 Farooq Riaz
Farooq Riaz Jing Zhang
Jing Zhang Fan Pan
Fan Pan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 01 February 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1274474
This article is part of the Research TopicUnraveling Tumor Metastasis: Investigating the Tumor Microenvironment and Novel In-vitro Models for Understanding Cancer ProgressionView all articles
Metastatic disease, a leading and lethal indication of deaths associated with tumors, results from the dissemination of metastatic tumor cells from the site of primary origin to a distant organ. Dispersion of metastatic cells during the development of tumors at distant organs leads to failure to comply with conventional treatments, ultimately instigating abrupt tissue homeostasis and organ failure. Increasing evidence indicates that the tumor microenvironment (TME) is a crucial factor in cancer progression and the process of metastatic tumor development at secondary sites. TME comprises several factors contributing to the initiation and progression of the metastatic cascade. Among these, various cell types in TME, such as mesenchymal stem cells (MSCs), lymphatic endothelial cells (LECs), cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs), T cells, and tumor-associated macrophages (TAMs), are significant players participating in cancer metastasis. Besides, various other factors, such as extracellular matrix (ECM), gut microbiota, circadian rhythm, and hypoxia, also shape the TME and impact the metastatic cascade. A thorough understanding of the functions of TME components in tumor progression and metastasis is necessary to discover new therapeutic strategies targeting the metastatic tumor cells and TME. Therefore, we reviewed these pivotal TME components and highlighted the background knowledge on how these cell types and disrupted components of TME influence the metastatic cascade and establish the premetastatic niche. This review will help researchers identify these altered components’ molecular patterns and design an optimized, targeted therapy to treat solid tumors and restrict metastatic cascade.
Metastasis, a key term delineating the progression and spread of malignancies to the distant and surrounding tissues and organs, is known for the majority of cancer-related morbidities and mortalities (1). In general, tumor cells evade the primary tumor site during the process of metastasis and ultimately disseminate to farther and distant sites through the circulatory system (2). A huge number of cancer patients develop advanced metastasis, which provokes the terminal stage of diseases and shows resistance to the currently available therapeutic remedies. These characteristics of metastasis make it a lethal hallmark of tumors (3). Formerly, metastasis was regarded as a progressive stage of the tumor, frequently happening during the tumor progression phase (4); nevertheless, previous observations verified the incidence of metastasis often at the early stage of oncogenesis. This concludes that metastasis can arise at earlier and later stages of tumorigenesis. However, the two stages signify diverse pathogenesis, e.g., cells contributing to the early metastasis carry truncal mutations, while cells causing late metastasis indicate subclonal mutations (5).
It has been implicated that malignant cells lead to the failure of vital organs and account for the overwhelming majority (up to 90%) of cancer-related deaths rather than primary cancers (6). Despite the fact that metastasis is the major reason for impaired tumor therapy and increased mortality, it has not been investigated well. Mounting evidence suggested that a huge amount of tumor cells is unleashed in the circulatory system in tumor patients; however, melanoma-related in-vivo studies indicated that the number of metastasized cells is <0.1% of tumors (7).
The dispersion of tumor cells detached from their primary tumor sites and their ultimate tendency to originate secondary tumors in distant organs requires a multistep, extremely inefficient, and complex but deadly approach which is termed as an invasion-metastasis cascade (8–10). This classical cascade of events implies a series of basic steps, including the invasion of primary cancer cells into adjacent tissues locally; hematogenous transit involving the intravasation of these tumor cells into the bloodstream with improved survival rate; extravasation and arrest of tumor cells into the distant tissues’ parenchyma through vascular walls; origination of micro-metastatic colonies into the tissue parenchyma; and the colonization involving the successive growth of microscopic colonies into apparent and clinically observable metastatic lesions (11). These events have assisted in rationalizing this complex series of biological processes, which are obligatory for the progression of a specific malignancy toward the explicit metastasis (11).
The oncogenic transformation of tumor cells relies on the interaction of several factors which regulate the perforation of a tight regulatory network and attain a compatible microenvironment that ultimately expedites the oncogenic transformations and metastasis (12). Nevertheless, a detailed pathogenetic mechanism revealing the primary tumor’s formation still lacks the biological understanding of metastatic diseases. Besides, several breakthroughs in unifying our understanding of tumor behavior and diverse types of metastases have emerged. It is evident that compared to primary tumor cells, metastasis-initiating cells frequently display very diverse phenotypic and transcriptomic features (13). These properties make metastatic tumors resistant to the currently available conventional remedies effective (14). During the last 2 decades, a wide range of emerging anti-oncogenic drugs considerably extended the patients’ survival rate, with a 5-year survival rate of <20% among cancer patients with stage IV tumors (15). Despite substantial efforts, a clear knowledge of the underlying mechanisms involving the metastatic process and the variation defining the abilities of various cancer cells that establish metastases are still elusive. Therefore, urgent attention should be paid to understanding the pathogenesis of metastasis, thereby advancing molecular therapies to target metastasis. In this chapter, we summarized the latest developments in understanding metastasis to provide a comprehensive review of the role of the tumor microenvironment (TME), including the cellular and extracellular TME, in affecting metastasis to help researchers identify effective anti-metastatic therapy.
Metastasis is characterized by the separation and local invasion of tumor cells from the primary site to the colonization and growth of metastatic cells at the secondary sites (11). The interaction of tumor cells with their adjacent stromal cells commences during the initial stage of cancer development. It continues with the onset of primary tumor growth, followed by invasion, intravasation, and colonization at the secondary site (11) (Figure 1). With time, it has become evident that TME contributes to tumor development at the secondary site by modulating the metastatic cascade (16). Investigations exhibit that TME is a complex entity that is comprised of a variety of components, including tumor-infiltrating immune cells, cancer-associated fibroblasts (CAFs), adipose cells, endothelial cells, extracellular matrix (ECM), and neuroendocrine cells (17). TME aggressively contributes to procuring cancer hallmarks, such as maintaining the growth rate of cancer cells, hindering cell mortality, enhancing the process of angiogenesis, avoiding immune destruction, triggering invasion and metastasis, and activating pro-tumor inflammation (18). These contributions of TME make it a striking therapeutic intervention.
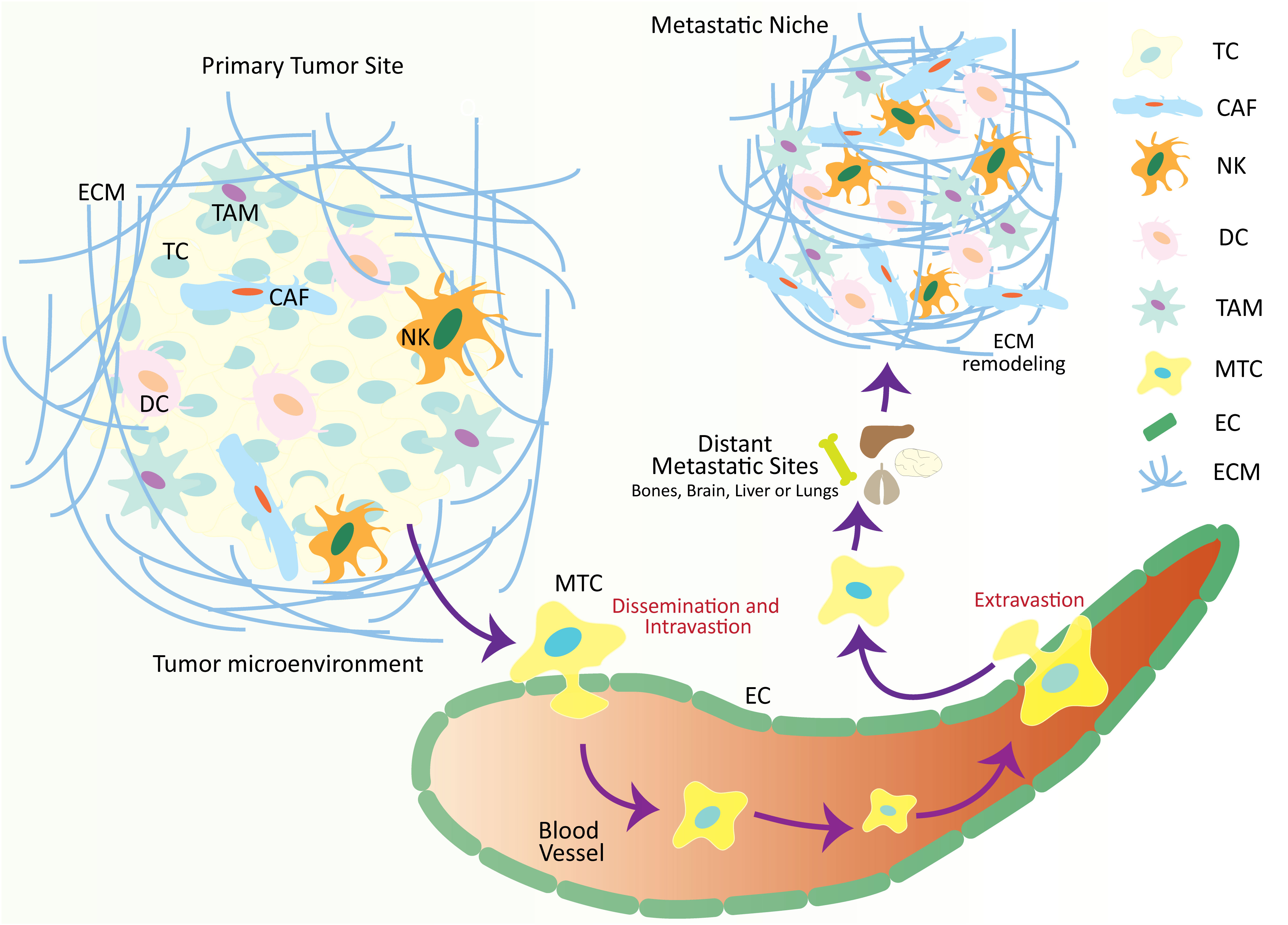
Figure 1 Schematic illustration over-viewing the physio-pathological processes of the metastatic cascade. The tumor microenvironment is comprised of numerous cellular and non-cellular factors that are involved in the process of tumor progression and metastasis. These factors initiate a series of cellular processes collectively called a metastatic cascade. During the metastatic cascade, metastatic tumor cells escape from their primary tumor site, enter the circulation, and translocate and thrive at a secondary tumor site in distant organs where a metastatic niche already exists, with favorable conditions for metastatic tumor development. TC, tumor cell; CAF, cancer-associated fibroblasts; NK, natural killer cell; DC, dendritic cell; TAM, tumor-associated macrophage; MTC, metastatic tumor cell; EC, epithelial cell; ECM, extracellular matrix.
Mesenchymal stem cells (MSCs), also known as multipotent stromal cells, have the tremendous ability of self-renewal and the potential to differentiate themselves into other cell types, e.g., adipocytes, osteoblasts, and chondrocytes (19). These MSCs reside within the tumors where they significantly impact the TME development and function, thereby playing various roles at different stages of cancer progression. It is evident that MSCs influence the tumor cells by inducing their metastatic and invasive properties at the primary tumor site (20, 21), and aid in establishing a metastatic niche at the site of distant tumors (22). Meanwhile, MSCs also express their ability to differentiate and migrate into CAFs, especially in the tumor stroma, and induce the metastasis and growth of colon cancer by ameliorating the invasiveness, motility, and angiogenesis of tumor cells (23).
Multiple mechanisms have been reported by which MSCs exert their indispensable roles in promoting metastasis. It is believed that MSCs excrete TGF-β, which enhances the migratory and invasive capability of tumor cells (24). Besides TGF-β secretion, MSCs are the major source of exosome production (25). MSCs-derived exosomes facilitate metastasis by interacting with the tumor cells and influencing their migration and growth (26). A recent study utilizing the breast cancer cell line MCF7 determines that MSC-derived exosomes enhance the migration ability of tumor cells by inducing the target genes of the WNT signaling pathway, i.e., Axin2 and Dkk1, and increasing the level of β-catenin (27). Meanwhile, another study reported that the co-culture of MCF-7 cells with MSCs enhances the migration through the ER-SDF-1/CXCR4 axis (28). Besides, MSCs derived from bone marrow (BM) also influence the migratory ability of breast cancer cells by modulating the CXCR2 receptor (29). In lung cancer, MSCs are recognized to impact non-small cell lung cancer (NSCLC) cell metastasis. NSCLC leads to transcriptional alterations in MSCs, which enhances the factors involved in epithelial-mesenchymal transition (EMT) and metastasis, for instance, MMP-9. In short, MSCs activate the ABL tyrosine kinases during the progression of primary lung cancer, which is necessary for the increased expression of MSC-dependent MMP-9 (30).
Aryl hydrocarbon receptor (AhR) has been known to regulate the activity of immune cells (31) and tumor development through EMT modulation by transforming epithelial cells towards malignancy form (32). It was investigated that overexpression of AhR activates the EMT markers and enhances cell motility, invasion, and migration (32). Similarly, it was also found that overexpression of AhR via FICZ elevates the EMT markers and cell migration in triple-negative breast cancer (33). Also, the elevated level of AhR is associated with lymph node metastases (34) and/or poor prognosis in inflammatory breast and esophageal squamous cell carcinomas (ESCC) (34, 35). In the ESCC tumor microenvironment, modulation of AhR by using AhR activating ligand 3,3′-diindolylmethane decreased the levels of Vimentin and Slug along with reduction in the RhoA/ROCK1 signaling, which ultimately restricted COX2/PGE2 pathway, secretion of prostaglandin E2, EMT, cell migration and metastasis (35–37).
MSCs-derived exosomes carry numerous non-coding RNAs, mRNAs, and proteins (26). A study exploring the influence of MSC-derived exosomes on cancer metastasis suggested that MSC-derived exosomes collected from gastric tumors indulge the progression of gastric cancer through the MSC-derived exosomal miRNAs (38). Precisely, the elevated level of MSC-derived exosomal miR-221 during the pathogenesis of gastric cancer exerts a significant impact on the exacerbated lymphatic metastasis, tumor-node-metastasis stage, and local invasion (38, 39). Similarly, the exosomes secreted from MSC provoke several cellular processes, including EMT, and strengthen the invasion and migration of gastric tumor cells through the protein kinase B signaling pathway (40). Likewise, BM-derived MSCs also produce exosomes that stimulate multiple myeloma cell migration. These BM-MSC exosomes were suggested to embrace several cytokines, which ultimately participate in the expansion of numerous survival-related pathways, including Akt, p53, p38, and c-Jun N-terminal kinase (41). MSCs interact with and trigger M2 macrophages at distant metastatic sites to enhance the EMT and metastasis capability of tumor cells (42). Alternatively, MSCs in the TME directly modulate the levels of secretory CCL7 and TGF-β through KLF5/CXCL5 (43). Meanwhile, at the primary metastatic site, CAF-derived PAI-1 is an important factor promoting the metastasis cascade (44). Overall, it is evident that MSCs are the key players in metastatic cancer, and further comprehensive studies defining the interaction of tumor cells and MSCs are necessary to develop new anti-tumor drugs.
The primary path of tumor dissemination is the lymphatic vessel (LV) located within the TME. Compared to normal blood vessels, LV is leaky (45). Increasingly studies have demonstrated the crucial role of lymphatic endothelial cells (LECs), which formulates the lining of LV, in the TME. It was revealed that LECs impact tumor progression and metastasis by significantly regulating the tumor cell-derived immune response within the TME (46, 47). The development of metastatic tumors at the distant site is triggered by the interaction of LECs and tumor cells (48). There are several factors that are secreted by the LECs to influence the impact of LECs on the various receptors in tumor cells (49). To recruit the tumor cells, LECs produce chemoattractants, i.e., CCL21 and SDF-1, which link to the chemokine receptors CCR7 and CXCR4 expressed in tumor cells (50). Meanwhile, distant tumors also produce multiple factors to facilitate the progression of the metastatic cascade, including recruitment, extravasation, and outgrowth, by conditioning the LECs. Among these factors, IL-6 is secreted by the tumor cells that activates the STAT3 in LECs and ultimately upregulates the VEGF expression (51), which is a necessary element for lymph angiogenesis (52). Moreover, this IL-6-dependent increase of VEGF expression in tumor cells is linked with the higher HIF-1α in LECs (52). Overall, it can be proposed that tumor-secreted factors directly influence the onset of lymphatic metastases. Further investigation regarding the interaction of LECs and tumor cells will be beneficial in treating cancer metastasis.
Cancer-associated fibroblasts (CAFs) are the primary cells that play an important role in TME (53). CAFs generally exert their roles in a variety of cellular processes, including cell stemness, cell proliferation cell differentiation, cell apoptosis, cell migration, and ECM remodeling. These cellular processes ultimately influence the biological behaviors of tumors, including tumorigenesis, tumor progression, proliferation, recurrence, immunity, tumor immunity, angiogenesis, energy metabolism, and metastasis (54, 55). Within solid tumors, CAFs consist of the most profound stromal components. These stromal components are biomarkers to easily distinguish CAFs from other cell subtypes. These markers include α-smooth muscle actin (α-SMA), platelet-derived growth factor receptor (PDGFRS), vimentin, fibroblast activation protein (FAP), integrin β1 (CD29), and podoplanin (56–58). It is evident that tumor cells activate fibroblasts in a series of events; for instance, during the early stages of tumor progression, tumor cells recruit fibroblasts, which undergo transformation towards the CAFs, and ultimately lead to performing their function in maintaining and reshaping the TME by inducing tissue repair; thereby play an anti-cancer role (58). It is noteworthy that activation of CAFs leads to the production of numerous signaling molecules which are used by tumor cells as growth factors for their proliferation and survival; thus, CAFs favor the growth of tumors and survival of cancer cells, and promote the recruitment and transformation of other cell types within the TME (58, 59). Meanwhile, CAFs produce MMPs, release collagen and fibronectin, and enhance VEGF expression to assist ECM remodeling (60–64). The CAFs-dependent ECM remodeling triggers the local invasion of tumor cells, and facilitates the onset of distant metastasis by providing essential survival conditions (65). To further assist the progression of metastatic tumors, several CAFs undergo reactivation at distant metastatic sites where they produce stromal components, e.g., periostin and tenascin, through various mechanisms that facilitate the colonization of tumor cells (66). It has been validated that a vigorous and aggressive cancer form can be observed if hypoxia-related factors, such as collapsed blood vessels and enhanced mechanical stress, combine with CAFs-triggered proliferative and pro-survival processes in tumor cells (67, 68).
A recent study revealed a new subtype of CAFs, namely, CAF-S1 in the microenvironment, which performs immunosuppressive functions by attracting and enhancing the growth, differentiation, activation, and survival of CD4+ CD25+ T cells (57). Additionally, increased CAF-S1 cells oblige the metastatic breast cancer to the distant organs through CDH11/osteoblast cadherin in patients with smaller (<2cm) primary tumors (69). Meanwhile, CAF-derived periostin endorses the tumor progression, metastasis, and cancer-stem cells (CSC) phenotype through the canonical Wnt/β-catenin axis in the head and neck squamous cell cancer (70). Another study pointed out that elevated YAP1 in CAFs increases the growth of tumor epithelial cells in prostate cancer and originates the secondary metastasis (71). CAF is also known to regulate the TGF-β pathway, which participates in the cancer progression through different processes, including tumor cell invasion, migration, proliferation, and ultimately metastasis (72). A study investigating hepatocellular carcinoma (HCC) progression showed that elevation of TGF-β1 increased the expression of connective tissue growth factor (CTGF). Moreover, the TGF-β receptor inhibitor enhanced the CTGF expression. It suppressed the proliferation of CAF=, thereby decreasing the tumor growth and indicating that targeting TGF-β can be an anti-metastatic therapy (73). Intriguingly, a recent study showed that CAFs dictate tumor cells to endorse migration by transferring mitochondria (74). Conversely, considering the role of IL1 signaling in shaping CAF heterogeneity, it was elucidated that pro-tumorigenic CAFs are characterized by IL1R1hi expression, which is positively allied with immunosuppressive TME and metastasis (75). Overall, CAFs play a critical role in the metastatic cascade, and it is the need of the hour to understand the role of CAF-related activation pathways in metastatic tumors.
Macrophages exhibit diverse phenotypic and functional heterogeneity and play notable roles in regulating the immune response (76). Macrophages participating in the TME are referred as TAMs. These TAMs are widely spread among various tumors (77), representing miscellaneous functions in response to TME-related signals from cancer and stromal cells (78). TAMs play a significant role in drug resistance and promote tumor development, invasion, and metastasis (79). It was suggested that the plasticity of macrophages largely depends on their functional diversity, which is modulated by TME-associated molecules and signals (79). Comprehensive reports have demonstrated that the increased density and localization of TAMs, linked with the pathogenesis of several cancers and poor clinical outcomes, play a critical role in the TME (80).
Even though limited studies have focused on the role of TAMs in metastasis, a recent investigation highlighted that activation of TAMs produces proteolytic enzymes and soluble factors, including matrix metalloproteinases (MMPs), which directly execute substantial effects promoting metastasis (81). Similarly, M2 cells are one of the principal cells that initiate cancer development in both primary and secondary sites. M2 contributes to the process of metastasis by regulating the angiogenesis, breakdown, and deposition of the basement membrane, recruiting leukocytes, and suppressing the immune system (82, 83). M2 macrophages assist the migration of detached cancer cells or cancer stromal cells by destroying the matrix membrane of endothelial cells through the secretion of cathepsins, serine proteases, MMPs, and decomposition of numerous extracellular matrix components, including collagen (84, 85).
Despite the weak antigen-presenting capability, TAMs transform into M2-like phenotypes to support cancer development and metastasis (86). TAMs also secret numerous inflammatory cytokines, including IL-17 and IL-23, to induce tumor-derived inflammation, which sequentially propels tumor growth (87). Likewise, the upregulation of TAM-derived IL-6 strengthens the inflammatory responses and promotes HCC initiation and development (88). During the metastasis, TAMs and cancer cells develop a symbiotic relationship by releasing the epidermal growth factor (EGF) and colony-stimulating factor 1 (CSF-1), respectively. The interaction of these two factors leads to the migration of tumor cells (89).
TAMs, as a vital component of dissemination and metastasis, affect almost all stages of the metastatic cascade by interaction with tumor cells, ECM, and numerous other components of the immune system, particularly in lung tumors (90, 91). TAMs are a significant contributor to bone-metastatic prostate cancer. Tumor cells and TAMs interact to sustain androgen deprivation resistance during bone metastatic disease. TAMs exert androgen deprivation resistance through activation of activin A, which ultimately leads to elevated fibronectin (FN1), FN1-integrin alpha 5 (ITGA5), and tyrosine kinase Src (SRC) network in prostate cancer cells (92). Overall, TAMs exert protumoral functions, and TAMs-specific therapy may emerge as a striking therapeutic selection to prevent metastasis.
Mounting proof demonstrates that myeloid-derived suppressor cells (MDSCs) are crucial to every metastasis stage. While the immune-suppressive activity of MDSCs plays a vital role in developing the metastatic niche (93, 94), these cells also use various strategies that promote metastases. Clinical evidence strongly suggests a potential role for myeloid-derived suppressor cells (MDSCs) in the metastatic process. In non-small cell lung cancer (NSCLC) patients, the presence of circulating CD14+HLA-DRlow monocytic MDSCs (M-MDSCs) was associated with extra-thoracic metastases (95). Similarly, an escalation in lymph node metastases among breast cancer patients correlated with an increase in indoleamine 2,3-dioxygenase (IDO)-expressing CD45+CD33+CD14−CD15− MDSCs within breast cancer tissue (96). Furthermore, a surge in circulating polymorphonuclear MDSCs (PMN-MDSCs) marked by CD11b+CD14-CD15+ and M-MDSCs in melanoma patients was linked to the initiation of metastases and reduced survival (97, 98).
The recruitment of MDSCs to the tumor site or the pre-metastatic microenvironment involves a complex interplay of chemokines and chemokine receptors. Existing research demonstrates that chemokines such as CXCL1, CXCL2, and CXCL5 play a pivotal role in attracting MDSCs to both the tumor site and the pre-metastatic microenvironment (99–101). The chemokine receptors CXCR2 and CXCR4 primarily recruit neutrophils, or PMN (polymorphonuclear)-MDSCs, to the premetastatic niches (102, 103). It has recently been demonstrated that, in contrast to naive neutrophils, bone marrow-derived neutrophils from early stages of cancer display higher levels of uncontrolled migration, higher levels of OXPHOS and glycolysis, and enhanced synthesis of ATP. These neutrophils also lack the immunosuppressive capability that is characteristic of MDSCs (104). Immunosuppressive bone marrow PMN-MDSCs from individuals with advanced cancer exhibit thorough variations in the cancer-specific neutrophils, suggesting the role of neutrophils in early tumor dissemination (105). Significantly, these cells demonstrated robust spontaneous migration, suggesting a potential for more effective movement toward non-inflammatory tissues compared to PMN-MDSCs or control neutrophils (93). It is conceivable that upon reaching their destination, these cells may undergo a transformation into PMN-MDSCs; however, this aspect has not been scientifically explored yet. PMN-MDSCs present in premetastatic niches may contribute to tumor cell escape through the inhibition of immune cells, induction of matrix remodeling, and stimulation of angiogenesis, all of which facilitate tumor cell engraftment (93). In a recent study, Li et al. reported an alternative regulatory mechanism of tumor metastasis, wherein neutrophils accumulated neutral lipids by suppressing the activity of adipose triglyceride lipase in a breast cancer model (106). The specific deletion of adipose triglyceride lipase in neutrophils was demonstrated to reduce metastasis in mice, highlighting the pivotal role of this enzyme in the process. It has also been established that lipid transfer from neutrophils to cancer cells promotes metastasis (106). According to the authors, these neutrophils exhibited a suppressive effect on NK cells, suggesting the potential identification of these cells as PMN-MDSCs.
Several recent investigations have shed light on the role of MDSCs in the EMT process. EMT is a phenomenon observed in certain tumor cells, wherein polarized epithelial cells undergo a transformation, losing their epithelial markers and adopting mesenchymal characteristics, facilitating their ability to spread, invade organs, and metastasize (107). In a notable study, Abastado et al. demonstrated the involvement of PMN-MDSCs in the RET transgenic mice model of spontaneous melanoma. PMN-MDSCs were attracted to the tumor site, and upon entering, they generated transforming growth factor-beta (TGF-β) and hepatocyte growth factor (HGF). These factors induced the initial melanoma cells to undergo EMT. Remarkably, reducing PMN-MDSCs in mice resulted in a decrease in EMT and subsequently reduced metastatic lesions (101). This research underscores the significant impact of PMN-MDSCs on promoting EMT and the subsequent metastatic process.
By augmenting the population of cancer stem cells or inducing a more stem-like state in cancer cells, myeloid-derived suppressor cells (MDSCs) actively promote cancer dissemination. The accumulation of Lin− CD45+ CD33+ MDSCs was correlated with diminished survival rates in both metastatic and non-metastatic ovarian cancer patients. Direct interaction between ovarian tumor cells and MDSCs resulted in the conversion of these cells into stem cells. The elevated levels of microRNA-101 in ovarian malignancies, targeting CtBP2—a co-repressor of stem cell genes—were identified as the mechanistic basis for this phenomenon. Moreover, when human ovarian tumor cells were co-cultured with MDSCs before inoculation into immunodeficient mice, there was an increased number of metastatic lesions in the liver and lungs and enhanced engraftment (108). In the context of pancreatic cancer, myeloid-derived suppressor cells of the monocytic subtype (M-MDSCs) directly stimulated the proliferation of aldehyde dehydrogenase-1+ (ALDH1) pancreatic cancer stem cells in a pancreatic cancer model. Similar outcomes were achieved using human CD14+ HLA-DR− M-MDSCs (109). This underscores the significant role of MDSCs in fostering the expansion of cancer stem cells, contributing to the progression and metastasis of cancer.
Although the role of MDSCs in modulating the immune response in various physiological and pathological settings, a substantial body of research supporting the pro-metastatic activity of MDSCs, a recent study has unveiled a surprising functional flexibility in MDSCs, revealing their potential to prevent metastasis in specific circumstances. In both non-metastatic and metastatic prostate and breast cancers, MDSCs were observed to accumulate in the pre-metastatic location of the lungs (110). In breast cancer, stress promotes the splenic MDSCs to form the pre-metastatic niche through the modulation of TAM/CXCL1 signaling (111). MDSCs derived from non-metastatic tumors exhibited elevated levels of thrombospondin-1 (TSP-1), a potent anti-angiogenic matrix protein, and demonstrated the ability to prevent metastasis. Prosaposin, a robust inducer of TSP-1, was identified as being released by non-metastatic tumors. Intriguingly, an amino acid peptide mimic of prosaposin was found to be sufficient to up-regulate TSP-1 in MDSCs in vivo, preventing tumor metastasis (110). On the other hand, the simultaneous implementation of ferroptosis induction and MDSC blockade restricts primary liver tumors and their metastases susceptible to immune checkpoint inhibition (112). This innovative approach challenges the conventional understanding of MDSCs in metastasis-promoting actions. Further research is imperative to corroborate the involvement of MDSCs in metastasis across diverse tumor models.
The quantity and quality of immune infiltrates in distinct metastases have been correlated with the progression of each individual lesion. Notably, regressing and stable metastases exhibited heightened infiltration of CD8+ and CD4+ T cells, along with T cell clonal expansion, in a patient with ovarian cancer undergoing chemotherapy (113). Conversely, concurrently growing metastatic tumors in the same individual displayed T-cell exclusion. Remarkably, both regressing and stable metastases demonstrated elevated expression levels of CXCL9, a chemokine implicated in T-cell trafficking (113). This observation underscores the interconnectedness of immune infiltration and treatment response, highlighting potential variations across metastatic lesions within a single patient. It is proposed that the efficacy of CD8+ T cell anti-tumor immunity plays a critical role in eradicating disseminated tumor cells (DTCs) in mouse melanoma and breast cancer models, as evidenced by promoting metastasis with T cell depletion (114, 115). Consequently, DTCs must withstand the assault from CD8+ T cells to develop into metastases successfully. Tumor cells can enhance the expression of programmed death-ligand 1 (PD-L1), a pivotal immunological checkpoint, as a mechanism to evade destruction by CD8+ T lymphocytes. A subset of circulating tumor cells during dissemination is responsible for seeding metastases. While research is ongoing, elevated PD-L1 expression in these cells among lung cancer patients is associated with an unfavorable prognosis (116, 117). Similarly, colorectal cancer metastases exhibit higher PD-L1 expression than the original lesions (118).
DTCs expressing PD-L1 may evade elimination by CD8+ T cells, establishing metastatic spread. In the case of pancreatic cancer, tumor cells can disseminate to the liver and remain dormant until conditions favorable for metastatic expansion arise. Notably, adaptive immunity exerts a selective pressure on DTCs that reach the liver, leading to the destruction of the majority of DTCs. However, quiescent cancer cells lacking Major Histocompatibility Complex class I (MHC class I) can evade T cell assault (119). Antigen presentation and CD8+ T cell recognition are contingent on tumor cells expressing MHC class I; therefore, DTCs with insufficient MHC class I evade identification and elimination by CD8+ T lymphocytes. As previously reported, the immune evasion mechanism of latent DTCs is akin to that of quiescent epithelial stem cells. This suggests that metastasis-initiating cells (MICs) may adopt immune evasive abilities similar to those observed in tissue stem cells (120). It has been investigated that elevated infiltration of CD8+ memory T cells restricts the metastatic invasion at earlier stages (121, 122), thus associating the CD8+ memory T cells with better survival in colorectal cancer patients (122). Accordingly, our recent study found that deletion of HIF1α reduces the B16 melanoma tumor growth and expands the population of effector memory function of CD8+ T cells (123, 124). This suggests the positive correlation of HIF1α with the CD8+ T cell exhaustion, exerting oncogenic roles and exacerbating tumor metastasis.
According to reports, CD4+ T lymphocytes with specificity for tumors can create pre-metastatic niches in bone by secreting receptor activators of nuclear factor-κB ligand (RANKL). This secretion, in turn, increases osteoclastogenesis and promotes the metastasis of breast cancer cells to the bone (125). Notably, the conditioned media from 4T1 breast cancer cells induces the expression of chemokine CCL22 in the lung stroma, subsequently elevating Treg levels in the pre-metastatic lung (126). Targeting Treg is an effective anti-cancer therapy as the role of Tregs, particularly the tissue-specific Tregs, in the progression of cancer and cancer metastasis is well studied (127–129). Accordingly, our previous investigation illustrates the elevated intra-tumoral Treg frequency in HIF1α depleted mice. At the same time, combinational therapy using HIF1α inhibitors and Treg inhibitors synergistically boosted anti-tumor immunity and decreased tumor metastasis (123).
The differentiation of naïve CD4+ T cells into Th1 or Th2 cells, which drive inflammatory and anti-inflammatory responses, respectively, plays a role in promoting cancer growth, compounded by the systemic inhibition exerted by tumor-specific Tregs. An imbalance in Th1 and Th2 differentiation has diminished pulmonary immune surveillance (130). For example, oxygen-sensing proteins expressed by T cells can reduce the lung Th1 immune response, which is crucial for maintaining an immunoregulatory state that prevents unnecessary inflammation in response to benign foreign antigens. Conversely, suppressing Th1 immunity in favor of Th2 immunity creates an immune-permissive environment that encourages metastatic colonization (130). Furthermore, the myeloid cell populations in the pre-metastatic niche have been reported to promote the differentiation of T cells into anti-inflammatory Th2 cells (130, 131). This underscores the collaborative efforts of various lymphoid cell populations in establishing a favorable environment that inhibits immune surveillance mediated by T and NK cells, ultimately promoting the spread of metastases. As this is an emerging study area, further investigation is warranted to elucidate the significance of non-myeloid cells and their interactions with myeloid cells in forming pre-metastatic niches.
Natural Killer (NK) cells are crucial in protecting the body against cancer by eliminating cells lacking MHC class I. DTCs must evade NK cell surveillance to survive and spread (132, 133). A balanced signaling mechanism intricately regulates the ability of NK cells to destroy tumor cells. NK inhibitory receptors recognize MHC class I, while activating receptors, such as NKG2D, bind to ligands on tumor cells. NKG2D ligands, like UL16-binding proteins, can undergo altered expression in cancer through processes such as microRNA modulation, transcriptional regulation by factors like IFNγ, DNA methylation, or low histone acetylation (134–136). Additionally, metalloproteinases can cleave NKG2D ligands from the cell surface or secrete them in exosomes, thereby diminishing NK cell assault on cancer cells (136–139).
Quiescent DTCs have been described as downregulating ligands for NK cell activating receptors, providing a mechanism to evade NK cell assault in lung and breast malignancies, particularly those expressing Sox2 and Sox9 and adopting stem cell-like characteristics (140). Moreover, overexpression of Sox9 in metastatic lung tumor cells by upregulating MHC class I is a protective shield against NK destruction (141). Notably, groups of DTCs exhibit more excellent resistance to NK cell-mediated clearance than individual DTCs, possibly due to their ability to suppress ligands activating NK receptors (142). Furthermore, tumor cell clusters may attract additional immunosuppressive myeloid cells in the metastatic niche, potentially thwarting NK cell activity (143, 144). These findings highlight that the regulation of DTCs by NK cells may depend on cancer cell characteristics, including their proliferative status, and may evolve as micro- and macrometastases.
The extracellular matrix (ECM) is responsible for performing various primary activities mediating the fate and function of both normal and tumor cells (145). ECM is generally organized by combining numerous matricellular-associated, fibrous, and proteoglycan proteins (146). ECM is regarded as a dynamic regulatory process that involves a tight regulation of organization, posttranslational modifications, and composition of ECM, which in turn supervise the biochemical and mechanical properties of ECM (147).
In normal physiological circumstances, the stroma cells and parenchymal cells interact with each other by secreting molecular messengers into the microenvironment, by cell-cell contact and communication, or by exerting multiple biophysical and biochemical cues through ECM (148). This interaction results in maintaining tissue homeostasis and establishing niches that bear the characteristics of a distinct microenvironment to ease the growth and survival of specialized cells, including stem cells (149). However, under diseased conditions, normal tissue homeostasis is disrupted, which leads to an interruption of the established niches. Generally, disease conditions lead to the development of tumor-causing genetic lesions from the transition of parenchymal cells, which results in the remodeling of the tissue microenvironment, including the cellular components and ECM (150). These tissue microenvironment modifications strongly impact the disease’s pathogenesis and progression (150).
Tumor progression, including the invasive and metastatic properties of tumors, is highly associated with EMT. Procuring a mesenchymal phenotype is highly dependent on the increased motility of tumor cells, enhanced survival of tumor cells, and elevated expression of enzymes involved in ECM remodeling, such as MMPs. All these changes are necessary for tumor cells to cross the basement membrane, endorse their interaction with ECM, and finally, the intravasation and longevity within vessels (151). Meanwhile, activation and expansion of fibroblasts, elevation in crosslinking and remodeling of ECM, enhanced angiogenesis, and chronic inflammation lead to the stiffness of tissue stroma and tumor desmoplasia (152). Furthermore, tissue stiffness and tumor desmoplasia occur when the tissue exhibits abnormal homeostasis and elevated levels of ECM proteins, such as type I collagen, matricellular proteins, proteoglycans, hyaluronic acid, tenascin, and fibronectin (153). Excessive ECM production indicates ECM stiffness and tumor progression (154). It is evident that a highly aggressive cell line of breast cancer, compared to a less aggressive breast cancer cell line, exhibited increased collagen content and cross-linking, which ultimately enhanced the ECM stiffness (155). During tumor progression, the stiffness and cross-linking of ECM are tightly regulated by TGFβ, which exerts substantial influence on the adjacent fibroblasts and other stromal cells. A myofibroblast phenotype in prostate cancers showed increased levels of smooth muscle α-actin, tenascin, vimentin, and collagen I, along with an increase in TGF-β1 within prostatic intraepithelial neoplasia (156). TGF-β1, therefore, encourages the stromal fibroblasts’ contraction and consequent ECM stiffness, which is linked with tumor aggressiveness (157, 158). Conclusively, ECM stiffness induces TGF-β-dependent EMT and stimulates a basal-like cancer cell phenotype which encourages the metastatic tumor [91], whereas reducing the ECM stiffness and crosslinking alleviates tumor metastasis (159, 160). Similarly, tumor ECM is also known to increase tumor cell motility (161). It was evident that TGFβ stimulates the CAFs in the colon, which secretes various factors, such as tenascin C and hepatocyte growth factor (HGF), to play influential roles in the invasion of colon cancer cells (162). In contrast, fibroblast-derived ECM in the TME directs cancer cells to stick to the cancerous matrices and participates in the metastatic cascade (163).
Tumor cells display few sense transmembrane receptors, e.g., syndecans, discoidin domain receptors (DDRs), and integrins, to sense the ECM mechanical and biochemical properties (164). Activation of these receptors by recognizing the specific motifs within the ECM activates downstream signaling pathways to impact the tumor cell behavior (164). Elevation in stiffened ECM and the crosstalk between integrins and other receptors can activate the Rho GTPase activity, aiding in tumor cell migration. Briefly, stimulation of RhoA GTPase activates phosphorylation of ROCK which ultimately induces the contraction of actomyosin (165). Meanwhile, activation of Rho GTPase through integrins is involved transcription of multiple genes which are vital in tumor growth, motility, differentiation and survival (165, 166). Besides that, collagen also induces the integrins which initiate a cascade of events comprising the recruitment and activation of molecules involved in focal adhesion, such as Talin, FAK, Rho and Ras. These focal adhesion molecules promote the progression of tumors, and lead to the contraction of tumors cells (167, 168). Increased cross-linking and stiffness of ECM also promotes the FAK phosphorylation, and enhances the cancer progression; however, decrease in tissue tension and ECM stiffens minimizes the FAK activity and lowers the invasion and metastasis of tumor cells (160, 169). Similarly, it was studied that ECM stiffness also activates the PI3K signaling which is involved in various cellular processes, including metastasis (170, 171).
The primary effectors of Hippo signaling pathways YAP and TAZ are known to be involved in regulating several cellular and biological processes, including cell proliferation, tumor metastasis, tissue homeostasis, cell differentiation, immune regulation, and tumor microenvironment (172, 173). During the activation of the Hippo signaling pathway, transmembrane cadherin FAT recruits the cascade of Ser/Thr kinase, which phosphorylates the YAP/TAZ and establishes a 14-3-3 binding site. Binding for 14-3-3 with YAP/TAZ hinders the transcription of YAP/TAZ target genes by confiscating this complex in the cytoplasm and hindering its translocation in the nucleus (174). Interestingly, integrins-mediated Rho GTPase-ROCK regulates the YAP in Hippo pathway-independent manner (175, 176), thus associating the gene transcription and mechanotransduction with promoting cell growth (177). Moreover, matrix stiffness induces the integrin cluster and Twist1 detachment from G3BP1 in the cytosol. This translocates the Twist1 into the nucleus, where it performs a critical role in driving EMT (178). Meanwhile, ECM stiffness influences the YAP phosphorylation and nuclear localization to affect cell growth (179). Collectively, ECM, collagen cross-linking and stiffness modulate the numerous keys signaling pathways involved in cell growth, differentiation, migration, invasion, survival, and metastasis (148, 164) (Figure 2).
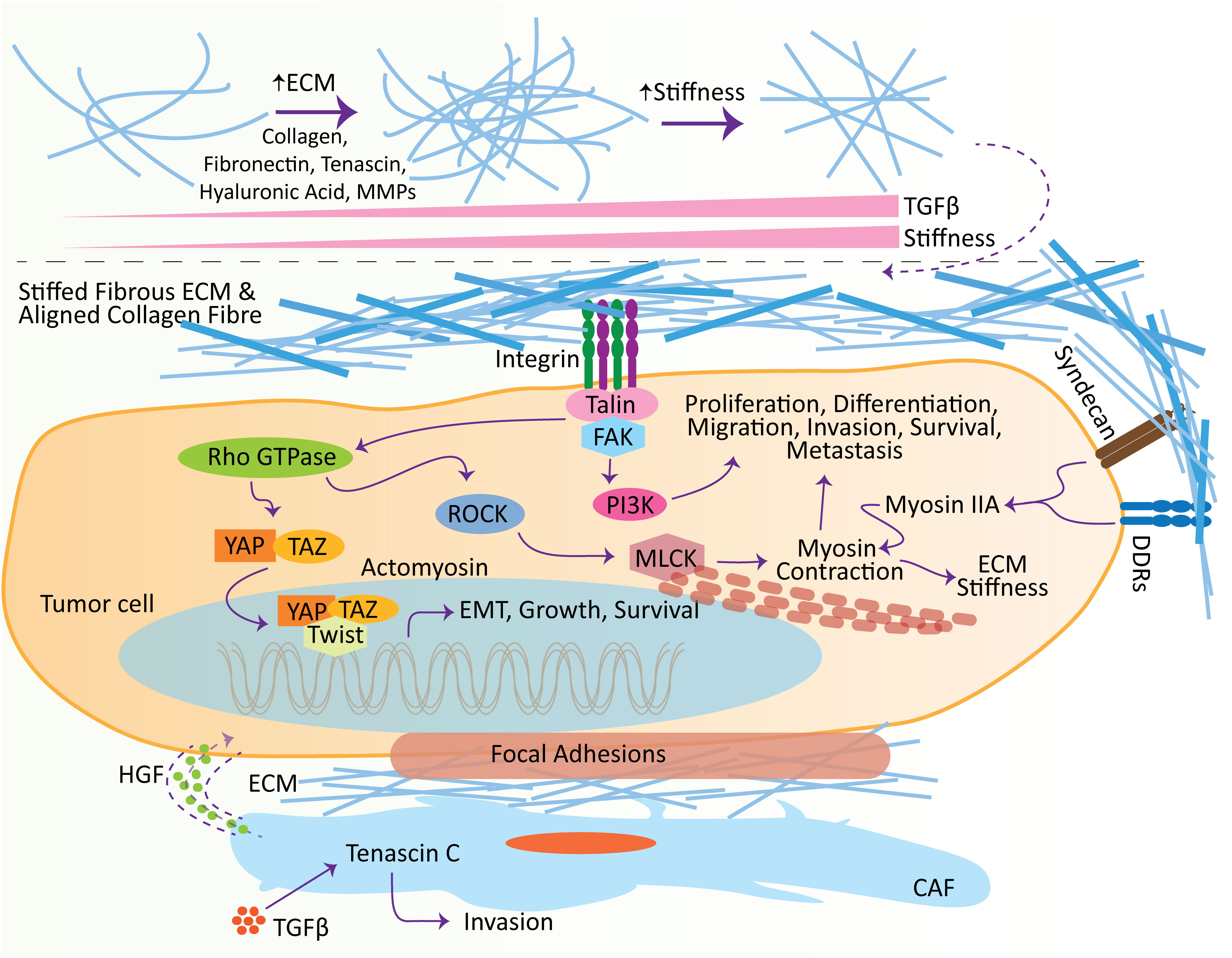
Figure 2 Effect of extracellular matrix (ECM) in promoting metastasis. ECM influences several hallmarks of cancer at different stages, including initiation and metastasis. With the increase in TGFβ, ECM molecules undergo remodeling, which leads to the stiffness of the ECM. Stiffens ECM binds to receptors, e.g., syndecans, DDRs, and integrins, located on the surface of tumor cells to induce intracellular pathways. Crosstalk between ECM ligands and integrins potentiates the Talin, which subsequently activates the focal adhesion kinase (FAK) to induce the adhesion complexes’ assembly, e.g., PI3K-Akt signaling pathway, which plays vital roles in the tumor cell proliferation, differentiation, migration, invasion, survival, and metastasis. Meanwhile, the crosstalk of integrin and growth factor receptors also activates the Rho GTPase. Rho GTPase consequently induces the ROCK, elevating the phosphorylation of MLCK and promoting actomyosin contraction. Furthermore, ECM ligand interaction with DDRs and syndecans induces cell migration and contraction by shortlisting numerous molecules, including myosin IIA. Additionally, ECM stiffness promotes the adhesion signaling integrin clustering and translocates Twist1 and YAP into the nucleus to induce EMT. Similarly, TGFβ encourages the level of Tenascin C in the CAFs, which is involved in cell invasion. These CAFs excrete HGFs to establish the niches in tumor cells. CAF, cancer-associated fibroblasts; TGFβ, transforming growth factor-β; ECM, extracellular matrix; HGF, hepatocyte growth factor; DDR, discoidin domain receptor; MMP, matrix metalloproteinase; FAK, focal adhesion kinase.
Oxygen, an essential factor obligatory for oxidative metabolism and adenosine 5’-triphosphate production, maintains normal tissue homeostasis and developmental processes in cells. The metazoans are using the hypoxic signaling pathway to regulate oxygen homeostasis (180). Hypoxia is an oxygen-deprived condition regarded as a crucial factor in TME. Hypoxia regulates numerous hallmarks of cancer, such as immune evasion, EMT, invasion, angiogenesis, stemness, and metastasis (3, 181). During the hypoxic conditions, hypoxia-inducible factors (HIFs), HIF-1 and HIF-2, activate numerous genes involved in cell apoptosis, differentiation, proliferation, angiogenesis, erythropoiesis, metabolism, and glucose uptake (180, 182). Increasing evidence denoted that tumor initiation, progression, and metastasis are positively correlated with HIF signaling (183).
It was assessed that impaired oxygen homeostasis, in terms of an imbalance between oxygen delivery and oxygen consumption, can cause regions of hypoxia and/or anoxia in 50 to 60% of solid tumors (184). In the settings of TME, abnormal tumor vasculature comprising the distended and leaky capillaries leads to an impaired oxygen supply (185). Meanwhile, the overwhelming population of cancerous cells and infiltrating immune cells increases the oxygen consumption (186). It was verified that oxygen-deprived cells show more aggressiveness and invasiveness with enhanced metastasizing ability. For example, inoculation of hypoxia-induced multiple myeloma cancer cells in mice displayed a faster dissemination rate to the bone marrow than normoxic cells (187, 188). Similarly, an elevated amount of lymph node metastases was observed in the cervical carcinoma disease model, which was exposed to acute hypoxia (189). Likewise, acute hypoxia-induced lung metastases in sarcoma tumor-bearing mice (190). Furthermore, clinical evidence also advocates that hypoxia is highly related with resistance to metastasis, radiotherapy and chemotherapy, poor patient survival, and the activation of HIF pathways (191–193).
EMT is a well-known physiological process active during embryogenesis and tissue regeneration (194). Besides, EMT also plays a critical role in the progression and development of tumorogenesis, especially in hematologic malignancies (188) and numerous solid tumors (195). It was noted that TGF-β and hypoxia act as a master regulator to promote EMT, which further activates numerous downstream transcriptional factors, including Twist, Slug, TCF3, Snail, ZEB1, and Smads (188, 196, 197). The underlying mechanism for hypoxia significantly impacted the EMT, which in turn changes the expression profile of multiple genes and affects the invasive and migratory capability of tumor cells (198). In brief, hypoxia-induced EMT decreases the expression of genes involved in epithelial regulation, for instance, β-catenin and E-cad (199), and elevates the expression of genes involved in mesenchymal-regulation, for instance, CXCR4 (187, 188), SMA, vimentin (200) and N-cad (201).
Under the influence of an oxygen-deprived microenvironment, hypoxia-inducible factors (HIFs) primarily regulate the expression of genes. Generally, HIFα and HIFβ subunits of the heterodimeric HIF complex mediate the HIF-dependent transcriptional regulation by binding to the hypoxia-responsive element (HRE) located within the promoter of target genes (202). It was found that the inactivation of HIF1α and HIF2α selectively revokes metastasis without affecting the formation of primary melanoma, suggesting the prime role of HIF signaling in the development of metastatic tumors and shaping the metastatic niches (203). Similarly, hypoxia and HIF signaling regulate lysyl oxidase (LOX) levels and ECM protein. It was noted that the upregulation of LOX is associated with distant metastasis and increased invasiveness of hypoxic cancer cells through cell-to-matrix adhesion and focal adhesion kinase (204). Meanwhile, LOX, secreted by primary hypoxic tumors, plays an essential role in forming a premetastatic niche by crosslinking with collagen IV and recruiting CD11b+ myeloid cells (205). Carbonic anhydrase IX (CAIX) is a downstream gene of HIF1α signaling, induced under hypoxic conditions and is widely available at the tumor site. It was investigated that CAIX induces the invasion and survival of tumor cells by regulating the intracellular and extracellular pH (206). Furthermore, CAIX was also associated with reduced metastasis and tumor volume in tumor models of breast cancer (206). Additionally, C-X-C chemokine receptor type 4 (CXCR4) is activated in tumor cells under oxygen-deprived conditions. This CXCR4 performs a crucial function in cell trafficking (207, 208). SDF-1 acts as a ligand for the CXCR4. Recent studies exhibited that metastatic malignant cells that express an elevated level of CXCR4 are also enriched with SDF-1, and targeting the CXCR4/SDF-1 axis can significantly disrupt the metastatic cascade (187, 209).
A new term, intermittent or cyclic hypoxia, is registered in multiple studies that temporarily describe the shutting down of inefficient vasculature in tumors. This could result in hypoxic and reoxygenation periodic cycles within cancer cells (210, 211). During intermittent hypoxia and hypoxia-induced EMT, HIF1α serves a central role in enhancing the aggressiveness of tumor cells by regulating numerous genes involved in EMT (212, 213). In the tumor, endothelial and hepatic cells, TGFβ and HIF1α activate each other (214–217). Moreover, HIF1α promotes breast cancer progression by modulating the TGFβ1/SMAD3 axis (218). Also, a study conducted using pancreatic cancer cells indicates that HIF-1α regulates the EMT and invasion of cancer cells through hedgehog signaling (219). Furthermore, HIF1α also drives the EMT in hepatocellular and prostate carcinoma through Wnt/β-catenin signaling (220, 221). Interestingly, in prostate cancer and pancreatic cancer, HIF1α exerts its physiological role on EMT indirectly through FoxM1 signaling (222) and the PAFAH1B2 gene (223), respectively. Similarly, HIF1α interacts with integrin-linked kinase (ILK) by establishing a mediating loop and inducing ILK expression to enhance EMT in prostate and breast cancer cells (224).
Another important function of HIF1α is that it is involved in mediating the expression of multiple immunosuppressive molecules, which are associated with the cancer cells’ development, progression, and metastasis, and possibly ignite the EMT in a TGF-β-dependent manner (225, 226). Induction of EMT retaliates by provoking numerous immune-regulatory effects, including the apoptosis of natural killer (NK) and T-cells but an increase in B cells and Tregs population (227). Similarly, as mentioned earlier, stromal cells regulate cell proliferation, adhesion, and survival by easing the development and dissemination of tumors. Hypoxia affects the production of multiple factors, such as SCF, LOX, SDF-1, VEGF, PDGF, ANGPTL-4, and Ang2, by inducing stromal cells. These factors influence lymphangiogenesis and the formation of new blood vessels. Meanwhile, SDF-1, derived from stromal cells, entices the tumor cells to facilitate metastasis (228, 229). Together, these studies indicate that HIF1α endorses numerous transcriptional factors and modulates various signaling pathways to play a central regulatory role in the hypoxia-induced EMT and metastatic cascade (Figure 3).
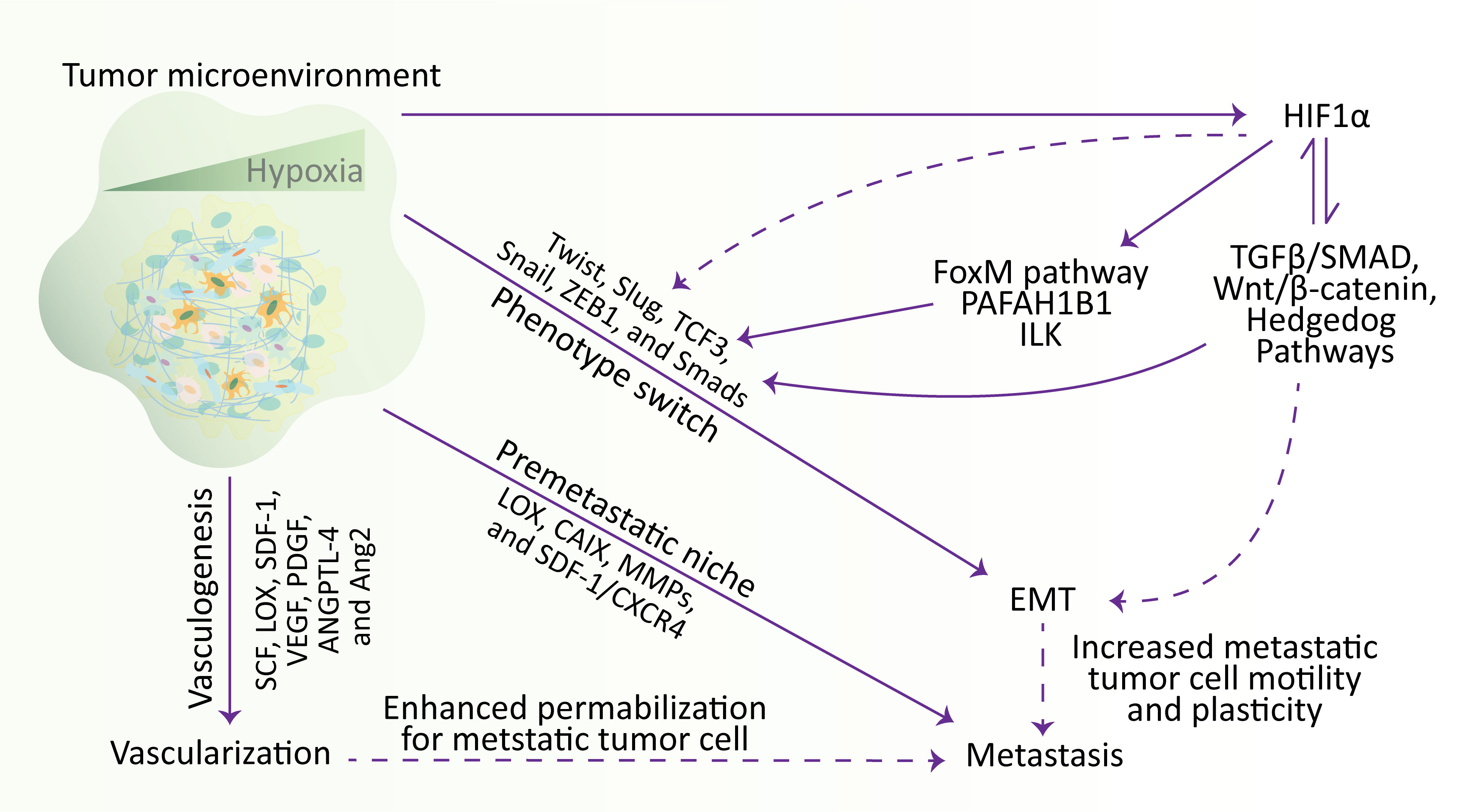
Figure 3 Hypoxia regulates the metastatic cascade. In the TME, hypoxia stimulates EMT, metastasis, and vascularization. Briefly, hypoxia stimulates the vascularization via influencing SCF, LOX, SDF-1, VEGF, PDGF, ANGPTL-4, and Ang2 genes, participates in metastasis by establishing a metastatic niche and modulating the expression of LOX, CAIX, MMPs, and SDF-1/CXCR4, and increase the expression of Twist, Slug, TCF3, Snail, ZEB1, and Smads transcription factors to affect the mobility and plasticity of tumor cells to undergo EMT. Meanwhile, hypoxia activates the HIF1α, which induces the transcription factors involved in EMT through various signaling pathways. Additionally, hypoxia-regulated EMT and vascularization also participate in metastasis by increasing cell motility and plasticity and enhancing permeabilization for metastatic tumor cells, respectively. EMT, epithelial-to-mesenchymal transition; HIF, hypoxia-inducible factor.
Host microbes are crucial regulators in modulating the susceptibility to the development and progression of tumors. These microbes employ their functions markedly through various indirect mechanisms, including their metabolites and influencing the immune system at proximal or distant tumor sites (230, 231). Various reports have indicated that tumor-resident microbes are highly associated with the increased risk of cancer progression (232) and cancer metastasis (233). Meanwhile, microbiota also influences the efficiency of immunotherapy and chemotherapy (234). Gastrointestinal tumors normally metastasize to the lungs, liver, and regional lymph nodes. In this process, gut-microbiota initiates metastatic cascade and promotes tumor growth with restricted response to anti-tumor therapy (235). How microbiota initiates the metastatic cascade has not yet been cleared. An investigation urged that the bacteria may exert a crucial role in the TME as they can infect the tumor cells at the primary site. Infection of tumor cells with bacteria is maintained till the colonization of tumor cells at distant metastatic sites, indicating the stability of the microbiome throughout the progression of the tumor from primary site to the metastatic site (236). In compliance with this, a recent study determined that numerous intracellular bacteria can undergo circulation with tumor cells during the metastatic cascade and play critical roles in metastatic colonization. Briefly, the researchers found that tumor-intracellular microbes modulate the actin cytoskeleton in the host cells and promote the overall survival and viability of metastatic tumor cells against the fluid shear and tear-out stress during the process of circulation (237).
The gut microenvironment, particularly in disease conditions, is enormously complex and comprised of countless bacteria and their products/metabolites (238). Sudden changes in the gut microflora considerably impact the colorectal cancer (CRC) microenvironment, which may also influence the progression and recurrence of colon cancer. Notably, reduced Bacteroides and enhanced Clostridial numbers may improve CRC-liver metastasis (239). Genetic ablation of p53, a tumor suppressor, induces colon cancer, promotes its progression, and increases intestinal permeability. This helps in the development of an inflammatory microenvironment in an NF-κB-dependent manner, which enhances the EMT and dissemination of metastatic tumor cells to the lymph nodes (240). In normal circumstances, gut microbiota has the ability to reduce the expression of p53 through post-translational modifications, transcriptional inhibition, and protein degradation (241).
Interestingly, the majority of the metastatic tumors in CRC models are observed in the liver. This is due to the continuous exposure of the liver to the gut microbial products/metabolites through enterohepatic circulation and portal vein, which extends the direct connection between the gut and liver, particularly during liver metastasis (242, 243). Gut microbiota establishes an immune suppressive hepatic microenvironment by regulating the inflammatory cytokines, such as IFN-γ, TNF-α, IL17, IL12 and IL6, which facilitate the development of hepatic metastatic tumor (244, 245). In parallel, gut microbiota influence the bile acid metabolism to regulate the population of intrahepatic natural killer T (NKT) cells through the regulation of CXCR16 in hepatic sinusoidal cells, which ultimately affects hepatic metastasis (244).
Kynurenine (Kyn) emerges as the primary product in the tryptophan metabolic pathway, regulated by IDO and TDO2 in tumor cells. Kyn has been shown to activate the AhR, initiating autocrine and paracrine functions that impact the anti-tumor immune response and tumor cell survival and migration (246). Its involvement in the induction of Slug expression inhibits E-cadherin, a crucial regulator of cell adhesion. Additionally, the AhR ligand, dibenzo-p-dioxin (TCDD), has been found to enhance the expression and function of MMP9 across various cancers, including melanoma, urothelial, prostate, and gastric cancer cells (247). AhR has also promoted EMT induced by polychlorinated biphenyls in HCC cells (247, 248). In the context of renal cell carcinoma, the study investigated the impact of AhR on EMT development. The findings revealed elevated AhR expression in RCC through the gut microbiota-derived tryptophan metabolite Kyn promoted the migration and invasion of 786-O cells while inhibiting cell death, suggesting a role for AhR in these processes (249).
In CRC, Fusobacterium nucleatum has been studied extensively. F. nucleatum elevates the invasion, proliferation, and recurrence of CRC through the stimulation of pro-inflammatory cytokines (250, 251). It was reported that F. nucleatum enhances the development of metastatic hepatic tumors in CRC by blocking the activity of NK cells (252), restricting the activity of cytotoxic T cells (253), inducing the pro-inflammatory pathways (254), and prompting the overall mucosa-associated inflammation (255). Remarkably, F. nucleatum is translocated to the hepatic metastatic tumor, signifying that F. nucleatum may assist the metastatic tumor cell migration, colonization, and proliferation at distant tumor sites during CRC (236).
In addition, Escherichia coli also interrupts the enterohepatic vessels to establish the premetastatic niche and endorses the recruitment of APCs for a pro-inflammatory environment suitable for colonizing metastatic tumor cells (256). E. coli has been implicated in an elevated risk of colon cancer due to its ability to synthesize colibactin, a genotoxic strain of bacteria. Colibactin induces dysbiosis in the gut microbiota, leading to DNA double-strand breaks and activating the Wnt/β-catenin and NF-κB pathways. This genotoxic effect, coupled with inflammation of the colonic mucosa, creates an environment conducive to cell proliferation, thus promoting the development of colon cancer (257, 258). In addition to E. coli, the gut microbiota harbors Fragilysin, another factor associated with colon cancer. Fragilysin binds to epithelial receptors in the colon, triggering the NF-κB pathway. This activation, in turn, enhances colon cell division, growth, and induces DNA damage (259, 260). Notably, Fragilysin is also implicated in the cleavage of E-cadherin, leading to the deregulation of the Wnt/β-catenin signaling pathway. This process further contributes to cell division and activates c-MYC, emphasizing its role in promoting the development of colon cancer (259–261). These studies prove the role of gut microbiota in the progression of tumors and the development of metastatic tumors at distant sites (Figure 4). Meanwhile, specific experiments do not validate how the microbiome influences cancer metastasis. Thus, additional studies are required to investigate the detailed molecular mechanism of the microbiome regulating the pre-metastatic niche development and the migration and colonization of metastatic tumor cells.
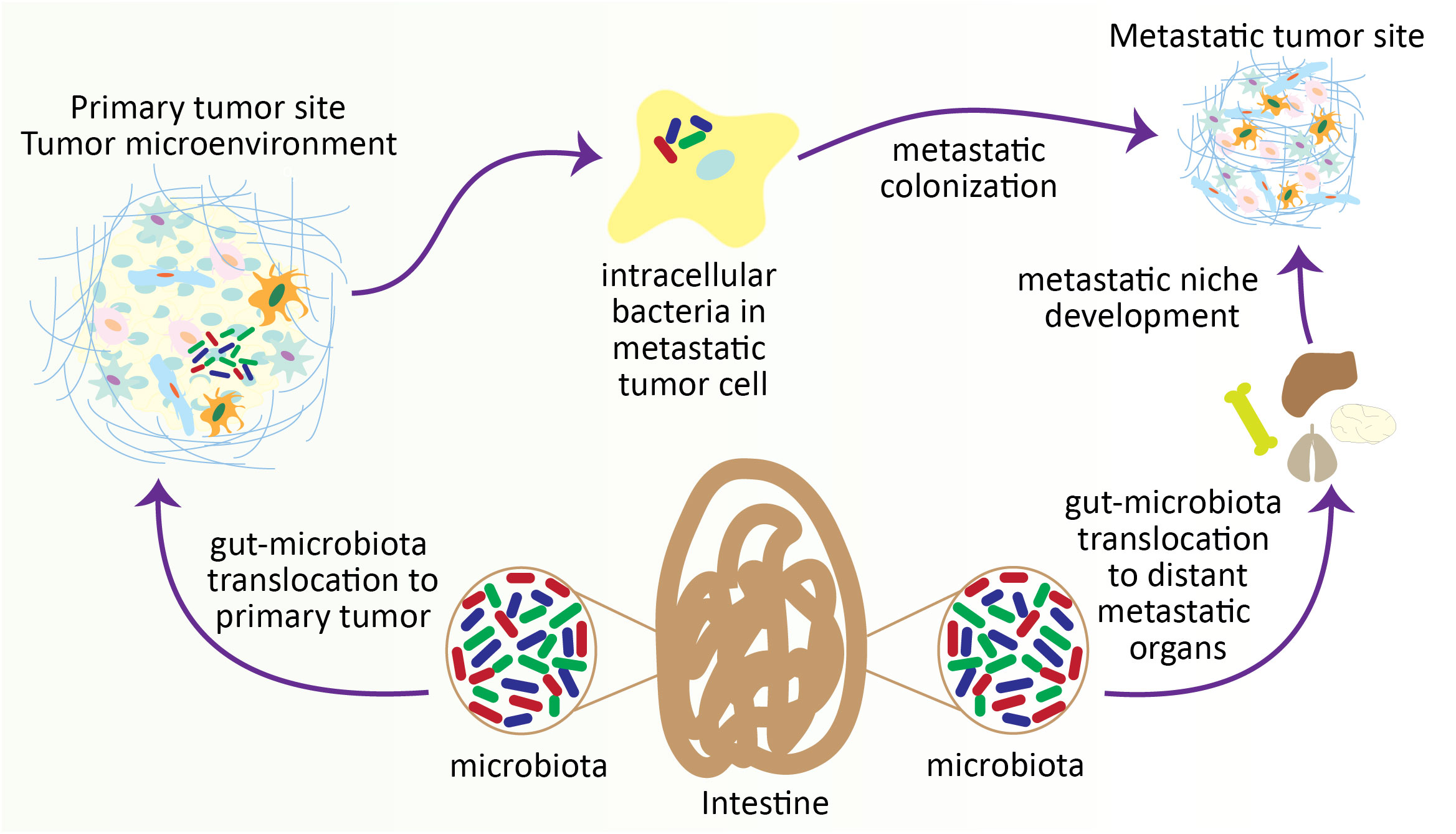
Figure 4 Gut microbiota contributes to colonizing metastatic tumor cells in distant organs. During the progression of tumors, particularly in intestinal tumors, the gut microbiota translocates to the distant metastatic organ and develops a metastatic niche. On the other hand, gut microbiota also penetrates the metastatic tumor cells within the TME and helps the colonization of metastatic tumor cells at the metastatic site.
The circadian clock regulating the circadian rhythm is an evolutionarily conserved mechanism that modulates ample molecular, cellular, and physiological processes (262). It is a 24-hour intrinsic clock that helps our brain regulate the time, being active and resting by retorting the changes in environmental light. Recent advances in cancer research have directed the role of circadian rhythm in cancer progression and metastasis by regulating various cellular processes (263, 264). Circadian rhythm plays a critical role in the metastatic cascade by influencing the dissemination, circulation, and intravasation of metastatic tumor cells, which was initially documented by understanding the role of melatonin (a hormone that acts as a time cue) in the tumor progression and metastasis (265–267). For instance, in a spontaneous mouse model of mammary carcinoma, severe disruption of the circadian rhythm elevates the dissemination and metastasis of tumor cells (Figure 5). In brief, disruption of circadian rhythm aggravates the stemness and tumor-initiating potential of tumor cells, thus diminishing the antitumor immunity (268). Similarly, disruption of the circadian cycle affects distant lymph node metastasis in a transplantation-arbitrated breast carcinoma without distressing the tumor growth at the primary site (269).
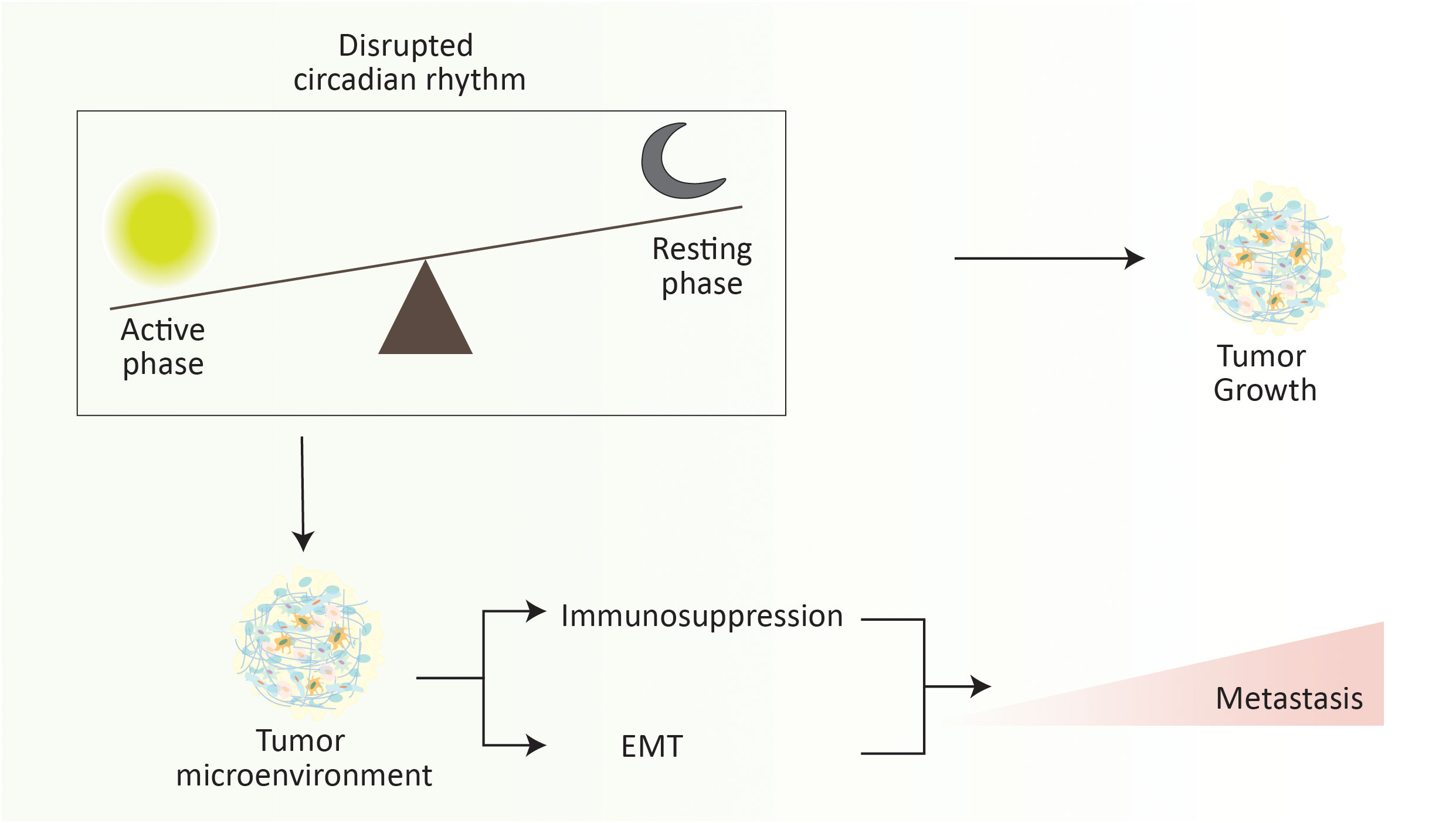
Figure 5 Disruption of circadian rhythm elevates the metastasis progression. Abrupt changes in the active phase and resting phase of an individual significantly increase the metastasis rate. It is noteworthy that the overall rate of metastasis increases during the resting phase. Briefly, disruption of the circadian rhythm promotes the immunosuppressive tumor microenvironment and EMT to elevate the risk of cancer metastasis.
Even though the role of circadian dynamics in the progression of tumor metastasis is poorly understood and has been linked to epidemiological studies (270–272), several experimental studies have discussed the role of circadian rhythm in the metastatic cascade (263, 273). Experimentally, it was validated that intravasation of circulatory tumor cells at distant metastatic sites is highly dependent on regulating the circadian clock, as metastatic breast tumors were increased in both human and animal breast cancer during sleep (273). Similarly, the fluctuation of daily circadian rhythm alters the population of circulatory tumor cells in multiple myeloma (274), and prostate cancer (275). However, the time when these metastatic tumor cells reach the peak is controversial. Bmal1, a circadian clock-related gene, regulates TGF-β expression by targeting PAI-1. It was noted that reduced Baml1 increases TGF-β functions by enhancing the plasmin production, thus leading to tumor metastasis (276).
A recent report has shown that the number of single circulatory tumor cells and the circulatory tumor cell cluster cell exacerbate at the resting phase of circadian rhythm along with an increased rate of intravasation (263). Meanwhile, the rate of metastasis due to heterotypic clusters, such as tumor cells with neutrophils or fibroblasts, increases during the active phase (263). This was previously verified in melanoma cancer, which progressed toward lung metastasis resulting from a circadian pattern. The researchers established that the lungs had more metastatic lesions during the resting phase of the circadian rhythm. Meanwhile, it was also indicated that the accumulation of neutrophils in the lungs aided in developing a metastatic niche in the lung in the resting phase, which supported the localization of metastatic tumor cells (277). In addition, the resting phase also aggravates the development of lung metastasis in a model of breast cancer and also demonstrates the role of circadian rhythm in the development of premetastatic niche and localization of metastatic tumor cell at distant metastatic site (273).
While there is considerable knowledge about circadian rhythms, understanding how they function in various disease states remains limited. Disruption of circadian clocks has been linked to accelerated tumor growth rates and a higher incidence of cancer (278, 279). Recent reports suggest that cancer cells exploit their circadian cycles to promote metastasis through disruption (273). BAML1, an important component of circadian rhythm, has been implicated in stimulating human cancer cell growth, migration, survival, and invasion. The intricate interplay between circadian rhythm and cancer metastasis involves various molecular mechanisms. One such mechanism is the PAI-1-TGF-β-myoCAF-dependent pathway, which promotes metastasis when the clock component BMAL1 is disrupted in mice (276). In colorectal cancer, BMAL1 plays a pivotal role in stimulating the migration and invasion of cancer cells. This effect is mediated through the upregulation of c-Myc expression, a process dependent on the activation of ERK and JNK signaling pathways. The dysregulation of BMAL1 in this context enhances the invasive potential of colorectal cancer cells, contributing to the metastatic cascade (280). Another facet of the circadian regulation of metastasis involves the downregulation of BMAL1 by miR-494-3p in hepatocellular carcinoma. This downregulation results in the upregulation of GPAM-mediated lipid biosynthesis, fostering the growth and metastasis of hepatocellular carcinoma cells (281). Furthermore, in breast cancer, the circadian protein BMAL1 emerges as a regulator of metastasis by influencing the expression of matrix metalloproteinase9 (MMP9). The increased expression of MMP9, orchestrated by BMAL1, enhances the invasive potential of breast cancer cells, contributing to their ability to invade and metastasize (282). Similarly, the downregulation of the circadian rhythm regulator HLF (Hepatic Leukemia Factor) has been identified as a critical factor contributing to the promotion of multiple-organ distant metastases in NSCLC. This phenomenon is mediated through the PPAR/NF-κB signaling pathway (283). Collectively, these findings illuminate the significant role of circadian rhythm, particularly the circadian protein BMAL1, in orchestrating molecular events that impact cancer metastasis. The modulation of key signaling pathways and regulatory networks underscores the complexity of the circadian clock’s influence on the metastatic behavior of various cancer types. The core circadian clock transcription factors (TFs) exhibit dual regulatory roles in influencing the induction of EMT. The expression of EMT-TFs, enhancement of stemness, upregulation of EMT markers, and acquisition of EMT-specific cellular characteristics are intricately linked to the downregulation of PER2 in breast epithelial and cancer cell lines (284). Conversely, another study highlights a contrasting effect, revealing that the downregulation of BMAL1 strengthens the epithelial state of colorectal cancer cells. This downregulation results in reduced cell motility, invasion, and drug resistance, suggesting a suppressive role of BMAL1 in maintaining an epithelial phenotype in colorectal cancer (285). Moreover, disturbances in circadian rhythm leading to lower melatonin levels are associated with EMT in rats. Additionally, higher RSK2 overexpression is linked to increased metastatic dissemination of transplanted breast cancer cells. These observations suggest a potential connection between circadian rhythm disruption, melatonin levels, and RSK2 expression in influencing the induction of EMT and subsequent metastatic processes (286).
As previously known, various genes are involved in the tight regulation of the circadian rhythm (287). Experimental studies have stipulated the role of circadian rhythm in tumor progression and metastasis development through genetic modulation of these genes (288). For instance, genetic ablation of Per1 and Per2 declined the development of hepatic metastatic tumors in mice injected with colon cancer cells. This signifies that hampering the circadian rhythm, particularly in TME, may constrain the colonization of metastatic tumor cells by elevating the tumor-suppressive microenvironment (289).
Therapeutic strategies to treat metastatic cancer: The current therapeutic landscape for metastatic disease employs three primary systemic approaches: chemotherapy, targeted therapy, and immunotherapy, often employed in combination. Targeted therapy, which focuses on drugs targeting tumor-driving oncoproteins, has shown substantial improvements in outcomes across various cancers (290). However, cytotoxic chemotherapy remains a cornerstone in metastatic treatment, particularly for cancer subtypes where targeted options are limited. While targeted therapy has demonstrated success, mutation-specific therapy represents another avenue with the potential for striking but often transient responses to tumors. Unfortunately, these drugs frequently contribute to the growth of tumor subclones carrying drug-resistant mutations or capable of evading specific pathways and secretomes. The ongoing efforts in research utilizing patient biospecimens are swiftly identifying mechanisms of resistance, offering valuable insights and potential avenues for advancements in treatment strategies (290–292). For instance, in NSCLC, where first-generation tyrosine kinase inhibitors (TKIs) like erlotinib and gefitinib improved overall survival in metastatic disease but showed limited efficacy in the adjuvant setting (293, 294). In contrast, the third-generation TKI osimertinib, targeting the drug-resistant EGFRT790M mutant, improved survival rates in both adjuvant and metastatic settings (295–297). This underscores the critical need for the development of more effective drugs targeting subclonal disease-resistance mutations, ideally at an earlier stage in the progression of the disease.
Efforts focused on reducing immunosuppressive cross-talk between immune cells, cancer cells, and other components of the metastatic tumor microenvironment hold significant promise (298, 299). The recent breakthrough of combination multi-kinase inhibitor treatment with ICI in metastatic tumors that were normally immune resistant demonstrates the potential of this strategy (300). Liver metastasis-directed treatments are gaining popularity, encompassing various interventions such as hepatic artery infusion chemotherapy, radiofrequency ablation, and embolization (301). In addition to addressing CRC liver metastasis, recent randomized phase 2 studies have demonstrated promising outcomes in individuals with oligometastatic malignancies. In these cases, consolidative radiation therapy is administered to eliminate residual cancer cells in the tumor bed following the surgical removal of the primary tumor. This combined approach has shown increased overall survival in certain patient populations, showcasing the potential of a multimodal strategy in managing oligometastatic disease (302).
A study found that both epithelial cells and immune cells with malignancies not generally linked with microbial interaction, such as those of the breast, ovary, bone, and brain, had a diverse intratumoral microbiome (303). Fusobacterium strains have been found to be present in both primary and metastatic CRC and breast cancer, as well as to cause metastasis (236, 304). Antibiotic therapy to lower Fusobacterium load lowered metastatic load in mouse xenografts (236). The makeup and variety of the gut microbiome have been linked to ICI response in metastatic malignancies (305, 306). The bulk of the human microbiome’s metabolites, processes, and therapeutic targets have yet to be discovered. Microbes that attack certain tissue types or attach to cancer cell receptors can also function as drug-delivery vehicles (307). In summary, the evolving landscape of metastatic disease treatment emphasizes the importance of advancing therapeutic strategies, developing more effective drugs, and addressing resistance mechanisms to improve overall patient outcomes.
Metastasis is acknowledged as one of the leading causes of death among cancer patients; thus, extra attention is being paid to understanding the detailed molecular mechanisms underlying metastasis. TME is one of the critical modulators of metastasis. TME comprises numerous cell types with altered co-factors that contribute to initiating the metastatic cascade and establishing the metastatic niche, ultimately promoting tumor invasion, colonization, and growth at the secondary site. Among these factors, circadian disruption, hypoxia, gut microbiota, and ECM are the prime contributors to tumor progression and metastasis. However, a few studies illustrate an in-depth mechanism of how these TME factors influence the metastatic cascade and the TME. It should be taken into consideration that the microenvironment at the distant tumor site differs from the microenvironment at the primary site. For instance, a new investigation found that TME in the secondary site in the brain was different from the site of origination in the breast (308, 309). Likewise, it was concluded that the tissue of the primary tumor site, e.g., melanoma, breast, or lung, directs the pattern of TME to be established at the distant tumor site and decides the TME-dependent regulation of metastatic tumor growth (309). Moreover, metastatic-tumor cells further promote metastasis by escorting their primary soil from their original site to the secondary site. However, the ECM composition at the secondary site also exerts moderate effects on the composition and components of the pre-metastatic niche environment (22, 310, 311). Meanwhile, a higher degree of inconsistency has been reported in the deposition and stiffness of ECM within an individual tumor (312). However, it has not been studied why the ECM heterogeneity varies and how it impacts the tumor progression.
Coupling ECM remodeling with ECM stiffness is a leading factor contributing to the rapid progression and development of tumors at secondary sites. Stiffened ECM initiates mechanotransduction signals that induce the excretion of MMPs from stromal and tumor cells (313), improving the reorganization and degradation of ECM (314). Therefore, stiffness and mechanotransduction-associated remodeling of ECM is regarded as a dynamic process in cancer which is crucial for immune evasion, tumor angiogenesis, induction of CAFs, migration, invasion of tumor cells, and cancer metastasis. However, further investigations are necessary to examine the molecular mechanisms underpinning ECM stiffness and remodeling-dependent modulation of the tumor microenvironment, tumor metastasis, and immune surveillance.
Conversely, hypoxia exerts a critical role in the progression of cancer metastasis. It is evident that both earlier and later stages of metastasis are being affected by hypoxia in the TME (186). Meanwhile, hypoxia also influences immune regulation by regulating the immune escaping through the promotion of tumor resistance and immune suppression (315). At the primary tumor site, HIF-mediated signaling regulates angiogenesis, EMT, invasion, and migration to facilitate tumor metastasis. Numerous targets of HIF signaling that mediate the invasion and metastasis can serve as potential targets to restrict the growth and development of metastatic diseases. Interestingly, it has been found that hypoxia and circadian disruption also cooperate with each other to enhance invasion and metastasis (316). Furthermore, gut microbiota act as a major influencer on hypoxia and circadian disruption (317, 318). In summary, this review highlights the role of cellular and non-cellular factors, beyond cytokines, influencing cancer metastasis. In the meantime, various advanced investigations have highlighted the divergent mechanisms through which gut microbiota, circadian disruption, ECM and hypoxia influence the progression of metastatic tumors at secondary sites. It is therefore important to identify the new molecular mechanism, therapeutic targets and biomarkers that could assist in diagnosing and treating metastatic disease.
FR: Data curation, Writing – original draft, Writing – review & editing. JZ: Writing – review & editing. FP: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study is supported by the National Key R&D Program of China (2021YFC2400500 and 2022YFC2403000), the Shenzhen Science and Technology Program (KQTD20210811090115019), the National Natural Science Foundation of China (Grant 32170925), the Shenzhen Science and Technology Program (JCYJ2022081800807016), and the start-up fund of SIAT, CAS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Christofori G. New signals from the invasive front. Nature (2006) 441(7092):444–50. doi: 10.1038/nature04872
2. Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev oncogenesis (2013) 18(1-2):43–73. doi: 10.1615/critrevoncog.v18.i1-2.40
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
4. Hu Z, Curtis C. Looking backward in time to define the chronology of metastasis. Nat Commun (2020) 11(1):3213. doi: 10.1038/s41467-020-16995-y
5. Graham TA, Shibata D. Navigating the path to distant metastasis. Nat Genet (2020) 52(7):642–3. doi: 10.1038/s41588-020-0660-z
6. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med (2006) 12(8):895–904. doi: 10.1038/nm1469
7. Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol (1998) 153(3):865–73. doi: 10.1016/s0002-9440(10)65628-3
8. Fidler IJ. The pathogenesis of cancer metastasis: the 'Seed and soil' Hypothesis revisited. Nat Rev Cancer (2003) 3(6):453–8. doi: 10.1038/nrc1098
9. Talmadge JE, Fidler IJ. Aacr centennial series: the biology of cancer metastasis: historical perspective. Cancer Res (2010) 70(14):5649–69. doi: 10.1158/0008-5472.Can-10-1040
10. Yeung KT, Yang J. Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol (2017) 11(1):28–39. doi: 10.1002/1878-0261.12017
11. Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell (2006) 127(4):679–95. doi: 10.1016/j.cell.2006.11.001
12. Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct Target Ther (2020) 5(1):28. doi: 10.1038/s41392-020-0134-x
13. Celià-Terrassa T, Kang Y. Distinctive properties of metastasis-initiating cells. Genes Dev (2016) 30(8):892–908. doi: 10.1101/gad.277681.116
14. Brabletz T, Lyden D, Steeg PS, Werb Z. Roadblocks to translational advances on metastasis research. Nat Med (2013) 19(9):1104–9. doi: 10.1038/nm.3327
15. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: Cancer J Clin (2020) 70(1):7–30. doi: 10.3322/caac.21590
16. Neophytou CM, Panagi M, Stylianopoulos T, Papageorgis P. The role of tumor microenvironment in cancer metastasis: molecular mechanisms and therapeutic opportunities. Cancers (Basel) (2021) 13(9):2053. doi: 10.3390/cancers13092053
17. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol (2020) 30(16):R921–R5. doi: 10.1016/j.cub.2020.06.081
18. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther (2021) 221:107753. doi: 10.1016/j.pharmthera.2020.107753
19. Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol (2008) 8(9):726–36. doi: 10.1038/nri2395
20. Chen J, Ji T, Wu D, Jiang S, Zhao J, Lin H, et al. Human mesenchymal stem cells promote tumor growth via mapk pathway and metastasis by epithelial mesenchymal transition and integrin α5 in hepatocellular carcinoma. Cell Death Dis (2019) 10(6):425. doi: 10.1038/s41419-019-1622-1
21. Frisbie L, Buckanovich RJ, Coffman L. Carcinoma-associated mesenchymal stem/stromal cells: architects of the pro-tumorigenic tumor microenvironment. Stem Cells (Dayton Ohio) (2022) 40(8):705–15. doi: 10.1093/stmcls/sxac036
22. Duda DG, Duyverman AMMJ, Kohno M, Snuderl M, Steller EJA, Fukumura D, et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci USA (2010) 107(50):21677–82. doi: 10.1073/pnas.1016234107
23. Shinagawa K, Kitadai Y, Tanaka M, Sumida T, Kodama M, Higashi Y, et al. Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int J Cancer (2010) 127(10):2323–33. doi: 10.1002/ijc.25440
24. Rodini CO, Gonçalves da Silva PB, Assoni AF, Carvalho VM, Okamoto OK. Mesenchymal stem cells enhance tumorigenic properties of human glioblastoma through independent cell-cell communication mechanisms. Oncotarget (2018) 9(37):24766–77. doi: 10.18632/oncotarget.25346
25. Yeo RW, Lai RC, Zhang B, Tan SS, Yin Y, Teh BJ, et al. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv Drug Deliv Rev (2013) 65(3):336–41. doi: 10.1016/j.addr.2012.07.001
26. Zhou J, Tan X, Tan Y, Li Q, Ma J, Wang G. Mesenchymal stem cell derived exosomes in cancer progression, metastasis and drug delivery: A comprehensive review. J Cancer (2018) 9(17):3129–37. doi: 10.7150/jca.25376
27. Lin R, Wang S, Zhao RC. Exosomes from human adipose-derived mesenchymal stem cells promote migration through wnt signaling pathway in a breast cancer cell model. Mol Cell Biochem (2013) 383(1-2):13–20. doi: 10.1007/s11010-013-1746-z
28. Rhodes LV, Antoon JW, Muir SE, Elliott S, Beckman BS, Burow ME. Effects of human mesenchymal stem cells on er-positive human breast carcinoma cells mediated through er-sdf-1/cxcr4 crosstalk. Mol Cancer (2010) 9:295. doi: 10.1186/1476-4598-9-295
29. Halpern JL, Kilbarger A, Lynch CC. Mesenchymal stem cells promote mammary cancer cell migration in vitro via the cxcr2 receptor. Cancer Lett (2011) 308(1):91–9. doi: 10.1016/j.canlet.2011.04.018
30. Gu JJ, Hoj J, Rouse C, Pendergast AM. Mesenchymal stem cells promote metastasis through activation of an abl-mmp9 signaling axis in lung cancer cells. PloS One (2020) 15(10):e0241423. doi: 10.1371/journal.pone.0241423
31. Riaz F, Pan F, Wei P. Aryl hydrocarbon receptor: the master regulator of immune responses in allergic diseases. Front Immunol (2022) 13:1057555. doi: 10.3389/fimmu.2022.1057555
32. Brooks J, Eltom S E. Malignant transformation of mammary epithelial cells by ectopic overexpression of the aryl hydrocarbon receptor. Curr Cancer Drug Targets (2011) 11(5):654–69. doi: 10.2174/156800911795655967
33. Stanford EA, Wang Z, Novikov O, Mulas F, Landesman-Bollag E, Monti S, et al. The role of the aryl hydrocarbon receptor in the development of cells with the molecular and functional characteristics of cancer stem-like cells. BMC Biol (2016) 14(1):1–22. doi: 10.1186/s12915-016-0240-y
34. Mohamed HT, Gadalla R, El-Husseiny N, Hassan H, Wang Z, Ibrahim SA, et al. Inflammatory breast cancer: activation of the aryl hydrocarbon receptor and its target cyp1b1 correlates closely with wnt5a/B-β-catenin signalling, the stem cell phenotype and disease progression. J Adv Res (2019) 16:75–86. doi: 10.1016/j.jare.2018.11.006
35. Zhu P, Yu H, Zhou K, Bai Y, Qi R, Zhang S. 3, 3′-diindolylmethane modulates aryl hydrocarbon receptor of esophageal squamous cell carcinoma to reverse epithelial-mesenchymal transition through repressing rhoa/rock1-mediated cox2/pge2 pathway. J Exp Clin Cancer Res (2020) 39(1):1–18. doi: 10.1186/s13046-020-01618-7
36. Zhu P, Zhou K, Lu S, Bai Y, Qi R, Zhang S. Modulation of aryl hydrocarbon receptor inhibits esophageal squamous cell carcinoma progression by repressing cox2/pge2/stat3 axis. J Cell Commun Signal (2020) 14:175–92. doi: 10.1007/s12079-019-00535-5
37. Gilkes DM, Xiang L, Lee SJ, Chaturvedi P, Hubbi ME, Wirtz D, et al. Hypoxia-inducible factors mediate coordinated rhoa-rock1 expression and signaling in breast cancer cells. Proc Natl Acad Sci (2014) 111(3):E384–E93. doi: 10.1073/pnas.1321510111
38. Wang M, Zhao C, Shi H, Zhang B, Zhang L, Zhang X, et al. Deregulated micrornas in gastric cancer tissue-derived mesenchymal stem cells: novel biomarkers and a mechanism for gastric cancer. Br J Cancer (2014) 110(5):1199–210. doi: 10.1038/bjc.2014.14
39. Liu K, Li G, Fan C, Diao Y, Wu B, Li J. Increased expression of microrna-221 in gastric cancer and its clinical significance. J Int Med Res (2012) 40(2):467–74. doi: 10.1177/147323001204000208
40. Gu H, Ji R, Zhang X, Wang M, Zhu W, Qian H, et al. Exosomes derived from human mesenchymal stem cells promote gastric cancer cell growth and migration via the activation of the akt pathway. Mol Med Rep (2016) 14(4):3452–8. doi: 10.3892/mmr.2016.5625
41. Wang J, Hendrix A, Hernot S, Lemaire M, De Bruyne E, Van Valckenborgh E, et al. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood (2014) 124(4):555–66. doi: 10.1182/blood-2014-03-562439
42. Li W, Zhang X, Wu F, Zhou Y, Bao Z, Li H, et al. Gastric cancer-derived mesenchymal stromal cells trigger M2 macrophage polarization that promotes metastasis and emt in gastric cancer. Cell Death Dis (2019) 10(12):918. doi: 10.1038/s41419-019-2131-y
43. Xu Z, Gao H, Zhang Y, Feng W, Miao Y, Xu Z, et al. Ccl7 and tgf-β Secreted by mscs play opposite roles in regulating crc metastasis in a klf5/cxcl5-dependent manner. Mol Ther J Am Soc Gene Ther (2022) 30(6):2327–41. doi: 10.1016/j.ymthe.2022.03.005
44. Wei WF, Zhou HL, Chen PY, Huang XL, Huang L, Liang LJ, et al. Cancer-associated fibroblast-derived pai-1 promotes lymphatic metastasis via the induction of endomt in lymphatic endothelial cells. J Exp Clin Cancer Res CR (2023) 42(1):160. doi: 10.1186/s13046-023-02714-0
45. Alitalo A, Detmar M. Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene (2012) 31(42):4499–508. doi: 10.1038/onc.2011.602
46. Lee E, Pandey NB, Popel AS. Pre-treatment of mice with tumor-conditioned media accelerates metastasis to lymph nodes and lungs: A new spontaneous breast cancer metastasis model. Clin Exp metastasis (2014) 31(1):67–79. doi: 10.1007/s10585-013-9610-9
47. Swartz MA, Lund AW. Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nat Rev Cancer (2012) 12(3):210–9. doi: 10.1038/nrc3186
48. Lee E, Pandey NB, Popel AS. Lymphatic endothelial cells support tumor growth in breast cancer. Sci Rep (2014) 4:5853. doi: 10.1038/srep05853
49. Chen ST, Pan TL, Juan HF, Chen TY, Lin YS, Huang CM. Breast tumor microenvironment: proteomics highlights the treatments targeting secretome. J Proteome Res (2008) 7(4):1379–87. doi: 10.1021/pr700745n
50. Albrecht I, Christofori G. Molecular mechanisms of lymphangiogenesis in development and cancer. Int J Dev Biol (2011) 55(4-5):483–94. doi: 10.1387/ijdb.103226ia
51. Lee E, Fertig EJ, Jin K, Sukumar S, Pandey NB, Popel AS. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat Commun (2014) 5:4715. doi: 10.1038/ncomms5715
52. Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol (2004) 5(1):74–80. doi: 10.1038/ni1013
53. Brichkina A, Polo P, Sharma SD, Visestamkul N, Lauth M. A quick guide to caf subtypes in pancreatic cancer. Cancers (Basel) (2023) 15(9):2614. doi: 10.3390/cancers15092614
54. Knops AM, South A, Rodeck U, Martinez-Outschoorn U, Harshyne LA, Johnson J, et al. Cancer-associated fibroblast density, prognostic characteristics, and recurrence in head and neck squamous cell carcinoma: A meta-analysis. Front Oncol (2020) 10:565306. doi: 10.3389/fonc.2020.565306
55. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer (2021) 20(1):131–. doi: 10.1186/s12943-021-01428-1
56. Puré E, Blomberg R. Pro-tumorigenic roles of fibroblast activation protein in cancer: back to the basics. Oncogene (2018) 37(32):4343–57. doi: 10.1038/s41388-018-0275-3
57. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell (2018) 33(3):463–79.e10. doi: 10.1016/j.ccell.2018.01.011
58. LeBleu VS, Kalluri R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis Models Mech (2018) 11(4):dmm029447. doi: 10.1242/dmm.029447
59. Heneberg P. Paracrine tumor signaling induces transdifferentiation of surrounding fibroblasts. Crit Rev Oncol/Hematol (2016) 97:303–11. doi: 10.1016/j.critrevonc.2015.09.008
60. Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for rhogtpases in leading and following cells. Nat Cell Biol (2007) 9(12):1392–400. doi: 10.1038/ncb1658
61. Eck SM, Côté AL, Winkelman WD, Brinckerhoff CE. Cxcr4 and matrix metalloproteinase-1 are elevated in breast carcinoma-associated fibroblasts and in normal mammary fibroblasts exposed to factors secreted by breast cancer cells. Mol Cancer Res MCR (2009) 7(7):1033–44. doi: 10.1158/1541-7786.Mcr-09-0015
62. Gonzalez-Avila G, Sommer B, Mendoza-Posada DA, Ramos C, Garcia-Hernandez AA, Falfan-Valencia R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit Rev Oncol/Hematol (2019) 137:57–83. doi: 10.1016/j.critrevonc.2019.02.010
63. Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol (2017) 216(11):3799–816. doi: 10.1083/jcb.201704053
64. Räsänen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res (2010) 316(17):2713–22. doi: 10.1016/j.yexcr.2010.04.032
65. Tang X, Hou Y, Yang G, Wang X, Tang S, Du YE, et al. Stromal mir-200s contribute to breast cancer cell invasion through caf activation and ecm remodeling. Cell Death Differ (2016) 23(1):132–45. doi: 10.1038/cdd.2015.78
66. Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med (2011) 17(7):867–74. doi: 10.1038/nm.2379
67. Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature (2011) 481(7379):85–9. doi: 10.1038/nature10694
68. DuFort CC, DelGiorno KE, Hingorani SR. Mounting pressure in the microenvironment: fluids, solids, and cells in pancreatic ductal adenocarcinoma. Gastroenterology (2016) 150(7):1545–57.e2. doi: 10.1053/j.gastro.2016.03.040
69. Bonneau C, Eliès A, Kieffer Y, Bourachot B, Ladoire S, Pelon F, et al. A subset of activated fibroblasts is associated with distant relapse in early luminal breast cancer. Breast Cancer Res BCR (2020) 22(1):76. doi: 10.1186/s13058-020-01311-9
70. Yu B, Wu K, Wang X, Zhang J, Wang L, Jiang Y, et al. Periostin secreted by cancer-associated fibroblasts promotes cancer stemness in head and neck cancer by activating protein tyrosine kinase 7. Cell Death Dis (2018) 9(11):1082. doi: 10.1038/s41419-018-1116-6
71. Shen T, Li Y, Zhu S, Yu J, Zhang B, Chen X, et al. Yap1 plays a key role of the conversion of normal fibroblasts into cancer-associated fibroblasts that contribute to prostate cancer progression. J Exp Clin Cancer Res CR (2020) 39(1):36. doi: 10.1186/s13046-020-1542-z
72. Shi X, Young CD, Zhou H, Wang X. Transforming growth factor-β Signaling in fibrotic diseases and cancer-associated fibroblasts. Biomolecules (2020) 10(12):1666. doi: 10.3390/biom10121666
73. Mazzocca A, Fransvea E, Dituri F, Lupo L, Antonaci S, Giannelli G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatol (Baltimore Md) (2010) 51(2):523–34. doi: 10.1002/hep.23285
74. Goliwas KF, Libring S, Berestesky E, Gholizadeh S, Schwager SC, Frost AR, et al. Mitochondrial transfer from cancer-associated fibroblasts increases migration in aggressive breast cancer. J Cell Sci (2023) 136(14):jcs260419. doi: 10.1242/jcs.260419
75. Koncina E, Nurmik M, Pozdeev VI, Gilson C, Tsenkova M, Begaj R, et al. Il1r1(+) cancer-associated fibroblasts drive tumor development and immunosuppression in colorectal cancer. Nat Commun (2023) 14(1):4251. doi: 10.1038/s41467-023-39953-w
76. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov (2018) 17(12):887–904. doi: 10.1038/nrd.2018.169
77. Ngambenjawong C, Gustafson HH, Pun SH. Progress in tumor-associated macrophage (Tam)-targeted therapeutics. Adv Drug Deliv Rev (2017) 114:206–21. doi: 10.1016/j.addr.2017.04.010
78. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141(1):39–51. doi: 10.1016/j.cell.2010.03.014
79. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol (2019) 19(6):369–82. doi: 10.1038/s41577-019-0127-6
80. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
81. Wei Y, Zhao Q, Gao Z, Lao X-M, Lin W-M, Chen D-P, et al. The local immune landscape determines tumor pd-L1 heterogeneity and sensitivity to therapy. J Clin Invest (2019) 129(8):3347–60. doi: 10.1172/JCI127726
82. Wang HW, Joyce JA. Alternative activation of tumor-associated macrophages by il-4: priming for protumoral functions. Cell Cycle (Georgetown Tex) (2010) 9(24):4824–35. doi: 10.4161/cc.9.24.14322
83. Caux C, Ramos RN, Prendergast GC, Bendriss-Vermare N, Ménétrier-Caux C. A milestone review on how macrophages affect tumor growth. Cancer Res (2016) 76(22):6439–42. doi: 10.1158/0008-5472.Can-16-2631
84. Annamalai RT, Turner PA, WFt C, Levi B, Kunkel S, Stegemann JP. Harnessing macrophage-mediated degradation of gelatin microspheres for spatiotemporal control of bmp2 release. Biomaterials (2018) 161:216–27. doi: 10.1016/j.biomaterials.2018.01.040
85. Hao S, Meng J, Zhang Y, Liu J, Nie X, Wu F, et al. Macrophage phenotypic mechanomodulation of enhancing bone regeneration by superparamagnetic scaffold upon magnetization. Biomaterials (2017) 140:16–25. doi: 10.1016/j.biomaterials.2017.06.013
86. Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C, et al. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int J Dev Biol (2011) 55(7-9):861–7. doi: 10.1387/ijdb.113371dl
87. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-linked barrier defects and microbial products drive il-23/il-17-mediated tumour growth. Nature (2012) 491(7423):254–8. doi: 10.1038/nature11465
88. Kong L, Zhou Y, Bu H, Lv T, Shi Y, Yang J. Deletion of interleukin-6 in monocytes/macrophages suppresses the initiation of hepatocellular carcinoma in mice. J Exp Clin Cancer Res CR (2016) 35(1):131. doi: 10.1186/s13046-016-0412-1
89. Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res (2004) 64(19):7022–9. doi: 10.1158/0008-5472.Can-04-1449
90. Güç E, Pollard JW. Redefining macrophage and neutrophil biology in the metastatic cascade. Immunity (2021) 54(5):885–902. doi: 10.1016/j.immuni.2021.03.022
91. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov (2022) 21(11):799–820. doi: 10.1038/s41573-022-00520-5
92. Li XF, Selli C, Zhou HL, Cao J, Wu S, Ma RY, et al. Macrophages promote anti-androgen resistance in prostate cancer bone disease. J Exp Med (2023) 220(4):e20221007. doi: 10.1084/jem.20221007
93. Wang Y, Ding Y, Guo N, Wang S. Mdscs: key criminals of tumor pre-metastatic niche formation. Front Immunol (2019) 10:172. doi: 10.3389/fimmu.2019.00172
94. Sánchez-León ML, Jiménez-Cortegana C, Silva Romeiro S, Garnacho C, de la Cruz-Merino L, García-Domínguez DJ, et al. Defining the emergence of new immunotherapy approaches in breast cancer: role of myeloid-derived suppressor cells. Int J Mol Sci (2023) 24(6):5208. doi: 10.3390/ijms24065208
95. Huang A, Zhang B, Wang B, Zhang F, Fan KX, Guo YJ. Increased cd14(+)Hla-dr (-/low) myeloid-derived suppressor cells correlate with extrathoracic metastasis and poor response to chemotherapy in non-small cell lung cancer patients. Cancer Immunol Immunother CII (2013) 62(9):1439–51. doi: 10.1007/s00262-013-1450-6
96. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through ido expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol (Baltimore Md 1950) (2013) 190(7):3783–97. doi: 10.4049/jimmunol.1201449
97. Achberger S, Aldrich W, Tubbs R, Crabb JW, Singh AD, Triozzi PL. Circulating immune cell and microrna in patients with uveal melanoma developing metastatic disease. Mol Immunol (2014) 58(2):182–6. doi: 10.1016/j.molimm.2013.11.018
98. Weide B, Martens A, Zelba H, Stutz C, Derhovanessian E, Di Giacomo AM, et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: comparison with regulatory T cells and ny-eso-1- or melan-a-specific T cells. Clin Cancer Res an Off J Am Assoc Cancer Res (2014) 20(6):1601–9. doi: 10.1158/1078-0432.ccr-13-2508
99. Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Müller M, et al. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med (2010) 207(7):1453–64. doi: 10.1084/jem.20091474
100. Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, et al. A cxcl1 paracrine network links cancer chemoresistance and metastasis. Cell (2012) 150(1):165–78. doi: 10.1016/j.cell.2012.04.042
101. Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PloS Biol (2011) 9(9):e1001162. doi: 10.1371/journal.pbio.1001162
102. Wang D, Sun H, Wei J, Cen B, DuBois RN. Cxcl1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res (2017) 77(13):3655–65. doi: 10.1158/0008-5472.can-16-3199
103. Seubert B, Grünwald B, Kobuch J, Cui H, Schelter F, Schaten S, et al. Tissue inhibitor of metalloproteinases (Timp)-1 creates a premetastatic niche in the liver through sdf-1/cxcr4-dependent neutrophil recruitment in mice. Hepatol (Baltimore Md) (2015) 61(1):238–48. doi: 10.1002/hep.27378
104. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21(8):485–98. doi: 10.1038/s41577-020-00490-y
105. Patel S, Fu S, Mastio J, Dominguez GA, Purohit A, Kossenkov A, et al. Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol (2018) 19(11):1236–47. doi: 10.1038/s41590-018-0229-5
106. Li P, Lu M, Shi J, Gong Z, Hua L, Li Q, et al. Lung mesenchymal cells elicit lipid storage in neutrophils that fuel breast cancer lung metastasis. Nat Immunol (2020) 21(11):1444–55. doi: 10.1038/s41590-020-0783-5
107. Gao D, Vahdat LT, Wong S, Chang JC, Mittal V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res (2012) 72(19):4883–9. doi: 10.1158/0008-5472.can-12-1223
108. Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microrna101 and suppressing the corepressor ctbp2. Immunity (2013) 39(3):611–21. doi: 10.1016/j.immuni.2013.08.025
109. Panni RZ, Sanford DE, Belt BA, Mitchem JB, Worley LA, Goetz BD, et al. Tumor-induced stat3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother CII (2014) 63(5):513–28. doi: 10.1007/s00262-014-1527-x
110. Catena R, Bhattacharya N, El Rayes T, Wang S, Choi H, Gao D, et al. Bone marrow-derived gr1+ Cells can generate a metastasis-resistant microenvironment via induced secretion of thrombospondin-1. Cancer Discov (2013) 3(5):578–89. doi: 10.1158/2159-8290.cd-12-0476
111. Zheng Y, Wang N, Wang S, Zhang J, Yang B, Wang Z. Chronic psychological stress promotes breast cancer pre-metastatic niche formation by mobilizing splenic mdscs via tam/cxcl1 signaling. J Exp Clin Cancer Res (2023) 42(1):1–21. doi: 10.1186/s13046-023-02696-z
112. Conche C, Finkelmeier F, Pešić M, Nicolas AM, Böttger TW, Kennel KB, et al. Combining ferroptosis induction with mdsc blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut (2023) 72(9):1774–82. doi: 10.1136/gutjnl-2022-327909
113. Jiménez-Sánchez A, Memon D, Pourpe S, Veeraraghavan H, Li Y, Vargas HA, et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell (2017) 170(5):927–38.e20. doi: 10.1016/j.cell.2017.07.025
114. Piranlioglu R, Lee E, Ouzounova M, Bollag RJ, Vinyard AH, Arbab AS, et al. Primary tumor-induced immunity eradicates disseminated tumor cells in syngeneic mouse model. Nat Commun (2019) 10(1):1430. doi: 10.1038/s41467-019-09015-1
115. Eyles J, Puaux A-L, Wang X, Toh B, Prakash C, Hong M, et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest (2010) 120(6):2030–9. doi: 10.1172/JCI42002
116. Kloten V, Lampignano R, Krahn T, Schlange T. Circulating tumor cell pd-L1 expression as biomarker for therapeutic efficacy of immune checkpoint inhibition in nsclc. Cells (2019) 8(8):809. doi: 10.3390/cells8080809
117. Cheng Y, Wang T, Lv X, Li R, Yuan L, Shen J, et al. Detection of pd-L1 expression and its clinical significance in circulating tumor cells from patients with non-small-cell lung cancer. Cancer Manage Res (2020) 12:2069–78. doi: 10.2147/CMAR.S245425
118. Wang HB, Yao H, Li CS, Liang LX, Zhang Y, Chen YX, et al. Rise of pd-L1 expression during metastasis of colorectal cancer: implications for immunotherapy. J Dig Dis (2017) 18(10):574–81. doi: 10.1111/1751-2980.12538
119. Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, Yan R, et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science (2018) 360(6394):eaao4908. doi: 10.1126/science.aao4908
120. Agudo J, Park ES, Rose SA, Alibo E, Sweeney R, Dhainaut M, et al. Quiescent tissue stem cells evade immune surveillance. Immunity (2018) 48(2):271–85.e5. doi: 10.1016/j.immuni.2018.02.001
121. Tallón de Lara P, Castañón H, Sterpi M, van den Broek M. Antimetastatic defense by cd8(+) T cells. Trends Cancer (2022) 8(2):145–57. doi: 10.1016/j.trecan.2021.10.006
122. Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med (2005) 353(25):2654–66. doi: 10.1056/NEJMoa051424
123. Wei P, Kou W, Riaz F, Zhang K, Fu J, Pan F. Combination therapy of hifα Inhibitors and treg depletion strengthen the anti-tumor immunity in mice. Eur J Immunol (2023) 53(12):2250182. doi: 10.1002/eji.202250182
124. Riaz F, Wei P, Pan F. Inhibiting hypoxia-inducible factor in T cells strengthens effector-memory function in cd8+ T cells. J Cell Mol Immunol (2023) 2(1):31–4. doi: 10.46439/immunol.2.021
125. Monteiro AC, Leal AC, Gonçalves-Silva T, Mercadante AC, Kestelman F, Chaves SB, et al. T Cells induce Pre-Metastatic Osteolytic disease and Help Bone Metastases Establishment in a Mouse Model of Metastatic Breast Cancer. PloS One (2013) 8(7):e68171. doi: 10.1371/journal.pone.0068171
126. Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, et al. Breast cancer lung metastasis requires expression of chemokine receptor ccr4 and regulatory T cells. Cancer Res (2009) 69(14):5996–6004. doi: 10.1158/0008-5472.can-08-4619
127. Riaz F, Wei P, Pan F. Fine-tuning of regulatory T cells is indispensable for the metabolic steatosis-related hepatocellular carcinoma: A review. Front Cell Dev Biol (2022) 10:949603. doi: 10.3389/fcell.2022.949603
128. Riaz F, Huang Z, Pan F. Targeting post-translational modifications of foxp3: A new paradigm for regulatory T cell-specific therapy. Front Immunol (2023) 14. doi: 10.3389/fimmu.2023.1280741
129. Huppert LA, Green MD, Kim L, Chow C, Leyfman Y, Daud AI, et al. Tissue-specific tregs in cancer metastasis: opportunities for precision immunotherapy. Cell Mol Immunol (2022) 19(1):33–45. doi: 10.1038/s41423-021-00742-4
130. Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, et al. Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell (2016) 166(5):1117–31.e14. doi: 10.1016/j.cell.2016.07.032
131. Yan HH, Pickup M, Pang Y, Gorska AE, Li Z, Chytil A, et al. Gr-1+ Cd11b+ Myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res (2010) 70(15):6139–49. doi: 10.1158/0008-5472.CAN-10-0706
132. López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by nk cells. Cancer Cell (2017) 32(2):135–54. doi: 10.1016/j.ccell.2017.06.009
133. Vyas M, Requesens M, Nguyen TH, Peigney D, Azin M, Demehri S. Natural killer cells suppress cancer metastasis by eliminating circulating cancer cells. Front Immunol (2023) 13:1098445. doi: 10.3389/fimmu.2022.1098445
134. Duan S, Guo W, Xu Z, He Y, Liang C, Mo Y, et al. Natural killer group 2d receptor and its ligands in cancer immune escape. Mol Cancer (2019) 18(1):1–14. doi: 10.1186/s12943-019-0956-8
135. Bui JD, Carayannopoulos LN, Lanier LL, Yokoyama WM, Schreiber RD. Ifn-dependent down-regulation of the nkg2d ligand H60 on tumors. J Immunol (2006) 176(2):905–13. doi: 10.4049/jimmunol.176.2.905
136. Carrette F, Vivier E. Nkg2a blocks the anti-metastatic functions of natural killer cells. Cancer Cell (2023) 41(2):232–4. doi: 10.1016/j.ccell.2023.01.008
137. Zingoni A, Cecere F, Vulpis E, Fionda C, Molfetta R, Soriani A, et al. Genotoxic stress induces senescence-associated adam10-dependent release of nkg2d mic ligands in multiple myeloma cells. J Immunol (2015) 195(2):736–48. doi: 10.4049/jimmunol.1402643
138. Raneros AB, Suarez-Álvarez B, López-Larrea C. Secretory pathways generating immunosuppressive nkg2d ligands: new targets for therapeutic intervention. Oncoimmunology (2014) 3:e28497. doi: 10.4161/onci.28497
139. Sun D, Wang X, Zhang H, Deng L, Zhang Y. Mmp9 mediates mica shedding in human osteosarcomas. Cell Biol Int (2011) 35(6):569–74. doi: 10.1042/CBI20100431
140. Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic latency and immune evasion through autocrine inhibition of wnt. Cell (2016) 165(1):45–60. doi: 10.1016/j.cell.2016.02.025
141. Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallée V-P, et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat Med (2020) 26(2):259–69. doi: 10.1038/s41591-019-0750-6
142. Komura N, Mabuchi S, Yokoi E, Kozasa K, Kuroda H, Sasano T, et al. Comparison of clinical utility between neutrophil count and neutrophil–lymphocyte ratio in patients with ovarian cancer: A single institutional experience and a literature review. Int J Clin Oncol (2018) 23:104–13. doi: 10.1007/s10147-017-1180-4
143. Lo HC, Xu Z, Kim IS, Pingel B, Aguirre S, Kodali S, et al. Resistance to natural killer cell immunosurveillance confers a selective advantage to polyclonal metastasis. Nat Cancer (2020) 1(7):709–22. doi: 10.1038/s43018-020-0068-9
144. Nakamura K, Smyth MJ. Immunoediting of cancer metastasis by nk cells. Nat Cancer (2020) 1(7):670–1. doi: 10.1038/s43018-020-0081-z
145. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci (2010) 123(Pt 24):4195–200. doi: 10.1242/jcs.023820
146. Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: A multiscale deconstruction. Nat Rev Mol Cell Biol (2014) 15(12):771–85. doi: 10.1038/nrm3902
147. Kai F, Drain AP, Weaver VM. The extracellular matrix modulates the metastatic journey. Dev Cell (2019) 49(3):332–46. doi: 10.1016/j.devcel.2019.03.026
148. Poltavets V, Kochetkova M, Pitson SM, Samuel MS. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Front Oncol (2018) 8:431. doi: 10.3389/fonc.2018.00431
149. Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim Biophys Acta (2014) 1840(8):2506–19. doi: 10.1016/j.bbagen.2014.01.010
150. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi: 10.1038/nm.3394
151. Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev (2013) 27(20):2192–206. doi: 10.1101/gad.225334.113
152. Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep (2014) 15(12):1243–53. doi: 10.15252/embr.201439246
153. Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, et al. Human breast cancer invasion and aggression correlates with ecm stiffening and immune cell infiltration. Integr Biol (Camb) (2015) 7(10):1120–34. doi: 10.1039/c5ib00040h
154. Hinz B. The extracellular matrix and transforming growth factor-β1: tale of a strained relationship. Matrix Biol J Int Soc Matrix Biol (2015) 47:54–65. doi: 10.1016/j.matbio.2015.05.006
155. Liverani C, Mercatali L, Cristofolini L, Giordano E, Minardi S, Porta GD, et al. Investigating the mechanobiology of cancer cell-ecm interaction through collagen-based 3d scaffolds. Cell Mol Bioeng (2017) 10(3):223–34. doi: 10.1007/s12195-017-0483-x
156. Tuxhorn JA, Ayala GE, Smith MJ, Smith VC, Dang TD, Rowley DR. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin Cancer Res an Off J Am Assoc Cancer Res (2002) 8(9):2912–23.
157. Campbell BH, Agarwal C, Wang JH. Tgf-beta1, tgf-beta3, and pge(2) regulate contraction of human patellar tendon fibroblasts. Biomech Model Mechanobiol (2004) 2(4):239–45. doi: 10.1007/s10237-004-0041-z
158. Lu P, Weaver VM, Werb Z. The extracellular matrix: A dynamic niche in cancer progression. J Cell Biol (2012) 196(4):395–406. doi: 10.1083/jcb.201102147
159. Bondareva A, Downey CM, Ayres F, Liu W, Boyd SK, Hallgrimsson B, et al. The lysyl oxidase inhibitor, beta-aminopropionitrile, diminishes the metastatic colonization potential of circulating breast cancer cells. PloS One (2009) 4(5):e5620. doi: 10.1371/journal.pone.0005620
160. Pickup MW, Laklai H, Acerbi I, Owens P, Gorska AE, Chytil A, et al. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-β-deficient mouse mammary carcinomas. Cancer Res (2013) 73(17):5336–46. doi: 10.1158/0008-5472.Can-13-0012
161. van Helvert S, Storm C, Friedl P. Mechanoreciprocity in cell migration. Nat Cell Biol (2018) 20(1):8–20. doi: 10.1038/s41556-017-0012-0
162. De Wever O, Nguyen QD, Van Hoorde L, Bracke M, Bruyneel E, Gespach C, et al. Tenascin-C and sf/hgf produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through rhoa and rac. FASEB J Off Publ Fed Am Societies Exp Biol (2004) 18(9):1016–8. doi: 10.1096/fj.03-1110fje
163. Rafaeva M, Jensen ARD, Horton ER, Zornhagen KW, Strøbech JE, Fleischhauer L, et al. Fibroblast-derived matrix models desmoplastic properties and forms a prognostic signature in cancer progression. Front Immunol (2023) 14:1154528. doi: 10.3389/fimmu.2023.1154528
164. Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S, et al. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target Ther (2021) 6(1):153–. doi: 10.1038/s41392-021-00544-0
165. Kai F, Laklai H, Weaver VM. Force matters: biomechanical regulation of cell invasion and migration in disease. Trends Cell Biol (2016) 26(7):486–97. doi: 10.1016/j.tcb.2016.03.007
166. Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and β-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell (2011) 19(6):776–91. doi: 10.1016/j.ccr.2011.05.008
167. Shi Q, Boettiger D. A novel mode for integrin-mediated signaling: tethering is required for phosphorylation of fak Y397. Mol Biol Cell (2003) 14(10):4306–15. doi: 10.1091/mbc.e03-01-0046
168. Lawson CD, Burridge K. The on-off relationship of rho and rac during integrin-mediated adhesion and cell migration. Small GTPases (2014) 5:e27958–e. doi: 10.4161/sgtp.27958
169. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell (2009) 139(5):891–906. doi: 10.1016/j.cell.2009.10.027
170. Rubashkin MG, Cassereau L, Bainer R, DuFort CC, Yui Y, Ou G, et al. Force engages vinculin and promotes tumor progression by enhancing pi3k activation of phosphatidylinositol (3,4,5)-triphosphate. Cancer Res (2014) 74(17):4597–611. doi: 10.1158/0008-5472.CAN-13-3698
171. Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting pi3k in cancer: mechanisms and advances in clinical trials. Mol Cancer (2019) 18(1):26. doi: 10.1186/s12943-019-0954-x
172. Yu F-X, Zhao B, Guan K-L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell (2015) 163(4):811–28. doi: 10.1016/j.cell.2015.10.044
173. Ni X, Tao J, Barbi J, Chen Q, Park BV, Li Z, et al. Yap is essential for treg-mediated suppression of antitumor immunity. Cancer Discov (2018) 8(8):1026–43. doi: 10.1158/2159-8290.Cd-17-1124
174. Chen YA, Lu CY, Cheng TY, Pan SH, Chen HF, Chang NS. Ww domain-containing proteins yap and taz in the hippo pathway as key regulators in stemness maintenance, tissue homeostasis, and tumorigenesis. Front Oncol (2019) 9:60. doi: 10.3389/fonc.2019.00060
175. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of yap/taz in mechanotransduction. Nature (2011) 474(7350):179–83. doi: 10.1038/nature10137
176. Xu Z, Orkwis JA, Harris GM. Cell shape and matrix stiffness impact schwann cell plasticity via yap/taz and rho gtpases. Int J Mol Sci (2021) 22(9):4821. doi: 10.3390/ijms22094821
177. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of yap and taz in physiology and disease. Nat Rev Mol Cell Biol (2017) 18(12):758–70. doi: 10.1038/nrm.2017.87
178. Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a twist1-G3bp2 mechanotransduction pathway. Nat Cell Biol (2015) 17(5):678–88. doi: 10.1038/ncb3157
179. Dobrokhotov O, Samsonov M, Sokabe M, Hirata H. Mechanoregulation and pathology of yap/taz via hippo and non-hippo mechanisms. Clin Transl Med (2018) 7(1):23–. doi: 10.1186/s40169-018-0202-9
180. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell (2012) 148(3):399–408. doi: 10.1016/j.cell.2012.01.021
181. Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer (1955) 9(4):539–49. doi: 10.1038/bjc.1955.55
182. Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol (2014) 9:47–71. doi: 10.1146/annurev-pathol-012513-104720
183. Hayashi Y, Yokota A, Harada H, Huang G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1α in cancer. Cancer Sci (2019) 110(5):1510–7. doi: 10.1111/cas.13990
184. Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer metastasis Rev (2007) 26(2):225–39. doi: 10.1007/s10555-007-9055-1
185. Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res (1998) 58(7):1408–16.
186. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science (2016) 352(6282):175–80. doi: 10.1126/science.aaf4405
187. Azab AK, Hu J, Quang P, Azab F, Pitsillides C, Awwad R, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood (2012) 119(24):5782–94. doi: 10.1182/blood-2011-09-380410
188. Muz B, de la Puente P, Azab F, Ghobrial IM, Azab AK. Hypoxia promotes dissemination and colonization in new bone marrow niches in waldenström macroglobulinemia. Mol Cancer Res MCR (2015) 13(2):263–72. doi: 10.1158/1541-7786.Mcr-14-0150
189. Cairns RA, Hill RP. Acute hypoxia enhances spontaneous lymph node metastasis in an orthotopic murine model of human cervical carcinoma. Cancer Res (2004) 64(6):2054–61. doi: 10.1158/0008-5472.can-03-3196
190. Cairns RA, Kalliomaki T, Hill RP. Acute (Cyclic) hypoxia enhances spontaneous metastasis of kht murine tumors. Cancer Res (2001) 61(24):8903–8.
191. Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ (2008) 15(4):678–85. doi: 10.1038/cdd.2008.21
192. Schindl M, Schoppmann SF, Samonigg H, Hausmaninger H, Kwasny W, Gnant M, et al. Overexpression of hypoxia-inducible factor 1alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2002) 8(6):1831–7.
193. Yamamoto Y, Ibusuki M, Okumura Y, Kawasoe T, Kai K, Iyama K, et al. Hypoxia-inducible factor 1alpha is closely linked to an aggressive phenotype in breast cancer. Breast Cancer Res Treat (2008) 110(3):465–75. doi: 10.1007/s10549-007-9742-1
194. Acloque H, Thiery JP, Nieto MA. The physiology and pathology of the emt. Meeting on the epithelial-mesenchymal transition. EMBO Rep (2008) 9(4):322–6. doi: 10.1038/embor.2008.30
195. Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, et al. Pten loss and ras/mapk activation cooperate to promote emt and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res (2012) 72(7):1878–89. doi: 10.1158/0008-5472.Can-11-3132
196. Tian M, Neil JR, Schiemann WP. Transforming growth factor-β and the hallmarks of cancer. Cell signalling (2011) 23(6):951–62. doi: 10.1016/j.cellsig.2010.10.015
197. Jiang J, Tang YL, Liang XH. Emt: A new vision of hypoxia promoting cancer progression. Cancer Biol Ther (2011) 11(8):714–23. doi: 10.4161/cbt.11.8.15274
198. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol (2006) 7(2):131–42. doi: 10.1038/nrm1835
199. Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/lef-1 signaling pathway in induction of emt. Cell Biol Int (2002) 26(5):463–76. doi: 10.1006/cbir.2002.0901
200. Manotham K, Tanaka T, Matsumoto M, Ohse T, Inagi R, Miyata T, et al. Transdifferentiation of cultured tubular cells induced by hypoxia. Kidney Int (2004) 65(3):871–80. doi: 10.1111/j.1523-1755.2004.00461.x
201. Hsu M, Andl T, Li G, Meinkoth JL, Herlyn M. Cadherin repertoire determines partner-specific gap junctional communication during melanoma progression. J Cell Sci (2000) 113(Pt 9):1535–42. doi: 10.1242/jcs.113.9.1535
202. Kaelin WG Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the hif hydroxylase pathway. Mol Cell (2008) 30(4):393–402. doi: 10.1016/j.molcel.2008.04.009
203. Hanna SC, Krishnan B, Bailey ST, Moschos SJ, Kuan PF, Shimamura T, et al. Hif1α and hif2α Independently activate src to promote melanoma metastases. J Clin Invest (2013) 123(5):2078–93. doi: 10.1172/jci66715
204. Erler JT, Giaccia AJ. Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res (2006) 66(21):10238–41. doi: 10.1158/0008-5472.Can-06-3197
205. Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell (2009) 15(1):35–44. doi: 10.1016/j.ccr.2008.11.012
206. Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase ix inhibitors. Cancer Res (2011) 71(9):3364–76. doi: 10.1158/0008-5472.Can-10-4261
207. Azab AK, Azab F, Blotta S, Pitsillides CM, Thompson B, Runnels JM, et al. Rhoa and rac1 gtpases play major and differential roles in stromal cell-derived factor-1-induced cell adhesion and chemotaxis in multiple myeloma. Blood (2009) 114(3):619–29. doi: 10.1182/blood-2009-01-199281
208. Azab AK, Runnels JM, Pitsillides C, Moreau A-S, Azab F, Leleu X, et al. Cxcr4 inhibitor amd3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood (2009) 113(18):4341–51. doi: 10.1182/blood-2008-10-186668
209. Azab AK, Weisberg E, Sahin I, Liu F, Awwad R, Azab F, et al. The influence of hypoxia on cml trafficking through modulation of cxcr4 and E-cadherin expression. Leukemia (2013) 27(4):961–4. doi: 10.1038/leu.2012.353
210. Saxena K, Jolly MK. Acute vs. Chronic vs. Cyclic hypoxia: their differential dynamics, molecular mechanisms, and effects on tumor progression. Biomolecules (2019) 9(8):339. doi: 10.3390/biom9080339
211. Matsumoto S, Yasui H, Mitchell JB, Krishna MC. Imaging cycling tumor hypoxia. Cancer Res (2010) 70(24):10019–23. doi: 10.1158/0008-5472.Can-10-2821
212. Saxena K, Jolly MK, Balamurugan K. Hypoxia, partial emt and collective migration: emerging culprits in metastasis. Transl Oncol (2020) 13(11):100845–. doi: 10.1016/j.tranon.2020.100845
213. Balamurugan K. Hif-1 at the crossroads of hypoxia, inflammation, and cancer. Int J Cancer (2016) 138(5):1058–66. doi: 10.1002/ijc.29519
214. Copple BL. Hypoxia stimulates hepatocyte epithelial to mesenchymal transition by hypoxia-inducible factor and transforming growth factor-beta-dependent mechanisms. Liver Int Off J Int Assoc Study Liver (2010) 30(5):669–82. doi: 10.1111/j.1478-3231.2010.02205.x
215. Nishi H, Nakada T, Hokamura M, Osakabe Y, Itokazu O, Huang LE, et al. Hypoxia-inducible factor-1 transactivates transforming growth factor-beta3 in trophoblast. Endocrinology (2004) 145(9):4113–8. doi: 10.1210/en.2003-1639
216. Zhang H, Akman HO, Smith EL, Zhao J, Murphy-Ullrich JE, Batuman OA. Cellular response to hypoxia involves signaling via smad proteins. Blood (2003) 101(6):2253–60. doi: 10.1182/blood-2002-02-0629
217. McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of phd2 expression. J Biol Chem (2006) 281(34):24171–81. doi: 10.1074/jbc.M604507200
218. Peng J, Wang X, Ran L, Song J, Luo R, Wang Y. Hypoxia-inducible factor 1α Regulates the transforming growth factor β1/smad family member 3 pathway to promote breast cancer progression. J Breast Cancer (2018) 21(3):259–66. doi: 10.4048/jbc.2018.21.e42
219. Lei J, Ma J, Ma Q, Li X, Liu H, Xu Q, et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol Cancer (2013) 12:66. doi: 10.1186/1476-4598-12-66
220. Jiang YG, Luo Y, He DL, Li X, Zhang LL, Peng T, et al. Role of wnt/beta-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1alpha. Int J Urol Off J Japanese Urological Assoc (2007) 14(11):1034–9. doi: 10.1111/j.1442-2042.2007.01866.x
221. Zhang Q, Bai X, Chen W, Ma T, Hu Q, Liang C, et al. Wnt/β-catenin signaling enhances hypoxia-induced epithelial-mesenchymal transition in hepatocellular carcinoma via crosstalk with hif-1α Signaling. Carcinogenesis (2013) 34(5):962–73. doi: 10.1093/carcin/bgt027
222. Tang C, Liu T, Wang K, Wang X, Xu S, He D, et al. Transcriptional regulation of foxm1 by hif−1α Mediates hypoxia−Induced emt in prostate cancer. Oncol Rep (2019) 42(4):1307–18. doi: 10.3892/or.2019.7248
223. Ma C, Guo Y, Zhang Y, Duo A, Jia Y, Liu C, et al. Pafah1b2 is a hif1a target gene and promotes metastasis in pancreatic cancer. Biochem Biophys Res Commun (2018) 501(3):654–60. doi: 10.1016/j.bbrc.2018.05.039
224. Chou CC, Chuang HC, Salunke SB, Kulp SK, Chen CS. A novel hif-1α-integrin-linked kinase regulatory loop that facilitates hypoxia-induced hif-1α Expression and epithelial-mesenchymal transition in cancer cells. Oncotarget (2015) 6(10):8271–85. doi: 10.18632/oncotarget.3186
225. Li Y, Patel SP, Roszik J, Qin Y. Hypoxia-driven immunosuppressive metabolites in the tumor microenvironment: new approaches for combinational immunotherapy. Front Immunol (2018) 9:1591. doi: 10.3389/fimmu.2018.01591
226. Terry S, Savagner P, Ortiz-Cuaran S, Mahjoubi L, Saintigny P, Thiery JP, et al. New insights into the role of emt in tumor immune escape. Mol Oncol (2017) 11(7):824–46. doi: 10.1002/1878-0261.12093
227. Ricciardi M, Zanotto M, Malpeli G, Bassi G, Perbellini O, Chilosi M, et al. Epithelial-to-mesenchymal transition (Emt) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br J Cancer (2015) 112(6):1067–75. doi: 10.1038/bjc.2015.29
228. Azab AK, Sahin I, Moschetta M, Mishima Y, Burwick N, Zimmermann J, et al. Cxcr7-dependent angiogenic mononuclear cell trafficking regulates tumor progression in multiple myeloma. Blood (2014) 124(12):1905–14. doi: 10.1182/blood-2014-02-558742
229. Semenza GL. Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis. Oncogene (2013) 32(35):4057–63. doi: 10.1038/onc.2012.578
230. Xavier JB, Young VB, Skufca J, Ginty F, Testerman T, Pearson AT, et al. The cancer microbiome: distinguishing direct and indirect effects requires a systemic view. Trends Cancer (2020) 6(3):192–204. doi: 10.1016/j.trecan.2020.01.004
231. Park EM, Chelvanambi M, Bhutiani N, Kroemer G, Zitvogel L, Wargo JA. Targeting the gut and tumor microbiota in cancer. Nat Med (2022) 28(4):690–703. doi: 10.1038/s41591-022-01779-2
232. Liu Y, Baba Y, Ishimoto T, Gu X, Zhang J, Nomoto D, et al. Gut microbiome in gastrointestinal cancer: A friend or foe? Int J Biol Sci (2022) 18(10):4101–17. doi: 10.7150/ijbs.69331
233. Fu A, Yao B, Dong T, Cai S. Emerging roles of intratumor microbiota in cancer metastasis. Trends Cell Biol (2022) 33(7):583–93. doi: 10.1016/j.tcb.2022.11.007
234. Chrysostomou D, Roberts LA, Marchesi JR, Kinross JM. Gut microbiota modulation of efficacy and toxicity of cancer chemotherapy and immunotherapy. Gastroenterology (2023) 164(2):198–213. doi: 10.1053/j.gastro.2022.10.018
235. Roy S, Trinchieri G. Microbiota: A key orchestrator of cancer therapy. Nat Rev Cancer (2017) 17(5):271–85. doi: 10.1038/nrc.2017.13
236. Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, et al. Analysis of fusobacterium persistence and antibiotic response in colorectal cancer. Science (2017) 358(6369):1443–8. doi: 10.1126/science.aal5240
237. Fu A, Yao B, Dong T, Chen Y, Yao J, Liu Y, et al. Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell (2022) 185(8):1356–72.e26. doi: 10.1016/j.cell.2022.02.027
238. Kim SH, Lim YJ. The role of microbiome in colorectal carcinogenesis and its clinical potential as a target for cancer treatment. Intestinal Res (2022) 20(1):31–42. doi: 10.5217/ir.2021.00034
239. Li Y, Wang S, Sun Y, Xu W, Zheng H, Wang Y, et al. Apple Polysaccharide Protects Icr Mice against Colitis Associated Colorectal Cancer through the Regulation of Microbial Dysbiosis. Carbohydr polymers (2020) 230:115726. doi: 10.1016/j.carbpol.2019.115726
240. Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I, Begus-Nahrmann Y, et al. Loss of P53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell (2013) 23(1):93–106. doi: 10.1016/j.ccr.2012.11.014
241. Zaika AI, Wei J, Noto JM, Peek RM. Microbial regulation of P53 tumor suppressor. PloS Pathog (2015) 11(9):e1005099. doi: 10.1371/journal.ppat.1005099
242. Sethi V, Kurtom S, Tarique M, Lavania S, Malchiodi Z, Hellmund L, et al. Gut microbiota promotes tumor growth in mice by modulating immune response. Gastroenterology (2018) 155(1):33–7.e6. doi: 10.1053/j.gastro.2018.04.001
243. Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and tlr4. Cancer Cell (2012) 21(4):504–16. doi: 10.1016/j.ccr.2012.02.007
244. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via nkt cells. Science (2018) 360(6391):eaan5931. doi: 10.1126/science.aan5931
245. Ma X, Zhou Z, Zhang X, Fan M, Hong Y, Feng Y, et al. Sodium butyrate modulates gut microbiota and immune response in colorectal cancer liver metastatic mice. Cell Biol Toxicol (2020) 36(5):509–15. doi: 10.1007/s10565-020-09518-4
246. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature (2011) 478(7368):197–203. doi: 10.1038/nature10491
247. Li L, Wang T, Li S, Chen Z, Wu J, Cao W, et al. Tdo2 promotes the emt of hepatocellular carcinoma through kyn-ahr pathway. Front Oncol (2020) 10:562823. doi: 10.3389/fonc.2020.562823
248. Song L, Guo L, Li Z. Molecular mechanisms of 3, 3′ 4, 4′, 5-pentachlorobiphenyl-induced epithelial-mesenchymal transition in human hepatocellular carcinoma cells. Toxicol Appl Pharmacol (2017) 322:75–88. doi: 10.1016/j.taap.2017.03.003
249. Dai G, Chen X, He Y. The gut microbiota activates ahr through the tryptophan metabolite kyn to mediate renal cell carcinoma metastasis. Front Nutr (2021) 8:712327. doi: 10.3389/fnut.2021.712327
250. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its fada adhesin. Cell Host Microbe (2013) 14(2):195–206. doi: 10.1016/j.chom.2013.07.012
251. Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor-κb, and up-regulating expression of microrna-21. Gastroenterology (2017) 152(4):851–66.e24. doi: 10.1053/j.gastro.2016.11.018
252. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the fap2 protein of fusobacterium nucleatum to human inhibitory receptor tigit protects tumors from immune cell attack. Immunity (2015) 42(2):344–55. doi: 10.1016/j.immuni.2015.01.010
253. Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD, et al. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res (2011) 21(10):1616–25. doi: 10.1101/gr.122705.111
254. Yin H, Miao Z, Wang L, Su B, Liu C, Jin Y, et al. Fusobacterium nucleatum promotes liver metastasis in colorectal cancer by regulating the hepatic immune niche and altering gut microbiota. Aging (2022) 14(4):1941–58. doi: 10.18632/aging.203914
255. Engevik MA, Danhof HA, Ruan W, Engevik AC, Chang-Graham AL, Engevik KA, et al. Fusobacterium nucleatum secretes outer membrane vesicles and promotes intestinal inflammation. mBio (2021) 12(2):e02706-20. doi: 10.1128/mBio.02706-20
256. Bertocchi A, Carloni S, Ravenda PS, Bertalot G, Spadoni I, Lo Cascio A, et al. Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell (2021) 39(5):708–24.e11. doi: 10.1016/j.ccell.2021.03.004
257. Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B, et al. Colonization of the human gut by E. Coli and colorectal cancer risk. Clin Cancer Res (2014) 20(4):859–67. doi: 10.1158/1078-0432.CCR-13-1343
258. Sun J, Hobert ME, Duan Y, Rao AS, He T-C, Chang EB, et al. Crosstalk between nf-κb and β-catenin pathways in bacterial-colonized intestinal epithelial cells. Am J Physiology-Gastrointestinal Liver Physiol (2005) 289(1):G129–G37. doi: 10.1152/ajpgi.00515.2004
259. Cheng WT, Kantilal HK, Davamani F. The mechanism of bacteroides fragilis toxin contributes to colon cancer formation. Malaysian J Med sciences: MJMS (2020) 27(4):9. doi: 10.21315/mjms2020.27.4.2
260. Sears CL, Geis AL, Housseau F. Bacteroides fragilis subverts mucosal biology: from symbiont to colon carcinogenesis. J Clin Invest (2014) 124(10):4166–72. doi: 10.1172/JCI72334
261. Wu S, Rhee K-J, Zhang M, Franco A, Sears CL. Bacteroides fragilis toxin stimulates intestinal epithelial cell shedding and Γ-secretase-dependent E-cadherin cleavage. J Cell Sci (2007) 120(11):1944–52. doi: 10.1242/jcs.03455
262. Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet (2017) 18(3):164–79. doi: 10.1038/nrg.2016.150
263. Diamantopoulou Z, Gvozdenovic A, Aceto N. A new time dimension in the fight against metastasis. Trends Cell Biol (2023) 33(9):736–48. doi: 10.1016/j.tcb.2023.02.002
264. Okazaki F, Matsunaga N, Okazaki H, Azuma H, Hamamura K, Tsuruta A, et al. Circadian clock in a mouse colon tumor regulates intracellular iron levels to promote tumor progression. J Biol Chem (2016) 291(13):7017–28. doi: 10.1074/jbc.M115.713412
265. Reiter RJ, Sharma R, Tan DX, Huang G, de Almeida Chuffa LG, Anderson G. Melatonin modulates tumor metabolism and mitigates metastasis. Expert Rev Endocrinol Metab (2023) 18(4):321–36. doi: 10.1080/17446651.2023.2237103
266. Talib WH, Alsayed AR, Abuawad A, Daoud S, Mahmod AI. Melatonin in cancer treatment: current knowledge and future opportunities. Molecules (Basel Switzerland) (2021) 26(9):2506. doi: 10.3390/molecules26092506
267. Liu C, Guo L, Fu C. Circadian-rhythm-regulating hormones: key factors to regulate breast cancer metastasis via circulating tumor cells. MedComm (2022) 3(4):e189. doi: 10.1002/mco2.189
268. Hadadi E, Taylor W, Li XM, Aslan Y, Villote M, Rivière J, et al. Chronic circadian disruption modulates breast cancer stemness and immune microenvironment to drive metastasis in mice. Nat Commun (2020) 11(1):3193. doi: 10.1038/s41467-020-16890-6
269. Numata M, Hirano A, Yamamoto Y, Yasuda M, Miura N, Sayama K, et al. Metastasis of breast cancer promoted by circadian rhythm disruption due to light/dark shift and its prevention by dietary quercetin in mice. J circadian rhythms (2021) 19:2. doi: 10.5334/jcr.203
270. Blakeman V, Williams JL, Meng QJ, Streuli CH. Circadian clocks and breast cancer. Breast Cancer Res BCR (2016) 18(1):89. doi: 10.1186/s13058-016-0743-z
271. Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol (2017) 3(4):524–48. doi: 10.1001/jamaoncol.2016.5688
272. Hansen J, Stevens RG. Case-control study of shift-work and breast cancer risk in danish nurses: impact of shift systems. Eur J Cancer (Oxford Engl 1990) (2012) 48(11):1722–9. doi: 10.1016/j.ejca.2011.07.005
273. Diamantopoulou Z, Castro-Giner F, Schwab FD, Foerster C, Saini M, Budinjas S, et al. The metastatic spread of breast cancer accelerates during sleep. Nature (2022) 607(7917):156–62. doi: 10.1038/s41586-022-04875-y
274. Paiva B, Paino T, Sayagues JM, Garayoa M, San-Segundo L, Martín M, et al. Detailed characterization of multiple myeloma circulating tumor cells shows unique phenotypic, cytogenetic, functional, and circadian distribution profile. Blood (2013) 122(22):3591–8. doi: 10.1182/blood-2013-06-510453
275. Zhu X, Suo Y, Fu Y, Zhang F, Ding N, Pang K, et al. In vivo flow cytometry reveals a circadian rhythm of circulating tumor cells. Light Sci Appl (2021) 10(1):110. doi: 10.1038/s41377-021-00542-5
276. Wu J, Jing X, Du Q, Sun X, Holgersson K, Gao J, et al. Disruption of the clock component bmal1 in mice promotes cancer metastasis through the pai-1-tgf-β-myocaf-dependent mechanism. Adv Sci (Weinheim Baden-Wurttemberg Germany) (2023) 10(24):e2301505. doi: 10.1002/advs.202301505
277. Casanova-Acebes M, Nicolás-Ávila JA, Li JL, García-Silva S, Balachander A, Rubio-Ponce A, et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J Exp Med (2018) 215(11):2778–95. doi: 10.1084/jem.20181468
278. Filipski E, King VM, Li X, Granda TG, Mormont M-C, Liu X, et al. Host circadian clock as a control point in tumor progression. J Natl Cancer Institute (2002) 94(9):690–7. doi: 10.1093/jnci/94.9.690
279. Sancar A, Van Gelder RN. Clocks, cancer, and chronochemotherapy. Science (2021) 371(6524):eabb0738. doi: 10.1126/science.abb0738
280. Shan L, Zheng W, Bai B, Hu J, Lv Y, Chen K, et al. Bmal1 promotes colorectal cancer cell migration and invasion through erk-and jnk-dependent C-myc expression. Cancer Med (2023) 12(4):4472–85. doi: 10.1002/cam4.5129
281. Yang Y, Yang T, Zhao Z, Zhang H, Yuan P, Wang G, et al. Down-regulation of bmal1 by mir-494-3p promotes hepatocellular carcinoma growth and metastasis by increasing gpam-mediated lipid biosynthesis. Int J Biol Sci (2022) 18(16):6129. doi: 10.7150/ijbs.74951
282. Wang J, Li S, Li X, Li B, Li Y, Xia K, et al. Circadian protein bmal1 promotes breast cancer cell invasion and metastasis by up-regulating matrix metalloproteinase9 expression. Cancer Cell Int (2019) 19:1–12. doi: 10.1186/s12935-019-0902-2
283. Chen J, Liu A, Lin Z, Wang B, Chai X, Chen S, et al. Downregulation of the circadian rhythm regulator hlf promotes multiple-organ distant metastases in non-small cell lung cancer through ppar/nf-κb signaling. Cancer Lett (2020) 482:56–71. doi: 10.1016/j.canlet.2020.04.007
284. Hwang-Verslues WW, Chang PH, Jeng YM, Kuo WH, Chiang PH, Chang YC, et al. Loss of Corepressor Per2 under Hypoxia up-Regulates Oct1-Mediated Emt Gene Expression and Enhances Tumor Malignancy. PNAS (2013) 110:12331–6. doi: 10.1073/pnas.1222684110
285. Zhang Y, Devocelle A, Desterke C, de Souza LEB, Hadadi É, Acloque H, et al. Bmal1 knockdown leans epithelial–mesenchymal balance toward epithelial properties and decreases the chemoresistance of colon carcinoma cells. Int J Mol Sci (2021) 22(10):5247. doi: 10.3390/ijms22105247
286. Mao L, Summers W, Xiang S, Yuan L, Dauchy RT, Reynolds A, et al. Melatonin represses metastasis in her2-postive human breast cancer cells by suppressing rsk2 expression. Mol Cancer Res (2016) 14(11):1159–69. doi: 10.1158/1541-7786.MCR-16-0158
287. Patke A, Young MW, Axelrod S. Molecular mechanisms and physiological importance of circadian rhythms. Nat Rev Mol Cell Biol (2020) 21(2):67–84. doi: 10.1038/s41580-019-0179-2
288. Xuan W, Khan F, James CD, Heimberger AB, Lesniak MS, Chen P. Circadian regulation of cancer cell and tumor microenvironment crosstalk. Trends Cell Biol (2021) 31(11):940–50. doi: 10.1016/j.tcb.2021.06.008
289. Shaashua L, Mayer S, Lior C, Lavon H, Novoselsky A, Scherz-Shouval R. Stromal expression of the core clock gene period 2 is essential for tumor initiation and metastatic colonization. Front Cell Dev Biol (2020) 8:587697. doi: 10.3389/fcell.2020.587697
290. Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature (2019) 575(7782):299–309. doi: 10.1038/s41586-019-1730-1
291. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to raf inhibitors. Nat Med (2013) 19(11):1401–9. doi: 10.1038/nm.3392
292. Sabnis AJ, Bivona TG. Principles of resistance to targeted cancer therapy: lessons from basic and translational cancer biology. Trends Mol Med (2019) 25(3):185–97. doi: 10.1016/j.molmed.2018.12.009
293. Wu Y-L, Zhong W, Wang Q, Mao W, Xu S-T, Wu L, et al. Ctong1104: adjuvant gefitinib versus chemotherapy for resected N1-N2 nsclc with egfr mutation—Final overall survival analysis of the randomized phase iii trial 1 analysis of the randomized phase iii trial. Am Soc Clin Oncol (2020) 38:9005. doi: 10.1200/JCO.2020.38.15_suppl.900
294. Jassem J. Adjuvant egfr tyrosine kinase inhibitors in egfr-mutant non-small cell lung cancer: still an investigational approach. Trans Lung Cancer Res (2019) 8(Suppl 4):S387–s90. doi: 10.21037/tlcr.2019.09.02
295. Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum-pemetrexed in egfr T790m-positive lung cancer. N Engl J Med (2017) 376(7):629–40. doi: 10.1056/NEJMoa1612674
296. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated egfr-mutated advanced non-small-cell lung cancer. N Engl J Med (2018) 378(2):113–25. doi: 10.1056/NEJMoa1713137
297. Herbst RS, Tsuboi M, John T, Grohé C, Majem M, Goldman JW, et al. Osimertinib as adjuvant therapy in patients (Pts) with stage ib–iiia egfr mutation positive (Egfrm) nsclc after complete tumor resection: adaura. J Clin Oncol (2020) 38(18_suppl):LBA5–LBA. doi: 10.1200/JCO.2020.38.18_suppl.LBA5
298. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. Tgfβ Drives immune evasion in genetically reconstituted colon cancer metastasis. Nature (2018) 554(7693):538–43. doi: 10.1038/nature25492
299. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. Tgfβ Attenuates tumour response to pd-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554(7693):544–8. doi: 10.1038/nature25501
300. Fukuoka S, Hara H, Takahashi N, Kojima T, Kawazoe A, Asayama M, et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: an open-label, dose-escalation, and dose-expansion phase ib trial (Regonivo, epoc1603). J Clin Oncol Off J Am Soc Clin Oncol (2020) 38(18):2053–61. doi: 10.1200/jco.19.03296
301. Clark ME, Smith RR. Liver-directed therapies in metastatic colorectal cancer. J gastrointestinal Oncol (2014) 5(5):374–87. doi: 10.3978/j.issn.2078-6891.2014.064
302. Palma DA, Olson R, Harrow S, Gaede S, Louie AV, Haasbeek C, et al. Stereotactic ablative radiotherapy versus standard of care palliative treatment in patients with oligometastatic cancers (Sabr-comet): A randomised, phase 2, open-label trial. Lancet (London England) (2019) 393(10185):2051–8. doi: 10.1016/s0140-6736(18)32487-5
303. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science (2020) 368(6494):973–80. doi: 10.1126/science.aay9189
304. Parhi L, Alon-Maimon T, Sol A, Nejman D, Shhadeh A, Fainsod-Levi T, et al. Breast cancer colonization by fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat Commun (2020) 11(1):3259. doi: 10.1038/s41467-020-16967-2
305. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of pd-1-based immunotherapy against epithelial tumors. Science (2018) 359(6371):91–7. doi: 10.1126/science.aan3706
306. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-pd-1 immunotherapy in melanoma patients. Science (2018) 359(6371):97–103. doi: 10.1126/science.aan4236
307. Forbes NS, Coffin RS, Deng L, Evgin L, Fiering S, Giacalone M, et al. White paper on microbial anti-cancer therapy and prevention. J Immunother Cancer (2018) 6(1):78. doi: 10.1186/s40425-018-0381-3
308. Ogiya R, Niikura N, Kumaki N, Yasojima H, Iwasa T, Kanbayashi C, et al. Comparison of immune microenvironments between primary tumors and brain metastases in patients with breast cancer. Oncotarget (2017) 8(61):103671–81. doi: 10.18632/oncotarget.22110
309. Cacho-Díaz B, García-Botello DR, Wegman-Ostrosky T, Reyes-Soto G, Ortiz-Sánchez E, Herrera-Montalvo LA. Tumor microenvironment differences between primary tumor and brain metastases. J Transl Med (2020) 18(1):1–. doi: 10.1186/s12967-019-02189-8
310. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. Vegfr1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature (2005) 438(7069):820–7. doi: 10.1038/nature04186
311. O'Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, et al. Vegf-a and tenascin-C produced by S100a4+ Stromal cells are important for metastatic colonization. Proc Natl Acad Sci USA (2011) 108(38):16002–7. doi: 10.1073/pnas.1109493108
312. Rombouts K, Carloni V. The fibrotic microenvironment as a heterogeneity facet of hepatocellular carcinoma. Fibrogenesis Tissue Repair (2013) 6(1):17. doi: 10.1186/1755-1536-6-17
313. Jiang Y, Zhang H, Wang J, Liu Y, Luo T, Hua H. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J Hematol Oncol (2022) 15(1):34–. doi: 10.1186/s13045-022-01252-0
314. Hua H, Li M, Luo T, Yin Y, Jiang Y. Matrix metalloproteinases in tumorigenesis: an evolving paradigm. Cell Mol Life Sci CMLS (2011) 68(23):3853–68. doi: 10.1007/s00018-011-0763-x
315. Palazon A, Goldrath AW, Nizet V, Johnson RS. Hif transcription factors, inflammation, and immunity. Immunity (2014) 41(4):518–28. doi: 10.1016/j.immuni.2014.09.008
316. Jensen LD. The circadian clock and hypoxia in tumor cell de-differentiation and metastasis. Biochim Biophys Acta (2015) 1850(8):1633–41. doi: 10.1016/j.bbagen.2014.10.025
317. Bishehsari F, Voigt RM, Keshavarzian A. Circadian rhythms and the gut microbiota: from the metabolic syndrome to cancer. Nat Rev Endocrinol (2020) 16(12):731–9. doi: 10.1038/s41574-020-00427-4
Keywords: metastasis, metastatic cascade, premetastatic niche, tumor microenvironment, hypoxia, extracellular matrix, circadian rhythm, gut-microbiota
Citation: Riaz F, Zhang J and Pan F (2024) Forces at play: exploring factors affecting the cancer metastasis. Front. Immunol. 15:1274474. doi: 10.3389/fimmu.2024.1274474
Received: 08 August 2023; Accepted: 19 January 2024;
Published: 01 February 2024.
Edited by:
Kihoon Nam, University of Missouri, United StatesReviewed by:
Sandra Cascio, University of Pittsburgh, United StatesCopyright © 2024 Riaz, Zhang and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fan Pan, ZmFuLnBhbkBzaWF0LmFjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.