 Zhaoxiang Song
Zhaoxiang Song Xiangyu Li
Xiangyu Li- Department of Gastroenterology, The First Affiliated Hospital of Zhengzhou University, ZhengZhou, China
Objectives: Previous studies have confirmed a link between specific inflammatory cytokines and inflammatory bowel disease (IBD), but the causal relationship between them is not completely clear. This Mendelian Randomization (MR) study aims to evaluate the causal relationship between 18 inflammatory cytokines and inflammatory bowel disease.
Method: Two-sample Mendelian randomization utilized genetic variances associated with IBD from two extensive publicly available genome-wide association studies (GWAS) (Crohn’s Disease (CD): 12,194 cases and 28,072 controls; Ulcerative Colitis (UC): 12,336 cases and 33,609 controls). The data of inflammatory cytokines was acquired from a GWAS including 8,293 healthy participants. We used inverse variance weighted method, MR-Egger, weighted median, simple model and weighted model to evaluate the causal relationship between inflammatory cytokines and IBD. Sensitivity analysis includes heterogeneity and pleiotropy analysis to evaluate the robustness of the results.
Results: The findings indicated suggestive positive associations between Interleukin-13 (IL-13) and macrophage migration inhibitory factor (MIF) with CD (odds ratio, OR: 1.101, 95%CI: 1.021-1.188, p = 0.013; OR: 1.134, 95%CI: 1.024-1.255, p = 0.015). IL-13 also displayed a significant positive correlation with UC (OR: 1.099, 95%CI: 1.018-1.186, p = 0.016). Stem cell factor (SCF) was suggested to be associated with the development of both CD and UC (OR: 1.032, 95%CI: 0.973-1.058, p = 0.012; OR: 1.038, 95%CI: 1.005-1.072, p = 0.024).
Conclusion: This study proposes that IL-13 may be a factor correlated with the etiology of IBD (CD and UC), while MIF just be specifically associated with CD. Additionally, SCF appears more likely to be involved in the downstream development of IBD (CD and UC).
1 Introduction
Inflammatory bowel disease (IBD) is a nonspecific immune-mediated, chronic recurrent gastrointestinal disease and can be subcategorized into Crohn’s disease (CD), ulcerative colitis (UC), and idiopathic colitis (1). The global incidence of UC is on the rise, and it is projected that by 2030, the prevalence rate among Western populations will reach 1%. This poses a significant burden on both global health and the economy (2, 3). Until now, a comprehensive understanding of the etiology and pathogenesis of UC has eluded researchers. The factors implicated include genetic susceptibility, compromised gut mucosal barriers, environmental influences such as increased hygiene standards, urban living, dietary elements, and the dysregulation between gut microbiota and mucosal immunity, any of which may contribute to the onset of UC (4). The primary focus of numerous studies has been on investigating the involvement of immune responses in the pathogenesis of IBD (5, 6). With prolonged activation of the immune system within the intestinal mucosa, it promotes the release of various biomarkers, such as cytokines, including interleukin, chemokine and tumor necrosis factor (7). In canine IBD, there is an initiation of a pro-inflammatory pathway leading to Th cell differentiation, primarily driven by microbial dysbiosis. This imbalance in the microbial community stimulates the generation of mainly pro-inflammatory factors, notably IL-1b. Furthermore, mutations in pattern recognition receptors (PRRs), for example Toll-like receptor 5 (TLR5), heighten the response to flagellin. With dysbiosis characterized by increased Enterobacteriaceae, flagellin expression intensifies, enhancing the mucosa’s pro-inflammatory reactions. Consequently, the inflammatory cytokines induce structural changes in epithelial cells, notably increasing permeability due to augmented leakage through tight junctions. This increased permeability establishes a vicious cycle, allowing more bacteria to breach the mucosal barrier, perpetuating the self-reinforcing cycle of inflammation (8). In CD, there’s a higher expression of Th1 cell-related cytokines, particularly IFN-γ and IL-2, compared to both UC and individuals without the condition. Conversely, mucosal cells in UC exhibit a tendency to produce Th2-type cytokines like IL-5 and IL-13 (9–12). Multiple studies have indicated an elevated synthesis of Th17 cell-related cytokines, such as IL-17a and IL-17F, by mucosal T cells in both CD and UC (13). Meanwhile, a systematic review showed that six chemokines, including CCL2 (MCP-1), CCL11 (EOTAXIN), CCL26 (EOTAXIN-3), CXCL1 (GROa), CXCL8 (IL-8) and CXCL10 (IP-10), as biomarkers of CD activity are controversial (14). Though some observational studies have endeavored to clarify the connections between inflammatory cytokines and IBD. The conclusions derived from these investigations might be influenced by unforeseen confounding factors or reverse causation, complicating the establishment of definite causal correlations.
Mendelian randomization (MR) is recognized as the analytical method that infers the causal impact of an exposure on an outcome by utilizing genetic variations in non-experimental data (15). As alleles are randomly assigned during meiosis, Mendelian randomization (MR) has the capacity to minimize traditional confounding variables and reverse causation, thereby offering improved evidence for causal inference (16). Conducting a two-sample MR analysis permits researchers to assess the associations between the instrumental variables and both exposure and outcome across two distinct population samples, thereby improving the test’s applicability and effectiveness (17). In this study, we first extracted valid genetic variants from the published genome-wide association study (GWAS) summary data of 18 inflammatory cytokines in order to investigate their associations with IBD, and then the direction of causation was further explored by reversing the exposures and outcomes.
2 Methods
2.1 Study design
The foundation of this study relies on a genetic association database derived from GWAS summary datasets (https://gwas.mrcieu.ac.uk/). Multiple single-nucleotide polymorphisms (SNPs) were chosen to represent genetic variability and used as instrumental variables in a two-sample MR analysis. Three primary hypotheses were established as outlined below (Figure 1): 1. The instrumental variables have a direct association with the exposure; 2. The instrumental variables are not influenced by any confounding variables; 3. Genetic variants solely impact outcomes through their effect on exposure (18). MR analysis was employed to evaluate the bidirectional causal connections between inflammatory cytokines and IBD, encompassing both UC and CD.
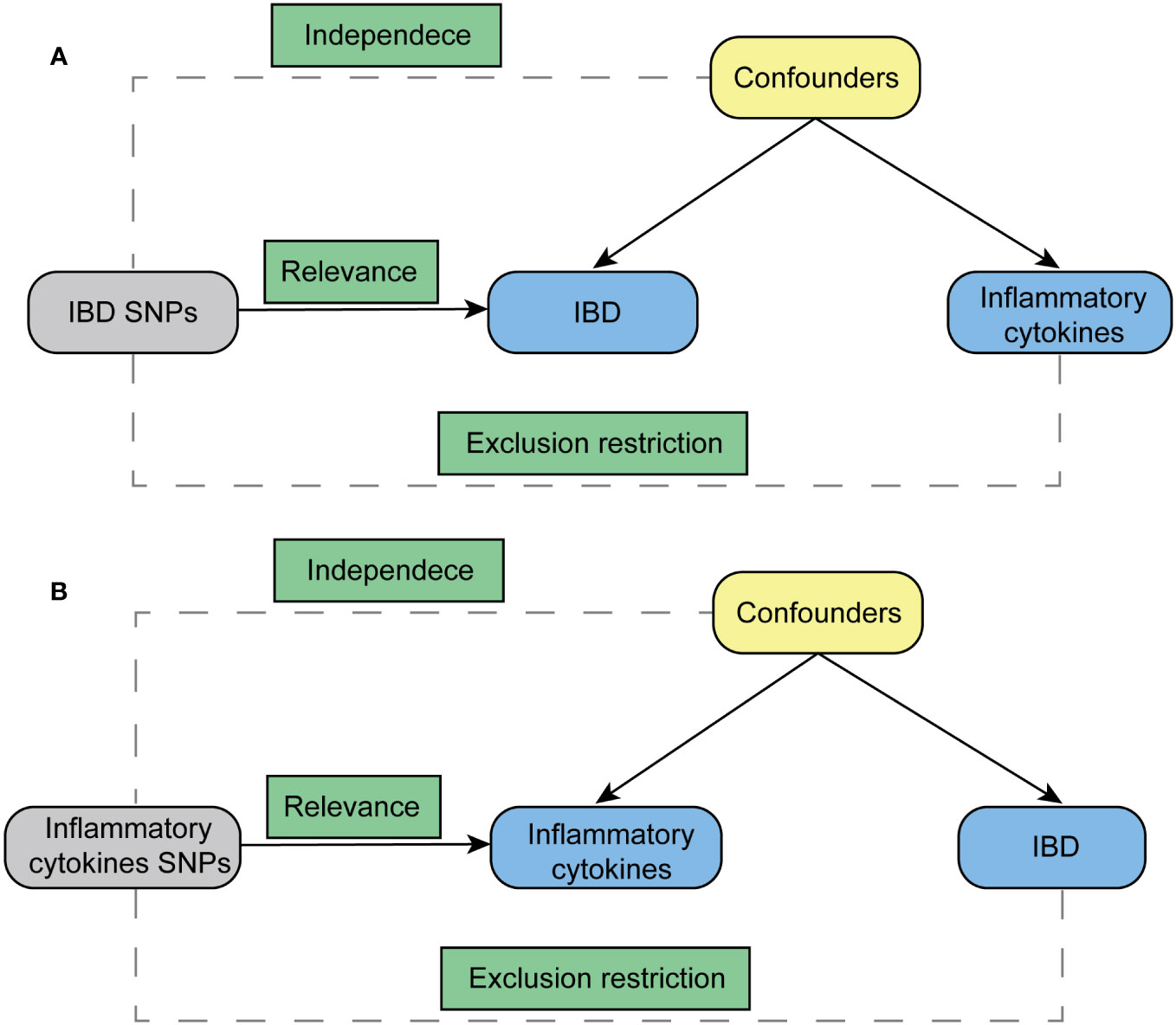
Figure 1 Diagram for key assumptions of MR analyses. (A) IBD SNPs were used as the genetic instruments to investigate the causal effect of IBD on inflammatory cytokines. (B) Genetic instruments in the form of inflammatory cytokine SNPs were utilized to explore the causal relationship between inflammatory cytokines and IBD. The lines with arrows signify the association of genetic instruments (SNPs) with the exposure, affecting the outcome solely through the exposure. Meanwhile, dashed lines represent the independence of the genetic instruments (SNPs) from any confounding variables concerning the outcomes. IBD stands for inflammatory bowel disease.
2.2 Data sources
Both datasets utilized in this MR analysis were sourced from publicly available summarized GWAS data. GWAS data of IBD, containing CD and UC, came from a meta-analysis study. 12194 CD cases (28072 healthy controls), and 12336 UC cases (33609 healthy controls) were available in this data set, with corresponding GWAS IDs of ebi-a-GCST004132 and ebi-a-GCST004133, respectively. For inflammatory cytokines, the data was from the study providing genome variant associations with cytokines and growth factors in 8,293 Finnish individuals. This study combined the results from The Cardiovascular Risk in Young Finns Study (YFS) and FINRISK surveys. The average participant ages are 37 years for YFS study and 60 years for FINRISK survey. There would be no overlap in population selection between the exposure group and the outcome group.
2.3 Instrumental variable selection
Initially, we established the genome-wide significance threshold as p < 5 × 10-8 to pinpoint highly associated SNPs linked with IBD and inflammatory cytokines. However, due to the limited number of identified SNPs for certain inflammatory cytokines when they were considered as the exposure, a higher cutoff (p < 5 × 10-6) was adopted. Next, for the purpose of evading linkage disequilibrium, we conducted SNP clumping (kb = 10,000, r2 = 0.001). Palindromic SNPs were omitted due to uncertainty regarding their alignment in the same orientation for both exposure and outcome in the GWAS of systemic inflammatory regulators. Finally, we assessed the potency of each SNP utilizing the F-statistic, which integrates the extent and accuracy of the genetic impact on the trait: F = R2(N - 2)/(1 - R2), where R2 signifies the proportion of the trait’s variance elucidated by the SNP, and N denotes the sample size of the GWAS encompassing SNPs associated with the trait (19). The R2 values were estimated using the formula R2 = 2×EAF×(1 - EAF)×β2. The effect allele frequency (EAF) of the SNP is denoted as EAF, and β represents the estimated effect of the SNP on the trait. SNPs with an F-statistic less than 10 were excluded, as an F-statistic greater than 10 indicated ample strength, ensuring the credibility of the SNPs.
2.4 Statistical analysis
Main MR analysis was conducted using the inverse variance weighted (IVW) method. In the MR analysis, multiplicative random effects were applied when utilizing more than three SNPs or in cases of heterogeneity. Other MR methodologies employed to verify result consistency encompassed the weighted median, MR-Egger, simple mode, and weighted mode. Heterogeneity among SNPs was evaluated using the Cochran Q test analysis of IVW and MR-Egger. The MR-Egger intercept test served to identify potential horizontal pleiotropy (version 4.2.2).
3 Results
3.1 Influence of 18 inflammatory cytokines on IBD
The outcome of the MR analysis indicated a significant positive correlation between genetically predicted IL-13 (OR: 1.101; 95%CI: 1.021-1.188; p = 0.013) and macrophage migration inhibitory factor (MIF) (OR: 1.134; 95%CI: 1.024-1.255; p = 0.015) with CD (Figure 2A). IL13 (OR: 1.099; 95%CI: 1.018-1.186; p = 0.016) exhibited a notable positive correlation with UC (Figure 2B). Figure 3 displays the scatter plots and funnel plots depicting the Mendelian randomization analyses for IL13 and MIF in IBD. The results from sensitivity analysis indicated that the MR-Egger regression analysis indicated no presence of horizontal pleiotropy, while the Cochran Q test demonstrated the absence of heterogeneity among IVs (Supplementary Table S1). Details of the SNPs are also presented in Supplementary Table S1. While the forest plots and leave-one-out sensitivity analyses of all suggestively significant regulators are presented in Supplementary Figure S1.
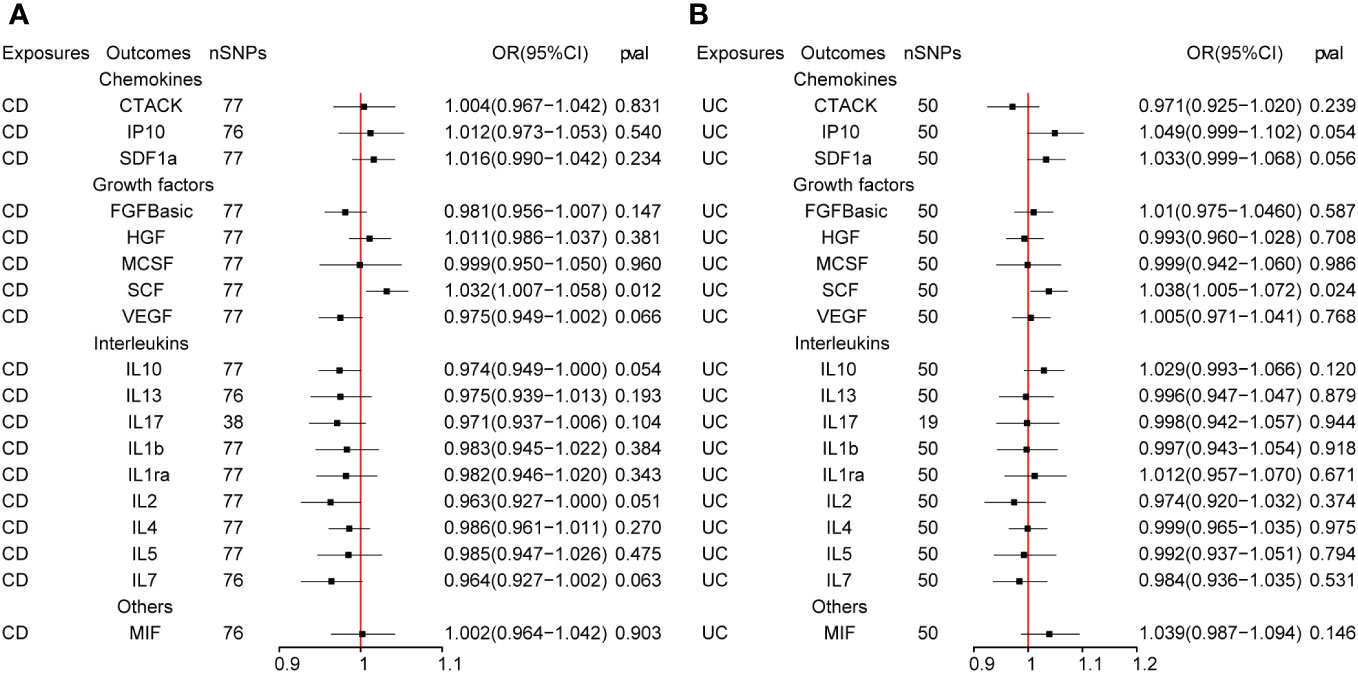
Figure 2 (A, B) The presented figures represent Mendelian randomization estimates illustrating the causal impacts of CD and UC on inflammatory cytokines. The estimates are displayed as OR and 95% CIs derived from bidirectional Mendelian randomization analyses. OR, odds ratio. 95% CI, 95% confidence interval.
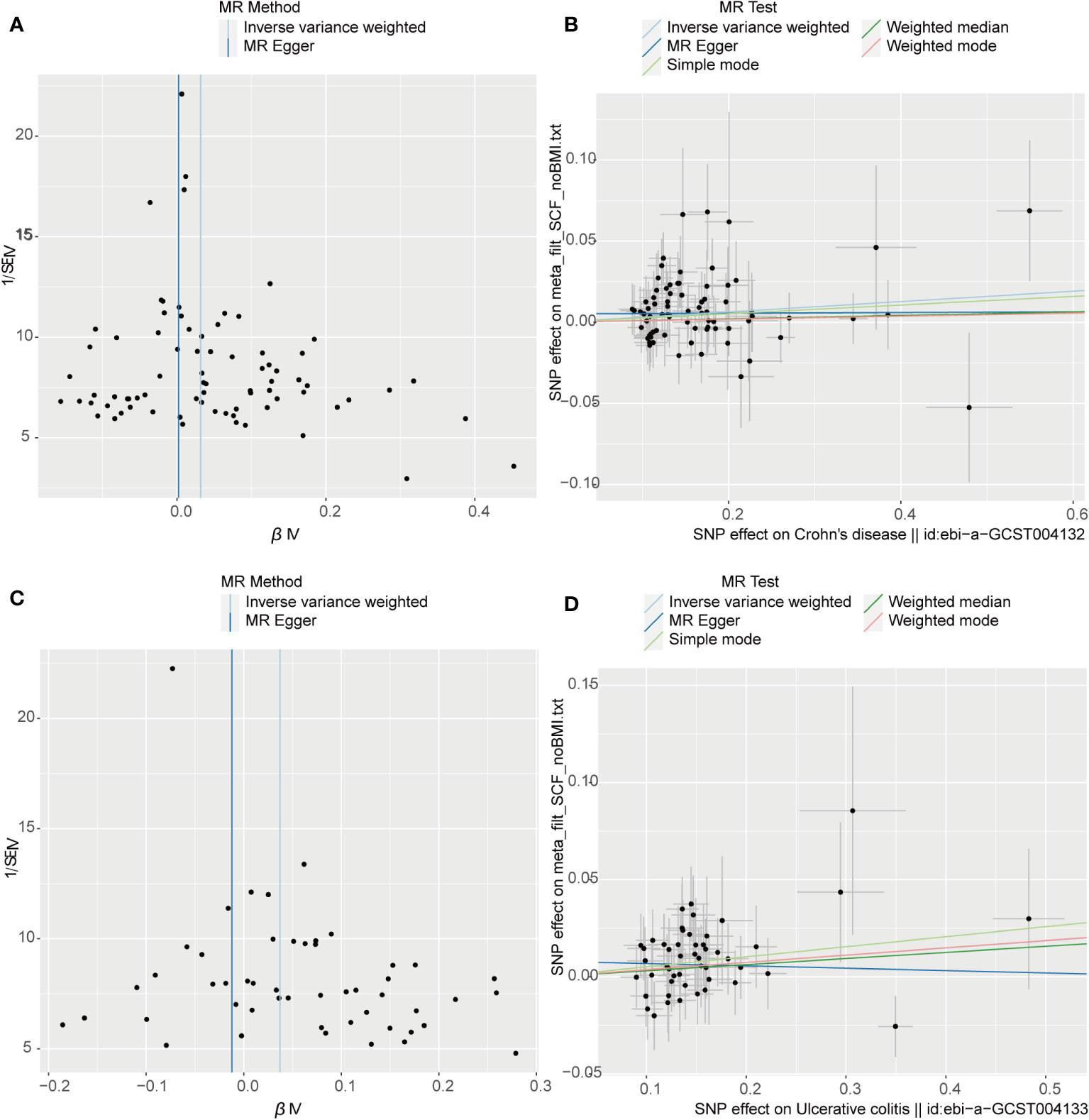
Figure 3 Visual aids like scatter plots and funnel plots were employed to illustrate the causal effects of CD and UC on inflammatory cytokines. (A, C) The funnel plots depict the inverse variance weighted MR estimates of single-nucleotide polymorphisms associated with CD and UC against cytokines versus 1/standard error (1/SEIV). (B, D) Black dots display individual inverse variance (IV) associations with the risk of CD and UC versus individual IV associations with cytokines. The 95%CI of odd ratio for each IV is shown by vertical and horizontal lines. The slope of the lines represents the estimated causal effect of the MR methods.
3.2 Influence of IBD on 18 inflammatory cytokines
Figure 4 presents the outcomes from the reverse MR analysis regarding the causality between IBD and inflammatory cytokines. The results obtained from the IVW method suggested a correlation between an increased level of Stem cell factor (SCF) and CD (OR: 1.032; 95% CI: 1.007-1.058; p = 0.012). Meanwhile, UC was also suggestively correlated with an elevated level of SCF (OR: 1.060; 95% CI: 1.006-1.118; p = 0.028). The scatter plots and funnel plots of SCF are displayed in Figure 5. The results from sensitivity analysis indicated that the MR-Egger regression analysis indicated no presence of horizontal pleiotropy, while the Cochran Q test demonstrated the absence of heterogeneity among IVs (Supplementary Table S1). Supplementary Figure S2 includes forest plots and leave-one-out sensitivity analyses for all regulators that showed suggestive significance.
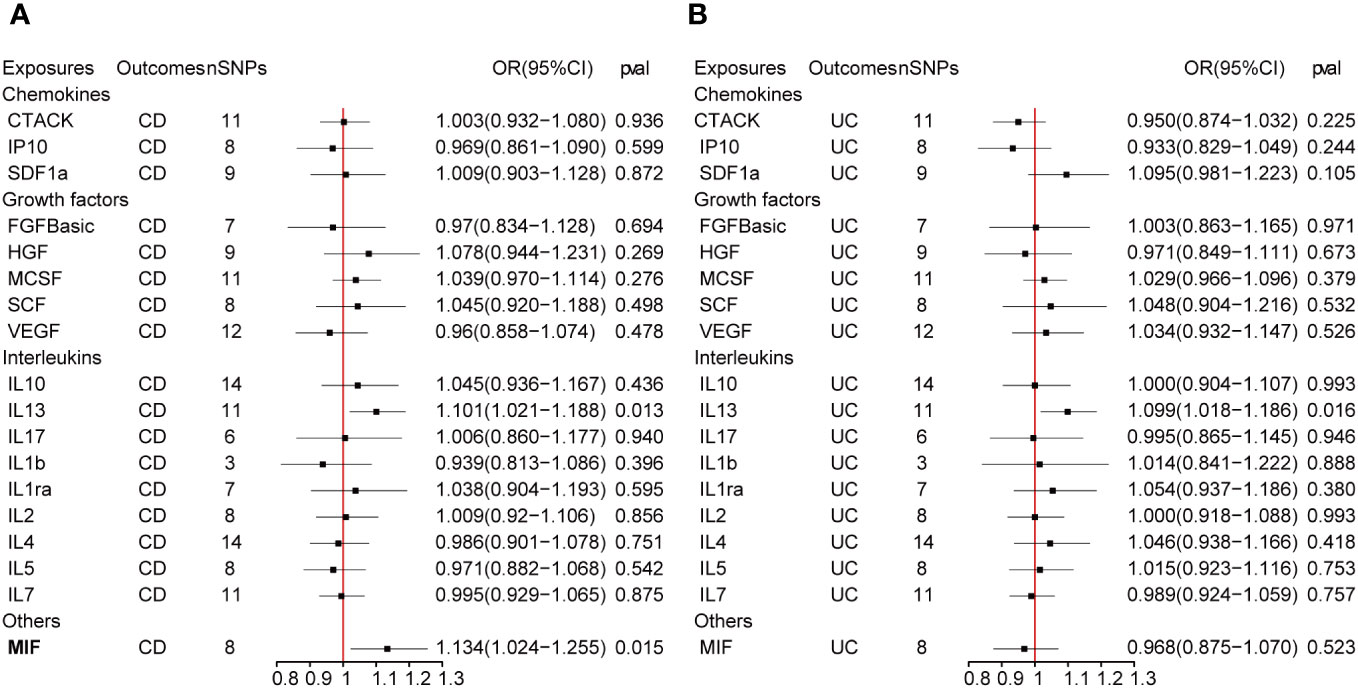
Figure 4 (A, B) represent mendelian randomized estimates of the causal effect of the ILs and chemokines on CD and UC. Estimates are presented as odds ratios (ORs) and 95% CIs from bidirectional mendelian randomization analyses.
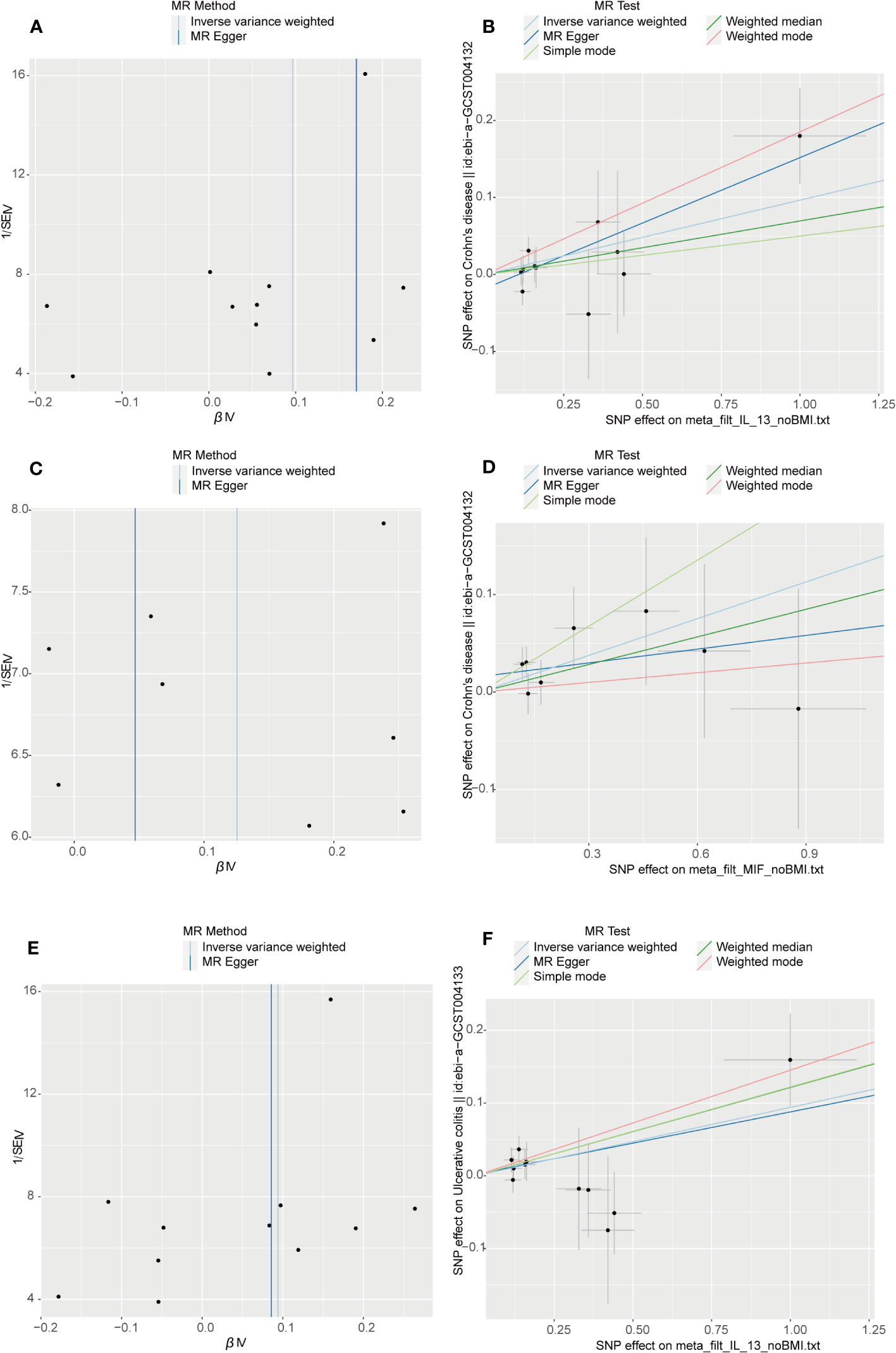
Figure 5 Visual aids like scatter plots and funnel plots were employed to illustrate the causal effects of inflammatory cytokines on CD and UC. (A, C, E) The funnel plots illustrate the inverse variance weighted MR estimates of each cytokine single-nucleotide polymorphism with CD and UC against 1/standard error (1/SEIV). (B, D, F) Black dots display individual inverse variance (IV) associations with the risk of cytokines versus individual IV associations with CD and UC. Vertical and horizontal lines depict the 95% confidence intervals (CI) of the odds ratio for each IV. The slope of these lines indicates the estimated causal effect determined by the MR methods.
4 Discussion
A recent Mendelian randomized study delved into the causal connections between five interleukins, six chemokines, and IBD. The findings indicated significant positive correlations of IL-16, IL-18, and CXCL10 with IBD, contrasting with IL-12p70 and CCL23, which showed significant negative correlations. Additionally, IL-16 and IL-18 suggested an increased risk of UC, while CXCL10 hinted at an increased risk of CD (20).
We expanded the number of inflammatory cytokines (which includes ILs, chemokines, growth factors and others) and explored the causal relationship between more inflammatory factors and IBD. To understand the causal relationship between IBD and inflammatory cytokines, we used publicly aggregated GWAS data for two-way MR analysis. In the forward MR analysis, elevated levels of IL-13 and MIF were associated with increased risk of CD, whereas IL-13 was also linked to an increased risk of UC. In our reverse MR analysis, CD and UC were suggestively associated with elevated levels of SCF.
Differentiating between CD and UC predominantly depends on the localization of inflammatory lesions and the specific cytokine involvement in their pathogenesis. CD manifests as a segmental, transmural disorder that can impact any segment of the gastrointestinal tract, while UC is identified by superficial, continuous mucosal ulcers restricted to the colon. Dysregulation between pro- and anti-inflammatory cytokines is widely acknowledged in both CD and UC (21). In CD, an association exists with a T helper type 1 (Th1) and T helper type 17 (Th17) immune response (22), leading to the secretion of diverse pro-inflammatory cytokines, such as IFNγ/IL12 and IL23/IL17, which encompass IL18, IL2, IL1, IL21, and IL22. Conversely, UC demonstrates a distinct Th17 and an altered Th2 cytokine profile, characterized by IL13 and IL5. Moreover, both Th1 and Th2 cells, alongside macrophages in both types of IBD, contribute to the production of IL6 and tumor necrosis alpha (TNFα) (23). Genetic polymorphisms in cytokine and cytokine receptor genes may significantly impact the progression of the inflammatory cascade, potentially elevating the susceptibility to developing IBD.
IL-13 is a typical Th2 cytokine produced from CD-1-reactive NKT cells, and its secretion mediates epithelial barrier dysfunction. An increase of IL-13 in lymphocytes of the lamina propria in the affected area of UC represented a significant role of the Th2 immune response in UC pathogenesis. Furthermore, IL-13 was responsible for impairment of mucosal permeability that resulted in epithelial barrier damage, and There were alterations observed in the tight junctions of intestinal epithelial cells. In patients with UC, IL-13 showed a significant increase within apoptotic cells and the corresponding apoptotic area (The Th2 colitis model, Oxazolone colitis, resembling ulcerative colitis, is mediated through IL-13-producing NK-T cells). In the lamina propria’s mononuclear cell culture, IL-13 heightened ion flux, leading to alterations in cellular tight junctions. IL-13 exerted an influence on mucosal repair, artificially reducing the rate of mucosal repair by 30% upon its addition to the mucosal lesions (24). Two papers published in 2004 and 2005 reported that ex vivo stimulated lamina propria T cells, obtained from resected specimens of UC patients, exhibited heightened protein levels of IL-13 compared to individuals with CD and those who were healthy (24, 25). The inflammatory infiltrate of TNF and IL-13 triggers epithelial-to-mesenchymal transition and upregulation of matrix metalloproteinases, resulting in tissue remodeling and the formation of fistulas (26–28). In a Polish population, the presence of IL13 -1112 CT (rs1800925) genotypes indicated an increased likelihood of both IBD and UC occurrence (29).
Initially identified as a factor released by T cells, macrophage migration inhibitory factor (MIF) inhibits the random migration of macrophages (30). Later investigations disclosed that MIF acts as a pro-inflammatory factor., which has important roles in various chronic inflammatory diseases and immune disorders, including UC (31). Some studies showed the capacity of MIF to induce increased functional capacity of DC, and to produce IL-1b and IL-8 from monocytes and DC, indicate a role of MIF in the induction and/or perpetuation of the inflammatory environment in UC and CD (32).
Stem cell factor, also recognized as SCF, KIT-ligand, or steel factor, is a pleiotropic cytokine that governs regulatory impacts on inflammation, tissue remodeling, and fibrosis by binding to its receptor c-KIT (33, 34). SCF is extensively recognized for its role in governing the survival, proliferation, migration, and differentiation of hematopoietic progenitors, melanocytes, and germ cells. Recent investigations have indicated the expression of SCF in dermal and intestinal epithelial cells (35–37). Reports have indicated elevated SCF expression in the inflamed mucosa of individuals with IBD, including SCF248 (a 248 amino acid cleavable form) (38, 39).
Within the cascade of inflammatory events leading to the development of IBD, the involvement of inflammatory cytokines is intricate and potentially interactive. However, MR analysis can isolate their individual impacts and assess the relationship between IBD and these cytokines solely from a genetic standpoint. Nevertheless, our study faces limitations. Primarily, our findings stem from statistical analysis; further validation through extensive basic and clinical research is imperative. Additionally, while restricting the study to individuals of European descent diminishes population structure bias, it could constrain the generalizability of our findings to other populations.
5 Conclusion
The publicly available data information from the GWAS database was sourced and analyzed in this study to evaluate the causal relationship between IBD and ILs, and IBD and chemokines, by bidirectional MR analysis. Our results have shown that levels of IL-13 increase the risk of CD and UC, while MIF increase the risk of CD. CD and UC were suggestively correlated with an elevated level of SCF. The underlying mechanism behind these outcomes remains unclear, necessitating further investigation to substantiate our findings.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: https://doi.org/10.5523/bris.3g3i5smgghp0s2uvm1doflkx9x.
Author contributions
ZS: Writing – review & editing. XL: Writing – review & editing. JX: Writing – original draft. FH: Writing – original draft. NW: Writing – original draft. YH: Writing – review & editing. JY: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work is supported by the key Scientific Research Project Plan of Henan Province Colleges and Universities in Department of Education of Henan Province (Grant Number 23A320020).
Acknowledgments
The authors sincerely thank related investigators for sharing the GWAS summary statistics included in this study. We thank Yimeng Deng from the bottom of our heart for her contribution to manuscript checking and spelling revision.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1327879/full#supplementary-material
Abbreviations
MR, Mendelian randomization; IBD, Inflammatory bowel disease; CD, Crohn’s disease; UC, Ulcerative colitis; GWAS, Genome-wide association study; SNPs, Single Nucleotide Polymorphisms; IVs, Instrumental variables; IVW, Inverse variance weighted; IL13, Interleukin-13; MIF, Macrophage migration inhibitory factor; SCF, Stem cell factor.
References
1. Nakase H, Uchino M, Shinzaki S, Matsuura M, Matsuoka K, Kobayashi T, et al. Evidence-based clinical practice guidelines for inflammatory bowel disease 2020. J Gastroenterology. (2021) 56(6):489–526. doi: 10.1007/s00535-021-01784-1
2. Ananthakrishnan AN, Kaplan GG, Ng SC. Changing global epidemiology of inflammatory bowel diseases: sustaining health care delivery into the 21st century. Clin Gastroenterol Hepatology. (2020) 18(6):1252–60. doi: 10.1016/j.cgh.2020.01.028
3. Alatab S, Sepanlou SG, Ikuta K, Vahedi H, Bisignano C, Safiri S, et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatology. (2020) 5(1):17–30. doi: 10.1016/s2468-1253(19)30333-4
4. Armstrong H, Alipour M, Valcheva R, Bording-Jorgensen M, Jovel J, Zaidi D, et al. Host immunoglobulin G selectively identifies pathobionts in pediatric inflammatory bowel diseases. Microbiome (2019) 7(1):1. doi: 10.1186/s40168-018-0604-3
5. Zhang Y-Z. Inflammatory bowel disease: Pathogenesis. World J Gastroenterology. (2014) 20(1):91–9. doi: 10.3748/wjg.v20.i1.91
6. Sewell GW, Kaser A. Interleukin-23 in the pathogenesis of inflammatory bowel disease and implications for therapeutic intervention. J Crohn’s Colitis (2022) 16(Supplement_2):ii3–ii19. doi: 10.1093/ecco-jcc/jjac034
7. Velikova TV, Miteva L, Stanilov N, Spassova Z, Stanilova SA. Interleukin-6 compared to the other Th17/Treg related cytokines in inflammatory bowel disease and colorectal cancer. World J Gastroenterology. (2020) 26(16):1912–25. doi: 10.3748/wjg.v26.i16.1912
8. Kopper JJ, Iennarella-Servantez C, Jergens AE, Sahoo DK, Guillot E, Bourgois-Mochel A, et al. Harnessing the biology of canine intestinal organoids to heighten understanding of inflammatory bowel disease pathogenesis and accelerate drug discovery: A one health approach. Front Toxicol (2021) 3:773953. doi: 10.3389/ftox.2021.773953
9. Tan CXW, Brand HS, de Boer NKH, Forouzanfar T. Gastrointestinal diseases and their oro-dental manifestations: Part 2: Ulcerative colitis. Br Dental J (2017) 222(1):53–7. doi: 10.1038/sj.bdj.2017.37
10. de Vries SAG, Tan CXW, Bouma G, Forouzanfar T, Brand HS, de Boer NK. Salivary function and oral health problems in Crohn’s disease patients. Inflammatory Bowel Diseases. (2018) 24(6):1361–7. doi: 10.1093/ibd/izy017
11. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol (2014) 15(1):30–44. doi: 10.1038/nri3785
12. Grössner-Schreiber B, Fetter T, Hedderich J, Kocher T, Schreiber S, Jepsen S. Prevalence of dental caries and periodontal disease in patients with inflammatory bowel disease: a case–control study. J Clin Periodontology. (2006) 33(7):478–84. doi: 10.1111/j.1600-051X.2006.00942.x
13. Brito F, FCd B, Zaltman C, Pugas Carvalho AT, De Vasconcellos Carneiro AJ, Fischer RG, et al. Prevalence of periodontitis and DMFT index in patients with Crohn’s disease and ulcerative colitis. J Clin Periodontology. (2008) 35(6):555–60. doi: 10.1111/j.1600-051X.2008.01231.x
14. Mello JDC, MGomes LE, Silva JF, Siqueira NSN, Pascoal LB, Martinez CAR. The role of chemokines and adipokines as biomarkers of Crohn’s disease activity: a systematic review of the literature. Am J Transl Res (2021) 13(8):8561–74.
15. Wootton RE, Richmond RC, Stuijfzand BG, Lawn RB, Sallis HM, Taylor GMJ, et al. Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: a Mendelian randomisation study. psychol Med (2019) 50(14):2435–43. doi: 10.1017/s0033291719002678
16. Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol (2015) 30(7):543–52. doi: 10.1007/s10654-015-0011-z
17. Hartwig FP, Davies NM, Hemani G, Davey Smith G. Two-sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int J Epidemiol (2016) 45(6):1717–26. doi: 10.1093/ije/dyx028
18. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. Bmj (2018) 362:k601. doi: 10.1136/bmj.k601
19. Palmer TM, Lawlor DA, Harbord RM, Sheehan NA, Tobias JH, Timpson NJ, et al. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res (2011) 21(3):223–42. doi: 10.1177/0962280210394459
20. Fang GJ, Kong FZ, Zhang HQ, Huang B, Zhang JF, Zhang XL. Association between inflammatory bowel disease and interleukins, chemokines: a two-sample bidirectional mendelian randomization study. Front Immunol (2023) 14:1168188. doi: 10.3389/fimmu.2023.1168188
21. Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol (1996) 157(3):1261–70.
22. Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut (2003) 52(1):65–70. doi: 10.1136/gut.52.1.65
23. Daniele F, Francesco P. What is the role of cytokines and chemokines in IBD? Inflammatory Bowel Diseases. (2008) 14:S117–8. doi: 10.1002/ibd.20677
24. Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology (2005) 129(2):550–64. doi: 10.1016/j.gastro.2005.05.002
25. Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest (2004) 113(10):1490–7. doi: 10.1172/jci19836
26. Scharl M, Frei S, Pesch T, Kellermeier S, Arikkat J, Frei P, et al. Interleukin-13 and transforming growth factor β synergise in the pathogenesis of human intestinal fistulae. Gut (2013) 62(1):63–72. doi: 10.1136/gutjnl-2011-300498
27. Frei SM, Pesch T, Lang S, Weber A, Jehle E, Vavricka SR, et al. A role for tumor necrosis factor and bacterial antigens in the pathogenesis of Crohn’s disease–associated fistulae. Inflammatory Bowel Diseases. (2013) 19(13):2878–87. doi: 10.1097/01.Mib.0000435760.82705.23
28. Sun J, Frei SM, Hemsley C, Pesch T, Lang S, Weber A, et al. The role for dickkopf-homolog-1 in the pathogenesis of Crohn’s disease-associated fistulae. PloS One (2013) 8(11):e78882. doi: 10.1371/journal.pone.0078882
29. Magyari L, Kovesdi E, Sarlos P, Javorhazy A, Sumegi K, Melegh B. Interleukin and interleukin receptor gene polymorphisms in inflammatory bowel diseases susceptibility. World J Gastroenterology. (2014) 20(12):3208–22. doi: 10.3748/wjg.v20.i12.3208
30. David JR. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc Natl Acad Sci USA (1966) 56(1):72–7. doi: 10.1073/pnas.56.1.72
31. Lue H, Kleemann R, Calandra T, Roger T, Bernhagen J. Macrophage migration inhibitory factor (MIF): mechanisms of action and role in disease. Microbes Infect (2002) 4(4):449–60. doi: 10.1016/s1286-4579(02)01560-5
32. Murakami H, Akbar SM, Matsui H, Horiike N, Onji M. Macrophage migration inhibitory factor activates antigen-presenting dendritic cells and induces inflammatory cytokines in ulcerative colitis. Clin Exp Immunol (2002) 128:504–10. doi: 10.1046/j.1365-2249.2002.01838.x
33. Flanagan J, Leder P. The kit ligand: a cell surface molecule altered in steel mutant fibroblasts. Cell (1990) 63:185–94. doi: 10.1016/0092-8674(90)90299-t
34. Zsebo K, Williams D, Geissler E, Broudy V, Martin F, Atkins H, et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell (1990) 63(1):213–24. doi: 10.1016/0092-8674(90)90302-u
35. Carter EL, O’Herrin S, Woolery C, Jack Longley B. Epidermal stem cell factor augments the inflammatory response in irritant and allergic contact dermatitis. J Invest Dermatol (2008) 128(7):1861–3. doi: 10.1038/sj.jid.5701247
36. Longley BJ, Morganroth GS, Tyrrell L, Ding TG, Anderson DM, Williams DE, et al. Altered metabolism of mast-cell growth factor (c-kit ligand) in cutaneous mastocytosis. N Engl J Med (1993) 328(18):1302–7. doi: 10.1056/NEJM199305063281803
37. Schmitt M, Schewe M, Sacchetti A, Feijtel D, van de Geer WS, Teeuwssen M, et al. Paneth cells respond to inflammation and contribute to tissue regeneration by acquiring stem-like features through SCF/c-kit signaling. Cell Rep (2018) 24(9):2312–2328.e2317. doi: 10.1016/j.celrep.2018.07.085
38. Comar M, Secchiero P, De Lorenzo E, Martelossi S, Tommasini A, Zauli G. JCV+ Patients with Inflammatory Bowel Disease show elevated plasma levels of MIG and SCF. Inflammatory Bowel Diseases. (2012) 18(6):1194–6. doi: 10.1002/ibd.22953
Keywords: inflammatory cytokines, biomarkers, inflammatory bowel disease, Mendelian randomization, GWAS
Citation: Song Z, Li X, Xie J, Han F, Wang N, Hou Y and Yao J (2024) Associations of inflammatory cytokines with inflammatory bowel disease: a Mendelian randomization study. Front. Immunol. 14:1327879. doi: 10.3389/fimmu.2023.1327879
Received: 25 October 2023; Accepted: 27 December 2023;
Published: 15 January 2024.
Edited by:
Ashish Patel, Hemchandracharya North Gujarat University, IndiaReviewed by:
Shaikhul Islam, Bangladesh Agricultural Research Council, BangladeshDipak Kumar Sahoo, Iowa State University, United States
Jigna G. Tank, Saurashtra University, India
Copyright © 2024 Song, Li, Xie, Han, Wang, Hou and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianning Yao, Smlhbm5pbmd5YW9AMTYzLmNvbQ==