
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 12 March 2024
Sec. NK and Innate Lymphoid Cell Biology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1230735
 Doua Kamyan1†
Doua Kamyan1† Maya Hassane1†
Maya Hassane1† Alanood Alnaqbi1†
Alanood Alnaqbi1† Abdul-Kader Souid2†
Abdul-Kader Souid2† Zakeya Al Rasbi1†
Zakeya Al Rasbi1† Abeer Al Tahrawi3†
Abeer Al Tahrawi3† Mariam Al Shamsi1*†
Mariam Al Shamsi1*†Background: Ozanimod (RPC1063) is an immunomodulator that has been recently approved by the FDA (2020) for the treatment of relapsing-remitting multiple sclerosis (RRMS). It is a selective agonist of the sphingosine-1-phophate receptors 1 and 5, expressed on naïve and central memory T and B cells, as well as natural killer (NK) cells, and is involved in lymphocyte trafficking. Oral administration of ozanimod was reported to result in rapid and reversible reduction in circulating lymphocytes in multiple sclerosis (MS) patients, however, only minimal effect on NK cells was observed. In this study, we sought to investigate the effect of ozanimod on NK cells and assess whether they play any role in ozanimod-induced remission in experimental autoimmune encephalomyelitis (EAE), the animal model of MS.
Methods: Active EAE induction was done in C57BL/6 female mice, followed by daily oral treatment with ozanimod (0.6mg/kg) starting at disease onset (score 1). Flow cytometry of blood and CNS was performed 24 hours after the last oral dose of ozanimod treatment in diseased mice. Histological analysis of lumbar spinal cord was performed for evaluating the level of inflammation and demyelination. Depletion of peripheral NK cells was done using anti-NK1.1 mouse antibody (mAb) at day 5 post-EAE induction.
Results: Ozanimod was effective in reducing the clinical severity of EAE and reducing the percentage of autoreactive CD4+ and CD8+ T cells along with significant inhibition of lymphocyte infiltration into the spinal cord, accompanied by reversed demyelination. Furthermore, ozanimod treatment resulted in a significant increase in the frequency of total NK cells in the blood and CNS along with upregulation of the activating receptor NKG2D on CD27low/- NK cell subset in the CNS. The effectiveness of ozanimod treatment in inhibiting the progression of the disease was reduced when NK cells were depleted using anti-NK1.1 mAb.
Conclusion: The current study demonstrated that ozanimod treatment significantly improved clinical symptoms in EAE mice. Ozanimod and anti-NK1.1 mAb appear to function in opposition to one another. Collectively, our data suggest that ozanimod-mediated remission is associated with an increased percentage of total NK cells and CD27low/- NK cells expressing the activating receptor, NKG2D in the CNS.
Multiple sclerosis (MS) is a chronic inflammatory autoimmune neurodegenerative disease of the central nervous system (CNS) which leads, in many cases, to irreversible disability. Globally, approximately 2.8 million people live with MS, with onset between 20 and 40 years of age, and women being two times more likely to develop the disease than men (1–3). Relapsing-remitting (RR) MS is the most common subtype of MS and constitutes 85–90% of young patients. RRMS manifests as unpredictable and acute attacks (relapses) causing deterioration of CNS functions over time, followed by partial or complete recovery (remissions) (4, 5). The disease onset is thought to be initiated by the activation of autoreactive naive CD4+ T cells against components of the CNS in the periphery and subsequent infiltration of the activated lymphocytes into the CNS through disrupted blood brain barrier (BBB). This is also thought to be followed by expansion of local and infiltrating peripheral leukocytes and their contribution to the inflammatory milieu within the CNS, resulting in demyelination and axonal damage (6, 7). MS has been studied using different animal models, with experimental autoimmune encephalomyelitis (EAE) being the most commonly employed murine model to understand MS pathogenesis and possible therapeutic targets (8).
The sphingolipid sphingosine-1-phosphate (S1P) is known for its important function in regulating immunity through affecting trafficking, differentiation, and survival of immune cells. Expression of S1PRs on lymphocytes is essential for regulation of their homing and migration through the primary and secondary lymphoid organs, as well as inflamed sites (9). There are different S1PRs expressed by lymphocytes such as T cells (S1PR1 and S1PR4), B cells (S1PR1, S1PR2, S1PR3, and S1PR4), while NK cells express low levels of S1PR1 and higher levels of S1PR5 (10). In addition, S1PRs are expressed at high levels on CNS cells, including neurons, oligodendrocytes, astrocytes, and microglia, enabling S1PRs agonists to exert different effects within the CNS (11). S1P agonists such as fingolimod and the recently FDA approved drug, ozanimod, have been reported to result in significant amelioration in the severity of MS and EAE, mainly attributed to interfering with infiltration of activated autoreactive T lymphocytes expressing S1PR1 into the CNS (12, 13). Even though the S1P modulators are agonists of the receptor, binding to the S1PRs results in internalization of the receptor-modulator complex into the lymphocytes, preventing their egress from the lymph nodes and therefore acting as an effective antagonist (14, 15). While fingolimod (FTY720), the first approved sphingosine-1-phosphate receptor (S1PR) modulator for treatment of MS, targets cells expressing S1P receptors 1, 3, 4 and 5 (12), ozanimod is more selective, only targeting S1PR1 and S1PR5 (16). Due to its selectivity, ozanimod offers a better risk-benefit profile compared to fingolimod, making treatment with ozanimod safer and more effective (13).
It is noteworthy to mention that treatment with S1PR agonists (ozanimod) also is associated with a significant decrease of circulating lymphocytes counts in a dose-dependent manner. However, rapid lymphocytes count recovery was observed after discontinuation of the treatment because of its short half-life of 19 hours, allowing for once-daily dosing (9, 13).
NK cells are large granulated innate lymphoid cells (ILCs), best known for their ability to identify and induce apoptosis in virus-infected cells and cancer cells (17). Majority of mature NK cells in humans or mice consistently display NK group 2, member D (NKG2D), CD161, NK-cell protein 46 (NKp46), and CD122. Nonetheless, variations in the expression of Ly49 family members, CD127, CD27, and KLRG1 (killer-cell lectin-like receptor subfamily G, member 1) in mice, as well as KIRs (killer-cell immunoglobulin-like receptors), CD56, and CD16 in humans, indicate diversity within the mature NK-cell population (18). Based on the expression of CD56 marker, human NK cells are subdivided into two major cell subsets, CD56dim and CD56bright. The CD56dim NK cell subset represents the mature cytotoxic subset that makes up to 90% of the total circulating NK cells. In contrast, the immature CD56bright NK cell subset is known for its efficiency in cytokines production and represents around 10% of the NK cell pool in the periphery. NK cells in C57BL/6 mice express NK1.1 and CD27, therefore, they can be subdivided into CD27high and CD27low/-, representing the equivalent of human CD56bright and CD56dim NK cell subsets, respectively (18–22). It has been suggested that the two subsets of NK cells, CD27high and CD27low/-, play different roles in the disease progression and at different stages (23). It is noteworthy that the number of circulating CD56bright NK cells in MS patients was suggested as an indicator for therapeutic efficacy since in a number of immunomodulatory treatments (e.g. daclizumab and dimethyl fumarate), the level of CD56bright NK cells was found to be increased. This increase was also accompanied with improved cytotoxic activity towards autoreactive CD4+ T cells and reduced disease severity (24–28). On the contrary, treatment of MS patients with fingolimod was reported to be associated with a decrease in the number of circulating CD56bright NK cells (29, 30), and a significant increase in an aged and less functional phenotype (CD56dim CD94low mature and less functional NK cells) (28).
It has not yet been studied how treatment with the selective S1PR modulator, ozanimod, affects NK cells and their subsets. Aside from ozanimod’s reported effects on other lymphocytes (CD4+, CD8+ T cells, and B cells), we are also interested in whether it is associated with an expansion in a specific NK cell subset.
In the present study, we demonstrated that oral administration of ozanimod is effective in reducing the number of relapses and suppressing the disease progression in EAE C57BL/6 mice through markedly decreasing number of infiltrating immune cells and reversing demyelination. Further, we investigated the effect of ozanimod on NK cells and their activation status. Our findings suggest that ozanimod increases the frequency of NK cells in the blood and CNS and enhances activation of CD27low/- NK cell subset in the CNS, which might be associated with improvements in motor function and overall EAE disability.
A total of 67 C57BL/6 female mice were used at 10 to13 weeks of age. Mice were housed at 22°C with 60% humidity and 12-h light-dark cycles. Rodent chow and filtered water were provided. Wet rodent chow and filtered water were made always accessible to mice with all disease scores.
EAE was induced in C57BL/6 female mice by injecting them with 200µg of MOG35-55 (myelin oligodendrocyte glycoprotein 35-55; AnaSpec, USA) peptide emulsified in incomplete Freund’s adjuvant (IFA) (Sigma, St Louis, MO, USA) containing 3mg/ml inactivated Mycobacterium tuberculosis H37Ra (Becton, Dickinson and company, Sparks, MD 21152 USA) subcutaneously at the back on day 0, followed by intraperitoneal injections of 500ng of pertussis toxin (Sigma, St Louis, MO, USA) on days 0 and 2. Control mice received a subcutaneous injection of IFA at the back. Mice were weighed daily and monitored for disease progression using the standard clinical scoring system of paralytic EAE: score 0, no disease; 1, flaccid tail; 2, hind limbs weakness and wobbly gait; 3, paralysis of hind limbs with residual mobility in both legs; 4, both hind legs completely paralyzed and partial front limb paralysis; 5, moribund or death (7). Wet food pellets were provided inside the cage for mice with advanced disease scores (score 3 and above).
Following disease induction, mice were randomly enrolled into different groups (7-12 mice per group). At the first sign of disease, flaccid tail, diseased mice received daily administration of ozanimod (OZ) (RPC1063) (SelleckChem) at 0.6mg/kg diluted in 5% DMSO, 5% Tween-20 and 90% 0.1N HCl by oral gavage (15) for up to 21 days. The control group was orally treated with the diluent of ozanimod (5% DMSO, 5% Tween-20 and 90% 0.1N HCl).
Mice were euthanized by intraperitoneal injection of ketamine (87.5 mg/kg) and xylazine (12.5mg/kg) (Ilium, Smithfield, NSW, Australia) followed by intracardiac perfusion with PBS. Spinal cord was removed, and the lumbar region (L1-L5) was excised and fixed in 10% formalin overnight at room temperature (RT). A series of paraffin-embedded 7 and 10μm thick lumbar spinal cord tissue sections were cut and deparaffinized in xylene followed by hydration in gradual decreasing concentrations of ethanol. H&E staining was performed on the 7μm tissue sections to assess mononuclear cells infiltration using H&E staining kit (IHC World, USA) following the manufacturer’s protocol. Spinal cord sections of 10μm were stained with Luxol fast blue (LFB) to assess the level of demyelination in diseased mice. Staining was performed using LFB staining kit (IHC World, USA) and following manufacturer’s guidance.
Interpretational analysis of the H&E and LFB stained sections was done in a blinded manner. Inflammatory score was evaluated using the following reporting criteria: 0, no inflammation; 1, cellular infiltration only in the perivascular areas and meninges; 2, mild cellular infiltration (less than one-third part of the white matter is infiltrated with inflammatory cells); 3, moderate cellular infiltration (more than one-third part of the white matter is infiltrated with inflammatory cells); and 4, infiltration of inflammatory cells is observed in the entire white matter (7). The assessment of demyelination was done by estimating the percentage of the demyelinated area. Cresyl violet stained neurons with violet and LFB solution stained myelin with blue-green color, loss of blue-green stain in the white matter is an indication of demyelination.
Brain and spinal cord were removed from euthanized and PBS perfused mice and were cut into small pieces that were mechanically dissociated by syringe plunger and filtered through a 70μm cell strainer. Cell suspension was transferred into a 50ml conical tube, topped with PBS and centrifuged at 1400 rpm for 10 min at 4°C. The cell pellet was resuspended in 20ml PBS + 30% Percoll (GE Healthcare) and was overlaid with 10ml PBS + 70% Percoll and centrifuged at 2800 rpm for 30 min at RT without brake. The myelin on top of the tube was removed and mononuclear cells in the 30–70% Percoll interface were collected, transferred to a 50ml conical tube and washed twice with PBS. Cells were resuspended in 1-2ml of FACS buffer (PBS, 0.5% BSA, and 0.1% NaN3), centrifuged and incubated with FACS antibodies for 25 min at 4°C protected from light.
Cardiac blood was collected in an EDTA tube. 1ml of red blood cell lysis buffer was added per 100μl of blood and was incubated for 5 min at RT. Reaction was stopped by adding 20ml PBS followed by centrifugation at 1400 rpm for 10 min at 4°C. Cell pellet was washed once with PBS then resuspended in 1-2ml of FACS buffer. Cells were then incubated with the antibodies for 25 min at 4°C protected from light.
Antibodies against mouse CD45 (BV605-conjugated, clone 30-F11), CD4 (BV785-conjugated, clone RM4-5), CD8a (APCCY7-conjugated, clone 53-6.7), CD3 (BV421-conjugated, clone 17A2), NK1.1 (FITC-conjugated, clone PK136), CD27 (BV510-conjugated, clone LG.3A10), NKG2A (PE-conjugated, clone 16A11), NKG2D (APC-conjugated, clone CX5) and appropriate isotype controls were purchased from BioLegend (SanDiego, CA, USA). Gating strategies used in this study are presented in Supplementary Figure 1. Data were collected using BD FACSCelesta™ Cell Analyzer from BD Biosciences (Germany) and analyzed using FlowJo-v 10.8.1 (FlowJo, LLC, Ashland, OR, USA). Anti-NK1.1 mAb (clone PK136) and its appropriate isotype control were purchased from BioXcell, USA.
In vivo depletion of NK cells was achieved through i.p. administration of 100µg of anti-NK1.1 mAb or IgG2a isotype control (BioXcell, USA) at day 5 post-EAE induction. Injection was repeated with lowered dose (50µg) of anti-NK1.1 mAb or IgG2a isotype control every 5 days until the end of the experiment (21 days after disease onset) (29).
All statistical analysis were performed using GraphPad Prism V10. Briefly, all data were tested for normality using Shapiro-Wilk test. For non-normally distributed data, a non-parametric test (Mann-Whitney U test) was used. Statistical significance was defined as p- values ≤ 0.05. Data are represented as mean ± SD.
The study was approved by the UAE University Animal Research Ethics Committee (Ref. ERA_2021_7326).
RR-EAE was successfully induced in female C57BL/6 mice through s.c. administration of MOG35-55 peptide emulsified in IFA. Average time for EAE onset in all diseased groups was between days 12 and 15, while peak was between days 18 and 24 (scores 2 and 3) (Figure 1A) in comparison to control mice which maintained a clinical score of 0 throughout the study duration (36 days).
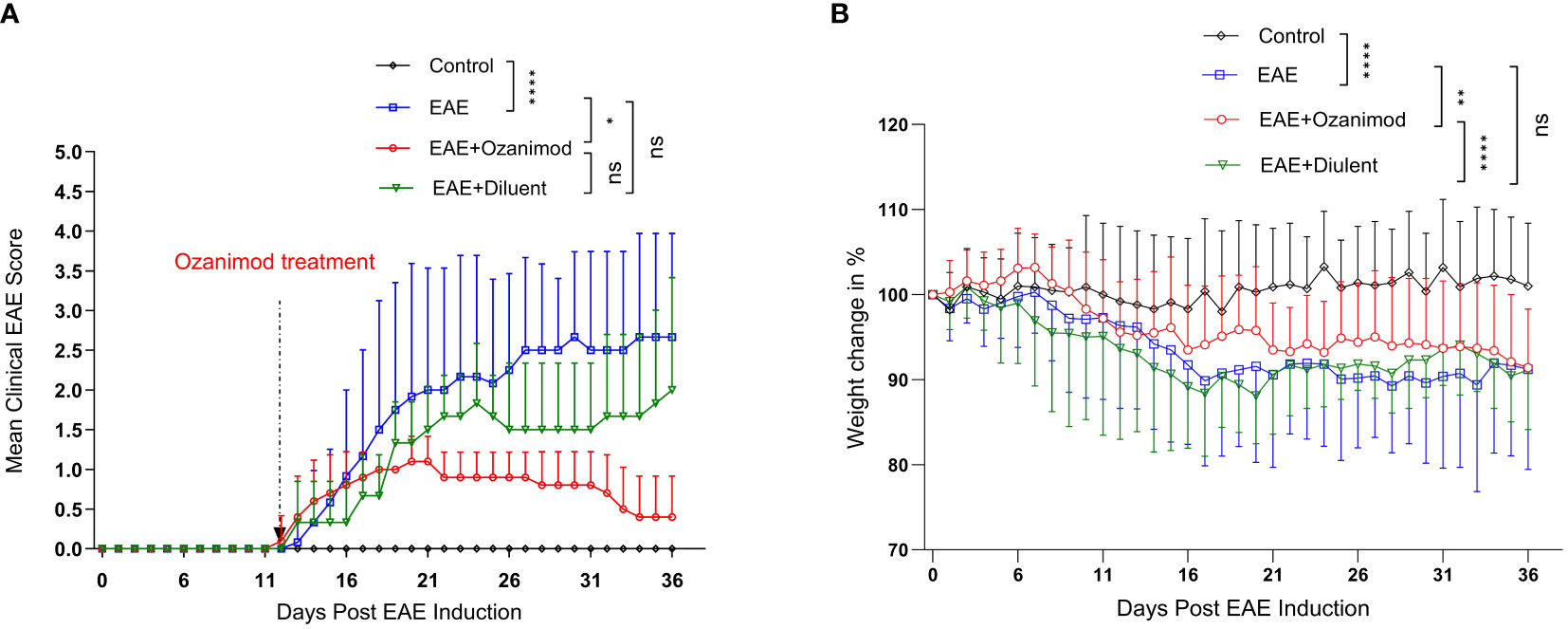
Figure 1 Therapeutic effect of ozanimod (RPC 1063) on the clinical score and body weight of EAE mice. (A) The clinical scores show a statistical difference between ozanimod-treated and untreated diseased mice. (B) The weight changes indicate a significant inhibition on further body weight loss in ozanimod-treated EAE mice compared to untreated mice. Data pooled from two independent experiments are shown (8-12 mice per group). Clinical scores and body weights (Percentage of the weight divided by starting weight) are shown. Data are represented as mean ± SD using Mann-Whitney U test. ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, **** P ≤ 0.0001.
Ozanimod treatment was initiated at disease onset (score 1, limp tail), and carried on for 21 days. The therapeutic potential of ozanimod in halting EAE progression started to become evident at day 22 (mean maximum clinical score MMCS 0.9 ± 0.3) and continued throughout the treatment, by the end of which 60% of the mice were at scores 0 and 40% were at score 1. Disease severity in ozanimod-treated mice was significantly reduced compared to their diseased untreated counterparts (P=0.015). Furthermore, diseased mice from both EAE and EAE+diluent groups experienced relapses starting from day 25, whereas no relapses were observed in the ozanimod-treated group, indicating the effectiveness of ozanimod in preventing relapses (Figure 1A). Individual clinical scores of mice (Supplementary Figure 2) further highlight these findings.
Treatment with ozanimod significantly prevented further weight loss in comparison to the control diseased groups whether treated or not with ozanimod’s diluent (P=0.0001 and 0.001, respectively) (Figure 1B). Diseased mice groups reported significant reduction in the body weight compared to the control group (P<0.0001), with a reduction of 8.8%, and 8.9% in EAE and EAE+diluent groups, respectively, compared to EAE+ozanimod group (7.5%) (Figure 1B). Altogether, these data indicate that treatment with ozanimod significantly improved disease outcome by reducing disease severity and weight loss.
Assessing inflammation (infiltration of mononuclear cells) and demyelination as pathological signs of the disease in the spinal cord sections of control, EAE and EAE+ozanimod mice groups was achieved by analyzing histology sections stained with H&E and LFB. H&E staining showed significant increase in the infiltration mononuclear cells in the white matter and perivascular region of the spinal cords of EAE mice group (mean inflammatory score (MIS) =2.3) compared to control (MIS=0.4) and ozanimod-treated (MIS=0.1) groups (Figures 2A, B). LFB staining revealed a significant increase in the percentage of demyelination, as exhibited by the loss of LFB staining and vacuolization, in the EAE group (38.3%) compared to their control counterparts (10.5%). This percentage was significantly decreased in ozanimod-treated mice (5%) (Figures 2C, D). These results indicate effectiveness of ozanimod in reducing inflammation, presented by mononuclear cell infiltration, and demyelination in the spinal cords of diseased mice.
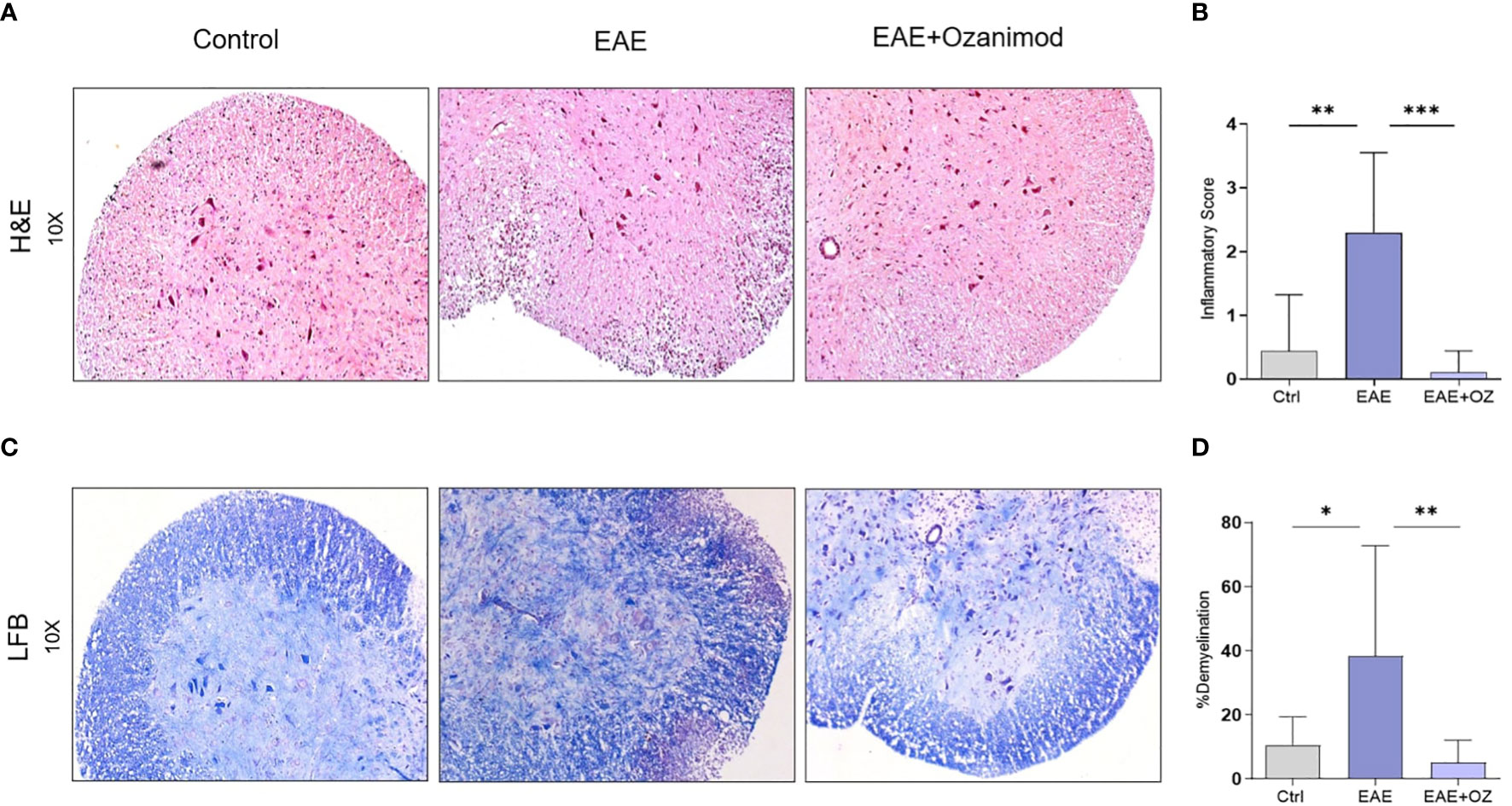
Figure 2 Histological sections of the lumbar region of the spinal cord obtained from control, EAE, and ozanimod-treated EAE mice. (A, B) H&E staining, performed to detect mononuclear cell infiltration level, shows a significant reduction in the given inflammatory score in ozanimod-treated EAE mice compared to untreated mice. (C, D) LFB staining, performed to detect the level of demyelination, indicates decreased demyelination in the white matter in ozanimod-treated EAE mice compared to those untreated as shown by reduced loss in the blue staining. Histological scores and percentages of demyelination are shown. Data are represented as mean ± SD using Mann-Whitney U test. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Changes in the frequency of CD4+ and CD8+ T cells in the blood and CNS were studied as part of confirming the therapeutic potential of ozanimod treatment. Flow cytometry analysis of blood revealed a significant reduction in the frequency of CD4+ T cells in ozanimod-treated EAE mice compared to their untreated counterparts (Figure 3A). Frequency of CD4+ T cells infiltrating the CNS increased significantly in EAE mice compared to control group (P=0.03) but remained unchanged following ozanimod treatment (Figure 3B). Additionally, there was a significant decrease in the frequency of CD8+ T cells in the blood and CNS in ozanimod-treated EAE mice as opposed to their untreated counterparts (Supplementary Figure 3). These results suggest that circulating CD4+ and CD8+ T cells are significantly affected by the immunomodulatory drug ozanimod.
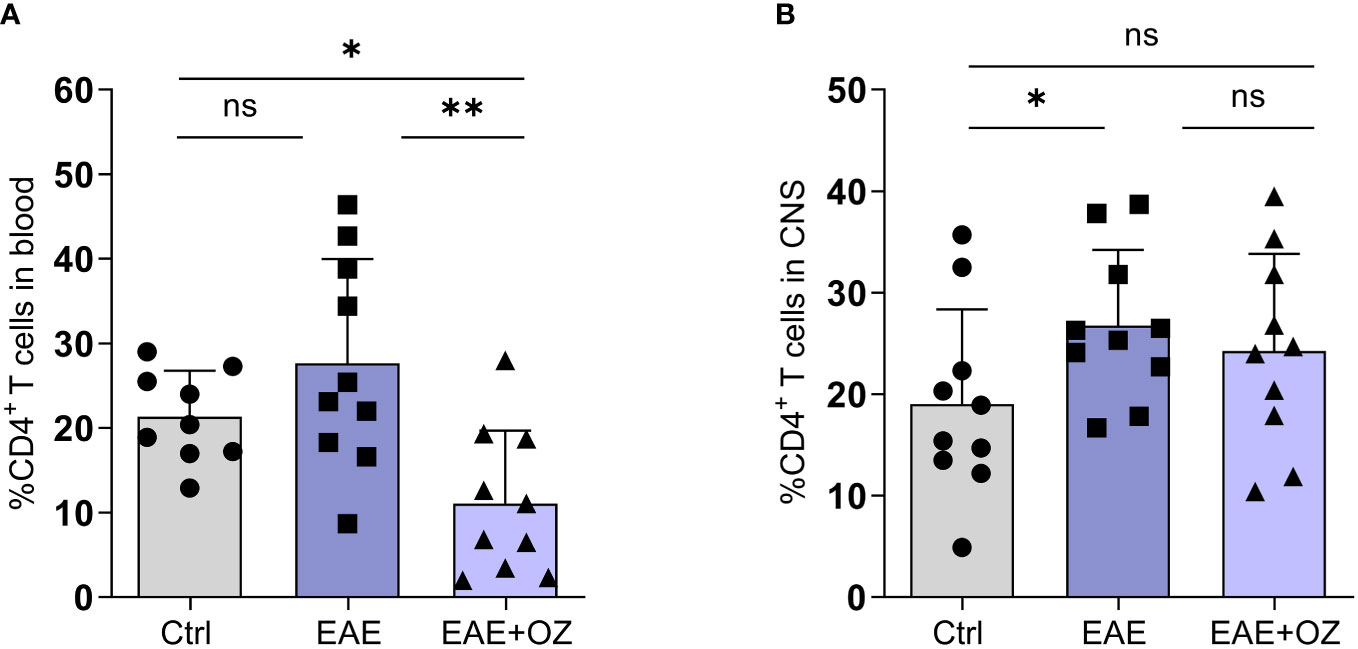
Figure 3 Frequencies of CD4+ T cells in the blood circulation and CNS of EAE mice. (A) Significant decrease in the frequency of circulating CD4+ T cells in ozanimod-treated EAE mice compared to those untreated. (B) No significant changes were detected in the frequency of CNS infiltrating CD4+ T cells in ozanimod-treated EAE mice compared to those untreated. Representative data from two independent experiments are shown (9-10 mice per group). Data are represented as mean ± SD using Mann-Whitney U test. ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01.
Given the contradictory findings reporting on the role of NK cells in EAE in the literature (23, 31, 32), we sought to assess their role in our model and how it is affected by ozanimod treatment. FACS analysis of total circulating and CNS NK cells frequencies showed a marked increase in the mean of peripheral NK cells frequency (37.83%) in mice treated with ozanimod compared to both control (7.60%, P<0.0001) and EAE (12.19%, P=0.0011) mice (Figure 4A). Similarly, the mean of CNS NK cells frequency was significantly higher in ozanimod-treated mice (38.69%) compared to control (13.53%, P<0.0001) and diseased untreated (16.09%, P<0.0001) groups (Figure 4B).
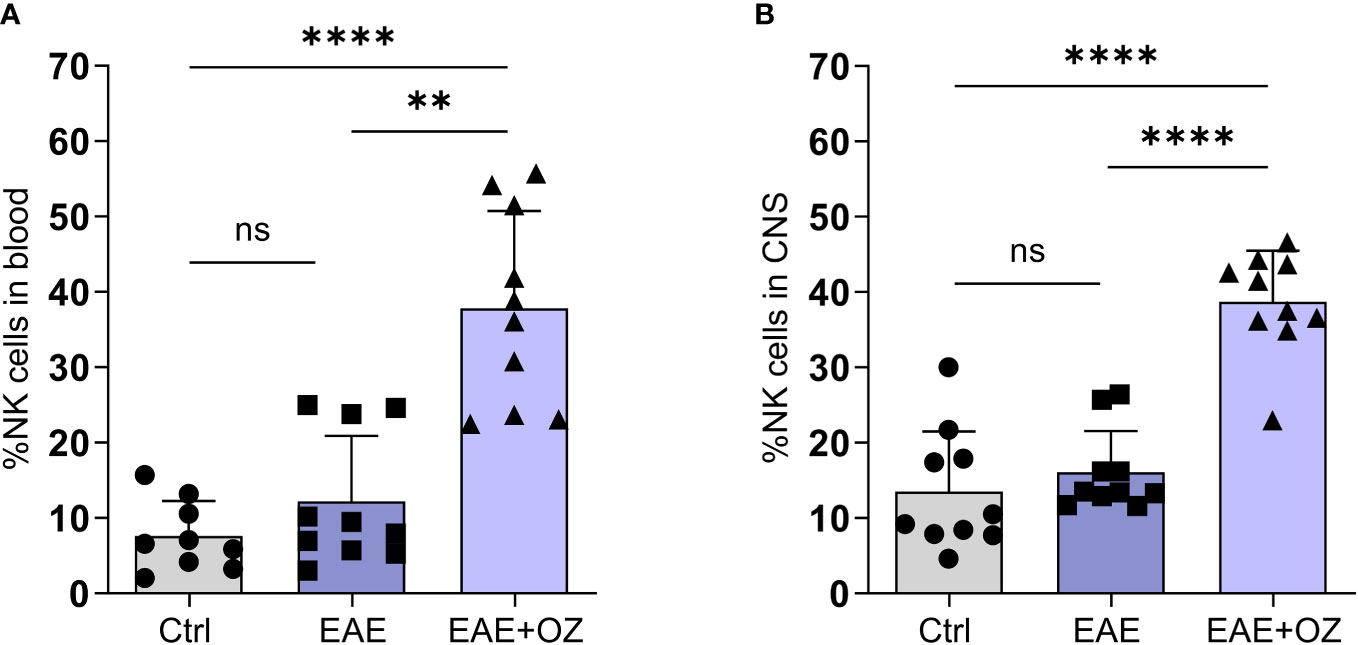
Figure 4 Frequencies of NK cells in the blood circulation and CNS of EAE mice. Treatment with ozanimod resulted in increased frequency of total (A) circulating NK cells and (B) CNS NK cells in EAE mice compared to untreated mice. Representative data from two independent experiments are shown (9-10 mice per group). Data are represented as mean ± SD using Mann-Whitney U test. ns P > 0.05, ** P ≤ 0.01, **** P ≤ 0.0001.
Upon investigating frequencies of NK cell subsets by assessing differential expression of CD27, no significant changes in the frequencies of both NK subsets (CD27high and CD27low/-) were detected in the blood or CNS whether treated or not with ozanimod (Supplementary Figure 4). These data indicate that ozanimod treatment affected total NK cells, but not one subpopulation over the other, by increasing frequencies of both subtypes in the circulation and in CNS.
Assessment of the expression levels of activating (NKG2D) and inhibitory (NKG2A) receptors on NK cells in the circulation and CNS revealed a significant increase in the expression of NKG2A on total circulating NK cells following treatment with ozanimod compared to their levels on NK cells of the control (P<0.0001) and diseased untreated (P=0.006) groups (Figure 5A). Treating diseased mice with ozanimod did not alter the level of expression of the activating receptor NKG2D on circulating NK cells (Figure 5B). However, ozanimod treatment resulted in a significant increase in the level of expression of the inhibitory receptor NKG2A on circulating CD27high NK cells when compared to diseased but untreated mice (P=0.018) (Supplementary Figure 5A). Furthermore, assessment of NKG2D on CD27high NK cells, as well as NKG2D and NKG2A expression on CD27low/- NK cell subsets in the circulation showed no significant changes in response to ozanimod treatment (Supplementary Figures 5B–D).
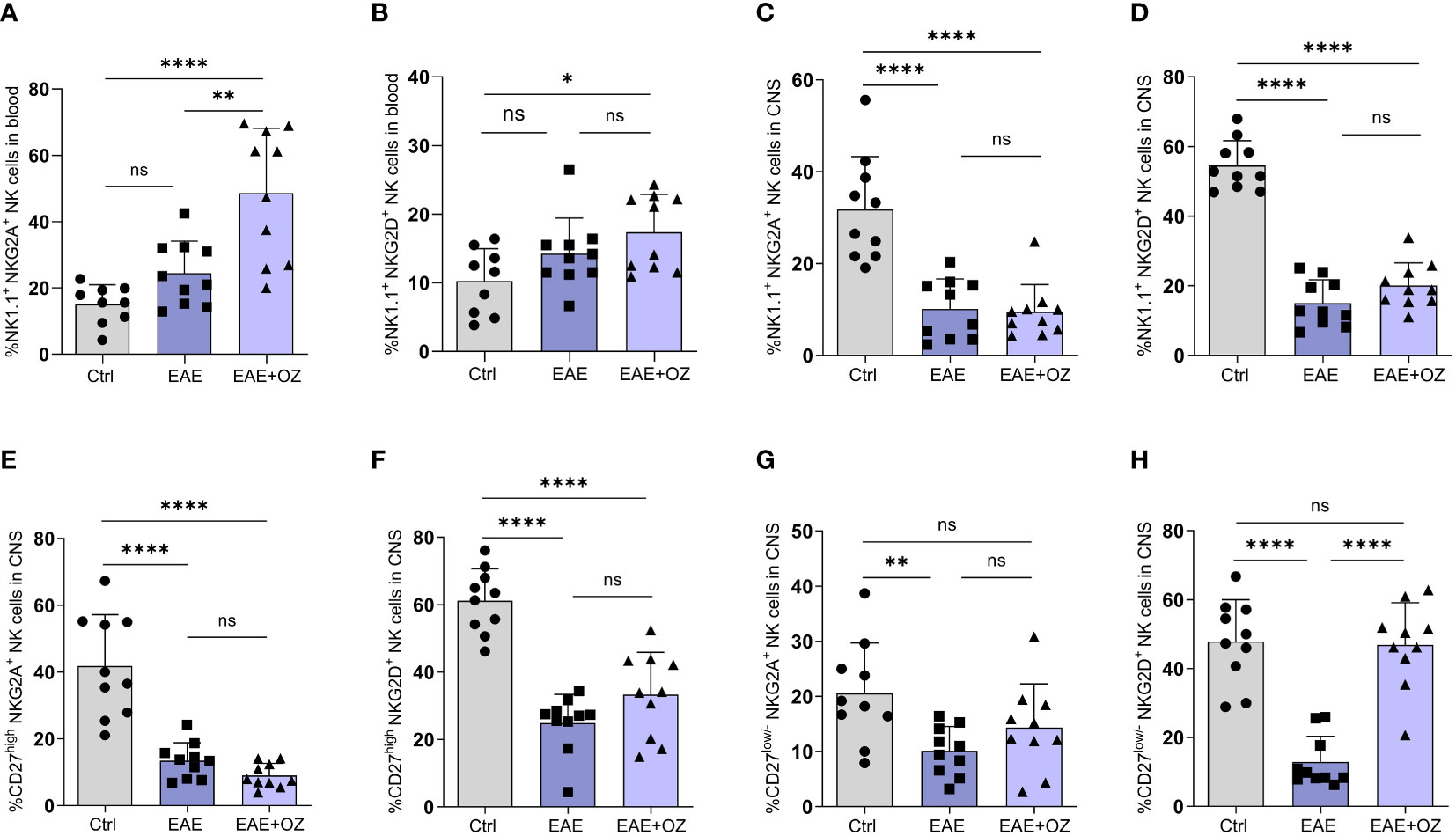
Figure 5 Frequencies of NK cell subsets (CD27high and CD27low) and their level of expression of activating (NKG2D) and inhibitory (NKG2A) markers in the circulation and CNS. (A) Significant increase in the expression of the inhibitory receptor, NKG2A, was detected compared to (B) insignificant change in the expression of the activation receptor, NKG2D, on NK cells in the blood of ozanimod-treated EAE mice compared to those diseased untreated. Insignificant change in the expression of (C) NKG2A and (D) NKG2D on NK cells in the CNS of EAE mice treated with ozanimod compared to those untreated. Insignificant changes in the level of expression of (E) NKG2A, and (F) NKG2D on CD27high NK cell subset in the CNS of EAE mice treated with ozanimod compared to untreated mice. (G) No detectable changes in the level of NKG2A compared to (H) significant increase in the expression of NKG2D on CD27low/- NK cell subset in the CNS of ozanimod-treated EAE mice compared to those untreated. Representative data from two independent experiments are shown (9-10 mice per group). Data are represented as mean ± SD using Mann-Whitney U test. ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, **** P ≤ 0.0001.
Expression of both receptors on total NK cells in the CNS did not seem to be affected following treatment with ozanimod compared to EAE mice (Figures 5C, D). However, while expression of both NKG2D and NKG2A on CD27high NK cell subset (Figures 5E, F), as well as the expression of NKG2A on CD27low/- NK cell subset (Figure 5G), was not affected by ozanimod treatment in comparison to diseased mice, expression of NKG2D was significantly increased (P <0.0001) (Figure 5H). In summary, the present findings suggest that ozanimod has little or no effect on peripheral NK cells activity, but can enhance the activity of NK cells in the CNS by selectively altering the expression of the activating receptor NKG2D on the surface of CD27low/- NK cell subset.
The role of NK cells in EAE and in response to treatment with ozanimod was further investigated by depleting NK cells in diseased mice. Successful depletion of circulating NK cells was confirmed by flow cytometry where levels of circulating NK cells in mice treated with anti-NK1.1 mAb showed significant decrease in their percentage (0.72%) compared to control mice (4.84%), which counts for ~85% depletion (Figure 6A). Our findings indicate that depleting NK cells at the pre-onset stage [5 days post-induction (dpi)] did not impact the disease onset compared to diseased mice treated with the isotype control, however, it significantly increased the severity of EAE (P=0.018) (Figure 6B). Furthermore, anti-NK1.1 mAb treated group exhibited higher clinical scores compared to the group with the combined treatment (ozanimod and anti-NK1.1 mAb) (P=0.011), whereas ozanimod treatment alone resulted in significantly lower clinical scores compared to the combined treatment (P=0.009) (Figure 6B). This suggests that NK cells play a significant role in maximizing ozanimod’s effectiveness against EAE.
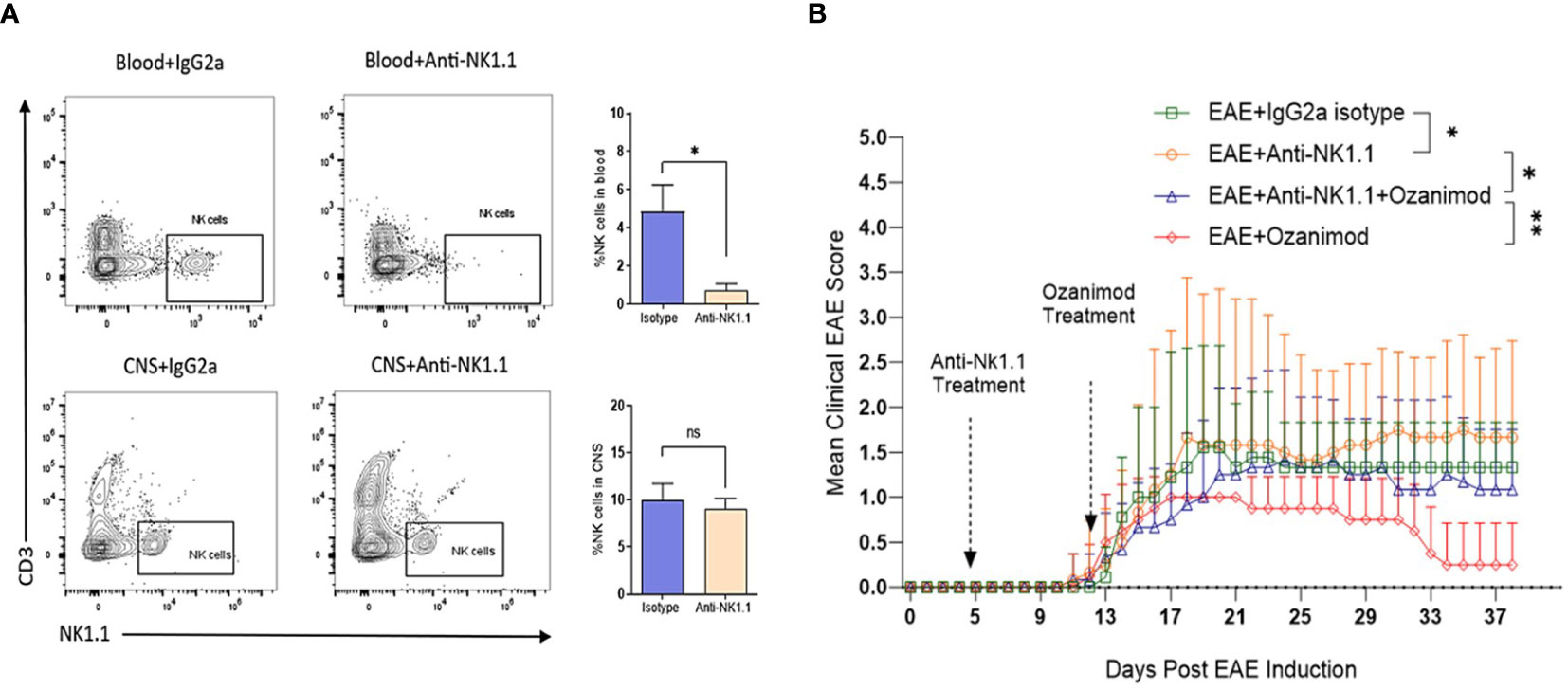
Figure 6 Effect of anti-NK1.1 mAb treatment on the clinical score and body weight of EAE mice. (A) Representative flow cytometry profiles of NK cells gated in the blood and CNS of 10-13 weeks old female C57BL/6 mice indicating successful depletion of NK cells in mice treated with anti-NK1.1 mAb in the blood compared to isotype control (85% depletion as represented by the bar graph). Treatment with the depleting antibody did not yield significant depletion of NK cells in the CNS. (B) Pre-onset depletion of NK cells using anti-NK1.1 mAb reduced the therapeutic effect of ozanimod as it almost failed to halt progress in the clinical score of EAE mice. Data from two independent experiments are shown (8-12 mice per group). Data are represented as mean ± SD using Mann-Whitney U test. ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01.
Impact of NK cells depletion on CD4+ and CD8+ T cells population in the CNS and the circulation was assessed alone or in combination with ozanimod treatment. Frequencies of circulating CD4+ T cells frequencies were markedly decreased in diseased mice depleted of NK cells and treated with ozanimod (6.88%) compared to their untreated counterparts (17.45%) (P=0.001) (Figure 7A). However, the depletion of NK cells, whether alone or combined with ozanimod, did not significantly change the frequencies of CD4+ T cells in the CNS (Figure 7B). Levels of CD8+ T cells did not change significantly between NK cell-depleted EAE groups, neither in the blood nor in the CNS (Supplementary Figure 6). This suggests that the effect of ozanimod on CD4+ and CD8+ T cells was not impacted by the depletion of NK cells.
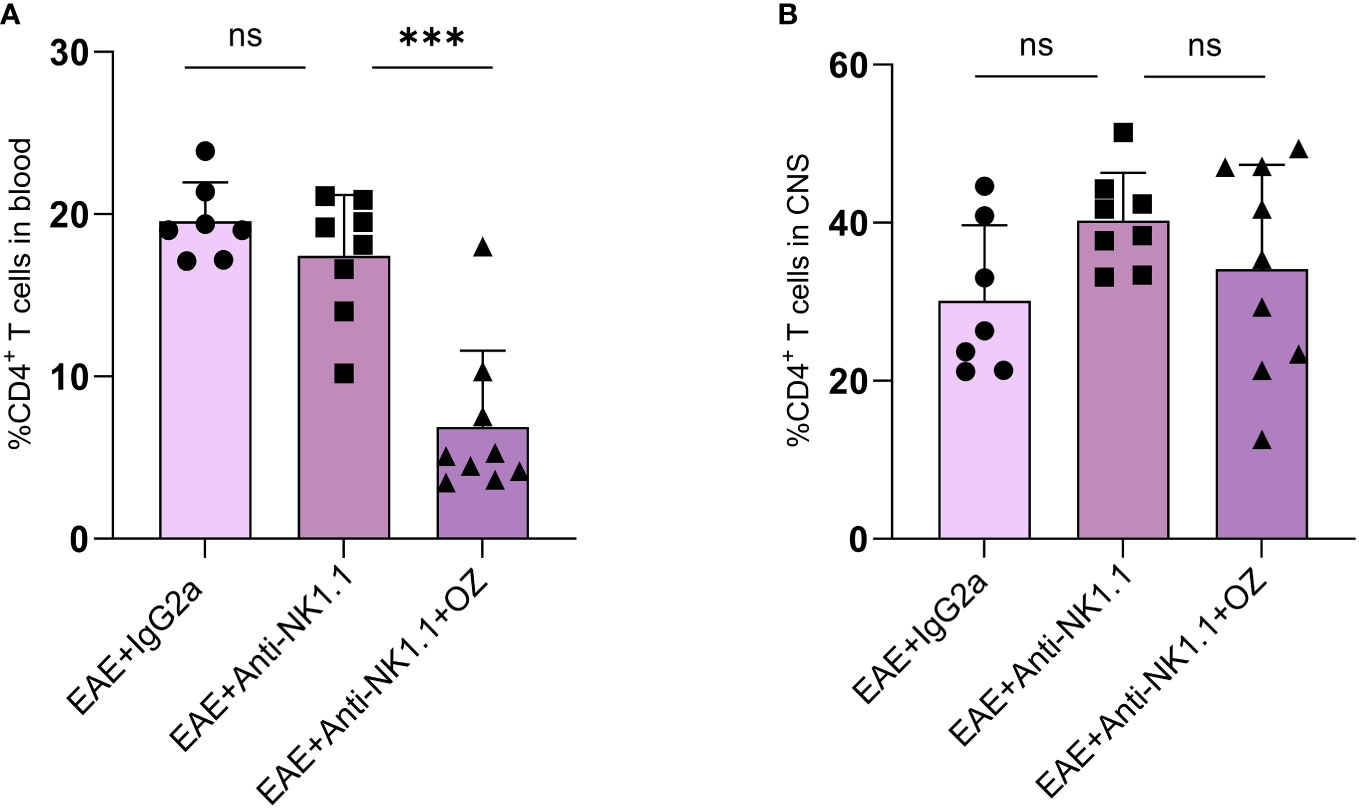
Figure 7 Percentage of CD4+ T cells in the blood circulation and CNS of EAE mice following depletion of NK cells using anti-NK1.1 mAb alone or combined with ozanimod treatment. (A) Combined treatment of anti-NK1.1 mAb and ozanimod in EAE mice resulted in a significant decrease in the percentage of circulating CD4+ T cells but had no effect on the frequency of CD4+ T cells in the CNS (B), compared to mice treated with anti-NK1.1 mAb alone. Data from two independent experiments are shown (7-10 mice per group). Data are represented as mean ± SD using Mann-Whitney U test. ns P > 0.05, *** P ≤ 0.001.
Circulating NK cells were successfully depleted in mice treated with anti-NK1.1 mAb compared to the control group (Figure 8A). Interestingly, diseased mice depleted of NK cells reported a marked increase in the frequency of NK cells following treatment with ozanimod (35.6%) compared to the untreated group (6.28%) (P=0.0002) (Figure 8A). These results may suggest an antagonistic effect of ozanimod on the depleting effect of anti-NK1.1 mAb on circulating NK cells. Notably, frequency of NK cells in the CNS did not significantly change among the different groups (Figure 8B). In addition, even though the antagonistic effect of ozanimod treatment on NK cells depletion was evident in the total circulating NK cells, it did not induce any significant changes in the percentage of NK cell subsets whether in the circulation or the CNS (Supplementary Figure 7).
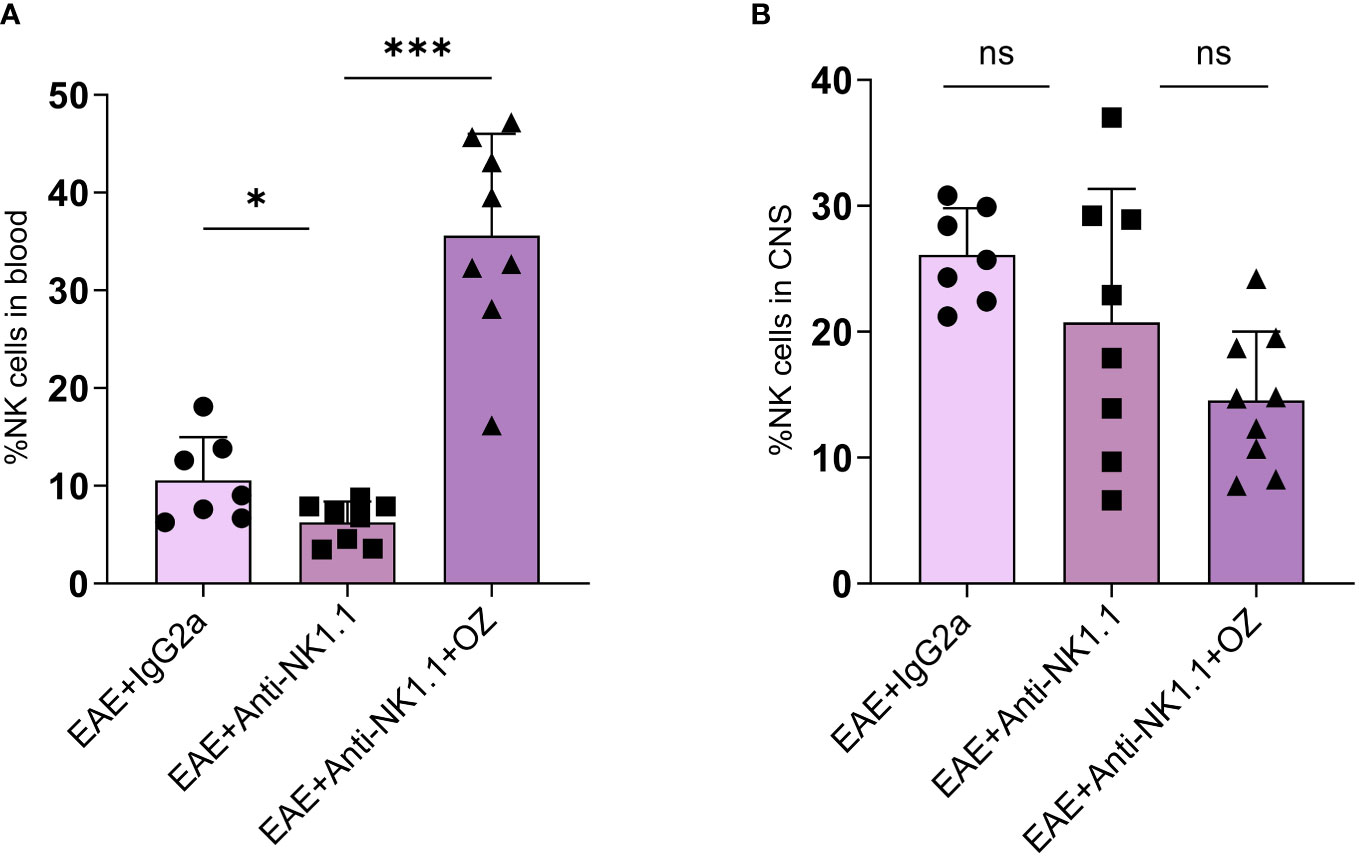
Figure 8 Percentage of NK cells in the blood and CNS of EAE mice treated with anti-NK1.1 mAb alone or combined with ozanimod. (A) Treatment with ozanimod following treatment with anti-NK1.1 mAb inhibited the depleting effect of anti-NK1.1 mAb. Combined treatment resulted in a significant increase in the percentage of NK cells in the blood compared to those treated only with anti-NK1.1 mAb (B) No significant changes in the percentage of NK cells in the CNS of EAE mice receiving the combined treatment of anti-NK1.1 mAb and ozanimod. Data from two independent experiments are shown (7-10 mice per group). Data are represented as mean ± SD using Mann-Whitney U test. ns P > 0.05, * P ≤ 0.05, *** P ≤ 0.001.
The expression of NKG2A and NKG2D on circulating NK cells did not show any significant change between EAE mice treated with anti-NK1.1 mAb and those treated with anti-NK1.1 mAb and ozanimod (Supplementary Figures 8A, B). The combined treatment of ozanimod and anti-NK1.1 mAb did not have any extra effect on NKG2A expression on CNS NK cells (Figure 9A) but significantly increased NKG2D expression (Figure 9B) compared to anti-NK1.1-treated mice. Furthermore, NKG2A and NKG2D expression on circulating CD27high NK cells significantly decreased with anti-NK1.1 treatment compared to the isotype control, remaining unchanged following ozanimod treatment (Supplementary Figures 8C, D). Anti-NK1.1 treatment decreased the expression of NKG2A and NKG2D on circulating CD27low/- NK cells compared to the isotype control, and the expression of NKG2D further decreased following the combined treatment (Supplementary Figures 8E, F). In contrast, ozanimod treatment increased the frequency of CNS CD27high NK cell subset expressing NKG2A and NKG2D compared to the group treated with anti-NK1.1 mAb (Figures 9C, D). NKG2A expression on CD27low/- NK cell subset in the CNS increased significantly in mice that received the combined treatment of anti-NK1.1 mAb and ozanimod compared to those that received anti-NK1.1 mAb only (P=0.002) (Figure 9E). Though, significantly reduced percentage of CNS CD27low/- NK cell subset expressing NKG2D was detected in diseased mice with combined treatment compared to the group treated with anti-NK1.1 mAb (P=0.013) (Figure 9F). These results indicate that the combined treatment of ozanimod and anti-NK1.1 mAb resulted mainly in an inhibition of the CD27low/- NK cell subset in the CNS, compared to mice treated with anti-NK1.1 mAb alone.
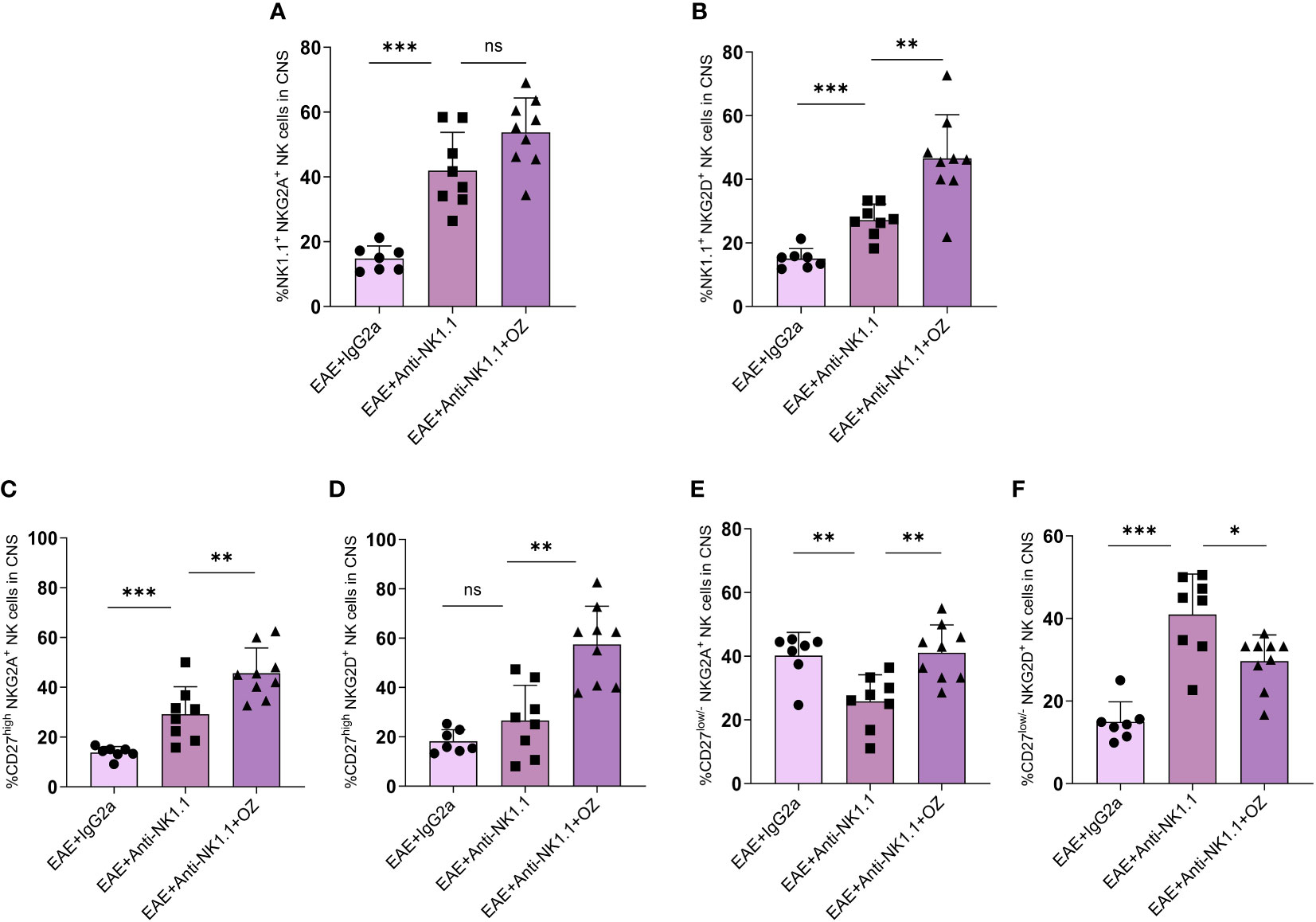
Figure 9 Expression levels of the activating receptor NKG2D and the inhibitory receptor NKG2A on total NK cells and their subsets in the CNS following treatment with anti-NK1.1 mAb alone or combined with ozanimod in EAE mice. (A) No detectable effect of the combined treatment of anti-NK1.1 and ozanimod on the expression of NKG2A on NK cells in the CNS compared to (B) a significant increase in the expression of NKG2D on NK cells, in comparison to mice treated with anti-NK1.1 mAb alone. Significant increase in the expression of (C) NKG2A and (D) NKG2D on CD27high NK cell subset in the CNS of mice treated with anti-NK1.1 mAb combined with ozanimod compared to those treated with anti-NK1.1 mAb alone. Significant (E) increase of NKG2A and (F) reduction of NKG2D expression on CD27low/- NK cell subset in the CNS of EAE mice following combined treatment with anti-NK1.1 mAb and ozanimod compared to mice treated with anti-NK1.1 mAb alone. Data from two independent experiments are shown (7-10 mice per group). Data are represented as mean ± SD using Mann-Whitney U test. ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Intensive investigations on the role of NK cells in MS/EAE by other researchers yielded contradictory findings. The focus of our current study was on investigating the effect of ozanimod treatment on disease progression and on NK cells in particular. Following our findings that ozanimod treatment affects NK cells and the expression of their activating and inhibitory receptors, further investigations were conducted to understand whether NK cells contribute to the therapeutic outcome of ozanimod treatment or not. Our findings on the therapeutic potentials of ozanimod on reducing clinical scores of EAE were in line with previously reported findings by Scott and his team (15) using two different doses of ozanimod (0.2mg/kg and 0.6mg/kg). We also showed that treatment with ozanimod was effective in alleviating inflammation and demyelination associated with advanced clinical scores in diseased mice.
Our results were also in agreement with previously reported findings of significantly reduced counts of circulating CD4+ and CD8+ T cells seen following treatment with ozanimod and other S1PR1 agonists (15, 33). Our results did not reveal any impact of the treatment on the frequencies of CNS-infiltrating activated CD4+ T cells (Figure 3B). These cells, identified as CCR7- lymphocytes, were previously documented as unaffected by S1PRs modulators (34–36), given the S1P-independent recirculation of activated lymphocytes (CCR7-) (12, 37). In addition, we did not look at the subsets of CD4+ T cells, which could explain why we did not see any significant change in the frequency of CNS infiltrating CD4+ T cells.
With regards to NK cells, our results have shown a significant increase of total NK cells following ozanimod treatment. It is worth mentioning that a similar observation was reported by Schwichtenberg et al. on the immunomodulator closely related to ozanimod, fingolimod (28), where frequencies of circulating NK cells were found to be markedly increased in MS patients treated with fingolimod. However, this was not mirrored by a matching increase in the percentage of either of the NK cell subsets (CD27high and CD27low/-). This may be explained by the fact that both immunomodulators are known to interact with S1PR1 and 5 on CCR7-expressing cells but both CCR7 and S1PRs are not co-expressed on NK cell subsets. While CD27low/- NK cell subset expresses high levels of S1PR-5 and low levels of CCR7 (27), CD27high NK cell subset expresses high levels of CCR7 and low levels of S1PR-5 (38, 39). Moreover, no changes were detected in the frequencies of circulating NK cells expressing NKG2D in diseased and ozanimod-treated mice, but a significant increase in the percentage of NK cells, particularly the CD27high NK cell subset, expressing NKG2A was observed, suggesting exhaustion status or suppression of circulating NK cells (Figures 5A, B; Supplementary Figure 5A). However, treatment with ozanimod particularly activated the CD27low/- NK cell subset in the CNS (Figure 5H). Nevertheless, combining ozanimod with anti-NK1.1 mAb resulted in a reversal of the expression of NKG2D receptor on this subset (Figure 9F). This reversal appears to be linked to the therapeutic potential of ozanimod, as indicated by our findings.
Following to the initially observed elevation in the frequency of NK cells in diseased mice, we investigated whether that increase plays a role towards the detected therapeutic outcome of ozanimod treatment. The findings from our study on depleting NK cells indicate that the efficacy of ozanimod treatment is reduced when combined with NK cells depleting antibody. Our findings confirmed a regulatory role of NK cells since their depletion yielded higher clinical scores (Figure 6B). This is consistent with the findings by others, where depletion of NK cells resulted in severe progression of EAE (29, 39, 40). In addition, our results showed that ozanimod alone improved the clinical score more efficiently than when it is combined with anti-NK1.1 mAb in EAE mice, suggesting that NK cells play a role in the ozanimod induced remission of EAE (Figure 6B).
Our findings also revealed that even though frequency of CNS NK cells did not change following peripheral NK cell depletion (Figure 8B), their activity status was altered as shown by increased expression of NKG2D on total NK cells following the combined treatment of anti-NK1.1 mAb and ozanimod. However, this increase was confirmed in the CD27high NK cell subset, whereas the CD27low/- NK cell subset exhibited lower levels (Figure 9). It is noteworthy that the severity of EAE was inversely proportional to the level of expression of NKG2D on CNS CD27low/- NK cell subset supporting the suggestion that upregulation of this receptor on CNS CD27low/- NK cell subset is crucial for improving disease outcome and maybe delaying the relapses in EAE mice. It is important to note that with the limited number of studies investigating the effect of ozanimod treatment in EAE, our findings serve to support the therapeutic potentials of the drug and report the novel finding of the role of NK cells in impacting the treatment. Though, future studies are needed to validate the findings and study the mechanism by which the two treatments counteract each other’s effect. Furthermore, exploring the mechanism by which ozanimod impacts the activation and inhibition receptors of NK cells would be intriguing.
In conclusion, the immunomodulatory drug ozanimod demonstrated significant disease ameliorating potentials in EAE. This therapeutic effect is believed to be associated with enhanced expression of NKG2D on CD27low/- NK cell subset in the CNS. Further, results from the NK cell depletion study confirm the crucial role of NK cells in boosting the efficacy of ozanimod treatment in reducing the severity of EAE.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was approved by Ethical approval committee, College of Medicine and Health Sciences, United Arab Emirates University. The study was conducted in accordance with the local legislation and institutional requirements.
AK-S and MS conceived and together with MH supervised the study. DK, MH, and AN performed the experiments. DK, MH, MS, AT, and ZR analyzed the data. DK and MH wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by a grant from the College of Medicine and Health Sciences, UAE University (Fund number: 31M470).
The authors wish to thank Mr. Mohammed Ashraf Al-Sbiei and Ms. Sumaya Zoughbor from the department of Medical Microbiology and Immunology and Mr. Hayate Abdul Qayume from the department of Anatomy for their great support in the practical work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1230735/full#supplementary-material
1. Rodi M, Dimisianos N, De Lastic AL, Sakellaraki P, Deraos G, Matsoukas J, et al. Regulatory cell populations in relapsing-remitting multiple sclerosis (RRMS) patients: effect of disease activity and treatment regimens. Int J Mol Sci (2016) 17(9):1398. doi: 10.3390/ijms17091398
2. Sparaco M, Bonavita S. The role of sex hormones in women with multiple sclerosis: From puberty to assisted reproductive techniques. Front Neuroendocrinol (2021) 60:100889. doi: 10.1016/j.yfrne.2020.100889
3. Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult Scler J (2020) 26(14):1816–21. doi: 10.1177/1352458520970841
4. Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol (2011) 164(4):1079–106. doi: 10.1111/j.1476-5381.2011.01302.x
5. Steinman L. Immunology of relapse and remission in multiple sclerosis. Annu Rev Immunol (2014) 32(1):257–81. doi: 10.1146/annurev-immunol-032713-120227
6. Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron (2018) 97(4):742–68. doi: 10.1016/j.neuron.2018.01.021
7. Sen MK, Almuslehi MSM, Shortland PJ, Coorssen JR, Mahns DA. Revisiting the pathoetiology of multiple sclerosis: has the tail been wagging the mouse? Front Immunol (2020) 11:2374. doi: 10.3389/fimmu.2020.572186
8. Burrows DJ, McGown A, Jain SA, De Felice M, Ramesh TM, Sharrack B, et al. Animal models of multiple sclerosis: From rodents to zebrafish. Mult Scler J (2019) 25(3):306–24. doi: 10.1177/1352458518805246
9. Chi H. Sphingosine 1-phosphate and immune regulation: trafficking and beyond. Trends Pharmacol Sci (2011) 32(1):16–24. doi: 10.1016/j.tips.2010.11.002
10. Blaho VA, Hla T. An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res (2014) 55(8):1596–608. doi: 10.1194/jlr.R046300
11. Healy LM, Antel JP. Sphingosine-1-phosphate receptors in the central nervous and immune systems. Curr Drug Targets (2016) 17(16):1841–50. doi: 10.2174/1389450116666151001112710
12. Chaudhry BZ, Cohen JA, Conway DS. Sphingosine 1-phosphate receptor modulators for the treatment of multiple sclerosis. Neurotherapeutics (2017) 14(4):859–73. doi: 10.1007/s13311-017-0565-4
13. Lassiter G, Melancon C, Rooney T, Murat AM, Kaye JS, Kaye AM, et al. Ozanimod to treat relapsing forms of multiple sclerosis: A comprehensive review of disease, drug efficacy and side effects. Neurol Int (2020) 12(3):89–108. doi: 10.3390/neurolint12030016
14. Park SJ, Im DS. Sphingosine 1-phosphate receptor modulators and drug discovery. Biomol Ther (2017) 25(1):80–90. doi: 10.4062/biomolther.2016.160
15. Scott FL, Clemons B, Brooks J, Brahmachary E, Powell R, Dedman H, et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br J Pharmacol (2016) 173(11):1778–92. doi: 10.1111/bph.13476
16. Surapaneni S, Yerramilli U, Bai A, Dalvie D, Brooks J, Wang X, et al. Absorption, metabolism, and excretion, in vitro pharmacology, and clinical pharmacokinetics of ozanimod, a novel sphingosine 1-phosphate receptor modulator. Drug Metab Dispos (2021) 49(5):405–19. doi: 10.1124/dmd.120.000220
17. Butler NS, Harty JT. A “memorable” NK cell discovery. Cell Res (2009) 19(3):277–8. doi: 10.1038/cr.2009.23
18. Kaur G, Trowsdale J, Fugger L. Natural killer cells and their receptors in multiple sclerosis. Brain (2013) 136(9):2657–76. doi: 10.1093/brain/aws159
19. Gianchecchi E, Delfino DV, Fierabracci A. Natural killer cells: potential biomarkers and therapeutic target in autoimmune diseases? Front Immunol (2021) 12:616853. doi: 10.3389/fimmu.2021.616853
20. Laroni A, Armentani E, Kerlero de Rosbo N, Ivaldi F, Marcenaro E, Sivori S, et al. Dysregulation of regulatory CD56(bright) NK cells/T cells interactions in multiple sclerosis. J Autoimmun (2016) 72:8–18. doi: 10.1016/j.jaut.2016.04.003
21. Poli A, Kmiecik J, Domingues O, Hentges F, Bléry M, CheKenya M, et al. NK cells in central nervous system disorders. J Immunol (2013) 190(11):5355–62. doi: 10.4049/jimmunol.1203401
22. Van Kaer L, Postoak JL, Wang C, Yang G, Wu L. Innate, innate-like and adaptive lymphocytes in the pathogenesis of MS and EAE. Cell Mol Immunol (2019) 16(6):531–9. doi: 10.1038/s41423-019-0221-5
23. Gao M, Yang Y, Li D, Ming B, Chen H, Sun Y, et al. CD27 natural killer cell subsets play different roles during the pre-onset stage of experimental autoimmune encephalomyelitis. Innate Immun (2016) 22(6):395–404. doi: 10.1177/1753425916658111
24. Marastoni D, Buriani A, Pisani AI, Crescenzo F, Zuco C, Fortinguerra S, et al. Increased NK cell count in multiple sclerosis patients treated with dimethyl fumarate: A 2-year longitudinal study. Front Immunol (2019) 10:1666. doi: 10.3389/fimmu.2019.01666
25. Martínez-Rodríguez JE, López-Botet M, Munteis E, Rio J, Roquer J, Montalban X, et al. Natural killer cell phenotype and clinical response to interferon-beta therapy in multiple sclerosis. Clin Immunol Orlando Fla (2011) 141(3):348–56. doi: 10.1016/j.clim.2011.09.006
26. Caruana P, Lemmert K, Ribbons K, Lea R, Lechner-Scott J. Natural killer cell subpopulations are associated with MRI activity in a relapsing-remitting multiple sclerosis patient cohort from Australia. Mult Scler Houndmills Basingstoke Engl (2017) 23(11):1479–87. doi: 10.1177/1352458516679267
27. Johnson TA, Evans BL, Durafourt BA, Blain M, Lapierre Y, Bar-Or A, et al. Reduction of the peripheral blood CD56(bright) NK lymphocyte subset in FTY720-treated multiple sclerosis patients. J Immunol Baltim Md 1950 (201) 187(1):570–9. doi: 10.4049/jimmunol.1003823
28. Schwichtenberg SC, Wisgalla A, Schroeder-Castagno M, Alvarez-González C, Schlickeiser S, Siebert N, et al. Fingolimod therapy in multiple sclerosis leads to the enrichment of a subpopulation of aged NK cells. Neurother J Am Soc Exp Neurother (2021) 18(3):1783–97. doi: 10.1007/s13311-021-01078-7
29. Hao J, Liu R, Piao W, Zhou Q, Vollmer TL, Campagnolo DI, et al. Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J Exp Med (2010) 207(9):1907–21. doi: 10.1084/jem.20092749
30. Huang D, Shi FD, Jung S, Pien GC, Wang J, Salazar-Mather TP, et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J (2006) 20(7):896–905. doi: 10.1096/fj.05-5465com
31. Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet (2017) 389(10076):1347–56. doi: 10.1016/S0140-6736(16)32388-1
32. Jiang W, Chai NR, Maric D, Bielekova B. Unexpected role for granzyme K in CD56bright NK cell-mediated immunoregulation of multiple sclerosis. J Immunol (2011) 187(2):781–90. doi: 10.4049/jimmunol.1100789
33. Aoki M, Aoki H, Ramanathan R, Hait NC, Takabe K. Sphingosine-1-phosphate signaling in immune cells and inflammation: roles and therapeutic potential. Mediators Inflamm (2016) 2016:8606878. doi: 10.1155/2016/8606878
34. Brinkmann V. FTY720 (fingolimod) in Multiple Sclerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol (2009) 158(5):1173–82. doi: 10.1111/j.1476-5381.2009.00451.x
35. Harris S, Tran JQ, Southworth H, Spencer CM, Cree BAC, Zamvil SS. Effect of the sphingosine-1-phosphate receptor modulator ozanimod on leukocyte subtypes in relapsing MS. Neurol Neuroimmunol Neuroinflamm (2020) 7(5). doi: 10.1212/NXI.0000000000000839
36. McGinley MP, Cohen JA. Sphingosine 1-phosphate receptor modulators in multiple sclerosis and other conditions. Lancet (2021) 398(10306):1184–94. doi: 10.1016/S0140-6736(21)00244-0
37. Berahovich RD, Lai NL, Wei Z, Lanier LL, Schall TJ. Evidence for NK cell subsets based on chemokine receptor expression. J Immunol Baltim Md 1950 (2006) 177(11):7833–40. doi: 10.4049/jimmunol.177.11.7833
38. Campbell JJ, Qin S, Unutmaz D, Soler D, Murphy KE, Hodge MR, et al. Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J Immunol Baltim Md 1950 (2001) 166(11):6477–82. doi: 10.4049/jimmunol.166.11.6477
39. Matsumoto Y, Kohyama K, Aikawa Y, Shin T, Kawazoe Y, Suzuki Y, et al. Role of natural killer cells and TCR gamma delta T cells in acute autoimmune encephalomyelitis. Eur J Immunol (1998) 28(5):1681–8. doi: 10.1002/(SICI)1521-4141(199805)28:05<1681::AID-IMMU1681>3.0.CO;2-T
Keywords: multiple sclerosis, experimental autoimmune encephalomyelitis, natural killer cells, ozanimod, sphingosine-1-phosphate, sphingosine-1-phosphate receptor
Citation: Kamyan D, Hassane M, Alnaqbi A, Souid A-K, Rasbi ZA, Tahrawi AA and Shamsi MA (2024) Ozanimod-mediated remission in experimental autoimmune encephalomyelitis is associated with enhanced activity of CNS CD27low/- NK cell subset. Front. Immunol. 15:1230735. doi: 10.3389/fimmu.2024.1230735
Received: 01 June 2023; Accepted: 30 January 2024;
Published: 12 March 2024.
Edited by:
Lan Wu, Vanderbilt University Medical Center, United StatesReviewed by:
Qiang Liu, Tianjin Medical University General Hospital, ChinaCopyright © 2024 Kamyan, Hassane, Alnaqbi, Souid, Rasbi, Tahrawi and Shamsi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariam Al Shamsi, bXNoYW1zaUB1YWV1LmFjLmFl
†ORCID: Doua Kamyan, orcid.org/0000-0003-0067-4243
Maya Hassane, orcid.org/0000-0002-7988-6240
Al Anood Al Naqbi, orcid.org/0000-0003-3036-0296
Abdul-Kader Souid, orcid.org/0000-0002-8562-4757
Zakeya Al Rasbi, orcid.org/0000-0001-9909-8658
Abeer Al Tahrawi, orcid.org/0009-0009-8973-3377
Mariam Al Shamsi, orcid.org/0000-0002-4976-3098
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.