
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 01 February 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1287459
This article is part of the Research Topic Novel Immunological Characteristics and Immunotherapeutic Targets in Pancreatic Cancer View all 5 articles
 Conner Hartupee1
Conner Hartupee1 Bolni Marius Nagalo2,3
Bolni Marius Nagalo2,3 Chiswili Y. Chabu4,5,6
Chiswili Y. Chabu4,5,6 Mulu Z. Tesfay2
Mulu Z. Tesfay2 Joycelynn Coleman-Barnett1,7John T. West7
Joycelynn Coleman-Barnett1,7John T. West7 Omeed Moaven1,7,8*
Omeed Moaven1,7,8*Pancreatic Ductal Adenocarcinoma (PDAC) is projected to become the 2nd leading cause of cancer-related deaths in the United States. Limitations in early detection and treatment barriers contribute to the lack of substantial success in the treatment of this challenging-to-treat malignancy. Desmoplasia is the hallmark of PDAC microenvironment that creates a physical and immunologic barrier. Stromal support cells and immunomodulatory cells face aberrant signaling by pancreatic cancer cells that shifts the complex balance of proper repair mechanisms into a state of dysregulation. The product of this dysregulation is the desmoplastic environment that encases the malignant cells leading to a dense, hypoxic environment that promotes further tumorigenesis, provides innate systemic resistance, and suppresses anti-tumor immune invasion. This desmoplastic environment combined with the immunoregulatory events that allow it to persist serve as the primary focus of this review. The physical barrier and immune counterbalance in the tumor microenvironment (TME) make PDAC an immunologically cold tumor. To convert PDAC into an immunologically hot tumor, tumor microenvironment could be considered alongside the tumor cells. We discuss the complex network of microenvironment molecular and cellular composition and explore how they can be targeted to overcome immuno-therapeutic challenges.
Pancreatic Ductal Adenocarcinoma (PDAC) is currently the 4th leading cause of cancer-related deaths in the United States (1). It is projected that by 2030 it will become the 2nd leading cause of cancer-related deaths (2). Although advancements are made in the treatment of resectable disease, this population makes up only 15-20% of new pancreatic cancer cases as most patients do not present until late into the disease course, at which point it is not resectable. Due to the nature of this disease, systemic treatment is an essential component of the multimodality treatment of pancreatic cancer (3). Unfortunately, primary and secondary resistance are very common in PDAC and there is an urgent need for developing therapeutic modalities with adequate oncologic benefit and durability. Immunotherapy has recently revolutionized the treatment of various cancers but repeat trials have shown PDAC is relatively unresponsive to such therapies.
There is a growing interest in focusing on the tumor microenvironment (TME) and its role in resistance to systemic therapies and as a target for more effective therapeutic modalities. Pancreatic adenocarcinoma, along with other solid tumors, is what is best described as an “immunologically cold” tumor. PDAC lacks excessive mutations that would allow the body’s immune system to recognize the tumor cells due to tumor specific antigens (4). Due to the limited immunogenicity of PDAC, this leads to insufficient cancer antigen presentation, resulting in a notably weak or even absent immune response and inadequate T-cell trafficking (5). Additionally, PDAC reprograms surrounding tissue and immune cells to create a dense, fibroblastic environment with limited anti-tumor immunity. Previous studies have evaluated the function and composition of the tumor microenvironment (TME) to better understand contributing factors to the therapeutic challenge of PDAC and to answer the question of why PDAC has poor immunogenicity. The PDAC TME includes a mixture of cancerous pancreatic cells, support cells, immune cells, and dense extracellular matrix. Even within this complex microenvironment, we see variability between one tumor’s molecular and genetic profile vs. another that results in differing clinical outcomes (6, 7). This heterogeneous system works in conjunction to provide an environment that remains relatively impermeable to treatment, adaptive to therapies, and supportive of cancerous spread.
Pancreatic cancer cells (PCC) arise from either ductal pancreas cells or acinar pancreas cells. Acinar cells perform the role of digestive enzyme production and ductal cells secrete bicarbonate ions and water needed to alkalize and activate the enzymatic secretions of the pancreas. Some debate remains as to the exact cell-of-origin but recent literature indicates the origin may vary based on the number and type of initial mutations seen with the tumor environment (8). The significance of the contextual difference in the cell-of-origin may eventually point us towards a better understanding of the biological function of these tumors and patient outcomes as seen with similar solid tumor cancers (9).
Formation of pancreatic cancer cells is believed to involve processes related to chronic inflammation, hypoxia, and the accumulation of repetitive mutations. These factors collectively contribute to the development of pancreatic intraepithelial neoplasia (PanIN). In the process of inflammation, an initial, acute inflammatory reaction drives the migration of neutrophils, natural killer cells, dendritic cells, and macrophages to the site of insult (10). The innate immune system in attempting to resolve the initial acute insult promotes a cascade of signaling that recruits the adaptive immune response along with stimulation of endogenous repair mechanisms to reestablish the structure and function of the surrounding tissue. When the acute inflammatory response begins to maladaptively produce a chronic inflammatory response, the combination of reactive oxygen species, continued tissue injury, and immunosuppressive functions of the chronic inflammation leads to an environment prone to oncogenic mutations (10).
Over time these pathologic insults will alter the genetic signature of the surround tissue and this neoplastic tissue accumulates a significant number of mutations that transforms lesions from a precancerous stage into cells more consistent with PDAC. Kirsten Rat Sarcoma (KRAS) presents as a consistent oncogenic mutation with inactivation of the GTPase activity of the KRAS protein complex leading to consistent activation and downstream signaling of key proliferative pathways like mTOR, NF-kB, and ERK (11). Further evidence supports that KRAS mutations are a key instigator in upregulation of glycolytic metabolic reprogramming and Sonic Hedge Hog (SHH) mediated activation of pancreatic stellate cells (12, 13).
Oncogenic KRAS mutation alone does not guarantee neoplastic formation, as cell populations positive for KRAS were observed to be non-proliferative within normal pancreatic tissue. These cells were instead signaled to cease proliferation through a mechanism known as oncogene-induced senescence. The physiologic role of this senescence is innately to protect against oncogenesis within these cells. This senescent state may be bypassed by proinflammatory mechanisms, stemming from pathologic events such as pancreatitis, that reactivate the proliferation of pre-neoplastic cells, enabling further mutagenesis (14). Senescent cell populations cease proliferation but remain as active components of the tissue microenvironment. Many of these senescent cells fall under what is termed senescence-associated secretory phenotype (SASP) and release a variety of pro-inflammatory molecules including growth factors, interleukins, cytokines, and proteases(15, 16). The intent of these signaling molecules is designed to awaken the immune system to eliminate said senescent cells; however, senescent cells release facts that act autonomously to accelerate cell proliferation and epithelial to mesenchymal transition (EMT). Additional mechanisms of cellular damage, including chemotherapeutics and radiotherapy, have been shown to induce a tumor-supportive senescence-like secretory phenotype in the TMEs, demonstrating therapy-resistant malignancies as these populations do not undergo apoptosis (17). A relatively new role within the neoplastic environment, senescent cells prior to oncogenesis and throughout a malignancy has been identified as a significant component of the TME (18).
Additional mutations, such as deactivating mutations in p53 lead to uninhibited collection of DNA damage and SMAD4 inactivation, leading to unregulated TGF-β signaling. Upregulation of TGF-β, a molecule present throughout the PDAC TME initially contributes to tumor suppression through apoptosis and cell-cycle arrest (19). Although the inactivation of SMAD4 and progression of PanIN leads to TGF-β functioning as a tumorigenic marker that promotes progression and EMT. These mutations function as the beginning of a neoplastic group of genes necessary but not always present within PDAC genetics. Additional precursor mutations of Her-2, p16, and BRCA2 have been detected within the PanINs as grading successively increases (20). Proto-oncogenic and oncogenic mutations combined with a reduction in response by the immune system to properly suppress precancerous cells leads to an environment of where normal regulatory mechanisms are compromised (21). Such precancerous cells inevitably alter the surrounding environment to isolate and support their growth concomitant with immune suppression to minimize interference. Part of this transition is directed by the signaling pathways evidenced in PCC mutations and signaling.
Noted to release a variety of chemokines and cytokines, PCCs play an essential role in the recruitment of support cells and manipulation of immune cells. PCCs directly signal pancreatic stellate cells (PSCs) for activation by means of key molecules such as transforming growth factor-beta (TGFβ1), platelet-derived growth factor (PDGF), and fibroblast growth factor-2 (FGF-2) leading to increased fibrogenesis by the PSCs (22). PCCs are shown to produce excess chemokines that are heavily involved in the activation of Cancer-associated fibroblasts, including CXCL1, -5, and -8 binding to fibroblasts’ CXCR2 receptors, leading to increased fibroblast activation and reprogramming (23). IL-1β, a traditionally pro-inflammatory cytokine, is shown to be released by PCCs in vivo early within tumorigenesis and is linked to recruitment and activation of both PSCs and immune suppressive cell types (24). The chemokines CSF-1 (also known as M-CSF), CSF-2 (also known as GM-CSF), and CSF-3 (also known as G-CSF) are also shown to be expressed by PCCs, leading to decreased concentrations of anti-tumor dendritic cells and increased concentrations of immune suppressive cells within the TME (25, 26). In large part, oncogenic mutations play a key role in upregulating the expression of proteins that allow PCCs to tailor the environment to reduce anti-tumor cell populations. Pre-cancerous cells that fail to express these proteins are potentially reduced by proper immune responses prior to becoming PanINs, hence, we see consistently high levels of KRAS and p53 mutations in PCC genetics.
Further, PCCs suppress immune components in PDAC TME via the release of signaling exosomes. PCCs release exosomes containing miRNA155 and miRNA1290, into cancer-associated fibroblasts(CAFs), leading to elevated fibrogenesis and resulting into highly fibrotic TME (27, 28). The fibrotic milieu generate a tissue mechanical landscape that promotes adaptation of PDAC cancer cells, contributing to robust tumorigenesis.
Low O2 saturation in the PDAC TME results in overexpression of hypoxia-inducible factors (HIFs). These proteins act as homeostatic transcription factors in a cascading response to counteract and adapt to the negative environmental stimuli the cells are in, producing VEGF, altering metabolic pathways to prioritize glycolysis, and upregulation of chemoresistant proteins like MDR1 and HGF (29, 30). Additionally, HIF1α/2α induces miRNA301a which leads to M2 macrophage polarization resulting in pro-tumorigenic downstream processes (31). NADPH Oxidase (NOX), a transmembrane protein with roles in multiple signaling pathways, is shown to be overexpressed in cancer cells and associated with poor clinical outcomes due to its role in ROS production and utilization in low oxygen environments (32). The cancer cells readily adapt to the hypoxic environment they create, and the additional metabolic changes further promote EMT and metastasis.
EMT, a key feature of malignant progression, is the loss of epithelial features and dedifferentiation to a mesenchymal phenotype. The mesenchymal phenotype is consistent with embryonic stem cells and supports migration and rapid proliferation. Down-regulation of the cell-to-cell adhesion marker, e-cadherin, along with upregulation of n-cadherin and vimentin denote EMT progression (33). Key transcription factors traditionally upregulated in PDAC mesenchymal cells includes SNAIL, SLUG, TWIST1, Zeb1, and 2 (34). Many of such transcription factors being linked to increased mobility and commonly observed in circulating tumor cells. Throughout the formation of the TME and malignant progression of the disease a series of complex interactions occur between the surrounding TME and PCCs much of which is discussed within this review. By understanding these cellular changes, we gain insights into how pancreatic cancer progresses. This knowledge and the current progress of research will continue to guide the development of strategies to halt or slow down the disease.
Pancreatic stellate cells function as a key component within all aspects of the pancreatic tumor microenvironment. Discovered in 1998 within rat pancreas, these cells work to maintain the proper tissue environment within the pancreas through insults and injury(35). Quiescent PSCs are characterized by high vitamin A, desmin, vimentin, and GFAP levels with little migration or proliferation (36). These cells maintain the extracellular matrix through protein production and basement membrane stabilization, and in times of physiologic stressors, may play a role in autoimmune regulation (37–39) Known triggers of PSCs include ethanol, high glucose, inflammation, reactive oxygen species, and the inflammatory markers produced in local pathologies (40). Transforming Growth Factor-Beta (TGF-β1), CTGF, PDGF, IL-1, TNF-A, AP-1, activin-a, and HIF-1 have been established as direct markers of activation, while PPAR-g, IL-10, SMAD7, and retinoic acid are markers of inhibition (40). PDAC is characterized by increased disproportionate expression of many activating markers through chronic inflammation and hypoxia resulting in the chronic and malignant activation of PSCs.
These activated PSCs function as the architects of extracellular matrix deposition and pancreatic tissue remodeling that is closer in nature to what is seen in pancreatic cancer. Activated PSCs are the primary contributor to the dense, fibrotic environment that supports the growth of pancreatic cancer cells. Transforming into a myofibroblast-like characteristic, PSCs produce and release a variety of ECM remodeling proteins including collagens, periostin, fibronectin, Matrix Metalloproteinases (MMPs) and a decreased release of Tissue Inhibitors of Metalloproteinases (TIMPS) (41). In healthy pancreas stroma, moderate production of these ECM proteins is balanced with appropriate breakdown and recycling of ECM tissue to allow moderation and turnover. The ratio of proteins deposited by PSCs becomes a determining factor in PDAC phenotypes translating to increased tumor progression (42). The disproportionate deposition of these fibrotic proteins in the PDAC TME promotes hydraulic compression 2-3x the normal pressure of the surrounding environment with a poorly perfused vascular supply (43, 44). The increased pressure and decreased vascular flow also lends itself to an environment less susceptible to local delivery of chemotherapeutic agents and antibody penetration (45, 46). The isolated environment developed by dysregulated protein deposition results in an unnatural encasement that isolates and protects the TME known as the desmoplastic reaction.
The role of PSCs in immune response modulation within the context of PDAC is notable alongside the fibrotic environment they create. PSCs exhibit a complicated influence on the immune landscape of the tumor microenvironment. Releasing Interleukin-6 (IL-6), which orchestrates the activation and migration of Myeloid-Derived Suppressor Cells (MDSCs) into the TME with additional consequent immunosuppression discussed further in the text (47). Additionally, activated PSCs contribute to immune regulation by releasing CXCL12, a factor responsible for sequestering CD8 T-cells, thereby hampering their inherent antitumor activity. Another aspect of PSC-mediated immune modulation involves the production of Galectin-1, a protein associated with diverse effects, including the induction of CD3+ T-cell apoptosis, release of TH2 cytokines, and evasion of immune cell responses (48, 49). Further research has underscored PSCs’ involvement in extracellular vesicle signaling, revealing the upregulation of CXCL10 in PSCs during co-culture with pancreatic cancer cells (PCCs), which suggests a crosstalk mechanism contributing to the recruitment of FoxP3+ Tregs (50). PSCs have a potentially significant influence over the initiation and maintenance of immune responses in PDAC.
The immunosuppressed shelter of the desmoplastic environment works in tandem with the role of PSCs in signaling support of PCCs. A combination of signaling molecules have been linked to increased growth of PDAC tumors along with further metastasis and chemoresistance that allows the PDAC TME to continue unchecked. A range of growth factors released by PSCs include TGF-β, Platelet-Derived Growth Factor (PDGF), and Vascular Endothelial Growth Factor (VEGF) (51). Along with growth factors, signaling pathways including WNT/B-Catenin are upregulated in PDAC cancer cells by PSC-bound glutamine synthetase when co-cultured (H. 52). Similarly, PDAC-triggered autophagy by GPT1 in PSCs provides an alanine fuel source to support metabolic pathways in PCCs, allowing growth and support for further spread (53).
Additionally, PSCs exert their impact on PCCs’ therapeutic responses, where the production of Hes1 by PSCs emerges as a determinant of chemoresistance via overactivation of the NOTCH signaling pathway, as elucidated by recent findings (54). Furthermore, the extracellular matrix (ECM) protein Periostin emerges as a key regulator, intricately linked to PDAC progression. Periostin, an ECM protein functioning in tissue remodeling, is found to be linked to increased proliferation and metastasis in PDAC, and when suppressed in spheroid models exhibited increased PDAC toxicity by Natural Killer Cells (55). Metastasis and further tumor progression are also benefited from PSCs due to other factors below.
PSCs play a key role in further carcinogenesis and evolution of PDAC. PSC-derived fibrosis and signaling pathways overexpressed by PSCs influence the EMT process as seen by previous research. Secretion of IL-6 and utilization of the STAT3/NRF2 pathway influences the PDAC EMT (56). Similar effects are seen with CD10+ PSCs explicit MMP3 release as a method of surrounding tissue breakdown to provide local tissue invasion (57). Kindlin-2 expression is also linked to PDAC progression through downregulating HOXB9 and E-cadherin, leading to increased metastasis (58). MiRNA-21 exosomal release by PSCs linked to enhancing Ras/ERK pathways and PDAC cancer cell migration (59).
Focused therapies targeting the role of PSCs in the PDAC TME are gaining traction as the library of evidence indicating their pro-tumorigenic role grows. Targeted inhibition of the pathways and molecules discussed previously have been referenced in the current literature, with some results more promising than others. Some limitations include the expandability from in vitro/in vivo models to clinical trials, which has shown limited success. One such article notes the promise of early intervention in the PanIN pathway with targeted inhibition may function as a curative approach; however, given the clinical presentation of late-stage disease makes such an approach currently infeasible.
Studies have shed light in therapeutic potential in repolarization of activated PSCs into a quiescent state. Vitamin A analogs have resulted in inactivation of PSCs in chronic pancreatitis that may protect the pancreatic tissue microenvironment (60). Calcipotriol, a vitamin D analog, has also been shown to reduce TGF-β and SMAD4 signaling with increased Meflin expression, assisted in stromal reprogramming with increased gemcitabine efficacy (61, 62). Similarly, BET inhibitors (JQ1/I-BET151) has been shown to induce PSC quiescence and reduce production of collagen 1 and PDAC fibrosis (63). The key to treatment may be in attenuation of overall PSC function rather than complete removal or targeted inhibition. Exploring which aspects of the ECM that PSCs produce and limiting the effects of over-expressed pathways may present a solution, but accessing and selectively targeting these pathways in clinical models remains a challenge.
In conjunction with PSCs, Cancer-Associated Fibroblasts (CAFs) are recognized as a primary contributor to the PDAC TME through generation of many of the extracellular matrix proteins seen. PSC differentiation into CAFs represents a portion of the suspected cells, approximately 15%, of origin involved in PDAC, with other proposed cells including endothelial cells, mesothelial cells, macrophages, stem cells, and a growing list of other cells (64–67). The significance of this literature suggests that malignant reprogramming through PCC signaling induces fibroblast-like phenotypes out of various cell lines depending on the location and context of PDAC.
Common markers for identifying CAFs include Collagen 1 Type A1 (COL1A1), Fibroblast activation Protein (FAP), Podoplanin (PDPN), and Decorin (DCN) (68, 69). Current literature categorizes CAFs into three main subsets, each with distinct markers and functions. A subset of CAFs that potentially presents a direct link to PSCs is Myofibroblastic CAFs (myCAFs), stimulated by TGF-β signaling, which cluster around PDAC cancer cells and are characterized by their expression of alpha-smooth muscle actin (a-SMA) and TGF-β signaling. These myCAFs are believed to contribute significantly to the dense extracellular matrix (ECM) observed in the TME. Inflammatory CAFs (iCAFs), on the other hand, are influenced by IL-1 signaling and are located at the periphery of the TME. These iCAFs exhibit signaling involving IL-6, IL-8, and CXCL1/2/9/12 and are thought to play a role in immunosuppression and tumorigenesis (70, 71). The component balance between the ratio of myCAFs and iCAFs has also been shown to indicate tumorigenesis. Increased TGF-β and SHH signaling increases the myCAF to iCAF ratio predicts slower tumor growth, while a higher iCAF to myCAF ratio leads to the opposite (72, 73).
Adding to this complexity, a subset known as complement-secreting CAFs (csCAFs), unique to the PDAC TME, exists in close proximity to PCCs. Expressing complement proteins C3, C7, CFB, CFD, CFH, and CFI, these csCAFs are thought to regulate complement activity and contribute to inflammation, but notably, these cells are in their highest concentration in early-stage PDAC with declining levels throughout its progression (74). A smaller subset of CAFs, known as antigen-presenting CAFs (apCAFs), express MHC II genes and induce CD4 T-cell activation, characterized by CD25 and CD69 expression, setting them apart from traditional antigen-presenting cells they lack CD80/CD86 (75). These apCAFs are believed to play a tumorigenic role in the production of regulatory T-cells (68). One other niche subset of CAFs includes metabolic CAFs (meCAFs). Phenotypically distinct from other CAFs, meCAFs present as a poor prognostic factor with a paradoxical increased responsiveness to immunotherapy (76). PLA2G2A is associated with higher CD8+ T-cells silencing but looser “hot” TME, PLA2G2A may function as a potential immunotherapeutic target in this regard (77).
CAF functions extend to impacting tumor metastasis within the PDAC TME through cell-cell interactions. Heparin Sulfate Proteoglycan 2 (HSPG2) exhibits increased expression in the context of paracrine signaling between p53+ PCCs and CAFs (78). HSPG2, or perlecan, is a complex proteoglycan present in the ECM. It displays the ability to separate cell layers and, when broken down, can additionally engage in growth factor signaling(79, 80). CAFs’ role in remodeling the ECM and facilitating tumor cell migration is evidenced by the upregulation of focal adhesion kinase (FAK) in these cells. FAK’s elevated presence aids in the active reconstructing of the ECM and promotes the tumor cell migration of differentiated cancer phenotypes (81).
The role of CAFs extends to cancer stem cell (CSC) support and chemoresistance, with specific subsets contributing to these processes. Notably, CD10+GPR77+ and CD44+ CAFs, when co-cultured with cancer stem cells and PDAC cancer cells, exhibit increased tumorigenesis, chemoresistance, and disease recurrence (82, 83). Demonstrating the cooperative role of CAFs with PDAC cancer cells in tumor progression and chemotherapeutic resistance, the TME must be considered from the perspective of the malignant redefining the role of benign pathways.
Appropriate therapeutic approaches demand evaluation of the cause-effect role that upstream pathways like myCAF knockouts and SHH pathways have on PDAC outcomes. SHH inhibition, leading to increased ratio of iCAFs to myCAFs, led to shorter overall survival in patient treatment (84). This may indicate the undue reliance on SHH signaling by fibrotic CAF phenotypes with a resulting increase in iCAFs that promotes tumorigenesis. Inhibition of iCAF activation through targeted measures via CXCL12 and CXCR4 inhibition increases the immune response in PDAC (85, 86). Unspecified targeting of these activated support cells will lead to unforeseen consequences for the patient if additional consideration for the effect these individual subsets have on the impact of PDAC outcomes. The variability in function and origin within this cell type implicates the role of signaling molecules and crosstalk with PCCs as major players in activation. Next steps should continue investigating the balancing mechanism of these effector proteins to determine the optimal strategy for successful clinical applications.
The pancreatic tumor is primarily composed of a desmoplastic, scar-like capsule that is a result of extracellular matrix (ECM) production and turnover by both PCCs and resident stromal cells. Included in this environment is a variety of proteins secreted by supporting cells with malignant reprogramming. Although once an appropriate response to inflammatory signaling and tissue injury, TME remodeling becomes an imbalance of indefinite fibrosis, alterations in ECM protein, and immune suppression. Such remodeling involves changes in protein deposition, post-translational modifications of the protein deposited, and breakdown of tissues surrounding the TME (87). Components of the desmoplastic environment that play a role in the malignancy of PDAC include the various proteins, the hypoxic environment, and the high pressure that innately resists outside intervention.
The capability of cancer cells and the tumor microenvironment to expand into the surrounding tissue and begin its malignant expansion has largely pointed back to the function of angiogenesis (88). Previous literature established the role of angiogenesis in solid tumor models as bridging the gap between hyperplastic tissue and neoplastic tissue as an indicator of reprogramming non-malignant cell populations for cancer survival. Different models of angiogenesis vary between recruitment of endothelial cells for neovascularization, cellular reprogramming to utilize undifferentiated cancer cells to mimic vessel function, and malignant spread that preserves the non-malignant tissue known as vascular co-option (89). PDAC differentiates itself from other solid tumor malignancies due to its characteristic nature of overall decreased vascularity. Desmoplasia with the abundance of fibroblast/CAFs and the dense ECM produced by PSCs lead to compression of microvasculature in the microenvironment and overall hypovascularity of the TME. Pancreatic cancer cells and pancreatic stellate cells produce hallmark transcription factors known to be involved in angiogenesis: namely NFκB, Sp, STAT3, and HIF-1α (90). This increased angiogenic signaling stops short of complete vascular perfusion as prior studies investigating blood flow through pancreatic tumors have shown significantly less perfusion through the TME vs surrounding pancreatic tissue with overall increased metabolism (91). Recent literature has suggested microvilli branching off of microvessels may be a unique supply within the peripheral of the PDAC TME for metabolic fuel unlike less dense tumor microenvironments (92).
Systematic review of clinical trials investigating anti-VEGF treatments found no clinically meaningful benefit in targeting angiogenic factors that have seen previous success in other solid tumor malignancies (93). Alternatively, preclinical models investigating “normalization” or reestablishing vascular supply to the hypoxic PDAC TME have shown increased efficacy in traditional chemotherapeutic agents as a means of increasing delivery and decreasing the hypoxic environment, a known mechanism of chemoresistance and tumorigenesis (94). Given the physiologic nature of the PDAC’s vascular supply, further investigation that looks at disrupting the fibrotic stroma with concurrent immunologic therapies may prove to have greatest therapeutic benefit moving forward.
The ECM is not strictly tumor promoting as previous studies that indiscriminately target myofibroblast cells led to increased metastasis (95, 96). Therefore, a combination of proteoglycans, collagen fiber, and other support structures that make up the ECM play a dual role of inhibiting and promoting depending on the individual molecule. Further investigation into the ECM with proteomic data reveals that the PDAC TME comprises upwards of 300 proteins with a mixture of collagens, glycoproteins, proteoglycans, ECM regulators, ECM-affiliated, and secreted factors (97). Within this composition lies the variability between normal pancreatic tissue and PDAC in the relative expression of ECM proteins. From early-stage PanIN to later stages of PDAC, there is a relative shift in the exact proteins expressed within the ECM (97). Distinct proteins and their prognostic associations within the ECM are continually studied to understand the role of ECM signaling and physical mechanisms in the PDAC TME.
Decorin expression, a small leucine-rich proteoglycan (SLRP), is generally linked with good overall outcomes with its detection in the extracellular matrix. Expressed by PSCs and to some extent PCCs, Decorin is shown to be downregulated in relative concentration compared to other ECM proteins secondary to TGF-β1 signaling by PCCs (98). Decorin possesses the role of a tyrosine kinase inhibitor for various extracellular signals that modulate the overgrowth of surrounding tissue (99). Examples of such modulation include anti-angiogenic, anti-metastatic, pro-inflammatory, and anti-proliferative properties (100–103). As such, decorin has become a predictive marker of clinical outcomes as lower intra-tumoral concentrations are correlated with poorer patient survival (104). Although previous literature indicated that decorin expression may be elevated in PDAC, it was further shown the isoform with anti-tumor properties was not present in large quantities within the desmoplastic environment.
Lumican, an SLRP present in the ECM, has been associated with prolonged survival in post-surgical patients, primarily attributed to its poorly understood anti-tumorigenic effects (105). Similar to Decorin, Lumican, although produced in excess due to the increased ECM production, is decreased by TGFβ1 signaling by PCCs (98). Evidence suggests that it inhibits the signaling of EGFR through extracellular binding with resultant downregulation of EGFR/ERK pathway to reduce PDAC cell proliferation and increase apoptosis (106). Notably, Li et al. observed that when Lumican was completely knocked down in in vivo models, it led to enhanced proliferation (106). Interestingly, overexpression or exogenous administration of excess Lumican did not significantly affect proliferation (107). Furthermore, Lumican has been found to exhibit suppressive and promotional roles in various other tumor types, making it a subject of ongoing investigation to pinpoint its precise function in PDAC (99).
In typical tissue environments, glycosaminoglycans (GAGs) constitute a large portion of the ECM. GAGs routinely bind large volumes of water molecules to structure the ECM and allow diffusion of materials. Hyaluronan, a common GAG molecule that composes a sizable portion of a stable ECM, is overexpressed in the TME to the point that the GAG to water ratio becomes oversaturated and the fluid-like environment becomes a higher pressure, gel-like environment (43). This increased hydrostatic pressure promotes a surrounding area of minimal poor vascular growth and present vascular collapse leading to additional hypoxia. HAS1 and HAS2, the genes that produce Hyaluronan, are highly expressed in iCAF cells as determined through single-cell RNA sequencing (68). Interestingly, a primary driver of the NF-κB pathway, IRAK4, is shown to directly promote the expression of HAS2 in both PCCs and CAFs exposed to PDAC conditioned media (108). On top of increased hyaluronan production, HAS2 upregulation dramatically reduced CD8 T-cell presence within the PDAC TME, this helps illustrate the role immunosuppressive role of hyaluronan. Previous interventions analyzing direct targeting of Hyaluronan in the TME led to significant improvements in overall survival in mouse trials as it increased perfusion and decreased interstitial fluid pressure allowing penetration of gemcitabine (5). Although short-term destruction of hyaluronan leads to temporary therapeutic improvements in the fibrotic TME; without continual treatment, production from PCCs and CAFs result in resurgence of hyaluronan and interstitial fluid pressure to before-treatment values (109). Administrative strategies and therapeutic timing may need to be reconsidered as Phase III clinical trials as late as 2020 examining the use of PEGPH20, a long-acting hyaluronidase enzyme, in conjunction with Nab-Paclitaxel plus Gemcitabine found no significant increase in overall survival (110). Possible limitations within this trial and our understanding of targeting such proteins may be the metastatic course of the disease, as the greatest impact from such therapies may be seen earlier in the disease process.
Although previously addressed as playing a more significant role within the PDAC TME, Matrix Metalloproteases (MMPs) have seen less investigation and research in recent years. The function of the MMP family is overarchingly to act as a protease within the extracellular environment to degrade varying ECM materials with further subclassification based on the targeted proteins. The major subsets based on targeted substrate include collagenases, gelatinases, stromelysins, matrilysins, membrane-type MMPs, and other MMPs (111). Although the purpose is to break down protein, MMPs additionally function in tissue remodeling, cellular differentiation, and proliferation (112). Among the MMPs, 2, 7, and 14 have routine support within the literature as pro-tumorigenic effects on the PDAC TME with some additional debate of MMP-9. MMP-2 and MMP-9 are within the gelatinase category, with MMP-2 known to be activated by TNF-α and TNF-β with expression from endothelial cells. MMP-9, although elevated in PDAC progression and believed to activate TGF-β signaling, has historically been the target of anti-invasive therapies (113) Clinical trials that targeted MMP-9 noticed counterintuitive responses with poorer overall survival. A phase III clinical trial in 2003 comparing a broad MMP inhibitor, BAY 12-9566, saw a lower median overall survival compared to gemcitabine in 277 patients (114). Additional pre-clinical research investigating systemic knockout models of MMP-9 witnessed secondary effects of poorer immune response leading to increased overall tumor invasiveness. Consequently, selective inhibition within the PDAC TME may remain beneficial, but systemic administration of a non-targeted inhibitor has proven to be deleterious. Evolving therapeutic modalities discussed later may provide the opportunity for such selective delivery that allows targeting proteins with vital normal tissue functions similar to MMP-9.
MMP-14, also known as MT1-MMP, is a Membrane-type Metalloproteinase expressed on fibroblasts and cancer cells. A major effect established by MMPs within the TME is their use in tumor invasion through the breakdown of surrounding tissue, particularly the role of MMP-14 and its membrane expression directly linked to invadopodia function and metastasis (115). Additionally MMP-14 is shown to activate TGFβ-1 by means of direct activation and enzymatically releasing TGF-binding protein-1 in the ECM (116, 117). The direct impact of increased TGF-β being increased collage production and PCC metastatic potential within the PDAC TME (118, 119). Expression of intracellular Metalloproteinase CT Binding Protein-1 protein and extracellular Tissue Inhibitor of Metalloproteinase-2 have demonstrated a reduction in invadopodia formation (115, 120). These proteins offer potential therapeutic targets in delivery mechanisms that allow local expression and inhibition rather than the general administration of exogenous proteins that lack specificity. Previous clinical therapies investigating inhibition of MMPs resulted in no significant benefit; similarly, due to difficulties in targeting particular MMPs and the potential need for early PDAC stage application as a potential time of efficacy for such therapies.
Additional ECM proteins with an established role within the PDAC TME but still require further investigation include the following discussed. Asporin, another SLRP present within the ECM, is released by both PSCs and pancreatic cancer cells. It is known to activate signaling through the CD44/NFκB pathway, promoting EMT and metastasis in pancreatic cancer (121). An additional SLRP of interest, Keratocan, is overexpressed in the PDAC TME and has been linked to a poorer prognosis and concurrent downregulation of p53 (107). The presence of Meflin, produced by cancer-associated fibroblasts, has been associated with additional regulation of PDAC tumorigenesis, yet further exploration is needed to fully understand its impact and potential use in therapeutic intervention. (62). These ECM proteins, although possessing established roles, require deeper investigation to understand their implications with the landscape of PDAC progression.
Further consideration of the role inflammation plays in PDAC oncogenesis points to the existence of MDSCs as a major player within the TME. Lacking in in vivo data for the initial transition of pancreatic tissue to PDAC, it is understood that an early, increased presence of MDSCs is indicative of oncogenesis (122). In the event of an injury to the pancreas, signaling molecules such as CSF-1 and CSF-2 will induce differentiation of hematopoietic stem cells into immature myeloid cells (IMC) and migrate to the site of injury. This recruitment’s initial intention is to provide precursor innate immune cells for additional support in the inflammatory process. The chronic inflammatory environment that functions as a precursor to the PDAC TME, serves to upregulate the generation and migration of MDSCs to the tumor environment as part of the inherent purpose of MDSCs to suppress inflammation. Such environmental signaling includes IL-6/IL-4, CSF-2, COX2, S100A8/9, IL-1β, and IFN-γ. A combination of both IL-6 and CSF-2 not only recruit MDSCs but induce miRNA generation to prevent them from further differentiation (123). Early evidence of primary tumor removal resulted in decreased circulating levels of IMCs as our early understanding investigated the role of recruitment of these anti-inflammatory immune cells (124). Although promising, transcriptomic analyses of lung cancer populations have exposed the limitations in evaluating blood samples as correlates of TME cell populations as many of the TME-specific populations are potentially differentiated once reaching the site of malignancy (125). Although they display numerous characteristics similar to other innate immune cells, MDSCs in humans are identified by surface receptors including CD33, CD11, and Ly6c (126). Two major MDSCs have been noted in the pancreatic TME, including the granulocytic MDSCs (PMN-MDSC) or Monocytic MDSCs (M-MDSC). Besides surface markers, M-MDSCs will feature CD14/15 where otherwise absent on PMN-MDSCs, and path of origin being their differences, the primary role each plays in the TME is based on their specific secretory molecules. M-MDSCs will primarily exhibit increased NO production while PMN-MDSCs will produce ROS, both of which reduce T-cell activity (127). Although there has been additional investigation into specific functions of each cell type, the overarching role of MDSCs remains the focus of this review.
Within the PDAC TME, exosomes released by malignant cells and malignantly programmed cells induce and upregulate transcription factors within MDSCs that further regulate and suppress the immune response(128). Both NF-kB and STAT3 have shown levels of upregulation within this context(129). Due to these mechanisms, MDSCs are shown to express elevated levels of arginase 1(ARG-1), an enzyme directly targeting L-arginine, and IDO-1/TDO, an enzyme targeting L-tryptophan to produce kynurenine. These two enzymes in combination work towards depletion of amino acids key in proliferation which indirectly suppress T-cell proliferation and potentially tumor proliferation (130). Additional effects of ROS and RNS production has shown to include decreased reactivity of MHC 1 molecules binding ability with cytotoxic T-cells in tumor cell populations (131). Additionally, MDSCs are known to release IL-10 and TGF-β for regulatory T-cell activation via CD40 (132). Receptor for advanced glycation end products (RAGE), a MHC class III receptor involved in recruitment and pro-longed survival of MDSCs, is shown to have increased expression PCCs and TME surrounding MDSCs. (133, 134). This receptor which responds to inflammatory signaling aims to sustain and recruit the MDSC population, prolonging their immunosuppressive effects on the TME. This along with continual recruitment of new MDSCs by the CXCR4/CXCL12 signaling pathway promotes indefinite immunosuppressive regulation by the PDAC TME (126). Decreased recruitment of MDSCs to the PDAC TME is showing a promising angle in novel therapeutic strategies by inhibiting the signaling pathways key in MDSC recruitment. As it stands, MDSCs serve as a major barrier in immunotherapies designed to utilize the body’s own immune system as they provide significant local-regional immune suppression.
Macrophages are demonstrated to be the frontline response within the immune system to instances of infection and injury (135). Monocytes make their way to the site of inflammation via VCAM-1/VLA-4 binding and CCR2/CCL2 migration (136). In addition to migratory monocytes, resident tissue macrophages simultaneously respond to initial insults. Alluded to earlier within this review, we discussed the balance and resolution of inflammation and how loss of such balance leads to a lack of resolution and the formation of chronic inflammation. M1 and M2 macrophages strike a homeostatic balance between pro-inflammatory and anti-inflammatory mechanisms based on cellular signaling and cytokine production. Within the early stages of an inflammatory process, M1 macrophages, induced by inflammatory markers such as IFN-γ and CSF-2, will release IL-1β, IL-12, IL-6, IFNα/β, and TNF-α in order to magnify the immune response (137). Over time as the initial infection subsides and repair mechanisms are prioritized, M2 cells will begin to dominate with the resolution of the infection while expressing immune suppressing and remodeling factors such as VEGF, IL-4, TGF-β, and IL-10 (138).
In the context of the PDAC TME, monocytes that are brought to the site maintain a similar initial response of differentiation and immunologic reaction to the pathology, but TME signaling provides an environment susceptible to imbalance. Cytokines and chemokines released by the TME such as CXCL8 binding CXCR1/2 on monocytes result in consistent recruitment for additional macrophages (139). A key chemokine mentioned previously, CCL2, is upregulated in the context of hypoxic environments secondary to HIF-1α promotion which is continuously produced within TME to recruit macrophages (140). These nonspecific monocytes and macrophages become polarized to favor the role of immunosuppressive M2s due to PCC signaling molecules such as IL-13 and TGF-β with most of these simultaneously suppressing M1 macrophage activation due to the nature of the macrophage’s polarity (141, 142). M2 macrophages are also shown to aid in metastatic properties of PDAC as previous literature indicates the role of granulin release by metastatic macrophages as associated with hepatic stellate cells fibroblastic activation and satellite TME formation (143). CSF-1 secretion by the TME directly affects repolarizing TAM M1s into M2s as inhibition of the receptor pathway in cancer cell models reduced the M2 to M1 decreased (144). The tumor microenvironment while prioritizing the formation of immunosuppressive M2s will also actively suppress the role M1s have in pro-inflammatory signaling. The general presence of TAMs within the TME points to the role of malignant transformation and rewiring of the inflammatory process in PDAC. Although CD163+/206+ are markers specific to M2 macrophages and is indicated in worse overall survival; CD68+, a non-specific marker of macrophage presence, is also associated with poor overall survival, speaking to the heterogeneity of the macrophage population (145). Proper targeting of macrophages cells within the TME has previously shown a promising role in potential therapeutics with clinical trials and research focused on this process ongoing.
In the complex makeup of the TME, neutrophils assume a pivotal role, with their recruitment being partially mediated by CXCR1 and CXCR2 receptors on neutrophils (146). These receptors are activated by multiple signaling molecules including CXCL-1, CXCL-2, CXCL-5, CXCL-8, and IL-8 released by various constituents of this environment, including cancer cells, immune cells, and cancer-associated fibroblasts. Supporting this recruitment, CSF-3 and IL-17 are involved in facilitating the migration and proliferation of neutrophils, contributing to their presence at the tumor site (147, 148). Upon reaching the TME, neutrophils undergo distinct activation pathways. IFN-β signaling prompts the activation of N1 neutrophils, specialized cells that release critical CD8-T-cell activating signals such as TNF-α, CCL-3, CXCL-9, and CXCL-10 (149, 150). These signals help bolster the immune response and enhance the cytotoxic activity of CD8-T-cells.
Conversely, IL-35 and TGF-β, primarily released by regulatory T-cells (Tregs), trigger the activation of N2 neutrophils. Unlike N1 neutrophils, N2 neutrophils possess immune-suppressive characteristics. They release molecules such as CCL2/3/4/17, Neutrophil Elastase, MMP9, and elevated arginase, contributing to immunosuppression and TME remodeling that fosters tumor progression. These pro-tumor neutrophils exhibit altered gene expression, upregulating genes like VEGFA, PLAU, LGALS3, PDE4D, and LDHA (151). PLAU, for example, is significant for encoding urokinase plasminogen activator, an upstream activator of the c-MET pathway in cancer cells (152). The TME’s unique conditions, including tissue hypoxia and ER stress, can also influence neutrophils to differentiate towards pro-tumor subtypes. In addition, the PDAC TME provides signaling, including IL-17, that activates a cellular function called neutrophil extracellular trap (NET) in which granular webs are released into the ECM (153). NETs work in a host’s defense system to trap pathologic organisms as part of the innate immune system, but research shows NETs are triggered within the PDAC TME and implicated in tumor metastasis and functioning as a physical barrier in anti-tumor immune responses (154, 155). Although the contributions of neutrophils to the PDAC TME and their role in immunosuppression has remained been debated in previous literature, a clearer understanding of the mechanisms and signaling involved has challenged this notion. Like other support cells in the PDAC TME, recruitment and unbalanced differentiation into tumor-promoting phenotypes may be the mechanism with greatest therapeutic potential.
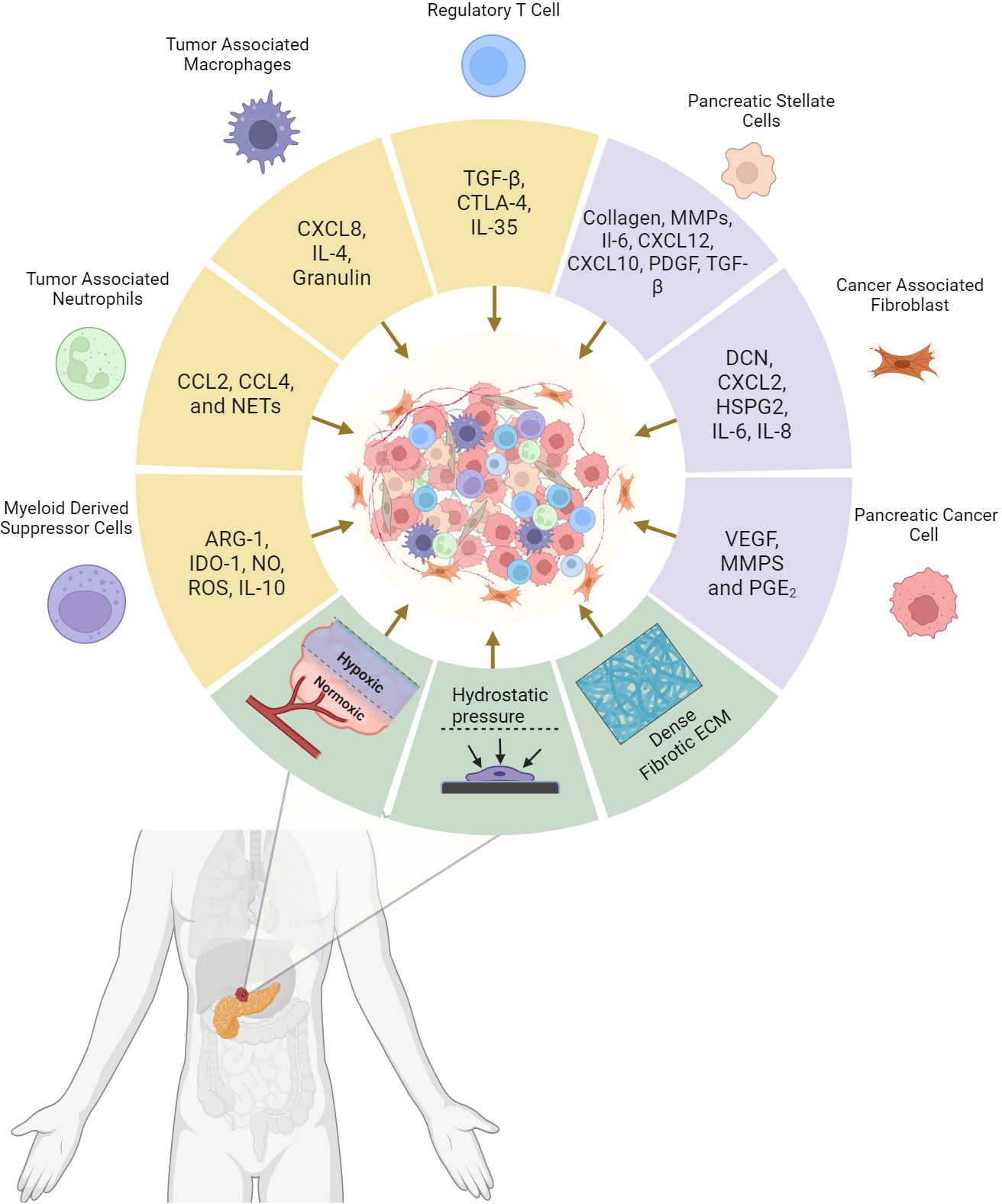
Figure 1 Pancreatic Ductal adenocarcinoma tumor microenvironment (TME) composition and the driving factors of tumor desmpolasia. Cytokines and tumorigenic signaling molecules are released by cancer cells, support cells, and immune cells that subsequently promotes the desmoplastic structure. In addition, mechanical elements stimulate these cells to perpetuate the environment and enhance further tumor growth. The resulting physical barrier and immune counterbalance are the critical factors responsible for systemic and immunotherapeutic resistance in PDAC. Created with BioRender.com.
Regulatory T-cells function as a safeguard in normal inflammation. Regulatory T-cells act as a part of the natural immunosuppression meant to minimize the risk of autoimmune diseases and other overactive immune responses (156). The immunoregulatory role of Tregs in the TME limits the efficacy of the immune system in attacking patient’s cancer cells. Regulatory T-cells are defined by their presence of the transcription factor FoxP3, IL-2Receptor (CD25), and CTLA-4 with direct roles in down-regulation of CD80/86 antigen-presenting cells and reduction of the immune response (157).
Tregs are a factory of general immunosuppression through a variety of mechanisms. Consistently present even within early stage PanIN, regulatory T-cells are examined within proximity of the tumor cell environment with the additional presence of M2 macrophages (158). In addition to CTLA-4, regulatory T-cells comprise part of the cellular populations that release TGF-β for later stage immunosuppression. In addition to TGF-β, regulatory T-cells demonstrate the release of IL-35 and IL-10 as mechanisms of potent immunosuppression (159). Combined inhibition of both TGF-β and PD-1 shows a direct increase in cytotoxic T-cells, emphasizing the effects Tregs and other secretory cells have in the TME (160). Although a simple expectation of removing Tregs from the TME seems like first response to this, evidence of early knockout of Treg cells related to tumor progression rather than reduction. This in part was seen due to compensatory accumulation of other CD4+ immune suppressing T-cells and increased fibroblast-linked chemokines shown to recruit myeloid derived suppressing cells (158). Rather, mitigation and inhibition of Treg’s role in recruitment and migration of MDSCs through CCR1 inhibition showed promise in reducing carcinogenesis. In support of this, MDSCs are shown directly communicating with Tregs through physical contact to promote tumor progression potentially through the B7-H1 pathway, but recent evidence suggests that MDSCs and Tregs work synergistically through bidirectional recruitment and tumor-specific signaling (161, 162). In therapeutic applications, one of the largest insights gained in recent years refers back to the effect of low-dose cyclophosphamide on depletion of Tregs in the context of the TME, yielding stronger response to immunotherapy. Similar measures focusing on the targeting of upstream pathways that result in Treg recruitment or inactivation will serve as a basis for future points of investigation.
The culmination of malignant reprogramming, cellular interactions, and structural malconformations produces the fibrous, dense tissue in pancreatic adenocarcinoma. The physiologic insults and aberrant signaling of pancreatic cancer cells, combined with the unsuccessful resolution of inflammation, is the initiating factor for fibroblastic activation and programming. PSC and CAF deposition of collagen, hyaluronan, and other ECM proteins have a combined utility in most physical characteristics of desmoplasia (Figure 1). The dense collagen matrices and increased interstitial pressures present a physical barrier for bioavailability of cytotoxic immune responses and therapeutic agents. For such metabolic demands with minimal perfusion, hypoxic environments promoting ROS formation, cellular autophagy, and transcriptional upregulation of HIF consequently lead to additional therapeutic resistance. Although primarily functioning in the ECM, previously mentioned proteins such as Asporin, Keratocan, and Periostin show direct influence on PDAC cancer cell tumorigenesis by demonstrating roles in membrane receptor binding and transcriptional regulation. The combination of high pressure, poor perfusion, and dense fibrosis presents a desmoplastic environment that encourages pancreatic cancer cells to search for greener pastures. Survival pressures and interactions from the extracellular matrix combined with paracrine signaling from support cells amplify the drive for cancer associated EMT. This combined with the supportive role of signaling growth factors from activated fibroblasts, compounds the effects on tumor growth. Inflammatory markers enhance these effects while MDSCs, Tregs, and M2 macrophages simultaneously suppress Effector T-cells, Dendritic Cells, and CD8 Cytotoxic T-cells from targeting and killing tumor cells. Strategies for targeting the desmoplastic stroma have shown promise, with a greater focus on reducing pressure, increase vascularization, and increase the diffusive capacity of the ECM.
Among emerging therapies, Chimeric Antigen Receptor T-cell (CAR-T) therapy has long received a level of interest since its inception in 1993 (163). The technology utilizes the patient’s immune system by introducing synthetic antigen receptors capitalizing on antibody binding sites with the T-cell intracellular signaling pathways. The benefit of CAR-T therapies is that they are not subject to immune-evasive effects that cancer cells routinely exhibit. The foundation for effective CAR-T-cell therapies, exemplified by the anti-CD19 drug for B-cell ALL, Tisagenlecleucel, is targeting unique receptor proteins predominantly expressed in cancer cells (164). A major barrier to CAR-T-cell therapies in PDAC patients includes the lack of uniquely defining receptor proteins that would consistently represent PDAC. Current PDAC CAR-T therapies have focused on CEA, EpCAM, Mesothelin, and CD133, but PDAC’s immune exhaustion mechanisms and the fibroblastic barrier to tumor cell exposure have proven to be limitations in CAR-T application (165). A Phase I clinical trial investigating the use of CAR-T cells programmed to bind Mesothelin in chemotherapy-refractory PDAC patients noted such results with limited detection of CAR-T transcripts within the TME following administration, although minimal toxicity was noted (166). Additional approaches to CAR-T cell administration have shown benefit through loco-regional administration of therapy rather than intravenous with marked increase in therapeutic response in preclinical models with further exploration following in phase I clinical trials. (167)(NCT01373047). In addition to challenging targets, adverse events relating to the induction of some novel immune therapies include cytokine release syndrome, seen with rapid activation of cytokine signaling cascades. Generational changes in CAR-T technology have focused on increasing cytokine response and support signaling to maximize the response in recognizing cancer epitopes and activating the immune response while minimizing these adverse events (168). Current clinical trials are beginning to utilize these newer generations of CAR-T-cell therapy and hope to address such limitations while investigating combined therapeutic strategies that utilize CAR-T-cell therapy with complementary immunotherapies.
Bispecific T-cell engaging (BiTE) antibody therapy, serves the purpose of activating the host immune response against specific cellular receptors that are either unique to or over-expressed on cancer cells. An example of an over-expressed receptor is EPCAM, an intercellular signaling receptor commonly seen on epithelial cells for cell-cell adhesion and communication. This over-expression is particularly notable in pancreatic cancer (169). MT-110 and Catumaxomab are two BiTEs designed for EPCAM, CD3 T-cell, and immune system activation against openly available EPCAM molecules to bind to (170). A phase 1 trial of MT-110 or soliton showed an unfortunately high rate (95%) of transfusion-related adverse events that warranted discontinuation of the investigative medicine (171). Besides off-target effects limiting applicability, most BiTE therapies target cell-surface proteins with limited efficacy for intracellular markers (172). Even while selecting patients with elevated levels of the designed target marker, IGF-1R, those in the phase II clinical trial for Istiratumab were not found to benefit meaningfully from the therapy in combination with standard of care (173). New research investigating HSV oncolytic viruses designed with Claudin 18.2 BITE targeting antibodies allow production of tumor-specific antibodies within the historically immunologically cold tumor microenvironment (174). Potential avenues worth exploring may be the combination of these novel immunologic therapies to maximize efficacy while minimizing off-target effects.
Vaccine therapy focuses on stimulating the host’s immune system to antigens uniquely expressed or upregulated within the PDAC TME. Tumor vaccine therapies involves a variety of vehicles, antigens, and genetic information to relay novel epitopes to the adaptive immune system. Vaccine methods include cell-, peptide-, protein-, DNA-, exosome-, and micro-organism-based. Such therapeutic methods although promising within early preclinical research and clinical trials, proved largely unresponsive in most phase II trials to-date (175). The largest phase III pancreatic cancer vaccine trial to date investigated the use of telomerase peptide vaccine in 1062 patients with no significant benefit in overall survival compared to standard of care alone (176). Evolving as a modern therapy in part due to the implementation of the COVID-19 vaccine, mRNA vaccines provide the schematics for protein expression with local reconstruction presenting the desired end product (177). Cancer mRNA vaccine therapy derives from using a patient’s tumor cell’s transcriptome, the mRNA present within the pancreatic cancer cells, to develop novel targets unique to the patient’s cancer. The benefits of mRNA vaccines over previous vaccines goes back to the large-scale utilization and manufacturing experienced during the COVID-19 pandemic. The limitations of modern research include our limited understanding of all potential targets within the PDAC TME. Some limitations in this therapy type go back to the immunosuppressive environment with a lack of immune system invasion through the PDAC TME. A recent trial utilizing personalized mRNA Neoantigen vaccines in addition to anti-PD-L1 (atezolizumab) and conventional chemotherapy in the adjuvant setting induced substantial T cell activity and demonstrated an increased recurrence-free survival in the phase I feasibility trial; although this patient population was limited to resected tumors and up to 20 neoepitopes within each vaccine (178). Based on these successful results, a phase 2 study is currently recruiting patients.
Bypassing the local immune suppression within pancreatic cancer remains a hurdle of this therapy as activated cytotoxic T-cells still must confront the TME, but methods of enhancing such therapies are a simultaneously evolving field. Codelivery with cytokine mRNA, such as IL-12, IL-15, IFN-α, or other immunostimulatory molecules, directly to tumor sites or in circulation with neoantigen mRNA may prove to be an alternative method to overcome these immunosuppressive barriers (179, 180).
Another emerging therapeutic approach involves the use of antibodies targeting of cancer-specific membrane molecules combined with a linker molecule and cytotoxic agent (181). These antibody-drug conjugates (ADCs) have been actively developed over the last two decades with FDA approval for Brentuximab Vedotin and Ado-trastuzumab emtansine for use in Hodgkin Lymphoma and breast cancer respectively (182, 183). Similar drugs are under investigation in PDAC with developmental ADCs targeting Anti-glypican-1, HER3, Claudin18.2, and CEACAM6 (184–187). A Phase II clinical trial investigating the use of Anetumab-Ravtansine, an ADC targeting mesothelin expressing pancreatic tumors, in 14 patients evaluated for toxicity and progression free survival. Progression free survival was a median of 63 days and approximately 11 of experienced serious adverse events (188). Some limitations in the past with this therapeutic mechanism included late onset-drug toxicity and “bystander killing” by the effects of the cytotoxic chemical component acting on surrounding normal cells. Although a benefit of this unintended effect may be the untargeted destruction of tumor-associated support cells and tumor-suppressive immune cells that populate the environment. In addition, due to the dense, desmoplastic stroma of the PDAC TME, limitations in efficacy may derive from regulations in antibody drugs properly reaching most cancer cells (189). New clinical trials are investigating a combination of treatments along with ADCs to maximize their therapeutic efficacy. Anetumab-Ravtansine combined with Gemcitabine and Nivolumab was showing signs of stable disease in a progressing Phase Ib trial with a now expanded investigation into the responding treatment regimen (190). Improvements have helped to reduce these effects with continued efforts to implement a potentially effective drug delivery mechanism that has shown promise with other solid tumors. Given the potential for potent targeted cytotoxicity, this avenue of research may benefit greatly from stromal modifying agents allowing for improved delivery to cancer cells.
Additional therapeutic trials are investigating the use of patient-derived tumor invading lymphocytes (TIL) in combination with autologous pancreatic cancer cell exposure for neoepitope generation (191). A current phase II trial is looking at approximately 330 metastatic cancer patients is investigating the use of TILs with concurrent use of immunotherapy targeting anti-PD1 to maximize the immune response (NCT01174121). Previous cases of TIL therapy showcase targeting of tumor-specific mutations that are unique to the patient’s PDAC cells. One example includes TIL directed towards KRAS G12D oncogene with conditioning immune modulating infusions tocilizumab and cyclophosphamide before administration of TIL therapy. The patient demonstrated metastatic regression and physiologic response to the therapy but ultimately succumbed to the disease (192). Engineering one’s immune system as the therapeutic model provides excellent potential for personalized medicine with targeting potentials, but immune evasion and inherent “cold” environment of PDAC requires additional means to potentiate this therapy.
Given PDAC’s immunosuppressive environment as discussed previously, novel therapies that target recruitment and activation of the immune system and breakdown of the physical barriers may play a key role in future treatments. Lytic viruses designed specifically to target cancer cells have become a recent field of interest for oncologic research. T-VEC, a strain of HSV designed to target advanced melanoma, was approved by the FDA in 2015 (193). Alongside tumor-specific targeting with genetic engineering, oncolytic viruses wield the potential to serve as a carrier of genetically significant proteins that may assist in combating the TME (194). An adenovirus loaded with Relaxin, CSF-2, and IL-12 variants demonstrated a reduced PDAC TME with a concurrent increase in immunoresponsiveness (195). Relaxin (RLX), an exogenous peptide hormone, has previously shown evidence of expression during the physiological reduction of fibrotic structures (196). Targeting the tumor microenvironment (TME) directly with relaxin nanoparticles has demonstrated the improved therapeutic efficacy of Gemcitabine within in vivo models (197). Research trials utilizing VCN-01, an adenovirus expressing PH20, and Pelareorep, a reovirus, in disrupting the TME and awakening the immune response are underway as this novel therapy continues to evolve (198, 199). Moving forward, dual targeting of the PDAC TME and pancreatic cancer cells may prove efficacious in maximizing the utility of this novel therapy and overcoming therapeutic challenges of PDAC. Our group and others are investigating this strategy to optimize PDAC response to immune oncology approaches and other systemic treatments. In addition to viruses, efforts are underway to leverage the tumor-targeting capability of some bacteria to convert PDAC into an immunologically hot milieu. Notably, a genetically attenuated strain of salmonella typhimurium expressing IL-2, Saltikva, recently received FDA fast-track designation for the treatment of metastatic pancreatic cancers following phase 2 clinical studies (NCT01486329), highlighting new opportunities ahead for bacteria-based cancer therapeutics.
PDAC remains a lethal disease due to many factors that prevent adequate delivery to, and targeting of, pancreatic cancer cells. Current literature highlights the evident role the TME has in the initiation, progression, and treatment intractability of PDAC. The cellular components of the TME, combined with the extracellular environment and signaling pathways with surrounding tissues, shed light on the intricate complexity of how PDAC develops and the complex nature of resistance to innate and external anti-tumorigenic mechanisms. As the field evolves to better understand the dynamic interactions within the TME, novel therapeutic strategies of drug delivery and unique targets will unravel this disease. Nevertheless, critical gaps in our knowledge demand further research to reveal the precise mechanisms underlying TME-tumor interactions, taking into consideration the heterogeneity that characterizes both the tumor and the TME. Integrating -omics data, advanced imaging techniques, and sophisticated in vitro and in vivo models will advance our understanding of these intricate relationships. Novel therapeutic modalities will be refined based on more comprehensive understanding of mechanisms involved with primary and secondary resistance to systemic and immuno-oncology approaches. Moreover, future directions will be more focused on multimodality precision approaches, targeting different aspects of tumors, including cancer cells, microenvironment, and immune system.
CH: Writing – original draft. BN: Writing – review & editing. CC: Writing – review & editing. MT: Writing – review & editing. JC-B: Writing – review & editing. JW: Writing – review & editing. OM: Conceptualization, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was financially supported by grant: NIGMS/NIH 5P20GM121288-07.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Siegel RL, Siegel R.L., Miller KD, Wagle NS, Jemal A. Cancer statistic. CA: A Cancer J Clin (2023) 73(1):17–48. doi: 10.3322/caac.21763
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res (2014) 74(11):2913–21. doi: 10.1158/0008-5472.CAN-14-0155
3. Grossberg AJ, Chu LC, Deig CR, Fishman EK, Hwang WL, Maitra A, et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA: Cancer J Clin (2020) 70(5):375–403. doi: 10.3322/caac.21626
4. Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature (2017) 551(7681):512–6. doi: 10.1038/nature24462
5. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 21(3):418–29. doi: 10.1016/j.ccr.2012.01.007
6. Kalimuthu N, Wilson GW, Grant RC, Seto M, O’Kane G, Vajpeyi R, et al. Morphological classification of pancreatic ductal adenocarcinoma that predicts molecular subtypes and correlates with clinical outcome. Gut (2020) 69(2):317–28. doi: 10.1136/gutjnl-2019-318217
7. Hwang WL, Jagadeesh KA, Guo JA, Hoffman HI, Yadollahpour P, Reeves JW, et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat Genet (2022) 54(8):1178–91. doi: 10.1038/s41588-022-01134-8
8. Flowers BM, Xu H, Mulligan AS, Hanson KJ, Seoane JA, Vogel H, et al. Cell-of-origin influences pancreatic cancer subtype. Cancer Discovery (2021) 11(3):660–77. doi: 10.1158/2159-8290.CD-20-0633
9. Blanpain C. Tracing the cellular origin of cancer. Nat Cell Biol (2013) 15(2):126–34. doi: 10.1038/ncb2657
10. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduction Targeted Ther (2021) 6(1):1–46. doi: 10.1038/s41392-021-00658-5
11. Mann KM, Ying H, Juan J, Jenkins NA, Copeland NG. KRAS-related proteins in pancreatic cancer. Pharmacol Ther (2016) 168:29–42. doi: 10.1016/j.pharmthera.2016.09.003
12. Tape CJ, Ling S, Dimitriadi M, McMahon KM, Worboys JD, Leong HS, et al. Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell (2016) 165(4):910–20. doi: 10.1016/j.cell.2016.03.029
13. Halbrook CJ, Lyssiotis CA. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell (2017) 31(1):5–19. doi: 10.1016/j.ccell.2016.12.006
14. Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernández-Porras I, Cañamero M, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell (2011) 19(6):728–39. doi: 10.1016/j.ccr.2011.05.011
15. Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PloS Biol (2008) 6(12):e301. doi: 10.1371/journal.pbio.0060301
16. Cortesi M, Zanoni M, Pirini F, Tumedei MM, Ravaioli S, Rapposelli IG, et al. Pancreatic cancer and cellular senescence: tumor microenvironment under the spotlight. Int J Mol Sci (2022) 23(1):254. doi: 10.3390/ijms23010254
17. De Blander H, Morel A-P, Senaratne AP, Ouzounova M, Puisieux A. Cellular plasticity: A route to senescence exit and tumorigenesis. Cancers (2021) 13(18):4561. doi: 10.3390/cancers13184561
18. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery (2022) 12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
19. Ahmed S, Bradshaw A-D, Gera S, Dewan MZ, Xu R. The TGF-β/smad4 signaling pathway in pancreatic carcinogenesis and its clinical significance. J Clin Med (2017) 6(1):5. doi: 10.3390/jcm6010005
20. Hruban RH, Wilentz RE, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol (2000) 156(6):1821–5. doi: 10.1016/S0002-9440(10)65054-7
21. Pandha H, Rigg A, John J, Lemoine N. Loss of expression of antigen-presenting molecules in human pancreatic cancer and pancreatic cancer cell lines. Clin Exp Immunol (2007) 148(1):127–35. doi: 10.1111/j.1365-2249.2006.03289.x
22. Bachem MG, Schünemann M, Ramadani M, Siech M, Beger H, Buck A, et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology (2005) 128(4):907–21. doi: 10.1053/j.gastro.2004.12.036
23. Ijichi H, Chytil A, Gorska AE, Aakre ME, Bierie B, Tada M, et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest (2011) 121(10):4106–17. doi: 10.1172/JCI42754
24. Das S, Shapiro B, Vucic EA, Vogt S, Bar-Sagi D. Tumor cell–derived IL1β Promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res (2020) 80(5):1088–101. doi: 10.1158/0008-5472.CAN-19-2080
25. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell (2012) 21(6):836. doi: 10.1016/j.ccr.2012.04.024
26. Meyer MA, Baer JM, Knolhoff BL, Nywening TM, Panni RZ, Su X, et al. Breast and pancreatic cancer interrupt IRF8-dependent dendritic cell development to overcome immune surveillance. Nat Commun (2018) 9(1):1250. doi: 10.1038/s41467-018-03600-6
27. Pang W, Su J, Wang Y, Feng H, Dai X, Yuan Y, et al. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci (2015) 106(10):1362–9. doi: 10.1111/cas.12747
28. Masamune A, Yoshida N, Hamada S, Takikawa T, Nabeshima T, Shimosegawa T. Exosomes derived from pancreatic cancer cells induce activation and profibrogenic activities in pancreatic stellate cells. Biochem Biophys Res Commun (2018) 495(1):71–7. doi: 10.1016/j.bbrc.2017.10.141
29. Büchler P, Büchler P, Reber HA, Büchler M, Shrinkante S, Büchler MW, Friess H, et al. Hypoxia-inducible factor 1 regulates vascular endothelial growth factor expression in human pancreatic cancer. Pancreas (2003) 26(1):56–64. doi: 10.1097/00006676-200301000-00010
30. Shi X, Wang M, Zhang Y, Guo X, Liu M, Zhou Z, et al. Hypoxia activated HGF expression in pancreatic stellate cells confers resistance of pancreatic cancer cells to EGFR inhibition. eBioMedicine (2022) 86. doi: 10.1016/j.ebiom.2022.104352
31. Wang X, Luo G, Zhang K, Cao J, Huang C, Jiang T, et al. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kγ to Promote Pancreatic Cancer Metastasis. Cancer Res (2018) 78(16):4586–98. doi: 10.1158/0008-5472.CAN-17-3841
32. Lu W, Hu Y, Chen G, Chen Z, Zhang H, Wang F, et al. Novel role of NOX in supporting aerobic glycolysis in cancer cells with mitochondrial dysfunction and as a potential target for cancer therapy. PloS Biol (2012) 10(5):e1001326. doi: 10.1371/journal.pbio.1001326
33. Yang J, Liu Y, Liu S. The role of epithelial-mesenchymal transition and autophagy in pancreatic ductal adenocarcinoma invasion. Cell Death Dis (2023) 14(8):1–14. doi: 10.1038/s41419-023-06032-3
34. Greco L, Rubbino F, Laghi L. Epithelial to mesenchymal transition as mechanism of progression of pancreatic cancer: from mice to men. Cancers (2022) 14(23):5797. doi: 10.3390/cancers14235797
35. Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut (1998) 43(1):128–33. doi: 10.1136/gut.43.1.128
36. Xue R, Jia K, Wang J, Yang L, Wang Y, Gao L, et al. A rising star in pancreatic diseases: pancreatic stellate cells. Front Physiol (2018) 9:754. doi: 10.3389/fphys.2018.00754
37. Bachem MG, Zhou Z, Zhou S, Siech M. Role of stellate cells in pancreatic fibrogenesis associated with acute and chronic pancreatitis. J Gastroenterol Hepatol (2006), S92–6. doi: 10.1111/j.1440-1746.2006.04592.x
38. Means AL. Pancreatic stellate cells: small cells with a big role in tissue homeostasis. Lab Invest (2013) 93(1):4–7. doi: 10.1038/labinvest.2012.161
39. Zhou Y, Wang H, Zhou J, Qiu S, Cai T, Li H, et al. Vitamin A and its multi-effects on pancreas: recent advances and prospects. Front Endocrinol (2021) 12:620941 (Accessed: 10 July 2023. doi: 10.3389/fendo.2021.620941
40. Jin G, Hong W, Guo Y, Bai Y, Chen B. Molecular mechanism of pancreatic stellate cells activation in chronic pancreatitis and pancreatic cancer. J Cancer (2020) 11(6):1505–15. doi: 10.7150/jca.38616
41. Ahmad RS, Eubank TD, Lukomski S, Boone BA. Immune cell modulation of the extracellular matrix contributes to the pathogenesis of pancreatic cancer. Biomolecules (2021) 11(6):901. doi: 10.3390/biom11060901
42. Berchtold S, Grünwald B, Krüger A, Reithmeier A, Hähl T, Cheng T, et al. Collagen type V promotes the Malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett (2015) 356(2 Pt B):721–32. doi: 10.1016/j.canlet.2014.10.020
43. DuFort CC, DelGiorno KE, Hingorani SR. Mounting pressure in the microenvironment: fluids, solids, and cells in pancreatic ductal adenocarcinoma. Gastroenterology (2016) 150(7):1545–1557.e2:doi: 10.1053/j.gastro.2016.03.040
44. Itoh Y, Takehara Y, Kawase T, Terashima K, Ohkawa Y, Hirose Y, et al. Feasibility of magnetic resonance elastography for the pancreas at 3T. J Magnetic Resonance Imaging (2016) 43(2):384–90. doi: 10.1002/jmri.24995
45. Sakai S, Iwata C, Tanaka HY, Cabral H, Morishita Y, Miyazono K, et al. Increased fibrosis and impaired intratumoral accumulation of macromolecules in a murine model of pancreatic cancer co-administered with FGF-2. J Controlled Release (2016) 230:109–15. doi: 10.1016/j.jconrel.2016.04.007
46. Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight (2019) 4(7):e126853. doi: 10.1172/jci.insight.126853
47. Mace TA, Bloomston M, Lesinski GB. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res (2013) 73(10):3007–18. doi: 10.1158/0008-5472.CAN-12-4601
48. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells: A viable target for reducing immunosuppression in the tumor microenvironment. Oncoimmunology (2013) 2(7):e24891. doi: 10.4161/onci.24891
49. Wu Y, Zhang C, Guo M, Hu W, Qiu Y, Li M, et al. Targeting pancreatic stellate cells in chronic pancreatitis: Focus on therapeutic drugs and natural compounds. Front Pharmacol (2022) 13:1042651. doi: 10.3389/fphar.2022.1042651
50. Lunardi S, Jamieson NB, Lim SY, Griffiths KL, Carvalho-Gaspar M, Al-Assar O, et al. IP-10/CXCL10 induction in human pancreatic cancer stroma influences lymphocytes recruitment and correlates with poor survival. Oncotarget (2014) 5(22):11064–80. doi: 10.18632/oncotarget.2519
51. Thomas D, Radhakrishnan P. Pancreatic stellate cells: the key orchestrator of the pancreatic tumor microenvironment. In: Birbrair A, editor. Tumor microenvironment: non-hematopoietic cells. Cham: Springer International Publishing (Advances in Experimental Medicine and Biology (2020). p. 57–70. doi: 10.1007/978-3-030-37184-5_5
52. Liu H, Zhang H, Liu X, Guo W, Liu Q, Chen L, et al. Pancreatic stellate cells exploit Wnt/β-catenin/TCF7-mediated glutamine metabolism to promote pancreatic cancer cells growth. Cancer Lett (2023) 555:216040. doi: 10.1016/j.canlet.2022.216040
53. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature (2016) 536(7617):479–83. doi: 10.1038/nature19084
54. Cao F, Li J, Sun H, Liu S, Cui Y, Li F. HES 1 is essential for chemoresistance induced by stellate cells and is associated with poor prognosis in pancreatic cancer. Oncol Rep (2015) 33(4):1883–9. doi: 10.3892/or.2015.3789
55. Karakas D, Erkisa M, Akar RO, Akman G, Senol EY, Ulukaya E. Targeting periostin expression makes pancreatic cancer spheroids more vulnerable to natural killer cells. Biomedicines (2023) 11(2):270. doi: 10.3390/biomedicines11020270
56. Wu YS, Chung I, Wong WF, Masamune A, Sim MS, Looi CY. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim Biophys Acta (BBA) - Gen Subj (2017) 1861(2):296–306. doi: 10.1016/j.bbagen.2016.10.006
57. Ikenaga N, Ohuchida K, Mizumoto K, Cui L, Kayashima T, Morimatsu K, et al. CD10+ Pancreatic stellate cells enhance the progression of pancreatic cancer. Gastroenterology (2010) 139(3):1041–1051.e8. doi: 10.1053/j.gastro.2010.05.084
58. Yoshida N, Masamune A, Hamada S, Kikuta K, Takikawa T, Motoi F, et al. Kindlin-2 in pancreatic stellate cells promotes the progression of pancreatic cancer. Cancer Lett (2017) 390:103–14. doi: 10.1016/j.canlet.2017.01.008
59. Ma Q, Wu H, Xiao Y, Liang Z, Liu T. Upregulation of exosomal microRNA−21 in pancreatic stellate cells promotes pancreatic cancer cell migration and enhances Ras/ERK pathway activity. Int J Oncol (2020) 56(4):1025–33. doi: 10.3892/ijo.2020.4986
60. Chronopoulos A, Robinson B, Sarper M, Cortes E, Auernheimer V, Lachowski D, et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun (2016) 7:12630. doi: 10.1038/ncomms12630
61. Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell (2014) 159(1):80–93. doi: 10.1016/j.cell.2014.08.007
62. Mizutani Y, Kobayashi H, Iida T, Asai N, Masamune A, Hara A, et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res (2019) 79(20):5367–81. doi: 10.1158/0008-5472.CAN-19-0454
63. Kumar K, DeCant BT, Grippo PJ, Hwang RF, Bentrem DJ, Ebine K, et al. BET inhibitors block pancreatic stellate cell collagen I production and attenuate fibrosis in vivo. JCI Insight (2017) 2(3):e88032. doi: 10.1172/jci.insight.88032
64. Rynne-Vidal A, Jiménez-Heffernan JA, Fernández-Chacón C, López-Cabrera M, Sandoval P. The mesothelial origin of carcinoma associated-fibroblasts in peritoneal metastasis. Cancers (2015) 7(4):1994–2011. doi: 10.3390/cancers7040872
65. Iwamoto C, Ohuchida K, Shinkawa T, Okuda S, Otsubo Y, Okumura T, et al. Bone marrow-derived macrophages converted into cancer-associated fibroblast-like cells promote pancreatic cancer progression. Cancer Lett (2021) 512:15–27. doi: 10.1016/j.canlet.2021.04.013
66. Helms EJ, Berry MW, Chaw RC, DuFort CC, Sun D, Onate MK, et al. Mesenchymal lineage heterogeneity underlies nonredundant functions of pancreatic cancer-associated fibroblasts. Cancer Discovery (2022) 12(2):484–501. doi: 10.1158/2159-8290.CD-21-0601
67. Lavie D, Ben-Shmuel A, Erez N, Scherz-Shouval R. Cancer-associated fibroblasts in the single-cell era. Nat Cancer (2022) 3(7):793–807. doi: 10.1038/s43018-022-00411-z
68. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discovery (2019) 9(8):1102–23. doi: 10.1158/2159-8290.CD-19-0094
69. McAndrews KM, Chen Y, Darpolor KJ, Zheng X, Yang S, Carstens J, et al. Identification of functional heterogeneity of carcinoma-associated fibroblasts with distinct IL6-mediated therapy resistance in pancreatic cancer. Cancer Discovery (2022) 12(6):1580–97. doi: 10.1158/2159-8290.CD-20-1484
70. Biffi G. Tracing the origin of fibroblasts in pancreatic cancer. Cell Mol Gastroenterol Hepatol (2020) 10(3):645–6. doi: 10.1016/j.jcmgh.2020.06.008
71. Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev (2021) 101(1):147–76. doi: 10.1152/physrev.00048.2019
72. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discovery (2019) 9(2):282–301. doi: 10.1158/2159-8290.CD-18-0710
73. Steele NG, Biffi G, Kemp SB, Zhang Y, Drouillard D, Syu L, et al. Inhibition of hedgehog signaling alters fibroblast composition in pancreatic cancer. Clin Cancer Research: Off J Am Assoc Cancer Res (2021) 27(7):2023–37. doi: 10.1158/1078-0432.CCR-20-3715
74. Chen K, Wang Q, Li M, Guo H, Liu W, Wang F, et al. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma Malignant progression. eBioMedicine (2021) 66. doi: 10.1016/j.ebiom.2021.103315
75. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med (2017) 214(3):579–96. doi: 10.1084/jem.20162024
76. Wang Y, Liang Y, Xu H, Zhang X, Mao T, Cui J, et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discovery (2021) 7(1):1–17. doi: 10.1038/s41421-021-00271-4
77. Ge W, Yue M, Lin R, Zhou T, Xu H, Wang Y, et al. PLA2G2A+ cancer-associated fibroblasts mediate pancreatic cancer immune escape via impeding antitumor immune response of CD8+ cytotoxic T cells. Cancer Lett (2023) 558:216095. doi: 10.1016/j.canlet.2023.216095
78. Vennin C, Mélénec P, Rouet R, Nobis M, Cazet AS, Murphy KJ, et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat Commun (2019) 10(1):3637. doi: 10.1038/s41467-019-10968-6
79. Farach-Carson MC, Warren CR, Harrington DA, Carson DD. Border patrol: Insights into the unique role of perlecan/heparan sulfate proteoglycan 2 at cell and tissue borders. Matrix Biol (2014) 34:64–79. doi: 10.1016/j.matbio.2013.08.004
80. Grindel B, Li Q, Arnold R, Petros J, Zayzafoon M, Muldoon M, et al. Perlecan/HSPG2 and matrilysin/MMP-7 as indices of tissue invasion: tissue localization and circulating perlecan fragments in a cohort of 288 radical prostatectomy patients. Oncotarget (2016) 7:10433–47. doi: 10.18632/oncotarget.11976
81. Zaghdoudi S, Decaup E, Belhabib I, Samain R, Cassant-Sourdy S, Rochotte J, et al. FAK activity in cancer-associated fibroblasts is a prognostic marker and a druggable key metastatic player in pancreatic cancer. EMBO Mol Med (2020) 12(11):e12010. doi: 10.15252/emmm.202012010
82. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10+GPR77+ Cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell (2018) 172:841–56. doi: 10.1016/j.cell.2018.01.009
83. Schuth S, Le Blanc S, Krieger TG, Jabs J, Schenk M, Giese NA, et al. Patient-specific modeling of stroma-mediated chemoresistance of pancreatic cancer using a three-dimensional organoid-fibroblast co-culture system. J Exp Clin Cancer Res (2022) 41(1):312. doi: 10.1186/s13046-022-02519-7
84. Catenacci DVT, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, et al. Randomized phase ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol (2015) 33(36):4284–92. doi: 10.1200/JCO.2015.62.8719
85. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci (2013) 110(50):20212–7. doi: 10.1073/pnas.1320318110
86. Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med (2020) 26(6):878–85. doi: 10.1038/s41591-020-0880-x
87. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun (2020) 11:5120. doi: 10.1038/s41467-020-18794-x
88. Folkman J. Angiogenesis. Annu Rev Med (2006) 57(1):1–18. doi: 10.1146/annurev.med.57.121304.131306
89. Ribatti D, Pezzella F. Overview on the different patterns of tumor vascularization. Cells (2021) 10(3):639. doi: 10.3390/cells10030639
90. Li S, Xu H-X, Wu C-T, Wang W-Q, Jin W, Gao H-L, et al. Angiogenesis in pancreatic cancer: current research status and clinical implications. Angiogenesis (2019) 22(1):15–36. doi: 10.1007/s10456-018-9645-2
91. Komar G, Kauhanen S, Liukko K, Seppänen M, Kajander S, Ovaska J, et al. Decreased blood flow with increased metabolic activity: A novel sign of pancreatic tumor aggressiveness. Clin Cancer Res (2009) 15(17):5511–7. doi: 10.1158/1078-0432.CCR-09-0414
92. Hexige S, Ardito-Abraham CM, Wu Y, Wei Y, Fang Y, Han X, et al. Identification of novel vascular projections with cellular trafficking abilities on the microvasculature of pancreatic ductal adenocarcinoma. J Pathol (2015) 236(2):142–54. doi: 10.1002/path.4506
93. Lu Z, Weniger M, Jiang K, Boeck S, Zhang K, Bazhin A, et al. Therapies targeting the tumor stroma and the VEGF/VEGFR axis in pancreatic ductal adenocarcinoma: a systematic review and meta-analysis. Targeted Oncol (2018) 13(4):447–59. doi: 10.1007/s11523-018-0578-x
94. Gilles M-E, Maione F, Cossutta M, Carpentier G, Caruana L, Di Maria S, et al. Nucleolin targeting impairs the progression of pancreatic cancer and promotes the normalization of tumor vasculature. Cancer Res (2016) 76(24):7181–93. doi: 10.1158/0008-5472.CAN-16-0300
95. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
96. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell (2014) 25(6):735–47. doi: 10.1016/j.ccr.2014.04.021
97. Tian C, Clauser KR, Öhlund D, Rickelt S, Huang Y, Gupta M, et al. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc Natl Acad Sci (2019) 116(39):19609–18. doi: 10.1073/pnas.1908626116
98. Köninger J, Giese T, di Mola FF, Wente MN, Esposito I, Bachem MG, et al. Pancreatic tumor cells influence the composition of the extracellular matrix. Biochem Biophys Res Commun (2004) 322(3):943–9. doi: 10.1016/j.bbrc.2004.08.008
99. Appunni S, Anand V, Khandelwal M, Gupta N, Rubens M, Sharma A. Small Leucine Rich Proteoglycans (decorin, biglycan and lumican) in cancer. Clinica Chimica Acta (2019) 491:1–7. doi: 10.1016/j.cca.2019.01.003
100. Santra M, Eichstetter I, Iozzo RV. An anti-oncogenic role for decorin: down-regulation of erbB2 leads to growth suppression and cytodifferentiation of mammary carcinoma cells *. J Biol Chem (2000) 275(45):35153–61. doi: 10.1074/jbc.M006821200
101. Buraschi S, Pal N, Tyler-Rubinstein N, Owens RT, Neill T, Iozzo RV. Decorin antagonizes met receptor activity and down-regulates β-catenin and myc levels. J Biol Chem (2010) 285(53):42075–85. doi: 10.1074/jbc.M110.172841
102. Yao T, Zhang C, Gong M, Zhang M, Wang L, Ding W. Decorin-mediated inhibition of the migration of U87MG glioma cells involves activation of autophagy and suppression of TGF-β signaling. FEBS Open Bio (2016) 6(7):707–19. doi: 10.1002/2211-5463.12076
103. Sainio AO, Järveläinen HT. Decorin-mediated oncosuppression – a potential future adjuvant therapy for human epithelial cancers. Br J Pharmacol (2019) 176(1):5–15. doi: 10.1111/bph.14180
104. Hong X, Yang Z, Wang M, Wang L, Xu Q. Reduced decorin expression in the tumor stroma correlates with tumor proliferation and predicts poor prognosis in patients with I–IIIA non-small cell lung cancer. Tumor Biol (2016) 37(12):16029–38. doi: 10.1007/s13277-016-5431-1
105. Li X, Truty MA, Kang Y, Chopin-Laly X, Zhang R, Roife DJ, et al. Extracellular lumican inhibits pancreatic cancer cell growth and is associated with prolonged survival after surgery. Clin Cancer research: an Off J Am Assoc Cancer Res (2014) 20(24):6529–40. doi: 10.1158/1078-0432.CCR-14-0970
106. Li X, Kang Y, Roife D, Lee Y, Pratt M, Perez MR, et al. Prolonged exposure to extracellular lumican restrains pancreatic adenocarcinoma growth. Oncogene (2017) 36(38):5432–8. doi: 10.1038/onc.2017.125
107. Gao H, Qian R, Ren Q, Zhang L, Qin W, Zhou C, et al. The upregulation of keratocan promotes the progression of human pancreatic cancer. Mol Cell Toxicol (2023). doi: 10.1007/s13273-023-00342-5
108. Somani V, Zhang D, Dodhiawala P, Lander V, Liu X, Kang L-I, et al. IRAK4 signaling drives resistance to checkpoint immunotherapy in pancreatic ductal adenocarcinoma. Gastroenterology (2022) 162:2047–62. doi: 10.1053/j.gastro.2022.02.035
109. Maloney E, DuFort CC, Provenzano PP, Farr N, Carlson MA, Vohra R, et al. Non-invasive monitoring of stromal biophysics with targeted depletion of hyaluronan in pancreatic ductal adenocarcinoma. Cancers (2019) 11, 772. doi: 10.3390/cancers11060772
110. Van Cutsem E, Tempero MA, Sigal D, Oh D-Y, Fazio N, Macarulla T, et al. Randomized phase III trial of pegvorhyaluronidase alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J Clin Oncol (2020) 38(27):3185–94. doi: 10.1200/JCO.20.00590
111. Mittal R, Patel AP, Debs LH, Nguyen D, Patel K, Grati M, et al. Intricate functions of matrix metalloproteinases in physiological and pathological conditions. J Cell Physiol (2016) 231(12):2599–621. doi: 10.1002/jcp.25430
112. Laronha H, Caldeira J. Structure and function of human matrix metalloproteinases. Cells (2020) 9(5):1076. doi: 10.3390/cells9051076
113. Tian M, Cui Y-Z, Song G-H, Zong M-J, Zhou X-Y, Chen Y, et al. Proteomic analysis identifies MMP-9, DJ-1 and A1BG as overexpressed proteins in pancreatic juice from pancreatic ductal adenocarcinoma patients. BMC Cancer (2008) 8(1):241. doi: 10.1186/1471-2407-8-241
114. Moore M, Hamm J, Dancey J, Eisenberg PD, Dagenais M, Fields A, et al. Comparison of gemcitabine versus the matrix metalloproteinase inhibitor BAY 12-9566 in patients with advanced or metastatic adenocarcinoma of the pancreas: A phase III trial of the national cancer institute of Canada clinical trials group. J Clin Oncol (2003) 21(17):3296–302. doi: 10.1200/JCO.2003.02.098
115. Qiang L, Cao H, Chen J, Weller SG, Krueger EW, Zhang L, et al. Pancreatic tumor cell metastasis is restricted by MT1-MMP binding protein MTCBP-1. J Cell Biol (2019) 218(1):317–32. doi: 10.1083/jcb.201802032
116. Tatti O, Vehviläinen P, Lehti K, Keski-Oja J. MT1-MMP releases latent TGF-β1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp Cell Res (2008) 314(13):2501–14. doi: 10.1016/j.yexcr.2008.05.018
117. Nguyen H-L, Kadam P, Helkin A, Cao K, Wu S, Samara GJ, Zhang Q, et al. MT1-MMP activation of TGF-β Signaling enables intercellular activation of an epithelial-mesenchymal transition program in cancer. Curr Cancer Drug Targets (2016) 16(7):618–30. doi: 10.2174/1568009616666160216125634
118. Krantz SB, Shields MA, Dangi-Garimella S, Cheon EC, Barron MR, Hwang RF, et al. MT1-MMP Cooperates with KrasG12D to Promote Pancreatic Fibrosis through Increased TGF-β Signaling. Cancer Res. (2011) 9, 1294–304. doi: 10.1158/1541-7786.MCR-11-0023
119. Shields MA, Dangi-Garimella S, Krantz SB, Bentrem DJ, Munshi HG. Pancreatic cancer cells respond to type i collagen by inducing snail expression to promote membrane type 1 matrix metalloproteinase-dependent collagen invasion *. J. Biol. Chem. (2011) 286, 10495–504. doi: 10.1074/jbc.M110.195628
120. Benzing C, Lam H, Tsang CM, Rimmer A, Arroyo-Berdugo Y, Calle Y, et al. TIMP-2 secreted by monocyte-like cells is a potent suppressor of invadopodia formation in pancreatic cancer cells. BMC Cancer (2019) 19(1):1214. doi: 10.1186/s12885-019-6429-z
121. Wang L, Wu H, Wang L, Zhang H, Lu J, Liang Z, et al. Asporin promotes pancreatic cancer cell invasion and migration by regulating the epithelial-to-mesenchymal transition (EMT) through both autocrine and paracrine mechanisms. Cancer Lett (2017) 398:24–36. doi: 10.1016/j.canlet.2017.04.001
122. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
123. Zhang Z, Huang X, Wang E, Huang Y, Yang R. Suppression of mll1-complex by stat3/cebpβ-induced miR-21a/21b/181b maintains the accumulation, homeostasis, and immunosuppressive function of polymorphonuclear myeloid-derived suppressor cells. J Immunol (Baltimore Md. (2020) 204(12):3400–15. doi: 10.4049/jimmunol.2000230
124. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer1. J Immunol (2001) 166(1):678–89. doi: 10.4049/jimmunol.166.1.678
125. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity (2019) 50(5):1317–1334.e10. doi: 10.1016/j.immuni.2019.03.009
126. Youn J-I, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukocyte Biol (2012) 91(1):167–81. doi: 10.1189/jlb.0311177
127. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species1. J Immunol (2004) 172(2):989–99. doi: 10.4049/jimmunol.172.2.989
128. Javeed N, Gustafson MP, Dutta SK, Lin Y, Bamlet WR, Oberg AL, et al. Immunosuppressive CD14+HLA-DRlo/neg monocytes are elevated in pancreatic cancer and “primed” by tumor-derived exosomes. Oncoimmunology (2017) 6(1):e1252013. doi: 10.1080/2162402X.2016.1252013
129. Trovato R, Fiore A, Sartori S, Canè S, Giugno R, Cascione L, et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J ImmunoTherapy Cancer (2019) 7(1):255. doi: 10.1186/s40425-019-0734-6
130. Mondanelli G, Iacono A, Allegrucci M, Puccetti P, Grohmann U. Immunoregulatory interplay between arginine and tryptophan metabolism in health and disease. Front Immunol (2019) 10:1565. doi: 10.3389/fimmu.2019.01565
131. Lu T, Ramakrishnan R, Altiok S, Youn J-I, Cheng P, Celis E, et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest (2011) 121(10):4015–29. doi: 10.1172/JCI45862
132. Ugel S, Sanctis FD, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest (2015) 125(9):3365–76. doi: 10.1172/JCI80006
133. Sparvero LJ, Asafu-Adjei D, Kang R, Tang D, Amin N, Im J, et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands, and their role in Cancer and Inflammation. J Trans Med (2009) 7(1):17. doi: 10.1186/1479-5876-7-17
134. Vernon PJ, Loux TJ, Schapiro NE, Kang R, Muthuswamy R, Kalinski P, et al. The receptor for advanced glycation end-products promotes pancreatic carcinogenesis and accumulation of myeloid-derived suppressor cells. J Immunol (Baltimore Md.: 1950) (2013) 190(3):1372–9. doi: 10.4049/jimmunol.1201151
135. Hirayama D, Iida T, Nakase H. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int J Mol Sci (2017) 19(1):92. doi: 10.3390/ijms19010092
136. Yang Z, Xie Z, Wan J, Yi B, Xu T, Shu X, et al. Current trends and research hotspots in pancreatic stellate cells: A bibliometric study. Front Oncol (2022) 12:896679 (Accessed: 6 July 2023. doi: 10.3389/fonc.2022.896679
137. Yang S, Liu Q, Liao Q. Tumor-associated macrophages in pancreatic ductal adenocarcinoma: origin, polarization, function, and reprogramming. Front Cell Dev Biol (2021) 8:607209. doi: 10.3389/fcell.2020.607209
138. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. Functional differentiation. Front Immunol (2014) 5:514. doi: 10.3389/fimmu.2014.00514
139. Zhang M, Ding G, Zhou L, Shen T, Xu X, Zhao T, et al. Interferon gamma inhibits CXCL8-induced proliferation and migration of pancreatic cancer bxPC-3 cell line via a rhoGDI2/rac1/NF-κB signaling pathway. J Interferon Cytokine Res (2018) 38(9):413–22. doi: 10.1089/jir.2018.0070
140. Li N, Li Y, Li Z, Huang C, Yang Y, Lang M, et al. Hypoxia inducible factor 1 (HIF-1) recruits macrophage to activate pancreatic stellate cells in pancreatic ductal adenocarcinoma. Int J Mol Sci (2016) 17(6):799. doi: 10.3390/ijms17060799
141. Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences1. J Immunol (2005) 175(1):342–9. doi: 10.4049/jimmunol.175.1.342
142. Liou G-Y, Bastea L, Fleming A, Döppler H, Edenfield BH, Dawson DW, et al. The presence of interleukin-13 at pancreatic ADM/panIN lesions alters macrophage populations and mediates pancreatic tumorigenesis. Cell Rep (2017) 19(7):1322–33. doi: 10.1016/j.celrep.2017.04.052
143. Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol (2016) 18(5):549–60. doi: 10.1038/ncb3340
144. Quail D, Joyce J. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi: 10.1038/nm.3394
145. Yu M, Guan R, Hong W, Zhou Y, Lin Y, Jin H, et al. Prognostic value of tumor-associated macrophages in pancreatic cancer: a meta-analysis. Cancer Manage Res (2019) 11:4041–58. doi: 10.2147/CMAR.S196951
146. Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut (2018) 67(6):1112–23. doi: 10.1136/gutjnl-2017-313738
147. Wengner AM, Pitchford SC, Furze RC, Rankin SM. The coordinated action of G-CSF and ELR + CXC chemokines in neutrophil mobilization during acute inflammation. Blood (2008) 111(1):42–9. doi: 10.1182/blood-2007-07-099648
148. Wu L, Awaji M, Saxena S, Varney ML, Sharma B, Singh RK. IL-17–CXC chemokine receptor 2 axis facilitates breast cancer progression by up-regulating neutrophil recruitment. Am J Pathol (2020) 190(1):222–33. doi: 10.1016/j.ajpath.2019.09.016
149. Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest (2010) 120(4):1151–64. doi: 10.1172/JCI37223
150. Andzinski L, Kasnitz N, Stahnke S, Wu C-F, Gereke M, von Köckritz-Blickwede M, et al. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer (2016) 138(8):1982–93. doi: 10.1002/ijc.29945
151. Wang S, Zheng Y, Yang F, Zhu L, Zhu X-Q, Wang Z-F, et al. The molecular biology of pancreatic adenocarcinoma: translational challenges and clinical perspectives. Signal Transduction Targeted Ther (2021) 6(1):1–23. doi: 10.1038/s41392-021-00659-4
152. Hosen SMZ, Uddin MdN, Xu Z, Buckley BJ, Perera C, Pang TCY, et al. Metastatic phenotype and immunosuppressive tumour microenvironment in pancreatic ductal adenocarcinoma: Key role of the urokinase plasminogen activator (PLAU). Front Immunol (2022) 13:1060957. doi: 10.3389/fimmu.2022.1060957
153. Zhang Yu, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, et al. Interleukin-17–induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med (2020) 217(12):e20190354. doi: 10.1084/jem.20190354
154. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science (2018) 361(6409):eaao4227. doi: 10.1126/science.aao4227
155. Teijeira Á., Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity (2020) 52(5):856–871.e8. doi: 10.1016/j.immuni.2020.03.001
156. Mota Reyes C, Demir E, Çifcibaşı K, Istvanffy R, Friess H, Demir IE. Regulatory T cells in pancreatic cancer: of mice and men. Cancers (2022) 14(19):4582. doi: 10.3390/cancers14194582
157. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci (2019) 110(7):2080–9. doi: 10.1111/cas.14069
158. Zhang Y, Lazarus J, Steele NG, Yan W, Lee H-J, Nwosu ZC, et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discovery (2020) 10(3):422–39. doi: 10.1158/2159-8290.CD-19-0958
159. Liu X, Xu J, Zhang B, Liu J, Liang C, Meng Q, et al. The reciprocal regulation between host tissue and immune cells in pancreatic ductal adenocarcinoma: new insights and therapeutic implications. Mol Cancer (2019) 18(1):184. doi: 10.1186/s12943-019-1117-9
160. Principe DR, Park A, Dorman MJ, Kumar S, Viswakarma N, Rubin J, et al. TGFβ Blockade augments PD-1 inhibition to promote T-cell–mediated regression of pancreatic cancer. Mol Cancer Ther (2019) 18(3):613–20. doi: 10.1158/1535-7163.MCT-18-0850
161. Fujimura T, Ring S, Umansky V, Mahnke K, Enk AH. Regulatory T cells stimulate B7-H1 expression in myeloid-derived suppressor cells in ret melanomas. J Invest Dermatol (2012) 132(4):1239–46. doi: 10.1038/jid.2011.416
162. Siret C, Collignon A, Silvy F, Robert S, Cheyrol T, André P, et al. Deciphering the crosstalk between myeloid-derived suppressor cells and regulatory T cells in pancreatic ductal adenocarcinoma. Front Immunol (2020) 10:3070. doi: 10.3389/fimmu.2019.03070
163. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci United States America (1993) 90(2):720–4. doi: 10.1073/pnas.90.2.720
164. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
165. DeSelm C, Tano ZE, Varghese AM, Adusumilli PS. CAR T-cell therapy for pancreatic cancer. J Surg Oncol (2017) 116(1):63–74. doi: 10.1002/jso.24627
166. Beatty GL, O’Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology (2018) 155(1):29–32. doi: 10.1053/j.gastro.2018.03.029
167. Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Trans Med (2014) 6(261):261ra151–261ra151. doi: 10.1126/scitranslmed.3010162
168. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer (2019) 120(1):26–37. doi: 10.1038/s41416-018-0325-1
169. Castillo J, Bernard V, San Lucas FA, Allenson K, Capello M, Kim DU. Surfaceome profiling enables isolation of cancer-specific exosomal cargo in liquid biopsies from pancreatic cancer patients. Ann Oncol (2018) 29(1):223–9. doi: 10.1093/annonc/mdx542
170. Cioffi M, Dorado J, Baeuerle PA, Heeschen C. EpCAM/CD3-Bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin Cancer Research: Off J Am Assoc Cancer Res (2012) 18(2):465–74. doi: 10.1158/1078-0432.CCR-11-1270
171. Kebenko M, Goebeler M-E, Wolf M, Hasenburg A, Seggewiss-Bernhardt R, Ritter B, et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE®) antibody construct, in patients with refractory solid tumors. OncoImmunology (2018) 7(8):e1450710. doi: 10.1080/2162402X.2018.1450710
172. Xu G, Luo Y, Wang H, Wang Y, Liu B, Wei J. Therapeutic bispecific antibodies against intracellular tumor antigens. Cancer Lett (2022) 5380:215699. doi: 10.1016/j.canlet.2022.215699
173. Kundranda M, Gracian AC, Zafar SF, Meiri E, Bendell J, Algül H, et al. Randomized, double-blind, placebo-controlled phase II study of istiratumab (MM-141) plus nab-paclitaxel and gemcitabine versus nab-paclitaxel and gemcitabine in front-line metastatic pancreatic cancer (CARRIE). Ann Oncol (2020) 31(1):79–87. doi: 10.1016/j.annonc.2019.09.004
174. Liu S, Li F, Deng L, Ma Q, Lu W, Zhao Z, et al. Claudin18.2 bispecific T cell engager armed oncolytic virus enhances antitumor effects against pancreatic cancer. Mol Ther - Oncolytics (2023) 30:275–85. doi: 10.1016/j.omto.2023.08.011
175. Huang X, Zhang G, Tang T-Y, Gao X, Liang T-B. Personalized pancreatic cancer therapy: from the perspective of mRNA vaccine. Military Med Res (2022) 9(1):53. doi: 10.1186/s40779-022-00416-w
176. Middleton G, Silcocks P, Cox T, Valle J, Wadsley J, Propper D, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol (2014) 15(8):829–40. doi: 10.1016/S1470-2045(14)70236-0
177. Shin MD, Shukla S, Chung YH, Beiss V, Chan SK, Ortega-Rivera OA, et al. COVID-19 vaccine development and a potential nanomaterial path forward. Nat Nanotechnology (2020) 15(8):646–55. doi: 10.1038/s41565-020-0737-y
178. Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature (2023) 618(7963):144–50. doi: 10.1038/s41586-023-06063-y
179. Hotz C, Wagenaar TR, Gieseke F, Bangari DS, Callahan M, Cao H, et al. Local delivery of mRNA-encoded cytokines promotes antitumor immunity and tumor eradication across multiple preclinical tumor models. Sci Trans Med (2021) 13(610):eabc7804. doi: 10.1126/scitranslmed.abc7804
180. Huff AL, Jaffee EM, Zaidi N. Messenger RNA vaccines for cancer immunotherapy: progress promotes promise. J Clin Invest (2022) 132(6):e156211. doi: 10.1172/JCI156211
181. Lambert JM, Berkenblit A. Antibody–drug conjugates for cancer treatment. Annu Rev Med (2018) 69(1):191–207. doi: 10.1146/annurev-med-061516-121357
182. Senter PD, Sievers EL. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat Biotechnol (2012) 30(7):631–7. doi: 10.1038/nbt.2289
183. Lambert JM, Chari RVJ. Ado-trastuzumab emtansine (T-DM1): an antibody–drug conjugate (ADC) for HER2-positive breast cancer. J Medicinal Chem (2014) 57(16):6949–64. doi: 10.1021/jm500766w
184. Tian B, Wong WY, Hegmann E, Gaspar K, Kumar P, Chao H. Production and characterization of a camelid single domain antibody–urease enzyme conjugate for the treatment of cancer. Bioconjugate Chem (2015) 26(6):1144–55. doi: 10.1021/acs.bioconjchem.5b00237
185. Bourillon L, Bourgier C, Gaborit N, Garambois V, Llès E, Zampieri A, et al. An auristatin-based antibody-drug conjugate targeting HER3 enhances the radiation response in pancreatic cancer. Int J Cancer (2019) 145(7):1838–51. doi: 10.1002/ijc.32273
186. Nishigaki T, Takahashi T, Serada S, Fujimoto M, Ohkawara T, Hara H, et al. Anti-glypican-1 antibody–drug conjugate is a potential therapy against pancreatic cancer. Br J Cancer (2020) 122(9):1333–41. doi: 10.1038/s41416-020-0781-2
187. Spisek R. 2P SOT102, a novel CLDN18.2-targeting antibody-drug conjugate for gastric and pancreatic cancer with a wide range of the tumor target expression. ESMO Open (2023) 8(1). doi: 10.1016/j.esmoop.2023.101196
188. Chokshi S, Hochster HS. An open-label, phase II study of intravenous anetumab ravtansine, an anti-mesothelin antibody drug conjugate, in pretreated mesothelin-expressing advanced pancreatic cancer. J Clin Oncol (2018) 36(4_suppl):TPS540–0. doi: 10.1200/JCO.2018.36.4_suppl.TPS540
189. Sorbara M, Cordelier P, Bery N. Antibody-based approaches to target pancreatic tumours. Antibodies (2022) 11(3):47. doi: 10.3390/antib11030047
190. Spiliopoulou P, Kasi A, Abushahin LI, Cardin DB, Lenz H-J, Dayyani F, et al. Phase Ib study of anetumab ravtansive in combination with immunotherapy or immunotherapy plus chemotherapy in mesothelin-enriched advanced pancreatic adenocarcinoma: NCI10208. J Clin Oncol (2022) 40(16_suppl):4136–6. doi: 10.1200/JCO.2022.40.16_suppl.4136
191. Meng Q, Valentini D, Rao M, Moro CF, Paraschoudi G, Jäger E, et al. Neoepitope targets of tumour-infiltrating lymphocytes from patients with pancreatic cancer. Br J Cancer (2019) 120(1):97–108. doi: 10.1038/s41416-018-0262-z
192. Leidner R, Silva NS, Huang H, Sprott D, Zheng C, Shih Y-P, et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. New Engl J Med (2022) 386(22):2112–9. doi: 10.1056/NEJMoa2119662
193. Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res (2016) 22(5):1048–54. doi: 10.1158/1078-0432.CCR-15-2667
194. Moaven O, Mangieri CW, Stauffer JA, Anastasiadis PZ, Borad MJ. Evolving role of oncolytic virotherapy: challenges and prospects in clinical practice. JCO Precis Oncol (2021) 5):432–41. doi: 10.1200/PO.20.00395
195. Jung B-K, Ko HY, Kang H, Hong J, Ahn HM, Na Y, et al. Relaxin-expressing oncolytic adenovirus induces remodeling of physical and immunological aspects of cold tumor to potentiate PD-1 blockade. J Immunotherapy Cancer (2020) 8(2):e000763. doi: 10.1136/jitc-2020-000763
196. Bathgate R, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev (2013) 93(1):405–80. doi: 10.1152/physrev.00001.2012
197. Mardhian DF, Storm G, Bansal R, Prakesh J. Nano-targeted relaxin impairs fibrosis and tumor growth in pancreatic cancer and improves the efficacy of gemcitabine in vivo. J Controlled Release (2018) 290:1–10. doi: 10.1016/j.jconrel.2018.09.031
198. Mahalingam D, Wilkinson GA, Eng KH, Fields P, Raber P, Moseley JL, et al. Pembrolizumab in combination with the oncolytic virus pelareorep and chemotherapy in patients with advanced pancreatic adenocarcinoma: A phase ib study. Clin Cancer Res (2020) 26(1):71–81. doi: 10.1158/1078-0432.CCR-19-2078
Keywords: pancreatic adenocarcinoma, tumor microenvironment, PDAC TME, tumor immune microenvironment, immune therapeutics, cancer immunology
Citation: Hartupee C, Nagalo BM, Chabu CY, Tesfay MZ, Coleman-Barnett J, West JT and Moaven O (2024) Pancreatic cancer tumor microenvironment is a major therapeutic barrier and target. Front. Immunol. 15:1287459. doi: 10.3389/fimmu.2024.1287459
Received: 01 September 2023; Accepted: 04 January 2024;
Published: 01 February 2024.
Edited by:
Husain Yar Khan, Wayne State University, United StatesReviewed by:
Sushil Kumar, Oregon Health and Science University, United StatesCopyright © 2024 Hartupee, Nagalo, Chabu, Tesfay, Coleman-Barnett, West and Moaven. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Omeed Moaven, b21vYXZlQGxzdWhzYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.