 Seyedeh Nasim Mirbahari1,2
Seyedeh Nasim Mirbahari1,2 Miles Da Silva3,4†
Miles Da Silva3,4† Abril Ixchel Muñoz Zúñiga5,6Nika Kooshki Zamani5,6Gabriel St-Laurent5,6
Abril Ixchel Muñoz Zúñiga5,6Nika Kooshki Zamani5,6Gabriel St-Laurent5,6 Mehdi Totonchi2*
Mehdi Totonchi2* Taha Azad5,6*
Taha Azad5,6*- 1Faculty of Sciences and Advanced Technologies in Biology, University of Science and Culture, Academic Center for Education, Culture and Research (ACECR), Tehran, Iran
- 2Department of Genetics, Reproductive Biomedicine Research Center, Royan Institute for Reproductive Biomedicine, Academic Center for Education, Culture and Research (ACECR), Tehran, Iran
- 3Department of Microbiology and Immunology, University of British Colombia, Vancouver, BC, Canada
- 4Ottawa Hospital Research Institute, The Ottawa Hospital, Ottawa, ON, Canada
- 5Department of Microbiology and Infectious Diseases, Université de Sherbrooke, Sherbrooke, QC, Canada
- 6Centre de Recherche du CHUS, Sherbrooke, QC, Canada
In recent years, oncolytic viruses have emerged as promising agents for treating various cancers. An oncolytic virus is a non-pathogenic virus that, due to genetic manipulation, tends to replicate in and cause lysis of cancerous cells while leaving healthy cells unaffected. Among these viruses, vaccinia virus is an attractive platform for use as an oncolytic platform due to its 190 Kb genome with a high capacity for encoding therapeutic payloads. Combining oncolytic VV therapy with other conventional cancer treatments has been shown to be synergistic and more effective than monotherapies. Additionally, OVV can be used as a vector to deliver therapeutic payloads, alone or in combination with other treatments, to increase overall efficacy. Here, we present a comprehensive analysis of preclinical and clinical studies that have evaluated the efficacy of oncolytic vaccinia viruses in cancer immunotherapy. We discuss the outcomes of these studies, including tumor regression rates, overall survival benefits, and long-term responses. Moreover, we provide insights into the challenges and limitations associated with oncolytic vaccinia virus- based therapies, including immune evasion mechanisms, potential toxicities, and the development of resistance.
Introduction
In recent years, innovative approaches have emerged for developing new biological therapies for cancer treatment, including CAR-T cell therapy and oncolytic viruses (OV), either alone or in combination with traditional treatments. OVs have shown remarkable efficacy against different types of cancers for over three decades (1).
Oncolytic viruses execute their anticancer activity through a multifaceted interplay of intricate mechanisms designed to selectively target and eliminate cancer cells. Upon administration, OVs specifically recognize and infect tumor cells. This specificity is achieved through a variety of genetic manipulations that exploit cancer cells inherent vulnerabilities. One primary mechanism involves the direct lysis of infected cancer cells, where viral replication within the tumor leads to cell lysis, causing the release of progeny viruses and intracellular contents (2, 3). Concurrently, the viral infection triggers immunogenic cell death, promoting the release of danger signals and facilitating the activation of antitumor immune responses. The immune system, now recruited and activated, recognizes and eliminates both infected and non-infected neighboring cancer cells, creating a systemic antitumor immune response (4). Furthermore, oncolytic viruses can induce a cascade of immunomodulatory effects within the tumor microenvironment, enhancing the recruitment and activation of immune cells, such as cytotoxic T lymphocytes and natural killer cells (5). OVs can also disrupt tumor vasculature, impeding blood supply to the tumor and contributing to the antitumor effect (6). In addition of its primary function, OVs can lead to expression of therapeutic transgene in the tumor microenvironment (TME) following infection of tumor cells. This feature increased the oncolytic potential and therefore is important for future combination of oncolytic therapy and immunotherapy. These mechanisms allow scientists to hijack the immune system to create OV-elicited antitumor immunity. Relative contributions of each mechanism depend on the nature and type of cancer cell, the characteristics of the viral vector, and interactions between the virus, tumor microenvironment and host immune system. These multifaceted mechanisms collectively contribute to the selective elimination of cancer cells by oncolytic viruses, presenting a promising avenue for innovative and targeted cancer immunotherapies.
OV therapy implicates different mechanisms, like interactions between tumor cells, viruses, and the immune system. The anti-tumor activity of the OVs can be identified in two categories, the killing of tumor cells, which is determined by the expression of the receptor in the cell surface, and the host cell’s anti-viral response. The other mechanism benefits the generation of a proinflammatory tumor microenvironment, boosting systematic anti-tumor immunity (7).
Several approaches have been developed to engineer viruses to make them oncolytic, including the downregulation of viral genes necessary for replication in healthy cells, the use of tissue or tumor-specific promoters. It has been reported that OVs replicate more efficiently within tumor cells due to a tumor cell’s deficiency in antiviral type I interferon signaling (8). Overall, oncolytic vaccinia virus (9) can infect healthy and tumors cells, but genetic modifications could make them selective for replication in tumor cells. Non-invasive imaging techniques, such as reporter transgenes, allow the tracking and tumor specificity evaluation of OVs (10). OVV has shown promising results in preclinical, phase-1, -2 and -3 clinical trials, and has a long history of safety (11–13). OVVs can infect tumor cells and replicate constantly, therefore causing oncolysis. By infecting tumor cells, tumor-associates molecular patterns are exposed, including antigens, DAMPs, PAMPs, and cytokines. These elements thereafter activate the immune responses in the tumor microenvironment. Furthermore, these patterns stimulate innate and adaptative immunity (14). However, treatment response may vary depending on the differential baseline immunological profile of each patient (15). Table 1 lists the OVVs currently in clinical trials, their specific genetic modifications, and any therapeutic combinations. The vaccinia virus (9) is an enveloped virus with a linear dsDNA genome of approximately 190 Kb containing approximately 250 protein-coding genes. It was the first safe and effective human vaccine, ultimately eradicating smallpox (13). Since VV replicates entirely in the cytoplasm of infected cells, there is no concern about the possibility of intra-nuclear mutagenesis. A wide range of cells are considered to be hosts of vaccinia. The virus enters host cells by an endocytic process through the cell membrane (35, 36). VV can express up to 50 kb of external DNA and can express multiple therapeutic genes simultaneously. Vaccinia has a rapid and lytic replication cycle that triggers a robust immune response and inflammation in the host (37). The host immune system easily controls the infection, however, in case of uncontrolled virus growth, antiviral treatments such as ST-246 (tecovirimat) and cidofovir exist (38, 39). During VV maturation, newly assembled immature virions are enveloped in a membrane to form infectious intracellular mature virions (IMV), which are further enveloped in a double membrane layer to form intracellular enveloped viruses (IEVs) (40). The outermost IEV membrane layer then fuses with the cytoplasmic membrane to release the virion from the host cell. Tecovirimat inhibits the VP37 protein, which is needed for IMV membrane envelopment to form IEV. The availability of tecovirimat supports the development of OVV therapies by providing an option to treat severe adverse effects of OVVs or reduce adverse effects of higher dose OVV use (41).
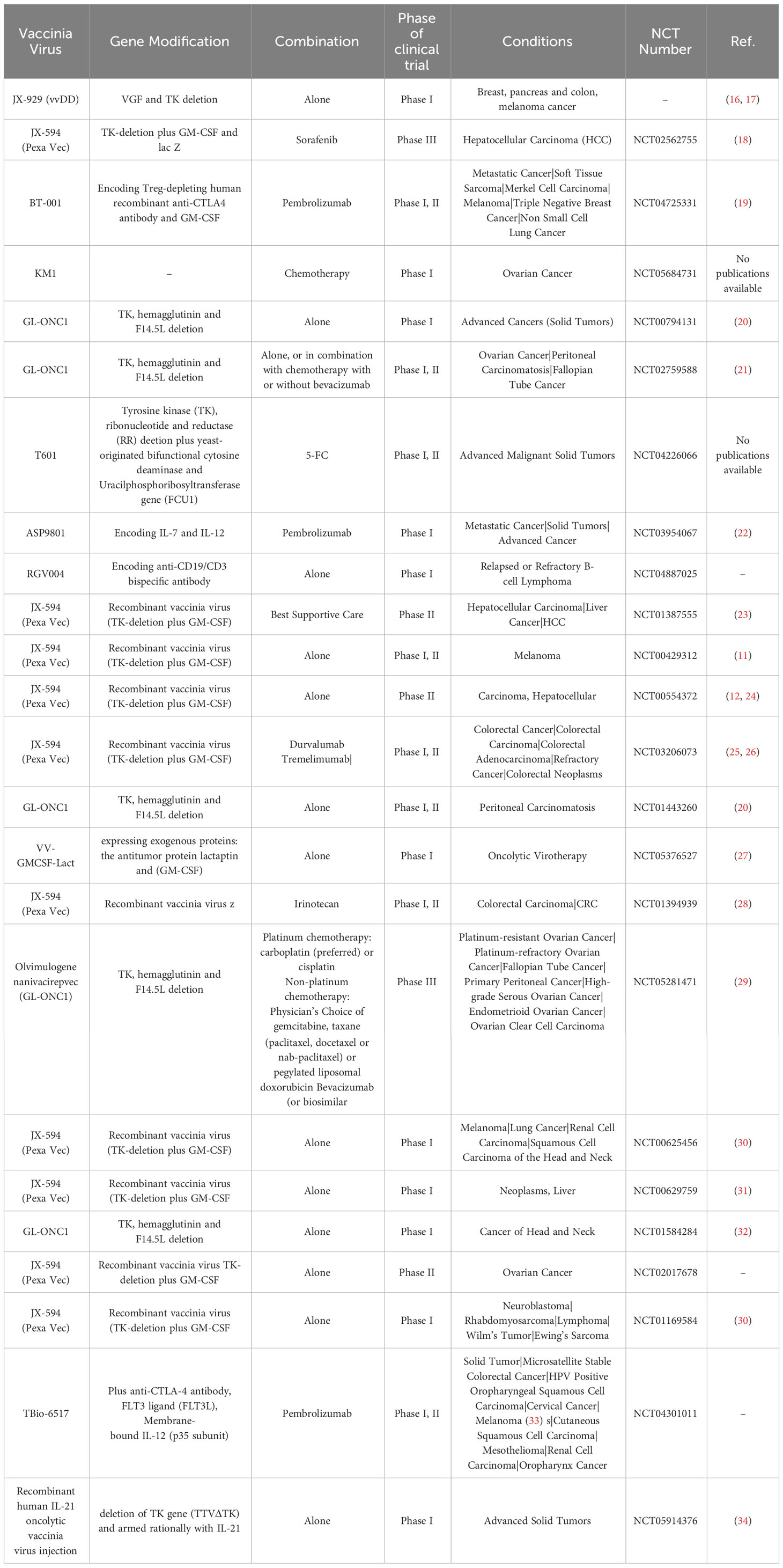
Table 1 List of OVVs (alone or in combination with other treatments) in different phases of clinical trial.
Recent advancements in molecular biology have made VV an attractive candidate for engineering as an oncolytic agent (40). With the genetic diversity of tumor types and their resistance to routine drugs, monotherapies have limited success in treating many cancer types. Combination methods can be more effective and have reinforcing results on treatment, but finding these combinations can be challenging. Figure 1 illustrates the different types of cancer treatments that have demonstrated synergistic effects with OVV therapy. This review will summarize recent applications of OVV treatment combinations and discuss their potential for improving the response to treatment.
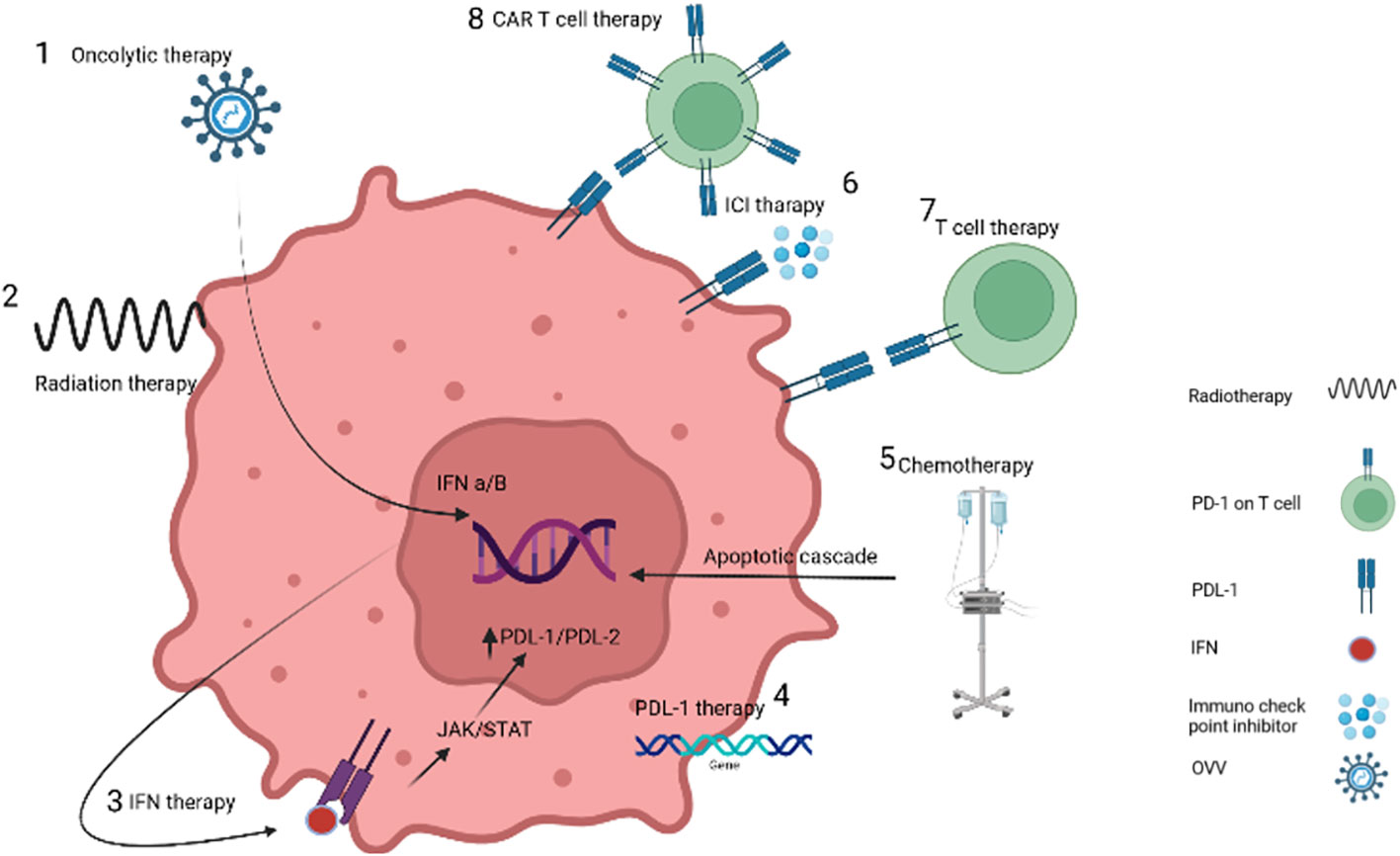
Figure 1 Targeting cancer cells by different treatment methods. 1. OVVs increase the expression of interferon by entering cancer cells. 2. Radiotherapy causes damage to the DNA of the tumor cell, while it does not damage the DNA of the vaccinia virus due to the difference in the time of injection. 3. Interferon therapy recruit immune cells. 4. PDL-1 protein injection or gene insertion increases the response to immunotherapy methods. 5. Chemotherapy destroys cancer cells by causing inflammation. 6. Blockage of immunochecking inhibitors such as anti-PD-1 causes the recognition of receptors by T-cells. 7 and 8 T-cells and CAR-T cells also destroy cancer cells by binding receptors.
OVVs as a next generation of immunotherapeutic to remodel TME
Immune checkpoint inhibitor antibody therapy
Immune checkpoint molecules are proteins found on the surface of cells that help regulate the immune system’s response to foreign invaders, such as cancer cells. These molecules act as “checkpoints’’, either activating or inhibiting the immune response. Some cancer cells can evade the immune system by exploiting these inhibitory checkpoint molecules, allowing them to avoid detection by lymphocytes and thus grow and spread. Immune checkpoint inhibitors (ICIs) are a class of drugs that target these checkpoint molecules to restore the immune system’s ability to attack cancer cells. Currently, ICIs are used as a treatment for certain types of cancer, such as melanoma and lung cancer. Several inhibitory checkpoint molecules have been identified, including CTLA-4, PD-1, CD80/86, MHC II, Galectin-3, FGL1, CD112, CD155, HVEM, and Ceacam-1, known as interferon (IFN) signaling-stimulated genes (ISGs) (42–45). Even though ICIs have shown promise in the treatment of different types of cancer, monotherapy rarely offers long-term benefits for most patients. Despite the capacity of CAR-T cell therapy to enhance T-cells to target cancer cells which is efficacious in treating hematologic malignancies, its ability to treat solid tumors is limited (14). Therefore, combining OVs with ICIs is a common way to increase treatment effectiveness, as both therapies work to relieve the tumor’s immunosuppressive environment. Additionally, OV infection triggers an anticancer immune response, increasing therapy efficacy (46–51). Various ICIs have been developed, with each targeting different checkpoint molecules. Examples include CTLA-4 inhibitors like ipilimumab, PD-1 inhibitors such as nivolumab and pembrolizumab, and LAG-3 inhibitors like MEDI3039, which block the activity of the respective proteins found on the surface of T-cells (52).
Programmed death receptor (PD-1), also known as CD279, is a type 1 transmembrane protein encoded by the PDCD1 gene of the CD28 immunoglobulin superfamily (53). PD-1 is 288 amino acids long and has a single Ig variable-type (IgV) extracellular domain, a transmembrane domain, and a cytoplasmic domain (54). It is mainly expressed in activated CD4+ T-cells, CD8+ T-cells, natural killer T-cells, B-cells, macrophages, dendritic cells (DCs) and monocytes. Its expression is induced by the T- or B-cell receptor pathway and enhanced by the stimulation of tumor necrosis factor (55). Naive T and B-cells barely express PD-1 (56). The biological functions of PD-1 rely on two ligands: PD-L1 (also known as B7-H1 or CD274) and PD-L2 (also known as B7-H2 or CD273). PD-L1 is constitutively expressed on T-cells, B-cells, DCs, cancer cells, macrophages and others, and is further upregulated by activated pro-inflammatory cytokines (57). PD-1/PD-L1 interaction is mainly responsible for the immune escape of cancers. When PD-L1 binds to PD-1, two tyrosines of the immune-receptor tyrosine-based switch motif (ITSM) region of PD-1’s intracellular domain is phosphorylated. These phosphorylated tyrosines then phosphorylate the B-cell receptor (BCR), and SHP-2, a phosphatase, binds to the C-terminal of PD-1. Subsequently, phosphorylated SHP-2 dephosphorylates the BCR, leading to impaired Ca2+ ion generation and long-term growth arrest (58). In normal cells, PD-1/PD-L1 binding prevents the overstimulation of T-cells and maintains immune tolerance to antigens, preventing the development of autoimmune diseases. In cancer cells, the binding of PD-L1 to PD-1 generates negative signals, which induce T-cell apoptosis and reduce immunocompetence, aiding cancer cells in avoiding recognition by the immune system. Additionally, activation of the PD-1/PD-L1 pathway negatively affects the differentiation of effector T-cells (59), memory T-cells (60), regulatory T-cells (Treg), and exhausted T-cells (61), greatly reducing the effect of T-cells on tumor cells (60). Interestingly, PD-L1 carried in exosomes can be transported to remote regions of the body via the circulatory system. This immigrated PD-L1 inhibits T-cell activity remotely before reaching metastatic lesions (62). Blocking the binding of PD-L1 to PD-1 inhibits these negative consequences, thereby preserving T-cell function and their ability to kill cancer cells.
A present-day study demonstrates the strengthening of systemic anti-tumor effects in various preclinical tumor models, with a vaccinia virus that coexpresses a PD-L1 inhibitor and GM-CSF (VV-iPDL1/GM). This proposal effectively suppressed PD-L1 expression in tumor and immune cells which therefore increased infiltration and activation of tumor-specific T-cells. These react against neoantigen epitopes derived from mutations, which results in a successful rejection of both virus-injected and distant tumors (63).
Today, ICI immunotherapy shows promise in treating various solid tumor cancers and hematological malignancies, with durable responses and long-term survival benefits (64, 65). Blocking ICIs can be combined with OVV therapy using two strategies: anti-ICI antibody genes can be inserted into the OVV genome, which is then categorized as an immuno-oncolytic virus, or patients can receive an injection of anti-ICIs antibodies before or after OVV therapy. Immuno-oncolytic viruses will be discussed further in the next section. Table 2 displays different ICI with OVV combination therapy studies, some applying the gene insertion technique while others utilize antibody injections. In a 2022 study, a regimen of 4 different therapies was designed, which included an OVV known as GLV-1h68, melphalan, TNFα, and intra-peritoneal injection of recombinant PD-1 protein, followed by completion with radiotherapy. The combination of these therapies recruited CD4+ and CD8+ T-cells and significantly altered the TME (69). In another study, OVV mpJX-594 (mpJX) was intravenously injected alone or together with doses of PD-1 into functional and metastatic pancreatic neuroendocrine tumors (PanNETs). Simultaneous injection of PD-1 with mpJX had a synergistic effect on the secretion of NK, CD8+ T-cell, leading to apoptosis and inhibition of the proliferation of pancreatic tumor cells. This therapeutic combination increased survival and anti-metastatic activity in liver metastasis model mice (76).
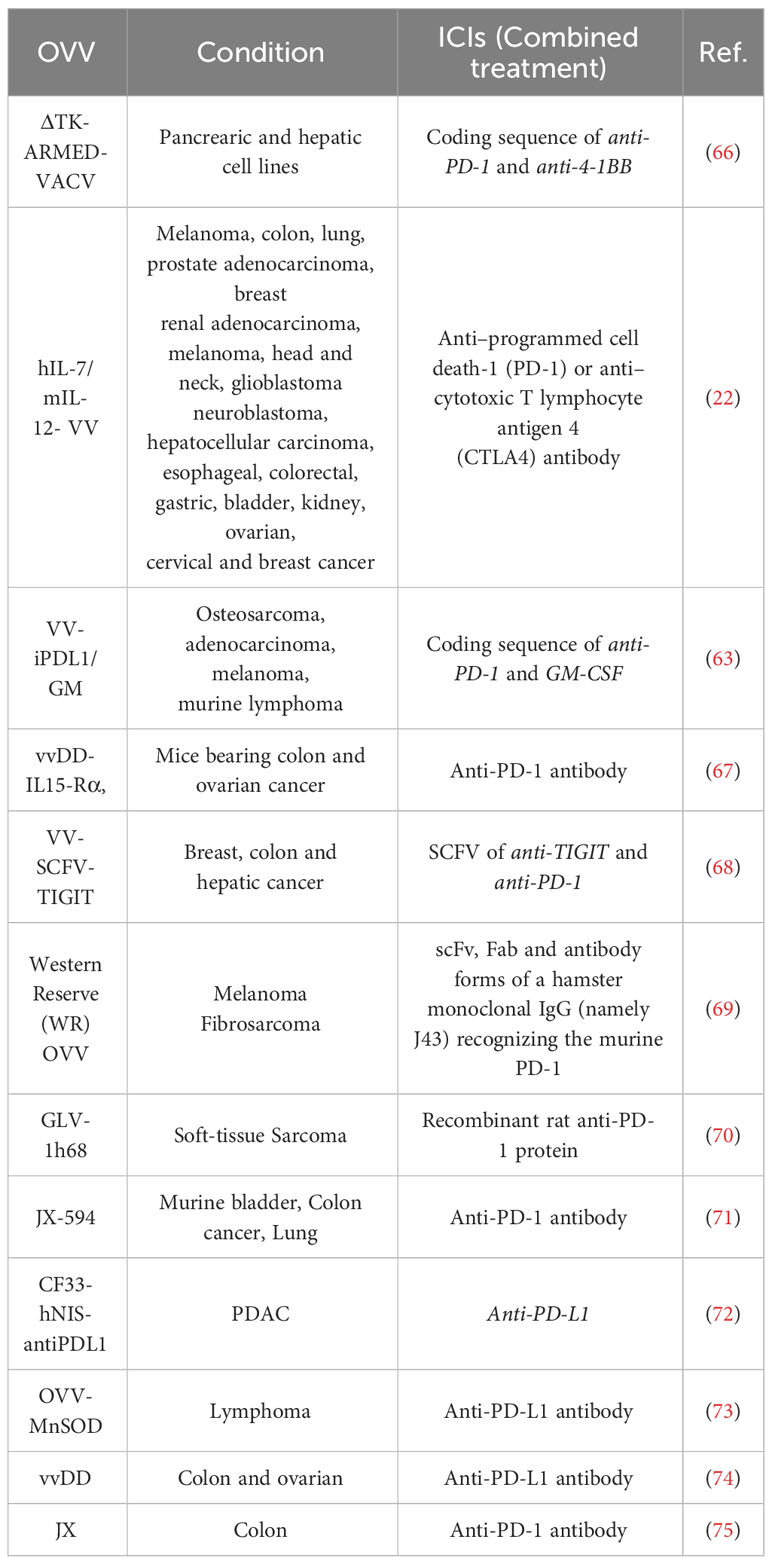
Table 2 OVV in combination with ICIs in different studies.
A similar study suggested the use of mJX-594 (JX), a vaccinia virus equipped with GM-CSF (cytokine), as an approach to remodel the tumor microenvironment and augment sensitivity to αPD-1 and/or αCTLA-4 immunotherapy. The administration of JX resulted in changes in the tumor microenvironment as indicated by the infiltration of T-cells into the tumor and the high expression of genes associated with immunity. It not only increases the infiltration of CD8+ T-cells within the tumor but also enhances abscopal effects in distant tumors. Combining intratumoral JX with systemic αPD-1 or αCTLA-4 enhanced the anticancer immune response. Notably, the strongest anticancer immunity was exhibited when including JX, αPD-1, and αCTLA4, causing complete tumor regression and prolonged overall survival (77).
ICI immuno-oncolytic virotherapy
Oncolytic viruses can be genetically engineered to create immuno-oncolytic viruses (IOVs), themselves encoding immunomodulatory factors (78). IOVs can be used for the localized expression of immune-modulating target proteins, obviating the need for separate immunotherapy following virotherapy. One type of immuno-oncolytic virotherapy involves the insertion of the anti-PD-L1 antibody (anti-PD-L1) expression cassette into an OV. This OV then both directly mediates cancer killing and induces the production of the anti-PD-L1 antibody. This anti-PD-L1 decreases immune inhibition, enhancing the treatment’s oncolytic efficacy by improving anti-tumor immune cell function (79).
Figure 2 illustrates the variety of ICI ligands on tumor cells and antigen-presenting cells (APCs), along with their cognate receptors on T-cells. The higher the ICI expression on the tumor cell’s surface, the greater the chance of response to immunotherapy treatments. ICIs that target the PD-1/PD-L1 axis have been effective in solid tumors, such as non-small cell lung cancer (NSCLC), with high levels of PD-L1 expression and active lymphocytic infiltrates (80). Unfortunately, ICIs that depend on single targets have failed in pancreatic ductal adenocarcinoma (PDAC) patients, even those with tumors overexpressing PD-L1. Low antigen recognition limited cytotoxic T-cell recruitment, and aberrant signaling of co-stimulatory immune checkpoints have been identified as critical barriers to the success of immunotherapies in PDAC (81). CF33-hNIS-antiPDL1 is an OV genetically engineered using the potent chimeric orthopox virus backbone CF33 to contain the human Sodium Iodide Symporter (hNIS) and anti-PD-L1. Unmodified CF33 has previously been demonstrated to be safe and well-tolerated at doses several magnitudes lower than other OVs currently in preclinical and clinical studies in pancreatic cancers by the authors (82). CF33-hNIS-anti-PD-L1 was shown to lyse PDAC cells in a dose-dependent manner, achieving >90% cell killing by day three. Infected cells were shown to produce bioactive anti-PD-L1, which blocked the PD-1/PD-L1 interaction. In vivo, a single dose of the engineered virus both reduced tumor burden and prolonged the survival of treated mice (72).
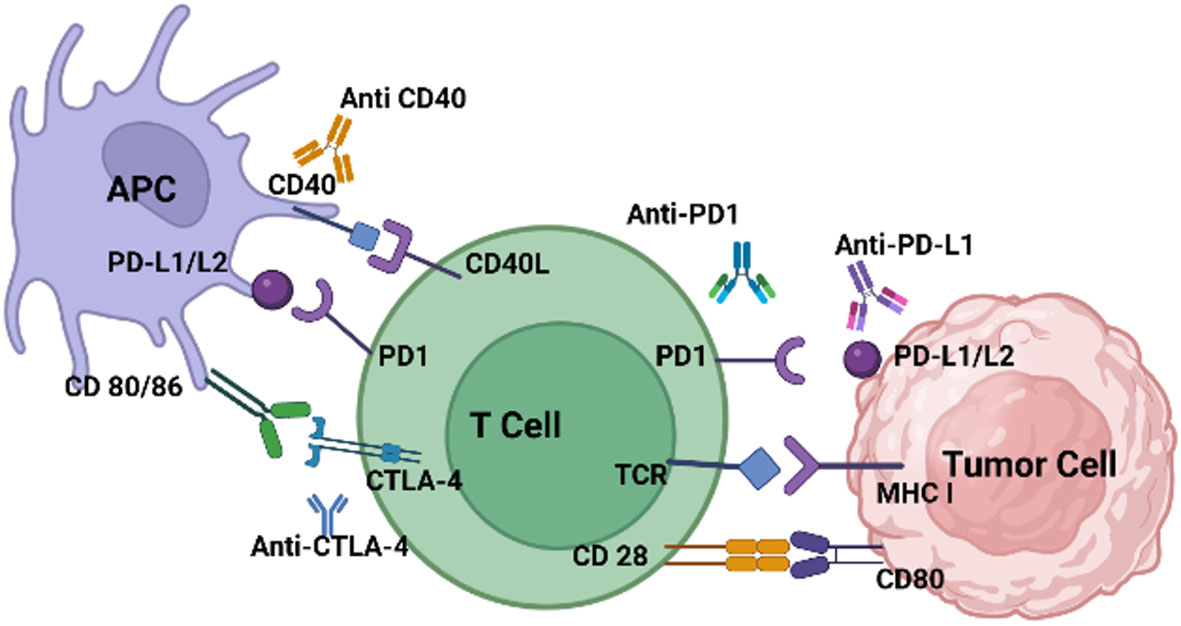
Figure 2 T-cells are inhibited when immune-checkpoints bind to ligands on tumor cells and APCs. PD-L1/L2, CD40, CD80/86, are expressed on tumor cells or APCs. Their expression is induced and maintained by many factors. PD-1, CTLA-4, CD40L and CD28 are expressed on exhausted effector T-cells. CTLA-4, which interacts with its ligands B7-1/CD80 and B7-2/CD86, or PD-1, binding to its ligand PD-L1, generate inhibitory signals, dampening T-cell immune responses. These receptors, CTLA-4 and PD-1, serve as targets for ICIs like anti-CTLA-4, PD-1, and PD-L1 aiming to decrease immune cell inhibition.
Researchers inserted two genes encoding the anti-PD-1 antibody (anti-PD-1) and the anti-human-tumor necrosis factor receptor superfamily member 9 (TNFRSF9 also known as CD134 or 4-1BB), into an OV named ΔTK-ARMED-VACV. ΔTK-ARMED-VACV was shown to have tumor-specific cytotoxic properties and the antibody genes were encoded without affecting viral replication. In an in vivo study, ΔTK-ARMED-VACV inhibited tumor growth and increased IFN-γ in treated mice (66). T-cell immunoglobulin and ITIM domain (TIGIT) is an immune checkpoint that inhibits the immune system’s activity. In 2021, Zue et al. designed VV-scFv-TGIT, an OVV encoding a single-chain variable fragment (scFv) targeting TIGIT. VV-scFv-TIGIT’s anti-tumor efficacy was explored alone and in concert with PD-1 or lymphocyte-activation gene 3 (LAG-3) blockades. Injection of VV-scFv-TIGIT into mouse models showed that this virus specifically replicates in tumor cells and converts cold to hot tumors by recruiting CD8+ T-cells to the site. The VV-scFv-TIGIT group expressed IFN-γ, TNF-α, Grant B, IL-6, and IL-10 more than the wild-type VV or PBS control groups. VV-scFv-TIGIT increase PD-L1 expression on the surface of tumor cells which can increase tumor immunogenicity and response to immunotherapy. PD-1 or PD-L1 can be inserted into OVV to induce ICI overexpression, and anti-PD-1, anti-PD-L1, or their associated antibodies can block the interaction of ICI ligand and receptor to allow T-cells to remain active to remove cancer cells more effectively (Figure 3). In mice, encoding 3 forms of mPD-1 binders including whole antibody (mAb), fragment antigen-binding (83), or single-chain variable fragment (scFv) into Western Reserve (WR) oncolytic vaccinia virus improved the oncolysis ability of this virus. Importantly, intra-tumoral injection of this virus increased mAB secretion 1900 times compared to subcutaneous injection (69).
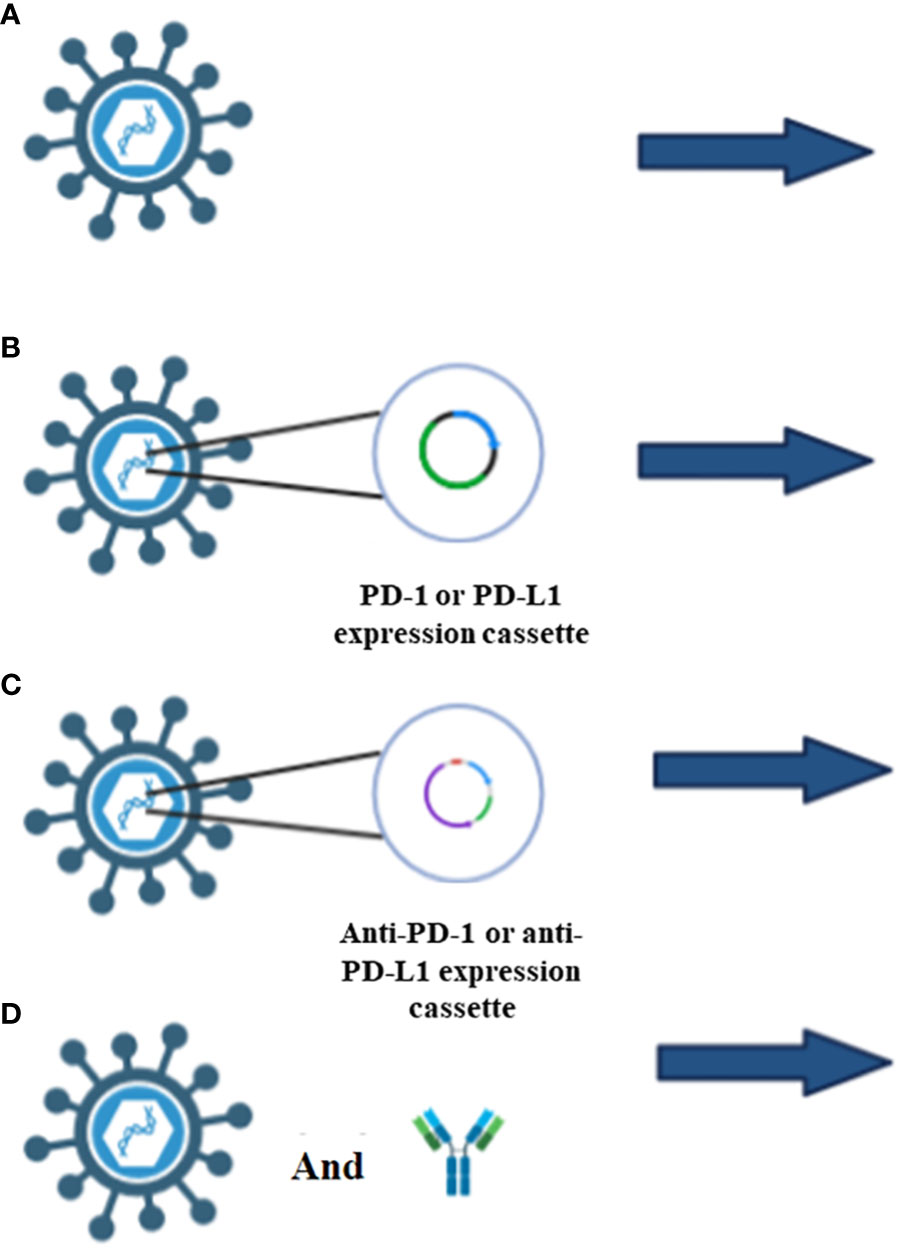
Figure 3 Different types of ICIs applications. (A) OVV injection alone increases the ICIs expression which is due to the increase of interferon γ levels through the JAK/STAT pathway. (B) Insertion of PD-1 or PD-L1 in OVV induce the overexpression of ICIs which increase the success rate of immunotherapy. (C) Insertion of anti-PD-1 or anti-PD-L1 antibody, block the interaction of ICIs with T-cells, therefore t-cell can inhibit the tumor cells. (D) Injection of OVV and anti-PD-1 antibody or anti-PD-L1 antibody in a designed regimen not only recruit immune cells, but also inhibit the interaction of ICIs with T-cells.
Cytokine-expressing OVs
Cytokines play a pivotal role in orchestrating the immune response against tumors, with various interleukins contributing to the elimination of cancerous cells. Interleukin-2 (IL-2) is a potent cytokine that stimulates the proliferation and activation of cytotoxic T-cells and natural killer cells, enhancing their ability to recognize and eliminate tumor cells (11). rVVDD-hIL2 is a genetically modified oncolytic vaccinia virus armed with human interleukin-2 (hIL2) (9). This recombinant virus effectively infects and kills tumor cells while expressing increasing levels of hIL2, indicating its potential as a promising vector for cancer treatment. The insertion of hIL2 does not compromise viral replication capacity, highlighting the therapeutic potential of this approach (84).
Interleukin-12 (IL-12) promotes the differentiation of T-cells into cytotoxic T lymphocytes (CTLs) and enhances their anti-tumor activity. Oncolytic vaccinia virus delivering tethered IL-12 converts “cold” tumors, characterized by a scarcity of T-cell tumor infiltration, into “hot” tumors, with an abundance of tumor-infiltrating T-cells, enhancing immune responses without systemic toxicity and cytokine release syndrome (85). An oncolytic vaccinia virus incorporating hIL-7 and mIL-12 genes exhibited antitumor activity by increasing the inflammatory immune status and rendered the tumors susceptible to immune checkpoint blockade. The activation of immune responses was observed both in treated tumors and untreated distant tumors found on the animal’s other flank. These encouraging outcomes, validated in humanized mice with human cancer cells, underscore the potential for further investigation in individuals with non-inflamed solid tumors (22).
IL-15 supports the development and function of NK cells and memory T-cells, contributing to sustained anti-tumor immunity. In one study, OVVs were developed from the VV Lister strain from the Institute of Virus Preparation, Moscow, Russia (LIVP) that express interleukin-15 (IL-15) or its receptor subunit alpha (IL-15Rα). Using murine colon and breast carcinoma models, the oncolytic activity of these new variants was evaluated, for each virus alone and both in combination. Shakiba et al. demonstrate that the combination of these recombinant variants promoted the generation of the IL-15/IL-15Rα complex. In vivo, mice bearing syngeneic 4T1 tumors had significant tumor regression and increased survival following treatment with the combination of LIVP-IL15-RFP and LIVP-IL15Ra-RFP, lymphocytes were recruited to tumor sites, and no harmful effects were observed in the liver or spleen (40).
The IL-1 cytokine family, including IL-36α, IL-36β, and IL-36γ, although less studied in the context of tumor immunity, has been implicated in inflammation and may have potential consequences for modulating the immune response against cancer. IL-36γ has previously been shown to promote interferon-γ (IFN-γ) production, increase type 1 immune responses in the TME, and promote antitumor immune responses (86). Several VV genes have been implicated in mediating the function of IL-1 family members, making these cytokines attractive targets when modulating OVV immunogenicity (54). These genes include A46R, a putative IL-1 antagonist, B13R, also known as SPI-2, inhibiting the enzyme converting pro-IL-1β to IL-1β, and B15R, a soluble IL-1β receptor (35, 63). Yang et al. developed an OVV expressing IL-36γ (IL-36γ -OV).
An OVV expressing IL-36γ (IL-36γ-OV) developed by Yang et al. showed significant therapeutic efficacy in multiple murine tumor models. Additionally, IL-36γ-OV modulated the TME, promoting dendritic cell and lymphocyte infiltration and reducing myeloid-derived suppressor cells and M2-like tumor-associated macrophages, into the tumor (87).
Thus, these engineered viruses encoding various cytokines not only induce direct oncolysis but are able modulate the tumor microenvironment, and may lead to increased infiltration of immune cells, reduced immunosuppressive elements, and increased differentiation of T-cells into effector cells. The combination of OVs with diverse cytokines showcases a multifaceted approach, harnessing both viral oncolysis and cytokine-mediated immunomodulation, holding great potential for advancing cancer treatment strategies.
IFN therapy
The introduction of IFN-γ genes into OVVs is strategically designed to enhance the OVVs’ oncolytic potential. This genetic modification aims to amplify the anti-tumor effects of OVVs, capitalizing on the immunomodulatory properties of interferon-beta (IFN-γ). Interferons play a crucial role in the body’s innate immune response, and IFN-γ, in particular, has been recognized for its ability to regulate immune functions and exhibit antiviral and anti-tumor activities (88). IFN-γ genes inserted into OVVs can increase the anti-tumor effects but also increase virus inactivation in non-cancerous cells. Preclinical testing of this combination therapy used the VV B18R deletion mutant as a backbone for the expression of IFN-γ. This newly engineered OVV showed IFN-dependent cancer selectivity and efficacy in both tumor cells and tumor-associated vascular endothelial cells in vitro, and tumor targeting and efficacy in in vivo mouse models (89).
Chemotherapy
Combining vaccinia virotherapy with chemotherapy agents is a promising approach to cancer treatment. For example, a combination of VV with paclitaxel produced a synergistic effect mediated by type I interferon, secreted soon after infection, and high-mobility group protein B1, secreted after cell death (90). The sorafenib and VV combination has shown promising anti-tumor results in some models and patient trials (31, 91, 92). In a mouse model of lung adenocarcinoma, CPA and GLV-1h68 had synergistic anti-tumor effects. In a study of human colorectal adenocarcinoma, OVV synergized with irinotecan to decrease tumor cell viability. Combination therapy significantly improved survival over either monotherapy. Cyclophosphamide (CPA), and GLV-1h68 have synergistic anti-tumor effects on PC14PE6-RFP xenografts. Protein profiling of tumor lysates for the untreated, CPA-, GLV-1h68- and combination treatments showed that host-derived pro-inflammatory cytokines and chemokines, such as eotaxin, MIP-1β, MCP-1, MCP-3, MCP-5, TNF-α, and MPO, are upregulated in tumor tissues after viral infection and combination therapy OVVs may synergizes with irinotecan (CPT-11) in human colorectal adenocarcinoma. This combination therapy significantly decreased tumor cell viability compared to each method used as a monotherapy. It is hypnotized that the sequence of administration of chemotherapy and virotherapy influences the interactions between these two treatment methods (93).
Many cancer cells secrete high levels of apoptosis-inhibiting proteins (IAPs) and escape from apoptosis in this way. Mitochondrial protein Smac inhibits IAPs, such as XIAP, and the activity of caspases, thereby inducing apoptosis in cancer cells. In the oncolytic virus VV-Smac, the Smac gene is inserted. When used in combination with chemotherapy, in addition to causing the Smac protein’s expression, VV-Smac reduced cancer cells’ survival (94).
The therapeutic combination of mpJX-594, a replication-competent VV, and sunitinib increases vascular pruning, leakage and recruitment of CD8+ T-cells; and decreases the invasion and metastasis of cancer cells. This synergy functions with sunitinib reducing tumor vasculature, subsequently cutting tumor angiogenesis, which triples the effect of the mpJX-594 compared to virus therapy alone (95).
Doxorubicin-resistant ovarian cancer cells (A2780-R), are resistant to oncolysis by OVV. The mechanism of this resistance is inhibition of OVV replication by STAT3 protein kinase inhibitors in resistant cells. To overcome this 2-sided resistance, combination therapy of OVV and trametinib was used, which significantly reduced xenograft tumor growth (96). In a case report study, pretreatment of a person resistant to chemotherapy with the OVV GL-ONC1 laparoscopically and a combination of chemo drugs (paclitaxel, carboplatin, bevacizumab) increased the response to treatment (29). In patient who have had surgery as the only treatment, without prior chemotherapy, the injection of a single dose of OVV can increase the response to treatment by secreting IFNα and other chemokines (97). Table 3 shows different combinations of OVV strains with chemotherapy drugs.
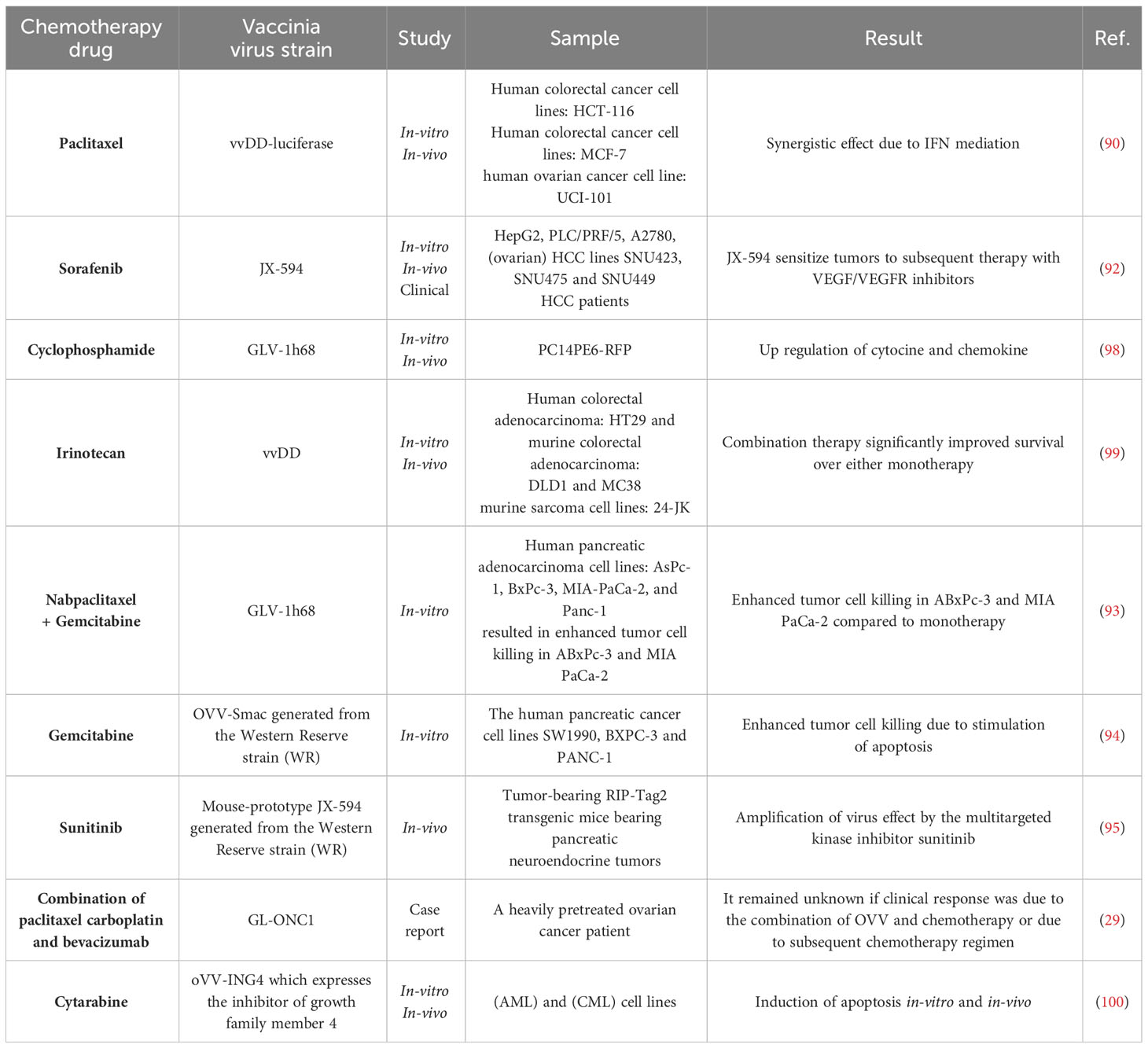
Table 3 Chemotherapy drugs in combination with OVV therapy.
Direct delivery of chemotherapy drugs to a specific site can be achieved using the isolated limb perfusion (ILP) technique. This method involves the insertion of a cannula into the blood vessels leading to the tumor area, isolation of the organ from systemic circulation through the use of a tourniquet, and high-dose chemotherapy administration. Standard ILP treatment employs melphalan with tumor necrosis factor alpha (TNF-α) and has been used to treat advanced extremity sarcoma and in-transit melanoma (101). The ILP of GLV-1h68 results significant viral deposition in the tumor with evidence of viral replication within the tumor. At the same time, the virus cannot damage healthy cells. The combination of GLV-1h68 with melphalan ILP intensified the effect of virus treatment (102).
Radio therapy
Radiotherapy has a well-established role in local disease control and may be given either pre- or post-operatively. Combining radiotherapy and OVs has demonstrated potential in cancer treatment. The synergistic effects of radiotherapy and OVV therapy can be seen in many studies. Expression data obtained from a combinatorial regimen of radiotherapy and OVV therapy demonstrated that this synergy is not due to increased viral replication. Rather, it is mediated through the induction of intrinsic apoptosis. One study demonstrated that GLV-1h68 therapy downregulated the anti-apoptotic BCL-2 proteins MCL-1 and BCL-XL as well as downstream inhibitors of apoptosis, resulting in cleavage of effector caspases 3 and 7. In the ILP rat model, the combination of OV and radiotherapy significantly delayed tumor growth and prolonged survival compared to single-agent therapy (103). The efficacy of OVV and radiotherapy in an in vitro assessment of head and neck cancer was shown to be dose- and time-dependent. In CD-1 nude mice, this same combination induced caspase activity and increased long-term regression (104). A combination of the OVV GLV-1h151 and radiotherapy in AsPC-1, a human pancreatic adenocarcinoma cell line, xenografts in mice inhibited tumor growth and had no toxicity to the mice (105). Combining radiotherapy and OVV increased necrosis and apoptosis in tumors and the ensuing release of DAMPs. This combination enhanced the in vivo anti-tumor effect and increased splenic CD4+Ki-67+ helper and CD8+Ki-67+ cytotoxic T lymphocytes, and tumor-infiltrating CD3+CD4+ helper and CD3+CD8+ cytotoxic T lymphocytes. Conversely, tumor-infiltrating regulatory T-cells were decreased using this same combination of OVV and high-dose hypofractionated stereotactic body radiotherapy (106). In a first-phase trial of intravenous OVV GL-ONC1 with cisplatin and radiotherapy, the 19 patients with advanced head and neck cancer included had no disease progression for 30 months following treatment, with an overall survival rate of 74.4% reported. However, due to the intravenous injection of therapeutics, virus delivery to the tumor was challenging, and some patients reported grade 1, 2, and 3 side effects (32). Since VV has a DNA genome, there is concern that it may be destroyed or modified following radiotherapy. To study this effect, Wilkinson et al. injected doses of GLV-1h68 by ILP to rats and then treated them with external beam radiation therapy (EBRT). This study shows that not only is the DNA of GLV-1h68 resistant to EBRT, but this combination has additive and possibly synergistic effect (103). The reason for this result in this study could be that the harmful effects of radiation on the virus were controlled in terms of time. It seems that prescribing a suitable program of virus injection and radiotherapy or any other combined treatment can not only limit the effects of the two methods on each other, but also can have more oncolysis effects.
CAR-T cells therapy
Chimeric antigen receptor (CAR) T-cell therapy has recently transformed the treatment of refractory hematological cancers, including acute lymphoblastic leukemia and chronic lymphocytic leukemia. CAR-T cell therapy entails the in vitro design, modification, and amplification of T-cells such that they recognize tumor cell surface antigens using the T-cell surface transduced CAR structure. This allows these CAR-T cells to move into the tumor microenvironment of “hot” tumors and to kill cells displaying the cognate antigen. In 2017, the FDA approved CD-19-specific CAR-T cells to treat refractory B-cell lymphomas (107). Despite this advance, in patients with multiple solid tumors, only minor and transient responses were observed, perhaps due to poor tumor penetration and impaired function of these T-cells in the “cold” tumor environment. OVs are capable of potential synergy with CAR-T cell therapies as they can promote the migration, proliferation, and activation of T-cells, particularly when they are engineered to deliver immunostimulatory cytokines, T-cell attracting chemokines, or immune checkpoint-targeting molecules (108).
T-cell trafficking depends on the correct combination of cell-secreted chemokines and chemokine receptors on the effector cell. Often, tumors produce only nominal amounts of chemokines, which result in the immunologically “cold” tumor phenotype with very few effectors successfully reaching the tumor (109). Modified OVV (VV.CXCL11) which is produced by inserting the CXCR3 ligand on CXCL11 increase T-cell trafficking into tumors. VV.CXCL11 has the ability to recruit total and antigen-specific T-cells into the TME. As well, VV.CXCL11 significantly enhanced the anti-tumor efficacy of recruited T-cells as compared to the direct delivery of CXCL11 by CAR-T cells (108, 109). Similarly, engineered OVV (OV19t) that express a non-signaling, truncated CD19 (CD19t) protein, enable CAR-T cell to target the infected cells. In vitro, OV19t infection of tumor cells result in de novo cell surface CD19 expression prior to the virus-mediated lysis of tumor cells, with co-cultured CD19-CAR-T cells secreting cytokines and exhibiting potent cytolytic activity against infected tumor cells. In vivo, OV19t aided tumor control following the administration of CD19-Car-T-cells and local immunity against tumor cells with tumor infiltration of both endogenous and injected CAR-T cells. The CAR-T cell-mediated tumor killing also caused OV19t to be released from dying cells which propagated tumor expression of CD19t, allowing greater viral spread and a more widespread response (110).
OVV armed with bi-specific T-cell engagers
Bi-specific T-cell engagers (BiTEs) are a type of immunotherapy designed to enhance the body’s immune response against cancer cells. These molecules are engineered to simultaneously bind to T-cells and cancer cells, bringing them into close proximity. This dual binding allows for the formation of the immunological synapse where activated T-cells can secrete perforin and other granzymes leasing to cancer cell lysis (111). OVVs can be engineered to encode BiTEs. T-cell engager CD3-scFv provides an alternative approach for the engagement of T-cells for cancer immunotherapies. In 2014, Yu et al. constructed one such VV encoding a BiTE consisting of a scFv for CD3 and one specific for EphA2, a tumor cell surface antigen, named EphA2-TEA-VV. In vitro analyses revealed similar replication and cancer cell lysis when compared to the GFP-expressing double-deleted WR control. Similarly, EphA2-TEA-VV directed T-cells towards cancer cells, activated T-cells through secretion of IFN-γ, IL-2, and binding to CD3, and induced bystander killing of noninfected tumor cells. In vivo analysis of an A549 NSCLC xenograft model in SCID-Bg mice demonstrated EphA2-TEA-VVs potent antitumor effect compared to the GFP control. Treatment with both EphA2-TEA-VV and unstimulated peripheral blood mononuclear cells from healthy donors synergized to have stronger antitumor effects compared to each treatment used as a monotherapy. Consequently, this suggests that arming oncolytic VVs with T-cell engagers has the potential to improve therapy potency by boosting T-cell activation and promoting broader destruction of tumor cells, including those not infected, offering a promising strategy for advancing oncolytic virus therapy in cancer treatment (112).
Yu et al.’s team also created an oncolytic vaccinia virus armed with a BiTE targeting murine CD3 and fibroblast activation protein (FAP), known as mFAP-TEA-VV. Their primary goal was to address safety concerns linked to FAP-targeted immunotherapies related to off-target activation, and which have shown significant antitumor effects like (112). Similarly, to EphA2-TEA-VV, mFAP-TEA-VV demonstrated similar replication ability and oncolysis as an unmodified VV control, and was also capable of triggering bystander killing of uninfected FAP-expressing cells in co-culture assays with murine T-cells. In an immunocompetent B16-F10 model in vivo, mFAP-TEA-VV increased viral load within the tumor and has strong antitumor effects compared to the VV control. Notably, the enhanced spread of the mFAP-TEA-VV virus was associated with the degradation of the tumor stroma, indicating a correlation between improved viral distribution and the destruction of the supportive tissue around the tumor (113).
The “double-deleted” Western Reserve vaccinia virus (vvDD), was engineered to express an anti-FAB/CD3 BiTE. This demonstrated improved viral replication and promoted the infiltration of active T-cells in the melanoma syngeneic models, tumor regression was proven. Furthermore, anti-FAP/CD3 BiTE-expressing Ad ICOVIR15K showed better antitumor responses and intratumoral T-cells. In addition, aside from the BiTE approach, VV that encoded anti-FAP expressed moderate regressions in tumor growth compared with unarmed VV. However, a notable reduction of cancer associated fibroblasts (CAF) was observed. This moderate regression in antitumoral activity can be caused using a cytotoxic FAP-blocking antibody over the cytotoxic anti-FAP BiTE. The FAP-blocking antibodies did allow the CAF to regrow and restore the TME (114).
In 2022, Wei et al. developed VV-EpCAM BiTE to enhance antitumor immunity in solid tumors by modulating the immunosuppressive TME. This recombinant virus secretes a BiTE against epithelial cell adhesion molecule (EpCAM), a solid tumor-associated antigen, and CD3. VV-EpCAM BiTE effectively infected and lysed cancer cells. The binding of EpCAM-expressing malignant cells and CD3ϵ on T-cells, mediated by the secrete EpCAM BiTE, activated T-cells and led to the release of IFN-γ and IL-2. When administered intratumorally, VV-EpCAM BiTE significantly improved antitumor activity, particularly in tumors with high EpCAM expression. This treatment also increased TME immune cell infiltration, reduced CD8+ T-cell exhaustion, and enhanced T-cell-mediated immune activation (115).
Lei et al. engineered an oncolytic vaccinia virus expressing CD19-specific BiTE (OVV-CD19BiTE). When compared to the non-engineered type, OVV-CD19BiTE can recruit more CD3, CD8, and naïve CD8 T subpopulations to tumor tissues, while retaining similar ability to replicate in and lyse tumor cells (116).
Photodynamic therapy
Photodynamic therapy (PDT), is a clinically approved therapy for solid tumors based on the photo-activation of a tumor localizing agent, the photosensitizer (60), resulting in the generation of cytotoxic singlet oxygen (1O2) [37 (60). In this treatment modality, a PS is combined with an immune protein to allow PS delivery to cancer cells. When light is applied, the PS kills cancer cells and causes the generation of an immune response which can lead to further cancerous cell death. The second-generation, chlorin-based 2-(1-hexyloxyethyl)-2-devinyl pyropheophorbide-a (HPPH) sensitizer exhibits favorable photophysical and pharmacokinetic properties in clinical trials, as compared to the porphyrin-based photosynthesizer Photofrin (117). In 2004 pre-clinical studies, HPPH-PDT was shown to result in varying patterns of vascular, cellular and inflammatory responses with long-term tumor control rates depending on the specific treatment regimen employed (118). HPPH-sensitized PDT may have therapeutic activity in combination with a TK- and VGF-deleted VV. This VV expressed enhanced green fluorescent protein, EGFP, used as a marker to track NXS2 neuroblastomas established in syngeneic mics and human FaDu xenografts in athymic nude mice. The combination of PDT and OVV was the most effective at controlling primary and metastatic tumor growth compared to either treatment used as a monotherapy. One dosage combination even caused a decrease in tumor volume in both NXS2 and FaDu tumors within six weeks of treatment. Since PDT leads to the disruption of tumor vascularization, the virus titer in the tumor was higher in the combination than in oncolytic monotherapy, which indicates that these two treatment methods combined synergistically and did not negatively impact each other (118).
RNA therapy
RNA therapy is a type of therapeutic method based on RNA molecules, in which RNAs are used to manipulate the expression and activity of target molecules. The first RNA-based drug was used in the 1990s, during which mRNAs were injected into mice to target protein production. Recently, micro RNAs, single-stranded mature miRNAs of ~22 nucleotides, have been used to treat diseases, especially cancer. In this method, miRNAs act as oncomiR by targeting tumor suppressor genes, or as tumor suppressor by targeting oncogenes. miRNAsbind to complementary sequences in the 3’-untranslated region (UTR) of a messenger RNA (mRNA), so they can repress gene expression post-transcriptionally. Therefore, by combing miRNA therapy and OVV therapy, the virus’ pathogenic genes can be removed to increase the safety profile of the OV therapy. For example, B5R protein downregulation mediated by let-7a miRNA was shown to decrease viral pathogenicity and impair the oncolytic activity of VV. This let-7a miRNA-regulated VV (MRVV) is capable of selectively replicating and inducing oncolysis of tumor cells while displaying no toxicity to healthy cells. B5R expression and MRVV replication depended on infected cells’ endogenous let-7a expression level. The oncolytic potentials of the B5R-negative LC16m8Δ and B5R-positive LC16mO viruses were compared in mouse models with human cancer xenografts using intratumoral administration of the MRVV. While both viruses decreased tumor size 18 days post-infection, LC16mO-treated mice died or were sacrificed on days 21-28 following severe viral toxicity symptoms, including weight loss and pock lesions on tails, paws, faces, and other areas of the body, while LC16m8Δ mice did not exhibit these same symptoms. This group showed that this transgene insertion did not affect let-7a-regulated oncolytic activity, MRVV replication was inhibited in normal tissues, and MRVV reduces viral pathogenicity while maintaining oncolytic activity (119).
OVV therapy was shown to be more effective in paclitaxel-resistant KFTX ovarian cancer cells than KFlow paclitazel-sensitive cells. The long non-coding RNA (lncRNA) urothelial carcinoma-associated 1 (UCA1) is overexpressed in paclitaxel-resistant cells, and UCA1 expression was shown to correlate to the oncolytic effects of OVV in various primary ovarian cancer cell lines. This suggests that UCA1 is involved in regulating OVV’s oncolytic effects. UCA1 also enhanced the cell-to-cell spread of OVV via the activation of Cdc42, a Rho GTPase, resulting in better therapeutic outcomes in ovarian cancer (120). Similarly, it has been shown that UCA1 significantly increases OVV cell-to-cell spread in colorectal cancer. UCA1 inhibited miR-18a and miR-182, promoting the Cdc42 activation that enhanced OVV spread via filopodia formation (120).
Hormone therapy
Aldosterone is a type of mineralocorticoid hormone that controls the balance of water and salts in the kidneys by retaining sodium and releasing potassium from the body. In the phase I hepatoma clinical trial of JX-594, an OVV derived from the Wyeth strain, Park et al. noted viral replication in most patients, with remarkably more detected in cancer patients with severe ascites and peripheral edema. Ascites is the abnormal accumulation of fluid within the peritoneal cavity that occurs due to advanced cancer. This abdominal fluid sequestration leads to further fluid retention by the kidneys due to the stimulatory effects of hormones such as aldosterone, which can cause further peripheral edema. It is subsequently assumed that aldosterone levels increase in these patients and may increase the proliferation of tumor cell lines (31). In another study, viral replication significantly increased with simultaneous JX-594 and aldosterone treatment in A2780, PC-3, and HepG2 cells but not U2OS cells. The differential timing of aldosterone treatment altered viral replication differently in each cell line. The aldosterone-receptor inhibitor spironolactone inhibited JX-594 entry across the cell’s plasma membrane. Similarly, viral entry was significantly decreased by treatment with 5-N-ethyl-N-isopropylamiloride (EIPA), an Na+/HA+-exchange inhibitor, but restored following aldosterone treatment (121). These findings suggest interactions between OVV therapy and hormone levels such that synergy may be observed between oncolytic virotherapy and hormone therapies.
Shielding OVVs from the innate and adaptive humoral immunity
One limitation of the therapeutic use of OVs is viral inhibition by the immune system. A range of different OV delivery methods have been explored, including both intratumoral injection and systemic delivery. Intratumorally injected OVs have demonstrated success but allow only the treatment of easily accessible solid tumors, relying on viral replication within the TME and subsequent dissemination to distant sites to treat metastases. This replication and dissemination is often ineffective due to immune responses against the OV. Systemic OV delivery allows the treatment of both primary tumors and any metastases, whether overt or undiagnosed, and is an attractive option for treating metastatic and inaccessible solid tumors and blood cancers. Vaccine-based OVs induce a strong immune response within the tumor to aid in killing tumor targets, but this same immune response may sometimes interfere with the virus’ oncolytic activity. A primary focus of OV research is thus how to protect the therapeutic virus from the patient’s immune system (122). During an infection, viral entry into cells increases intracellular interferon secretion, leading to inflammation. The secretion of innate immune cells, such as NK cells and phagocytes, increases during the inflammatory response. Concurrently, viral antigens enter lymph nodes and, with the help of antigen-presenting cells such as DCs, activate the adaptive immune response of B and T-cells. Activated B-cells differentiate into plasma cells that secrete antibodies that disrupt the viral life cycle by preventing entry into susceptible cells. T-cells directly attack and destroy the virus through cytotoxic effects. Similarly, systemic VV delivery is limited by pre-existing immunity. For OVV, as worldwide immunization with VV was undertaken during the eradication of smallpox, many individuals who are currently developing cancer present with immunity to this virus.
Complement is a crucial component of the innate immune system’s first line of defense, targeting foreign pathogens for opsonization, neutralization, phagocytosis, and clearance from the circulatory system. In some cancer patients, neutralizing antibodies due to smallpox vaccination can limit the efficacy of OV therapy. Natural barriers in the blood, including these antibodies and complement, will likely limit the efficacy of repeated intravenous OV administrations. Depleting complement proteins using cobra venom factor (CVF) improved OV delivery and infection of tumors in rats (123) Additionally, a combinatory strategy of complement inhibition and OV therapy in cynomolgus macaques demonstrated enhanced tumor control at early time points (124).
Given the promising results of existing oncolytic therapies and research, it is clear that a balance must be struck between antiviral and anti-tumor immunity. This balance may be created by injecting sufficiently high initial amounts of the virus, combining OV therapy with other immunotherapies, and methods that shield the virus from the immune system, since increasing OV doses is not a good option as it may cause symptoms associated with viral disease in patients, especially those already immunocompromised at the start of treatment (125). Local intratumoral virus injection can largely overcome this problem and deliver the virus to the tumor cells before being targeted by the immune system. Below, various methods are discussed for keeping the OVV from the immune system.
Coatings
One way to protect the therapeutic virus from the immune system is to use physical shields. These physical protectors can be cell-derived nanovesicles, liposomes, or chemical polymers. Polylactic-co-glycolic acid (PLGA) is a biocompatible and biodegradable synthetic material widely used in studies of nanotechnology and drug delivery. PLGA nanofiber has been used as a stent for the targeted delivery of an OVV to a CRC tumor and was studied using in vitro cell cultures and in an in vivo animal tumor xenograft., In CT-26 cells, the infectivity and anti-tumor activity of OVVs released from the PLGA membrane were preserved, with cell viability decreasing in a dose-dependent manner. Similarly, this study showed that the embedded virus was continuously released over 48 hours (126). The use of virus coatings, however, may have limitations that impede use. For example, coatings may prevent OVs from accessing and affecting tumor cells and present barriers to commercialization due to greatly increased costs (127).
Cellular carriers
Another way to protect viruses from the immune system is via the use of cell carriers. In this method, carriers are loaded with the virus to allow their undetected transport throughout the body. One example where the use of cell carriers may be suitable for the delivery of OVs is in brain cancers. Due to the presence of the blood-brain barrier, OVV has difficulty accessing brain tumors. However, the influx of blood and cerebrospinal fluid into the brain following tumor resection can be hijacked, such that, using cell carriers, oncolytic virotherapeutics are delivered to the brain. These cell carriers can be derived from a variety of cells. For example, they may come from stem cells such as mesenchymal stromal cells (MSCs). Extracted MSCs from adipose tissue, called adipose tissue-derived stem cells (ADSCs), were infected with the VV LIVP strain and co-cultured with blood mononuclear cells. The viability of these cells was then checked in vitro. The results show that co-culture of OVV with these mesenchymal cell carriers (the ADSCs) increased OVV efficacy (128). These cell carriers face limitations to their use in clinical studies due to allogeneic responses from NK cells and adaptive T-cells, and since therapeutic efficiency may be reduced due to the removal of the ADSCs by the patient’s immune cells. Cell carriers may also be derived from solid tumors. Due to potential risks, injecting these types of carriers into patients has yet to progress into clinical trials. In addition to taking advantage of both OVV and T-cells or CAR-T cells, these cells can also be considered as carriers for the virus. Injecting T-cells and human HER2-CAR-T cells with a double deleted vaccinia, vvDD-GFP to mouse showed that combining T-cell and OVV therapies causes the successful and precise delivery of the OVV to the tumor site without decreasing the individual effect of each therapy (129). Additionally, these T-cell carriers do not face the same limitations as other cell carriers, such as the elimination by the immune system. Because engineered T-cells recognize surface-expressed tumor antigens independent of antigen processing or MHC presentation, using CARs avoids the restrictions faced by TCR-activated T-cells (130).
Recent clinical trial results of different OVVs
In recent years, many different OVVs have shown promising results in clinical trials, such as JX-929, GL-ONC1, JX-594, and others (Table 1). JX929 is based on the Western Reserve strain of vaccinia virus and has been engineered to selectively replicate in tumor cells by modifying the VGK and TK genes (16). This OVV has been tested first for intratumoral injection and then for intravenous injection in humans against different types of solid tumors for phase I clinical trials (16, 17). Results have shown that this OV can be administered safely, no dose limiting toxicities was observed and the virus is replicating selectively in tumor tissue (16, 17). However, clinical benefits from the administration of this virus cannot be confirmed at this moment, further experiments need to be done. The GL-ONC1 is a Lister strain OVV, it incorporated multiple gene modification such as the TK and the hemagglutinin genes (20). This OVV has been tested for intraperitoneal and intravenous injection either alone or in combination with other treatments. When tested alone or in combination with chemotherapy or with radiation, no dose limiting toxicities and little adverse effects were observed (20, 29, 32). Currently into phase III clinical trial, preliminary results of GL-ONC1 in combination with chemotherapy suggest anti-tumor activity following the infection of tumor cells, but the increased efficiency cannot be confirmed to be caused by the OV, further analysis needed to be done to validate the impact of GL-ONC1 (29). The double recombinant VV “VV-GMCSF-Lact”, which is a Lister strain based VV, is currently into phase I of clinical trial. This OVV is expressing lactaptin protein and human granulocyte-macrophage colony-stimulating factor (GM-CSF). Preliminary results indicate that this OV is a potential treatment against chemo resistant tumors (27). There is also the recombinant human IL-21 oncolytic vaccinia virus (hV01) with deletion of TK and VGF gene that is currently into phase I clinical trial. Preliminary results shown potential benefits of intratumoral expression IL-21 on TME (34). Finally, one of the most tested OVV is JX-594 also known as PexaVec. It is based on Wyeth vaccine strain of VV, it has been engineered to express GM-CSF and to selectively replicate in tumor cells by deletion of TK gene. This OV has been tested against many types of solid tumors with either intratumoral injection or intravenous infusion and as a monotherapy or in combination with other treatments (11, 12, 18, 23–26, 28, 30, 31). As monotherapy, there is a multiple clinical trial, either in phase I or II. Results suggest a potential antitumor activity offered by the OV in addition to a safe administration into the patient, the administration was well tolerated (11, 12, 23, 24, 30, 31). Few groups are now testing this OV in combination with agents like irinotecan, Durvalumab and Tremelimumab (28, 131). The first one is a chemotherapy agents and the other two are antibodies that target different immune checkpoint modulator interactions. To this date, the combination of PexaVec with immune checkpoint therapy indicates that there is no additional toxicity observed and seems to be well tolerated. In addition, the combination of treatments shows evidence that it can cause immune changes in the TME (26, 131). In general, the different clinical trials that have been done or are ongoing at this moment show that there is a lot of work to do to validate the benefits of the OVV and to optimize these treatments in order to increase the long-lasting influence and their efficacy as well as their safety. Even if there is no dose limiting toxicities observed, a number of low adverse effects have been reported, it would be interesting to see if these OVV can be optimized in order to get lower adverse effects.
Concluding remarks and perspectives
Currently, oncolytic viruses are a promising area of cancer immunotherapy research. Gene editing technologies allow researchers to modify viruses such that they are only capable of replicating in cancerous cells while sparing healthy cells. These viruses can release tumor antigens to stimulate the patient’s immune response and simultaneously deliver a therapeutic payload. While oncolytic viruses exhibit promising anticancer potential, certain limitations exist when employing them as monotherapy. One notable constraint is the variability in treatment response among patients, influenced by the diverse immunological profiles and genetic characteristics of individual tumors (132). Moreover, tumor cells may develop resistance to oncolytic viruses over time, compromising the sustained efficacy of monotherapeutic approaches. Acknowledging the dual role of tumor vasculature, some strategies aim to collapse it for anti-cancer effects, while others focus on normalizing it for enhanced OV dissemination (6). Additionally, the host immune system may mount a robust response against the oncolytic virus, leading to its clearance before achieving optimal antitumor effects. Addressing these inherent challenges faced by oncolytic virus monotherapies thus becomes imperative in creating successful treatment strategies for patient. Combining OVs with traditional therapies such as chemotherapy or radiation can create synergistic effects, leveraging the strengths of each approach to overcome individual limitations. Furthermore, the combination of oncolytic viruses with immunomodulators, including immune checkpoint inhibitors, can augment the immune response against cancer cells, fostering a more sustained and potent antitumor effect. These combinational strategies offer a comprehensive and personalized approach to cancer treatment with improved efficacy and broader applicability. Combination OV treatments have yet to be approved but various are undergoing clinical and pre-clinical trials, with the aim of increasing response rates via additivity or synergy between treatments. This review has summarized the main oncolytic vaccinia virus combinations currently under investigation and potential explanations for this synergy. Continuing research is required to elucidate the mechanisms underlying the efficacy of these treatments to allow for their approval. Additionally, research may be expanded to study the effects of OVV with other secondary treatments including different types of chemotherapy drugs and immune therapies such as cytokines. Further research on delivery methods must also be undertaken to allow for the greatest response rates while minimizing off-target side-effects and toxicity, as well as investigating OVs for other types of cancers. The promising combination studies provide necessary evidence for continuing OV research in the hopes of having high response rates and becoming a more mainstream treatment option for patients.
In conclusion, the field of oncolytic virus therapy for cancer treatment is rapidly advancing, and combination treatments hold great promise for improving response rates and overall patient outcomes. The use of gene editing technologies has enabled the development of viruses that can specifically target cancer cells, thus avoiding harm to healthy cells. This review has highlighted the current state of oncolytic vaccinia virus combination treatments under investigation, and potential mechanisms behind the observed synergy.
Despite the encouraging results, further research is required to demonstrate the efficacy of these treatments and secure their approval for clinical use. Studies on the combination of OVs with other secondary treatments, such as chemotherapy drugs and immune therapies, should also be undertaken to further optimize treatment outcomes. Additionally, the delivery methods of oncolytic viruses must be optimized to maximize response rates while minimizing off-target side-effects and toxicity.
The future of oncolytic virus immunotherapy for cancer treatment is bright, and it holds the potential to become a mainstream treatment option for patients. As the field continues to evolve, we can expect new discoveries and advancements that will further increase response rates and improve patient outcomes. Further research and investment in this area will be crucial to realizing the full potential of oncolytic virus therapy for cancer treatment.
In light of the above, it can be concluded that the oncolytic virus therapy holds great promise as a cancer treatment option, and the combination treatments offer a particularly promising avenue for future research. The next steps should be focused on demonstrating the efficacy of these treatments and optimizing delivery methods to bring these treatments to the forefront of cancer care.
Author contributions
TA: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. SM: Conceptualization, Formal Analysis, Writing – original draft, Writing – review & editing. MD: Conceptualization, Writing – original draft, Writing – review & editing. AZ: Writing – original draft, Writing – review & editing. NK: Conceptualization, Writing – original draft, Writing – review & editing. GS: Writing – original draft, Writing – review & editing. MT: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the grant the Canadian Cancer Society.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Andreansky S, Soroceanu L, Flotte ER, Chou J, Markert JM, Gillespie GY, et al. Evaluation of genetically engineered herpes simplex viruses as oncolytic agents for human Malignant brain tumors. Cancer Res. (1997) 57:1502–9.
2. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discovery. (2015) 14:642–62. doi: 10.1038/nrd4663
3. Shalhout SZ, Miller DM, Emerick KS, Kaufman HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol. (2023) 20:160–77. doi: 10.1038/s41571-022-00719-w
4. Lemos de Matos A, Franco LS, McFadden G. Oncolytic viruses and the immune system: the dynamic duo. Mol Ther Methods Clin Dev. (2020) 17:349–58. doi: 10.1016/j.omtm.2020.01.001
5. Wang L, Chard Dunmall LS, Cheng Z, Wang Y. Remodeling the tumor microenvironment by oncolytic viruses: beyond oncolysis of tumor cells for cancer treatment. J immunotherapy Cancer. (2022) 10:e004167. doi: 10.1136/jitc-2021-004167
6. Santry LA, van Vloten JP, Knapp JP, Matuszewska K, McAusland TM, Minott JA, et al. Tumour vasculature: Friend or foe of oncolytic viruses? Cytokine Growth Factor Rev. (2020) 56:69–82. doi: 10.1016/j.cytogfr.2020.07.007
7. Zhang B, Wang X, Cheng P. Remodeling of tumor immune microenvironment by oncolytic viruses. Front Oncol. (2021) 10:561372. doi: 10.3389/fonc.2020.561372
8. Zheng M, Huang J, Tong A, Yang H. Oncolytic viruses for cancer therapy: barriers and recent advances. Mol Ther - Oncolytics. (2019) 15:234–47. doi: 10.1016/j.omto.2019.10.007
9. Kelly CM, Antonescu CR, Bowler T, Munhoz R, Chi P, Dickson MA, et al. Objective response rate among patients with locally advanced or metastatic sarcoma treated with talimogene laherparepvec in combination with pembrolizumab: A phase 2 clinical trial. JAMA Oncol. (2020) 6:402–8. doi: 10.1001/jamaoncol.2019.6152
10. Rojas JJ, Thorne SH. Theranostic potential of oncolytic vaccinia virus. Theranostics. (2012) 2:363–73. doi: 10.7150/thno.3724
11. Mastrangelo MJ, Maguire HC Jr., Eisenlohr LC, Laughlin CE, Monken CE, McCue PA, et al. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. (1999) 6:409–22. doi: 10.1038/sj.cgt.7700066
12. Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. (2013) 19:329–36. doi: 10.1038/nm.3089
13. Guo ZS, Lu B, Guo Z, Giehl E, Feist M, Dai E, et al. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J immunotherapy Cancer. (2019) 7:6. doi: 10.1186/s40425-018-0495-7
14. Li M, Zhang M, Ye Q, Liu Y, Qian W. Preclinical and clinical trials of oncolytic vaccinia virus in cancer immunotherapy: a comprehensive review. Cancer Biol Med. (2023) 20:646–61. doi: 10.20892/j.issn.2095-3941.2023.0202
15. West EJ, Scott KJ, Tidswell E, Bendjama K, Stojkowitz N, Lusky M, et al. Intravenous oncolytic vaccinia virus therapy results in a differential immune response between cancer patients. Cancers. (2022) 14:2181. doi: 10.3390/cancers14092181
16. Zeh HJ, Downs-Canner S, McCart JA, Guo ZS, Rao UN, Ramalingam L, et al. First-in-man study of western reserve strain oncolytic vaccinia virus: safety, systemic spread, and antitumor activity. Mol Ther J Am Soc Gene Ther. (2015) 23:202–14. doi: 10.1038/mt.2014.194
17. Downs-Canner S, Guo ZS, Ravindranathan R, Breitbach CJ, O'Malley ME, Jones HL, et al. Phase 1 study of intravenous oncolytic poxvirus (vvDD) in patients with advanced solid cancers. Mol Ther J Am Soc Gene Ther. (2016) 24:1492–501. doi: 10.1038/mt.2016.101
18. Abou-Alfa GK, Galle PR, Chao Y, Brown KT, Heo J, Borad MJ, et al. PHOCUS: A phase 3 randomized, open-label study comparing the oncolytic immunotherapy Pexa-Vec followed by sorafenib (SOR) vs SOR in patients with advanced hepatocellular carcinoma (HCC) without prior systemic therapy. J Clin Oncol. (2016) 34:TPS4146–TPS. doi: 10.1200/JCO.2016.34.15_suppl.TPS4146
19. Semmrich M, Marchand J-B, Fend L, Rehn M, Silvestre N, Mårtensson L, et al. 594 BT-001, an oncolytic vaccinia virus armed with a Treg-depleting human recombinant anti-CTLA4 antibody and GM-CSF to target the tumor microenvironment. J immunotherapy Cancer. (2020) 8:A356–A. doi: 10.1136/jitc-2020-SITC2020.0594
20. Lauer UM, Schell M, Beil J, Berchtold S, Koppenhöfer U, Glatzle J, et al. Phase I study of oncolytic vaccinia virus GL-ONC1 in patients with peritoneal carcinomatosis. Clin Cancer Res an Off J Am Assoc Cancer Res. (2018) 24:4388–98. doi: 10.1158/1078-0432.ccr-18-0244
21. Lauer U, Zimmermann M, Sturm J, Koppenhoefer U, Bitzer M, Malek NP, et al. Phase I/II clinical trial of a genetically modified and oncolytic vaccinia virus GL-ONC1 in patients with unresactable, chemotherapy-resistant peritoneal carcinomatosis. J Clin Oncol. (2013) 31:3098. doi: 10.1200/jco.2013.31.15_suppl.3098
22. Nakao S, Arai Y, Tasaki M, Yamashita M, Murakami R, Kawase T, et al. Intratumoral expression of IL-7 and IL-12 using an oncolytic virus increases systemic sensitivity to immune checkpoint blockade. Sci Trans Med. (2020) 12:eaax7992. doi: 10.1126/scitranslmed.aax7992
23. Moehler M, Heo J, Lee HC, Tak WY, Chao Y, Paik SW, et al. Vaccinia-based oncolytic immunotherapy Pexastimogene Devacirepvec in patients with advanced hepatocellular carcinoma after sorafenib failure: a randomized multicenter Phase IIb trial (TRAVERSE). Oncoimmunology. (2019) 8:1615817. doi: 10.1080/2162402x.2019.1615817
24. Breitbach CJ, Arulanandam R, De Silva N, Thorne SH, Patt R, Daneshmand M, et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. (2013) 73:1265–75. doi: 10.1158/0008-5472.can-12-2687
25. Rajani KR, Vile RG. Harnessing the power of onco-immunotherapy with checkpoint inhibitors. Viruses. (2015) 7:5889–901. doi: 10.3390/v7112914
26. Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res an Off J Am Assoc Cancer Res. (2016) 22:1048–54. doi: 10.1158/1078-0432.ccr-15-2667
27. Kochneva G, Sivolobova G, Tkacheva A, Grazhdantseva A, Troitskaya O, Nushtaeva A, et al. Engineering of double recombinant vaccinia virus with enhanced oncolytic potential for solid tumor virotherapy. Oncotarget. (2016) 7:74171–88. doi: 10.18632/oncotarget.12367
28. Park SH, Breitbach CJ, Lee J, Park JO, Lim HY, Kang WK, et al. Phase 1b trial of biweekly intravenous pexa-vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus in colorectal cancer. Mol Ther J Am Soc Gene Ther. (2015) 23:1532–40. doi: 10.1038/mt.2015.109
29. Mori KM, Giuliano PD, Lopez KL, King MM, Bohart R, Goldstein BH. Pronounced clinical response following the oncolytic vaccinia virus GL-ONC1 and chemotherapy in a heavily pretreated ovarian cancer patient. Anti-cancer Drugs. (2019) 30:1064–6. doi: 10.1097/cad.0000000000000836
30. Cripe TP, Ngo MC, Geller JI, Louis CU, Currier MA, Racadio JM, et al. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol Ther J Am Soc Gene Ther. (2015) 23:602–8. doi: 10.1038/mt.2014.243
31. Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. (2008) 9:533–42. doi: 10.1016/s1470-2045(08)70107-4
32. Mell LK, Brumund KT, Daniels GA, Advani SJ, Zakeri K, Wright ME, et al. Phase I trial of intravenous oncolytic vaccinia virus (GL-ONC1) with cisplatin and radiotherapy in patients with locoregionally advanced head and neck carcinoma. Clin Cancer Res an Off J Am Assoc Cancer Res. (2017) 23:5696–702. doi: 10.1158/1078-0432.Ccr-16-3232
33. Jacobs BL, Langland JO, Kibler KV, Denzler KL, White SD, Holechek SA, et al. Vaccinia virus vaccines: past, present and future. Antiviral Res. (2009) 84:1–13. doi: 10.1016/j.antiviral.2009.06.006
34. Chen T, Ding X, Liao Q, Gao N, Chen Y, Zhao C, et al. IL-21 arming potentiates the anti-tumor activity of an oncolytic vaccinia virus in monotherapy and combination therapy. J immunotherapy Cancer. (2021) 9:e001647. doi: 10.1136/jitc-2020-001647
35. Smith GL, Benfield CTO, Maluquer de Motes C, Mazzon M, Ember SWJ, Ferguson BJ, et al. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol. (2013) 94:2367–92. doi: 10.1099/vir.0.055921-0
36. Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. (2009) 9:64–71. doi: 10.1038/nrc2545
37. Moss B. Vaccinia virus: a tool for research and vaccine development. Sci (New York NY). (1991) 252:1662–7. doi: 10.1126/science.2047875
38. Cono J, Casey CG, Bell DM. Smallpox vaccination and adverse reactions. Guidance for clinicians. MMWR Recommendations Rep Morbidity mortality weekly Rep Recommendations Rep. (2003) 52:1–28.
39. Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, et al. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus Challenge. J Virol. (2005) 79:13139–49. doi: 10.1128/jvi.79.20.13139-13149.2005
40. Shakiba Y, Vorobyev PO, Yusubalieva GM, Kochetkov DV, Zajtseva KV, Valikhov MP, et al. Oncolytic therapy with recombinant vaccinia viruses targeting the interleukin-15 pathway elicits a synergistic response. Mol Ther oncolytics. (2023) 29:158–68. doi: 10.1016/j.omto.2023.05.002
41. Russo AT, Grosenbach DW, Chinsangaram J, Honeychurch KM, Long PG, Lovejoy C, et al. An overview of tecovirimat for smallpox treatment and expanded anti-orthopoxvirus applications. Expert Rev anti-infective Ther. (2021) 19:331–44. doi: 10.1080/14787210.2020.1819791
42. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/coc.0000000000000239
43. Carenza C, Calcaterra F, Oriolo F, Di Vito C, Ubezio M, Della Porta MG, et al. Costimulatory molecules and immune checkpoints are differentially expressed on different subsets of dendritic cells. Front Immunol. (2019) 10:1325. doi: 10.3389/fimmu.2019.01325
44. He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. (2020) 30:660–9. doi: 10.1038/s41422-020-0343-4
45. Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer. (2019) 18:155. doi: 10.1186/s12943-019-1091-2
46. Oh CM, Chon HJ, Kim C. Combination immunotherapy using oncolytic virus for the treatment of advanced solid tumors. Int J Mol Sci. (2020) 21:7743. doi: 10.3390/ijms21207743
47. Chiu M, Armstrong EJL, Jennings V, Foo S, Crespo-Rodriguez E, Bozhanova G, et al. Combination therapy with oncolytic viruses and immune checkpoint inhibitors. Expert Opin Biol Ther. (2020) 20:635–52. doi: 10.1080/14712598.2020.1729351
48. Ren Y, Miao JM, Wang YY, Fan Z, Kong XB, Yang L, et al. Oncolytic viruses combined with immune checkpoint therapy for colorectal cancer is a promising treatment option. Front Immunol. (2022) 13:961796. doi: 10.3389/fimmu.2022.961796
49. Sivanandam V, LaRocca CJ, Chen NG, Fong Y, Warner SG. Oncolytic viruses and immune checkpoint inhibition: the best of both worlds. Mol Ther oncolytics. (2019) 13:93–106. doi: 10.1016/j.omto.2019.04.003
50. Hwang JK, Hong J, Yun CO. Oncolytic viruses and immune checkpoint inhibitors: preclinical developments to clinical trials. Int J Mol Sci. (2020) 21:8627. doi: 10.3390/ijms21228627
51. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
52. Wojtukiewicz MZ, Rek MM, Karpowicz K, Górska M, Polityńska B, Wojtukiewicz AM, et al. Inhibitors of immune checkpoints-PD-1, PD-L1, CTLA-4-new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer metastasis Rev. (2021) 40:949–82. doi: 10.1007/s10555-021-09976-0
53. Wang M, Liu Y, Cheng Y, Wei Y, Wei X. Immune checkpoint blockade and its combination therapy with small-molecule inhibitors for cancer treatment. Biochim Biophys Acta Rev Cancer. (2019) 1871:199–224. doi: 10.1016/j.bbcan.2018.12.002
54. Zak KM, Grudnik P, Magiera K, Dömling A, Dubin G, Holak TA. Structural biology of the immune checkpoint receptor PD-1 and its ligands PD-L1/PD-L2. Structure (London Engl 1993). (2017) 25:1163–74. doi: 10.1016/j.str.2017.06.011
55. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. (2007) 8:239–45. doi: 10.1038/ni1443
56. Tarhini AA, Zahoor H, Yearley JH, Gibson C, Rahman Z, Dubner R, et al. Tumor associated PD-L1 expression pattern in microscopically tumor positive sentinel lymph nodes in patients with melanoma. J Trans Med. (2015) 13:319. doi: 10.1186/s12967-015-0678-7
57. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
58. Seto T, Sam D, Pan M. Mechanisms of primary and secondary resistance to immune checkpoint inhibitors in cancer. Med Sci (Basel Switzerland). (2019) 7:14. doi: 10.3390/medsci7020014
59. Malekshah OM, Chen X, Nomani A, Sarkar S, Hatefi A. Enzyme/prodrug systems for cancer gene therapy. Curr Pharmacol Rep. (2016) 2:299–308. doi: 10.1007/s40495-016-0073-y
60. Efremova M, Rieder D, Klepsch V, Charoentong P, Finotello F, Hackl H, et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat Commun. (2018) 9:32. doi: 10.1038/s41467-017-02424-0
61. MacNeil DE, Bensoussan HJ, Autexier C. Telomerase regulation from beginning to the end. Genes. (2016) 7:64. doi: 10.3390/genes7090064
62. Theodoraki MN, Yerneni SS, Hoffmann TK, Gooding WE, Whiteside TL. Clinical significance of PD-L1(+) exosomes in plasma of head and neck cancer patients. Clin Cancer Res an Off J Am Assoc Cancer Res. (2018) 24:896–905. doi: 10.1158/1078-0432.Ccr-17-2664
63. Wang G, Kang X, Chen KS, Jehng T, Jones L, Chen J, et al. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat Commun. (2020) 11:1395. doi: 10.1038/s41467-020-15229-5
64. Daskivich TJ, Belldegrun A. Words of wisdom. Re: Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. Eur Urol. (2015) 67:816–7. doi: 10.1016/j.eururo.2014.12.052
65. Liu B, Song Y, Liu D. Recent development in clinical applications of PD-1 and PD-L1 antibodies for cancer immunotherapy. J Hematol Oncol. (2017) 10:174. doi: 10.1186/s13045-017-0541-9
66. Shi Z, Liu B, Huang C, Xie W, Cen Y, Chen L, et al. An oncolytic vaccinia virus armed with anti-human-PD-1 antibody and anti-human-4-1BB antibody double genes for cancer-targeted therapy. Biochem Biophys Res Commun. (2021) 559:176–82. doi: 10.1016/j.bbrc.2021.04.078
67. Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma C, Ravindranathan R, et al. Superagonist IL-15-armed oncolytic virus elicits potent antitumor immunity and therapy that are enhanced with PD-1 blockade. Mol Ther J Am Soc Gene Ther. (2018) 26:2476–86. doi: 10.1016/j.ymthe.2018.07.013
68. Zuo S, Wei M, Xu T, Kong L, He B, Wang S, et al. An engineered oncolytic vaccinia virus encoding a single-chain variable fragment against TIGIT induces effective antitumor immunity and synergizes with PD-1 or LAG-3 blockade. J immunotherapy Cancer. (2021) 9:e002843. doi: 10.1136/jitc-2021-002843
69. Kleinpeter P, Fend L, Thioudellet C, Geist M, Sfrontato N, Koerper V, et al. Vectorization in an oncolytic vaccinia virus of an antibody, a Fab and a scFv against programmed cell death -1 (PD-1) allows their intratumoral delivery and an improved tumor-growth inhibition. Oncoimmunology. (2016) 5:e1220467. doi: 10.1080/2162402x.2016.1220467
70. Smith HG, Mansfield D, Roulstone V, Kyula-Currie JN, McLaughlin M, Patel RR, et al. PD-1 blockade following isolated limb perfusion with vaccinia virus prevents local and distant relapse of soft-tissue sarcoma. Clin Cancer Res an Off J Am Assoc Cancer Res. (2019) 25:3443–54. doi: 10.1158/1078-0432.Ccr-18-3767
71. Jeon YH, Lee N, Yoo J, Won S, Shin SK, Kim KH, et al. Oncolytic vaccinia virus augments T cell factor 1-positive stem-like CD8(+) T cells, which underlies the efficacy of anti-PD-1 combination immunotherapy. Biomedicines. (2022) 10:805. doi: 10.3390/biomedicines10040805
72. Woo Y, Zhang Z, Yang A, Chaurasiya S, Park AK, Lu J, et al. Novel chimeric immuno-oncolytic virus CF33-hNIS-antiPDL1 for the treatment of pancreatic cancer. J Am Coll Surgeons. (2020) 230:709–17. doi: 10.1016/j.jamcollsurg.2019.12.027
73. Lou J, Dong J, Xu R, Zeng H, Fang L, Wu Y, et al. Remodeling of the tumor microenvironment using an engineered oncolytic vaccinia virus improves PD-L1 inhibition outcomes. Bioscience Rep. (2021) 41:BSR20204186. doi: 10.1042/bsr20204186
74. Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun. (2017) 8:14754. doi: 10.1038/ncomms14754
75. Lee YS, Lee WS, Kim CW, Lee SJ, Yang H, Kong SJ, et al. Oncolytic vaccinia virus reinvigorates peritoneal immunity and cooperates with immune checkpoint inhibitor to suppress peritoneal carcinomatosis in colon cancer. J immunotherapy Cancer. (2020) 8:e000857. doi: 10.1136/jitc-2020-000857
76. Inoue M, Kim M, Inoue T, Tait M, Byrne T, Nitschké M, et al. Oncolytic vaccinia virus injected intravenously sensitizes pancreatic neuroendocrine tumors and metastases to immune checkpoint blockade. Mol Ther oncolytics. (2022) 24:299–318. doi: 10.1016/j.omto.2021.12.016
77. Chon HJ, Lee WS, Yang H, Kong SJ, Lee NK, Moon ES, et al. Tumor microenvironment remodeling by intratumoral oncolytic vaccinia virus enhances the efficacy of immune-checkpoint blockade. Clin Cancer Res an Off J Am Assoc Cancer Res. (2019) 25:1612–23. doi: 10.1158/1078-0432.ccr-18-1932
78. Kana SI, Essani K. Immuno-oncolytic viruses: emerging options in the treatment of colorectal cancer. Mol diagnosis Ther. (2021) 25:301–13. doi: 10.1007/s40291-021-00517-7
79. Ballesteros-Briones MC, Martisova E, Casales E, Silva-Pilipich N, Buñuales M, Galindo J, et al. Short-term local expression of a PD-L1 blocking antibody from a self-replicating RNA vector induces potent antitumor responses. Mol Ther J Am Soc Gene Ther. (2019) 27:1892–905. doi: 10.1016/j.ymthe.2019.09.016
80. Onoi K, Chihara Y, Uchino J, Shimamoto T, Morimoto Y, Iwasaku M, et al. Immune checkpoint inhibitors for lung cancer treatment: A review. J Clin Med. (2020) 9:1362. doi: 10.3390/jcm9051362
81. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. (2017) 170:1109–19.e10. doi: 10.1016/j.cell.2017.08.027
82. O’Leary MP, Choi AH, Kim S-I, Chaurasiya S, Lu J, Park AK, et al. Novel oncolytic chimeric orthopoxvirus causes regression of pancreatic cancer xenografts and exhibits abscopal effect at a single low dose. J Trans Med. (2018) 16:110. doi: 10.1186/s12967-018-1483-x
83. Erices JI, Bizama C, Niechi I, Uribe D, Rosales A, Fabres K, et al. Glioblastoma microenvironment and invasiveness: new insights and therapeutic targets. Int J Mol Sci. (2023) 24:7047. doi: 10.3390/ijms24087047
84. Liu L, Li H, Xu Q, Wu Y, Chen D, Yu F. Antitumor activity of recombinant oncolytic vaccinia virus with human IL2. Open Med (Warsaw Poland). (2022) 17:1084–91. doi: 10.1515/med-2022-0496
85. Ge Y, Wang H, Ren J, Liu W, Chen L, Chen H, et al. Oncolytic vaccinia virus delivering tethered IL-12 enhances antitumor effects with improved safety. J immunotherapy Cancer. (2020) 8(1):e000710. doi: 10.1136/jitc-2020-000710
86. Wang X, Zhao X, Feng C, Weinstein A, Xia R, Wen W, et al. IL-36γ Transforms the tumor microenvironment and promotes type 1 lymphocyte-mediated antitumor immune responses. Cancer Cell. (2015) 28:296–306. doi: 10.1016/j.ccell.2015.07.014
87. Yang M, Giehl E, Feng C, Feist M, Chen H, Dai E, et al. IL-36γ-armed oncolytic virus exerts superior efficacy through induction of potent adaptive antitumor immunity. Cancer immunology immunotherapy CII. (2021) 70:2467–81. doi: 10.1007/s00262-021-02860-4
88. Borden EC. Interferons α and β in cancer: therapeutic opportunities from new insights. Nat Rev Drug Discovery. (2019) 18:219–34. doi: 10.1038/s41573-018-0011-2
89. Kirn DH, Wang Y, Le Boeuf F, Bell J, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PloS Med. (2007) 4:e353. doi: 10.1371/journal.pmed.0040353
90. Huang B, Sikorski R, Kirn DH, Thorne SH. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the IFN response and HMGB1. Gene Ther. (2011) 18:164–72. doi: 10.1038/gt.2010.121
91. Jo HH, Chun SJ, Yoo J-J, Lee MH, Kim SG, Kim YS. Early experience of oncolytic virus injection combined with sorafenib in a patient with advanced hepatocellular carcinoma and portal vein thrombosis. J Liver Cancer. (2020) 20:177–82. doi: 10.17998/jlc.20.2.177
92. Heo J, Breitbach CJ, Moon A, Kim CW, Patt R, Kim MK, et al. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol Ther J Am Soc Gene Ther. (2011) 19:1170–9. doi: 10.1038/mt.2011.39
93. Binz E, Berchtold S, Beil J, Schell M, Geisler C, Smirnow I, et al. Chemovirotherapy of pancreatic adenocarcinoma by combining oncolytic vaccinia virus GLV-1h68 with nab-paclitaxel plus gemcitabine. Mol Ther oncolytics. (2017) 6:10–21. doi: 10.1016/j.omto.2017.04.001
94. Chen W, Fan W, Ru G, Huang F, Lu X, Zhang X, et al. Gemcitabine combined with an engineered oncolytic vaccinia virus exhibits a synergistic suppressive effect on the tumor growth of pancreatic cancer. Oncol Rep. (2019) 41:67–76. doi: 10.3892/or.2018.6817
95. Kim M, Nitschké M, Sennino B, Murer P, Schriver BJ, Bell A, et al. Amplification of oncolytic vaccinia virus widespread tumor cell killing by sunitinib through multiple mechanisms. Cancer Res. (2018) 78:922–37. doi: 10.1158/0008-5472.Can-15-3308
96. Lee S, Yang W, Kim DK, Kim H, Shin M, Choi KU, et al. Inhibition of MEK-ERK pathway enhances oncolytic vaccinia virus replication in doxorubicin-resistant ovarian cancer. Mol Ther oncolytics. (2022) 25:211–24. doi: 10.1016/j.omto.2022.04.006
97. Samson A, West EJ, Carmichael J, Scott KJ, Turnbull S, Kuszlewicz B, et al. Neoadjuvant intravenous oncolytic vaccinia virus therapy promotes anticancer immunity in patients. Cancer Immunol Res. (2022) 10:745–56. doi: 10.1158/2326-6066.cir-21-0171
98. Hofmann E, Weibel S, Szalay AA. Combination treatment with oncolytic Vaccinia virus and cyclophosphamide results in synergistic antitumor effects in human lung adenocarcinoma bearing mice. J Trans Med. (2014) 12:197. doi: 10.1186/1479-5876-12-197
99. Ottolino-Perry K, Acuna SA, Angarita FA, Sellers C, Zerhouni S, Tang N, et al. Oncolytic vaccinia virus synergizes with irinotecan in colorectal cancer. Mol Oncol. (2015) 9:1539–52. doi: 10.1016/j.molonc.2015.04.009
100. Peng J, Wang S, Fan W, Li S, Wu Y, Mou X, et al. Synergistic suppression effect on tumor growth of acute myeloid leukemia by combining cytarabine with an engineered oncolytic vaccinia virus. OncoTargets Ther. (2018) 11:6887–900. doi: 10.2147/ott.S172037
101. Smith HG, Cartwright J, Wilkinson MJ, Strauss DC, Thomas JM, Hayes AJ. Isolated limb perfusion with melphalan and tumour necrosis factor α for in-transit melanoma and soft tissue sarcoma. Ann Surg Oncol. (2015) 22 Suppl 3:S356–61. doi: 10.1245/s10434-015-4856-x
102. Pencavel TD, Wilkinson MJ, Mansfield DC, Khan AA, Seth R, Karapanagiotou EM, et al. Isolated limb perfusion with melphalan, tumour necrosis factor-alpha and oncolytic vaccinia virus improves tumour targeting and prolongs survival in a rat model of advanced extremity sarcoma. Int J Cancer. (2015) 136:965–76. doi: 10.1002/ijc.29059
103. Wilkinson MJ, Smith HG, McEntee G, Kyula-Currie J, Pencavel TD, Mansfield DC, et al. Oncolytic vaccinia virus combined with radiotherapy induces apoptotic cell death in sarcoma cells by down-regulating the inhibitors of apoptosis. Oncotarget. (2016) 7:81208–22. doi: 10.18632/oncotarget.12820
104. Mansfield D, Pencavel T, Kyula JN, Zaidi S, Roulstone V, Thway K, et al. Oncolytic Vaccinia virus and radiotherapy in head and neck cancer. Oral Oncol. (2013) 49:108–18. doi: 10.1016/j.oraloncology.2012.07.019
105. Dai MH, Liu SL, Chen NG, Zhang TP, You L, QZ F, et al. Oncolytic vaccinia virus in combination with radiation shows synergistic antitumor efficacy in pancreatic cancer. Cancer Lett. (2014) 344:282–90. doi: 10.1016/j.canlet.2013.11.007
106. Chen WY, Chen YL, Lin HW, Chang CF, Huang BS, Sun WZ, et al. Stereotactic body radiation combined with oncolytic vaccinia virus induces potent anti-tumor effect by triggering tumor cell necroptosis and DAMPs. Cancer Lett. (2021) 523:149–61. doi: 10.1016/j.canlet.2021.09.040
107. Mullard A. FDA approves first CAR T therapy. Nat Rev Drug Discovery. (2017) 16:669–70. doi: 10.1038/nrd.2017.196
108. Shi T, Song X, Wang Y, Liu F, Wei J. Combining oncolytic viruses with cancer immunotherapy: establishing a new generation of cancer treatment. Front Immunol. (2020) 11:683. doi: 10.3389/fimmu.2020.00683
109. Moon EK, Wang L-CS, Bekdache K, Lynn RC, Lo A, Thorne SH, et al. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology. (2018) 7:e1395997. doi: 10.1080/2162402X.2017.1395997
110. Park AK, Fong Y, Kim S-I, Yang J, Murad JP, Lu J, et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci Trans Med. (2020) 12:eaaz1863. doi: 10.1126/scitranslmed.aaz1863
111. Tian Z, Liu M, Zhang Y, Wang X. Bispecific T cell engagers: an emerging therapy for management of hematologic Malignancies. J Hematol Oncol. (2021) 14:75. doi: 10.1186/s13045-021-01084-4
112. Yu F, Wang X, Guo ZS, Bartlett DL, Gottschalk SM, Song XT. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther J Am Soc Gene Ther. (2014) 22:102–11. doi: 10.1038/mt.2013.240
113. Yu F, Hong B, Song X-T. A T-cell engager-armed oncolytic vaccinia virus to target the tumor stroma. Cancer Trans Med. (2017) 3:122. doi: 10.4103/ctm.ctm_13_17
114. Wan PK, Ryan AJ, Seymour LW. Beyond cancer cells: Targeting the tumor microenvironment with gene therapy and armed oncolytic virus. Mol Ther J Am Soc Gene Ther. (2021) 29:1668–82. doi: 10.1016/j.ymthe.2021.04.015
115. Wei M, Zuo S, Chen Z, Qian P, Zhang Y, Kong L, et al. Oncolytic vaccinia virus expressing a bispecific T-cell engager enhances immune responses in EpCAM positive solid tumors. Front Immunol. (2022) 13:1017574. doi: 10.3389/fimmu.2022.1017574
116. Lei W, Ye Q, Hao Y, Chen J, Huang Y, Yang L, et al. CD19-targeted BiTE expression by an oncolytic vaccinia virus significantly augments therapeutic efficacy against B-cell lymphoma. Blood Cancer J. (2022) 12:35. doi: 10.1038/s41408-022-00634-4
117. Bellnier DA, Greco WR, Loewen GM, Nava H, Oseroff AR, Pandey RK, et al. Population pharmacokinetics of the photodynamic therapy agent 2-[1-hexyloxyethyl]-2-devinyl pyropheophorbide-a in cancer patients. Cancer Res. (2003) 63:1806–13.
118. Gil M, Bieniasz M, Seshadri M, Fisher D, Ciesielski M, Chen Y, et al. Photodynamic therapy augments the efficacy of oncolytic vaccinia virus against primary and metastatic tumours in mice. Br J Cancer. (2011) 105:1512–21. doi: 10.1038/bjc.2011.429
119. Hikichi M, Kidokoro M, Haraguchi T, Iba H, Shida H, Tahara H, et al. MicroRNA regulation of glycoprotein B5R in oncolytic vaccinia virus reduces viral pathogenicity without impairing its antitumor efficacy. Mol Ther J Am Soc Gene Ther. (2011) 19:1107–15. doi: 10.1038/mt.2011.36
120. Horita K, Kurosaki H, Nakatake M, Kuwano N, Oishi T, Itamochi H, et al. lncRNA UCA1-mediated Cdc42 signaling promotes oncolytic vaccinia virus cell-to-cell spread in ovarian cancer. Mol Therapy-Oncolytics. (2019) 13:35–48. doi: 10.1016/j.omto.2019.03.003
121. Lee HJ, Rho J, Gui SR, Kim MK, Lee YK, Lee YS, et al. Effect of aldosterone on the amplification of oncolytic vaccinia virus in human cancer lines. Korean J Hepatol. (2011) 17:213–9. doi: 10.3350/kjhep.2011.17.3.213
122. Ferguson MS, Lemoine NR, Wang Y. Systemic delivery of oncolytic viruses: hopes and hurdles. Adv Virol. (2012) 2012:805629. doi: 10.1155/2012/805629
123. Wakimoto H, Ikeda K, Abe T, Ichikawa T, Hochberg FH, Ezekowitz RAB, et al. The complement response against an oncolytic virus is species-specific in its activation pathways. Mol Ther. (2002) 5:275–82. doi: 10.1006/mthe.2002.0547
124. Evgin L, Acuna SA, Tanese de Souza C, Marguerie M, Lemay CG, Ilkow CS, et al. Complement inhibition prevents oncolytic vaccinia virus neutralization in immune humans and cynomolgus macaques. Mol Ther J Am Soc Gene Ther. (2015) 23:1066–76. doi: 10.1038/mt.2015.49
125. Goossens M, Pauwels K, Willemarck N, Breyer D. Environmental risk assessment of clinical trials involving modified vaccinia virus Ankara (MVA)-based vectors. Curr Gene Ther. (2013) 13:413–20. doi: 10.2174/156652321306140103221941
126. Badrinath N, Jeong YI, Woo HY, Bang SY, Kim C, Heo J, et al. Local delivery of a cancer-favoring oncolytic vaccinia virus via poly (lactic-co-glycolic acid) nanofiber for theranostic purposes. Int J pharmaceutics. (2018) 552:437–42. doi: 10.1016/j.ijpharm.2018.10.020
127. Francini N, Cochrane D, Illingworth S, Purdie L, Mantovani G, Fisher K, et al. Polyvalent diazonium polymers provide efficient protection of oncolytic adenovirus enadenotucirev from neutralizing antibodies while maintaining biological activity in vitro and in vivo. Bioconjugate Chem. (2019) 30:1244–57. doi: 10.1021/acs.bioconjchem.9b00189
128. Draganov DD, Santidrian AF, Minev I, Nguyen D, Kilinc MO, Petrov I, et al. Delivery of oncolytic vaccinia virus by matched allogeneic stem cells overcomes critical innate and adaptive immune barriers. J Trans Med. (2019) 17:100. doi: 10.1186/s12967-019-1829-z
129. VanSeggelen H, Tantalo DG, Afsahi A, Hammill JA, Bramson JL. Chimeric antigen receptor-engineered T cells as oncolytic virus carriers. Mol Ther oncolytics. (2015) 2:15014. doi: 10.1038/mto.2015.14
130. Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. (2009) 21:215–23. doi: 10.1016/j.coi.2009.02.009
131. Monge C, Xie C, Myojin Y, Coffman K, Hrones DM, Wang S, et al. Phase I/II study of PexaVec in combination with immune checkpoint inhibition in refractory metastatic colorectal cancer. J immunotherapy Cancer. (2023) 11:e005640. doi: 10.1136/jitc-2022-005640
Keywords: oncolytic virus, oncolytic vaccinia virus, combination therapy, cancer therapy, immunotherapy
Citation: Mirbahari SN, Da Silva M, Zúñiga AIM, Kooshki Zamani N, St-Laurent G, Totonchi M and Azad T (2024) Recent progress in combination therapy of oncolytic vaccinia virus. Front. Immunol. 15:1272351. doi: 10.3389/fimmu.2024.1272351
Received: 03 August 2023; Accepted: 29 February 2024;
Published: 13 March 2024.
Edited by:
Zong Sheng Guo, University at Buffalo, United StatesReviewed by:
Qibin Liao, Shenzhen Third People’s Hospital, ChinaLance Hellman, Nevada State College, United States
Copyright © 2024 Mirbahari, Da Silva, Zúñiga, Kooshki Zamani, St-Laurent, Totonchi and Azad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mehdi Totonchi, bS50b3RvbmNoaUByb3lhbmluc3RpdHV0ZS5vcmc=; Taha Azad, dGFoYS5hemFkQHVzaGVyYnJvb2tlLmNh
†Present address: Miles Da Silva, Basic and Molecular Epidemiology of Gastrointestinal Disorders Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Irans