
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 29 February 2024
Sec. Alloimmunity and Transplantation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1315283
 Beatrice Claudia Cianciotti1
Beatrice Claudia Cianciotti1 Zulma Irene Magnani1†
Zulma Irene Magnani1† Alessia Ugolini1†
Alessia Ugolini1† Barbara Camisa1,2
Barbara Camisa1,2 Ivan Merelli3
Ivan Merelli3 Valentina Vavassori4
Valentina Vavassori4 Alessia Potenza1Antonio Imparato1Francesco Manfredi1Danilo Abbati1
Alessia Potenza1Antonio Imparato1Francesco Manfredi1Danilo Abbati1 Laura Perani5
Laura Perani5 Antonello Spinelli5Eric Shifrut6,7,8
Antonello Spinelli5Eric Shifrut6,7,8 Fabio Ciceri9,10
Fabio Ciceri9,10 Luca Vago10,11
Luca Vago10,11 Raffaella Di Micco12Luigi Naldini4,10Pietro Genovese4,13
Raffaella Di Micco12Luigi Naldini4,10Pietro Genovese4,13 Eliana Ruggiero1*†
Eliana Ruggiero1*† Chiara Bonini1,10*†
Chiara Bonini1,10*†Background: In adoptive T cell therapy, the long term therapeutic benefits in patients treated with engineered tumor specific T cells are limited by the lack of long term persistence of the infused cellular products and by the immunosuppressive mechanisms active in the tumor microenvironment. Exhausted T cells infiltrating the tumor are characterized by loss of effector functions triggered by multiple inhibitory receptors (IRs). In patients, IR blockade reverts T cell exhaustion but has low selectivity, potentially unleashing autoreactive clones and resulting in clinical autoimmune side effects. Furthermore, loss of long term protective immunity in cell therapy has been ascribed to the effector memory phenotype of the infused cells.
Methods: We simultaneously redirected T cell specificity towards the NY-ESO-1 antigen via TCR gene editing (TCRED) and permanently disrupted LAG3, TIM-3 or 2B4 genes (IRKO) via CRISPR/Cas9 in a protocol to expand early differentiated long-living memory stem T cells. The effector functions of the TCRED-IRKO and IR competent (TCRED-IRCOMP) cells were tested in short-term co-culture assays and under a chronic stimulation setting in vitro. Finally, the therapeutic efficacy of the developed cellular products were evaluated in multiple myeloma xenograft models.
Results: We show that upon chronic stimulation, TCRED-IRKO cells are superior to TCRED-IRCOMP cells in resisting functional exhaustion through different mechanisms and efficiently eliminate cancer cells upon tumor re-challenge in vivo. Our data indicate that TIM-3 and 2B4-disruption preserve T-cell degranulation capacity, while LAG-3 disruption prevents the upregulation of additional inhibitory receptors in T cells.
Conclusion: These results highlight that TIM-3, LAG-3, and 2B4 disruptions increase the therapeutic benefit of tumor specific cellular products and suggest distinct, non-redundant roles for IRs in anti-tumor responses.
● T cell exhaustion is a dysfunctional status dampening T cell effector functions and is driven by multiple inhibitory receptors. In this study we show that the disruption of three major inhibitory receptors (TIM-3 LAG-3 and 2B4) increases the antitumor activity of antigen-specific T cells. Our study will be pivotal in designing new tailored and more effective cancer immunotherapy approaches.
Adoptive T cell therapy (ACT) represents one of the most recent advances in biomedical research, and largely relies on the ability of T lymphocytes to recognize and kill their targets and to persist as memory cells. The transfer of genes encoding for high-affinity tumor-reactive T cell receptor (TCR) or chimeric antigen receptors (CAR) efficiently redirect T cell specificity towards tumor-associated and tumor-specific antigens (1–8). These living drugs display a number of distinctive qualities, compared to conventional chemical compounds, including the ability to respond to their target through expansion and contraction cycles and the capacity to persist and mediate long-term clinical effects (9). The efficacy of ACT largely relies on high-avidity cellular products. The application of genome editing technologies to T cells opened up a wide range of therapeutic opportunities for ACT. For instance, TCR gene editing allows to completely redirect T cell specificity, thus maximizing the tumor-specific TCR expression and the overall avidity and specificity of the cellular products (10, 11).
Several preclinical and clinical observations indicate that the ability of ACT to mediate prolonged clinical responses is associated with the long-term persistence of the infused T-cell products (12). Accordingly, manufacturing protocols favoring the generation of T-cell products endowed with an early differentiation, central memory T cells/stem cell memory T cells (TCM/TSCM) functional phenotype display superior anti-tumor activity, proliferation capacity, and long-term survival when compared to effectors (13). However, in cancer, chronic antigen stimulation and signals from the immunosuppressive tumor microenvironment cooperate in inducing T cell exhaustion, a dysfunctional differentiation status, that reduces and, in some cases, nullifies the therapeutic effect of ACT. While from an evolutionary perspective T cell exhaustion is beneficial to dampen excessive tissue damage, in cancer it represents a major immune evasion mechanism. Exhausted T cells display sustained upregulation of inhibitory receptors (IRs), including PD-1, CTLA-4, LAG-3, TIM-3, 2B4, CD39, CD160, BTLA and TIGIT, in the absence of co-stimulatory receptors. IR triggering results in a progressive and hierarchical loss of effector functions, desensitization of TCR, metabolic deregulation, altered expression of transcription factors required for effector functions and failure to acquire a memory phenotype (14–16).
Immune checkpoint blockade (ICB) reinvigorates exhausted cells and reactivates protective tumor-specific T cell responses (17, 18) and, although the clinical use of ICBs has shown considerable benefits, some drawbacks limit its broad applicability. First, the efficacy of ICBs is restricted to immunogenic tumors and even within those cancer types, only 20% of patients achieves durable responses as a substantial fraction of treated patients shows lack of initial responses (i.e. primary resistance) or develops failure of response overtime (i.e. acquired resistance) (19). Several mechanisms have been ascribed to ICB failure. Among them, the upregulation of alternative immune checkpoints by TILs upon ICB therapies in hot-immunogenic tumors suggests a non-redundant role of IRs (20, 21). Furthermore, the widespread action of ICBs on the entire T-cell repertoire is responsible for several immune-related adverse events (irAEs), reported in 90% of treated patients, with incidence of severe (grade 3-5) irAEs ranging from 20 to 60% (19, 22). Strategies to tailor inhibitory receptor disruption in tumor-specific cellular products have been proposed to overcome these limitations, but are mainly focused on PD-1- or CTLA-4- disrupted cells tested in short-term anti-tumor responses (23–31). Here, we selected TIM-3, LAG-3 and 2B4, three inhibitory receptors frequently expressed by T cells in several cancer types and involved in resistance to immunotherapy (32, 33), and we exploited the multiplexicity of CRISPR/Cas9 with lentiviral vectors to simultaneously redirect T cell specificity and disrupt genes encoding for TIM-3, LAG-3, or 2B4, within a protocol able to expand long-living early differentiated TSCM (34). With this approach, we investigated the relative contribution of distinct IR knocked-out in tumor specific cellular products chronically challenged by multiple myeloma cells.
Peripheral blood mononuclear cells (PBMCs) were collected from healthy donors, after written informed consent, according to the San Raffaele Scientific Institutional Ethical Committee guidelines. The PBMCs were then isolated by Ficoll-Hypaque gradient separation (Lymphoprep; Fresenius) and the T cells were cultured as previously described (34). Briefly, PBMC harvested from healthy donors were activated using anti-CD3/anti-CD28-coated magnetic beads (ClinExVivo CD3/CD28; Invitrogen) and maintained at a concentration of 106 cells/mL in complete X-vivo supplemented with IL-7 and IL-15 (5ng/mL each). Two days after stimulation, T cells were electroporated with ribonucleoprotein (RNP) complexes (consisting of purified Spy Cas9 nuclease duplexed with synthetic gRNAs specific for the indicated genes) simultaneously using the Lonza Nucleofector 4D Electroporation System. The day after, T cells were transduced with a lentiviral vector (LV) encoding for the NY-ESO1-specific TCR. At day 6 post stimulation, beads were removed from culture and at day 18 tested for phenotype and function.
Myeloma cell lines (MM.1s and U266) were grown with RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin and 1% glutamine (Lonza). Adherent MM.1s were detached using TrypLE Express enzyme (Gibco). To obtain HLA-A2pos NY-ESO-1pos luciferasepos MM1.s cells, wild-type MM1.s were transduced with 3 different LVs encoding for HLA-A2, NY-ESO-1 and ΔLNGFR, luciferase and GFP. Transduced cells were FACS-sorted to obtain a pure population (> 98%) of HLA-A2pos NY-ESO-1pos luciferasepos cells (MM1.s A2pos ESO-1pos). HLA-A2, ΔLNGFR, and GFP transgene expressions were quantified by flow cytometry. In selected experiments, for IR ligand evaluations, cell lines were stimulated with IFNγ (600IU/mL) for 48 hours. Galectin-9 and HMGB1 secretions in cell culture medium were quantified using Galectin-9 ELISA kit (RayBiotech) or HMGB1 ELISA kit (IBL International) according to the manufacturer’s instructions. CD48 and HLA- DR expressions were quantified by flow cytometry upon staining with fluorochrome conjugated monoclonal antibodies. Cells were counted every 3-4 days by Trypan blue dye exclusion.
The gRNAs sequences used for IR and TCR disruption were as follows: TRAC: 5’- GAGAATCAAAATCGGTGAATAGG-3’; TIM-3: 5’- GAGTCACATTCTCTATGGTCAGG-3’; LAG-3: 5’- CACCGCGGCGCGGTACTCGCCGG-3’; 2B4: 5’- AGTTGAGAAACCCCGCCTACAGG-3’. The PAM sequence is indicated in bold. Commercial crRNA (Alt-R® CRISPR-Cas9 crRNA, IDT) and tracrRNA (Alt-R® CRISPR-Cas9 tracrRNA, IDT) were mixed in equimolar concentrations and heated at 95°C for 5 min to form a duplex with a final 100 μM concentration. crRNA:tracrRNA duplex and Cas9 enzyme were assembled in a 120:104 pmol ratio for 20 minutes at RT according to the manufacturer’s instructions. For each RNP targeting a single gene, 1 uL of Enhancer (100 uM; Alt-R® Electroporation Enhancer, IDT) was added to the reaction. Stimulated human CD3+ cells were electroporated 48 hours after stimulation. Two million cells/condition were resuspended in P3 primary buffer (Lonza) and assembled RNPs targeting one or more coding sequences of selected genes were simultaneously added to the cell suspension. Cells were electroporated using Nucleofector 4D (Lonza) according to manufacturer’s instructions; 24 hours later, the electroporated cells were counted and transduced with a previously validated lentiviral vector encoding for an HLA-A2 restricted NY-ESO-1157-165 specific TCR. The expression of the tumor-specific TCR was evaluated by flow cytometry through anti-Vβ13.1 antibody (FITC, Beckman Coulter) or with an APC-conjugated dextramer (Immudex).
Targeted T lymphocytes were collected at least one week after genomic manipulation to evaluate the frequency of non-homologous end joining (NHEJ) at each targeted genomic locus. DNA was extracted through QIAamp DNA Mini or Micro kits (Qiagen), depending on the number of cells and according to the manufacturer’s instructions. NHEJ assessment in manipulated cells was performed using ddPCR NHEJ custom Genome Edit Detection Assays (Bio-Rad) to amplify the TIM-3, LAG-3, 2B4 and TRAC targeted loci, according to the manufacturer’s instructions, with the only exception being the time and temperature of annealing/extension, 3 minutes and 59.4°C, respectively. Data were acquired using QuantaSoft Software and analyzed with QuantaSoft Analysis Pro Software.
Killing assays were performed by co-colturing multiple myeloma cells for 3 days with TCRED-IRKO or TCRED-IRCOMP T cells at decreasing effector:target (E:T) ratio. Wild type HLA-A2negNY-ESO-1neg MM.1s cell line and unedited, untransduced T lymphocytes (UT) were used as controls. After three days, cells were analyzed by flow cytometry. U266 cells were selected as CD138+ cells; HLA-A2posNY-ESO-1pos MM1.s were selected as GFP+ cells, whereas wild type MM.1s were selected as CD38+ cells. The number of residual target cells were counted according to the number of cells acquired at flow cytometry and the total well volume.
Cytokine production was analyzed 24 hours after antigen-specific stimulation. A panel of 13 different cytokines was quantified in supernatants of co-colture assays using LEGENDplex human Th panel (13-plex, Biolegend) according to the manufacturer’s instruction, and the data were analyzed using LEGENDplex data analysis software (Vigentech).
For degranulation assays, target and effector cells were co-coltured for 6 hrs in a 1:1 E:T ratio and CD107a+ CD3+ T cells were quantified by flow cytometry. Medium, HLA-A2negNY-ESO-1neg MM1.s wild-type cells and unedited, untransduced T lymphocytes (UT) were used as negative controls. T cells stimulated with 50 ng/ml PMA (Sigma-Aldrich) and 1mg/mL ionomycin (Sigma-Aldrich) were used as positive control. In chronic stimulation experiments, effector cells were stimulated daily with target cells in a 10:1 E:T ratio. After 15-21 days, effector and target cells were co-coltured to evaluate killing, degranulation and cytokine production. In all functional assays performed after chronic stimulation, effector:target ratio were calculated considering the viable number of CD3+ T cells at end of chronic stimulation. Absence of residual target cells after chronic stimulation was ensured by flow cytometry.
Twenty-four hours after co-culture with HLA-A2pos NY-ESO-1pos MM1.s cell line (effector: target ratio = 1:1), T cells were collected for RNA extraction. Total RNA was extracted with RNeasy mini kit (QIAGEN) according to manufacturer’s instructions. RNA was quantified with The Qubit 2.0 fluorometer (ThermoFisher) and its quality was assessed by Agilent RNA ScreenTape system (Agilent). RNA library preparations and sequencing reactions were conducted at GENEWIZ, LLC. (South Plainfield, NJ, USA). SMART-Seq v4 Ultra Low Input Kit for Sequencing was used for full-length cDNA synthesis and amplification (Clontech, Mountain View, CA) and Illumina Nextera XT library was used for sequencing library preparation. Briefly, cDNA was fragmented and adaptor was added using Transposase, followed by limited-cycle PCR to enrich and add index to the cDNA fragments. The final library was assessed with Agilent TapeStation. The sequencing libraries were multiplexed and clustered on a flowcell. After clustering, the flowcell was loaded on the Illumina HiSeq instrument according to manufacturer’s instructions. The samples were sequenced using a 2x150 Paired End (PE) configuration. Image analysis and base calling were conducted by the HiSeq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq was converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mis-match was allowed for index sequence identification.
Raw single-end reads quality control was determined using FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and read trimming was performed using Trim Galore software (https://doi.org/10.5281/zenodo.5127899) to remove residual adapters and low-quality sequences. Trimmed reads were aligned against the human reference genome (GRCh38) using STAR (35) with standard input parameters and only uniquely mapped reads were considered for downstream analyses. Reads were assigned to genes with the featureCounts tool (36) using the GENCODE primary assembly v.35 gene transfer file (GTF) as reference annotation for the genomic features. Gene count matrices were then processed by the R/Bioconductor differential gene expression analysis packages DESeq2 (37) following standard workflows. In particular, a paired analysis was set up, modelling gene counts using the following design formula: ~donor + condition. Genes with adjusted p-values less than 0.1 were considered differentially expressed.
A functional enrichment analysis was performed on the lists of significantly upregulated and downregulated DEGs. Enrichment analysis was performed considering the MSigDB database (C5: Ontology Gene Sets) using the enricher function from the R/Bioconductor package clusterProfiler (38) (v 3.8.1). Enriched terms with an adjusted p-value < 0.05 were considered statistically significant. Volcano plots were generated using the R package ggplot2 (https://ggplot2.tidyverse.org) and have been used to display RNA-seq results plotting the statistical significance (adjusted p-value) versus the magnitude of change (fold change). Heatmaps were generated using the R package pheatmap (https://CRAN.R-project.org/package=pheatmap). UpSet analysis was performed using UpSetR shiny app (39).
The protocol was approved by the Institutional Animal Care and Use Committee (IACUC). For the U266 mouse models, 6-8 weeks old female NOD/Scid gamma mice (NSG, Charles-River Italia) received sub-lethal total body irradiation (150 rads) and were then infused intravenously with 10x106 luciferasepos U266 cells (U266 dluc). Tumor growth was monitored by total body bioluminescent imaging (BLI) with IVIS SpectrumCT System. Briefly, mice were injected intraperitoneally with 150mg luciferin/kg 10 minutes before BLI. BLI image analysis was performed by measuring the total flux (photons/seconds) within regions with lesions. Images were acquired and analyzed with Living Image 4.4. Upon specific tumor engraftment (according to the experimental settings), mice were treated with different doses of engineered or untransduced T lymphocytes. High-tumor burden was defined as total flux > 106. Human chimerism and activation of T cells on peripheral blood were assessed weekly by flow cytometry. Mice were monitored weekly and in the presence of limping or weight loss greater than 5%, they were sacrificed. At sacrifice, spleen and bone marrow were harvested and extensive phenotypic, activation and exhaustion analysis of human T cells was performed by flow cytometry. In the U266 re-challenge mouse model, 10x106 U266 dluc were infused intravenously in mice that showed an anti-tumor response at day 14 after T cell treatment.
Live/Dead Fixable Violet Dead cell stain kit or DAPI were used to exclude dead cells prior to surface staining (Thermo-Fisher). For intracellular staining, anti-CD107a antibody (eBioscience) was added to cell culture. Cells were stained with surface antibodies, fixed and permeabilized with FIX & PERM Cell Fixation & Permeabilization Kit (ThermoFisher), according to the manufacturer’s instructions.
For phospho-flow experiments, T cells were stimulated with 5mM of H2O2 for 8 minutes. After stimulation, cells were fixed with 200μL of Fixation Buffer (BD CytofixTM Buffer) at 37°C for 10 minutes. Fixed cells were then washed in PBS with 1% FBS and permeabilized with Permeabilization Buffer (BD PhosflowTM Perm Buffer III) on ice for 45 minutes. Permeabilized cells were then washed, resuspended in PBS with 1% FBS and stained with Pacific Blue-conjugated anti-phospho-ERK and PerCP-conjugated anti-CD3 antibodies for 45 minutes on ice. After staining, cells were washed in PBS with 1% FBS and samples analyzed by BD FACS Canto II flow cytometer.
For in vivo experiments, 50-100 μL of whole blood were stained with fluorochrome conjugated monoclonal antibody specific for mouse CD45, human CD45, 2B4, CD3, CD8, CTLA-4, PD-1, TIM-3, LAG-3, HLA-DR, CD45RA, CD62L. After surface staining, red blood cells were lysed with ACK (Ammonium-Chloride-Potassium) buffer for 10 minutes at room temperature. Human T cells were counted on peripheral blood using Flow Count fluorescent beads (Beckman Coulter), according to manufacturer’s instruction. For phenotype analysis in chronic antigen stimulation experiments, stimulated T cells were stained with fluorochrome conjugated monoclonal antibodies specific for CD3, CD8, CD4, LAG-3, CD45RA, CD62L, HLA-DR, CTLA4, PD-1, TIM-3, 2B4, TIGIT, CD160, CD39.
For each experiment, dead cells were excluded by DAPI positive staining. Data were acquired using a BD FACS Canto II, LSR Fortessa or Symphony (BD Biosciences) and analyzed with FlowJo version 10 software (TreeStar). Memory subsets were identified using CD45RA and CD62L surface markers, as previously described (40). Briefly, stem cell memory cells (TSCM) were gated as CD45RA+ CD62L+ T cells; central memory cells (TCM) were gated as CD45RA- CD62L+ T cells; effector memory cells (TEM) were gated as CD45RA- CD62L- T cells; CD45RA-expressing effector memory cells (TEMRA) were gated as CD45RA+ CD62L- cells.
All antibodies used for flow cytometry are listed in Supplementary Table 1.
Flow Cytometry Standard (FCS) files were analyzed using FlowJo software (ver.10) to remove doublets and dead cells and to isolate CD3+ T cells. Down sampling was performed to export 3000 CD3+ events/samples. Subsequent clustering analysis was performed using a public available pipeline (41–43). The k value was set to 30 for clustering analysis.
Statistical analyses were performed with Prism 9 (GraphPad Software). The Mann-Whitney test was used when comparing two independent groups. A two-way ANOVA was used when comparing variables across two or more normally distributed subsets. Linear regression analysis was used to model the linear relationships between analyzed variables. For all comparisons, two-sided P values were used and p < 0.05 was considered statistically significant.
To generate TSCM redirected against tumor antigens and resistant to exhaustion signals, we combined the multiplexing capacity of CRISPR/Cas9 with the efficiency of lentiviral vectors for the simultaneous TCR genetic redirection and IR genetic ablation following a protocol that favors the expansion of early differentiated T cells (Figure 1A). We designed and tested multiple sgRNAs targeting TRAC, TIM-3 (HAVCR2), LAG-3 and 2B4 (CD244) loci and selected those with the highest gene disruption efficiencies (Supplementary Figure 1A). CRISPR/Cas9-RNPs targeting TRAC and one selected IR locus (TIM-3, LAG-3 or 2B4) were simultaneously delivered by electroporation in activated T cells. Since CD3 requires an intact TCR to translocate to cell surface while TIM-3 and LAG-3 are expressed at moderate to high levels by more than 80% of activated CD3+ T cells in the first 6 days after stimulation (Supplementary Figures 1B, C), we quantified the efficiency of TRAC, TIM-3 and LAG-3 disruption by flow cytometry. Upon double gene disruption, we observed a median of 94.7% and 92.7% of CD3NEG-TIM-3NEG and CD3NEG-LAG-3NEG cells respectively (Figure 1B).
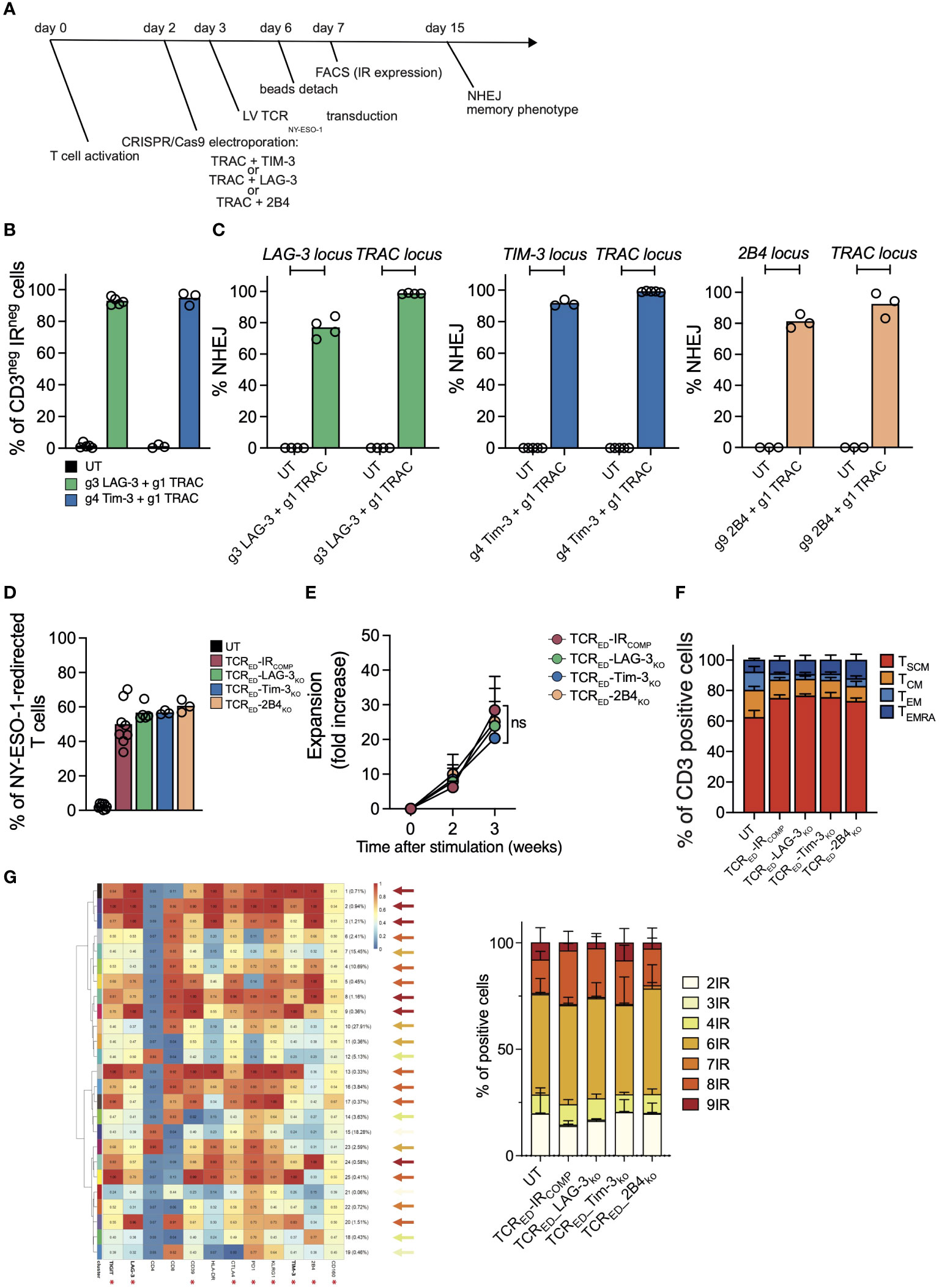
Figure 1 CRISPR-Cas9-mediated TIM-3, LAG-3 or 2B4 disruption does not alter the expansion capacity nor the differentiation phenotype of TCRED T cells. (A) Schematic representation of the protocol for the generation of TIM-3-, LAG-3, or 2B4-disrupted TCR-edited (TCRED) T cells. (B) Percentages of CD3negLAG-3neg (green bar; N=5) or CD3negTIM-3neg (blue bar; N=3) live cells measured by flow cytometry in T cells electroporated with CRISPR/Cas9 targeting LAG-3 or TIM-3 in combination with CRISPR/Cas9 targeting TRAC. Unmanipulated cells (UT, black bars) are shown as control. (C) Percentages of NHEJ at LAG-3 (green bar; N=4), TIM-3 (blue bar; N=3/5), and 2B4 (light orange bar; N=3) loci in UT cells (black bar; N=3/5) and in T cells electroporated with CRISPR/Cas9 targeting the specific inhibitory receptor and the TRAC locus. (D) Frequency of cells redirected against NY-ESO-1 upon lentiviral transduction of unmanipulated cells (UT, black bar; N=8) or in T cells electroporated with CRISPR/Cas9 targeting TRAC alone (TCRED-IRCOMP, red bar; N=8) or in combination with gRNAs targeting LAG-3 (TCRED-LAG-3KO, green bar; N=5), TIM3 (TCRED-TIM-3KO, blue bar; N=3) or 2B4 (TCRED-2B4KO, light orange bar; N=3). Transduction efficiency was measured by flow cytometry upon HLA-A*02:01-NY-ESO-1157-165 dextramer staining. Fold increase (E) and memory phenotype (F) of inhibitory receptor (IR) competent (TCRED-IRCOMP; N=5), TCRED-LAG-3KO (N=4), TCRED-TIM-3KO (N=3) and TCRED-2B4KO (N=3) T cells at day +21. g1, g3, g4, g9 refer to gRNAs designed to disrupt the indicated genes. (G) Meta-cluster composition of TCRED or TCRED-IRKO T cells (left). Arrows colors indicate the number of IR co-expressed in each cluster according to the color code indicated on the right. Percentages of cells co-expressing from 2 to 9 of the IRs marked with an asterisk, in TCRED-IRCOMP (N=3) or TCRED-IRKO (N=3) cells are shown on the right. Data are shown as mean ± SEM of 3-8 biological replicates; ns= not statistically significant.
By quantifying NHEJ events at each targeted locus with ddPCR, we observed a median efficiency of 2B4 genetic disruption of 81.53% and we confirmed high efficiency of knock-out at the LAG-3, TIM3 loci (76.8% and 91.6% respectively) in double TRAC-IR knock-out cells (Figure 1C).
TCRNEG-IRKO and TCRNEG cells competent for IR expression (TCRNEG-IRCOMP) were transduced with a lentiviral vector encoding for an HLA-A2-restricted-NY-ESO-1157-165 specific TCR (4) to obtain a pool of TCR edited cells (TCRED-IRCOMP, TCRED-LAG-3KO, TCRED-TIM-3KO, TCRED-2B4KO) specific for the same tumor antigen. As shown in Figure 1D, transduction efficiency, measured by flow cytometry, was higher than 50% in all cellular products. Of note, TCRED-IRCOMP and TCRED-IRKO T cells displayed similar expansion kinetics (Figure 1E) and were equally enriched in early differentiated memory cells at the end of the expansion (Figure 1F). Gene modifications (IR disruption and NY-ESO-1 specific TCR expression) and memory phenotype were similar in CD4+ and CD8+ T-cell subsets present in our cellular products (Supplementary Figures 2A–D). In addition, exploiting unsupervised high-dimensional analysis we could assess the percentage of cells co-expressing from 2 to 9 different IR among TIGIT, LAG-3, CD39, CTLA4, PD1, KLRG1, TIM3, 2B4, and CD160. Noticeably, no differences were observed in IR co-expression in our cellular products, with approximately 25% of cells expressing ≤ 4 IR (Figure 1G).
Overall, these data demonstrate that we can efficiently generate high numbers of early differentiated T cells redirected against a tumor antigen and permanently devoid of one inhibitory receptor. Importantly, we showed that the disruption of LAG-3, TIM-3 or 2B4 does not impair the expansion capacity nor the memory phenotype of edited CD4+ and CD8+ T cells in vitro.
Upon antigen recognition, cancer cells foster inhibitory signals through the upregulation of IR ligands and the pattern of IR engagement displays both inter-tumor and intra-tumor variability. To investigate if multiple myeloma cells engage TIM-3-, LAG-3- and 2B4-mediated pathways, we analyzed the expression of IR ligands in two multiple myeloma cell lines (U266 and MM1.s) in both resting conditions and in pro-inflammatory conditions. We quantified the release in cell culture medium of Gal-9 and HMGB1 (TIM-3 ligands) and of FGL-1 (LAG-3 ligand), the flipping of phosphatidyl-serin (TIM-3 triggering signal) in the outer layer of the cell membrane, the membrane bound Gal-9 and CEACAM (TIM-3 ligands) and the upregulation of MHC-II (LAG-3 ligand) and CD48 (2B4 ligand). Both MM1.s and U266 cell lines expressed all tested IR ligands, albeit at varying levels. Of note, the level of IR ligands remained stable upon IFN-γ exposure (Supplementary Figures 3A, B). To test the effect of TIM-3, LAG-3 and 2B4 disruption in tumor-specific T cells upon antigen recognition on cancer cells expressing IR ligands, we challenged TCRED-IRCOMP and TCRED-IRKO T cells with either U266 (a cell line naturally expressing HLA-A2 and NY-ESO-1) or MM1.s transduced to express HLA-A2 and NY-ESO-1 antigen and luciferase (MM1.s A2posESO-1pos, Supplementary Figures 3C, D). In a short-term co-culture assay, we observed that the disruption of TIM-3, LAG-3, and 2B4 does not impact the killing efficiency nor degranulation ability of T cells (Figures 2A–D), with the exception of a slight increase in MM1.s A2posESO-1pos killing observed in all TCRED-IRKO cells at limiting effector:target ratio. Interestingly, the analysis of secreted pro-inflammatory cytokines and effector molecules showed that TIM-3-, LAG-3-, and 2B4-disrupted TCRED T cells produce higher amounts of IL-2, TNFα, sFasL and perforin compared to TCRED-IRCOMP T cells when tested with the two different multiple myeloma models (Figures 2E, F; Supplementary Figure 4A). TCRED-IRCOMP and all TCRED-IRKO cells produced equal and high amount of IFNγ (Supplementary Figure 4B). These results indicate that the absence of TIM-3, LAG-3, or 2B4 in tumor-specific T cells does not affect their killing capacity nor degranulation ability while induces an increased production of pro-inflammatory cytokines and molecules.
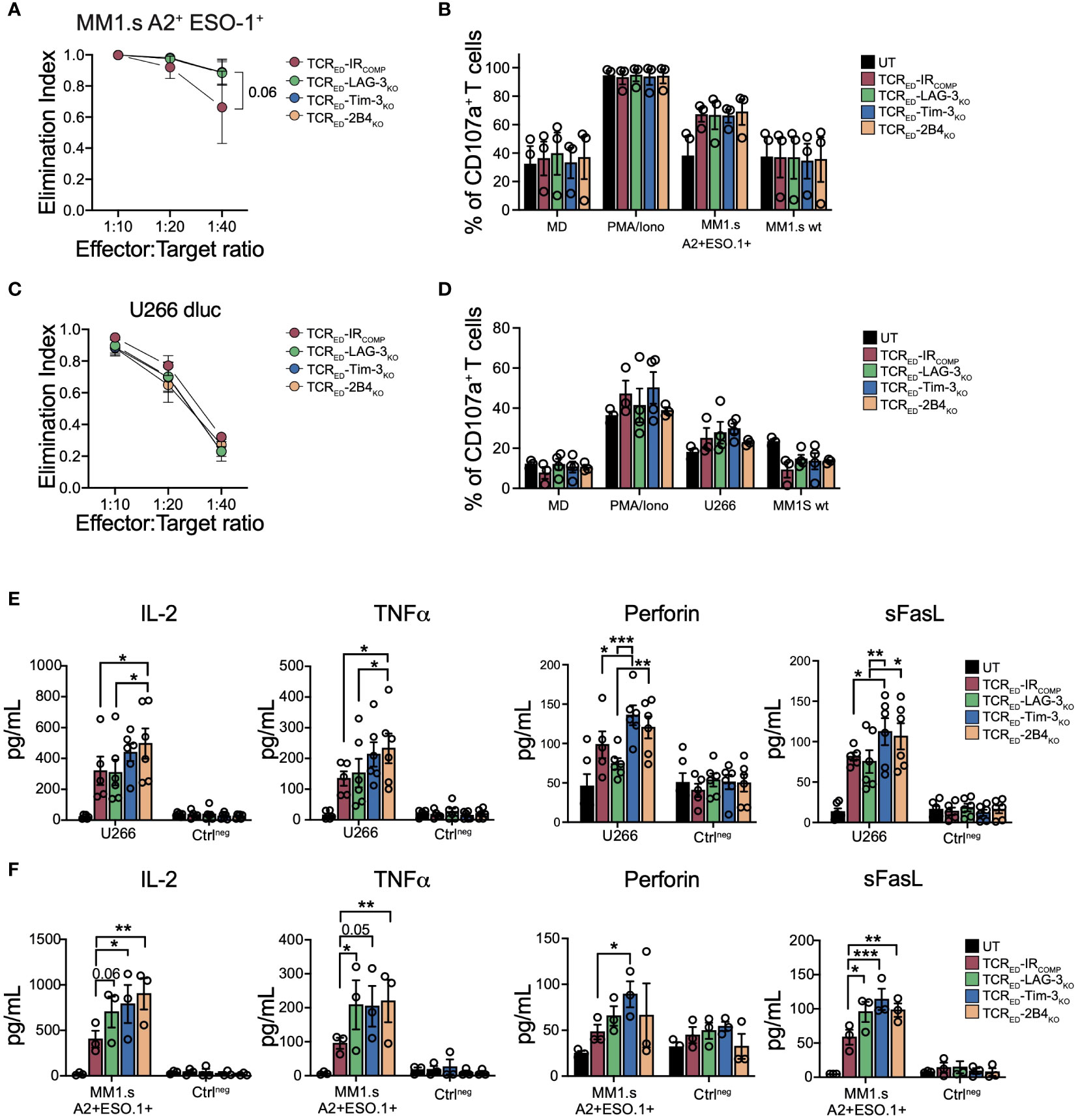
Figure 2 IR disruption increases the production of effector molecules in TCR gene edited T cells. Elimination index (A, C) and degranulation (B, D) of TCRED-IRCOMP (red), TCRED-LAG-3KO (green), TCRED-TIM-3KO (blue) and TCRED-2B4KO (light orange) T cells co-cultured with U266 or HLA-A2neg NY-ESO-1neg MM1.s (MM1.s A2+ESO-1+) or HLA-A2neg NY-ESO-1neg MM1.s (MM1.s wt). In panel (B, D) only medium (MD) and PMA/Ionomycin (PMA/iono) were used as controls. Degranulation of untransduced (UT, black bars) cells is shown. (E, F) Quantification of IL-2, TNFα, perforin, and sFasL produced by TCRED-IRCOMP (red bars), TCRED-LAG-3KO (green bars), TCRED-TIM-3KO (blue bars), and TCRED-2B4KO (light orange bars) T cells or UT T cells (black bars) upon exposure to HLA-A2pos NY-ESOpos U266 cells (E) or MM1.s A2posESO-1pos cells (F). Negative controls (Ctrlneg) indicate exposure of effector cells to medium or wild-type MM1.s cells. Data are shown as mean ± SEM of 3-6 biological replicates; (A, B, F) TCRED-IRCOMP (N=3), TCRED-LAG-3KO (N=3), TCRED-TIM-3KO (N=3) and TCRED-2B4KO (N=3) UT (N=3); (C, D) TCRED-IRCOMP (N=3), TCRED-LAG-3KO (N=4), TCRED-TIM-3KO (N=4) and TCRED-2B4KO (N=3) UT (N=4); (E) TCRED-IRCOMP (N=5), TCRED-IRKO and UT (N=6)). *: p value <0.05; **: p value < 0.01; ***: p value < 0.001. Elimination index=1-[number of living target cells co-cultured with redirected T cells/number of living target cells co-cultured with UT cells].
To shed lights on the mechanisms underlying the functional advantage of TCRED-IRKO cells compared to IRCOMP counterparts, we compared the transcriptional profiles of our cellular products upon 24 hrs co-culture with MM1.s A2posESO-1pos multiple myeloma cells or left unstimulated. First, we analyzed differentially upregulated or downregulated genes in each TCRED-IRKO vs TCRED-IRCOMP cells after stimulation with cancer cells. Volcano plot of the differentially expressed genes revealed 183 differentially expressed genes in TCRED-LAG-3KO vs TCRED-IRCOMP cells (Figure 3A). On the contrary, in the same experimental condition, only 92 and 11 genes were differentially regulated in TCRED-TIM-3KO and TCRED-2B4KO cells respectively, compared with TCRED-IRCOMP cells (Supplementary Figures 5A, B). Among the 92 differentially expressed genes (DEGs) in TCRED-TIM-3KO T cells, ~1/3 of the DEGs is represented by long non coding RNA (lncRNA) transcripts, suggesting that the lack of IR signaling might reshape lncRNA pattern (44). Gene ontology analysis of the biological process of the 183 DEGs identified in stimulated TCRED-LAG-3KO cells revealed a significant enrichment in glycolytic processes, ADP and NAD metabolism, and nucleotide metabolism, thus suggesting a possible metabolic switch in LAG-3-disrupted antigen specific T cells (Figure 3B). To further shed light on the effects of antigen stimulation on our cells, we analyzed the under and overexpressed genes observed in each TCRED, IRCOMP and IRKO cellular product upon stimulation with cancer cells. In this analysis, we observed 464 genes differentially expressed in resting vs activated TCRED-IRCOMP T cells. Strikingly, this analysis confirmed a significant transcriptional change in TCRED-LAG-3KO T cells, with 926 differentially expressed genes after antigen stimulation. On the contrary, only a few DEGs emerged in TCRED-TIM-3KO T cells and TCRED-2B4KO T cells (Figure 3C). Pathway enrichment analysis of the 174 DEGs identified in stimulated TCRED-2B4KO cells, using Gene Ontology-Biological Process as reference, revealed the enrichment of pathways involved in response to pro-inflammatory stimuli, DNA-replication and protein secretion (Supplementary Figure 5C). We then focused on genes uniquely up or downregulated in each different cellular product upon antigen exposure.
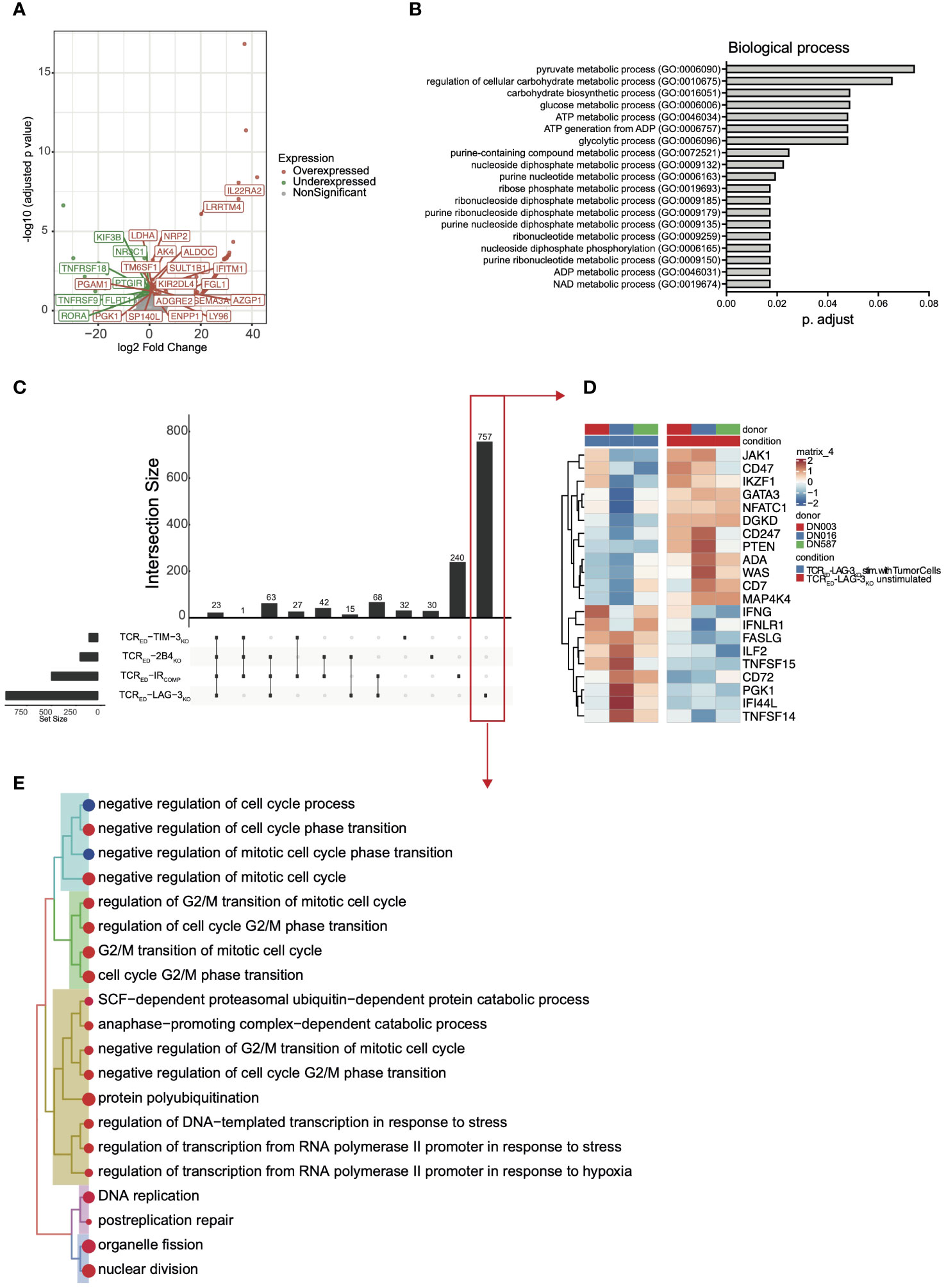
Figure 3 TCRED-IRKO cells display a unique transcriptional profile upon target recognition. Volcano plot showing significant down- (green) and up- (red) regulated genes (A) and curated Gene ontology terms (biological process-(B) in stimulated TCRED-LAG-3KO vs stimulated TCRED-IRCOMP cells. (C) UpSet analysis showing shared (connected dots in the lower panel) or unique (not-connected dots in the lower panel) differentially expressed genes in stimulated vs unstimulated TCRED-IRCOMP, TCRED-LAG-3KO, TCRED-TIM-3KO, TCRED-2B4KO cells. (D) Heatmap showing the level of expression of selected genes among the 757 uniquely differentially expressed genes in TCRED-LAG-3KO upon target recognition (red box in panel (C). (E) Treeplot of Gene Ontology (Biological Process) terms enriched uniquely in TCRED-LAG-3KO upon target recognition (red box in panel (C)). Data are shown for 3 biological replicates for each cellular product, generated from 3 matched healthy donors. MM1.s A2posESO-1pos multiple myeloma cells were used as antigen source for T cell stimulation.
A total of 757 genes were uniquely differentially expressed in TCRED-LAG-3KO cells (Figure 3C red box and Figure 3D). Among these, genes associated with T cell activation and effector functions (FASLG, TNFSF15, CD72, PGK1, TNFSF14, IFNG, ILF2) and IFNg response genes (IFI44L, IFNLR1) were significantly upregulated only in TCRED-LAG3KO T cells. Moreover, genes dampening T cell response (IKZF1, CD74 and DGKD (45, 46)) were significantly downregulated exclusively in TCRED-LAG-3KO T cells. Among the downregulated genes, transcription factors (TFs)/TCR-signaling mediators, which are physiologically downregulated upon TCR engagement to preserve homeostatic T cell response (47, 48) (PTEN, GATA3, ADA, WAS, MAP4K4, NFATC1, CD274, JAK1), emerged in TCRED-LAG-3KO T cells (Figure 3D). We then performed a pathway enrichment analyses, using Gene Ontology -Biological Process as reference, of the 757 unique DEGs in TCRED-LAG-3KO cells to identify biological processes that were differentially regulated upon antigen recognition by this cellular product. Genes involved in cell cycle, DNA-replication, nucleotide metabolism were significantly upregulated in stimulated TCRED-LAG3KO cells (Figure 3E), thus suggesting that, upon target recognition by T cells, LAG-3 disruption might boost the induction of DNA replication. To verify whether TCR triggering induces stronger activation in TCRED-LAG-3KO cells compared to other cellular products, we quantified the phosphorylation of ERK induced by TCR triggering, in our cells (49, 50). According to our hypothesis, we observed a significantly increased phospho-ERK upon activation only in TCRED-LAG-3KO T cells compared to TCRED-IRCOMP cells (Supplementary Figure 5D). Overall, transcriptomic analysis suggests a different mechanism underlying the effects of LAG-3, TIM-3, and 2B4 genetic loss in T cell products and show that LAG-3 disruption has a major impact on the transcriptional profile of T cells.
To model T cell exhaustion and verify whether the lack of TIM-3-, LAG-3- or 2B4 overcome the loss of function of tumor-specific T cells, we adapted a chronic stimulation protocol (51–53) and daily stimulated TCRED-IRCOMP and TCRED-IRKO T cells with U266 or MM1.s A2posESO-1pos cancer cells, and we assessed the anti-tumor response 2 to 3 weeks later (Figure 4A). In these stressed conditions, TCRED-TIM-3KO and TCRED-2B4KO lymphocytes displayed a higher degranulation capacity than TCRED-IRCOMP counterparts, when challenged with U266 (Figure 4B) or MM1.s A2posESO-1pos cell lines (Figure 4C), indicating that the lack of TIM-3 and 2B4 in tumor-specific T cells sustains prolonged effector functions and overcomes exhaustion upon chronic antigen exposure. Despite ligands expression on U266 and MM1.s tumor cells, LAG-3 disruption didn’t appear to impact on TCRED function in these assays. Of notice, no significant differences were observed in IL-2, perforin, sFASL nor TNFα secretion in the different T cell products (Supplementary Figure 6).
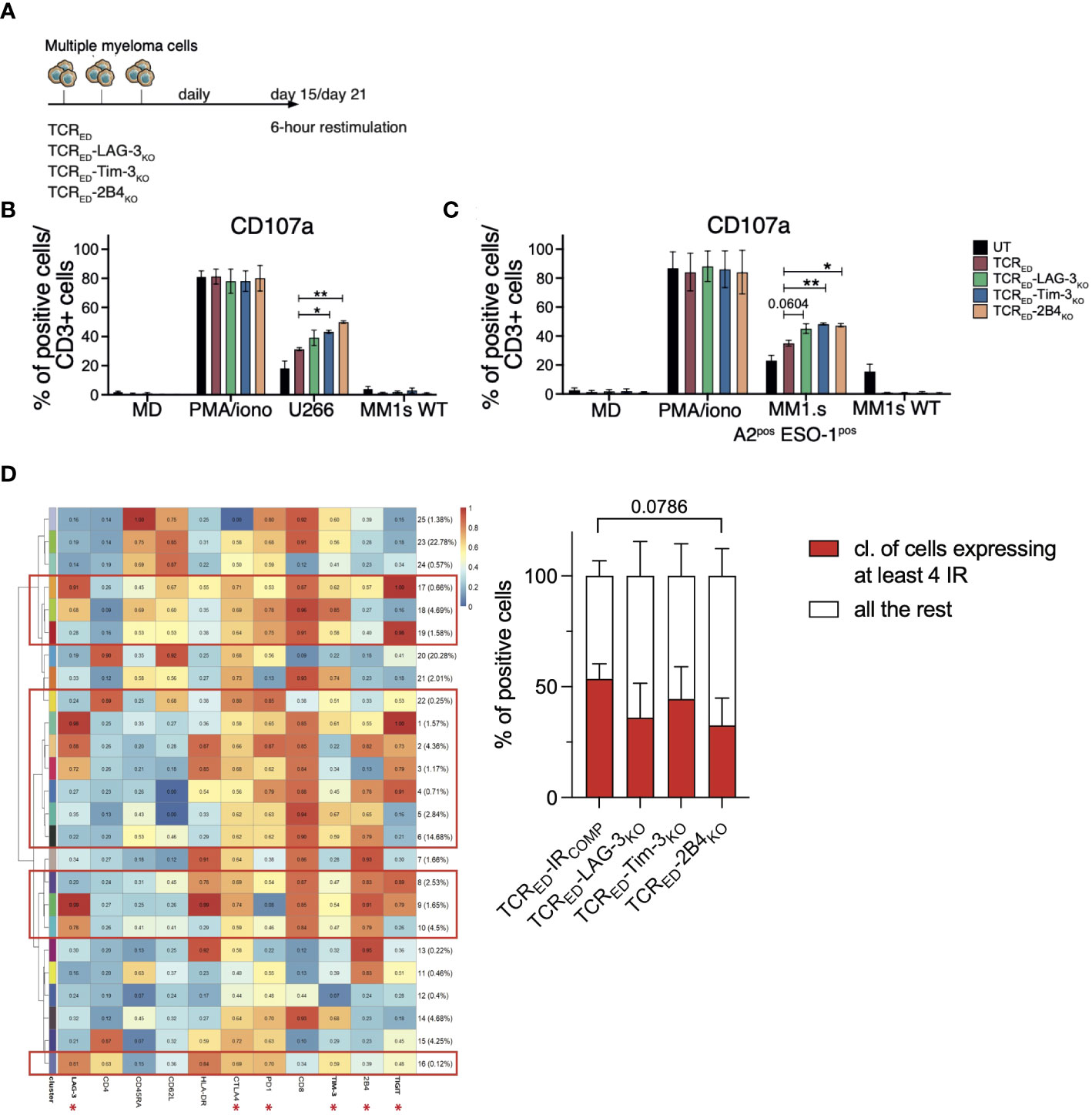
Figure 4 TCRED-IRKO cells outperform TCRED-IRCOMP T cells upon chronic antigen stimulation. (A) Schematic representation of the T cell exhaustion experiment. (B, C) Percentages of degranulating (measured as CD107a+) CD3+ T cells after 6 hours of restimulation of TCRED-IRCOMP (red bars), TCRED-LAG-3KO (green bars), TCRED-TIM-3KO (blue bars), and TCRED-2B4KO (light orange bars) or unmanipulated (UT, black bars) T cells challenged for 15-21 days with U266 or MM1.s A2posESO-1pos tumor cells. Only medium, PMA/Ionomycin and HLA-A2negNY-ESO-1neg MM1.s wt cells were used as controls (N=3). (D) Meta-cluster composition of TCRED-IRCOMP and TCRED-IRKO T cells phenotyped after 18 days of daily stimulation with MM1.s A2posESO-1pos cells (left). The red squares indicate clusters displaying the co-expression of at least 4 IRs among the ones indicated with an asterisk. On the right, the percentages of TCRED-IRCOMP and TCRED-IRKO T cells co-expressing ≥ 4 IRs at the end of chronic antigen stimulation is shown. Data are shown as mean ± SEM of 3 biological replicates for each cellular product.*: p value <0.05; **: p-value < 0.01; ***: p-value < 0.001; ****: p-value < 0.0001.
To further investigate how the lack of IR-mediated signaling may affect chronically stimulated tumor-specific T cells, we phenotyped TCRED-IRKO cells and IR competent counterparts at the end of chronic stimulation with MM1.s A2posESO-1pos. Unsupervised analysis was used to identify the clusters of events based on the co-expression of IRs, and, in particular, the percentage of cells expressing more than 4 IRs among LAG-3, CTLA4, PD-1, TIM-3, 2B4, and TIGIT was calculated on each T cell product. TCRED-IRKO cells display a reduced, though not significant, number of IRs on the cell surface, compared to TCRED-IRCOMP counterparts (Figure 4D). These observations suggest that the disruption of IRs might prevent the upregulation of additional inhibitory receptors upon chronic antigenic stimulation and, overall, indicate that the disruption of different IRs affects distinct T cell effector functions.
To verify in vivo the advantages of the lack of IRs, we set up a model in which mice with high tumor burden were treated with a limited doses of tumor-specific T cells. In this setting, cancer cells represent a continuous source of T cell activation that could potentially trigger T cell exhaustion, similarly to what occurs in cancer patients. Considering the high expression of LAG-3 ligands measured in U266 cells (Supplementary Figure 3) with this model we initially compared TCRED-IRCOMP vs TCRED-LAG3KO T cells. 10x106 luciferasepos U266 cells, expressing high levels of LAG-3 ligands, were injected in sub-lethally irradiated NSG mice. When high-tumor burden was detected by total body bioluminescence imaging (BLI), mice were treated with two different doses (106 or 5x106) of either TCRED-LAG-3KO or TCRED-IRCOMP effector cells. Control mice were left untreated (nil) or treated with untransduced cells (UT, 5x106 dose) (Supplementary Figure 7A). The lowest dose of lymphocytes was not sufficient for controlling tumor growth independently from the infused cellular product. Instead, 5x106 TCRED-IRCOMP T cells induced a partial tumor control, while 5x106 TCRED-LAG-3KO T cells completely eradicated the tumor (Supplementary Figure 7B). The frequencies of circulating human lymphocytes increased with the dose infused (Supplementary Figure 7C). A similar growth kinetic was observed with both TCRED cellular products, while untransduced cells, although ineffective in controlling the disease, further expanded as observed in xeno-GvHD models (54, 55). Interestingly, a significant correlation was detected between the tumor burden and the expansion of TCRED-LAG-3KO T cells, but not with TCRED-IRCOMP T cells, suggesting a dose-dependent tumor killing in the absence of LAG-3 (Supplementary Figure 7D).
Although not sufficient for tumor eradication, TCRED-LAG-3KO circulating in mice treated with the lowest T cell dose maintained a higher activation profile than TCRED-IRCOMP cells (Supplementary Figure 7E), indicating a preserved ability to respond upon prolonged antigen exposure. Accordingly, at sacrifice, 3 weeks after T-cell infusion, a higher proportion of TCRED-IRCOMP cells infiltrating the bone marrow co-expressed multiple IRs compared to TCRED-LAG-3KO cells (Supplementary Figure 7F), indicating that LAG-3 disruption in T cells redirected against NY-ESO-1 curtails the upregulation of other inhibitory receptors, even in stressed conditions of tumor outgrowth.
Interestingly, although U266 cells expressed lower levels of TIM3 and 2B4 ligands than MM1s cells in vitro (Supplementary Figure 3), when we analyzed the profile of U266 cells harvested from mice, we observed an upregulation of CD48, HLA-DR and CEACAM (Supplementary Figures 8A–D) and we measured detectable levels of HMGB1 and Galectin-9 in the serum (Supplementary Figure 8E). Thus, we reasoned that U266 could represent an appropriate model to compare all our TCRED-IRKO cellular products.
To verify whether the lack of LAG-3, TIM-3, or 2B4-mediated signaling in TCRED T cells prevents functional exhaustion and promotes immunological memory, the anti-tumor responses of TCRED-IRCOMP, TCRED-LAG-3KO, TCRED-TIM-3KO and TCRED-2B4KO cells were compared in a tumor rechallenge model. Sub-lethally irradiated NSG mice were infused with luciferasepos U266 cells and treated with low doses (1x106/mouse) of engineered (TCRED-IRCOMP, TCRED-LAG-3KO, TCRED-TIM-3KO, TCRED-2B4KO T cells) or untransduced cells in a minimal residual disease setting (total flux <= 106) (Figure 5A). As shown in Figure 5B, Supplementary Figure 9A, all edited cellular products completely eradicated the tumors in 2 weeks, while animals receiving untransduced lymphocytes displayed a massive tumor growth. We then rechallenged all mice treated with edited lymphocytes with 10x106 U266 cells. Impressively, all TCRED-IRKO cells, but not TCRED-IRCOMP cells, were able to completely eradicate tumor cells (Figure 5C, Supplementary Figure 9A), thus demonstrating enhanced T cell fitness in vivo. Despite a similar kinetic of T cell expansion observed with all engineered effectors upon T-cell infusion (Figure 5D), after tumor rechallenge, TCRED-LAG-3KO T cells displayed the highest and most consistent expansion profile (Figure 5E), in accordance with their transcriptional profile. Interestingly, at sacrifice, a significantly higher proportion of activated CD3+ cells were present in the spleens of all TCRED-IRKO treated mice compared to TCRED-IRCOMP treated mice (Supplementary Figure 9B). In addition, human T cells displayed a similar differentiation phenotype, dominated by effector cells but with a measurable proportion of early memory (TSCM and TCM) lymphocytes (Supplementary Figure 9C). Of interest, the majority of BM-T cells remained negative for each genetically disrupted IR (Figure 5F), indicating the absence of a counterselection of IR edited cellular products in this experimental setting.
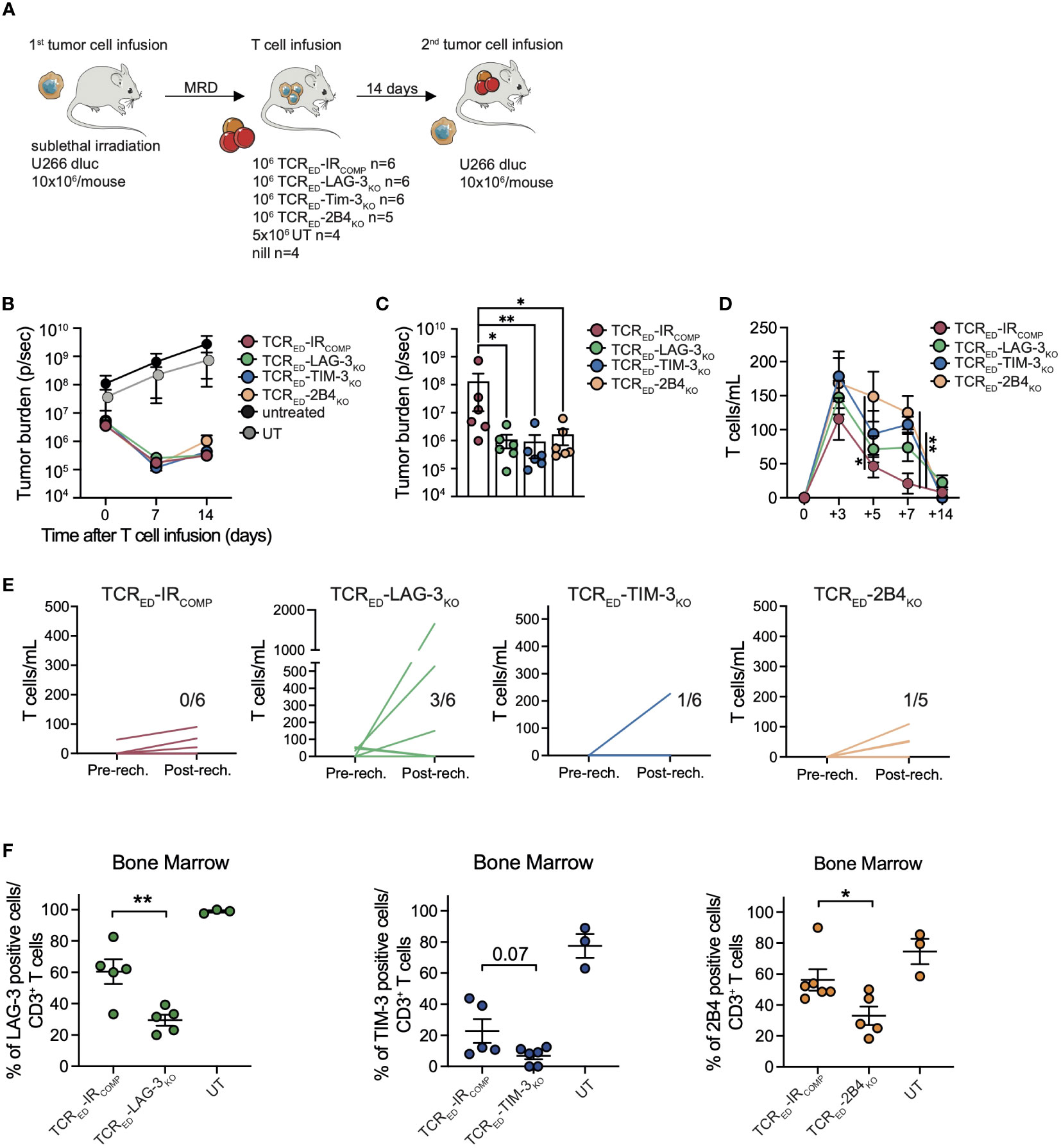
Figure 5 TCRED-IRKO cells outperform TCRED-IRCOMP T cells against multiple myeloma in vivo. (A) Schematic representation of in vivo rechallenge experiment with U266 multiple myeloma cells. Tumor burden evaluated by total body bioluminescence (BLI) during (B) the first anti-tumor response and (C) after re-challenge with U266, in mice treated with low dose of TCRED-IRCOMP (red lines), TCRED-LAG-3KO (green lines), TCRED-TIM-3KO (blue lines), TCRED-2B4KO (orange lines) T cells. In panel B tumor bearing mice left untreated (black) or injected with untransduced lymphocytes (grey) are shown. Absolute quantification of circulating CD3+ cells before (D) and after (E) tumor rechallenge. In panel E each line represents a single mouse and the number in each graph indicates the number of mice with human CD3 total cells/mL higher than 100. (F) Frequency of human T cells expressing TIM-3 (blue), LAG-3 (green) and 2B4 (orange) in the bone marrow of treated mice at sacrifice. Data are shown as mean ± SEM. N. of treated and analyzed animals per group are shown in (A). *: p value <0.05; **: p value < 0.01; ***: p value < 0.001.
Overall, our data indicate that LAG-3, TIM-3, or 2B4 disruption not only prevents the upregulation of additional inhibitory molecules and enhances the cytotoxic activity upon chronic stimulations, but also supports a prolonged anti-tumor activity.
The long-term efficacy of adoptive T cell therapy with TCR gene-edited or CAR-T cells is still limited by T cell exhaustion and by limited persistence of infused cellular products. Here, we aimed at overcoming these hurdles by developing innovative cellular products redirected against a tumor-antigen, endowed with long-term persistence potential and resistant to inhibitory pathways.
In cancer patients, tumor-specific T cells are chronically stimulated by tumor antigens in an immunosuppressive tumor microenvironment (TME) and acquire an exhausted phenotype. Loss of effector functions in exhausted cells is triggered by different pathways downstream to multiple inhibitory receptors that are expressed on the cell surface, including TIM-3, LAG-3 and 2B4. To foster immune escape, cancer cells upregulate IR ligands and exploit these inhibitory pathways to dampen T cell responses. Checkpoint inhibitors revert T cell exhaustion by restoring T cell proliferation and pro-inflammatory cytokine production, thus rescuing effective anti-tumor responses. Although the use of ICBs in clinical studies induces prolonged tumor-free survival in a fraction of treated patients, their use is often limited by the occurrence of severe autoimmune related adverse events. This secondary unwanted reaction is mainly due to the lack of specificity of ICB, which may unleash autoreactive T cells. To overcome these limitations, we simultaneously disrupted the endogenous TCR α chain in combination with TIM-3, LAG-3 or 2B4 and we generated TCRED-IRKO cells by transduction with a lentiviral vector encoding for a HLA-A2 restricted NY-ESO-1 specific TCR (56). With our optimized biotechnological tools, we were able to obtain highly efficient gene disruption and transduction.
Although chromosomal translocations derived from bi- or tri-genic modifications have been reported in clinical trials with genetically engineered cells (23, 57), their frequency was comparable to that observed in healthy individuals (58–60) and no secondary malignant transformation associated to translocations was reported in these trials (23, 57). While long-term follow up of treated patients will provide further insights into the safety profile of CRISPR/Cas9 nucleases, other alternative gene editing tools that do not require DNA double strand break induction, such as the recently developed base editing and prime editing platforms, could be used to further reduce the risk of translocations when editing multiple genes. In our study, we employed a high-fidelity version of Cas9, and expect to detect low numbers of off-targets in our cellular products. Although specificity will need to be evaluated before clinical implementation, no differences in the expansion capacity were detected in vitro or in vivo among different engineered T cells, indicating that no significant perturbation of T cell fitness occurs upon multiple gene disruptions.
Durable clinical responses strongly rely on the persistence of adoptively transferred cells. TSCM and TCM cells are endowed with self-renewal, high-proliferative potential and superior long-term persistence, thus representing ideal subsets for ACT (61–65). Here, we combined genetic engineering with a stimulation and culture protocol that supports the expansion of TSCM/TCM cells (34), and we observed that multiple gene disruptions by CRISPR/Cas9 in combination with TCR gene editing is feasible in TSCM cells without altering their phenotype, expansion, or proliferative capacities. Notably, our results indicate that TCRED-IRKO cells retain an early differentiated phenotype, thus suggesting that TIM-3, LAG-3 and 2B4 disruptions do not impact the memory differentiation of edited cells. Accordingly, a measurable proportion of TSCM cells could be also quantified in the bone marrow of mice treated with our engineered cellular products.
The disruption of inhibitory receptors in tumor-specific T cells has been recently proposed (23–31, 66). In these studies, the authors abrogated the expression of PD-1, CTLA-4, LAG-3, A2AR or CD39 encoding genes in engineered T cells. While a slight increase in survival was shown in mice treated with IR-disrupted cells, conclusive results on the impact of each IR disruption on tumor-redirected T cells in tumor control upon chronic stimulation are lacking. Furthermore, a comparative analysis of cellular products devoid of single IR has not been reported. In a clinical trial with PD-1 disrupted cells edited for T cell specificity, the frequency of cells with edits in the PDCD1 locus decreased from 25% to 5% of the cells expressing the transgenic TCR at 4 months after infusion, consistently with mouse studies of chronic infection where PD-1 deficient T cells proved less able to establish memory (23). However, recent reports challenged this hypothesis and demonstrated in a murine model long-term persistence and function of murine adoptively transferred antigen-specific T cells with genetically ablated PD-1 expression (67).
The TCRED-IRKO cells developed in the present study provide a unique platform to compare the impact of different IRs and proliferative capacity of human T-cell based products. A single antigen specificity allowed the assessment of antitumor activity without the confounding factors of unspecific or allo-reactivities or xeno-reactivity, and also permitted to shed light on TIM-3-, LAG-3-, or 2B4-mediated exhaustion mechanisms in antigen-specific human T cells. Furthermore, the evaluation of cytotoxicity in short-term assays showed that TCRED-TIM-3KO, TCRED-LAG-3KO, and TCRED-2B4KO cells display comparable killing activity to TCRED-IRCOMP cells. These results indicate that IR-disrupted T cells retain their killing capacity, thus proving that TIM-3, LAG-3, or 2B4 are not required on T cells for effective effector functions.
Although originally interpreted as a homogeneous state of T cell dysfunction, T cell exhaustion appears today as a progressive developmental path, characterized by different cell states, with heterogeneous functional phenotypic profiles (68, 69). In line with this notion, in our study, effects of IRKO in our cells were heterogenous, and amplified upon increase in the complexity of the experimental conditions. In short term culture, TCRED-IRKO cells were similar to TCRED-IRCOMP cells in killing efficiency and degranulation ability, but with increased capacity to secrete pro-inflammatory cytokines. Upon chronic antigen stimulation, TCRED-TIM-3KO and TCRED-2B4KO lymphocytes showed a higher degranulation capacity than TCRED-IRCOMP counterparts. Finally, upon in vivo tumor rechallenge, all TCRED-IRKO proved superior to TCRED-IRCOMP counterparts in killing cancer cells.
Although IRs have been extensively investigated in infectious and cancer models, the role of each individual inhibitory receptor has not been compared in tumor-specific cellular products. This side-by-side comparison would be pivotal in the development of tailored and effective cellular products for cancer patients. Here, although a similar positive effect was detected in all TCRED-IRKO cells, upon chronic in vitro antigen stimulation and in vivo, data indicate that the disruption of TIM-3, LAG-3, or 2B4 might affect different T cell effector functions. Gene expression analysis uncovered enrichment of genes involved in proliferation and cell cycle upon LAG-3 disruption, and of genes associated with effector response in 2B4-disrupted T cells. Interestingly, the few DEGs identified in TCRED-TIM-3KO T cells, were highly enriched for long non coding RNA (lncRNA) transcripts. This observation is in line with a recent report showing specific lncRNA regulating TIM-3 signaling in TILs (44) and supports the notion that inhibitory receptors dampen T cell responses through distinct mechanisms.
Chronic stimulation with low Ag burden has been reported to model T cell exhaustion in vitro (51–53). The disruption of TIM-3 and 2B4 increases the degranulation capacity of NY-ESO-1 specific T cells upon chronic stimulation with multiple myeloma cells. Since exhausted T cells often co-express several IRs, multiple IR gene disruption might be beneficial. Based on our in vitro and in vivo observations, and based on the results of trascriptomic data, we propose LAG-3/TIM-3 and LAG-3/2B4 kock-out combinations, to be prioritized based on the frequency and relevance of TIM-3 and 2B4 in the selected diseases.
In a high tumor burden setting, designed to foster T cell exhaustion, we observed that TCRED-LAG-3KO cells infiltrating the bone marrow in conditions of antigen persistence display a reduced induction of compensatory inhibitory molecules (CTLA-4, PD-1 and TIM-3), thus further supporting our in vitro findings. In the same model, high doses of TCRED-LAG-3KO cells mediated a significantly higher anti-tumor activity than TCRED-IRCOMP cells.
The final aim of our study was to reveal whether IR disruptions in our cellular products provide resistance to T-cell exhaustion and promote functional immunological memory. To this aim, the anti-tumor responses of TCRED-IRCOMP, TCRED-LAG-3KO, TCRED-TIM-3KO and TCRED-2B4KO cells were compared in a tumor rechallenge model. Upon tumor control in mice, we challenged T cells in vivo with a second infusion of tumor cells. Only TCRED-IRKO cells were able to completely eradicate the second tumor engraftment, indicating their ability to resist functional exhaustion. The enhanced fitness of TCRED-IRKO cells was also reflected by the higher proportion of activated IRKO tumor-specific T cells infiltrating the bone marrow compared to TCRED-IRCOMP cells, at sacrifice. Interestingly, in accordance with the transcriptional findings, TCRED-LAG-3KO T cells displayed the highest and most consistent expansion profile, after tumor rechallenge, further confirming a unique mechanism of action for LAG3KO cellular products.
Limitations of our study include the lack of stable and reliable surface markers to quantify by flow cytometry IR-disrupted cells and the fact that immunodeficient mice models cannot recapitulate the entire tumor microenvironment. Additional information may be gained by future investigation of IRKO in additional tumor models, additional immunodeficient models and in immunocompetent mice. Our study provides a novel and unique platform to test the impact of single IR disruptions on antigen-specific cellular products and to shed lights on their mechanisms of action in clinically relevant models. In future developments, the fine-tuning of multiple IR disruptions tailored for specific tumor types and able to foster active proliferation in long-living memory cells will ultimately generate better cellular products for disease-specific adoptive T cell therapy approaches.
The data presented in the study are deposited in the Gene Expression Omnibus (GEO) repository, accession number GSE256035.
The studies involving humans were approved by San Raffaele Scientific Institutional Ethical Committee. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from primarily isolated as part of your previous study for which ethical approval was obtained. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. The animal study was approved by Institutional Animal Care and Use Committee (IACUC). The study was conducted in accordance with the local legislation and institutional requirements.
BCC: Conceptualization, Formal analysis, Funding acquisition, Investigation, Validation, Visualization, Writing – original draft. ZIM: Investigation, Methodology, Validation, Visualization, Resources, Writing – review & editing. AU: Investigation, Methodology, Validation, Visualization, Resources, Writing – review & editing. BC: Investigation, Resources, Methodology, Writing – review & editing. IM: Data curation, Formal Analysis, Writing – review & editing. ES: Data curation, Formal Analysis, Writing – review & editing. LP: Investigation, Writing – review & editing. AS: Investigation, Writing – review & editing. AP: Conceptualization, Formal Analysis, Investigation Visualization, Writing – review & editing. VV: Conceptualization, Formal Analysis, Investigation, Writing – review & editing. AI: Conceptualization, Formal Analysis, Investigation, Writing – review & editing. FM: Formal Analysis, Software, Writing – review & editing. DA: Formal Analysis, Software, Writing – review & editing. LV: Conceptualization, Writing – review & editing. RDM: Conceptualization, Writing – review & editing. FC: Conceptualization, Writing – review & editing. LN: Conceptualization, Writing – review & editing. PG: Conceptualization, Writing – review & editing. ER: Conceptualization, Formal Analysis, Funding acquisition, Project administration, Supervision, Visualization, Writing – review & editing. CB: Conceptualization, Formal Analysis, Funding acquisition, Project administration, Supervision, Visualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by AIRC (Ig 18458 and Ig 24965) and AIRC 5xMille, Rif. 22737, Italian Ministry of Research and University (PRIN 2015NZWsec; PRIN 2017WC8499), Italian Ministry of Health (Research project on CAR-T cells for hematological malignancies and solid tumors and RF-2019-12370243). BCC has been supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) fellowship. This work was partially supported by Ministero della Salute (Ricerca Finalizzata Grant Cianciotti et al. 25 No. GR-2016-02364847) to ER Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License, were used to create figures.
The authors thank all the members of Experimental Hematology Unit and Innovative Immunotherapies Unit for the help during the experiments and for the scientific discussion. The authors also thank prof. D. Busch for the scientific support during the project.
CB, ER, ZM, BC, AP, LV, FC, PG, LN and BCC are inventors on different patents on cancer immunotherapy and genetic engineering. CB has been member of Advisory Board and Consultant for Molmed, Intellia, TxCell, Novartis, GSK, Allogene, Kite/Gilead, Miltenyi, Kiadis, Evir, Janssen and received research support from Molmed s.p.a and Intellia Therapeutics. LV received royalties and research support from GEN-DX and research support from Moderna Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1315283/full#supplementary-material
1. Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. (2013) 39:49–60. doi: 10.1016/j.immuni.2013.07.002
2. Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. (1999) 163:507–13. doi: 10.4049/jimmunol.163.1.507.
3. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Sci (80-). (2006) 314:126–9. doi: 10.1126/science.1129003.
4. Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol. (2005) 174:4415–23. doi: 10.4049/jimmunol.174.7.4415.
5. Hughes MS, Yu YYL, Dudley ME, Zheng Z, Robbins PF, Li Y, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther. (2005) 16:457–72. doi: 10.1089/hum.2005.16.457.
6. Roszkowski JJ, Lyons GE, Kast WM, Yee C, Van Besien K, Nishimura MI. Simultaneous generation of CD8+ and CD4+ melanoma-reactive T cells by retroviral-mediated transfer of a single T-cell receptor. Cancer Res. (2005) 65:1570–6. doi: 10.1158/0008-5472.CAN-04-2076.
7. Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent anittumor. Blood. (2013) 122:3461–73. doi: 10.1182/blood-2013-04-493361.
8. Schultz LM, Davis KL, Baggott C, Chaudry C, Marcy AC, Mavroukakis S, et al. Phase 1 study of CD19/CD22 bispecific chimeric antigen receptor (CAR) therapy in children and young adults with B cell acute lymphoblastic leukemia (ALL). Blood. (2018) 132(supplement 1):898. doi: 10.1182/blood-2018-99-117445.
9. Weber EW, Maus MV, Mackall CL. The emerging landscape of immune cell therapies. Cell. (2020) 181:46–62. doi: 10.1016/j.cell.2020.03.001
10. Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. (2012) 18:807–15. doi: 10.1038/nm.2700.
11. Mastaglio S, Genovese P, Magnani Z, Ruggiero E, Landoni E, Camisa B, et al. NY-ESO-1 TCR single edited stem and central memory T cells to treat multiple myeloma without graft-versus-host disease. Blood. (2017) 130:606–18. doi: 10.1182/blood-2016-08-732636.
12. Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: Building the ultimate antitumour T cell. Nat Rev Cancer. (2012) 12:671–84. doi: 10.1038/nrc3322.
13. Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD81 memory stem cells for the treatment of human B-cell Malignancies. Blood. (2016) 128:519–28. doi: 10.1182/blood-2015-11-683847.
14. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6.
15. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Sci (80-). (1995) 270:985–8. doi: 10.1126/science.270.5238.985.
16. Fuertes Marraco SA, Neubert NJ, Verdeil G, Speiser DE. Inhibitory receptors beyond T cell exhaustion. Front Immunol. (2015) 6:1–14. doi: 10.3389/fimmu.2015.00310.
17. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated Melanoma. N Engl J Med. (2015) 373:23–34. doi: 10.1056/NEJMoa1504030.
18. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus Ipilimumab in advanced melanoma. N Engl J Med. (2013) 369:122–33. doi: 10.1056/NEJMoa1302369.
19. Morad G, Helmink BA, Sharma P, Wargo JA. LL Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. (2021) 184(21):5309–37. doi: 10.1016/j.cell.2021.09.020
20. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. (2010) 207:2175–86. doi: 10.1084/jem.20100637.
21. Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. (2010) 107:14733–8. doi: 10.1073/pnas.1009731107.
22. June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. (2017) 23:540–7. doi: 10.1038/nm.4321.
23. Stadtmauer EA, Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, et al. CRISPR-engineered T cells in patients with refractory cancer. (2020) 7365:1–20. doi: 10.1126/science.aba7365Vdoi: 10.1126/science.aba7365
24. Choi BD, Yu X, Castano AP, Darr H, Henderson DB, Bouffard AA, et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J Immunother Cancer. (2019) 7:1–8. doi: 10.1186/s40425-019-0806-7.
25. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-Tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. (2017) 7:1–10. doi: 10.1038/s41598-017-00462-8.
26. Giuffrida L, Sek K, Henderson MA, Lai J, Chen AXY, Meyran D, et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat Commun. (2021) 12:3236. doi: 10.1038/s41467-021-23331-5.
27. Guo X, Jiang H, Shi B, Zhou M, Zhang H, Shi Z, et al. Disruption of PD-1 enhanced the anti-tumor activity of chimeric antigen receptor T cells against hepatocellular carcinoma. Front Pharmacol. (2018) 9:1–15. doi: 10.3389/fphar.2018.01118.
28. Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med. (2017) 11:554–62. doi: 10.1007/s11684-017-0543-6.
29. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. (2017) 23:2255–66. doi: 10.1158/1078-0432.CCR-16-1300.
30. Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. (2019) 68:365–77. doi: 10.1007/s00262-018-2281-2
31. Zhang W, Shi L, Zhao Z, Du P, Ye X, Li D, et al. Disruption of CTLA-4 expression on peripheral blood CD8 + T cell enhances anti-tumor efficacy in bladder cancer. Cancer Chemother Pharmacol [Internet]. (2019) 83:911–20. doi: 10.1007/s00280-019-03800-x
32. Malaer JD, Marrufo AM, Porunelloor AM. 2B4 (CD244, SLAMF4) and CS1 (CD319, SLAMF7) in systemic lupus erythematosus and cancer. Clin Immunol. (2019) 204:50–6. doi: 10.1016/j.clim.2018.10.009.
33. Roy D, Gilmour C, Patnaik S, Wang LL. Combinatorial blockade for cancer immunotherapy: targeting emerging immune checkpoint receptors. Front Immunol. (2023) 14:1264327. doi: 10.3389/fimmu.2023.1264327
34. Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 instruct the generation ofhuman memory stem T cells from naive precursors.Blood. (2013) 121:573–85. doi: 10.1182/blood-2012-05-431718.
35. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. (2013) 29:15–21. doi: 10.1093/bioinformatics/bts635
36. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. (2014) 30:923–30. doi: 10.1093/bioinformatics/btt656
37. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol [Internet]. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
38. Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. (2021) 2:100141. doi: 10.1016/j.xinn.2021.100141.
39. Lex A, Gehlenborg N, Strobelt H, Vuillemot R, Pfister H. UpSet: visualization of intersecting sets. IEEE Trans Vis Comput Graph. (2014) 20:1983–92. doi: 10.1109/TVCG.2014.2346248.
40. Cianciotti BC, Ruggiero E, Campochiaro C, Oliveira G, Magnani ZI, Baldini M, et al. CD4 + memory stem T cells recognizing citrullinated epitopes are expanded in patients with Rheumatoid Arthritis and sensitive to TNF-α blockade. Arthritis Rheumatol. (2019) 72(4):565–75. doi: 10.1002/art.41157
41. Manfredi F, Abbati D, Cianciotti BC, Stasi L, Potenza A, Ruggiero E, et al. Flow cytometry data mining by cytoChain identifies determinants of exhaustion and stemness in TCR-engineered T cells. Eur J Immunol. (2021) 51(8):1992–2005. doi: 10.1002/eji.202049103.
42. Puccio S, Grillo G, Alvisi G, Scirgolea C, Galletti G, Maria E, et al. CRUSTY: a versatile web platform for the rapid analysis and visualization of high-dimensional flow cytometry data. Nat Commun. (2023) 14:5102. doi: 10.1038/s41467-023-40790-0.
43. Brummelman J, Haftmann C, Núñez NG, Alvisi G, Mazza EMC, Becher B, et al. Development, application and computational analysis of high-dimensional fluorescent antibody panels for single-cell flow cytometry. Nat Protoc. (2019) 14:1946–69. doi: 10.1038/s41596-019-0166-2.
44. Ji J, Yin Y, Ju H, Xu X, Liu W, Fu Q, et al. Long non-coding RNA Lnc-Tim3 exacerbates CD8 T cell exhaustion via binding to Tim-3 and inducing nuclear translocation of Bat3 in HCC. Cell Death Dis. (2018) 9:478. doi: 10.1038/s41419-018-0528-7.
45. O’Brien S, Thomas RM, Wertheim GB, Zhang F, Shen H, Wells AD. Ikaros imposes a barrier to CD8+ T cell differentiation by restricting autocrine IL-2 production. J Immunol. (2014) 192:5118–29. doi: 10.4049/jimmunol.1301992.
46. Mangarin L, Liu C, Zappasodi R, Holland P, Wolchok J, Merghoub T, et al. Tired and hungry: a potential role for CD47 in T cell exhaustion. In: Regular and young investigator award abstracts. BMJ Publishing Group Ltd (2020). p. A304.2–A305. doi: 10.1136/jitc-2020-SITC2020.0493
47. Esen E, Sergin I, Jesudason R, Himmels P, Webster JD, Zhang H, et al. MAP4K4 negatively regulates CD8 T cell-mediated antitumor and antiviral immunity. Sci Immunol. (2020) 5(45):eaay2245. doi: 10.1126/sciimmunol.aay2245.
48. Newton RH, Turka LA. Regulation of T cell homeostasis and responses by pten. Front Immunol. (2012) 3:151/abstract. doi: 10.3389/fimmu.2012.00151/abstract
49. Guy CS, Vignali KM, Temirov J, Bettini ML, Overacre AE, Smeltzer M, et al. Distinct TCR signaling pathways drive proliferation and cytokine production in T cells. Nat Immunol. (2013) 14:262–70. doi: 10.1038/ni.2538.
50. Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res. (2014) 2:70–9. doi: 10.1158/2326-6066.CIR-13-0160.
51. Nelson N, Lopez-Pelaez M, Palazon A, Poon E, de la Roche M, Barry S, et al. A cell-engineered system to assess tumor cell sensitivity to CD8 + T cell-mediated cytotoxicity. Oncoimmunology. (2019) 8:1–10. doi: 10.1080/2162402X.2019.1599635
52. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092.
53. Ambrose C, Su L, Wu L, Dufort FJ, Sanford T, Birt A, et al. Anti-CD19 CAR T cells potently redirected to kill solid tumor cells. PloS One. (2021) 16:e0247701. doi: 10.1371/journal.pone.0247701
54. Kaneko S, Mastaglio S, Bondanza A, Ponzoni M, Sanvito F, Aldrighetti L, et al. IL-7 and IL-15 allow the generation of suicide gene–modified alloreactive self-renewing central memory human T lymphocytes. Blood. (2009) 113:1006–15. doi: 10.1182/blood-2008-05-156059.
55. Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. (2010) 16:565–70. doi: 10.1038/nm.2128.
56. Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin Cancer Res. (2015) 21:1019–27. doi: 10.1158/1078-0432.CCR-14-2708.
57. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. C A N C E R Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Science translational medicine. (2020) 9(374):eaaj2013. doi: 10.1126/scitranslmed.aaj2013vvv
58. Bäsecke J, Griesinger F, Trümper L, Brittinger G. Leukemia- and lymphoma-associated genetic aberrations in healthy individuals. Ann Hematol. (2002) 81:64–75. doi: 10.1007/s00277-002-0427-x
59. Ismail SI, Naffa RG, Yousef AF, Ghanim MT. Incidence of bcr−abl fusion transcripts in healthy individuals. Mol Med Rep. (2014) 9:1271–6. doi: 10.3892/mmr.2014.1951/abstract
60. Chiarle R. Translocations in normal B cells and cancers: insights from new technical approaches. Adv Immunol. (2013) 117:39–71. doi: 10.1016/B978-0-12-410524-9.00002-5.
61. Fuertes Marraco SA, Soneson C, Cagnon L, Gannon PO, Allard M, Maillard SA, et al. Long-lasting stem cell-like memory CD8+ T cells with a naïve-like profile upon yellow fever vaccination. Sci Transl Med. (2015) 7(282):282ra48. doi: 10.1126/scitranslmed.aaa3700.
62. Oliveira G, Ruggiero E, Stanghellini MTL, Cieri N, D’Agostino M, Fronza R, et al. Tracking genetically engineered lymphocytes long-term reveals the dynamics of t cell immunological memory. Sci Transl Med. (2015) 7:1–14. doi: 10.1126/scitranslmed.aac8265.
63. Biasco L, Scala S, Basso Ricci L, Dionisio F, Baricordi C, Calabria A, et al. In vivo tracking of T cells in humans unveils decade-long survival and activity of genetically modified T memory stem cells. Sci Transl Med. (2015) 7:273ra13. doi: 10.1126/scitranslmed.3010314.
64. Cieri N, Oliveira G, Greco R, Forcato M, Taccioli C, Cianciotti B, et al. Generation of human memory stem T cells after haploidentical T-replete hematopoietic stem cell transplantation. Blood. (2015) 125:2865–74. doi: 10.1182/blood-2014-11-608539.
65. Roberto A, Castagna L, Zanon V, Bramanti S, Crocchiolo R, McLaren JE, et al. Role of naive-derived T memory stem cells in T-cell reconstitution following allogeneic transplantation. Blood. (2015) 125:2855–64. doi: 10.1182/blood-2014-11-608406.
66. Potenza A, Balestrieri C, Spiga M, Albarello L, Pedica F, Manfredi F, et al. Revealing and harnessing CD39 for the treatment of colorectal cancer and liver metastases by engineered T cells. GUT. (2023) 72:1887–903. doi: 10.1136/gutjnl-2022-328042.
67. Dötsch S, Svec M, Schober K, Hammel M, Wanisch A, Gökmen F, et al. Long-term persistence and functionality of adoptively transferred antigen-specific T cells with genetically ablated PD-1 expression. Proc Natl Acad Sci. (2023) 120(10):e2200626120. doi: 10.1073/pnas.2200626120
68. Gilles JR, Globig AM, Kaech SM, Wherry EJ. CD8+ T cells in the cancer-immunity cycle. Immunity. (2023) 56:2231–53. doi: 10.1016/j.immuni.2023.09.005
Keywords: TCR - T cell receptor, adoptive T cell immunotherapy, inhibitory receptor, genome editing, CRISPR/Cas9
Citation: Cianciotti BC, Magnani ZI, Ugolini A, Camisa B, Merelli I, Vavassori V, Potenza A, Imparato A, Manfredi F, Abbati D, Perani L, Spinelli A, Shifrut E, Ciceri F, Vago L, Di Micco R, Naldini L, Genovese P, Ruggiero E and Bonini C (2024) TIM-3, LAG-3, or 2B4 gene disruptions increase the anti-tumor response of engineered T cells. Front. Immunol. 15:1315283. doi: 10.3389/fimmu.2024.1315283
Received: 10 October 2023; Accepted: 05 February 2024;
Published: 29 February 2024.
Edited by:
Hans-Jochem Kolb, Kolb Consulting UG, GermanyReviewed by:
Sabrina N. Copsel, Makana Therapeutics Inc., United StatesCopyright © 2024 Cianciotti, Magnani, Ugolini, Camisa, Merelli, Vavassori, Potenza, Imparato, Manfredi, Abbati, Perani, Spinelli, Shifrut, Ciceri, Vago, Di Micco, Naldini, Genovese, Ruggiero and Bonini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Bonini, Ym9uaW5pLmNoaWFyYUBoc3IuaXQ=; Eliana Ruggiero, cnVnZ2llcm8uZWxpYW5hQGhzci5pdA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.