 Jingyu Xu
Jingyu Xu Ling Yu
Ling Yu Surong Ye
Surong Ye Zitong Ye
Zitong Ye Luyi Yang
Luyi Yang Xiaoxi Xu
Xiaoxi Xu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 February 2024
Sec. Microbial Immunology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1254516
This article is part of the Research Topic Overcoming Challenges in Microbial Immunology: 2022 View all 10 articles
There exists a bidirectional relationship between oral health and general well-being, with an imbalance in oral symbiotic flora posing a threat to overall human health. Disruptions in the commensal flora can lead to oral diseases, while systemic illnesses can also impact the oral cavity, resulting in the development of oral diseases and disorders. Porphyromonas gingivalis and Fusobacterium nucleatum, known as pathogenic bacteria associated with periodontitis, play a crucial role in linking periodontitis to accompanying systemic diseases. In periodontal tissues, these bacteria, along with their virulence factors, can excessively activate the host immune system through local diffusion, lymphatic circulation, and blood transmission. This immune response disruption contributes to an imbalance in osteoimmune mechanisms, alveolar bone resorption, and potential systemic inflammation. To restore local homeostasis, a deeper understanding of microbiota–host interactions and the immune network phenotype in local tissues is imperative. Defining the immune network phenotype in periodontal tissues offers a promising avenue for investigating the complex characteristics of oral plaque biofilms and exploring the potential relationship between periodontitis and associated systemic diseases. This review aims to provide an overview of the mechanisms underlying Porphyromonas gingivalis- and Fusobacterium nucleatum-induced alveolar bone resorption, as well as the immunophenotypes observed in host periodontal tissues during pathological conditions.
Oral health is an indispensable element of general health and well-being ensuring the fulfillment of basic daily human functions. However, according to the 2015 Global Burden of Disease (GBD) study, about 3.5 billion people worldwide suffer from oral conditions (1). The pronounced global prevalence and severity of oral diseases have sparked significant concern among the public. These progressive chronic clinical diseases affect the teeth and various tissues within the oral cavity. Dental caries, periodontal diseases, oral mucosal diseases, and oral cancer are the main types of oral diseases, exhibiting high prevalence and severe adverse prognosis for individuals, communities, and society (2).
Beyond their prevalence and public concern, oral diseases are believed to have bidirectional associations with systemic health (3–7). Simultaneous or sequential occurrences of oral diseases and systemic diseases (8–19), such as gastrointestinal, immune, cardiovascular, and nervous system diseases, have been reported. Moreover, the tight relationship between human microbial communities and human health has drawn significant interest from researchers, with the oral microbiome considered to play a vital role in oral diseases and the connection between oral and general well-being.
Oral pathogens colonize the surfaces of different habitats within the oral cavity and form functional groups with pathogenic roles. Typical representatives of these groups include Porphyromonas gingivalis and Fusobacterium nucleatum. These two periodontal pathogens can disrupt bone homeostasis by excessively activating host immune responses. The resulting microbial–host interaction-induced local inflammation may spread throughout the body, leading to systemic diseases. In this review, we elucidate the mechanisms behind Porphyromonas gingivalis-and Fusobacterium nucleatum-induced bone resorption, construct the immune defense phenotypes of the human body against the invasion of oral pathogenic microorganisms, and further explore the interaction between oral microbial communities and the host.
The oral cavity is an open system where microbes are ingested with every breath, meal, and drink, colonizing through close contact with other humans, animals, or the physical environment. It provides a habitat for microbes, with suitable temperature, humidity, and nutrition. Despite there being millions of microbial species on Earth, only approximately 760 have been identified as major oral residents (20). In typical oral ecology, there are only 296 species-level microbial taxa (21), which are collectively referred to as the human oral microbiota (22). Alongside planktonic forms, the oral microbiota tends to assemble into complex spatial structures and form symbiotic communities to adapt to environmental changes and maintain microbial community and host homeostasis.
Microbial dysbiosis is generally considered a state that mediates the associations between microbiota patterns and disease states (23). As the oral cavity is an open ecosystem, oral microbial homeostasis is often challenged by many factors, such as genetics, gender, habitat, age, diet, living habits, and environment. Long-term nongenetic factors may cause genetic variation, resulting in dramatic changes in the structure of the bacterial flora (24).
The bidirectional association between oral microbial dysbiosis and general disease states might occur three distinct manners (25). Oral bacteria and their products can be transferred into the circulatory system via open or closed foci, such as inflammatory and ruptured epithelium or infection around the root apices. This transfer can cause transient bacteremia, resulting in systemic inflammation and metabolic and functional disorders (6, 26). Bacterial products, such as gingipains secreted by the typical periodontal pathogen P. gingivalis, have the potential to promote such pathological processes by degrading tight junction proteins, not only in periodontal tissues but also in vascular endothelial cells (27).
Oral pathogens can also be disseminated through non-hematogenous processes. Routes such as oro-pharyngeal or oro-digestive pathways may lead to ectopic colonization in the gut, disrupting the local microbial composition, triggering inflammation, compromising the intestinal mucosal barrier function, and inducing systemic diseases (28–31). An imbalance in gut homeostasis can promote the colonization of oral bacteria in the intestines (32–34). In addition, immune cells and factors responsive to oral pathobionts in the gut or other parts of the body can migrate to the oral cavity, exacerbating oral inflammatory conditions like periodontitis (35, 36). These processes illustrate the probability of an oral-systemic axis that regulates human health and disease conditions.
Microbes in the oral cavity are not uniformly distributed. Only a few dozen species are abundant and constitute the core of the oral microbial community, whereas others are less abundant (37, 38). Heterogeneous colonization of oral microorganisms can be attributed to the uniqueness of oral niches, including the saliva, tongue, oral mucosa, mineralized tooth surfaces, and periodontal tissues (22). The spatial organization of oral microbes is in a state of dynamic equilibrium, maintained by opposing forces such as salivary flow, microbial adhesion, shedding and colonization, and crucially, microbe–microbe and microbe–host interactions (25, 39, 40). The microbiome colonizing the surface of mineralized teeth exists in the form of biofilms. Depending on their composition, nutritional background, ecological site anatomy, and antigen and immune exposure, plaques can be classified as subepithelial or subgingival (41, 42). Microorganisms within plaque biofilms rarely live independently; instead, they interact to form different functional groups and cooperate as ecological guilds to perform higher physiological functions (43). This applies to periodontal pathogens in subgingival plaques, where dominant species in the periodontal ecological guilds can determine the overall function of the group or play a crucial auxiliary role.
The role of periodontal pathogens in amplifying systemic inflammation and organ dysfunction has recently been established in several systemic diseases (44–46), such as inflammatory bowel disease (IBD), stroke, chronic renal diseases, cardiovascular diseases (47–49), diabetes (47), pneumonia, meningitis, rheumatoid arthritis (47, 50), cognitive disorders (51), as well as poor pregnancy outcomes (52, 53) and cancer (54). Porphyromonas gingivalis, a keystone periodontal pathogen, can ferment amino acids and grow deep in the glucose-poor periodontal pocket. P. gingivalis also invades gingival tissues and epithelial cells, promoting cell proliferation and causing epithelial radicular proliferation, which is a typical manifestation of periodontitis. The interaction between P. gingivalis and local host immune responses can have two contrasting outcomes, speculated to be related to the concentration of P. gingivalis and its virulence factors, mainly lipopolysaccharide (LPS), fimbriae, and gingipains. The concentration of virulence factors is high in the superficial layer, leading to immune escape, and low in the deep layer, resulting in a pro-inflammatory response that increases nutrient (heme) requirements (55). P. gingivalis employs unique and complex pathogenic mechanisms. These include strong invasive properties to allow it to enter the circulatory system, induce cell apoptosis, initiate oxidative stress, influence the host innate immune response by inducing dysfunction in neutrophils and macrophages, and facilitate the expression of acute phase proteins and numerous pro-inflammatory cytokines (52). Furthermore, P. gingivalis has the ability to regulate the innate immune response, ensuring the growth, colonization, and invasion of other opportunist and symbiont bacteria such as F. nucleatum, Firmicutes, C. rectus, Streptococci, Staphylococci, Enterobacteriaceae, Prevotella, Hemophilus parainfluenza, and Dialister (56–58). The dysbiotic microbiome induced by P. gingivalis is inherently resilient and can be stably transferred and easily restored even after antibiotic therapy is discontinued (59), making the local and systemic disease conditions triggered by P. gingivalis difficult to cure.
The obligate anaerobes Fusobacterium nucleatum, another core member of dental plaque, is believed to play a significant role in plaque maturation and dental plaque diversity (60). Its ability to co-cluster with various taxa serves as a physical bridge between early and late colonization of dental plaque organisms (60). Other hypotheses suggest that F. nucleatum acts as an indicator of establishing an anaerobic microenvironment and promoting plaque maturation (61–63), and has long been considered an initiating factor in periodontal disease. F. nucleatum tend to synergistically aggravate periodontitis and other systemic diseases when combined with P. gingivalis (52). However, despite being recognized as a periodontal pathogen, recent studies on F. nucleatum mostly discuss its role in tumorigenesis and immune evasion, with relatively few studies linking it to periodontal bone destruction.
The involvement of P. gingivalis and F. nucleatum in bone remodeling has always been a concern because of periodontitis. Periodontitis is a chronic inflammatory disease of the mouth that primarily develops from gingivitis. The accumulation of subgingival biofilm drives the progression from gingivitis to periodontitis, leading to the loss of periodontal supporting tissues. This progression occurs through continuous and complex interactions between the subgingival biofilm and the host’s immune response (21, 64, 65). Different clinical phenotypes of periodontitis have been associated with oral flora exhibiting different characteristics (66). While commensal gut microbes also have the capacity to regulate osteoimmune processes in the alveolar bone (67), P. gingivalis and F. nucleatum, which are oriented toward the commensal oral microbiota, have been shown to independently contribute to alveolar bone remodeling, separate from the systemic microbiome (39).
Pathogenic bacteria flourish in the gingival sulcus owing to their immune resistance, and their secretion of virulence factors or parasitic behavior can stimulate the immune response in the gingival tissues. This immune response effectively transmits virulence signals to the bone marrow cavity, leading to enhanced bone marrow hematopoiesis (39), which is an important pathway for immune cell generation. Under the dual stimulation of dysregulated bacterial flora and an excessive immune response, the homeostasis of alveolar bone tissue is unbalanced. To further demonstrate the roles of P. gingivalis and F. nucleatum in bone resorption, it is necessary to briefly review the mechanisms of osteoimmunology and the key regulatory axis of bone homeostasis, the receptor activator of nuclear factor-kappa B ligand (RANKL)–receptor activator of nuclear factor-kappa B (RANK)–osteoprotegerin (OPG) axis.
The term ‘osteoimmunology,’ coined by Arron and Choi in 2000 (68), refers to the field that investigates the interactions between immune cells and bone cells. These interactions mediate skeletal development, modification, and homeostasis under both physiological and pathophysiological conditions. Both innate and adaptive immune cells participate in bone turnover through direct contact or expressing a range of immune molecules, such as cytokines, chemokines, and immunoglobulins.
Recently, a research group provided a cellular atlas of specific oral mucosal positions in health and disease conditions, revealing a distinct stromal–immune responsive axis that dysregulates under inflammatory conditions. This axis may be capable of mediating periodontal osseous tissues homeostasis (69). The major cell types within healthy gingival tissues include epithelial cells, endothelial cells, fibroblasts, and immune cells. Within healthy gingival tissues, the immune category can be divided into five major clusters: T, NK, B/plasma, granulocyte, and myeloid cells, with T cells being the most numerous. T cells in gingival tissues can be subdivided into αβ CD4+T, TH17, mucosal-associated invariant T (MAIT), αβ CD8+T, γδ T, Treg, and NKT cells. The second largest population was myeloid linages, including neutrophils—which dominated this compartment—macrophages (Mφ), and myeloid dendritic cells (mDC). This result suggests that neutrophil-mediated innate immune responses are activated even when the periodontium is healthy. Sustained and highly coordinated neutrophil chemotaxis from the gingival vessels to the healthy gingival sulcus constitutes one of the major protective mechanisms against colonization by pathogenic microorganisms (65). Proper neutrophil monitoring targeting dental plaque biofilms has a dual benefit, conferring resistance to microbial colonization in periodontal tissues while maintaining an appropriate microbial composition for normal periodontal tissues function (70).
The epithelial and stromal cells present in the oral mucosa exhibit inflammation-related antimicrobial defense functions and can express transcriptional signatures of periodontitis inflammation and recruitment factors for neutrophils (69). This may be one of the reasons for the significantly elevated proportion of neutrophils in the oral mucosa. Stromal and immune cells can interact with each other through the expression of periodontitis susceptibility genes, becoming potential drivers of periodontal inflammation and immune cell over-recruitment, ultimately forming the basis of destructive hyperreactive immune responses (69).
Under healthy conditions, alveolar bone homeostasis is maintained by neutrophil-mediated innate immunity and T cell-mediated adaptive immunity. The cells and molecules involved stimulate bone remodeling cells, such as osteoblasts, osteoclasts, and their precursors, regulating their generation, development, function, and survival, ultimately maintaining bone homeostasis.
Bone homeostasis is maintained by the coordinated action of mesenchymal-lineage-derived bone-forming osteoblasts and myeloid-lineage-derived bone-resorbing osteoclasts (71). Osteoclasts resorb osseous tissues by secreting hydrogen ions and lytic enzymes, while osteoblasts support mineralization by secreting unmineralized bone matrix and non-collagenous proteins (72).
Osteoclasts originate from monocyte–macrophage precursor cells, which are originally differentiated from HSCs. Studies have demonstrated that M1 macrophages contribute to osteoclastogenesis (73–75) under pathogenesis, and immature dendritic cells can develop into osteoclasts mediated by RANKL–RANK signaling (76, 77). Macrophage colony-stimulating factor (M-CSF) activates its cognate receptor c-Fms, inducing the expression of RANK on pre-osteoclasts (78), and consequently, induces the expression of NFATc1, a transcription factor that results in osteoclast proliferation and differentiation (79–82). Dendritic cell-specific transmembrane protein (DC-STAMP) (83, 84) and osteoclast stimulatory transmembrane protein (OC-STAMP) (85) are crucial for osteoclast maturation in a RANKL-dependent manner. RANKL-induced expression of the integrin-β3 subunit guarantees the αVβ3-mediated cell adhesion, which can seal certain podosomes, providing a critical microenvironment for osteoclast physiological functions such as motility and bone degradation/resorption (40–42). The secretion of cathepsin K, tartrate-resistant acid phosphatase (TRAP), and proteolytic enzymes occurs via the NFATc1-mediated RANKL signaling pathway (40–42).
Osteoblasts are mesenchymal lineage-originated osteogenic cells that eventually become bone-lining cells or osteocytes. Osteoblast differentiation and function are regulated by the transcription factors osterix and activating transcription factor 4 (ATF4), with the support of WNT, bone morphogenetic protein (BMP), fibroblast growth factor (FGF), insulin-like growth factor (IGF) signaling,. A recent study revealed that RANKL contributes to the osteogenic direction of bone marrow mesenchymal stromal cell (MSC) differentiation (86), indicating that membrane-bound RANKL, as a member of TNF superfamily, possesses the capability to act as a receptor for vesicular RANK derived from mature osteoclasts (87) or apoptotic bodies (88), performing reverse signaling from osteoclasts to osteoblasts and contributing to osteogenesis, and consequently promote the coupling of bone resorption and formation (87). This specific function is regarded as bidirectional signaling transportation, which might be closely related to the intracellular proline-rich motif (87). Given that membrane-bound RANKL is an easy-clustering-featured molecular and the clustering of this type of receptor was proven to induce cell activation (89–93), the accumulation and clustering of RANKL seems to be the critical mechanism triggering RANKL reverse signaling (94). Vesicular RANK binding to RANKL activates osteoblasts and promotes osteogenesis through mammalian target of rapamycin complex 1 (mTORC1) signaling and Runt-related transcription factor 2 (Runx2) activation (87). However, OPG, as a competing receptor for RANKL, cannot stimulate osteoblast activation owing to its characteristic of disturbing RANKL clustering (94).
Thus, the RANKL–RANK–OPG axis produces essential signals that mediate intercellular communication in osteoclast–osteoblast coupling by regulating effector gene expression that drives cell proliferation, differentiation, maturation, function, and survival (Figure 1).
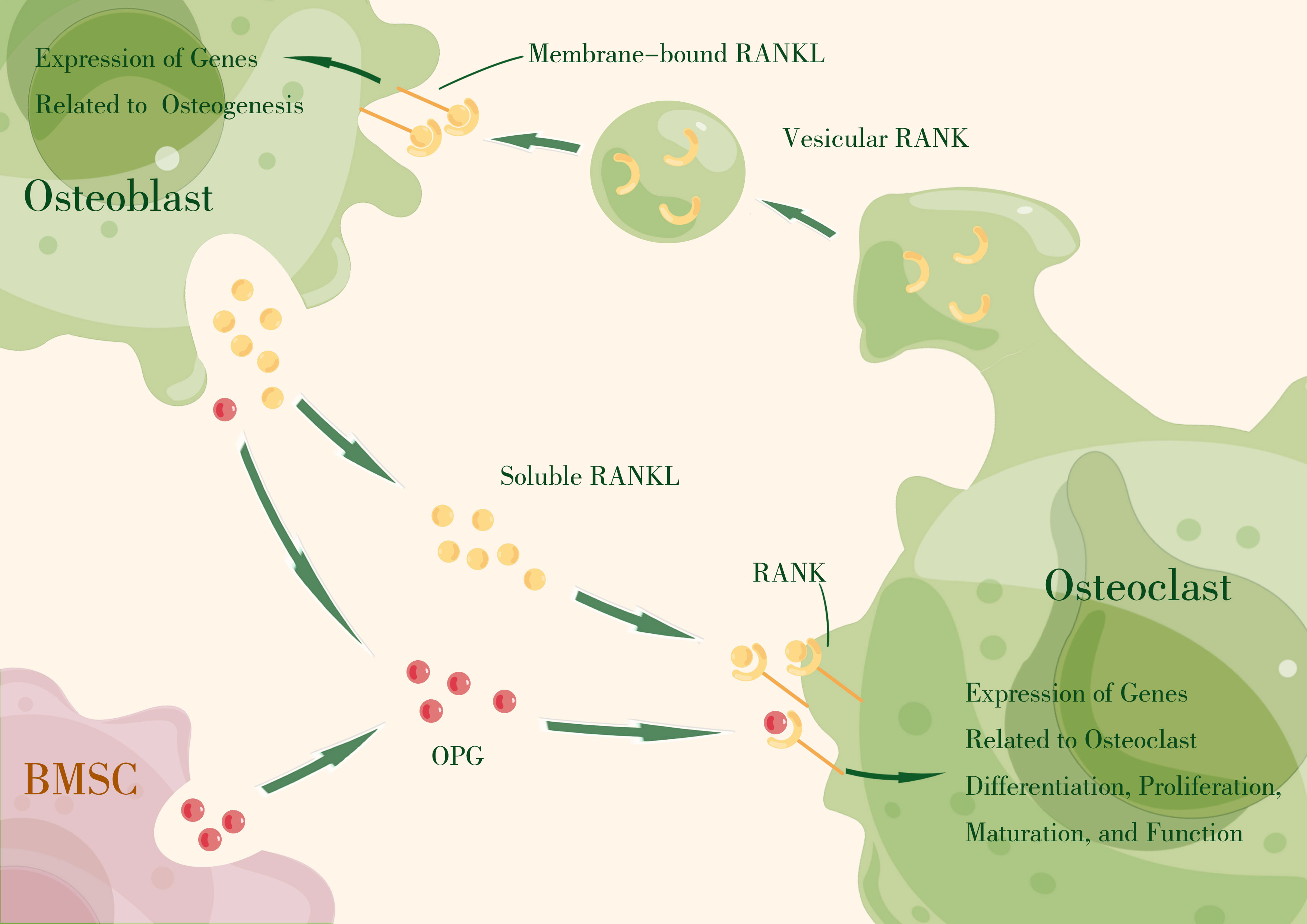
Figure 1 The RANKL–RANK axis produces essential signals that mediate intercellular communication in osteoclast–osteoblast coupling by regulating effector gene expression that drives cell proliferation, differentiation, maturation, function, and survival. OPG, primarily expressed by bone marrow stromal cells (BMSCs) and osteoblasts, acts as a decoy receptor, competitively binding RANKL to block RANKL–RANK interaction. (By Figdraw).
The receptor activator of nuclear factor-kappa B ligand (RANKL) and the receptor activator of nuclear factor-kappa B (RANK) were first discovered during the study of T-cell activation, and were found to be essential regulators of T cell and DC activation, thereby influencing T cell-mediated immune responses (95, 96). Subsequently, their critical role in osteoclast differentiation and bone remodeling was revealed (97, 98). RANKL, along with other biological mediators, regulates osteoclast differentiation, and under pathological conditions, it directly upregulates the expression of pro-osteoclastic cytokines and indirectly signals stromal-osteoblastic cells (99–101). Simultaneously, independent research groups identified RANKL as the osteoclast differentiation factor (ODF) from mouse myelo-monocytic cell lines and bone marrow-derived stromal cell lines (102, 103). Similarly, RANK was identified as the osteoclast differentiation factor receptor from mouse macrophage-like cell line (104, 105). Additionally, OPG was discovered to be an inhibitor of osteoclast differentiation (106, 107). These findings laid the foundation for understanding the regulatory effect of the RANKL–RANK–OPG axis in bone homeostasis.
RANKL, encoded by the tumor necrosis factor superfamily member 11 (TNFSF11) gene, is a type II homotrimeric membrane protein. It is produced by a variety of cell types, including osteoblasts, osteocytes, bone stromal cells, and immune cells within skeletal tissues. RANKL exists in three isoforms, with RANKL1 and RANKL2 being membrane-bound forms (108) that can be converted to soluble forms through proteolytic shedding (109, 110). RANKL3 lacks a transmembrane domain and is considered a soluble form (108). The membrane-bound form of RANKL can basically fulfill the function of this protein, but the soluble form contributes to physiological bone remodeling (111).
RANK, encoded by the tumor necrosis factor receptor superfamily member 11a (TNFRSF11A) gene, is a type I membrane receptor mainly expressed by hematopoietic cells, but also by osteoclasts and their precursors (78). It can also be detected on the surface of mesenchymal stem cells (86, 112). The intracellular domain of RANK contains a binding site for TNF receptor-associated factor (TRAFs) (113), which regulates the expression of genes associated with osteoclast function through the TRAF pathway (114).
OPG, encoded by the tumor necrosis factor receptor superfamily member 11b (TNFRSF11B) gene, is a member of the TNFR superfamily. It is primarily expressed by bone marrow stromal cells and osteoblasts, but can also be expressed in B cells, DCs, and follicular DCs. OPG exists only in its secreted molecular form and acts as a decoy receptor, competitively binding RANKL to block RANKL–RANK interaction (106). Local OPG is considered more crucial for skeletal and immune homeostasis compared to circulating OPG (115). In addition to RANK and OPG, LGR4 has been identified as a third competitive receptor that negatively regulates osteoclastogenesis through the GSK3-β signaling pathway by restraining NFATc1 expression (116). However, the binding affinity between RANKL and LGR4 is thought to be lower than that between RANKL and OPG, making OPG the main inhibitor of RANKL–RANK signaling (117).
The RANKL–RANK–OPG axis is a crucial signaling pathway for maintaining bone homeostasis through osteoblast-osteoclast coupling, with the concentration of soluble RANKL playing the key role. Disruptions in this pathway, caused by various stimulatory signals targeting RANKL secretion, can lead to an imbalance in bone homeostasis and contribute to pathogenic bacteria-induced bone resorption. In the following section, we will explore the virulence factors of major periodontal pathogens and their abilities to interfere with RANKL secretion through specific pathways.
P. gingivalis and F. nucleatum possess various virulence factors that contribute to their pathogenicity. These factors play a significant role in the development and progression of periodontal disease. In recent years, there has been increased research interest in the role of bacterial extracellular vesicles (BEVs) in the pathogenic mechanisms of these microorganisms. We will explore these separately.
P. gingivalis, an opportunistic pathogen and member of Socransky’s red complex, produces several virulence factors that induce detrimental effects on the host. The main virulence factors of P. gingivalis are LPS, fimbriae, and gingipains, which are crucial for the survival and metabolism of the bacterium.
LPS is an outer membrane component of gram-negative bacteria. It interacts with host cells, triggering a series of intracellular signaling events. LPS molecules consist of core polysaccharides, O-antigens, and lipid A; the latter two in P. gingivalis are highly diverse regions that confer antigenic differences and alter the interaction with pattern recognition receptors (PRRs), mainly TLR2, TLR4, and CD14. The disparity in LPS molecules depends on microenvironmental conditions (117) and sometimes leads to opposing immunological actions, immune evasion, or pro-inflammatory responses. This demonstrates that by manipulating the host immune activities, P. gingivalis can ensure its adaptation and survival (118, 119).
P. gingivalis LPS stimulates bone resorption in experimental models and activates various cell types, including mono-macrophages, endothelial cells, and epithelial cells, leading to the release of pro-inflammatory mediators and triggering immunoinflammatory reactions in the host tissues (120, 121). In vitro studies have also shown that P. gingivalis LPS increases the expression of pro-inflammatory cytokines in monocytes and macrophages, promoting bone resorption. In vivo, P. gingivalis LPS can activate mono-macrophages, endothelial cells, and epithelial cells through pathogen-associated molecular pattern (PAMP)-PRR recognition, resulting in the activation of cell signaling pathways like NF-kB and MAPK. These pathways ultimately stimulate the synthesis and release of IL-1, IL-6, TNF-α, NO, and other inflammatory mediators, contributing to a series of immunoinflammatory reactions in host tissues. In vitro, P. gingivalis LPS has also been proven to increase the expression of pro-inflammatory cytokines, such as IL-1, IL-6, IL-8, TNF-α, and IL-18, in monocytes and macrophages (122–125). These pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α, have been shown to stimulate bone remodeling cells and influence the RANKL–RANK–OPG axis, thereby promoting bone resorption.
Fimbriae are slender filamentous protrusions on the surface of P. gingivalis that that play a role in adherence and have pro-inflammatory capabilities (126–128). These fimbriae can stimulate signal generation through either TLR2 or TLR4, activating two distinct intercellular pathways. This activation leads to the production of pro-inflammatory factors and matrix metalloproteinases (MMPs), including TNF-α, IL-1, IL-6, IL-8, and MMP-9 (129, 130). Fimbriae also promote the expression of cell adhesins such as ICAM-1 (131). Moreover, fimbriae can interact with and activate the binding capacity of Complement Receptor 3 (CR3) through “inside-out” signaling (132, 133), facilitating the internalization of P. gingivalis by macrophages and reducing IL-12 production, which may inhibit bacterial clearance (133). Notably, fimbriae play a significant role in inducing bone destruction in experimental periodontitis models (134), and may be a target for immunotherapy aimed at reduce bone resorption (135, 136).
Gingipains, a series of cysteine proteinases generated by P. gingivalis, can be categorized into two types: arginine-specific (Arg-X) and lysine-specific (Lys-X) gingipains (137, 138). These gingipains can be present either on the cell surface or secreted in a soluble form. They are considered vital virulence factors of P. gingivalis but exhibit contradictory effects on innate immunity. On one hand, gingipains can activate protease-activated receptors (PARs) and act as pro-inflammatory stimulators and enhancers (139, 140) in neutrophils (141), gingival fibroblasts, gingival epithelial cells (142) and T-cells (143). They stimulate the production of IL-6 in oral epithelial cells (142) and IL-8 in gingival fibroblasts (144), and promote the recruitment of polymorphonuclear neutrophils (PMNs) through complement system activation (145, 146). On the other hand, gingipains can hinder the host immunity by cleaving several TCRs (147) and proteolytically inactivating factors such as IFN-γ, IL-4, IL-5, and IL-12 (148–151), even reducing bacterial opsonization (152) to cause increased resistance to bactericidal activity in P. gingivalis. Apart from manipulating host immunity, gingipains have also been shown to facilitate the adherence and invasion of fibroblasts and gingival epithelial cells (153–155), as well as increase vascular permeability and hemin availability in periodontal tissues, creating favorable conditions for P. gingivalis growth (156).
F. nucleatum, a member of the Socransky’s orange complex, is a symbiont, opportunistic pathogen, and oncobacterium (157–159). Several virulence factors of F. nucleatum have been characterized, including FadA (160–164), which regulates adhesion and invasiveness; the heat-shock protein GroEL, which triggers host inflammatory factors (161); the endotoxin LPS, which activates NLRP3 and induces the release of inflammatory cytokines such as IL-1β (165); the metabolite butyric acid, which promotes the production of reactive oxygen species (ROS) and induces apoptosis of histocytes and immune cells (166); and multiple outer membrane adhesins (167) that can mediate the adhesion and coaggregation with various oral microbiota species, including Streptococcus gordonii (168), Streptococcus sanguis (169), Streptococcus mutans (170, 171), Staphylococcus aureus (172), P. gingivalis (173–177), and Candida albicans (178, 179). These virulence factors contribute to the expression of certain virulence factors, promote the formation and stability of plaque biofilm, and mediate the adhesion to immune cells (167).
F. nucleatum possesses various adhesins, which can be categorized into two types: amino acid inhibitors (e.g., RadD, CmpA, Aid1, FomA) associated with coaggregation with gram-positive bacteria, and lactose inhibitors (e.g., Fap2) associated with gram-negative bacteria. Coaggregation between F. nucleatum and P. gingivalis is mediated not only by a variety of adhesins but also by the capsular polysaccharide (CPS) and LPS, resulting in increased expression of virulence factors and altered energy metabolism in both species (180).
FadA is the most representative virulence factor of F. nucleatum, playing a crucial role in the adhesion and invasion of host cells. FadA exists in two forms: secretory and non-secretory. These two forms work together to regulate the adhesion and invasion of F. nucleatum. Through the interaction of the secretory autonomous transporter RadD and membrane occupation and recognition nexus protein 2 (MORN2) (181), F. nucleatum can invade gingival epithelial cells by binding to epithelial cadherin (E-cadherin). FadA can also help interact with the intracellular receptor retinoic acid-inducible gene I (RIG-I), activating the NF-κB signaling pathway to induce inflammatory responses and cause periodontal tissues destruction. Furthermore, F. nucleatum can promote epithelial–mesenchymal transition of gingival epithelial cells, up-regulating Snail-1 expression, down-regulating E-cadherin expression, and disrupting the integrity of the gingival epithelium. This promotes the invasion of pathogenic bacteria into deeper periodontal tissues (182). Recent research has discovered that F. nucleatum can secrete FadA-containing outer membrane vesicles (OMVs) which stimulate inflammatory bone loss in RA via the FadA–Rab5a–YB-1 axis in macrophages (183), and may have similar effects in periodontitis.
BEVs are spherical nanostructures encapsulated in bacterial lipid bilayers. They range in size from 20 to 300 nm and contain various functional active substances secreted by bacteria, including bacterial virulence factors and sRNA (184). Since the first discovery of extracellular vesicles in Vibrio cholerae in 1967 (185), BEVs have been considered an important mode of physiological and pathological functions in bacteria. They facilitate bidirectional communication between bacteria–bacteria and bacteria–cells, in addition to direct contact (186), and play crucial roles in bacterial colonization, survival, inflammation, pathogenesis, and regulation of host metabolism and immunity (187–194). At present, the field of cancer-related research believes that BEVs in the tumor microenvironment can be used as a new target for the diagnosis and monitoring of tumors and related diseases (195). Although research on BEVs in the oral pathological microenvironment is limited, these vesicles have the potential to provide valuable insights into the pathogenesis and pathological state of oral diseases, as well as the development of more efficient treatment methods.
The interaction between the oral microbiome (including living bacteria, virulence factors, and BEVs) and human immunity, known as the oral host–microbial interactome, promotes homeostasis under healthy conditions. The commensal microbiota educates and facilitates the immune system (196), imprinting innate and adaptive immunity memory to mount rapid and effective resistance against massive PAMP invasion. However, this immune memory can lead to overreactions and become a major cause of tissue destruction, including periodontal bone loss (6).
Studies have shown that dental biofilm plaque-induced bone loss in the periodontal tissues has an ‘effective radius of action’ known as the range of effectiveness. This range typically spans from 0.5 mm to 2.7 mm, with 2.5 mm being the precise measure (197–199). The constant distance between the base of the gingival groove and the alveolar crest, known as the biological width, is approximately 2 mm, falling within the range of effectiveness. This indicates that antigens and virulence factors present in biofilm plaque can traverse the epithelial barrier of the gingival tissues and penetrate the underlying connective tissues. Consequently, this triggers the release of paracrine signaling molecules, thereby affecting the balance of alveolar bone remodeling (65, 200). Research has demonstrated that the stimulation of PAMPs derived from subgingival plaque can elicit characteristic activation signals of bone marrow hematopoiesis, indicating the generation of immune cells derived from the myeloid lineage and the activation of associated immune responses (39). Meanwhile, innate immune cells present in the gums can uptake bacterial antigens from subgingival plaque and migrate to adjacent cervical lymph nodes, where they present antigens to activate the adaptive immune response. As a result, cytokines and immune cells, including T cells and memory T cells, may disseminate to the local gum tissues or even the entire body through the circulatory or lymphatic system (39, 201).
The oral microbial–host interactome can also transmit signals that extend beyond local tissues and contribute to the development of extra-oral comorbidities by initiating systemic inflammation or ectopic colonization in distant parts of the digestive tract (28, 36, 51, 196). Interestingly, a recent study suggested that the majority of healthy individuals do not exhibit detectable microbes in their blood, and even when a few species are detected, the microbial community patterns differ among various samples, with no apparent correlation between microbial species and the phenotype of healthy individuals (202). This implies that local disruption of the mucosal barrier serves as the initial step towards systemic comorbidities. Transient bacteremia facilitates the dissemination of microorganisms, such as oncobacteria, along with their virulence factors, to susceptible sites, thereby initiating or exacerbating disease progression at multiple sites. On a positive note, the microbial profile of gingival tissues in pathological conditions holds potential for aiding the diagnosis and treatment of extra-oral complications through blood microbial detection.
Under pathophysiological conditions, the subsets of immune cells that exist in a healthy state, such as T/NK, B/plasma, and granulocyte/myeloid cells, do not undergo significant changes in their overall categories. However, there are alterations in their proportions, particularly an increase in neutrophils and plasma cells (69).
The oral mucosal surface constantly faces microbial challenges, and neutrophils play a crucial role in maintaining alveolar bone homeostasis through innate immunity (203). Gingivitis is characterized by decreased neutrophils and bone activation factors, suggesting protective responses of the gingival tissues and bone during inflammation (66). However, as gingivitis progresses to periodontitis, there is an excessive inflammatory response leading to an increase in the number of neutrophils in local tissues. The quantity of neutrophils in the gingival tissues is more closely associated with the health or disease status of the periodontal tissues rather than their bactericidal function, which can be compensated by innate immune cells such as macrophages (204). Numerous studies have shown a positive correlation between the number of neutrophils in gingival tissues and the severity of periodontitis (205–207). In chronic periodontitis, dysfunctions in chemotactic accuracy, increased recruitment, and prolonged survival of neutrophils contribute to their extensive infiltration in periodontal tissues (204, 208, 209). These spontaneous hyperreactive neutrophils release various inflammatory factors (such as TNF, IL-1β, and IL-8), cytotoxic mediators, matrix metalloproteinases, and RANKL, which worsen periodontal tissues damage and bone resorption (210–213). Neutrophils can also migrate to the lymph nodes, where they interact with DCs to regulate antigen presentation and activate adaptive immunity (214). In the presence of CCL20, neutrophils can induce Th17 recruitment to inflamed tissues (215). They also promote B cell survival, proliferation, and differentiation into plasma cells by secreting B lymphocyte stimulator (BLyS) and a proliferation-inducing ligand (APRIL) (216, 217). Excessive neutrophils contribute to the progression of periodontitis and skeletal tissues destruction by initiating periodontal tissue lesions, exacerbating immune responses, and secreting local inflammatory factors and osteoclast-related factors. However, neutrophils deficiency in gingival tissues can also lead to periodontitis (218–220). Animal experiments have shown that impaired neutrophils recruitment associated with leukocyte adhesion deficiency Type I leads to increased periodontal inflammation, bone loss, and abnormal expression of IL-17 (221). This phenomenon might be related to a homeostasis mechanism of neutrophils recruitment, clearance, and generation, known as ‘neutrostat,’ which involves the IL-23–IL-17–granulocyte-colony stimulating factor (G-CSF) negative feedback loop (222). Impaired neutrophils recruitment results in unrestricted expression of IL-23, IL-17, and G-CSF in local tissues, leading to excessive inflammation and tissue damage (221).
Plasma cells are also significantly increased in patients with periodontitis compared to that in healthy individuals. The majority of plasma cells express IgG, while a minority express IgA (69). IgG is the main force in humoral immunity; it undergoes opsonization and antibody-dependent cell-mediated cytotoxicity (ADCC), and can activate the complement system through the classical pathway. These autoimmune responses may be the main factors contributing to periodontal destruction. Plasma cells may play a role in neutrophils recruitment by binding to the IgGFcR on the surface of neutrophils.
B cells have a dual role in periodontitis-related bone loss, which may depend on the activated B cell type. Certain B cell subsets exacerbate the severity of periodontal bone loss. In addition to IgG- and IgA-generated B cells, IgD- and IgM-generated B cells can also be associated with bacteria-induced periodontal bone loss, possibly through RANKL expression (223, 224). Memory B cells can promote osteoclast differentiation and maturation by expressing RANKL and various pro-inflammatory factors, such as TNF, IL-6 and IL-1β, and by increasing Th1 and Th17 production (225–228). Recent studies have highlighted the role of B cell activating factor (BAFF) in promoting periodontitis development by enhancing inflammatory conditions and macrophages activity (229). Conversely, regulatory B cells, also known as B10 cells, can reduce bone loss by upregulating IL-10 expression and downregulating IL-17 and RANKL expression (230–232).
In addition to neutrophils and B cells, T cells can be activated by antigens from P. gingivalis and F. nucleatum via TCR recognition and can differentiate into various subsets. Under pathologic conditions, T cells can affect bone remodeling by directly increasing the expression of pro-osteoclastic cytokines such as RANKL or indirectly signaling stromal–osteoblastic cells (99–101). Among all T cell subsets, γδ T cells (233); regulatory T cells (Treg) (234, 235); and helper T cells (Th, also known as CD4+T cells), including Th1 and Th2 (236, 237), Th9 and Th22 (238), Th17 (239), may be more closely associated with alveolar bone resorption (237).
Th1 and Th2 cells have been implicated in bone resorption in periodontitis, although the specific mechanisms have not been fully elucidated. The presence or absence of Th1 and Th2 cells may both contribute to bone resorption (240–243). The Th1/Th2 ratio was historically considered an important factor in evaluating the degree of bone resorption in periodontitis, as Th1 cells were believed to mediate the establishment of early periodontitis lesions, while Th2 cells gradually became quantitatively dominant as periodontitis progressed (236, 237). Interestingly, Th2 cells can promote the transformation of B cells into plasma cells by secreting IL-4, which may explain the increased proportion of plasma cells in gingival tissues under pathological conditions.
Current studies on bone remodeling have shifted focus from the Th1/Th2 balance to the Th17/Treg paradigm (67, 244, 245). Th17 has been closely associated with periodontitis and bone loss since their discovery (246, 247), and recent studies have shown that dysbiotic microbiomes activate Th17 cells to mediate oral mucosal immunopathology and periodontitis-induced bone destruction (239). Previous studies have demonstrated increased levels of Th17 and IL-17 in gingivitis and periodontitis (248–252), which are not necessarily related to the active or inactive stage of periodontitis (253–255). IL-23, a cytokine that promotes Th17 differentiation, is also highly expressed along with IL-17 in periodontitis (248, 253). Th17 cells, derived from naive T cells (also called Th0 cells) after stimulation by antigen presentation or pro-inflammatory factors (such as IL-1β, IL-6, and IL-23), are the primary source of IL-17 and can express other pro-inflammatory factors such as IL-21, IL-22, and TNF (256, 257). Although IL-17 may not directly act on the RANKL-macrophage colony-stimulating factor (M-CSF)-osteoclast culture system (258), it can promote osteoclastogenesis through the expression of RANKL mediated by osteoblastic cells (259). Interestingly, Th17-related neutrophil mobilization in gingival tissues can inhibit P. gingivalis-induced periodontal bone loss (260, 261), and IL-17 receptor α-deficient mice show reduced cytokine-dependent recruitment of neutrophils and increased bone resorption (262, 263), indicating that Th17 cells also possess bone-protective potential through neutrophil mobilization. Tregs, a subset of CD4+CD25+Foxp3+ T cells with anti-inflammatory and homeostatic functions, can secrete IL-10, IL-12, and TGF to achieve negative immune regulation. The presence of Tregs in periodontitis may represent a compensatory mechanism to mitigate excessive tissue damage caused by immune responses (237). Some studies have found that Tregs can improve pathological bone resorption through the CCR4–CCL22 pathway (234, 235). However, Tregs are highly plastic and can lose their immunosuppressive ability in chronic periodontitis (264). They may also differentiate into Th17 cells during the mid-stage of periodontitis (265). Therefore, the Th17/Treg ratio is a reasonable parameter to evaluate the dysbiotic microbiome-mediated periodontal inflammation status to a certain extent.
In addition to T cells, B cells, and neutrophils, other immune cells may also play a role in bone remodeling. NK cells in rheumatoid arthritis can promote osteoclastogenesis by expressing RANKL and M-CSF (266) and inhibiting osteoblast generation through a pro-apoptotic pathway (267). The degree of mast cell degranulation in chronic periodontitis is proportional to the severity of periodontal disease (268, 269), possibly owing to their ability to secrete IL-17 (270) and indirectly increase RANKL expression through IL-33 secretion (271). DCs synthesize and secrete a series of cytokines to increase RANKL expression (272–274) and immature DCs can differentiate into mature osteoclasts through the RANKL–RANK–M-CSF axis (76, 77, 274). Macrophages are recognized as immune cells that are closely related to osteoclasts. Macrophages are homologous to osteoclasts, as mentioned earlier, and in vivo, macrophages are able to participate in osteoclastogenesis through the RANKL–RANK–OPG axis, with the assistance of M-CSF.
Oral health and systemic status are intertwined, with lesions in one affecting the other. Failure to address this cycle can lead to the progression of systemic diseases. The oral microbial community plays a crucial role in oral health, and any disruption in the ecological guilds can contribute to the development of oral diseases, including periodontal disease and the subsequent loss of periodontal hard tissues, which poses a significant threat to oral and systemic health.
The imbalance in osteoblast–osteoclast coupling, mediated by the RANKL–RANK–OPG axis, is at the core of alveolar bone remodeling disruption. Excessive immunity activation triggered by host–microbe interactions appears to be the primary reason for this imbalance. Key members in certain ecological guilds, such as P. gingivalis and F. nucleatum, drive periodontal inflammation with. Virulence factors from these pathogens activate the host immune system through local diffusion, lymphatic pathways, and blood transmission. Under chronic inflammatory conditions, continuous host–microbe interactions lead to an exaggerated immune response, resulting in periodontal tissues destruction and alveolar bone resorption (Figure 2).
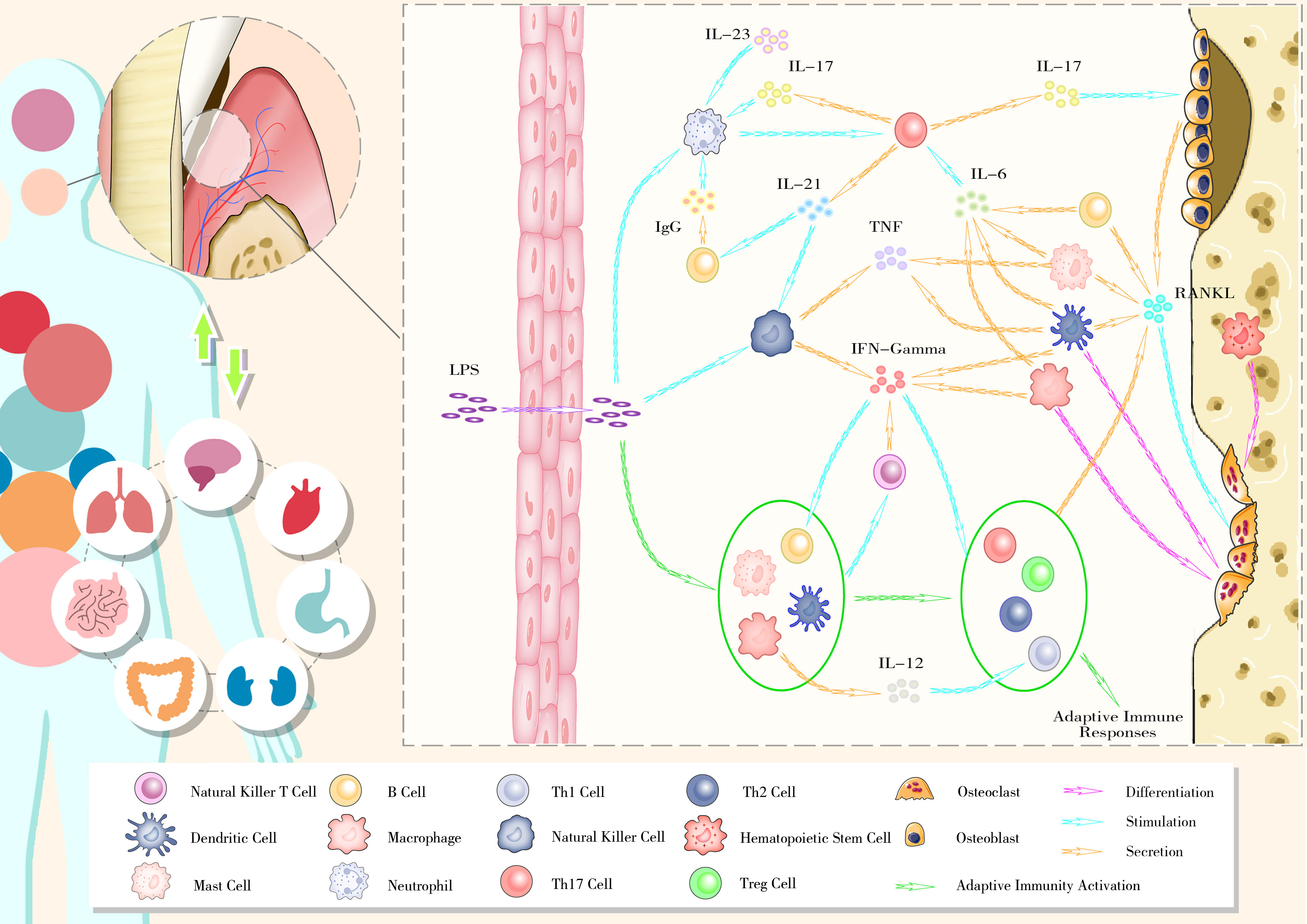
Figure 2 The bidirectional relationship between oral health and general well-being and the gingival immunophenotype of periodontitis.
Osteoimmunity involves intricate interactions between immune cells and molecules. The excessive osteoimmune response activated by the major functional microbiota associated with periodontitis cannot be solely attributed to changes in the proportion or function of individual immune cells. The phenotypes of the periodontal immune network must be established by studying the local and systemic immune status in the context of periodontal inflammation. Conversely, periodontal immunophenotypes reflect the characteristics of local ecological guilds. While the human body’s immunological characteristics are relatively clear and specific compared to the complexity of symbiotic microbial communities, the application of immunophenotypes holds promise as a straightforward method to evaluate the stability of plaque biofilms and study the symbiotic network of complex ecological guilds.
Although significant progress has been made in understanding the local immunophenotype of periodontitis and the role of pathogenic microorganisms, there are still gaps to be filled. Detailed investigations are needed to interpret the pathogenic effects of periodontal microorganisms. While the role of P. gingivalis in promoting alveolar bone resorption is well-established, there is limited research on the role of F. nucleatum, which has been recently focused as an oncobacterium in gastrointestinal tumors but not as a periodontal pathogen in alveolar bone resorption. Additionally, as the vital effect of extracellular vesicles gradually come into sight, the contents, secretion characteristics, and roles of P. gingivalis and F. nucleatum vesicles in bone remodeling are yet to be clarified. Furthermore, the contribution of other members within the periodontal pathogenic ecological guilds to alveolar bone resorption remains to be clarified.
Although the blood of healthy individuals is typically considered sterile, the presence of BEVs is a possibility. These vesicles may participate in immune system education in healthy individuals. However, once susceptible disease sites emerge, BEVs could potentially contribute to disease development even before the mucosal barrier is destroyed. Exploring the existence, content, and functions of vesicles in the blood of healthy individuals is an important area of investigation. Another aspect that remains to be elucidated is the immunophenotype of periodontitis. It is imperative to clarify the interaction network of immune cells and molecules in the disease state, identify the main functional groups, and screen characteristic high-expression cell phenotypes.
Maintenance of periodontal bone homeostasis is crucial in oral treatments that rely on physiological bone remodeling, such as periodontal therapy, orthodontic treatment, and implant restoration. How to block the progression of periodontitis, and restore lost bone, accelerate orthodontic effects by regulating bone remodeling, and how to reduce peri-implantitis to increase the success rate of implant surgery are all research focuses as well as difficulties in stomatology. However, these treatments often introduce various external stimuli to the teeth and periodontal tissues, resulting in oral hygiene challenges and disturbances to the periodontal microenvironment. To achieve optimal therapeutic outcomes, researchers should not simply focus on regulating the function of osteoblasts or osteoclasts, but aim to correct the unbalanced periodontal microenvironment and restore it to a healthy physiological state. By addressing these factors, some of the aforementioned clinical problems may find solutions.
Enabling patients to aesthetics and function healthfully is the fundamental principle of stomatology research. Modern medicine demands that dental practitioners not only control patients’ oral health during the short-term treatment and follow-up, but also maintain their lifelong well-being, which aligns with the WHO’s ‘8020’ goal, striving for improved oral health for the overall benefit of humanity.
JX: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. LiY: Resources, Writing – review & editing. SY: Resources, Writing – review & editing. ZY: Resources, Writing – review & editing. LuY: Funding acquisition, Supervision, Writing – review & editing. XX: Conceptualization, Supervision, Writing – review & editing.
This study was funded by the Jilin Provincial Health and Health Technology Innovation Program “Study on the correlation of crown and root morphology of anterior teeth based on genetic algorithm” (2020J050).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kassebaum NJ, Smith AGC, Bernabé E, Fleming TD, Reynolds AE, Vos T, et al. Global, regional, and national prevalence, incidence, and disability-adjusted life years for oral conditions for 195 countries, 1990–2015: a systematic analysis for the global burden of diseases, injuries, and risk factors. J Dent Res (2017) 96:380–7. doi: 10.1177/0022034517693566
2. Peres MA, Macpherson LMD, Weyant RJ, Daly B, Venturelli R, Mathur MR, et al. Oral diseases: a global public health challenge. Lancet (2019) 394:249–60. doi: 10.1016/S0140-6736(19)33016-8
3. Botelho J, MaChado V, Leira Y, Proença L, Chambrone L, Mendes JJ. Economic burden of periodontitis in the United States and Europe: an updated estimation. J Periodontol (2022) 93:373–9. doi: 10.1002/JPER.21-0111
4. Bastos JL, Celeste RK, Paradies YC. Racial inequalities in oral health. J Dent Res (2018) 97:878–86. doi: 10.1177/0022034518768536
5. GBD 2017 Oral Disorders Collaborators, Bernabe E, Marcenes W, Hernandez CR, Bailey J, Abreu LG, et al. Global, regional, and national levels and trends in burden of oral conditions from 1990 to 2017: a systematic analysis for the global burden of disease study 2017. J Dent Res (2020) 99:362–73. doi: 10.1016/S0140-6736(18)32279-7
6. Hajishengallis G, Chavakis T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol (2021) 21:426–40. doi: 10.1038/s41577-020-00488-6
7. Meurman JH, Bascones-Martinez A. Oral infections and systemic health – more than just links to cardiovascular diseases. Oral Hlth Prev Dent (2021) 19:441–8. doi: 10.3290/j.ohpd.b1993965
8. Jorth P, Turner KH, Gumus P, Nizam N, Buduneli N, Whiteley M. Metatranscriptomics of the human oral microbiome during health and disease. mBio (2014) 5:e01012–14. doi: 10.1177/0022034518761644
9. Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science (2017) 358:359–65. doi: 10.1126/science.aan4526
10. Blod C, Schlichting N, Schülin S, Suttkus A, Peukert N, Stingu CS, et al. The oral microbiome—the relevant reservoir for acute pediatric appendicitis? Int J Colorectal Dis (2018) 33:209–18. doi: 10.1007/s00384-017-2948-8
11. Fardini Y, Chung P, Dumm R, Joshi N, Han YW. Transmission of diverse oral bacteria to murine placenta: evidence for the oral microbiome as a potential source of intrauterine infection. Infect Immun (2010) 78:1789–96. doi: 10.1128/IAI.01395-09
12. Kuczynski J, Lauber CL, Walters WA, Parfrey LW, Clemente JC, Gevers D, et al. Experimental and analytical tools for studying the human microbiome. Nat Rev Genet (2011) 13:47–58. doi: 10.1038/nrg3129
13. Ling Z, Liu X, Cheng Y, Jiang X, Jiang H, Wang Y, et al. Decreased diversity of the oral microbiota of patients with Hepatitis B virus-induced chronic liver disease: a pilot project. Sci Rep (2015) 5:17098. doi: 10.1038/srep17098
14. Lira-Junior R, Boström EA. Oral-gut connection: one step closer to an integrated view of the gastrointestinal tract? Mucosal Immunol (2018) 11:316–8. doi: 10.1038/mi.2017.116
15. Peters BA, Wu J, Pei Z, Yang L, Purdue MP, Freedman ND, et al. Oral microbiome composition reflects prospective risk for esophageal cancers. Cancer Res (2017) 77:6777–87. doi: 10.1158/0008-5472.CAN-17-1296
16. Plaza-Díaz J, Ruiz-Ojeda F, Gil-Campos M, Gil A. Immune-mediated mechanisms of action of probiotics and synbiotics in treating pediatric intestinal diseases. Nutrients (2018) 10:42. doi: 10.3390/nu10010042
17. Reddy RM, Weir WB, Barnett S, Heiden BT, Orringer MB, Lin J, et al. Increased variance in oral and gastric microbiome correlates with esophagectomy anastomotic leak. Ann Thorac Surg (2018) 105:865–70. doi: 10.1016/j.athoracsur.2017.08.061
18. Roszyk E, Puszczewicz M. Role of human microbiome and selected bacterial infections in the pathogenesis of rheumatoid arthritis. Reumatologia (2017) 55:242–50. doi: 10.5114/reum.2017.71641
19. Zarco MF, Vess TJ, Ginsburg GS. The oral microbiome in health and disease and the potential impact on personalized dental medicine: the oral microbiome. Oral Dis (2012) 18:109–20. doi: 10.1111/j.1601-0825.2011.01851.x
20. Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner ACR, Yu WH, et al. The human oral microbiome. J Bacteriol (2010) 192:5002–17. doi: 10.1128/JB.00542-10
21. Kilian M, Chapple IL, Hannig M, Marsh PD, Meuric V, Pedersen AM, et al. The oral microbiome – an update for oral healthcare professionals. Br Dent J (2016) 221:657–66. doi: 10.1038/sj.bdj.2016.865
22. The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature (2012) 486:207–14. doi: 10.1038/nature11234
23. Hooks KB, O’Malley MA. Dysbiosis and its discontents. mBio (2017) 8:e01492–17. doi: 10.1128/mBio.01492-17
24. Gao L, Xu T, Huang G, Jiang S, Gu Y, Chen F. Oral microbiomes: more and more importance in oral cavity and whole body. Protein Cell (2018) 9:488–500. doi: 10.1007/s13238-018-0548-1
25. Hajishengallis G, Lamont RJ, Koo H. Oral polymicrobial communities: assembly, function, and impact on diseases. Cell Host Microbe (2023) 31:528–38. doi: 10.1016/j.chom.2023.02.009
27. Nonaka S, Kadowaki T, Nakanishi H. Secreted gingipains from Porphyromonas gingivalis increase permeability in human cerebral microvascular endothelial cells through intracellular degradation of tight junction proteins. Neurochem Int (2022) 154:105282. doi: 10.1016/j.neuint.2022.105282
28. Kitamoto S, Nagao-Kitamoto H, Jiao Y, Gillilland MG, Hayashi A, Imai J, et al. The intermucosal connection between the mouth and gut in commensal pathobiont-driven colitis. Cell (2020) 182:447–462.e14. doi: 10.1016/j.cell.2020.05.048
29. Kato T, Yamazaki K, Nakajima M, Date Y, Kikuchi J, Hase K, et al. Oral administration of Porphyromonas gingivalis alters the gut microbiome and serum metabolome. mSphere (2018) 3:e00460–18. doi: 10.1128/mSphere.00460-18
30. Xing T, Liu Y, Cheng H, Bai M, Chen J, Ji H, et al. Ligature induced periodontitis in rats causes gut dysbiosis leading to hepatic injury through SCD1/AMPK signalling pathway. Life Sci (2022) 288:120162. doi: 10.1016/j.lfs.2021.120162
31. Chen B, Lin W, Li Y, Bi C, Du L, Liu Y, et al. Roles of oral microbiota and oral-gut microbial transmission in hypertension. J Adv Res (2023) 43:147–61. doi: 10.1016/j.jare.2022.03.007
32. Chen B, Jia X, Xu J, Zhao L, Ji J, Wu B, et al. An autoimmunogenic and proinflammatory profile defined by the gut microbiota of patients with untreated systemic lupus erythematosus. Arthritis Rheumatol (2021) 73:232–43. doi: 10.1002/art.41511
33. Schirmer M, Denson L, Vlamakis H, Franzosa EA, Thomas S, Gotman NM, et al. Compositional and temporal changes in the gut microbiome of pediatric ulcerative colitis patients are linked to disease course. Cell Host Microbe (2018) 24:600–610.e4. doi: 10.1016/j.chom.2018.09.009
34. Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med (2019) 25:968–76. doi: 10.1038/s41591-019-0458-7
35. Nagao Ji, Kishikawa S, Tanaka H, Toyonaga K, Narita Y, Negoro-Yasumatsu K, et al. Pathobiont-responsive Th17 cells in gut-mouth axis provoke inflammatory oral disease and are modulated by intestinal microbiome. Cell Rep (2022) 40:111314. doi: 10.1016/j.celrep.2022.111314
36. Li X, Wang H, Yu X, Saha G, Kalafati L, Ioannidis C, et al. Maladaptive innate immune training of myelopoiesis links inflammatory comorbidities. Cell (2022) 185:1709–1727.e18. doi: 10.1016/j.cell.2022.03.043
37. Eren AM, Borisy GG, Huse SM, Mark Welch JL. Oligotyping analysis of the human oral microbiome. Proc Natl Acad Sci USA (2014) 111:E2875–84. doi: 10.1073/pnas.1409644111
38. Mark Welch JL, Dewhirst FE, Borisy GG. Biogeography of the oral microbiome: the site-specialist hypothesis. Ann Rev Microbiol (2019) 73:335–58. doi: 10.1146/annurev-micro-090817-062503.38
39. Hathaway-Schrader JD, Aartun JD, Poulides NA, Kuhn MB, McCormick BE, Chew ME, et al. Commensal oral microbiota induces osteoimmunomodulatory effects separate from systemic microbiome in mice. JCI Insight (2022) 7:e140738. doi: 10.1172/jci.insight.140738
40. de Albuquerque JB, Altenburger LM, Abe J, von Werdt D, Wissmann S, Magdaleno JM, et al. Microbial uptake in oral mucosa–draining lymph nodes leads to rapid release of cytotoxic CD8+ T cells lacking a gut-homing phenotype. Sci Immunol (2022) 7:eabf1861. doi: 10.1126/sciimmunol.abf1861
41. Bik EM, Long CD, Armitage GC, Loomer P, Emerson J, Mongodin EF, et al. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J (2010) 4:962–74. doi: 10.1038/ismej.2010.30
42. Simón-Soro Á, Tomás I, Cabrera-Rubio R, Catalan MD, Nyvad B, Mira A. Microbial geography of the oral cavity. J Dent Res (2013) 92:616–21. doi: 10.1177/0022034513488119
43. Wu G, Zhao N, Zhang C, Lam YY, Zhao L. Guild-based analysis for understanding gut microbiome in human health and diseases. Genome Med (2021) 13:22. doi: 10.1186/s13073-021-00840-y
44. Kapila YL. Oral health’s inextricable connection to systemic health: special populations bring to bear multimodal relationships and factors connecting periodontal disease to systemic diseases and conditions. Periodontol 2000 (2021) 87:11–6. doi: 10.1111/prd.12398
45. Genco RJ, Sanz M. Clinical and public health implications of periodontal and systemic diseases: An overview. Periodontol 2000 (2020) 83:7–13. doi: 10.1111/prd.12344
46. Peng X, Cheng L, You Y, Tang C, Ren B, Li Y, et al. Oral microbiota in human systematic diseases. Int J Oral Sci (2022) 14:14. doi: 10.1038/s41368-022-00163-7
47. Lagervall M, Jansson L, Bergström J. Systemic disorders in patients with periodontal disease: systemic disorders and periodontal diesease. J Clin Periodontol (2003) 30:293–9. doi: 10.1034/j.1600-051x.2003.00325.x
48. Carrizales-Sepúlveda EF, Ordaz-Farías A, Vera-Pineda R, Flores-Ramírez R. Periodontal disease, systemic inflammation and the risk of cardiovascular disease. Heart Lung Circ (2018) 27:1327–34. doi: 10.1016/j.hlc.2018.05.102
49. Arsiwala LT, Mok Y, Yang C, Ishigami J, Selvin E, Beck JD, et al. Periodontal disease measures and risk of incident peripheral artery disease: the atherosclerosis risk in communities (ARIC) Study. J Periodontol (2022) 93:943–53. doi: 10.1002/JPER.21-0342
50. Jung ES, Choi YY, Lee KH. Relationship between rheumatoid arthritis and periodontal disease in Korean adults: data from the sixth korea national health and nutrition examination survey, 2013 to 2015. J Periodontol (2019) 90:350–7. doi: 10.1002/JPER.18-0290
51. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv (2019) 5:eaau3333. doi: 10.1126/sciadv.aau3333
52. Chopra A, Radhakrishnan R, Sharma M. Porphyromonas gingivalis and adverse pregnancy outcomes: a review on its intricate pathogenic mechanisms. Crit Rev Microbiol (2020) 46:213–36. doi: 10.1080/1040841X.2020.1747392
53. Daalderop LA, Wieland BV, Tomsin K, Reyes L, Kramer BW, Vanterpool SF, et al. Periodontal disease and pregnancy outcomes: overview of systematic reviews. JDR Clin Transl Res (2018) 3:10–27. doi: 10.1177/2380084417731097
54. Zhang Y, Ren X, Hu T, Cheng R, Bhowmick NA. The relationship between periodontal disease and breast cancer: from basic mechanism to clinical management and prevention. Oral Hlth Prev Dent (2023) 21:49–60. doi: 10.3290/j.ohpd.b3904343
55. Bostanci N, Belibasakis GN. Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol Lett (2012) 333:1–9. doi: 10.1111/j.1574-6968.2012.02579.x
56. Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med (2014) 6:237ra65. doi: 10.1126/scitranslmed.3008599
57. Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol (2012) 10:717–25. doi: 10.1038/nrmicro2873
58. Queiros da Mota V, Prodhom G, Yan P, Hohlfheld P, Greub G, Rouleau C. Correlation between placental bacterial culture results and histological chorioamnionitis: a prospective study on 376 placentas. J Clin Pathol (2013) 66:243–8. doi: 10.1136/jclinpath-2012-201124
59. Payne MA, Hashim A, Alsam A, Joseph S, Aduse-Opoku J, Wade WG, et al. Horizontal and vertical transfer of oral microbial dysbiosis and periodontal disease. J Dent Res (2019) 98:1503–10. doi: 10.1177/0022034519877150
60. Kolenbrander PE, Palmer RJ Jr, Rickard AH, Jakubovics NS, Chalmers NI, Diaz PI. Bacterial interactions and successions during plaque development. Periodontol 2000 (2006) 42:47–79. doi: 10.1111/j.1600-0757.2006.00187.x
61. Mark Welch JL, Ramírez-Puebla ST, Borisy GG. Oral microbiome geography: micron-scale habitat and niche. Cell Host Microbe (2020) 28:160–8. doi: 10.1016/j.chom.2020.07.009
62. Mark Welch JL, Rossetti BJ, Rieken CW, Dewhirst FE, Borisy GG. Biogeography of a human oral microbiome at the micron scale. Proc Natl Acad Sci USA (2016) 113:E791–800. doi: 10.1073/pnas.1522149113
63. Zijnge V, van Leeuwen MBM, Degener JE, Abbas F, Thurnheer T, Gmür R, et al. Oral biofilm architecture on natural teeth. PloS One (2010) 5:e9321. doi: 10.1371/journal.pone.0009321
64. Meyle J, Chapple I. Molecular aspects of the pathogenesis of periodontitis. Periodontol 2000 (2015) 69:7–17. doi: 10.1111/prd.12104
65. Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol (2010) 8:481–90. doi: 10.1038/nrmicro2337
66. Bamashmous S, Kotsakis GA, Kerns KA, Leroux BG, Zenobia C, Chen D, et al. Human variation in gingival inflammation. Proc Natl Acad Sci USA (2021) 118:1. doi: 10.1073/pnas.2012578118
67. Hathaway-Schrader JD, Carson MD, Gerasco JE, Warner AJ, Swanson BA, Aguirre JI, et al. Commensal gut bacterium critically regulates alveolar bone homeostasis. Lab Invest (2022) 102:363–75. doi: 10.1038/s41374-021-00697-0
69. Williams DW, Greenwell-Wild T, Brenchley L, Dutzan N, Overmiller A, Sawaya AP, et al. Human oral mucosa cell atlas reveals a stromal-neutrophil axis regulating tissue immunity. Cell (2021) 184:4090. doi: 10.1016/j.cell.2021.05.013
70. Darveau RP. The oral microbial consortium’s interaction with the periodontal innate defense system. DNA Cell Biol (2009) 28:389–95. doi: 10.1089/dna.2009.0864
71. Boyce BF. Advances in the regulation of osteoclasts and osteoclast functions. J Dent Res (2013) 92:860–7. doi: 10.1177/0022034513500306
72. Florencio-Silva R, Rodrigues da Silva Sasso G, Sasso-Cerri E, Simões MJ, Cerri PS. Biology of bone tissue: structure, function, and factors that influence bone cells. BioMed Rese Int (2015) 2015:421746–17. doi: 10.1155/2015/421746
73. Yu T, Zhao L, Huang X, Ma C, Wang Y, Zhang J, et al. Enhanced activity of the macrophage M1/M2 phenotypes and phenotypic switch to M1 in periodontal infection. J Periodontol (2016) 87:1092–102. doi: 10.1902/jop.2016.160081
74. Lam RS, O'Brien-Simpson NM, Lenzo JC, Holden JA, Brammar GC, Walsh KA, et al. Macrophage depletion abates Porphyromonas gingivalis-induced alveolar bone resorption in mice. J Immunol (2014) 193:2349–62. doi: 10.4049/jimmunol.1400853
75. Huang CB, Alimova Y, Ebersole JL. Macrophage polarization in response to oral commensals and pathogens. Pathog Dis (2016) 74:ftw011. doi: 10.1093/femspd/ftw011
76. Speziani C, Rivollier A, Gallois A, Coury F, Mazzorana M, Azocar O, et al. Murine dendritic cell transdifferentiation into osteoclasts is differentially regulated by innate and adaptive cytokines. Eur J Immunol (2007) 37:747–57. doi: 10.1002/eji.200636534
77. Alnaeeli M, Penninger JM, Teng YA. Immune interactions with CD4+ T cells promote the development of functional osteoclasts from murine CD11c+ dendritic cells. J Immunol (2006) 177:3314–26. doi: 10.4049/jimmunol.177.5.3314
78. Arai F, Miyamoto T, Ohneda O, Inada T, Sudo T, Brasel K, et al. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med (1999) 190:1741–54. doi: 10.1084/jem.190.12.1741.78
79. Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys (2008) 473:139–46. doi: 10.1016/j.abb.2008.03.018
80. Feng X, Teitelbaum SL. Osteoclasts: new insights. Bone Res (2013) 1:11–26. doi: 10.4248/BR201301003
81. Nakashima T, Takayanagi H. New regulation mechanisms of osteoclast differentiation. Ann N Y Acad Sci (2011) 1240:E13–8. doi: 10.1111/j.1749-6632.2011.06373.x
82. Tanaka S, Takahashi N, Udagawa N, Tamura T, Akatsu T, Stanley ER, et al. Macrophage colony-stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J Clin Invest (1993) 91:257–63. doi: 10.1172/JCI116179
83. Kukita T, Wada N, Kukita A, Kakimoto T, Sandra F, Toh K, et al. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J Exp Med (2004) 200:941–6. doi: 10.1084/jem.20040518
84. Yagi M, Miyamoto T, Sawatani Y, Iwamoto K, Hosogane N, Fujita N, et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med (2005) 202:345–51. doi: 10.1084/jem.20050645
85. Yang M, Birnbaum MJ, MacKay CA, Mason-Savas A, Thompson B, Odgren PR. Osteoclast stimulatory transmembrane protein (OC-STAMP), a novel protein induced by RANKL that promotes osteoclast differentiation. J Cell Physiol (2008) 215:497–505. doi: 10.1002/jcp.21331
86. Schena F, Menale C, Caci E, Diomede L, Palagano E, Recordati C, et al. Murine rankl-/- mesenchymal stromal cells display an osteogenic differentiation defect improved by a RANKL-expressing lentiviral vector. Stem Cells (2017) 35:1365–77. doi: 10.1002/stem.2574
87. Ikebuchi Y, Aoki S, Honma M, Hayashi M, Sugamori Y, Khan M, et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature (2018) 561:195–200. doi: 10.1038/s41586-018-0482-7
88. Stepan JJ, Alenfeld F, Boivin G, Feyen JHM, Lakatos P. Mechanisms of action of antiresorptive therapies of postmenopausal osteoporosis. Endocr Regul (2003) 37:225–38.
89. Kanazawa K, Kudo A. Self-assembled RANK induces osteoclastogenesis ligand-independently. J Bone Miner Res (2005) 20:2053–60. doi: 10.1359/JBMR.050706
90. Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell (1995) 80:213–23. doi: 10.1016/0092-8674(95)90404-2
91. Kosmides AK, Necochea K, Hickey JW, Schneck JP. Separating T cell targeting components onto magnetically clustered nanoparticles boosts activation. Nano Lett (2018) 18:1916–24. doi: 10.1021/acs.nanolett.7b05284
92. Erickson-Miller CL, DeLorme E, Tian SS, Hopson CB, Stark K, Giampa L, et al. Discovery and characterization of a selective, nonpeptidyl thrombopoietin receptor agonist. Exp Hematol (2005) 33:85–93. doi: 10.1016/j.exphem.2004.09.006
93. Boger DL, Goldberg J. Cytokine receptor dimerization and activation: prospects for small molecule agonists. Bioorg Med Chem (2001) 9:557–62. doi: 10.1016/s0968-0896(00)00276-5
94. Sone E, Noshiro D, Ikebuchi Y, Nakagawa M, Khan M, Tamura Y, et al. The induction of RANKL molecule clustering could stimulate early osteoblast differentiation. Biochem Biophys Res Commun (2019) 509:435–40. doi: 10.1016/j.bbrc.2018.12.093
95. Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem (1997) 272:25190–4. doi: 10.1074/jbc.272.40.25190
96. Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature (1997) 390:175–9. doi: 10.1038/36593
97. Pacifici R. T cells: critical bone regulators in health and disease. Bone (2010) 47:461–71. doi: 10.1016/j.bone.2010.04.611
98. Weitzmann MN, Ofotokun I. Physiological and pathophysiological bone turnover - role of the immune system. Nat Rev Endocrinol (2016) 12:518–32. doi: 10.1038/nrendo.2016.91
99. Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature (1999) 402:304–9. doi: 10.1038/46303
100. Wang R, Zhang L, Zhang X, Moreno J, Celluzzi C, Tondravi M, et al. Regulation of activation-induced receptor activator of NF-kappaB ligand (RANKL) expression in T cells. Eur J Immunol (2002) 32:1090–8. doi: 10.1002/1521-4141(200204)32:4<1090::AID-IMMU1090>3.0.CO;2-P
101. Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature (1999) 397:315–23. doi: 10.1038/16852.101
102. Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA (1998) 95:3597–602. doi: 10.1073/pnas.95.7.3597
103. Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell (1998) 93:165–76. doi: 10.1016/s0092-8674(00)81569-x
104. Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Yano K, et al. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem Biophys Res Commun (1998) 253:395–400. doi: 10.1006/bbrc.1998.9788
105. Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A, Timms E, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci USA (1999) 96:3540–5. doi: 10.1073/pnas.96.7.3540
106. Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell (1997) 89:309–19. doi: 10.1016/s0092-8674(00)80209-3
107. Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology (1998) 139:1329–37. doi: 10.1210/endo.139.3.5837
108. Ikeda T, Kasai M, Utsuyama M, Hirokawa K. Determination of three isoforms of the receptor activator of nuclear factor-kappaB ligand and their differential expression in bone and thymus. Endocrinology (2001) 142:1419–26. doi: 10.1210/endo.142.4.8070
109. Mabbott NA, Donaldson DS, Ohno H, Williams IR, Mahajan A. Microfold (M) cells: important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol (2013) 6:666–77. doi: 10.1038/mi.2013.30
110. Darnay BG, Ni J, Moore PA, Aggarwal BB. Activation of NF-kappaB by RANK requires tumor necrosis factor receptor-associated factor (TRAF) 6 and NF-kappaB-inducing kinase. Identification of a novel TRAF6 interaction motif. J Biol Chem (1999) 274:7724–31. doi: 10.1074/jbc.274.12.7724
111. Xiong J, Cawley K, Piemontese M, Fujiwara Y, Zhao H, Goellner JJ, et al. Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat Commun (2018) 9:2909. doi: 10.1038/s41467-018-05244-y
112. Chen X, Zhi X, Wang J, Su J. RANKL signaling in bone marrow mesenchymal stem cells negatively regulates osteoblastic bone formation. Bone Res (2018) 6:34. doi: 10.1038/s41413-018-0035-6
113. Wong BR, Josien R, Lee SY, Vologodskaia M, Steinman RM, Choi Y. The TRAF family of signal transducers mediates NF-kappaB activation by the TRANCE receptor. J Biol Chem (1998) 273:28355–9. doi: 10.1074/jbc.273.43.28355
114. Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J (2001) 20:1271–80. doi: 10.1093/emboj/20.6.1271
115. Tsukasaki M, Asano T, Muro R, Huynh NC, Komatsu N, Okamoto K, et al. OPG production matters where it happened. Cell Rep (2020) 32:108124. doi: 10.1016/j.celrep.2020.108124
116. Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G, et al. LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat Med (2016) 22:539–46. doi: 10.1038/nm.4076
117. Al-Qutub MN, Braham PH, Karimi-Naser LM, Liu X, Genco CA, Darveau RP. Hemin-dependent modulation of the lipid A structure of Porphyromonas gingivalis lipopolysaccharide. Infect Immun (2006) 74:4474–85. doi: 10.1128/IAI.01924-05
118. Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, et al. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immun (2004) 72:5041–51. doi: 10.1128/IAI.72.9.5041-5051.2004
119. Hajishengallis G. Porphyromonas gingivalis-host interactions: open war or intelligent guerilla tactics? Microbes Infect (2009) 11:637–45. doi: 10.1016/j.micinf.2009.03.009
120. Nishida E, Hara Y, Kaneko T, Ikeda Y, Ukai T, Kato I. Bone resorption and local interleukin-1alpha and interleukin-1beta synthesis induced by Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis lipopolysaccharide. J Periodontal Res (2001) 36:1–8. doi: 10.1034/j.1600-0765.2001.00637.x
121. Chiang CY, Kyritsis G, Graves DT, Amar S. Interleukin-1 and tumor necrosis factor activities partially account for calvarial bone resorption induced by local injection of lipopolysaccharide. Infect Immun (1999) 67:4231–6. doi: 10.1128/IAI.67.8.4231-4236.1999
122. Bostanci N, Allaker R, Johansson U, Rangarajan M, Curtis MA, Hughes FJ, et al. Interleukin-1alpha stimulation in monocytes by periodontal bacteria: antagonistic effects of Porphyromonas gingivalis. Oral Microbiol Immunol (2007) 22:52–60. doi: 10.1111/j.1399-302X.2007.00322.x
123. Hamedi M, Belibasakis GN, Cruchley AT, Rangarajan M, Curtis MA, Bostanci N. Porphyromonas gingivalis culture supernatants differentially regulate interleukin-1beta and interleukin-18 in human monocytic cells. Cytokine (2009) 45:99–104. doi: 10.1016/j.cyto.2008.11.005
124. Zhou Q, Desta T, Fenton M, Graves DT, Amar S. Cytokine profiling of macrophages exposed to Porphyromonas gingivalis, its lipopolysaccharide, or its FimA protein. Infect Immun (2005) 73:935–43. doi: 10.1128/IAI.73.2.935-943.2005
125. Bostanci N, Allaker RP, Belibasakis GN, Rangarajan M, Curtis MA, Hughes FJ, et al. Porphyromonas gingivalis antagonises Campylobacter rectus induced cytokine production by human monocytes. Cytokine (2007) 39:147–56. doi: 10.1016/j.cyto.2007.07.002
126. Amano A, Nakagawa I, Okahashi N, Hamada N. Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. J Periodontal Res (2004) 39:136–42. doi: 10.1111/j.1600-0765.2004.00719.x
127. Hajishengallis G, Wang M, Liang S, Triantafilou M, Triantafilou K. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc Natl Acad Sci USA (2008) 105:13532–7. doi: 10.1073/pnas.0803852105
128. Lamont RJ, Jenkinson HF. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev (1998) 62:1244–63. doi: 10.1128/MMBR.62.4.1244-1263.1998
129. Jotwani R, Eswaran SV, Moonga S, Cutler CW. MMP-9/TIMP-1 imbalance induced in human dendritic cells by Porphyromonas gingivalis. FEMS Immunol Med Microbiol (2010) 58:314–21. doi: 10.1111/j.1574-695X.2009.00637.x
130. Takahashi Y, Davey M, Yumoto H, Gibson FC 3rd, Genco CA. Fimbria-dependent activation of pro-inflammatory molecules in Porphyromonas gingivalis infected human aortic endothelial cells. Cell Microbiol (2006) 8:738–57. doi: 10.1111/j.1462-5822.2005.00661.x
131. Hajishengallis G, Wang M, Liang S. Induction of distinct TLR2-mediated proinflammatory and proadhesive signaling pathways in response to Porphyromonas gingivalis fimbriae. J Immunol (2009) 182:6690–6. doi: 10.4049/jimmunol.0900524
132. Wang M, Shakhatreh MA, James D, Liang S, Nishiyama S, Yoshimura F, et al. Fimbrial proteins of Porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J Immunol (2007) 179:2349–58. doi: 10.4049/jimmunol.179.4.2349
133. Hajishengallis G, Shakhatreh MA, Wang M, Liang S. Complement receptor 3 blockade promotes IL-12-mediated clearance of Porphyromonas gingivalis and negates its virulence in vivo. J Immunol (2007) 179:2359–67. doi: 10.4049/jimmunol.179.4.2359
134. Jotwani R, Cutler CW. Fimbriated Porphyromonas gingivalis is more efficient than fimbria-deficient P. gingivalis in entering human dendritic cells in vitro and induces an inflammatory Th1 effector response. Infect Immun (2004) 72:1725–32. doi: 10.1128/IAI.72.3.1725-1732.2004
135. Malek R, Fisher JG, Caleca A, Stinson M, van Oss CJ, Lee JY, et al. Inactivation of the Porphyromonas gingivalis fimA gene blocks periodontal damage in gnotobiotic rats. J Bacteriol (1994) 176:1052–9. doi: 10.1128/jb.176.4.1052-1059.1994
136. Sharma A, Honma K, Evans RT, Hruby DE, Genco RJ. Oral immunization with recombinant Streptococcus gordonii expressing Porphyromonas gingivalis FimA domains. Infect Immun (2001) 69:2928–34. doi: 10.1128/IAI.69.5.2928-2934.2001
137. Curtis MA, Aduse-Opoku J, Rangarajan M. Cysteine proteases of Porphyromonas gingivalis. Crit Rev Oral Biol Med (2001) 12:192–216. doi: 10.1177/10454411010120030101
138. Guo Y, Nguyen KA, Potempa J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontol 2000 (2010) 54:15–44. doi: 10.1111/j.1600-0757.2010.00377.x
139. Holzhausen M, Cortelli JR, da Silva VA, Franco GC, Cortelli SC, Vergnolle N. Protease-activated receptor-2 (PAR(2)) in human periodontitis. J Dent Res (2010) 89:948–53. doi: 10.1177/0022034510373765
140. Fagundes JA, Monoo LD, Euzébio Alves VT, Pannuti CM, Cortelli SC, Cortelli JR, et al. Porphyromonas gingivalis is associated with protease-activated receptor-2 upregulation in chronic periodontitis. J Periodontol (2011) 82:1596–601. doi: 10.1902/jop.2011.110073
141. Lourbakos A, Chinni C, Thompson P, Potempa J, Travis J, Mackie EJ, et al. Cleavage and activation of proteinase-activated receptor-2 on human neutrophils by gingipain-R from Porphyromonas gingivalis. FEBS Lett (1998) 435:45–8. doi: 10.1016/s0014-5793(98)01036-9
142. Lourbakos A, Potempa J, Travis J, D'Andrea MR, Andrade-Gordon P, Santulli R, et al. Arginine-specific protease from Porphyromonas gingivalis activates protease-activated receptors on human oral epithelial cells and induces interleukin-6 secretion. Infect Immun (2001) 69:5121–30. doi: 10.1128/IAI.69.8.5121-5130.2001
143. Belibasakis GN, Bostanci N, Reddi D. Regulation of protease-activated receptor-2 expression in gingival fibroblasts and Jurkat T cells by Porphyromonas gingivalis. Cell Biol Int (2010) 34:287–92. doi: 10.1042/CBI20090290
144. Oido-Mori M, Rezzonico R, Wang PL, Kowashi Y, Dayer JM, Baehni PC, et al. Porphyromonas gingivalis gingipain-R enhances interleukin-8 but decreases gamma interferon-inducible protein 10 production by human gingival fibroblasts in response to T-cell contact. Infect Immun (2001) 69:4493–501. doi: 10.1128/IAI.69.7.4493-4501.2001
145. Wingrove JA, DiScipio RG, Chen Z, Potempa J, Travis J, Hugli TE. Activation of complement components C3 and C5 by a cysteine proteinase (gingipain-1) from Porphyromonas (Bacteroides) gingivalis. J Biol Chem (1992) 267:18902–7.
146. Imamura T, Banbula A, Pereira PJ, Travis J, Potempa J. Activation of human prothrombin by arginine-specific cysteine proteinases (Gingipains R) from Porphyromonas gingivalis. J Biol Chem (2001) 276:18984–91. doi: 10.1074/jbc.M006760200
147. Kitamura Y, Matono S, Aida Y, Hirofuji T, Maeda K. Gingipains in the culture supernatant of Porphyromonas gingivalis cleave CD4 and CD8 on human T cells. J Periodontal Res (2002) 37:464–8. doi: 10.1034/j.1600-0765.2002.01364.x
148. Yun PL, Decarlo AA, Collyer C, Hunter N. Hydrolysis of interleukin-12 by Porphyromonas gingivalis major cysteine proteinases may affect local gamma interferon accumulation and the Th1 or Th2 T-cell phenotype in periodontitis. Infect Immun (2001) 69:5650–60. doi: 10.1128/IAI.69.9.5650-5660.2001
149. Yun PL, DeCarlo AA, Hunter N. Modulation of major histocompatibility complex protein expression by human gamma interferon mediated by cysteine proteinase-adhesin polyproteins of Porphyromonas gingivalis. Infect Immun (1999) 67:2986–95. doi: 10.1128/IAI.67.6.2986-2995.1999
150. Yun PL, DeCarlo AA, Collyer C, Hunter N. Modulation of an interleukin-12 and gamma interferon synergistic feedback regulatory cycle of T-cell and monocyte cocultures by Porphyromonas gingivalis lipopolysaccharide in the absence or presence of cysteine proteinases. Infect Immun (2002) 70:5695–705. doi: 10.1128/IAI.70.10.5695-5705.2002
151. Tam V, O'Brien-Simpson NM, Chen YY, Sanderson CJ, Kinnear B, Reynolds EC. The RgpA-Kgp proteinase-adhesin complexes of Porphyromonas gingivalis inactivate the Th2 cytokines interleukin-4 and interleukin-5. Infect Immun (2009) 77:1451–8. doi: 10.1128/IAI.01377-08
152. Schenkein HA, Fletcher HM, Bodnar M, Macrina FL. Increased opsonization of a prtH-defective mutant of Porphyromonas gingivalis W83 is caused by reduced degradation of complement-derived opsonins. J Immunol (1995) 154:5331–7.
153. Grenier D, Roy E, Mayrand D. Modulation of Porphyromonas gingivalis proteinase activity by suboptimal doses of antimicrobial agents. J Periodontol (2003) 74:1316–9. doi: 10.1902/jop.2003.74.9.1316
154. Chen T, Nakayama K, Belliveau L, Duncan MJ. Porphyromonas gingivalis gingipains and adhesion to epithelial cells. Infect Immun (2001) 69:3048–56. doi: 10.1128/IAI.69.5.3048-3056.2001
155. Andrian E, Grenier D, Rouabhia M. In vitro models of tissue penetration and destruction by Porphyromonas gingivalis. Infect Immun (2004) 72:4689–98. doi: 10.1128/IAI.72.8.4689-4698.2004
156. Imamura T, Potempa J, Pike RN, Travis J. Dependence of vascular permeability enhancement on cysteine proteinases in vesicles of Porphyromonas gingivalis. Infect Immun (1995) 63:1999–2003. doi: 10.1128/iai.63.5.1999-2003.1995
157. Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL Jr. Microbial complexes in subgingival plaque. J Clin Periodontol (1998) 25:134–44. doi: 10.1111/j.1600-051x.1998.tb02419.x
158. Carrouel F, Viennot S, Santamaria J, Veber P, Bourgeois D. Quantitative molecular detection of 19 major pathogens in the interdental biofilm of periodontally healthy young adults. Front Microbiol (2016) 7:840. doi: 10.3389/fmicb.2016.00840
159. Brennan CA, Garrett WS. Fusobacterium nucleatum - symbiont, opportunist and oncobacterium. Nat Rev Microbiol (2019) 17:156–66. doi: 10.1038/s41579-018-0129-6
160. Bakken V, Aarø S, Hofstad T, Vasstrand EN. Outer membrane proteins as major antigens of Fusobacterium nucleatum. FEMS Microbiol Immunol (1989) 1:473–83. doi: 10.1111/j.1574-6968.1989.tb02438.x
161. Lee HR, Jun HK, Kim HD, Lee SH, Choi BK. Fusobacterium nucleatum GroEL induces risk factors of atherosclerosis in human microvascular endothelial cells and ApoE(-/-) mice. Mol Oral Microbiol (2012) 27:109–23. doi: 10.1111/j.2041-1014.2011.00636.x
162. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe (2013) 14:195–206. doi: 10.1016/j.chom.2013.07.012
163. Kumar A, Thotakura PL, Tiwary BK, Krishna R. Target identification in Fusobacterium nucleatum by subtractive genomics approach and enrichment analysis of host-pathogen protein-protein interactions. BMC Microbiol (2016) 16:84. doi: 10.1186/s12866-016-0700-0
164. Kapatral V, Anderson I, Ivanova N, Reznik G, Los T, Lykidis A, et al. Genome sequence and analysis of the oral bacterium Fusobacterium nucleatum strain ATCC 25586. J Bacteriol (2002) 184:2005–18. doi: 10.1128/JB.184.7.2005-2018.2002
165. Hung SC, Huang PR, Almeida-da-Silva CLC, Atanasova KR, Yilmaz O, Ojcius DM. NLRX1 modulates differentially NLRP3 inflammasome activation and NF-κB signaling during Fusobacterium nucleatum infection. Microbes Infect (2018) 20:615–25. doi: 10.1016/j.micinf.2017.09.014
166. Chang MC, Chen YJ, Lian YC, Chang BE, Huang CC, Huang WL, et al. Butyrate stimulates histone H3 acetylation, 8-isoprostane production, RANKL expression, and regulated osteoprotegerin expression/secretion in MG-63 osteoblastic cells. Int J Mol Sci (2018) 19:4071. doi: 10.3390/ijms19124071
167. Kaplan CW, Ma X, Paranjpe A, Jewett A, Lux R, Kinder-Haake S, et al. Fusobacterium nucleatum outer membrane proteins Fap2 and RadD induce cell death in human lymphocytes. Infect Immun (2010) 78:4773–8. doi: 10.1128/IAI.00567-10
168. Shokeen B, Park J, Duong E, Rambhia S, Paul M, Weinberg A, et al. Role of FAD-I in fusobacterial interspecies interaction and biofilm formation. Microorganisms (2020) 8:70. doi: 10.3390/microorganisms8010070
169. Guo L, Shokeen B, He X, Shi W, Lux R. Streptococcus mutans SpaP binds to RadD of Fusobacterium nucleatum ssp. polymorphum Mol Oral Microbiol (2017) 32:355–64. doi: 10.1111/omi.12177
170. Lima BP, Shi W, Lux R. Identification and characterization of a novel Fusobacterium nucleatum adhesin involved in physical interaction and biofilm formation with Streptococcus gordonii. Microbiologyopen (2017) 6:e00444. doi: 10.1002/mbo3.444
171. Lima BP, Hu LI, Vreeman GW, Weibel DB, Lux R. The oral bacterium Fusobacterium nucleatum binds Staphylococcus aureus and alters expression of the staphylococcal accessory regulator sarA. Microb Ecol (2019) 78:336–47. doi: 10.1007/s00248-018-1291-0
172. Park J, Shokeen B, Haake SK, Lux R. Characterization of Fusobacterium nucleatum ATCC 23726 adhesins involved in strain-specific attachment to Porphyromonas gingivalis. Int J Oral Sci (2016) 8:138–44. doi: 10.1038/ijos.2016.27
173. Rosen G, Sela MN. Coaggregation of Porphyromonas gingivalis and Fusobacterium nucleatum PK 1594 is mediated by capsular polysaccharide and lipopolysaccharide. FEMS Microbiol Lett (2006) 256:304–10. doi: 10.1111/j.1574-6968.2006.00131.x
174. Tefiku U, Popovska M, Cana A, Zendeli-Bedxeti L, Recica B, Spasovska-Gjorgovska A, et al. Determination of the role of fusobacterium nucleatum in the pathogenesis in and out the mouth. Pril (Makedon Akad Nauk Umet Odd Med Nauki) (2020) 41:87–99. doi: 10.2478/prilozi-2020-0026
175. Coppenhagen-Glazer S, Sol A, Abed J, Naor R, Zhang X, Han YW, et al. Fap2 of Fusobacterium nucleatum is a galactose-inhibitable adhesin involved in coaggregation, cell adhesion, and preterm birth. Infect Immun (2015) 83:1104–13. doi: 10.1128/IAI.02838-14
176. Liu PF, Shi W, Zhu W, Smith JW, Hsieh SL, Gallo RL, et al. Vaccination targeting surface FomA of Fusobacterium nucleatum against bacterial co-aggregation: Implication for treatment of periodontal infection and halitosis. Vaccine (2010) 28:3496–505. doi: 10.1016/j.vaccine.2010.02.047
177. Wu T, Cen L, Kaplan C, Zhou X, Lux R, Shi W, et al. Cellular Components Mediating Coadherence of Candida albicans and Fusobacterium nucleatum. J Dent Res (2015) 94:1432–8. doi: 10.1177/0022034515593706
178. Bor B, Cen L, Agnello M, Shi W, He X. Morphological and physiological changes induced by contact-dependent interaction between Candida albicans and Fusobacterium nucleatum. Sci Rep (2016) 6:27956. doi: 10.1038/srep27956
179. Ali Mohammed MM, Pettersen VK, Nerland AH, Wiker HG, Bakken V. Label-free quantitative proteomic analysis of the oral bacteria Fusobacterium nucleatum and Porphyromonas gingivalis to identify protein features relevant in biofilm formation. Anaerobe (2021) 72:102449. doi: 10.1016/j.anaerobe.2021.102449
180. Umaña A, Sanders BE, Yoo CC, Casasanta MA, Udayasuryan B, Verbridge SS, et al. Utilizing whole Fusobacterium genomes to identify, correct, and characterize potential virulence protein families. J Bacteriol (2019) 201:e00273-19. doi: 10.1128/JB.00273-19
181. Abdulkareem AA, Shelton RM, Landini G, Cooper PR, Milward MR. Potential role of periodontal pathogens in compromising epithelial barrier function by inducing epithelial-mesenchymal transition. J Periodont Res (2018) 53:565–74. doi: 10.1111/jre.12546
182. Hong M, Li Z, Liu H, Zheng S, Zhang F, Zhu J, et al. Fusobacterium nucleatum aggravates rheumatoid arthritis through FadA-containing outer membrane vesicles. Cell Host Microbe (2023) 31:798–810.e7. doi: 10.1016/j.chom.2023.03.018
183. Díaz-Garrido N, Badia J, Baldomà L. Microbiota-derived extracellular vesicles in interkingdom communication in the gut. J Extracell Vesicles (2021) 10:e12161. doi: 10.1002/jev2.12161
184. Chatterjee SN, Das J. Electron microscopic observations on the excretion of cell-wall material by Vibrio cholerae. J Gen Microbiol (1967) 49:1. doi: 10.1099/00221287-49-1-1
185. Ñahui Palomino RA, Vanpouille C, Costantini PE, Margolis L. Microbiota–host communications: Bacterial extracellular vesicles as a common language. PloS Pathog (2021) 17:e1009508. doi: 10.1371/journal.ppat.1009508
186. Zafar H, Saier MH. Gut Bacteroides species in health and disease. Gut Microbes (2021) 13:1–20. doi: 10.1080/19490976.2020.1848158
187. Lee EY, Bang JY, Park GW, Choi DS, Kang JS, Kim HJ, et al. Global proteomic profiling of native outer membrane vesicles derived from Escherichia coli. Proteomics (2007) 7:3143–53. doi: 10.1002/pmic.200700196
188. Hu R, Lin H, Wang M, Zhao Y, Liu H, Min Y, et al. Lactobacillus reuteri-derived extracellular vesicles maintain intestinal immune homeostasis against lipopolysaccharide-induced inflammatory responses in broilers. J Anim Sci Biotechnol (2021) 12:25. doi: 10.1186/s40104-020-00532-4
189. Joshi B, Singh B, Nadeem A, Askarian F, Wai SN, Johannessen M, et al. Transcriptome profiling of staphylococcus aureus associated extracellular vesicles reveals presence of small RNA-cargo. Front Mol Biosci (2021) 7:566207. doi: 10.3389/fmolb.2020.566207
190. Stanton BA. Extracellular vesicles and host–pathogen interactions: a review of inter-kingdom signaling by small noncoding RNA. Genes (Basel) (2021) 12:1010. doi: 10.3390/genes12071010
191. Bäuerl C, Coll-Marqués JM, Tarazona-González C, Pérez-Martínez G. Lactobacillus casei extracellular vesicles stimulate EGFR pathway likely due to the presence of proteins P40 and P75 bound to their surface. Sci Rep (2020) 10:19237. doi: 10.1038/s41598-020-75930-9
192. Lee J, Lee EY, Kim SH, Kim DK. Staphylococcus aureus extracellular vesicles carry biologically active β-lactamase. Antimicrob Agents chemother (2013) 57:2589–95. doi: 10.1128/AAC.00522-12
193. Lee J, Kim SH, Choi DS, Lee JS. Proteomic analysis of extracellular vesicles derived from Mycobacterium tuberculosis. Proteomics (Weinheim) (2015) 15:3331–7. doi: 10.1002/pmic.201500037
194. Northrop-Albrecht EJ, Taylor WR, Huang BQ, Kisiel JB, Lucien F. Assessment of extracellular vesicle isolation methods from human stool supernatant. J Extracell Vesicles (2022) 11:e12208. doi: 10.1002/jev2.12208
195. Freire M, Nelson KE, Edlund A. The oral host–microbial interactome: an ecological chronometer of health? Trends Microbiol (2021) 29:551–61. doi: 10.1016/j.tim.2020.11.004
196. Garant PR, Cho MI. Histopathogenesis of spontaneous periodontal disease in conventional rats.: I. Histometric and histologic study. J Periodontal Res (1979) 14:297–309. doi: 10.1111/j.1600-0765.1979.tb00794.x
197. Waerhaug J. The angular bone defect and its relationship to trauma from occlusion and downgrowth of subgingival plaque. J Clin Periodontol (1979) 6:61–82. doi: 10.1111/j.1600-051x.1979.tb02185.x
198. Page RC, Schroeder HE. Periodontitis in man and other animals. S. Karger AG, (1982). p. 199. doi: 10.1159/isbn.978-3-318-05027-1
199. Curtis MA, Zenobia C, Darveau RP. The relationship of the oral microbiotia to periodontal health and disease. Cell Host Microbe (2011) 10:302–6. doi: 10.1016/j.chom.2011.09.008
200. Aoyagi T, Sugawara-Aoyagi M, Yamazaki K, Hara K. Interleukin 4 (IL-4) and IL-6-producing memory T-cells in peripheral blood and gingival tissues in periodontitis patients with high serum antibody titers to Porphyromonas gingivalis. Oral Microbiol Immunol (1995) 10:304–10. doi: 10.1111/j.1399-302x.1995.tb00159.x
201. Tan CCS, Ko KKK, Chen H, Liu J, Loh M. No evidence for a common blood microbiome based on a population study of 9,770 healthy humans. Nat Microbiol (2023) 8:973–85. doi: 10.1038/s41564-023-01350-w
202. Hooper LV. Bacterial contributions to mammalian gut development. Trends Microbiol (2004) 12:129–34. doi: 10.1016/j.tim.2004.01.001
203. Hajishengallis G, Korostoff JM. Revisiting the Page & Schroeder model: the good, the bad and the unknowns in the periodontal host response 40 years later. Periodontol 2000 (2017) 75:116–51. doi: 10.1111/prd.12181
204. Landzberg M, Doering H, Aboodi GM, Tenenbaum HC, Glogauer M. Quantifying oral inflammatory load: oral neutrophil counts in periodontal health and disease. J Periodontal Res (2015) 50:330–6. doi: 10.1111/jre.12211
205. Lee W, Aitken S, Sodek J, McCulloch CA. Evidence of a direct relationship between neutrophil collagenase activity and periodontal tissue destruction in vivo : role of active enzyme in human periodontitis. J Periodontal Res (1995) 30:23–33. doi: 10.1111/j.1600-0765.1995.tb01249.x
206. Hernández M, Gamonal J, Tervahartiala T, Mäntylä P, Rivera O, Dezerega A, et al. Associations between matrix metalloproteinase-8 and -14 and myeloperoxidase in gingival crevicular fluid from subjects with progressive chronic periodontitis: a longitudinal study. J Periodontol (2010) 81:1644–52. doi: 10.1902/jop.2010.100196
207. Lakschevitz FS, Aboodi GM, Glogauer M. Oral neutrophil transcriptome changes result in a pro-survival phenotype in periodontal diseases. PloS One (2013) 8:e68983. doi: 10.1371/journal.pone.0068983
208. Roberts HM, Ling MR, Insall R, Kalna G, Spengler J, Grant MM, et al. Impaired neutrophil directional chemotactic accuracy in chronic periodontitis patients. J Clin Periodontol (2015) 42:1–11. doi: 10.1111/jcpe.12326
209. Cooper PR, Palmer LJ, Chapple IL. Neutrophil extracellular traps as a new paradigm in innate immunity: friend or foe? Periodontol 2000 (2013) 63:165–97. doi: 10.1111/prd.12025
210. Sima C, Glogauer M. Neutrophil dysfunction and host susceptibility to periodontal inflammation: current state of knowledge. Curr Oral Health Rep (2014) 1:95–103. doi: 10.1007/s40496-014-0015-x
211. Ryder MI. Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontol 2000 (2010) 53:124–37. doi: 10.1111/j.1600-0757.2009.00327.x
212. Chapple IL, Matthews JB. The role of reactive oxygen and antioxidant species in periodontal tissue destruction. Periodontol 2000 (2007) 43:160–232. doi: 10.1111/j.1600-0757.2006.00178.x.213
213. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11:519–31. doi: 10.1038/nri3024
214. Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, Costantini C. et al, Evidence for a cross-talk between human neutrophils and Th17 cells. Blood (2010) 115:335–43. doi: 10.1182/blood-2009-04-216085
215. Huard B, McKee T, Bosshard C, Durual S, Matthes T, Myit S, et al. APRIL secreted by neutrophils binds to heparan sulfate proteoglycans to create plasma cell niches in human mucosa. J Clin Invest (2008) 118:2887–95. doi: 10.1172/JCI33760
216. Scapini P, Carletto A, Nardelli B, Calzetti F, Roschke V, Merigo F, et al. Proinflammatory mediators elicit secretion of the intracellular B-lymphocyte stimulator pool (BLyS) that is stored in activated neutrophils: Implications for inflammatory diseases. Blood (2005) 105:830–7. doi: 10.1182/blood-2004-02-0564
217. Schmidt S, Moser M, Sperandio M. The molecular basis of leukocyte recruitment and its deficiencies. Mol Immunol (2013) 55:49–58. doi: 10.1016/j.molimm.2012.11.006
218. Nussbaum G, Shapira L. How has neutrophil research improved our understanding of periodontal pathogenesis? J Clin Periodontol (2011) 38 Suppl 11:49–59. doi: 10.1111/j.1600-051X.2010.01678.x
219. Larjava H, Koivisto L, Heino J, Häkkinen L. Integrins in periodontal disease. Exp Cell Res (2014) 325:104–10. doi: 10.1016/j.yexcr.2014.03.010
220. Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med (2014) 6:229ra40. doi: 10.1126/scitranslmed.3007696
221. Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity (2005) 22:285–94. doi: 10.1016/j.immuni.2005.01.011
222. Baker PJ, Boutaugh NR, Tiffany M, Roopenian DC. B cell IgD deletion prevents alveolar bone loss following murine oral infection. Interdiscip Perspect Infect Dis (2009) 2009:864359. doi: 10.1155/2009/864359
223. Oliver-Bell J, Butcher JP, Malcolm J, MacLeod MKL, Adrados Planell A, Campbell L, et al. Periodontitis in the absence of B cells and specific anti-bacterial antibody. Mol Oral Microbiol (2015) 30:160–9. doi: 10.1111/omi.12082
224. Han Y, Jin Y, Miao Y, Shi T, Lin X. Switched memory B cells promote alveolar bone damage during periodontitis: An adoptive transfer experiment. Int Immunopharmacol (2018) 62:147–54. doi: 10.1016/j.intimp.2018.07.003
225. Donati M, Liljenberg B, Zitzmann NU, Berglundh T. B-1a cells and plasma cells in periodontitis lesions. J Periodontal Res (2009) 44:683–8. doi: 10.1111/j.1600-0765.2008.01178.x
226. Mahanonda R, Champaiboon C, Subbalekha K, Sa-Ard-Iam N, Rattanathammatada W, Thawanaphong S, et al. Human memory B cells in healthy gingiva, gingivitis, and periodontitis. J Immunol (2016) 197:715–25. doi: 10.4049/jimmunol.1600540
227. Han Y, Jin Y, Miao Y, Shi T, Lin X. Improved RANKL expression and osteoclastogenesis induction of CD27+CD38– memory B cells: A link between B cells and alveolar bone damage in periodontitis. J Periodont Res (2019) 54:73–80. doi: 10.1111/jre.12606
228. Wang L, Zhang T, Zhang Z, Wang Z, Zhou YJ, Wang Z. B cell activating factor regulates periodontitis development by suppressing inflammatory responses in macrophages. BMC Oral Health (2021) 21:426. doi: 10.1186/s12903-021-01788-6
229. Hu Y, Yu P, Yu X, Hu X, Kawai T, Han X. IL-21/anti-Tim1/CD40 ligand promotes B10 activity in vitro and alleviates bone loss in experimental periodontitis in vivo. Biochim Biophys Acta Mol Basis Dis (2017) 1863:2149–57. doi: 10.1016/j.bbadis.2017.06.001
230. Cherukuri A, Ding Q, Sharma A, Mohib K. Rothstein DM.Regulatory and effector B cells: a new path toward biomarkers and therapeutic targets to improve transplant outcomes? Clin Lab Med (2019) 39:15–29. doi: 10.1016/j.cll.2018.10.011
231. Shi T, Jin Y, Miao Y, Wang Y, Zhou Y, Lin X. IL-10 secreting B cells regulate periodontal immune response during periodontitis. Odontology (2020) 108:350–7. doi: 10.1007/s10266-019-00470-2
232. Barel O, Aizenbud Y, Tabib Y, Jaber Y, Leibovich A, Horev Y, et al. γδ T cells differentially regulate bone loss in periodontitis models. J Dent Res (2022) 101:428–36. doi: 10.1177/00220345211042830
233. Glowacki AJ, Yoshizawa S, Jhunjhunwala S, Vieira AE, Garlet GP, Sfeir C, et al. Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via recruitment of regulatory lymphocytes. Proc Natl Acad Sci USA (2013) 110:18525–30. doi: 10.1073/pnas.1302829110
234. Araujo-Pires AC, Vieira AE, Francisconi CF, Biguetti CC, Glowacki A, Yoshizawa S, et al. IL-4/CCL22/CCR4 axis controls regulatory T-cell migration that suppresses inflammatory bone loss in murine experimental periodontitis. J Bone Miner Res (2015) 30:412–22. doi: 10.1002/jbmr.2376
235. Gemmell E, Seymour GJ. Immunoregulatory control of Th1/Th2 cytokine profiles in periodontal disease. Periodontol 2000 (2004) 35:21–41. doi: 10.1111/j.0906-6713.2004.003557.x
236. Hathaway-Schrader JD, Novince CM. Maintaining homeostatic control of periodontal bone tissue. Periodontol 2000 (2021) 86:157–87. doi: 10.1111/prd.12368
237. Díaz-Zúñiga J, Melgar-Rodríguez S, Rojas L, Alvarez C, Monasterio G, Carvajal P, et al. Increased levels of the T-helper 22-associated cytokine (interleukin-22) and transcription factor (aryl hydrocarbon receptor) in patients with periodontitis are associated with osteoclast resorptive activity and severity of the disease. J Periodontal Res (2017) 52:893–902. doi: 10.1111/jre.12461
238. Dutzan N, Kajikawa T, Abusleme L, Greenwell-Wild T, Zuazo CE, Ikeuchi T, et al. A dysbiotic microbiome triggers T H 17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med (2018) 10:eaat0797. doi: 10.1126/scitranslmed.aat0797
239. Cheng J, Liu J, Shi Z, Xu D, Luo S, Siegal GP, et al. Interleukin-4 inhibits RANKL-induced NFATc1 expression via STAT6: A novel mechanism mediating its blockade of osteoclastogenesis. J Cell Biochem (2011) 112:3385–92. doi: 10.1002/jcb.23269
240. Alayan J, Ivanovski S, Farah CS. Alveolar bone loss in T helper 1/T helper 2 cytokine-deficient mice. J Periodontal Res (2007) 42:97–103. doi: 10.1111/j.1600-0765.2006.00920.x
241. Kohara H, Kitaura H, Fujimura Y, Yoshimatsu M, Morita Y, Eguchi T, et al. IFN-γ directly inhibits TNF-α-induced osteoclastogenesis in vitro and in vivo and induces apoptosis mediated by Fas/Fas ligand interactions. Immunol Lett (2011) 137:53–61. doi: 10.1016/j.imlet.2011.02.017
242. Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature (2000) 408:600–5. doi: 10.1038/35046102
243. Cafferata EA, Jerez A, Vernal R, Monasterio G, Pandis N, Faggion CM Jr. The therapeutic potential of regulatory T lymphocytes in periodontitis: A systematic review. J Periodont Res (2019) 54:207–17. doi: 10.1111/jre.12629
244. Guo M, Liu H, Yu Y, Zhu X, Xie H, Wei C, et al. Lactobacillus rhamnosus GG ameliorates osteoporosis in ovariectomized rats by regulating the Th17/Treg balance and gut microbiota structure. Gut Microbes (2023) 15:2190304. doi: 10.1080/19490976.2023.2190304
245. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6:1133–41. doi: 10.1038/ni1261
246. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6:1123–32. doi: 10.1038/ni1254
247. Allam JP, Duan Y, Heinemann F, Winter J, Götz W, Deschner J, et al. IL-23-producing CD68+ macrophage-like cells predominate within an IL-17-polarized infiltrate in chronic periodontitis lesions: Macrophage-like cells in chronic periodontitis. J Clin Periodontol (2011) 38:879–86. doi: 10.1111/j.1600-051X.2011.01752.x
248. Beklen A, Ainola M, Hukkanen M, Gürgan C, Sorsa T, Konttinen YT. MMPs, IL-1, and TNF are regulated by IL-17 in periodontitis. J Dent Res (2007) 86:347–51. doi: 10.1177/154405910708600409
249. Honda T, Aoki Y, Takahashi N, Maekawa T, Nakajima T, Ito H, et al. Elevated expression of IL-17 and IL-12 genes in chronic inflammatory periodontal disease. Clin Chim Acta (2008) 395:137–41. doi: 10.1016/j.cca.2008.06.003
250. Cardoso CR, Garlet GP, Crippa GE, Rosa AL, Júnior WM, Rossi MA, et al. Evidence of the presence of T helper type 17 cells in chronic lesions of human periodontal disease. Oral Microbiol Immunol (2009) 24:1–6. doi: 10.1111/j.1399-302X.2008.00463.x
251. Adibrad M, Deyhimi P, Ganjalikhani Hakemi M, Behfarnia P, Shahabuei M, Rafiee L. Signs of the presence of Th17 cells in chronic periodontal disease: Presence of Th17 cells in periodontal disease. J Periodontal Res (2012) 47:525–31. doi: 10.1111/j.1600-0765.2011.01464.x
252. Ohyama H, Kato-Kogoe N, Kuhara A, Nishimura F, Nakasho K, Yamanegi K, et al. The involvement of IL-23 and the Th17 pathway in periodontitis. J Dent Res (2009) 88:633–8. doi: 10.1177/0022034509339889
253. Sun L, Girnary M, Wang L, Jiao Y, Zeng E, Mercer K, et al. IL-10 dampens an IL-17–mediated periodontitis-associated inflammatory network. J Immunol (2020) 204:2177–91. doi: 10.4049/jimmunol.1900532
254. Dutzan N, Gamonal J, Silva A, Sanz M, Vernal R. Over-expression of forkhead box P3 and its association with receptor activator of nuclear factor- κ B ligand, interleukin (IL) -17, IL-10 and transforming growth factor- β during the progression of chronic periodontitis. J Clin Periodontol (2009) 36:396–403. doi: 10.1111/j.1600-051X.2009.01390.x
255. Stockinger B, Omenetti S. The dichotomous nature of T helper 17 cells. Nat Rev Immunol (2017) 17:535–44. doi: 10.1038/nri.2017.50
256. Dutzan N, Konkel JE, Greenwell-Wild T, Moutsopoulos NM. Characterization of the human immune cell network at the gingival barrier. Mucosal Immunol (2016) 9:1163–72. doi: 10.1038/mi.2015.136
257. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med (2006) 203:2673–82. doi: 10.1084/jem.20061775
258. Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest (1999) 103:1345–52. doi: 10.1172/JCI5703
259. Yu JJ, Ruddy MJ, Wong GC, Sfintescu C, Baker PJ, Smith JB, et al. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor–dependent signals. Blood (2007) 109:3794–802. doi: 10.1182/blood-2005-09-010116
260. Yu JJ, Ruddy MJ, Conti HR, Boonanantanasarn K, Gaffen SL. The interleukin-17 receptor plays a gender-dependent role in host protection against Porphyromonas gingivalis -induced periodontal bone loss. Infect Immun (2008) 76:4206–13. doi: 10.1128/IAI.01209-07
261. Settem RP, Honma K, Nakajima T, Phansopa C, Roy S, Stafford GP, et al. A bacterial glycan core linked to surface (S)-layer proteins modulates host immunity through Th17 suppression. Mucosal Immunol (2013) 6:415–26. doi: 10.1038/mi.2012.85
262. Settem RP, Honma K, Sharma A. Neutrophil mobilization by surface-glycan altered Th17-skewing bacteria mitigates periodontal pathogen persistence and associated alveolar bone loss. PloS One (2014) 9:e108030. doi: 10.1371/journal.pone.0108030
263. Okui T, Aoki Y, Ito H, Honda T, Yamazaki K. The presence of IL-17 + /FOXP3 + double-positive cells in periodontitis. J Dent Res (2012) 91:574–9. doi: 10.1177/0022034512446341
264. Tsukasaki M, Komatsu N, Nagashima K, Nitta T, Pluemsakunthai W, Shukunami C, et al. Host defense against oral microbiota by bone-damaging T cells. Nat Commun (2018) 9:701. doi: 10.1038/s41467-018-03147-6
265. Pridgeon C, Lennon GP, Pazmany L, Thompson RN, Christmas SE, Moots RJ. Natural killer cells in the synovial fluid of rheumatoid arthritis patients exhibit a CD56bright, CD94bright, CD158negative phenotype. Rheumatol (Oxford) (2003) 42:870–8. doi: 10.1093/rheumatology/keg240
266. Takeda H, Kikuchi T, Soboku K, Okabe I, Mizutani H, Mitani A, et al. Effect of IL-15 and natural killer cells on osteoclasts and osteoblasts in a mouse coculture. Inflammation (2014) 37:657–69. doi: 10.1007/s10753-013-9782-0
267. Huang S, Lu F, Chen Y, Huang B, Liu M. Mast cell degranulation in human periodontitis. J Periodontol (2013) 84:248–55. doi: 10.1902/jop.2012.120066
268. Parachuru VPB, Coates DE, Milne TJ, Rich AM, Seymour GJ. FoxP3+ regulatory T cells, interleukin 17 and mast cells in chronic inflammatory periodontal disease. J Periodont Res (2018) 53:622–35. doi: 10.1111/jre.12552
269. Köseoğlu S, Hatipoğlu M, Sağlam M, Enhoş Ş, Esen HH. Interleukin-33 could play an important role in the pathogenesis of periodontitis. J Periodont Res (2015) 50:525–34. doi: 10.1111/jre.12235
270. Chauhan D, Singh AV, Brahmandam M, Carrasco R, Bandi M, Hideshima T, et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: a therapeutic target. Cancer Cell (2009) 16:309–23. doi: 10.1016/j.ccr.2009.08.019
271. Sawant A, Ponnazhagan S. Role of plasmacytoid dendritic cells in breast cancer bone dissemination. OncoImmunology (2013) 2:e22983. doi: 10.4161/onci.22983
272. Alnaeeli M, Teng YT. Dendritic cells differentiate into osteoclasts in bone marrow microenvironment in vivo. Blood (2009) 113:264–5. doi: 10.1182/blood-2008-09-180836
273. Kaplan CW, Lux R, Haake SK, Shi W. The Fusobacterium nucleatum outer membrane protein RadD is an arginine-inhibitable adhesin required for inter-species adherence and the structured architecture of multispecies biofilm. Mol Microbiol (2009) 71:35–47. doi: 10.1111/j.1365-2958.2008.06503.x
Keywords: oral host-microbial interactome, oral-systemic axis, periodontitis, Porphyromonas gingivalis, Fusobacterium nucleatum, bacterial extracellular vesicles, alveolar bone resorption
Citation: Xu J, Yu L, Ye S, Ye Z, Yang L and Xu X (2024) Oral microbiota–host interaction: the chief culprit of alveolar bone resorption. Front. Immunol. 15:1254516. doi: 10.3389/fimmu.2024.1254516
Received: 07 July 2023; Accepted: 02 February 2024;
Published: 22 February 2024.
Edited by:
Sylvie Bertholet, GSK Vaccines, United StatesReviewed by:
Fernando Gómez-Chávez, National Polytechnic Institute (IPN), MexicoCopyright © 2024 Xu, Yu, Ye, Ye, Yang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luyi Yang, bHl5YW5nQGpsdS5lZHUuY24=; Xiaoxi Xu, eGlhb3hpX3h1MDFAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.