 Katherine Figarella
Katherine Figarella Jieun Kim
Jieun Kim Wei Ruan
Wei Ruan Tingting Mills
Tingting Mills Holger Klaus Eltzschig
Holger Klaus Eltzschig Xiaoyi Yuan
Xiaoyi Yuan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 19 January 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1328565
The human respiratory and circulatory systems collaborate intricately to ensure oxygen delivery to all cells, which is vital for ATP production and maintaining physiological functions and structures. During limited oxygen availability, hypoxia-inducible factors (HIFs) are stabilized and play a fundamental role in maintaining cellular processes for hypoxia adaptation. First discovered during investigations of erythropoietin production regulation, HIFs influence physiological and pathological processes, including development, inflammation, wound healing, and cancer. HIFs promote extracellular adenosine signaling by enhancing adenosine generation and receptor signaling, representing an endogenous feedback mechanism that curbs excessive inflammation, supports injury resolution, and enhances hypoxia tolerance. This is especially important for conditions that involve tissue hypoxia, such as acute respiratory distress syndrome (ARDS), which globally poses significant health challenges without specific treatment options. Consequently, pharmacological strategies to amplify HIF-mediated adenosine production and receptor signaling are of great importance.
Acute lung injury caused by various insults, including pneumonia, sepsis, and trauma, can aggravate and lead to a more severe condition known as acute respiratory distress syndrome (ARDS). The acute onset of respiratory insufficiency, bilateral lung infiltrates on chest imaging, and severe progressive hypoxemia (low oxygen levels in the blood) are key characteristics of ARDS (1). Its pathophysiology is complex and multifactorial, involving direct and indirect lung injury (2). Direct lung injury can be caused by factors such as aspiration of gastric contents, inhalation of toxic gases, or viral and bacterial infections. Indirect lung injury can be caused by systemic factors such as sepsis, trauma, or pancreatitis, which can generate an inflammatory response that ultimately affects the lung (Figure 1). The initial insult to the lung induces the activation of neutrophils and macrophages, which release proinflammatory cytokines and chemokines (3). This leads to increased permeability of the alveolar-capillary barrier, allowing protein-rich fluid to leak into the alveolar space and impairing gas exchange. The initial inflammatory response is accompanied by abnormalities in surfactant function, which can further impair gas exchange. Surfactant is a mixture of lipids and proteins that lines the alveoli and helps maintain alveolar stability and prevent collapse (4). The combination of inflammation, impaired surfactant function, and increased alveolar-capillary barrier permeability causes a progressive decline in oxygenation and respiratory function.
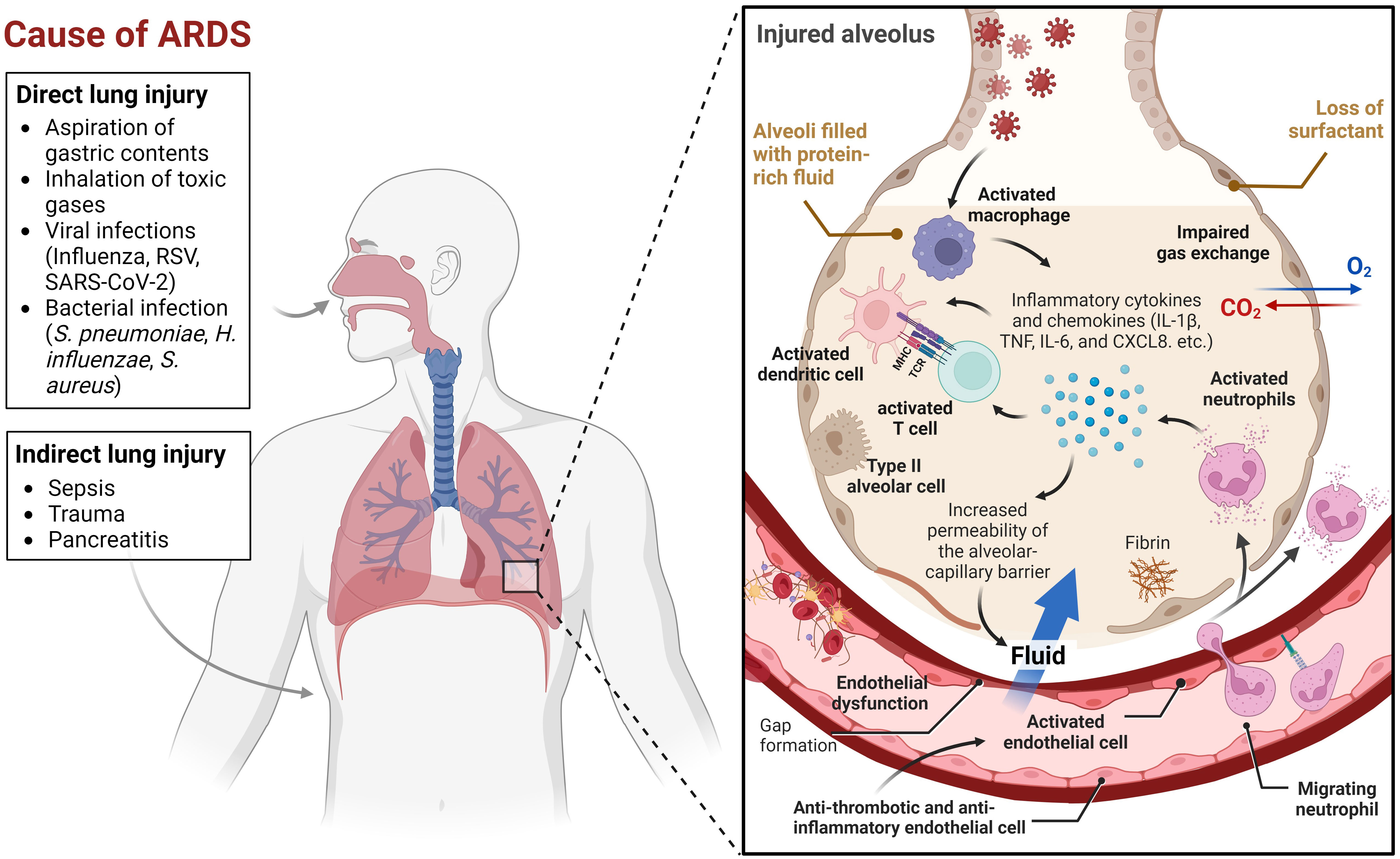
Figure 1 Multifactorial events leading to Acute Respiratory Distress Syndrome (ARDS). This figure illustrates the many factors that can contribute to ARDS. Lung injury can be caused by a variety of direct or indirect insults, or a combination of both. When an initial insult occurs, neutrophils and macrophages are activated, leading to the release of proinflammatory cytokines and chemokines. Some of the key chemokines and cytokines implicated in ARDS include interleukin-1β (IL-1β), tumor necrosis factor (TNF), interleukin-6 (IL-6), and CXCL8 (3). These molecules play a crucial role in the recruitment and activation of immune cells, such as neutrophils, macrophages, dendritic cells, and T cells, which contribute to the inflammatory response in ARDS. Endothelial cells (ECs) coordinate the immune and hemostatic response by shifting from their normal anti-thrombotic and anti-inflammatory phenotype to an activated state of endothelial dysfunction. This shift is a critical host response to infection and injury and is involved in ARDS pathogenesis. The inflammatory response increases the permeability of the alveolar-capillary barrier, which causes protein-rich fluid to leak into the alveolar space, resulting in impaired gas exchange. Meanwhile, abnormalities in surfactant function further exacerbate the disruption of gas exchange. This combination of factors, including inflammation, impaired surfactant function, and increased alveolar-capillary barrier permeability, leads to a progressive decline in oxygenation and respiratory function. The visual representation of this complex process was created using BioRender.com.
Treatment for ARDS primarily focuses on supporting the patient while the lungs heal and addressing the underlying cause of the condition. It includes measures such as mechanical ventilation with low tidal volumes, prone positioning, and administration of supplemental oxygen (5). In some cases, extracorporeal membrane oxygenation (ECMO) may temporarily support oxygenation (5). The therapeutic interventions used to manage the disease are summarized in Table 1. Although there have been improvements in providing support and care, the mortality rate of ARDS is still high, ~40% (12). There is still no targeted treatment for ARDS. Thus, a better understanding of the pathophysiology of the complex multifactorial ARDS may lead to the development of novel therapeutic strategies to improve outcomes for patients with this devastating condition.
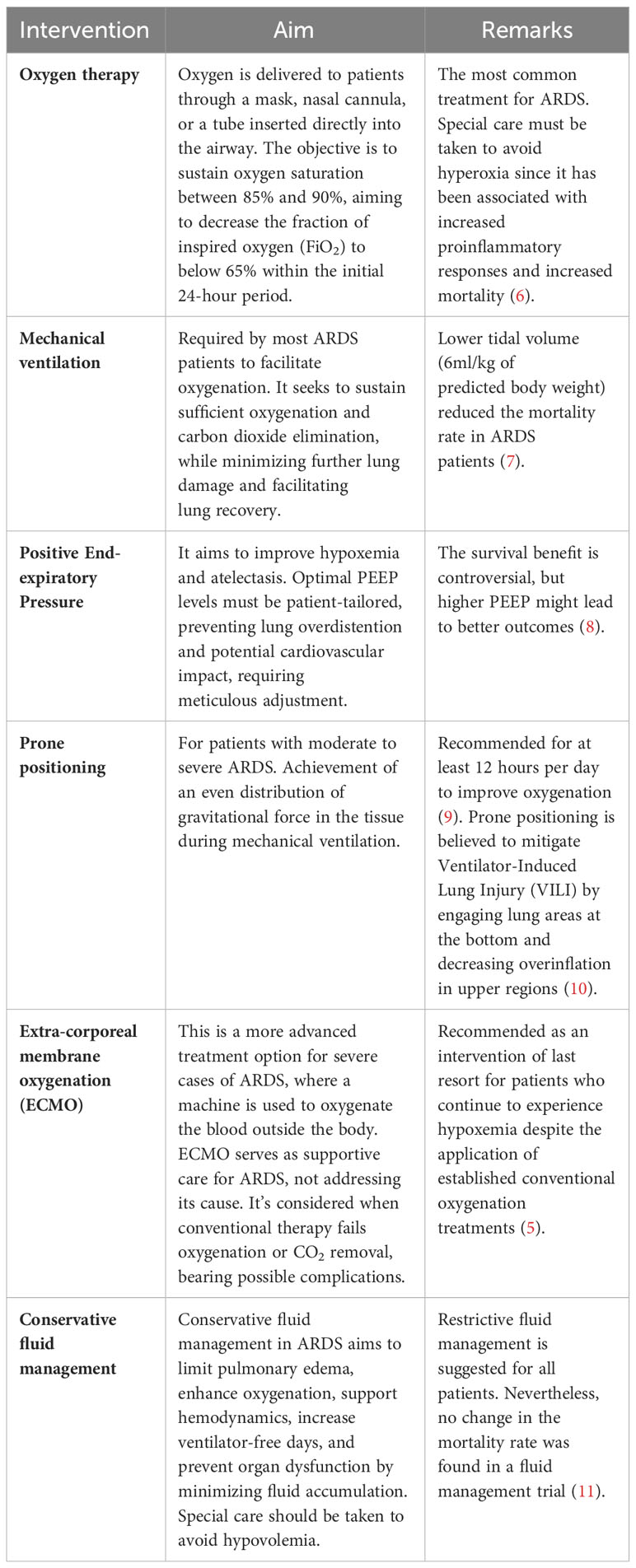
Table 1 Therapeutical interventions applied to patients with ARDS.
Hypoxia is a hallmark of ARDS and is known to play a central role in the pathophysiology of the disease. Hypoxia can initially be an inflammatory stimulus that promotes proinflammatory responses and disrupts tissue barriers (13). The molecular mechanism involves the activation of NF-κB, a transcription factor that mediates the expression of proinflammatory genes (14). However, stabilizing hypoxia-inducible factors (HIFs), a group of transcription factors responsible for regulating the expression of numerous genes that promote cellular adaptation and survival, triggers anti-inflammatory responses to provide endogenous protection. Notably, the extracellular adenosine pathway, augmented during ARDS, -as evidenced by the upregulation of enzymes involved in adenosine production and the downregulation of genes that act in the termination of the adenosine signaling pathway-, is driven by HIFs and results in mitigating excessive inflammatory responses and reinforcing barrier function during ARDS (15–18). This highlights the importance of studying the hypoxia-adenosine axis during ARDS to optimize treatment strategies and improve patient outcomes. This review delves into the sophisticated relationship between hypoxia and adenosine signaling, encompassing adenosine generation, receptors, and metabolism. We will also address the upregulation and stabilization of hypoxia-inducible factors in ARDS. Finally, we will examine the diverse preclinical studies and current trials for clinical strategies to treat acute lung injury through therapeutic targeting of the hypoxia-adenosine axis.
Reduced oxygen availability in tissues or organs plays a significant role in various human diseases (19, 20), including inflammatory conditions such as ARDS. HIF is stabilized during hypoxia via various mechanisms (21, 22). There are two primary components of HIF: the alpha subunits (HIFα), encompassing the oxygen-dependent HIF-1α, HIF-2α, and HIF-3α, and the constitutive beta subunit (HIF1β). The stability and activity of HIFs are tightly regulated by a family of oxygen-sensing enzymes called prolyl hydroxylase domain (PHDs). Under normoxic conditions (normal oxygen levels), PHDs hydroxylate specific proline residues on the HIFα subunit. This modification allows the von Hippel-Lindau tumor suppressor protein (pVHL) to recognize and bind to the HIFα proteins. As a result, the HIFα proteins are labeled for degradation by the proteasome (23). Conversely, PHD activity is inhibited under hypoxic conditions, allowing HIFα to accumulate and translocate into the nucleus, which binds to the HIF1β subunit. This complex binds to hypoxia-responsive elements (HRE) within the promoter region of target genes, activates their transcription, and enables cellular adaptation and survival during hypoxia (24). Three PHD isoforms exist, namely, PHD1, PHD2, and PHD3, each with distinct roles regulating HIFs (25). All three isoforms are sensitive to oxygen concentrations; however, the oxygen-dependent inhibition pattern differs among them. When the oxygen concentrations fall below 120 μmol/L (65 mmHg), which is lower than physiological levels for most tissues and would be considered hypoxic, PHD1, PHD2, and PHD3 are inhibited (26). In ARDS, the oxygen concentration can drop below 60 mmHg and in the lungs the induction of hypoxia-triggered genes occur at approximately 50–60 mmHg (27). PHD1 regulates HIF-2α-mediated transcription and has been linked to cellular metabolism, proliferation, and apoptosis (28). PHD2 is the main oxygen sensor and plays a crucial role in regulating HIF-1α under normoxic conditions, while PHD3 is the most relevant HIF-1α-regulating isoform in hypoxia (25, 29). The PHD isoforms have different expression patterns, tissue distribution, subcellular localization, and abilities to hydroxylate HIF-α subunits (26).
HIFs and PHDs play critical roles in various physiological and pathophysiological processes in the lung. In ARDS, hypoxia signaling activates adenosine signaling to inhibit the inflammatory response in the lungs (30). Some examples of HIFs target genes in this pathway, which harbor HRE in their promoter, are the adenosine-producing enzyme CD73, the adenosine transporters ENT1 and ENT2, and the adenosine receptors A2AAR and A2BAR. Moreover, HIFs can upregulate in response to hypoxia the expression of genes involved in angiogenesis (such as VEGF and NOS, among others) (31), leading to the development of new blood vessels in the lung. This can help to increase oxygen delivery to hypoxic tissues and promote tissue repair. Furthermore, HIFs can regulate gene expression in the immune response, including cytokines, chemokines, and adhesion molecules (32). This can help to recruit immune cells to the site of injury or infection in the lung, allowing them to clear invading pathogens and promote tissue repair. Inhibition of PHDs is beneficial in animal models of acute lung injury, likely due to the activation of HIFs and the subsequent upregulation of genes involved in tissue repair and anti-inflammatory signaling (33). Another study demonstrated that enhancing glycolysis by PHD inhibition protected alveolar epithelial cells from acute lung injury (34). Additionally, the HIF PHD inhibitor Roxadustat was observed to stabilize HIF-1α and have regenerative potential in a mouse model of hyperoxia-induced lung injury (35). Moreover, in endotoxin-mediated injury, the endothelial adherens junction integrity improved due to the enhancement of vascular endothelial protein tyrosine phosphatase (VE-PTP) via HIF-2α activation (36). These studies point out the potential therapeutic benefits of enhancing HIF in the context of acute lung injury.
Changes in cellular metabolism, such as modifications in glycolysis and fatty acid oxidation (FAO), have become crucial mechanisms in various pathological conditions, including inflammation (37). Recent research presents compelling evidence that carbohydrate metabolites and intermediates, beyond their traditional role as an energy source, have significant functions in controlling inflammatory processes and the potential to alleviate lung inflammation (38). HIF-1α stabilization was shown to play a protective role by regulating glucose metabolism in alveolar epithelial cells, ultimately reducing lung inflammation and pulmonary edema (38). Mice with alveolar epithelial cell-specific Hif1a deletion showed increased lung inflammation. Succinate, a tricarboxylic acid cycle intermediate, has been shown to have a protective role in alveolar epithelial cells during ARDS by enhancing HIF-1α levels (39). The succinate-mediated HIF stabilization takes place through distinct mechanisms. Firstly, succinate induces the production of Reactive Oxygen Species (ROS) by inhibiting succinate dehydrogenase (SDH), consequently stabilizing HIF. This stabilization occurs via ROS-mediated oxidation of Fe2+, an essential cofactor for PHD activity, thus limiting its function (40). Additionally, succinate can traverse from mitochondria to the cytosol through specific transporters. Excessive cytosolic succinate impedes PHD activity, resulting in HIF stabilization and activation, an occurrence called ‘pseudohypoxia’ (41). In an in vitro setting, the addition of succinate reduced epithelial inflammation following stretch. In a mouse model featuring inducible alveolar-epithelial Sdha deletion (Sdhaloxp/loxp/SPC-CreER mice), several favorable outcomes were observed, including diminished lung inflammation, improved integrity of the alveolar barrier and mitigated histological changes towards injury. This aligns with the notion that succinate plays a functional role in stabilizing HIF. Notably, Sdhaloxp/loxp/SPC-CreER mice exhibited heightened HIF-1α levels in response to hypoxia or acute lung injury (ALI) (39). These findings suggest that targeting succinate and its related pathways may have potential therapeutic benefits in ARDS.
Sepsis is one of the causes leading to ARDS. Thiel et al. investigated the role of HIF-1α in T cells under hypoxic conditions in the context of sepsis and found that deleting the HIF-1α gene in T cells improved survival in septic mice (42). This was attributed to preventing T-cell inhibition in hypoxic inflamed tissues. Moreover, T lymphocyte development and activity have been shown to be influenced by hypoxia (43). For instance, cytotoxic T lymphocytes (CTLs) development is delayed at 2.5% oxygen compared to 20%. However, the development of CTLs at 2.5% oxygen is more sustained, and the CTLs are much more lytic. The results indicate that targeting HIF-1α in T cells could be a therapeutic strategy for improving survival in septic conditions.
Adenosine is a naturally occurring molecule in the body that plays a significant role in various physiological processes. It is produced in all cells of the body, mainly by the breakdown of adenosine triphosphate (ATP) and other sources like NAD+ or through hydrolysis of S-adenosyl homocysteine (44, 45). Adenosine synthesis can occur inside or on the cell surface (46). The latter has been associated with cellular stress and the release of ATP from inside cells into the extracellular space, either through leakage or controlled release pathways involving connexin and pannexin channels or ATP-binding cassette transporters (47, 48). In the ARDS scenario, extracellular adenosine produced mainly by endothelial cells, alveolar epithelial cells, alveolar macrophages, and neutrophils provides protective effects (49). In the extracellular space, the classical pathway for generating adenosine consists of the breakdown of extracellular ATP through the action of ectonucleotidases, such as CD39 (also known as ectonucleoside triphosphate diphosphohydrolase 1) and CD73 (also known as 5′-nucleotidase) (Figure 2). Ectonucleotidases are tethered to the plasma membrane and possess their active sites facing the extracellular milieu, facilitating the enzymatic breakdown of nucleotides present outside the cell (50). This spatial orientation enables these enzymes to participate in the modulation of extracellular nucleotide concentrations, thereby influencing purinergic signaling pathways. CD39 is an enzyme found on cell membranes, and it catalyzes the hydrolysis of extracellular ATP, resulting in the production of extracellular ADP and AMP. Then, extracellular AMP is dephosphorylated into adenosine by CD73, an enzyme anchored by glycophosphatidylinositol, and can also exist in a soluble form (51). Extracellular AMP levels are additionally modulated by secreted or membrane-associated enzymes such as ecto-adenylate kinase (ecto-AK) and ecto-nucleoside diphosphate kinase (ecto-NDPK) (52). These enzymes regulate the conversion of AMP back to extracellular ATP.
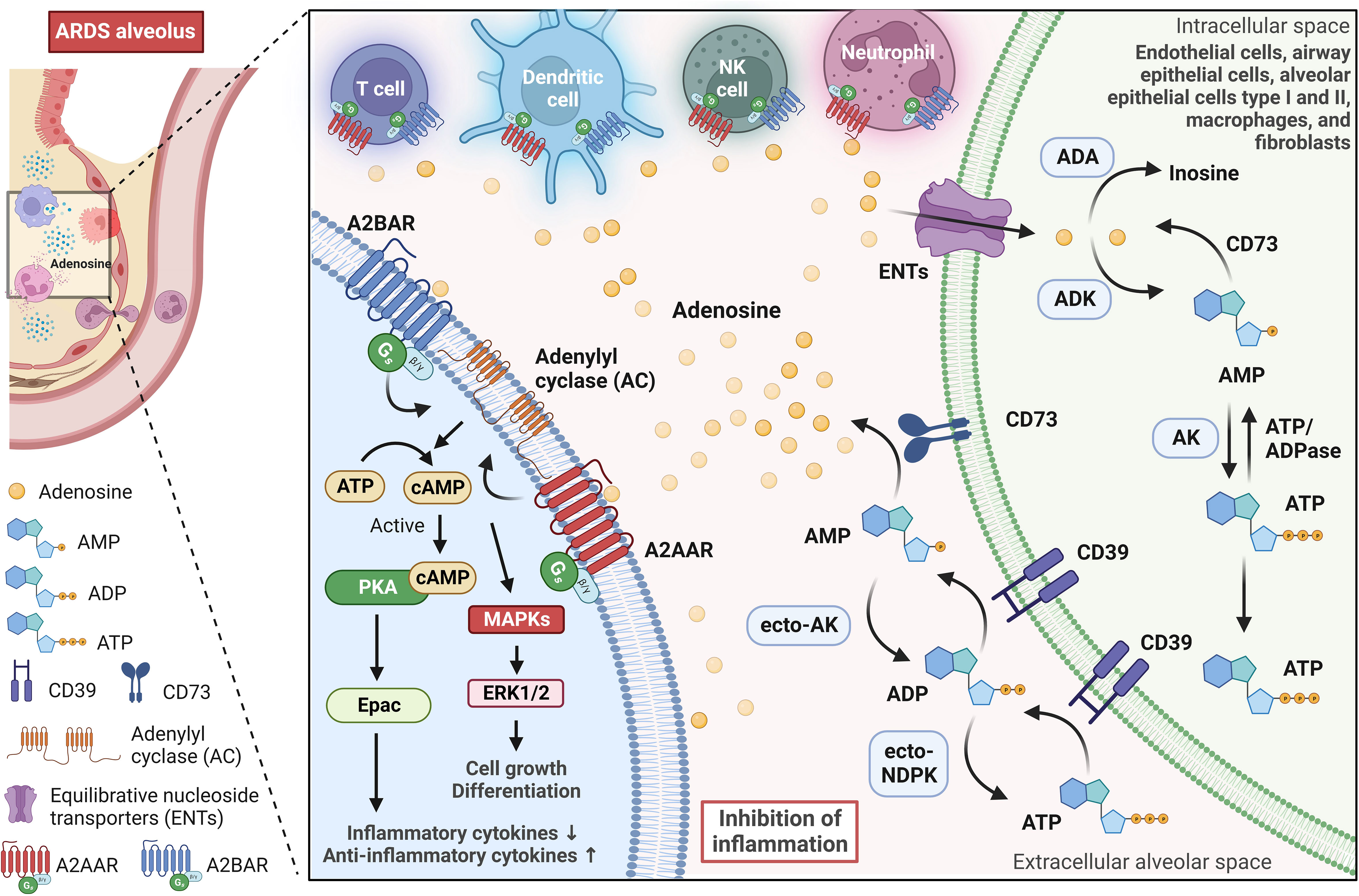
Figure 2 Adenosine signaling pathway in the alveolar microenvironment during ARDS. Adenosine is synthesized on the surface of cells, especially during stressful situations when ATP is released into the extracellular alveolar space. In the lungs, adenosine-producing cells (green cell on the right) involve endothelial cells, airway epithelial cells, alveolar epithelial cells type I and II, macrophages, and fibroblasts. The extracellular ATP is broken down by ectonucleotidases CD39 and CD73, which leads to the production of extracellular ADP, AMP, and finally, adenosine. Further regulation of extracellular AMP and ADP levels is carried out by enzymes such as adenylate kinase (ecto-AK) and nucleoside diphosphate kinase (ecto-NDPK), which convert AMP back to extracellular ATP. In the alveolar environment, extracellular adenosine primarily activates G-protein-coupled adenosine receptors A2AAR and A2BAR. These receptors are expressed in alveolar epithelial cells and various immune and inflammatory cells, and they play a significant role in modulating immune responses. A2AAR and A2BAR signal via a Gs protein and activate adenylyl cyclase, leading to an increase in intracellular cyclic adenosine monophosphate (cAMP), which in turn, activates protein kinase A (PKA) and the exchange protein directly activated by cAMP (Epac). PKA and Epac pathways suppress inflammatory cytokine production while strongly elevating anti-inflammatory cytokines. Furthermore, A2AAR and A2BAR can also influence the activation of mitogen-activated protein kinases (MAPKs), such as ERK1/2, which are involved in various cellular activities, including cell growth and differentiation. Equilibrative nucleoside transporters (primarily ENT2) regulate extracellular adenosine levels in the lung by mediating its uptake from the extracellular space into cells. Intracellular adenosine is metabolized into inosine by adenosine deaminase (ADA) or AMP by adenosine kinase (ADK). This image was created using BioRender.com.
Extracellular adenosine has a short half-life and exerts tissue function by stimulating four G-protein-coupled adenosine receptors: A1AR, A2AAR, A2BAR, and A3AR (53). Adenosine receptors are conservative among vertebrate species (54). Mice engineered with targeted deletions of each of these receptors exist and have played a pivotal role in elucidating the physiological and pathological functions associated with these receptors. Under normal physiological conditions, adenosine concentrations are adequate to activate A1AR, A2AAR, and A3AR, mainly if these receptors are abundantly expressed (46). In contrast, A2BAR requires higher concentrations of adenosine to achieve significant activation, concentrations that are typically found in more extreme or pathological situations. The adenosinergic pathway plays an essential role in modulating immune and inflammatory responses in the body (55). Adenosine can act as an immunosuppressive metabolite, mainly activating A2AAR and A2BAR. This activation initiates the cAMP/PKA/CREB pathway, which is instrumental in suppressing immune cells and helping tumors evade the immune system (56). Moreover, adenosine inhibits the release of anti-tumor cytokines by phosphorylating CREB in T cells (57). Adenosine also has a significant impact on regulatory T cells (Tregs), as it increases their population and enhances their immune regulatory functions (58–60). Adenosine can also affect dendritic cells (DCs), stimulating a more tolerogenic phenotype (53).An autocrine action of adenosine has been reported in T regulatory cells (Tregs) via CD73 and A2AAR, which is fundamental for their ability to suppress innate immune responses (48). In models of kidney ischemia-reperfusion injury, the absence of CD73 or A2AAR in Tregs renders them ineffective in preventing injury (61). Moreover, adenosine’s autocrine loops have also been demonstrated in neutrophils, modulating migration towards chemoattractants (62), and in tumor environments, contributing to immunosuppression in CD73+CD4+ effector T cells (63). Extracellular adenosine signaling is regulated by equilibrative nucleoside transporters (ENT1 and ENT2) that mediate adenosine uptake from the extracellular milieu to the intracellular room. ENT1, widely present in human and rodent tissues, predominantly regulates adenosine signaling during hypoxia (64). In contrast, ENT2 is ubiquitous but highly expressed in tissues like vascular endothelium and skeletal muscle; it has lower adenosine affinity but transports various nucleosides (65). Adenosine is then metabolized intracellularly into inosine by adenosine deaminase (ADA) or AMP by adenosine kinase (ADK). In immune function, adenosine plays a central modulatory role in inflammatory responses in both physiological and pathological conditions.
Over the past decade, investigators have identified multiple genes under the regulatory influence of HIFs, which govern the modulation of extracellular adenosine metabolism and signaling (66). HIF-driven adenosine metabolism and signaling play a pivotal role in safeguarding tissues during conditions such as myocardial injury (67), inflammatory bowel disease (68), and acute lung injury (69). Both HIF-1α and HIF-2α are involved in adenosine pathway, and many of their targets overlap since both recognize the same consensus DNA-binding element in their target gene promoter region (70). Thus, their functional role is given by their tissue/cell-specific expression profile (71). For instance, HIF-1α has been shown to orchestrate adenosine efflux in hepatocellular carcinoma (72), while HIF-2α has been found to promote angiogenesis through A2AAR signaling in pulmonary endothelial cells (71). Moreover, HIFs further influence gene expression in adenosine-related pathways. ENT1 and ENT2 promoters, along with CD73, A2AAR, and A2BAR, contain hypoxia response elements (HREs), making them transcriptional targets of HIFs. During ARDS, hypoxia signaling prominently affects lung endothelial cells (LECs), alveolar epithelial cells (AECs), infiltrating leukocytes, and monocytes. LECs and AECs, lining blood vessels and alveoli, respectively, are pivotal in regulating inflammatory lung injury and repair (73, 74). Infiltrating leukocytes and monocytes also undergo hypoxia-driven changes, influencing the inflammatory response within the lungs (75). HIFs 1 and 2 orchestrate these cellular responses, modulating a spectrum of target genes that regulate critical processes, including cell survival, differentiation, migration, and proliferation (75, 76).
Inflammation or hypoxia triggers in various cell types the release of adenosine precursor molecules, mainly ATP and ADP, into the extracellular space through mechanisms such as vesicular exocytosis, opening of conductive pathways or through the action of connexins and pannexins (77–79), but also due to the cell membrane damage (80). ATP can activate proinflammatory responses by binding to the purinergic ionotropic P2X7 receptor and eliciting NLRP3 inflammasome activation (81). Hypoxia-induced enzymatic processes convert ATP and ADP into adenosine, shifting the signaling balance from proinflammatory to anti-inflammatory. Research examining the impact of hypoxia on gene transcription has unveiled that under such circumstances, CD39 undergoes an increase in transcription with dependence on an SP1-mediated pathway (82). Likewise, CD73 transcriptional induction occurs via HIF-1α binding to the CD73 promoter, resulting in elevated CD73 transcript and protein levels (83, 84). Interestingly, Sp1 and HIF-1α transcription factors regulate their activities reciprocally (85). Sp1 induces HIF1A gene transcription, while HIF-1α binds to SP1 gene promoter, upregulating Sp1 expression and influencing cellular adaptation to low oxygen levels (86). In regulatory T cells, which, among others, are responsible for regulating excessive inflammatory responses, an upregulation of CD73 expression driven by HIF-1α stabilization was demonstrated to potentiate their immunosuppressive effects via adenosine on effector T cells (87, 88). Deletion of CD39 and CD73 was shown to exacerbate acute lung injury in mechanically ventilated mice (89). Treatment with soluble CD39 and CD73 compounds showed promise in attenuating mechanical ventilation-induced lung injury and improving survival, suggesting a therapeutic strategy for non-infectious lung pathologies.
It is important to note that in cancer, the concentration of adenosine is elevated in the tumor microenvironment (TME) due to the high expression of the adenosine-generating ectoenzyme CD73 by tumor cells. This can lead to the suppression of immune responses against the tumor. By supplementing oxygenation, the hypoxic conditions in the TME can be reduced, which in turn improves the efficacy of anti-PD-1 therapy (90). These findings suggest that supplemental oxygenation could be a potential strategy to enhance the effectiveness of immunotherapies for cancer treatment.
Adenosine receptors are expressed in various cell types throughout the body, mainly but not limited to epithelial, endothelial, nerve, muscle, and immune cells, including neutrophils and macrophages. Factors like hypoxia, cytokines, and neuronal activity regulate their expression across various cell types (46, 91). Comparative studies revealed selective HIF-1α dependent induction of A2BAR in various tissues, including the lung, during inflammation or hypoxia (92). A2BAR is a crucial connection between hypoxia and adenosine signaling in cases of acute lung damage. In a normobaric hypoxia (8% O2, 92% N2) mouse model, Eckle et al. revealed that A2bar-/- mice had significantly higher vascular permeability in comparison to A1ar-/-, A2aar-/-, or A3ar-/- mice (93). This indicates that A2BAR plays a specific role in endothelial leakage in response to hypoxia. Consequently, the A2BAR inhibitor PSB1115, caused significant pulmonary edema in hypoxia-exposed C57BL/6 mice, and the highly selective agonist BAY 60-6583, significantly attenuated hypoxia-induced pulmonary edema (93). The same results were obtained in ventilator-induced lung injury and endotoxin-induced lung injury (94, 95). In a study by Hoegl et al., the protective role of alveolar epithelial A2BAR in acute lung injury (ALI) was investigated (96). This study highlighted that the expression of A2BAR in alveolar epithelial cells contributes to mitigating ALI. Mice with specific A2bar deletion in alveolar epithelial cells exhibited detrimental effects similar to those observed in whole-body A2BAR knockout mice during ALI. Aerosol treatment with BAY 60-6583 in a two-hit model (LPS + VILI) was found to improve lung injury and alveolar fluid clearance, emphasizing the protective role of A2BAR expressed by alveolar epithelial cells in ALI. This targeted approach for delivering compounds directly to alveolar epithelial cells through inhalation holds promise for minimizing potential systemic side effects. These findings reveal a role for A2BAR signaling in the alveolar-capillary barrier during ARDS and implicate A2BAR agonist as a potential therapeutic target.
A2AAR is also transcriptionally induced in pulmonary endothelial cells during hypoxia through HIF-2α-controlled pathways (71). Its activation has been extensively studied for its potent anti-inflammatory properties. In a groundbreaking study, Ohta and Sitkovsky discovered that A2AAR is crucial in reducing inflammation and preventing tissue damage in vivo (97). The investigation uncovered a mechanism of hypoxia-driven adenosine-mediated immunosuppression, whereby an increase in the level of extracellular adenosine triggers intracellular signaling pathways that result in the downregulation of inflammation and protection from tissue damage through the activation of A2AAR. Research conducted by Reece et al. demonstrated that A2AAR activation during early reperfusion in a pig lung transplantation model reduced lung inflammation and preserved lung function, suggesting its therapeutic potential (98). A specific A2AAR agonist, ATL202, was found effective in reducing immune cell recruitment and cytokine production in a mouse model of acute lung injury (ALI) (99). Notably, A2AAR on myeloid cells appeared to play a role in controlling immune cell recruitment. Adenosine-mediated activation of A2AAR suppresses the secretion of the pro-inflammatory molecule TNF-α in macrophages (100, 101). A recent study has shown that vessel-associated macrophages initiate PMN migration during inflammation by stimulating endothelial cells to form ICAM-1 “hot spots” that support PMN transendothelial migration. Macrophage-derived TNF-α activates the endothelial TNFR2 axis, which is essential for this mechanism (102). Therefore, activating A2AAR by adenosine reduces TNF-α levels, thus suppressing immune cell recruitment. Additionally, the activation of peroxisome proliferator-activated receptor-γ (PPARγ) was shown to reduce lung injury in an A2AAR-dependent manner, and the combined application of PPARγ and A2AAR agonists exhibited a synergistic effect in suppressing inflammation and improving lung function (103). Post-treatment with CGS21680, an A2AAR agonist, also reduced neutrophil infiltration and lung tissue damage in a mouse model of pleurisy (104). These findings collectively suggest that A2AAR agonists hold promise for treating various inflammatory lung diseases.
The termination of adenosine signaling is also regulated by hypoxia. Studies utilizing radiolabeled adenosine demonstrate a decelerated extracellular adenosine uptake in hypoxic conditions. This was coupled with a notable reduction in the expression of ENT1 and ENT2, prominent adenosine transporters within vascular endothelial and epithelial cells (105, 106). HIF-1α plays a pivotal role in modulating the expression of ENT1 and ENT2, and the hypoxia-induced downregulation of these transporters mitigates inflammatory responses. Consequently, the absence of HIF-1α in epithelial cells leads to an elevation in ENT expression. Studies revealed that in the lung, mainly ENT2 mediates the termination of the adenosine signaling and that inhibition of ENT2 results in lung protection via A2BAR activation (107, 108). Similar results were observed in intestinal inflammation, where repression or deletion of epithelial ENT2 favors the resolution of inflammation through A2BAR signaling (109). Interestingly, ENT1 was shown to be associated with adenosine termination in myocytes and erythrocytes and ENT1 inhibition with cardioprotection and faster acclimatization to high altitudes, respectively (110, 111). Furthermore, hypoxia exerts inhibitory effects on adenosine kinase (AK) through HIF-1α-mediated transcriptional repression (112). This inhibition results in an augmented release of extracellular adenosine and a consequential reduction in vascular leakage during hypoxic conditions. Mechanistically, HIFs can act indirectly through additional molecules that may exert repressive effects on members of the adenosine signaling pathway. Such is the case of microRNAs (miRNAs), which are short non-coding RNAs involved in gene expression through post-transcriptional processes (113–115). Nowadays, hypoxia-regulated miRNAs, collectively known as hypoxamirs, are recognized as pivotal components in the cellular response to hypoxia (116). Some examples of miRNAs upregulated by hypoxia that target members of the adenosine signaling pathway are miR-30b (targets CD73) (117), miR-146b (targets ADA) (118), miR-155 (targets HIF-1α itself, proposed as negative-feedback loop during prolonged hypoxia) (119). Altogether, hypoxia orchestrates adenosine synthesis, signaling, and termination via hypoxia-dependent induction of genes constrained in the adenosine signaling pathway.
Under hypoxic conditions, various HIF dependent molecular signals can modulate the adenosine signaling pathway. An example of such a signal is netrin-1 (NTN1), classified as a member of neuronal guidance proteins involved in axon guidance and cell migration. NTN1 is expressed in multiple tissues, including the heart muscle, brain, adrenal gland, kidney, liver, lung, and others. At the cellular level, besides glial cells that display the highest levels, it is expressed in cardiomyocytes, epithelial, endothelial, and germ cells (available from proteinatlas.org). Recent studies also demonstrated its anti-inflammatory properties in atherosclerotic cardiovascular disease, inflammatory bowel disease, and acute lung injury (120–123). Hypoxia increases extracellular adenosine levels and elevates NTN1 levels through direct transcriptional induction of its gene expression (124). Intriguingly, NTN1 appears to potentiate signaling through the adenosine receptor A2BAR, inhibiting leukocyte migration and serving as a robust limiting factor during situations of oxygen scarcity and inflammation (55, 125). By binding to A2BAR, NTN1 increases intracellular cAMP levels, which mediates its effects (124). The precise mechanism underlying netrin-1’s augmentation of extracellular adenosine signaling remains elusive. Nonetheless, we can speculate that this interaction may represent a mechanism through which A2BAR is stimulated, even if adenosine levels decrease below the concentration required to activate A2BAR. This could, therefore, help prevent tissue injury following episodes of hypoxia and ischemia, and assist inflamed tissue in returning to its usual state of balance.
The balance between oxygen supply and demand is essential during physiological cellular activities. As previously referred, hypoxia stabilizes HIFs and affects the expression of many genes that are significant in ARDS (75). Studies have provided compelling evidence that HIF and adenosine signaling are tightly linked to modulating inflammatory responses during lung injury (69, 71, 126, 127). For this reason, researchers have made significant efforts to intervene in the hypoxia-adenosine axis to develop targeted treatments for ARDS (Figure 3). In this section, we summarize ongoing clinical trials targeting the hypoxia-adenosine axis to treat ARDS and other lung-related diseases (Table 2).
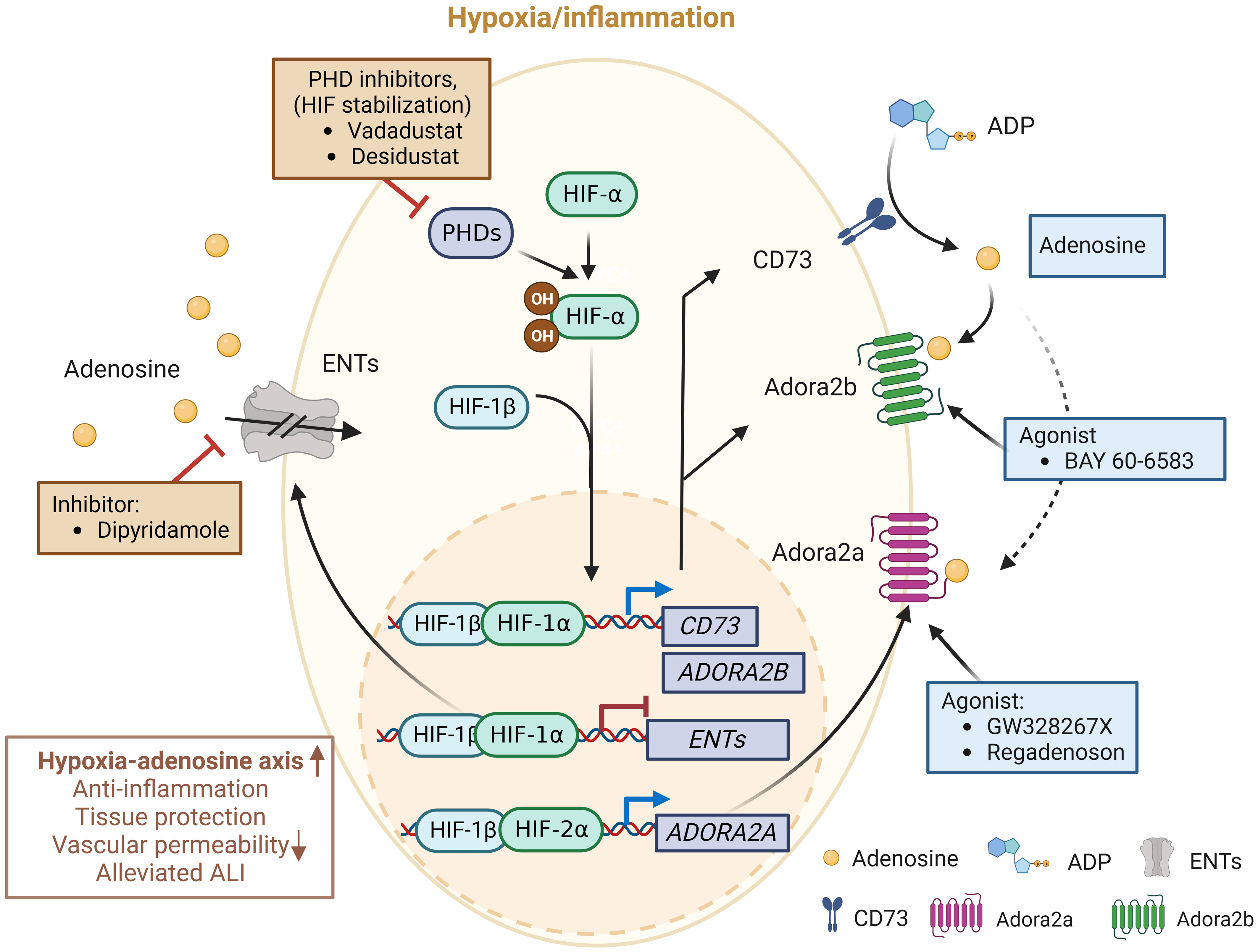
Figure 3 Molecules in clinical trials aimed at modulating the hypoxia-adenosine axis. Currently, interventions are being evaluated to regulate inflammatory responses and develop treatments for various lung diseases, including ARDS, by targeting different stages of the adenosine signaling pathway. Molecules in blue boxes represent stimulators, while molecules in red boxes are inhibitors. This image was created using BioRender.com.
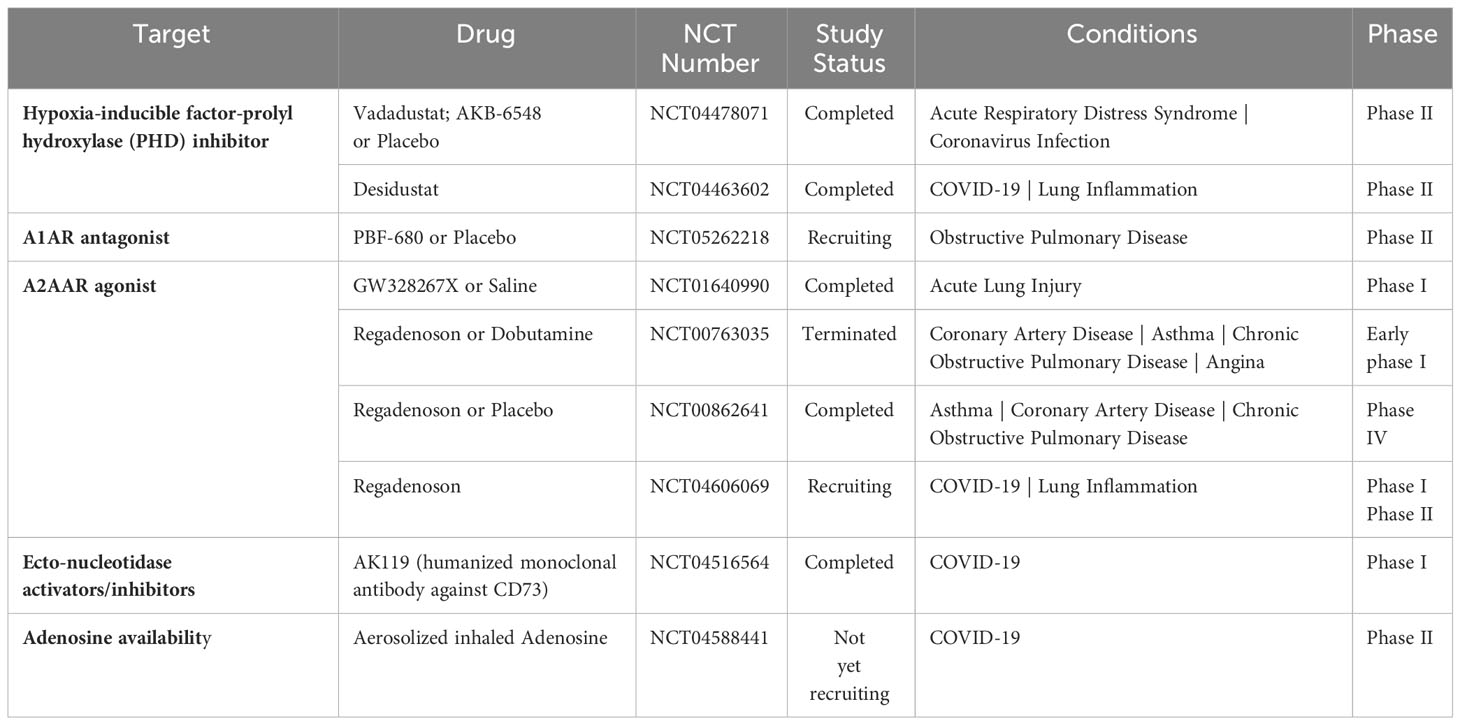
Table 2 Therapeutic approaches for lung disease targeting the hypoxia-adenosine axis.
HIF-PHD inhibitors (HIF-PHIs) are an encouraging new category of drugs that can impact the adenosine signaling pathway. These molecules are designed to inhibit HIF-PHDs, stabilizing HIFs under normoxic conditions (128). Among these inhibitors, Daprodustat (GSK-1278863) is administered orally and was the first to receive approval for treating anemia in patients with dialysis-dependent chronic kidney disease (CKD) by the Food and Drug Administration in the US. Other HIF-PHIs, including Roxadustat (FG-4592), Vadadustat (AKB-6548), Molidustat (BAY 85-3934), Enarodustat (JTZ-951), and Desidustat (ZYAN1), have already completed or are presently engaged in various clinical trials for their potential future applications (129). Several HIF-PHIs are approved for clinical usage in Japan and the European Union (129). Recently, HIF-PHIs have been recognized as potential novel treatment approaches for clinical trials involving COVID-19 patients (130). For example, Vadadustat (NCT04478071) and Desidustat (NCT04463602) are currently being evaluated in a phase II clinical trial with the expectation of preventing the onset of ARDS in COVID-19 patients who are hospitalized (131, 132). The pharmacological HIF activators could potentially be effective in enhancing HIF-driven extracellular adenosine signaling in COVID-19 patients, which may help provide tissue protection during adenosine-dependent immunosuppression (133, 134). Finally, conservative oxygen therapy could also serve as an approach to promote HIF stabilization during ARDS (135). However, the optimal oxygenation target in ARDS patients for best clinical outcomes will need to be determined in future clinical studies.
Evidence has shown that oxygenation can exacerbate inflammatory reactions, contributing to tissue damage and the development of acute respiratory distress (ARDS) (136). This response has been linked to reduced levels of endogenous adenosine, which usually activates A2AAR on immune-competent cells, mainly pulmonary invariant natural killer T (iNKT) cells, upon injury (137). Interestingly, similar effects could be partially replicated by using an A2AAR agonist. As a result, a proposal emerged suggesting that oxygen therapy should be administered combined with an A2AAR agonist (136). This idea aligns with the notion that A2AAR agonists could be valuable in preventing various forms of ischemia-reperfusion damage, a concept supported by substantial experimental evidence (138).
A2AAR has been extensively researched in recent decades because of its potential for different therapeutic uses. In particular, the expression of A2AAR is widespread in the lungs, including on bronchial epithelial cells and immune cells (139). In this regard, selective A2AAR agonists have been suggested to have a significant impact on airway inflammation and injury to lung tissue (140). Among the A2AAR agonists, GW328267X is a potent and selective agonist for A2AAR and a competitive antagonist at the adenosine A(3) receptor (141). However, two subsequent clinical trials were discontinued due to poor efficacy against allergic rhinitis (intranasal) (142) and asthma (inhaled) (143). The A2AAR agonist Regadenoson, among vasodilatory agents, is recognized for its effectiveness in promoting near-maximal coronary hyperemia without inducing bronchospasm in patients with coronary artery disease who have advanced lung disease (144). Certainly, over the past decade, there has been an increasing amount of evidence that supports the safety of regadenoson for patients with asthma and COPD, showing no variations in lung function parameters (145–147). Furthermore, following the outbreak of COVID-19, it was observed that a cytokine storm, a phenomenon where an excessive release of cytokines occurs due to an overreaction to the virus, could potentially be the primary cause of respiratory failure, which is seemingly the leading cause of death among severe COVID-19 patients (148). Hence, investigations conjectured that regulating the occurrence of the cytokine storm may assist in alleviating symptoms and aiding recovery in severe COVID-19 patients. A Phase I/II clinical trial enrolled participants to examine the hypothesis that regadenoson facilitates clinical improvement and enhances survival rates in COVID-19 patients when compared to patients who are undergoing a placebo-control (NCT04606069), the trial was recently completed but results are not available yet. Conversely, pentoxifylline, an A2AAR antagonist (149), has also shown anti-inflammatory effects (149–152), showcasing the complexity of therapeutic targeting of A2AAR in inflammatory diseases. Despite encouraging experimental data, the absence of A2BAR agonist testing in patients, particularly compounds like BAY-60-6583, remains a missed opportunity, with reasons for exclusion from clinical ARDS trials unclear.
Preclinical data indicates that extracellular CD73‐mediated adenosine production exhibits a protective role in non-infectious lung pathologies. However, it has been hypothesized that during SARS-CoV-2 infections, inhibition of CD73 may increase extracellular ATP, which may raise the production of IFN-β and trigger antiviral responses. A first-in-human (FIH), phase I, placebo-controlled, single ascending dose study to evaluate the safety, tolerability, pharmacokinetics, and immunogenicity of AK119, a humanized monoclonal antibody targeting the CD73, was conducted with the future aim of treating COVID-19 patients (NCT04516564) (153). Interestingly, alterations in the CD39/CD73 axis of T cells have been associated with SARS-CoV-2-induced ARDS. For example, higher frequencies of CD39+ memory T effector cells and lower frequencies of CD73+ T reg cells were found in severe compared to mild COVID-19 patients (154, 155), while its functional contribution remains unclear. Taken together, therapeutic targeting of CD73 for ARDS is still in its infancy.
Initial studies on the potential therapeutic effects of inhaled adenosine in patients in Italy and the US with severe COVID-19 demonstrated a significant improvement in oxygenation, reduced inflammation, and improved clinical outcomes in most treated patients (156, 157). While the study provides promising initial findings, it is based on fewer cases. An upcoming clinical trial, the ARCTIC trial, will use aerosolized inhaled adenosine treatment in patients with ARDS caused by COVID-19 (NCT04588441). This phase II study aims to evaluate adenosine’s effectiveness in mitigating the excessive lung inflammation observed in COVID-19, as evidenced by improvements in lung function, resolution of clinically significant lung function indicators, and reduction in systemic inflammation and coagulation markers. The results from this trial will be instrumental in determining the functional role of extracellular adenosine during COVID-19 ARDS.
The regulation of immune cell function in hypoxic and normoxic conditions is crucial, and hypoxia-inducible factors (HIFs) play a significant role in this process. HIFs are transcription factors that assist immune cells in adapting to hypoxic environments, such as infected or inflamed tissues. HIF-α stabilization can also occur during immunity and inflammation under normoxia, regulating metabolism and directly affecting immune genes. HIFs have been linked to numerous inflammatory diseases and immunosuppression in tumors. The differences in HIF-1α and HIF-2α functioning between cell types and environments are a challenging factor for therapeutic targeting. The connection between adenosine and signaling processes during hypoxic conditions is significant. Adenosine has the potential to impact oxygen availability and demand in diverse ways, both short-term and long-term, for instance, by promoting the development of new blood vessels (31). One intriguing aspect of adenosine’s function is its ability to regulate multiple facets of inflammation. It mitigates neutrophil-driven inflammation by curbing their recruitment, adhesion to endothelial cells, and reactive oxygen species production. It modulates cytokine production, notably inhibiting TNF-α and IL-12. Adenosine also suppresses the inflammatory activation of macrophages and fosters the recruitment of immature dendritic cells to sites of inflammation (the latter via A1AR and A3AR). Additionally, it influences regulatory T-cell function via A2AAR in hypoxic environments, contributing to the overall regulation of inflammation, especially within hypoxic conditions, triggering endothelial adenosine upregulation (53, 158, 159). Furthermore, the precise roles of adenosine receptors can vary depending on timing and the specific tissues involved. Their expression varies across tissues, leading to specific physiological responses. They have different affinities for adenosine, which modulate their responses under different conditions. These receptors also couple to distinct signal transduction pathways, further diversifying their impact. Prolonged exposure to agonists can trigger receptor desensitization, reducing its responsiveness (160). Adenosine receptor function is dynamic and context-dependent, and it is shaped by various factors such as hypoxia, ischemia, and circadian rhythms, elucidating their significance across different physiological and pathological states (161). Forthcoming studies in this matter should focus on aspects like a) exploring the correlation between hypoxia and adenosine, encompassing adenosine generation, its receptors, and processing, in acute and chronic lung ailments; b) investigating existing strategies to target the hypoxia-adenosine pathway therapeutically in various disease contexts; c) appraising the complexity of hypoxic-adenosinergic responses in the pathophysiology of ARDS and the development of lung fibrosis; d) studying the role of adenosine and HIFs in initiating vascular remodeling processes that contribute to the pathogenesis of ARDS; e) understanding cell-specific functions to develop targeted therapies for local lung delivery.
Results of investigations in the past decades highlight the fact that there is a reasonable rationale to evaluate the potential of adenosine-targeting therapies in ARDS. The large amount of ongoing clinical trials attests to this. A critical factor to consider when designing clinical trials is the clinical heterogeneity of ARDS. In about one-fifth of the patients that develop ARDS, the cause of lung injury is multifactorial and not due to a unique event. Moreover, mixed cases presenting direct and indirect components of lung injury can happen and should be analyzed carefully. Overall, the role of HIF in regulating inflammation and tissue repair in the lung is complex and context-dependent. Higher comprehension of the mechanisms governing hypoxia and adenosine signaling within the pulmonary context holds the potential to pave the way for innovative therapeutic approaches in the realm of ARDS.
Future research in ARDS should prioritize understanding its inherent heterogeneity to delineate distinct phenotypes based on sub-patient groups (e.g., septic ARDS, bacterial pneumonia, etc.) and facilitate targeted clinical trials (162). Optimizing established interventions including lung protective mechanical ventilation and prone positioning remains crucial (163). Establishing diagnostic protocols to identify treatable diseases within the ARDS diagnosis, evaluating the impact of secondary hits, and discerning patients with unresolved ARDS are pivotal (164). Given their higher mortality risk, it is imperative to explore novel therapeutic targets for older patients (165). Additionally, assessing the potential of therapeutic interventions developed for COVID-19 ARDS in other illness-induced ARDS and re-evaluating past research interventions, such as the prone position, are essential for refining ARDS management (166). Including different models in preclinical studies to verify findings will also be helpful. Collectively, these research avenues will enhance our comprehension of ARDS, optimize existing treatments, and unravel novel therapeutic avenues to improve patient outcomes.
KF: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. JK: Writing – original draft, Writing – review & editing. WR: Visualization, Writing – review & editing. TM: Writing – review & editing. HE: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. XY: Conceptualization, Funding acquisition, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. National Institute of Health Grants R01HL154720, R01HL154720-03S1, R01HL165748, R01HL169519, R01DK122796, T32GM135118 and Department of Defense Grant W81XWH2110032 to HE; sponsored contract through Akebia Therapeutics, Inc. to HE. National Institute of Health Grants R01HL155950, R01HL169519, and Parker B. Francis Fellowship to XY, sponsored contract through Akebia Therapeutics, Inc. to XY. Natural Science Foundation of Hunan Province Grant 018JJ3736, Young Talent Foundation of Hunan Province Grant 2021RC3034, and 2022 International Anesthesia Research Society Mentored Research Award to WR. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Figures are created with BioRender.com.
HE and XY received research funding through a contract between Akebia Therapeutics and UTHealth to support a clinical trial on the effect of vadadustat in hospitalized COVID-19 patients NCT04478071.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute respiratory distress syndrome: The Berlin definition. JAMA (2012) 307:2526–33. doi: 10.1001/jama.2012.5669
2. Ciara MS, Julie A MD. Bastarache, clinical and biological heterogeneity in ARDS: direct versus indirect lung injury. Clin Chest Med (2014) 35:639–53. doi: 10.1016/j.ccm.2014.08.004
3. Moldoveanu B, Otmishi P, Jani P, Walker J, Sarmiento X, Guardiola J, et al. Inflammatory mechanisms in the lung. J Inflamm Res (2008) 2:1–11.
4. Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med (2010) 61:105–19. doi: 10.1146/annurev.med.60.041807.123500
5. Williams GW, Berg NK, Reskallah A, Yuan X, Eltzschig HK. Acute respiratory distress syndrome: contemporary management and novel approaches during COVID-19. Anesthesiology (2021) 134:270–82. doi: 10.1097/ALN.0000000000003571
6. Helmerhorst HJF, Roos-Blom MJ, Van Westerloo DJ, De Jonge E. Association between arterial hyperoxia and outcome in subsets of critical illness: A systematic review, meta-analysis, and meta-regression of cohort studies. Crit Care Med (2015) 43:1508–19. doi: 10.1097/CCM.0000000000000998
7. Network A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for ALI and the ARDS. N Engl J Med (2000) 342:1301–8. doi: 10.1056/NEJM200005043421801
8. Guo L, Xie J, Huang Y, Pan C, Yang Y, Qiu H, et al. Higher PEEP improves outcomes in ARDS patients with clinically objective positive oxygenation response to PEEP: A systematic review and meta-analysis. BMC Anesthesiol (2018) 18:172. doi: 10.1186/s12871-018-0631-4
9. Guérin C, Reignier J, Richard J-C, Beuret P, Gacouin A, Boulain T, et al. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med (2013) 368:2159–68. doi: 10.1056/NEJMoa1214103
10. Scholten EL, Beitler JR, Prisk GK, Malhotra A. Treatment of ARDS with prone positioning. Chest (2017) 151:215–24. doi: 10.1016/j.chest.2016.06.032
11. The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome Clinical Trials Network, Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med (2006) 354:2564–75. doi: 10.1056/NEJMoa062200
12. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA (2016) 315:788–800. doi: 10.1001/jama.2016.0291
13. Ruan W, Eltzschig HK, Yuan X. Hypoxia-stabilized RIPK1 promotes cell death. Nat Cell Biol (2023) 25:921–2. doi: 10.1038/s41556-023-01176-y
14. Bartels K, Grenz A, Eltzschig HK. Hypoxia and inflammation are two sides of the same coin. Proc Natl Acad Sci USA (2013) 110:18351–2. doi: 10.1073/pnas.1318345110
15. Le TT, Berg NK, Harting MT, Li X, Eltzschig HK, Yuan X. Purinergic signaling in pulmonary inflammation. Front Immunol (2019) 10:1633. doi: 10.3389/fimmu.2019.01633
16. Li X, Berg NK, Mills T, Zhang K, Eltzschig HK, Yuan X. Adenosine at the interphase of hypoxia and inflammation in lung injury. Front Immunol (2020) 11:604944. doi: 10.3389/fimmu.2020.604944
17. Ruan W, Ma X, Bang IH, Liang Y, Muehlschlegel JD, Tsai KL, et al. The hypoxia-adenosine link during myocardial ischemia-reperfusion injury. Biomedicines (2022) 10:1939. doi: 10.3390/biomedicines10081939
18. Yuan X, Ferrari D, Mills T, Wang Y, Czopik A, Doursout MF, et al. Editorial: purinergic signaling and inflammation. Front Immunol (2021) 12:699069. doi: 10.3389/fimmu.2021.699069
19. Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med (2011) 364:656–65. doi: 10.1056/NEJMra0910283
20. Eltzschig HK. Prolonging cellular life after hypoxic death. N Engl J Med (2022) 387:2089–91. doi: 10.1056/NEJMcibr2210456
21. Eltzschig HK, Eckle T. Ischemia and reperfusion-from mechanism to translation. Nat Med (2011) 17:1391–401. doi: 10.1038/nm.2507
22. Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci USA (1991) 88:5680–4. doi: 10.1073/pnas.88.13.5680
23. Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell (2001) 107:43–54. doi: 10.1016/s0092-8674(01)00507-4
24. Yuan X, Lee JW, Bowser JL, Neudecker V, Sridhar S, Eltzschig HK. Targeting hypoxia signaling for perioperative organ injury. Anesth Analg (2018) 126:308–21. doi: 10.1213/ANE.0000000000002288
25. Appelhoffl RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem (2004) 279:38458–65. doi: 10.1074/jbc.M406026200
26. Karuppagounder SS, Ratan RR. Hypoxia-inducible factor prolyl hydroxylase inhibition: Robust new target or another big bust for stroke therapeutics. J Cereb Blood Flow Metab (2012) 32:1347–61. doi: 10.1038/jcbfm.2012.28
27. Capellier G, Barrot L, Winizewski H. Oxygenation target in acute respiratory distress syndrome. J Intensive Med (2023) 3:220–7. doi: 10.1016/j.jointm.2023.03.002
28. Kennel KB, Burmeister J, Schneider M, Taylor CT. The PHD1 oxygen sensor in health and disease. J Physiol (2018) 596:3899–913. doi: 10.1113/JP275327
29. Swain L, Wottawa M, Hillemann A, Beneke A, Odagiri H, Terada K, et al. Prolyl-4-hydroxylase domain 3 (PHD3) is a critical terminator for cell survival of macrophages under stress conditions. J Leukoc Biol (2014) 96:365–75. doi: 10.1189/jlb.2HI1013-533R
30. Suresh MV, Balijepalli S, Solanki S, Aktay S, Choudhary K, Shah YM, et al. Hypoxia-inducible factor 1α and its role in lung injury: adaptive or maladaptive. Inflammation (2023) 46:491–508. doi: 10.1007/s10753-022-01769-z
31. Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis: good and evil. Genes Cancer (2011) 2:1117–33. doi: 10.1177/1947601911423654
32. Taylor CT, Doherty G, Fallon PG, Cummins EP. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J Clin Invest (2016) 126:3716–24. doi: 10.1172/JCI84433
33. Nagamine Y, Tojo K, Yazawa T, Takaki S, Baba Y, Goto T, et al. Inhibition of prolyl hydroxylase attenuates fas ligand-induced Apoptosis and lung injury in mice. Am J Respir Cell Mol Biol (2016) 55:878–88. doi: 10.1165/rcmb.2015-0266OC
34. Tojo K, Tamada N, Nagamine Y, Yazawa T, Ota S, Goto T. Enhancement of glycolysis by inhibition of oxygen-sensing prolyl hydroxylases protects alveolar epithelial cells from acute lung injury. FASEB J (2018) 32:2258–68. doi: 10.1096/fj.201700888R
35. Huang LT, Chou HC, Chen CM. Roxadustat attenuates hyperoxia-induced lung injury by upregulating proangiogenic factors in newborn mice. Pediatr Neonatol (2021) 62:369–78. doi: 10.1016/j.pedneo.2021.03.012
36. Gong H, Rehman J, Tang H, Wary K, Mittal M, Chatturvedi P, et al. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest (2015) 125:652–64. doi: 10.1172/JCI77701
37. Deberardinis RJ, Thompson CB. Cellular metabolism and disease: What do metabolic outliers teach us? Cell (2012) 148:1132–44. doi: 10.1016/j.cell.2012.02.032
38. Eckle T, Brodsky K, Bonney M, Packard T, Han J, Borchers CH, et al. HIF1A reduces acute lung injury by optimizing carbohydrate metabolism in the alveolar epithelium. PloS Biol (2013) 11:e1001665. doi: 10.1371/journal.pbio.1001665
39. Vohwinkel CU, Coit EJ, Burns N, Elajaili H, Hernandez-Saavedra D, Yuan X, et al. Targeting alveolar-specific succinate dehydrogenase A attenuates pulmonary inflammation during acute lung injury. FASEB J (2021) 35:e21468. doi: 10.1096/fj.202002778R
40. Atallah R, Olschewski A, Heinemann A. Succinate at the crossroad of metabolism and angiogenesis: roles of SDH, HIF1α and SUCNR1. Biomedicines (2022) 10:3089. doi: 10.3390/biomedicines10123089
41. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell (2005) 7:77–85. doi: 10.1016/j.ccr.2004.11.022
42. Thiel M, Caldwell CC, Kreth S, Kuboki S, Chen P, Smith P, et al. Targeted deletion of HIF-1alpha gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PloS One (2007) 2:e853. doi: 10.1371/journal.pone.0000853
43. Caldwell CC, Kojima H, Lukashev D, Armstrong J, Farber M, Apasov SG, et al. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol (2001) 167:6140–9. doi: 10.4049/jimmunol.167.11.6140
44. Deussen A, Lloyd HGE, Schrader J. Contribution of S-adenosylhomocysteine to cardiac adenosine formation. J Mol Cell Cardiol (1989) 21:773–82. doi: 10.1016/0022-2828(89)90716-5
45. Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, et al. A CD38/CD203A/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. OncoImmunology (2013) 2:e26246. doi: 10.4161/onci.26246
46. Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ (2007) 14:1315–23. doi: 10.1038/sj.cdd.4402132
47. Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol (2020) 17:611–29. doi: 10.1038/s41571-020-0382-2
48. Eltzschig HK. Extracellular adenosine signaling in molecular medicine. J Mol Med (Berl) (2013) 91(2):141–6. doi: 10.1007/s00109-013-0999-z
49. Eckle T, Koeppen M, Eltzschig HK. Role of extracellular adenosine in acute lung injury. Physiol (Bethesda Md.) (2009) 24:298–306. doi: 10.1152/physiol.00022.2009
50. Robson SC, Sévigny J, Zimmermann H. The E-NTPDase family of ectonucleotidases: Structure function relationships and pathophysiological significance. Purinergic Signal (2006) 2:409–30. doi: 10.1007/s11302-006-9003-5
51. Zimmermann H. 5’-Nucleotidase: Molecular structure and functional aspects. Biochem J (1992) 285:345. doi: 10.1042/bj2850345
52. Donaldson SH, Picher M, Boucher RC. Secreted and cell-associated adenylate kinase and nucleoside diphosphokinase contribute to extracellular nucleotide metabolism on human airway surfaces. Am J Respir Cell Mol Biol (2002) 26:209–15. doi: 10.1165/ajrcmb.26.2.4650
53. Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov (2008) 7:759–70. doi: 10.1038/nrd2638
54. Fredholm BB, Ijzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev (2001) 53:527–52.
55. Yuan X, Mills T, Doursout MF, Evans SE, Vidal Melo MF, Eltzschig HK. Alternative adenosine Receptor activation: The netrin-Adora2b link. Front Pharmacol (2022) 13:944994. doi: 10.3389/fphar.2022.944994
56. Sun C, Wang B, Hao S. Adenosine-A2A receptor pathway in cancer immunotherapy. Front Immunol (2022) 13:837230. doi: 10.3389/fimmu.2022.837230
57. Bai Y, Zhang X, Zheng J, Liu Z, Yang Z, Zhang X. Overcoming high level adenosine-mediated immunosuppression by DZD2269, a potent and selective A2aR antagonist. J Exp Clin Cancer Res: CR (2022) 41:302. doi: 10.1186/s13046-022-02511-1
58. Antonioli L, Fornai M, Blandizzi C, Pacher P, Haskó G. Adenosine signaling and the immune system: When a lot could be too much. Immunol Lett (2019) 205:9–15. doi: 10.1016/j.imlet.2018.04.006
59. Vigano S, Alatzoglou D, Irving M, Ménétrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-Cell function. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00925
60. Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol (2014) 5:304. doi: 10.3389/fimmu.2014.00304
61. Kinsey GR, Huang L, Jaworska K, Khutsishvili K, Becker DA, Ye H, et al. Autocrine adenosine signaling promotes regulatory T cell-mediated renal protection. J Am Soc Nephrol: JASN (2012) 23:1528–37. doi: 10.1681/ASN.2012010070
62. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol (2011) 11:201–12. doi: 10.1038/nri2938
63. Gourdin N, Bossennec M, Rodriguez C, Vigano S, Machon C, Jandus C, et al. Autocrine adenosine regulates tumor polyfunctional CD73(+)CD4(+) effector T cells devoid of immune checkpoints. Cancer Res (2018) 78:3604–18. doi: 10.1158/0008-5472.CAN-17-2405
64. Löffler M, Morote-Garcia JC, Eltzschig SA, Coe IR, Eltzschig HK. Physiological roles of vascular nucleoside transporters. Arterioscler Thrombosis Vasc Biol (2007) 27:1004–13. doi: 10.1161/ATVBAHA.106.126714
65. Naes SM, Ab-Rahim S, Mazlan M, Abdul Rahman A. Equilibrative nucleoside transporter 2: properties and physiological roles. BioMed Res Int (2020) 2020:5197626. doi: 10.1155/2020/5197626
66. Bowser JL, Lee JW, Yuan X, Eltzschig HK. The hypoxia-adenosine link during inflammation. J Appl Physiol (1985) (2017) 123:1303–20. doi: 10.1152/japplphysiol.00101.2017
67. Eckle T, Hartmann K, Bonney S, Reithel S, Mittelbronn M, Walker LA, et al. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat Med (2012) 18:774–82. doi: 10.1038/nm.2728
68. Bowser JL, Phan LH, Eltzschig HK. The hypoxia-adenosine link during intestinal inflammation. J Immunol (2018) 200:897–907. doi: 10.4049/jimmunol.1701414
69. Eckle T, Kewley EM, Brodsky KS, Tak E, Bonney S, Gobel M, et al. Identification of hypoxia-inducible factor HIF-1A as transcriptional regulator of the A2B adenosine receptor during acute lung injury. J Immunol (2014) 192:1249–56. doi: 10.4049/jimmunol.1100593
70. Hu C-J, Wang L-Y, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α in hypoxic gene regulation. Mol Cell Biol (2003) 23:9361–74. doi: 10.1128/MCB.23.24.9361-9374.2003
71. Ahmad A, Ahmad S, Glover L, Miller SM, Shannon JM, Guo X, et al. Adenosine A2A receptor is a unique angiogenic target of HIF-2α in pulmonary endothelial cells. Proc Natl Acad Sci USA (2009) 106:10684–9. doi: 10.1073/pnas.0901326106
72. Cheu JWS, Chiu DKC, Kwan KKL, Yang C, Yuen VWH, Goh CC, et al. Hypoxia-inducible factor orchestrates adenosine metabolism to promote liver cancer development. Sci Adv (2023) 9:eade5111. doi: 10.1126/sciadv.ade5111
73. Evans CE. Hypoxia-inducible factor signaling in inflammatory lung injury and repair. Cells (2022) 11:183. doi: 10.3390/cells11020183
74. McClendon J, Jansing NL, Redente EF, Gandjeva A, Ito Y, Colgan SP, et al. Hypoxia-inducible factor 1α Signaling promotes repair of the alveolar epithelium after acute lung injury. Am J Pathol (2017) 187:1772–86. doi: 10.1016/j.ajpath.2017.04.012
75. Mirchandani AS, Jenkins SJ, Bain CC, Sanchez-Garcia MA, Lawson H, Coelho P, et al. Hypoxia shapes the immune landscape in lung injury and promotes the persistence of inflammation. Nat Immunol (2022) 23:927–39. doi: 10.1038/s41590-022-01216-z
76. Poth JM, Brodsky K, Ehrentraut H, Grenz A, Eltzschig HK. Transcriptional control of adenosine signaling by hypoxia-inducible transcription factors during ischemic or inflammatory disease. J Mol Med (2013) 91:183–93. doi: 10.1007/s00109-012-0988-7
77. Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP release by inflammatory cells. Int J Mol Sci (2018) 19:1222. doi: 10.3390/ijms19041222
78. Kirby BS, Sparks MA, Lazarowski ER, Lopez Domowicz DA, Zhu H, McMahon TJ. Pannexin 1 channels control the hemodynamic response to hypoxia by regulating O(2)-sensitive extracellular ATP in blood. Am J Physiol Heart Circulatory Physiol (2021) 320:H1055–h1065. doi: 10.1152/ajpheart.00651.2020
79. Fitz JG. Regulation of cellular ATP release. Trans Am Clin Climatological Assoc (2007) 118:199–208.
80. Eltzschig HK, Weissmüller T, Mager A, Eckle T. Nucleotide metabolism and cell-cell interactions. Cell-Cell Interactions: Methods Protoc (2006) 341:73–87. doi: 10.1385/1-59745-113-4:73
81. Gombault A, Baron L, Couillin I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol (2012) 3:414. doi: 10.3389/fimmu.2012.00414
82. Eltzschig HK, Kö hler D, Eckle T, Kong T, Robson SC, Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood (2009) 113:224–32. doi: 10.1182/blood-2008-06-165746
83. Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: Role of ectonucleotidases and adenosine A 2B receptors. J Exp Med (2003) 198:783–96. doi: 10.1084/jem.20030891
84. Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, et al. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med (2004) 200:1395–405. doi: 10.1084/jem.20040915
85. Koizume S, Miyagi Y. Diverse mechanisms of sp1-dependent transcriptional regulation potentially involved in the adaptive response of cancer cells to oxygen-deficient conditions. Cancers (2015) 8:2. doi: 10.3390/cancers8010002
86. Kimura K, Jackson TLB, Huang RCC. Interaction and collaboration of SP1, HIF-1, and MYC in regulating the expression of cancer-related genes to further enhance anticancer drug development. Curr Issues Mol Biol (2023) 45:9262–83. doi: 10.3390/cimb45110580
87. Jarvis LB, Rainbow DB, Coppard V, Howlett SK, Georgieva Z, Davies JL, et al. Therapeutically expanded human regulatory T-cells are super-suppressive due to HIF1A induced expression of CD73. Commun Biol (2021) 4:1186. doi: 10.1038/s42003-021-02721-x
88. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol (2006) 177:6780–6. doi: 10.4049/jimmunol.177.10.6780
89. Eckle T, Füllbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, et al. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol (2007) 178:8127–37. doi: 10.4049/jimmunol.178.12.8127
90. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Trans Med (2015) 7:277ra30. doi: 10.1126/scitranslmed.aaa1260
91. St Hilaire C, Carroll SH, Chen H, Ravid K. Mechanisms of induction of adenosine receptor genes and its functional significance. J Cell Physiol (2009) 218:35–44. doi: 10.1002/jcp.21579
92. Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J (2006) 20:2242–50. doi: 10.1096/fj.06-6419com
93. Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood (2008) 111:2024–35. doi: 10.1182/blood-2007-10-117044
94. Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest (2008) 118:3301–15. doi: 10.1172/JCI34203
95. Schingnitz U, Hartmann K, MacManus CF, Eckle T, Zug S, Colgan SP, et al. Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol (2010) 184:5271–9. doi: 10.4049/jimmunol.0903035
96. Hoegl S, Brodsky KS, Blackburn MR, Karmouty-Quintana H, Zwissler B, Eltzschig HK. Alveolar epithelial A2B adenosine receptors in pulmonary protection during acute lung injury. J Immunol (2015) 195:1815–24. doi: 10.4049/jimmunol.1401957
97. Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature (2001) 414:916–20. doi: 10.1038/414916a
98. Reece TB, Ellman PI, Maxey TS, Crosby IK, Warren PS, Chong TW, et al. Adenosine A2A receptor activation reduces inflammation and preserves pulmonary function in an in vivo model of lung transplantation. J Thorac Cardiovasc Surg (2005) 129:1137–43. doi: 10.1016/j.jtcvs.2004.11.042
99. Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J Immunol (2007) 179:1254–63. doi: 10.4049/jimmunol.179.2.1254
100. Kreckler LM, Gizewski E, Wan TC, Auchampach JA. Adenosine suppresses lipopolysaccharide-induced tumor necrosis factor-alpha production by murine macrophages through a protein kinase A- and exchange protein activated by cAMP-independent signaling pathway. J Pharmacol Exp Ther (2009) 331:1051–61. doi: 10.1124/jpet.109.157651
101. Chen H, Yang D, Carroll SH, Eltzschig HK, Ravid K. Activation of the macrophage A2b adenosine receptor regulates tumor necrosis factor-alpha levels following vascular injury. Exp Hematol (2009) 37:533–8. doi: 10.1016/j.exphem.2009.02.001
102. Ren X, Manzanares LD, Piccolo EB, Urbanczyk JM, Sullivan DP, Yalom LK, et al. Macrophage-endothelial cell crosstalk orchestrates neutrophil recruitment in inflamed mucosa. J Clin Invest (2023) 133:e170733. doi: 10.1172/JCI170733
103. He X, Hu JL, Li J, Zhao L, Zhang Y, Zeng YJ, et al. A feedback loop in PPARγ-adenosine A2A receptor signaling inhibits inflammation and attenuates lung damages in a mouse model of LPS-induced acute lung injury. Cell Signal (2013) 25:1913–23. doi: 10.1016/j.cellsig.2013.05.024
104. Impellizzeri D, Di Paola R, Esposito E, Mazzon E, Paterniti I, Melani A, et al. CGS 21680, an agonist of the adenosine (A 2A) receptor, decreases acute lung inflammation. Eur J Pharmacol (2011) 668:305–16. doi: 10.1016/j.ejphar.2011.06.049
105. Eltzschig HK, Abdulla P, Hoffman E, Hamilton KE, Daniels D, Schönfeld C, et al. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med (2005) 202:1493–505. doi: 10.1084/jem.20050177
106. Morote-Garcia JC, Rosenberger P, Nivillac NMI, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology (2009) 136:607–18. doi: 10.1053/j.gastro.2008.10.037
107. Eckle T, Hughes K, Ehrentraut H, Brodsky KS, Rosenberger P, Choi DS, et al. Crosstalk between the equilibrative nucleoside transporter ENT2 and alveolar Adora2b adenosine receptors dampens acute lung injury. FASEB J (2013) 27:3078–89. doi: 10.1096/fj.13-228551
108. Wang W, Chen NY, Ren D, Davies J, Philip K, Eltzschig HK, et al. Enhancing extracellular adenosine levels restores barrier function in acute lung injury through expression of focal adhesion proteins. Front Mol Biosci (2021) 8. doi: 10.3389/fmolb.2021.636678
109. Aherne CM, Collins CB, Rapp CR, Olli KE, Perrenoud L, Jedlicka P, et al. Coordination of ENT2-dependent adenosine transport and signaling dampens mucosal inflammation. JCI Insight (2018) 3:e121521. doi: 10.1172/jci.insight.121521
110. Ruan W, Li J, Choi S, Ma X, Liang Y, Nair R, et al. Targeting myocardial equilibrative nucleoside transporter ENT1 provides cardioprotection by enhancing myeloid Adora2b signaling. JCI Insight (2023) 8:e166011. doi: 10.1172/jci.insight.166011
111. Song A, Zhang Y, Han L, Yegutkin GG, Liu H, Sun K, et al. Erythrocytes retain hypoxic adenosine response for faster acclimatization upon re-ascent. Nat Commun (2017) 8:14108. doi: 10.1038/ncomms14108
112. Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-l-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood (2008) 111:5571–80. doi: 10.1182/blood-2007-11-126763
113. Ju C, Wang M, Tak E, Kim B, Emontzpohl C, Yang Y, et al. Hypoxia-inducible factor-1alpha-dependent induction of miR122 enhances hepatic ischemia tolerance. J Clin Invest (2021) 131:e140300. doi: 10.1172/JCI140300
114. Lee TJ, Yuan X, Kerr K, Yoo JY, Kim DH, Kaur B, et al. Strategies to modulate microRNA functions for the treatment of cancer or organ injury. Pharmacol Rev (2020) 72:639–67. doi: 10.1124/pr.119.019026
115. Lee LK, Medzikovic L, Eghbali M, Eltzschig HK, Yuan X. The role of microRNAs in acute respiratory distress syndrome and sepsis, from targets to therapies: A narrative review. Anesth Analg (2020) 131:1471–84. doi: 10.1213/ANE.0000000000005146
116. Nallamshetty S, Chan SY, Loscalzo J. Hypoxia: A master regulator of microRNA biogenesis and activity. Free Radical Biol Med (2013) 64:20–30. doi: 10.1016/j.freeradbiomed.2013.05.022
117. Wang N, Xiang X, Chen K, Liu P, Zhu A. Targeting of NT5E by miR-30b and miR-340 attenuates proliferation, invasion and migration of gallbladder carcinoma. Biochimie (2018) 146:56–67. doi: 10.1016/j.biochi.2017.10.027
118. Fulzele S, El-Sherbini A, Ahmad S, Sangani R, Matragoon S, El-Remessy A, et al. MicroRNA-146b-3p regulates retinal inflammation by suppressing adenosine deaminase-2 in diabetes. BioMed Res Int (2015) 2015:846501. doi: 10.1155/2015/846501
119. Bruning U, Cerone L, Neufeld Z, Fitzpatrick SF, Cheong A, Scholz CC, et al. MicroRNA-155 promotes resolution of hypoxia-inducible factor 1alpha activity during prolonged hypoxia. Mol Cell Biol (2011) 31:4087–96. doi: 10.1128/MCB.01276-10
120. Aherne CM, Collins CB, Masterson JC, Tizzano M, Boyle TA, Westrich JA, et al. Neuronal guidance molecule netrin-1 attenuates inflammatory cell trafficking during acute experimental colitis. Gut (2012) 61:695–705. doi: 10.1136/gutjnl-2011-300012
121. Grenz A, Clambey E, Eltzschig HK. Hypoxia signaling during intestinal ischemia and inflammation. Curr Opin Crit Care (2012) 18:178–85. doi: 10.1097/MCC.0b013e3283514bd0
122. Mutz C, Mirakaj V, Vagts DA, Westermann P, Waibler K, König K, et al. The neuronal guidance protein netrin-1 reduces alveolar inflammation in a porcine model of acute lung injury. Crit Care (2010) 14:R189. doi: 10.1186/cc9301
123. Schlegel M, Sharma M, Brown EJ, Newman AAC, Cyr Y, Afonso MS, et al. Silencing myeloid netrin-1 induces inflammation resolution and plaque regression. Circ Res (2021) 129:530–46. doi: 10.1161/CIRCRESAHA.121.319313
124. Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, et al. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol (2009) 10:195–202. doi: 10.1038/ni.1683
125. Berg NK, Li J, Kim B, Mills T, Pei G, Zhao Z, et al. Hypoxia-inducible factor-dependent induction of myeloid-derived netrin-1 attenuates natural killer cell infiltration during endotoxin-induced lung injury. FASEB J (2021) 35:e21334. doi: 10.1096/fj.202002407R
126. Philip K, Mills TW, Davies J, Chen NY, Karmouty-Quintana H, Luo F, et al. HIF1A up-regulates the ADORA2B receptor on alternatively activated macrophages and contributes to pulmonary fibrosis. FASEB J (2017) 31:4745–58. doi: 10.1096/fj.201700219R
127. Zhong H, Belardinelli L, Maa T, Zeng D. Synergy between A2B adenosine receptors and hypoxia in activating human lung fibroblasts. Am J Respir Cell Mol Biol (2005) 32:2–8. doi: 10.1165/rcmb.2004-0103OC
128. Kim SY, Yang EG. Recent advances in developing inhibitors for hypoxia-inducible factor prolyl hydroxylases and their therapeutic implications. Molecules (2015) 20:20551–68. doi: 10.3390/molecules201119717
129. Li J, Haase VH, Hao CM. Updates on hypoxia-inducible factor prolyl hydroxylase inhibitors in the treatment of renal anemia. Kidney Dis (Basel) (2023) 9:1–11. doi: 10.1159/000527835
130. Del Vecchio L, Locatelli F. Hypoxia response and acute lung and kidney injury: Possible implications for therapy of COVID-19. Clin Kidney J (2020) 13:494–9. doi: 10.1093/ckj/sfaa149
131. NCT04463602, Desidustat in the Management of COVID-19 Patients (2020). Available at: https://clinicaltrials.gov/show/NCT04463602.
132. NCT04478071, Vadadustat for the Prevention and Treatment of Acute Respiratory Distress Syndrome (ARDS) in Hospitalized Patients With Coronavirus Disease 2019 (COVID-19) (2020). Available at: https://clinicaltrials.gov/show/NCT04478071.
133. Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: Coordination by extracellular nucleotide metabolism. Blood (2004) 104:3986–92. doi: 10.1182/blood-2004-06-2066
134. Grenz A, Zhang H, Weingart J, Von Wietersheim S, Eckle T, Schnermann J, et al. Lack of effect of extracellular adenosine generation and signaling on renal erythropoietin secretion during hypoxia. Am J Physiol - Renal Physiol (2007) 293:F1501–11. doi: 10.1152/ajprenal.00243.2007
135. Liang Y, Ruan W, Jiang Y, Smalling R, Yuan X, Eltzschig HK. Interplay of hypoxia-inducible factors and oxygen therapy in cardiovascular medicine. Nat Rev Cardiol (2023) 20:723–37. doi: 10.1038/s41569-023-00886-y
136. Thiel M, Chouker A, Ohta A, Jackson E, Caldwell C, Smith P, et al. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PloS Biol (2005) 3:e174. doi: 10.1371/journal.pbio.0030174
137. Nowak-Machen M, Schmelzle M, Hanidziar D, Junger W, Exley M, Otterbein L, et al. Pulmonary natural killer T cells play an essential role in mediating hyperoxic acute lung injury. Am J Respir Cell Mol Biol (2013) 48:601–9. doi: 10.1165/rcmb.2012-0180OC
138. Lappas CM, Sullivan GW, Linden J. Adenosine A2A agonists in development for the treatment of inflammation. Expert Opin Investig Drugs (2005) 14:797–806. doi: 10.1517/13543784.14.7.797
139. Haskó G, Pacher P. A2A receptors in inflammation and injury: lessons learned from transgenic animals. J Leukoc Biol (2008) 83:447–55. doi: 10.1189/jlb.0607359
140. Spicuzza L, Di Maria G, Polosa R. Adenosine in the airways: Implications and applications. Eur J Pharmacol (2006) 533:77–88. doi: 10.1016/j.ejphar.2005.12.056
141. Bevan N, Butchers PR, Cousins R, Coates J, Edgar EV, Morrison V, et al. Pharmacological characterisation and inhibitory effects of (2R,3R,4S,5R)-2-(6-amino-2-{[(1S)-2-hydroxy-1-(phenylmethyl)ethyl]amino} -9H-purin-9-yl)-5-(2-ethyl-2H-tetrazol-5-yl)tetrahydro-3,4-furandiol, a novel ligand that demonstrates both adenosine A2A receptor agonist and adenosine A3 receptor antagonist activity. Eur J Pharmacol (2007) 564:219–25. doi: 10.1016/j.ejphar.2007.01.094
142. Rimmer J, Peake HL, Santos CMC, Lean M, Bardin P, Robson R, et al. Targeting adenosine receptors in the treatment of allergic rhinitis: A randomized, double-blind, placebo-controlled study. Clin Exp Allergy (2007) 37:8–14. doi: 10.1111/j.1365-2222.2006.02546.x
143. Luijk B, Van Den Berge M, Kerstjens HAM, Postma DS, Cass L, Sabin A, et al. Effect of an inhaled adenosine A2A agonist on the allergen-induced late asthmatic response. Allergy: Eur J Allergy Clin Immunol (2008) 63:75–80. doi: 10.1111/j.1398-9995.2007.01557.x
144. Henzlova MJ, Duvall WL, Einstein AJ, Travin MI, Verberne HJ. ASNC imaging guidelines for SPECT nuclear cardiology procedures: Stress, protocols, and tracers. J Nucl Cardiol (2016) 23:606–39. doi: 10.1007/s12350-015-0387-x
145. Leaker BR, O’Connor B, Hansel TT, Barnes PJ, Meng L, Mathur VS, et al. Safety of regadenoson, an adenosine A2A receptor agonist for myocardial perfusion imaging, in mild asthma and moderate asthma patients: A randomized, double-blind, placebo-controlled trial. J Nucl Cardiol (2008) 15:329–36. doi: 10.1016/j.nuclcard.2008.02.009
146. Prenner BM, Bukofzer S, Behm S, Feaheny K, McNutt BE. A randomized, double-blind, placebo-controlled study assessing the safety and tolerability of regadenoson in subjects with asthma or chronic obstructive pulmonary disease. J Nucl Cardiol (2012) 19:681–92. doi: 10.1007/s12350-012-9547-4
147. Thomas GS, Tammelin BR, Schiffman GL, Marquez R, Rice DL, Milikien D, et al. Safety of regadenoson, a selective adenosine A2A agonist, in patients with chronic obstructive pulmonary disease: A randomized, double-blind, placebo-controlled trial (RegCOPD trial). J Nucl Cardiol (2008) 15:319–28. doi: 10.1016/j.nuclcard.2008.02.013
148. Montazersaheb S, Hosseiniyan Khatibi SM, Hejazi MS, Tarhriz V, Farjami A, Ghasemian Sorbeni F, et al. COVID-19 infection: an overview on cytokine storm and related interventions. Virol J (2022) 19:1–15. doi: 10.1186/s12985-022-01814-1
149. Konrad FM, Neudeck G, Vollmer I, Ngamsri KC, Thiel M, Reutershan J. Protective effects of pentoxifylline in pulmonary inflammation are adenosine receptor A2A dependent. FASEB J (2013) 27:3524–35. doi: 10.1096/fj.13-228122
150. Abdel-Salam OM, Baiuomy AR, El-Shenawy SM, Arbid MS. The anti-inflammatory effects of the phosphodiesterase inhibitor pentoxifylline in the rat. Pharmacol Res (2003) 47:331–40. doi: 10.1016/S1043-6618(03)00002-1
151. Doherty GM, Jensen JC, Alexander HR, Buresh CM, Norton JA. Pentoxifylline suppression of tumor necrosis factor gene transcription. Surgery (1991) 110:192–8.
152. Han J, Thompson P, Beutler B. Dexamethasone and pentoxifylline inhibit endotoxin-induced cachectin/tumor necrosis factor synthesis at separate points in the signaling pathway. J Exp Med (1990) 172:391–4. doi: 10.1084/jem.172.1.391
153. Zhong T, Huang Z, Pang X, Chen N, Kwek K, Wynne C, et al. 476 AK119, a humanized anti-CD73 monoclonal antibody, as Immunotherapy for COVID-19. BMJ Specialist Journals (2020) 8:A292. doi: 10.1136/jitc-2020-SITC2020.0476
154. Díaz-García E, García-Tovar S, Alfaro E, Zamarrón E, Mangas A, Galera R, et al. Role of CD39 in COVID-19 severity: dysregulation of purinergic signaling and thromboinflammation. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.847894
155. Dorneles GP, Teixeira PC, da Silva IM, Schipper LL, Santana Filho PC, Rodrigues Junior LC, et al. Alterations in CD39/CD73 axis of T cells associated with COVID-19 severity. J Cell Physiol (2022) 237:3394–407. doi: 10.1002/jcp.30805
156. Correale P, Caracciolo M, Bilotta F, Conte M, Cuzzola M, Falcone C, et al. Therapeutic effects of adenosine in high flow 21% oxygen aereosol in patients with Covid19-pneumonia. PloS One (2020) 15:e0239692. doi: 10.1371/journal.pone.0239692
157. Spiess BD, Sitkovsky M, Correale P, Gravenstein N, Garvan C, Morey TE, et al. Case report: can inhaled adenosine attenuate COVID-19? Front Pharmacol (2021) 12:676577. doi: 10.3389/fphar.2021.676577
158. Haskó G, Cronstein B. Regulation of inflammation by adenosine. Front Immunol (2013) 4:85. doi: 10.3389/fimmu.2013.00085
159. Linden J. New insights into the regulation of inflammation by adenosine. J Clin Invest (2006) 116:1835–7. doi: 10.1172/JCI29125
160. van Galen PJ, Stiles GL, Michaels G, Jacobson KA. Adenosine A1 and A2 receptors: structure–function relationships. Medicinal Res Rev (1992) 12:423–71. doi: 10.1002/med.2610120502
161. Jagannath A, Varga N, Dallmann R, Rando G, Gosselin P, Ebrahimjee F, et al. Adenosine integrates light and sleep signalling for the regulation of circadian timing in mice. Nat Commun (2021) 12:2113. doi: 10.1038/s41467-021-22179-z
162. Peck TJ, Hibbert KA. Recent advances in the understanding and management of ARDS. F1000Research (2019) 8:Rev–1959. doi: 10.12688/f1000research.20411.1
163. Griffiths MJD, McAuley DF, Perkins GD, Barrett N, Blackwood B, Boyle A, et al. Guidelines on the management of acute respiratory distress syndrome. BMJ Open Respir Res (2019) 6:e000420. doi: 10.1136/bmjresp-2019-000420
164. Bos LDJ, Artigas A, Constantin JM, Hagens LA, Heijnen N, Laffey JG, et al. Precision medicine in acute respiratory distress syndrome: workshop report and recommendations for future research. Eur Respir Rev (2021) 30:200317. doi: 10.1183/16000617.0317-2020
165. Huang X, Zhang X, Machireddy N, Evans CE, Trewartha SD, Hu G, et al. Endothelial FoxM1 reactivates aging-impaired endothelial regeneration for vascular repair and resolution of inflammatory lung injury. Sci Trans Med (2023) 15:eabm5755. doi: 10.1126/scitranslmed.abm5755
Keywords: hypoxia, adenosine, ARDS, inflammation, lung injury
Citation: Figarella K, Kim J, Ruan W, Mills T, Eltzschig HK and Yuan X (2024) Hypoxia-adenosine axis as therapeutic targets for acute respiratory distress syndrome. Front. Immunol. 15:1328565. doi: 10.3389/fimmu.2024.1328565
Received: 30 October 2023; Accepted: 03 January 2024;
Published: 19 January 2024.
Edited by:
Lei Xi, Virginia Commonwealth University, United StatesReviewed by:
Erika M. Palmieri, National Cancer Institute at Frederick (NIH), United StatesCopyright © 2024 Figarella, Kim, Ruan, Mills, Eltzschig and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyi Yuan, eGlhb3lpLnl1YW5AdXRoLnRtYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.