 Rodrigo V. Motta
Rodrigo V. Motta Emma L. Culver
Emma L. Culver- 1Translational Gastroenterology and Liver Unit, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom
- 2Department of Gastroenterology and Hepatology, Oxford University Hospitals NHS Foundation Trust, Oxford, United Kingdom
Immunoglobulins are an essential part of the humoral immune response. IgG4 antibodies are the least prevalent subclass and have unique structural and functional properties. In this review, we discuss IgG4 class switch and B cell production. We review the importance of IgG4 antibodies in the context of allergic responses, helminth infections and malignancy. We discuss their anti-inflammatory and tolerogenic effects in allergen-specific immunotherapy, and ability to evade the immune system in parasitic infection and tumour cells. We then focus on the role of IgG4 autoantibodies and autoantigens in IgG4-autoimmune diseases and IgG4-related disease, highlighting important parallels and differences between them. In IgG4-autoimmune diseases, pathogenesis is based on a direct role of IgG4 antibodies binding to self-antigens and disturbing homeostasis. In IgG4-related disease, where affected organs are infiltrated with IgG4-expressing plasma cells, IgG4 antibodies may also directly target a number of self-antigens or be overexpressed as an epiphenomenon of the disease. These antigen-driven processes require critical T and B cell interaction. Lastly, we explore the current gaps in our knowledge and how these may be addressed.
1 Introduction
Antibodies are the fundamental component of humoral immune responses, also known as antibody-dependent responses. These molecules can recognise and neutralise pathogens, either by binding to molecular antigens and directly preventing their pathogenic effect, or by opsonising these pathogens and triggering effector functions, such as the complement system, antibody-dependent cell-mediated cytotoxicity and antibody-dependent cellular phagocytosis.
Five different classes of antibodies, or immunoglobulins (Ig) are found in humans, namely IgM, IgD, IgA, IgE and IgG. IgM and IgD are involved in primary adaptive humoral immunity and can be found in mature naïve B cells (1), which undergo specialisation and a class switch towards long lived B cells and plasmablasts that produce IgA, IgE and IgG (2, 3). IgA is mainly involved in mucosal humoral immunity. IgE is one of the main components of allergic reactions. IgG is part of several different processes both in health and disease.
IgG has four subclasses (i.e., IgG1, IgG2, IgG3 and IgG4) which form part of the immune response. Under normal conditions, IgG1-3 can fixate complement and opsonise pathogens, whilst IgG4 only activates complement under special circumstances, at high antibody and antigen concentrations. IgG and its subclasses are involved in autoimmune processes, such as rheumatoid arthritis, systemic lupus erythematous, ANCA-associated vasculitides, as well as IgG4-autoimmune diseases (IgG4-AID) and IgG4-related disease (IgG4-RD).
In this review, we describe the structure and function of IgG4 antibodies, discuss the role of this class in allergy, helminth infections, malignancies, and autoimmune diseases. A greater emphasis is placed on IgG4-related disease and the role of IgG4 antibodies in the pathogenesis of the disease.
2 Structure and function of IgG4 antibodies
Immunoglobulin G has four subclasses, with IgG4 representing up to 5% of the total IgG concentration (4). Their structure is based on 2 heavy and 2 light chains bound together by disulphide bridges. There are interactions between the light and heavy chains, as well as a connection between the two heavy chains at the hinge region. This region is important because it gives structural flexibility to the molecule. Both the light and heavy chains have antigen-binding sites, the variable region (VL and VH), and areas responsible for the effector function of the antibodies, called constant region (CL and CH1, CH2, CH3, CH4). Furthermore, these molecules can be divided by function, the “Fragment, antigen binding” (Fab) region and the “Fragment, crystallised” (Fc) region, which is responsible for the effector function (5). In the CH2 domain, a switch of proline to serine at position 228 in the hinge of IgG4 facilitates Fab-arm exchange (Figure 1). Schuurman, Aalberse and colleagues observed that this phenomenon is marked by the dissociation of the two heavy chains and recombination of two random IgG4 monomers (heavy and light chains) to form a bispecific dimer (6). Even though this new molecule might recognise two different antigens, it is functionally monovalent, and cannot form large immune complexes (7–9). The Fc portion in IgG4 antibodies have a high affinity for antigens and FcγIIb receptors but low affinity for Fcγ stimulatory receptors and cannot tipically activate the classical complement pathway (10). These characteristics hinder Fc mediated effector functions and prevent further sensitisation of the immune system (8, 11–13). There are, however, situations where IgG4 molecules can activate the complement pathway. The first is via recruiting mannose-binding lectins and activating the lectin pathway, which has been observed in anti-PLA2R membranous nephropathy (14). The second is through mutations of the Fc region, enabling these molecules to form hexamers and bind to C1q (15). These molecules can also activate the complement system using the classical pathway when there is high antigen density and high antibody concentration (16). Finally, Oskam, Rispens and collaborators showed that in opposition to previous evidence, the heavy chains of IgG4 molecules can dissociate and interact with the Fc region of other IgGs. It is not clear whether this mechanism plays a role in the pathogenesis of autoimmune diseases, but it modulates the affinity of IgG1 and IgG2 for C1q (17). Hence, IgG4 has anti-inflammatory properties, and it is involved both in health, such as the development of tolerance in allergies (18), as well as disease, such as curbing immune responses against helminth infections (19) (Table 1).
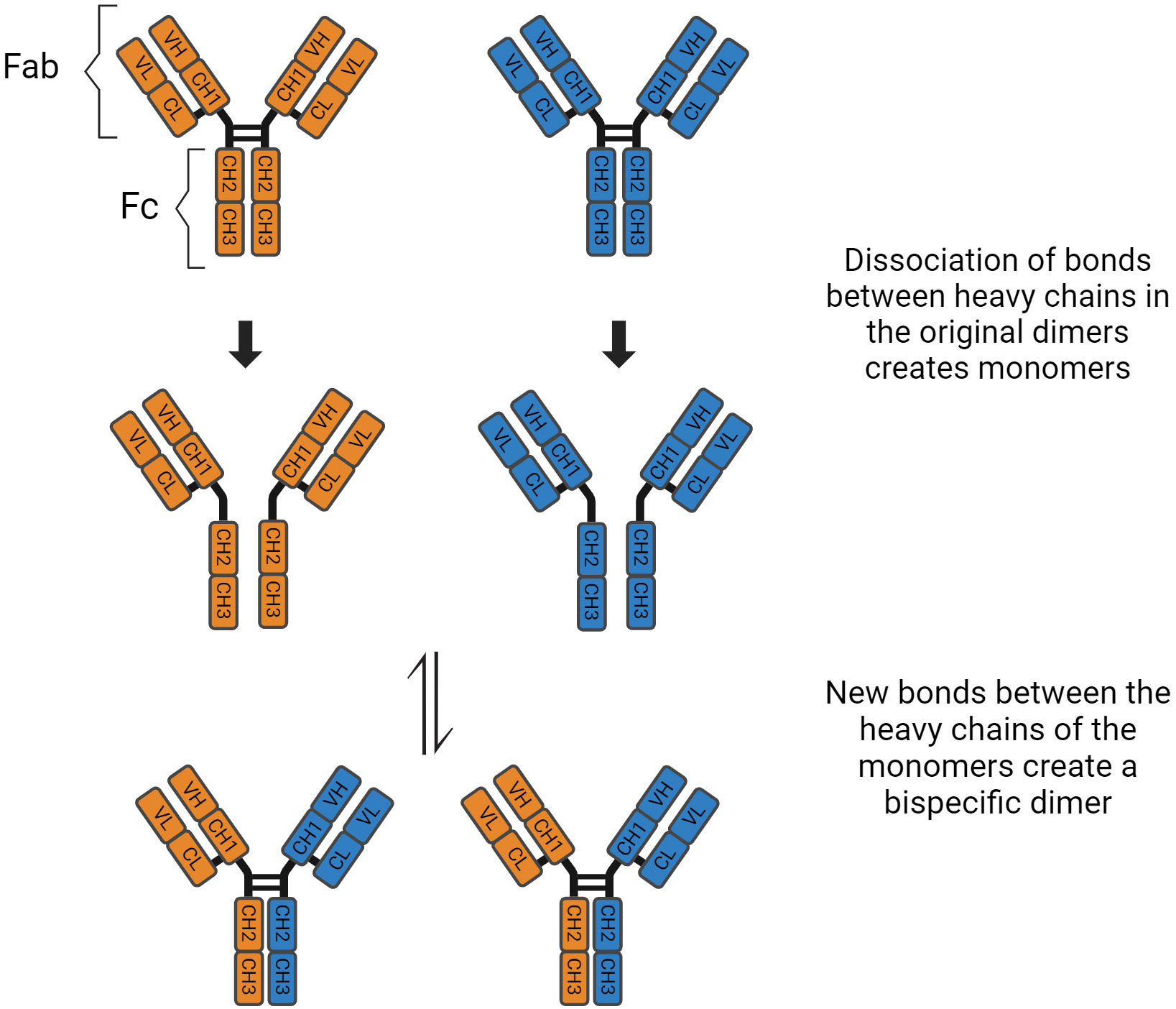
Figure 1 Fab arm exchange in IgG4 antibodies.
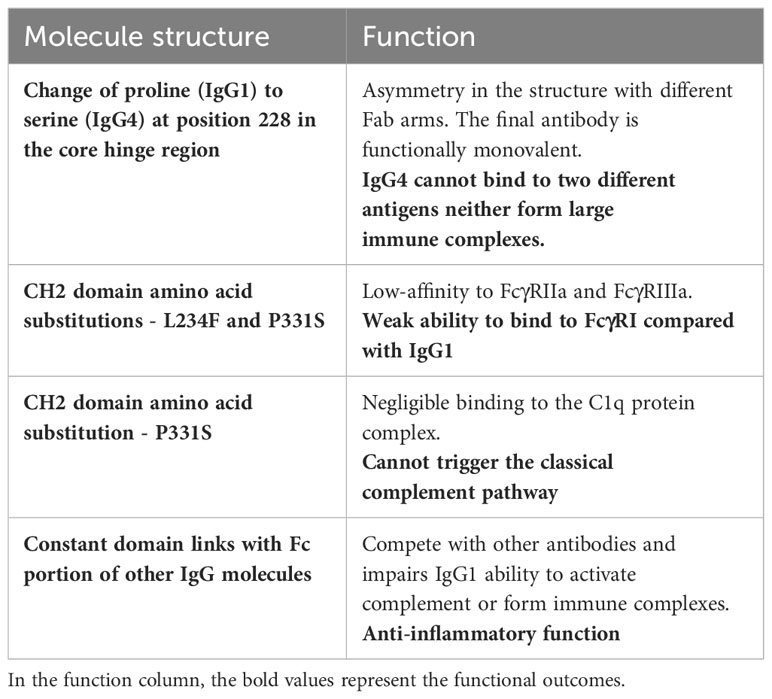
Table 1 IgG4 antibodies: structural characteristics and functional consequences.
3 Class switch recombination and somatic hypermutation
When exposed to antigens, naïve B cells become antibody secreting B cells via the extrafollicular and follicular pathways, which are complementary. The initial response to antigens is predominantly driven by the extrafollicular path (20, 21). It involves T follicular helper (Tfh) cells driving the differentiation of naïve B cells into short lived plasmablasts, which will maintain an immune response for up to a week after the trigger (20, 22).
To create an immune response based on IgG4, B cells must undergo class switch recombination (CSR) and somatic hypermutation (SHM) (11, 23, 24). These processes are hallmarks of follicular responses, which take part in germinal centres (GC) of lymph nodes and the spleen. In the follicular pathway, antibody secreting B cells develop high affinity and specificity and become either memory B cells or long-lived plasma cells (LLPC). The GCs are divided in dark and light zones, with functional differences. The dark zone promotes proliferation of B cells, as well as CSR and SHM (25). CSR is marked by deletional recombination in the heavy chain gene that leads to a change in Ig class, thus changing effector functions of the antibodies, while preserving the affinity for specific antigens. Given its mechanism, switch in Ig class follows a sequential pattern from IgM/IgD to IgG3, IgG1 and IgA1, called proximal classes. A secondary switch then gives rise to distal classes such as IgG2, IgG4 and IgA2 (23, 26, 27) and has been attributed to stimuli from T helper 2 (Th2) cells. Interleukin (IL) 4 and IL-13 are responsible for skewing the antibody production to IgG4 and stimuli involving IL-10 and/or IL-21 lead to selection of IgG1 antibodies (28). After these clones have developed high-affinity and specificity, they migrate into the light zone. There, they will undergo a selection according to interactions with Tfh cells and follicular dendritic cells (29). Once the highly specific clones are selected, they leave the GC and become LLPC or migrate to the bone marrow as memory B cells.
4 IgG4 in allergic reactions
IgG4 antibodies have an important role in the development of tolerance in atopic patients. It has been extensively shown that beekeepers develop an IgG4-based response to bee venom associated with reduction of symptoms after exposure to the toxin (30, 31), and the same event has been described in other cases of occupational exposure (32, 33). Furthermore, allergen-specific immunotherapy is based on the development of IgG4 antibodies against allergens through regular and incremental exposure to allergenic antigens (34). IgG4 competitively binds to allergens, thus hindering the formation of IgE-antigen immune complexes (35, 36). Furthermore, it also binds to FcγRIIb, an inhibitory Fcγ receptor, preventing the degranulation of mast cells and the cascade that would lead to allergic responses (37–39).
On the other hand, IgG4 antibodies are also involved in the maintenance of the disease in cases of eosinophilic oesophagitis (EoE) and eosinophilic chronic rhinosinusitis (40–42). The most robust evidence that this class is involved in disease comes from studies associating it with symptoms in patients with EoE. During food avoidance tests, patients referred less symptoms and biopsies showed reduction of IgG4 deposition (43). In addition, histological levels of IgG4 were found to be significantly lower during remission when compared to samples collected during disease activity (44). For a detailed review on the topic, please see (45).
5 IgG4 in helminthic infections
Helminthic infections are another scenario where IgG4 antibodies show their tolerogenic effects. Patients with acute parasitic infection usually show a balanced Th1/Th2 immune response, with a predominance of IFN-γ over IL-10, and a higher count of peripheral eosinophils (46). If the pathogen fails to evade the host response, this leads to parasite clearance and termination of the infection.
Individuals with chronic infection, however, show an immune response with a Th2 profile, where IL-10 plays a critical modulatory role in skewing antibody production towards IgG4 (46). A response based on parasite-specific IgG4 antibodies, then, dampens IgE-mediated immune response and perpetuates the infection. Asymptomatic patients show a restricted response to parasite antigens, while those with severe chronic disease have lower levels of serum IgG4 antibodies and show signs of organ damage (46). Studies also show that individuals with a high parasite burden have higher levels of parasite-specific IgG4 than patients with a lower parasite burden (47, 48). The immunomodulatory effects of helminthic infections are also seen in the response to allergens, where patients with chronic parasitic infection show lower levels of IgE-reactivity to dust antigens (49).
6 IgG4 in malignancies
Antitumour humoral responses can prevent malignancies from developing further and even annihilate them by triggering humoral and cell-mediated immune responses. Some neoplasms induce class switch of anti-tumour antibodies to the IgG4 class, leading to immune escape and IgG4-mediated tumour-tolerogenic responses (50). Melanoma and pancreatic ductal adenocarcinoma cells induce a modified Th2 immune response around their microenvironment, triggering B cells to undergo antibody class switch towards IgG4 (51, 52). These antibodies will then competitively bind to cancer antigens and damp anti-tumour immune responses. Higher serum levels of IgG4 are associated with fewer cytotoxic T cells (53) and with a worse prognosis in patients with cholangiocarcinoma and melanoma (50, 54).
A meta-analysis reported that patients with IgG4-RD have an increased risk of clonal B cell lymphoma and pancreatic malignancy when compared with a matched general population (55). The mechanisms driving this increased risk remain speculative and include 1) chronic inflammation-induced carcinogenesis; 2) IgG4-related tumour-induced immune escape; 3) paraneoplastic phenomenon, as most malignancies detected distant from the site of disease activity. The class switch towards IgG4 induced by some malignancies may also play a role in the initiation of IgG4-RD. K-ras codon gene mutations associated with gastrointestinal cancer have been reported in patients with IgG4-related pancreatitis (56). These mutations are also associated with cellular transformation and genetic instability (57, 58).
7 IgG4 in IgG4-autoimmune diseases
Huijbers et al. were the first to collectively describe IgG4 autoimmune diseases (IgG4-AID) as a spectrum of conditions characterised by autoantibody responses against a known antigen (59). Evidence shows that these diseases are caused by IgG4 antibodies recognising autoantigens, although not all antigens have been validated. The diagnosis of IgG4-AID depends on high level of suspicion and identification of IgG4 autoantibodies against the antigen(s) specific to each disease (60). Koneczny proposed a classification system based on evidence of antibodies against extracellular antigens, and direct pathogenic mechanism of IgG4 antibodies observed in vitro and validated in animal models through passive transfer (61). Although 29 antigens have been described in IgG4-AID (Table 2), only the six antigens have met all three Koneczny criteria of IgG4 pathogenicity (61). For a thorough review on IgG4-AID and autoantigens involved in these disorders, please read (62).
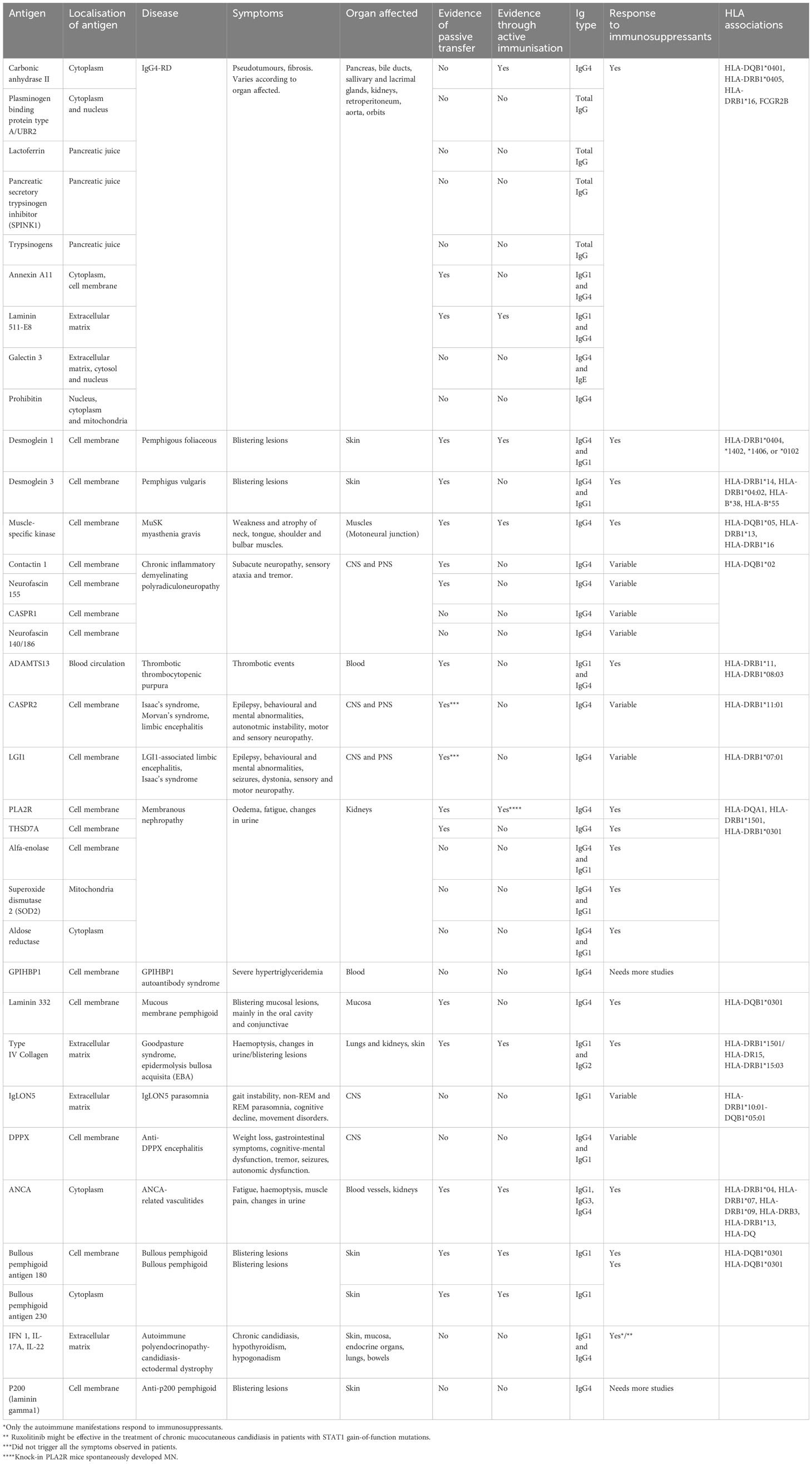
Table 2 Comparison between antigens, symptoms, animal models, HLA associations and responsiveness to immunosuppression between IgG4-AID and IgG4-RD.
7.1 Desmoglein 1 (Dsg1) and 3 (Dsg3):
Desmoglein 1 and 3 are transmembrane proteins found in keratinocytes. Anti-Dsg1 IgG4 is responsible for the development of pemphigus foliaceous (PF), while anti-Dsg3 IgG4 antibodies, and in some cases anti-Dsg1 IgG4, cause pemphigus vulgaris. These antibodies target epitopes in EC1 and EC2 domains, including in fogo selvagem (FS), an endemic PF specific to Brazil (63), and in the endemic PF found in Tunisia (64). Qian and Peng observed a cross-link between anti-LJM11, a protein present in the saliva of sand flies (L. longipalpis), and anti-Dsg1 antibodies in patients with FS and healthy controls from the same region (65, 66). The difference being that antibodies from the healthy population recognised the EC5 domain of Dsg1 rather than EC1 and EC2 (63, 65–67). Studies investigating the Tunisian population found that endemic PF patients and healthy controls from the same area had anti-Dsg1 IgG and demonstrated a cross-link between these antibodies and the salivary extract from P. papatasi (68–70). The antibodies in healthy Tunisians, instead of targeting the epitopes involved in endemic PF, bound to epitopes in the EC3, EC4 and EC5 of Dsg1 (64) and were mostly IgG1, IgG2 and IgG3 (71)
7.2 Muscle-specific kinase (MuSK):
MuSK is a tyrosine kinase involved in the transduction of electrical signals at the neuromuscular junction and is involved in MuSK myasthenia gravis (MG) (72). Anti-MuSK IgG4 antibodies often target the Ig-like 1 domain of the protein (73–75), which has important functional implications. This region contains the site where lipoprotein receptor-related protein 4 (Lrp4) binds to MuSK and activates the kinase, which causes clustering of acetylcholine receptors (AchR) at the synapse. By competitively binding to that domain, IgG4 antibodies block Lrp4-dependent activation of MuSK (76, 77), thus impairing migration of AchR to the neuromuscular junction. Other IgG subclasses might also be involved in MuSK MG. Huijbers et al. observed that valency affects the outcomes of antibody binding to MuSK and may, in fact, lead to activation of the kinase (78). Furthermore, patients with MuSK-MG have higher levels of IgG4, but they also have higher levels of IgG1 when compared to controls, which does not corroborate the theory that these patients have overall responses biased towards IgG4 (79).
7.3 Contactin 1 (CNTN1):
Contactin 1 binds with CNTN1-associated protein 1 (Caspr1) and neurofascin 155 (NF155), forming an axo-glial complex that is involved in paranode architecture and maintenance of myelin in axons (80). Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a IgG4-AID, and one of the possible antigens in this disorder is CNTN1. Anti-CNTN1 IgG4 antibodies recognise the IgC2 domains of CNTN1 and block its interaction with NF155, which leads to impaired cell aggregation and changes in paranodal architecture (81). Patients may present with other autoantibodies and have different clinical syndromes. Those presenting antibodies against neurofascin 186 (NF186), for instance, may present with renal impairment given both CNTN1 and NF186 are found in podocytes (62, 82).
7.4 Neurofascin 155 (NF155):
Neurofascin 155 is expressed by myelinating glial cells and, alongside Caspr1 and CNTN1, is part of the septate-like junction in paranodes found both in the central and peripherical nervous systems. Animal studies have confirmed the pathogenicity of anti-NF155 antibodies. Manso and colleagues demonstrated that intrathecal infusion of IgG4 from anti-NF155 CIDP patients induced similar symptoms in previously healthy mice (83). Early studies identified that these antibodies recognise the Fn3 and Fn4 domains of NF155, which does not participate in the interaction with CNTN1, and suggested a possible blocking mechanism that would cause the clinical presentation (84). Nevertheless, Manso et al. found that anti-NF155 IgG4 is associated with aggregation of the antigen and induces its depletion. They could not determine which mechanism was responsible for the depletion of surface NF155 and suggested that it might have to do with proteolysis of the antigen or molecular instability (83, 85).
7.5 ADAMTS13:
ADAM metallopeptidase with thrombospondin type 1 motif 13 (ADAMTS13) is a protease found in the blood circulation. It is responsible for the proteolytic cleavage of the multimeric form of von Willebrand factor (vWf) and ensuring normal haemostasis (86). Anti-ADAMTS13 IgG4 antibodies and vWf recognise the spacer-domain of the antigen, thus creating a competition for binding with ADAMTS13 (87). Autoimmune thrombotic thrombocytopenic purpura (TTP) is marked by an initial response based on IgG1 antibodies against ADAMTS13. The chronic antigenic stimulation then promotes class switch towards IgG4, which perpetuates the disease (88). The accumulation of vWf causes platelet aggregation and formation of microthrombi, which leads to the characteristic phenotype of microangiopathic haemolytic anaemia.
8 IgG4 in IgG4-related disease
IgG4-related disease (IgG4-RD) is a chronic relapsing immune-mediated fibro-inflammatory disorder and its hallmark is the presence of IgG4 antibodies in the sites affected (89). The diverse organ involvement in this disease is linked by a unique histopathology; a lymphoplasmacytic infiltrate rich in IgG4+ plasma blasts and CD4+ T cells, storiform fibrosis, and obliterative phlebitis (90).
There are currently three theories on the role of IgG4 antibodies in IgG4-RD: 1) IgG4 antibodies directly cause the disease by targeting self-antigens; 2) IgG4-RD patients have an immune response inherently biased towards production of IgG4 antibodies to any stimulus; 3) IgG4 is only present to modulate an immune response based on different pathways. It continues to be elusive whether the overexpression of these antibodies is an epiphenomenon of the inflammatory reaction or has a causal role in disease pathology.
8.1 Do IgG4 antibodies directly cause disease by targeting self-antigens?
Next-generation sequencing studies yield evidence to support that the pathogenesis of IgG4-RD is an antigen-driven process mediated by B and T cell interactions (91). Circulating B cells are predominantly IgG4+ B cells with extensive V-region mutations affecting both hypervariable and framework regions, suggesting signs of antibody maturation (91, 92). Mattoo et al. observed expansion of CD19+CD27+CD20-CD38hi plasmablasts, which are oligoclonally restricted as shown by analysis of immunoglobulin heavy V and J regions (92). Naïve B cells then undergo de novo recruitment during disease relapse. In patients who have a disease flare after treatment with rituximab, activated B cells and plasmablasts have increased somatic hypermutation when compared to before B cell depleting therapy, suggesting a possible driver for the autoreactive pathogenic process (92).
Differences in B cell phenotypes may be clinically important, as demonstrated by Li and colleagues (93). Patients with active IgG4-RD had increased plasmablasts (CD19+CD24-CD38hi, CD19+CD27hiCD38hi, and CD19+IgD-CD38hi) and reduced numbers of CD19+ B cells, IgD+CD38+/- naïve B cells and CD24hiCD38hi B regulatory cells (Breg) when compared to controls (93). Cluster analysis revealed that patients with a high count of plasmablast and memory B cells, low count of naïve B cells and Bregs had a higher disease burden and were predominantly male (93). This highlights the importance of antigen-specific LLPC in the pathogenesis of IgG4-RD. Whilst Rituximab (anti-CD20) reduces the number of short-lived B cells expressing CD20, memory B cells and LLPC lack this surface protein and can evade depletion, contributing to the relapse of disease.
Analysis of IgG4-RD lesions showed that tissue CD4+ T cells are clonally expanded with a cytolytic phenotype, expressing granzyme A, perforin and SLAMF7+ (SLAM family member 7) (94), and secreting IL-1β, TGF-β and IFN-γ, cytokines associated with fibrosis (94, 95). Drugs that deplete B cells lead to profound clinical remission and significantly reduce serum plasmablasts and CD4+ cytotoxic cell populations. Therefore, the excellent response to B cell depletion support that these cells are involved in the pathophysiology of IgG4-RD (93, 96, 97). Overall, this evidence suggests that T-dependent B cell activation is essential in the pathogenesis of IgG4-RD. For a thorough review of the roles of different T cells in IgG4-RD, please see (91).
HLA-mediated antigen presentation occurs in a number of autoimmune diseases, such as DRB1*15 in multiple sclerosis, DRB1*04 in rheumatoid arthritis and DQB1*02 and DQB1*03 in coeliac disease. A genome-wide association study in 857 Japanese IgG4-RD patients suggested that the HLA-DRB1*04:05 allele was important in the development of IgG4-RD (98). Indeed, mice expressing this risk allele develop autoimmune pancreatitis (99). This allele is also associated with type 1 diabetes and Crohn’s disease (100, 101). Furthermore, the single nucleotide polymorphism (SNP) rs134097 found in FcγRIIb was strongly associated with IgG4-RD, as well as with two phenotypes of the disease (pancreato-biliary and Mikulicz), number of swollen organs and serum IgG4 level at diagnosis. This SNP might play a critical role in IgG4-RD because its locus directly impacts on the expression of FcyRIIb, a receptor involved in the elimination of self-reactive B cells.
8.1.1 Autoantigens in IgG4-RD
Many studies suggest a role of autoantigens in patients with IgG4-RD. Neonatal mice that received passive transfer of purified IgG1 and IgG4 from patients with active IgG4-RD showed evidence of damage to salivary glands and the pancreas (102). Several autoantibodies have been identified in the context of IgG4-related autoimmune pancreatitis, such as lactoferrin, carbonic anhydrase, pancreas secretory trypsin inhibitor, amylase-alpha, heat shock protein and plasminogen-binding protein. However, many of these lacked sensitivity and specificity for those with other organ manifestations (103).
8.1.1.1 Helicobacter pylori antigens
Human carbonic anhydrase II (CAII) is an enzyme found in the cytoplasm of pancreatic ductal epithelial cells, as well as in kidney tubules, gallbladder and glial cells. Given the discovery of antibodies against H. pylori in the sera of patients with IgG4-related pancreatitis and Sjogren’s syndrome (104–106), it was proposed that H. pylori infection could be driving the disease through molecular mimicry with CAII (107). Frulloni and colleagues identified that, in fact, immunoglobulins from IgG4-RD patients targeted plasminogen-binding protein type A (PBP), also an antigen found in H. pylori. This protein shares sequence homology with a human protein, ubiquitin protein ligase E3 component n-recognin 2 (UBR2). The group proceeded to show that 19 (95%) of their patients with IgG4-related pancreatitis had antibodies against PBP and similar results were replicated with UBR2 (108). A large study by Culver et al., however, did not replicate the findings. Among 69 IgG4-RD patients and 51 controls with autoimmune or inflammatory diseases, authors found similar T and B cell reactivity against PBP between groups (109). Jesnowski and colleagues investigated the presence of conserved sequences of H. pylori in the pancreatic tissue and juice of patients with IgG4-related pancreatitis and pancreatic cancer via nested PCR and couldn’t identify any signal of H. pylori DNA (110), corroborating previous findings that the bacterium is unlikely to be directly involved in the pathogenesis of IgG4-related pancreatitis.
8.1.1.2 Acinar antigens
Lactoferrin (LF), pancreatic secretory trypsinogen inhibitor (SPINK1) and trypsinogens have been identified as possible antigens in IgG4-related pancreatitis (111, 112) and antibodies against the last two proteins have been reported in different cohorts of patients with the disease. Löhr and colleagues showed that patients with chronic IgG4-related pancreatitis have a severely reduced population of acinar cells in histopathological studies. The presence of antibodies against trypsinogens was replicated in an animal model of the disease, thus corroborating this finding (112). Nevertheless, they could not differentiate between subtypes of autoimmune pancreatitis and could not explain why acinar cells resume production of digestive enzymes during remission of IgG4-related pancreatitis.
8.1.1.3 Annexin A11
Annexin A11 is part of a family of calcium-dependent phospholipid-binding proteins. It is found in the cytosol of cholangiocytes, pancreatic duct cells and islands of Langerhans as well as other tissues. Hubers and colleagues (113) identified IgG4 antibodies against annexin A11 in 9 (18%) patients with IgG4-related pancreatitis and cholangitis, whilst not in controls with other pancreatobiliary diseases, including malignancies. Reinforcing this finding, the location where this antigen was identified corresponded to the pattern of injury in pancreatobiliary disease (114). It was also found in patients with IgG4-related salivary involvement, suggesting that annexin A11 is not specific to pancreatobiliary involvement.
The ‘biliary bicarbonate umbrella’ is responsible for protecting human cholangiocytes from hydrophobic bile acid influx (115). Herta et al. reported that annexin A11 is necessary so that this defensive mechanism can be adequate. Antibodies against annexin A11 inhibited the function of this self-antigen, thus the authors speculate that this may impair the biliary bicarbonate umbrella and facilitate bile duct damage in patients with IgG4-related cholangitis (116).
Lastly, Hubers et al. demonstrated that IgG4 antibodies isolated from patients with IgG4-RD compete with IgG1 for binding to annexin A11, thus reinforcing that the first has an anti-inflammatory role in the disease pathogenesis (113). These results have been replicated in the passive transfer experiment with neonatal mice, where IgG from IgG4-RD patients induced a similar pattern of pancreatic and salivary lesion. Shiokawa and colleagues observed that both IgG1 and IgG4 antibodies could trigger the injury. The first, however, had its activity dampened by simultaneous injection of IgG4 antibodies (117).
8.1.1.4 Laminin 511-E8
Laminin is a component of the extracellular matrix (ECM) of tissues, and integrin alfa-6-beta-4 is a cellular adhesion molecule that binds to laminins in the ECM. A Japanese group identified antibodies against laminin 511-E8 in 51% (26/51) of patients with IgG4-related pancreatitis and in only 1.6% of healthy volunteers (118). Sixteen percent (4/25) of those who did not have antibodies to laminin, had antibodies against its ligand, integrin alpha-6-beta-1.
Passive transfer and active immunisation animal models confirmed that mice that received human sera from patients with IgG4-related pancreatitis and those immunized with human laminin 511-E8 developed an immune response responsible for injury in pancreatic tissue and salivary glands in the same pattern as observed in IgG4-RD (119). Regardless of serum concentration of IgG4, half of these patients had specific IgG1 antibodies whilst only one patient had IgG4 against laminin 511-E8. Furthermore, only pancreatic and salivary gland injury was demonstrated.
Shiokawa observed that anti-laminin 511-E8 antibodies decreased upon treatment with glucocorticoids in an equivalent manner as other biomarkers of activity (119) (e.g., serum IgG4 concentration and pancreatic imaging). These results have not been validated externally. In contrast, In a US IgG4-RD cohort, the positivity rate for laminin 511-E8 antibody was similar between IgG4-RD (7%), disease controls, and healthy volunteers (118).
8.1.1.5 Galectin-3
Galectin-3 is a cytoplasmatic b-galactoside-binding lectin identified in systemic fibroproliferative conditions, such as systemic sclerosis and pulmonary fibrosis (120). In a US cohort of 121 IgG4-RD patients, using immunoaffinity chromatography and mass spectrometry of plasmablast clones, 34 (28%) were positive for IgG4-specific anti-galectin-3 antibody while almost none of the 45 disease controls with interstitial pulmonary fibrosis and 50 healthy volunteers showed similar results (121). Furthermore, IgE-specific anti-galectin-3 antibodies were also detected but other IgG subclasses had little to no reactivity among the participants (121).
Perugino et al. used a galectin level threshold above 10.25ng/mL, which is independently associated with all-cause mortality in systemic sclerosis (122), to divide their cohort into two groups; those with higher levels had 64% positivity for IgG4 anti-galectin-3 antibodies, whilst those with lower levels had 23% positivity (121). Moreover, the authors reported a correlation between the presence of these antibodies and lymphadenopathy in IgG4-RD, as well as a trend between higher serum concentration of galectin-3 and increased IgG4-RD Responder Index of disease activity, number of organs and multi-organ involvement.
A Japanese group used proteomic analysis to demonstrate a 13-fold increased expression of galectin-3 in the pancreas of patients with IgG4-related pancreatitis compared with heathy pancreatic tissue (123). Galectin-3 is expressed by different cell lines in affected organs in IgG4-RD, including pancreas, bile ducts, salivary glands, kidney, lung, aorta and retroperitoneum, supporting a role in tissue fibrosis in IgG4-RD (124).
8.1.1.6 Prohibitin
Prohibitins are ubiquitously expressed and are important in critical cell processes. Prohibitin 1 is involved in transcription, apoptosis and mitochondrial protein folding (125), while prohibitin 2 is essential in mitochondrial homeostasis and autophagy (126). Du and colleagues identified a protein that has 8 unique peptides matched to human prohibitin, and also shares 40% of sequence similarity, using affinity purification of patient serum and mass spectrometry (127). Seventy-three percent (65/89) of patients with IgG4-RD had antibodies against prohibitin, whilst only 13% with Sjogren’s syndrome and 1.4% of healthy volunteers showed positivity. Moreover, the anti-prohibitin antibody was observed in patients with IgG4-related pancreatic disease, salivary disease, retroperitoneal fibrosis, and other organ involvement (127).
8.1.2 Role of autoantigens as biomarkers in IgG4-RD
Currently, there is no evidence that measuring autoantigens can help to diagnose IgG4-RD, detect response to treatment or predict early signs of a disease flare. There are discrepancies between cohorts on the relative frequency of each autoantigen; Liu and colleagues assessed for the presence of autoantigens in a US IgG4-RD cohort and found 7% positivity for laminin 511-E8, 10% for prohibitin, 12% for annexin A11 and 28% for galectin-3 (118). The authors also observed that there was no clinically meaningful difference between patients with and without the presence of one autoantibody. Nevertheless, patients with positivity for more than one antibody had a more severe presentation, usually with higher levels of inflammatory biomarkers (e.g., total IgG and subclasses, C-reactive protein, and complement consumption) as well as greater risk of visceral organ involvement (118). This is in keeping with findings in other autoimmune diseases.
We can speculate that it is not just one autoantigen that dominates in IgG4-RD, and that the presence of multiple autoantigens may promote a larger breach of B cell tolerance and correlate with a more aggressive inflammatory disease phenotype. Another possible explanation for the wide variety of autoantibodies found in IgG4-RD might be that, in fact, it is not a single disease, but rather a spectrum of disorders characterised by a similar response to different stimuli. Further studies need to demonstrate a single trigger for the disease and/or identify mechanisms to justify why an autoantigen is not involved in all four phenotypes of the disease.
8.2 Do patients with IgG4-RD have a biased immune response towards the production of IgG4 against any stimulus?
Another theory behind the development of IgG4-RD is that patients have an inherently biased immune response and preferentially produce IgG4. IgG4 antibody has an important role in the development of tolerance after chronic exposure to allergens (34), and an association between allergy and/or atopy and IgG4-RD has been described by many groups (128–133). There are different definitions for allergy and atopy, which creates a variance in the prevalence of these conditions reported in patients with IgG4-RD (134). Studies observed between 18 and 76% of patients with IgG4-RD have allergy (130–133) and between 14 and 46% have atopy (129, 135, 136). A predominance of type 2 immunity has been reported in such patients. There is an abundance of IL-4 and IL-13, which drive CSR towards IgG4 and IgE production, as well as activation of eosinophils (137). Some studies have suggested a phenotype that is more treatment refractory and aggressive in those with allergy/atopy, higher IgE and IgG4 level and peripheral blood eosinophil counts.
Our group has shown that antigens derived from food and animals elicited a polyclonal IgG4 response in patients with IgG4-RD. These food antigens included egg white and yolk, milk, banana, peanut, rice, wheat, and animal cat dander. Indeed, high serum levels of IgG4 in this setting may reflect a defective regulation of the overall immune response (138).
We have also shown that IgG4-RD may be associated with prolonged contact with occupational antigens. Exposure to solvents, industrial and metal dusts, automotive’s pigments and oils was described in up to 61% of patients in two independent cohorts of IgG4-related sclerosing cholangitis in the Netherlands and the UK (139). Blue collar workers were further identified as a risk factor for developing disease in a case-controlled study (140). No single contaminant was identified, which raises the question whether prolonged contact with a number of environmental antigens trigger the disease and drive the IgG4 response. There is also emerging data on the role of asbestos in IgG4-related retroperitoneal fibrosis, and tobacco smoking conferring an increased risk of developing the disease.
An IgG4 class switch response and production of excessive IgG4 antibodies in the disease is likely determined by the immune cell milieu. During active IgG4-RD, restricted clones of B cells and plasmablasts proliferate through a Tfh2 cell-dependent pathway. Interleukin (IL)-21 producing Tfh2 lymphocytes promote somatic hypermutation in B cells inside GC, while IL-4 producing Tfh2 cells are involved in CSR (141–143). Regulatory T cells and IL-10 are also involved in the making of an IgG4-based response (144, 145).
8.3 Are IgG4 antibodies trying to curb an overactive inflammation caused by a different trigger in IgG4-RD?
Finally, we need to consider whether IgG4 antibodies are an epiphenomenon and actually are trying to control inflammation based on a different mechanism. Mouse models confirmed that total IgG from patients with IgG4-RD caused a similar pancreatic injury in the animals (113, 117). Shiokawa and colleagues analysed differences between the pathogenic effect of IgG1 and IgG4 of patients with IgG4-RD when these antibodies were injected in mice (117). When injected separately, both classes caused pancreatic injury and IgG4 also caused salivary damage, but interestingly, when these antibodies were injected simultaneously, IgG4 competed with IgG1 and significantly reduced its binding to pancreatic tissue. Thus, there may be a competitive effect of producing excess IgG4 anitbodies to dampen an inflammatory process that precedes them.
Against this theory is the evidence that IgG4 antibodies in patients with IgG4-RD are highly specific and have high affinity for their target given that circulating B cells are in their majority IgG4+ B cells with extensive V-region mutations (91, 92). These findings suggest that these antibodies come from B cells/plasma cells that have undergone rounds of maturation rather than an inflammatory response, thus making the case that IgG4-RD behaves more like an autoimmune disease instead of an inflammatory disorder.
9 Parallels between IgG4-AID and IgG4-RD
The antigens proposed for IgG4-RD are ubiquitous and found both in the cytosol and the extra cellular matrix. Nevertheless, with the exception of laminin 511-E8, they fall short of meeting either Witebsky or Koneczny criteria for autoimmunity (61, 146). This brings into question whether these antibodies are the drivers of IgG4-RD. There is evidence that antibodies can penetrate the plasma membrane and recognise intracellular antigens (147–149). In fact, many entry mechanisms have been studied, such as interaction of basic residues with a negatively charged cell membrane (150), Fc-receptor mediated entry (151), endocytosis (147, 152, 153), and nucleoside transporters (154). Among the six IgG4-AID that meet IgG4 autoimmunity criteria, however, all the antigens involved in their pathogenesis are found in the cell membrane or free in the blood circulation (62).
Another important consideration when analysing the role of autoantigens in IgG4-RD is the clinical presentation of the disease. Regardless of phenotype (i.e., pancreatobiliary, head and neck, retroperitoneum and aorta, and systemic), the hallmarks of IgG4-RD are proliferative and/or fibrotic lesions in the organs affected. These findings are diametrically opposed to those from IgG4-AID, where symptoms are driven by IgG4 antibodies blocking antigen function, such as in CNTN1-associated CIDP (155). Histological examination of IgG4-RD lesions shows a massive invasion of IgG4+ B cells and plasmablasts and extensive (storiform) fibrosis (156). Furthermore, CD4+ and CD8+ cytotoxic T lymphocytes are also part of the immune response found in organs with active inflammation and peripheral blood (157), suggesting that the interaction between T and B cells play a critical role in the damage caused by IgG4-RD.
10 Current research gaps in IgG4-RD
Despite recent developments in IgG4-RD research, there are essential questions without an answer. One of the most important ones is why and how patients develop it. Previous studies reported an association of HLA-DRB1 and FCGR2B with IgG4-RD, including with phenotypical characteristics (98), and two studies observed a relation between HLA genes, particularly HLA-DQB1*04:01", HLA-DRB1*04:05 and HLA-DRB1*16 in patients with autoimmune pancreatitis type 1 and 2 (158, 159). Moreover, it is also important to look at B cell specialisation and how it can impact humoral immune responses favouring IgG4 expression. As in IgG4-AID, it is paramount to identify whether naturally occurring antigen(s) is(are) driving the immune response in IgG4-RD and the evidence surrounding self-antigens needs to be validated in external cohorts. We must consider the possibility that, such as IgG4-AID, IgG4-RD is a spectrum of diseases rather than a single entity. Finally, the increased expression of IgG4 antibodies and the presence of IgG4+ plasma cells in sites with IgG4-RD inflammation highlights the importance of clarifying the mechanisms that favour class switch towards IgG4 rather than other IgG subclasses. This knowledge is crucial to understand the pathogenesis of IgG4-RD (160).
Author contributions
RVM: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. EC: Conceptualization, Data curation, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The National Institute for Health Research (NIHR) Biomedical Research Centre (BRC) Oxford supports ELC for work in inflammation and cancer.
Conflict of interest
EC: consults for Horizon Therapeutics, Zenus BioPharma and Sanofi for IgG4-RD. EC receives funding from the National Institute for Health Research NIHR Biomedical Research Centre BRC Oxford, Oxford Hospitals Charitable Grant and Research Capability Fund.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Klein U, Rajewsky K, Küppers R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med (1998) 188:1679–89. doi: 10.1084/jem.188.9.1679
2. Brinkmann V, Müller S, Heusser CH. T cell dependent differentiation of human B cells: direct switch from IgM to IgE, and sequential switch from IgM via IgG to IgA production. Mol Immunol (1992) 29:1159–64. doi: 10.1016/0161-5890(92)90051-x
3. Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol (2009) 10:1292–9. doi: 10.1038/ni.1814
4. Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med (2012) 366:539–51. doi: 10.1056/NEJMra1104650
5. Schroeder HW, Cavacini L. Structure and function of immunoglobulins. J Allergy Clin Immunol (2010) 125:S41–52. doi: 10.1016/j.jaci.2009.09.046
6. Schuurman J, Van Ree R, Perdok GJ, Van Doorn HR, Tan KY, Aalberse RC. Normal human immunoglobulin G4 is bispecific: it has two different antigen-combining sites. Immunology (1999) 97:693–8. doi: 10.1046/j.1365-2567.1999.00845.x
7. van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martínez-Martínez P, Vermeulen E, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science (2007) 317:1554–7. doi: 10.1126/science.1144603
8. Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Exp Allergy (2009) 39:469–77. doi: 10.1111/j.1365-2222.2009.03207.x
9. Angal S, King DJ, Bodmer MW, Turner A, Lawson AD, Roberts G, et al. A single amino acid substitution abolishes the heterogeneity of chimeric mouse/human (IgG4) antibody. Mol Immunol (1993) 30:105–8. doi: 10.1016/0161-5890(93)90432-b
10. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood (2009) 113:3716–25. doi: 10.1182/blood-2008-09-179754
11. James LK, Bowen H, Calvert RA, Dodev TS, Shamji MH, Beavil AJ, et al. Allergen specificity of IgG(4)-expressing B cells in patients with grass pollen allergy undergoing immunotherapy. J Allergy Clin Immunol (2012) 130:663–670.e3. doi: 10.1016/j.jaci.2012.04.006
12. Van der Zee S, Aalberse RC. IgG4 and hyposensitization. N Engl Reg Allergy Proc (1987) 8:389–91. doi: 10.2500/108854187778999667
13. van der Zee JS, van Swieten P, Aalberse RC. Inhibition of complement activation by IgG4 antibodies. Clin Exp Immunol (1986) 64:415–22.
14. Yang Y, Wang C, Jin L, He F, Li C, Gao Q, et al. IgG4 anti-phospholipase A2 receptor might activate lectin and alternative complement pathway meanwhile in idiopathic membranous nephropathy: an inspiration from a cross-sectional study. Immunol Res (2016) 64:919–30. doi: 10.1007/s12026-016-8790-1
15. Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science (2014) 343:1260–3. doi: 10.1126/science.1248943
16. Prodjinotho UF, Hoerauf A, Adjobimey T. IgG4 antibodies from patients with asymptomatic bancroftian filariasis inhibit the binding of IgG1 and IgG2 to C1q in a Fc-Fc-dependent mechanism. Parasitol Res (2019) 118:2957–68. doi: 10.1007/s00436-019-06451-2
17. Oskam N, Damelang T, Streutker M, Ooijevaar-de Heer P, Nouta J, Koeleman C, et al. Factors affecting IgG4-mediated complement activation. Front Immunol (2023) 14:1087532. doi: 10.3389/fimmu.2023.1087532
18. Navas A, Ruiz-Leon B, Serrano P, Martí M, Espinazo ML, Blanco N, et al. Natural and induced tolerance to hymenoptera venom: A single mechanism? Toxins (Basel) (2022) 14:426. doi: 10.3390/toxins14070426
19. Hagel I, Cabrera M, Buvat E, Gutiérrez L, Santaella C, Borges R, et al. Antibody responses and resistance against Ascaris lumbricoides infection among Venezuelan rural children: the influence of ethnicity. J Trop Pediatr (2008) 54:354–6. doi: 10.1093/tropej/fmn032
20. Elsner RA, Shlomchik MJ. Germinal center and extrafollicular B cell responses in vaccination, immunity, and autoimmunity. Immunity (2020) 53:1136–50. doi: 10.1016/j.immuni.2020.11.006
21. Weisel F, Shlomchik M. Memory B cells of mice and humans. Annu Rev Immunol (2017) 35:255–84. doi: 10.1146/annurev-immunol-041015-055531
22. Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature (2008) 453:667–71. doi: 10.1038/nature06890
23. Stavnezer J, Guikema JEJ, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol (2008) 26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248
24. Aalberse RC, van der Gaag R, van Leeuwen J. Serologic aspects of IgG4 antibodies. I. Prolonged immunization results in an IgG4-restricted response. J Immunol (1983) 130:722–6. doi: 10.4049/jimmunol.130.2.722
25. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell (2000) 102:553–63. doi: 10.1016/s0092-8674(00)00078-7
26. Jackson KJL, Wang Y, Collins AM. Human immunoglobulin classes and subclasses show variability in VDJ gene mutation levels. Immunol Cell Biol (2014) 92:729–33. doi: 10.1038/icb.2014.44
27. Kitaura K, Yamashita H, Ayabe H, Shini T, Matsutani T, Suzuki R. Different somatic hypermutation levels among antibody subclasses disclosed by a new next-generation sequencing-based antibody repertoire analysis. Front Immunol (2017) 8:389. doi: 10.3389/fimmu.2017.00389
28. Unger P-PA, Lighaam LC, Vermeulen E, Kruithof S, Makuch M, Culver EL, et al. Divergent chemokine receptor expression and the consequence for human IgG4 B cell responses. Eur J Immunol (2020) 50:1113–25. doi: 10.1002/eji.201948454
29. Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, Nussenzweig MC. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood (2012) 120:2240–8. doi: 10.1182/blood-2012-03-415380
30. Matysiak J, Matysiak J, Bręborowicz A, Kycler Z, Dereziński P, Kokot ZJ. Immune and clinical response to honeybee venom in beekeepers. Ann Agric Environ Med (2016) 23:120–4. doi: 10.5604/12321966.1196866
31. García-Robaina JC, de la Torre-Morín F, Vazquez-Moncholi C, Fierro J, Bonnet-Moreno C. The natural history of Apis-specific IgG and IgG4 in beekeepers. Clin Exp Allergy (1997) 27:418–23. doi: 10.1111/j.1365-2222.1997.tb00727.x
32. Matsui EC, Diette GB, Krop EJM, Aalberse RC, Smith AL, Curtin-Brosnan J, et al. Mouse allergen-specific immunoglobulin G and immunoglobulin G4 and allergic symptoms in immunoglobulin E-sensitized laboratory animal workers. Clin Exp Allergy (2005) 35:1347–53. doi: 10.1111/j.1365-2222.2005.02331.x
33. Jones M, Jeal H, Schofield S, Harris JM, Shamji MH, Francis JN, et al. Rat-specific IgG and IgG₄ antibodies associated with inhibition of IgE-allergen complex binding in laboratory animal workers. Occup Environ Med (2014) 71:619–23. doi: 10.1136/oemed-2014-102119
34. Jarkvist J, Salehi C, Akin C, Gülen T. Venom immunotherapy in patients with clonal mast cell disorders: IgG4 correlates with protection. Allergy (2020) 75:169–77. doi: 10.1111/all.13980
35. James LK, Shamji MH, Walker SM, Wilson DR, Wachholz PA, Francis JN, et al. Long-term tolerance after allergen immunotherapy is accompanied by selective persistence of blocking antibodies. J Allergy Clin Immunol (2011) 127:509–16. doi: 10.1016/j.jaci.2010.12.1080
36. Shamji MH, Kappen J, Abubakar-Waziri H, Zhang J, Steveling E, Watchman S, et al. Nasal allergen-neutralizing IgG4 antibodies block IgE-mediated responses: Novel biomarker of subcutaneous grass pollen immunotherapy. J Allergy Clin Immunol (2019) 143:1067–76. doi: 10.1016/j.jaci.2018.09.039
37. Burton OT, Logsdon SL, Zhou JS, Medina-Tamayo J, Abdel-Gadir A, Noval Rivas M, et al. Oral immunotherapy induces IgG antibodies that act through FcγRIIb to suppress IgE-mediated hypersensitivity. J Allergy Clin Immunol (2014) 134:1310–1317.e6. doi: 10.1016/j.jaci.2014.05.042
38. Uermösi C, Zabel F, Manolova V, Bauer M, Beerli RR, Senti G, et al. IgG-mediated down-regulation of IgE bound to mast cells: a potential novel mechanism of allergen-specific desensitization. Allergy (2014) 69:338–47. doi: 10.1111/all.12327
39. Kanagaratham C, Derakhshan T, El Ansari YS, Furiness KN, Hollers E, Keldsen M, et al. IgG:FcγRIIb signals block effector programs of IgE:FcϵRI-activated mast cells but spare survival pathways. J Allergy Clin Immunol (2023) 152:453–68. doi: 10.1016/j.jaci.2023.03.027
40. Clayton F, Fang JC, Gleich GJ, Lucendo AJ, Olalla JM, Vinson LA, et al. Eosinophilic esophagitis in adults is associated with IgG4 and not mediated by IgE. Gastroenterology (2014) 147:602–9. doi: 10.1053/j.gastro.2014.05.036
41. Koyama T, Kariya S, Sato Y, Gion Y, Higaki T, Haruna T, et al. Significance of IgG4-positive cells in severe eosinophilic chronic rhinosinusitis. Allergol Int (2019) 68:216–24. doi: 10.1016/j.alit.2018.09.002
42. Rosenberg CE, Mingler MK, Caldwell JM, Collins MH, Fulkerson PC, Morris DW, et al. Esophageal IgG4 levels correlate with histopathologic and transcriptomic features in eosinophilic esophagitis. Allergy (2018) 73:1892–901. doi: 10.1111/all.13486
43. Hoofien A, Papadopoulou A, Gutiérrez-Junquera C, Martínez Gómez MJ, Domínguez-Ortega G, Oudshoorn J, et al. Sustained remission of eosinophilic esophagitis following discontinuation of dietary elimination in children. Clin Gastroenterol Hepatol (2020) 18:249–251.e1. doi: 10.1016/j.cgh.2019.03.008
44. Quinn L, Nguyen B, Menard-Katcher C, Spencer L. IgG4+ cells are increased in the gastrointestinal tissue of pediatric patients with active eosinophilic gastritis and duodenitis and decrease in remission. Dig Liver Dis (2023) 55:53–60. doi: 10.1016/j.dld.2022.08.020
45. Qin L, Tang L-F, Cheng L, Wang H-Y. The clinical significance of allergen-specific IgG4 in allergic diseases. Front Immunol (2022) 13:1032909. doi: 10.3389/fimmu.2022.1032909
46. Caldas IR, Campi-Azevedo AC, Oliveira LFA, Silveira AMS, Oliveira RC, Gazzinelli G. Human schistosomiasis mansoni: immune responses during acute and chronic phases of the infection. Acta Trop (2008) 108:109–17. doi: 10.1016/j.actatropica.2008.05.027
47. Figueiredo JP, Oliveira RR, Cardoso LS, Barnes KC, Grant AV, Carvalho EM, et al. Adult worm-specific IgE/IgG4 balance is associated with low infection levels of Schistosoma mansoni in an endemic area. Parasite Immunol (2012) 34:604–10. doi: 10.1111/pim.12001
48. Adjobimey T, Hoerauf A. Induction of immunoglobulin G4 in human filariasis: an indicator of immunoregulation. Ann Trop Med Parasitol (2010) 104:455–64. doi: 10.1179/136485910X12786389891407
49. Resende SD, Magalhães FC, Rodrigues-Oliveira JL, Castro VN, Souza CSA, Oliveira EJ, et al. Modulation of allergic reactivity in humans is dependent on schistosoma mansoni parasite burden, low levels of IL-33 or TNF-α and high levels of IL-10 in serum. Front Immunol (2018) 9:3158. doi: 10.3389/fimmu.2018.03158
50. Harada K, Shimoda S, Kimura Y, Sato Y, Ikeda H, Igarashi S, et al. Significance of immunoglobulin G4 (IgG4)-positive cells in extrahepatic cholangiocarcinoma: molecular mechanism of IgG4 reaction in cancer tissue. Hepatology (2012) 56:157–64. doi: 10.1002/hep.25627
51. Karagiannis P, Gilbert AE, Josephs DH, Ali N, Dodev T, Saul L, et al. IgG4 subclass antibodies impair antitumor immunity in melanoma. J Clin Invest (2013) 123:1457–74. doi: 10.1172/JCI65579
52. Hinz S, Pagerols-Raluy L, Oberg H-H, Ammerpohl O, Grüssel S, Sipos B, et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res (2007) 67:8344–50. doi: 10.1158/0008-5472.CAN-06-3304
53. Kimura Y, Harada K, Nakanuma Y. Pathologic significance of immunoglobulin G4-positive plasma cells in extrahepatic cholangiocarcinoma. Hum Pathol (2012) 43:2149–56. doi: 10.1016/j.humpath.2012.03.001
54. Karagiannis P, Villanova F, Josephs DH, Correa I, Van Hemelrijck M, Hobbs C, et al. Elevated IgG4 in patient circulation is associated with the risk of disease progression in melanoma. Oncoimmunology (2015) 4:e1032492. doi: 10.1080/2162402X.2015.1032492
55. Yu T, Wu Y, Liu J, Zhuang Y, Jin X, Wang L. The risk of Malignancy in patients with IgG4-related disease: a systematic review and meta-analysis. Arthritis Res Ther (2022) 24:14. doi: 10.1186/s13075-021-02652-2
56. Kamisawa T, Horiguchi S-I, Hayashi Y, Yun X, Yamaguchi T, Tsuruta K, et al. K-ras mutation in the major duodenal papilla and gastric and colonic mucosa in patients with autoimmune pancreatitis. J Gastroenterol (2010) 45:771–8. doi: 10.1007/s00535-010-0211-y
57. Jinesh GG, Sambandam V, Vijayaraghavan S, Balaji K, Mukherjee S. Molecular genetics and cellular events of K-Ras-driven tumorigenesis. Oncogene (2018) 37:839–46. doi: 10.1038/onc.2017.377
58. Zhao B, Kimura W, Futakawa N, Muto T, Kubota K, Harihara Y, et al. p53 and p21/Waf1 protein expression and K-ras codon 12 mutation in carcinoma of the papilla of Vater. Am J Gastroenterol (1999) 94:2128–34. doi: 10.1111/j.1572-0241.1999.01309.x
59. Huijbers MG, Querol LA, Niks EH, Plomp JJ, van der Maarel SM, Graus F, et al. The expanding field of IgG4-mediated neurological autoimmune disorders. Eur J Neurol (2015) 22:1151–61. doi: 10.1111/ene.12758
60. Rispens T, Huijbers MG. The unique properties of IgG4 and its roles in health and disease. Nat Rev Immunol (2023) 23(11):763–78. doi: 10.1038/s41577-023-00871-z
61. Koneczny I. A new classification system for igG4 autoantibodies. Front Immunol (2018) 9:97. doi: 10.3389/fimmu.2018.00097
62. Koneczny I. Update on IgG4-mediated autoimmune diseases: New insights and new family members. Autoimmun Rev (2020) 19:102646. doi: 10.1016/j.autrev.2020.102646
63. Li N, Aoki V, Hans-Filho G, Rivitti EA, Diaz LA. The role of intramolecular epitope spreading in the pathogenesis of endemic pemphigus foliaceus (fogo selvagem). J Exp Med (2003) 197:1501–10. doi: 10.1084/jem.20022031
64. Toumi A, Saleh MA, Yamagami J, Abida O, Kallel M, Masmoudi A, et al. Autoimmune reactivity against precursor form of desmoglein 1 in healthy Tunisians in the area of endemic pemphigus foliaceus. J Dermatol Sci (2013) 70:19–25. doi: 10.1016/j.jdermsci.2013.02.002
65. Qian Y, Jeong JS, Maldonado M, Valenzuela JG, Gomes R, Teixeira C, et al. Cutting Edge: Brazilian pemphigus foliaceus anti-desmoglein 1 autoantibodies cross-react with sand fly salivary LJM11 antigen. J Immunol (2012) 189:1535–9. doi: 10.4049/jimmunol.1200842
66. Peng B, Temple BR, Yang J, Geng S, Culton DA, Qian Y. Identification of a primary antigenic target of epitope spreading in endemic pemphigus foliaceus. J Autoimmun (2021) 116:102561. doi: 10.1016/j.jaut.2020.102561
67. Diaz LA, Prisayanh P, Qaqish B, Temple BR, Aoki V, Hans-Filho G, et al. A lutzomyia longipalpis salivary protein induces cross-reactive antibodies to pemphigus autoantigen desmoglein 1. J Invest Dermatol (2020) 140:2332–2342.e10. doi: 10.1016/j.jid.2020.02.041
68. Kallel Sellami M, Ben Ayed M, Mouquet H, Drouot L, Zitouni M, Mokni M, et al. Anti-desmoglein 1 antibodies in Tunisian healthy subjects: arguments for the role of environmental factors in the occurrence of Tunisian pemphigus foliaceus. Clin Exp Immunol (2004) 137:195–200. doi: 10.1111/j.1365-2249.2004.02493.x
69. Abida O, Kallel-Sellami M, Joly P, Ben Ayed M, Zitouni M, Masmoudi A, et al. Anti-desmoglein 1 antibodies in healthy related and unrelated subjects and patients with pemphigus foliaceus in endemic and non-endemic areas from Tunisia. J Eur Acad Dermatol Venereol (2009) 23:1073–8. doi: 10.1111/j.1468-3083.2009.03265.x
70. Zaraa I, Boussoffara T, Ben Ahmed M, Marzouki S, Ben Hassouna N, Sellami MK, et al. Exposure to Phlebotomus papatasi and/or Leishmania major: possible etiologic link to Tunisian pemphigus. J Invest Dermatol (2012) 132:479–82. doi: 10.1038/jid.2011.291
71. Kallel Sellami M, Zitouni M, Tombari W, Ben Ayed M, Abida O, Laadhar L, et al. Anti-desmoglein-1 antibodies are prevalent in Tunisian patients with hydatidosis and leishmaniasis. Br J Dermatol (2007) 156:591–3. doi: 10.1111/j.1365-2133.2006.07687.x
72. Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med (2001) 7:365–8. doi: 10.1038/85520
73. McConville J, Farrugia ME, Beeson D, Kishore U, Metcalfe R, Newsom-Davis J, et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol (2004) 55:580–4. doi: 10.1002/ana.20061
74. Huijbers MG, Vink A-FD, Niks EH, Westhuis RH, van Zwet EW, de Meel RH, et al. Longitudinal epitope mapping in MuSK myasthenia gravis: implications for disease severity. J Neuroimmunol (2016) 291:82–8. doi: 10.1016/j.jneuroim.2015.12.016
75. Takamori M, Nakamura T, Motomura M. Antibodies against Wnt receptor of muscle-specific tyrosine kinase in myasthenia gravis. J Neuroimmunol (2013) 254:183–6. doi: 10.1016/j.jneuroim.2012.09.001
76. Koneczny I, Cossins J, Waters P, Beeson D, Vincent A. MuSK myasthenia gravis IgG4 disrupts the interaction of LRP4 with MuSK but both IgG4 and IgG1-3 can disperse preformed agrin-independent AChR clusters. PloS One (2013) 8:e80695. doi: 10.1371/journal.pone.0080695
77. Huijbers MG, Zhang W, Klooster R, Niks EH, Friese MB, Straasheijm KR, et al. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc Natl Acad Sci U.S.A. (2013) 110:20783–8. doi: 10.1073/pnas.1313944110
78. Huijbers MG, Vergoossen DL, Fillié-Grijpma YE, van Es IE, Koning MT, Slot LM, et al. MuSK myasthenia gravis monoclonal antibodies: Valency dictates pathogenicity. Neurology(R) neuroimmunology Neuroinflamm (2019) 6:e547. doi: 10.1212/NXI.0000000000000547
79. Vergoossen DLE, Ruiter AM, Keene KR, Niks EH, Tannemaat MR, Strijbos E, et al. Enrichment of serum IgG4 in MuSK myasthenia gravis patients. J Neuroimmunol (2022) 373:577978. doi: 10.1016/j.jneuroim.2022.577978
80. Faivre-Sarrailh C, Devaux JJ. Neuro-glial interactions at the nodes of Ranvier: implication in health and diseases. Front Cell Neurosci (2013) 7:196. doi: 10.3389/fncel.2013.00196
81. Labasque M, Hivert B, Nogales-Gadea G, Querol L, Illa I, Faivre-Sarrailh C. Specific contactin N-glycans are implicated in neurofascin binding and autoimmune targeting in peripheral neuropathies. J Biol Chem (2014) 289:7907–18. doi: 10.1074/jbc.M113.528489
82. Sistani L, Rodriguez PQ, Hultenby K, Uhlen M, Betsholtz C, Jalanko H, et al. Neuronal proteins are novel components of podocyte major processes and their expression in glomerular crescents supports their role in crescent formation. Kidney Int (2013) 83:63–71. doi: 10.1038/ki.2012.321
83. Manso C, Querol L, Lleixà C, Poncelet M, Mekaouche M, Vallat J-M, et al. Anti-Neurofascin-155 IgG4 antibodies prevent paranodal complex formation. vivo. J Clin Invest (2019) 129:2222–36. doi: 10.1172/JCI124694
84. Ng JKM, Malotka J, Kawakami N, Derfuss T, Khademi M, Olsson T, et al. Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology (2012) 79:2241–8. doi: 10.1212/WNL.0b013e31827689ad
85. Dutta DJ, Woo DH, Lee PR, Pajevic S, Bukalo O, Huffman WC, et al. Regulation of myelin structure and conduction velocity by perinodal astrocytes. Proc Natl Acad Sci U.S.A. (2018) 115:11832–7. doi: 10.1073/pnas.1811013115
86. Zheng XL. ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu Rev Med (2015) 66:211–25. doi: 10.1146/annurev-med-061813-013241
87. Ferrari S, Mudde GC, Rieger M, Veyradier A, Kremer Hovinga JA, Scheiflinger F. IgG subclass distribution of anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost (2009) 7:1703–10. doi: 10.1111/j.1538-7836.2009.03568.x
88. Sinkovits G, Szilágyi Á, Farkas P, Inotai D, Szilvási A, Tordai A, et al. Concentration and subclass distribution of anti-ADAMTS13 igG autoantibodies in different stages of acquired idiopathic thrombotic thrombocytopenic purpura. Front Immunol (2018) 9:1646. doi: 10.3389/fimmu.2018.01646
89. Masaki Y, Umehara H. [IgG4-related disease-the diagnostic confusion and how to avoid it]. Nihon Rinsho Meneki Gakkai Kaishi (2009) 32:478–83. doi: 10.2177/jsci.32.478
90. Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al. International consensus guidance statement on the management and treatment of igG4-related disease. Arthritis Rheumatol (2015) 67:1688–99. doi: 10.1002/art.39132
91. Maillette de Buy Wenniger LJ, Doorenspleet ME, Klarenbeek PL, Verheij J, Baas F, Elferink RPO, et al. Immunoglobulin G4+ clones identified by next-generation sequencing dominate the B cell receptor repertoire in immunoglobulin G4 associated cholangitis. Hepatology (2013) 57:2390–8. doi: 10.1002/hep.26232
92. Mattoo H, Mahajan VS, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol (2014) 134:679–87. doi: 10.1016/j.jaci.2014.03.034
93. Li J, Liu Z, Zhang P, Lin W, Lu H, Peng Y, et al. Peripheral B-cell immunophenotyping identifies heterogeneity in igG4-related disease. Front Immunol (2021) 12:747076. doi: 10.3389/fimmu.2021.747076
94. Mattoo H, Mahajan VS, Maehara T, Deshpande V, Della-Torre E, Wallace ZS, et al. Clonal expansion of CD4(+) cytotoxic T lymphocytes in patients with IgG4-related disease. J Allergy Clin Immunol (2016) 138:825–38. doi: 10.1016/j.jaci.2015.12.1330
95. Maehara T, Mattoo H, Ohta M, Mahajan VS, Moriyama M, Yamauchi M, et al. Lesional CD4+ IFN-γ+ cytotoxic T lymphocytes in IgG4-related dacryoadenitis and sialoadenitis. Ann Rheum Dis (2017) 76:377–85. doi: 10.1136/annrheumdis-2016-209139
96. Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis (2015) 74:1171–7. doi: 10.1136/annrheumdis-2014-206605
97. Lanzillotta M, Della-Torre E, Wallace ZS, Stone JH, Karadag O, Fernández-Codina A, et al. Efficacy and safety of rituximab for IgG4-related pancreato-biliary disease: A systematic review and meta-analysis. Pancreatology (2021) 21:1395–401. doi: 10.1016/j.pan.2021.06.009
98. Terao C, Ota M, Iwasaki T, Shiokawa M, Kawaguchi S, Kuriyama K, et al. IgG4-related disease in the Japanese population: a genome-wide association study. Lancet Rheumatol (2019) 1:e14–22. doi: 10.1016/S2665-9913(19)30006-2
99. Freitag TL, Cham C, Sung H-H, Beilhack GF, Durinovic-Belló I, Patel SD, et al. Human risk allele HLA-DRB1*0405 predisposes class II transgenic Ab0 NOD mice to autoimmune pancreatitis. Gastroenterology (2010) 139:281–91. doi: 10.1053/j.gastro.2010.03.038
100. Thomson G, Valdes AM, Noble JA, Kockum I, Grote MN, Najman J, et al. Relative predispositional effects of HLA class II DRB1-DQB1 haplotypes and genotypes on type 1 diabetes: a meta-analysis. Tissue Antigens (2007) 70:110–27. doi: 10.1111/j.1399-0039.2007.00867.x
101. Arimura Y, Isshiki H, Onodera K, Nagaishi K, Yamashita K, Sonoda T, et al. Characteristics of Japanese inflammatory bowel disease susceptibility loci. J Gastroenterol (2014) 49:1217–30. doi: 10.1007/s00535-013-0866-2
102. Overdijk MB, Verploegen S, Ortiz Buijsse A, Vink T, Leusen JHW, Bleeker WK, et al. Crosstalk between human IgG isotypes and murine effector cells. J Immunol (2012) 189:3430–8. doi: 10.4049/jimmunol.1200356
103. Smyk DS, Rigopoulou EI, Koutsoumpas AL, Kriese S, Burroughs AK, Bogdanos DP. Autoantibodies in autoimmune pancreatitis. Int J Rheumatol (2012) 2012:940831. doi: 10.1155/2012/940831
104. Nishimori I, Yamamoto Y, Okazaki K, Morita M, Onodera M, Kino J, et al. Identification of autoantibodies to a pancreatic antigen in patients with idiopathic chronic pancreatitis and Sjögren’s syndrome. Pancreas (1994) 9:374–81. doi: 10.1097/00006676-199405000-00015
105. Kountouras J, Zavos C, Chatzopoulos D. A concept on the role of Helicobacter pylori infection in autoimmune pancreatitis. J Cell Mol Med (2005) 9:196–207. doi: 10.1111/j.1582-4934.2005.tb00349.x
106. Guarneri F, Guarneri C, Benvenga S. Helicobacter pylori and autoimmune pancreatitis: role of carbonic anhydrase via molecular mimicry? J Cell Mol Med (2005) 9:741–4. doi: 10.1111/j.1582-4934.2005.tb00506.x
107. Kountouras J, Zavos C, Gavalas E, Tzilves D. Challenge in the pathogenesis of autoimmune pancreatitis: potential role of helicobacter pylori infection via molecular mimicry. Gastroenterology (2007) 133:368–9. doi: 10.1053/j.gastro.2007.05.044
108. Frulloni L, Lunardi C, Simone R, Dolcino M, Scattolini C, Falconi M, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med (2009) 361:2135–42. doi: 10.1056/NEJMoa0903068
109. Culver EL, Smit WL, Evans C, Sadler R, Cargill T, Makuch M, et al. No evidence to support a role for Helicobacter pylori infection and plasminogen binding protein in autoimmune pancreatitis and IgG4-related disease in a UK cohort. Pancreatology (2017) 17:395–402. doi: 10.1016/j.pan.2017.04.002
110. Jesnowski R, Isaksson B, Möhrcke C, Bertsch C, Bulajic M, Schneider-Brachert W, et al. Helicobacter pylori in autoimmune pancreatitis and pancreatic carcinoma. Pancreatology (2010) 10:462–6. doi: 10.1159/000264677
111. Asada M, Nishio A, Uchida K, Kido M, Ueno S, Uza N, et al. Identification of a novel autoantibody against pancreatic secretory trypsin inhibitor in patients with autoimmune pancreatitis. Pancreas (2006) 33:20–6. doi: 10.1097/01.mpa.0000226881.48204.fd
112. Löhr J-M, Faissner R, Koczan D, Bewerunge P, Bassi C, Brors B, et al. Autoantibodies against the exocrine pancreas in autoimmune pancreatitis: gene and protein expression profiling and immunoassays identify pancreatic enzymes as a major target of the inflammatory process. Am J Gastroenterol (2010) 105:2060–71. doi: 10.1038/ajg.2010.141
113. Hubers LM, Vos H, Schuurman AR, Erken R, Oude Elferink RP, Burgering B, et al. Annexin A11 is targeted by IgG4 and IgG1 autoantibodies in IgG4-related disease. Gut (2018) 67:728–35. doi: 10.1136/gutjnl-2017-314548
114. Zamboni G, Lüttges J, Capelli P, Frulloni L, Cavallini G, Pederzoli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch (2004) 445:552–63. doi: 10.1007/s00428-004-1140-z
115. Hohenester S, Wenniger LM de B, Paulusma CC, van Vliet SJ, Jefferson DM, Elferink RPO, et al. A biliary HCO3- umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology (2012) 55:173–83. doi: 10.1002/hep.24691
116. Herta T, Kersten R, Chang J-C, Hubers L, Go S, Tolenaars D, et al. Role of the IgG4-related cholangitis autoantigen annexin A11 in cholangiocyte protection. J Hepatol (2022) 76:319–31. doi: 10.1016/j.jhep.2021.10.009
117. Shiokawa M, Kodama Y, Kuriyama K, Yoshimura K, Tomono T, Morita T, et al. Pathogenicity of IgG in patients with IgG4-related disease. Gut (2016) 65:1322–32. doi: 10.1136/gutjnl-2015-310336
118. Liu H, Perugino CA, Ghebremichael M, Wallace ZS, Montesi SB, Stone JH, et al. Disease severity linked to increase in autoantibody diversity in igG4-related disease. Arthritis Rheumatol (2020) 72:687–93. doi: 10.1002/art.41140
119. Shiokawa M, Kodama Y, Sekiguchi K, Kuwada T, Tomono T, Kuriyama K, et al. Laminin 511 is a target antigen in autoimmune pancreatitis. Sci Transl Med (2018) 10(453):eaaq0997. doi: 10.1126/scitranslmed.aaq0997
120. Hirani N, MacKinnon AC, Nicol L, Ford P, Schambye H, Pedersen A, et al. Target inhibition of galectin-3 by inhaled TD139 in patients with idiopathic pulmonary fibrosis. Eur Respir J (2021) 57(5):2002559. doi: 10.1183/13993003.02559-2020
121. Perugino CA, AlSalem SB, Mattoo H, Della-Torre E, Mahajan V, Ganesh G, et al. Identification of galectin-3 as an autoantigen in patients with IgG4-related disease. J Allergy Clin Immunol (2019) 143:736–745.e6. doi: 10.1016/j.jaci.2018.05.011
122. Faludi R, Nagy G, Tőkés-Füzesi M, Kovács K, Czirják L, Komócsi A. Galectin-3 is an independent predictor of survival in systemic sclerosis. Int J Cardiol (2017) 233:118–24. doi: 10.1016/j.ijcard.2016.12.140
123. Salah A, Yoshifuji H, Ito S, Kitagori K, Kiso K, Yamada N, et al. High expression of galectin-3 in patients with igG4-related disease: A proteomic approach. Patholog Res Int (2017) 2017:9312142. doi: 10.1155/2017/9312142
124. Li L, Li J, Gao J. Functions of galectin-3 and its role in fibrotic diseases. J Pharmacol Exp Ther (2014) 351:336–43. doi: 10.1124/jpet.114.218370
125. Liu H, Fan H, He P, Zhuang H, Liu X, Chen M, et al. Prohibitin 1 regulates mtDNA release and downstream inflammatory responses. EMBO J (2022) 41:e111173. doi: 10.15252/embj.2022111173
126. Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy (2020) 16:3–17. doi: 10.1080/15548627.2019.1603547
127. Du H, Shi L, Chen P, Yang W, Xun Y, Yang C, et al. Prohibitin is involved in patients with igG4 related disease. PloS One (2015) 10:e0125331. doi: 10.1371/journal.pone.0125331
128. Culver EL, Sadler R, Bateman AC, Makuch M, Cargill T, Ferry B, et al. Increases in igE, eosinophils, and mast cells can be used in diagnosis and to predict relapse of igG4-related disease. Clin Gastroenterol Hepatol (2017) 15:1444–1452.e6. doi: 10.1016/j.cgh.2017.02.007
129. Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy (2014) 69:269–72. doi: 10.1111/all.12320
130. Gao Y, Zheng M, Cui L, Chen N, Wang Y-N, Zhan Y-T, et al. IgG4-related disease: association between chronic rhino-sinusitis and systemic symptoms. Eur Arch Otorhinolaryngol (2018) 275:2013–9. doi: 10.1007/s00405-018-5013-5
131. Zhang X, Zhang P, Li J, He Y, Fei Y, Peng L, et al. Different clinical patterns of IgG4-RD patients with and without eosinophilia. Sci Rep (2019) 9:16483. doi: 10.1038/s41598-019-52847-6
132. Saeki T, Kobayashi D, Ito T, Tamura M, Yoshikawa S, Yamazaki H. Comparison of clinical and laboratory features of patients with and without allergic conditions in IgG4-related disease: A single-center experience in Japan. Mod Rheumatol (2018) 28:845–8. doi: 10.1080/14397595.2017.1416891
133. Sanders S, Fu X, Zhang Y, Perugino CA, Wallwork R, Della-Torre E, et al. Lifetime allergy symptoms in igG4-related disease: A case-control study. Arthritis Care Res (Hoboken) (2022) 74:1188–95. doi: 10.1002/acr.24545
134. D’Astous-Gauthier K, Ebbo M, Chanez P, Schleinitz N. Implication of allergy and atopy in IgG4-related disease. World Allergy Organ J (2023) 16:100765. doi: 10.1016/j.waojou.2023.100765
135. Della-Torre E, Germanò T, Ramirez GA, Dagna L, Yacoub MR. IgG4-related disease and allergen-specific immunotherapy. Ann Allergy Asthma Immunol (2020) 124:631–3. doi: 10.1016/j.anai.2020.03.024
136. Mattoo H, Della-Torre E, Mahajan VS, Stone JH, Pillai S. Circulating Th2 memory cells in IgG4-related disease are restricted to a defined subset of subjects with atopy. Allergy (2014) 69:399–402. doi: 10.1111/all.12342
137. Michailidou D, Schwartz DM, Mustelin T, Hughes GC. Allergic aspects of igG4-related disease: implications for pathogenesis and therapy. Front Immunol (2021) 12:693192. doi: 10.3389/fimmu.2021.693192
138. Culver EL, Vermeulen E, Makuch M, van Leeuwen A, Sadler R, Cargill T, et al. Increased IgG4 responses to multiple food and animal antigens indicate a polyclonal expansion and differentiation of pre-existing B cells in IgG4-related disease. Ann Rheum Dis (2015) 74:944–7. doi: 10.1136/annrheumdis-2014-206405
139. de Buy Wenniger LJM, Culver EL, Beuers U. Exposure to occupational antigens might predispose to IgG4-related disease. Hepatology (2014) 60:1453–4. doi: 10.1002/hep.26999
140. Hubers LM, Schuurman AR, Buijs J, Mostafavi N, Bruno MJ, Vermeulen RCH, et al. Blue-collar work is a risk factor for developing IgG4-related disease of the biliary tract and pancreas. JHEP Rep (2021) 3:100385. doi: 10.1016/j.jhepr.2021.100385
141. Cargill T, Makuch M, Sadler R, Lighaam LC, Peters R, van Ham M, et al. Activated T-follicular helper 2 cells are associated with disease activity in igG4-related sclerosing cholangitis and pancreatitis. Clin Transl Gastroenterol (2019) 10:e00020. doi: 10.14309/ctg.0000000000000020
142. Bélanger S, Crotty S. Dances with cytokines, featuring TFH cells, IL-21, IL-4 and B cells. Nat Immunol (2016) 17:1135–6. doi: 10.1038/ni.3561
143. Weinstein JS, Herman EI, Lainez B, Licona-Limón P, Esplugues E, Flavell R, et al. TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol (2016) 17:1197–205. doi: 10.1038/ni.3554
144. Lighaam LC, Rispens T. The immunobiology of immunoglobulin G4. Semin Liver Dis (2016) 36:200–15. doi: 10.1055/s-0036-1584322
145. Akiyama M, Suzuki K, Yasuoka H, Kaneko Y, Yamaoka K, Takeuchi T. Follicular helper T cells in the pathogenesis of IgG4-related disease. Rheumatol (Oxford) (2018) 57:236–45. doi: 10.1093/rheumatology/kex171
146. Witebsky E, Rose NR, Terplan K, Paine JR, Egan RW. Chronic thyroiditis and autoimmunization. J Am Med Assoc (1957) 164:1439–47. doi: 10.1001/jama.1957.02980130015004
147. Douglas J N. Antibodies to an intracellular antigen penetrate neuronal cells and cause deleterious effects. J Clin Cell Immunol (2013) 04:157ra142. doi: 10.4172/2155-9899.1000134
148. Hansen JE, Chan G, Liu Y, Hegan DC, Dalal S, Dray E, et al. Targeting cancer with a lupus autoantibody. Sci Transl Med (2012) 4(157):157ra142. doi: 10.1126/scitranslmed.3004385
149. Noble PW, Young MR, Bernatsky S, Weisbart RH, Hansen JE. A nucleolytic lupus autoantibody is toxic to BRCA2-deficient cancer cells. Sci Rep (2014) 4:5958. doi: 10.1038/srep05958
150. Song Y-C, Sun G-H, Lee T-P, Huang JC, Yu C-L, Chen C-H, et al. Arginines in the CDR of anti-dsDNA autoantibodies facilitate cell internalization via electrostatic interactions. Eur J Immunol (2008) 38:3178–90. doi: 10.1002/eji.200838678
151. Lisi S, Sisto M, Soleti R, Saponaro C, Scagliusi P, D’Amore M, et al. Fcgamma receptors mediate internalization of anti-Ro and anti-La autoantibodies from Sjögren’s syndrome and apoptosis in human salivary gland cell line A-253. J Oral Pathol Med (2007) 36:511–23. doi: 10.1111/j.1600-0714.2007.00563.x
152. Choi D-K, Bae J, Shin S-M, Shin J-Y, Kim S, Kim Y-S. A general strategy for generating intact, full-length IgG antibodies that penetrate into the cytosol of living cells. MAbs (2014) 6:1402–14. doi: 10.4161/mabs.36389
153. Jang JY, Jeong JG, Jun HR, Lee SC, Kim JS, Kim YS, et al. A nucleic acid-hydrolyzing antibody penetrates into cells via caveolae-mediated endocytosis, localizes in the cytosol and exhibits cytotoxicity. Cell Mol Life Sci (2009) 66:1985–97. doi: 10.1007/s00018-009-9179-2
154. Hansen JE, Tse C-M, Chan G, Heinze ER, Nishimura RN, Weisbart RH. Intranuclear protein transduction through a nucleoside salvage pathway. J Biol Chem (2007) 282:20790–3. doi: 10.1074/jbc.C700090200
155. Endmayr V, Tunc C, Ergin L, De Rosa A, Weng R, Wagner L, et al. Anti-neuronal igG4 autoimmune diseases and igG4-related diseases may not be part of the same spectrum: A comparative study. Front Immunol (2021) 12:785247. doi: 10.3389/fimmu.2021.785247
156. Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Zhang L, et al. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol (2006) 4:1010–6. doi: 10.1016/j.cgh.2006.05.017
157. Lu C, Li S, Qing P, Zhang Q, Ji X, Tang Z, et al. Single-cell transcriptome analysis and protein profiling reveal broad immune system activation in IgG4-related disease. JCI Insight (2023) 8(17):e167602. doi: 10.1172/jci.insight.167602
158. Ota M, Katsuyama Y, Hamano H, Umemura T, Kimura A, Yoshizawa K, et al. Two critical genes (HLA-DRB1 and ABCF1)in the HLA region are associated with the susceptibility to autoimmune pancreatitis. Immunogenetics (2007) 59:45–52. doi: 10.1007/s00251-006-0178-2
159. Goni E, Regel I, Mahajan UM, Amodio A, De Marchi G, Beyer G, et al. HLA-DRB1∗16 and -DQB1∗05 alleles are strongly associated with autoimmune pancreatitis in a cohort of hundred patients. Pancreatology (2022) 22:466–71. doi: 10.1016/j.pan.2022.03.015
Keywords: IgG4, IgG4-RD, autoantibody, antigen, autoimmunity
Citation: Motta RV and Culver EL (2024) IgG4 autoantibodies and autoantigens in the context of IgG4-autoimmune disease and IgG4-related disease. Front. Immunol. 15:1272084. doi: 10.3389/fimmu.2024.1272084
Received: 03 August 2023; Accepted: 25 January 2024;
Published: 16 February 2024.
Edited by:
Hani S. Mousa, University of Cambridge, United KingdomReviewed by:
Maartje G. Huijbers, Leiden University Medical Center (LUMC), NetherlandsMarco Lanzillotta, Vita-Salute San Raffaele University, Italy
Inga Koneczny, Medical University of Vienna, Austria
Copyright © 2024 Motta and Culver. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emma L. Culver, ZW1tYS5jdWx2ZXJAbmhzLm5ldA==