactive oxygen species (ROS), malondialdehyde, and improved mitochondrial function in neurons (115) (Figure 3) (Table 1).
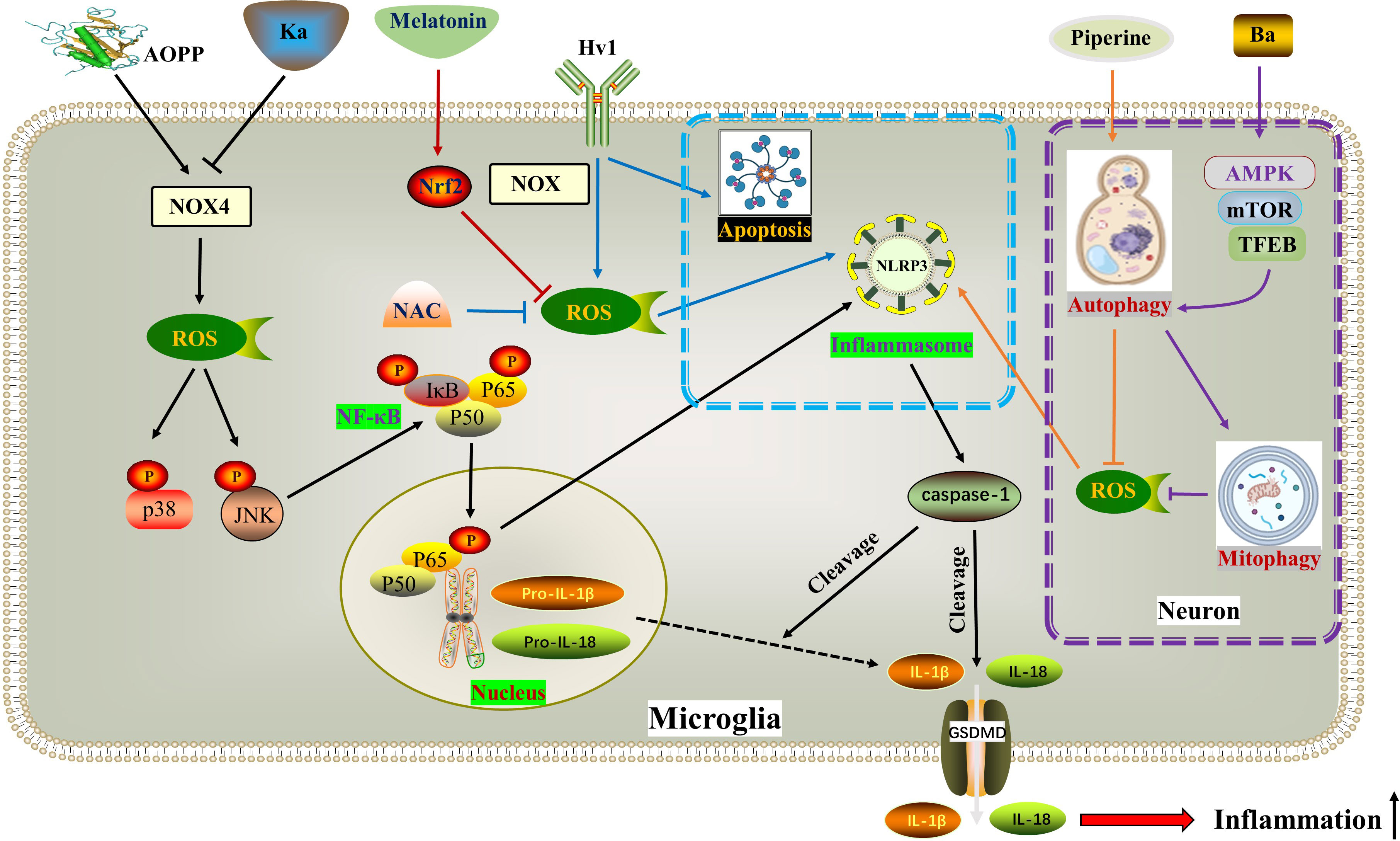
Figure 3 ROS in pyroptosis. AOPP increases NOX4-mediated ROS production, thereby phosphorylating and activating p38 and JNK as well as downstream NF-κB, ultimately activating NLRP3-dependent pyroptosis. Ka exhibits opposite effects through the same pathway. Hv1 accelerates neuronal NLRP3-mediated pyroptosis and apoptosis by promoting ROS production in microglia. Piperine reduces ROS formation and NLRP3/GSDMD-mediated pyroptosis by activating autophagy. BA activates macroautophagy through the AMPK-mTOR-TFEB pathway, thereby reducing ROS accumulation by enhancing mitophagy, thereby inhibiting pyroptosis. Melatonin inhibits the generation of ROS and the activation of NLRP3 inflammasome by activating the Nrf2/ARE signaling pathway. * The blue dotted box indicates the effect of Hv1-mediated microglial ROS production on neuronal pyroptosis and apoptosis. The purple dotted box shows autophagy and pyroptosis in neurons. AOPPs, Advanced oxidation protein products; BA, Betulinic acid; Hv1, The microglial voltagegated proton channel; Ka, Kaempferol; NOX4, Nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidases.
The concept of ferroptosis, which was first proposed in 2012 (116), is a novel model of regulated cell death (RCD) characterized by specific changes in cell morphology, such as mitochondrial atrophy and reduction or disappearance of mitochondrial cristae (117). Ferroptosis is a process in which the accumulation of lethal lipid reactive oxygen species (ROS) is dependent on iron and involves uninhibited lipid peroxidation induced by iron metabolism. Iron homeostasis in the nervous system is tightly regulated by various ferritins, including iron regulatory proteins (IRPs), divalent metal transporter 1 (DMT1), transferrin receptors (TfRs), ferritin, and iron transporter 1 (Fpn1) (118). Excessive iron in cells leads to mitochondrial damage and the generation of excessive ROS through the Fenton reaction, which reacts with polyunsaturated fatty acids (PUFAs) in the lipid membrane, causing lethal lipid peroxidation (119–121). The extrinsic pathway of ferroptosis involves the inhibition of cell membrane transport proteins or the activation of proteins involved in iron ion transport, while the intrinsic pathway involves the inhibition of intracellular antioxidant enzyme systems (122). For example, reduced activity of the antioxidant enzyme GPx4 fails to suppress intracellular ROS production, resulting in lipid peroxidation (116). Further research has identified several genes, including Ptgs2, Gpx4, and Fsp1, that regulate ferroptosis. Ptgs2 encodes cyclooxygenase-2 (COX-2) and is significantly upregulated during ferroptosis (123).
In the acute phase of spinal cord injury (SCI), the blood-spinal cord barrier and blood vessels are destroyed, resulting in an increase in iron concentration at the site of injury. This leads to a vicious positive cycle of iron overload, excessive production of reactive oxygen species (ROS), and ferroptosis, which worsens the loss of neurological function after SCI. Therefore, reducing iron overload is crucial in breaking this harmful pattern (124, 125). Current evidence suggests that several drugs show potential for the treatment of SCI, including the iron chelator deferoxamine, the ferroptosis agent SRS 16-86, zinc and proanthocyanidins, and the Nrf2 pathway activator carnosic acid (60, 126–129). Interestingly, drug delivery to the central nervous system has been hindered by the blood-brain barrier and blood-spinal cord barrier. Some studies have found that oral iron chelators, like deferiferrin, have limited or no effect on improving motor function after SCI (61). However, there are iron chelators like hydroxybenzylethylenediamine (HBED) that can cross the blood-brain barrier, and minocycline, an antibiotic with iron-chelating properties. Both of these substances can alleviate neurological damage by reducing microglial activation (130, 131).
Motor neuron death following spinal cord injury (SCI) can result in atrophy of the primary motor cortex. Zhou Feng et al. (123) conducted a study which revealed a significant increase in iron deposition in motor cortex neurons of both SCI patients and rats. This iron deposition triggered the accumulation of ROS and led to neuronal ferroptosis. However, the researchers found that the use of an iron chelator called DFO, a ROS inhibitor called NAC, and a ferroptosis inhibitor called Fer-1 successfully reversed neuronal ferroptosis. Further investigations indicated that the release of NO from activated microglia after SCI resulted in an increased expression of IRP1, DMT1, and TfR1, while ferritin expression decreased. The inhibition of microglial activation by minocycline and the inhibition of NO synthase by L-NAME were both found to reverse neuronal ferroptosis and promote the recovery of motor nerve function. Therefore, therapeutic approaches aimed at balancing iron metabolism hold promise for the treatment of SCI.
Trehalose, a non-reducing disaccharide with antioxidant effects, has shown promise in improving the prognosis of various neurological diseases, including Huntington’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis (62, 63, 132). In a study by Fangyi Gong et al. (133), it was found that trehalose effectively reduced oxidative stress and ROS generation after spinal cord injury (SCI). This reduction was accompanied by an increase in GSH and Gpx4 content, as well as a decrease in MDA and Acsl4 levels, neuronal degeneration, and iron accumulation. The mechanism behind this effect involves the activation of the NRF2/HO-1 signaling pathway, which inhibits neuronal cell ferroptosis. Additionally, trehalose has been found to reduce neuroinflammation after SCI, possibly by suppressing ROS and subsequent ferroptosis.
TFEB, a member of the microphthalmia-associated transcription factor family, plays a crucial role in the oxidative stress process (134, 135). In a study, Astragaloside IV (AS-IV) was found to mitigate H2O2-induced damage to PC12 cells by inhibiting the generation of ROS and promoting TFEB expression, thereby preventing ferroptosis. The protective effects of AS-IV were reversed when TFEB was knocked down (136). The integrity of white matter bundles is vital for motor function recovery after SCI (137), as well as for neuronal communication and cognitive function improvement (138). Oligodendrocytes, which wrap around axons, form the basic structure of white matter bundles. Due to their high content of unsaturated fatty acids, oligodendrocytes are vulnerable to iron toxicity. Research by Shengli Hu demonstrated that both hepcidin and ferrostatin-1 can inhibit ferroptosis in OPCs by reducing ROS production. However, they differ in their mechanisms: hepcidin down-regulates the expression of DMT1 and TfR1, while ferrostatin-1 negatively modulates the expression of IREB2 and PTGS2 (139, 140). Hepcidin, a peptide hormone primarily secreted by the liver, can also reduce iron uptake and release by astrocytes and neurons by decreasing the expression of Fpn1, TfR1, and DMT1 (141–143) (Figure 4) (Table 1).
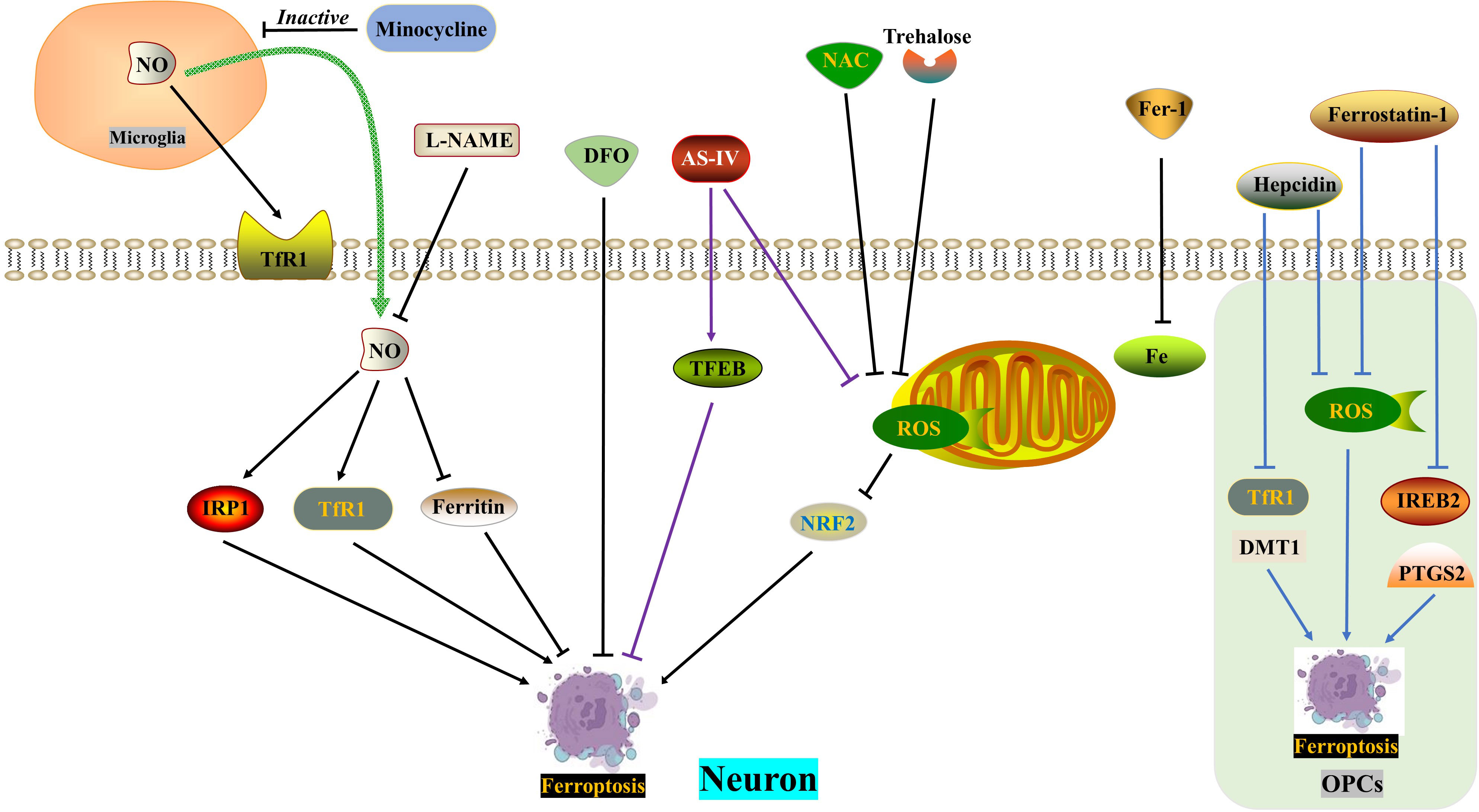
Figure 4 ROS in ferroptosis. DFO, NAC and Fer-1 reverse SCI-induced neuronal ferroptosis, respectively. SCI induces increased NO release from microglia and exacerbates neuronal ferroptosis, which is reversed by minocycline and L-NAME. Trehalose inhibits neuronal ferroptosis through the ROS/NRF2 pathway. AS-IV alleviates neuronal ferroptosis by inhibiting ROS generation and promoting TFEB expression. Hepcidin ameliorates ferroptosis in OPCs by repressing ROS level and DMT1/TfR1 expression. Ferrostatin-1 reduces ferroptosis in OPCs by inhibiting ROS generation and IREB2/PTGS2 level. AS-IV, Astragaloside IV; DFO, Deferoxamine, an iron chelator; DMT1, Divalent metal transporter 1; FAC, Ferric ammonium citrate; Fer-1, Ferrostatin-1, a ferroptosis inhibitor; IRP1, Iron regulatory protein 1; L-NAME, an NO synthase inhibitor; NAC, N-acetylcysteine, a ROS inhibitor; NO, Nitric oxide; OPCs, oligodendrocyte progenitor cells; ROS, Reactive oxygen species; TfR1, Ransferrin receptor1.
In order to effectively reduce oxidative stress and eliminate excess ROS, researchers are focusing on developing new carriers or materials such as exosomes, hydrogels, and nanomaterials. Exosomes are nanoscale extracellular vesicles with a particle size of 50-150nm. They play a paracrine role by transmitting genetic material information, including non-coding RNA, mRNA, and proteins. Due to their nanometer size and membrane permeability, exosomes are considered a promising approach for treating central nervous system diseases (144–146). One study showed that the application of exosomes derived from microvascular endothelial cells (USP13) alleviated SCI injury by improving mitochondrial damage and reducing ROS production (147). Hydrogels, which have good biocompatibility, hydrophilicity, and controlled drug release, also have unique advantages in SCI treatment. However, their use for SCI treatment is limited due to the high fragility of the spinal cord, as hydrogels with high hardness cannot be used. Yibo Ying et al. (148) utilized TPA and Laponite to develop hydrogels that exhibit shear thinning capabilities. They conducted experiments to demonstrate that these hydrogels can synergistically impede ferroptosis by enhancing vascular function and regulating iron metabolism. Additionally, some researchers have successfully prepared a polylipoic acid-sodium lipoate hydrogel (PLAS) with desirable mechanical properties, easy injectability, and sufficient adhesion. Animal experiments have revealed that PLAS can effectively eliminate cellular ROS, thereby promoting recovery from spinal cord injury (149).
Nanoparticles offer significant advantages in terms of size manipulation, chemical and physical properties. Researchers often modify the size and surface of nanoparticles to minimize biological clearance, evade immune defense systems, prolong the duration of action, and enhance drug release. Selenium nanoparticles have gained significant attention as potential carriers for antioxidant drugs. In a study by Siyuan Rao et al. (150), a multifunctional functionalized selenium nanoparticle loaded with tetramethylpyrazine (TMP)/monosialotetrahexosylganglioside (GM1) (SeNPs@GM1/TMP) was designed and synthesized. The researchers found that SeNPs@GM1/TMP effectively reduced the excessive production of ROS by inhibiting the activation of p53 and MAPKs pathways, thereby preventing mitochondrial dysfunction and promoting functional recovery after SCI. Another experiment conducted by the same research team involved loading astragalus polysaccharides (APS) and tanshinone IIA (TSIIA) on SeNPs (TSIIA@SeNPs-APS), which also demonstrated effective ROS inhibition and protection of neuronal cells (151). Additionally, a chitosan-modified hollow manganese dioxide nanoparticle system loaded with resveratrol (RESV) was found to effectively reduce ROS, MDA, and SOD levels, while increasing glutathione (GSH) levels (152). This reduction in inflammation and neuronal apoptosis after SCI was observed. Similarly, loading RES into plasma complex component functionalized manganese-doped silica nanoparticles (PMMSN) carrier (PMMSN-RES) also led to reduced ROS, MDA, and inflammatory factors (TNF-α, IL-1β, and IL-6) after SCI, thereby promoting neuronal survival and restoring spinal cord functionality in mice (153). In another study by Haifei Shi et al. (154), the mesoporous pores and hydrophobic benzene ring structure of polydopamine (mPDA) were utilized to effectively encapsulate and sustainably release rapamycin (Rapa) (mPDA@Rapa). In vitro experiments demonstrated that these nanoparticles inhibited excessive ROS production and reduced spinal cord inflammation. Furthermore, some scholars have developed a highly biologically active iridium complex (IrFPHtz) that can directly target SOD1, upregulating its expression and eliminating excessive ROS production induced by SCI (155). The continuous advancement of nanomaterial technology allows for ongoing exploration of the ROS scavenging ability and antioxidant effect of nanomaterials in the treatment of spinal cord injury (151, 156, 157). These innovative materials hold great potential for the treatment of SCI and may become promising candidates for clinical practice in the future.
SCI imposes a significant financial burden on patients and caregivers, with estimated lifetime care costs ranging from $1.6 million to $4.8 million. It is widely accepted that inflammation plays a role in the progression of SCI and subsequent neurological dysfunction. ROS and RNS are early events in mechanical damage in SCI. The excessive production of ROS, such as •OH, H2O2, and •O2−, during secondary injury surpasses the body’s antioxidant capacity, leading to increased oxidative stress and regulated cell death (RCD). Excessive activation of iNOS results in the overproduction of NO, which can react with •O2− to form peroxynitrite (PN: ONOO−), further promoting DNA and lipid peroxidation (158). The cascade of oxygen radical reactions begins with the production of O2•−, which reacts with nitric oxide (•NO) radicals to form the highly reactive oxidant peroxynitrite (PN: ONOO−) and the by-product hydroxyl radical. SOD can quickly catalyze the decomposition of O2•− into H2O2 and oxygen. Subsequently, PN decomposition leads to the formation of highly reactive cytotoxic free radicals, including nitrogen dioxide (•NO2) and carbonate radicals (•CO3). These PN-derived free radicals induce oxidative damage to proteins, lipids, and nucleic acids (159, 160), impairing mitochondrial respiration and Ca2+ buffering capacity (161). Additionally, PN products can stimulate lipid peroxidation. The formation of peroxynitrite and lipid peroxidation irreversibly impair neuronal membrane lipid and protein function, leading to subsequent ion homeostasis imbalance, glutamate-mediated excitotoxicity, mitochondrial respiratory failure, and microvascular damage (7). Therefore, compounds that inhibit the formation of ROS and RNS or scavenge ROS and RNS are effective strategies for the treatment of SCI.
Currently, there is no FDA-approved strategy for effectively improving SCI. Given the detrimental impact of oxidative stress and inflammation on SCI progression, reducing ROS levels and oxidative stress is generally considered a fundamental treatment approach (162). As mentioned earlier, SCI triggers various forms of RCD, such as apoptosis, autophagy, pyroptosis, and ferroptosis, due to excessive accumulation of free radicals, mitochondrial dysfunction, glutamate release, and calcium overload. There is intricate crosstalk between signaling pathways involved in these types of RCD. Considering the pivotal role of ROS in the inflammatory cascade following SCI, antioxidants have emerged as a key area of research for scavenging ROS, reducing oxidative stress, and alleviating RCD. Endogenous antioxidants include SOD, CAT, GPx-1, thiol antioxidants (glutathione, α-lipoic acid), uric acid, and coenzyme Q10 (CoQ10) (163, 164). Exogenous antioxidants, including vitamin C (ascorbic acid), vitamin A, carotenoids, and polyphenols have promising applications in central nervous system trauma and neurological degenerative diseases by regulating RCD of nerve cells and glial cells (165–169). Additionally, there are synthetic antioxidants like butylated hydroxyanisole, butylated hydroxytoluene, propyl gallate, and tert-butylhydroquinone. However, drug delivery challenges, particularly effectively penetrating the blood-spinal cord barrier and blood-brain barrier, and being taken up by target cells, are major concerns (170). The responsibility of weighing the benefit-risk balance of a drug lies with the clinician. Edaravone, a free radical scavenger, is currently the only FDA-approved antioxidant treatment drug, used for early-stage amyotrophic lateral sclerosis in the United States and Japan. However, its benefit is minimal in advanced cases, leading to the withdrawal of Radicava (the active substance of Edaravone) from marketing authorization in Europe (171). To overcome the limitations of natural and synthetic antioxidants, exosome therapy and new nanomaterials offer new possibilities for antioxidant treatment, providing more efficient drug delivery and greater antioxidant effects (172, 173). Cuproptosis, PANoptosome, and endoplasmic reticulum stress are also hot topics in this research field. In conclusion, the improvement of antioxidant therapy and the discovery of new antioxidants for alleviating ROS-induced RCD continue to provide more targets for SCI treatment.
ZY: Data curation, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. BW: Writing – review & editing, Resources, Software, Visualization. GG: Investigation, Methodology, Project administration, Resources, Writing – review & editing. JY: Investigation, Methodology, Project administration, Resources, Software, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We acknowledge financial support from the Special Funds for Science Development of the Clinical Teaching Hospitals of Jiangsu Vocational College of Medicine (Key Project, 20229103), Jiangsu Provincial Health Commission Project (LGY2020052, 2022-208), Nanjing Health Science and Technology Development Special Fund Project (ZKX22061 and YKK22222), Nanjing Medical University Kangda College Scientific Research Development Fund Project (KD2023KYJJ230) and Lianyungang Science and Technology Bureau Project (SF2118). Applied research project unit of geriatric medicine clinical technology in Jiangsu Province(LD2022001) and Management project of Jinling Hospital Affiliated to Nanjing University (YYQN2021097).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lipinski M, Wu J, Faden A, Sarkar C. Function and mechanisms of autophagy in brain and spinal cord trauma. Antioxidants Redox Signaling (2015) 23(6):565–77. doi: 10.1089/ars.2015.6306
2. Global, regional, and national burden of neurological disorders, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol (2019) 18(5):459–80. doi: 10.1016/s1474-4422(18)30499-x
3. Ma Y, Li P, Ju C, Zuo X, Li X, Ding T, et al. Photobiomodulation attenuates neurotoxic polarization of macrophages by inhibiting the notch1-HIF-1α/NF-κB signalling pathway in mice with spinal cord injury. Front Immunol (2022) 13:816952. doi: 10.3389/fimmu.2022.816952
4. Chen N, Sung C, Wen Z, Chen C, Feng C, Hung H, et al. Therapeutic effect of platelet-rich plasma in rat spinal cord injuries. Front Neurosci (2018) 12:252. doi: 10.3389/fnins.2018.00252
5. Guo W, Qi Z, Yu L, Sun T, Qu W, Liu Q, et al. Melatonin combined with chondroitin sulfate ABC promotes nerve regeneration after root-avulsion brachial plexus injury. Neural regeneration Res (2019) 14(2):328–38. doi: 10.4103/1673-5374.244796
6. Dimitrijevic M, Danner S, Mayr W. Neurocontrol of movement in humans with spinal cord injury. Artif organs. (2015) 39(10):823–33. doi: 10.1111/aor.12614
7. Bains M, Hall E. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta (2012) 1822(5):675–84. doi: 10.1016/j.bbadis.2011.10.017
8. Fatima G, Sharma V, Das S, Mahdi A. Oxidative stress and antioxidative parameters in patients with spinal cord injury: implications in the pathogenesis of disease. Spinal cord. (2015) 53(1):3–6. doi: 10.1038/sc.2014.178
9. Visavadiya N, Patel S, VanRooyen J, Sullivan P, Rabchevsky A. Cellular and subcellular oxidative stress parameters following severe spinal cord injury. Redox Biol (2016) 8:59–67. doi: 10.1016/j.redox.2015.12.011
10. Xiong Y, Rabchevsky A, Hall E. Role of peroxynitrite in secondary oxidative damage after spinal cord injury. J neurochemistry. (2007) 100(3):639–49. doi: 10.1111/j.1471-4159.2006.04312.x
11. Sohn H, Hwang J, Ryu J, Kim J, Park S, Park J, et al. Simvastatin protects ischemic spinal cord injury from cell death and cytotoxicity through decreasing oxidative stress: in vitro primary cultured rat spinal cord model under oxygen and glucose deprivation-reoxygenation conditions. J orthopaedic Surg Res (2017) 12(1):36. doi: 10.1186/s13018-017-0536-9
12. Zhang W, Cheng L, Hou Y, Si M, Zhao Y, Nie L. Plumbagin protects against spinal cord injury-induced oxidative stress and inflammation in wistar rats through nrf-2 upregulation. Drug Res (2015) 65(9):495–9. doi: 10.1055/s-0034-1389950
13. Stinghen A, Chillon J, Massy Z, Boullier A. Differential effects of indoxyl sulfate and inorganic phosphate in a murine cerebral endothelial cell line (bEnd. 3). Toxins (2014) 6(6):1742–60. doi: 10.3390/toxins6061742
14. Lee I, Lin H, Huang T, Tseng C, Cheng H, Huang W, et al. Anti-Inflammatory Effect of Resveratrol Derivatives via the Downregulation of Oxidative-Stress-Dependent and c-Src Transactivation EGFR Pathways on Rat Mesangial Cells. Antioxidants (Basel Switzerland). (2022) 11(5). doi: 10.3390/antiox11050835
15. Ayala A, Muñoz M, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell longevity. (2014) 2014:360438. doi: 10.1155/2014/360438
16. Anwar M, Al Shehabi T, Eid A. Inflammogenesis of secondary spinal cord injury. Front Cell Neurosci (2016) 10:98. doi: 10.3389/fncel.2016.00098
17. Jia Z, Zhu H, Li J, Wang X, Misra H, Li Y. Oxidative stress in spinal cord injury and antioxidant-based intervention. Spinal cord. (2012) 50(4):264–74. doi: 10.1038/sc.2011.111
18. Wang S, Wang M, Wang M, Tian Y, Sun X, Sun G, et al. Bavachinin Induces Oxidative Damage in HepaRG Cells through p38/JNK MAPK Pathways. Toxins (2018) 10(4). doi: 10.3390/toxins10040154
19. Hu X, Wang Y, Zhao Y, Chi C, Wang B. Lophius litulonAntioxidant peptides from the protein hydrolysate of monkfish () muscle: purification, identification, and cytoprotective function on hepG2 cells damage by HO. Mar Drugs (2020) 18(3). doi: 10.3390/md18030153
20. Mueller-Buehl A, Tsai T, Hurst J, Theiss C, Peters L, Hofmann L, et al. Reduced Retinal Degeneration in an Oxidative Stress Organ Culture Model through an iNOS-Inhibitor. Biology (2021) 10(5). doi: 10.3390/biology10050383
21. Cohen G. Caspases: the executioners of apoptosis. Biochem J (1997), 1–16. doi: 10.1042/bj3260001
22. Fernandes R, Cotter T. Apoptosis or necrosis: intracellular levels of glutathione influence mode of cell death. Biochem Pharmacol (1994) 48(4):675–81. doi: 10.1016/0006-2952(94)90044-2
23. Oda T, Sadakata N, Komatsu N, Muramatsu T. Specific efflux of glutathione from the basolateral membrane domain in polarized MDCK cells during ricin-induced apoptosis. J Biochem (1999) 126(4):715–21. doi: 10.1093/oxfordjournals.jbchem.a022508
24. Das S, Kohr M, Dunkerly-Eyring B, Lee D, Bedja D, Kent O, et al. Divergent Effects of miR-181 Family Members on Myocardial Function Through Protective Cytosolic and Detrimental Mitochondrial microRNA Targets. J Am Heart Assoc (2017) 6(3). doi: 10.1161/jaha.116.004694
25. Siddiqi F, Majumder S, Thai K, Abdalla M, Hu P, Advani S, et al. The histone methyltransferase enzyme enhancer of zeste homolog 2 protects against podocyte oxidative stress and renal injury in diabetes. J Am Soc Nephrology: JASN. (2016) 27(7):2021–34. doi: 10.1681/asn.2014090898
26. Qiu Y, Zhao Z, Chen Q, Zhang B, Yang C. MiR-495 regulates cell proliferation and apoptosis in HO stimulated rat spinal cord neurons through targeting signal transducer and activator of transcription 3 (STAT3). Ann Trans Med (2021) 9(6):461. doi: 10.21037/atm-21-102
27. Wei Z, Liu J, Xie H, Wang B, Wu J, Zhu Z. MiR-122-5p mitigates inflammation, reactive oxygen species and SH-SY5Y apoptosis by targeting CPEB1 after spinal cord injury via the PI3K/AKT signaling pathway. Neurochemical Res (2021) 46(4):992–1005. doi: 10.1007/s11064-021-03232-1
28. Li M, Hasiqiqige, Huan Y, Wang X, Tao M, Jiang T, et al. Calycosin ameliorates spinal cord injury by targeting Hsp90 to inhibit oxidative stress and apoptosis of nerve cells. J Chem neuroanatomy. (2023) 127:102190. doi: 10.1016/j.jchemneu.2022.102190
29. Zhan J, Li X, Luo D, Yan W, Hou Y, Hou Y, et al. Polydatin attenuates OGD/R-induced neuronal injury and spinal cord ischemia/reperfusion injury by protecting mitochondrial function via nrf2/ARE signaling pathway. Oxid Med Cell longevity. (2021) 2021:6687212. doi: 10.1155/2021/6687212
30. Zhang S, Wu D, Wang J, Wang Y, Wang G, Yang M, et al. Stress protein expression in early phase spinal cord ischemia/reperfusion injury. Neural regeneration Res (2013) 8(24):2225–35. doi: 10.3969/j.issn.1673-5374.2013.24.002
31. Li D, Shao N, Moonen J, Zhao Z, Shi M, Otsuki S, et al. ALDH1A3 coordinates metabolism with gene regulation in pulmonary arterial hypertension. Circulation (2021) 143(21):2074–90. doi: 10.1161/circulationaha.120.048845
32. Diskin C, Ryan T, O’Neill L. Modification of proteins by metabolites in immunity. Immunity (2021) 54(1):19–31. doi: 10.1016/j.immuni.2020.09.014
33. Shakespear M, Iyer A, Cheng C, Das Gupta K, Singhal A, Fairlie D, et al. Lysine deacetylases and regulated glycolysis in macrophages. Trends Immunol (2018) 39(6):473–88. doi: 10.1016/j.it.2018.02.009
34. Qian X, Li X, Shi Z, Xia Y, Cai Q, Xu D, et al. PTEN suppresses glycolysis by dephosphorylating and inhibiting autophosphorylated PGK1. Mol Cell (2019) 76(3):516–27.e7. doi: 10.1016/j.molcel.2019.08.006
35. Bollong M, Lee G, Coukos J, Yun H, Zambaldo C, Chang J, et al. A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signalling. Nature (2018) 562(7728):600–4. doi: 10.1038/s41586-018-0622-0
36. Zheng Q, Maksimovic I, Upad A, David Y. Non-enzymatic covalent modifications: a new link between metabolism and epigenetics. Protein Cell (2020) 11(6):401–16. doi: 10.1007/s13238-020-00722-w
37. Cuadrado A, Rojo A, Wells G, Hayes J, Cousin S, Rumsey W, et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug discovery. (2019) 18(4):295–317. doi: 10.1038/s41573-018-0008-x
38. Baird L, Yamamoto M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol Cell Biol (2020) 40(13). doi: 10.1128/mcb.00099-20
39. Bellezza I, Giambanco I, Minelli A, Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta Mol Cell Res (2018) 1865(5):721–33. doi: 10.1016/j.bbamcr.2018.02.010
40. Xu T, Gao P, Huang Y, Wu M, Yi J, Zhou Z, et al. Git1-PGK1 interaction achieves self-protection against spinal cord ischemia-reperfusion injury by modulating Keap1/Nrf2 signaling. Redox Biol (2023) 62:102682. doi: 10.1016/j.redox.2023.102682
41. Zou P, Zhang X, Zhang R, Chai X, Zhao Y, Li E, et al. Blockage of ERCC6 alleviates spinal cord injury through weakening apoptosis, inflammation, senescence, and oxidative stress. Front Mol biosciences. (2022) 9:853654. doi: 10.3389/fmolb.2022.853654
42. Takahashi T, Sato Y, Yamagata A, Goto-Ito S, Saijo M, Fukai S. Structural basis of ubiquitin recognition by the winged-helix domain of Cockayne syndrome group B protein. Nucleic Acids Res (2019) 47(7):3784–94. doi: 10.1093/nar/gkz081
43. Proietti-De-Santis L, Balzerano A, Prantera G. CSB: an emerging actionable target for cancer therapy. Trends cancer. (2018) 4(3):172–5. doi: 10.1016/j.trecan.2018.01.005
44. Liu L, Zhou J, Wang Y, Qi T, Wang Z, Chen L, et al. Imatinib inhibits oxidative stress response in spinal cord injury rats by activating Nrf2/HO-1 signaling pathway. Exp Ther Med (2020) 19(1):597–602. doi: 10.3892/etm.2019.8270
45. Hochhaus A, Saglio G, Hughes T, Larson R, Kim D, Issaragrisil S, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia (2016) 30(5):1044–54. doi: 10.1038/leu.2016.5
46. Lu K, Cho C, Liang C, Chen S, Liliang P, Wang S, et al. Inhibition of the MEK/ERK pathway reduces microglial activation and interleukin-1-beta expression in spinal cord ischemia/reperfusion injury in rats. J Thorac Cardiovasc surgery. (2007) 133(4):934–41. doi: 10.1016/j.jtcvs.2006.11.038
47. Kundi S, Bicknell R, Ahmed Z. The role of angiogenic and wound-healing factors after spinal cord injury in mammals. Neurosci Res (2013) 76:1–9. doi: 10.1016/j.neures.2013.03.013
48. Zhao X, Li Z, Liang S, Li S, Ren J, Li B, et al. Different epidermal growth factor receptor signaling pathways in neurons and astrocytes activated by extracellular matrix after spinal cord injury. Neurochemistry Int (2019) 129:104500. doi: 10.1016/j.neuint.2019.104500
49. Mruthyunjaya S, Manchanda R, Godbole R, Pujari R, Shiras A, Shastry P. Laminin-1 induces neurite outgrowth in human mesenchymal stem cells in serum/differentiation factors-free conditions through activation of FAK-MEK/ERK signaling pathways. Biochem Biophys Res Commun (2010) 391(1):43–8. doi: 10.1016/j.bbrc.2009.10.158
50. Zhang H, Xiang D, Liu X, Xiang L. PPARα agonist relieves spinal cord injury in rats by activating Nrf2/HO-1 via the Raf-1/MEK/ERK pathway. Aging (2021) 13(22):24640–54. doi: 10.18632/aging.203699
51. Zhao X, Sun J, Yuan Y, Lin S, Lin J, Mei X. Zinc promotes microglial autophagy through NLRP3 inflammasome inactivation via XIST/miR-374a-5p axis in spinal cord injury. Neurochemical Res (2022) 47(2):372–81. doi: 10.1007/s11064-021-03441-8
52. Liu T, Ma X, Ouyang T, Chen H, Xiao Y, Huang Y, et al. Efficacy of 5-aminolevulinic acid-based photodynamic therapy against keloid compromised by downregulation of SIRT1-SIRT3-SOD2-mROS dependent autophagy pathway. Redox Biol (2019) 20:195–203. doi: 10.1016/j.redox.2018.10.011
53. Zhou L, Yao M, Tian Z, Liu S, Song Y, Ye J, et al. Muscone suppresses inflammatory responses and neuronal damage in a rat model of cervical spondylotic myelopathy by regulating Drp1-dependent mitochondrial fission. J neurochemistry. (2020) 155(2):154–76. doi: 10.1111/jnc.15011
54. Zhou K, Zheng Z, Li Y, Han W, Zhang J, Mao Y, et al. TFE3, a potential therapeutic target for Spinal Cord Injury via augmenting autophagy flux and alleviating ER stress. Theranostics (2020) 10(20):9280–302. doi: 10.7150/thno.46566
55. Liu Z, Yao X, Sun B, Jiang W, Liao C, Dai X, et al. Pretreatment with kaempferol attenuates microglia-mediate neuroinflammation by inhibiting MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. Free Radical Biol Med (2021) 168:142–54. doi: 10.1016/j.freeradbiomed.2021.03.037
56. Bock F, Tait S. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol (2020) 21(2):85–100. doi: 10.1038/s41580-019-0173-8
57. Smith R, Walsh C. Ion channel functions in early brain development. Trends neurosciences. (2020) 43(2):103–14. doi: 10.1016/j.tins.2019.12.004
58. Jiang L, Yuan F, Yin X, Dong J. Responses and adaptations of intervertebral disc cells to microenvironmental stress: a possible central role of autophagy in the adaptive mechanism. Connective Tissue Res (2014) 55:311–21. doi: 10.3109/03008207.2014.942419
59. Wu L, Wu G, Akhavan Sharif M, Baker A, Jia Y, Fahey F, et al. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat Neurosci (2012) 15(4):565–73. doi: 10.1038/nn.3059
60. Cheng J, Xu T, Xun C, Guo H, Cao R, Gao S, et al. Carnosic acid protects against ferroptosis in PC12 cells exposed to erastin through activation of Nrf2 pathway. Life Sci (2021) 266:118905. doi: 10.1016/j.lfs.2020.118905
61. Sauerbeck A, Schonberg D, Laws J, McTigue D. Systemic iron chelation results in limited functional and histological recovery after traumatic spinal cord injury in rats. Exp neurology. (2013) 248:53–61. doi: 10.1016/j.expneurol.2013.05.011
62. Argüelles J. Physiological roles of trehalose in bacteria and yeasts: a comparative analysis. Arch Microbiol (2000) 174(4):217–24. doi: 10.1007/s002030000192
63. Tanaka M, Machida Y, Niu S, Ikeda T, Jana N, Doi H, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med (2004) 10(2):148–54. doi: 10.1038/nm985
64. Yin Z, Gong G, Wang X, Liu W, Wang B, Yin J. The dual role of autophagy in periprosthetic osteolysis. Front Cell Dev Biol (2023) 11:1123753. doi: 10.3389/fcell.2023.1123753
65. Gong G, Wan W, Liu X, Yin J. Apelin-13, a regulator of autophagy, apoptosis and inflammation in multifaceted bone protection. Int immunopharmacology. (2023) 117:109991. doi: 10.1016/j.intimp.2023.109991
66. Ichimiya T, Yamakawa T, Hirano T, Yokoyama Y, Hayashi Y, Hirayama D, et al. Autophagy and autophagy-related diseases: A review. Int J Mol Sci (2020) 21(23). doi: 10.3390/ijms21238974
67. Ariosa A, Lahiri V, Lei Y, Yang Y, Yin Z, Zhang Z, et al. A perspective on the role of autophagy in cancer. Biochim Biophys Acta Mol basis disease. (2021) 1867(12):166262. doi: 10.1016/j.bbadis.2021.166262
68. Sullivan P, Krishnamurthy S, Patel S, Pandya J, Rabchevsky A. Temporal characterization of mitochondrial bioenergetics after spinal cord injury. J neurotrauma. (2007) 24(6):991–9. doi: 10.1089/neu.2006.0242
69. Guicciardi M, Leist M, Gores G. Lysosomes in cell death. Oncogene (2004) 23(16):2881–90. doi: 10.1038/sj.onc.1207512
70. Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol neurobiology. (2015) 35(5):615–21. doi: 10.1007/s10571-015-0166-x
71. Yin J, Yin Z, Wang B, Zhu C, Sun C, Liu X, et al. Angiopoietin-1 protects spinal cord ischemia and reperfusion injury by inhibiting autophagy in rats. Neurochemical Res (2019) 44(12):2746–54. doi: 10.1007/s11064-019-02893-3
72. Yin Z, Gong G, Zhu C, Wang B, Sun C, Liu X, et al. Angiopoietin-1 protects neurons by inhibiting autophagy after neuronal oxygen-glucose deprivation/recovery injury. Neuroreport (2020) 31(11):825–32. doi: 10.1097/wnr.0000000000001491
73. Wen X, Klionsky D. At a glance: A history of autophagy and cancer. Semin Cancer Biol (2020) 66:3–11. doi: 10.1016/j.semcancer.2019.11.005
74. Li Q, Gao S, Kang Z, Zhang M, Zhao X, Zhai Y, et al. Rapamycin enhances mitophagy and attenuates apoptosis after spinal ischemia-reperfusion injury. Front Neurosci (2018) 12:865. doi: 10.3389/fnins.2018.00865
75. Wu C, Chen H, Zhuang R, Zhang H, Wang Y, Hu X, et al. Betulinic acid inhibits pyroptosis in spinal cord injury by augmenting autophagy via the AMPK-mTOR-TFEB signaling pathway. Int J Biol Sci (2021) 17(4):1138–52. doi: 10.7150/ijbs.57825
76. Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin H, et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol (2019) 26:101254. doi: 10.1016/j.redox.2019.101254
77. Chu C. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol disease. (2019) 122:23–34. doi: 10.1016/j.nbd.2018.07.015
78. Salech F, Ponce D, SanMartín C, Rogers N, Henríquez M, Behrens M. Cancer imprints an increased PARP-1 and p53-dependent resistance to oxidative stress on lymphocytes of patients that later develop alzheimer’s disease. Front Neurosci (2018) 12:58. doi: 10.3389/fnins.2018.00058
79. Anjum A, Yazid M, Fauzi Daud M, Idris J, Ng A, Selvi Naicker A, et al. Spinal cord injury: pathophysiology, multimolecular interactions, and underlying recovery mechanisms. Int J Mol Sci (2020) 21(20). doi: 10.3390/ijms21207533
80. Galluzzi L, Vitale I, Aaronson S, Abrams J, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death differentiation. (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
81. Chasapis C, Ntoupa P, Spiliopoulou C, Stefanidou M. Recent aspects of the effects of zinc on human health. Arch toxicology. (2020) 94(5):1443–60. doi: 10.1007/s00204-020-02702-9
82. Kitamura H, Morikawa H, Kamon H, Iguchi M, Hojyo S, Fukada T, et al. Toll-like receptor-mediated regulation of zinc homeostasis influences dendritic cell function. Nat Immunol (2006) 7(9):971–7. doi: 10.1038/ni1373
83. Hu H, Xia N, Lin J, Li D, Zhang C, Ge M, et al. Zinc Regulates Glucose Metabolism of the Spinal Cord and Neurons and Promotes Functional Recovery after Spinal Cord Injury through the AMPK Signaling Pathway. Oxid Med Cell longevity. (2021) 2021:4331625. doi: 10.1155/2021/4331625
84. Jiang D, Yang X, Ge M, Hu H, Xu C, Wen S, et al. Zinc defends against Parthanatos and promotes functional recovery after spinal cord injury through SIRT3-mediated anti-oxidative stress and mitophagy. CNS Neurosci Ther (2023) 29(10):2857–72. doi: 10.1111/cns.14222
85. Harris K, Morosky S, Drummond C, Patel M, Kim C, Stolz D, et al. RIP3 regulates autophagy and promotes coxsackievirus B3 infection of intestinal epithelial cells. Cell Host Microbe (2015) 18(2):221–32. doi: 10.1016/j.chom.2015.07.007
86. Nakashima A, Cheng S, Kusabiraki T, Motomura K, Aoki A, Ushijima A, et al. Endoplasmic reticulum stress disrupts lysosomal homeostasis and induces blockade of autophagic flux in human trophoblasts. Sci Rep (2019) 9(1):11466. doi: 10.1038/s41598-019-47607-5
87. Liu S, Yao S, Yang H, Liu S, Wang Y. Autophagy: Regulator of cell death. Cell Death disease. (2023) 14(10):648. doi: 10.1038/s41419-023-06154-8
88. Denton D, Kumar S. Autophagy-dependent cell death. Cell Death differentiation. (2019) 26(4):605–16. doi: 10.1038/s41418-018-0252-y
89. Prajapati P, Dalwadi P, Gohel D, Singh K, Sripada L, Bhatelia K, et al. Enforced lysosomal biogenesis rescues erythromycin- and clindamycin-induced mitochondria-mediated cell death in human cells. Mol Cell Biochem (2019) 461:23–36. doi: 10.1007/s11010-019-03585-w
90. Tilija Pun N, Jang W, Jeong C. Role of autophagy in regulation of cancer cell death/apoptosis during anti-cancer therapy: focus on autophagy flux blockade. Arch pharmacal Res (2020) 43(5):475–88. doi: 10.1007/s12272-020-01239-w
91. Rasheduzzaman M, Park S. Antihypertensive drug-candesartan attenuates TRAIL resistance in human lung cancer via AMPK-mediated inhibition of autophagy flux. Exp Cell Res (2018) 368(1):126–35. doi: 10.1016/j.yexcr.2018.04.022
92. Liu Z, Yao X, Jiang W, Li W, Zhu S, Liao C, et al. Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. J neuroinflammation. (2020) 17(1):90. doi: 10.1186/s12974-020-01751-2
93. Yin J, Yin Z, Lai P, Liu X, Ma J. Pyroptosis in periprosthetic osteolysis. Biomolecules (2022) 12(12). doi: 10.3390/biom12121733
94. Yin J, Gong G, Wan W, Liu X. Pyroptosis in spinal cord injury. Front Cell Neurosci (2022) 16:949939. doi: 10.3389/fncel.2022.949939
95. Rathinam V, Vanaja S, Fitzgerald K. Regulation of inflammasome signaling. Nat Immunol (2012) 13(4):333–42. doi: 10.1038/ni.2237
96. van Bruggen R, Köker M, Jansen M, van Houdt M, Roos D, Kuijpers T, et al. Human NLRP3 inflammasome activation is Nox1-4 independent. Blood (2010) 115(26):5398–400. doi: 10.1182/blood-2009-10-250803
97. Deng J, Fang L, Zhu X, Zhou B, Zhang T. A CC-NBS-LRR gene induces hybrid lethality in cotton. J Exp botany. (2019) 70(19):5145–56. doi: 10.1093/jxb/erz312
98. Yu Y, Sun X, Gu J, Yu C, Wen Y, Gao Y, et al. Deficiency of DJ-1 ameliorates liver fibrosis through inhibition of hepatic ROS production and inflammation. Int J Biol Sci (2016) 12(10):1225–35. doi: 10.7150/ijbs.15154
99. Liu G, Fan G, Guo G, Kang W, Wang D, Xu B, et al. FK506 attenuates the inflammation in rat spinal cord injury by inhibiting the activation of NF-κB in microglia cells. Cell Mol neurobiology. (2017) 37(5):843–55. doi: 10.1007/s10571-016-0422-8
100. Kushairi N, Phan C, Sabaratnam V, David P, Naidu M. Hericium erinaceusLion’s mane mushroom, (Bull.: fr.) pers. Suppresses HO-induced oxidative damage and LPS-induced inflammation in HT22 hippocampal neurons and BV2 microglia. Antioxidants (Basel Switzerland). (2019) 8(8). doi: 10.3390/antiox8080261
101. Mony L, Berger T, Isacoff E. A specialized molecular motion opens the Hv1 voltage-gated proton channel. Nat Struct Mol Biol (2015) 22(4):283–90. doi: 10.1038/nsmb.2978
102. Coe D, Poobalasingam T, Fu H, Bonacina F, Wang G, Morales V, et al. Loss of voltage-gated hydrogen channel 1 expression reveals heterogeneous metabolic adaptation to intracellular acidification by T cells. . JCI Insight (2022) 7(10). doi: 10.1172/jci.insight.147814
103. Mir F, Jha S. Locus coeruleus acid-sensing ion channels modulate sleep-wakefulness and state transition from NREM to REM sleep in the rat. Neurosci bulletin. (2021) 37(5):684–700. doi: 10.1007/s12264-020-00625-0
104. Ramsey I, Ruchti E, Kaczmarek J, Clapham D. Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc Natl Acad Sci United States America. (2009) 106(18):7642–7. doi: 10.1073/pnas.0902761106
105. Li T, Zhao J, Xie W, Yuan W, Guo J, Pang S, et al. Specific depletion of resident microglia in the early stage of stroke reduces cerebral ischemic damage. J neuroinflammation. (2021) 18(1):81. doi: 10.1186/s12974-021-02127-w
106. Li X, Yu Z, Zong W, Chen P, Li J, Wang M, et al. Deficiency of the microglial Hv1 proton channel attenuates neuronal pyroptosis and inhibits inflammatory reaction after spinal cord injury. J neuroinflammation. (2020) 17(1):263. doi: 10.1186/s12974-020-01942-x
107. Bai Z, Liu W, He D, Wang Y, Yi W, Luo C, et al. Protective effects of autophagy and NFE2L2 on reactive oxygen species-induced pyroptosis of human nucleus pulposus cells. Aging (2020) 12(8):7534–48. doi: 10.18632/aging.103109
108. Jiang C, Jiang L, Li Q, Liu X, Zhang T, Dong L, et al. Acrolein induces NLRP3 inflammasome-mediated pyroptosis and suppresses migration via ROS-dependent autophagy in vascular endothelial cells. Toxicology (2018) 410:26–40. doi: 10.1016/j.tox.2018.09.002
109. Zhang H, Wu C, Yu D, Su H, Chen Y, Ni W. Piperine attenuates the inflammation, oxidative stress, and pyroptosis to facilitate recovery from spinal cord injury via autophagy enhancement. Phytotherapy research: PTR. (2023) 37(2):438–51. doi: 10.1002/ptr.7625
110. Holloway P, Gillespie S, Becker F, Vital S, Nguyen V, Alexander J, et al. Sulforaphane induces neurovascular protection against a systemic inflammatory challenge via both Nrf2-dependent and independent pathways. Vasc Pharmacol (2016) 85:29–38. doi: 10.1016/j.vph.2016.07.004
111. Buendia I, Michalska P, Navarro E, Gameiro I, Egea J, León R. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol Ther (2016) 157:84–104. doi: 10.1016/j.pharmthera.2015.11.003
112. Ahmadi Z, Ashrafizadeh M. Melatonin as a potential modulator of Nrf2. Fundam Clin Pharmacol (2020) 34(1):11–9. doi: 10.1111/fcp.12498
113. Fernández-Ortiz M, Sayed R, Fernández-Martínez J, Cionfrini A, Aranda-Martínez P, Escames G, et al. Melatonin/nrf2/NLRP3 connection in mouse heart mitochondria during aging. Antioxidants (Basel Switzerland) (2020) 9(12). doi: 10.3390/antiox9121187
114. Fan C, Feng J, Tang C, Zhang Z, Feng Y, Duan W, et al. Melatonin suppresses ER stress-dependent proapoptotic effects via AMPK in bone mesenchymal stem cells during mitochondrial oxidative damage. Stem Cell Res Ther (2020) 11(1):442. doi: 10.1186/s13287-020-01948-5
115. Wang H, Wang H, Huang H, Qu Z, Ma D, Dang X, et al. Melatonin attenuates spinal cord injury in mice by activating the nrf2/ARE signaling pathway to inhibit the NLRP3 inflammasome. Cells (2022) 11(18). doi: 10.3390/cells11182809
116. Dixon S, Lemberg K, Lamprecht M, Skouta R, Zaitsev E, Gleason C, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
117. Chen X, Li J, Kang R, Klionsky D, Tang D. Ferroptosis: machinery and regulation. Autophagy (2021) 17(9):2054–81. doi: 10.1080/15548627.2020.1810918
118. Rouault T. Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat Rev Neurosci (2013) 14(8):551–64. doi: 10.1038/nrn3453
119. Sumneang N, Siri-Angkul N, Kumfu S, Chattipakorn S, Chattipakorn N. The effects of iron overload on mitochondrial function, mitochondrial dynamics, and ferroptosis in cardiomyocytes. Arch Biochem biophysics. (2020) 680:108241. doi: 10.1016/j.abb.2019.108241
120. Cao J, Dixon S. Mechanisms of ferroptosis. Cell Mol Life sciences: CMLS. (2016) 73:2195–209. doi: 10.1007/s00018-016-2194-1
121. Miotto G, Rossetto M, Di Paolo M, Orian L, Venerando R, Roveri A, et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol (2020) 28:101328. doi: 10.1016/j.redox.2019.101328
122. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol (2021) 18(5):280–96. doi: 10.1038/s41571-020-00462-0
123. Feng Z, Min L, Chen H, Deng W, Tan M, Liu H, et al. Iron overload in the motor cortex induces neuronal ferroptosis following spinal cord injury. Redox Biol (2021) 43:101984. doi: 10.1016/j.redox.2021.101984
124. Zhang Y, Fan B, Pang Y, Shen W, Wang X, Zhao C, et al. Neuroprotective effect of deferoxamine on erastininduced ferroptosis in primary cortical neurons. Neural regeneration Res (2020) 15(8):1539–45. doi: 10.4103/1673-5374.274344
125. Pai Kotebagilu N, Reddy Palvai V, Urooj A. Ex vivo antioxidant activity of selected medicinal plants against fenton reaction-mediated oxidation of biological lipid substrates. Biochem Res Int (2015) 2015:728621. doi: 10.1155/2015/728621
126. Yao X, Zhang Y, Hao J, Duan H, Zhao C, Sun C, et al. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural regeneration Res (2019) 14(3):532–41. doi: 10.4103/1673-5374.245480
127. Zhang Y, Sun C, Zhao C, Hao J, Zhang Y, Fan B, et al. Ferroptosis inhibitor SRS 16-86 attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury. Brain Res (2019) 1706:48–57. doi: 10.1016/j.brainres.2018.10.023
128. Zhou H, Yin C, Zhang Z, Tang H, Shen W, Zha X, et al. Proanthocyanidin promotes functional recovery of spinal cord injury via inhibiting ferroptosis. J Chem neuroanatomy. (2020) 107:101807. doi: 10.1016/j.jchemneu.2020.101807
129. Ge M, Tian H, Mao L, Li D, Lin J, Hu H, et al. Zinc attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury by activating Nrf2/GPX4 defense pathway. CNS Neurosci Ther (2021) 27(9):1023–40. doi: 10.1111/cns.13657
130. Khalaf S, Ahmad A, Chamara K, Doré S. Unique properties associated with the brain penetrant iron chelator HBED reveal remarkable beneficial effects after brain trauma. J neurotrauma. (2018) 36(1):43–53. doi: 10.1089/neu.2017.5617
131. Zhang L, Xiao H, Yu X, Deng Y. Minocycline attenuates neurological impairment and regulates iron metabolism in a rat model of traumatic brain injury. Arch Biochem biophysics. (2020) 682:108302. doi: 10.1016/j.abb.2020.108302
132. Du J, Liang Y, Xu F, Sun B, Wang Z. Trehalose rescues Alzheimer’s disease phenotypes in APP/PS1 transgenic mice. J Pharm Pharmacol (2013) 65(12):1753–6. doi: 10.1111/jphp.12108
133. Gong F, Ge T, Liu J, Xiao J, Wu X, Wang H, et al. Trehalose inhibits ferroptosis via NRF2/HO-1 pathway and promotes functional recovery in mice with spinal cord injury. Aging (2022) 14(7):3216–32. doi: 10.18632/aging.204009
134. Kawakami A, Fisher D. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab investigation; J Tech Methods pathology. (2017) 97(6):649–56. doi: 10.1038/labinvest.2017.9
135. Tsunemi T, Ashe T, Morrison B, Soriano K, Au J, Roque R, et al. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Trans Med (2012) 4(142):142ra97. doi: 10.1126/scitranslmed.3003799
136. Zhou Y, Li L, Mao C, Zhou D. in vitroAstragaloside IV ameliorates spinal cord injury through controlling ferroptosis in HO-damaged PC12 cells. Ann Trans Med (2022) 10(21):1176. doi: 10.21037/atm-22-5196
137. Mekhail M, Almazan G, Tabrizian M. Oligodendrocyte-protection and remyelination post-spinal cord injuries: a review. Prog neurobiology. (2012) 96(3):322–39. doi: 10.1016/j.pneurobio.2012.01.008
138. Zhao Y, Wu J, Li D, Liu J, Chen W, Hou Z, et al. Human ESC-derived immunity- and matrix- regulatory cells ameliorated white matter damage and vascular cognitive impairment in rats subjected to chronic cerebral hypoperfusion. Cell proliferation. (2022) 55(5):e13223. doi: 10.1111/cpr.13223
139. Shi J, Xue X, Yuan L, He G, Jiang Z, Wang L, et al. Amelioration of white matter injury through mitigating ferroptosis following hepcidin treatment after spinal cord injury. Mol neurobiology. (2023) 60(6):3365–78. doi: 10.1007/s12035-023-03287-x
140. Ge H, Xue X, Xian J, Yuan L, Wang L, Zou Y, et al. Ferrostatin-1 alleviates white matter injury via decreasing ferroptosis following spinal cord injury. Mol neurobiology. (2022) 59(1):161–76. doi: 10.1007/s12035-021-02571-y
141. Qian Z, Ke Y. Hepcidin and its therapeutic potential in neurodegenerative disorders. Medicinal Res Rev (2020) 40(2):633–53. doi: 10.1002/med.21631
142. Du F, Qian Z, Luo Q, Yung W, Ke Y. Hepcidin suppresses brain iron accumulation by downregulating iron transport proteins in iron-overloaded rats. Mol neurobiology. (2015) 52(1):101–14. doi: 10.1007/s12035-014-8847-x
143. Du F, Qian C, Qian Z, Wu X, Xie H, Yung W, et al. Hepcidin directly inhibits transferrin receptor 1 expression in astrocytes via a cyclic AMP-protein kinase A pathway. Glia (2011) 59(6):936–45. doi: 10.1002/glia.21166
144. Liu W, Wang Y, Gong F, Rong Y, Luo Y, Tang P, et al. Exosomes derived from bone mesenchymal stem cells repair traumatic spinal cord injury by suppressing the activation of A1 neurotoxic reactive astrocytes. J neurotrauma. (2019) 36(3):469–84. doi: 10.1089/neu.2018.5835
145. Hessvik N, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life sciences: CMLS. (2018) 75(2):193–208. doi: 10.1007/s00018-017-2595-9
146. Chevillet J, Kang Q, Ruf I, Briggs H, Vojtech L, Hughes S, et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc Natl Acad Sci United States America. (2014) 111(41):14888–93. doi: 10.1073/pnas.1408301111
147. Ge X, Zhou Z, Yang S, Ye W, Wang Z, Wang J, et al. Exosomal USP13 derived from microvascular endothelial cells regulates immune microenvironment and improves functional recovery after spinal cord injury by stabilizing IκBα. Cell bioscience. (2023) 13(1):55. doi: 10.1186/s13578-023-01011-9
148. Ying Y, Huang Z, Tu Y, Wu Q, Li Z, Zhang Y, et al. A shear-thinning, ROS-scavenging hydrogel combined with dental pulp stem cells promotes spinal cord repair by inhibiting ferroptosis. Bioactive materials. (2023) 22:274–90. doi: 10.1016/j.bioactmat.2022.09.019
149. Du J, Wang F, Li J, Yang Y, Guo D, Zhang Y, et al. Green polymer hydrogels from a natural monomer with inherent antioxidative capability for efficient wound healing and spinal cord injury treatment. Biomaterials science. (2023) 11(10):3683–94. doi: 10.1039/d3bm00174a
150. Rao S, Lin Y, Du Y, He L, Huang G, Chen B, et al. Designing multifunctionalized selenium nanoparticles to reverse oxidative stress-induced spinal cord injury by attenuating ROS overproduction and mitochondria dysfunction. J materials Chem B (2019) 7(16):2648–56. doi: 10.1039/c8tb02520g
151. Rao S, Lin Y, Lin R, Liu J, Wang H, Hu W, et al. Traditional Chinese medicine active ingredients-based selenium nanoparticles regulate antioxidant selenoproteins for spinal cord injury treatment. J nanobiotechnology. (2022) 20(1):278. doi: 10.1186/s12951-022-01490-x
152. Li Y, Zou Z, An J, Wu Q, Tong L, Mei X, et al. Chitosan-modified hollow manganese dioxide nanoparticles loaded with resveratrol for the treatment of spinal cord injury. Drug delivery. (2022) 29(1):2498–512. doi: 10.1080/10717544.2022.2104957
153. Jiang X, Liu X, Yu Q, Shen W, Mei X, Tian H, et al. Functional resveratrol-biodegradable manganese doped silica nanoparticles for the spinal cord injury treatment. Materials Today Bio. (2022) 13:100177. doi: 10.1016/j.mtbio.2021.100177
154. Shi H, Jin L, Li J, Liang K, Li X, Ye Z, et al. Mesoporous polydopamine nanoparticles for sustained release of rapamycin and reactive oxygen species scavenging to synergistically accelerate neurogenesis after spinal cord injury. J materials Chem B (2022) 10(33):6351–9. doi: 10.1039/d2tb00841f
155. Ji Z, Gao G, Ma Y, Luo J, Zhang G, Yang H, et al. Highly bioactive iridium metal-complex alleviates spinal cord injury via ROS scavenging and inflammation reduction. Biomaterials (2022) 284:121481. doi: 10.1016/j.biomaterials.2022.121481
156. Ma D, Shen H, Chen F, Liu W, Zhao Y, Xiao Z, et al. Inflammatory microenvironment-responsive nanomaterials promote spinal cord injury repair by targeting IRF5. Advanced healthcare materials. (2022) 11(23):e2201319. doi: 10.1002/adhm.202201319
157. Ma J, Li J, Wang X, Li M, Teng W, Tao Z, et al. GDNF-loaded polydopamine nanoparticles-based anisotropic scaffolds promote spinal cord repair by modulating inhibitory microenvironment. Advanced healthcare materials. (2023) 12(8):e2202377. doi: 10.1002/adhm.202202377
158. Tran A, Warren P, Silver J. The biology of regeneration failure and success after spinal cord injury. Physiol Rev (2018) 98(2):881–917. doi: 10.1152/physrev.00017.2017
159. Beckman J. Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res toxicology. (1996) 9(5):836–44. doi: 10.1021/tx9501445
160. Radi R. Peroxynitrite reactions and diffusion in biology. Chem Res toxicology. (1998) 11(7):720–1. doi: 10.1021/tx980096z
161. Bringold U, Ghafourifar P, Richter C. Peroxynitrite formed by mitochondrial NO synthase promotes mitochondrial Ca2+ release. Free Radical Biol Med (2000) 29:343–8. doi: 10.1016/s0891-5849(00)00318-x
162. Coyoy-Salgado A, Segura-Uribe J, Guerra-Araiza C, Orozco-Suárez S, Salgado-Ceballos H, Feria-Romero I, et al. The importance of natural antioxidants in the treatment of spinal cord injury in animal models: an overview. Oxid Med Cell longevity. (2019) 2019:3642491. doi: 10.1155/2019/3642491
163. He L, He T, Farrar S, Ji L, Liu T, Ma X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol biochemistry: Int J Exp Cell physiology biochemistry Pharmacol (2017) 44(2):532–53. doi: 10.1159/000485089
164. Lee K, Cha M, Lee B. Neuroprotective effect of antioxidants in the brain. Int J Mol Sci (2020) 21(19). doi: 10.3390/ijms21197152
165. Jakubczyk K, Kałduńska J, Dec K, Kawczuga D, Janda K. Antioxidant properties of small-molecule non-enzymatic compounds. Polski merkuriusz lekarski: Organ Polskiego Towarzystwa Lekarskiego. (2020) 48(284):128–32.
166. Kiokias S, Proestos C, Oreopoulou V. Effect of natural food antioxidants against LDL and DNA oxidative changes. Antioxidants (Basel Switzerland). (2018) 7(10). doi: 10.3390/antiox7100133
167. Rizvi S, Raza S, Ahmed F, Ahmad A, Abbas S, Mahdi F. The role of vitamin e in human health and some diseases. Sultan Qaboos Univ Med J (2014) 14(2):e157–65.
168. Manochkumar J, Doss C, El-Seedi H, Efferth T, Ramamoorthy S. The neuroprotective potential of carotenoids in vitro and in vivo. Phytomedicine: Int J phytotherapy phytopharmacology (2021) 91:153676. doi: 10.1016/j.phymed.2021.153676
169. Pandey K, Rizvi S. Plant polyphenols as dietary antioxidants in human health and disease. Oxid Med Cell longevity. (2009) 2(5):270–8. doi: 10.4161/oxim.2.5.9498
170. Taghvaei M, Jafari S. Application and stability of natural antioxidants in edible oils in order to substitute synthetic additives. J Food Sci technology. (2015) 52(3):1272–82. doi: 10.1007/s13197-013-1080-1
171. Breiner A, Zinman L, Bourque P. Edaravone for amyotrophic lateral sclerosis: barriers to access and lifeboat ethics. CMAJ: Can Med Assoc J = J l’Association medicale canadienne. (2020) 192(12):E319–E20. doi: 10.1503/cmaj.191236
172. Li M, Al-Jamal K, Kostarelos K, Reineke J. Physiologically based pharmacokinetic modeling of nanoparticles. ACS nano. (2010) 4(11):6303–17. doi: 10.1021/nn1018818
Keywords: spinal cord injury, ROS, apoptosis, autophagy, ferroptosis, pyroptosis
Citation: Yin Z, Wan B, Gong G and Yin J (2024) ROS: Executioner of regulating cell death in spinal cord injury. Front. Immunol. 15:1330678. doi: 10.3389/fimmu.2024.1330678
Received: 31 October 2023; Accepted: 08 January 2024;
Published: 23 January 2024.
Edited by:
Juan Pablo de Rivero Vaccari, University of Miami, United StatesReviewed by:
Nataliya Pidkovka, South College, United StatesCopyright © 2024 Yin, Wan, Gong and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Yin, eWluamlhbmRvY0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.