 Mehakpreet K. Thind1,2,3
Mehakpreet K. Thind1,2,3 Holm H. Uhlig4,5,6Michael Glogauer7,8
Holm H. Uhlig4,5,6Michael Glogauer7,8 Nades Palaniyar2,9,10Celine Bourdon2,3
Nades Palaniyar2,9,10Celine Bourdon2,3 Agnes Gwela3,11
Agnes Gwela3,11 Christina L. Lancioni3,12
Christina L. Lancioni3,12 James A. Berkley3,11,13Robert H. J. Bandsma1,2,3,14,15*
James A. Berkley3,11,13Robert H. J. Bandsma1,2,3,14,15* Amber Farooqui2,3,16*
Amber Farooqui2,3,16*- 1Department of Nutritional Sciences, Faculty of Medicine, University of Toronto, Toronto, ON, Canada
- 2Translational Medicine Program, The Hospital for Sick Children, Toronto, ON, Canada
- 3The Childhood Acute Illness & Nutrition Network (CHAIN), Nairobi, Kenya
- 4Translational Gastroenterology Unit, Experimental Medicine, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom
- 5Department of Paediatrics, University of Oxford, Oxford, United Kingdom
- 6Biomedical Research Centre, University of Oxford, Oxford, United Kingdom
- 7Faculty of Dentistry, University of Toronto, Toronto, ON, Canada
- 8Department of Dental Oncology and Maxillofacial Prosthetics, Princess Margaret Cancer Centre, University Health Network, Toronto, ON, Canada
- 9Laboratory Medicine and Pathobiology, Faculty of Medicine, University of Toronto, Toronto, ON, Canada
- 10Institute of Medical Sciences, Faculty of Medicine, University of Toronto, Toronto, ON, Canada
- 11Kenya Medical Research Institute (KEMRI)/Wellcome Trust Research Programme, Centre for Geographic Medicine Research, Kilifi, Kenya
- 12Department of Pediatrics, Oregon Health and Science University, Portland, OR, United States
- 13Centre for Tropical Medicine and Global Health, University of Oxford, Oxford, United Kingdom
- 14Laboratory of Pediatrics, Center for Liver, Digestive, and Metabolic Diseases, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
- 15Division of Gastroenterology, Hepatology, and Nutrition, The Hospital for Sick Children, Toronto, ON, Canada
- 16Omega Laboratories Inc, Mississauga, ON, Canada
Neutrophils are the most abundant innate immune cells. Multiple mechanisms allow them to engage a wide range of metabolic pathways for biosynthesis and bioenergetics for mediating biological processes such as development in the bone marrow and antimicrobial activity such as ROS production and NET formation, inflammation and tissue repair. We first discuss recent work on neutrophil development and functions and the metabolic processes to regulate granulopoiesis, neutrophil migration and trafficking as well as effector functions. We then discuss metabolic syndromes with impaired neutrophil functions that are influenced by genetic and environmental factors of nutrient availability and usage. Here, we particularly focus on the role of specific macronutrients, such as glucose, fatty acids, and protein, as well as micronutrients such as vitamin B3, in regulating neutrophil biology and how this regulation impacts host health. A special section of this review primarily discusses that the ways nutrient deficiencies could impact neutrophil biology and increase infection susceptibility. We emphasize biochemical approaches to explore neutrophil metabolism in relation to development and functions. Lastly, we discuss opportunities and challenges to neutrophil-centered therapeutic approaches in immune-driven diseases and highlight unanswered questions to guide future discoveries.
1 Introduction
Phagocytic cells have evolved as a key defense line against sterile and microbial insults (1). Neutrophils are the most abundant terminally differentiated effector innate immune cell line in the bone marrow (BM) and peripheral blood. They rapidly localize to sites of infection to implement immediate and effective immune responses for pathogen clearance, and resolution of acute inflammatory responses. In fact, the anti-microbial functions of neutrophils promote an environment that is unfavourable to pathogens at the expense of tissue integrity therefore neutrophil numbers are tightly regulated. Due to their relatively short and variable half-life across tissues (2), neutrophils are constantly replenished from proliferative BM precursors. Differentiation through successive stages maintains their homeostatic levels and ensures their immediate availability to counter invading pathogens (Figure 1). Consequently, most hematopoietic stem cells (HSCs) and hematopoietic stem/progenitor cells (HSPCs) in the BM, that give rise to all blood cells through intermediaries, are committed to the production of neutrophils (3, 4). Neutrophils participate in the capture and destruction of invading microorganisms through chemotaxis, phagocytosis, degranulation, reactive oxygen species (ROS) production, formation of neutrophil extracellular traps (NETs), and production of cytokines and other inflammatory mediators upon pathogen recognition [reviewed in (5, 6)]. In this regard, neutrophil activation and subsequent stimulation of specific cell-surface receptors from a broad receptor repertoire (>30) controls neutrophil antimicrobial functions. Defects in neutrophil development, migration, function and clearance increases susceptibility to infection, inflammation and organ dysfunction in multiple organisms (7–10).
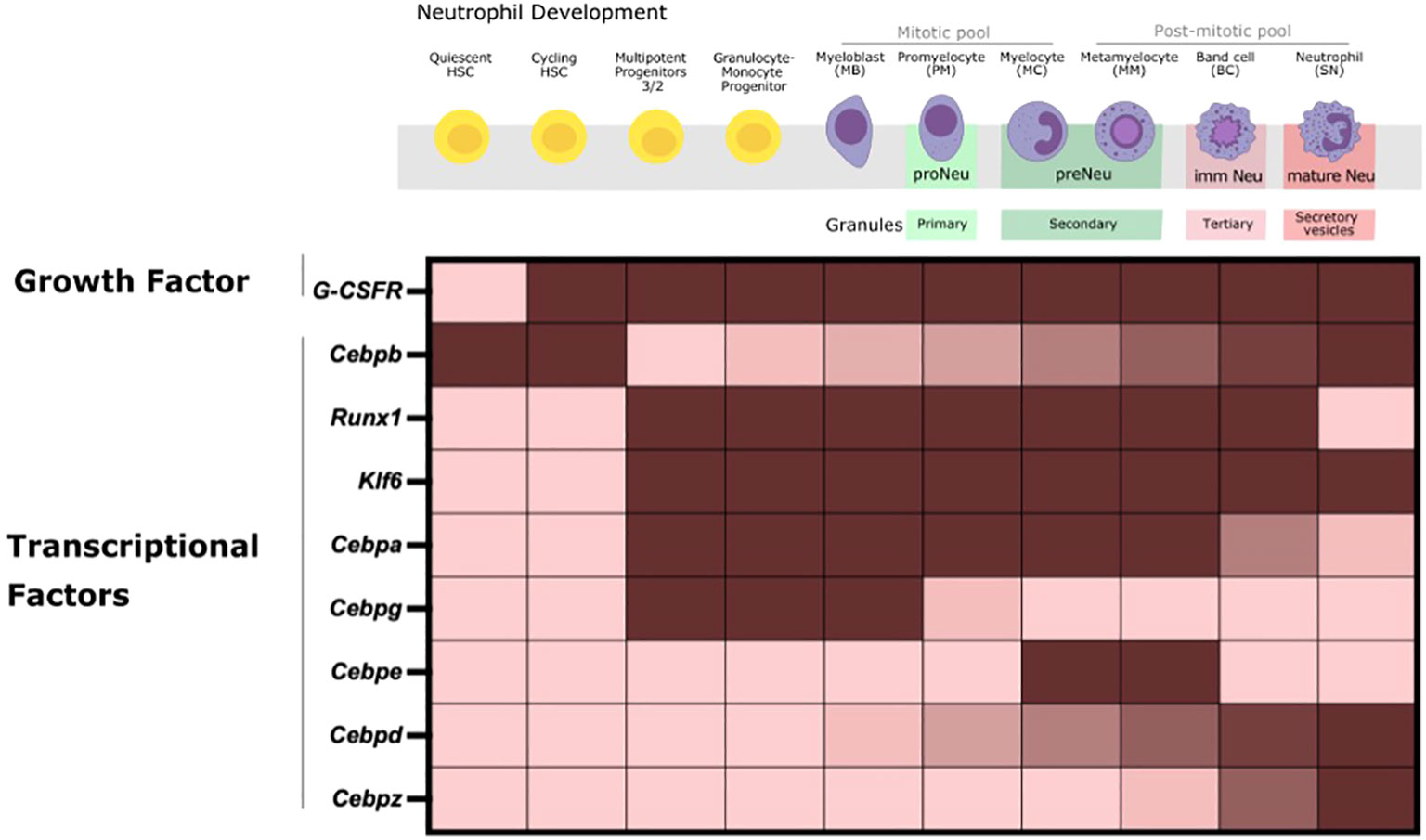
Figure 1 Neutrophil Production in the BM during steady-state happens through multiple successive stages. HSCs proliferate and differentiate into oligopotent committed myeloid progenitors (CMP, GMP) and sequentially into unipotent early neutrophil committed and proliferative progenitors that ultimately give rise to mature neutrophils with the full range of effector functions. Cell- intrinsic regulation of the expression of a multitude of TFs, as depicted here, is crucial to tightly regulate the expression of stage-specific granules (primary, secondary, tertiary and secretory vesicles that contain specific effector proteins) and genes for neutrophil commitment, proliferation, differentiation and maturation. Fully differentiated mature neutrophils then exit the BM to enter into the circulation and tissues under steady-state conditions.
2 Neutrophil biology: the basics
2.1 Neutrophil development from the bone marrow, the circulation and tissues
HSCs and HSPCs in the BM proliferate and differentiate into granulocyte-monocyte progenitors (GMPs). Under the control of granulocyte colony- stimulating factor (G-CSF), produced by macrophages, and endothelial cells (ECs), these GMPs commit to neutrophil generation (granulopoiesis) (11, 12). A pool of hematopoietic progenitors and neutrophil precursors also exists outside the BM, including in the spleen, lung and peripheral circulation, where they differentiate into mature and functionally competent neutrophils to allow acceleration of antimicrobial responses in infected tissues. Notably, neutrophil phenotype reprogramming occurs in tissues regardless of origin (2, 9, 13). During the early phase of an infection and inflammatory response, a rapid increase in de novo neutrophil release and production with the appearance of immature neutrophils in the peripheral blood (known as left-shift), termed “emergency” granulopoiesis, helps to meet the enhanced demand for neutrophils as these cells are consumed in large quantities to rapidly counteract bacterial invasion (3, 14–16). This process occurs at the expense of lymphopoiesis and has been reviewed elsewhere in greater detail [reviewed in (14, 17, 18)]. Briefly, conserved microbial elements known as pathogen-associated molecular patterns and host tissue derived damage-associated molecular patterns are recognized by pattern recognition receptors such as, toll-like receptors (TLRs) which are transmembrane proteins, on the surface of multiple immune and non-immune cell types leading to immune cell activation (19, 20). This causes subsequent cytokine and chemokine production and signalling through a common pathway involving nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) activation (19). NF-kB, consisting of RelA (p65) and p50, activated downstream of TLRs, which upon phosphorylation by mitogen-activated protein kinases (MAPKs), promote its translocation to the nucleus and leads to robust transcriptional expression of cytokine & chemokine genes including G-CSF, and an array of additional immune genes (19). Dimerization of granulocyte colony-stimulating factor receptor (G-CSFR) by G-CSF, expressed on BM neutrophil precursors (i.e. metamyelocytes and onwards), activates downstream signal transduction pathways [reviewed in (21)]. This regulates HSCs proliferation and differentiation, and directs their commitment and that of early neutrophil progenitors (proNeu1 and proNeu2) (4), and precursors, towards the myeloid lineage (3, 22). This aids in the production of mature neutrophils in order to maintain homeostatic levels under basal and emergency conditions (21, 23). However, G-CSF and G-CSFR independent mechanisms to produce mature granulocytes also exist. This includes IL6-mediated (4) pathways, as well as through intracellular signaling cascades, epigenetic landscapes, and transcriptional networks, such as CCAAT/enhancer-binding proteins (Cebps), to activate specific and tightly regulated gene expression programs for neutrophil differentiation (2). Additionally, there is a role of different nutrient sources, namely glucose, and amino and fatty acids, for HSC lineage specification and commitment (24–26).
As myeloid precursors pass through the various stages of differentiation and maturation to become mature neutrophils in the BM, they undergo morphological changes (nuclear segmentation), increase chemotactic responsiveness and acquire features necessary for microbicidal activity (Figure 1). Neutrophil classification based on nuclear morphology, single-cell RNA sequencing (scRNA-seq), and surface marker expression has allowed assessment of mouse and human neutrophil ontogeny, phenotypic heterogeneity and mapping the developmental continuum of cell fate hierarchies (5, 27, 28). The granulopoietic niches of neutrophils within the BM are divided into proliferative mitotic, post-mitotic and the mature neutrophil pool. Neutrophils are released into systemic circulation as a result of differential expression of C-X-C chemokine receptor type 4 and 2 (CXCR4 and CXCR2, respectively) (14, 29, 30). Directional cues from the chemokine stromal cell-derived factor 1, also known as CXC motif chemokine ligand 12 (CXCL12), produced by BM stromal cells, i.e., CXCL12-abundant reticular cells (CAR cells), regulate the CXCL12/CXCR4 chemokine/surface receptor signaling axis for the retention of neutrophils in the BM (31). In contrast, the upregulation of CXC motif chemokine ligand 2 and 1 (CXCL2 and CXCL1, respectively) and CXCR-2 receptor on BM ECs and neutrophils respectively allows for the recruitment of these cells into the circulation, and into naïve and inflamed tissues through activation of downstream signaling pathways (32).
2.2 Neutrophil interactions in the tissue microenvironment
2.2.1 Neutrophil crosstalk with innate and adaptive immune cells
It is well accepted that macrophages are responsible for the clearance of apoptotic neutrophils at the end of their lifecycle and resolve inflammation (33). In fact, studies report neutrophils that have physiologically “aged” in the circulation (CD62LloCXCR4hi neutrophils) to be eliminated by macrophages in the BM and tissues. This process also maintains the rhythmic egress of neutrophils into the circulation (30, 34). Similarly, in self-limited lipopolysaccharide (LPS)-induced peritonitis, BM-derived Resolvin D4, a pro-resolving lipid-derived mediator, increases BM-macrophage efferocytosis of apoptotic neutrophils to aid in the resolution infectious inflammation (35). In a mouse model of acetaminophen-induced acute liver injury, neutrophils have crucial functions in liver repair. They do so by promoting the phenotypic conversion of pro-inflammatory Ly6ChiCX3CR1lo monocytes/macrophages to pro-resolving reparative Ly6CloCX3CR1hi macrophages mediated through neutrophil nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase ROS production (36). Neutrophils also support monocyte and macrophage recovery in blood, BM and spleen following genotoxic injury as well T helper 17 cells and macrophage recruitment and priming in a mouse model of atherosclerosis (2, 37). Importantly here, both macrophage or neutrophil-mediated cytokine production leads to immune cell activation and priming for an exaggerated immune response. Therefore the interaction between both is crucial in infectious and inflammatory disease outcomes. These concepts are reviewed elsewhere in greater details (38–40).
A common phenomenon in chronological ageing known as reverse transendothelial migration (rTEM), where neutrophil exhibit retrograde mobility within ECs junctions and re-enter the vascular lumen. This process is shown to be mast cell-derived CXCL1 dependent (32, 41, 42). Here, the rTEM neutrophils are of noxious phenotype capable of inducing remote organ damage in acutely inflamed aged tissues. In fact, multiple studies also show neutrophil recruitment in physiology to be mast cell dependent (43–46). The impact of neutrophils on T cells in various disease contexts (47–52) is also summarized in excellent reviews (39, 40, 53–55).
2.2.2 Neutrophil crosstalk with non-immune cells
Neutrophils’ interaction with non-immune cells is crucial to regulate their biology. In this regard, BM CAR cells, a population of mesenchymal stem cells, produce CXCL12 that mediates the retention of neutrophils in the BM through CXCR4 ligation (31). Platelet-neutrophil interaction are also implicated in homeostasis and inflammation (40, 54, 56–59). Neutrophil and ECs interaction allows for unidirectional migration through venular walls (54, 60, 61). Most importantly, aberrant neutrophil-endothelial interactions are implicated in a wide range of inflammatory diseases that relate to neutrophil influx and tissue damage (62–64). Similarly, neutrophil-epithelial crosstalk is involved in the maintenance of the epithelial-lined organs where uncontrolled neutrophil processes contribute to pathogenesis of diseases (65–68).
2.3 Mechanisms implicated in neutrophil-mediated immunity
Acute and chronic inflammatory and infectious immune responses are marked with heightened immune cell recruitment, and dramatic shifts in tissue and systemic metabolism including nutrient depletion, hypoxia and the generation of large quantities of reactive nitrogen and oxygen intermediates (69). There is an heightened interest in identifying the role of unique metabolites and metabolic pathways in immunoregulation, a field termed immunometabolism, ranging from energy metabolism to the modulation of signalling pathways and post-translational modifications (70–74). Traditionally, neutrophils are thought to be purely glycolytic and the role of mitochondria, a central organelle for energy homeostasis and metabolic control, is believed to be minimal, for neutrophil function (75). However, this view has been challenged, as their critical role as first line defenders requires high metabolic plasticity to respond to environmental cues and regulate innate immune responses (76).
2.3.1 Overview of neutrophil metabolism
Circulating neutrophils primarily rely on glycolysis and the pentose phosphate pathway (PPP), both of which take place within the cytosol, as their preferred metabolic strategy to fuel phagocytosis, ROS production, and NET formation. However, fatty acid oxidation (FAO), tricarboxylic acid (TCA) cycle, and oxidative phosphorylation (OXPHOS) that occur within the mitochondria, are also undoubtedly crucial under both steady-state and inflammatory conditions (77–80). Although immunometabolism (70, 81) and metabolic plasticity is increasingly understood, minimal data is available on neutrophil metabolic reprogramming under diverse nutritional, metabolic and pathologic conditions. This review is therefore focused on key neutrophil functions and their dependence on different key metabolic pathways in experimental animals and human cohorts. We also emphasize approaches used to study immunometabolism and how different nutrient environments contribute to neutrophil biology.
3 Metabolic programmes for granulopoiesis
Neutrophil production is tightly regulated by intrinsic and extrinsic cellular factors via a number of transcription factors (TFs) that regulate subsequent stages of neutrophil development. The hierarchical expression and activation of tightly defined TFs is required for lineage specification of HSPCs and proper commitment and differentiation of myeloid precursors into mature neutrophils in the BM (Figure 1) (9, 31). TFs are largely regulated at transcriptional, translational, and posttranslational levels. Consequently, transcriptional control, isoform usage, phosphorylation, and acetylation of TFs are crucial for the proper activation of gene regulatory mechanisms that orchestrate the differentiation of HSCs into committed cells.
3.1 Transcriptional regulation of granulopoiesis
TF such as Cebps and others work in a combinatorial manner to orchestrate the transcriptional networks for neutrophil lineage commitment, proliferation, differentiation and functional responses. The expression of these TFs is also correlated with stage-specific granule expression across the neutrophil lineage and therefore antimicrobial capacity (4, 23, 82, 83). Changes in its expression can significantly affect downstream genes that impact the neutrophil development and function trajectory (84).
Cepba binds to promoters of myeloid-related genes to activate myeloid-lineage gene expression program and repress non-myeloid lineage genes in hematopoietic progenitors of multi-lineage potential in the BM. The combinatorial expression of Cebpa, Gata1 and low levels of PU.1 are required at the GMP stage for granulopoiesis initiation, and expression of primary granule proteins, and Cebpa deletion is shown to skew progenitors towards lymphopoieses (31, 81, 85, 86). Similarly, Cebpg is a pro-proliferative factor that is particularly required by myeloblasts, promyelocytes and myelocytes. Cebpe further drives differentiation at myelocytes, and metamyelocytes and contributes to the expression of secondary granules (4, 31). Here, Cebpe deletion leads to an accumulation of GMPs (4, 31). Expression of Cebpb, Cebpd, and PU.1 is found in the most mature neutrophil precursors and in peripheral blood granulocytes. However, studies in Cebpb deficient mice do not observe defects in steady-state granulopoiesis, but rather Cebpb is shown to be crucial for “emergency” granulopoiesis (86, 87). On the contrary, Cebpd and PU.1 are involved in the expression of tertiary granules such as matrix metalloproteinase-9 (Mmp9) and CXCL2 at the band cell stage for the terminal differentiation of neutrophils (31, 81). The expression of Cebpz is prominent from band cell onwards with peak levels in the most mature neutrophil precursors and in peripheral blood granulocytes (31, 81). Additionally, Runx1 and Klf6 are shown to modulate neutrophil maturation (9).
Altogether, co- expression of specific TFs within specific subpopulations serves to drive early lineage specification towards distinct progenitors. These TFs also function during the later developmental stages to drive differentiation and maturation for optimal antimicrobial defense. Newer studies focus solely on the global gene expression patterns of these central TFs to drive neutrophil ontogeny through bulk and scRNA-seq to assess transcriptional programs across neutrophil subsets (31, 81, 88). However, the post-transcriptional modifications and mechanisms that enable these transcriptional signals to regulate coordinated neutrophil differentiation remain incompletely understood. Nevertheless, there is some evidence on the role of changes in genome accessibility, master transcriptional regulators and metabolites for controlling these TFs and subsequently immune responses, some of which are discussed in more detail below (9, 23, 23, 73).
3.2 Metabolic regulation of the transcriptional regulatory networks
An intricate link between cellular metabolism, transcription, and signalling pathways directly and indirectly supports the regulation of genes involved in cellular differentiation and cellular processes (73, 81, 84, 89). It is well-accepted that the mitochondria, through TCA metabolites, control chromatin modifications, DNA methylation, and post-translational modifications of proteins to alter their function. However, how specific metabolites regulate TFs for neutrophil development has not been described previously (73, 90). Enhancing intracellular nicotinamide adenine dinucleotide (NAD+) levels, through Vitamin B3 supplementation, is found to support sirtuin-1, Cebpa, Cebpb, G-CSF and G-CFSr expression for neutrophilic differentiation and migration in CD34+ hematopoietic progenitor cells, the promyelocytic leukemia cell line HL-60, and in primary bone marrow CD34+ cells from severe congenital neutropenic patients (91). Glucose metabolism has also been shown to regulate Cebpb activity for adipogenic gene expression and differentiation of preadipocytes to adipocytes (92). Glucose can induce nicotinamide mononucleotide adenylyltransferase (NMNAT)-2 expression, an enzyme for NAD+ biosynthesis, and reduce nuclear poly (ADP-ribose) polymerase (PARP)-1 enzymatic activity with Cebpb. Conversely, glucose deprivation reduces NMNAT-2 levels, increases the enzymatic activity of PARP-1 and reduces the binding of Cebpb to target gene promoters thereby preventing adipogenesis (92). At the same time, activity of mammalian target of rapamycin complex (mTORC)-1 controls Cebpb expression and its activity is inhibited under caloric restriction (93). The mechanism of Cebpb regulation by glucose, NAD+, and mTORC1 remains unknown. The Vitamin D receptor and Retinoic Acid Receptor in human acute myeloid leukemia cell lines (HL60, and KG1 cells) is also shown to regulate the expression of Cebpa, Cebpb and subsequently Cebpe for granulocytic differentiation (88). While these studies examine the direct and indirect roles of metabolites for Cebp regulation in the adipose, liver tissues and specific cell lines, the metabolic pathways regulating these TFs in the hypoxic BM microenvironment for neutrophil development remains an open avenue for future research. A study of autophagy Atg7 deficient neutrophils demonstrated that the metabolic program does not influence the transcriptional program in neutrophil precursors where these precursors are morphologically and functionally immature (7). Therefore, further studies are needed to identify the parallel roles of transcriptional regulators and metabolic programming, if any, for cellular differentiation. Interestingly, glycolytic and mitochondrial metabolites are shown to control chromatin modifications, and DNA methylation in physiology and disease and may be relevant in this context (73, 94).
3.3 Metabolic programming of cellular differentiation
3.3.1 Metabolism as a key determinant of hematopoietic stem cell fate
Differentiation of HSCs into fully functional mature neutrophils requires considerable energy for extensive cytoplasmic and nuclear remodelling (95) (Figure 2). Autophagy decreases mitochondrial mass in quiescent HSCs for their self-renewal capacity. As a result, quiescent HSCs primarily rely on anaerobic glycolysis to maintain stem cell quiescence and self-renewal in the low oxygen niches of the BM. Increased peroxisome proliferator-activated receptor (PPAR) activation for mitochondrial biogenesis, mitochondrial ROS, and FAO in cycling HSCs demarcates asymmetric division, one daughter with stem cell features and one committed progenitor, and further regulates HSC maintenance (25, 71, 78, 96–100). Therefore, the metabolic programs of HSCs balance self-renewal and commitment. Similarly, a lower mitochondrial mass and mitochondrial activity and increased transcriptional activation of the TF hypoxia-inducible factor 1-alpha (HIF-1a) to drive aerobic glycolysis by Meis1 in quiescent HSCs, allows for a long-term reconstitution capacity of these cells in transplantation experiments. Here, increased mitochondrial biogenesis is associated with exit from quiescence (25, 26, 97, 98, 101). In fact, flow cytometric analysis of the metabolic profile of HSCs using tetramethylrhodamine methyl ester (TMRM) shows only 6%-9% of total BM cells with low mitochondrial potential (TMRMlo) but this population contains more than 80% of quiescent HSCs (98). Interestingly, TMRMlo quiescent HSCs cultured for 5 days under differentiation-inducing conditions (with cytokines SCF, Flt3, IL-3 and IL-6) and carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) revert to a state of self-renewal. TMRMhi quiescent HSCs are however unable to revert to functional stem cells (25). Additionally, proteomic analyses highlights the role of increased translation of mitochondrial transcription factor A (TFAM) in proerythrocytes and its regulation of downstream genes associated with mitochondrial metabolism for proper erythrocyte differentiation (102). Similarly, to improve the efficacy of hematopoietic cell transplantation, where limited numbers of HSCs are present, transplantation of CD34+ HSCs with peroxisome proliferator-activated receptor γ (PPARG)-specific small hairpin RNA (shRNA) can promote expansion of HSCs and HSPCs. This is through enhanced glycolysis and HSC self-renewal, conversely inhibition of glycolysis and enhancement in PPARG suppresses this expansion (95, 103). These studies highlight the essential role of nutrients and their associated signaling pathways on the self-renewal, differentiation and lineage commitment potential of HSCs.
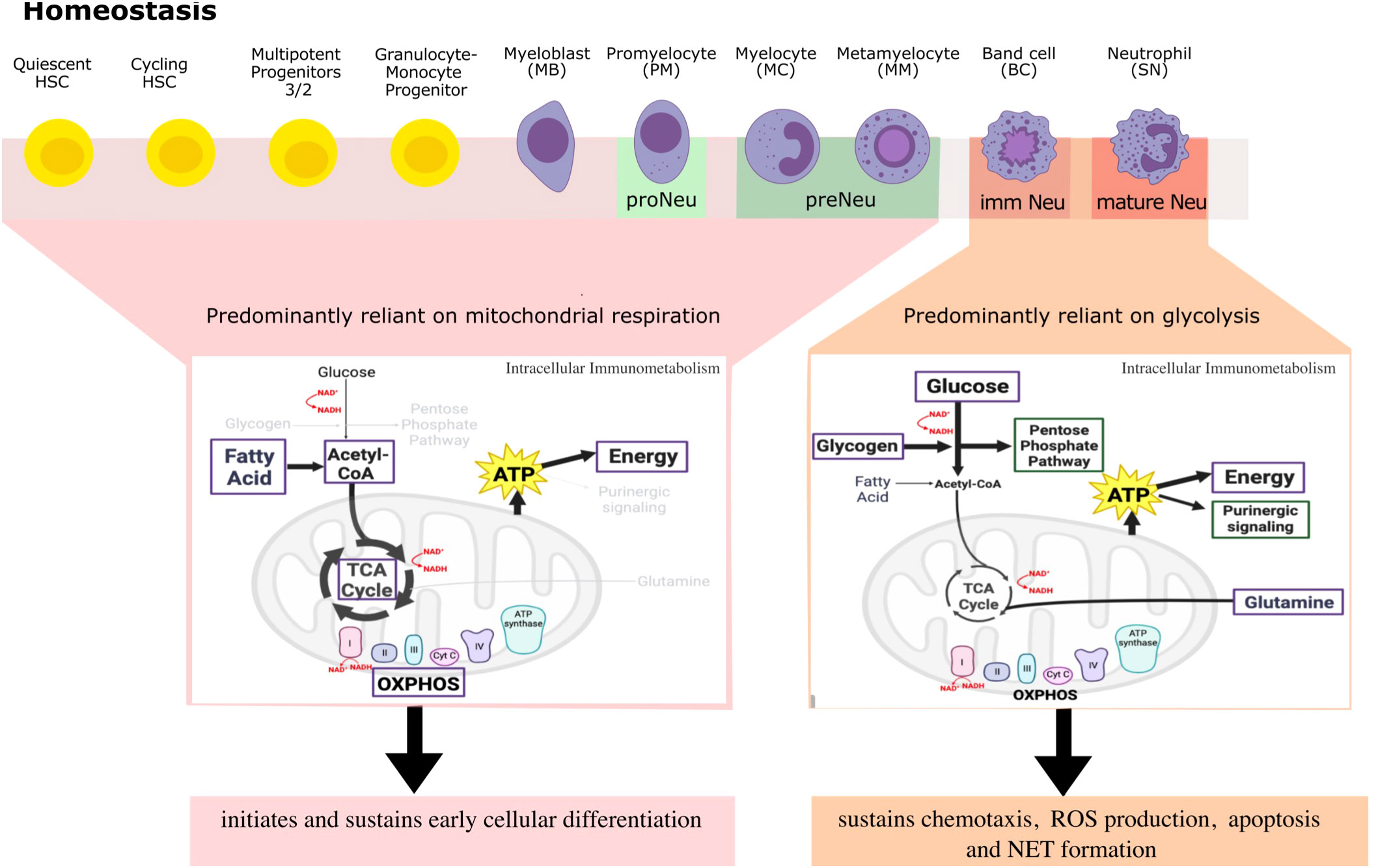
Figure 2 A simplified overview of differential metabolites and metabolic pathways that neutrophils engage in throughout differentiation and beyond to regulate their large repertoire of immune responses in steady-state. Here, neutrophil committed progenitors and early precursors engage in fatty acid oxidation, TCA cycle and OXPHOS for ATP generation that sustains neutrophil differentiation programs. Indeed, higher mitochondrial activity is indicative of a more immature neutrophil phenotype. Mature neutrophils on the other hand, rely primarily on glycolysis and little mitochondrial respiration to regulate and sustain ATP production for effector functions and for autocrine purinergic signaling. Metabolic requirements of neutrophils are not ‘black and white’ in inflammatory conditions, but rather a continuum of these processes works together to regulate an optimal response, a perspective that requires more investigation. Highlighted in purple are metabolites used for energy production and green are pathways used by metabolites to regulate neutrophil functions.
3.3.2 Metabolism as a master regulator of neutrophil state
The metabolic state also shapes committed progenitor cells. Expression of type III receptor tyrosine kinase ckit, in early committed and immature neutrophil progenitors in the BM requires enhanced mitochondrial function, i.e., oxidative phosphorylation, to promote rapid symmetric differentiation. This further highlights the essential role of increased mitochondrial activity for HSC cell fate decisions under steady-state (71, 104). Similarly, transcriptomics and proteomics has shown that mitochondrial function is essential in the early stages of granulopoiesis for lineage commitment and the initiation of neutrophil differentiation in ckit+ neutrophils. Mitochondrial function is key for the differentiation in ckit+ HSCs from free fatty acid utilisation (104–106). Complex III of the electron transport chain may be responsible for ATP generation in early myeloid precursors in the BM in this context (47, 105). Similarly, shRNA-mediated knockdown of alanine, serine, cysteine transporter 2 (ASCT2) glutamine transporter in CD34+progenitors, upregulates CD11b myeloid marker, highlighting the skewness of the progenitors towards the myeloid lineage that happens irrespective of glucose and glutamine metabolism (71). Adenylate kinase 2 (AK2) regulates the homeostasis of mitochondrial adenine nucleotides (ADP, ATP and AMP) by catalyzing the transfer of high-energy phosphate in the mitochondrial intermembrane space, once the HSCs commit to a specific lineage pathway. AK2 deficiency results in impaired proliferation and differentiation in granulocyte precursor cells shown using the HL-60 promyelocytes cell line, through impairment in oxidative phosphorylation for energy metabolism (107). In fact, even though enhancement of glycolysis is evident by accumulation of pyruvate and lactate, neutrophil differentiation is hindered due to incomplete mitochondrial activity by hematopoietic progenitors that is required to meet the greater energy demand. This suggests an indispensable role of mitochondrial metabolism in neutrophil differentiation (106, 107). This is relevant for reticular dysgenesis, an inherited immune deficiency disorder caused by AK2 deficiency. As neutrophils differentiate and become mature neutrophils, there is a decline in mitochondrial number and activity with a change in mitochondrial morphology from a more tubular phenotype to round to regulate spontaneous cell death (108–110).
3.3.3 Role of autophagy for mitochondrial metabolism in neutrophil differentiation
Autophagy and mitochondrial respiration are critical determinants of neutrophil differentiation. Autophagy is a conserved cellular recycling process, involving several conserved autophagy-related genes (Atg), that enables the degradation of cytoplasmic content in lysosomes for diverse cellular processes. In fact, autophagy (regulated by Atg5 or Atg7) degrades lipid droplets, providing free fatty acids used by the mitochondria through the TCA cycle and oxidative phosphorylation for ATP production at the myeloblast and myelocyte stage for terminal neutrophil differentiation (Figure 2) (7). As a result, higher oxygen consumption rate (OCR) is evident in BM neutrophils. Here, microfluidic gene expression analysis with the Fluidigm-Biomark array reveals that all 15 glycolytic pathway genes analyzed are downregulated while the mitochondrial content simultaneously undergoes a 2-fold increase during normal neutrophil differentiation (47, 111). This is further validated by targeted metabolomics. In Atg7 deficient myeloblast cells, ECAR (measure of glycolysis, extracellular flux assay) and lipid accumulation, assessed through lipidomics, is increased, and mitochondrial ATP production is decreased. As a result, the expression of Ldha is also increased to generate lactate and NAD+ to keep glycolysis going but the ATP levels and differentiation is not restored to wild-type amounts (7). In the same study, administration of either free fatty acids or pyruvate is able to restore energy metabolism and differentiation, supporting that mitochondrial respiration is essential for early neutrophil differentiation where energy requirements are the highest (7). Furthermore, deletion of Atg7 at the GMP stage also leads to failure of neutrophil differentiation and an accumulation of immature forms which are functionally defective. Deletion of autophagy genes at later development stages results in a different phenotype where mature neutrophil are primarily expressed (7, 112). This reflects the need to carefully consider the developmental stages where autophagy genes are knocked down as it may have differential effect on neutrophil maturation.
Conflicting data indicates that since neutrophil mitochondria have decreased protein expression of some complexes, they may not be fully involved in ATP production but rather in maintaining functional responses through the mitochondrial membrane potential () (105, 113). In fact, studies show that inhibition of complex III in both glucose-rich and glucose-depleted environments, completely diminishes but without an effect on ATP production. This suggests a role for mitochondria that extends beyond oxidative phosphorylation and ATP production for neutrophil development (105, 109).
4 Metabolism driven regulation of neutrophil migration
4.1 Neutrophil mobilization and trafficking
Neutrophil mobility is essential for cell recruitment to sites of inflammation and infection. Chemotaxis, is the ability of neutrophils to sense gradients, polarize and directionally migrate within a chemotactic gradient field to access invading pathogens [reviewed in (114, 115)]. TFs KLF6 and RUNX1 are shown to be valuable for controlling genes for neutrophil migration into inflamed sites (9). Interestingly, “swarming”, tightly regulated by exosomes and lipid leukotriene 4 (LTB4), is also the coordinated feedforward movement of many neutrophils to accumulate in large numbers in inflamed and infected tissues in early stages of infection for optimal bacterial clearance (116–118). This is followed by migration arrest at later stages of infection to avoid uncontrolled aggregation at inflamed sites through G protein-coupled receptor kinase 2 (GRK2)-mediated G protein-coupled receptors (GPCR) desensitization, and prostaglandin E2 (PGE2) synthesis to promote anti-inflammatory programs for tissue repair (119, 120). Resolvin D4 also mediates this migration arrest (35).
In contrast, transendothelial migration (TEM) defines neutrophil migration across the endothelium to both inflamed and naïve sites, albeit at lower levels. This is dependent on receptor ligand interactions of adhesins, integrins and selectins. Adhesion molecules i.e., chemokines, integrin ligands and selectins on the endothelium, bind to G-protein-coupled chemokine receptors, integrins (Mac1-CD11b/CD18), and selectin ligands on the surface of neutrophils. These molecules tightly regulate rolling, adhesion and transmigration, i.e., paracellular (~96% of the time) vs. transcellular into tissues, as has been well defined in excellent reviews [reviewed in (113)]. Additionally, rTEM, re-entry of activated neutrophils from inflamed sites into the circulation, due to overexpression of CXCL1 at endothelial junctions and CXCR2 desensitization in mice also controls inflammatory responses (41, 42).
4.2 Metabolic regulation of neutrophil trafficking
Metabolic stress is a hallmark of several conditions including trauma and infection. Neutrophils isolated from trauma patients have enhanced markers of mitochondrial damage, i.e., mitochondrial-derived damage-associated molecular patterns, inflammasome activation, and reduced expression of CXCR2, thereby impaired chemotaxis (121, 122). Studies further show that upon stimulation of neutrophil-like HL-60 cell line with N-formyl-methionyl-leucyl-phenylalanine (fMLP), which activates formyl peptide receptors (FPRs) expressed on these cells, mitochondria with high localize to cell protrusions and mitochondrial-derived ATP is released into the extracellular spaces. This serves as an autocrine messenger to amplify chemotaxis signals through activation of P2Y2R-mediated mTOR signaling at the leading edge, rather than as a direct energy source (123). In fact, two phases of purinergic signaling are suggested: an initial burst of ATP release that is driven by mitochondrial ATP and a second phase that involves glycolytic ATP production to maintain chemotaxis (Figure 3) (123). In this study, the fluorescent ATP probe 2-2Zn(II) anchored on cell membrane of live neutrophils reveals an increase in extracellular ATP production by the mitochondria immediately upon stimulation of cells with fMLP (123). Similarly, pretreatment with carbonyl cyanide 3-chlorophenylhydrazone (CCCP) or FCCP for 2 hours shows a direct relation between loss of , detected by increased green fluorescence of JC-1 probe, rounded cell shape and the inhibition of chemotaxis (75). Studies in zebrafish also show that neutrophils depend on for chemotaxis and that neutrophil-specific disruption of mitochondria in vivo is associated with inhibited motility, with an inability to reduce ROS species, and cell apoptosis (Figure 3) (124, 125). Similarly, hyperglycemic conditions in cancer provide microenvironments that promote metastasis of tumor-associated neutrophils and impair mobilization of antitumor neutrophils, resulting in poor prognosis that is seen for these patients. This shows that migration is selective based on the dominant neutrophil subset in the disease state and their preferred fuel of choice (126).
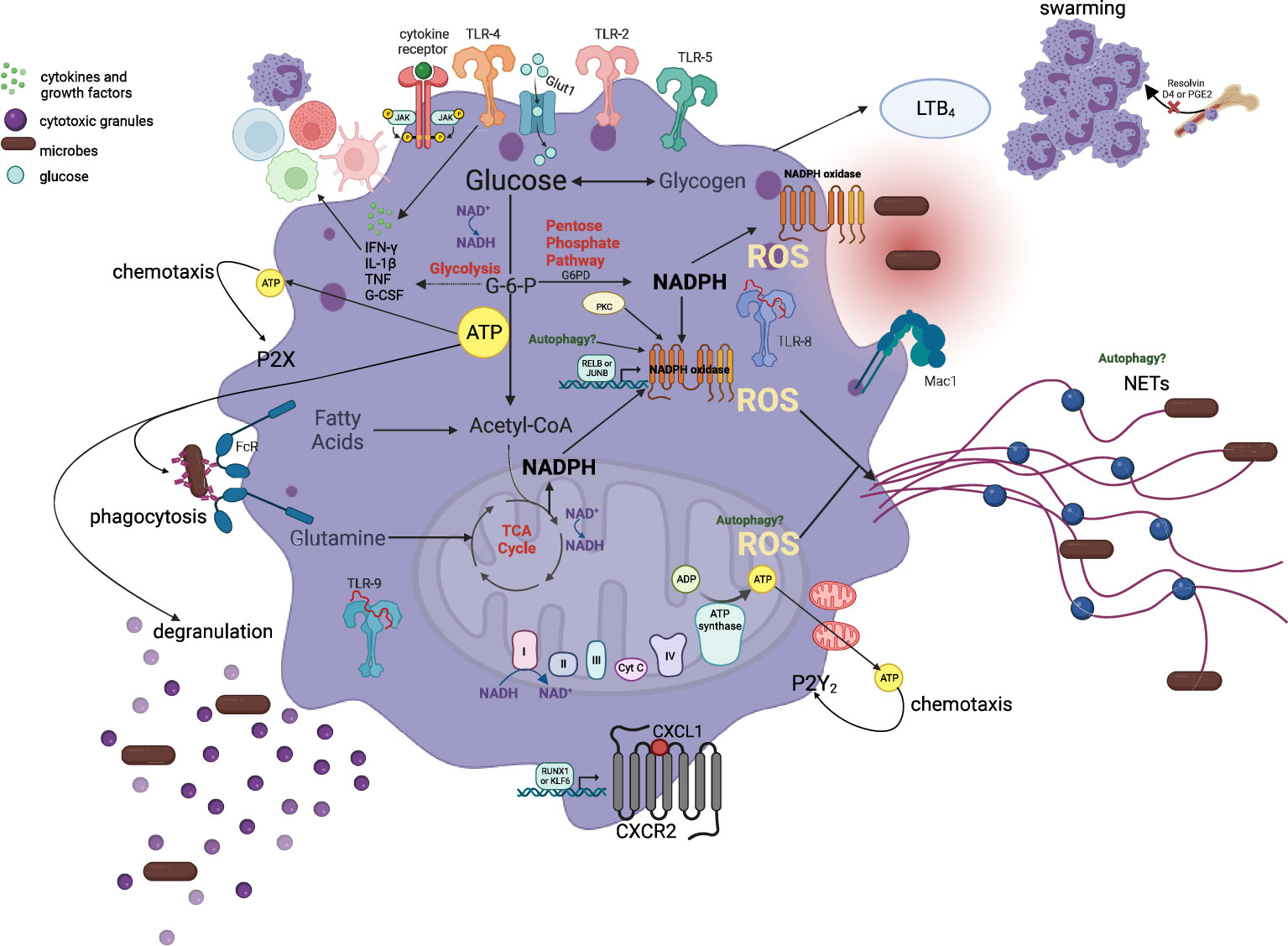
Figure 3 Metabolic pathways involved in the maintenance of neutrophil effector functions. Neutrophils rely primarily on glycolysis and very little mitochondrial respiration to regulate and sustain ATP production for chemotaxis, phagocytosis, cytokine expression, ROS production, degranulation, and NET formation in the circulation and peripheral tissues. An initial burst of mitochondrial-derived ATP release and a second phase that involves glycolytic ATP production maintains the autocrine purinergic signaling for chemotaxis. Glucose uptake and glycolysis regulates phagocytosis and degranulation while PPP is indispensable for NADPH generation, which is oxidized by the NADPH oxidase for ROS generation and eventually NETs. Additionally, mitochondrial-derived ROS can also enhance NET production. NADH/NAD ratio is crucial for the maintenance of glycolytic and mitochondrial ATP production and thus functional properties of neutrophils. Multiple metabolites, as depicted in this figure, including glycogen, and glutamine, are also capable of entering these metabolic pathways to regulate and sustain neutrophil responses in tissues. This metabolic flexibility of neutrophils allows them to coordinate optimal effector responses in tissues with diverse substrate availability.
5 Modulation of metabolism regulates NADPH-oxidase dependent functions
5.1 Overview of NADPH-oxidase dependent antimicrobial functions
As myeloid precursors become mature neutrophils, they acquire features necessary for optimal microbicidal activity including stage-specific granule and NADPH oxidase proteins (Figure 1), the most abundant proteins in mouse and human neutrophils and the main complex for neutrophil ROS production, respectively (31, 81, 127, 128). In fact, primary [cathepsin G (CTSG), myeloperoxidase (MPO), elastase (ELANE)], secondary [lactoferrin (LTF), neutrophil gelatinase-associated lipocalin (NGAL), cathelicidin (CAMP)], and tertiary (MMP9, lipocalin 2 (LCN2), CD11b/CD18, CXCL2) granules and secretory vesicles (β2 integrins), are well-defined granules that carry distinct types of cargo as mentioned. These highly toxic granule proteins are released into the phagocytic vacuole or extracellularly in a ‘formed-first-released-last’ model to help destroy invading pathogens (69, 128). In this context, the strength of the stimulus initiates the release of granules, both intracellularly and extracellularly, with the secretory vesicles being mobilized first and primary granules secreted at the end in response to multiple inflammatory mediators (129).
The NADPH oxidase (NOX) is a multicomponent electron-transfer complex that generates superoxide and other ROS into the phagosome or extracellularly for microbial killing events (23). This complex is comprised of five subunits that are expressed at different stages during neutrophil differentiation and packaged into the granules. More specifically, p22, p40, and Rac1/2 have stable gene expression levels throughout all stages, whereas gp91 has maximal gene expression at the metamyelocyte and band cell stages, and p47 and p67 are upregulated in the metamyelocytes and have the maximal gene expression at the mature stages (23). Consequently, studies show that while all NOX subunits are expressed by the metamyelocyte stage, peak levels of ROS are only reached at the end of differentiation (23). Interestingly, RELB and JUNB are important TFs for the expression of these genes for NADPH ROS generation (9). In steady-state, the membrane-bound (b558 - gp91, p22) and cytosolic components (p47, p67, p40, Rac1/2) do not interact with each other. Upon phagocyte activation in mature neutrophils, consumption of oxygen is increased, and these subunits are phosphorylated by protein kinase C (PKC). The cytosolic components migrate almost instantly to the phagocyte membrane where they assemble with the flavocytochrome b558 to form the active enzyme. In this regard, NADPH-oxidase mediated ROS production accounts for majority of neutrophil ROS production (127). Excessive ROS production can also be damaging to the host cells therefore, balanced levels of ROS are crucial for pathogen clearance and restoration of tissue homeostasis.
5.2 Metabolic reprogramming for pathogen killing via NADPH oxidase ROS production
In this section, we aim to summarize the literature as it relates to metabolic requirements for neutrophil ROS generation.
5.2.1 Pentose phosphate pathway
When glucose enters cells, it is phosphorylated by hexokinase generating glucose-6-phosphate (G6P). G6P can flow through two different pathways: the glycolytic pathway, generating fructose-6-phosphate that is metabolized to lactate, to produce ATP, or the pentose phosphate pathway (PPP) that converts NADP+ to NADPH (Figure 3) (130–132). The latter is especially required for NADPH oxidase dependent ROS generation upon cell activation, that occurs with infectious and inflammatory stimuli, to enable high NADPH yield (132). Upon neutrophil activation here, PPP intermediates accumulate profoundly while many intermediates in glycolysis increase to a lesser extent, indicative of metabolic adaptation by neutrophils (132). NADPH oxidase, the primary source of neutrophil ROS production, oxidizes cytosolic NADPH to reduce molecular oxygen to the superoxide anion (O2-) that can metabolize into more potent ROS for effective pathogen clearance (133, 134). The role of PPP for NADPH and subsequent NADPH-oxidase ROS production has been validated. In this study, two chemical inhibitors LDC7559, and NA-11, are both shown to activate the glycolytic enzyme phosphofructokinase-1 liver type (PFKL), the main phosphofructokinase-1 in immune cells, and dampen flux through the PPP in human peripheral blood neutrophils. This inhibits NADPH-oxidase ROS production, NETosis, efficient bacterial killing and tissue damage in an in vitro model of acute respiratory distress syndrome (ARDS) (130, 132).
5.2.2 Glycolysis and the electron transport chain
Lectins localized on the bacterial surface bind to carbohydrate-containing epitopes (fucose and mannose glycans) on neutrophil surface to induce intracellular ROS production (135). In addition, neutrophils respond to disseminated candidiasis through enhanced glucose uptake and glycolysis. Glycolysis is elevated through an accumulation of glucose transporter 1 (Glut1) on cell membrane to regulate phagocytosis, ROS production, and fungi elimination (136). Here, neutrophil-specific deletion of Glut1 does not affect neutrophil development, but rather antifungal activity and the control of systemic fungal infection through compromised glucose uptake and glycolysis (136). Moreover, BM derived neutrophils (ckit+) appear to be dependent on both glucose metabolism and mitochondrial function for NADPH oxidase dependent ROS production (47, 104). In these studies, inhibition of glycolysis results in a substantial increase in mitochondrial oxygen consumption rate (OCR – measure of ROS production). Inhibition of mitochondria following restricted glucose utilization, completely abolishes OCR (47). On the contrary, inhibition of mitochondrial function in primary murine BM neutrophils with rotenone and antimycin A (complex I and III inhibitors, respectively) alone does not affect early ROS (47). However, reductions in the late phase of the response are seen, suggesting that while the mitochondria is not required for the rapid initiation of neutrophil responses, they may be required to sustain these responses (47). Furthermore, the same study provides evidence that when glycolysis is inhibited by 2- deoxy-D-glucose (2DG) as in cancer, the mitochondria can prioritize ROS production by sustaining levels of NADPH through fatty acid metabolism (Figure 3) in the immature, ckit+/CXCR2+ BM neutrophil phenotype (31, 47, 134, 135). This study also highlights the role of neutrophil phenotypic and functional heterogeneity for the regulation of disease state outcomes.
Other studies show that mitochondria-derived ROS (mitoROS) by the electron transport chain can indirectly regulate neutrophil activation by promoting degranulation and activation of NADPH oxidase upon cell stimulation with fMLP. This emphasizes that on its own mitoROS is inefficient for neutrophil functional responses and that NADPH oxidase is required for efficient microbial killing (137, 138). Inhibition of complex I or III of the respiratory chain upregulates mitochondria ROS production, diminishes LPS-stimulated neutrophils, and reduces severity of LPS-induced acute lung injury in mice (139, 140).
5.2.3 Metabolic perturbations implicated in NADPH-oxidase dependent ROS production
Mitochondrial dysfunction plays an important role in metabolic diseases such as diabetes. Autophagy is shown to be important for neutrophil function as neutrophils from streptozotocin-induced diabetic rats where high incident of infections is prevalent, show autophagy impairment (141). Consequently, reduced ROS production, depolarization of , low ATP content, and high content of cleaved caspase 3 (apoptosis marker) after phorbol myristate acetate (PMA) stimulation is seen. This is consistent with findings where Atg7 and Atg5 knockout mice show impaired degranulation of primary, secondary and tertiary granules and NADPH oxidase ROS (137, 141). Similarly, in another study, phagocytosis and bacterial killing is reduced in Atg7 and Atg5 deficient neutrophils (7). This suggests that while mTOR activity, which regulates glycolysis, is enhanced in diabetic rats, optimal neutrophil functions are achieved through mitochondrial metabolism. Here, treatment with metformin and induction of autophagy with AMP-activated protein kinase (AMPK) improves neutrophil responses (111, 125). In acute myocardial infarction (MI), the negative consequences, i.e., tissue damage and heart failure, are attributed in part to upregulation of neutrophil mitochondrial ROS production, and p47 phosphorylation with energy derived from glycolysis (142). However, treatment with arjunolic acid (AA), an antioxidant, reduces ROS via the inhibition of p47 and reduced mitochondrial and glycolytic oxidative burst activity to alleviate the negative consequences of MI (142). In vivo models of acute lung injury show that both metformin (used frequently in the treatment of diabetes) and rotenone, can inhibit complex I of the electron transport chain (ETC). This inhibition increases intracellular ROS production by mitochondria, despite obvious reductions in ATP levels, and diminishes production of pro-inflammatory cytokines to mitigate disease severity (134). The mechanism through which neutrophils can use alternative fuel sources in diverse altered nutrient conditions remains to be explored further.
6 Metabolic requirements for NET formation
6.1 Overview of NETs
Within neutrophils, the ROS released by the NADPH oxidase complex may also induce the formation of neutrophil extracellular traps (NETs). NETs are decondensed chromatin strands of nuclear and mitochondrial DNA (mtDNA), decorated with antimicrobial molecules from primary granules (MPO, NE). These are released into the extracellular space to effectively trap and eliminate large pathogens, such as Candida albicans hyphae, S. aureus, K. pneumoniae, and P. gingivalis to name a few, but not small or single microbes, to prevent dissemination of these pathogens to systemic organs (143, 144). NETs generally result in neutrophil death, but NADPH oxidase-independent mechanisms, i.e., mitoROS-dependent, non-lytic form of NET formation also exist, for the degradation of gram-negative and positive bacteria (145). The pathway choice is largely dependent on the specific agonists encountered by the neutrophils (146). Pathogen size sensing by dectin-1 on neutrophils and NE translocation to the phagosome or nucleus, promotes phagocytosis and NET release with small, and large microbes, respectively, for efficient pathogen clearance, minimal tissue damage, and host survival. Lack of dectin-1 or Mpo in mice leads to increased NET formation and local and systemically disseminated C. albicans infection (144). However, the origin and mechanism of the DNA scaffold in in vivo NETs in bacterial infections remains debated. Transcription factor and kinase (p38 MAPK) activity, peptidylarginine deiminase 4 (PAD4) mediated histone citrullination or ROS-mediated DNA oxidation and repair machinery and resulting chromatin decondensation for NET formation may be important in homeostasis and inflammatory disease states (146–149). In cancer settings, NET formation drives metastases and during sterile conditions, increased NETs are indicative of autoimmune disease states (80, 150). The presence of ‘adherent’ NETs, extruded DNA adhering to the vasculature, by “fresh” neutrophils enhances vascular damage in mice (10). Therefore, despite their role in host defense, excessive NET formation is associated with pathology. As such, blunted NET formation may help to protect mouse and human lungs from exacerbated injury in pneumonia (10).
6.2 Metabolic requirements for pathogen killing via NETs
NET formation is strictly dependent on glucose through the glycolytic pathway [Figure 3; (136)]. In vitro assays show that neutrophils stimulated with 100 nM PMA in glucose-free media exhibit chromatin decondensation within a few hours, resembling the early stages of NET formation, but no NET release. The addition of glucose at a later time point allows for NET formation to occur within 10 minutes (151). Addition of glycolysis inhibitor 2-DG also completely inhibits NET release, while the addition of oligomycin also inhibits the formation of NETs through an undefined mechanism (151). Both NADPH oxidase ROS and autophagy play debated roles in chromatin decondensation for NET formation (152). For example, in trauma patients the increased AMPK activity modulates autophagy and impairs aerobic glycolysis and NETosis causing neutrophil dysfunction and adverse clinical outcomes (121, 153). The role of mTOR and H1F-1a as regulators of NET formation upon LPS stimulation is identified but the mechanism through which H1F-1a regulates NET formation remains to be explored (153). In this case, pharmacological and genetic H1F-1a knockdown reduces NETosis (153). Furthermore, both NADPH-oxidase dependent and independent NET formation by human and murine blood neutrophils are shown to depend on glycolysis dependent lactate formation where inhibition of lactate dehydrogenase (LDH) activity inhibits NETosis (154). The role of glycans, including heparan sulfate, and enhanced glycolysis for NETosis has also been studied extensively (155, 156). The findings presented here suggest that glycolysis, regulated through multiple pathways, plays an undisputed role in regulating maximal neutrophil responses. Here, neutrophil dysfunction following acute trauma is a result of reduced glucose uptake and metabolism. On the contrary, studies show that immature low-density neutrophils (iLDNs; neutrophil precursors) are able to promote breast cancer liver metastasis by executing NETosis under glucose-deprived conditions through glutamate and proline catabolism for mitochondrial-dependent ATP production (80). Furthermore, mitochondrial, optic atrophy type-1 (OPA1), appears indispensable for NOX-independent NET release and bacterial killing through indirect regulation of glycolysis (Figure 3) (81, 113, 153). Other agonists known to induce NET formation such as PMA produced similar effects as NETs of mtDNA origin, but the mechanism is unknown.
7 Expression of inflammatory mediators by neutrophils
7.1 Overview of cytokine and chemokine generation by neutrophils
Expression of cytokines and chemokines by myeloid and non-hematopoietic cells in response to bacterial components via TLR/NF-kB and MyD88 signaling drives neutrophil activation, and inflammation. Neutrophils themselves are capable of generating pro-inflammatory cytokines such as IL1a, IL1β, TNF and IL6 and the chemokines CCL2 and CXCL2, in response to stimulation (2, 9, 128). The TF JUNB is identified as a major regulator of the expression of these mediators (9). The aberrant production of these mediators can also drive pathological destruction of tissues (9, 41, 42).
7.2 Metabolic requirement for cytokine and chemokine generation by neutrophils
Studies examining the metabolic requirements for cytokine and chemokine generation by neutrophils in homeostasis and disease states are currently limited. In sepsis, neutrophils are crucial components of the innate immune response and the cytokine storm that characterizes the acute phase response, and this process requires glycolysis (157–159). Inhibition of glycolysis with metformin and rapamycin diminishes survival of mice with sepsis caused by C. albicans, lowers ex vivo cytokine production (of TNF and IL-1) and increases fungal growth. Therefore, pro-inflammatory cytokine production appears to be a glycolysis driven process whereby stimulated mature neutrophils in inflamed tissues directly generate these inflammatory mediators (Figure 3) (9). In cancer, heterogenous neutrophil subsets indirectly influence cytokine production. In fact, a more glycolytic neutrophil (ckit-CXCR2+ phenotype) is unable to induce T cell death and inhibit interferon-γ (IFN-γ) production by T-cells, thereby enhancing anti-tumor immunity (47). On the contrary, oxidative neutrophils (ckit+CXCR2- phenotype) in the tumor microenvironment suppress anti-tumor immunity through IFN-γ (47).
8 Inherited metabolic disorders that affect neutrophil function
A total of 485 inborn errors of immunity (IEI) with altered molecular, cellular, and immunological mechanisms contribute to our understanding of the inheritable monogenic defects for immunological disorders. IEI are categorized (160) by the International Union of Immunological Societies Expert Committee. Here, particular gene defects, characterized as combined immunodeficiencies, and congenital defects of phagocyte number or function, relate specially to neutropenia, neutrophil dysfunction and recurrent infections. We do not discuss all the inborn neutropenia and neutrophil disorders in this article. We only focus on those that link inherited metabolic defects with neutrophil dysregulation as metabolic processes are critical for neutrophils’ functional fitness. Neutrophils are capable of glycogen cycling through glycogenesis and gluconeogenesis for energy production and maintenance of effector functions (161). Here, the enzyme glucose-6 phosphatase catalytic subunit 3 (G6PC3) hydrolyzes glucose-6-phosphate to generate glucose for glycolysis and downstream metabolic processes. Neutrophils from hypoglycemic patients with glycogen storage disease type Ib (GSD-Ib) and G6PC3 deficiency in severe congenital neutropenia have impaired ROS, killing defects, and recurrent infection (162–164). This emphasizes the indispensable role of glycolysis and PPP for neutrophil effector functions and infection outcomes. Here, dietary glucose and gene therapy holds therapeutic potential to restore neutrophil dysfunction (165). Similarly, deficiency of any of the oxidase components, due to mutations of autosomal or X-linked recessive genes encoding the 5 components, and the resultant inability to oxidize NADPH results in chronic granulomatous disease (CGD). Neutrophils from these patients migrate and phagocytose normally but fail to generate an NAPDH-oxidase dependent oxidative burst with the resultant failure of intracellular killing leading to recurrent bacterial and fungal infections, including pneumonia (134, 145). The literature on these inherited disease states is reviewed more extensively elsewhere (166) which readers are referred to. Mitochondrial respiration is known to maintain neutrophil differentiation and functions. Pearson syndrome is a rare mitochondrial disorder accompanied with large-scale mtDNA deletions, severe defects in erythroid and myeloid precursor cells, neutropenia, and both severe and fatal infections (167, 168). This emphasizes the previously underappreciated role of mitochondria in neutrophil development and control of infection outcomes. Additionally, 3-Methylglutaconic aciduria due to CLPB, an enzyme for leucine degradation, deficiency is also associated with mitochondrial dysfunction and altered neutrophil differentiation (160). Patients with mutated OPA1 develop autosomal dominant optic atrophy (ADOA) and neutrophils from these patients have a reduced ability for extracellular DNA released and impaired ability for pathogen killing due to impaired complex I activity, and limited NAD+ for glycolytic ATP production (113).
9 Regulation of neutrophil functions in various nutrient environmental conditions
Nutrition orchestrates cellular metabolism. Fluctuations in nutrient availability, i.e. nutrient excess and deficiency can modulate metabolic processes and consequently cause hyperactivation or immunosuppression of neutrophils associated with diverse inflammatory disease states (Figure 4). Inadequate and inefficacious immune responses are common in more affluent and developed countries with excess nutrient intake (171). Nutrient deficiency is associated with exaggerated inflammation and impaired resolution of inflammation (172, 173).
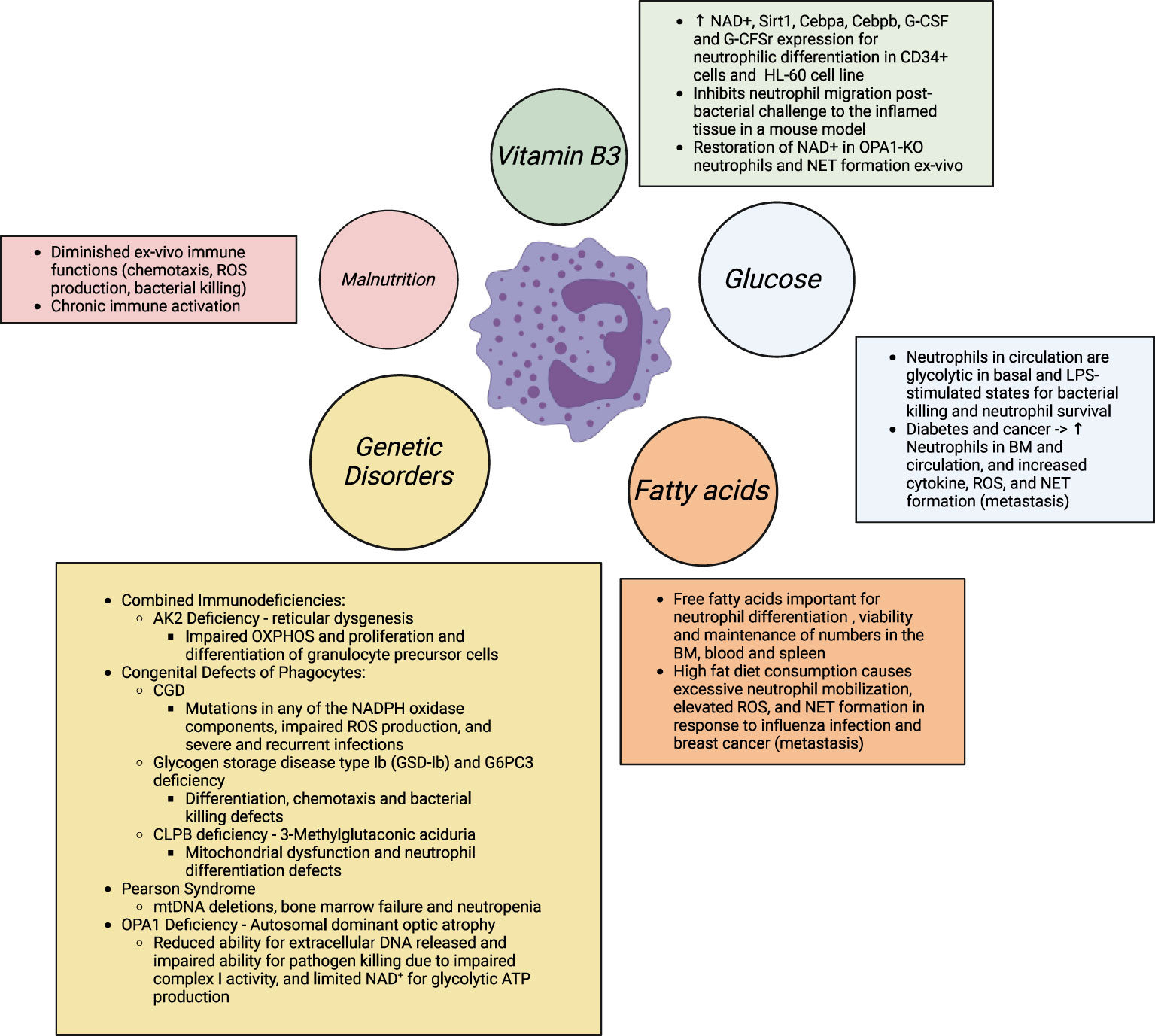
Figure 4 Neutrophil differentiation and functions are disrupted with altered nutrient availability, as well altered usage in multiple genetic diseases. In obesity and associated co-morbidities (diabetes, cancer, chronic inflammation), excess nutrient availability, i.e., glucose and free fatty acids, has shown to prime neutrophils for elevated ROS and pro-inflammatory cytokine production, degranulation, and NETosis for elevated tissue damage. Studies further show that elevated NETs and the metabolically flexible neutrophil subset in obese hosts (immature low- density neutrophils; iLDNs) makes the host more prone to breast cancer cell lung and liver metastasis and adverse outcomes in cancer (8, 169, 170). In contrast, in malnutrition, defined with scarce substrate availability, especially of amino acids, neutrophils are unable to kill pathogens and therefore, have a sustained hyperinflammatory response that leads to morbidity and mortality in this vulnerable population. Similarly, in a myriad of inherited immunodeficiency diseases, altered substrate utilization also predisposes the host to severe recurrent and fatal infections. The IEI classification is based on published literature (156). Supplementation with NAD+ precursors may provide therapeutic benefit in these conditions of neutrophil dysfunction.
9.1 Micronutrient-dependent regulation of neutrophils in homeostasis and inflammation
9.1.1 Overview of micronutrients involved in neutrophil biology
Different micronutrients relate to neutrophil biology in homeostasis and disease states (174), in particular, Vitamin C (175–178), iron (179–181), zinc (182–185), Vitamin D (186, 187) and Vitamin A (188, 189). In general, a deficiency in any one of these micronutrients is associated with dysregulated neutrophil functions, particularly neutrophil recruitment, degranulation, ROS formation and NETosis, resulting in higher susceptibility to recurrent and severe infections. We particularly focus on the rapidly evolving field of nicotinamide and nicotinic riboside, which are both forms of Vitamin B3 and central mediators of metabolic processes, for the regulation of neutrophil biology.
9.1.2 Overview of NAD and vitamin B3 metabolism
Nicotinamide adenine dinucleotide (NAD+) is an essential cofactor that plays an indispensable role in key redox reactions and serves as a substrate for PARPs, sirtuins, CD38, ADP-ribosyl-transferases, sterile alpha and TIR-motif-containing protein 1, and RNA polymerases. For its role in oxidation-reduction reactions, it accepts electrons from glycolysis, FAO and TCA intermediates to form NADH and feeds them into complex I of the electron transport chain for OXPHOS. As such, NAD+/NADH are essential regulators of many cellular processes. Despite its fast and dynamic metabolism, NAD+ concentration reflects the balance between NAD+ consumption and synthesis from the de novo, Preiss-Handler and salvage pathways. The de novo biosynthesis of NAD results from the breakdown of tryptophan mostly in the liver (190). However, the major pathway of NAD+ biosynthesis is the salvage pathway. Here, NAD+ is converted to nicotinamide (NAM), a form of vitamin B3, and reconverted to NAD+ using the enzyme nicotinamide phosphoribosyltransferase (NAMPT). Nicotinamide riboside (NR), another form of vitamin B3, can also enter the salvage pathway through NR kinase (Nrk1/2)-mediated phosphorylation of NR into nicotinamide mononucleotide (NMN) (99). Progressive decline in NAD+ homeostasis and mitochondrial function are common hallmarks of ageing and disease pathologies including neutrophil dysfunction and hyperinflammation. While some studies report beneficial effects of NAD+ precursors such as NR and NAM to counter age-related functional decline in murine models, others report no such effects in human subjects [reviewed in (191)]. We focus on the role of NAD metabolism in neutrophil biology.
9.1.3 NAD and vitamin B3 metabolism in neutrophils
The treatment of hematopoietic progenitor (CD34+) cells in healthy individuals with either G-CSF or NAM increases intracellular levels of both NAMPT and NAD+. Neutrophilic differentiation is also induced through NAD+-dependent sirtuin-1 activation, subsequent binding and activation of Cebpa and Cebpb, and, ultimately, upregulation of G-CSF synthesis and G-CSF receptor expression (91). NAMPT expression is highest in promyelocytes and NAMPT is specifically localized to the mitochondria (91, 192). This suggests that neutrophil precursors express functional mitochondria for NAD+ conversion from NAM and therefore neutrophil differentiation regulation via the mechanism that is proposed (91). However, no study has studied this mechanism in closer detail.
NR supplementation is shown to improve HSC and progenitor function by increasing mitochondrial clearance in HSCs. This finding is surprising since multiple publications consistently show an increase in mitochondrial fitness with NR treatment (99, 193, 194). Nevertheless, no study has yet examined how NAD+ precursor treatment in committed progenitors affects differentiation where mitochondrial function is preferred to sustain increased ATP demands. While an increase in granulocytic differentiation is shown in steady-state through the involvement of NAMPT, NAD+, G-CSF and G-CSFR upon NAM treatment, the role of NAM on NAD+ in regulating neutrophil homeostasis in steady-state and emergency granulopoiesis needs clarification. Oral NR supplementation at a dose of 400 mg/kg/day for 8 weeks shows an alleviation of the BM HSC ageing phenotype, that is normally dominated by a significant expansion of the HSC pool and differentiation skewed towards the myeloid lineage and increased metabolic activity of HSCs. NR promotes molecular and mitochondrial changes in the aged BM that resembles the young HSC state including improved NAD/NADH ratio, reduction in the frequency of myeloid and lymphoid progenitors, improvement in age-deregulated HSC genes, and a reduction in the metabolic potential of aged HSCs at the transcriptional level accompanied with increased sirtuin-3, for mitophagy. Here, there is a modest restoration of the ageing HSC phenotype with NR but no effect of NR is observed in the differentiated myeloid cells (26). Moreover, increased extracellular NAD+ in inflammatory conditions can help delay mitochondrial-dependent neutrophil apoptosis at inflamed sites (169). This is dependent on NAD+ inhibiting the degradation of Mcl-1 (anti-apoptotic), suppressing Bax translocation to the mitochondria, attenuating the dissipation of mitochondrial membrane potential and cytochrome C release from mitochondria into the cytosol and supressing caspase-9 and -3 activation (169). The role of administrating 1000 mg/kg NAM in attenuating exacerbated neutrophil recruitment to inflamed tissues in murine models has also been studied although the downstream molecules regulating this process has not been determined (195, 196). NAM and nicotinic acid effectively inhibit neutrophil migration when administered twice, i.e., 30 minutes before and 1 hour after bacterial insult while single administration after bacterial challenge fails to prevent neutrophil recruitment into the mouse pleural cavity (195, 196). In mice with OPA1 gene mutations where NAD+ is reduced, treatment with NMN rescues systemic NAD+ levels. This in turn helps to indirectly regulate NET formation following neutrophil activation (Figure 3) (113).
9.2 Glucose requirements in neutrophil biology
Neutrophils function in metabolically challenged environments. Human peripheral blood neutrophils are primarily glycolytic in both basal and LPS-stimulated states (161). With glucose deprivation, these cells can maintain their intracellular energy homeostasis provided their glycolytic pathway is active due to increased glycogen stores in these cells (161). Interestingly, neutrophils express enzymes required for active glycogenesis and gluconeogenesis that enables this metabolic flexibility in glucose deprivation and other metabolically challenged environments. Through 13C labeling, glutamine is identified as the substrate in gluconeogenesis, where inhibition of these processes and associated enzymes impairs bacterial killing and neutrophil survival, and promotes systemic spread of bacteria in mice (161).
9.2.1 Role of glucose in obesity-associated comorbidities
Hyperglycemia, as is commonly in diabetic mice and humans, is directly associated with an increase in BM and circulating neutrophils with an increased expression of the calprotectin subunit S100A8, and extracellular ROS production (197). Similarly, incubation of neutrophils from healthy donors with high glucose media or neutrophils from diabetic mice produces more PAD4 and NETs for an impaired wound healing response (198). Additional dysregulated functions of neutrophils in type 1 and 2 diabetes have been reviewed elsewhere (199). The mechanisms by which increased glucose availability alters neutrophil biology, for the heightened inflammatory response and tissue damage has not yet been addressed. Persistent elevation of glucose levels in diabetes and polyol and hexosamine pathway may be involved but this has not been studied mechanistically in the forementioned studies and should be the focus of future studies.
9.3 Fatty acid metabolism in neutrophils
Endogenous fatty acid synthesis and utilization is crucial for the maintenance of mature neutrophils. Dietary lipid intake in neutrophil-specific fatty acid synthase (FAS) knockout mice can partially reverse the aberrant phenotypes of FAS deficiency, reduced neutrophil viability and numbers in the BM, blood and spleen (200). Similarly, in Atg7 deficient murine myeloblasts, pyruvate treatment or exogenous free fatty acids (linolenic acid or a mix of unsaturated and saturated free fatty acids) alone is sufficient to restore normal glucose metabolism and rescue the defective neutrophil differentiation.
9.3.1 Role of free fatty acids in obesity and associated comorbidities
High fat consumption is linked to poor outcomes of influenza infection in BALB/c mice fed a high-fat diet (HFD) for 18-weeks due to elevated H2O2 concentration and NET formation (201). Additionally, mice on the HFD for 15 weeks injected with breast cancer (BC) cells have lung neutrophilia, and increased lung vascular permeability compared to mice fed an isocaloric low-fat diet. This is attributed to higher NADPH-oxidase dependent ROS, granule proteins, pro-inflammatory cytokines and NETosis by lung neutrophils from obese hosts. Therefore, BC cells in the lungs (lung metastasis) of obese mice are increased compared to lean mice. In this regard, depletion of neutrophils with anti-Ly6G antibody in obese mice reduces lung permeability and BC cell extravasation (8, 202). Similarly, obese human subjects with elevated BMI (35–68 kg/m2) and serum triglycerides have elevated chemotaxis and superoxide generation in unstimulated and stimulated neutrophils compared with lean controls. In this case, phagocytosis is not affected (203).
9.4 Protein and amino acid metabolism for neutrophils’ biological processes
The role of key amino acids such as glutamine on neutrophil biology has been discussed above. In this section, we discuss the impact of perturbed amino acid and protein homeostasis for neutrophil biology. In chronological aging, alterations in protein intake and anabolic metabolism, especially of the essential branched-chain amino acid, leucine, is associated with age-related progressive loss of muscle mass, osteoporosis and frailty, and can be overcome with a higher level of protein intake (204–206). In this context, increased neutrophil numbers and degranulation, reduced coordinated neutrophil migration, phagocytosis, ROS production and NET generation, and a heightened pro-inflammatory state contributes to loss of skeletal muscle mass [reviewed in (207–209)]. Particularly, an expansion of neutrophils with a capacity for reverse transmigration and increased remote organ damage has been identified (41). Additionally, various basic leucine zipper TFs constitute the transcriptional network for the neutrophil life cycle of which, leucine content is a major determinant. However, no study to date has yet explored how dysregulation of specific amino acids, such as leucine, influences neutrophil biology in aged tissues. A randomized clinical trial has assessed the effects of whey protein and leucine ingestion post-exercise on neutrophil functions, which are attenuated during intensive and prolonged endurance exercise, during 6 days of intense cycling in 12 male cyclists (210). Leucine-enriched whey protein ingestion improves neutrophil oxidative burst post-exercise, to prime neutrophils for host immunity and tissue repair (210). Additionally, the effects of a hypocaloric Mediterranean diet (MD) and two high protein diets, with (HPW) and without (HP) whey protein supplementation, on body composition, lipid profiles, inflammation and muscle-damage blood indices in overweight, sedentary, young participants has been assessed (211). Neutrophil-mediated inflammation and muscle-damage is increased in HP and HPW compared to the MD group. No other aspects of neutrophil activity or functions are studied here (211). The association between dysregulated tryptophan-kynurenine metabolism and neutrophils in obesity is also summarized extensively (212).
9.4.1 Nutrient deficiencies as contributors to neutrophil dysfunction
Earlier, we focus on excess intake of macronutrients as they relate to co-morbidities of obesity including diabetes, cancer, and chronic inflammation. Multiple excellent articles and reviews have already been published on this in great detail that readers are referred to for additional learning [reviewed in (213–216)]. The section below is focused on inadequate nutrient intake as it relates to undernutrition and neutrophil perturbations, on which very little has been published to date.
In undernutrition, referred to as malnutrition in this section, inadequate intake of protein and calories, and micronutrient deficiencies predisposes children to diminished immune functions and chronic immune activation that increases susceptibility to infections and multiple organ failure [reviewed in (173, 217–219)]. There are limited studies performing functional assays of immune cells in low-and-middle-income countries in children with varying forms of malnutrition. Some longitudinal studies compare immune cell function in children with severe malnutrition at admission, and during recovery with exclusion of HIV-positive children. Even more limited studies in childhood malnutrition suggest some impairments in peripheral blood neutrophil function. Here, in vitro assays show reduced chemotaxis in children with severe malnutrition but no infection (220, 221). In another context, Leishmania donovani, a parasite that causes visceral leishmaniasis, in children with malnutrition reduces in vitro neutrophil and monocyte TEM after PMA stimulation as determined indirectly by flow cytometry using CD62L (222). The same study also shows reduced ROS production in these cells as determined by Dihydrorhodamine 123, but the study did not stratify by the degree of malnutrition in the sample (222). In children with severe malnutrition but no infection, in vitro microbicidal defects are shown to occur with S. aureus, E. coli and C. albicans in mononuclear and polymorphonuclear cells, while the postphagocytic morphological events, including vacuole formation and degranulation, are normal (220, 221). The data on phagocytosis is inconsistent, with some reporting unaffected phagocytosis whist others observing reduced phagocytosis especially when infection is present (217, 219, 223–225). Similarly, consensus points to impaired ROS production determined through nitroblue tetrazolium reduction assay and impaired bacterial killing capacity in children with severe malnutrition with or without infection (217, 224–227). It is unknown from these assays however whether NADPH or mitochondrial ROS production is hindered in malnutrition. These studies are conducted in different geographical sites, and differing disease states which may underlie the inconsistencies in findings. While the data suggests that not all functions are affected to the same extent, these studies only show associations and not the causal pathways linking malnutrition to impaired functions and clinical outcomes. Mice fed a protein-free diet have less neutrophils in infected lungs and this reduction could be related to impaired granulopoiesis, but the mechanism is not elucidated (224, 228). On the contrary, increased immature neutrophils are found in the blood defined through electron microscopy and flow cytometry in human cohorts while others report no change in total blood leukocyte count (220, 226, 229).
9.5 Therapeutic strategies for neutrophil recovery in nutritional deficiencies and excess
Anorexia, characterized by phenotypes and increased susceptibility to bacterial pathogens observed in undernutrition, is also an evolutionary conserved common response to infectious diseases. Immunological changes reported in anorexia are reviewed elsewhere in greater detail (230). Here, the refeeding syndrome is a serious complication of anorexia treatment that results from excessive nutrient supplementation resulting in rapid hormonal and metabolic disturbances and high morbidity and mortality (170, 231). In fact, diminished ROS production during the initial period of refeeding (11-40 days of hospitalization) in patients with anorexia that return to normal values to that of healthy control subjects with an extended period of refeeding is commonly seen (232). A fiber-rich diet in this refeeding regime may be sufficient to restore efficient immunity (233).
In conditions of nutrient excess, inadequate and inefficacious immune responses also underlie poor infection outcomes, as discussed above. This includes exacerbated neutrophil mobilization, and ROS and NET formation that prolongs inflammation in obesity (8, 216). Here, mice fed a mild calorie restricted diet regimen had an improved pulmonary anti-mycobacterial host response with reduced bacterial load, and lung immunopathology (172). In addition, adiponectin, a hormone that is depleted in obesity, repletion in obese mice and humans regulates neutrophil oxidative burst through its anti-inflammatory properties (234). Clinically approved drugs for type 2 diabetes including pioglitazone (235), a PPARɣ agonist and metformin, can induce adiponectin in mice and humans to regulate the exacerbated inflammatory responses in this context. How these therapeutics directly target immune cell metabolism remains to be studied further, but modulation of autophagy and mitochondrial metabolism may be important here (236). Gout is a debilitating chronic inflammatory arthritis, exaggerated by age and diet-induced lipotoxicity. Here, a ketogenic diet ameliorates neutrophil IL-1β secretion by increasing levels of β-hydroxybutyrate and inactivating NLRP3 inflammasome to protect the mice against inflammation (237, 238). Through its histone deacetylase inhibitory activity and therefore gene expression, β-hydroxybutyrate also regulates neutrophil-mediated immunity (233, 239). As a result, this therapeutic strategy is also relevant for other inflammatory diseases related to nutrient excess that are driven by chronic neutrophil activation.
10 Concluding remarks & perspectives
In the last decade, progress has been made to discern the metabolic requirements and flexibility of neutrophils that extends beyond glycolysis using traditional and novel immunometabolism approaches. Importantly, we discuss in this review from evidence in humans and mouse models reviewed here that neutrophils rely on the activity of multiple metabolic pathways to fulfill their energy requirements throughout their life stages. Hindrance in these processes, substrate utilization and breakdown, disrupts optimal neutrophil biology. Here in steady-state, neutrophil committed progenitors rely on fatty acid oxidation and mitochondrial ATP generation for differentiation while mature neutrophils require both mitochondrial and non-mitochondrial sources for energy production. In inflammatory states including obesity, and undernutrition, the ability to sense and reprogram metabolism based on nutrient availability also marks the response in these conditions. In these contexts, heterogenous neutrophils populations with different energy requirements may be recruited to mediate the inflammatory response and clinical outcomes, although this remains to be explored further. By further understanding the unique role of metabolism, although not ‘black and white’ for neutrophil biology, we will be better able to therapeutically modulate these pathways for better outcomes in inflammatory disease states.
11 Outstanding questions
Despite advancement in the understanding of neutrophil metabolism with newer technologies, much more work needs to be done to uncover the molecular mechanisms coordinating the neutrophil lifecycle and antimicrobial functions before we fully understand neutrophil metabolism for its modulation in different disease contexts. Here, we provide few examples of key areas where additional work is needed.
11.1 HSC and neutrophil development in the BM
Although HSCs are early determinants of neutrophil commitment, we still do not know how the cellular and metabolic components of HSCs interact, either directly or indirectly, to regulate neutrophil development and functions under steady-state and stress conditions. The mechanism linking NAD+ supplementation with regulation of TFs for neutrophil differentiation and maturation (Cebpa, Cebpb) has not been addressed to date and remains to be studied. Autophagy, mitochondrial respiration, and their link with gene regulation remains to be studied in the human neutrophil developmental trajectory.
11.2 Neutrophil migration
Newer technologies provide a better understanding of the metabolic requirements for efficient neutrophil migration in healthy tissues. Therefore, investigating neutrophil migration behaviour in inflammatory pathology and metabolic challenge may have implications in the resolution of human disease where neutropenia or neutrophilia is common. The metabolic requirements for TEM and swarming also remain unexplored and require further investigation.
11.3 NADPH oxidase ROS production
There is evidence that glycolysis regulates NADPH oxidase activity, but we still do not understand how glycolytic activity regulates NADPH oxidase subunits’ activation and the mechanism that connects reduced glucose utilization to enhanced mitochondrial capacity in inflammatory disease states. Here, the role of metabolites in controlling chromatin modifications, DNA methylation, and post-translational modifications of proteins for the determination of cell fate and function will be relevant, but this has never been explored for neutrophil biology. The advent in the integration of multiple single cell ‘omics’ enables for such research and might be worth exploring in this context. Additionally, the mechanism of the interplay between neutrophil metabolism, differentiation and ROS-mediated functions requires further investigation in homeostasis and disease states to aid in the development of targeted therapeutics.
11.4 NET formation
It was highlighted in the text that NET formation requires autophagy and while autophagy may play a role in removing damaged mitochondria, mitophagy, the exact mechanism through which autophagy regulates NET formation remains unclear and requires further clarification. Accumulation of lactate and NETs may partially explain disease severity and adverse outcomes associated with sepsis, however how lactate can trigger NETosis remains elusive (154). Although H1F-1a regulates NETosis, optimal NETosis also requires NADPH oxidase ROS, which is decreased under hypoxia due to limited oxygen availability. It would therefore be interesting to explore the extent to which H1F-1a can help recover NETosis in hypoxia where NADPH oxidase ROS is limited. Additionally, the role of mitoROS for NOX-independent vital NET release in this mechanism remains disputed and requires more exploration.
11.5 Neutrophil crosstalk with other cells
Studies are needed to examine metabolic programming involved directly in neutrophil communication with immune and non-immune cells irrespective of the soluble mediators that have often been discussed previously.
11.6 Neutrophils in altered nutritional environments
As decline in NAD+ has been implicated in many diseases, future studies must investigate the impact of changes in NAD+ availability on innate immune responses, specifically that of neutrophils and the mechanisms involved in the dysregulation. Additionally, studies must investigate the mechanistic pathway regulating granulopoiesis and neutrophil functions with the supplementation of NAD+ and its precursors’. Studies have also not yet addressed how free fatty acids regulate epigenetic modifications to drive neutrophil differentiation, irrespective of mitochondrial ATP generation. The molecular mechanism through which excess glucose and metabolic rewiring in diabetes promotes neutrophilia and exacerbates inflammatory disease requires further exploration in mice and humans. This is crucial in the context of diverse tissue microenvironments where excess circulating glucose is accompanied by overt pathologies in multiple organ systems. Similarly, it is not known if correction of circulating glucose levels in these conditions eliminates neutrophil dysfunction and associated pathologies. Additionally, how excess glucose alters neutrophil phenotype and function needs to be studied further. Future research needs to focus on the exploration of novel neutrophil-centered treatments to resolve inflammation and improve patient outcomes in conditions of excess glucose availability. The causative mechanism of fatty acid excess on neutrophil functional diversity and inflammation has not been identified. Studies mentioned in the Role of Free Fatty Acids in Obesity and associated Comorbidities section suggest that although free fatty acids are crucial for normal neutrophil functions, excess substrate availability promotes the emergence of functionally distinct neutrophils that can promote aberrant disease states. More mechanistic studies are needed to evaluate the effects of varying quantities of fatty acid consumption on neutrophil biology. The existing studies with the existent pre-clinical models do not accurately represent phenotypic changes associated with severe malnutrition where multiple physiological changes are observed therefore the translational ability of these studies is questioned. This warrants the need to develop pre-clinical models that are representative of real-life in which to modulate metabolism and improve neutrophilic processes. Additionally, most of the studies conducted in children with malnutrition have major limitations: small sample size, lack of appropriate control groups, i.e., adequately nourished children from high-income counties as controls with no overt infection, and several forms of malnutrition. These studies are also limited by ethical constraints and methodological limitations, i.e., tissue sampling and immunological techniques. The mechanisms exacerbating impaired neutrophil functions in response to malnutrition and infection remains unexplained since observational studies can only determine associations, and not causality (145, 217). In this case, genetic approaches to decipher proposed pathways have not been undertaken and therefore causation cannot be determined. It is also impossible to delineate from these studies whether neutrophil dysfunction is a cause or consequence of malnutrition when infection is present. Additionally, the impact of specific metabolites and metabolic perturbations has not been closely explored in neutrophils from children with malnutrition especially since multiple metabolic derangements exist in this context and the role of limited substrate availability in this case also requires further exploration. Impaired autophagic flux and mitochondrial dysfunction has been associated with hepatic steatosis and gut barrier dysfunction in a mouse model of severe malnutrition (240–242). Differences in blood metabolomic profiles of children with severe malnutrition that died, especially an increase in metabolites of mitochondria-related bioenergetic pathways has also been identified (243, 244). Therefore, the role of mitochondria for neutrophil-associated catastrophe in the malnourished host requires further exploration. Additionally, the impact of specific micronutrient deficiencies on neutrophil biology has not been studied in detail in malnutrition. The impact of refeeding in malnutrition on additional neutrophil functions beyond ROS production and how it contributes to the refeeding complications is not well known. The role of changes in specific macro and micronutrient composition with refeeding and their impact on neutrophil biology is unknown and requires further exploration. It is well-accepted that mitochondrial function is lost as neutrophils mature and heterogeneity in neutrophil metabolic programming exists between different tissues and within neutrophil subsets. As such, uncovering neutrophil metabolic adaptations in inflammatory disease states and across heterogenous neutrophil subsets may help in targeting these pathways to improve long-term outcomes. Similarly, the role that the metabolic switch plays at inflammatory/infectious sites during metabolic disorders for outcome determination requires further investigation. We believe these studies will help novice and expert researchers in the field expand our understanding of the unique role of metabolism in steering neutrophil biology, a field that has largely remained neglected.
Author contributions
MT: Conceptualization, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing, Project administration. HU: Writing – review & editing. MG: Writing – review & editing. NP: Writing – review & editing. CB: Writing – review & editing. AG: Writing – review & editing. CL: Writing – review & editing. JB: Writing – review & editing. RB: Writing – review & editing, Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Resources. AF: Conceptualization, Investigation, Project administration, Supervision, Writing – review & editing, Validation, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This article is supported by Canadian Institutes of Health Research (CIHR156307) funding to RHJB. HHU is supported by the Leona M. and Harry B. Helmsley Charitable Trust and by the NIHR Oxford Biomedical Research Centre, University of Oxford.
Acknowledgments
All figures were created using BioRender software.
Conflict of interest
HU received research support or consultancy fees from Janssen, Eli Lilly, UCB Pharma, GSK, BMS/Celgene, MiroBio, OMass, and Mestag. AF is employed by Omega Laboratories Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Leiding JW. Neutrophil evolution and their diseases in humans. Front Immunol (2017) 8:1009. doi: 10.3389/fimmu.2017.01009
2. Ballesteros I, Rubio-Ponce A, Genua M, Lusito E, Kwok I, Fernández-Calvo G, et al. Co-option of neutrophil fates by tissue environments. Cell (2020) 183(5):1282–97. doi: 10.1016/j.cell.2020.10.003
3. Mann M, Mehta A, de Boer CG, Kowalczyk MS, Lee K, Haldeman P, et al. Heterogeneous responses of hematopoietic stem cells to inflammatory stimuli are altered with age. Cell Rep (2018) 25(11):2992–3005. doi: 10.1101/163402
4. Kwok I, Becht E, Xia Y, Ng M, Teh YC, Tan L, et al. Combinatorial single-cell analyses of granulocyte-monocyte progenitor heterogeneity reveals an early uni-potent neutrophil progenitor. J Clean Prod (2020) 53(2):303–318.e5. doi: 10.1016/j.immuni.2020.06.005
5. Hidalgo A, Chilvers ER, Summers C, Koenderman L. The neutrophil life cycle. Trends Immunol (2019) 40(7):584–97. doi: 10.1016/j.it.2019.04.013
6. Németh T, Sperandio M, Mócsai A. Neutrophils as emerging therapeutic targets. Nat Rev Drug Discovery (2020) 19:253–75. doi: 10.1038/s41573-019-0054-z
7. Riffelmacher T, Clarke A, Richter FC, Stranks A, Pandey S, Danielli S, et al. Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity (2017) 27(3):466–80. doi: 10.1016/j.immuni.2017.08.005
8. McDowell SAC, Luo RBE, Arabzadeh A, Doré S, Bennett NC, Breton V, et al. Neutrophil oxidative stress mediates obesity-associated vascular dysfunction and metastatic transmigration. Nat Cancer (2021) 2(5):545–62. doi: 10.1038/s43018-021-00194-9
9. Khoyratty TE, Ai Z, Ballesteros I, Eames HL, Mathie S, Martín-Salamanca S, et al. Distinct transcription factor networks control neutrophil-driven inflammation. Nat Immunol (2021) 22:1093–1106. doi: 10.1038/s41590-021-00968-4
10. Adrover JM, Aroca-Crevillén A, Crainiciuc G, Ostos F, Rojas-Vega Y, Rubio-Ponce A, et al. Programmed ‘disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat Immunol (2020) 21:135–44. doi: 10.1038/s41590-019-0571-2
11. Rosales C. Neutrophil: A cell with many roles in inflammation or several cell types? Front Physiol (2018) 9. doi: 10.3389/fphys.2018.00113
12. Takehara M, Seike S, Sonobe Y, Bandou H, Yokoyama S, Takagishi T, et al. Clostridium perfringens α-toxin impairs granulocyte colony-stimulating factor receptor-mediated granulocyte production while triggering septic shock. Commun Biol (2019) 2. doi: 10.1038/s42003-019-0280-2
13. Crainiciuc G, Palomino-Segura M, Molina-Moreno M, Sicilia J, Aragones DG, Li JLY, et al. Behavioural immune landscapes of inflammation. Nature (2022) 601(7893):415–21. doi: 10.1038/s41586-021-04263-y
14. Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol (2014) 14:302–14. doi: 10.1038/nri3660
15. Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K, et al. Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell (2017) 21(2):225–40. doi: 10.1016/j.stem.2017.06.013
16. Sato A, Kamio N, Yokota A, Hayashi Y, Tamura A, Miura Y, et al. C/EBPβ isoforms sequentially regulate regenerating mouse hematopoietic stem/progenitor cells. Blood Adv (2020) 4(14):3343–56. doi: 10.1182/bloodadvances.2018022913
17. Paudel S, Ghimire L, Jin L, Jeansonne D, Jeyaseelan S. Regulation of emergency granulopoiesis during infection. Front Immunol (2022) 13:961601. doi: 10.3389/fimmu.2022.961601
18. Malengier-Devlies B, Metzemaekers M, Wouters C, Proost P, Matthys P. Neutrophil homeostasis and emergency granulopoiesis: the example of systemic juvenile idiopathic arthritis. Front Immunol (2021) 12:766620. doi: 10.3389/fimmu.2021.766620
19. Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell (2020) 180(6):1044–66. doi: 10.1016/j.cell.2020.02.041
20. Boettcher S, Gerosa RC, Radpour R, Bauer J, Ampenberger F, Heikenwalder M, et al. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood (2014) 124(9):1393–403. doi: 10.1182/blood-2014-04-570762
21. Dwivedi P, Greis KD. Granulocyte colony-stimulating factor receptor signaling in severe congenital neutropenia, chronic neutrophilic leukemia, and related Malignancies. Exp Hematol (2017) 46:9–20. doi: 10.1016/j.exphem.2016.10.008
22. Zhao JL, Ma C, O’Connell RM, Mehta A, Diloreto R, Heath JR, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell (2014) 14(4):445–59. doi: 10.1016/j.stem.2014.01.007
23. Grassi L, Pourfarzad F, Ullrich S, Merkel A, Were F, Carrillo-de-Santa-Pau E, et al. Dynamics of transcription regulation in human bone marrow myeloid differentiation to mature blood neutrophils. Cell Rep (2018) 24(10):2784–94. doi: 10.1016/j.celrep.2018.08.018
24. Ito K, Ito K. Hematopoietic stem cell fate through metabolic control. Exp Hematol (2018) 64:1–11. doi: 10.1016/j.exphem.2018.05.005
25. Vannini N, Girotra M, Naveiras O, Nikitin G, Campos V, Giger S, et al. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat Commun (2016) 7. doi: 10.1038/ncomms13125
26. Sun X, Cao B, Naval-Sanchez M, Pham T, Sun YBY, Williams B, et al. Nicotinamide riboside attenuates age-associated metabolic and functional changes in hematopoietic stem cells. Nat Commun (2021) 12(1):2665. doi: 10.1038/s41467-021-22863-0
27. Yvan-Charvet L, Ng LG. Granulopoiesis and neutrophil homeostasis: A metabolic, daily balancing act. Trends Immunol (2019) 40(7):598–612. doi: 10.1016/j.it.2019.05.004
28. Kim MH, Yang D, Kim M, Kim SY, Kim D, Kang SJ. A late-lineage murine neutrophil precursor population exhibits dynamic changes during demand-adapted granulopoiesis. Sci Rep (2017) 7. doi: 10.1038/srep39804
29. Borregaard N. Neutrophils, from marrow to microbes. Immunity (2010) 33(5):657–70. doi: 10.1016/j.immuni.2010.11.011
30. Adrover JM, del Fresno C, Crainiciuc G, Cuartero MI, Casanova-Acebes M, Weiss LA, et al. A neutrophil timer coordinates immune defense and vascular protection. Immunity (2019) 50(2):390–402. doi: 10.1016/j.immuni.2019.11.001
31. Evrard M, Kwok IWH, Chong SZ, Teng KWW, Becht E, Chen J, et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity (2018) 48:364–79. doi: 10.1016/j.immuni.2018.02.002
32. Girbl T, Lenn T, Perez L, Rolas L, Barkaway A, Thiriot A, et al. Distinct compartmentalization of the chemokines CXCL1 and CXCL2 and the atypical receptor ACKR1 determine discrete stages of neutrophil diapedesis. Immunity (2018) 49(6):1062–1076.e6. doi: 10.1016/j.immuni.2018.09.018
33. Mylvaganam S, Freeman SA. The resolution of phagosomes. Immunol Rev (2023) 319:45–64. doi: 10.1111/imr.13260
34. Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chèvre R, A-González N, et al. XRhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell (2013) 153(5):1025–35. doi: 10.1016/j.cell.2013.04.040
35. Libreros S, Nshimiyimana R, Lee B, Serhan CN. Infectious Neutrophil deployment is regulated by Resolvin D4. Blood (2023) 142:589–606. doi: 10.1182/blood.2022019145
36. Yang W, Tao Y, Wu Y, Zhao X, Ye W, Zhao D, et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat Commun (2019) 10(1):1076. doi: 10.1038/s41467-019-09046-8
37. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science (2015) 349(6245):316–20. doi: 10.1126/science.aaa8064
38. Bouchery T, Harris N. Neutrophil–macrophage cooperation and its impact on tissue repair. Immunol Cell Biol (2019) 97(3):289–98. doi: 10.1111/imcb.12241
39. Lowe DM, Redford PS, Wilkinson RJ, O’Garra A, Martineau AR. Neutrophils in tuberculosis: friend or foe? Trends Immunol (2012) 33(1):14–25. doi: 10.1016/j.it.2011.10.003
40. Scapini P, Cassatella MA. Social networking of human neutrophils within the immune system. Blood (2014) 124(5):710–9. doi: 10.1182/blood-2014-03-453217
41. Barkaway A, Rolas L, Joulia R, Bodkin J, Lenn T, Owen-Woods C, et al. Age-related changes in the local milieu of inflamed tissues cause aberrant neutrophil trafficking and subsequent remote organ damage. Immunity (2021) 54(7):1494–510.e7. doi: 10.1016/j.immuni.2021.04.025
42. Owen-Woods C, Joulia R, Barkaway A, Rolas L, Ma B, Nottebaum AF, et al. Local microvascular leakage promotes trafficking of activated neutrophils to remote organs. J Clin Invest (2020) 130(5):2301–18. doi: 10.1172/JCI133661
43. Sahu SK, Mittal SK, Foulsham W, Li M, Sangwan VS, Chauhan SK. Mast cells initiate the recruitment of neutrophils following ocular surface injury. Invest Ophthalmol Vis Sci (2018) 59(5):1732–40. doi: 10.1167/iovs.17-23398
44. Pimentel TA, Sampaio ALF, D’Acquisto F, Perretti M, Oliani SM. An essential role for mast cells as modulators of neutrophils influx in collagen-induced arthritis in the mouse. Lab Invest (2011) 91(1):33–42. doi: 10.1038/labinvest.2010.140
45. De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood (2013) 121(24):4930–7. doi: 10.1182/blood-2013-02-486217
46. Dudeck J, Kotrba J, Immler R, Hoffmann A, Voss M, Alexaki VI, et al. Directional mast cell degranulation of tumor necrosis factor into blood vessels primes neutrophil extravasation. Immunity (2021) 54(3):468–83.e5. doi: 10.1016/j.immuni.2020.12.017
47. Rice CM, Davies LC, Subleski JJ, Maio N, Gonzalez-Cotto M, Andrews C, et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat Commun (2018) 9. doi: 10.1038/s41467-018-07505-2
48. Wang H, Zhang H, Wang Y, Brown ZJ, Xia Y, Huang Z, et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol (2021) 75(6):1271–83. doi: 10.1016/j.jhep.2021.07.032
49. Kaltenmeier C, Yazdani HO, Morder K, Geller DA, Simmons RL, Tohme S. Neutrophil extracellular traps promote T cell exhaustion in the tumor microenvironment. Front Immunol (2021) 12:785222. doi: 10.3389/fimmu.2021.785222
50. Cui C, Chakraborty K, Tang XA, Zhou G, Schoenfelt KQ, Becker KM, et al. Neutrophil elastase selectively kills cancer cells and attenuates tumorigenesis. Cell (2021) 184(12):3163–77.e21. doi: 10.1016/j.cell.2021.04.016
51. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature (2015) 522(7556):345–8. doi: 10.1038/nature14282
52. Parackova Z, Bloomfield M, Klocperk A, Sediva A. Neutrophils mediate Th17 promotion in COVID-19 patients. J Leukoc Biol (2021) 109(1):73–6. doi: 10.1002/JLB.4COVCRA0820-481RRR
53. Bert S, Nadkarni S, Perretti M. Neutrophil-T cell crosstalk and the control of the host inflammatory response. Immunol Rev (2023) 314(1):36–49. doi: 10.1111/imr.13162
54. Segal BH, Giridharan T, Suzuki S, Khan ANH, Zsiros E, Emmons TR, et al. Neutrophil interactions with T cells, platelets, endothelial cells, and of course tumor cells. Immunol Rev (2023) 314(1):13–35. doi: 10.1111/imr.13178
55. Okeke EB, Uzonna JE. The pivotal role of regulatory T cells in the regulation of innate immune cells. Front Immunol (2019) 10:680. doi: 10.3389/fimmu.2019.00680
56. Mandel J, Casari M, Stepanyan M, Martyanov A, Deppermann C. Beyond hemostasis: platelet innate immune interactions and thromboinflammation. Int J Mol Sci (2022) 23(7):3868. doi: 10.3390/ijms23073868
57. Hirsch J, Uzun G, Zlamal J, Singh A, Bakchoul T. Platelet-neutrophil interaction in COVID-19 and vaccine-induced thrombotic thrombocytopenia. Front Immunol (2023) 14:1186000. doi: 10.3389/fimmu.2023.1186000
58. Gauer JS, Ajjan RA, Ariëns RAS. Platelet–neutrophil interaction and thromboinflammation in diabetes: considerations for novel therapeutic approaches. J Am Heart Assoc (2022) 11(20):e027071. doi: 10.1161/JAHA.122.027071
59. Torres LS, Hidalgo A. Neutrophils as drivers of vascular injury in sickle cell disease. Immunol Rev (2023) 314(1):302–12. doi: 10.1111/imr.13146
60. Filippi MD. Neutrophil transendothelial migration: updates and new perspectives. Blood (2019) 133(20):2149–58. doi: 10.1182/blood-2018-12-844605
61. Wang Y, Liu Y. Neutrophil-induced liver injury and interactions between neutrophils and liver sinusoidal endothelial cells. Inflammation (2021) 44(4):1246–62. doi: 10.1007/s10753-021-01442-x
62. Martín-Fernández M, Tamayo-Velasco Á, Aller R, Gonzalo-Benito H, Martínez-Paz P, Tamayo E. Endothelial dysfunction and neutrophil degranulation as central events in sepsis physiopathology. Int J Mol Sci (2021) 22(12):6272. doi: 10.3390/ijms22126272
63. Margraf A, Lowell CA, Zarbock A. Neutrophils in acute inflammation: current concepts and translational implications. Blood (2022) 139(14):2130–44. doi: 10.1182/blood.2021012295
64. Ren X, Manzanares LD, Piccolo EB, Urbanczyk JM, Sullivan DP, Yalom LK, et al. Macrophage–endothelial cell crosstalk orchestrates neutrophil recruitment in inflamed mucosa. J Clin Invest (2023) 133(15):e170733. doi: 10.1172/JCI170733
65. Kang L, Fang X, Song YH, He ZX, Wang ZJ, Wang SL, et al. Neutrophil–epithelial crosstalk during intestinal inflammation. Cell Mol Gastroenterol Hepatol (2022) 14(6):1257–67. doi: 10.1016/j.jcmgh.2022.09.002
66. Brazil JC, Parkos CA. Pathobiology of neutrophil–epithelial interactions. Immunol Rev (2016) 273(1):94–111. doi: 10.1111/imr.12446
67. Campbell EL, Kao DJ, Colgan SP. Neutrophils and the inflammatory tissue microenvironment in the mucosa. Immunol Rev (2016) 273(1):112–20. doi: 10.1111/imr.12456
68. Colgan SP. Neutrophils and inflammatory resolution in the mucosa. Resolut Inflamm (2015) 27(3):177–83. doi: 10.1016/j.smim.2015.03.007
69. Lodge KM, Cowburn AS, Li W, Condliffe AM. The impact of hypoxia on neutrophil degranulation and consequences for the host. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21041183
70. O’Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol (2016) 16:553–65. doi: 10.1038/nri.2016.70
71. Oburoglu L, Tardito S, Fritz V, De Barros SC, Merida P, Craveiro M, et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell (2014) 15(2):169–84. doi: 10.1016/j.stem.2014.10.009
72. Voss K, Hong HS, Bader JE, Sugiura A, Lyssiotis CA, Rathmell JC. A guide to interrogating immunometabolism. Nat Rev Immunol (2021) 21(10):637–52. doi: 10.1038/s41577-021-00529-8
73. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun (2020) 11(1):102. doi: 10.1038/s41467-019-13668-3
74. Kedia-Mehta N, Finlay DK. Competition for nutrients and its role in controlling immune responses. Nat Commun (2019) 10(1):2123. doi: 10.1038/s41467-019-10015-4
75. Fossati G, Moulding DA, Spiller DG, Moots RJ, White MRH, Edwards SW. The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J Immunol (2003) 170(4):1964–72. doi: 10.4049/jimmunol.170.4.1964
76. Kim TS, Jin YB, Kim YS, Kim S, Kim JK, Lee HM, et al. SIRT3 promotes antimycobacterial defenses by coordinating mitochondrial and autophagic functions. Autophagy (2019) 15(8):1356–75. doi: 10.1080/15548627.2019.1582743
77. Song J, Herrmann JM, Becker T. Quality control of the mitochondrial proteome. Nat Rev Mol Cell Biol (2020) 22:54–70. doi: 10.1038/s41580-020-00300-2
78. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol (2020) 21:363–83. doi: 10.1038/s41580-020-0230-3
79. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res (2018) 122:877–902. doi: 10.1161/CIRCRESAHA.117.311401
80. Hsu BE, Tabariès S, Johnson RM, Andrzejewski S, Senecal J, Lehuédé C, et al. Immature low-density neutrophils exhibit metabolic flexibility that facilitates breast cancer liver metastasis. Cell Rep (2019) 27(13):3902–15. doi: 10.1016/j.celrep.2019.05.091
81. Strauss L, Guarneri V, Gennari A, Sica A. Implications of metabolism-driven myeloid dysfunctions in cancer therapy. Cell Mol Immunol (2020) 18:829–41. doi: 10.1038/s41423-020-00556-w
82. Giladi A, Paul F, Herzog Y, Lubling Y, Weiner A, Yofe I, et al. Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat Cell Biol (2018) 20(7):836–46. doi: 10.1038/s41556-018-0121-4
83. Lawrence SM, Corriden R, Nizet V. The ontogeny of a neutrophil: mechanisms of granulopoiesis and homeostasis. Microbiol Mol Biol Rev (2018) 82:e00057–17. doi: 10.1128/MMBR.00057-17
84. Tolomeo M, Grimaudo S. The “janus” role of C/EBPs family members in cancer progression. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21124308
85. Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell (2015) 163(7):1663–77. doi: 10.1016/j.cell.2015.11.013
86. Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno SI, Imanishi J, et al. C/EBPβ is required for “emergency” granulopoiesis. Nat Immunol (2006) 7:732–39. doi: 10.1038/ni1354
87. Coffelt SB, Wellenstein MD, De Visser KE. Neutrophils in cancer: Neutral no more. Nat Rev Cancer (2016) 16:431–46. doi: 10.1038/nrc.2016.52
88. Marchwicka A, Marcinkowska E. Regulation of expression of CEBP genes by variably expressed vitamin D receptor and retinoic acid receptor α in human acute myeloid leukemia cell lines. Int J Mol Sci (2018) 19(7):1918. doi: 10.3390/ijms19071918
89. Li X, Egervari G, Wang Y, Berger SL, Lu Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Biol (2018) 19(9):563–78. doi: 10.1038/s41580-018-0029-7
90. Matilainen O, Quirós PM, Auwerx J. Mitochondria and epigenetics – crosstalk in homeostasis and stress. Trends Cell Biol (2017) 27(6):453–63. doi: 10.1016/j.tcb.2017.02.004
91. Skokowa J, Lan D, Thakur BK, Wang F, Gupta K, Cario G, et al. NAMPT is essential for the G-CSF-induced myeloid differentiation via a NAD+-sirtuin-1-dependent pathway. Nat Med (2009) 15. doi: 10.1055/s-0029-1222684
92. Ryu KW, Nandu T, Kim J, Challa S, DeBerardinis RJ, Lee Kraus W. Metabolic regulation of transcription through compartmentalized NAD+ biosynthesis. Science (2018) 360. doi: 10.1126/science.aan5780
93. Zidek LM, Ackermann T, Hartleben G, Eichwald S, Kortman G, Kiehntopf M, et al. Deficiency in mTORC 1-controlled C/ EBP β - mRNA translation improves metabolic health in mice. EMBO Rep (2015) 16:1022–36. doi: 10.15252/embr.201439837
94. Zasłona Z, O’Neill LAJ. Cytokine-like roles for metabolites in immunity. Mol Cell (2020) 78(5):814–23. doi: 10.1016/j.molcel.2020.04.002
95. Nakamura-Ishizu A, Ito K, Suda T. Hematopoietic stem cell metabolism during development and aging. Dev Cell (2020) 54:239–55. doi: 10.1016/j.devcel.2020.06.029
96. Harris JM, Esain V, Frechette GM, Harris LJ, Cox AG, Cortes M, et al. Glucose metabolism impacts the spatiotemporal onset and magnitude of HSC induction in vivo. Blood (2013) 121:2483–93. doi: 10.1182/blood-2012-12-471201
97. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML-PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med (2012) 18:1350–8. doi: 10.1038/nm.2882
98. Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell (2010) 7(3):380–90. doi: 10.1016/j.stem.2010.07.011
99. Vannini N, Campos V, Girotra M, Trachsel V, Rojas-Sutterlin S, Tratwal J, et al. The NAD-booster nicotinamide riboside potently stimulates hematopoiesis through increased mitochondrial clearance. Cell Stem Cell (2019) 24(3):405–418.e7. doi: 10.1016/j.stem.2019.02.012
100. Yamashita M, Dellorusso PV, Olson OC, Passegué E. Dysregulated haematopoietic stem cell behaviour in myeloid leukaemogenesis. Nat Rev Cancer (2020) 20:365–82. doi: 10.1038/s41568-020-0260-3
101. Batsivari A, Haltalli MLR, Passaro D, Pospori C, Lo Celso C, Bonnet D. Dynamic responses of the haematopoietic stem cell niche to diverse stresses. Nat Cell Biol (2020) 22:7–17. doi: 10.1038/s41556-019-0444-9
102. Liu X, Zhang Y, Ni M, Cao H, Signer RAJ, Li D, et al. Regulation of mitochondrial biogenesis in erythropoiesis by mTORC1-mediated protein translation. Nat Cell Biol (2017) 19:626–38. doi: 10.1038/ncb3527
103. Guo B, Huang X, Lee MR, Lee SA, Broxmeyer HE. Antagonism of PPAR-γ 3 signaling expands human hematopoietic stem and progenitor cells by enhancing glycolysis. Nat Med (2018) 24:360–67. doi: 10.1038/nm.4477
104. Huang Z, Bin RH, Xian L, Chen W, Jiang S, Song A, et al. The stem cell factor/Kit signalling pathway regulates mitochondrial function and energy expenditure. Nat Commun (2014) 5. doi: 10.1038/ncomms5282
105. van Raam BJ, Sluiter W, de Wit E, Roos D, Verhoeven AJ, Kuijpers TW. Mitochondrial membrane potential in human neutrophils is maintained by complex III activity in the absence of supercomplex organisation. PloS One (2008) 3:e2013. doi: 10.1371/journal.pone.0002013
106. Tanimura A, Miyoshi K, Horiguchi T, Hagita H, Fujisawa K, Noma T. Mitochondrial activity and unfolded protein response are required for neutrophil differentiation. Cell Physiol Biochem (2018) 47:1936–50. doi: 10.1159/000491464
107. Six E, Lagresle-Peyrou C, Susini S, De Chappedelaine C, Sigrist N, Sadek H, et al. AK2 deficiency compromises the mitochondrial energy metabolism required for differentiation of human neutrophil and lymphoid lineages. Cell Death Dis (2015) 6:e1856. doi: 10.1038/cddis.2015.211
108. Hoogendijk AJ, Pourfarzad F, Aarts CEM, Tool ATJ, Hiemstra IH, Grassi L, et al. Dynamic transcriptome-proteome correlation networks reveal human myeloid differentiation and neutrophil-specific programming. Cell Rep (2019) 29(8):2505–2519.e4. doi: 10.1016/j.celrep.2019.10.082
109. Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW. Functional characterization of mitochondria in neutrophils: A role restricted to apoptosis. Cell Death Differ (2004) 11:143–53. doi: 10.1038/sj.cdd.4401320
110. Injarabian L, Devin A, Ransac S, Marteyn BS. Neutrophil metabolic shift during their lifecycle: Impact on their survival and activation. Int J Mol Sci (2020) 21:287. doi: 10.3390/ijms21010287
111. Riffelmacher T, Richter FC, Simon AK. Autophagy dictates metabolism and differentiation of inflammatory immune cells. Autophagy (2018) 14:199–206. doi: 10.1080/15548627.2017.1362525
112. Rožman S, Yousefi S, Oberson K, Kaufmann T, Benarafa C, Simon HU. The generation of neutrophils in the bone marrow is controlled by autophagy. Cell Death Differ (2015) 22:445–56. doi: 10.1038/cdd.2014.169
113. Amini P, Stojkov D, Felser A, Jackson CB, Courage C, Schaller A, et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat Commun (2018) 9. doi: 10.1038/s41467-018-05387-y
114. Wang X, Chen D. Purinergic regulation of neutrophil function. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00399
115. SenGupta S, Parent CA, Bear JE. The principles of directed cell migration. Nat Rev Mol Cell Biol (2021) 22(8):529–47. doi: 10.1038/s41580-021-00366-6
116. Pulsifer AR, Vashishta A, Reeves SA, Wolfe JK, Palace SG, Proulx MK, et al. Redundant and cooperative roles for Yersinia pestis yop effectors in the inhibition of human neutrophil exocytic responses revealed by gain-of-function approach. Infect Immun (2020) 88(3):e00909–19. doi: 10.1128/IAI.00909-19
117. Sung BH, Parent CA, Weaver AM. Extracellular vesicles: Critical players during cell migration. Dev Cell (2021) 56(13):1861–74. doi: 10.1016/j.devcel.2021.03.020
118. Hopke A, Scherer A, Kreuzburg S, Abers MS, Zerbe CS, Dinauer MC, et al. Neutrophil swarming delays the growth of clusters of pathogenic fungi. Nat Commun (2020) 11(1):2031. doi: 10.1038/s41467-020-15834-4
119. Kienle K, Glaser KM, Eickhoff S, Mihlan M, Knöpper K, Reátegui E, et al. Neutrophils self-limit swarming to contain bacterial growth in vivo. Science (2021) 372(6548):eabe7729. doi: 10.1126/science.abe7729
120. Coombs C, Georgantzoglou A, Walker HA, Patt J, Merten N, Poplimont H, et al. Chemokine receptor trafficking coordinates neutrophil clustering and dispersal at wounds in zebrafish. Nat Commun (2019) 10(1):5166. doi: 10.1038/s41467-019-13107-3
121. Hazeldine J, Dinsdale RJ, Harrison P, Lord JM. Traumatic injury and exposure to mitochondrial-derived damage associated molecular patterns suppresses neutrophil extracellular trap formation. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00685
122. Li H, Itagaki K, Sandler N, Gallo D, Galenkamp A, Kaczmarek E, et al. Mitochondrial damage-associated molecular patterns from fractures suppress pulmonary immune responses via formyl peptide receptors 1 and 2. J Trauma Acute Care Surg (2015) 78:272–81. doi: 10.1097/TA.0000000000000509
123. Bao Y, Ledderose C, Seier T, Graf AF, Brix B, Chong E, et al. Mitochondria regulate Neutrophil activation by generating ATP for Autocrine Purinergic signaling. J Biol Chem (2014) 289(39):26794–803. doi: 10.1074/jbc.M114.572495
124. Zhou W, Cao L, Jeffries J, Zhu X, Staiger CJ, Deng Q. Neutrophil-specific knockout demonstrates a role for mitochondria in regulating neutrophil motility in zebrafish. DMM Dis Models Mech (2018) 11. doi: 10.1242/dmm.033027
125. Park DW, Zmijewski JW. Mitochondrial dysfunction and immune cell metabolism in sepsis. Infection Chemother (2017) 49:10–21. doi: 10.3947/ic.2017.49.1.10
126. Fainsod-Levi T, Gershkovitz M, Völs S, Kumar S, Khawaled S, Sagiv JY, et al. Hyperglycemia impairs neutrophil mobilization leading to enhanced metastatic seeding. Cell Rep (2017) 21(9):2384–92. doi: 10.1016/j.celrep.2017.11.010
127. Xie X, Shi Q, Wu P, Zhang X, Kambara H, Su J, et al. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat Immunol (2020) 21(9):1119–33. doi: 10.1101/792200
128. Cassatella MA, Östberg NK, Tamassia N, Soehnlein O. Biological roles of neutrophil-derived granule proteins and cytokines. Trends Immunol (2019) 40:648–64. doi: 10.1016/j.it.2019.05.003
129. Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, et al. Neutrophils: New insights and open questions. Sci Immunol (2018) 3(30). doi: 10.1126/sciimmunol.aat4579
130. Amara N, Cooper MP, Voronkova MA, Webb BA, Lynch EM, Kollman JM, et al. Selective activation of PFKL suppresses the phagocytic oxidative burst. Cell (2021) 184(17):4480–4494.e15. doi: 10.1016/j.cell.2021.07.004
131. Azevedo EP, Rochael NC, Guimarães-Costa AB, De Souza-Vieira TS, Ganilho J, Saraiva EM, et al. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-Acetate-induced neutrophil extracellular trap (NET) formation. J Biol Chem (2015) 290(36):22174–83. doi: 10.1074/jbc.M115.640094
132. Britt EC, Lika J, Giese MA, Schoen TJ, Seim GL, Huang Z, et al. Switching to the cyclic pentose phosphate pathway powers the oxidative burst in activated neutrophils. Nat Metab (2022) 4(3):389–403. doi: 10.1038/s42255-022-00550-8
133. Braidy N, Berg J, Clement J, Khorshidi F, Poljak A, Jayasena T, et al. Role of nicotinamide adenine dinucleotide and related precursors as therapeutic targets for age-related degenerative diseases: rationale, biochemistry, pharmacokinetics, and outcomes. Antioxid Redox Signal (2019) 30:251–94. doi: 10.1089/ars.2017.7269
134. El-Benna J, Dang PMC, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: Role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol (2008) 30:279–89. doi: 10.1007/s00281-008-0118-3
135. Sanchez Klose FP, Dahlstrand Rudin A, Bergqvist L, Scheffler JM, Jönsson K, Islander U, et al. The Pseudomonas aeruginosa lectin LecB modulates intracellular reactive oxygen species production in human neutrophils. Eur J Immunol (2023) 2350623. doi: 10.1002/eji.202350623
136. Li DD, Jawale CV, Zhou C, Lin L, Trevejo-Nunez GJ, Rahman SA, et al. Fungal sensing enhances neutrophil metabolic fitness by regulating antifungal Glut1 activity. Cell Host Microbe (2022) 30(4):530–44.e6. doi: 10.1016/j.chom.2022.02.017
137. Bhattacharya A, Wei Q, Shin JN, Abdel Fattah E, Bonilla DL, Xiang Q, et al. Autophagy is required for neutrophil-mediated inflammation. Cell Rep (2015) 12(11):1731–39. doi: 10.1016/j.celrep.2015.08.019
138. Vorobjeva N, Prikhodko A, Galkin I, Pletjushkina O, Zinovkin R, Sud’ina G, et al. Mitochondrial reactive oxygen species are involved in chemoattractant-induced oxidative burst and degranulation of human neutrophils in vitro. Eur J Cell Biol (2017) 96(3):254–65. doi: 10.1016/j.ejcb.2017.03.003
139. Zmijewski JW, Lorne E, Zhao X, Tsuruta Y, Sha Y, Liu G, et al. Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury. Am J Respir Crit Care Med (2008) 178(2):168–79. doi: 10.1164/rccm.200710-1602OC
140. Zmijewski JW, Lorne E, Banerjee S. Abraham E. Participation of mitochondrial respiratory complex III in neutrophil activation and lung injury. Am J Physiol - Lung Cell Mol Physiol (2009) 296(4):L624–34. doi: 10.1152/ajplung.90522.2008
141. Kuwabara WMT, Curi R, Alba-Loureiro TC. Autophagy is impaired in neutrophils from streptozotocin-induced diabetic rats. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00024
142. Miriyala S, Chandra M, Maxey B, Day A, St. Clair DK, Panchatcharam M. Arjunolic acid ameliorates reactive oxygen species via inhibition of p47phox-serine phosphorylation and mitochondrial dysfunction. Int J Biochem Cell Biol (2015) 68:70–7. doi: 10.1016/j.biocel.2015.08.015
143. Boeltz S, Amini P, Anders HJ, Andrade F, Bilyy R, Chatfield S, et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differentiation (2019) 26:395–408. doi: 10.1038/s41418-018-0261-x
144. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol (2014) 15(11):1017–25. doi: 10.1038/ni.2987
145. Balamayooran G, Batra S, Fessler MB, Happel KI, Jeyaseelan S. Mechanisms of neutrophil accumulation in the lungs against bacteria. Am J Respir Cell Mol Biol (2010) 43(1):5–16. doi: 10.1165/rcmb.2009-0047TR
146. Azzouz D, Khan MA, Sweezey N, Palaniyar N. Two-in-one: UV radiation simultaneously induces apoptosis and NETosis. Cell Death Discovery (2018) 4(1):51. doi: 10.1038/s41420-018-0048-3
147. Azzouz D, Palaniyar N. Mitochondrial ROS and base excision repair steps leading to DNA nick formation drive ultraviolet induced-NETosis. Front Immunol (2023) 14:1198716. doi: 10.3389/fimmu.2023.1198716
148. Azzouz D, Khan MA, Palaniyar N. ROS induces NETosis by oxidizing DNA and initiating DNA repair. Cell Death Discovery (2021) 7(1):113. doi: 10.1038/s41420-021-00491-3
149. Azzouz D, Palaniyar N. ROS and DNA repair in spontaneous versus agonist-induced NETosis: Context matters. Front Immunol (2022) 13:1033815. doi: 10.3389/fimmu.2022.1033815
150. Carmona-Rivera C, Carlucci PM, Goel RR, James E, Brooks SR, Rims C, et al. Neutrophil extracellular traps mediate articular cartilage damage and enhance cartilage component immunogenicity in rheumatoid arthritis. JCI Insight (2020) 5(13):e139388. doi: 10.1172/jci.insight.139388
151. Rodríguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MMB, López-Villegas EO, Sánchez-García FJ. Metabolic requirements for neutrophil extracellular traps formation. Immunology (2015) 145(2):213–24. doi: 10.1111/imm.12437
152. Remijsen Q, Vanden BT, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res (2011) 21:290–304. doi: 10.1038/cr.2010.150
153. McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, et al. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood (2012) 120:3118–25. doi: 10.1182/blood-2012-01-405993
154. Awasthi D, Nagarkoti S, Sadaf S, Chandra T, Kumar S, Dikshit M. Glycolysis dependent lactate formation in neutrophils: A metabolic link between NOX-dependent and independent NETosis. Biochim Biophys Acta - Mol Basis Di (2019) 1865(12). doi: 10.1016/j.bbadis.2019.165542
155. Bornhöfft KF, Galuska SP. Glycans as modulators for the formation and functional properties of neutrophil extracellular traps: used by the forces of good and evil. Front Immunol (2019) 10:959. doi: 10.3389/fimmu.2019.00959
156. Krysa SJ, Allen LAH. Metabolic reprogramming mediates delayed apoptosis of human neutrophils infected with Francisella tularensis. Front Immunol (2022) 13:836754. doi: 10.3389/fimmu.2022.836754
157. Liu TF, Vachharajani VT, Yoza BK, McCall CE. NAD+ -dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem (2012) 287(31):25758–69. doi: 10.1074/jbc.M112.362343
158. An R, Zhao L, Xu J, Xi C, Li H, Shen G, et al. Resveratrol alleviates sepsis−induced myocardial injury in rats by suppressing neutrophil accumulation, the induction of TNF−α and myocardial apoptosis via activation of Sirt1. Mol Med Rep (2016) 14(6):5297–303. doi: 10.3892/mmr.2016.5861
159. Cheng SC, Scicluna BP, Arts RJW, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol (2016) 17(4):11. doi: 10.1038/ni.3398
160. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol (2022) 42(7):1473–507. doi: 10.1007/s10875-022-01289-3
161. Sadiku P, Willson JA, Ryan EM, Sammut D, Coelho P, Watts ER, et al. Neutrophils fuel effective immune responses through gluconeogenesis and glycogenesis. Cell Metab (2021) 33(2):411–23.e4. doi: 10.1016/j.cmet.2020.11.016
162. McKinney C, Ellison M, Briones NJ, Baroffio A, Murphy J, Tran AD, et al. Metabolic abnormalities in G6PC3-deficient human neutrophils result in severe functional defects. Blood Adv (2020) 4(23):5888–901. doi: 10.1182/bloodadvances.2020002225
163. Dai R, Lv G, Li W, Tang W, Chen J, Liu Q, et al. Altered functions of neutrophils in two Chinese patients with severe congenital neutropenia type 4 caused by G6PC3 mutations. Front Immunol (2021) 12:699743. doi: 10.3389/fimmu.2021.699743
164. Jun HS, Weinstein DA, Lee YM, Mansfield BC, Chou JY. Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib. Blood (2014) 123(18):2843–53. doi: 10.1182/blood-2013-05-502435
165. Sim SW, Weinstein DA, Lee YM, Jun HS. Glycogen storage disease type Ib: role of glucose-6-phosphate transporter in cell metabolism and function. FEBS Lett (2020) 594(1):3–18. doi: 10.1002/1873-3468.13666
166. Kumar S, Dikshit M. Metabolic insight of neutrophils in health and disease. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.02099
167. Yoshimi A, Ishikawa K, Niemeyer C, Grünert SC. Pearson syndrome: a multisystem mitochondrial disease with bone marrow failure. Orphanet J Rare Dis (2022) 17(1):379. doi: 10.1186/s13023-022-02538-9
168. Tadiotto E, Maines E, Degani D, Balter R, Bordugo A, Cesaro S. Bone marrow features in Pearson syndrome with neonatal onset: A case report and review of the literature. Pediatr Blood Cancer (2018) 65(4):e26939. doi: 10.1002/pbc.26939
169. Pliyev BK, Ivanova AV, Savchenko VG. Extracellular NAD+ inhibits human neutrophil apoptosis. Apoptosis (2014) 19:5810593. doi: 10.1007/s10495-013-0948-x
170. Bhutta ZA, Berkley JA, Bandsma RHJ, Kerac M, Trehan I, Briend A. Severe childhood malnutrition. Nature reviews. Dis Primers (2017) 3. doi: 10.1038/nrdp.2017.67
171. Collins N, Belkaid Y. Control of immunity via nutritional interventions. Immunity (2022) 55(2):210–23. doi: 10.1016/j.immuni.2022.01.004
172. Palma C, Rocca CL, Gigantino V, Aquino G, Piccaro G, Silvestre DD, et al. Caloric restriction promotes immunometabolic reprogramming leading to protection from tuberculosis. Cell Metab (2021) 33(2):300–18.e12. doi: 10.1016/j.cmet.2020.12.016
173. Ibrahim MK, Zambruni M, Melby CL, Melby PC. Impact of childhood malnutrition on host defense and infection. Clin Microbiol Rev (2017) 30. doi: 10.1128/CMR.00119-16
174. Shao YR, Xu DY, Lin J. Nutrients and rheumatoid arthritis: From the perspective of neutrophils. Front Immunol (2023) 14:1113607. doi: 10.3389/fimmu.2023.1113607
175. Bozonet SM, Carr AC. The role of physiological vitamin c concentrations on key functions of neutrophils isolated from healthy individuals. Nutrients (2019) 11. doi: 10.3390/nu11061363
176. Wolvers DAW, Van Herpen-Broekmans WMR, Logman MHGM, van der Wielen RPJ, Albers R. Effect of a mixture of micronutrients, but not of bovine colostrum concentrate, on immune function parameters in healthy volunteers: A randomized placebo-controlled study. Nutr J (2006) 5. doi: 10.1186/1475-2891-5-28
178. Liugan M, Carr AC. Vitamin c and neutrophil function: Findings from randomized controlled trials. Nutrients (2019) 11. doi: 10.3390/nu11092102
179. Ni S, Yuan Y, Kuang Y, Li X. Iron metabolism and immune regulation. Front Immunol (2022) 13:816282. doi: 10.3389/fimmu.2022.816282
180. Cassat JE, Skaar EP. Iron in infection and immunity. Cell Host Microbe (2013) 13(5):509–19. doi: 10.1016/j.chom.2013.04.010
181. Ganz T, Nemeth E. Iron homeostasis in host defence and inflammation. Nat Rev Immunol (2015) 15(8):500–10. doi: 10.1038/nri3863
182. Maares M, Haase H. Zinc and immunity: An essential interrelation. Arch Biochem Biophys (2016) 611:58–65. doi: 10.1016/j.abb.2016.03.022
183. Bonaventura P, Benedetti G, Albarède F, Miossec P. Zinc and its role in immunity and inflammation. Autoimmun Rev (2015) 14(4):277–85. doi: 10.1016/j.autrev.2014.11.008
184. Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients (2017) 9(6):624. doi: 10.3390/nu9060624
185. Vrieling F, Stienstra R. Obesity and dysregulated innate immune responses: impact of micronutrient deficiencies. Trends Immunol (2023) 44(3):217–30. doi: 10.1016/j.it.2023.01.003
186. Mailhot G, White JH. Vitamin D and immunity in infants and children. Nutrients (2020) 12(5):1233. doi: 10.3390/nu12051233
187. Carlberg C. Vitamin D and its target genes. Nutrients (2022) 14(7):1354. doi: 10.3390/nu14071354
188. Stephensen CB. VITAMIN A, INFECTION, AND IMMUNE FUNCTION*. Annu Rev Nutr (2001) 21(1):167–92. doi: 10.1146/annurev.nutr.21.1.167
189. Stephensen CB, Lietz G. Vitamin A in resistance to and recovery from infection: relevance to SARS-CoV2. Br J Nutr (2021) 126(11):1663–72. doi: 10.1017/S0007114521000246
190. Xie N, Zhang L, Gao W, Huang C, Huber PE, Zhou X, et al. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther (2020) 5(1):227. doi: 10.1038/s41392-020-0116-z
191. Chini CCS, Zeidler JD, Kashyap S, Warner G, Chini EN. Evolving concepts in NAD+ metabolism. Cell Metab (2021) 33(6):1076–87. doi: 10.1016/j.cmet.2021.04.003
192. Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, et al. Nutrient-sensitive mitochondrial NAD+ Levels dictate cell survival. Cell (2007) 130(6):1095–107. doi: 10.1016/j.cell.2007.07.035
193. Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab (2012) 15(6):838–47. doi: 10.1016/j.cmet.2012.04.022
194. Cantó C, Menzies KJ, Auwerx J. NAD+ Metabolism and the control of energy homeostasis: A balancing act between mitochondria and the nucleus. Cell Metab (2015) 22(1):31–53. doi: 10.1016/j.cmet.2015.05.023
195. Ferreira RG, Matsui TC, Godin AM, Gomides LF, Pereira-Silva PEM, Duarte IDG, et al. Neutrophil recruitment is inhibited by nicotinamide in experimental pleurisy in mice. Eur J Pharmacol (2012) 685:198–204. doi: 10.1016/j.ejphar.2012.04.014
196. Ferreira RG, Matsui TC, Gomides LF, Godin AM, Menezes GB, De Matos Coelho M, et al. Niacin inhibits carrageenan-induced neutrophil migration in mice. Naunyn Schmiedebergs Arch Pharmacol (2013) 386:533–40. doi: 10.1007/s00210-013-0854-3
197. Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab (2013) 17(5):695–708. doi: 10.1016/j.cmet.2013.04.001
198. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med (2015) 21(7):815–9. doi: 10.1038/nm.3887
199. Dowey R, Iqbal A, Heller SR, Sabroe I. Prince LR. A bittersweet response to infection in diabetes; targeting neutrophils to modify inflammation and improve host immunity. Front Immunol (2021) 12:678771. doi: 10.3389/fimmu.2021.678771
200. Lodhi IJ, Wei X, Yin L, Feng C, Adak S, Abou-Ezzi G, et al. Peroxisomal lipid synthesis regulates inflammation by sustaining neutrophil membrane phospholipid composition and viability. Cell Metab (2015) 21(1):51–64. doi: 10.1016/j.cmet.2014.12.002
201. Moorthy AN, Tan KB, Wang S, Narasaraju T, Chow VT. Effect of high-fat diet on the formation of pulmonary neutrophil extracellular traps during influenza pneumonia in BALB/c mice. Front Immunol (2016) 7:289–9. doi: 10.3389/fimmu.2016.00289
202. Quail DF, Olson OC, Bhardwaj P, Walsh LA, Akkari L, Quick ML, et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat Cell Biol (2017) 19(8):974–87. doi: 10.1038/ncb3578
203. Brotfain E, Hadad N, Shapira Y, Avinoah E, Zlotnik A, Raichel L, et al. Neutrophil functions in morbidly obese subjects. Clin Exp Immunol (2015) 181(1):156–63. doi: 10.1111/cei.12631
204. Baum JI, Kim IY, Wolfe RR. Protein consumption and the elderly: what is the optimal level of intake? Nutrients (2016) 8(6):359. doi: 10.3390/nu8060359
205. Paddon-Jones D, Rasmussen BB. Dietary protein recommendations and the prevention of sarcopenia. Curr Opin Clin Nutr Metab Care (2009) 12(1):86–90. doi: 10.1097/MCO.0b013e32831cef8b
206. Han Q, Lin S, He F, Zhang R, Xie X, Qing F, et al. A high neutrophil to lymphocyte ratio is associated with poor nutritional status in chronic kidney disease patients. Br J Nutr (2022) 128(10):1990–6. doi: 10.1017/S000711452100516X
207. Hazeldine J, Lord JM. Innate immunesenescence: underlying mechanisms and clinical relevance. Biogerontology (2015) 16(2):187–201. doi: 10.1007/s10522-014-9514-3
208. Bartleson JM, Radenkovic D, Covarrubias AJ, Furman D, Winer DA, Verdin E. SARS-CoV-2, COVID-19 and the aging immune system. Nat Aging (2021) 1(9):769–82. doi: 10.1038/s43587-021-00114-7
209. Zhang X, Li H, He M, Wang J, Wu Y, Li Y. Immune system and sarcopenia: Presented relationship and future perspective. Exp Gerontol (2022) 164:111823. doi: 10.1016/j.exger.2022.111823
210. Nelson AR, Jackson L, Clarke J, Stellingwerff T, Broadbent S, Rowlands DS. Effect of post-exercise protein–leucine feeding on neutrophil function, immunomodulatory plasma metabolites and cortisol during a 6-day block of intense cycling. Eur J Appl Physiol (2013) 113(9):2211–22. doi: 10.1007/s00421-013-2650-7
211. Feidantsis K, Methenitis S, Ketselidi K, Vagianou K, Skepastianos P, Hatzitolios A, et al. Comparison of short-term hypocaloric high-protein diets with a hypocaloric Mediterranean diet: Effect on body composition and health-related blood markers in overweight and sedentary young participants. Nutrition (2021) 91–92:111365. doi: 10.1016/j.nut.2021.111365
212. Obesity and Lipotoxicity (2023). Available at: https://books-scholarsportal-info.myaccess.library.utoronto.ca/en/read?id=/ebooks/ebooks3/springer/2017-08-17/5/9783319483825#page=509.
213. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol (2020) 17(6):327–40. doi: 10.1038/s41569-019-0326-7
214. Soehnlein O, Steffens S, Hidalgo A, Weber C. Neutrophils as protagonists and targets in chronic inflammation. Nat Rev Immunol (2017) 17(4):248–61. doi: 10.1038/nri.2017.10
215. Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol (2015) 15(2):104–16. doi: 10.1038/nri3793
216. Herrero-Cervera A, Soehnlein O, Kenne E. Neutrophils in chronic inflammatory diseases. Cell Mol Immunol (2022) 19(2):177–91. doi: 10.1038/s41423-021-00832-3
217. Bourke CD, Jones KDJ, Prendergast AJ. Current understanding of innate immune cell dysfunction in childhood undernutrition. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.01728
218. Bourke CD, Berkley JA, Prendergast AJ. Immune dysfunction as a cause and consequence of malnutrition. Trends Immunol (2016) 37(6):386–98. doi: 10.1016/j.it.2016.04.003
219. Rytter MJH, Kolte L, Briend A, Friis H, Christensen VB. The immune system in children with malnutrition - A systematic review. PLoS One (2014) 9:e105017. doi: 10.1371/journal.pone.0105017
220. Schopfer K, Douglas SD. Fine structural studies of peripheral blood leucocytes from children with kwashiorkor: morphological and functional properties. Br J Haematol (1976) 32(4):573–8. doi: 10.1111/j.1365-2141.1976.tb00961.x
221. Schopfer K, Douglas SD. Neutrophil function in children with kwashiorkor. J Lab Clin Med (1976) 88:450–61.
222. Kumar V, Bimal S, Singh SK, Chaudhary R, Das S, Lal C, et al. Leishmania donovani: Dynamics of L. donovani evasion of innate immune cell attack due to malnutrition in visceral leishmaniasis. Nutrition (2014) 30(4):449–58. doi: 10.1016/j.nut.2013.10.003
223. Harris MC, Douglas SD, Lee JC, Ziegler MM, Gerdes JS, Polin RA. Diminished polymorphonuclear leukocyte adherence and chemotaxis following protein-calorie malnutrition in newborn rats. Pediatr Res (1987) 21(6):542–6. doi: 10.1203/00006450-198706000-00007
224. Herrera M, Salva S, Villena J, Barbieri N, Marranzino G, Alvarez S. Dietary supplementation with lactobacilli improves emergency granulopoiesis in protein-malnourished mice and enhances respiratory innate immune response. PLoS One (2014) 9(4):1–13. doi: 10.1371/journal.pone.0090227
225. Nayak KC, Sethi AS, Aggarwal TD, Chadda VS, Kumar KK. Bactericidal power of neutrophils in protein calorie malnutrition. Indian J Pediatr (1989) 56:371–7. doi: 10.1007/BF02722303
226. Takele Y, Adem E, Getahun M, Tajebe F, Kiflie A, Hailu A, et al. Malnutrition in healthy individuals results in increased mixed cytokine profiles, altered neutrophil subsets and function. PLoS One (2016) 11:e0157919. doi: 10.1371/journal.pone.0157919
227. Shousha S. Nitro blue tetrazolium test in children with kwashiorkor with a comment on the use of latex particles in the test. J Clin Pathol (1972) 25(6):494–7. doi: 10.1136/jcp.25.6.494
228. Salva S, Alvarez S. The role of microbiota and immunobiotics in granulopoiesis of immunocompromised hosts. Front Immunol (2017) 8:507. doi: 10.3389/fimmu.2017.00507
229. Jose DG, Shelton M, Tauro GP, Belbin R, Hosking CS. Deficiency of immunological and phagocytic function in aboriginal children with protein calorie malnutrition. Med J Aust (1975) 2(18):699–705. doi: 10.5694/j.1326-5377.1975.tb106221.x
230. Słotwińska SM, Słotwiński R. Immune disorders in anorexia. Cent Eur J OfnbspImmunol (2017) 42(3):294–300. doi: 10.5114/ceji.2017.70973
231. Doig GS, Simpson F, Heighes PT, Bellomo R, Chesher D, Caterson ID, et al. Restricted versus continued standard caloric intake during the management of refeeding syndrome in critically ill adults: a randomised, parallel-group, multicentre, single-blind controlled trial. Lancet Respir Med (2015) 3(12):943–52. doi: 10.1016/S2213-2600(15)00418-X
232. Vaisman N, Tabachnik E, Hahn T, Voet H, Guy N. Superoxide production during refeeding in patients with anorexia nervosa. Metabolism (1992) 41(10):1097–9. doi: 10.1016/0026-0495(92)90292-I
233. Siracusa F, Schaltenberg N, Kumar Y, Lesker TR, Steglich B, Liwinski T, et al. Short-term dietary changes can result in mucosal and systemic immune depression. Nat Immunol (2023) 24:1473–86. doi: 10.1038/s41590-023-01587-x
234. Chedid P, Hurtado-Nedelec M, Marion-Gaber B, Bournier O, Hayem G, Gougerot-Pocidalo MA, et al. Adiponectin and its globular fragment differentially modulate the oxidative burst of primary human phagocytes. Am J Pathol (2012) 180(2):682–92. doi: 10.1016/j.ajpath.2011.10.013
235. Miyazaki Y, Mahankali A, Wajcberg E, Bajaj M, Mandarino LJ, DeFronzo RA. Effect of pioglitazone on circulating adipocytokine levels and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab (2004) 89(9):4312–9. doi: 10.1210/jc.2004-0190
236. Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, et al. Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca2+ and AMPK/SIRT1. Nature (2010) 464(7293):1313–9. doi: 10.1038/nature08991
237. Goldberg EL, Asher JL, Molony RD, Shaw AC, Zeiss CJ, Wang C, et al. β-hydroxybutyrate deactivates neutrophil NLRP3 inflammasome to relieve gout flares. Cell Rep (2017) 18(9):2077–87. doi: 10.1016/j.celrep.2017.02.004
238. Chen Y, Ouyang X, Hoque R, Garcia-Martinez I, Yousaf MN, Tonack S, et al. β-Hydroxybutyrate protects from alcohol-induced liver injury via a Hcar2-cAMP dependent pathway. J Hepatol (2018) 69(3):687–96. doi: 10.1016/j.jhep.2018.04.004
239. Luda KM, Longo J, Kitchen-Goosen SM, Duimstra LR, Ma EH, Watson MJ, et al. Ketolysis drives CD8+ T cell effector function through effects on histone acetylation. Immunity (2023) 56(9):2021–2035.e8. doi: 10.1016/j.immuni.2023.07.002
240. Hu G, Ling C, Chi L, Thind MK, Furse S, Koulman A, et al. The role of the tryptophan-NAD + pathway in a mouse model of severe malnutrition induced liver dysfunction. Nat Commun (2022) 13(1):7576. doi: 10.1038/s41467-022-35317-y
241. Maghraby MK, Li B, Chi L, Ling C, Benmoussa A, Provost P, et al. Extracellular vesicles isolated from milk can improve gut barrier dysfunction induced by malnutrition. Sci Rep (2021) 11(1):7635. doi: 10.1038/s41598-021-86920-w
242. van Zutphen T, Ciapaite J, Bloks VW, Ackereley C, Gerding A, Jurdzinski A, et al. Malnutrition-associated liver steatosis and ATP depletion is caused by peroxisomal and mitochondrial dysfunction. J Hepatol (2016) 65(6):1198–208. doi: 10.1016/j.jhep.2016.05.046
243. Wen B, Njunge JM, Bourdon C, Gonzales GB, Gichuki BM, Lee D, et al. Systemic inflammation and metabolic disturbances underlie inpatient mortality among ill children with severe malnutrition. Sci Adv (2022) 8(7):eabj6779. doi: 10.1126/sciadv.abj6779
Keywords: metabolic reprogramming, neutrophil differentiation, glycolysis, immune mediated diseases, autophagy, mitochondrial respiration
Citation: Thind MK, Uhlig HH, Glogauer M, Palaniyar N, Bourdon C, Gwela A, Lancioni CL, Berkley JA, Bandsma RHJ and Farooqui A (2024) A metabolic perspective of the neutrophil life cycle: new avenues in immunometabolism. Front. Immunol. 14:1334205. doi: 10.3389/fimmu.2023.1334205
Received: 06 November 2023; Accepted: 15 December 2023;
Published: 08 January 2024.
Edited by:
Sue Tsai, University of Alberta, CanadaReviewed by:
Maria Pina Mollica, University of Naples Federico II, ItalyNathan K. Archer, Johns Hopkins University, United States
Copyright © 2024 Thind, Uhlig, Glogauer, Palaniyar, Bourdon, Gwela, Lancioni, Berkley, Bandsma and Farooqui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert H. J. Bandsma, cm9iZXJ0LmJhbmRzbWFAc2lja2tpZHMuY2E=; Amber Farooqui, YWZhcm9vcXVpQG9tZWdhbGFicy5uZXQ=