 Jiyoung Lee
Jiyoung Lee J.-H. James Ou
J.-H. James Ou- Department of Molecular Microbiology and Immunology, University of Southern California, Keck School of Medicine, Los Angeles, CA, United States
The interplay between autophagy and host innate immunity has been of great interest. Hepatitis C virus (HCV) impedes signaling pathways initiated by pattern-recognition receptors (PRRs) that recognize pathogens-associated molecular patterns (PAMPs). Autophagy, a cellular catabolic process, delivers damaged organelles and protein aggregates to lysosomes for degradation and recycling. Autophagy is also an innate immune response of cells to trap pathogens in membrane vesicles for removal. However, HCV controls the autophagic pathway and uses autophagic membranes to enhance its replication. Mitophagy, a selective autophagy targeting mitochondria, alters the dynamics and metabolism of mitochondria, which play important roles in host antiviral responses. HCV also alters mitochondrial dynamics and promotes mitophagy to prevent premature cell death and attenuate the interferon (IFN) response. In addition, the dysregulation of the inflammasomal response by HCV leads to IFN resistance and immune tolerance. These immune evasion properties of HCV allow HCV to successfully replicate and persist in its host cells. In this article, we discuss HCV-induced autophagy/mitophagy and its associated immunological responses and provide a review of our current understanding of how these processes are regulated in HCV-infected cells.
Introduction
Despite significant advances in its treatment and the availability of efficacious direct-acting antiviral drugs (DAAs), hepatitis C virus (HCV) remains prevalent around the world with approximately 58 million chronic HCV carriers and 1.5 million new infections every year. Chronic HCV infection can lead to severe liver diseases including steatosis, cirrhosis and hepatocellular carcinoma (HCC). HCV is a positive-stranded RNA virus with a genome size of approximately 9600 nucleotides. Its RNA genome is packaged in an icosahedral capsid, which is further surrounded by a lipid envelope (1). HCV infection of hepatocytes is initiated by receptor-mediated endocytosis followed by the fusion of the viral envelope with the endocytic membrane and the release of its RNA genome into the cytosol. The RNA genome of HCV then directs the synthesis of viral proteins using the internal ribosome entry site (IRES) located near its 5’-end, producing a polyprotein that is roughly 3000 amino acids in length. Viral and host proteases proteolytically cleave the HCV polyprotein, giving rise to ten viral proteins, which are the core protein, E1 and E2 envelope proteins, the p7 viroporin, and nonstructural proteins NS2, NS3, NS4A, NS4B, NS5A and NS5B.
Although cells in response to HCV infection can generate antiviral responses, HCV has developed means to evade these antiviral responses to establish persistent infection. Approximately 75-85% of patients infected by HCV fail to clear the virus and become chronically infected. Autophagy can remove intracellular microbial pathogens and is a part of the antiviral responses. However, HCV exploits autophagy to overcome this host antiviral response. In this article, we will discuss the interaction between HCV-induced autophagy and the host innate immune responses. We hope this article will provide not only the information for understanding the HCV-host interaction to assist the development of HCV vaccines, which are not yet available, but also the information for understanding how cells may respond to other viral infections.
Evasion of host innate immune response by HCV
Pattern-recognition receptors (PRRs)
PRRs play important roles in triggering anti-microbial responses. They recognize pathogen-associated molecular patterns (PAMPs) and, upon binding to PAMPs, will trigger anti-microbial responses. There are three classes of PRRs, which are the retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs). RLRs, including RIG-I, melanoma differentiation antigen 5 (MDA5) and the laboratory of genetics and physiology 2 (LGP2), are RNA helicases that recognize RNA PAMPs (2). RIG-I and MDA5 recognize double-stranded RNA (dsRNA). Although the specific PAMPs that they detect are different, their activation by HCV RNA can both lead to the production of interferons (IFNs) (3, 4). RIG-I recognizes the 5’-PPP and the 3’ poly U/UC sequence of the HCV genomic RNA (5). Once activated, the caspase activation and recruitment domain (CARD) of RIG-I interacts with the mitochondrial adaptor protein MAVS (also known as IPS-1, VISA or Cardif) on mitochondrial outer membranes. MAVS then further recruits TRAF3 and TRAF6 and subsequently activates the downstream TANK-binding kinase 1 (TBK1). TBK1 phosphorylates and activates Interferon Regulatory Factor 3 (IRF3), whose homodimerization and translocation to the nucleus activates the IFN genes. LGP2, which lacks the CARD domain, binds to the foreign RNA and synergizes with MDA5 to induce innate immune signaling (2).
HCV proteins and their effects on interferon signaling pathways
Hepatocytes infected by HCV produce type I and type III IFNs and upregulate the expression of IFN-stimulated genes (ISGs). Type I IFN-α and IFN-β bind to the heterodimer of IFN-α receptor (IFNAR) 1 and 2, type II IFN-γ binds to the heterodimeric complex of IFN-γ receptor (IFNGR) 1 and 2 and type III IFN-λ binds to the heterodimeric complex of IFN-λ receptor (IFNLR) and IL-10Rβ. Type I and III IFN signaling is mediated by Janus kinases (JAKs) and STAT proteins and induces the expression of ISGs including ISG15, 2’,5’-oligoA synthase (OAS), protein kinase RNA-activated (PKR), the GTPase myxovirus resistance 1 (Mx1) and ribonuclease L (RNase L), to name a few (6). Nonetheless, HCV proteins can dampen the host IFN response to enhance its replication (Figure 1). NS3/4A, the viral serine protease complex of HCV, plays a crucial role in hampering the antiviral signaling of IFNs. NS3/4A cleaves MAVS, the adaptor protein of the RIG-I signaling (7, 8). The introduction of a point mutation at cysteine-508 (Cys-508) to prevent MAVS from being cleaved by NS3/4A restored the IFN production in HCV-infected cells or cells containing an HCV subgenomic RNA replicon (4, 9). The mitochondria-associated membranes (MAMs) at the endoplasmic reticulum (ER)-mitochondria contact site serve as the site for MAVS signaling (Figure 1) (8). Also, NS3/4A can cleave peroxisome-associated MAVS and disrupt RIG-I signaling initiated from peroxisomes (10). In addition, NS3/4A can cleave the TIR-domain containing adapter-inducing IFN-β (TRIF), an adaptor protein of the TLR3 signaling pathway (Figure 1) (10, 11). HCV protein NS4B was also reported to induce the degradation of TRIF via caspase-8 (12). Other HCV proteins like core, E2, NS4B, and NS5A had also been shown to be involved in the resistance to IFNs by inducing the suppressor of the cytokine signaling proteins 1 and 3 (SOCS1 and 3), sequestering STING to inhibit the activation of TBK1 and sequestering MyD88 to inhibit TLR signaling [for more details, see reviews (13–15)]. In 293T cells, NS3/4A bound to TBK1 and interfered with its association with IRF3 (16). PKR, a dsRNA-activated kinase, is an antiviral protein that is also induced by IFNs. HCV NS5B has been shown to induce the activation of PKR and the phosphorylation and inactivation of its downstream effector eIF2α, a protein translation initiation factor, to suppress the production of IFNs, ISGs and MHC class I (Figure 1) (17–20).
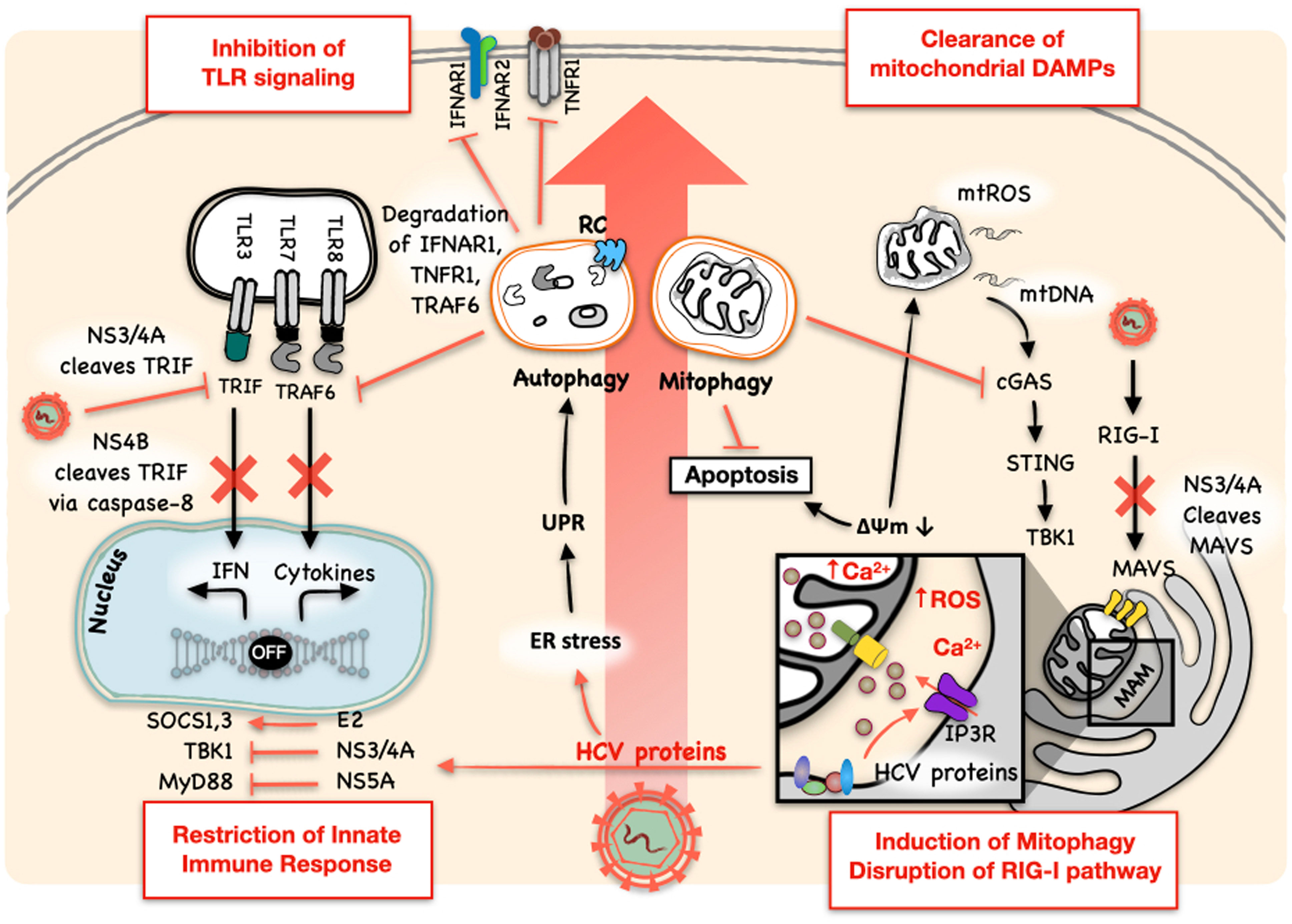
Figure 1 HCV inhibits the innate immune response. HCV proteins can directly inhibit the innate immune response. NS3/4A plays multiple roles in blocking the host innate immune response. It cleaves MAVS on MAM to block the RIG-I signaling pathway and TRIF to inhibit the TLR3 signaling pathway. NS4B can also disrupt TLR3-mediated IFN signaling by inducing the degradation of TRIF via caspase-8. Other HCV proteins had also been shown to regulate and restrict the production of IFNs and their antiviral signaling: E2 induces SOCS1 and SOCS3; NS3/4A sequesters STING to inhibit the activation of TBK1; NS5A sequesters MyD88 to inhibit TLR signaling; and NS5B induces and activates PKR to inhibit the expression of IFNs and ISGs. HCV also usurps the antiviral activity of autophagy and uses it for its own replication. It not only utilizes the autophagic membranes as the sites for its RNA replication and the production of infectious HCV particles, but also manipulates autophagy to regulate type I IFN response by promoting the autophagic degradation of IFNAR1, TNFR1, and TRAF6, important signal transducers for the activation of NF-κB and the expression of pro-inflammatory cytokines. HCV-induced mitophagy also regulates innate immune response by clearing mitochondrial DAMPs, namely mtROS and mtDNA, and damaged mitochondria themselves, which otherwise would activate the cGAS/STING/TBK1 pathway to induce the expression of IFNs. Mitophagy also inhibits premature apoptotic cell death and promotes HCV persistence. RC: HCV RNA replication complex. The mitochondria-associated membrane (MAM) is boxed and enlarged in the inset to reveal the Ca2+ transporter IP3R.
HCV-induced autophagy and innate immune response
HCV-induced autophagy
Autophagy is a catabolic process by which cells deliver damaged organelles and dysfunctional cellular components to lysosomes for degradation. In the early stage of autophagy, a crescent membrane structure termed phagophore appears in the cytoplasm. The subsequent expansion of the membranes of phagophores leads to the formation of an enclosed double-membrane structure termed autophagosomes. Autophagosomes fuse with lysosomes to form autolysosomes. The cargoes of autophagosomes are then digested by lysosomal enzymes in autolysosomes for recycling. Autophagy is important for maintaining cellular homeostasis. It is closely associated with the growth and energy state of the cell, and the activation of mTOR suppresses the initiation of autophagy. mTOR regulates cell growth and metabolism, integrates the cellular nutrient state with environmental signals and controls the initiation of autophagy by regulating the activation of AMPK, the AMP sensor. HCV induces autophagy via multiple pathways, including the expression of viral proteins, nutrient exploitation, alteration of cellular metabolism and production of reactive oxygen species (ROS). Interaction of HCV NS3 with immunity-related GTPase family M protein (IRGM) is sufficient to induce autophagy (21). HCV triggers the activation of both IRGM and early autophagy initiator Unc-51-like kinase 1 (ULK1), which are required for membrane remodeling and Golgi fragmentation, to initiate autophagy (22). NS4B can also bind to Beclin-1, hVps34 and Rab5 to initiate autophagy [for a detailed review, see (23)].
As mentioned above, intracellular events like the ER stress and the generation of ROS from mitochondria in response to HCV infection can also induce autophagy. HCV-induced ER stress stimulates the unfolded protein response (UPR), which is triggered by the activation of the inositol-requiring enzyme 1 (IRE1), the activating transcription factor 6 (ATF6) and the double-stranded RNA-activated protein kinase-like ER kinase (PERK) (24). The UPR attenuates protein synthesis, promotes chaperone-mediated protein folding and facilitates the degradation of misfolded proteins to alleviate the ER stress (23). The silencing of IRE1, ATF6 or PERK to disrupt the UPR, which also disrupts HCV-induced autophagy, or the silencing of autophagy-related proteins Atg5, Atg7 and Atg12 suppressed the replication of HCV, indicating that the UPR and autophagy benefit HCV replication (24–27) (Figure 1). The importance of autophagy in HCV infection can be partially attributed to the fact that autophagic membranes, where lipid rafts rich in cholesterol and sphingolipids are also localized, can serve as the sites for the assembly of the HCV RNA replication complex (24, 28–30). ROS is a byproduct of mitochondrial aerobic respiration. The enhanced generation of ROS or oxidative stress was observed in cell cultures infected by HCV and in the liver of chronic hepatitis C patients (31–33). The elevation of ROS prompted the phosphorylation of the sequestosome protein p62 at Ser349, which is implicated in the initiation of autophagy (33). p62 binds to poly-ubiquitinated proteins for their sequestration in autophagosomes for autophagic degradation. Although ROS has antiviral potential as demonstrated in the studies of different viruses, HCV as well as human immunodeficiency virus type 1 (HIV-1), influenza virus and dengue virus exploits ROS-induced autophagy for their genome replication and immune suppression (33–35).
The term xenophagy depicts the process by which intracellular microbial pathogens are removed by autophagy. The removal of viruses by autophagy is termed virophagy (36, 37). These terms describe autophagy as a host antiviral response that clears intracellular viral particles (37). However, HCV can usurp the antiviral activity of autophagy and use it to benefit its own replication (24, 38) (Figure 1). First, HCV can use autophagic membranes as the sites for its RNA replication (24, 27, 30). Second, HCV induces Rubicon to delay the maturation of autophagosomes to maximize its RNA replication (24, 39) (see below). Third, HCV promotes the interaction between its envelope protein E2 and apolipoprotein E (ApoE), which is important for enhancing the infectivity of HCV, using autophagic membranes (29, 40). Fourth, autophagy suppresses type I IFN response (for detailed reviews, see (41, 42)), and the silencing of UPR- or autophagy-related genes increased the expression of ISGs and reduced HCV replication (25). Finally, HCV dampens the innate immune response by stimulating the autophagic degradation of TRAF6, an adaptor protein important for the activation of nuclear factor-κB (NF-κB) and the expression of pro-inflammatory cytokines (24, 34, 42), as well as the autophagic degradation of TNFR1 and IFNAR1, which are important for the IFN response (43, 44).
HCV-induced autophagy and IFN response
The interplay between HCV-induced autophagy and the host immune response has attracted a lot of attention (45, 46). Under normal conditions, cells maintain a basal autophagy activity to regulate energy and nutrient states and remove damaged organelles and protein aggregates to maintain cellular homeostasis. Numerous disorders like heart diseases, neurodegenerative diseases and tumors are associated with the impairment of autophagy, which leads to disruption of many intracellular processes. Among them is innate immune responses (47). Autophagy also regulates innate immune responses in HCV-infected cells. Although there were conflicting results regarding whether the inhibition of autophagic initiation affected the type 1 IFN pathway in HCV-infected cells (25, 46), the silencing of RUN Domain Beclin-1-interacting and cysteine-rich domain-containing protein (Rubicon) stimulated the IFN response. Rubicon plays a critical role in the control of maturation of autophagosomes (48, 49). It can bind to the complex of UV radiation resistance-associated gene (UVRAG) and class III phosphatidylinositol-3-kinase (PI3KC3) to suppress the maturation of autophagosomes (48). It can also sequester UVRAG from class C vacuolar protein sorting (C-Vps)-homotypic fusion and protein sorting (HOPS) complex to inhibit the activation of Rab7 and the fusion between autophagosomes and lysosomes (49). HCV induces the expression of Rubicon in the early stage of infection to prevent the maturation of autophagosomes, leading to the accumulation of autophagosomes to enhance HCV RNA replication (39, 46). However, the inhibition of this late-stage of autophagy, which prevents the autophagic protein degradation, also activates the innate immune response (46) (Figure 2). The silencing of autophagic proteins like LC3B to prevent the formation of autophagosomes or the treatment with chloroquine to inhibit autophagic protein degradation enhances IFN-β production and IFN signaling in HCV-infected cells (25). In addition, Chandra et al. showed that IFNAR1 was degraded by autophagy induced by HCV infection (43). The use of thapsigargin (TG) to induce ER stress or the inhibition of mTOR downregulated IFNAR1, and the inhibition of autophagy by silencing Atg7 or PERK upregulated the expression of IFNAR1 in the presence of HCV. As a result, the alleviation of the ER stress or the inhibition of autophagy using chemical inhibitors or siRNA enhanced viral clearance in cell cultures treated with both IFN-α and ribavirin (RBV) (43). These results demonstrated an important role of autophagic protein degradation, the late stage of autophagy, in the control of IFN response in HCV-infected cells. In contrast to HCV, it was shown that the silencing of Rubicon with shRNA, which promoted the maturation of autophagosomes, enhanced IFN-β and IL-6 production and inhibited the replication of H1N1 influenza virus and vesicular stomatitis virus (VSV) (50).
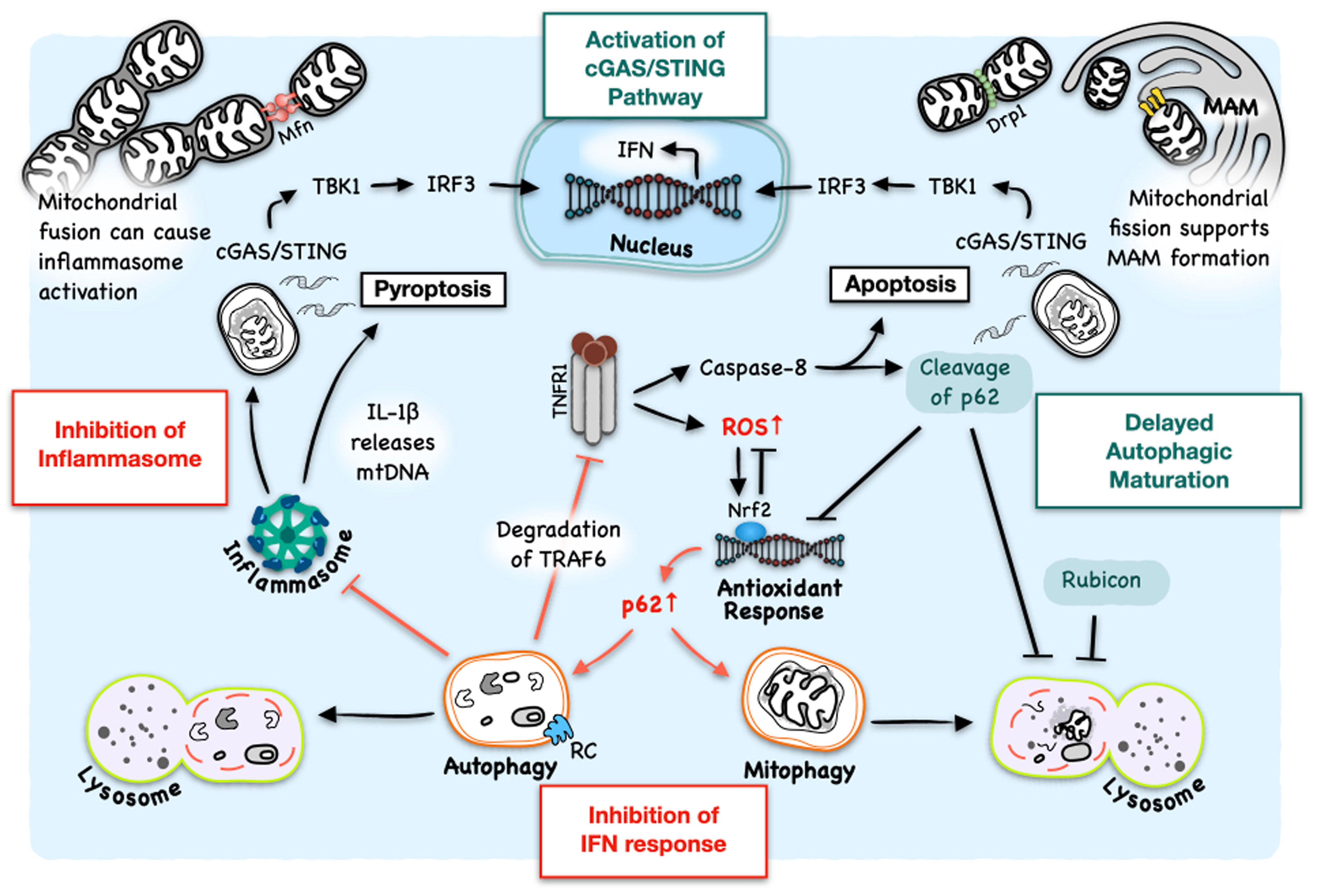
Figure 2 Interplay between HCV-induced autophagy and host innate immune response. HCV-induced autophagy removes DAMPs to inhibit the formation of inflammasomes and hence pyroptosis. HCV-induced autophagic degradation of TNFR1 also inhibits the activation of caspase-8 and apoptosis. However, host cells in response to HCV infection also express TNF-α and IFNAR2 (also see Figure 1). TNF-α at a low concentration causes the generation of ROS to initiate autophagy. However, it, at a high concentration, can also put a halt on the completion of autophagy via caspase-8-mediated cleavage of p62, ultimately activating the cGAS/STING/TBK1 pathway and apoptosis. TNF-α-induced cleavage of p62 also disrupts Nrf2 signaling, which is activated by HCV to induce the expression of antioxidant genes to alleviate oxidative stress. HCV also delays the maturation of autophagosomes by inducing the expression of Rubicon for the accumulation of autophagosomes to support its RNA replication, which can also lead to the accumulation of DAMPs. Mitochondrial dynamics are closely related to the regulation of innate immune response. On the other hand, mitochondrial fusion supports inflammasome activation and pro-survival pathways that, if prolonged, can contribute to tumorigenesis. On the other hand, mitochondrial fission, which is coupled with mitophagy and promoted by HCV, supports the formation of MAM which can serve as the platform to mediate IFN response. RC: HCV replication complex.
HCV-induced autophagy and antioxidant response
NF-E2-related factor 2 (Nrf2) activates antioxidant gene expression in response to oxidative stress. The E3 ubiquitin ligase Kelch-like ECH-associated protein 1 (Keap1) associates with Nrf2 to keep it in an inactive state. The killer inhibitory receptor (KIR) domain of p62 allows p62 to bind to Keap1 to free Nrf2. This allows Nrf2 to move into the nucleus to bind to the antioxidant response element (ARE) to induce the expression of antioxidant genes (51). Since the p62 promoter contains the ARE, the activated Nrf2, in turn, induces the expression of p62 (52). On the other way around, Keap1, by binding to p62, prevents p62 from interacting with LC3 and delivering cargoes to autophagosomes for autophagic degradation. This can impede the autophagic removal of dysfunctional mitochondria under oxidative stress and activate the cGAS/STING pathway to lead to the production of IFN (more details below). In the meantime, p62 can associate with Raptor to activate mTOR complex 1 (mTORC1) and promote the pro-survival pathway (53). The activation of Nrf2 in response to HCV infection is mediated by the mitogen-activated protein kinase (MAPK) p38 and JAK, and the knockdown of Nrf2 inhibits the phosphorylation of the kinase Akt and its downstream effector Bad, suggesting a role of Nrf2 in the survival of HCV-infected cells and their possible oncogenic transformation (54). Indeed, the accumulation of p62 was observed in multiple cancers including tumor tissues positive for HCV (55, 56).
HCV-induced autophagy and inflammasome
Inflammasome is a multiprotein complex composed of an NLR such as NLRP3, the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) and the effector protease caspase-1. It can be activated by PAMPs, damage-associated molecular patterns (DAMPs), and toxins (57). The assembly of inflammasomes leads to the activation of caspase-1, which then cleaves IL-1β and IL-18 to produce their mature form for release from cells. The formation of inflammasomes also triggers pyroptosis, a regulated form of cell death associated with a high inflammation state. Caspase-1 also cleaves Gasdermin D (GSDMD) to produce GSDMD-C and GSDMD-N. GSDMD-N oligomerizes to form membrane pores on the plasma membrane for the release of IL-1β and IL-18 and eventually leads to pyroptosis (57, 58).
In response to HCV infection, the NLRP3 inflammasome is activated in hepatocytes and macrophages, leading to the release of IL-1β (59, 60). Elevated serum level of IL-1β was observed in chronic HCV patients as well (61). HCV can also enter Kupffer cells, the resident macrophages of the liver, and trigger the TLR7-mediated inflammasome activation and the release of IL-1β (61). The expression of HCV proteins, the increase of ROS, the induction of ER stress and other organelle stresses caused by HCV infection may exacerbate the inflammasome response (59). Lipid accumulation was shown to activate the NLRP3 inflammasome as well (59, 62, 63). The HCV core protein can also stimulate IL-1β production from macrophages, and this process requires HCV core-induced Ca2+ mobilization and phospholipase C activation (59, 61).
Autophagy, via lysosomal degradation, eliminates the activators of inflammasomes, such as inflammasome components, cytokines, mitochondrial ROS (mtROS), mitochondrial DNA (mtDNA) and damaged mitochondria. Thus, autophagy negatively regulates the NLRP3 inflammasome to prevent excessive inflammation, which can lead to the development of cancer (64). HCV induces the expression of Rubicon to inhibit the maturation of autophagosomes and disrupt the autophagic flux at as early as 6 hours post-infection in cell cultures (39). It is conceivable that this disruption of autophagic flux plays a role in the induction of inflammasomes and pyroptosis, which are detected in cells 2-3 days after HCV infection (60). HCV also induces the expression of NLRP3, an important component of inflammasomes, and the silencing of NLRP3 inhibits HCV-induced Golgi fragmentation, which is a possible source of autophagosomal membranes in HCV-infected cells (65). Thus, there appears to be a two-way crosstalk between autophagy and inflammasomes in HCV-infected cells.
HCV-induced inflammasome and innate immune response
The prompt removal of cytotoxic agents by autophagy protects cells from dangers, and the insufficient induction of autophagy or delay in autophagic degradation can cause hyperinflammatory responses (66). HCV delays the maturation of autophagosomes for autophagosomal membranes to accumulate to support the replication of its genome. However, the inflammation may arise due to the delay in the autophagic clearing of DAMPs. Aarreberg et al. found that IL-1β could induce the release of mtDNA into the cytosol to activate cyclic GMP-AMP synthase (cGAS), a cytosolic DNA sensor (Figure 2). This led to the production of the second messenger cyclic GMP-AMP (cGAMP) and the activation of STING followed by the activation of IRF3 and NF-κB to induce the expression of IFNs and proinflammatory cytokines (67). Type I IFNs, in turn, can repress the NLRP1- and NLRP3-dependent IL-1β production via the STAT1 transcription factor, which can also induce the expression of IL-10 to activate STAT3 through an autocrine mechanism to suppress the expression of pro-IL-1α and pro-IL-1β (68). Nitric Oxide (NO) induction or inducible nitric oxide synthase (iNOS) deficiency induced by TNF-α or IFN-γ suppresses inflammasome activation, preventing prolonged inflammation (69, 70). The mutually exclusive relationship between inflammasome and IFN production is well depicted by Banerjee et al. (71). They showed that inflammasome- and caspase-1-mediated cleavage of GSDMD and the subsequent formation of GSDMD pores led to K+ efflux and negatively regulated the IFN-β production, and GSDMD knockout enhanced the activation of TBK1 and IRF3 in response to intracellular poly(dA:dT) or Francisella novicida infection (71). In HCV-infected cells, Wallace et al. observed that pyroptosis preceded apoptosis, and the depletion of NLRP3 increased the activation of caspase-3, an executioner caspase of apoptosis (60) (Figure 2), most likely due to the increase in innate immune signaling (72). Interestingly, caspase-3 knockout also decreased the activation of caspase-1, suggesting a possible role of caspase-3 in the initiation of pyroptosis (60). The continuous interplays between IL-1β and IFN in the microenvironment of HCV-infected cells may contribute to immune tolerance, IFN resistance, an extended inflammation state and the development of liver cirrhosis and HCC (73, 74) (Figure 2). This may also be the reason why the interferon therapy does not generate sustained virological response in the great majority of HCV patients (75).
HCV-induced mitophagy and innate immune response
HCV-induced mitophagy and immune response
Maintaining a healthy pool of mitochondria is critical for cellular function and survival. Mitochondria undergo constant fusion and fission via guanosine triphosphatases (GTPases)-dependent activities. Mitofusins (Mfn1 and Mfn2), fusogenic transmembrane GTPases located on the mitochondrial outer membrane, bridge two adjacent mitochondria to promote mitochondrial fusion. Dynamin-related protein 1 (Drp1) and its receptor mitochondrial fission factor (Mff) promote mitochondrial fission by forming helical oligomers of Drp1 that encircle the mitochondrial outer membrane. Healthy mitochondria maintain a low Ca2+ level in their matrix where matrix dehydrogenases and oxidative phosphorylation produce ATP. Mitochondrial dysfunction can cause a reduction in the mitochondrial membrane potential (ΔΨm) due to uncontrolled production of ROS or the mitochondrial Ca2+ influx. The decrease in ΔΨm leads to the opening of the mitochondrial permeability transition pore (MPTP), through which mtDNA and cytochrome c are released (76). Dysfunctional mitochondria and the accumulation of DAMPs eventually lead to the disruption of ATP production and promote anaerobic glycolysis (77). Ca2+ overload can also lead to mitochondrial fragmentation, the disruption of ATP synthesis, and apoptosis (78, 79).
Mitochondria-associated membranes (MAMs), the contact sites of ER and mitochondria, create microdomains with high flux of Ca2+ where the transportation of Ca2+ occurs (Figure 1). Ca2+ goes through ER-resident calcium transporters (RyR/IP3R) and mitochondrial voltage-dependent anion channel type 1 (VDAC 1) on the outer membrane of mitochondria (OMM) and the calcium uniporter (MCU) on the inner membrane of mitochondria (IMM). Mitochondrial fusion leads to the loss of MAMs where MAVS relays the RIG-I signaling, and failure in maintaining MAMs can compromise innate immunity. Instead, the fusion of dysfunctional mitochondria can lead to the activation of inflammasome (80) via the accumulation of mtROS and mtDNA within the prolonged network of damaged mitochondria (77). Ichinohe et al. showed that endogenous NLRP3 was associated with Mfns after the infection of influenza virus or encephalomyocarditis virus (EMCV) in the LPS-primed bone marrow-derived macrophages (BMMs) (80). The treatment with carbonyl cyanide m-chlorophenyl hydrazone (CCCP), a protonophore that dissipates ΔΨm, or the knockdown of Mfn2 decreased the secretion of IL-1β (80).
Mitochondrial fission precedes mitophagy, a selective autophagy that removes damaged mitochondria for degradation (77). Failure in clearance of damaged mitochondria will lead to the accumulation of DAMPs, particularly mtDNA, to trigger the transcription and translation of inflammatory cytokines (77). The immune-evoking signals like cytochrome c, ROS and mtDNA from damaged mitochondria can also stimulate host responses. During HCV infection, MAMs can provide a site for the replication of the viral genome (81). HCV proteins present in the MAMs cause mitochondrial Ca2+ overload followed by local production of ROS and trigger the collapse of ΔΨm that leads to apoptosis, eliminating HCV-infected cells (82). Although it was noted that HCV-induced ROS can play a role in restricting the replication of HCV (83–85), mitochondrial fission and mitophagy induced by HCV infection in cell cultures or in patients or by the expression of HCV protein NS5A alone can overcome the antiviral effect of ROS and ROS-induced apoptosis (86–89). By removing damaged mitochondria along with mitochondrial DAMPs, mitophagy plays a critical role in preventing cell death caused by damaged mitochondria and mtROS. Evidence showed that HCV could upregulate mitophagy-related proteins including Drp1, the Drp1 receptor Mff, Parkin and PINK1 (86, 87), and the inhibition of PINK1 or Parkin decreased HCV replication, suggesting a positive role of mitophagy in HCV infection (86). PINK1 is a protein kinase that phosphorylates and recruits Parkin, an E3 ubiquitin ligase, to the outer membrane of mitochondria. Parkin can then ubiquitinate mitochondrial outer membrane proteins to trigger mitophagy.
MAMs also serve as the sites where immune signaling complexes form to control the viral infection. In the case of influenza virus, the fusion state of mitochondria in A549 cells, a human lung cancer cell line, was associated with the translocation of Drp1 from the mitochondria to the cytosol after viral infection (90). MAMs restored after the fragmentation of mitochondria by Mito-C, a pro-fission compound, increased IFN production for viral clearance (90). Similarly, the silencing of MFN2 enhanced the IFN-β promoter activity and reduced the permissiveness of cells to HCV infection (8). It is of note that the depletion of Drp1 suppressed HCV release with an increase in innate immune response and apoptosis (87).
The antagonistic relationship between mitophagy and apoptosis in the context of immune response was recently confirmed by Kim et al. who found that HCV-induced mitochondrial fission coincided with the attenuation of apoptosis (87). The authors added that the increase in caspase-3 activation and mitochondria-mediated apoptosis was linked to the increase in innate immune signaling (87). The pro-survival and proliferation nature of mitophagy was established as Drp1-mediated mitochondrial fission was shown to deplete the tumor suppressor p53 and increase the phosphorylation of the retinoblastoma (Rb) protein, resulting in the G1/S cell cycle progression and tumorigenesis (91–93). Plus, under the hypoxic condition, nuclear p53 represses the expression of Bcl-2 interacting protein 3 (BNIP3) thereby attenuating mitophagy (94). BNIP3 induces mitophagy by triggering mitochondrial membrane depolarization (94).
Intriguingly, evidence showed that the delay in autophagy could prime cells for apoptosis. In CD14+ cells, IFN-α impaired autophagic degradation of mitochondria, resulting in the accumulation of mtDNA and ultimately the STING-mediated immune response (95). In HCV-infected cells, the inhibition of autophagosomal maturation by the upregulation of Rubicon also resulted in the accumulation of damaged mitochondria (39), which then activated the IFN pathway (46), and led to both apoptosis and pyroptosis (96).
TNF alpha-induced mitophagy and innate immune response
TLR7 and TLR8 can sense HCV RNA in the endosome and induce the expression of tumor necrosis factor-α (TNF-α) to suppress HCV replication (44, 97). TNF-α signaling can activate NF-κB and IRFs, leading to the production of pro-inflammatory cytokines and IFNs, respectively. TNF-α induced by HCV is necessary for type I IFN signaling in HCV-infected cells, as the silencing of TNF-α or its receptor TNFR1 abrogates the expression of IFNAR2 and desensitizes HCV-infected cells to type I IFNs (44, 98).
TNFR1 exists in two temporally and spatially distinct signaling complexes: TNFR1 complex I (TNFR1-CI) and II (TNFR1-CII) (99, 100). TNFR1-CI, formed on the plasma membrane, recruits RIPK1, TRADD, TRAF2 and the cellular inhibitor of apoptosis protein (cIAP) 1 and 2. cIAP proteins add K63, K11 and K48 poly-ubiquitin chains to themselves and RIPK1. This further recruits linear ubiquitin chain assembly complex (LUBAC) to add M1 linear polyubiquitin chain to RIPK1 and fully activates TNFR1-CI and NF-kB. TNFR1-CII is formed when RIPK1 becomes deubiquitinated and released from TNFR1-CI. TNFR1-CII, assembled after the internalization of TNFR1, recruits FADD, caspase-8 and FLICE-like inhibitory protein (FLIP). The degradation of cIAPs and the activation of caspase-8 initiate the apoptotic pathway. In the case of caspase inhibition, RIPK3 associates with RIPK1 to induce necroptosis. Zhao et al. reported that the cleavage of p62 by caspase-8, a key protease in extrinsic apoptotic signaling initiated from death receptors including TNFR1, prevented p62 from participating in autophagy (101) (Figure 2). The mutation of D329, the caspase-8 cleavage site in p62, to histidine (D329H) or glycine (D329G) rendered p62 resistant to caspase-8 and inhibited caspase-8-induced apoptosis. This cleaved p62 lacks the KIR domain and can no longer bind to Keap1, which associates with Nrf2 to inhibit its activity (101). Thus, the activity of caspase-8 on p62 shuts off the oxidative stress response and at the same time inhibits further expression of p62 and autophagic degradation. As mentioned above, the association of Keap1 with p62 attenuates autophagic degradation and in the meantime activates Nrf2-mediated oxidative stress response (51). An interesting biphasic effect of TNF-α on the Keap1/Nrf2 pathway had been observed, with TNF-α activating and promoting the nuclear localization of Nrf2 at low concentrations and impairing the antioxidant signaling of Nrf2 to result in severe oxidative stress and cell death at high concentrations (102). The activation of Nrf2 in response to HCV was mediated by the p38 MAPK and JAK, and the silencing of Nrf2 inhibited the phosphorylation of Akt and its downstream effector Bad, implicating Nrf2 in the survival of HCV-infected cells and their potential oncogenic transformation (54). As discussed above, the inhibition of autophagic degradation of mitochondria under oxidative stress can activate the cGAS/STING pathway to induce the expression of IFNs. Oroxylin A is a flavonoid compound isolated from Scutellaria baicalensis. It has antioxidant, anti-inflammatory and anti-tumor activities (101). The apoptosis of liver tumor cells induced by oroxylin A is dependent on p62-mediated activation of caspase-8, which cleaves p62 at D329 to remove its KIR domain (101). The cleavage of p62 by caspase-8 down-regulated Nrf2 and reduced the oxidative stress response (Figure 2). However, it is noteworthy that HCV fights the action of TNF-α and TNFR1 signaling by inducing the autophagic degradation of TNFR1 (44). It should be noted as well that TNF-α had also been shown, through a paracrine mechanism, to disrupt the tight junctions and promote the entry of HCV into polarized hepatocytes, hence promoting HCV infection (103). In Huh 7.5 cells, HCV-induced autophagic degradation of IFN-α receptor, IFNAR1 rendered the infected cells resistant to IFN treatment (43).
Concluding remarks
Autophagy, a process used by the cell to maintain its homeostasis, also plays important roles in antiviral responses. Besides its role in virophagy, autophagy influences other innate immune pathways including PRR signaling, the formation of inflammasomes, and the expression of pro-inflammatory cytokines and IFNs. HCV-induced autophagy affects all these intracellular antiviral responses.
HCV induces autophagy/mitophagy to promote its replication. Mitophagy is a quality control mechanism used by the cell to maintain a healthy pool of mitochondria, which, if damaged, can trigger cell death. To cope with the HCV-associated damage in mitochondria, HCV induces mitophagy to remove dysfunctional mitochondria to prevent premature cell death. Rubicon is induced by HCV to delay the maturation of autophagosomes, causing the accumulation of autophagosomal membranes, which support the HCV genome replication (23). This delay in autophagic degradation can also actuate type I IFN signaling (46). In this regard, TNF-α-induced cleavage of p62 by caspase-8 may have the same effect as Rubicon in delaying autophagosome maturation and on IFN signaling.
Mitochondrial fission and fusion (i.e., mitochondrial dynamics) also affect HCV replication. Mitophagy is often coupled with mitochondrial fission. HCV-induced mitochondrial fission correlates with the attenuation of apoptosis (87). Interestingly, the inhibition of mitochondrial fission affected the secretion of progeny viruses (87), indicating a possible role of mitophagy in the release of infectious viral particles. The perturbation of mitochondrial metabolism can desensitize HCV-infected cells to IFN-α. These immune-antagonizing activities of HCV explain why most HCV patients fail to eradicate this virus and eventually develop severe liver diseases and why IFN therapies did not generate sustained response in the majority of patients infected by HCV.
In patients, HCV can induce inflammasomes to produce mature IL-1β, mainly by Kupffer cells. IL-1β induces the release of mtDNA that can activate cGAS/STING pathway to produce IFNs. This expression of IFNs in turn inhibits the formation of inflammasomes to repress further generation of the mature IL-1β. By inducing autophagic degradation of the components of inflammasomes, HCV hampers the inflammasome formation. This interplay between IL-1β and IFN likely plays an important role in HCV persistence and pathogenesis.
HCV-induced mitochondrial oxidative stress triggers Nrf2 activation. p62, an adaptor molecule important for autophagic/mitophagic protein degradation, binds to Keap1 to activate the Nrf2-mediated antioxidant response (56). The Nrf2 activation in turn promotes the expression of p62, thereby enforcing the antioxidant response and mitophagy, which helps to remove immune-evoking DAMPs and promote the survival of infected cells, potentially inducing tumorigenesis.
There is a constant battle between viruses and their host cells for survival. One such battleground is the control of the autophagic pathway. HCV has developed remarkable mechanisms to control this pathway not only to promote its replication but also to attenuate the host innate immune response. The successful suppression of expression of cytokines and IFNs and their signaling pathways plays a critical role for HCV to establish chronic infection in the great majority of patients that it infects. The prolonged perturbation of the autophagic pathway by HCV likely plays an important role in HCV pathogenesis. The current DAAs, which target HCV NS3 protease, NS5A and/or NS5B polymerase to inhibit HCV replication, can also alleviate the long-term effect of HCV on this important cellular pathway to restore the health of hepatocytes.
Author contributions
JL: Writing – original draft, Writing – review & editing. J-HO: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by NIH grants AI129540, AI148304 and DK094652.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, et al. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol (2006) 80:6964–72. doi: 10.1128/JVI.00024-06
2. Sung PS, Shin EC. Interferon response in hepatitis C virus-infected hepatocytes: issues to consider in the era of direct-acting antivirals. Int J Mol Sci (2020) 21(7):2583. doi: 10.3390/ijms21072583
3. Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity (2006) 25:373–81. doi: 10.1016/j.immuni.2006.08.007
4. Cao X, Ding Q, Lu J, Tao W, Huang B, Zhao Y, et al. MDA5 plays a critical role in interferon response during hepatitis C virus infection. J Hepatol (2015) 62:771–8. doi: 10.1016/j.jhep.2014.11.007
5. Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature (2008) 454:523–7. doi: 10.1038/nature07106
6. Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol (2008) 8:559–68. doi: 10.1038/nri2314
7. Foy E, Li K, Sumpter R Jr., Loo YM, Johnson CL, Wang C, et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U.S.A. (2005) 102:2986–91. doi: 10.1073/pnas.0408707102
8. Horner SM, Liu HM, Park HS, Briley J, Gale M Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U.S.A. (2011) 108:14590–5. doi: 10.1073/pnas.1110133108
9. Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U.S.A. (2005) 102:17717–22. doi: 10.1073/pnas.0508531102
10. Ferreira AR, Magalhaes AC, Camoes F, Gouveia A, Vieira M, Kagan JC, et al. Hepatitis C virus NS3-4A inhibits the peroxisomal MAVS-dependent antiviral signalling response. J Cell Mol Med (2016) 20:750–7. doi: 10.1111/jcmm.12801
11. Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U.S.A. (2005) 102:2992–7.
12. Liang Y, Cao X, Ding Q, Zhao Y, He Z, Zhong J. Correction: Hepatitis C virus NS4B induces the degradation of TRIF to inhibit TLR3-mediated interferon signaling pathway. PloS Pathog (2018) 14:e1007455. doi: 10.1371/journal.ppat.1007455
13. Kumthip K, Maneekarn N. The role of HCV proteins on treatment outcomes. Virol J (2015) 12:217. doi: 10.1186/s12985-015-0450-x
14. Wong MT, Chen SS. Emerging roles of interferon-stimulated genes in the innate immune response to hepatitis C virus infection. Cell Mol Immunol (2016) 13:11–35. doi: 10.1038/cmi.2014.127
15. Ferreira AR, Ramos B, Nunes A, Ribeiro D. Hepatitis C virus: evading the intracellular innate immunity. J Clin Med (2020) 9(3):790. doi: 10.3390/jcm9030790
16. Otsuka M, Kato N, Moriyama M, Taniguchi H, Wang Y, Dharel N, et al. Interaction between the HCV NS3 protein and the host TBK1 protein leads to inhibition of cellular antiviral responses. Hepatology (2005) 41:1004–12. doi: 10.1002/hep.20666
17. Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe (2009) 6:513–22. doi: 10.1016/j.chom.2009.11.004
18. Arnaud N, Dabo S, Maillard P, Budkowska A, Kalliampakou KI, Mavromara P, et al. Hepatitis C virus controls interferon production through PKR activation. PloS One (2010) 5:e10575. doi: 10.1371/journal.pone.0010575
19. Kang W, Sung PS, Park SH, Yoon S, Chang DY, Kim S, et al. Hepatitis C virus attenuates interferon-induced major histocompatibility complex class I expression and decreases CD8+ T cell effector functions. Gastroenterology (2014) 146:1351–60 e1-4. doi: 10.1053/j.gastro.2014.01.054
20. Suzuki R, Matsuda M, Shimoike T, Watashi K, Aizaki H, Kato T, et al. Activation of protein kinase R by hepatitis C virus RNA-dependent RNA polymerase. Virology (2019) 529:226–33. doi: 10.1016/j.virol.2019.01.024
21. Gregoire IP, Richetta C, Meyniel-Schicklin L, Borel S, Pradezynski F, Diaz O, et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PloS Pathog (2011) 7:e1002422. doi: 10.1371/journal.ppat.1002422
22. Hansen MD, Johnsen IB, Stiberg KA, Sherstova T, Wakita T, Richard GM, et al. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc Natl Acad Sci U.S.A. (2017) 114:E3462–71. doi: 10.1073/pnas.1616683114
23. Wang L, Ou JH. Hepatitis C virus and autophagy. Biol Chem (2015) 396:1215–22. doi: 10.1515/hsz-2015-0172
24. Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology (2008) 48:1054–61. doi: 10.1002/hep.22464
25. Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest (2011) 121:37–56. doi: 10.1172/JCI41474
26. Wang J, Kang R, Huang H, Xi X, Wang B, Wang J, et al. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy (2014) 10:766–84. doi: 10.4161/auto.27954
27. Fahmy AM, Labonte P. The autophagy elongation complex (ATG5-12/16L1) positively regulates HCV replication and is required for wild-type membranous web formation. Sci Rep (2017) 7:40351. doi: 10.1038/srep40351
28. Aizaki H, Lee KJ, Sung VM, Ishiko H, Lai MM. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology (2004) 324:450–61. doi: 10.1016/j.virol.2004.03.034
29. Sir D, Kuo CF, Tian Y, Liu HM, Huang EJ, Jung JU, et al. Replication of hepatitis C virus RNA on autophagosomal membranes. J Biol Chem (2012) 287:18036–43. doi: 10.1074/jbc.M111.320085
30. Kim JY, Wang L, Lee J, Ou JJ. Hepatitis C virus induces the localization of lipid rafts to autophagosomes for its RNA replication. J Virol (2017) 91(20):e00541-17. doi: 10.1128/JVI.00541-17
31. Waris G, Turkson J, Hassanein T, Siddiqui A. Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: role of STAT-3 in HCV replication. J Virol (2005) 79:1569–80. doi: 10.1128/JVI.79.3.1569-1580.2005
32. Ivanov AV, Bartosch B, Smirnova OA, Isaguliants MG, Kochetkov SN. HCV and oxidative stress in the liver. Viruses (2013) 5:439–69. doi: 10.3390/v5020439
33. Medvedev R, Ploen D, Spengler C, Elgner F, Ren H, Bunten S, et al. HCV-induced oxidative stress by inhibition of Nrf2 triggers autophagy and favors release of viral particles. Free Radic Biol Med (2017) 110:300–15. doi: 10.1016/j.freeradbiomed.2017.06.021
34. Chan ST, Lee J, Narula M, Ou JJ. Suppression of host innate immune response by hepatitis C virus via induction of autophagic degradation of TRAF6. J Virol (2016) 90:10928–35. doi: 10.1128/JVI.01365-16
35. Sander WJ, Fourie C, Sabiu S, O'neill FH, Pohl CH, O'neill HG. Reactive oxygen species as potential antiviral targets. Rev Med Virol (2022) 32:e2240. doi: 10.1002/rmv.2240
36. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol (2013) 13:722–37. doi: 10.1038/nri3532
37. Liang S, Wu YS, Li DY, Tang JX, Liu HF. Autophagy in viral infection and pathogenesis. Front Cell Dev Biol (2021) 9:766142. doi: 10.3389/fcell.2021.766142
38. Ait-Goughoulte M, Kanda T, Meyer K, Ryerse JS, Ray RB, Ray R. Hepatitis C virus genotype 1a growth and induction of autophagy. J Virol (2008) 82:2241–9. doi: 10.1128/JVI.02093-07
39. Wang L, Tian Y, Ou JH. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PloS Pathog (2015) 11:e1004764. doi: 10.1371/journal.ppat.1004764
40. Kim JY, Ou JJ. Regulation of apolipoprotein E trafficking by hepatitis C virus-induced autophagy. J Virol (2018) 92(14):e00211-18. doi: 10.1128/JVI.00211-18
41. Chan ST, Ou JJ. Hepatitis C virus-induced autophagy and host innate immune response. Viruses (2017) 9(8):224. doi: 10.3390/v9080224
42. Tian Y, Wang ML, Zhao J. Crosstalk between autophagy and type I interferon responses in innate antiviral immunity. Viruses (2019) 11(2):132. doi: 10.3390/v11020132
43. Chandra PK, Bao L, Song K, Aboulnasr FM, Baker DP, Shores N, et al. HCV infection selectively impairs type I but not type III IFN signaling. Am J Pathol (2014) 184:214–29. doi: 10.1016/j.ajpath.2013.10.005
44. Lee J, Tian Y, Chan ST, Kim JY, Cho C, Ou JH. TNF-alpha Induced by Hepatitis C Virus via TLR7 and TLR8 in Hepatocytes Supports Interferon Signaling via an Autocrine Mechanism. PloS Pathog (2015) 11:e1004937. doi: 10.1371/journal.ppat.1004937
45. Liu X, Zhang S, An L, Wu J, Hu X, Lai S, et al. Loss of Rubicon ameliorates doxorubicin-induced cardiotoxicity through enhancement of mitochondrial quality. Int J Cardiol (2019) 296:129–35. doi: 10.1016/j.ijcard.2019.07.074
46. Shiode Y, Hikita H, Tanaka S, Shirai K, Doi A, Sakane S, et al. Hepatitis C virus enhances Rubicon expression, leading to autophagy inhibition and intracellular innate immune activation. Sci Rep (2020) 10:15290. doi: 10.1038/s41598-020-72294-y
47. Jiang GM, Tan Y, Wang H, Peng L, Chen HT, Meng XJ, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer (2019) 18:17. doi: 10.1186/s12943-019-0944-z
48. Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol (2009) 11:385–96. doi: 10.1038/ncb1846
49. Sun Q, Westphal W, Wong KN, Tan I, Zhong Q. Rubicon controls endosome maturation as a Rab7 effector. Proc Natl Acad Sci U.S.A. (2010) 107:19338–43. doi: 10.1073/pnas.1010554107
50. Kim JH, Kim TH, Lee HC, Nikapitiya C, Uddin MB, Park ME, et al. Rubicon modulates antiviral type I interferon (IFN) signaling by targeting IFN regulatory factor 3 dimerization. J Virol (2017) 91:4. doi: 10.1128/JVI.00248-17
51. Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell (2013) 51:618–31. doi: 10.1016/j.molcel.2013.08.003
52. Bender D, Hildt E. Effect of hepatitis viruses on the Nrf2/Keap1-signaling pathway and its impact on viral replication and pathogenesis. Int J Mol Sci (2019) 20(18):4659. doi: 10.3390/ijms20184659
53. Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell (2011) 44:134–46. doi: 10.1016/j.molcel.2011.06.038
54. Burdette D, Olivarez M, Waris G. Activation of transcription factor Nrf2 by hepatitis C virus induces the cell-survival pathway. J Gen Virol (2010) 91:681–90. doi: 10.1099/vir.0.014340-0
55. Saito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K, et al. p62/Sqstm1 promotes Malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun (2016) 7:12030. doi: 10.1038/ncomms12030
56. Sanchez-Martin P, Saito T, Komatsu M. p62/SQSTM1: 'Jack of all trades' in health and cancer. FEBS J (2019) 286:8–23. doi: 10.1111/febs.14712
57. Wu X, Dong L, Lin X, Li J. Relevance of the NLRP3 inflammasome in the pathogenesis of chronic liver disease. Front Immunol (2017) 8:1728. doi: 10.3389/fimmu.2017.01728
58. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther (2021) 6:128. doi: 10.1038/s41392-021-00507-5
59. Negash AA, Olson RM, Griffin S, Gale M Jr. Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PloS Pathog (2019) 15:e1007593. doi: 10.1371/journal.ppat.1007593
60. Wallace HL, Wang L, Gardner CL, Corkum CP, Grant MD, Hirasawa K, et al. Crosstalk between pyroptosis and apoptosis in hepatitis C virus-induced cell death. Front Immunol (2022) 13:788138. doi: 10.3389/fimmu.2022.788138
61. Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PloS Pathog (2013) 9:e1003330. doi: 10.1371/journal.ppat.1003330
62. Abid K, Pazienza V, De Gottardi A, Rubbia-Brandt L, Conne B, Pugnale P, et al. An in vitro model of hepatitis C virus genotype 3a-associated triglycerides accumulation. J Hepatol (2005) 42:744–51. doi: 10.1016/j.jhep.2004.12.034
63. Piodi A, Chouteau P, Lerat H, Hezode C, Pawlotsky JM. Morphological changes in intracellular lipid droplets induced by different hepatitis C virus genotype core sequences and relationship with steatosis. Hepatology (2008) 48:16–27. doi: 10.1002/hep.22288
64. Biasizzo M, Kopitar-Jerala N. Interplay between NLRP3 inflammasome and autophagy. Front Immunol (2020) 11:591803. doi: 10.3389/fimmu.2020.591803
65. Daussy CF, Monard SC, Guy C, Munoz-Gonzalez S, Chazal M, Anthonsen MW, et al. The inflammasome components NLRP3 and ASC act in concert with IRGM to rearrange the golgi apparatus during hepatitis C virus infection. J Virol (2021) 95(3):e00826-20. doi: 10.1128/JVI.00826-20
66. Zuo H, Chen C, Sa Y. Therapeutic potential of autophagy in immunity and inflammation: current and future perspectives. Pharmacol Rep (2023) 75:499–510. doi: 10.1007/s43440-023-00486-0
67. Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale M Jr. Interleukin-1beta Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol Cell (2019) 74:801–815 e6. doi: 10.1016/j.molcel.2019.02.038
68. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity (2011) 34:213–23. doi: 10.1016/j.immuni.2011.02.006
69. Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res (2013) 23:201–12. doi: 10.1038/cr.2013.6
70. Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat Immunol (2013) 14:52–60. doi: 10.1038/ni.2474
71. Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, et al. Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity (2018) 49:413–426 e5. doi: 10.1016/j.immuni.2018.07.006
72. Lin XH, Qiu BQ, Ma M, Zhang R, Hsu SJ, Liu HH, et al. Suppressing DRP1-mediated mitochondrial fission and mitophagy increases mitochondrial apoptosis of hepatocellular carcinoma cells in the setting of hypoxia. Oncogenesis (2020) 9:67. doi: 10.1038/s41389-020-00251-5
73. Burke TP, Engstrom P, Chavez RA, Fonbuena JA, Vance RE, Welch MD. Inflammasome-mediated antagonism of type I interferon enhances Rickettsia pathogenesis. Nat Microbiol (2020) 5:688–96. doi: 10.1038/s41564-020-0673-5
74. Schwerk J, Negash A, Savan R, Gale M Jr. Innate immunity in hepatitis C virus infection. Cold Spring Harb Perspect Med (2021) 11(2):a036988. doi: 10.1101/cshperspect.a036988
75. Brzdek M, Zarebska-Michaluk D, Invernizzi F, Cilla M, Dobrowolska K, Flisiak R. Decade of optimizing therapy with direct-acting antiviral drugs and the changing profile of patients with chronic hepatitis C. World J Gastroenterol (2023) 29:949–66. doi: 10.3748/wjg.v29.i6.949
76. Guo Y, Gu R, Gan D, Hu F, Li G, Xu G. Mitochondrial DNA drives noncanonical inflammation activation via cGAS-STING signaling pathway in retinal microvascular endothelial cells. Cell Commun Signal (2020) 18:172. doi: 10.1186/s12964-020-00637-3
77. Geto Z, Molla MD, Challa F, Belay Y, Getahun T. Mitochondrial dynamic dysfunction as a main triggering factor for inflammation associated chronic non-communicable diseases. J Inflammation Res (2020) 13:97–107. doi: 10.2147/JIR.S232009
78. Krols M, Bultynck G, Janssens S. ER-Mitochondria contact sites: A new regulator of cellular calcium flux comes into play. J Cell Biol (2016) 214:367–70. doi: 10.1083/jcb.201607124
79. Strubbe-Rivera JO, Chen J, West BA, Parent KN, Wei GW, Bazil JN. Modeling the effects of calcium overload on mitochondrial ultrastructural remodeling. Appl Sci (Basel) (2021) 11(5):2071. doi: 10.3390/app11052071
80. Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc Natl Acad Sci U.S.A. (2013) 110:17963–8. doi: 10.1073/pnas.1312571110
81. Duponchel S, Monnier L, Molle J, Bendridi N, Alam MR, Gaballah A, et al. Hepatitis C virus replication requires integrity of mitochondria-associated ER membranes. JHEP Rep (2023) 5:100647. doi: 10.1016/j.jhepr.2022.100647
82. Scrima R, Piccoli C, Moradpour D, Capitanio N. Targeting endoplasmic reticulum and/or mitochondrial ca(2+) fluxes as therapeutic strategy for HCV infection. Front Chem (2018) 6:73. doi: 10.3389/fchem.2018.00073
83. Choi J, Lee KJ, Zheng Y, Yamaga AK, Lai MM, Ou JH. Reactive oxygen species suppress hepatitis C virus RNA replication in human hepatoma cells. Hepatology (2004) 39:81–9. doi: 10.1002/hep.20001
84. Choi J, Ou JH. Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am J Physiol Gastrointest Liver Physiol (2006) 290:G847–51. doi: 10.1152/ajpgi.00522.2005
85. Foo J, Bellot G, Pervaiz S, Alonso S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol (2022) 30:679–92. doi: 10.1016/j.tim.2021.12.011
86. Kim SJ, Syed GH, Siddiqui A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PloS Pathog (2013) 9:e1003285. doi: 10.1371/journal.ppat.1003285
87. Kim SJ, Syed GH, Khan M, Chiu WW, Sohail MA, Gish RG, et al. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci U.S.A. (2014) 111:6413–8. doi: 10.1073/pnas.1321114111
88. Jassey A, Liu CH, Changou CA, Richardson CD, Hsu HY, Lin LT. Hepatitis C virus non-structural protein 5A (NS5A) disrupts mitochondrial dynamics and induces mitophagy. Cells (2019) 8(4):290. doi: 10.3390/cells8040290
89. Cho DH, Kim JK, Jo EK. Mitophagy and innate immunity in infection. Mol Cells (2020) 43:10–22. doi: 10.14348/molcells.2020.2329
90. Pila-Castellanos I, Molino D, Mckellar J, Lines L, Da Graca J, Tauziet M, et al. Correction: Mitochondrial morphodynamics alteration induced by influenza virus infection as a new antiviral strategy. PloS Pathog (2021) 17:e1009485. doi: 10.1371/journal.ppat.1009485
91. Zhan L, Cao H, Wang G, Lyu Y, Sun X, An J, et al. Drp1-mediated mitochondrial fission promotes cell proliferation through crosstalk of p53 and NF-kappaB pathways in hepatocellular carcinoma. Oncotarget (2016) 7:65001–11. doi: 10.18632/oncotarget.11339
92. Liu K, Lee J, Kim JY, Wang L, Tian Y, Chan ST, et al. Mitophagy controls the activities of tumor suppressor p53 to regulate hepatic cancer stem cells. Mol Cell (2017) 68:281–292 e5. doi: 10.1016/j.molcel.2017.09.022
93. Vara-Perez M, Felipe-Abrio B, Agostinis P. Mitophagy in cancer: A tale of adaptation. Cells (2019) 8(5):493. doi: 10.3390/cells8050493
94. Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res (2012) 111:1208–21. doi: 10.1161/CIRCRESAHA.112.265819
95. Gkirtzimanaki K, Kabrani E, Nikoleri D, Polyzos A, Blanas A, Sidiropoulos P, et al. IFNalpha impairs autophagic degradation of mtDNA promoting autoreactivity of SLE monocytes in a STING-dependent fashion. Cell Rep (2018) 25:921–933 e5. doi: 10.1016/j.celrep.2018.09.001
96. Kofahi HM, Taylor NG, Hirasawa K, Grant MD, Russell RS. Hepatitis C virus infection of cultured human hepatoma cells causes apoptosis and pyroptosis in both infected and bystander cells. Sci Rep (2016) 6:37433. doi: 10.1038/srep37433
97. Lee J, Wu CC, Lee KJ, Chuang TH, Katakura K, Liu YT, et al. Activation of anti-hepatitis C virus responses via Toll-like receptor 7. Proc Natl Acad Sci U.S.A. (2006) 103:1828–33. doi: 10.1073/pnas.0510801103
98. Willemsen J, Neuhoff MT, Hoyler T, Noir E, Tessier C, Sarret S, et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep (2021) 37:109977. doi: 10.1016/j.celrep.2021.109977
99. de Almagro MC, Goncharov T, Newton K, Vucic D. Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination. Cell Death Dis (2015) 6:e1800. doi: 10.1038/cddis.2015.158
100. Gough P, Myles IA. Tumor necrosis factor receptors: pleiotropic signaling complexes and their differential effects. Front Immunol (2020) 11:585880. doi: 10.3389/fimmu.2020.585880
101. Zhao Y, Zhu Q, Bu X, Zhou Y, Bai D, Guo Q, et al. Triggering apoptosis by oroxylin A through caspase-8 activation and p62/SQSTM1 proteolysis. Redox Biol (2020) 29:101392. doi: 10.1016/j.redox.2019.101392
102. Shanmugam G, Narasimhan M, Sakthivel R, Kumar RR, Davidson C, Palaniappan S, et al. A biphasic effect of TNF-alpha in regulation of the Keap1/Nrf2 pathway in cardiomyocytes. Redox Biol (2016) 9:77–89. doi: 10.1016/j.redox.2016.06.004
Keywords: HCV, autophagy, mitophagy, interferons, STING, inflammasome, oxidative stress
Citation: Lee J and Ou J-HJ (2024) HCV-induced autophagy and innate immunity. Front. Immunol. 15:1305157. doi: 10.3389/fimmu.2024.1305157
Received: 14 November 2023; Accepted: 19 January 2024;
Published: 02 February 2024.
Edited by:
Chaofeng Han, Second Military Medical University, ChinaReviewed by:
Yunhao Tan, AbbVie (United States), United StatesRanjit Ray, Saint Louis University, United States
Srikanta Dash, Tulane University, United States
Copyright © 2024 Lee and Ou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J.-H. James Ou, amFtZXNvdUB1c2MuZWR1