 Xiaoting Zhang
Xiaoting Zhang Jinhai Liu
Jinhai Liu Xiaoming Deng
Xiaoming Deng- Faculty of Anesthesiology, Changhai Hospital, Naval Medical University, Shanghai, China
Coronavirus disease 2019 (COVID-19) is an infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Due to its high infectivity, the pandemic has rapidly spread and become a global health crisis. Emerging evidence indicates that endothelial dysfunction may play a central role in the multiorgan injuries associated with COVID-19. Therefore, there is an urgent need to discover and validate novel therapeutic strategies targeting endothelial cells. PIEZO1, a mechanosensitive (MS) ion channel highly expressed in the blood vessels of various tissues, has garnered increasing attention for its potential involvement in the regulation of inflammation, thrombosis, and endothelial integrity. This review aims to provide a novel perspective on the potential role of PIEZO1 as a promising target for mitigating COVID-19-associated endothelial dysfunction.
1 Introduction
Coronavirus disease 2019 (COVID-19) has become a major global health concern caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which primarily spreads through respiratory droplets (1). With the emergence of highly transmissible variants, the COVID-19 pandemic has caused a significant healthcare burden worldwide, affecting millions of people. SARS-CoV-2 infection can lead to various clinical manifestations, ranging from asymptomatic cases to life-threatening illnesses. In addition to respiratory symptoms, a variety of extrapulmonary manifestations have also been reported (2). Recent studies suggest that endothelial dysfunction and immunothrombosis are key pathogenic mechanisms in COVID-19 (3, 4). However, effective therapeutic management of COVID-19 patients remains challenging. Understanding the pathogenesis and pathophysiological mechanisms underlying COVID-19-associated endothelial dysfunction is crucial for identifying potential therapeutic targets.
Piezo1, a member of the Piezo ion channel family, is expressed in vascular endothelial cells where it senses mechanical stimuli induced by blood flow and blood pressure. Over the past decade, Piezo1 has been shown to function as both a physiological regulator and a pathogenic factor in endothelial cells by modulating endothelial functions and barrier integrity (5–9). Interventions targeting Piezo1 in endothelial cells have demonstrated its role in various diseases, including ventilator-induced lung injury, hypertension, atherosclerosis, and thrombosis. Human PIEZO1 and PIEZO2 genes are located in the 16q24.3 region of chromosome 16 and the 18p11.22-p11.21 region of chromosome 18, respectively (10). Notably, Sukumar et al. (11) found that single nucleotide polymorphisms (SNPs) within the PIEZO1 gene are more prevalent in SARS-CoV-2 infected individuals. This suggests that PIEZO1 merits further study as a potential therapeutic target for COVID-19. In this review, we discuss how SARS-CoV-2 infection damages the endothelium and propose that PIEZO1 dysregulation could contribute to COVID-19-associated endothelial dysfunction, highlighting it as a promising therapeutic candidate.
2 COVID-19-associated endothelial dysfunctions
The ongoing battle against SARS-CoV-2 necessitates a deeper understanding of the intricate interplay between the virus and the host immune system. Following infection, the host immune response is activated, leading to the release of inflammatory signaling molecules and the recruitment of T lymphocytes, monocytes, and neutrophils (12, 13). This sustained immune response, coupled with epithelial injury, disruption of the endothelial barrier, and activation of coagulation, collectively contribute to diverse forms of tissue damage (14–16). Notably, clinicians are increasingly concerned about the emergence of cardiovascular complications in COVID-19 patients (17–20). Mounting evidence indicates that hemodynamic dysfunction, endothelitis, vascular leakage, and thrombosis play pivotal roles in the multi-organ damage observed in organs such as the lungs, heart, kidneys, and intestines (21). Consequently, resolving COVID-19-associated endothelial dysfunction has become an urgent priority.
2.1 COVID-19-associated hemodynamics dysfunction
Angiotensin-converting enzyme 2 (ACE2) is a surface membrane protein that is expressed in multiple tissues (22). It is widely known as an important enzyme in the cardiovascular system, which converts angiotensin II (Ang II) into angiotensin (1−7), and negatively regulates the renin-angiotensin system. ACE2 and angiotensin (1−7) are essential for endothelial cell function as they inhibit the inflammatory response (23, 24). Angiotensin (1−7) also participates in the activation of endothelial nitric oxide synthase (eNOS), which is mainly responsible for nitric oxide (NO) production and plays an important role in vasodilation (25, 26). Therefore, loss of ACE2 can lead to NO imbalance and vascular dysfunction.
During COVID-19, ACE2 is the main entry receptor for SARS-CoV-2 (27, 28). The active site domains of ACE2 are exposed on the extracellular surface, and SARS-CoV-2 can thus bind through the spike (S) protein on its surface, following the lock and key mechanism. Upon binding, transmembrane protease serine 2 (TMPRSS2), a cellular protease, cleaves and primes the S protein to further mediate viral uptake (29, 30). The virus then starts self-replicating. However, high-affinity SARS-CoV-2 competes with Ang II to bind the ACE2 receptor, leading to the accumulation of Ang II and causing an imbalance in the Ang (1−7)/Ang II ratio (31).
The accumulation of Ang II, accompanied by SARS-CoV-2 infection, can activate a transmembrane metalloproteinase called ADAM17, which mediates the ectodomain shedding of ACE2 (32, 33). Elevated soluble ACE2 (sACE2) levels can be observed in the blood circulation of patients with COVID-19 (34). ADAM17 cleaves the extracellular juxtamembrane region of ACE2 and releases its catalytically active extracellular domain of ACE2 into the extracellular environment, which may ultimately facilitate viral entry (35, 36). Owing to the ubiquitous expression of ACE2, TMPRSS2, and ADAM17 in various tissues, SARS-CoV-2 infection leads to multi-organ injuries (37). Moreover, the accumulating Ang II binds to the angiotensin type 1 receptor (AT1 receptor) and induces the internalization and downregulation of ACE2 (38). With the replication of SARS-CoV-2, the expression of ACE2 on the cell surface is downregulated, which reduces its vasodilatory effect in the vascular system. Prolonged vasoconstriction results in increased endothelial dysfunction and inflammation, which leads to severe cardiovascular injury. In a recent study, it was found that a significant proportion of patients hospitalized due to COVID-19 exhibited the production of autoantibodies against Ang II. The presence of these autoantibodies was strongly correlated with poorer blood oxygenation, dysregulated blood pressure, and overall increased severity of the disease (39). Furthermore, some COVID-19 patients also developed autoantibodies against AT1 and ACE2, leading to heightened proinflammatory responses and greater disease severity (40, 41). These findings provide valuable insights into the quantification of autoantibodies targeting crucial molecules in the renin-angiotensin pathway, i.e., Ang II, AT1, and ACE2. Moreover, they highlight the significance of disturbances in vascular tone among COVID-19 patients.
2.2 COVID-19-associated endotheliitis and endothelial barrier breakdown
Endotheliitis and disruption of the endothelial barrier are crucial pathological mechanisms in COVID-19, contributing to increased vascular permeability and inflammation, which ultimately lead to multi-organ dysfunction (42). Electron microscopy observations have revealed that SARS-CoV-2 can directly infect endothelial cells (ECs), thereby potentially altering vascular homeostasis (21). In some COVID-19 patients, elevated levels of circulating ECs and soluble circulating endothelial derangement parameters have been detected, indicating EC apoptosis and breakdown of the endothelial barrier (43).
In severe cases of COVID-19, excessive production of pro-inflammatory cytokines, often referred to as a “cytokine storm,” contributes to the transition of the endothelial cell phenotype from a protective state to an inflammatory state. This transition is associated with increased vascular leakage, tissue damage, and immunothrombosis. As mentioned previously, hyperactivity of Ang II serves as a trigger for vasoconstriction and inflammation. Additionally, SARS-CoV-2 activates the NLRP3 inflammasome in monocytes, leading to the release of pro-inflammatory cytokines that further amplify endotheliitis (44). In severe cases, a delayed or impaired interferon type I response results in persistent viral load in the blood and exacerbates the inflammatory response (45).
SARS-CoV-2 infection can cause severe damage to the endothelial barrier. Activation of the NLRP3 inflammasome by SARS-CoV-2 increases the release of interleukin-1 beta (IL-1β), which suppresses cAMP formation and CREB-mediated transcription of VE-cadherin in ECs, consequently contributing to vascular leakage (46). Recent studies have shown that lactate induces vascular hyperpermeability by promoting cleavage and endocytosis of VE-cadherin through signaling via the lactate receptor GPR81 in endothelial cells (47). Lactate, as a major byproduct of glycolysis, has also been found to be elevated in severe COVID-19 patients (48, 49), indicating the involvement of metabolic changes in endothelial dysfunction. Furthermore, recruited neutrophils release excessive reactive oxygen species (ROS), which are exacerbated by reduced antioxidants due to viral infection, further intensifying endothelial injury (50).
2.3 COVID-19-associated immunothrombosis
Under homeostatic conditions, the glycocalyx and anticoagulants produced by ECs play a crucial role in preventing microvascular thrombosis and maintaining normal blood flow (51). However, SARS-CoV-2 infection disrupts vascular integrity, exposing thrombogenic basement membranes (52). Consequently, the activation of the coagulation cascade contributes to the development of immunothrombosis (53, 54).
Vascular damage and the inflammatory environment activate the tissue factor (TF) and extrinsic pathways. SARS-CoV-2 also triggers complement activation and elevates plasma TF levels (55–57). Neutrophil extracellular traps (NETs) are recognized as important mediators of tissue damage. The concentration of NETs is increased in samples from COVID-19 patients, including plasma, tracheal aspirate, and lung autopsy tissues. Neutrophils from COVID-19 patients release excessive NETs in vitro, and plasma from these patients triggers NET formation (58, 59). The enhanced release of NETs can activate the intrinsic coagulation pathway via factor XII. Neutrophil elastase and myeloperoxidase present in NETs cleave and inactivate natural anticoagulants, such as tissue factor pathway inhibitor and thrombomodulin (60, 61). Moreover, NETs promote platelet adhesion and activation (62). Complement cascade activation induces NETosis and platelet activation (63). Consequently, activated platelets, distorted neutrophils, aggregated NETs, and fibrin strands contribute to vascular occlusions. In response, fibrinolysis is activated, resulting in elevated levels of fibrin breakdown products.
Endothelial dysfunction is a prominent feature during the progression of COVID-19. This dysfunction leads to vasoconstriction, breakdown of the endothelial barrier, and a procoagulant state (Figure 1). Systemic impairment of microcirculatory function poses a significant threat to COVID-19 patients. Therefore, it is crucial to develop strategies that target endothelial cells, especially for patients with pre-existing endothelial conditions (e.g., smoking, hypertension, and diabetes), who are at a higher risk of mortality.

Figure 1 COVID-19-associated endothelial dysfunction.
(1) ACE2, the primary receptor for SARS-CoV-2, is a critical enzyme in the cardiovascular system responsible for the conversion of Ang II into angiotensin (1−7). In the context of COVID-19 infection, internalization and downregulation of ACE2 result in the accumulation of Ang II, thereby potentially contributing to the mediation of vasoconstriction and hemodynamic dysfunction. (2) SARS-CoV-2 infection triggers an immune response, characterized by the secretion of cytokines from recruited monocytes and neutrophils, subsequently leading to the induction of endotheliitis. The escalated oxidative stress, cytokine storm, and leukocyte adhesion to the endothelium contribute to severe endothelial barrier disruption. (3) Vascular damage and inflammatory states initiate the activation of tissue factor (TF) and the extrinsic coagulation pathway. The amplified release of neutrophil extracellular traps (NETs) can cleave and inactivate endogenous anticoagulants. Moreover, NETs play a pivotal role in facilitating platelet adhesion and activation. The aggregation of NETs, fibrin strands, deformed neutrophils, and activated platelets synergistically contribute to the genesis of vascular occlusions.
3 Mechanosensitive Piezo ion channels
Piezos, encompassing Piezo1 and Piezo2, are expressed in various tissues. Both channels are expressed on the cellular membrane and are encoded by the Piezo1 gene (family with sequence similarity 38A, Fam38A) and the Piezo2 gene (family with sequence similarity 38B, Fam38B), respectively (64). Mechanosensitive Piezo ion channels were initially identified as fundamental components of distinct mechanically activated cation channels in 2010 (65). The groundbreaking discoveries made by Ardem Patapoutian’s research group marked a significant milestone in this field, unveiling a realm of limitless possibilities. Subsequent extensive investigations have revealed an expanding repertoire of functions that extend beyond the realm of mechanotransduction processes. In recognition of his pioneering work in identifying these force receptors, Ardem Patapoutian was awarded the Nobel Prize in Physiology or Medicine in 2021.
3.1 Structure of Piezo1 ion channel
The core architecture of mouse Piezo1 consists of a homotrimeric complex exhibiting a propeller-like shape with a central cap, three peripheral blade-like structures, and three beams connecting the blades to the cap (66, 67). Additionally, the high-resolution three-dimensional cryo-electron microscopy revealed that each subunit of the Piezo1 homotrimeric complex encompasses 38 transmembrane (TM) segments, collectively summing up to 114 TM segments. In 2022, the mechanism of Piezo1 structural deformation in response to mechanical force within lipid bilayers was elucidated (68). The TM regions observed within the three-bladed structure of Piezo channels are unusually curved and shape a distinctive nano-bowl configuration. The researchers postulated a hypothesis that the application of force on the cell membrane may induce a flattening of the paddles, consequently resulting in an expansion of the membrane area. The findings presented in this study provide a comprehensive understanding of the intricate mechanistic pathway through which Piezo1 converts physical mechanical stimuli into bioelectric signals, consequently offering valuable insights into the mechanotransduction process.
3.2 Expression and functions of Piezo1 ion channel
Piezo1 represents a pivotal mechanically-activated cation channel that senses mechanical stimuli and plays critical roles in diverse physiological functions. Mechanically activated Piezo1 converts the mechanical stretch experienced by cardiomyocytes into intracellular Ca2+ and reactive oxygen species (ROS) signaling, thereby playing a key role in heart mechano-chemo transduction and the maintenance of normal cardiac function (69). In enterochromaffin cells, Piezo1 detects single-stranded RNA derived from the intestinal microbiota and facilitates serotonin production (70), which is relevant to bone and gut disorders. Moreover, Piezo1 is essential for vascular development, lymphatic development, red blood cell volume regulation, blood pressure control, arterial remodeling, iron metabolism, bone formation, and tissue inflammation (5, 71–75). Consequently, Piezo1 emerges as a potential therapeutic target for numerous diseases.
3.3 Unique features of Piezo1 compared to other MS channels
Several MS channels have been identified, including PIEZO, transient receptor potential (TRP) channels, epithelial sodium channels (ENaC), K2P channels, and others (76). Each of these channels has unique structures and mechanotransduction mechanisms that align with their specific biological functions. In comparison to other MS ion channels, Piezo1 exhibits unique properties and activation characteristics. As described above, Piezo1 exhibits activation in response to various mechanical stimuli, including external poking, stretching, shear stress, local membrane tension, and intracellular forces mediated by myosin-II traction (68). Transient receptor potential vanilloid 4 (TRPV4) can be activated by osmotic changes, membrane stretch, chemical ligands, and temperature changes (77–79). K2P channels respond to a range of stimuli, including protons, heat, stretch, diverse lipids, and pharmacological substances such as general anesthetics (80, 81). Among these MS channels, Piezo protein family has recently been established as the first bona fide mechano-gated cation channels in mammals, a groundbreaking discovery that has long been sought after (64). Piezo1 is characterized by its low-threshold, small-conductance, fast inactivation, and depolarization upon mechanical activation. It acts as a non-selective cationic channel that allows the permeation of sodium, potassium, and calcium ions (68, 82, 83). The activation of Piezo1 is notable for its exceptional mechanosensitivity, with a pressure of approximately -30 mmHg required for half-maximal activation and a lateral membrane tension of around 1.4 mN/m (64, 84). These values highlight the sensitivity of Piezo1 to mechanical stimuli. The unitary conductance, which measures the ability of a single opened channel to allow ion movement, is estimated to be about 20 pS for Piezo1 and 30 pS for Piezo2 (68, 85). As a mechanosensitive cation channel, Piezo1 is mainly permeable to calcium ions. Calcium plays a crucial role as a second messenger in numerous endothelial cell activities. Changes in intracellular calcium ion levels and ensuing signaling cascades are responsible for these processes. Under normal conditions, quiescent endothelial cells maintain low intracellular cytosolic free calcium concentrations. However, the initiation of calcium signaling can be induced by proinflammatory mediators, and elevated intracellular calcium concentrations can trigger diverse signaling pathways in both physiological and pathological conditions (68, 86). Upon opening, Piezo1 channels facilitate the entry of calcium ions, subsequently initiating downstream signaling pathways that are crucial for endothelial function and vascular homeostasis. Furthermore, studies have revealed that purified Piezo1 exhibits activation in an asymmetric bilayer but not in a symmetric bilayer, indicating its intrinsic sensitivity to membrane curvature (87). The distinctions in conductance and mechanical activation thresholds between Piezo1 and other mechanosensitive channels emphasize their unique contributions to physiological function.
3.4 Pharmacological modulators of Piezo1
Through high-throughput screening assays, specific agonists of Piezo1, such as Yoda1 and Jedi1/2, have been identified (88, 89). Jedi1/2 exhibit superior water solubility compared to Yoda1. Co-administration of Jedi1 and Yoda1 synergistically potentiate the Piezo1 poking currents, suggesting distinct mechanisms underlying their activation of Piezo1. However, currently known inhibitors of Piezo1 lack specificity, including ruthenium red (RR), gadolinium (Gd3+), and the peptide toxin GsMTx-4 (90). Given the elucidation of the molecular structure of Piezo1, we anticipate the emergence of additional ligand candidates targeting Piezo1 in the near future.
4 Potential roles of endothelial PIEZO1 in COVID-19
Endothelial cells serve as a critical interface between the circulatory system and surrounding tissues, playing a multitude of functions in physiological homeostasis and pathological conditions. These roles include regulating vascular permeability, modulating inflammatory responses, maintaining coagulation balance, and promoting angiogenesis (6, 91). The ability of endothelial cells to sense and respond to mechanical stimuli is crucial for maintaining vascular homeostasis. It is well-established that MS channels play a pivotal role in converting mechanical forces into electrochemical signals. Specifically, MS channels in endothelial cells are involved in various events such as vasodilation, vascular inflammation, and vascular permeability. Endothelial PIEZO1 holds promising potential in the treatment of COVID-19, particularly in maintaining systemic hemodynamic stability, endothelial barrier integrity, and muscle capillary density; however, it may also contribute to a procoagulant tendency under certain circumstances (Figure 2).
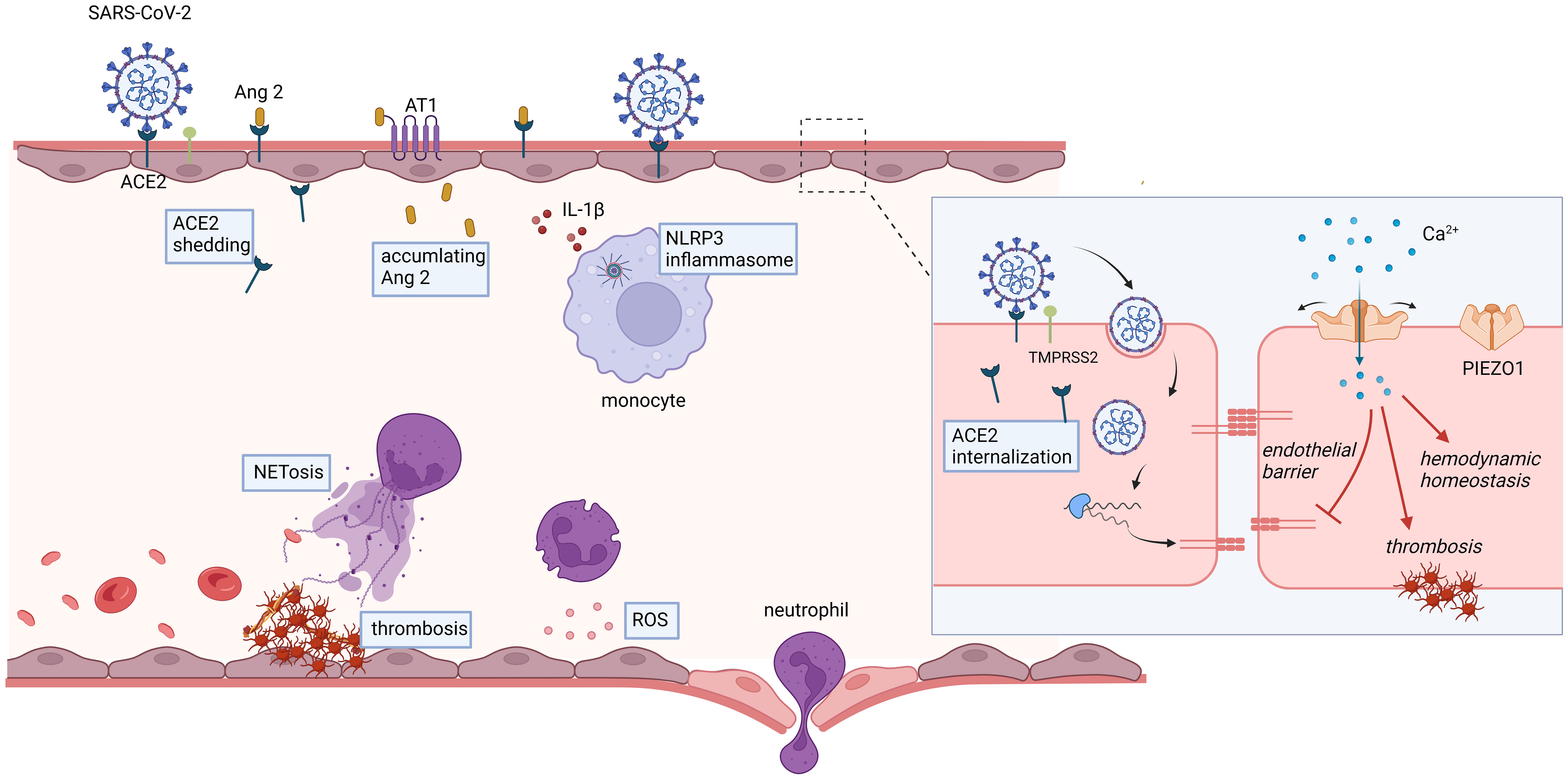
Figure 2 Potential roles of endothelial PIEZO1 in COVID-19.
4.1 Potential links between PIEZO1 and SARS-CoV-2 induced calcium disturbance
SARS-CoV-2 has been elucidated to interact with calcium channels, potentially leading to profound physiological perturbations (92, 93). The spike protein of SARS-CoV-2 features a critical region known as the spike protein receptor-binding domain (S-RBD), which exhibits a high affinity for human ACE-2 receptors and is crucial for viral entry (94, 95). S-RBD has been implicated in inducing an acute to prolonged increase in intracellular calcium concentration in human pulmonary arterial endothelial cells, which is associated with the activation and expression of Piezo1 and store-operated calcium channels (SOCC) (96). The SARS-CoV-2 induced persistent perturbation of intracellular calcium homeostasis, driven by the upregulation of Piezo1 and other calcium channels, may lead to elevated apoptosis and impairment of pulmonary vascular endothelial cells, contributing to the vascular complications observed in severe COVID-19 cases.
Additionally, the S-RBD disrupts intracellular calcium homeostasis and triggers cell apoptosis by engaging with the ACE2 receptor and formation of ACE2-bounded calcium channel clusters (96). The formation of clusters involving ACE2, Piezo1, and SOCC, induced by the S-RBD, suggests a complex interplay between the virus and the host’s calcium signaling system. These clusters enable the opening of Piezo1 and SOCC, and their formation is dependent on the S-RBD-ACE2 interaction and the presence of intracellular calcium. The use of KobA, which blocks the interaction between ACE2 and S-RBD, has been shown to significantly reduce the S-RBD-induced upregulation and activation of Piezo1, as well as baseline calcium levels. This indicates that the S-RBD’s interaction with ACE2 disrupts intracellular calcium homeostasis, leading to cell apoptosis and the formation of calcium channel clusters, which are critical for the virus’s pathogenic effects.
Collectively, the potential link between Piezo1 and SARS-CoV-2 induced calcium disturbance highlights the intricate relationship between viral mechanisms and host cellular responses. Although there is currently no direct evidence supporting Piezo1 as a receptor for SARS-CoV-2 infection, it is worth considering Piezo1 as a promising target to modulate calcium disturbance in COVID-19 associated endothelial dysfunction.
4.2 Potential role of PIEZO1 in maintaining hemodynamic homeostasis
The endothelium is consistently exposed to shear stress induced by blood flow and cyclic pressure, which are essential for sustaining hemodynamic stability. As a Ca2+-permeable non-selective cation channel, Piezo1 functions as a vital sensor of blood flow. Patapoutian et al. demonstrated that Piezos mediate neuronal sensing of blood pressure and the baroreceptor reflex (97). Additionally, Piezo1 serves as a key regulator of flow-induced ATP release and shear stress-induced NO production (98). Mice with inducible, endothelium-specific Piezo1 deficiency exhibited reduced endothelial nitric oxide synthase (eNOS) activity, resulting in impaired NO synthesis and the development of arterial hypertension. NO, as an essential vascular vasodilator, also possesses potent anti-inflammatory, anti-apoptotic, and anti-thrombotic properties. Clinical reports have indicated the potential benefits of inhaled NO in the treatment of COVID-19 (99, 100). Collectively, these studies suggest that PIEZO1 could represent a prospective target for regulating and preserving normal hemodynamics in COVID-19 by maintaining hemodynamic homeostasis.
4.3 Potential role of PIEZO1 in the maintenance of endothelial barrier
An autopsy study published in The New England Journal of Medicine in 2020 revealed three distinct pulmonary vascular features in patients with COVID-19, i.e., severe endothelial injury, widespread vascular thrombosis, and augmented intussusceptive angiogenesis in the lungs (101). Given its high expression in the lungs, Piezo1 might play an essential role in regulating inflammation and maintaining pulmonary endothelial integrity. A recent study demonstrated that endothelial Piezo1 promotes angiogenesis and facilitates bone fracture repair (102), suggesting its potential involvement in angiogenesis and vascular remodeling during early SARS-CoV-2 infection in the lungs.
Extravasation of leukocytes is a pivotal hallmark of the inflammatory response. A previous study has demonstrated that during the process of leukocyte transendothelial migration, blood flow shear stress intervenes, leading to an increase in membrane tension in vascular endothelial cells. This mechanical force is detected by Piezo1 ion channels, subsequently initiating intracellular calcium elevation and activating endothelial cells. Consequently, endothelial cell contraction ensues, resulting in barrier opening and ultimately facilitating leukocyte transmigration. Thus, Piezo1 functions as a critical mediator whereby vascular endothelial cells orchestrate leukocyte transmigration and contribute to the progression of vascular inflammation (103).
A recent preclinical study provides compelling evidence for the first time that a single exposure to the spike protein or receptor-binding domain of SARS-CoV-2 is sufficient to induce acute-to-prolonged damage to pulmonary vascular endothelium. This damage occurs through the upregulation and activation of Piezo1 and store-operated calcium channels, leading to increased intracellular calcium concentrations. Pharmacological inhibition of Piezo1 using GsMTx4 effectively prevents disruption of intracellular calcium homeostasis in endothelial cells. These findings strongly suggest that targeting Piezo1 could be a potential therapeutic strategy for mitigating S-RBD-induced pulmonary vascular damage, offering novel insights into the management of COVID-associated pulmonary vascular diseases and long-term complications in individuals with long COVID (96). It has been also reported to enhance lung endothelial barrier function and mitigate ventilator-induced lung injury by suppressing Src-induced VE-cadherin phosphorylation (7). Additionally, Piezo1-mediated mechanosensation is critical for innate immunity (5), as infiltrating monocytes recognize cyclic hydrostatic pressure in the lungs through Piezo1, triggering neutrophil-mediated bacterial clearance in mice. These findings collectively suggest that targeting PIEZO1 could represent a promising therapeutic approach for mitigating pulmonary vascular damage in COVID-19.
4.4 Potential role of PIEZO1 in the regulation of thrombosis
Piezo1 dysregulation has recently been identified in patients with type 2 diabetes mellitus (T2DM) through perturbational screening (9). Notably, increased Piezo1 activity in platelets, red blood cells, and neutrophils in T2DM leads to a prothrombotic cellular response. Inhibiting Piezo1 may offer a potential strategy for mitigating thrombosis, particularly in the context of hyperglycemia. Another study revealed that activated Piezo1 in platelets of hypertensive mice resulted in significant platelet activation, whereas Piezo1 inhibition improved mitochondrial dysfunction and platelet apoptosis (104). Patients with severe COVID-19 exhibit elevated markers of platelet apoptosis, including heightened depolarization of mitochondrial inner transmembrane potential, cytosolic Ca2+ concentration, and phosphatidylserine externalization (105). Targeting procoagulant platelets and platelet apoptosis may hold therapeutic significance for the treatment of COVID-19. Overall, PIEZO1 exhibits important potential in regulating COVID-19-associated thrombosis.
4.5 Potential role of PIEZO1 in the regulation of ventilator-induced lung injury
Ventilator-induced lung injury (VILI) is a critical concern in the field of intensive care, referring to the damage or worsening of lung function caused by mechanical ventilation. Mechanical ventilation is commonly required for patients with acute hypoxemic respiratory failure caused by SARS-CoV-2 infection. In severe cases of COVID-19 pneumonia, prolonged use of positive airway pressure (PAP) for more than 24 hours can lead to potential complications, including lung injury due to excessive tidal volume (106). Several mechanisms contribute to VILI, namely barotrauma, volutrauma, atelectrauma, and biotrauma (107). These mechanisms result in alveolar distention and injury, leading to increased alveolar permeability, edema in the alveoli and interstitium, alveolar hemorrhage, and the formation of hyaline membranes. These pathological changes collectively impair surfactant function and ultimately lead to alveolar collapse (108).
Mechanical ventilation applies cyclic and intermittent expansion and distension to the lungs, which in turn exerts mechanical forces on different lung cells via the mechanosensitive ion channel known as Piezo1. These stretching forces are also transmitted to endothelial cells, leading to increased tension on the underlying plasma membrane. Recent studies have primarily aimed to comprehend the involvement of Piezo1 in the progression of VILI. A study conducted by Malik et al. in 2019 found that long-term mechanical ventilation (38 h, 108 h, and 252 h) led to significantly reduced expression of Piezo1 in lung tissue compared to short-term ventilation (less than 1 h) (7). However, in a rat model, increased Piezo1 expression was observed in lung tissue following high-volume ventilation and cyclic stretch treatment. Suppression of Piezo1 activity using GSMTx4 alleviated VILI in rats, resulting in decreased edema, diminished protein leakage, attenuated systemic inflammation, and improved survival rates (109). In another rat model of acute respiratory distress syndrome (ARDS), high tidal volume ventilation during ARDS activated the Piezo1 channel and downstream calpain. Excessive mechanical stretch further exacerbated lung vascular hyperpermeability in ARDS rats, but pharmacological inhibition of calpain or Piezo1 knockdown prevented the disassembly of endothelial adherens junctions and improved endothelial barrier function (110).
Mechanical stretching during ventilation also affects pulmonary epithelial cells. In a murine model involving mechanical ventilation following acid aspiration-induced lung injury, Piezo1 played a pivotal role by mediating calcium influx and adenosine triphosphate (ATP) release in epithelial cells. Targeting epithelial Piezo1 may be a potential therapeutic strategy for preventing pulmonary fibrosis in ARDS patients undergoing mechanical ventilation (111). Furthermore, stretch-induced Piezo1 activation triggers the proteolytic activity of metalloproteinases ADAM10 and ADAM17 at the plasma membrane of primary human lung epithelial cells, resulting in the shedding of the epithelial junctional adhesion molecule-A (JAM-A) and affecting epithelial permeability (112).
Although there is limited research on MS ion channels in the context of COVID-19-associated endothelial dysfunction, a recent preclinical investigation has shown promising results regarding the pharmacological inhibition of Piezo1 using GsMTx4 (96). This inhibition helps maintain intracellular calcium homeostasis in endothelial cells and may hold potential for managing pulmonary vascular disorders associated with COVID-19 and long-term complications in individuals with long COVID. In short, mechanical ventilation can activate PIEZO1 in various lung cells, contributing to lung injury. It should be mentioned that these studies primarily focus on models with mechanical ventilation, highlighting the need for further investigations into lung injuries caused by COVID-19 or other diseases.
4.6 Potential role of PIEZO1 in the regulation of sarcopenia
Severe sarcopenia has been observed in certain patients with COVID-19. Contributing factors include immobilization, mechanical ventilation, systemic inflammation, malnutrition, and the targeting of ACE2 by SARS-CoV-2 in skeletal muscle (113, 114). As previously discussed, endothelial Piezo1 serves as a sensor for blood flow and activates eNOS to facilitate nitric oxide (NO) synthesis. Remarkably, NO suppresses thrombospondin-2, an inducer of endothelial cell apoptosis, which is predominantly expressed in muscle pericytes. Endothelial Piezo1 supports muscle capillary density and sustains physical activity through the endothelial cell-pericyte interaction within the muscle microenvironment (115). A recent study demonstrated that immobilization leads to reduced cytosolic calcium concentration in skeletal muscle cells, potentially due to Piezo1 downregulation. Acute disruption of Piezo1 promotes skeletal muscle atrophy involving KLF15 and IL-6 pathways (116). These findings indicate that activating endothelial PIEZO1 represents a promising therapeutic approach for COVID-19-associated sarcopenia.
4.7 Potential role of PIEZO1 in the regulation of long COVID
Long COVID is characterized by prolonged symptoms following COVID-19 and has become a significant global healthcare concern (117). The spectrum of symptoms observed in individuals with long COVID encompasses fatigue, brain fog, respiratory complications, and cardiovascular abnormalities (118). It is now widely recognized that the persistence of these symptoms is not solely attributable to viral persistence, but rather involves a complex interplay of factors including viral reservoir, systemic inflammation, endothelial dysfunction, and organ damage (119).
Endothelial dysfunction is one of the key features of long COVID and has been linked to thrombotic events and impaired oxygen delivery. The presence of microclots have been observed in both acute COVID-19 and long COVID (120, 121). Long-term alterations in the size and stiffness of blood cells have also been identified in individuals with long COVID, potentially leading to impaired oxygen delivery (122). In addition, patients with long COVID exhibited a persistent decrease in vascular density, particularly in small capillaries (123). Therefore, endotheliitis and coagulation have been recognized as two important targets for the multifaceted therapeutic approach to long COVID treatment (124, 125). Given the potential involvement of PIEZO1 in regulating vascular homeostasis, inflammation, endothelial barrier integrity, and thrombotic processes, targeting PIEZO1 may hold therapeutic promise for alleviating or preventing the symptoms associated with long COVID.
The endothelial PIEZO1 demonstrates significant therapeutic potential in the context of COVID-19 therapeutics, due to its crucial role in preserving systemic hemodynamic stability and ensuring the integrity of the endothelial barrier. Nevertheless, it may also elicit a procoagulant propensity under certain conditions.
5 Challenges in utilizing PIEZO1 as a therapeutic target for COVID-19
The role of PIEZO1 in endothelial dysfunction following SARS-CoV-2 infection is still not well understood, but its involvement in other inflammatory disorders suggests its potential as a target for clinical research. However, there is currently limited clinical evidence supporting PIEZO1 as a therapeutic target for COVID-19. In a study conducted in 2020 by Sukumar et al., the analysis of data from the UK Biobank revealed a significant association between COVID-19 fatality and three missense SNPs within the PIEZO1 gene, independent of established risk factors (11). It is worth noting that these SNPs affect amino acid residues in the unexplored proximal N-terminus region of the PIEZO1 protein. Furthermore, variations in the prevalence of these SNPs across different ethnic groups were observed through a comprehensive analysis of genomic sequences. These findings provide some evidence of PIEZO1’s potential contribution to COVID-19 fatality and its association with ethnic susceptibility.
Despite the promise of PIEZO1 as a therapeutic target for COVID-19, clinical research on its potential remains limited compared to basic scientific studies. Before considering its clinical application, several significant challenges need to be addressed. Firstly, the precise role of PIEZO1 in the pathogenesis of COVID-19 remains unclear, as the mechanisms underlying SARS-CoV-2-induced inflammation, thrombosis, and other complications are complex and multifaceted. Understanding the specific contributions of PIEZO1 in these processes is essential for targeted interventions. Additionally, careful examination of the off-target effects of PIEZO1 modulation is necessary. PIEZO1 is ubiquitously expressed in various tissues and cell types throughout the body, playing crucial roles in diverse physiological functions. Modulating PIEZO1 may inadvertently disrupt normal cellular processes, leading to unintended consequences. Developing safe and effective therapeutics that selectively target PIEZO1 poses another challenge. Small molecule inhibitors, gene silencing techniques, and other strategies must undergo rigorous testing to determine their specificity, bioavailability, and potential side effects.
6 Conclusion and prospective
In light of the ongoing presence and long-term impact of the COVID-19, it is crucial to prioritize reducing mortality and mitigating post-COVID syndrome. Recent evidence suggests that endothelial injury induced by SARS-CoV-2 infection plays a significant role in organ dysfunction. Therefore, it is imperative to comprehend the implications of COVID-19-associated endothelial dysfunction for the development of effective therapeutic strategies. Although investigations into PIEZO1 as a potential therapeutic target for COVID-19 remain limited, extensive research has elucidated the multifaceted biological functions of the PIEZO1 ion channel in vascular physiology. Notably, studies have shown a correlation between genetic variants of PIEZO1 and COVID-19 fatality. Preclinical investigations have also demonstrated that exposure to the S-RBD of SARS-CoV-2 can impair pulmonary vascular endothelium, activating Piezo1. Pharmacological inhibition of Piezo1 effectively prevents disruption of endothelial calcium homeostasis. Furthermore, basic research has revealed that regulating PIEZO1 can ameliorate endothelial dysfunction and improve VILI in animal models. These findings highlight the therapeutic potential of targeting PIEZO1 in mitigating COVID-19-associated endothelial dysfunction.
Future research should focus on unraveling the precise molecular mechanisms underlying PIEZO1-mediated endothelial dysfunction and identifying specific signaling pathways activated by PIEZO1. Additionally, exploring the safety and efficacy of PIEZO1 modulators through clinical trials will be essential for translating these findings into clinical practice. Further studies are warranted to validate endothelial PIEZO1 as a novel target for developing therapeutics against COVID-19. Additionally, investigating the contributions of other mechanosensitive calcium channels in COVID-19-associated endothelial dysfunction may provide further insights into the complex mechanotransduction pathways involved.
In summary, understanding the role of mechanosensitive ion channels, particularly PIEZO1, in COVID-19-associated endothelial dysfunction holds great promise for the development of novel therapeutic interventions aimed at restoring endothelial homeostasis and improving patient outcomes. Leveraging rational drug design and innovative endothelial-targeted nanoparticles, targeting PIEZO1 may offer a promising approach to promote vascular protection in the context of COVID-19.
Author contributions
XZ: Conceptualization, Writing – original draft. JL: Writing – review & editing. XD: Conceptualization, Supervision, Writing – review & editing. LB: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the Fund Project of Shanghai Science and Technology Committee Rising-Star Program (19QA1408500).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): A review. JAMA J Am Med Assoc. (2020) 324:782–93. doi: 10.1001/jama.2020.12839
2. Para K, Weiner E, Kupfer Y. A fatal extrapulmonary manifestation of COVID-19. Chest. (2020) 158:A929. doi: 10.1016/j.chest.2020.08.865
3. Liu N, Long H, Sun J, Li H, He Y, Wang Q, et al. New laboratory evidence for the association between endothelial dysfunction and COVID-19 disease progression. J Med Virol. (2022) 94:3112–20. doi: 10.1002/jmv.27693
4. Katsoularis I, Fonseca-Rodriguez O, Farrington P, Jerndal H, Lundevaller EH, Sund M, et al. Risks of deep vein thrombosis, pulmonary embolism, and bleeding after covid-19: nationwide self-controlled cases series and matched cohort study. Bmj. (2022) 377:e069590. doi: 10.1136/bmj-2021-069590
5. Solis AG, Bielecki P, Steach HR, Sharma L, Harman C, Yun S, et al. Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity. Nature. (2019) 573:69–74. doi: 10.1038/s41586-019-1485-8
6. Friedrich EE, Hong Z, Xiong S, Zhong M, Di A, Rehman J, et al. Endothelial cell Piezo1 mediates pressure-induced lung vascular hyperpermeability via disruption of adherens junctions. Proc Natl Acad Sci. (2019) 116:12980–85. doi: 10.1073/pnas.1902165116
7. Zhong M, Wu W, Kang H, Hong Z, Xiong S, Gao X, et al. Alveolar stretch activation of endothelial piezo1 protects adherens junctions and lung vascular barrier. Am J Respir Cell Mol Biol. (2020) 62:168–77. doi: 10.1165/rcmb.2019-0024OC
8. Geng J, Shi Y, Zhang J, Yang B, Wang P, Yuan W, et al. TLR4 signalling via Piezo1 engages and enhances the macrophage mediated host response during bacterial infection. Nat Commun. (2021) 12(1):3519. doi: 10.1038/s41467-021-23683-y
9. Zhu W, Guo S, Homilius M, Nsubuga C, Wright SH, Quan D, et al. PIEZO1 mediates a mechanothrombotic pathway in diabetes. Sci Transl Med. (2022) 14:k1707. doi: 10.1126/scitranslmed.abk1707
10. Qin L, He T, Chen S, Yang D, Yi W, Cao H, et al. Roles of mechanosensitive channel Piezo1/2 proteins in skeleton and other tissues. Bone Res. (2021) 9:44. doi: 10.1038/s41413-021-00168-8
11. Cheng CW, Deivasikamani V, Ludlow MJ, De Vecchis D, Kalli AC, Beech D, et al. Ethnically diverse mutations in PIEZO1 associate with SARS-CoV-2 positivity. medRxiv. (2020). doi: 10.1101/2020.06.01.20119651
12. Szabo PA, Dogra P, Gray JI, Wells SB, Connors TJ, Weisberg SP, et al. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity. (2021) 54:797–814. doi: 10.1016/j.immuni.2021.03.005
13. Meroni PL, Croci S, Lonati PA, Pregnolato F, Spaggiari L, Besutti G, et al. Complement activation predicts negative outcomes in COVID-19: The experience from Northen Italian patients. Autoimmun Rev. (2023) 22:103232. doi: 10.1016/j.autrev.2022.103232
14. Kumar A, Narayan RK, Prasoon P, Kumari C, Kaur G, Kumar S, et al. COVID-19 mechanisms in the human body-what we know so far. Front Immunol. (2021) 12:693938. doi: 10.3389/fimmu.2021.693938
15. Wan E, Zhang R, Mathur S, Yan V, Lai F, Chui C, et al. Post-acute sequelae of COVID-19 in older persons: multi-organ complications and mortality. J Travel Med. (2023) 30(5):taad082. doi: 10.1093/jtm/taad082
16. Nie X, Qian L, Sun R, Huang B, Dong X, Xiao Q, et al. Multi-organ proteomic landscape of COVID-19 autopsies. Cell. (2021) 184:775–91. doi: 10.1016/j.cell.2021.01.004
17. Wang W, Wang CY, Wang SI, Wei JC. Long-term cardiovascular outcomes in COVID-19 survivors among non-vaccinated population: A retrospective cohort study from the TriNetX US collaborative networks. Eclinicalmedicine. (2022) 53:101619. doi: 10.1016/j.eclinm.2022.101619
18. Xie Y, Xu E, Bowe B, Al-Aly Z. Long-term cardiovascular outcomes of COVID-19. Nat Med. (2022) 28:583–90. doi: 10.1038/s41591-022-01689-3
19. Brogi E, Marino F, Bertini P, Tavazzi G, Corradi F, Forfori F. Cardiac complications in patients with COVID-19: a systematic review. J Anesthesia Analgesia Crit Care. (2022) 2:1–35. doi: 10.1186/s44158-022-00046-7
20. Burn E, Duarte-Salles T, Fernandez-Bertolin S, Reyes C, Kostka K, Delmestri A, et al. Venous or arterial thrombosis and deaths among COVID-19 cases: a European network cohort study. Lancet Infect Dis. (2022) 22:1142–52. doi: 10.1016/S1473-3099(22)00223-7
21. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–18. doi: 10.1016/S0140-6736(20)30937-5
22. Li MY, Li L, Zhang Y, Wang XS. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty. (2020) 9:45. doi: 10.1186/s40249-020-00662-x
23. Simões E Silva AC, Silveira KD, Ferreira AJ, Teixeira MM. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol. (2013) 169:477–92. doi: 10.1111/bph.12159
24. Zhang YH, Zhang YH, Dong XF, Hao QQ, Zhou XM, Yu QT, et al. ACE2 and Ang-(1-7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflammation Res. (2015) 64:253–60. doi: 10.1007/s00011-015-0805-1
25. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. (2012) 33:829–37. doi: 10.1093/eurheartj/ehr304
26. Sampaio WO, Souza DSR, Faria-Silva R, Da MML, Schiffrin EL, Touyz RM. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension. (2007) 49:185–92. doi: 10.1161/01.HYP.0000251865.35728.2f
27. Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol. (2022) 23:3–20. doi: 10.1038/s41580-021-00418-x
28. Datta PK, Liu F, Fischer T, Rappaport J, Qin X. SARS-CoV-2 pandemic and research gaps: Understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy. Theranostics. (2020) 10:7448–64. doi: 10.7150/thno.48076
29. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-coV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80. doi: 10.1016/j.cell.2020.02.052
30. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-coV-2 spike glycoprotein. Cell. (2020) 181:281–92. doi: 10.1016/j.cell.2020.02.058
31. Tomasoni D, Italia L, Adamo M, Inciardi RM, Lombardi CM, Solomon SD, et al. COVID-19 and heart failure: from infection to inflammation and angiotensin II stimulation. Searching evidence New disease. Eur J Heart Fail. (2020) 22:957–66. doi: 10.1002/ejhf.1871
32. de Queiroz TM, Lakkappa N, Lazartigues E. ADAM17-mediated shedding of inflammatory cytokines in hypertension. Front Pharmacol. (2020) 11:1154. doi: 10.3389/fphar.2020.01154
33. Pedersen KB, Chodavarapu H, Porretta C, Robinson LK, Lazartigues E. Dynamics of ADAM17-mediated shedding of ACE2 applied to pancreatic islets of male db/db mice. Endocrinology. (2015) 156:4411–25. doi: 10.1210/en.2015-1556
34. Brevini T, Maes M, Webb GJ, John BV, Fuchs CD, Buescher G, et al. FXR inhibition may protect from SARS-CoV-2 infection by reducing ACE2. Nature. (2023) 615:134–42. doi: 10.1038/s41586-022-05594-0
35. Wang K, Gheblawi M, Nikhanj A, Munan M, MacIntyre E, O'Neil C, et al. Dysregulation of ACE (Angiotensin-converting enzyme)-2 and renin-angiotensin peptides in SARS-coV-2 mediated mortality and end-organ injuries. Hypertension. (2022) 79:365–78. doi: 10.1161/HYPERTENSIONAHA.121.18295
36. Kuba K, Imai Y, Ohto-Nakanishi T, Penninger JM. Trilogy of ACE2: a peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol Ther (Oxford). (2010) 128:119–28. doi: 10.1016/j.pharmthera.2010.06.003
37. Iwata-Yoshikawa N, Kakizaki M, Shiwa-Sudo N, Okura T, Tahara M, Fukushi S, et al. Essential role of TMPRSS2 in SARS-CoV-2 infection in murine airways. Nat Commun. (2022) 13:6100. doi: 10.1038/s41467-022-33911-8
38. Busse LW, Chow JH, McCurdy MT, Khanna AK. COVID-19 and the RAAS-a potential role for angiotensin II? Crit Care. (2020) 24:136. doi: 10.1186/s13054-020-02862-1
39. Briquez PS, Rouhani SJ, Yu J, Pyzer AR, Trujillo J, Dugan HL, et al. Severe COVID-19 induces autoantibodies against angiotensin II that correlate with blood pressure dysregulation and disease severity. Sci Adv. (2022) 8:eabn3777. doi: 10.1126/sciadv.abn3777
40. Rodriguez-Perez AI, Labandeira CM, Pedrosa MA, Valenzuela R, Suarez-Quintanilla JA, Cortes-Ayaso M, et al. Autoantibodies against ACE2 and angiotensin type-1 receptors increase severity of COVID-19. J Autoimmun. (2021) 122:102683. doi: 10.1016/j.jaut.2021.102683
41. Casciola-Rosen L, Thiemann DR, Andrade F, Trejo ZM, Hooper JE, Leonard EK, et al. IgM autoantibodies recognizing ACE2 are associated with severe COVID-19. Jci Insight. (2020) 7(9):e158362. doi: 10.1101/2020.10.13.20211664
42. Michalick L, Weidenfeld S, Grimmer B, Fatykhova D, Solymosi PD, Behrens F, et al. Plasma mediators in patients with severe COVID-19 cause lung endothelial barrier failure. Eur Respir J. (2020) 57:2002384. doi: 10.1183/13993003.02384-2020
43. Falcinelli E, Petito E, Becattini C, De Robertis E, Paliani U, Sebastiano M, et al. Role of endothelial dysfunction in the thrombotic complications of COVID-19 patients. J infection. (2021) 82:186–230. doi: 10.1016/j.jinf.2020.11.041
44. Vora SM, Lieberman J, Wu H. Inflammasome activation at the crux of severe COVID-19. Nat Rev Immunol. (2021) 21:694–703. doi: 10.1038/s41577-021-00588-x
45. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol. (2020) 5(49):eabd1554. doi: 10.1126/sciimmunol.abd1554
46. Xiong S, Hong Z, Huang LS, Tsukasaki Y, Nepal S, Di A, et al. IL-1beta suppression of VE-cadherin transcription underlies sepsis-induced inflammatory lung injury. J Clin Invest. (2020) 130:3684–98. doi: 10.1172/JCI136908
47. Khatib-Massalha E, Bhattacharya S, Massalha H, Biram A, Golan K, Kollet O, et al. Lactate released by inflammatory bone marrow neutrophils induces their mobilization via endothelial GPR81 signaling. Nat Commun. (2020) 11:3547. doi: 10.1038/s41467-020-17402-2
48. Velavan TP, Kieu LL, Kreidenweiss A, Gabor J, Krishna S, Kremsner PG. Longitudinal monitoring of lactate in hospitalized and ambulatory COVID-19 patients. Am J Trop Med Hyg. (2021) 104:1041–44. doi: 10.4269/ajtmh.20-1282
49. Vassiliou AG, Jahaj E, Ilias I, Markaki V, Malachias S, Vrettou C, et al. Lactate kinetics reflect organ dysfunction and are associated with adverse outcomes in intensive care unit patients with COVID-19 pneumonia: preliminary results from a GREEK single-centre study. Metabolites. (2020) 10(10):386. doi: 10.3390/metabo10100386
50. Laforge M, Elbim C, Frere C, Hemadi M, Massaad C, Nuss P, et al. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat Rev Immunol. (2020) 20:515–16. doi: 10.1038/s41577-020-0407-1
51. Uchimido R, Schmidt EP, Shapiro NI. The glycocalyx: a novel diagnostic and therapeutic target in sepsis. Crit Care. (2019) 23:16. doi: 10.1186/s13054-018-2292-6
52. Hashimoto R, Takahashi J, Shirakura K, Funatsu R, Kosugi K, Deguchi S, et al. SARS-CoV-2 disrupts respiratory vascular barriers by suppressing Claudin-5 expression. Sci Adv. (2022) 8:o6783. doi: 10.1126/sciadv.abo6783
53. Sciaudone A, Corkrey H, Humphries F, Koupenova M. Platelets and SARS-coV-2 during COVID-19: immunity, thrombosis, and beyond. Circ Res. (2023) 132:1272–89. doi: 10.1161/CIRCRESAHA.122.321930
54. Knight R, Walker V, Ip S, Cooper JA, Bolton T, Keene S, et al. Association of COVID-19 with major arterial and venous thrombotic diseases: A population-wide cohort study of 48 million adults in england and wales. Circulation. (2022) 146:892–906. doi: 10.1161/CIRCULATIONAHA.122.060785
55. Subrahmanian S, Borczuk A, Salvatore S, Fung KM, Merrill JT, Laurence J, et al. Tissue factor upregulation is associated with SARS-CoV-2 in the lungs of COVID-19 patients. J Thromb Haemost. (2021) 19:2268–74. doi: 10.1111/jth.15451
56. Wang J, Pendurthi UR, Yi G, Rao LVM. SARS-CoV-2 infection induces the activation of tissue factor–mediated coagulation via activation of acid sphingomyelinase. Blood. (2021) 138:344–49. doi: 10.1182/blood.2021010685
57. Lo MW, Amarilla AA, Lee JD, Albornoz EA, Modhiran N, Clark RJ, et al. SARS-CoV-2 triggers complement activation through interactions with heparan sulfate. Clin Transl Immunol. (2022) 11:e1413. doi: 10.1002/cti2.1413
58. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. (2020) 136:1169–79. doi: 10.1182/blood.2020007008
59. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. (2020) 217(12):e20201129. doi: 10.1084/jem.20201129
60. Leppkes M, Knopf J, Naschberger E, Lindemann A, Singh J, Herrmann I, et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. Ebiomedicine. (2020) 58:102925. doi: 10.1016/j.ebiom.2020.102925
61. Bonaventura A, Vecchie A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. (2021) 21:319–29. doi: 10.1038/s41577-021-00536-9
62. Zhang R, Sun C, Han Y, Huang L, Sheng H, Wang J, et al. Neutrophil autophagy and NETosis in COVID-19: perspectives. Autophagy. (2023) 19:758–67. doi: 10.1080/15548627.2022.2099206
63. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest. (2020) 130:6151–57. doi: 10.1172/JCI141374
64. Szczot M, Nickolls AR, Lam RM, Chesler AT. The form and function of PIEZO2. Annu Rev Biochem. (2021) 90:507–34. doi: 10.1146/annurev-biochem-081720-023244
65. Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. (2010) 330:55–60. doi: 10.1126/science.1193270
66. Saotome K, Murthy SE, Kefauver JM, Whitwam T, Patapoutian A, Ward AB. Structure of the mechanically activated ion channel Piezo1. Nature. (2018) 554:481–86. doi: 10.1038/nature25453
67. Zhao Q, Zhou H, Chi S, Wang Y, Wang J, Geng J, et al. Structure and mechanogating mechanism of the Piezo1 channel. Nature. (2018) 554:487–92. doi: 10.1038/nature25743
68. Jiang Y, Yang X, Jiang J, Xiao B. Structural designs and mechanogating mechanisms of the mechanosensitive piezo channels. Trends Biochem Sci. (2021) 46:472–88. doi: 10.1016/j.tibs.2021.01.008
69. Jiang F, Yin K, Wu K, Zhang M, Wang S, Cheng H, et al. The mechanosensitive Piezo1 channel mediates heart mechano-chemo transduction. Nat Commun. (2021) 12:869. doi: 10.1038/s41467-021-21178-4
70. Sugisawa E, Takayama Y, Takemura N, Kondo T, Hatakeyama S, Kumagai Y, et al. RNA sensing by gut piezo1 is essential for systemic serotonin synthesis. Cell. (2020) 182:609–24. doi: 10.1016/j.cell.2020.06.022
71. Wang L, You X, Lotinun S, Zhang L, Wu N, Zou W. Mechanical sensing protein PIEZO1 regulates bone homeostasis via osteoblast-osteoclast crosstalk. Nat Commun. (2020) 11:282. doi: 10.1038/s41467-019-14146-6
72. Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, et al. Piezo1 integration of vascular architecture with physiological force. Nature. (2014) 515:279–82. doi: 10.1038/nature13701
73. Lukacs V, Mathur J, Mao R, Bayrak-Toydemir P, Procter M, Cahalan SM, et al. Impaired PIEZO1 function in patients with a novel autosomal recessive congenital lymphatic dysplasia. Nat Commun. (2015) 6:8329. doi: 10.1038/ncomms9329
74. Ma S, Cahalan S, LaMonte G, Grubaugh ND, Zeng W, Murthy SE, et al. Common PIEZO1 allele in african populations causes RBC dehydration and attenuates plasmodium infection. Cell. (2018) 173:443–55. doi: 10.1016/j.cell.2018.02.047
75. Ma S, Dubin AE, Zhang Y, Mousavi SAR, Wang Y, Coombs AM, et al. A role of PIEZO1 in iron metabolism in mice and humans. Cell. (2021) 184:969–82. doi: 10.1016/j.cell.2021.01.024
76. Jin P, Jan LY, Jan YN. Mechanosensitive ion channels: structural features relevant to mechanotransduction mechanisms. Annu Rev Neurosci. (2020) 43:207–29. doi: 10.1146/annurev-neuro-070918-050509
77. Shibasaki K. TRPV4 activation by thermal and mechanical stimuli in disease progression. Lab Invest. (2020) 100:218–23. doi: 10.1038/s41374-019-0362-2
78. Liu L, Guo M, Lv X, Wang Z, Yang J, Li Y, et al. Role of transient receptor potential vanilloid 4 in vascular function. Front Mol Biosci. (2021) 8:677661. doi: 10.3389/fmolb.2021.677661
79. Fu S, Meng H, Inamdar S, Das B, Gupta H, Wang W, et al. Activation of TRPV4 by mechanical, osmotic or pharmaceutical stimulation is anti-inflammatory blocking IL-1β mediated articular cartilage matrix destruction. Osteoarthritis Cartilage. (2021) 29:89–99. doi: 10.1016/j.joca.2020.08.002
80. Zúñiga L, Cayo A, González W, Vilos C, Zúñiga R. Potassium channels as a target for cancer therapy: current perspectives. Onco Targets Ther. (2022) 15:783–97. doi: 10.2147/OTT.S326614
81. Honore E. The neuronal background K2P channels : focus on TREK1. Nature Reviews. Neuroscience. (2007) 8:251–61. doi: 10.1038/nrn2117
82. Douguet D, Honore E. Mammalian mechanoelectrical transduction: structure and function of force-gated ion channels. Cell. (2019) 179:340–54. doi: 10.1016/j.cell.2019.08.049
83. Delmas P, Parpaite T, Coste B. PIEZO channels and newcomers in the mammalian mechanosensitive ion channel family. Neuron. (2022) 110:2713–27. doi: 10.1016/j.neuron.2022.07.001
84. Lewis AH, Grandl J. Mechanical sensitivity of Piezo1 ion channels can be tuned by cellular membrane tension. Elife. (2015) 4:e12088. doi: 10.7554/eLife.12088
85. Coste B, Xiao B, Santos JS, Syeda R, Grandl J, Spencer KS, et al. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature. (2012) 483:176–81. doi: 10.1038/nature10812
86. Dalal PJ, Muller WA, Sullivan DP. Endothelial cell calcium signaling during barrier function and inflammation. Am J Pathol. (2020) 190:535–42. doi: 10.1016/j.ajpath.2019.11.004
87. Syeda R, Florendo MN, Cox CD, Kefauver JM, Santos JS, Martinac B, et al. Piezo1 channels are inherently mechanosensitive. Cell Rep. (2016) 17:1739–46. doi: 10.1016/j.celrep.2016.10.033
88. Syeda R, Xu J, Dubin AE, Coste B, Mathur J, Huynh T, et al. Chemical activation of the mechanotransduction channel Piezo1. Elife. (2015) 4:e07369. doi: 10.7554/eLife.07369
89. Wang Y, Chi S, Guo H, Li G, Wang L, Zhao Q, et al. A lever-like transduction pathway for long-distance chemical- and mechano-gating of the mechanosensitive Piezo1 channel. Nat Commun. (2018) 9:1300. doi: 10.1038/s41467-018-03570-9
90. Bae C, Sachs F, Gottlieb PA. The mechanosensitive ion channel Piezo1 is inhibited by the peptide GsMTx4. Biochemistry. (2011) 50:6295–300. doi: 10.1021/bi200770q
91. Lee HW, Xu Y, He L, Choi W, Gonzalez D, Jin SW, et al. Role of venous endothelial cells in developmental and pathologic angiogenesis. Circulation. (2021) 144:1308–22. doi: 10.1161/CIRCULATIONAHA.121.054071
92. Yang Y, Yang P, Huang C, Wu Y, Zhou Z, Wang X, et al. Inhibitory effect on SARS-CoV-2 infection of neferine by blocking Ca2+-dependent membrane fusion. J Med Virol. (2021) 93:5825–32. doi: 10.1002/jmv.27117
93. Gerasimenko JV, Petersen OH, Gerasimenko OV. SARS-coV-2 S protein subunit 1 elicits ca2+ Influx-dependent ca2+ Signals in pancreatic stellate cells and macrophages in situ. Funct (Oxford England). (2022) 3:zqac002. doi: 10.1093/function/zqac002
94. Liu X, Wang YL, Wu J, Qi J, Zeng Z, Wan Q, et al. Neutralizing aptamers block S/RBD-ACE2 interactions and prevent host cell infection. Angewandte Chemie (International Ed.). (2021) 60:10273–78. doi: 10.1002/anie.202100345
95. Ye F, Lin X, Chen Z, Yang F, Lin S, Yang J, et al. S19W, T27W, and N330Y mutations in ACE2 enhance SARS-CoV-2 S-RBD binding toward both wild-type and antibody-resistant viruses and its molecular basis. Signal Transduct Target Ther. (2021) 6:312–43. doi: 10.1038/s41392-021-00756-4
96. Yang K, Liu S, Yan H, Lu W, Shan X, Chen H, et al. SARS-CoV-2 spike protein receptor-binding domain perturbates intracellular calcium homeostasis and impairs pulmonary vascular endothelial cells. Signal Transduct Target Ther. (2023) 8:276. doi: 10.1038/s41392-023-01556-8
97. Zeng W, Marshall KL, Min S, Daou I, Chapleau MW, Abboud FM, et al. PIEZOs mediate neuronal sensing of blood pressure and the baroreceptor reflex. Sci (American Assoc Advancement Science). (2018) 362:464–67. doi: 10.1126/science.aau6324
98. Wang S, Chennupati R, Kaur H, Iring A, Wettschureck N, Offermanns S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J Clin Invest. (2016) 126:4527–36. doi: 10.1172/JCI87343
99. Safaee FB, Wiegand SB, Pinciroli R, Gianni S, Morais C, Ikeda T, et al. High concentrations of nitric oxide inhalation therapy in pregnant patients with severe coronavirus disease 2019 (COVID-19). Obstet Gynecol. (2020) 136:1109–13. doi: 10.1097/AOG.0000000000004128
100. Feng WX, Yang Y, Wen J, Liu YX, Liu L, Feng C. Implication of inhaled nitric oxide for the treatment of critically ill COVID-19 patients with pulmonary hypertension. Esc Heart Fail. (2021) 8:714–18. doi: 10.1002/ehf2.13023
101. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. New Engl J Med. (2020) 383:120–28. doi: 10.1056/NEJMoa2015432
102. Chen P, Zhang G, Jiang S, Ning Y, Deng B, Pan X, et al. Mechanosensitive Piezo1 in endothelial cells promotes angiogenesis to support bone fracture repair. Cell Calcium. (2021) 97:102431. doi: 10.1016/j.ceca.2021.102431
103. Wang S, Wang B, Shi Y, Moller T, Stegmeyer RI, Strilic B, et al. Mechanosensation by endothelial PIEZO1 is required for leukocyte diapedesis. Blood. (2022) 140:171–83. doi: 10.1182/blood.2021014614
104. Zhao W, Wei Z, Xin G, Li Y, Yuan J, Ming Y, et al. Piezo1 initiates platelet hyperreactivity and accelerates thrombosis in hypertension. J Thromb Haemost. (2021) 19:3113–25. doi: 10.1111/jth.15504
105. Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Häberle H, et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood. (2021) 137:1061–71. doi: 10.1182/blood.2020008762
106. Saegeman V, Cohen MC, Abasolo L, Rello J, Fernandez-Gutierrez B, Fernandez-Rodriguez A. Positive airway pressure longer than 24 h is associated with histopathological volutrauma in severe COVID-19 pneumonia-an ESGFOR based narrative case-control review. Ann Transl Med. (2022) 10:644. doi: 10.21037/atm-22-605
107. Karageorgos V, Proklou A, Vaporidi K. Lung and diaphragm protective ventilation: a synthesis of recent data. Expert Rev Respir Med. (2022) 16:375–90. doi: 10.1080/17476348.2022.2060824
108. Chen L, Xia HF, Shang Y, Yao SL. Molecular mechanisms of ventilator-induced lung injury. Chin Med J (Engl). (2018) 131:1225–31. doi: 10.4103/0366-6999.226840
109. Zhang Y, Jiang L, Huang T, Lu D, Song Y, Wang L, et al. Mechanosensitive cation channel Piezo1 contributes to ventilator-induced lung injury by activating RhoA/ROCK1 in rats. Respir Res. (2021) 22:250. doi: 10.1186/s12931-021-01844-3
110. Jiang L, Zhang Y, Lu D, Huang T, Yan K, Yang W, et al. Mechanosensitive Piezo1 channel activation promotes ventilator-induced lung injury via disruption of endothelial junctions in ARDS rats. Biochem Biophys Res Commun. (2021) 556:79–86. doi: 10.1016/j.bbrc.2021.03.163
111. Fang X, Li M, Wang Y, Zhang P, Sun M, Xu J, et al. Mechanosensitive ion channel Piezo1 mediates mechanical ventilation-exacerbated ARDS-associated pulmonary fibrosis. J Adv Res. (2023) 53:175–86. doi: 10.1016/j.jare.2022.12.006
112. Grannemann C, Pabst A, Honert A, Schieren J, Martin C, Hank S, et al. Mechanical activation of lung epithelial cells through the ion channel Piezo1 activates the metalloproteinases ADAM10 and ADAM17 and promotes growth factor and adhesion molecule release. Biomaterials Adv. (2023) 152:213516. doi: 10.1016/j.bioadv.2023.213516
113. Morley JE, Kalantar Zadeh K, Anker SD. COVID-19: a major cause of cachexia and sarcopenia? J Cachexia Sarcopenia Muscle. (2020) 11:863–65. doi: 10.1002/jcsm.12589
114. Soares MN, Eggelbusch M, Naddaf E, Gerrits KHL, van der Schaaf M, van den Borst B, et al. Skeletal muscle alterations in patients with acute Covid-19 and post-acute sequelae of Covid-19. J Cachexia Sarcopenia Muscle. (2022) 13:11–22. doi: 10.1002/jcsm.12896
115. Bartoli F, Debant M, Chuntharpursat-Bon E, Evans EL, Musialowski KE, Parsonage G, et al. Endothelial Piezo1 sustains muscle capillary density and contributes to physical activity. J Clin Invest. (2022) 132(5):e141775. doi: 10.1172/JCI141775
116. Hirata Y, Nomura K, Kato D, Tachibana Y, Niikura T, Uchiyama K, et al. A Piezo1/KLF15/IL-6 axis mediates immobilization-induced muscle atrophy. J Clin Invest. (2022) 132(10):1–13. doi: 10.1172/JCI154611
117. Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol. (2023) 21:133–46. doi: 10.1038/s41579-022-00846-2
119. Iwasaki A, Putrino D. Why we need a deeper understanding of the pathophysiology of long COVID. Lancet Infect Dis. (2023) 23:393–95. doi: 10.1016/S1473-3099(23)00053-1
120. Pretorius E, Vlok M, Venter C, Bezuidenhout JA, Laubscher GJ, Steenkamp J, et al. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc Diabetol. (2021) 20:1–172. doi: 10.1186/s12933-021-01359-7
121. Fang X, Wang Y, Xu J, He Y, Peng Z, Shang Y. Immunothrombosis in acute respiratory dysfunction of COVID-19. Front Immunol. (2021) 12:651545. doi: 10.3389/fimmu.2021.651545
122. Kubánková M, Hohberger B, Hoffmanns J, Fürst J, Herrmann M, Guck J, et al. Physical phenotype of blood cells is altered in COVID-19. Biophys J. (2021) 120:2838–47. doi: 10.1016/j.bpj.2021.05.025
123. Osiaevi I, Schulze A, Evers G, Harmening K, Vink H, Kümpers P, et al. Persistent capillary rarefication in long COVID syndrome. Angiogenesis (London). (2023) 26:53–61. doi: 10.1007/s10456-022-09850-9
124. Altmann DM, Whettlock EM, Liu S, Arachchillage DJ, Boyton RJ. The immunology of long COVID. Nat Rev Immunol. (2023) 23:618–34. doi: 10.1038/s41577-023-00904-7
Keywords: COVID-19, SARS-CoV-2, endothelial dysfunction, immunothrombosis, PIEZO1, therapeutic target.
Citation: Zhang X, Liu J, Deng X and Bo L (2024) Understanding COVID-19-associated endothelial dysfunction: role of PIEZO1 as a potential therapeutic target. Front. Immunol. 15:1281263. doi: 10.3389/fimmu.2024.1281263
Received: 22 August 2023; Accepted: 14 February 2024;
Published: 29 February 2024.
Edited by:
Patricia Pia Wadowski, Medical University of Vienna, AustriaReviewed by:
Peng Zhang, Institute of ENT and Shenzhen Key Laboratory of ENT, ChinaNhat Tu Le, Houston Methodist Research Institute, United States
You Shang, Huazhong University of Science and Technology, China
Copyright © 2024 Zhang, Liu, Deng and Bo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lulong Bo, YmFydGJvQHNtbXUuZWR1LmNu; Xiaoming Deng, ZGVuZ3BoZEBzbW11LmVkdS5jbg==