 Rana A. Estaleen
Rana A. Estaleen Christopher M. Reilly
Christopher M. Reilly Xin M. Luo
Xin M. Luo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 January 2024
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1330500
Systemic lupus erythematosus (SLE) is a systemic chronic disease initiated by an abnormal immune response to self and can affect multiple organs. SLE is characterized by the production of autoantibodies and the deposition of immune complexes. In regard to the clinical observations assessed by rheumatologists, several chemokines and cytokines also contribute to disease progression. One such chemokine and adhesion molecule is CX3CL1 (otherwise known as fractalkine). CX3CL1 is involved in cell trafficking and inflammation through recognition by its receptor, CX3CR1. The CX3CL1 protein consists of a chemokine domain and a mucin-like stalk that allows it to function both as a chemoattractant and as an adhesion molecule. In inflammation and specifically lupus, the literature displays contradictory evidence for the functions of CX3CL1/CX3CR1 interactions. In addition, the gut microbiota has been shown to play an important role in the pathogenesis of SLE. This review highlights current studies that illustrate the interactions of the gut microbiota and CX3CR1 in SLE.
Systemic lupus erythematosus (SLE), also known as lupus, is a chronic autoimmune disease that can affect multiple organs in the body (1, 2). The disease is caused by an abnormal autoimmune response (1, 3). Normally, the body’s immune system works to protect against foreign invaders. In SLE, the immune system becomes hyperactive, producing antibodies that attack normal tissues and organs, some of which include the skin, blood, heart, lungs, joints, kidneys, and brain (2, 3). SLE is diagnosed when a patient meets 4 out of 11 diagnostic criteria, established by the American College of Rheumatology and the European League Against Rheumatism. It is one of the most heterogeneous diseases treated by physicians, presenting challenges to the diagnosis as well as establishment of proper treatments (4). Lupus is characterized by “on” and “off” periods known as flares and remission, respectively. These are when the patient endures periods of illness and periods of wellness, respectively (3, 5). Phenotypic expression of lupus varies between individuals from different ethnicities (6), with incidence rates ranging between 20 and 200 cases per 100,000 persons. There is a higher prevalence in individuals of African, Asian, and Hispanic backgrounds or ancestries (2, 6). SLE affects both men and women; however, the disease is much more frequent among women than men (6), particularly in women of childbearing age who are diagnosed nine times more frequently than men (2). In more economically developed countries, the 5-year survival rate is over 95% in both adults and children; however, in less economically developed countries, the survival is significantly lower in both populations (7). Although the cause of SLE is unknown (3), there are some factors that influence the development and/or severity of SLE, such as lifestyle, environmental, genetic, epigenetic, hormonal, and immunoregulatory factors (Figure 1).
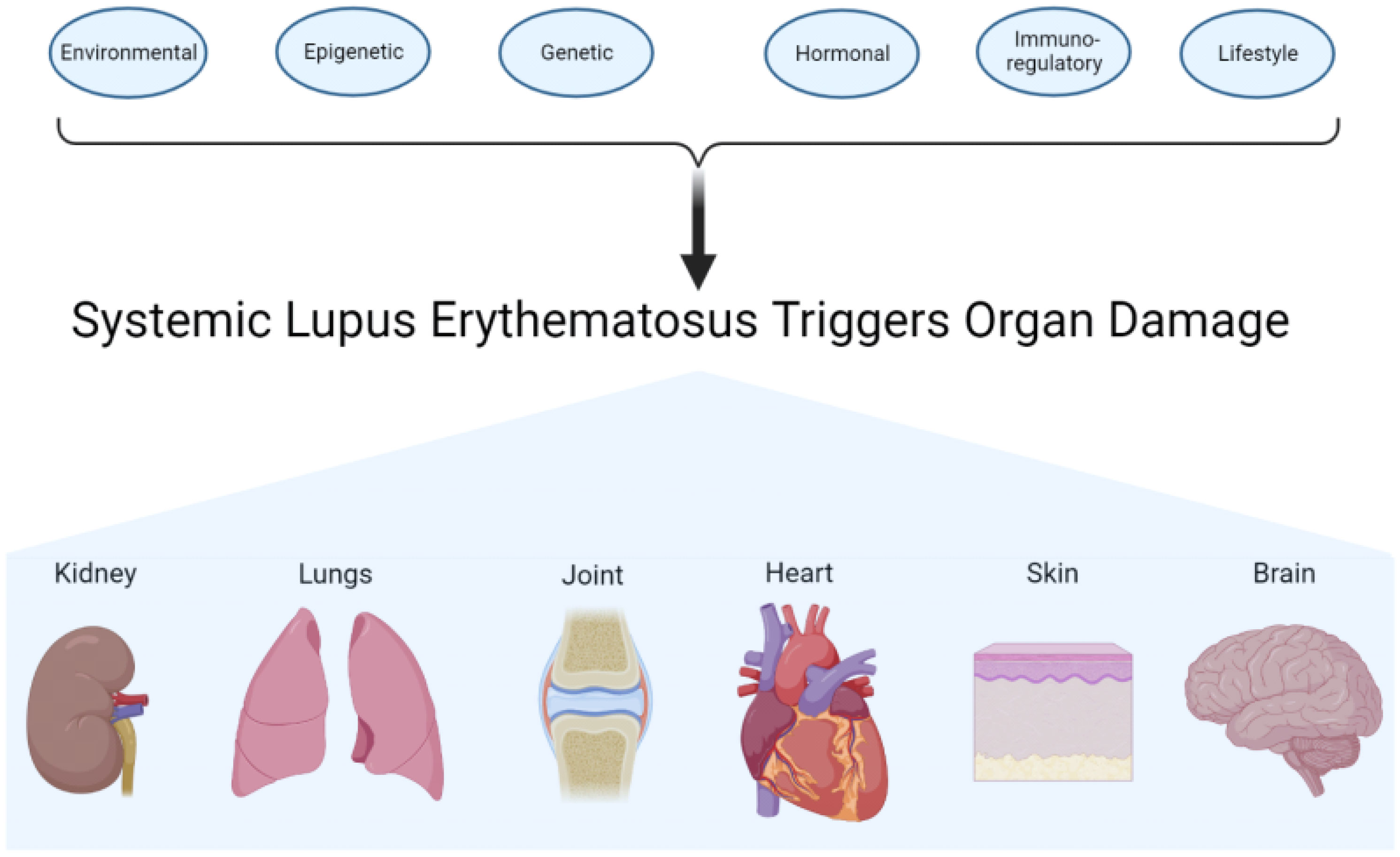
Figure 1 Contributing factors and target organs of systemic lupus erythematosus. Environmental, epigenetic, genetic, hormonal, immunoregulatory, and lifestyle factors work on the immune system. The actions of the different factors affect and may also damage different organs or tissues.
Chemokines are a group of molecules that recruit leukocyte subsets under homeostatic and pathological conditions (8). The chemokine ligand superfamily is divided into two subgroups: the CC chemokine family, which includes 28 members, and the CXC chemokine family, which includes 16 members. They interact with chemokine receptors expressed on the cell surface. Chemokine receptors are G-protein-coupled receptors and can promote target cells to adhere to the endothelium or direct their movement to their destination based on the concentration gradient of a given chemokine (9). Chemokine C-X3-C motif ligand 1, CX3CL1, also known as fractalkine, binds to its seven transmembrane G-protein-coupled receptor CX3CR1 (10). CX3CR1 was discovered, using fractalkine–alkaline phosphatase fusion protein in 1997, as a receptor with a high affinity for CX3CL1 that is expressed by and lymphocytes and monocytes (10).
CX3CL1 is produced by the renal tubular epithelium in humans (11), and it can be found on leukocytes, blood monocytes, phagocytes, and T cells in both humans and mice (12). CX3CL1 has a soluble form and a transmembrane form, which function to induce chemotaxis and adhesion of CX3CR1+ leukocytes, respectively (8). CX3CL1/CX3CR1 interaction has an antiapoptotic effect that sustains the survival of CX3CR1+ leukocytes (8, 13). CX3CR1 is found on several types of leukocytes. It has high expression on CD16+ natural killer cells, and its expression is upregulated by IL-2 in human CD4+ and CD8+ T cells (10).
In healthy individuals, CX3CR1 is required for atherogenesis and homeostasis of monocytes by promoting cell survival (13). It was reported that in the absence of the chemokine receptor or its ligand, fractalkine, there was a significant reduction in Gr1low blood monocytes levels under both steady-state and inflammatory conditions. This suggests that the interaction between CX3CL1 and CX3CR1 is an essential survival signal given that their absence would lead to monocyte death (13). Of note, Ly6G and Ly6C, previously referred to as Gr1, are markers of myeloid differentiation. Neutrophils express Ly6G and Ly6C; in addition, dendritic cells and subpopulations of lymphocytes and monocytes express Ly6C (14, 15). Gr1+ cells, under steady-state conditions, can be found in the bloodstream, contributing to immune surveillance (16). In inflammatory conditions, Gr1 cells have increased mobilization of Ly6C+ monocytes from bone marrow to bloodstream (16). This contributes to monocyte recruitment and migration to the kidney, which leads to kidney injury (17). In this aspect, the reduction of Gr1low blood monocytes in the absence of CX3CL1 and CX3CR1 interaction may be beneficial, although it is unclear if the decrease of monocytes in blood is partly due to increased recruitment to tissues. The latter, obviously, suggests a protective role of CX3CL1 and CX3CR1 interaction in blocking the mobilization of Gr1low monocytes from the blood to the kidney. Therefore, in the context of lupus, the absence of CX3CL1/CX3CR1 interaction may facilitate migration of Gr1+ inflammatory monocytes to the kidney, causing injury.
However, studies reported that an antagonist of CX3CL1 delayed the onset and slowed the progression of lupus nephritis in MRL/lpr mice (18), suggesting a detrimental role for CX3CL1/CX3CR1 interaction in lupus. On the other hand, we demonstrated that the treatment of Lactobacillus spp. attenuated splenomegaly and renal lymphadenopathy via a CX3CR1-dependent mechanism (19), suggesting that CX3CR1 may be used as a target for therapeutics. Moreover, CX3CR1 may locally prevent profibrotic macrophage retention, thus reducing kidney fibrosis (20). Additionally, CX3CL1 acts as a chemoattractant and adhesion molecule in glomerulonephritis (21). Table 1 illustrates a dichotomy in the literature concerning CX3CL1/CX3CR1 activation. These studies taken together show that the CX3CL1/CX3CR1 interaction can be considered to be a “double-edged sword” due to its involvement of both the pathogenesis and protection of renal diseases. Notably, some studies were performed in lupus-like mouse models and further research is needed for human lupus nephritis.
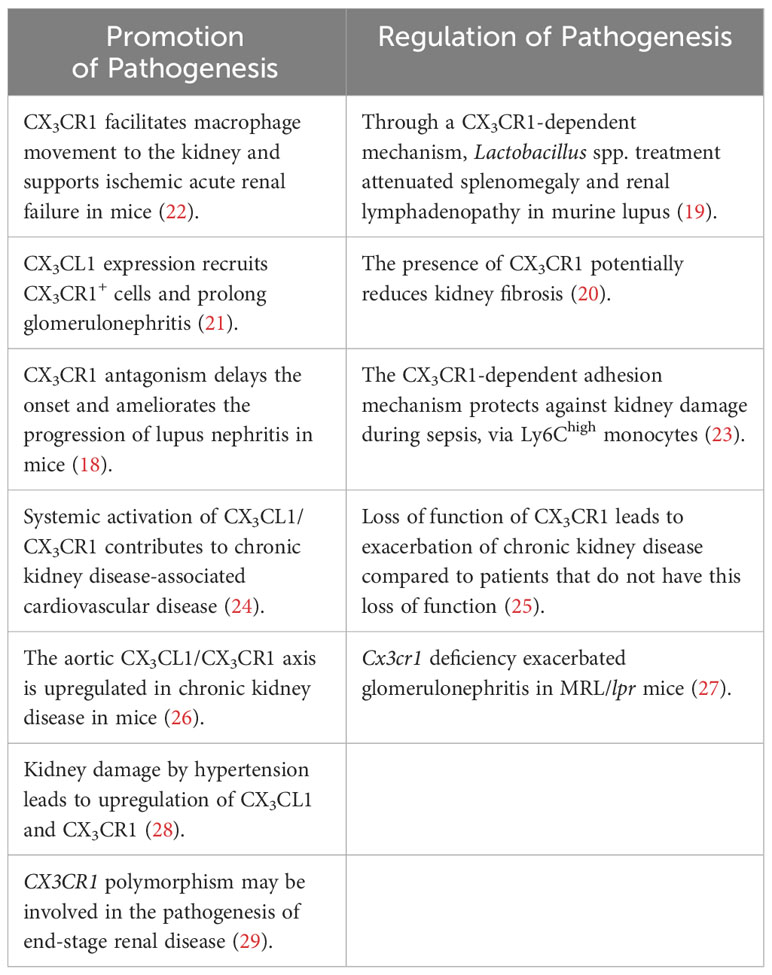
Table 1 Roles of CX3CL1/CX3CR1 in the kidney as pathogenesis promoters or regulators.
It has been recognized that a healthy gut microbiome contributes to the health of the host (30). Microbiota is the microorganisms’ entire population that colonizes a specific space, which includes archaea, bacteria, fungi, protozoans, and viruses (31). These microbes occupy different organs or systems such as the gut, mouth, skin, and vagina (32). The microbiota engages in many features of normal host physiology, from nutritional status to stress and behavioral responses. Moreover, they can contribute to many diseases and affect different organs (31). The immune system and bacteria have a meticulous relationship that maintains a balance to control inflammation (19). Gut microbiota dysbiosis has been reported in numerous autoimmune diseases (33), including antiphospholipid syndrome (APS) (34), inflammatory bowel disease (IBD) (35), rheumatoid arthritis (RA) (36), SLE (33), Sjogren’s syndrome (SS) (37), and systemic sclerosis (SSC) (38). Figure 2 illustrates different bacteria altered in autoimmune diseases.
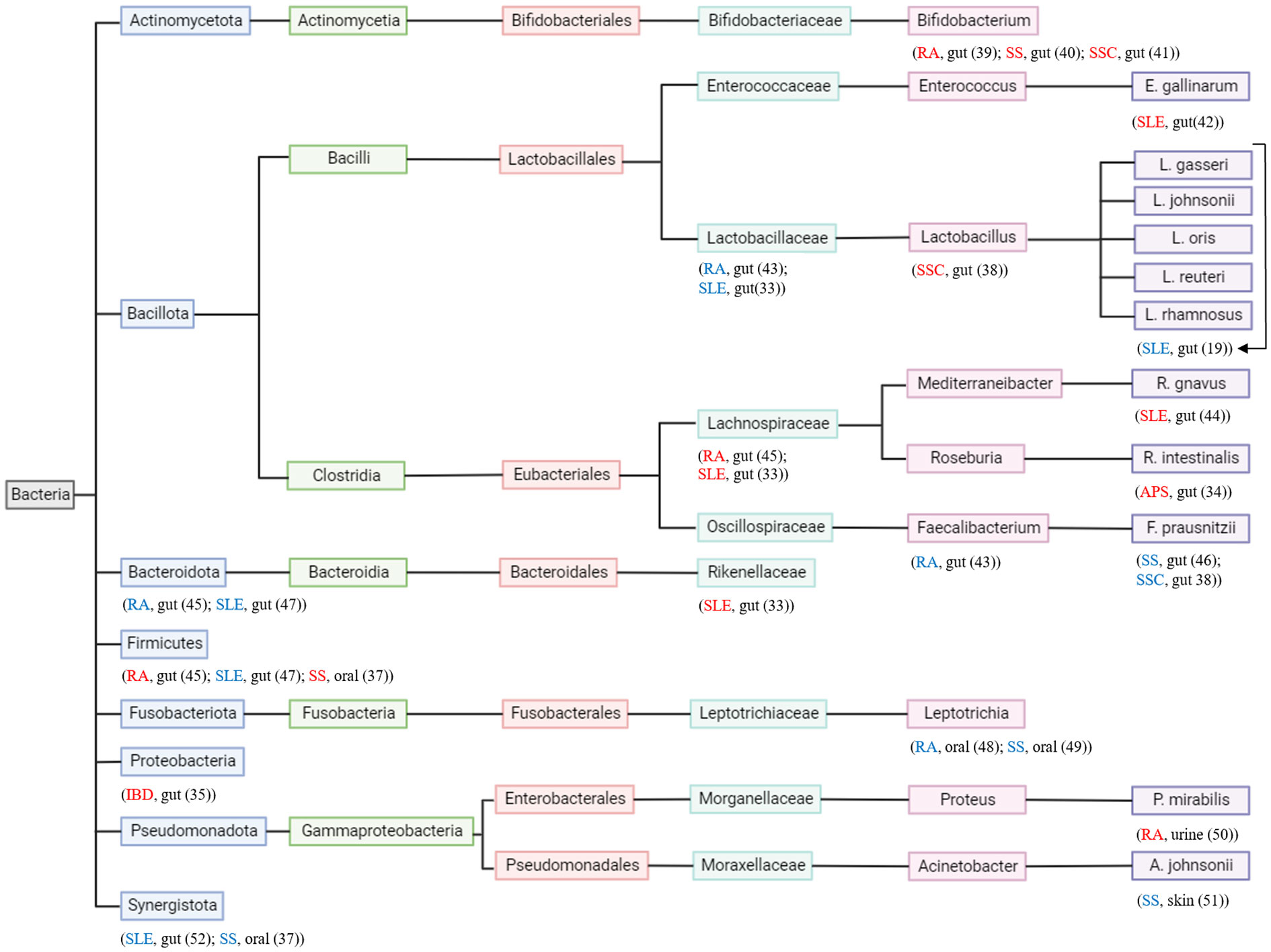
Figure 2 Phylogenetic tree of bacteria and autoimmune diseases. Each taxonomy level has a different color. Autoimmune disease and where in the body the bacteria is found are located under the studied bacterium. Autoimmune disease in “red” indicates the bacteria contributes to its pathogenesis and autoimmune disease in “blue” indicates the bacteria contributes to its attenuation.
A study of lupus nephritis linked disease activity to a gut commensal. Fecal microbiota of SLE patients was tested for pathobionts using 16S ribosomal RNA (rRNA) analyses, and it was found that SLE patients possessed a microbiome that had a decrease in species richness diversity as well as a reduction in taxonomic complexity (44). Ruminococcus (Blautia) gnavus was found to be significantly higher in SLE patients than in healthy individuals (44). R. gnavus was further investigated in a longitudinal analyses of lupus gut microbiota to study microbiota resilience and disease activity. R. gnavus was found to be expanded during high disease activity, and it was detected in almost half of patients during lupus nephritis disease flares. After whole-genome sequence analysis of R. gnavus, it was found that there were 34 genes that help adapt and expand within a host with inflammation (53). This rationalizes that these bacteria that are found expanded during disease flares possess the necessary tools to withstand inflammation.
Toll-like receptor (TLR) 7.1 transgenic (Tg) C57BL/6 mice were cohoused with wild-type littermates (WTLs). This revealed an enrichment of Lactobacillus, Desulfovibrio, and Rikenellaceae in TLR7.1 Tg mice. When WTL mice were colonized with microbiota containing these pathobionts from TLR7.1 Tg mice, they showed an increase in leaky gut with translocation of pathobionts (54). Thus, the enrichment of these bacterial communities exacerbates SLE pathogenesis. Moreover, fecal transfer from dysbiotic gut microbiota of triple congenic (TC) lupus-prone mice into germ-free congenic C57BL/6 mice induced autoimmune phenotypes when TC donor mice exhibited autoimmunity (55). Again, this shows the significance of how the gut microbiota affects SLE pathogenesis.
The role of gut microbiota in renal pathogenesis of SLE has not been widely investigated. We found that the gut microbiota of MRL/lpr lupus-prone mice has significant depletion of Lactobacillales as disease develops. A weekly treatment of a mixture of five Lactobacillus strains (Lactobacillus gasseri, L. johnsonii, L. oris, L. reuteri, and L. rhamnosus) attenuated lupus-like clinical signs, including splenomegaly and lymphadenopathy, and prolonged survival (19). Individually, the different strains did not have an effect, but the mixture of Lactobacillus spp. acted synergistically to attenuate lupus-like disease. Mechanistically, the mixture of Lactobacillus spp. increased the percentages of the effector memory T cells in the spleen and mesenteric lymph nodes while decreasing the percentages of the central memory T cells and double-negative T cells. The mentioned outcomes suggest that in order to attenuate lymphadenopathy and splenomegaly, Lactobacillus spp. may act on T cells (19). Further investigations will determine if the above results can be replicated in human SLE patients. Furthermore, Lactobacillus spp. may increase FOXP3-negative Tr1 cells in the spleen and mesenteric lymph nodes to control inflammation (19). The treatment of Lactobacillus reversed a “leaky” gut, which the MRL/lpr mice possessed. The treatment also promoted an anti-inflammatory environment by decreasing IL-6 and increasing IL-10 production in the gut. In the circulation, IL-10 was increased whereas IgG2a was decreased (the latter of which is believed to be a major immune deposit in the MRL/lpr kidney). Furthermore, T cells showed a Treg-Th17 balance toward the Treg phenotype in the kidney (2).
In another study, fecal microbiota transplantation from untreated active SLE female patients vs. healthy female patients to germ-free (GF) mice was investigated (56). SLE patients’ fecal microbiota caused GF mice to develop a series of lupus-like phenotypic features, including imbalanced cytokines, upregulation of SLE-related genes, autoimmune antibodies, and altered distribution of immune cells in mucosal and peripheral immune response. Importantly, these results depict a causal role of aberrant gut microbiota in influencing the pathogenesis of SLE (56). A 12-week investigation of the safety and efficacy of fecal microbiota transplantation to treat SLE patients was also explored (57). SLE patients were treated with oral encapsulated fecal microbiome from healthy donors. Upon completion of this study, it was concluded that fecal microbiota transplantation is safe and effective for SLE patients. Also, they found that fecal microbiota transplantation alters the gut microbiome and modifies the short-chain fatty acid metabolic profile in SLE patients (57).
There was a decrease in the Firmicutes/Bacteroidetes ratio in SLE patients (47). This balance between the two phyla in the human gut microbiota is dependent on the individual’s physiology. This ratio is important because dysbiosis of these two phyla in the intestines is associated with SLE (47). Another study has shown that the frequency of Synergistetes positivity correlated with the Firmicutes/Bacteroidetes ratio in healthy individuals but reduced in SLE patients’ fecal samples with an increase in anti-double stranded DNA (dsDNA) titers (52). This may suggest a protective role that the intestinal bacterium Synergistetes has on humoral immunity. Notably, many studies, including ours, have used 16S rRNA sequencing that can only be accurate at the genus level. Genus-level microbiota evaluation is likely insufficient to ideally link mechanisms of pathobionts to SLE disease. Metagenomic shotgun sequencing, which can reach species and even the strain level, will be more useful.
CX3CR1+ cells are present in tissues lining the intestine, and phagocytes expressing CX3CR1 can clear pathogenic bacteria from the gut lumen (58). When the chemokine is absent, the integrity of the intestinal barrier is compromised (12). This phenomenon results in an altered microbiome as well as endotoxemia and aggravation of the gut and liver inflammation (59, 60). This suggests that in SLE, CX3CR1 may play a protective role in clearing pathogenic gut bacteria and, in its absence, the gut lining may be compromised, which will cause a leaky gut and bacteria to interact with peripheral organs, consequently causing inflammation.
Using the MRL/lpr lupus nephritis model, we observed the enhancement of gut mucosal barrier with Lactobacillales treatment that resulted in less bacteria translocation across the intestinal epithelium (2). This led to reduced activation and migration of CX3CR1+ antigen-presenting cells to lymph nodes. Furthermore, the Lactobacillales treatment significantly reduced Cx3cr1 in the mesenteric lymph node, which would suggest that the treatment may reduce the migration of antigen-presenting cells to the mesenteric lymph node (2).
We have demonstrated that Cx3cr1-deficient mice have a noticeably different gut microbiota from Cx3cr1+/+ mice (27). The gut microbiota of Cx3cr1-deficient mice was corrected with Lactobacillus administration, and consequently, glomerulonephritis was reversed (27). This suggests that CX3CR1 plays an important role in glomerulonephritis in MRL/lpr mice through a gut microbiota-dependent mechanism. We demonstrated that the treatment of Lactobacillus spp. attenuated splenomegaly and renal lymphadenopathy via a CX3CR1-dependent mechanism (19), suggesting that CX3CR1 may be used as a target for therapeutics.
In C57BL/6 mice, the depletion of the gut microbiota via broad-spectrum antibiotics, which was reversible by fecal transplantation, decreased the levels of CX3CR1 in macrophages and bone marrow monocytes (61). This suggests that the gut microbiota plays a role in the induction of CX3CR1 in addition to activation of downstream pathways.
Figure 3 illustrates different mechanisms of action for CX3CR1 as it interacts with CX3CL1 and the gut microbiota in SLE.
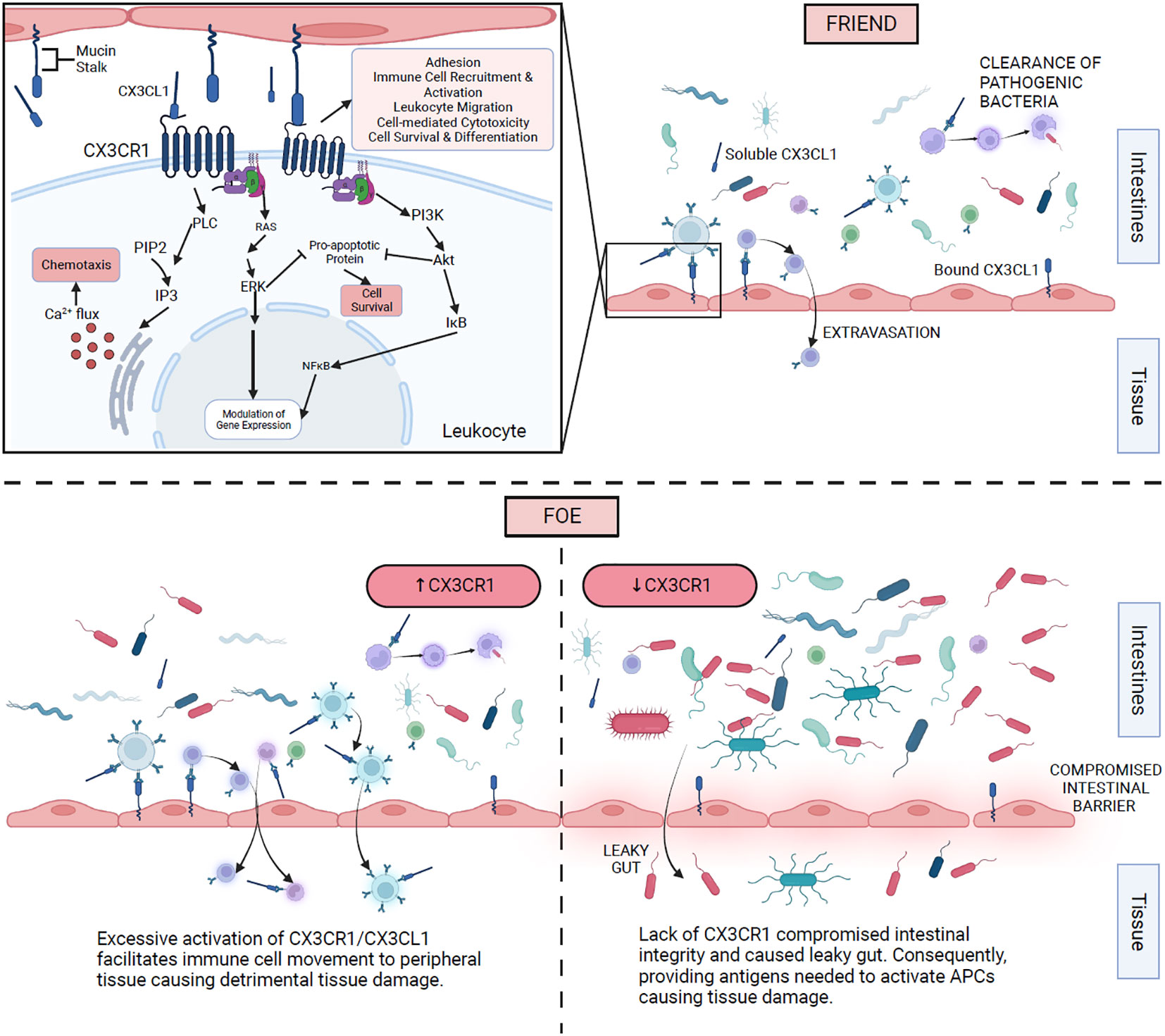
Figure 3 Proposed mechanisms of action of CX3CR1 and gut microbiota in SLE. The top right section shows CX3CR1 interaction with membrane-bound and soluble CX3CL1 on different immune cells. The top left section illustrates the intracellular mechanism of action once CX3CR1 is activated. The bottom right section expresses the consequences of the absence of CX3CR1. The bottom left section displays the effects of excessive activation of CX3CR1.
CX3CR1 has not been studied in context with the gut microbiota in human SLE. We speculate that a single loss-of-function nucleotide polymorphism of CX3CR1 in humans may result in an exacerbation of lupus nephritis. Further investigation of CX3CR1 is needed in order to replicate the mentioned findings in human SLE patients.
While the cause of SLE remains unknown, there are factors that can influence its severity, including environmental, epigenetic, genetic, hormonal, immunoregulatory, and lifestyle factors. When SLE is in the “on” phase of the disease, it may affect the kidney, lungs, joints, heart, skin, and/or brain. We and others have established that the gut microbiota is a causative factor in SLE instead of the result. Indeed, immune cells help maintain the gut barrier and protect against pathogens entering the body. The CX3CR1 receptor helps maintain the integrity of the gut barrier (12). The data are more in support of its protective properties in SLE. Finally, the gut microbiota and CX3CR1 interact to influence one another in various pathways.
The pathways by which the gut microbiota and CX3CR1 interact are not well studied. Investigations suggest that the gut microbial dysbiosis causing a leaky gut activates CX3CR1-expressing cells, which subsequently increase inflammation, thus causing aggregated spleen and/or kidney inflammation. It was reported that a single loss-of-function nucleotide polymorphism of CX3CR1 leads to exacerbation of chronic kidney disease compared to patients that do not have this loss of function (25). An altered gut microbiome may lead to lupus nephritis, and when combined with the loss of function of CX3CR1, it may increase the risk of renal failure.
The contradictions in Table 1 are pronounced studies that ought to be investigated further for a better understanding of CX3CL1/CX3CR1 interactions, particularly on how they can be used therapeutically. A potential treatment of the leaky gut in lupus is to agonize CX3CR1 in antigen-presenting cells, which may activate these cells to clear any bacteria leaking from the gut. Future investigations may shed light on whether CX3CR1 is a friend or foe in SLE. This may be addressed by blocking CX3CR1 or its ligand, CX3CL1, and assess disease progression. Another way to investigate the role of CX3CR1 is to have a conditional knockout in an SLE model and dissect the mechanism of action of CX3CR1 in a cell- or tissue-specific manner.
RE: Writing – original draft. CR: Writing – review & editing. XL: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. RE is supported by the Biomedical and Veterinary Sciences (BMVS) Graduate Program and the Office of Research and Graduate Studies, Virginia-Maryland College of Veterinary Medicine, Virginia Tech.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tsokos GC. Systemic lupus erythematosus. N Eng J Med (2011) 365(22):2110–21. doi: 10.1056/NEJMra1100359
2. Mu Q, Zhang H, Liao X, Lin K, Liu H, Edwards MR, et al. Control of lupus nephritis by changes of gut microbiota. Microbiome (2017) 5(1):73. doi: 10.1186/s40168-017-0300-8
3. Lahita RG. Introduction. In: Lahita RG, editor. Systemic lupus erythematosus, Fifth Edition. San Diego: Academic Press (2011). p. xvii–xix.
4. Dörner T, Furie R. Novel paradigms in systemic lupus erythematosus. Lancet (2019) 393(10188):2344–58. doi: 10.1016/S0140-6736(19)30546-X
5. Nakano M, Ayano M, Kushimoto K, Kawano S, Higashioka K, Inokuchi S, et al. Association of elevated serum soluble CD226 levels with the disease activity and flares of systemic lupus erythematosus. Sci Rep (2021) 11(1):16162. doi: 10.1038/s41598-021-95711-2
6. Pons-Estel GJ, Alarcón GS, Scofield L, Reinlib L, Cooper GS. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheumatol (2010) 39(4):257–68. doi: 10.1016/j.semarthrit.2008.10.007
7. Fanouriakis A, Tziolos N, Bertsias G, Boumpas DT. Update on the diagnosis and management of systemic lupus erythematosus. Ann Rheum Dis (2021) 80(1):14–25. doi: 10.1136/annrheumdis-2020-218272
8. Liao X, Pirapakaran T, Luo XM. Chemokines and chemokine receptors in the development of lupus nephritis. Mediators Inflamm (2016) 2016:6012715. doi: 10.1155/2016/6012715
9. Ransohoff RM. Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity (2009) 31(5):711–21. doi: 10.1016/j.immuni.2009.09.010
10. Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell (1997) 91(4):521–30. doi: 10.1016/S0092-8674(00)80438-9
11. Cockwell P, Calderwood JW, Brooks CJ, Chakravorty SJ, Savage CO. Chemoattraction of T cells expressing CCR5, CXCR3 and CX3CR1 by proximal tubular epithelial cell chemokines. Nephrol Dial Transplant (2002) 17(5):734–44. doi: 10.1093/ndt/17.5.734
12. von Vietinghoff S, Kurts C. Regulation and function of CX3CR1 and its ligand CX3CL1 in kidney disease. Cell Tissue Res (2021) 385(2):335–44. doi: 10.1007/s00441-021-03473-0
13. Landsman L, Bar-On L, Zernecke A, Kim KW, Krauthgamer R, Shagdarsuren E, et al. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood (2009) 113(4):963–72. doi: 10.1182/blood-2008-07-170787
14. Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol (2008) 83(1):64–70. doi: 10.1189/jlb.0407247
15. Park MY, Kim HS, Lee HY, Zabel BA, Bae YS. Novel CD11b(+)Gr-1(+)Sca-1(+) myeloid cells drive mortality in bacterial infection. Sci Adv (2020) 6(4):eaax8820. doi: 10.1126/sciadv.aax8820
16. Geissmann F, Auffray C, Palframan R, Wirrig C, Ciocca A, Campisi L, et al. Blood monocytes: distinct subsets, how they relate to dendritic cells, and their possible roles in the regulation of T-cell responses. Immunol Cell Biol (2008) 86(5):398–408. doi: 10.1038/icb.2008.19
17. Cao Y, Xu QXM, Liu C, Fu C. Role of spleen-derived CD11b+Gr-1+ cells in sepsis-induced acute kidney injury. Clin Invest Med (2020) 43(2):E24–34. doi: 10.25011/cim.v43i2.34189
18. Inoue A, Hasegawa H, Kohno M, Ito MR, Terada M, Imai T, et al. Antagonist of fractalkine (CX3CL1) delays the initiation and ameliorates the progression of lupus nephritis in MRL/lpr mice. Arthritis Rheumatol (2005) 52(5):1522–33. doi: 10.1002/art.21007
19. Cabana-Puig X, Mu Q, Lu R, Swartwout B, Abdelhamid L, Zhu J, et al. Lactobacillus spp. act in synergy to attenuate splenomegaly and lymphadenopathy in lupus-prone MRL/lpr mice. Front Immunol (2022) 13:923754. doi: 10.3389/fimmu.2022.923754
20. Engel DR, Krause TA, Snelgrove SL, Thiebes S, Hickey MJ, Boor P, et al. CX3CR1 reduces kidney fibrosis by inhibiting local proliferation of profibrotic macrophages. J Immunol (2015) 194(4):1628–38. doi: 10.4049/jimmunol.1402149
21. Ito Y, Kawachi H, Morioka Y, Nakatsue T, Koike H, Ikezumi Y, et al. Fractalkine expression and the recruitment of CX3CR1+ cells in the prolonged mesangial proliferative glomerulonephritis. Kidney Int (2002) 61(6):2044–57. doi: 10.1046/j.1523-1755.2002.00369.x
22. Oh DJ, Dursun B, He Z, Lu L, Hoke TS, Ljubanovic D, et al. Fractalkine receptor (CX3CR1) inhibition is protective against ischemic acute renal failure in mice. Am J Physiol Renal Physiol (2008) 294(1):F264–71. doi: 10.1152/ajprenal.00204.2007
23. Chousterman BG, Boissonnas A, Poupel L, Baudesson de Chanville C, Adam J, Tabibzadeh N, et al. Ly6Chigh Monocytes Protect against Kidney Damage during Sepsis via a CX3CR1-Dependent Adhesion Mechanism. J Am Soc Nephrol (2016) 27(3):792–803. doi: 10.1681/ASN.2015010009
24. Cormican S, Griffin MD. Fractalkine (CX3CL1) and its receptor CX3CR1: A promising therapeutic target in chronic kidney disease? Front Immunol (2021) 12:664202. doi: 10.3389/fimmu.2021.664202
25. Yadav AK, Kumar V, Jha V. Association of chemokine receptor CX3CR1 V249I and T280M polymorphisms with chronic kidney disease. Indian J Nephrol (2016) 26(4):275–9. doi: 10.4103/0971-4065.163426
26. Li C, Zhong X, Xia W, He J, Gan H, Zhao H, et al. The CX3CL1/CX3CR1 axis is upregulated in chronic kidney disease and contributes to angiotensin II-induced migration of vascular smooth muscle cells. Microvasc Res (2020) 132:104037. doi: 10.1016/j.mvr.2020.104037
27. Cabana-Puig X, Lu R, Geng S, Michaelis JS, Oakes V, Armstrong C, et al. CX(3)CR1 modulates SLE-associated glomerulonephritis and cardiovascular disease in MRL/lpr mice. Inflammation Res Off J Eur Histamine Res Soc [et al]. (2023) 72(5):1083–97. doi: 10.1007/s00011-023-01731-1
28. Shimizu K, Furuichi K, Sakai N, Kitagawa K, Matsushima K, Mukaida N, et al. Fractalkine and its receptor, CX3CR1, promote hypertensive interstitial fibrosis in the kidney. Hypertens Res (2011) 34(6):747–52. doi: 10.1038/hr.2011.23
29. Żywiec J, Gosek K, Kuźniewicz R, Górczyńska-Kosiorz S, Trautsolt W, et al. Association of CX3CR1 gene polymorphisms with fractalkine, fractalkine receptor, and C-reactive protein levels in patients with kidney failure. Int J Environ Res Public Health (2021) 18(4). doi: 10.3390/ijerph18042202
30. Jandhyala SM, Talukdar R, Subramanyam C, Vuyyuru H, Sasikala M, Nageshwar Reddy D. Role of the normal gut microbiota. World J Gastroenterol (2015) 21(29):8787–803. doi: 10.3748/wjg.v21.i29.8787
31. Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev (2010) 90(3):859–904. doi: 10.1152/physrev.00045.2009
32. De Luca F, Shoenfeld Y. The microbiome in autoimmune diseases. Clin Exp Immunol (2019) 195(1):74–85. doi: 10.1111/cei.13158
33. Luo XM, Edwards MR, Mu Q, Yu Y, Vieson MD, Reilly CM, et al. Gut microbiota in human systemic lupus erythematosus and a mouse model of lupus. Appl Environ Microbiol (2018) 84(4). doi: 10.1128/AEM.02288-17
34. Ruff WE, Vieira SM, Kriegel MA. The role of the gut microbiota in the pathogenesis of antiphospholipid syndrome. Curr Rheumatol Rep (2015) 17(1):472. doi: 10.1007/s11926-014-0472-1
35. Rizzatti G, Lopetuso LR, Gibiino G, Binda C, Gasbarrini A. Proteobacteria: A common factor in human diseases. BioMed Res Int (2017) 2017:9351507. doi: 10.1155/2017/9351507
36. Tobón GJ, Youinou P, Saraux A. The environment, geo-epidemiology, and autoimmune disease: Rheumatoid arthritis. J Autoimmun (2010) 35(1):10–4. doi: 10.1016/j.jaut.2009.12.009
37. Siddiqui H, Chen T, Aliko A, Mydel PM, Jonsson R, Olsen I. Microbiological and bioinformatics analysis of primary Sjogren's syndrome patients with normal salivation. J Oral Microbiol (2016) 8:31119. doi: 10.3402/jom.v8.31119
38. Andréasson K, Alrawi Z, Persson A, Jönsson G, Marsal J. Intestinal dysbiosis is common in systemic sclerosis and associated with gastrointestinal and extraintestinal features of disease. Arthritis Res Ther (2016) 18(1):278. doi: 10.1186/s13075-016-1182-z
39. Wang Q, Zhang SX, Chang MJ, Qiao J, Wang CH, Li XF, et al. Characteristics of the gut microbiome and its relationship with peripheral CD4(+) T cell subpopulations and cytokines in rheumatoid arthritis. Front Microbiol (2022) 13:799602. doi: 10.3389/fmicb.2022.799602
40. Mandl T, Marsal J, Olsson P, Ohlsson B, Andréasson K. Severe intestinal dysbiosis is prevalent in primary Sjögren's syndrome and is associated with systemic disease activity. Arthritis Res Ther (2017) 19(1):237. doi: 10.1186/s13075-017-1446-2
41. Volkmann ER, Chang YL, Barroso N, Furst DE, Clements PJ, Gorn AH, et al. Association of systemic sclerosis with a unique colonic microbial consortium. Arthritis Rheumatol (2016) 68(6):1483–92. doi: 10.1002/art.39572
42. Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science (2018) 359(6380):1156–61. doi: 10.1126/science.aar7201
43. Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, et al. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med (2016) 8(1):43. doi: 10.1186/s13073-016-0299-7
44. Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, et al. Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann Rheumatic Diseases (2019) 78(7):947–56. doi: 10.1136/annrheumdis-2018-214856
45. Rogier R, Evans-Marin H, Manasson J, van der Kraan PM, Walgreen B, Helsen MM, et al. Alteration of the intestinal microbiome characterizes preclinical inflammatory arthritis in mice and its modulation attenuates established arthritis. Sci Rep (2017) 7(1):15613. doi: 10.1038/s41598-017-15802-x
46. de Paiva CS, Jones DB, Stern ME, Bian F, Moore QL, Corbiere S, et al. Altered mucosal microbiome diversity and disease severity in Sjögren syndrome. Sci Rep (2016) 6:23561. doi: 10.1038/srep23561
47. Hevia A, Milani C, López P, Cuervo A, Arboleya S, Duranti S, et al. Intestinal dysbiosis associated with systemic lupus erythematosus. mBio (2014) 5(5):e01548–14. doi: 10.1128/mBio.01548-14
48. Lehenaff R, Tamashiro R, Nascimento MM, Lee K, Jenkins R, Whitlock J, et al. Subgingival microbiome of deep and shallow periodontal sites in patients with rheumatoid arthritis: a pilot study. BMC Oral Health (2021) 21(1):248. doi: 10.1186/s12903-021-01597-x
49. Sharma D, Sandhya P, Vellarikkal SK, Surin AK, Jayarajan R, Verma A, et al. Saliva microbiome in primary Sjögren's syndrome reveals distinct set of disease-associated microbes. Oral Dis (2020) 26(2):295–301. doi: 10.1111/odi.13191
50. Wilson C, Thakore A, Isenberg D, Ebringer A. Correlation between anti-Proteus antibodies and isolation rates of P. mirabilis in rheumatoid arthritis. Rheumatol Int (1997) 16(5):187–9. doi: 10.1007/BF01330294
51. Szymula A, Rosenthal J, Szczerba BM, Bagavant H, Fu SM, Deshmukh US. T cell epitope mimicry between Sjögren's syndrome Antigen A (SSA)/Ro60 and oral, gut, skin and vaginal bacteria. Clin Immunol (2014) 152(1-2):1–9. doi: 10.1016/j.clim.2014.02.004
52. López P, de Paz B, Rodríguez-Carrio J, Hevia A, Sánchez B, Margolles A, et al. Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients. Sci Rep (2016) 6:24072. doi: 10.1038/srep24072
53. Azzouz DF, Chen Z, Izmirly PM, Chen LA, Li Z, Zhang C, et al. Longitudinal gut microbiome analyses and blooms of pathogenic strains during lupus disease flares. Ann Rheumatic Diseases (2023) 82(10):1315–27. doi: 10.1136/ard-2023-223929
54. Zegarra-Ruiz DF, El Beidaq A, Iñiguez AJ, Lubrano Di Ricco M, Manfredo Vieira S, Ruff WE, et al. A diet-sensitive commensal lactobacillus strain mediates TLR7-dependent systemic autoimmunity. Cell Host Microbe (2019) 25(1):113–27.e6. doi: 10.1016/j.chom.2018.11.009
55. Choi SC, Brown J, Gong M, Ge Y, Zadeh M, Li W, et al. Gut microbiota dysbiosis and altered tryptophan catabolism contribute to autoimmunity in lupus-susceptible mice. Sci Transl Med (2020) 12(551). doi: 10.1126/scitranslmed.aax2220
56. Ma Y, Guo R, Sun Y, Li X, He L, Li Z, et al. Lupus gut microbiota transplants cause autoimmunity and inflammation. Clin Immunol (2021) 233:108892. doi: 10.1016/j.clim.2021.108892
57. Huang C, Yi P, Zhu M, Zhou W, Zhang B, Yi X, et al. Safety and efficacy of fecal microbiota transplantation for treatment of systemic lupus erythematosus: An EXPLORER trial. J Autoimmun (2022) 130:102844. doi: 10.1016/j.jaut.2022.102844
58. Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Sci (New York NY). (2005) 307(5707):254–8. doi: 10.1126/science.1102901
59. Schneider KM, Bieghs V, Heymann F, Hu W, Dreymueller D, Liao L, et al. CX3CR1 is a gatekeeper for intestinal barrier integrity in mice: Limiting steatohepatitis by maintaining intestinal homeostasis. Hepatology (2015) 62(5):1405–16. doi: 10.1002/hep.27982
60. Kim M, Galan C, Hill AA, Wu WJ, Fehlner-Peach H, Song HW, et al. Critical role for the microbiota in CX(3)CR1(+) intestinal mononuclear phagocyte regulation of intestinal T cell responses. Immunity (2018) 49(1):151–63.e5. doi: 10.1016/j.immuni.2018.05.009
Keywords: systemic lupus erythematosus, lupus, lupus nephritis, CX3CR1, gut microbiota, autoimmune disease, autoimmunity
Citation: Estaleen RA, Reilly CM and Luo XM (2024) A double-edged sword: interactions of CX3CL1/CX3CR1 and gut microbiota in systemic lupus erythematosus. Front. Immunol. 14:1330500. doi: 10.3389/fimmu.2023.1330500
Received: 30 October 2023; Accepted: 28 December 2023;
Published: 17 January 2024.
Edited by:
Chris Wincup, King’s College Hospital NHS Foundation Trust, United KingdomReviewed by:
Gregg Joshua Silverman, New York University, United StatesCopyright © 2024 Estaleen, Reilly and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin M. Luo, eGlubHVvQHZ0LmVkdQ==; Christopher M. Reilly, Q2hyZWlsbHlAdnQudmNvbS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.