 James R. Barnacle
James R. Barnacle Angharad G. Davis1,3
Angharad G. Davis1,3
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 09 January 2024
Sec. Microbial Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1326651
This article is part of the Research TopicAdvances in the Management of Tuberculosis MeningitisView all 5 articles
Tuberculous meningitis (TBM), the most severe form of tuberculosis, causes death in approximately 25% cases despite antibiotic therapy, and half of survivors are left with neurological disability. Mortality and morbidity are contributed to by a dysregulated immune response, and adjunctive host-directed therapies are required to modulate this response and improve outcomes. Developing such therapies relies on improved understanding of the host immune response to TBM. The historical challenges in TBM research of limited in vivo and in vitro models have been partially overcome by recent developments in proteomics, transcriptomics, and metabolomics, and the use of these technologies in nested substudies of large clinical trials. We review the current understanding of the human immune response in TBM. We begin with M. tuberculosis entry into the central nervous system (CNS), microglial infection and blood-brain and other CNS barrier dysfunction. We then outline the innate response, including the early cytokine response, role of canonical and non-canonical inflammasomes, eicosanoids and specialised pro-resolving mediators. Next, we review the adaptive response including T cells, microRNAs and B cells, followed by the role of the glutamate-GABA neurotransmitter cycle and the tryptophan pathway. We discuss host genetic immune factors, differences between adults and children, paradoxical reaction, and the impact of HIV-1 co-infection including immune reconstitution inflammatory syndrome. Promising immunomodulatory therapies, research gaps, ongoing challenges and future paths are discussed.
Tuberculous meningitis (TBM), the most severe form of tuberculosis, carries a mortality of over a quarter despite antibiotic therapy, and over half of survivors are left with neurological disability (1). In HIV-1 coinfection, mortality rates can approach 50% (2). In tuberculin-sensitised animals, death has been shown to occur after meningeal inoculation of dead bacilli (3) and it is recognised that a dysregulated and excessive host immune response contributes significantly to poor outcome. Host-directed therapies (HDT) are a promising tool to modulate this immune response and improve outcomes. Over the past 20 years the only evidence based adjunctive HDT in TBM has been corticosteroids (4). However, corticosteroids remain a blunt and poorly understood immunomodulator in TBM. Recently, studies have shown that corticosteroids may not reduce mortality or neurologic outcomes in people living with HIV (PLWH), and amongst HIV-uninfected patients, not all patients may benefit depending on their genotype (5, 6).
There is therefore an acute need for novel HDT and a comprehensive understanding of the host immune response to TBM is essential in developing new therapies or re-purposing existing ones. Challenges with accessing the site of disease and a lack of in vivo and in vitro models have been partially offset by recent developments in multiomics technology, allowing large amounts of data to be generated from limited clinical samples. This review examines recent advances in our understanding of human host immune responses in TBM beginning with Mycobacterium tuberculosis (M. tuberculosis) entry into the CNS, then moving to CNS barrier dysfunction, the innate and adaptive responses, and metabolomic and genetic aspects. It draws from the wider literature on tuberculosis and neurological infection to identify gaps and opportunities in TBM research. Promising HDT, future challenges and a path forward are discussed.
To understand the immunopathogenesis of TBM, physiological CNS immunity must be understood. The only immune cells found in the brain parenchyma in health are microglia, mast cells and macrophages (7). This immune privilege is maintained by the blood-brain barrier (BBB) and the brain-cerebrospinal fluid (CSF) barrier (BCSF) (Figure 1). The BBB is a collection of structural, transport and metabolic barriers maintained by endothelial cells bound together with tight junctions and adherens junctions lining the blood vessels, which are enveloped by pericytes and astrocytes (8). These endothelial cells have low rates of transcytosis, low numbers of leukocyte adhesion molecules and specific efflux transporters (9).
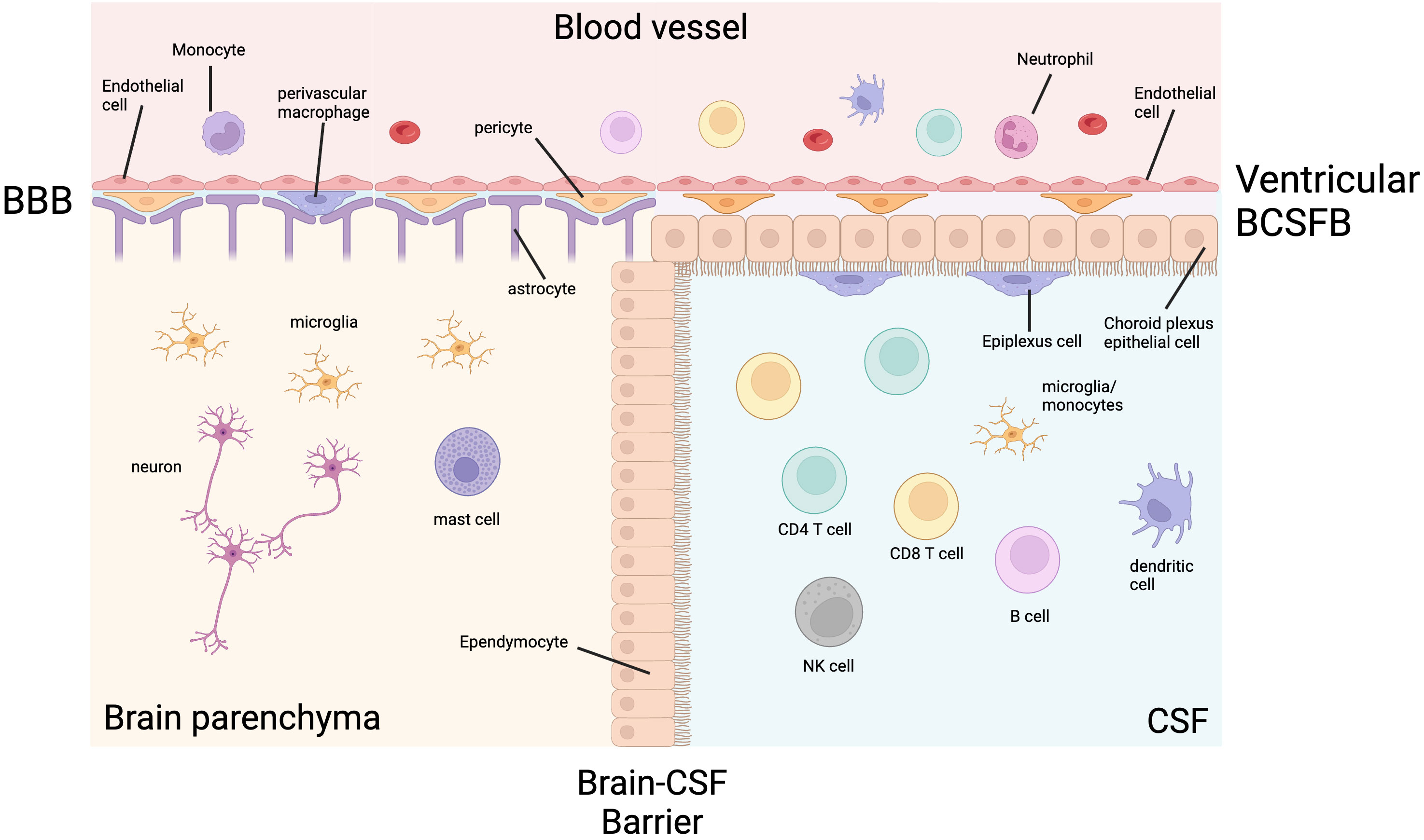
Figure 1 The ventricular CNS barriers in health. Cerebrospinal fluid (CSF) and the brain parenchyma are separated from the systemic blood circulation by the blood-brain barrier (BBB) and blood-CSF barrier (BCSFB). The BBB is made up of a luminal endothelial cell layer, a basement membrane, a perivascular space containing pericytes and perivascular macrophages, and layer of astrocytic foot processes. The ventricular BCSFB is made up of an endothelial layer, a stromal layer containing pericytes, choroid plexus epithelial cells, and epiplexus cells (macrophages). The brain parenchymal immune cells are limited to microglia, macrophages and mast cells. The CSF is less privileged and contains lymphocytes, as well as lower numbers of other immune cells. Created with BioRender.com.
CSF is less immune privileged than the brain parenchyma and has a more diverse immune cell composition physiologically, but it is still protected from the systemic circulation. In the ventricles, the BCSFB is formed by tight junctions between epithelial cells of the choroid plexus. Epiplexus macrophages, also known as Kolmer cells, adhere to the CSF side of the epithelial layer and have phagocytic and antigen-presenting properties. In the meningeal and cisternal subarachnoid space, blood vessels have a similar structure to the BBB, except they lack the surrounding pericytes and astrocytes. The subarachnoid space is separated from the dura by arachnoid barrier cells containing tight junctions forming a barrier between the fenestrated vasculature of the dura and the CNS (10, 11). Despite these robust defences, M. tuberculosis can enter the CNS and cause TBM, spinal arachnoiditis and tuberculomas.
TBM follows systemic infection with M. tuberculosis. After inhalation of aerosolised M. tuberculosis and infection of alveolar macrophages, a cytokine response induces migration of dendritic cells to local lymph nodes. Dendritic cells (DC) present antigen and stimulate T-helper 1 (Th1) cell differentiation. Alongside cytotoxic and regulatory functions, these differentiated Th1 cells release TNF, IFNγ and other cytokines at the site of infection which activate macrophages to contain M. tuberculosis and form granulomas.
In those with an inadequate primary immune response, or later immune compromise, control of M. tuberculosis in the lungs is lost and pulmonary or systemic spread may occur. The exact mechanism of M. tuberculosis infiltration into the CNS remains uncertain. It is thought that bacilli cross the BBB by transcytosis, paracytosis or within phagocytic immune cells (‘Trojan Horse’) following haematogenous spread (Figure 2). An in vitro study showed that M. tuberculosis was able to cross a model BBB significantly more efficiently than non-pathogenic M. smegmatis via actin rearrangement of EC (12). A murine model found that CNS invasion was attenuated in five M. tuberculosis mutants, including Rv0986, which forms a transporter thought to be involved in cell adhesion and entry, and Rv0931c which encodes a serine-threonine protein kinase thought to help M. tuberculosis adapt to unfavourable environments, such as the CNS (13). In a zebrafish model, M. marinum was able to cross the BBB within infected phagocytes, as well as via ESX-1-dependent invasion of EC (14). These studies suggest that M. tuberculosis may use a combination of methods and virulence factors to pass through the BBB and survive in the CNS environment.
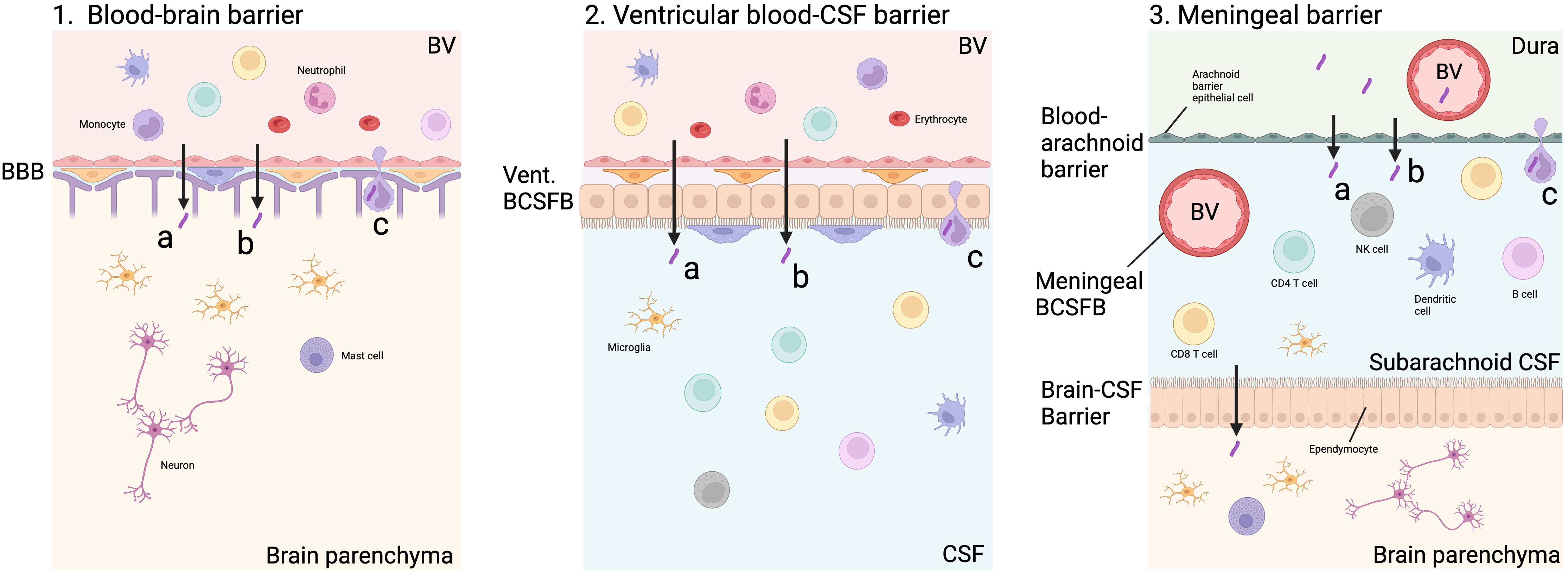
Figure 2 Possible routes of M. tuberculosis entry into CNS. M. tuberculosis may enter the brain parenchyma directly via capillary blood flow through the blood-brain barrier (BBB, Figure 2.1). This may be via paracytosis (A), transcytosis (B) or within an infected cell (C). M. tuberculosis may also pass through the blood-CSF barrier lining the ventricles, which contains an epithelial choroid plexus layer along with an endothelial layer through the same mechanisms (Figure 2.2). In the meninges, the blood-arachnoid barrier separates the unfiltered interstitial fluid and blood from the dura from the sub-arachnoid space within the CNS via an epithelial layer of arachnoid barrier cells. The arachnoid barrier cells are poorly vascular, and exchange is likely to be minimal. In addition, blood vessels (BV) passing through the subarachnoid space represent another blood-CSF barrier which does not contain a choroid plexus layer. M. tuberculosis in the subarachnoid space may be able to pass across the brain-CSF barrier formed by an ependymal layer into the brain parenchyma (Figure 2.3). Created with BioRender.com.
Following infiltration into the CNS, a leptomeningeal or cortical focus known as a ‘Rich focus’ is established. Rich and McCordock were sceptical of the link between the ‘Rich focus’ and miliary TB citing examples of TBM without miliary spread and cortical lesions that preceded miliary spread in post-mortems (15), however, it is now widely believed that the primary route of CNS infiltration is haematogenous, although bacillaemia may not always be recognised (16). The granulomatous lesion is likely to progress through a non-necrotising phase characterised by activated epithelioid macrophages, a necrotising gummatous phase with central necrosis and reticulin fibres, and finally a necrotising abscess with leukocytoclastic neutrophils which ruptures into the subarachnoid space (17). Unlike the brain parenchyma, subarachnoid blood vessels are not supported by astrocytic foot processes and are therefore more permeable to bacilli and immune cells, potentially explaining why TBM is most often localised to the leptomeninges and subpial cortex; deep parenchymal granulomas are rarely seen (17).
There is a lack of research and understanding about other CNS barriers that may be entry points for M. tuberculosis and immune cells. The initial point of M. tuberculosis entry may be across the ventricular BCSFB, meningeal BCSFB or arachnoid barrier, rather than the BBB (Figure 2) (18). Classically, the BCSFB is described as having a choroid plexus epithelial cell layer which is found predominantly, if not exclusively, lining the ventricles. However, in the meninges, blood vessels pass through the sub-arachnoid CSF creating another BCSFB (19). Another meningeal barrier is that of the epithelial arachnoid barrier cells. Compared to other layers, this layer is thought to play a minimal role in exchange because of its avascular nature and lower surface area (18). After the rupture of the initial granuloma, the leukocyte infiltration into the CSF seen in TBM is unlikely to come via the brain parenchyma following BBB infiltration, but rather across the BCSFB. These meningeal and cisternal barriers are poorly understood and have not been modelled in TBM. The factors influencing the permeability of these barriers may play an important part in immunopathology and should be investigated.
Microglia are the principal cells infected by M. tuberculosis (20). Direct infection of neurons and astrocytes may occur to a lesser extent (21). Given the established roles of both these latter cell types in the immune axis, this is likely to influence the host immune response in TBM. Microglia recognise M. tuberculosis via pattern recognition receptors (PRR) such as toll-like receptors (TLR). Internalisation of M. tuberculosis may be dependent on CD14, a monocyte differentiation antigen, demonstrated by a study which showed anti-CD14 monoclonal antibodies and soluble CD14 ligand reduced the uptake of non-opsonised bacilli by foetal microglia by 64% and 62% respectively (22), but this was not reproduced in human monocytes, monocyte-derived macrophages or alveolar macrophages. CD14 expression on these cells however increases in M. tuberculosis infection (23).
Activated microglia secrete cytokines which mediate immune cell recruitment and play an essential role in host defence but may also contribute to excess inflammation. Microglia infected with M. tuberculosis have been shown to secrete TNF, C-C motif chemokine ligand 2 (CCL2), CCL5, C-X-C motif chemokine ligand 10 (CXCL10), IL-1α, IL-10, IL-12p40, granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) in vitro, demonstrating the role of microglia in escalating the initial immune response through stimulation, activation and chemoattraction of other leukocytes (21, 24). The IFNγ-inducible chemokines CXCL9 and CXCL10 are secreted by microglia and macrophages, as well as astrocytes in the case of CXCL10, and are important in T cell and NK cell recruitment. CSF CXCL9 and CXCL10 levels are significantly increased in TBM compared to other neurological infections and decrease with treatment (25). The chemokines CCL2 and CCL5 recruit leukocytes to the CNS which may contribute to immunopathology, but CCL2 has also been shown to reduce neuronal death following N-methyl-D-aspartate (NMDA) challenge, reduce glutamate release from astrocytes, and protect neurons from oxygen-glucose deprivation in vitro (26, 27). The balance of these chemokines, and their subsequent activation of microglia may be important (20).
An influx of peripheral immune cells escalates the CNS host response to M. tuberculosis. Infiltration into the CNS is aided by increased BBB permeability in TBM. Several mediators of BBB dysfunction are implicated including matrix metalloproteases (MMP), vascular endothelial growth factor (VEGF), caspases, and cytokines (IFNγ, IL-1β, TNF, IL-8, IL-6), but the mechanisms of dysfunction are not fully understood (28).
MMP are zinc-containing proteases which degrade extracellular matrix (ECM), including the BBB and are inhibited by tissue inhibitors of metalloproteinases (TIMP). They are critical in tissue homeostasis and development. MMP activate immune mediators and promote leukocyte migration by modulating cytokine and chemokine activity. They are likely to exert a pathological positive feedback role by aiding the influx of neutrophils, monocytes and other leukocytes that promote BBB dysfunction and chemoattraction. This influx may lead to further MMP production, particularly from neutrophils, further driving dysfunction.
MMP-2, also known as gelatinase-A, is the only MMP present in significant amounts in the physiological CNS. In vitro models suggest that astrocytes and microglia secrete MMP-1, 3 and 9 in response to interactions with M. tuberculosis-infected monocytes, but not upon direct M. tuberculosis infection. This MMP expression is driven by cytokines including TNF and IL-1β, although in the case of MMP-2, TNF actually inhibits microglial secretion (29). Inhibition of MMP-3 and 9 results in suppression of inducible nitric oxide synthase (iNOS), IL-1β, IL-1Ra, TNF and IL-6 gene expression in microglia showing that MMP have pro-inflammatory effects on microglia (30).
MMP-9 is a collagenase secreted by astrocytes, microglia and neutrophils which breaks down type IV collagen and downregulates tight junction proteins ZO-1, claudin-5 and occludin between EC in the BBB (31). It is usually undetectable in the normal brain. CSF MMP-9 levels are associated with severity, neurological complications, and brain tissue damage in TBM (32). In addition, compared with aseptic meningitis and non-inflammatory controls, MMP-2 and 9 are raised, and levels remained higher in those TBM patients who developed late neurological complications (33). Adjunctive dexamethasone decreases CSF MMP-9 concentrations but this decline is not associated with improved outcome (34). PLWH who develop TBM-IRIS have higher blood concentrations of MMP-1, 3, 7 and 10 at the time of TBM diagnosis and ART commencement which rise even further at the point of developing TBM-IRIS symptoms (35). However, one study showed no correlation between CSF MMP-9 and outcome in HIV-uninfected TBM patients (36). In a paediatric TBM population, patients with increased CSF MMP-9 levels were twice as likely to have a better outcome (37). Myeloperoxidase, which is mostly expressed in neutrophils, may also contribute to BBB dysregulation by activating MMP-9 (38). Finally, there may be a role for MMP in recovery and remodelling depending on the stage of disease, especially in children (39).
VEGF-A is a potent endothelial growth factor involved in angiogenesis and endothelial permeability. It is a useful biomarker of disease in TBM and is thought to cause BBB dysfunction through its effects on the ECM and tight junctions. Plasma and CSF VEGF-A concentrations are significantly higher in TBM patients than controls although not associated with outcome (40). In a study looking for a CSF biosignature for TBM using proteomics in HIV-uninfected children, a combination of VEGF-A, IFNγ and myeloperoxidase had a sensitivity and specificity of 91.3% and 100% respectively compared to a heterogeneous non-TBM group, suggesting VEGF-A as an important contributor in TBM pathogenesis (41). Studies have also shown that VEGF-A increases MMP-9 activation (42).
Adhesion molecules intercellular adhesion molecule 1 (ICAM-1), and vascular cell adhesion molecule 1 (VCAM-1), P-selectin and E-selectin also play a role in BBB dysregulation and leukocyte transmigration and supernatant concentrations increased 25-, 54-, 44-, and 53-fold respectively in an M. tuberculosis-stimulated BBB in vitro model compared to controls in association with increased leukocyte and neutrophil transmigration (31). Manyelo et al. also reported a second biomarker combination which performed equally well in discriminating TBM and non-TBM, which included soluble ICAM-1, myeloperoxidase, CXCL8 (IL-8), and IFNγ (41), suggesting a key role for ICAM-1 in TBM pathology.
Whilst pericytes are known to be critical to the integrity of the BBB, little is understood about their role in BBB dysfunction in TBM. Pericytes cover up to 70% of the CNS surface of the BBB. A recent study used a human cerebral endothelial barrier model to show that paracrine cytokine signalling from monocyte-derived macrophages (MDM) acted on pericytes to enhance neutrophil transmigration via production of a repertoire of neutrophil chemokines (43), suggesting pericytes may be important in neutrophil CNS infiltration in TBM. They used MDM to model perivascular macrophages embedded in the BBB. The authors propose inhibition of atypical chemokine receptor 1 (ACKR1), which transcytoses C-C and C-X-C chemokines across the endothelium, to block the effect of pericyte-derived chemokines on neutrophils, rather than targeting the broad range of individual chemokines. The contribution of pericyte signalling to immunopathology in TBM is unknown, but such therapies may be a focused tool to control neutrophil influx.
Other CNS barriers including the ventricular and meningeal BCSFB, and the arachnoid barrier, have not been investigated in TBM, but may be important in pathology. In fact, one study which infected mice with pulmonary M. tuberculosis and examined their choroid plexus and BBB using electron microscopy at post-mortem found disruption of the BCSFB, but not the BBB (44).
The inflammatory response to lipopolysaccharide (LPS) and several bacteria, but not M. tuberculosis, has been studied using gene expression analysis in in vivo and in vitro models of the choroid plexus. LPS exposure is associated with increased expression of adhesion molecules, chemokines, and inflammatory miRNAs (19). Streptococcus suis disrupts the integrity of epithelial choroid plexus cells by downregulating tight junction proteins (TJP), increases adhesion molecule expression and demonstrates transepithelial migration within polymorphonuclear leukocytes (19, 45). Whether M. tuberculosis has a similar effect on the BCFSB remains to be explored.
In conclusion, signalling networks between infected microglia, astrocytes, endothelial cells, perivascular macrophages and pericytes lead to the release of cytokines, VEGF-A, MMP, and increased expression of surface adhesion molecules, leading to BBB dysfunction, chemotaxis and an influx of leukocytes into the CNS. These cellular interactions in TBM are poorly understood, as is the balance of their inflammatory mediators between pathology and protection and warrants further investigation. In addition, similar mechanisms of dysfunction have been seen at the choroid plexus in other bacteria but have not been investigated in M. tuberculosis. The roles of the meningeal BCSFB and arachnoid barrier in TBM immunopathology are unknown.
The role of the inflammasome has become increasingly studied in TBM and other forms of tuberculosis in recent years. Microglial, macrophage and DC TLR recognition of M. tuberculosis triggers the inflammasome pathway leading to pyroptosis and the activation of pro-inflammatory IL-1β and IL-18. Inflammasomes are divided into canonical and non-canonical. The canonical inflammasomes involve the effector caspase 1 and include NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3), AIM2 and NLRC4. NLRP3 is triggered by various damage-associated molecular patterns (DAMP), such as as adenosine triphosphate (ATP) from dying cells, and pathogen-associated molecular patterns (PAMP) including those found on M. tuberculosis, whereas AIM2 and NLRC4 are triggered by intracellular nucleic acids and Gram-negative secretion systems respectively (46).
The non-canonical inflammasome was first described in 2011 and is characterised by a caspase 4/5-dependent pathway in humans, and caspase 11 in mice. It was initially thought to be triggered by LPS only, which is not found in M. tuberculosis, but oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC), a DAMP released by dying cells, and the cyclic GMP-AMP synthase/stimulator of IFN genes (cGAS/STING) pathway, which recognises intracellular bacteria have now also been implicated (47). The different roles of caspases 4 and 5 remain poorly understood.
In TBM, a study comparing blood bulk RNA-Seq in paediatric TBM patients with healthy controls found significant upregulation of transcripts associated with PRR antigen recognition, inflammasome activation and IL-1 signalling (32). Another study compared adult HIV-associated TBM patients with and without immune reconstitution inflammatory syndrome (IRIS) and found enrichment of transcripts associated with canonical and non-canonical inflammasome activation in those with IRIS following anti-tubercular treatment (ATT) as well as significantly higher caspase 1 concentrations in the CSF (48).
Whilst inflammasome activation is associated with IRIS, it is likely that the inflammasome plays a protective as well as a detrimental role in TBM. Programmed cell death (PCD) kills M. tuberculosis along with the host cell, and M. tuberculosis inhibits NLRP3 inflammasome activation via PknF and Hip1, and the AIM2 inflammasome via an ESX-dependent mechanism in an effort to favour necrosis (49). However, lymphocyte-derived IFNγ inhibits the NLRP3 inflammasome in mice mediated by nitric oxide (NO) implying NO produced by the adaptive immunity modulates the innate inflammatory response (50).
There is conflicting evidence as to whether IL-1β plays a protective or detrimental role in TBM and a fine balance may be required. IL-1β is essential for the immune response against TB. IL-1 has been shown to confer host resistance by limiting excess type 1 IFN production via the induction of eicosanoids and helps regulate the containment and replication of bacilli (51). In an Indonesian HIV-uninfected TBM study, CSF IL-1β was elevated 44-fold compared to controls but did not predict mortality, nor did levels of IL-6, a downstream target of IL-1β. Interestingly, IL-1Ra, the receptor antagonist of IL-1, was increased 26-fold in TBM and closely correlated with IL-1β levels but did not predict mortality either. In addition, genetic loci known to regulate IL1B expression do not associate with mortality from TBM (52).
IL-1β production is not synonymous with canonical inflammasome activation; neutrophil-associated proteases including MMP-2, 3, and 9 can also activate IL-1β and IL-18 independently of the inflammasome (49). Another recently discovered microglial source of IL-1β is the non-canonical caspase-8-dependent inflammasome. Caspase 8 has been found to be important for IL-1β production in microglia (53). The importance of these non-inflammasome sources of IL-1β in TBM are unclear.
There is a clear role for neutrophils in the inflammatory response in TBM. Neutrophils are not found in the brain or CSF in healthy subjects and so neutrophil chemotaxis, infiltration, and activation may play a critical role in the dysregulated immune response in TBM. Once neutrophils have been recruited to the CNS, they phagocytose M. tuberculosis. Virulent M. tuberculosis initiates necrosis, and the proteases released can damage neighbouring cells and tissues. Macrophages phagocytose necrotic neutrophils and M. tuberculosis aiding further M. tuberculosis distribution and inflammation (54).
Higher pre-treatment CSF neutrophil counts associate with an increased likelihood of culturing M. tuberculosis and neurological events (55), but their association with mortality is variable. In Indonesia, higher CSF and blood neutrophil counts at diagnosis associated with mortality in TBM (56). Significantly increased whole blood transcripts and CSF concentrations of neutrophil-associated proteins including MMP, cathepsin G, lipocalin 2 and human neutrophil peptides 1-3 (α-defensins) are found in TBM-IRIS (48). S100A8, a neutrophil chemotactic protein, is differentially expressed in TBM compared to other forms of meningitis (57), and increased CSF levels of S100A8/9 accompany TBM-IRIS (35). Variations in the LTA4H genotype have been shown to predict outcomes in TBM. Leukotriene A4 hydrolase (LTA4H) converts LTA4 to LTB4, a potent neutrophil chemoattractant and pro-inflammatory eicosanoid. Patients with the pro-inflammatory TT genotype of LTA4H have higher CSF concentrations of IL-1β, IL-2 and IL-6 and benefit more from adjunctive corticosteroid therapy (5). A clinical trial stratifying corticosteroid treatment by LTA4H genotype in HIV-uninfected TBM patients is ongoing in Vietnam (NCT03100786).
As well as degranulation and phagocytosis, neutrophils activated by M. tuberculosis release neutrophil-extracellular traps (NETs), which consist of modified chromatin containing bactericidal proteins such as myeloperoxidase and neutrophil elastase designed to capture and kill bacteria. This release is part of NETosis, a regulated form of cell death, although a ‘vital’ NETosis has now been described allowing cell survival (58). NETs from M. tuberculosis-activated neutrophils triggered significant secretion of IL-6, TNF, IL-1β, and IL-10 by macrophages in vitro (59). NETs activate platelets and the complement pathway and may promote thrombosis in brain microvessels during TBM. More research is needed to understand the role of NETosis in TBM.
In summary, whilst not found in the healthy CNS, neutrophils play a central role in TBM immunopathology with neutrophil-associated proteins and transcripts characterising more severe phenotypes. Further research is required to understand the exact mechanism of this damage. Studies are limited by neutrophil loss ex vivo between the bedside and laboratory which can lead to bias in transcriptomic studies. In addition, the role of NETosis in inflammation and thrombosis in TBM deserves further attention.
As discussed above, the activation of microglia and the influx of leukocytes into the CNS in TBM results in cytokine release (Figure 3). Cytokines and their expression in cells can be easily measured in blood and CSF and have been well studied in TBM but for many cytokines, it is clear that labelling them protective or pathogenic is simplistic and they can play both roles depending on the cell, the disease course, the patient and the pathogen. In contrast to the hypothesis that death in TBM is due to hyperinflammation, in HIV-uninfected adults in Vietnam, patients who died had an attenuated inflammatory response with lower CSF cytokine concentrations (55). A systematic review of cytokine levels in lumbar CSF in TBM found that IFNγ, IL-13, and sIL-2R were increased in TBM compared with other causes of bacterial meningitis, whereas TNF, IL-1β, IL-1Ra, IL-8, IFNγ, sIL-2R, IL-13, and IL-17 were increased compared to viral or aseptic meningitis (60). Kwon et al. compared 10 pre-treatment definite and probable TBM patients with 45 patients with other neurological infections and found that CSF IL-12p40, IL-13, MIP-1α, sPD-1, and PD-L1 were significantly increased in TBM (61). sVCAM1, MMP-1, sRAGE, CXCL10 are higher in TBM patients with stroke compared with those without stroke, and other forms of meningitis in children (62).
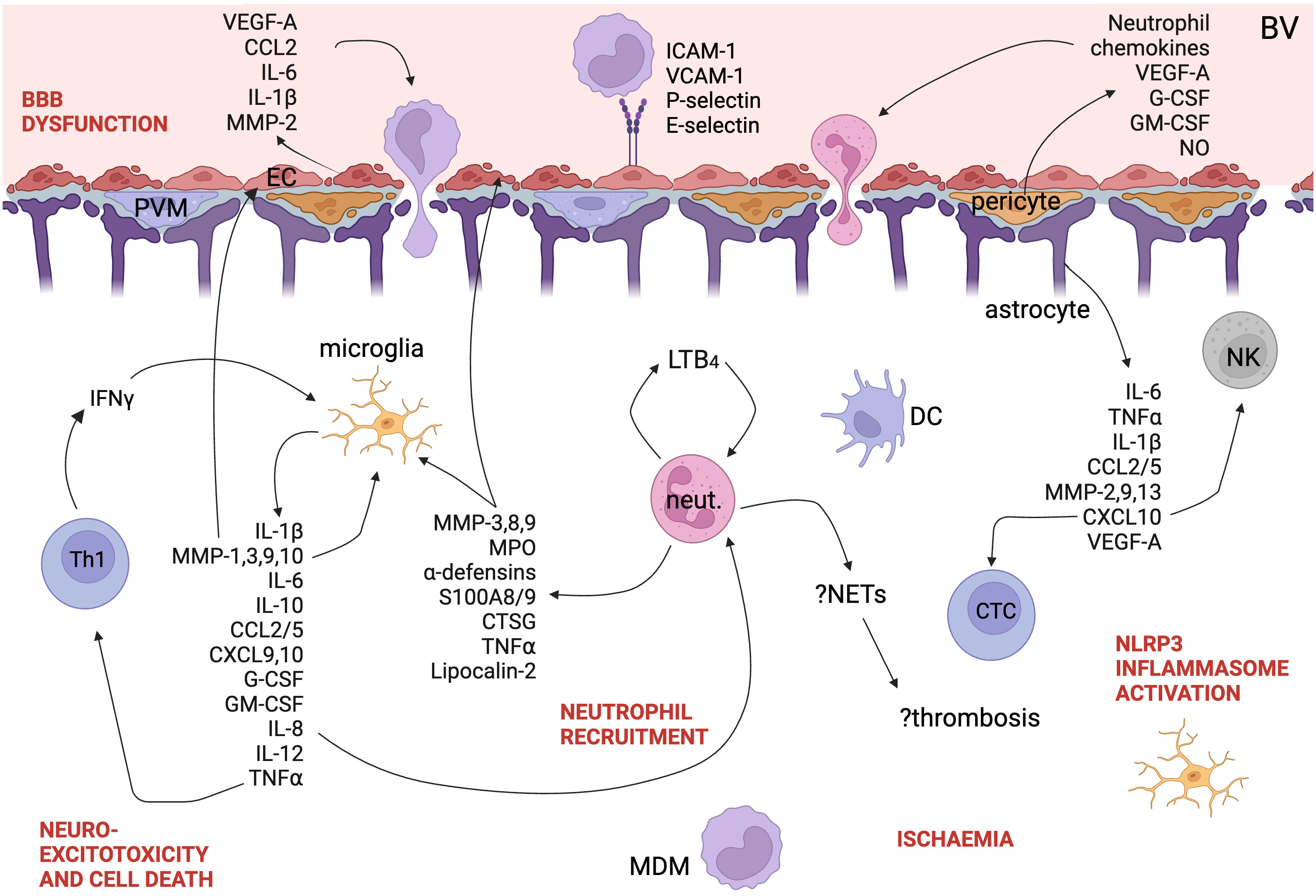
Figure 3 A schematic of the immune landscape of TBM. Cytokines, chemokines, and other proteins implicated in the pathogenesis of TBM are shown. Polymorphisms down- and up-regulating LT4B pathway are both associated with worse outcomes in TBM. Created with BioRender.com. EC, endothelial cell; DC, dendritic cell; MDM, monocyte-derived macrophage; Th1, Th1-helper cell; NETs, neutrophil extracellular traps; neut., neutrophil; LT4B, leukotriene 4B; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; PVM, perivascular macrophage; BBB, blood-brain barrier; BV, blood vessel; NK, natural killer cell; NETs, neutrophil extracellular traps; CTL, cytotoxic T lymphocyte; VEGF, vascular endothelial growth factor; CCL, C-C motif chemokine ligand; IL, interleukin; MMP, matrix metalloprotease; ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule; LTB4, leukotriene B4; IFN, interferon; CXCL, C-X-C motif chemokine ligand; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; TNF, tumour necrosis factor; CTSG, cathepsin G; MPO, myeloperoxidase; NLRP3, NACHT, LRR, and PYD domains-containing protein 3; NO, nitric oxide.
TNF is synthesised by several cells found within the inflamed CNS including microglia, macrophages, neutrophils, dendritic cells, lymphocytes, astrocytes and neurons. It has a protective role in the immune response to tuberculosis including granuloma formation and maintenance. TNF-/- mice are highly susceptible to TB dissemination, including to the brain, and early mortality (63). However, TNF is undoubtedly critical in the pathogenesis of TBM. TNF increases the permeability of the BBB, triggers further cytokine release, and correlates with severity in mouse models (64). It is found at high levels at the interface between the cellular and necrotic zones of TB granulomas in post-mortem studies (17). High CSF TNF levels with low IFNγ levels predict TBM-IRIS. Thalidomide, a TNF antagonist, downregulated TNF production and increased survival in a rabbit model of TBM (64). Optimal levels of TNF are likely to be determined by disease course and individual patient factors in TBM.
IFNγ is produced by NK cells, and CD8+ and CD4+ T cells, including γδ cells, and is essential to activate microglia and other macrophages to induce intracellular mycobacterial killing. IFNγ inhibits neutrophil accumulation and survival in M. tuberculosis-infected lung both directly and via inhibition of CD4+ T cell production of IL-17, which regulates neutrophil recruitment via S100A8/9 production (65, 66). The effect of IFNγ on neutrophil CNS recruitment alongside its macrophage and microglial activity may help explain why low CSF IFNγ concentrations independently predict death in PLWH with TBM (5, 67).
Other important cytokines in TBM include IL-6, IL-12, and IL-10. IL-6 is associated with a more severe presentation of TBM (67). Mice lacking the larger IL-12 subunit, p40, are more susceptible to mycobacterial infections, and inherited IL-12p40 deficiency in humans is associated with an increased risk of TB and failure of PBMC to produce IFNγ. IL-12 secretion by microglia, DC, neutrophils and CNS macrophages induces T helper 1 cell (Th1) differentiation of naïve T cells and stimulates IFNγ and TNF production by NK and T cells. IL-10 is produced by microglia upon TLR stimulation as well as neurons and has anti-inflammatory activity. IL-13 is produced by T cells and also has anti-inflammatory properties. Despite being significantly raised in TBM compared with bacterial, viral and aseptic meningitis (60), there is no literature investigating the role of IL-13 in TBM. Lower levels in bacterial meningitis could be explained by a neutrophil predominance. Given that IL-13 inhibits macrophage inflammatory cytokines and Th1 cells, an imbalance may contribute towards immunopathology in TBM.
The role of specialised pro-resolving mediators (SPM) and pro-inflammatory eicosanoids has been investigated in TBM (68). SPM are a recently discovered genus of lipid mediators which terminate inflammation and promote tissue repair and include lipoxins, resolvins, protectins and maresins. They can counter regulate the production of pro-inflammatory cytokines and eicosanoids, control leukocyte trafficking and phenotype and regulate the phagocytosis and killing of bacteria. Using metabolomics, Colas et al. found that pre-treatment concentrations of SPM and increased levels of pro-inflammatory eicosanoids including prostaglandins and cysteinyl leukotrienes in lumbar CSF significantly associated with mortality and higher British medical research council (BMRC) grades. Fatty acid substrates of SPM and enzyme activity were not reduced in more severe cases, and CSF white cell counts were similar implying that reduced SPM concentrations were due to differences in cell phenotype or population rather than absolute leukocyte numbers (68). It is known that different macrophages have distinct lipid mediator profiles (69) and further understanding of the cell populations responsible for the balance of SPM and eicosanoids may provide opportunities for novel HDT in TBM. Aspirin plays a role in the balance of these mediators in TBM; Mai et al. showed dose-dependent inhibition of thromboxane B2, the inactive metabolite of the rapidly degraded pro-inflammatory eicosanoid thromboxane A2, and upregulation of protectins in lumbar CSF in a study comparing 81mg and 1000mg aspirin daily with placebo. Thromboxane B2 levels were significantly lower in lumbar CSF at day 30 in the aspirin, 1000mg group compared to placebo (70). Phase 3 clinical trials investigating aspirin in TBM are ongoing.
The role of the adaptive immune response against TBM is well documented. CD137L expression by resident microglia activates infiltrating T cells. In addition, infiltrating neutrophils promote the activation of CD4+ T cells via DC. Whilst certain T cell phenotypes may contribute to excess inflammation, their positive role in reducing morbidity and mortality from TBM is clearer than other leukocytes such as neutrophils. TBM is characterised by a T cell-dominant lymphocytic CSF. Th1 cells are essential to activate macrophages and microglia via IFNγ, and a CSF IFNγ-release assay (IGRA) response is seen in TBM (71). In an Indonesian cohort of 160 HIV-uninfected TBM patients, there were reduced αβ cells, γδ cells, NK cells and MAIT cells in blood, but increased neutrophils and classical monocytes compared with pulmonary TB (PTB) and healthy controls. The T cell make-up of the CSF was 57% αβ, 13% NK, 1.5% γδ, 0.4% MAIT, 0.06% NKT revealing that important T cell subsets are found in the CSF of TBM and may play an unknown role in the host immune response. Higher levels of CSF αβ cells and NK cells were associated with better outcomes in TBM (72). Effector CD8+ and Vγ9/Vδ2 T cells have also been implicated in TBM (73, 74).
Although a robust T cell response is important, so is the nature of the response. Strongyloides stercoralis co-infection is significantly associated with reduced pro-inflammatory cytokine concentrations in the CSF before treatment and reduced neurological complications at three months in adult TBM. The Th2 immune response induced by Strongyloides stercoralis may non-specifically inhibit the pro-inflammatory Th1 response and protect the host from immunopathology (75).
Impaired T cell function is a general feature of TB which is also seen in TBM (76). A study examining blood bulk RNA transcripts of HIV-uninfected children with TBM found that T cell development and function were downregulated, along with a reduced in vitro T cell response to non-specific mitogen activation, which resolved with treatment (77). In addition, children with TBM have downregulated T cell activation and signalling in whole blood transcripts compared with healthy controls, but not in ventricular CSF versus other neurological infections (32). In a Korean study, soluble programmed death protein 1 (sPD-1) and programmed death ligand 1 (PD-L1) were increased in lumbar CSF in probable or definite TBM compared with other inflammatory or infectious neurological conditions (61). PD-1 is expressed on the cell surface and limits the activation and function of T cells. PD-L1 blocking increases CD8 T cell cytotoxicity against M. tuberculosis-infected macrophages, especially M1-polarised (78). However, anti-PD-1 monoclonal antibodies such as pembrolizumab also predispose to TB reactivation (79), and efforts to resolve impaired T cell function with PD-1/PD-L1 blockage should be approached with caution.
MicroRNAs (miRNAs) are highly conserved non-coding RNAs involved in the regulation of gene expression. The miR-29 family regulates the adaptive immune response, including T cell differentiation (80), and suppress IFNγ production by directly targeting IFNγ mRNA. In a mouse model with transgenic expression of a target which competes with endogenous miR-29 targets thereby reducing the effect of miR-29 expression, a greater IFNγ and Th1 response and lower Listeria monocytogenes burden were seen in infected mice suggesting miR-29 suppresses the immune response to intracellular bacteria. One study found that mi-R29a levels were increased in PBMC and lumbar CSF of children with TBM compared with healthy controls and were associated with markers of severity (81). Increased levels of mi-R29a may impair the adaptive response to TBM and mi-R29 could be a promising therapeutic target.
Overall, T cells are essential for a robust host response to TBM, and impaired T cell function may contribute to worse outcomes. However, characterising the role of specific T cell subsets such as MAIT, γδ and NKT cells has been limited by low CSF cell numbers, and the granularity of bulk RNA sequencing. The polarisation of T cells is likely to be important, as well as epigenetic influences.
There is very limited data on B cells in TBM. B cells are found in the CSF in health and TBM disease. B cells are necessary for granuloma formation and in active TB, frequencies are lower and B cells appear to be functionally impaired (82). In addition to antibody production, B cells produce cytokines and modulate the function of T cells, macrophages and neutrophils. Functional impairment of B cells may therefore have consequences for T cell activation (83), but how this may influence CNS infection is unknown.
Glutamate is a neuroexcitatory neurotransmitter, regulated by the glutamate-glutamine cycle, which acts on post-synaptic neurons, oligodendrocytes, glial cells, and astrocytes (84) (Figure 4). Glutamate concentrations are tightly controlled, and ambient levels are maintained mainly by astrocytes (85). Prolonged glutamate exposure leads to excitotoxic neuronal damage, which is exacerbated under conditions of oxidative or metabolic stress e.g., ischaemia (86). The balance between glutamate and γ-aminobutyric acid (GABA), a neuroinhibitory neurotransmitter, is crucial for neurologic homeostasis (87). In addition, glutamate and GABA influence, and are influenced by, CNS immunity. TNF inhibits glutamate re-uptake by astrocytes, potentially increasing ambient glutamate concentrations in the extracellular space and worsening excitotoxicity (88, 89). GABA has been shown to inhibit IL-1β production in macrophages (90), and this effect may occur in microglia. In addition, EC express NMDA receptors, and glutamate induced endothelial permeability in a rat model has been demonstrated via a NO and cyclic guanosine monophosphate (cGMP) signalling cascade which rearranged TJP away from cell-cell contact regions (91, 92).
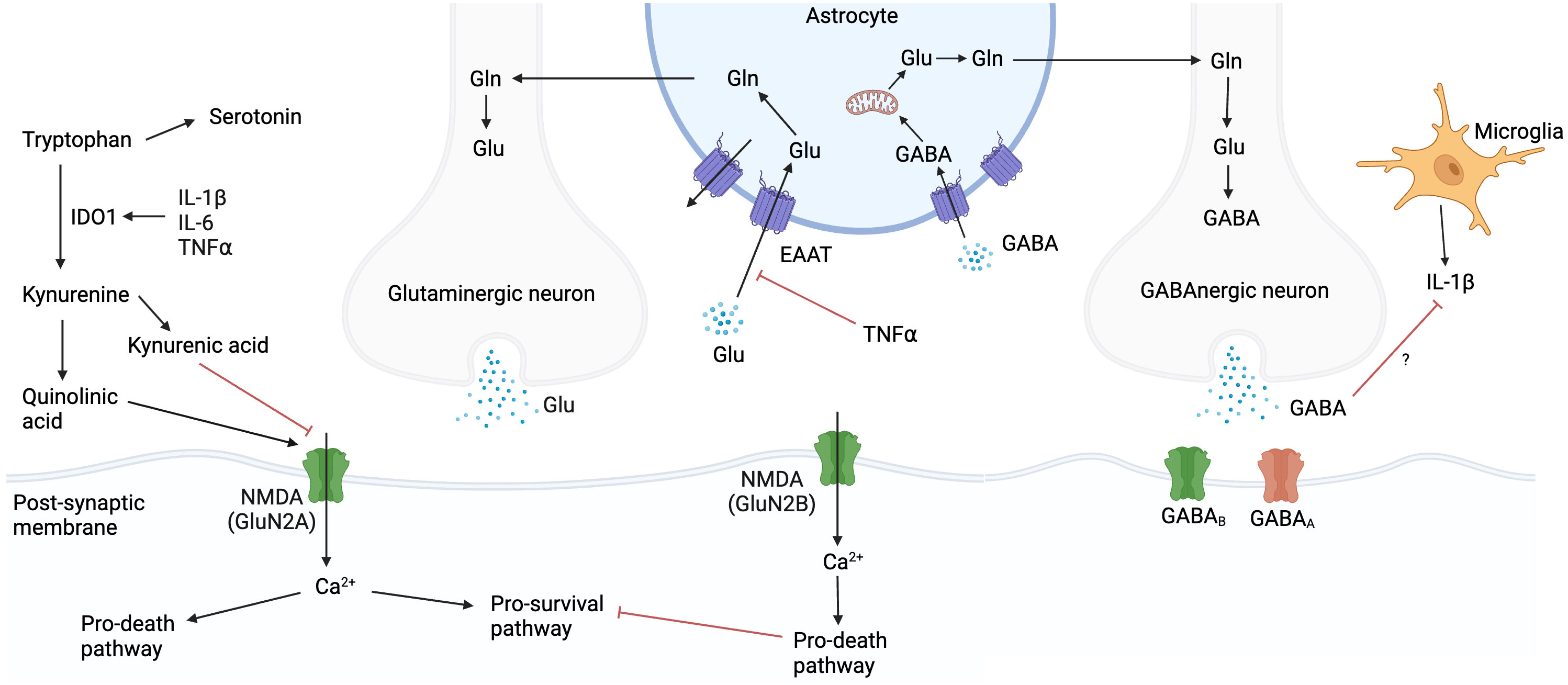
Figure 4 Glutamate, glutamine, and GABA cycle. Glutamate concentrations are tightly controlled, and ambient levels are maintained mainly by astrocytes. Prolonged glutamate exposure leads to neuronal damage, which is exacerbated under conditions of oxidative or metabolic stress. Tryptophan can be metabolised via the serotonin or kynurenine pathways. IL-1β, IL-6 and TNF upregulate IDO1 which favours the kynurenine pathway. Kynurenine can be metabolised to either quinolinic acid or kynurenic acid, which agonise or antagonise NMDA receptors respectively. Created with BioRender.com. Gln, glutamine; Glu, glutamate; GABA, γ-aminobutyric acid; IL, interleukin; NMDA, N-methyl-D-aspartate; TNF, tumour necrosis factor; Ca2+, calcium ion; IDO1, indoleamine 2,3-dioxygenase.
Glutamate-driven neuro-excitotoxicity underlies pathogenesis in a wide range of neurological conditions including Alzheimer’s disease and traumatic brain injury (93, 94). In the ventricular CSF of HIV-uninfected children with TBM, transcripts associated with glutamate release, post-synaptic NMDA receptor binding and calcium influx were enriched, suggesting a role for glutamate in TBM-associated neuro-excitotoxicity (32). A study in South Africa used hydrogen nuclear magnetic resonance (1H NMR) metabolomics to compare 18 metabolites in the lumbar CSF of HIV-uninfected paediatric TBM with non-meningitis controls and found glutamate and glutamine significantly elevated. The authors hypothesised that transporter function is reduced in both astrocytes and neurons in TBM (95).
Caution must be used when interpreting glutamate levels. HIV-1 affects glutamate concentrations in the brain and in studies of PLWH appropriate controls must be used (89). In addition, glutamate is actively cleared from the CSF both into the blood via choroid plexus epithelial cells, and into the brain via ependymal cells and so lumbar CSF may be a poor proxy for extracellular brain concentrations (96). In addition, whether glutamate is degraded as it descends the spinal tract is unknown. Magnetic resonance spectroscopy (MRS) is an established non-invasive method for direct detection of brain metabolites. Glutamate and GABA are amongst those metabolites detectable (97, 98) and MRS could allow the investigation of neurotransmitter concentrations in vivo, although it cannot always distinguish between glutamate and glutamine, or extracellular and intracellular levels, concentrations being about, 1000-fold higher in the latter.
The tryptophan pathway has been implicated in TBM pathology in recent years. Tryptophan, an essential amino acid, can be metabolised via the serotonin or kynurenine pathways. IL-1β, IL-6, IFNγ and TNF upregulate indoleamine 2,3-dioxygenase (IDO1) which induces the kynurenine pathway (99, 100). Kynurenine pathway metabolites have broad antibacterial properties although this is unlikely to be significant in TB infection (101). IDO1 may inhibit IL-17 expression and subsequent neutrophil migration in PTB (100). Blockage of IDO1 activity reduces the clinical manifestations of TB in macaques (102). The tryptophan pathway may play a role in neuroexcitotoxicity (Figure 4). Kynurenine can be metabolised to either quinolinic acid or kynurenic acid, which agonise or antagonise NMDA receptors respectively. Therefore, over induction of the quinolinic acid pathway may contribute to excitotoxicity. Higher lumbar CSF tryptophan levels correlate positively with mortality and negatively with CSF IFNγ in TBM (103, 104). In addition, CSF kynurenine metabolites correlate with CSF inflammation and markers of blood-CSF leakage, in both HIV-uninfected and PLWH. Induction of the kynurenine pathway may benefit the host by reducing the amino acid energy source for M. tuberculosis and triggering a kynurenine-induced macrophage response (103). In the CNS, IDO1 expression has been shown in macrophages, DC, microglia and choroid plexus epithelial cells (105, 106). The relative contributions of these cells to tryptophan homeostasis in TBM and the compartmental differences in concentrations of tryptophan and its metabolites are unclear.
The role of neurotransmitters in host damage and immunity in TBM is an exciting area. NMDA receptor antagonists have been unsuccessful in improving stroke outcomes due to poor tolerance at therapeutic doses, possibly because of their effects on pro-survival signalling in neurons. However, downstream inhibitors specifically targeting NMDA receptor pro-death pathways could be further investigated in TBM and are an area of active research in stroke (107).
Host genetic polymorphisms which associate with the risk of developing or dying from TBM can give us clues to the host immune response (Table 1). As mentioned above, a single nucleotide polymorphism (SNP) in the LTA4H gene has been associated with inflammatory cell recruitment, severe IRIS, survival and response to adjunctive corticosteroids which was reproduced in a large Vietnamese HIV-uninfected cohort (5, 119). This association was not seen however in an Indonesian study (56), but nonetheless may herald an era of more personalised therapy. Other polymorphisms have been associated with developing TBM include those encoding for mannose-binding lectin (MBL2), TLR2, TLR9, toll-like adaptor protein (TIRAP), toll-like receptor/IL-1R signalling (PKP3-SIGIRR-TMEM16J gene region), vitamin D receptor (VDR) and the lung protein mucin-5AC (MUC5AC) (108, 109, 111–116). Worse outcomes in TBM have been associated with polymorphisms in SPN which encodes for CD43, a surface glycoprotein found on immune cells involved in M. tuberculosis adhesion and pro-inflammatory cytokine release (117). These polymorphisms are mostly in genes responsible for the innate immune response.

Table 1 Polymorphisms associated with susceptibility to outcome in TBM.
Even within the same geographical region, identifying polymorphisms can be limited by genetic differences. This was demonstrated recently in a genome-wide association study (GWAS) of South African children with TBM versus healthy controls and PTB cases which did not find any associations with TBM susceptibility (120). The authors attribute this to the sample size, age differences and the ethnic heterogeneity of the study population.
Only a few studies have directly compared HIV-infected and HIV-uninfected patients in TBM. TBM in the context of HIV-1 infection has a distinct pathophysiology. HIV-1 is a risk factor for PTB, extrapulmonary TB, bacillaemia and consequently TBM. Poorer outcomes are seen in PLWH, who often present with occult disease and up to 50% can develop TBM-IRIS depending upon timing of ART. Although this population is the most immunosuppressed, the potential for immunomodulation with HDT is greatest given the higher mortality and more dysregulated immune response. A comprehensive understanding of the host immune response in PLWH is therefore paramount to developing novel treatment options.
HIV-1-associated TBM is characterised by increased bacillary load and CNS neutrophil counts. Mean CSF neutrophils and cytokines are higher in HIV-1 co-infection, especially in those with CD4 counts less than 150 cells/mm3 in whom increased death is also seen (5). In a review of cytokines in TBM, IL-1β, TNF, and IL-2 were increased in PLWH compared with HIV-uninfected patients (60). Higher neutrophils are associated with an increased likelihood of culturing M. tuberculosis in PLWH (55). As well as an impaired immune response, disruption of the BBB by HIV-1 may aid M. tuberculosis entry into the CNS and subsequent leukocyte infiltration in TBM through the HIV-1 proteins gp120, trans-activator of transcription (Tat), negative regulatory factor (Nef) and viral protein R (Vpr). These proteins also induce MMP production, cytokine release, chemotaxis, astrocyte disturbance and TJP downregulation (121).
The pathogenesis of IRIS is driven by M. tuberculosis load and may result from excessive neutrophil-mediated inflammation. High CSF neutrophil counts and M. tuberculosis culture positivity at presentation predict TBM-IRIS, as well as a combination of high CSF TNF and low IFNγ. Neutrophil mediators, in particular IL-17-induced S100A8/9, are associated with the development of TBM-IRIS. The same longitudinal study revealed that many of the hallmarks of TBM, including MMP, inflammasome markers (IL-1β, IL-18), IL-6 and neutrophils, are increased in TBM-IRIS compared to those who do not develop IRIS, suggesting a spectrum of severity rather than a discrete pathology (35). Another study looking at bulk RNA sequencing changes in the blood of PLWH who did and did not develop TBM-IRIS over time found significantly upregulated neutrophil- and inflammasome-associated transcripts in those who developed TBM-IRIS (48).
This review summarises the existing literature on the human host immune response in TBM. M. tuberculosis probably enters the CNS via a combination of transcytosis, paracytosis and immune cell invasion but the various CNS barriers have been poorly studied and modelled. Microglia trigger the initial inflammatory response by attracting, stimulating, and activating other leukocytes, which is followed by CNS barrier dysfunction mediated by cytokines, MMP, VEGF, adhesion molecules and pericytes. The inflammasome plays a key role in the innate response and increased CSF concentrations are associated with TBM and TBM-IRIS, but identifying HDT targets requires further understanding of the mechanisms of inflammasome activation in TBM. Neutrophils and their associated proteins are associated with poorer outcomes and TBM-IRIS. The contribution of NETosis is unknown. The balance of specialised pro-resolving mediators and pro-inflammatory eicosanoids is associated with mortality and controlling this balance may be a promising target for HDT beyond aspirin in the future. T cells are vital for the response to M. tuberculosis and impaired function is seen in TBM. MiR-29, a microRNA mediator of T cell differentiation and IFNγ production, associates with poorer outcomes. Glutamate concentrations are increased in lumbar CSF and targeting the glutamate-GABA cycle and tryptophan pathway may reduce neuro-excitotoxicity in TBM.
Host-directed therapy can be divided into drugs which dampen or augment host immunity. Given that excess inflammation drives morbidity in TBM, HDT is believed to be a necessary adjunct to ATT to dampen the immune response. Corticosteroids reduce acute mortality in HIV-uninfected patients (122), but despite these, mortality in TBM remains unacceptably high. In addition, a recent study investigating corticosteroids in TBM patients with advanced HIV found only a trend towards reduced mortality in those who received dexamethasone which did not reach significance, with similar incidences of neurologic disability and new neurologic events (6). This study throws into question the routine use of steroids in PLWH, potentially leaving no evidence-based adjuncts in this group. A better understanding of TBM would allow us to develop more selective HDT and improve outcomes.
Anti-TNF drugs may have a role in TBM but when to initiate them and in whom are important questions that need answering. The only randomised controlled trial (RCT) of an anti-TNF agent was high-dose thalidomide in a paediatric TBM population, which was stopped early due to adverse events (123). Despite that, there have been promising results with the use of infliximab and lower-dose thalidomide in paediatric and adult TBM in paradoxical reactions refractory to steroids (124, 125). A retrospective cohort study showed improved outcomes with up to three doses of 10mg/kg infliximab in patients with symptomatic tuberculomas, spinal cord involvement with paraparesis and optochiasmic arachnoiditis despite adequate ATT and corticosteroid therapy. The median duration from start of ATT and corticosteroid treatment was six months. The cohort only included one PLWH (126). A randomised trial is needed to investigate whether these improved outcomes with infliximab are seen more generally when administered alongside ATT from diagnosis.
In a meta-analysis of three RCT, aspirin has been shown to reduce new-onset stroke in TBM (HR 0.51, 95% CI: 0.29-0.87) but not mortality (127). Low-dose aspirin has anti-platelet activity alone, whereas high-dose aspirin has additional anti-inflammatory properties by inhibiting pro-inflammatory prostaglandins and thromboxane A2. Only two RCT included high-dose alongside low-dose aspirin, at different doses (1000mg/day and 100mg/kg/day) and in different populations (adults and children) (70, 128). Since the meta-analysis, an additional phase 2A study including high-dose aspirin at 1000mg/day demonstrated good safety (129). Existing studies in high-dose aspirin are underpowered to detect improvements in mortality but phase 3 trials are ongoing in adults (NCT04145258) and children (ISRCTN40829906) which may help to establish whether aspirin should be given routinely in TBM, and if so, at what dose.
Promising future HDT could include IDO1 blockers to downregulate the kynurenine pathway, NMDA receptor inhibitors, endothelial ACKR1 inhibitors to block chemokine transcytosis, anthracyclines as NETosis suppressing drugs (130), and small molecular inhibitors of the NLRP3 inflammasome pathway e.g., MCC950, glyburide, and sulforaphane (131), but these will require further research both in vitro and in a rabbit model. Ongoing studies of HDT in PTB include statins, tyrosine kinase inhibitors, metformin, phosphodiesterase inhibitors, and NSAIDS/COX2 inhibitors (132). These may be applicable to TBM and should be monitored closely.
Our understanding of the immunopathology in TBM remains limited and hampers the development of HDT. Several limitations to existing data, and future challenges in TBM research exist. In a small brain microdialysis study in paediatric TBM, brain extracellular fluid (ECF) cytokine levels were lower than ventricular CSF, which were in turn lower than lumbar CSF (133). In addition, Rohlwink et al. have demonstrated a compartmentalised response, with differences between blood, lumbar CSF, and ventricular CSF (32), all of which are likely to be proxies of varying accuracy of the immune response occurring primarily in the basal cisterns, brain leptomeninges and subpial border zone. Heterogeneities between PLWH and HIV-uninfected patients, and adults and children limit extrapolation. Existing data have come from bulk transcriptomics and proteomics, and the role of individual immune cell subsets remains unclear. The difficulty acquiring CSF samples has led to a reliance on blood which may be influenced by TB elsewhere, or other systemic confounders. The universal use of corticosteroids in the treatment of TBM, given for the very purpose of moderating the immune response, influences any data in a way that is poorly understood, and makes it challenging to find suitable controls. The poor sensitivity of diagnostic tools means relying on clinical criteria or missing the majority of cases. The Marais criteria classify TBM patient as ‘possible’, ‘probable’ or ‘definite’ (134). Patients classified as ‘possible’ will inevitably include non-TBM cases; sensitivity can be adjusted for improved specificity by excluding ‘possible’ cases, which can mean lower enrolment in clinical trials, especially in settings of high HIV-1 prevalence. Finally, a lack of in vivo and in vitro brain/BBB models, as well as containment level 3 (CL3) considerations, hinder pre-clinical research.
There has been notable progress in TBM research in recent years. Historical challenges have been partially overcome by recent developments in multiomics technologies. It is increasingly evident that host immunity goes beyond the recognised elements of the immune system as shown by the complex interactions of the neurovascular unit at the BBB, and the influence of the tryptophan pathway and glutamate-glutamine-GABA cycle. At a cellular and molecular level, areas of future research could include microglial and macrophage subsets, polarisation and ontogeny, inflammasome regulation, and the role of non-canonical inflammasomes, NK cells, NETosis, specialised pro-resolving mediators, neurotransmitters, and rarer T cell subsets. ScRNA-Seq with or without single-cell proteomics can be used to investigate these areas in addition to other ‘omics’ modalities. Adults, children, HIV-uninfected and PLWH must be proportionally represented given the acknowledged differences in their immune phenotypes. Longitudinal studies allow an insight into dynamic changes in the immune response, especially relevant in the context of paradoxical reactions and TBM-IRIS. The path forward must also maintain a translational focus ensuring that any advances made lead to rapid improvement in outcomes for patients diagnosed with this devastating disease.
JB: Writing – original draft. AD: Writing – review & editing. RW: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Francis Crick Institute (to JB, AD, and RW) which receives funding from Wellcome (CC2112), Cancer Research UK (CC2112) and the Medical Research Council (CC2112), the Wellcome Trust through core funding from the Wellcome Centre for Infectious Diseases Research in Africa (203135/Z/16/Z, to RW), the National Institutes of Health (R01145436, to RW), Meningitis Now (to RW and AD), and the EDCTP2 programme supported by the European Union (RIA2017T-2019, to RW).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wang MG, Luo L, Zhang Y, Liu X, Liu L, He JQ. Treatment outcomes of tuberculous meningitis in adults: a systematic review and meta-analysis. BMC Pulm Med (2019) 19:200. doi: 10.1186/s12890-019-0966-8
2. Marais S, Pepper DJ, Schutz C, Wilkinson RJ, Meintjes G. Presentation and outcome of tuberculous meningitis in a high HIV prevalence setting. PloS One (2011) 6(5):e20077. doi: 10.1371/journal.pone.0020077
3. Burn CG, Finley KH. The role of hypersensitivity in the productions of experimental meningitis: I. Experimental meningitis in tuberculous animals. J Exp Med (1932) 56(2):203–21. doi: 10.1084/jem.56.2.203
4. Thwaites GE, Bang ND, Dung NH, Quy HT, Oanh DTT, Thoa NTC, et al. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N Engl J Med (2004) 351(17):1741–51. doi: 10.1056/NEJMoa040573
5. Thuong NTT, Heemskerk D, Tram TTB, Thao LTP, Ramakrishnan L, Ha VTN, et al. Leukotriene A4 hydrolase genotype and HIV infection influence intracerebral inflammation and survival from tuberculous meningitis. J Infect Dis (2017) 215(7):1020–8. doi: 10.1093/infdis/jix050
6. Donovan J, Bang ND, Imran D, Nghia HDT, Burhan E, Huong DTT, et al. Adjunctive dexamethasone for tuberculous meningitis in HIV-positive adults. N Engl J Med (2023) 389(15):1357–67. doi: 10.1056/NEJMoa2216218
7. Arcuri C, Mecca C, Giambanco I, Donato R. Parenchymal and non-parenchymal immune cells in the brain: A critical role in regulating CNS functions. Int J Dev Neurosci (2019) 77:26–38. doi: 10.1016/j.ijdevneu.2019.04.005
8. Profaci CP, Munji RN, Pulido RS, Daneman R. The blood–brain barrier in health and disease: Important unanswered questions. J Exp Med (2020) 217(4):e20190062. doi: 10.1084/jem.20190062
9. Daneman R, Prat A. The blood–brain barrier. Cold Spring Harb Perspect Biol (2015) 7(1):a020412. doi: 10.1101/cshperspect.a020412
10. Derk J, Jones HE, Como C, Pawlikowski B, Siegenthaler JA. Living on the edge of the CNS: meninges cell diversity in health and disease. Front Cell Neurosci (2021) 15:703944. doi: 10.3389/fncel.2021.703944
11. Saunders NR, Habgood MD, Møllgård K, Dziegielewska KM. The biological significance of brain barrier mechanisms: help or hindrance in drug delivery to the central nervous system? F1000Research (2016) 5:313. doi: 10.12688/f1000research.7378.1
12. Jain SK, Paul-Satyaseela M, Lamichhane G, Kim KS, Bishai WR. Mycobacterium tuberculosis invasion and traversal across an in vitro human blood-brain barrier as a pathogenic mechanism for central nervous system tuberculosis. J Infect Dis (2006) 193(9):1287–95. doi: 10.1086/502631
13. Be NA, Lamichhane G, Grosset J, Tyagi S, Cheng QJ, Kim KS, et al. Murine model to study the invasion and survival of Mycobacterium tuberculosis in the central nervous system. J Infect Dis (2008) 198(10):1520–8. doi: 10.1086/592447
14. van Leeuwen LM, van der Kuip M, Youssef SA, de Bruin A, Bitter W, van Furth AM, et al. Modelling tuberculous meningitis in zebrafish using Mycobacterium marinum. Dis Model Mech (2014) 7(9):1111–22. doi: 10.1242/dmm.015453
15. Rich AR, McCordock HA. The pathogenesis of tuberculous meningitis. Bull John Hopkins Hosp (1933) 52:2–37.
16. Donald PR, Schaaf HS, Schoeman JF. Tuberculous meningitis and miliary tuberculosis: the Rich focus revisited. J Infect (2005) 50(3):193–5. doi: 10.1016/j.jinf.2004.02.010
17. Zaharie SD, Franken DJ, van der Kuip M, van Elsland S, de Bakker BS, Hagoort J, et al. The immunological architecture of granulomatous inflammation in central nervous system tuberculosis. Tuberculosis (2020) 125:102016. doi: 10.1016/j.tube.2020.102016
18. Kadry H, Noorani B, Cucullo L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS (2020) 17(1):69. doi: 10.1186/s12987-020-00230-3
19. Solár P, Zamani A, Kubíčková L, Dubový P, Joukal M. Choroid plexus and the blood–cerebrospinal fluid barrier in disease. Fluids Barriers CNS (2020) 17(1):35. doi: 10.1186/s12987-020-00196-2
20. Spanos JP, Hsu NJ, Jacobs M. Microglia are crucial regulators of neuro-immunity during central nervous system tuberculosis. Front Cell Neurosci (2015) 9:182. doi: 10.3389/fncel.2015.00182
21. Rock RB, Hu S, Gekker G, Sheng WS, May B, Kapur V, et al. Mycobacterium tuberculosis-induced cytokine and chemokine expression by human microglia and astrocytes: effects of dexamethasone. J Infect Dis (2005) 192(12):2054–8. doi: 10.1086/498165
22. Peterson PK, Gekker G, Hu S, Sheng WS, Anderson WR, Ulevitch RJ, et al. CD14 receptor-mediated uptake of nonopsonized Mycobacterium tuberculosis by human microglia. Infect Immun (1995) 63(4):1598–602. doi: 10.1128/iai.63.4.1598-1602.1995
23. Shams H, Wizel B, Lakey DL, Samten B, Vankayalapati R, Valdivia RH, et al. The CD14 receptor does not mediate entry of Mycobacterium tuberculosis into human mononuclear phagocytes. FEMS Immunol Med Microbiol (2003) 36(1–2):63–9. doi: 10.1016/S0928-8244(03)00039-7
24. Davis AG, Rohlwink UK, Proust A, Figaji AA, Wilkinson RJ. The pathogenesis of tuberculous meningitis. J Leukoc Biol (2019) 105(2):267–80. doi: 10.1002/JLB.MR0318-102R
25. Yang Q, Cai Y, Zhao W, Wu F, Zhang M, Luo K, et al. IP-10 and MIG are compartmentalized at the site of disease during pleural and meningeal tuberculosis and are decreased after antituberculosis treatment. Clin Vaccine Immunol CVI (2014) 21(12):1635–44. doi: 10.1128/CVI.00499-14
26. Bruno V, Copani A, Besong G, Scoto G, Nicoletti F. Neuroprotective activity of chemokines against N-methyl-D-aspartate or beta-amyloid-induced toxicity in culture. Eur J Pharmacol (2000) 399(2–3):117–21. doi: 10.1016/S0014-2999(00)00367-8
27. Madrigal JLM, Leza JC, Polak P, Kalinin S, Feinstein DL. Astrocyte-derived MCP-1 mediates neuroprotective effects of noradrenaline. J Neurosci (2009) 29(1):263–7. doi: 10.1523/JNEUROSCI.4926-08.2009
28. Yang R, Wang J, Wang F, Zhang H, Tan C, Chen H, et al. Blood–brain barrier integrity damage in bacterial meningitis: the underlying link, mechanisms, and therapeutic targets. Int J Mol Sci (2023) 24(3):2852. doi: 10.3390/ijms24032852
29. Green JA, Dholakia S, Janczar K, Ong CW, Moores R, Fry J, et al. Mycobacterium tuberculosis-infected human monocytes down-regulate microglial MMP-2 secretion in CNS tuberculosis via TNFα, NFκB, p38 and caspase 8 dependent pathways. J Neuroinflamm (2011) 8(1):46. doi: 10.1186/1742-2094-8-46
30. Woo MS, Park JS, Choi IY, Kim WK, Kim HS. Inhibition of MMP-3 or -9 suppresses lipopolysaccharide-induced expression of proinflammatory cytokines and iNOS in microglia. J Neurochem (2008) 106(2):770–80. doi: 10.1111/j.1471-4159.2008.05430.x
31. Brilha S, Ong CWM, Weksler B, Romero N, Couraud PO, Friedland JS. Matrix metalloproteinase-9 activity and a downregulated Hedgehog pathway impair blood-brain barrier function in an in vitro model of CNS tuberculosis. Sci Rep (2017) 7(1):16031. doi: 10.1038/s41598-017-16250-3
32. Rohlwink UK, Figaji A, Wilkinson KA, Horswell S, Sesay AK, Deffur A, et al. Tuberculous meningitis in children is characterized by compartmentalized immune responses and neural excitotoxicity. Nat Commun (2019) 10(1):3767. doi: 10.1038/s41467-019-11783-9
33. Lee KY, Kim EH, Yang WS, Ryu H, Cho SN, Lee BI, et al. Persistent increase of matrix metalloproteinases in cerebrospinal fluid of tuberculous meningitis. J Neurol Sci (2004) 220(1):73–8. doi: 10.1016/j.jns.2004.02.008
34. Green JA, Tran CTH, Farrar JJ, Nguyen MTH, Nguyen PH, Dinh SX, et al. Dexamethasone, cerebrospinal fluid matrix metalloproteinase concentrations and clinical outcomes in tuberculous meningitis. PloS One (2009) 4(9):e7277. doi: 10.1371/journal.pone.0007277
35. Marais S, Wilkinson KA, Lesosky M, Coussens AK, Deffur A, Pepper DJ, et al. Neutrophil-associated central nervous system inflammation in tuberculous meningitis immune reconstitution inflammatory syndrome. Clin Infect Dis (2014) 59(11):1638–47. doi: 10.1093/cid/ciu641
36. Mailankody S, Dangeti GV, Soundravally R, Joseph NM, Mandal J, Dutta TK, et al. Cerebrospinal fluid matrix metalloproteinase 9 levels, blood-brain barrier permeability, and treatment outcome in tuberculous meningitis. PloS One (2017) 12(7):e0181262. doi: 10.1371/journal.pone.0181262
37. Li YJ, Wilkinson KA, Wilkinson RJ, Figaji AA, Rohlwink UK. Elevated matrix metalloproteinase concentrations offer novel insight into their role in pediatric tuberculous meningitis. J Pediatr Infect Dis Soc (2020) 9(1):82–6. doi: 10.1093/jpids/piy141
38. Meli DN, Christen S, Leib SL. Matrix metalloproteinase-9 in pneumococcal meningitis: activation via an oxidative pathway. J Infect Dis (2003) 187(9):1411–5. doi: 10.1086/374644
39. Rohlwink UK, Walker NF, Ordonez AA, Li YJ, Tucker EW, Elkington PT, et al. Matrix metalloproteinases in pulmonary and central nervous system tuberculosis—A review. Int J Mol Sci (2019) 20(6):1350. doi: 10.3390/ijms20061350
40. Saghazadeh A, Rezaei N. Vascular endothelial growth factor levels in tuberculosis: A systematic review and meta-analysis. PloS One (2022) 17(5):e0268543. doi: 10.1371/journal.pone.0268543
41. Manyelo CM, Solomons RS, Snyders CI, Mutavhatsindi H, Manngo PM, Stanley K, et al. Potential of host serum protein biomarkers in the diagnosis of tuberculous meningitis in children. Front Pediatr (2019) 7:376. doi: 10.3389/fped.2019.00376
42. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol (2000) 2(10):737–44. doi: 10.1038/35036374
43. Gil E, Venturini C, Stirling D, Turner C, Tezera LB, Ercoli G, et al. Pericyte derived chemokines amplify neutrophil recruitment across the cerebrovascular endothelial barrier. Front Immunol (2022) 13:935798. doi: 10.3389/fimmu.2022.935798
44. Sánchez-Garibay C, Salinas-Lara C, Gómez-López MA, Soto-Rojas LO, Castillón-Benavides NK, Castillón-Benavides OJ, et al. Mycobacterium tuberculosis Infection Induces BCSFB Disruption but No BBB Disruption In Vivo: Implications in the Pathophysiology of Tuberculous Meningitis. Int J Mol Sci (2022) 23(12):6436. doi: 10.3390/ijms23126436
45. Wewer C, Seibt A, Wolburg H, Greune L, Schmidt MA, Berger J, et al. Transcellular migration of neutrophil granulocytes through the blood-cerebrospinal fluid barrier after infection with Streptococcus suis. J Neuroinflamm (2011) 8:51. doi: 10.1186/1742-2094-8-51
46. Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
47. Matikainen S, Nyman TA, Cypryk W. Function and regulation of noncanonical caspase-4/5/11 inflammasome. J Immunol (2020) 204(12):3063–9. doi: 10.4049/jimmunol.2000373
48. Marais S, Lai RPJ, Wilkinson KA, Meintjes G, O’Garra A, Wilkinson RJ. Inflammasome activation underlying central nervous system deterioration in HIV-associated tuberculosis. J Infect Dis (2017) 215(5):677–86. doi: 10.1093/infdis/jiw561
49. Rastogi S, Briken V. Interaction of mycobacteria with host cell inflammasomes. Front Immunol (2022) 13:791136. doi: 10.3389/fimmu.2022.791136
50. Mishra BB, Rathinam VAK, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat Immunol (2013) 14(1):52–60. doi: 10.1038/ni.2474
51. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature (2014) 511(7507):99–103. doi: 10.1038/nature13489
52. Koeken VACM, Ganiem AR, Dian S, Ruslami R, Chaidir L, Netea MG, et al. Cerebrospinal fluid IL-1β is elevated in tuberculous meningitis patients but not associated with mortality. Tuberculosis (2021) 126:102019. doi: 10.1016/j.tube.2020.102019
53. Zhang P, Zhang W, Lang Y, Qu Y, Chu F, Chen J, et al. Mass spectrometry-based metabolomics for tuberculosis meningitis. Clin Chim Acta (2018) 483:57–63. doi: 10.1016/j.cca.2018.04.022
54. Dallenga T, Repnik U, Corleis B, Eich J, Reimer R, Griffiths GW, et al. M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe (2017) 22(4):519–530.e3. doi: 10.1016/j.chom.2017.09.003
55. Thuong NTT, Vinh DN, Hai HT, Thu DDA, Nhat LTH, Heemskerk D, et al. Pretreatment cerebrospinal fluid bacterial load correlates with inflammatory response and predicts neurological events during tuberculous meningitis treatment. J Infect Dis (2019) 219(6):986. doi: 10.1093/infdis/jiy588
56. van Laarhoven A, Dian S, Ruesen C, Hayati E, Damen MSMA, Annisa J, et al. Clinical parameters, routine inflammatory markers, and LTA4H genotype as predictors of mortality among 608 patients with tuberculous meningitis in Indonesia. J Infect Dis (2017) 215(7):1029–39. doi: 10.1093/infdis/jix051
57. Huang M, Ding Z, Li W, Chen W, Du Y, Jia H, et al. Identification of protein biomarkers in host cerebrospinal fluid for differential diagnosis of tuberculous meningitis and other meningitis. Front Neurol (2022) 13:886040. doi: 10.3389/fneur.2022.886040
58. de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol (2019) 16:19–27. doi: 10.1038/s41423-018-0024-0
59. Braian C, Hogea V, Stendahl O. Mycobacterium tuberculosis-induced neutrophil extracellular traps activate human macrophages. J Innate Immun (2013) 5(6):591–602. doi: 10.1159/000348676
60. Saghazadeh A, Rezaei N. Central inflammatory cytokines in tuberculous meningitis: A systematic review and meta-analysis. J Interferon Cytokine Res (2022) 42(3):95–107. doi: 10.1089/jir.2021.0176
61. Kwon JS, Park JH, Kim JY, Cha HH, Kim MJ, Chong YP, et al. Diagnostic usefulness of cytokine and chemokine levels in the cerebrospinal fluid of patients with suspected tuberculous meningitis. Am J Trop Med Hyg (2019) 101(2):343–9. doi: 10.4269/ajtmh.18-0947
62. Manyelo CM, Solomons RS, Walzl G, Chegou NN. Tuberculous meningitis: pathogenesis, immune responses, diagnostic challenges, and the potential of biomarker-based approaches. J Clin Microbiol (2021) 59(3):e01771–20. doi: 10.1128/JCM.01771-20
63. Francisco NM, Hsu NJ, Keeton R, Randall P, Sebesho B, Allie N, et al. TNF-dependent regulation and activation of innate immune cells are essential for host protection against cerebral tuberculosis. J Neuroinflamm (2015) 12(1):125. doi: 10.1186/s12974-015-0345-1
64. Tsenova L, Sokol K, Freedman VH, Kaplan G. A combination of thalidomide plus antibiotics protects rabbits from mycobacterial meningitis-associated death. J Infect Dis (1998) 177(6):1563–72. doi: 10.1086/515327
65. Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med (2013) 188(9):1137–46. doi: 10.1164/rccm.201304-0803OC
66. Nandi B, Behar SM. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med (2011) 208(11):2251–62. doi: 10.1084/jem.20110919
67. Simmons CP, Thwaites GE, Quyen NTH, Torok E, Hoang DM, Chau TTH, et al. Pretreatment intracerebral and peripheral blood immune responses in Vietnamese adults with tuberculous meningitis: diagnostic value and relationship to disease severity and outcome. J Immunol (2006) 176(3):2007–14. doi: 10.4049/jimmunol.176.3.2007
68. Colas RA, Nhat LTH, Thuong NTT, Gómez EA, Ly L, Thanh HH, et al. Proresolving mediator profiles in cerebrospinal fluid are linked with disease severity and outcome in adults with tuberculous meningitis. FASEB (2019) 33(11):13028–39. doi: 10.1096/fj.201901590R
69. Werz O, Gerstmeier J, Libreros S, de la Rosa X, Werner M, Norris PC, et al. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat Commun (2018) 9(1):59. doi: 10.1038/s41467-017-02538-5
70. Mai NTH, Dobbs N, Phu NH, Colas RA, Thao LTP, Thuong NTT, et al. A randomised double blind placebo controlled phase 2 trial of adjunctive aspirin for tuberculous meningitis in HIV-uninfected adults. eLife (2018) 7:e33478. doi: 10.7554/eLife.33478
71. Shi F, Qiu X, Yu M, Huang Y. Tuberculosis-specific antigen stimulated and unstimulated interferon-γ for tuberculous meningitis diagnosis: A systematic review and meta-analysis. Wang MS editor. PloS One (2022) 17(8):e0273834. doi: 10.1371/journal.pone.0273834
72. van Laarhoven A, Dian S, van Dorp S, Purnama F, Koeken VACM, Diandini E, et al. Immune cell characteristics and cytokine responses in adult HIV-negative tuberculous meningitis: an observational cohort study. Sci Rep (2019) 9(1):884. doi: 10.1038/s41598-018-36696-3
73. Dieli F, Sireci G, Di Sano C, Champagne E, Fourniè JJ, Salerno JI. Predominance of Vgamma9/Vdelta2 T lymphocytes in the cerebrospinal fluid of children with tuberculous meningitis: reversal after chemotherapy. Mol Med Camb Mass (1999) 5(5):301–12.
74. Caccamo N, Meraviglia S, La Mendola C, Guggino G, Dieli F, Salerno A. Phenotypical and functional analysis of memory and effector human CD8 T cells specific for mycobacterial antigens. J Immunol (2006) 177(3):1780–5. doi: 10.4049/jimmunol.177.3.1780
75. Donovan J, Tram TTB, Phu NH, Hiep NTT, Van VTT, Mui DTH, et al. Influence of strongyloides stercoralis coinfection on the presentation, pathogenesis, and outcome of tuberculous meningitis. J Infect Dis (2022) 225(9):1653–62. doi: 10.1093/infdis/jiaa672
76. Russell SL, Lamprecht DA, Mandizvo T, Jones TT, Naidoo V, Addicott KW, et al. Compromised Metabolic Reprogramming Is an Early Indicator of CD8+ T Cell Dysfunction during Chronic Mycobacterium tuberculosis Infection. Cell Rep (2019) 29(11):3564–3579.e5. doi: 10.1016/j.celrep.2019.11.034
77. Hemingway C, Berk M, Anderson ST, Wright VJ, Hamilton S, Eleftherohorinou H, et al. Childhood tuberculosis is associated with decreased abundance of T cell gene transcripts and impaired T cell function. PloS One (2017) 12(11):e0185973. doi: 10.1371/journal.pone.0185973
78. Suarez GV, Melucci Ganzarain C del C, Vecchione MB, Trifone CA, Marín Franco JL, Genoula M, et al. PD-1/PD-L1 Pathway Modulates Macrophage Susceptibility to Mycobacterium tuberculosis Specific CD8+ T cell Induced Death. Sci Rep (2019) 9(1):187. doi: 10.1038/s41598-018-36403-2
79. Liu K, Wang D, Yao C, Qiao M, Li Q, Ren W, et al. Increased tuberculosis incidence due to immunotherapy based on PD-1 and PD-L1 blockade: A systematic review and meta-analysis. Front Immunol (2022) 13:727220. doi: 10.3389/fimmu.2022.727220
80. Liston A, Papadopoulou AS, Danso-Abeam D, Dooley J. MicroRNA-29 in the adaptive immune system: setting the threshold. Cell Mol Life Sci CMLS (2012) 69(21):3533–41. doi: 10.1007/s00018-012-1124-0
81. Pan D, Pan M, Xu YM. Mir-29a expressions in peripheral blood mononuclear cell and cerebrospinal fluid: Diagnostic value in patients with pediatric tuberculous meningitis. Brain Res Bull (2017) 130:231–5. doi: 10.1016/j.brainresbull.2017.01.013
82. Loxton AG. B cells and their regulatory functions during Tuberculosis: Latency and active disease. Mol Immunol (2019) 111:145–51. doi: 10.1016/j.molimm.2019.04.012
83. Joosten SA, Meijgaarden KEv, Nonno Fd, Baiocchini A, Petrone L, Vanini V, et al. Patients with tuberculosis have a dysfunctional circulating B-cell compartment, which normalizes following successful treatment. PloS Pathog (2016) 12(6):e1005687. doi: 10.1371/journal.ppat.1005687
84. Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev (1999) 51(1):7–61.
85. Featherstone DE. Intercellular glutamate signaling in the nervous system and beyond. ACS Chem Neurosci (2009) 1(1):4–12. doi: 10.1021/cn900006n
86. Gamir-Morralla A, López-Menéndez C, Medina M, Iglesias T. A novel Neuroprotection Target With Distinct Regulation in Stroke and Alzheimer’s Disease. In: Neuroprotection in Alzheimer’s Disease. Massachusetts, USA: Academic Press, Cambridge, (2017). p. 123–47. Available at: https://linkinghub.elsevier.com/retrieve/pii/B9780128036907000077.
87. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem (2006) 98(3):641–53. doi: 10.1111/j.1471-4159.2006.03913.x
88. Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, et al. Tumor necrosis factor α Inhibits glutamate uptake by primary human astrocytes: implications for pathogenesis of HIV-1 dementia. J Biol Chem (1996) 271(26):15303–6. doi: 10.1074/jbc.271.26.15303
89. Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, et al. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology (2003) 312(1):60–73. doi: 10.1016/S0042-6822(03)00181-8
90. Fu J, Han Z, Wu Z, Xia Y, Yang G, Yin Y, et al. GABA regulates IL-1β production in macrophages. Cell Rep (2022) 41(10):111770. doi: 10.1016/j.celrep.2022.111770
91. Vazana U, Veksler R, Pell GS, Prager O, Fassler M, Chassidim Y, et al. Glutamate-mediated blood-brain barrier opening: implications for neuroprotection and drug delivery. J Neurosci Off J Soc Neurosci (2016) 36(29):7727–39. doi: 10.1523/JNEUROSCI.0587-16.2016
92. Xhima K, Weber-Adrian D, Silburt J. Glutamate induces blood–brain barrier permeability through activation of N-methyl-D-aspartate receptors. J Neurosci (2016) 36(49):12296–8. doi: 10.1523/JNEUROSCI.2962-16.2016
93. Butterfield DA, Pocernich CB. The glutamatergic system and Alzheimer’s disease: therapeutic implications. CNS Drugs (2003) 17(9):641–52. doi: 10.2165/00023210-200317090-00004
94. Chamoun R, Suki D, Gopinath SP, Goodman JC, Robertson C. Role of extracellular glutamate measured by cerebral microdialysis in severe traumatic brain injury. J Neurosurg (2010) 113(3):564–70. doi: 10.3171/2009.12.JNS09689
95. van Zyl CDW, Loots DT, Solomons R, van Reenen M, Mason S. Metabolic characterization of tuberculous meningitis in a South African paediatric population using 1H NMR metabolomics. J Infect (2020) 81(5):743–52. doi: 10.1016/j.jinf.2020.06.078
96. Akanuma Si, Sakurai T, Tachikawa M, Kubo Y, Hosoya Ki. Transporter-mediated L-glutamate elimination from cerebrospinal fluid: possible involvement of excitatory amino acid transporters expressed in ependymal cells and choroid plexus epithelial cells. Fluids Barriers CNS (2015) 12(1):11. doi: 10.1186/s12987-015-0006-x
97. Holmes MJ, Robertson FC, Little F, Randall SR, Cotton MF, van der Kouwe AJW, et al. Longitudinal increases of brain metabolite levels in 5-10 year old children. PloS One (2017) 12(7):e0180973. doi: 10.1371/journal.pone.0180973
98. Robertson FC, Holmes MJ, Cotton MF, Dobbels E, Little F, Laughton B, et al. Perinatal HIV infection or exposure is associated with low N-acetylaspartate and glutamate in basal ganglia at age 9 but not 7 years. Front Hum Neurosci (2018) 12:145. doi: 10.3389/fnhum.2018.00145
99. Zhang YJ, Reddy MC, Ioerger TR, Rothchild AC, Dartois V, Schuster BM, et al. Tryptophan biosynthesis protects mycobacteria from CD4 T cell-mediated killing. Cell (2013) 155(6):1296–308. doi: 10.1016/j.cell.2013.10.045
100. Desvignes L, Ernst JD. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity (2009) 31(6):974–85. doi: 10.1016/j.immuni.2009.10.007
101. Narui K, Noguchi N, Saito A, Kakimi K, Motomura N, Kubo K, et al. Anti-infectious activity of tryptophan metabolites in the L-tryptophan-L-kynurenine pathway. Biol Pharm Bull (2009) 32(1):41–4. doi: 10.1248/bpb.32.41
102. Gautam US, Foreman TW, Bucsan AN, Veatch AV, Alvarez X, Adekambi T, et al. In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune-mediated control of Mycobacterium tuberculosis. Proc Natl Acad Sci USA (2018) 115(1):E62–71. doi: 10.1073/pnas.1711373114
103. Ardiansyah E, Avila-Pacheco J, Nhat LTH, Dian S, Vinh DN, Hai HT, et al. Tryptophan metabolism determines outcome in tuberculous meningitis: a targeted metabolomic analysis. eLife (2023) 12:e85307. doi: 10.7554/eLife.85307
104. van Laarhoven A, Dian S, Aguirre-Gamboa R, Avila-Pacheco J, Ricaño-Ponce I, Ruesen C, et al. Cerebral tryptophan metabolism and outcome of tuberculous meningitis: an observational cohort study. Lancet Infect Dis (2018) 18(5):526–35. doi: 10.1016/S1473-3099(18)30053-7
105. Adam RA, Tenenbaum T, Valentin-Weigand P, Laryea M, Schwahn B, Angelow S, et al. Porcine choroid plexus epithelial cells induce streptococcus suis bacteriostasis in vitro. Infect Immun (2004) 72(5):3084–7. doi: 10.1128/IAI.72.5.3084-3087.2004
106. Kwidzinski E, Bechmann I. IDO expression in the brain: a double-edged sword. J Mol Med (2007) 85(12):1351–9. doi: 10.1007/s00109-007-0229-7
107. Wu QJ, Tymianski M. Targeting NMDA receptors in stroke: new hope in neuroprotection. Mol Brain (2018) 11(1):15. doi: 10.1186/s13041-018-0357-8
108. Hoal-Van Helden EG, Epstein J, Victor TC, Hon D, Lewis LA, Beyers N, et al. Mannose-binding protein B allele confers protection against tuberculous meningitis. Pediatr Res (1999) 45(4):459–64. doi: 10.1203/00006450-199904010-00002
109. Rizvi I, Garg RK, Jain A, Malhotra HS, Singh AK, Prakash S, et al. Vitamin D status, vitamin D receptor and toll like receptor-2 polymorphisms in tuberculous meningitis: a case-control study. Infection (2016) 44(5):633–40. doi: 10.1007/s15010-016-0907-x
110. Zhao Y, Bu H, Hong K, Yin H, Zou YL, Geng SJ, et al. Genetic polymorphisms of CCL1 rs2072069 G/A and TLR2 rs3804099 T/C in pulmonary or meningeal tuberculosis patients. Int J Clin Exp Pathol (2015) 8(10):12608–20. doi: 10.1086/507907
111. Thuong NTT, Hawn TR, Thwaites GE, Chau TTH, Lan NTN, Quy HT, et al. A polymorphism in human TLR2 is associated with increased susceptibility to tuberculous meningitis. Genes Immun (2007) 8(5):422–8. doi: 10.1038/sj.gene.6364405
112. Graustein AD, Horne DJ, Arentz M, Bang ND, Chau TTH, Thwaites GE, et al. TLR9 gene region polymorphisms and susceptibility to tuberculosis in Vietnam. Tuberculosis (2015) 95(2):190–6. doi: 10.1016/j.tube.2014.12.009
113. Hawn TR, Dunstan SJ, Thwaites GE, Simmons CP, Thuong NT, Lan NTN, et al. A polymorphism in Toll-interleukin 1 receptor domain containing adaptor protein is associated with susceptibility to meningeal tuberculosis. J Infect Dis (2006) 194(8):1127–34. doi: 10.1086/507907
114. Dissanayeke SR, Levin S, Pienaar S, Wood K, Eley B, Beatty D, et al. Polymorphic variation in TIRAP is not associated with susceptibility to childhood TB but may determine susceptibility to TBM in some ethnic groups. PloS One (2009) 4(8):e6698. doi: 10.1371/journal.pone.0006698
115. Horne DJ, Randhawa AK, Chau TTH, Bang ND, Yen NTB, Farrar JJ, et al. Common polymorphisms in the PKP3-SIGIRR-TMEM16J gene region are associated with susceptibility to tuberculosis. J Infect Dis (2012) 205(4):586–94. doi: 10.1093/infdis/jir785
116. Sabo MC, Thuong NTT, Chang X, Ardiansyah E, Tram TTB, Hai HT, et al. MUC5AC genetic variation is associated with tuberculous meningitis cerebral spinal fluid cytokine responses and mortality. J Infect Dis (2023) 228(3):343–52. doi: 10.1093/infdis/jiad050
117. Campo M, Randhawa AK, Dunstan S, Farrar J, Caws M, Bang ND, et al. Common polymorphisms in the CD43 gene region are associated with tuberculosis disease and mortality. Am J Respir Cell Mol Biol (2015) 52(3):342–8. doi: 10.1165/rcmb.2014-0114OC
118. Feng WX, Mokrousov I, Wang BB, Nelson H, Jiao WW, Wang J, et al. Tag SNP polymorphism of CCL2 and its role in clinical tuberculosis in han chinese pediatric population. PloS One (2011) 6(2):e14652. doi: 10.1371/journal.pone.0014652
119. Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell (2012) 148(3):434–46. doi: 10.1016/j.cell.2011.12.023
120. Schurz H, Glanzmann B, Bowker N, Van Toorn R, Solomons R, Schoeman J, et al. Deciphering genetic susceptibility to tuberculous meningitis. Front Neurol (2022) 13:820168. doi: 10.3389/fneur.2022.820168
121. Atluri VSR, Hidalgo M, Samikkannu T, Kurapati KRV, Jayant RD, Sagar V, et al. Effect of human immunodeficiency virus on blood-brain barrier integrity and function: an update. Front Cell Neurosci (2015) 9:212. doi: 10.3389/fncel.2015.00212
122. Prasad K, Singh MB, Ryan H. Corticosteroids for managing tuberculous meningitis. Cochrane Database Syst Rev (2016) 2016(4):CD002244. doi: 10.1002/14651858.CD002244.pub4
123. Schoeman JF, Springer P, van Rensburg AJ, Swanevelder S, Hanekom WA, Haslett PAJ, et al. Adjunctive thalidomide therapy for childhood tuberculous meningitis: results of a randomized study. J Child Neurol (2004) 19(4):250–7. doi: 10.1177/088307380401900402
124. van Toorn R, Zaharie SD, Seddon JA, van der Kuip M, Marceline van Furth A, Schoeman JF, et al. The use of thalidomide to treat children with tuberculosis meningitis: A review. Tuberculosis (2021) 130:102125. doi: 10.1016/j.tube.2021.102125
125. Marais BJ, Cheong E, Fernando S, Daniel S, Watts MR, Berglund LJ, et al. Use of infliximab to treat paradoxical tuberculous meningitis reactions. Open Forum Infect Dis (2020) 8(1):ofaa604. doi: 10.1093/ofid/ofaa604
126. Manesh A, Gautam P, Kumar DSS, Mannam P, Jasper A, Gunasekaran K, et al. Effectiveness of adjunctive high-dose infliximab therapy to improve disability-free survival among patients with severe central nervous system tuberculosis: A matched retrospective cohort study. Clin Infect Dis (2023), ciad401. doi: 10.1093/cid/ciad401
127. Rohilla R, Shafiq N, Malhotra S. Efficacy and safety of aspirin as an adjunctive therapy in tubercular meningitis: A systematic review and meta-analysis. eClinicalMedicine (2021) 34:100819. doi: 10.1016/j.eclinm.2021.100819
128. Schoeman JF, Janse Van Rensburg A, Laubscher JA, Springer P. The role of aspirin in childhood tuberculous meningitis. J Child Neurol (2011) 26(8):956–62. doi: 10.1177/0883073811398132
129. Davis AG, Wasserman S, Stek C, Maxebengula M, Jason Liang C, Stegmann S, et al. A phase 2A trial of the safety and tolerability of increased dose rifampicin and adjunctive linezolid, with or without aspirin, for human immunodeficiency virus–associated tuberculous meningitis: the LASER-TBM trial. Clin Infect Dis (2023) 76(8):1412–22. doi: 10.1093/cid/ciac932
130. Khan MA, D’Ovidio A, Tran H, Palaniyar N. Anthracyclines suppress both NADPH oxidase- dependent and -independent NETosis in human neutrophils. Cancers (2019) 11(9):1328. doi: 10.3390/cancers11091328
131. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov (2020) 6(1):1–22. doi: 10.1038/s41421-020-0167-x
132. Wallis RS, O’Garra A, Sher A, Wack A. Host-directed immunotherapy of viral and bacterial infections: past, present and future. Nat Rev Immunol (2022). doi: 10.1038/s41577-022-00734-z
133. Loxton NW, Rohlwink UK, Tshavhungwe M, Dlamini L, Shey M, Enslin N, et al. A pilot study of inflammatory mediators in brain extracellular fluid in paediatric TBM. PloS One (2021) 16(3):e0246997. doi: 10.1371/journal.pone.0246997
Keywords: tuberculous meningitis (TBM), tuberculosis, Mycobacterium tuberculosis, extrapulmonary tuberculosis (EPTB), immune response, host-directed therapy, immunity
Citation: Barnacle JR, Davis AG and Wilkinson RJ (2024) Recent advances in understanding the human host immune response in tuberculous meningitis. Front. Immunol. 14:1326651. doi: 10.3389/fimmu.2023.1326651
Received: 23 October 2023; Accepted: 18 December 2023;
Published: 09 January 2024.
Edited by:
Vishwanath Venketaraman, Western University of Health Sciences, United StatesReviewed by:
Hong Yien Tan, Xiamen University, MalaysiaCopyright © 2024 Barnacle, Davis and Wilkinson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James R. Barnacle, amFtZXMuYmFybmFjbGVAY3JpY2suYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.