 Shaifali Tyagi1,2
Shaifali Tyagi1,2 Srikanth Sadhu3
Srikanth Sadhu3 Taruna Sharma1,2Abhijit Paul4
Taruna Sharma1,2Abhijit Paul4 Manitosh Pandey1
Manitosh Pandey1 Vaibhav Kumar Nain1,2
Vaibhav Kumar Nain1,2 Deepak Kumar Rathore3
Deepak Kumar Rathore3 Samrat Chatterjee4
Samrat Chatterjee4 Amit Awasthi3*
Amit Awasthi3* Amit Kumar Pandey1*
Amit Kumar Pandey1*- 1Mycobacterial Pathogenesis Laboratory, Translational Health Science and Technology Institute, Faridabad, Haryana, India
- 2Special Centre for Molecular Medicine, Jawaharlal Nehru University, New Delhi, India
- 3Immunobiology Laboratory, Translational Health Science and Technology Institute, Faridabad, Haryana, India
- 4Complex Analysis Laboratory, Translational Health Science and Technology Institute, Faridabad, Haryana, India
Mechanistic understanding of antibiotic persistence is a prerequisite in controlling the emergence of MDR cases in Tuberculosis (TB). We have reported that the cholesterol-induced activation of VapC12 ribonuclease is critical for disease persistence in TB. In this study, we observed that relative to the wild type, mice infected with ΔvapC12 induced a pro-inflammatory response, had a higher pathogen load, and responded better to the anti-TB treatment. In a high-dose infection model, all the mice infected with ΔvapC12 succumbed early to the disease. Finally, we reported that the above phenotype of ΔvapC12 was dependent on the presence of the TLR4 receptor. Overall, the data suggests that failure of a timely resolution of the early inflammation by the ΔvapC12 infected mice led to hyperinflammation, altered T-cell response and high bacterial load. In conclusion, our findings suggest the role of the VapC12 toxin in modulating the innate immune response of the host in ways that favor the long-term survival of the pathogen inside the host.
Introduction
COVID-19 has augmented the tuberculosis (TB) burden by disrupting the existing healthcare systems resulting in a delay in diagnosis and treatment (1). In spite of the availability of anti-tubercular therapy, its eradication still remains obscure (2). In addition, non-compliance to a prolonged treatment regimen fuels the development of multidrug and extensively drug-resistant TB posing blockades to global TB control (3). The virulence of Mycobacterium tuberculosis (M. tuberculosis) is highly transmissible primarily through the aerosolization of the respiratory secretions from the infected individuals. This together with a relatively low virulence has allowed this pathogen to successfully co-evolve with humans for centuries without causing any symptoms or death (4). We believe that the complex network of host-pathogen interaction, thus developed during evolution, defines TB in terms of its presentation either as an asymptomatic infection or an acute infection presented with clear symptoms. Nevertheless, both the pathogen and the host have gradually evolved mechanisms that would identify and neutralize any mutually directed threats.
Innate immunity is an indispensable arm of the host immune system mediated by phagocytes such as macrophages; bona fide host cells harboring the mycobacteria (5). The role of neutrophils which are highly motile innate immune cells has been shown in experimental models of TB pathogenesis (6–9). Following the M. tuberculosis infection, these are the primary cells to percolate the lungs and have an extremely intricate role in TB pathology (10). Neutrophils mediate their phagocytic activity by producing proteases, reactive oxygen and nitrogen intermediates and antimicrobial peptides (11). These immune cells are the most common cell type present in the sputum, bronchoalveolar fluid and cavities of active TB patients (10, 12). There is a negative correlation between their abundance in the blood of M. tuberculosis-infected patients and disease severity (13–16). On the contrary, some neutrophil subpopulations are also known to aid in providing a permissive replicating niche and nutrient reservoir for M. tuberculosis, indicating their role in promoting mycobacterial persistence (17, 18). The same has also been reported in M. tuberculosis infected rhesus macaques (19) and zebrafish (20). Growing evidence suggests the phenotypic and functional diversities of neutrophils which extend well beyond their conventional role as antimicrobial agent. Timely resolution of neutrophils at the site of infection dictates their fate as either protective or dysregulated immune cells and is crucial for the effective restoration of the excessive inflammatory response to homeostasis. The ineffective clearance of neutrophils leads to exacerbation of inflammatory response and perpetuation of tissue damage (21). Furthermore, the role of mycobacterial genes in shaping an appropriate neutrophil-mediated innate immune response and its timely resolution that affects the downstream adaptive immunity, remains largely unknown.
Previously, using the guinea pig model, we demonstrated that both lungs and spleen isolated from guinea pigs infected with M. tuberculosis lacking the VapC12 ribonuclease toxin were presented with higher bacterial burden and increased lung pathology relative to the wild type (WT) (22). We hypothesize that the VapC12 toxin in M. tuberculosis is critical for modulating the host immune response and hence the absence of this toxin in M. tuberculosis resulted in a severe form of the disease. To further our understanding of the role of the host immune system in imparting the above phenotype to the vapC12 deleted strain, in the current study, we performed comprehensive immune-profiling studies in mice infected with WT, ΔvapC12 and the complemented strains to identify host correlates of disease persistence. We found that in comparison to WT, the lungs of mice infected with the mutant strain were presented with higher bacterial load and enhanced pathology. Additionally, these mice showed an excess of macrophages, neutrophils and myeloid-derived suppressor cells (MDSCs) in the lungs with an alleviated T-cell immune response. In the survival study, performed with a high-dose infection model, while all the mice infected with WT strain survived none of the mice from the ▵vapC12 group survived 18 weeks post-infection. Additionally, lung transcriptome analysis revealed that the observed neutrophil accumulation was mirrored by increased S100A8 and S100A9 mRNA levels in the lungs infected with ΔvapC12 when compared with the WT strain. Interestingly, we found that the enhanced pathogenesis observed in the mice infected with ΔvapC12 was dependent on the presence of TLR-4 receptors. These results suggest that vapC12 ribonuclease mediated regulation of expression of pathogen-associated molecular patterns (PAMPs) in M. tuberculosis modulates host immune response which is essential for the long-term survival of the pathogen inside the host. A better understanding of the mechanism of generation and maintenance of antibiotic persistence in the context of host-pathogen interaction will go a long way in designing novel intervention strategies against tuberculosis. Host-directed therapies (HDT) targeting such pathways in combination with the existing anti-TB treatment can increase the efficacy of the current TB regimen.
Methods
Bacterial strains and culture conditions
All mycobacterial strains were grown and maintained in Middlebrook 7H9 broth and 7H11 agar (Difco Middlebrook 7H9 broth [catalog no. 271310] and 7H11 agar [catalog no. 283810]) and supplemented with 10% OADS (bovine serum albumin, oleic acid, and dextrose); 0.05% Tween was added in 7H9 broth to enhance bacterial growth. Bacterial enumeration by CFU was examined on 7H11 agar plates. The concentrations of antibiotics used were as follows: hygromycin B (50 µg/mL) and kanamycin (25 µg/mL).
Mouse infection study
Animal experiment protocols were reviewed and approved by the animal ethics committee of Translational Health Science and Technology Institute India [(IAEC/THSTI/183), (IAEC/THSTI/167)], Faridabad, India. Animal experiments were performed in accordance with guidelines provided by the Committee for the Control and Supervision of Experiments on Animals (CCSEA). Pathogen-free C57BL/6 and C3H/HeJ mice were obtained from Experimental Animal Facility, THSTI. As described earlier, briefly, prior to infection, mid-log phase cultures (O.D =0.8-1) of H37Rv (WT), ΔvapC12 and ΔvapC12:vapBC12 M. tuberculosis strains were harvested, washed and single-cell suspensions were prepared (23). Six to eight-week old mice were infected by aerosol exposure in the Glas-Col airborne infection apparatus with approximately 50-100 CFUs of different strains. Mice from each group (N = 5) were sacrificed at different time points as indicated in the experiments (Day 0, 2, 4 and 8 weeks post infection). The bacillary load was determined by removing lungs and spleen of infected mice and the serial dilution of the organ homogenates was plated on 7H11 agar plates supplemented with 10% OADS for bacterial enumeration. The lung sections were treated with 10% formalin and hematoxylin and eosin stained were used for histopathological analysis. A blind assessment strategy was employed for scoring the granulomas. For drug clearance study, after establishing bacterial infection till week 4, mice were treated with 10mg/kg RIF through oral gavaging till week 8. Also, a separate untreated group was kept as a control. Bacillary enumeration was done on week 4 and 8 by aseptically removing organs and homogenizing them. The homogenized organs were then serially diluted and plated on 7H11 agar (containing Polymyxin A, amphotericin B, vancomycin, trimethoprim, carbenicillin and cycloheximide mix).
Flow cytometry analysis
Mice were sacrificed at 2 and 4 week post-infection, and their lungs, spleen and LN (draining lymph node) were isolated. Single-cell suspension was prepared and stained (1 x 106 cells) with fluorescently labelled anti-mouse CD11b (eBioscience Invitrogen, USA), anti-mouse Gr1 (eBioscience Invitrogen, USA), anti-mouse F4/80-FITC (eBioscience Invitrogen, USA), anti-mouse CD68 (eBioscience Invitrogen, USA), anti-mouse CD206 (eBioscience Invitrogen, USA), anti-mouse CD80 (eBioscience Invitrogen, USA), anti-mouse NK1.1 (eBioscience Invitrogen, USA), anti-mouse CD3 (BD Biosciences USA), anti-mouse CD4 (BD Biosciences USA), anti-mouse CD8 (eBioscience Invitrogen, USA) and anti-mouse γδ-TCR (eBioscience Invitrogen, USA). Briefly, for surface staining, cells were stained for 20 minutes at room temperature, and were washed twice with PBS. Stained samples were then fixed for 20 minutes using 4% paraformaldehyde. Data was acquired on FACS Canto (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR, USA) (24, 25).
RNA sequencing
For transcriptome analysis, lung tissues of mice infected with different strains (4-week infected WT, ▵vapC12 and ΔvapC12:vapBC12) were homogenized by bead beating, and total RNA was isolated using RNeasy mini kit according to the manufacturers protocol (Qiagen catalog no. 74104). RNA was eluted in nuclease-free water, subjected to DNA removal using TURBO DNase. NEB Ultra II directional RNA-sequencing Library Prep kit protocol was used to prepare libraries for total RNA-Seq (NEB, catalog no. E7760L). An initial concentration of 500ng of total RNA was taken for the assay. mRNA molecules are captured using magnetic Poly(T) beads (NEB, catalog no. E7490L) following purification, the enriched mRNA was fragmented using divalent cations under elevated temperatures. The cleaved RNA fragments were copied into first-strand cDNA using reverse transcriptase. Second strand cDNA synthesis was performed using DNA Polymerase I and RNase H enzyme. The cDNA fragments were then subjected to a series of enzymatic steps that repair the ends, tails the 3 end with a single ‘A base, followed by ligation of the adapters. The adapter-ligated products were then purified and enriched using the following thermal conditions: initial denaturation 98°C for 30sec; 12 cycles of - 98°C for 10sec, 65°C for 75sec; final extension of 65°C for 5mins. PCR products are then purified and checked for fragment size distribution on Fragment Analyzer using HS NGS Fragment Kit (1-6000bp) (Agilent, catalog no. DNF-474-1000). Genes with low expression value (mean FPKM value<0.5) were treated as background (i.e. ‘noise) (26) and hence excluded from further analysis. After the noise removal, the gene expression values for each experimental condition were merged, and the final data comprised the FPKM value of 13796 genes. To assess the variation in the gene expression data, we performed the principal component analysis (PCA) using the prcomp package in R, which applies the singular value decomposition to the data matrix. For the comparison between gene expression profiles from mice infected lungs with WT and ▵vapC12 strain of M. tuberculosis, genes with a fold change of 2 and false discovery rate (FDR)-adjusted p-value <0.05 were considered differentially expressed. The Benjamin-Hochberg multiple hypothesis testing (27) was applied to obtain the FDR-adjusted p-value. Subsequently, the analyses were performed on the MATLAB and R software. The mafdr function in MATLAB was applied to obtain the FDR-adjusted p-value. PCA plot, volcano plot and dot plot were generated in R using ggplot2, enrichplot and gplots packages. Heatmaps were built using heatmap.2. Pathway enrichment analysis was performed in R using the clusterProfiler package.
Quantitative RT-PCR
Total RNAs from lungs were isolated using RNeasy Micro kit (Qiagen), and the cDNAs were synthesized by reverse transcriptase (RT) using the high capacity cDNA archive random priming kit (Applied Biosystems) according to the manufacturers recommended protocol. Real-time PCR was performed with 2 × SYBR mix (TaKaRa) in a QuantStudio-6 Flex Real-Time PCR System (Applied Biosystems) using the default run program. The relative expression of the target genes was calculated using the target threshold cycle value (Ct) and the 2−ΔΔCt method with GAPDH as the internal loading control (28). The primer sequences used for real time PCR are listed in Supplementary Table S1.
Depletion of neutrophils
Neutrophils were depleted using 200μg anti-mouse Ly6G (Bio X Cell) or isotype IgG (Bio X Cell). Briefly, in acute infections (less than 21 dpi), mice were given 200μg of anti-mouse Ly6G intra- peritoneally every other day between 10 and 20 dpi.
Statistical analysis
Each experiment was repeated at least three times independently and three biological replicates were used. Data were plotted as mean ± SEM. Students t-test (Mann-Whitney), two-way ANOVA and one-way ANOVA (Kruskal-Wallis) were performed wherever applicable by using Graph Pad Prism 7.0 software. For the survival curve, Kaplan Meier statistical analysis was performed. P-value < 0.05 was considered to be statistically significant.
Results
VapC12 ribonuclease toxin modulates growth, virulence and drug tolerance in M. tuberculosis
To replicate our earlier findings of the study performed on guinea pigs (22), we infected C57BL/6 mice with WT, vapC12 null mutant and complemented M. tuberculosis strains. We found a significantly higher bacterial load in the lungs and spleen of mice infected with vapC12 null strain as compared to the WT and complemented strains both at 4 and 8 weeks post-infection (Figures 1A, B). In comparison to the WT, gross examination of the lungs infected with the mutant strain revealed higher inflammation and enhanced tissue damage. Histological examination of the lungs infected with the WT strain at 8 weeks post-infection revealed a typical necrotic granuloma in the centre of the lung (40X) with epithelioid cells and lymphocytes in the granuloma (100X) (Figure 1C and Supplementary Figure 1A). In contrast, at a similar time point post infection, lung tissues of mice infected with the vapC12 null strain displayed a significantly higher number of granulomas (Figure 1D). Mice infected with complemented strain had a phenotype similar to those infected with WT strain (Figures 1C, D). Previously, using kill-curve experiments, we have demonstrated that the absence of the vapC12 gene in M. tuberculosis reduced the frequency of generation of drug-tolerant population in presence of rifampicin under in-vitro growth conditions (22). To replicate the above findings under disease conditions, we performed a drug clearance study using C57BL/6 mice infection model. We infected a group of mice with either WT and ▵vapC12 strain and estimated the rate of pathogen clearance by estimating bacterial load in infected organs at different time points (Figure 1E). As expected, 4 week post infection, the organs infected with ▵vapC12 had high bacterial load in comparison with the organs isolated from the WT infected mice (Figures 1F, G). Importantly, upon treatment with RIF and INH, the rate of decline in the bacterial load was higher in mice infected with ▵vapC12 strain relative to WT at 8 weeks post infection (Figures 1F, G). To account for the differences in the initial bacterial load (week 0), we had to normalize the data relative to week 0 between H37Rv and vapC12 mutant strains (Figures 1F, G). Importantly, upon treatment with RIF we observed a ~2 fold and ~7 fold increase in the killing of the pathogen in the lungs and spleen respectively in the mice infected with ▵vapC12 as compared to mice infected with the H37Rv (Figures 1F, G) whereas treatment with INH resulted in ~10 fold better killing in the lungs and spleen of the mice infected with ▵vapC12 relative of the H37Rv. These results show that the absence of vapC12 gene in M. tuberculosis decreased the frequency of generation of drug-tolerant population potentiating the efficiency of RIF and INH treatment in infected mice (Figures 1F, G). Since vapC12 deleted strain of M. tuberculosis demonstrated enhanced pathogenicity, we measured the estimated time of survival of WT and ▵vapC12 infected mice in a high dose infection model (500-1000 bacteria/lung) (Supplementary Figure 1B). Interestingly, we found that 16 weeks post-infection, all the mice (n=10) infected with the WT strain survived while none of the mice infected with the ▵vapC12 survive (n=10) survived (Figure 1H). Collectively, these data suggested that absence of vapC12 imparted hypervirulence phenotype and reduces the frequency of generation of persisters in mice infected with M. tuberculosis.
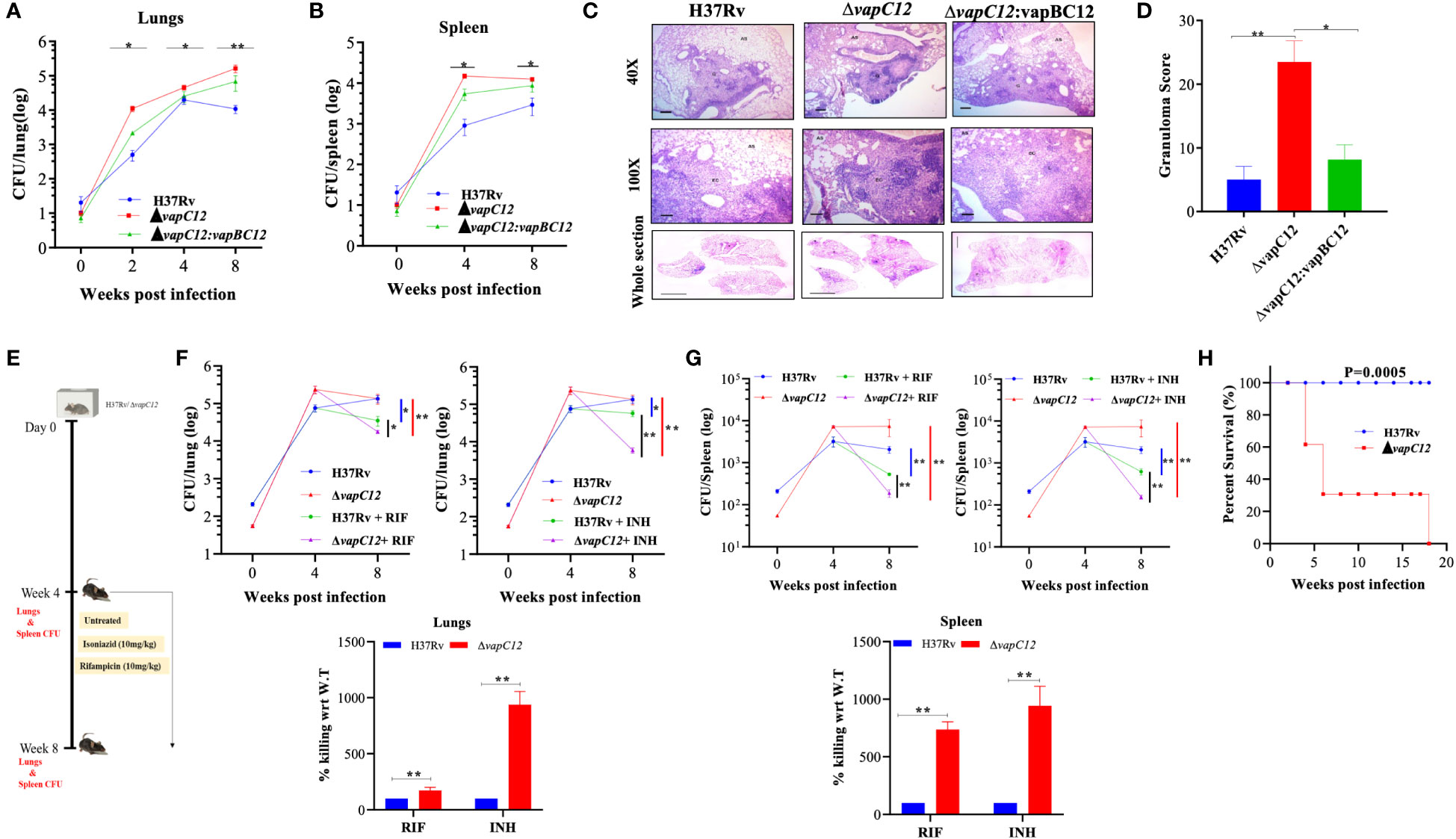
Figure 1 vapC12 mutant strain of M. tuberculosis modulates growth and drug tolerance. (A, B) Bacterial load in the (A) lungs or (B) spleen of mice infected with H37Rv, ΔvapC12, and ΔvapC12:vapBC12 strains of M. tuberculosis at designated time points, the lungs were homogenized in 2 ml of saline, and ten-fold serial dilutions of homogenates were plated on 7H11+OADC plates. Each group constituted five mice per time point. Data plotted represent the mean ± SEM. Significant differences observed between groups are indicated. Data were analyzed using the MannWhitney test with **P < 0.01 and *P <0.05. (C) Photomicrographs of H&E-stained (40× and 100×) and whole section image of lung sections from mice infected with different strains of M. tuberculosis at 8 weeks post infection. G=Granuloma, As=Alveolar Space, EC=Epithelioid Cells and L=Lymphocytes (D) Granuloma fraction of the lung tissue samples of mice infected with different strains of M. tuberculosis, based on the semi-quantitative estimation of the fraction of the lung tissue covered with granuloma. Data were analyzed using the MannWhitney test with *P < 0.05 and **P < 0.01. (E) Schematic representation of mice infection experiment. Mice (n = 5 per group at each time point) were infected with H37Rv and ΔvapC12. After the establishment of infection for 4 weeks, antibiotics (INH or RIF) were orally administered to the mice for the next 4 weeks. A parallel group of mice was left untreated (control). Bacterial loads were enumerated in the lungs (F) and spleens (G) at different time points post-infection as depicted by scatter plot. Each data point represents the log10 CFU of an infected animal in individual organs, and the error bar depicts the SEM for each group. Bar graph represents the percent killing of the vapC12 mutant strain with respect to WT at week 8 post-infection in the (F) lungs and (G) spleen. Data was normalized with the untreated group and plotted with respect to H37Rv (100%) on the y-axis. Data were analyzed using the MannWhitney test with **P < 0.01 and *P <0.05. (H) Percent survival plot of mice (n=20) infected with H37Rv and ΔvapC12 strains for 18 weeks post infection. Data were analyzed using the log-rank test with ***P value 0.0005.
VapC12 toxin modulates host immune response during M. tuberculosis infection
To understand the vapC12 mediated differences in the host immune response, we performed detailed immune-profiling studies on organs isolated from mice infected with WT, ▵vapC12 and complemented strains of M. tuberculosis at week 4 post-infection (Figure 2A). We estimated the innate immune cell population; monocytes, macrophages, MDSCs, neutrophils, and Natural Killer-T (NKT) cells in lungs and spleen isolated from infected mice (Figures 2B, C and Supplementary Figure 2A). A set of uninfected (UI) mice was included as a control group in all the experiments. We found that relative to the WT, both lungs and spleen isolated from mice infected with ▵vapC12 had a significantly lower percentage (~2%) of monocytes population (Figures 2B, C). In contrast, relative to the WT, organs isolated from the mice infected with the vapC12 mutant strain had a significantly higher percentages of macrophage, neutrophil and MDSCs (Figures 2B, C). The restoration of the mutant specific phenotype to the WT levels in the group of mice infected with the complemented strain further confirms the role of VapC12 toxin in modulating the host immunity. We also observed that the MDSCs population in the lungs was significantly elevated by ~3-fold in the mutant strain in comparison to the WT infected group (Figures 2B, C). Our findings corroborated the previous studies as the accumulation of MDSCs parallels disease progression in lung parenchyma and exacerbate disease by suppressing T-cell functions (29–32). Neutrophil infiltration in the lungs; which is a common feature of disease severity in TB patients (10) and susceptible mice (18, 33, 34) was found to be ~3-fold higher in the vapC12 mutant strain as compared to the WT and complemented strain (Figures 2B, C). Furthermore, the neutrophil population in the spleen isolated from vapC12 infected mice was also observed to be ~3 fold higher than in the WT (Figure 2C). Surprisingly, we found that the percentage of NKT cells in lungs and spleen isolated from vapC12 mutant infected mice were 2-fold and 3-fold elevated as compared to the organs isolated from WT infected mice (Figures 2D, E). It has been reported that individuals with pulmonary TB have lower number of NKT cells as compared to the healthy controls, indicating that they may have a protective role in TB infection (35, 36). Together, these data indicated that vapC12 mutant induces hyperinflammation by promoting the infiltration of myeloid cells in the lungs.
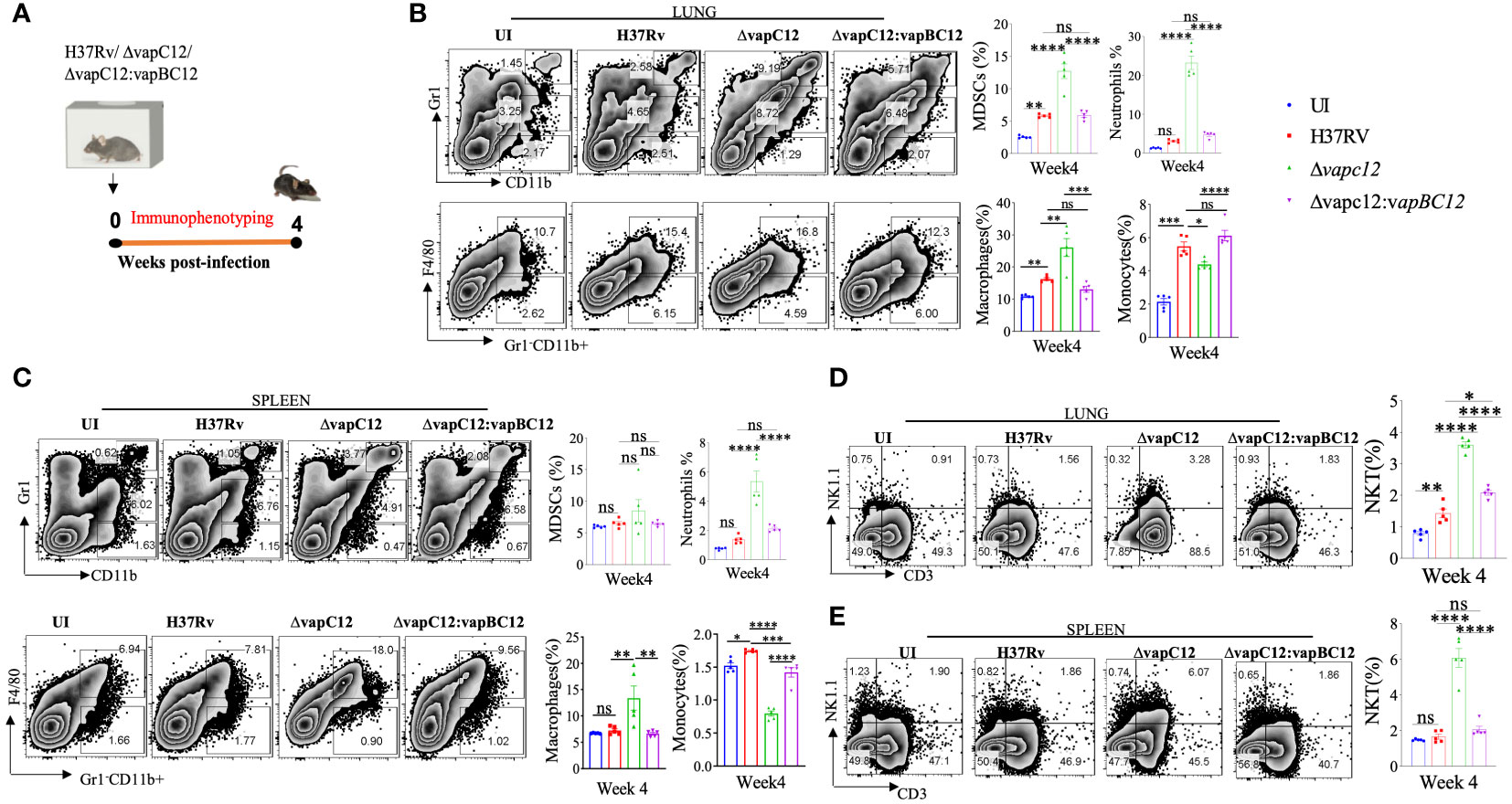
Figure 2 VapC12 toxin modulates host immune response during M. tuberculosis infection. (A) Schematic representation of the murine infection experiment. Mice (n =5 per group at each time point) were infected with H37Rv, ΔvapC12, and ΔvapC12:vapBC12. Immuno-profiling of lung infiltrated cells and splenocytes harvested at 4 weeks post infection. (B, C) Zebra plots and its representative bar graphs indicating the percentage frequency of monocytes (Gr1-CD11b+F4/80-), macrophages (Gr1-CD11b+F4/80+), MDSCs (CD11b+ Gr1+int) and neutrophils (CD11b+Gr-1high) cells harvested from (B) lungs and (C) spleen. Data are shown as mean ± SEM (n=5 animals per group). Data were analyzed using the one-way ANOVA followed by Tukeys multiple comparison test (*P <0.05, **P < 0.01, ***P<0.0005, ****P<0.0001). (D, E) Zebra plots and its representative bar graphs showing the percentage frequency of NKT (NK1.1+CD3+) cells harvested from (D) lungs and (E) spleen. Data are shown as mean ± SEM (n=5 animals per group). Data were analyzed using the one-way ANOVA followed by Tukeys multiple comparison test (*P <0.05, **P < 0.01, ***P<0.0005, ****P<0.0001).
VapC12 toxin regulates the polarization of macrophages during M. tuberculosis infection
The choice of effector mechanism selected by the host to clear infection is regulated by the type of cytokine response dictated by the initial innate immune response. The fate of Th1 and Th2 polarisation, ideal for intracellular and extracellular pathogens respectively, is effectively decided by the initial differentiation of the macrophages to either classical or alternate types. As an effective host immune escape strategy, the pathogen tries to manipulate the macrophage differentiation process in its own favor by subverting the host cell metabolic pathways (37). To investigate role of the vapC12 gene in manipulating macrophage differentiation we used antibodies against CD80 and CD206, classical markers for M1 and M2 respectively to quantify the relative M1/M2 population in the lungs of mice infected with different M. tuberculosis strains. We found a ~1.5-fold decrease in the frequency of M1 (CD80+) macrophages in lungs isolated from mice infected with vapC12 mutant as compared to the WT. On the contrary, we found higher frequency of M2 expressing macrophages in the lung tissue infected with ▵vapC12 (Figure 3A and Supplementary Figure 3A). Furthermore, the vapC12 mutant strain demonstrated a ~10% increase in M1 macrophages and a corresponding ~10% decline in M2 macrophages as compared to the WT (Figure 3A).The same was further validated by quantifying the transcripts of markers specific to either M1 or M2 macrophages. We estimated the transcripts levels of M1 [CD80 and CD86] and M2 [Arginase1 (Arg1), YM1 and Fizz1) specific markers by isolating RNA from the lungs of WT, vapC12 mutant infected and uninfected (UI) mice. As expected, we found ~6-fold (Fizz1), ~10-fold (YM1) and ~35-fold (Arg1) increase in the expression of M2 markers in the vapC12 mutant strain as compared to the WT strain (Figure 3B). In concordance with elevated levels of M2 markers, a clear upregulation in the gene expression levels of the Th2 related cytokines; IL 6 (****P<.0.0001) and IL10 (*P<.0.05) was also observed (Figure 3C). Surprisingly, the transcription signature associated with M1 and Th1 markers CD80, CD86, IFN-γ and TNF-α showed non-significant changes in their gene expression levels and a clear reduction in Th1-induced secretion of IL-2 was noted (Figures 3B, C). In context to T cell differentiation, we observed that the vapC12 mutant demonstrated an increased percentage of CD4+ T cells producing IFN-γ in both the lungs and spleen when compared to the WT. Conversely, there was a notable reduction in the IL17-producing T-cell population in the spleen of the vapC12 mutant compared to the WT, with no discernible impact observed in the lung (Supplementary Figure 3B). Overall, the data convincingly demonstrates that the ratio of M1/M2 (Th1/Th2) is significantly lower in the mice infected with ▵vapC12 as compared to the WT strain. In sum, these data suggest that levels of VapC12 ribonuclease help M. tuberculosis control the M1/M2 polarization modulating the Th1/Th2 pathway critical for regulating the disease outcome.
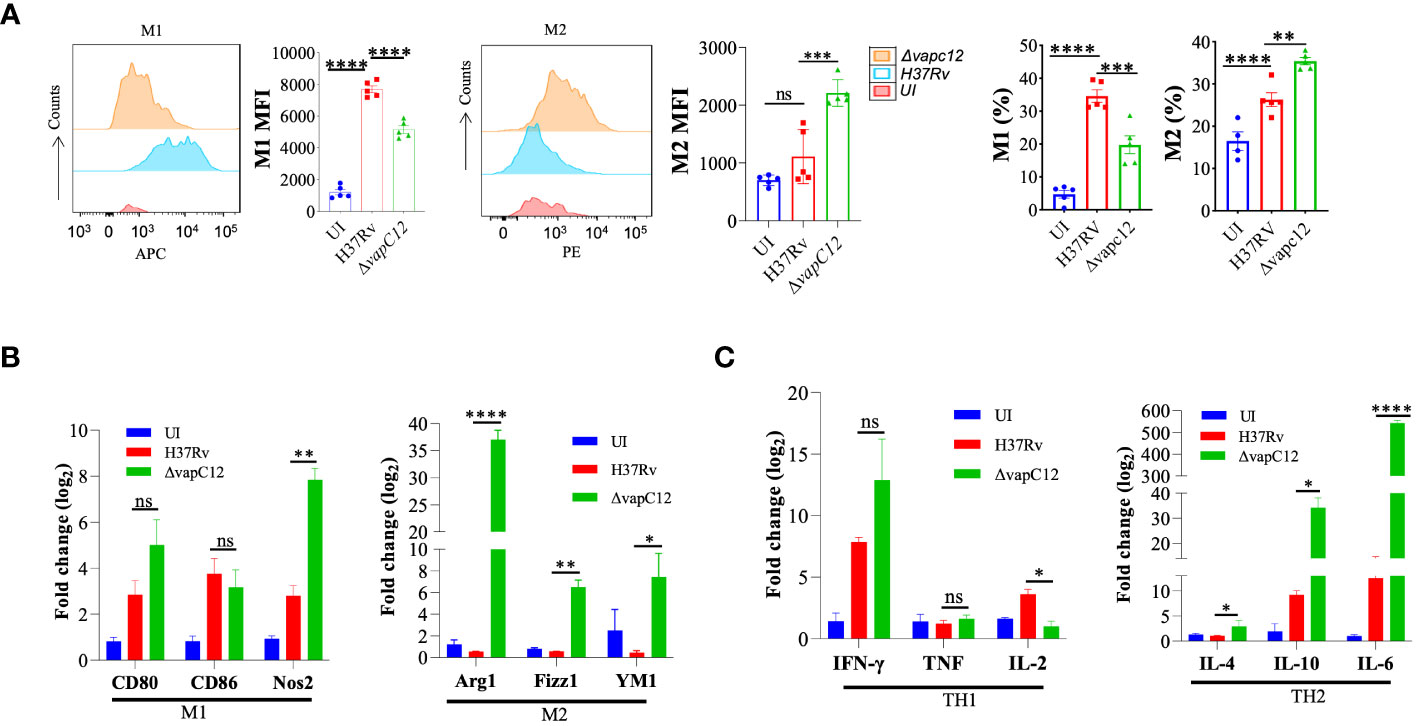
Figure 3 VapC12 toxin regulates the polarization of macrophages during M. tuberculosis infection. (A) Left panel M1: shows the histogram and bar graph of CD80 and right panel M2: shows the histogram and bar graph of CD206 in Gr1-CD11b+ F4/80+ cells by flow cytometry and bar graph represents MFI (Mean fluorescent index) and frequency (%). Data were analyzed using FlowJo software (tree star) and statistical calculation was presented as mean ± SEM, n = 5. ***P<0.0002, ****P < 0.0001. (B) Bar graph for mRNA expression of M1 (CD80 and CD86) and M2 (Arg1, YM1 and Fizz1) macrophage markers in the lung cells, data representative of triplicates *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (C) Bar graph for mRNA expression of TH1 (IFN-γ, TNF and IL-2) and TH2 (IL-4, IL-10 and IL-6) in the lung cells, data representative of triplicates *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
VapC12 is critical for induction of adequate T cell immune response during M. tuberculosis infection
In addition to myeloid cells, CD4+ and CD8+ T lymphocytes are the key players in the establishment of protective immunity against TB in mice. The changes in cellular phenotype coincided with the significant decrease in the adaptive immune markers i.e. CD4+ and CD8+ T lymphocytes in the organs isolated from the ▵vapC12 infected mice relative to the WT. In lungs harvested from mice infected with vapC12 mutant strain, we observed a significant 3-fold and 2-fold reduction in the percentage of CD4+ and CD8+ T-cells respectively, both with p<0.05 as compared to the WT infected mice (Figure 4A and Supplementary Figure 4A). Surprisingly, while the CD4+ T-cell profile in the spleen for both groups were identical to the lungs, the CD8+ T-cell remained unaltered (Figure 4B). As shown in Figure 4C, both CD4+ and CD8+ T-cell percentages was suppressed in the lymph nodes of vapC12 infected mice. Interestingly, in comparison to the WT, the secondary lymphoid organs isolated from the mice infected with ▵vapC12 harbored significantly higher percentage of γδ-T cell 4 weeks post-infection (Figures 4B, C) with no such differences observed in the lungs (Figure 4A). This data corroborates the previous findings which suggest the role of γδ-T cells in TB pathogenesis by inducing inflammation and granuloma formation (38). Thus, we conclude that the absence of VapC12 toxin in M. tuberculosis prevents the induction of adequate T-cell response in the lungs and lymphoid organs (lymph nodes and spleen) of infected mice.
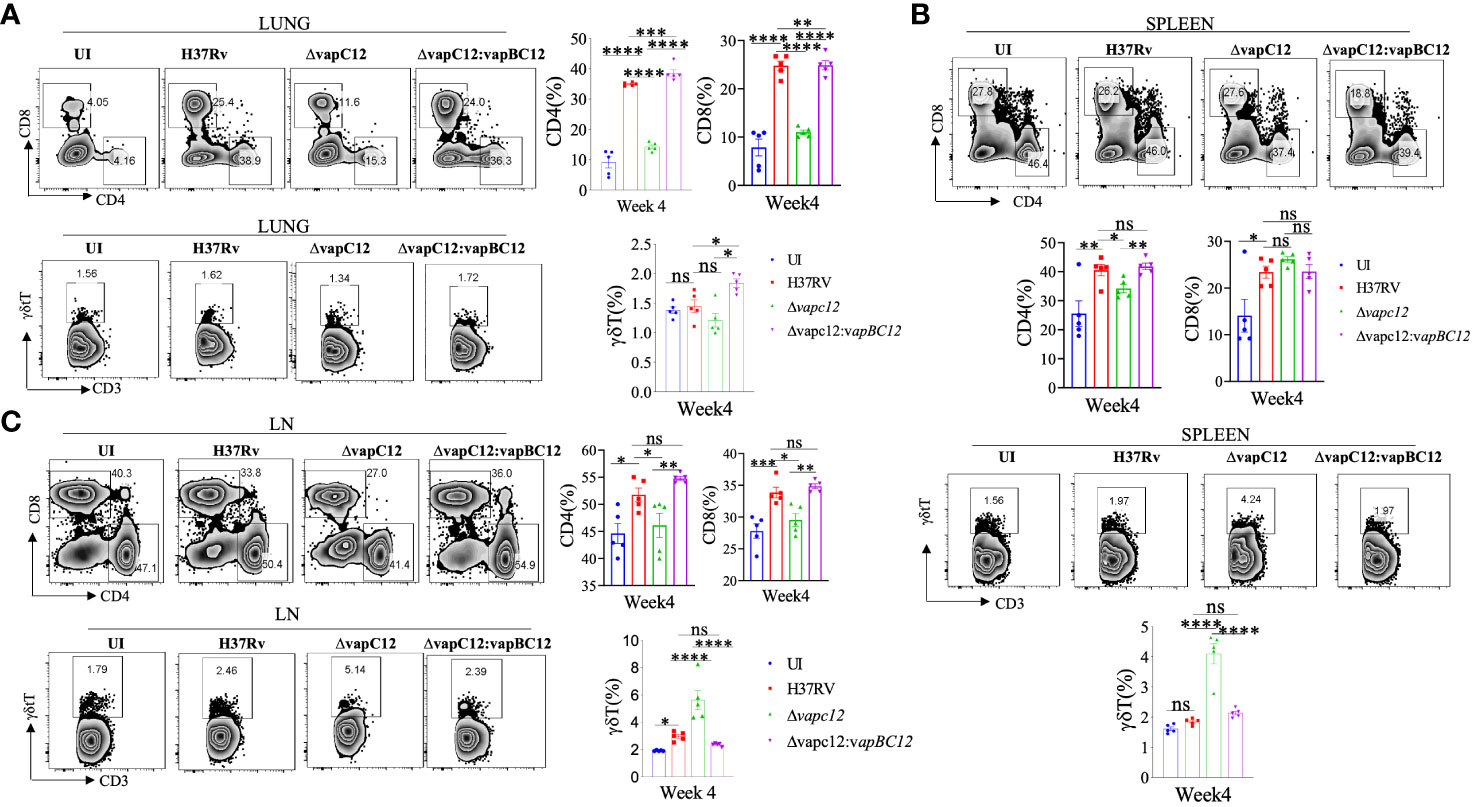
Figure 4 vapC12 is critical for induction of adequate T cell immune response during M. tuberculosis infection. (A-C) Zebra plots and its representative bar graphs indicating percentage frequency of CD4, CD8 and γδ+T cells in the (A) lung (B) spleen and (C) Lymph nodes. Data were analyzed using FlowJo and shown as mean ± SEM (n=5 animals per group). Data were analyzed using the one-way Anova test followed by Tukeys multiple comparison test (*P <0.05, **P < 0.01, ***p<0.0005, ****p<0.0001).
VapC12 is essential for the resolution of neutrophil-mediated inflammation in tuberculosis
To further delineate the role of host factors associated with the hypervirulent phenotype of the mutant strain, we analysed the RNA sequencing data of host genes from lung tissues of mice infected with WT, ▵vapC12 and complemented strain. The same was subjected to a detailed transcriptome analysis (Figure 5A). Initially, a principal component analysis (PCA) was performed to visualize the variations in the obtained expression profiles. Data displayed in the first two principal components (PCs) suggest that all three gene clusters are well separated from each other (Supplementary Figure 5A). To study the VapC12-mediated differential expression of the host genes, we compared the gene expression profiles of lung tissues from mice infected with the ▵vapC12 and the WT strains (Figure 5B). In total, 372 differentially expressed genes (DEGs) were obtained, out of which 82 and 290 genes were up- and down-regulated, respectively (Supplementary Figure 5B and Supplementary File S1). Pathway enrichment analysis of the upregulated genes revealed an enrichment in the processes related to neutrophil functions like neutrophil migration and neutrophil-mediated immunity (Figure 5C). We found that in comparison to the WT, infection with the ▵vapC12 strain enhanced the expression of genes that encode proteins implicated in neutrophil-mediated immunity in the host (Cxcl2, Trem1, S100A8/A9 and Ccl3) (Figure 5D). S100A8 and S100A9; cytosolic proteins of neutrophils are important for the recruitment of neutrophils to the granulomas as demonstrated in human and animal models of TB (39). A ~2.56 fold (log10) increase in the transcript levels of genes encoding S100a8 and S100a9 by the host infected with the vapC12 mutant strongly suggests an increase in neutrophil recruitment and accumulation in the lung tissues. Genes belonging to the inflammation-associated markers like matrix metalloproteases (Mmp8 and Mmp9), secreted by neutrophils that are known to be critical for the formation of granuloma and inducing inflammation during the acute stage of infection (7), were found to be significantly induced in the lung tissues of mice infected with the mutant strain. We also found an increase in the expression of Cxcr2 (22-fold), Cxcl1 (18-fold) and Cxcl2 (177-fold) genes which suggests enhanced neutrophil recruitment in lungs of mice infected with vapC12 mutant relative to the WT (40) (Supplementary File S2). Additionally, IL1a (30-fold) and IL1b (19-fold), the pro-inflammatory cytokine markers associated with TB (41) were also found to be significantly upregulated in the tissue isolated from mice infected with mutant strain as compared to the WT and complemented strain (Figure 5E). Interestingly, in comparison to the WT, the expression of cystatins such as Cstdc4 [3.2-fold (log10)], Stfa2l1 [2.8-fold (log10)] and Csta2 [2.26-fold (log10)] which are the endogenous inhibitors of the host antimicrobial proteins cathepsins (42)were also increased in lungs of mutant strain infected animals (Figure 5F). Simultaneously, peptidases which play an important role in pathogen killing by activating the antimicrobial peptides were found to be significantly reduced in the ▵vapC12 infected mice lungs (Figures 5G, H), setting off an unrestricted growth of M. tuberculosis leading to excessive inflammatory responses. Additionally, the observed reduction in the transcript level of genes associated with adaptive immune response correlates with our earlier findings that the ▵vapC12 infected mice fail to induce an effective adaptive immune response (Supplementary Figure 5C). Together, these observations suggest that ▵vapC12 proliferates and induces a proinflammatory response by modulating the transcript level of proteins that enhances neutrophil recruitment, IL-1 production, endopeptidase inhibitors and by suppressing the antimicrobial proteases and appropriate adaptive immune response inside the host.
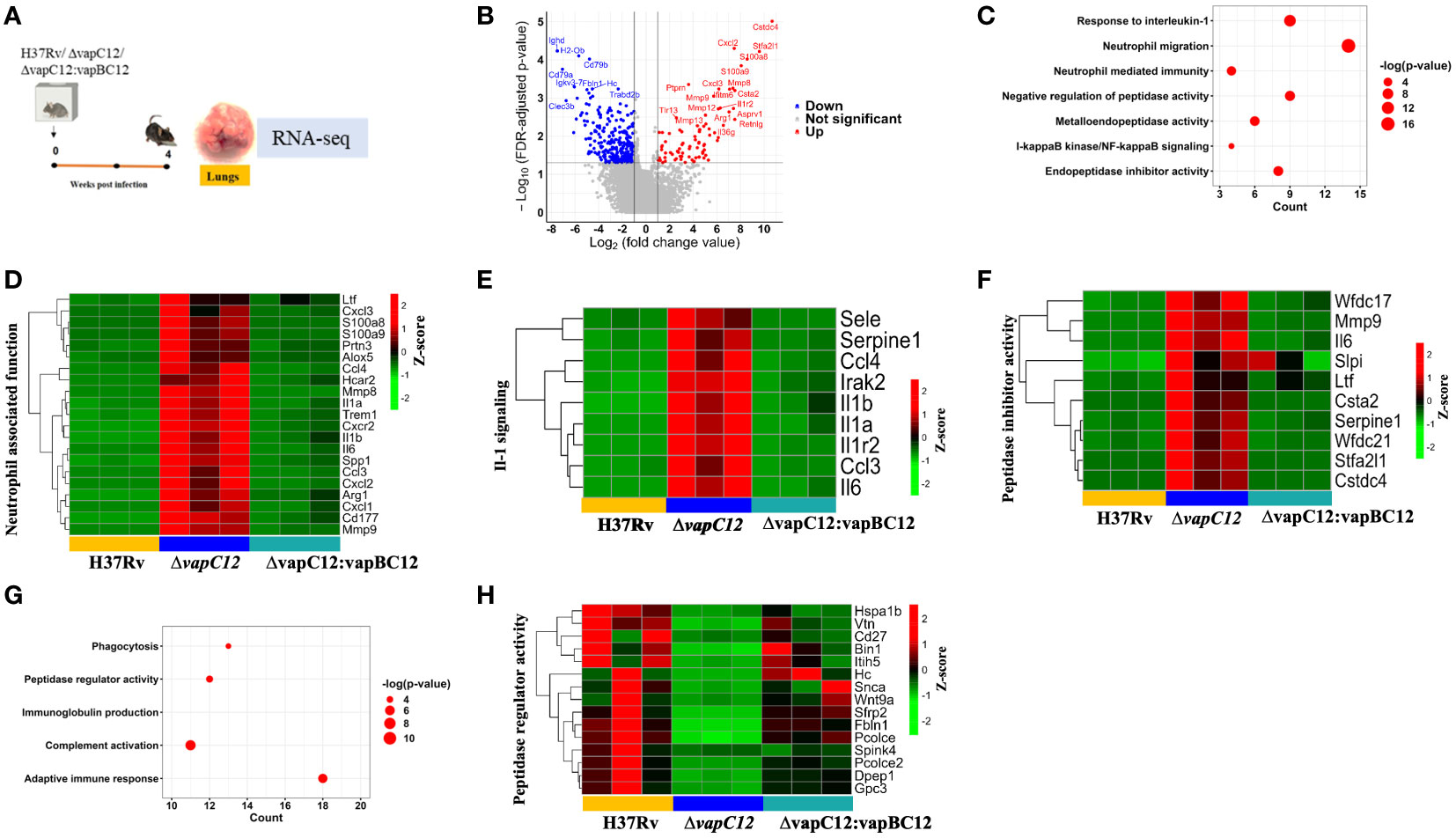
Figure 5 VapC12 is essential for the resolution of neutrophil-mediated inflammation in tuberculosis. (A) Pictorial representation of the process of animal experiment with mice. RNA isolated from lung tissues of mice (n=3) infected with WT, ▵vapC12 and complemented strain after 4 weeks of infection. (B) The volcano plot showing comparison of gene expression profiles from mice infected with parental (WT) and ▵vapC12 strain of M. tuberculosis. Differentially expressed genes (DEGs) were selected using the fold change cut-off value of 2 and FDR-adjusted p-value < 0.05. The up- and down-regulated genes are highlighted here in red and blue, respectively. (C) Dot plot of the gene ontology (GO) pathway enriched in upregulated genes. The x-axis represents the number of obtained upregulated genes for each pathway. (D) Heatmap representing the expression levels of the 21 upregulated genes involved in the neutrophil associated function. Data were Z-score transformed and the name of the genes are displayed on the right. The names of the corresponding infected strains for each sample are mentioned at the bottom of the figure. (E) Heatmap representing the expression levels of the 9 upregulated genes involved in the IL-1-mediated signaling. (F) Heatmap representing the expression levels of the 10 upregulated genes involved in the peptidase inhibitor activity. (G) Dot plot of the gene ontology (GO) pathway enriched in down-regulated genes. The x-axis represents the number of obtained upregulated genes for each pathway. (H) Heatmap representing the expression levels of the 15 downregulated genes involved in the peptidase regulator activity.
Depletion of neutrophils failed to restore the vapC12 mutant phenotype
As neutrophils are an abundant component of the innate immune system, their highly inflammatory actions have potentially severe consequences for host tissues (43). When the regulatory mechanisms responsible for clearing apoptotic neutrophils are compromised or if the underlying infection cannot be resolved, the actions of neutrophils may turn detrimental to the host (44, 45). As previously reported, neutrophils depletion can be achieved by the administration of 1A8 antibody (45). To confirm the role of neutrophils in ▵vapC12 induced hypervirulent phenotype we infected groups of C57BL/6 mice, with either 1A8 antibody or their isotype control (Figure 6A). Lungs treated with 1A8 antibody in vapC12 mutant resulted in significant decrease in the neutrophils population relative to the isotype group (Figure 6B). Additionally, 1A8 antibody treatment resulted in a significant decrease in the monocytes (~4%) and macrophages population (~5%) in the lungs (Figure 6B). Treatment with 1A8 antibody also resulted in a significant reduction (5%) in the population of CD4+ and CD8+ T cells in the lungs and spleen harvested from antibody-treated vapC12 mutant mice as compared to WT (Figure 6C). Surprisingly, we did not find any difference in the overall lung and spleen bacillary burden in mice infected with vapC12 mutant strain that were either treated with 1A8 antibody or the isotype control (Figure 6D). These data collectively suggest that the ▵vapC12 induced hypervirulence phenotype is independent of the increase in the infiltration of neutrophil observed in the lungs of ▵vapC12 infected mice.
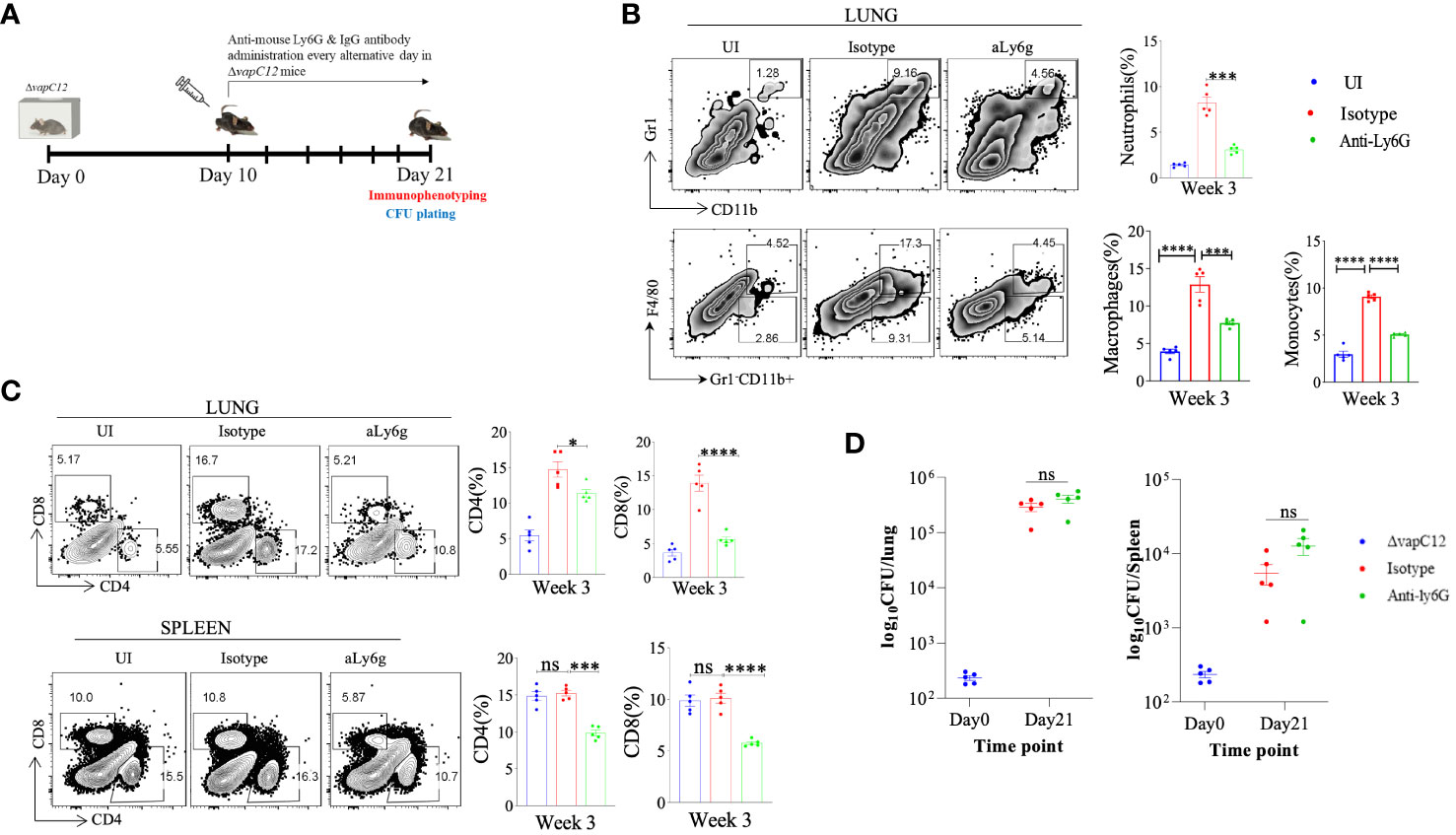
Figure 6 Neutrophil depletion failed to restore the vapC12 mutant phenotype to the wild type levels. (A) Schematic representation of antibody depletion experiment in mice. ΔvapC12 infected mice (n=5) were either treated with neutrophil depleting Anti-Ly6G or IgG control antibody every alternative day from day 10 to day 21 post-infection. Mice were sacrificed on day 21 for immunophenotyping & bacterial enumeration. (B) Zebra plots and its representative bar graph shows the percentage frequency of monocytes (Gr1-CD11b+F4/80-), macrophages (Gr1-CD11b+F4/80+),neutrophils (CD11b+Gr-1high) cells in the lungs. Data are shown as mean ± SEM (n=5 animals per group). Data were analyzed using the one-way ANOVA followed by Tukeys multiple comparison test (*P <0.05, **P < 0.01, ***P<0.0005, ****P<0.0001). (C) Zebra plots and its representative bar graphs indicating percentage frequency of CD4+ and CD8+ T cells in the lung and spleen. Data were analyzed using FlowJo and shown as mean ± SEM (n=5 animals per group). Data were analyzed using the one-way ANOVA test followed by Tukeys multiple comparison test (*P <0.05, **P < 0.01, ***p<0.0005, ****p<0.0001). (D) Scatter plot showing bacterial load in the lungs and spleen of mice infected with ΔvapC12 at designated time points, the lungs were homogenized in 2 ml of saline, and ten-fold serial dilutions of homogenates were plated on 7H11+OADC plates. Each group constituted five mice per time point. Data plotted represent the mean ± SEM. Significant differences observed between groups are indicated. Data were analyzed using the MannWhitney test with **P < 0.01 and *P <0.05.
VapC12 deletion induced hypervirulence in M. tuberculosis is dependent on TLR4
Neutrophil associated proteins, S100A8/A9 function as DAMPs (danger-associated molecular pattern) and can act as a ligand of TLR4 thus activating the innate immune responses (46). Since RNA-sequencing data showed that both S100A8/A9 neutrophil-associated markers were elevated in the lungs of vapC12 mutant, we hypothesized that TLR4 signaling could play an important role in ▵vapC12 mediated hypervirulent phenotype. To study the role of TLR4 signaling, we infected C3H/HeJ mice (carrying mutated TLR4) with both the WT and ▵vapC12 strains (Figure 7A). In parallel, we included a C57BL/6 mice group infected with WT and vapC12 mutant as a hypervirulent phenotype control. As expected, relative to the WT, we observed an increase in the bacterial load in the organs isolated from C57BL/6 mice infected with the ▵vapC12 strain (Supplementary Figure 6). Surprisingly, the above phenotype observed in the C57BL/6 mice was completely reversed in C3H/HeJ mice (Figures 7B, C). In comparison to the C57/BL6, C3H/HeJ mice infected with ▵vapC12 were presented with a ~10-fold & ~40-fold reduction in bacterial burden in the lungs & spleen respectively (Supplementary Figure 6A, B). Interestingly, the reduction in the bacterial burden observed in C3H/HeJ mice coincided with a significant reduction in the population of neutrophils in these organs (p < 0.05). Additionally, we also observed that the percentage of MDSCs and macrophage population from the lungs of ▵vapC12 infected TLR4 defective mice were restored to the WT levels (Figure 7D). Surprisingly, in comparison to the lungs, restoration of the adaptive immune response in ▵vapC12 infected TLR4 defective mice were specifically observed in the spleen (Figure 7E). These findings were in line with our bacterial enumeration data suggesting that TLR4 signaling pathway could play an important role in ▵vapC12 mediated hypervirulent phenotype observed in the mice.
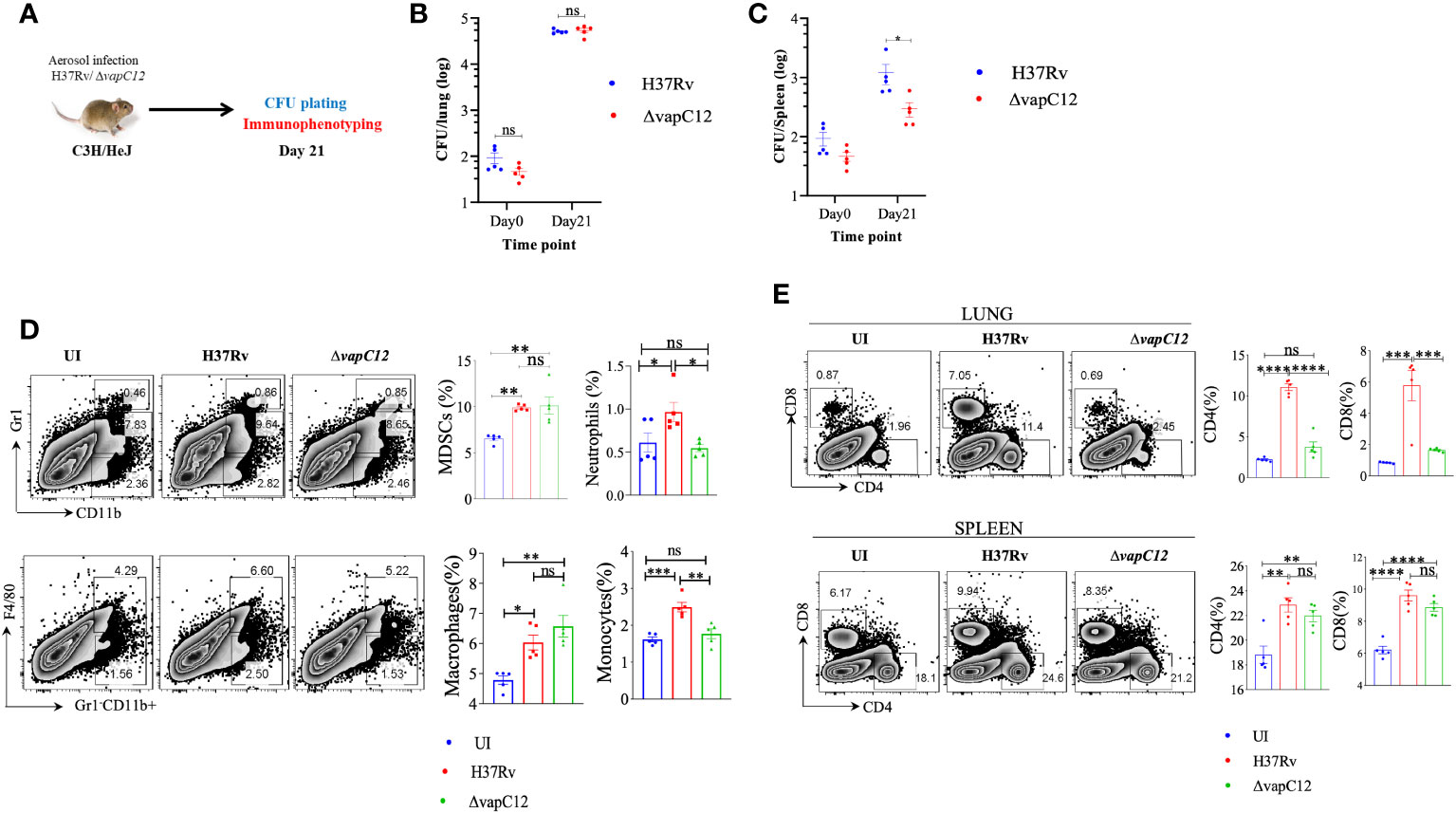
Figure 7 vapC12 deletion induced hypervirulence in M. tuberculosis is dependent on TLR4. (A) Schematic representation of the animal experiment. C3H/HeJ mice were aerosol infected with H37Rv and ΔvapC12. Mice were sacrificed on indicated time point for bacterial enumeration and immunophenotyping. (B, C) Scatter plot showing bacterial load in the (B) lungs and (C) spleen of mice infected with H37Rv and ΔvapC12 at indicated time points, the lungs were homogenized in 2 ml of saline, and ten-fold serial dilutions of homogenates were plated on 7H11+OADC plates. Each group constituted five mice per time point. Data plotted represent the mean ± SEM. Significant differences observed between groups are indicated. Data were analyzed using the MannWhitney test with **P < 0.01 and *P <0.05. (D) Zebra plot and its representative bar graphs showing the percentage frequency of monocytes (Gr1-CD11b+F4/80-), macrophages (Gr1-CD11b+F4/80+), MDSCs (CD11b+ Gr1+int) and neutrophils (CD11b+Gr-1high) cells in the lung. Data are shown as mean ± SEM (n=5 animals per group). Data were analyzed using the one-way ANOVA followed by Tukeys multiple comparison test (*P <0.05, **P < 0.01, ***P<0.0005, ****P<0.0001). (E) Zebra plots and its representative bar graphs indicating the percentage frequency of CD4+ and CD8+ T cells in the lung and spleen. Data were analyzed using FlowJo and shown as mean ± SEM (n=5 animals per group). Data were analyzed using the one-way ANOVA test followed by Tukeys multiple comparison test (*P <0.05, **P < 0.01, ***p<0.0005, ****p<0.0001).
Discussion
Earlier we reported that the vapC12 ribonuclease toxin targets proT tRNA and modulates the levels of various proline-rich PE-PGRS proteins (22). In the same study, we further demonstrated that organs isolated from guinea pigs infected with M. tuberculosis strain lacking vapC12 gene had higher bacterial load and were found to be more pathogenic measured in terms of the degree of inflammation and pathological lesions as compared to the WT strain (22). The current study was undertaken to further our understanding of the hypothesis that vapC12 mediated differential expression of antigenic proteins modulates host immune response. Due to easy availability of good genetic and immunological toolbox, we in the current study have decided to use mice as a surrogate host. Similar to the phenotype observed in guinea we found that the vapC12 deficient strain of M. tuberculosis demonstrated a hypervirulent phenotype marked by an increase in bacterial burden and enhanced pathological lesions. Subsequently we have also demonstrated that ▵vapC12 promoted the influx of pro-inflammatory myeloid cells at the site of infection resulting in robust transcription of S100A8/A9 proteins providing enhanced growth benefit to the mutant strain as compared to the WT. By inhibiting the S100A8/9-TLR4 axis we were able to mitigate the bacterial growth in vapC12 mutant by reducing the detrimental inflammatory immune response.
The killing of pathogens by macrophages is known to be the primary defence mechanism against intracellular pathogens. Like T-lymphocytes, macrophages also differentiate into M1 and M2 phenotypes depending on their microenvironment and amino acid metabolic shift (37). Arginine catabolism via iNOS leads to the production of L-citrulline and nitric oxide (NO), playing a predominant role in providing resistance against parasitic and bacterial infections. On the contrary, use of arginine as a substrate by Arg1 results in the generation of L-ornithine and urea and is known to promote disease activity (37). During M. tuberculosis infection, Arg1 stimulation in-vitro in macrophages through a TLR-dependent and STAT6-independent pathway leads to the inhibition of NO production. Conversely, lack of Arg1 was connected to elevated NO expression and improved control of mycobacterial infection (47). Arginine metabolism has been linked to infection as high arginase activity has been reported in lesions of leishmaniasis patients and human TB granulomas (48–50). Most of the available piece of evidence suggests that pathogens responsible for acute as well as chronic infections manipulate STAT6-PPARγ/δ pathways to avoid M1 polarization (51–55). While the WT M. tuberculosis induced polarization of macrophage towards M1, a skewed polarization towards M2 macrophages observed in mice infected with vapC12 deficient M. tuberculosis strain suggests that a vapC12-dependent protein expression actively influences the macrophage polarization in the host. As a consequence, there is an altered T-cell response activated in terms of Th1/Th2 by the host. This implies that M. tuberculosis actively modulates host immunity by differentially expressing its proteins and the resulting spatiotemporal expression of M. tuberculosis antigens critically regulate the host immune pathways which ultimately affects the disease progression and outcome. Contrary to the WT, failure of ▵vapC12 to prevent the accumulation of excessive neutrophils at the site of infection results in enhanced inflammation, M2 polarization and a Th2 type response which together creates an environment conducive for the growth of M. tuberculosis inside the host. In agreement with our observations, several reports have established that excessive neutrophil infiltration leads to tissue damage and exacerbation of inflammation which is detrimental to the host (56–59). Additionally, upregulation of the IL-1 cytokine signaling genes suggest increased levels of proinflammatory IL-1α and IL-1β cytokines in the lungs infected with vapC12 deficient M. tuberculosis strain. The dysregulation of the levels of IL-1 and IL-1 receptor antagonists cannot be ruled out.
Cathepsins carry out efficient innate and adaptive antibacterial functions, either directly by taking part in the death of invasive bacteria or by processing and presenting the antigen of infected bacteria to lymphocytes, which escalates the removal of the pathogen and triggers the development of long-lasting immunity (60). However, bacteria have the ability to manipulate cathepsin expression and proteolytic activity in order to benefit their own intracellular survival in macrophages and escape the host-mediated immune surveillance (60). M. tuberculosis avoids contact with cathepsins by modulating phagosome maturation, impairing the delivery of lysosomes to phagosomes in NF-κB dependent manner and by escaping from the phagosomes to the cytosol, thus facilitating its replication and survival (61–63). Cystatins, which are the inhibitors of cathepsins; the proteolytic enzymes involved in the control of M. tuberculosis infection were increased at the transcript level in the lungs of ▵vapC12 infected mice (42). In support of increased peptidase inhibitor activity, we also observed suppressed levels of peptidases in lung homogenates of ▵vapC12 infected mice. Our findings are also in concordance with previous reports that cathepsins play a central role in the immune system and hence its necessary to regulate its activity to maintain homeostasis within cells or tissues (64). In recent years, MDSCs which are a heterogeneous population of immunocytes from myeloid origin have also been found to be a safe harbor for M. tuberculosis proliferation (65, 66). As reported in recent studies, we have identified that the increase in MDSCs population mirrored alleviating protective T-cell response in the spleen of mice infected with the vapC12 mutant as compared to the WT strain infected mice (30, 67, 68). These results implicate that the influx of neutrophils along with MDSCs, IL-1 cytokines and endopeptidase inhibitors might be responsible for increased bacillary load and lung pathology in the mice infected with ▵vapC12 mutant.
S100A8/A9 proteins, members of the S100 family promote the recruitment of myeloid cells, specifically neutrophils, dendritic cells and monocytes to the lungs through the induction of inflammatory cytokines (39). A recent study has mechanistically demonstrated that the expression of these calprotectin is needed for the upregulation of CD11b integrin on neutrophils resulting in their accumulation during chronic TB infection (69). Indeed, our study revealed that S100A8/A9 levels corroborated accumulation of myeloid-derived inflammatory cells in the lungs of mice infected with vapC12 strain during TB disease progression. The RNA-sequencing data also showed increased expression of the marker gene Cxcr2 suggesting increased recruitment of myeloid cells at week 4 post-infection in the vapC12 mutant infected mice. These findings are consistent with the literature available on the function of S100A8/A9 in host defence mechanisms against bacterial and viral infections (39, 70).
S100A8/A9 plays a decisive role in innate immunity by binding to receptors such as TLR4 (46) and receptor for advanced glycation end products (RAGE), which activates multiple inflammatory pathways mediated by these pattern-recognition receptors (PRRs) (46, 71, 72). Finally, we also investigated the role of TLR4 in inducing S100A8/A9 mediated hyper inflammation in vapC12 mutant. By blocking the TLR4 signaling (C3He/HeJ mice) the bacillary burden in the lungs of vapC12 mice became comparable to that of the WT strain. Based on these findings, we believe that the absence of TLR4 in C3He/HeJ prevented the induction of PAMP-mediated inflammatory response. This resulted in the restoration of the growth of vapC12 mutant to that of the WT levels. The exact mechanism of S100A8/A9 overexpression and TLR4 signal activation induced in vapC12 mutant requires further investigation. Characterizing the differentially expressed secretory proteins of vapC12 mutant would further give us a better insight into mechanistically understanding the downstream processes.
In conclusion, we have studied how the absence of the ribonuclease toxin in M. tuberculosis greatly impacted the ability of the pathogen to dampen the early innate immune response. The uninterrupted migration of granulocytes and continuous expression of proinflammatory cytokines resulted in enhanced pathology. The resulting M2 mediated Th-2 response, generated in the mice infected with ▵vapC12 favored the replication and growth of M. tuberculosis. Overall our data suggest that VapC12 ribonuclease toxin-mediated differential expression of M. tuberculosis proteins help in the timely resolution of TLR4-mediated inflammation which is critical for disease persistence in TB (Figure 8).
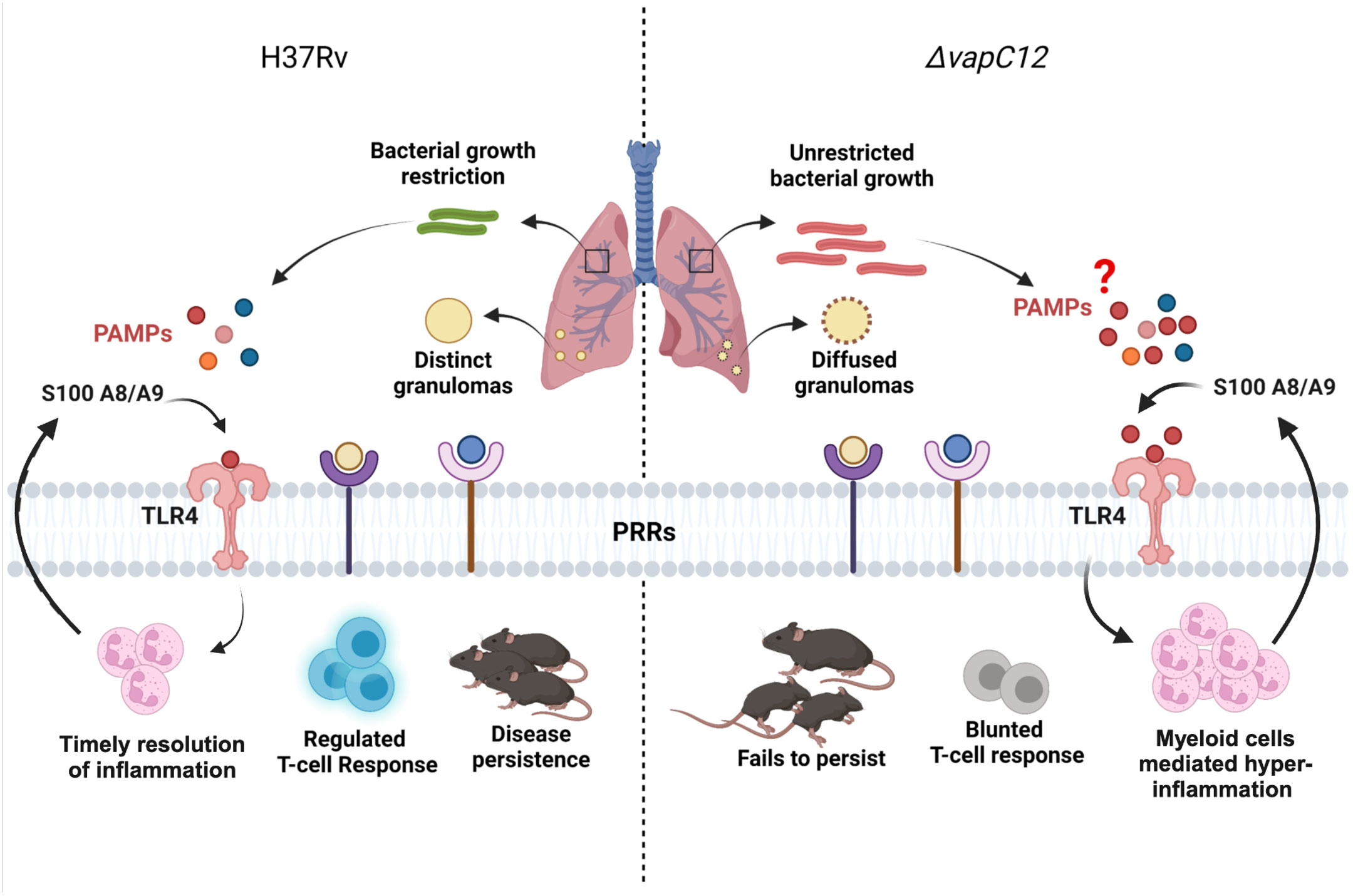
Figure 8 Model depicting the VapC12 toxin-mediated host immunomodulation during M. tuberculosis infection. Our findings demonstrate that the absence of ribonuclease toxin VapC12 favors unrestricted bacterial growth and exacerbated pathology. Additionally, vapC12 mutant hinders the ability of M. tuberculosis to control the initial influx of myeloid cells to the site of infection leading to continuous inflammation and blunted T-cell response. Further, myeloid cells mediated hyper-inflammation resulted in over activation of S100A8/A9-TLR4 signaling. Finally, vapC12 mediated expression of M. tuberculosis proteins is critical for disease persistence in tuberculosis. Illustrated using Biorender (www.biorender.com).
Data availability statement
The datasets presented in this study can be found in online repository - India Nucleotide Data Archive (INDA) maintained at Indian Biological Data Centre (IBDC), India. The accession number(s) can be found below: https://inda.rcb.ac.in/viaaccessionIDPRJEB63988.
Ethics statement
The animal study was approved by Institutional Animal Ethics Committee (IAEC), THSTI. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
ST: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing – original draft, Writing – review & editing. SS: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing – review & editing. TS: Data curation, Investigation, Methodology, Writing – review & editing. AP: Formal analysis, Methodology, Software, Writing – review & editing. MP: Investigation, Methodology, Writing – review & editing. VN: Investigation, Writing – review & editing. DR: Methodology, Software, Writing – review & editing. SC: Methodology, Software, Writing – review & editing. AA: Project administration, Resources, Writing – review & editing. AKP: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the India-Singapore grant by the Department of Science and Technology (DST), India, to AKP. (INT/Sin/P-08/2015). DST-SERB grant (CRG/2019/004250) from the Department of Science and Technology, Government of India, and intramural funding by THSTI to AKP are acknowledged. ST was supported by fellowship from UGC [19/06/2016(i)EU-V].
Acknowledgments
We are thankful to EAF and IDRF at THSTI for their support. We thank ILBS, New Delhi and histopathology facility at THSTI for histopathological analysis and Surjeet Yadav for lab maintenance. THSTI is acknowledged for providing all support for equipment and other infrastructure.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1302163/full#supplementary-material
References
2. Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, et al. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell. (2011) 145:39–53. doi: 10.1016/j.cell.2011.02.022.
3. Dartois VA, Rubin EJ. Anti-tuberculosis treatment strategies and drug development: challenges and priorities. Nat Rev Microbiol. (2022) 20:685–701. doi: 10.1038/s41579-022-00731-y.
4. Ehrt S, Schnappinger D, Rhee KY. Metabolic principles of persistence and pathogenicity in Mycobacterium tuberculosis. Nat Rev Microbiol. (2018) 16:496–507. doi: 10.1038/s41579-018-0013-4.
5. Bussi C, Gutierrez MG. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol Rev. (2019) 43:341–61. doi: 10.1093/femsre/fuz006.
6. Lombard R, Doz E, Carreras F, Epardaud M, Le Vern Y, Buzoni-Gatel D, et al. IL-17RA in non-hematopoietic cells controls CXCL-1 and 5 critical to recruit neutrophils to the lung of mycobacteria-infected mice during the adaptive immune response. PloS One. (2016) 11:e0149455. doi: 10.1371/journal.pone.0149455.
7. Kang DD, Lin Y, Moreno J-R, Randall TD, Khader SA. Profiling early lung immune responses in the mouse model of tuberculosis. PloS One. (2011) 6:e16161. doi: 10.1371/journal.pone.0016161.
8. Capuano SV III, Croix DA, Pawar S, Zinovik A, Myers A, Lin PL, et al. Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infection Immun. (2003) 71:5831–44. doi: 10.1128/IAI.71.10.5831-5844.2003.
9. Palmer MV, Wiarda J, Kanipe C, Thacker TC. Early pulmonary lesions in cattle infected via aerosolized Mycobacterium bovis. Veterinary Pathol. (2019) 56:544–54. doi: 10.1177/0300985819833454.
10. Eum S-Y, Kong J-H, Hong M-S, Lee Y-J, Kim J-H, Hwang S-H, et al. via LE, Barry III CE: Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest. (2010) 137:122–8. doi: 10.1378/chest.09-0903.
11. Gideon HP, Phuah J, Junecko BA, Mattila JT. Neutrophils express pro-and anti-inflammatory cytokines in granulomas from Mycobacterium tuberculosis-infected cynomolgus macaques. Mucosal Immunol. (2019) 12:1370–81. doi: 10.1038/s41385-019-0195-8.
12. Ozaki T, Nakahira S, Tani K, Ogushi F, Yasuoka S, Ogura T. Differential cell analysis in bronchoalveolar lavage fluid from pulmonary lesions of patients with tuberculosis. Chest. (1992) 102:54–9. doi: 10.1378/chest.102.1.54.
13. Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. (2010) 466:973–7. doi: 10.1038/nature09247.
14. Liu Q, Pan L, Han F, Luo B, Jia H, Xing A, et al. Proteomic profiling for plasma biomarkers of tuberculosis progression. Mol Med Rep. (2018) 18:1551–9. doi: 10.3892/mmr.
15. Lowe DM, Bandara AK, Packe GE, Barker RD, Wilkinson RJ, Griffiths CJ, et al. Neutrophilia independently predicts death in tuberculosis. Eur Respir J. (2013) 42:1752–7. doi: 10.1183/09031936.00140913.
16. Martineau AR, Newton SM, Wilkinson KA, Kampmann B, Hall BM, Nawroly N, et al. Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest. (2007) 117:1988–94. doi: 10.1172/JCI31097.
17. Lovewell RR, Baer CE, Mishra BB, Smith CM, Sassetti CM. Granulocytes act as a niche for Mycobacterium tuberculosis growth. Mucosal Immunol. (2021) 14:229–41. doi: 10.1038/s41385-020-0300-z.
18. Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, et al. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat Microbiol. (2017) 2:1–11. doi: 10.1038/nmicrobiol.2017.72.
19. Hansen SG, Zak DE, Xu G, Ford JC, Marshall EE, Malouli D, et al. Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat Med. (2018) 24:130–43. doi: 10.1038/nm.4473.
20. Yang C-T, Cambier C, Davis JM, Hall CJ, Crosier PS, Ramakrishnan L. Neutrophils exert protection in the early tuberculous granuloma by oxidative killing of mycobacteria phagocytosed from infected macrophages. Cell Host Microbe. (2012) 12:301–12. doi: 10.1016/j.chom.2012.07.009.
21. Filep JG. Targeting neutrophils for promoting the resolution of inflammation. Front Immunol. (2022) 1008. doi: 10.3389/fimmu.2022.866747.
22. Talwar S, Pandey M, Sharma C, Kutum R, Lum J, Carbajo D, et al. Role of VapBC12 toxin-antitoxin locus in cholesterol-induced mycobacterial persistence. M Syst. (2020) 5:e00855–00820. doi: 10.1128/mSystems.00855-20.
23. Pandey M, Talwar S, Bose S, Pandey AK. Iron homeostasis in Mycobacterium tuberculosis is essential for persistence. Sci Rep. (2018) 8:17359. doi: 10.1038/s41598-018-35012-3.
24. Sadhu S, Rizvi ZA, Pandey RP, Dalal R, Rathore DK, Kumar B, et al. Gefitinib results in robust host-directed immunity against Salmonella infection through proteo-metabolomic reprogramming. Front Immunol. (2021) 12:648710. doi: 10.3389/fimmu.2021.648710.
25. Sadhu S, Dalal R, Dandotiya J, Binayke A, Singh V, Tripathy MR, et al. IL-9 aggravates SARS-CoV-2 infection and exacerbates associated airway inflammation. Nat Commun. (2023) 14:4060. doi: 10.1038/s41467-023-39815-5.
26. Petryszak R, Burdett T, Fiorelli B, Fonseca NA, Gonzalez-Porta M, Hastings E, et al. Expression Atlas update—a database of gene and transcript expression from microarray-and sequencing-based functional genomics experiments. Nucleic Acids Res. (2014) 42:D926–32. doi: 10.1093/nar/gkt1270
27. Storey JD. A direct approach to false discovery rates. J R Stat Society: Ser B. (2002) 64:479–98. doi: 10.1111/1467-9868.00346.
28. Malik S, Sadhu S, Elesela S, Pandey RP, Chawla AS, Sharma D, et al. Transcription factor Foxo1 is essential for IL-9 induction in T helper cells. Nat Commun. (2017) 8:815. doi: 10.1038/s41467-017-00674-6.
29. Du Plessis N, Jacobs R, Gutschmidt A, Fang Z, van Helden PD, Lutz MB, et al. Phenotypically resembling myeloid derived suppressor cells are increased in children with HIV and exposed/infected with Mycobacterium tuberculosis. Eur J Immunol. (2017) 47:107–18. doi: 10.1002/eji.201646658.
30. Tsiganov EN, Verbina EM, Radaeva TV, Sosunov VV, Kosmiadi GA, Nikitina IY, et al. Gr-1dimCD11b+ immature myeloid-derived suppressor cells but not neutrophils are markers of lethal tuberculosis infection in mice. J Immunol. (2014) 192:4718–27. doi: 10.4049/jimmunol.1301365.
31. Kato K, Yamamoto K. Suppression of BCG cell wall-induced delayed-type hypersensitivity by BCG pre-treatment. II. Induction of suppressor T cells by heat-killed BCG injection. Immunology. (1982) 45:655.
32. Klimpel GR, Okada M, Henney CS. Inhibition of in vitro Cytotoxic Responses by BCG-Induced Macrophage-Like Suppressor Cells: II. Suppression Occurs at the Level of a “Helper” T Cell. J Immunol. (1979) 123:350–7. doi: 10.4049/jimmunol.123.1.350.
33. Nouailles G, Dorhoi A, Koch M, Zerrahn J, Weiner J, Fae KC, et al. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J Clin Invest. (2014) 124:1268–82. doi: 10.1172/JCI72030.
34. Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. (2015) 528:565–9. doi: 10.1038/nature16451.
35. Montoya CJ, Catano JC, Ramirez Z, Rugeles MT, Wilson SB, Landay AL. Invariant NKT cells from HIV-1 or Mycobacterium tuberculosis-infected patients express an activated phenotype. Clin Immunol. (2008) 127:1–6. doi: 10.1016/j.clim.2007.12.006.
36. Snyder-Cappione JE, Nixon DF, Loo CP, Chapman JM, Meiklejohn DA, Melo FF, et al. Individuals with pulmonary tuberculosis have lower levels of circulating CD1d-restricted NKT cells. J Infect Dis. (2007) 195:1361–4. doi: 10.1086/513567
37. Muraille E, Leo O, Moser M. TH1/TH2 paradigm extended: macrophage polarization as an unappreciated pathogen-driven escape mechanism? Front Immunol. (2014) 5:603. doi: 10.3389/fimmu.2014.00603.
38. Huang D, Shen Y, Qiu L, Chen CY, Shen L, Estep J, et al. Immune distribution and localization of phosphoantigen-specific Vγ2Vδ2 T cells in lymphoid and nonlymphoid tissues in Mycobacterium tuberculosis infection. Infection Immun. (2008) 76:426–36. doi: 10.1128/IAI.01008-07.
39. Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med. (2013) 188:1137–46. doi: 10.1164/rccm.201304-0803OC.
40. Niazi MK, Dhulekar N, Schmidt D, Major S, Cooper R, Abeijon C, et al. Lung necrosis and neutrophils reflect common pathways of susceptibility to Mycobacterium tuberculosis in genetically diverse, immune-competent mice. Dis Models Mech. (2015) 8:1141–53. doi: 10.1242/dmm.020867.
41. Silverio D, Goncalves R, Appelberg R, Saraiva M. Advances on the role and applications of interleukin-1 in tuberculosis. Mbio. (2021) 12:e03134–03121. doi: 10.1128/mBio.03134-21.
42. Anes E, Azevedo-Pereira JM, Pires D. Cathepsins and their endogenous inhibitors in host defense during mycobacterium tuberculosis and HIV infection. Front Immunol. (2021) 12:726984. doi: 10.3389/fimmu.2021.726984.
43. Gaffney E, Murphy D, Walsh A, Connolly S, Basdeo S, Keane J, et al. Defining the role of neutrophils in the lung during infection: Implications for tuberculosis disease. Front Immunol. (2022) 13:984293. doi: 10.3389/fimmu.2022.984293.
44. Witko-Sarsat V, Rieu P, Descamps-Latscha B, Lesavre P, Halbwachs-Mecarelli L. Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest. (2000) 80:617–53. doi: 10.1038/labinvest.3780067.
45. Olofsen PA, Stip MC, Jansen JM, Chan C, Nederend M, Tieland RG, et al. Effective, long-term, neutrophil depletion using a murinized anti-ly-6G 1A8 antibody. Cells. (2022) 11:3406. doi: 10.3390/cells11213406.
46. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, Van Zoelen MA, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. (2007) 13:1042–9. doi: 10.1038/nm1638.
47. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, et al. Toll-like receptorinduced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol. (2008) 9:1399–406. doi: 10.1038/ni.1671.
48. Pessanha AP, Martins RA, Mattos-Guaraldi AL, Vianna A, Moreira LO. Arginase-1 expression in granulomas of tuberculosis patients. FEMS Immunol Med Microbiol. (2012) 66:265–8. doi: 10.1111/j.1574-695X.2012.01012.x.
49. Abebe T, Hailu A, Woldeyes M, Mekonen W, Bilcha K, Cloke T, et al. Local increase of arginase activity in lesions of patients with cutaneous leishmaniasis in Ethiopia. PloS Negl Trop Dis. (2012) 6:e1684. doi: 10.1371/journal.pntd.0001684.
50. Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, et al. via LE Kirschner DE: Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol. (2013) 191:773–84. doi: 10.4049/jimmunol.1300113.
51. Abdullah Z, Geiger S, Nino-Castro A, Bottcher JP, Muraliv E, Gaidt M, et al. Lack of PPARγ in myeloid cells confers resistance to Listeria monocytogenes infection. PloS One. (2012) 7:e37349. doi: 10.1371/journal.pone.0037349.
52. Shirey KA, Cole LE, Keegan AD, Vogel SN. Francisella tularensis live vaccine strain induces macrophage alternative activation as a survival mechanism. J Immunol. (2008) 181:4159–67. doi: 10.4049/jimmunol.181.6.4159.
53. Xavier MN, Winter MG, Spees AM, den Hartigh AB, Nguyen K, Roux CM, et al. PPARγ-mediated increase in glucose availability sustains chronic Brucella abortus infection in alternatively activated macrophages. Cell Host Microbe. (2013) 14:159–70. doi: 10.1016/j.chom.2013.07.009.
54. Almeida PE, Carneiro AB, Silva AR, Bozza PT. PPARγ expression and function in mycobacterial infection: roles in lipid metabolism, immunity, and bacterial killing. PPAR Res. (2012). doi: 10.1155/2012/383829
55. Eisele NA, Ruby T, Jacobson A, Manzanillo PS, Cox JS, Lam L, et al. Salmonella require the fatty acid regulator PPARδ for the establishment of a metabolic environment essential for long-term persistence. Cell Host Microbe. (2013) 14:171–82. doi: 10.1016/j.chom.2013.07.010.
56. Condos R, Rom WN, Liu YM, Schluger NW. Local immune responses correlate with presentation and outcome in tuberculosis. Am J Respir Crit Care Med. (1998) 157:729–35. doi: 10.1164/ajrccm.157.3.9705044.
57. Keller C, Hoffmann R, Lang R, Brandau S, Hermann C, Ehlers S. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infection Immun. (2006) 74:4295–309. doi: 10.1128/IAI.00057-06.
58. Nandi B, Behar SM. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med. (2011) 208:2251–62. doi: 10.1084/jem.20110919.
59. Lowe DM, Redford PS, Wilkinson RJ, OGarra A, Martineau AR. Neutrophils in tuberculosis: friend or foe? Trends Immunol. (2012) 33:14–25. doi: 10.1016/j.it.2011.10.003.
60. Szulc-Dabrowska L, Bossowska-Nowicka M, Struzik J, Toka FN. Cathepsins in bacteria-macrophage interaction: defenders or victims of circumstance? Front Cell infection Microbiol. (2020) 10:601072. doi: 10.3389/fcimb.2020.601072.
61. Sharma T, Grover S, Arora N, Ehtesham NZ, Hasnain SE. PGRS domain of Rv0297 of Mycobacterium tuberculosis is involved in modulation of macrophage functions to favor bacterial persistence. Front Cell Infection Microbiol. (2020) 10:451. doi: 10.3389/fcimb.2020.00451.
62. Gutierrez MG, Mishra BB, Jordao L, Elliott E, Anes E, Griffiths G. NF-κB activation controls phagolysosome fusion-mediated killing of mycobacteria by macrophages. J Immunol. (2008) 181:2651–63. doi: 10.4049/jimmunol.181.4.2651.
63. Amaral EP, Riteau N, Moayeri M, Maier N, Mayer-Barber KD, Pereira RM, et al. Lysosomal cathepsin release is required for NLRP3-inflammasome activation by Mycobacterium tuberculosis in infected macrophages. Front Immunol. (2018) 9:1427. doi: 10.3389/fimmu.2018.01427.
64. Pires D, Marques J, Pombo JP, Carmo N, Bettencourt P, Neyrolles O, et al. Role of cathepsins in Mycobacterium tuberculosis survival in human macrophages. Sci Rep. (2016) 6:32247. doi: 10.1038/srep32247.
65. Knaul JK, Jorg S, Oberbeck-Mueller D, Heinemann E, Scheuermann L, Brinkmann V, et al. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am J Respir Crit Care Med. (2014) 190:1053–66. doi: 10.1164/rccm.201405-0828OC.
66. Agrawal N, Streata I, Pei G, Weiner J, Kotze L, Bandermann S, et al. Human monocytic suppressive cells promote replication of Mycobacterium tuberculosis and alter stability of in vitro generated granulomas. Front Immunol. (2018) 9:2417. doi: 10.3389/fimmu.2018.02417.
67. Magcwebeba T, Dorhoi A, du Plessis N. The emerging role of myeloid-derived suppressor cells in tuberculosis. Front Immunol. (2019) 10:917. doi: 10.3389/fimmu.2019.00917.
68. Parveen S, Lun S, Urbanowski ME, Cardin M, Shen J, Murphy JR, et al. Effective host-directed therapy for tuberculosis by depletion of myeloid-derived suppressor cells and related cells using a diphtheria toxin fusion protein. J Infect Dis. (2021) 224:1962–72. doi: 10.1093/infdis/jiab235.
69. Scott NR, Swanson RV, Al-Hammadi N, Domingo-Gonzalez R, Rangel-Moreno J, Kriel BA, et al. S100A8/A9 regulates CD11b expression and neutrophil recruitment during chronic tuberculosis. J Clin Invest. (2020) 130:3098–112. doi: 10.1172/JCI130546.
70. Guo Q, Zhao Y, Li J, Liu J, Yang X, Guo X, et al. Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID-19. Cell Host Microbe. (2021) 29:222–35:e224. doi: 10.1016/j.chom.2020.12.016.
71. Narumi K, Miyakawa R, Ueda R, Hashimoto H, Yamamoto Y, Yoshida T, et al. Proinflammatory proteins S100A8/S100A9 activate NK cells via interaction with RAGE. J Immunol. (2015) 194:5539–48. doi: 10.4049/jimmunol.1402301.
Keywords: tuberculosis, toxin-antitoxin, virulence, neutrophils, persistence, and inflammation
Citation: Tyagi S, Sadhu S, Sharma T, Paul A, Pandey M, Nain VK, Rathore DK, Chatterjee S, Awasthi A and Pandey AK (2024) VapC12 ribonuclease toxin modulates host immune response during Mycobacterium tuberculosis infection. Front. Immunol. 15:1302163. doi: 10.3389/fimmu.2024.1302163
Received: 26 September 2023; Accepted: 01 February 2024;
Published: 07 March 2024.
Edited by:
Rajiv Kumar, Banaras Hindu University, IndiaReviewed by:
Sadiya Parveen, Johns Hopkins University, United StatesMohd Saqib, Albany Medical College, United States
Copyright © 2024 Tyagi, Sadhu, Sharma, Paul, Pandey, Nain, Rathore, Chatterjee, Awasthi and Pandey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amit Awasthi, YWF3YXN0aGlAdGhzdGkucmVzLmlu; Amit Kumar Pandey, YW1pdHBhbmRleUB0aHN0aS5yZXMuaW4=