 Juan Guerrero-Mauvecin1
Juan Guerrero-Mauvecin1 Natalia Villar-Gómez1,2
Natalia Villar-Gómez1,2 Sandra Rayego-Mateos2,3
Sandra Rayego-Mateos2,3 Adrian M. Ramos1,2
Adrian M. Ramos1,2 Marta Ruiz-Ortega2,3,4
Marta Ruiz-Ortega2,3,4 Alberto Ortiz1,2,4,5
Alberto Ortiz1,2,4,5 Ana B. Sanz1,2*
Ana B. Sanz1,2*- 1Laboratorio de Nefrología Experimental, Instituto de Investigación Sanitaria-Fundación Jimenez Diaz (IIS-FJD), Universidad Autonoma de Madrid, Madrid, Spain
- 2Redes de Investigación Cooperativa Orientadas a Resultados en Salud (RICORS2040), Madrid, Spain
- 3Cellular Biology in Renal Diseases Laboratory, IIS-FJD-Universidad Autónoma, Madrid, Spain
- 4Department of Medicine, Universidad Autonoma de Madrid, Madrid, Spain
- 5Instituto Reina Sofia en Investigación en Nefrología (IRSIN), Madrid, Spain
Acute kidney injury (AKI) frequently occurs in patients with chronic kidney disease (CKD) and in turn, may cause or accelerate CKD. Therapeutic options in AKI are limited and mostly relate to replacement of kidney function until the kidneys recover spontaneously. Furthermore, there is no treatment that prevents the AKI-to-CKD transition. Regulated necrosis has recently emerged as key player in kidney injury. Specifically, there is functional evidence for a role of necroptosis, ferroptosis or pyroptosis in AKI and the AKI-to-CKD progression. Regulated necrosis may be proinflammatory and immunogenic, triggering subsequent waves of regulated necrosis. In a paradigmatic murine nephrotoxic AKI model, a first wave of ferroptosis was followed by recruitment of inflammatory cytokines such as TWEAK that, in turn, triggered a secondary wave of necroptosis which led to persistent kidney injury and decreased kidney function. A correct understanding of the specific forms of regulated necrosis, their timing and intracellular molecular pathways may help design novel therapeutic strategies to prevent or treat AKI at different stages of the condition, thus improving patient survival and the AKI-to-CKD transition. We now review key regulated necrosis pathways and their role in AKI and the AKI-to-CKD transition both at the time of the initial insult and during the repair phase following AKI.
1 Overview of AKI
Acute kidney injury (AKI) is defined by a rapid decline of renal function, resulting in increased serum creatinine levels or decreased urine output below certain thresholds (1, 2). AKI may be triggered by pre-renal, renal, and post-renal (urinary tract obstruction) causes. Renal causes like drugs, sepsis/shock and ischemia-reperfusion injury (IRI) that may lead to tubular cell death include from direct tubular toxicity to crystal-induced kidney injury (2, 3). Even though the loss of renal function is at least partially reversible in most patients who survive, the mortality rate of AKI remains high (over 50%) (4, 5). Moreover, AKI episodes favor the progression to chronic kidney disease (CKD), and CKD is a risk factor for AKI (6, 7). Cell death and inflammation play a key role in AKI. Systemic and local inflammation can cause tubular cell death and AKI, and dying tubular cells may trigger a secondary inflammatory response that may further amplify tubular cell death (8). Several pathways of regulated necrosis, such as necroptosis, ferroptosis and pyroptosis, have emerged as proinflammatory cell death pathways since dying cells release proinflammatory factors that amplify tissue injury (8, 9). A role for regulated necrosis has been observed in preclinical AKI induced by sepsis, IRI and nephrotoxicity, and there is evidence of its occurrence in humans (10–12). The desired outcome of an AKI episode is complete recovery of kidney structure and function, but in most cases, this does not occur. Tubular cells with sublethal damage can either completely recover their function and phenotype or, if regeneration is defective, they may evolve to a profibrotic phenotype, contributing to CKD progression (13). Now, we review the role of regulated necrosis pathways in inflammation and repair in AKI.
2 Adaptive and maladaptive repair after AKI: AKI and CKD progression
The AKI-to-CKD transition may be related to factors that depend on the nature and intensity of the stimulus or its interactions with the kidney tissue and the specific cellular niches affected. Mechanistically, all kinds of AKI centrally impact the tubular epithelium by producing cell stress and death. Thus, tubular cells are the epicenter from which damage expands to other areas of the kidney (14). Adaptive tissue repair after an AKI episode depends on an orderly balance between tubular cell death and the proliferation of survival cells that should maintain a healthy tubular cell phenotype. The persistence or recurrence of damaging stimuli shifts this balance towards increased cellular stress and a state of maladaptive repair characterized by ongoing cell death, persistent inflammation, and tissue fibrosis and aging, all of which contribute to chronicity. Extensive kidney cell death without recovery depletes the normal functional epithelia which is replaced by scar tissue and fibrosis. Therefore, consistent with the central role of cell death in promoting AKI initiation and the AKI-to-CKD transition, cell death is a primary therapeutic target to improve AKI outcomes.
3 Regulated necrosis pathways
Necroptosis, ferroptosis and pyroptosis are the main regulated necrosis pathways and they are interrelated.
3.1 Necroptosis
Necroptosis results in cell swelling, and membrane disruption and triggers an inflammatory response (15). The best characterized trigger is activation of TNF superfamily receptors by their ligands, recruiting TRADD, TRAF2 and RIPK1, among other proteins. In a pro-survival context, cellular inhibitors of apoptosis proteins (CIAPs) and the Linear Ubiquitin Chain Assembly Complex (LUBAC) drive the polyubiquitination of RIPK1, activating the NF-кB pathway, which promotes cell proliferation and survival (16, 17). On the contrary, when NF-kB activation is inhibited, RIPK1 is deubiquitinated and associates with FADD and procaspase-8, resulting in cleavage of procaspase-8, generating active caspase-8 that results in execution of apoptosis (18, 19). In the absence of caspase-8 activity, RIPK1 is not cleaved, and associates with RIPK3, resulting in transphosphorylation or autophosphorylation of RIPK3, and the formation of the necrosome, which phosphorylates MLKL, resulting in MLKL oligomerization and translocation to the plasma membrane (20). Membrane-bound pMLKL oligomers form pores that lead to calcium influx and the release of damage-associated patterns (DAMPs), that trigger an inflammatory response (21–24).
3.2 Ferroptosis
Ferroptosis is an iron-dependent regulated form of necrotic cell death characterized by excessive lipid peroxidation of organelle and cell membranes that causes their disruption, leading to cell death and the release of DAMPs that trigger an inflammatory response (15). Unlike other regulated forms of cell death, ferroptosis is not regulated by specific molecular mediators, but it depends on the redox balance and the cellular antioxidant defense. Key factors involved in triggering ferroptosis, include low levels of the antioxidant molecule glutathione (GSH), impaired GPX4 activity, imbalanced polyunsaturated fatty acid (PUFA) contents and iron (Fe) availability (25). Inhibition of the Xc- antiporter can trigger ferroptosis, since it allows cystine import into cells, which is indispensable for synthesis of GSH, a GPX4 cofactor (26). GPX4 activity is the main cellular antioxidant defense against ferroptosis and reduces lipid hydroperoxides (L-OOH) on their corresponding lipid alcohols (L-OH) (27, 28). Kidney tubular cells are especially dependent on GPX4 and acquired GPX4 deficiency triggers AKI (27). Other mediators of ferroptosis include PUFAs that can be esterified into membrane phospholipid (PL) and become oxidative susceptible species (PL-PUFAs). The activation and esterification of PUFAs are mediated by acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) (29–31). An increased content of these species could increase the susceptibility to ferroptosis since PL-PUFAs can be oxidized by lipoxygenases (LOX) forming lipid peroxides (32, 33). Another key regulator of ferroptosis is iron, which is necessary for LOX activity and can trigger Fenton reactions creating more lipid peroxides (34, 35). Ferroptosis may be propagated to adjacent tubular cells as a synchronized wave of cell death, where a single tubular cell may trigger teh death to the whole tubule (36). This synchronized wave of cell death seems to be propagated through calcium signals and might be stimulus-dependent, was observed in erastin-induced GSH depletion but not during GPX4 inhibition (37). Understanding the mechanisms that mediate the spread of ferroptosis through cell populations will help us to identify new ferroptosis inhibitors.
3.3 Pyroptosis
Pyroptosis is characterized by membrane rupture and pro-inflammatory effects. Gasdermins (GSDM) are the main effectors of pyroptosis. Activated GSDM inserts into cell membranes and forms pores leading to the release of cytokines, alarmins and DAMPs, cell membrane rupture and cell death (12). GSDM activation can be mediated by canonical and non-canonical pathways. The canonical pathway involves the activation of Toll-like receptors (TLRs), which induce the expression of inflammasome components and pro-inflammatory cytokines. In parallel, inflammasome sensors such as NLRP3 are activated by a variety of DAMPs and PAMPs and recruit adaptor proteins, CARD and pro-caspase-1, to form the inflammasome complex and activate caspase-1. Caspase 1 cleaves pro-IL-β, pro-IL-18, and GSDMD promoting membrane pore formation, cytokine release and lytic cell death (12, 38). In the non-canonical pathway, caspases-4, -5 and -11 are directly activated by intracellular lipopolysaccharide, independent of the inflammasome, and they consequently cleave GSDMD to execute pyroptosis, without IL-1β and IL-18 cleavage (39). Pyroptosis can interact with apoptosis since caspase-3 and -7 may activate GSDME and caspase-8 GSDMD (40, 41). Pyroptosis is associated with host antimicrobial defense, but it can also be involved in sterile diseases such as atherosclerosis and neurodegenerative diseases (42–44).
3.4 Molecular interactions between different forms of regulated necrosis and apoptosis
As recently reviewed, there are multiple interactions between different modalities of regulated necrosis and apoptosis, beyond the fact that regulated necrosis may recruit inflammation-driven cell death, that should be accounted for when designing therapeutic interventions (8). Examples include the several roles of caspases in cell death modalities ranging from apoptosis to necroptosis or pyroptosis, final common pathways such as the recruitment of NINJ1 to lyse the cell membrane and common protective mechanisms such as preservation of cell membrane integrity by ESCRT-III (8).
4 Regulated necrosis pathways and induction of AKI
Contrary to the old-fashioned assumption that apoptosis accounts for the majority of dying cells in AKI, much evidence has emerged in the last decade for a predominant role and contribution of regulated forms of necrotic cell death, in particular necroptosis and ferroptosis. Therefore, elucidating their relative contribution and interconnection in AKI will allow for the development of more precision targeted therapies. To this end, several studies have investigated the involvement of regulated necrosis pathways in experimental AKI through interference with the activity of key components of the molecular pathway.
In general, the role of ferroptosis and necroptosis in different models of AKI has been clearly demonstrated. During folic acid-induced nephrotoxic AKI (FA-AKI), a first wave cell death by ferroptosis induces an inflammatory response that triggers a secondary wave of cell death by necroptosis in which the inflammatory cytokine TWEAK activation of the Fn14 receptor is involved (45, 46) In rhabdomyolysis-induced kidney injury, ferroptosis appears to be the dominant pathway, as ferrostatin-1 improved renal function while the necroptosis inhibitor necrostatin-1 had no effect (47, 48). This makes sense, since myoglobin is a heme-containing protein, i.e., a source of excess iron. In both IRI-AKI and crystal nephropathy, targeting necroptosis, ferroptosis and mitochondrial permeability transition pore-regulated necrosis (MPTP-RN) were protective (36, 49–53). Moreover, necroptosis and ferroptosis may be interconnected in IRI-AKI, since MLKL-deficient mice subjected to renal IRI showed an earlier upregulation of ACSL4, a potential mediator of ferroptosis (54), supporting the notion that combined therapy may be more effective than targeting a single pathway. In murine cisplatin-AKI, deficiency of RIPK3 or MLKL resulted in improved renal function, pointing out necroptosis as a major mechanism of tubular cell death (55), but there may also be a link with other regulated necrotic pathways, as some studies suggest a role for ferroptosis (56–58). In sepsis-AKI, RIPK3 aggravated kidney injury in a MLKL-independent manner by promoting mitochondrial dysfunction via NOX4 upregulation, but the contribution of tubular cell death was not clearly demonstrated (59).
By contrast, the role of pyroptosis in AKI is controversial. Some studies have found that caspase-11 expression and cleavage of GSDMD or GSDME were increased in both IRI-AKI and cisplatin-AKI. Caspase-11-, GSDMD- or GSDME-deficient mice were protected from cisplatin-AKI, and specifically GSDME deficiency also ameliorated injury in IRI-AKI (60–62). In contrast, an independent group reported that in both GSDMD- and GSDME-deficient mice the severity of IRI-AKI, cisplatin-AKI and calcium oxalate-AKI were increased due to activation of necroptosis (63). In addition, whether tubular cells express pyroptosis proteins is disputed (60, 61, 63, 64). Further studies should clarify which cells activate pyroptosis in AKI, whether targeting pyroptosis is truly protective and which is the optimal way to target pyroptosis. Multicenter preclinical trials may help address these discrepancies (65).
Moreover, further research should address the in vivo relationships between different modes of regulated necrosis in different forms of AKI: which forms of cell death occur initially and how do they trigger similar or another form of cell death in neighboring cells (8), what is the impact of the nature and strength of the stimulus, along with the presence of co-stimulatory factors, on these dynamics (8), and above all, how these preclinical observations relate to the clinical situation, in which the time-course of injury is frequently unclear and different insults may pile up in the same patient at different time points following the initial injury. Clinical translation sorely needs soluble biomarkers of different modalities of regulated necrosis that allow a dynamic follow-up of ongoing types of regulated necrosis and their response to different therapeutic interventions.
5 Interaction between regulated necrosis and inflammation in AKI
During homeostatic and developmental scenarios, apoptosis-driven removal of damaged or unneeded cells regulates cell populations in the kidney and other organs. This process involves efferocytosis, a non-inflammatory mechanism where cell surface molecules (eat-me signals) are recognized by macrophages that engulf and clear apoptotic cells (66). In contrast, regulated necrosis pathways are characterized by the lack of early engulfment, formation of pores in cellular membranes, membrane lysis and the consequent release of DAMPs, which engage an inflammatory response that amplifies injury, in a process termed necroinflammation (67). DAMPs released during regulated necrosis may also be immunogenic and are thought to play a role in autoimmune diseases such as lupus nephritis. Ferroptosis has the most proinflammatory and immunogenic potential, since it both releases DAMPs and lipid peroxides but also propagates cell death in a synchronized manner (37), whereas necroptosis generates both pro-inflammatory cytokines such as IL-1β (68) and anti-inflammatory cytokines such as IL-33 and CXCL1 (68–70).
During AKI, damaged and dying kidney parenchymal cells release DAMPs, which can activate pattern recognition receptors such as TLRs or NOD-like receptors proteins (NLRPs) on kidney resident immune cells like dendritic cells and macrophages, as well as chemokines and cytokines, thus attracting and activating leucocytes and amplifying the inflammation (71). The resulting inflammation depends on the nature and the persistence of the stimulus, as well as the renal compartment that is affected (72). Likewise, the specific secondary mediators differ among different forms of regulated cell death, which contributes to the complexity and heterogeneity of their impact on kidney injury (73). For example, in FA-AKI, ferroptosis is activated at early time points, and in addition to inducing cell death, it also triggers the expression of proinflammatory mediators such as Fn14 (TWEAK receptor), which promote a second wave of cell death by necroptosis (45). Similarly, in both cisplatin- and IRI-AKI, MLKL and RIPK3 deficiency reduces necroptosis, and also the tubular expression of inflammatory cytokines, such as TNFα, that trigger necroptosis (55, 74). In this regard, a novel inhibitor of RIPK1, Cpd-71, prevented cell death and inflammation in cisplatin-AKI (75).
Conversely, inflammation can trigger cell death, as characterized for IRI-AKI, where prostaglandin activation of the E-prostanoid 3 receptor (EP3) in myeloid cells promotes the release of inflammatory cytokines that activate necroptosis and necroinflammation in tubular cells (76). Some stimuli activate both inflammation and cell death, as illustrated by cisplatin- and IRI-AKI, where the interaction of the gastrin-releasing peptide receptor with TLR4 in tubular cells activates STAT1 to promote the expression of MLKL and CCL2, leading to necroptosis and inflammation (77).
On the other hand, RIPK3 can promote kidney inflammation independently of necroptosis. In FA-AKI, RIPK3 deficiency reduced inflammation but not cell death at early time points when necroptosis had not yet been recruited as a key cell death pathway (78). RIPK3 also mediated kidney inflammation after systemic injection of TWEAK, a model of inflammation that does not cause kidney cell death or dysfunction (78). Additionally, in experimental sepsis, where inflammation plays a key role, RIPK3, but not MLKL. mediated kidney injury and dysfunction (59, 79).
Overall, during AKI, cell death and inflammation are interconnected pathways, and an in-depth knowledge of this connection is necessary to optimize therapeutic approaches to AKI.
6 Regulated necrosis and tissue repair after AKI
Apoptosis of tubular cells was widely investigated in the 20th and early 21st centuries as a mechanism of regulated cell death driving AKI and the AKI-to-CKD. However, none of the therapeutic approaches made it to the clinic and apoptosis may also lead to loss of other cell types or clear excess tubular cells generated by cell proliferation following injury as well as irreversibly damaged cells (80). Thus, whereas caspase-3 deficiency increased the severity of early IRI-AKI, probably by shifting cell death to necrosis, it reduced long-term renal damage by inhibiting endothelial apoptosis, vascular rarefaction, and fibrosis (81) (Figure 1A).
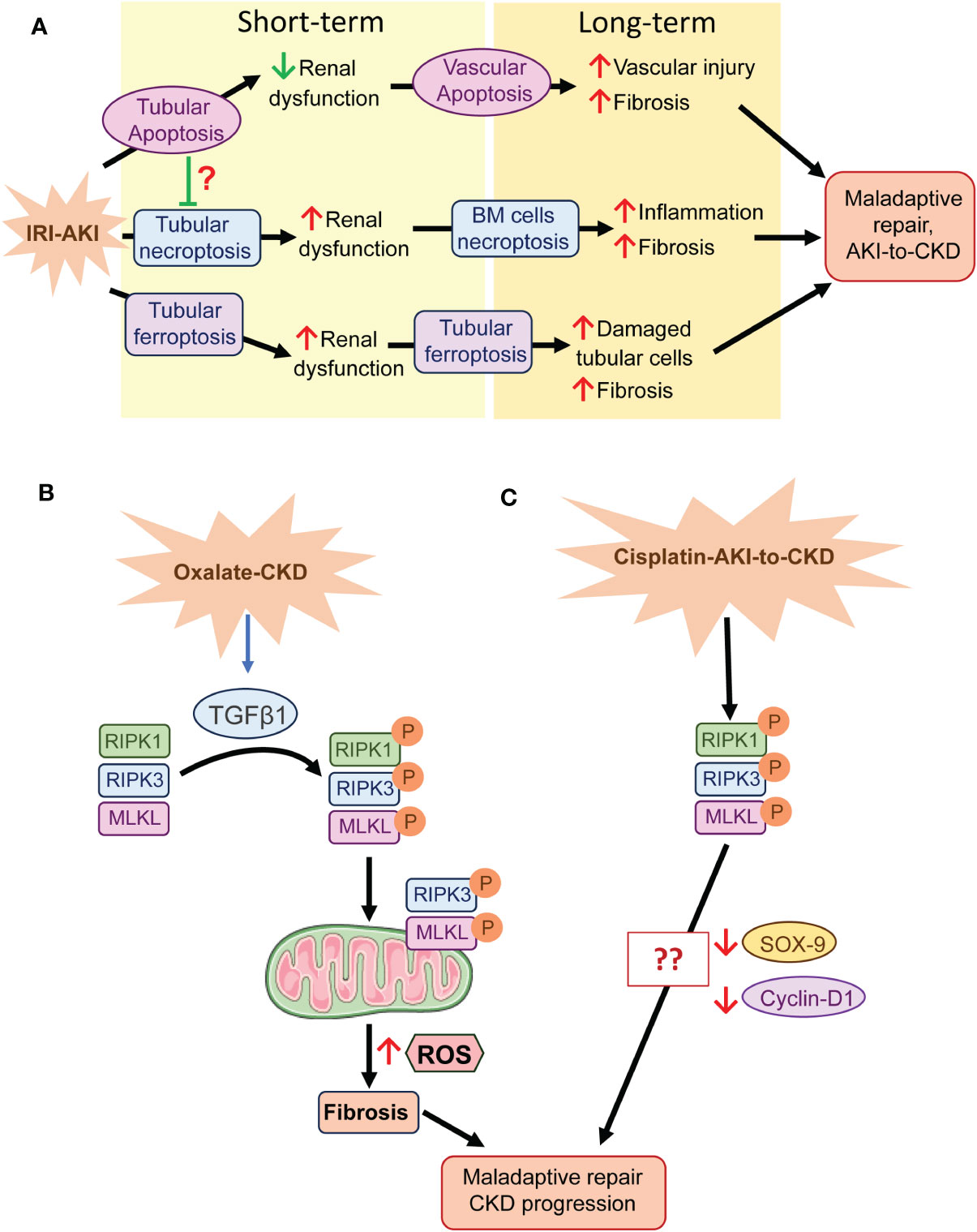
Figure 1 Regulated necrosis and tissue repair after AKI. (A) In IRI-AKI, several cell death pathways are activated. Apoptosis may play a beneficial role in the short term, probably regulating cellular homeostasis, but in the long term it favors fibrosis through cell death of perivascular cells. However, tubular necroptosis and ferroptosis play a detrimental role in both the short and long term, probably mediated by the necroinflammatory loop. (B) In oxalate CKD, the necrosome is activated by TGFβ1 and RIPK3 and MLKL translocate to the mitochondrial membrane where they promote ROS generation, fibrosis and consequent maladaptive repair and CKD progression. (C) The necrosome is activated in cisplatin-induced AKI-to-CKD transition, but its specific role in CKD progression is unknown, although, SOX-9 and cyclin-D1 downregulation may be involved. BM, Bone marrow.
In contrast, regulated necrosis, which is always considered pathological, may offer new therapeutic alternatives to treat AKI or the AKI-to-CKD transition (8). Several preclinical studies have associated regulated cell death with maladaptive repair following AKI (82). In IRI-AKI inhibition of the necroptotic pathway improved kidney function early in IRI-AKI and the progression to fibrosis in the long term, where bone marrow cells play a key role, suggesting that the inflammation associated to regulated necrosis favors the AKI-to-CKD transition (74). Single-cell transcriptomics showed that tubular cells undergoing maladaptive repair after prolonged ischemia presented an enrichment of ferroptosis and pyroptosis pathways, supporting their role in maladaptive repair (83). Another report, which combined a single-cell transcriptomic study with genetic approaches, demonstrated that chronic inflammation after IRI-AKI downregulated the gene expression of glutathione metabolism components triggering ferroptotic stress and identified GPX4 as a key coordinator of kidney repair and regeneration (84) (Figure 1A). Deficiency of IL-18, a component of NLRP3-inflammatory response, ameliorated the early phase of necroptosis as well as later tissue regeneration in murine FA-AKI (85).
In CKD patients and mice with oxalate-induced CKD increased renal levels of RIPK1, RIPK3, MLKL correlated with increased extracellular matrix (ECM) production and declining kidney function (86). Both pharmacological and genetic inhibition of RIPK3 diminished ECM accumulation in oxalate-induced CKD, adenine diet-induced renal fibrosis and, unilateral ureteral obstruction (86, 87), showing the involvement of necroptosis in kidney fibrosis. Moreover, profibrotic factors recruit RIPK3 and MLKL to mitochondria resulting in mitochondrial dysfunction and reactive oxygen species (ROS) production in murine fibroblasts stimulated with TGFβ1 and in oxalate-induced CKD (86) (Figure 1B).
Additionally, necroptosis was identified as a determinant pathway in the progression of CKD in cisplatin-AKI, although the specific mechanisms of necroptosis in this model were not described (88). Activation of specific transcriptional repair programs, including STAT3 and SOX-9 pathways, contribute to cell repair during regeneration following AKI (89–91). In murine cisplatin-AKI, 7-Hydroxycoumarin diminished renal necroptosis by modulating the RIPK1/RIPK3/MLKL pathway, and increased tissue repair through upregulation of cyclin D1 (92). Moreover, in cultured HK2 cells, SOX-9 deficiency reduced the beneficial effect of 7-Hydroxycoumarin against cisplatin cytotoxicity (92), suggesting a potential relationship between SOX-9, necroptosis, and tissue repair (Figure 1C).
Following AKI, damaged tubular cells may become senescent and display the secretory associated senescence phenotype (SASP), characterized by profibrotic and proinflammatory factors, which spread cellular senescence to neighboring cells and contribute to the propagation of kidney damage (13). Cellular communication network factor 2 (CCN2) is a component of the SASP that induces senescence in cultured tubular cells, is involved in experimental renal fibrosis following IRI- and FA-AKI and can activate the RIKP3/NLRP3 pathway in the acute phase of FA-AKI (93–95). In aging mice, FA-AKI is more severe compared to young mice, and this has been linked to cellular senescence related mechanisms, including increased expression of SASP components such as CCN2, and to upregulation of necroptosis cell-death pathways (96), suggesting a link between senescence and necroinflammation in renal injury. In this line, deficiency of IL-18, a component of NLRP3-inflammatory response, ameliorated the early phase of necroptosis as well as later tissue regeneration in murine FA-AKI (85).
The therapeutic effect of extracellular vesicles (EVs) on renal recovery after AKI has also been linked to modulation of regulated necrosis pathways (97–99). Exosomes from human umbilical cord-derived mesenchymal stem cells modulated necroptosis through miR-874-3p attenuating HK2 cell injury and enhancing repair following cisplatin stimulation (100). In murine LPS-induced AKI, treatment with adipose-derived EVs reduced renal inflammation and pyroptosis and promoted tubular cell repair through miR-21-5p/TLR4, blocking the NF-κB/NLRP3 pathway (101).
Overall, these results support a key role of regulated cell death in the regeneration and repair phase of AKI.
7 Conclusion
In conclusion, there is accumulating evidence for a role of different modalities of regulated necrosis in the initial phase of AKI, the amplification and persistence of injury following the initial insults, the repair phase from AKI and the AKI-to-CKD transition. A better understanding of the molecular mechanisms involved in different phases of AKI following diverse insults may identify novel therapeutic targets. However, clinical development should rely on the development of biomarkers that allow to monitor the activation of different modalities of regulated necrosis as well as their response to therapeutic interventions in humans (Figure 2).
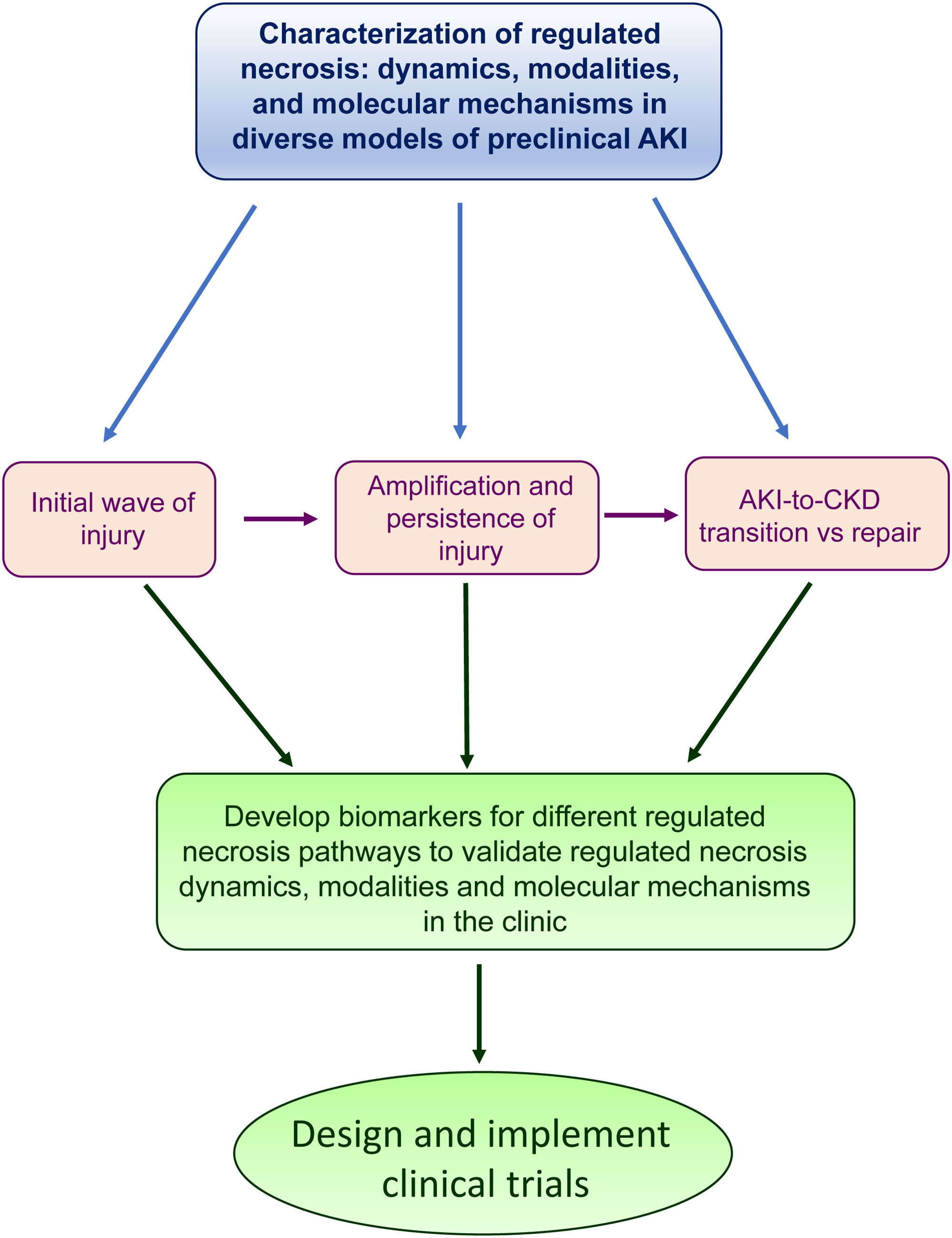
Figure 2 The path to clinical translation of findings in preclinical models. The findings described in preclinical models about the role of regulated necrosis in the different phases of AKI could help to develop novel biomarkers in the clinic.
Author contributions
JG: Writing – original draft, Writing – review & editing. NV: Writing – original draft, Writing – review & editing. SR: Writing – original draft, Writing – review & editing. AR: Writing – original draft, Writing – review & editing. MR: Writing – original draft, Writing – review & editing. AO: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. AS: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. ISCIII/Fondos FEDER (PI22/00469, PI22/00050, PI21/1453, PI19/00588, ERA-PerMed-JTC2022 (SPAREKID AC22/00027), RICORS program to RICORS2040 (RD21/0005/0001) funded by European Union – NextGenerationEU, Mecanismo para la Recuperación y la Resiliencia (MRR) and SPACKDc PMP21/00109. Sociedad Española de Nefrología, FRIAT, Comunidad de Madrid en Biomedicina P2022/BMD-7223, CIFRA_COR-CM. COST Action PERMEDIK CA21165, supported by COST (European Cooperation in Science and Technology) 2023-2027. PREVENTCKD Consortium Project ID: 101101220 Program: EU4H DG/Agency: HADEA. Salary support: RICORS2040 to NV-G, Ramon y Cajal program to ABS, MICIU to JG-M, Juan de la Cierva incorporación program (IJC2018-035187-I) to SR-M.
Conflict of interest
AO has received grants from Sanofi and consultancy or speaker fees or travel support from Adviccene, Alexion, Astellas, AstraZeneca, Amicus, Amgen, Boehringer Ingelheim, Fresenius Medical Care, GSK, Bayer, Sanofi-Genzyme, Menarini, Mundipharma, Kyowa Kirin, Lilly, Freeline, Idorsia, Chiesi, Otsuka, Novo-Nordisk, Sysmex and Vifor Fresenius Medical Care Renal Pharma and Spafarma and is Director of the Catedra UAM-Astrazeneca of chronic kidney disease and electrolytes. He has stock in Telara Farma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Levey AS, Eckardt KU, Dorman NM, Christiansen SL, Cheung M, Jadoul M, et al. Nomenclature for kidney function and disease: executive summary and glossary from a Kidney Disease: Improving Global Outcomes consensus conference. Clin Kidney J (2020) 13:485–93. doi: 10.1093/ckj/sfaa123
2. Kellum JA, Romagnani P, Ashuntantang G, Ronco C, Zarbock A, Anders HJ. Acute kidney injury. Nat Rev Dis Primers (2021) 7:52. doi: 10.1038/s41572-021-00284-z
3. Ronco C, Bellomo R, Kellum JA. Acute kidney injury. Lancet (2019) 394:1949–64. doi: 10.1016/S0140-6736(19)32563-2
4. Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet (2012) 380:756–66. doi: 10.1016/S0140-6736(11)61454-2
5. Bienholz A, Wilde B, Kribben A. From the nephrologist's point of view: diversity of causes and clinical features of acute kidney injury. Clin Kidney J (2015) 8:405–14. doi: 10.1093/ckj/sfv043
6. Rifkin DE, Coca SG, Kalantar-Zadeh K. Does AKI truly lead to CKD? J Am Soc Nephrol (2012) 23:979–84. doi: 10.1681/ASN.2011121185
7. Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol (2015) 26:1765–76. doi: 10.1681/ASN.2015010006
8. Sanz AB, Sanchez-Niño MD, Ramos AM, Ortiz A. Regulated cell death pathways in kidney disease. Nat Rev Nephrol (2023) 19:281–99. doi: 10.1038/s41581-023-00694-0
9. Tonnus W, Meyer C, Paliege A, Belavgeni A, von Mässenhausen A, Bornstein SR, et al. The pathological features of regulated necrosis. J Pathol (2019) 247:697–707. doi: 10.1002/path.5248
10. Kolbrink B, von Samson-Himmelstjerna FA, Murphy JM, Krautwald S. Role of necroptosis in kidney health and disease. Nat Rev Nephrol (2023) 19:300–14. doi: 10.1038/s41581-022-00658-w
11. Bayır H, Dixon SJ, Tyurina YY, Kellum JA, Kagan VE. Ferroptotic mechanisms and therapeutic targeting of iron metabolism and lipid peroxidation in the kidney. Nat Rev Nephrol (2023) 19:315–36. doi: 10.1038/s41581-023-00689-x
12. Elias EE, Lyons B, Muruve DA. Gasdermins and pyroptosis in the kidney. Nat Rev Nephrol (2023) 19:337–50. doi: 10.1038/s41581-022-00662-0
13. Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, Rodrigues-Diez RR. Targeting the progression of chronic kidney disease. Nat Rev Nephrol (2020) 16:269–88. doi: 10.1038/s41581-019-0248-y
14. Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol (2015) 11:264–76. doi: 10.1038/nrneph.2015.3
15. Belavgeni A, Meyer C, Stumpf J, Hugo C, Linkermann A. Ferroptosis and necroptosis in the kidney. Cell Chem Biol (2020) 27:448–62. doi: 10.1016/j.chembiol.2020.03.016
16. Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell (2009) 36:831–44. doi: 10.1016/j.molcel.2009.10.013
17. Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell (2008) 30:689–700. doi: 10.1016/j.molcel.2008.05.014
18. Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev (1999) 13:2514–26. doi: 10.1101/gad.13.19.2514
19. Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell (2008) 133:693–703. doi: 10.1016/j.cell.2008.03.036
20. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell (2012) 150:339–50. doi: 10.1016/j.cell.2012.06.019
21. Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep (2014) 7:971–81. doi: 10.1016/j.celrep.2014.04.026
22. Petrie EJ, Sandow JJ, Jacobsen AV, Smith BJ, Griffin MDW, Lucet IS, et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat Commun (2018) 9:2422. doi: 10.1038/s41467-018-04714-7
23. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol (2014) 16:55–65. doi: 10.1038/ncb2883
24. Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res (2014) 24:105–21. doi: 10.1038/cr.2013.171
25. Stern JE, Liu CL, Hwang SS, Dukhovny D, Diop H, Cabral H. Contributions to prematurity of maternal health conditions, subfertility, and assisted reproductive technology. Fertil Steril (2020) 114:828–36. doi: 10.1016/j.fertnstert.2020.03.036
26. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife (2014) 3:e02523. doi: 10.7554/eLife.02523
27. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol (2014) 16:1180–91. doi: 10.1038/ncb3064
28. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
29. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13:91–8. doi: 10.1038/nchembio.2239
30. Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun (2016) 478:1338–43. doi: 10.1016/j.bbrc.2016.08.124
31. Lee JY, Kim WK, Bae KH, Lee SC, Lee EW. Lipid metabolism and ferroptosis. Biol (Basel) (2021) 10:184. doi: 10.3390/biology10030184
32. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA (2016) 113:E4966–4975. doi: 10.1073/pnas.1603244113
33. Shintoku R, Takigawa Y, Yamada K, Kubota C, Yoshimoto Y, Takeuchi T, et al. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci (2017) 108:2187–94. doi: 10.1111/cas.13380
34. Shah R, Shchepinov MS, Pratt DA. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci (2018) 4:387–96. doi: 10.1021/acscentsci.7b00589
35. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
36. Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA (2014) 111:16836–41. doi: 10.1073/pnas.1415518111
37. Riegman M, Sagie L, Galed C, Levin T, Steinberg N, Dixon SJ, et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol (2020) 22:1042–8. doi: 10.1038/s41556-020-0565-1
38. Hadian K, Stockwell BR. The therapeutic potential of targeting regulated non-apoptotic cell death. Nat Rev Drug Discovery (2023) 22:723–42. doi: 10.1038/s41573-023-00749-8
39. Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
40. Demarco B, Ramos S, Broz P. Detection of gasdermin activation and lytic cell death during pyroptosis and apoptosis. Methods Mol Biol (2022) 2523:209–37. doi: 10.1007/978-1-0716-2449-4_14
41. Demarco B, Grayczyk JP, Bjanes E, Le Roy D, Tonnus W, Assenmacher CA, et al. Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci Adv (2020) 6:eabc3465. doi: 10.1126/sciadv.abc3465
42. Wang S, Moreau F, Chadee K. Gasdermins in innate host defense against. Front Immunol (2022) 13:900553. doi: 10.3389/fimmu.2022.900553
43. Zhang BL, Yu P, Su EY, Zhang CY, Xie SY, Yang X, et al. Inhibition of GSDMD activation by Z-LLSD-FMK or Z-YVAD-FMK reduces vascular inflammation and atherosclerotic lesion development in ApoE. Front Pharmacol (2023) 14:1184588. doi: 10.3389/fphar.2023.1184588
44. Yao J, Wang Z, Song W, Zhang Y. Targeting NLRP3 inflammasome for neurodegenerative disorders. Mol Psychiatry (2023). doi: 10.1038/s41380-023-02239-0
45. Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Niño MD, et al. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol (2017) 28:218–29. doi: 10.1681/ASN.2015121376
46. Martin-Sanchez D, Fontecha-Barriuso M, Carrasco S, Sanchez-Niño MD, Mässenhausen AV, Linkermann A, et al. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc Natl Acad Sci USA (2018) 115:4182–7. doi: 10.1073/pnas.1716578115
47. Stoppe C, Averdunk L, Goetzenich A, Soppert J, Marlier A, Kraemer S, et al. The protective role of macrophage migration inhibitory factor in acute kidney injury after cardiac surgery. Sci Transl Med (2018) 10:eaan4886. doi: 10.1126/scitranslmed.aan4886
48. Guerrero-Hue M, García-Caballero C, Palomino-Antolín A, Rubio-Navarro A, Vázquez-Carballo C, Herencia C, et al. Curcumin reduces renal damage associated with rhabdomyolysis by decreasing ferroptosis-mediated cell death. FASEB J (2019) 33:8961–75. doi: 10.1096/fj.201900077R
49. Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ (2016) 23:1565–76. doi: 10.1038/cdd.2016.46
50. Linkermann A, Bräsen JH, Darding M, Jin MK, Sanz AB, Heller JO, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA (2013) 110:12024–9. doi: 10.1073/pnas.1305538110
51. von Mässenhausen A, Tonnus W, Himmerkus N, Parmentier S, Saleh D, Rodriguez D, et al. Phenytoin inhibits necroptosis. Cell Death Dis (2018) 9:359. doi: 10.1038/s41419-018-0394-3
52. Mulay SR, Desai J, Kumar SV, Eberhard JN, Thomasova D, Romoli S, et al. Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat Commun (2016) 7:10274. doi: 10.1038/ncomms10274
53. Mulay SR, Honarpisheh MM, Foresto-Neto O, Shi C, Desai J, Zhao ZB, et al. Mitochondria permeability transition versus necroptosis in oxalate-induced AKI. J Am Soc Nephrol (2019) 30:1857–69. doi: 10.1681/ASN.2018121218
54. Müller T, Dewitz C, Schmitz J, Schröder AS, Bräsen JH, Stockwell BR, et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci (2017) 74:3631–45. doi: 10.1007/s00018-017-2547-4
55. Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, et al. A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol (2015) 26:2647–58. doi: 10.1681/ASN.2014080741
56. Hu Z, Zhang H, Yi B, Yang S, Liu J, Hu J, et al. VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis (2020) 11:73. doi: 10.1038/s41419-020-2256-z
57. Kim DH, Choi HI, Park JS, Kim CS, Bae EH, Ma SK, et al. Farnesoid X receptor protects against cisplatin-induced acute kidney injury by regulating the transcription of ferroptosis-related genes. Redox Biol (2022) 54:102382. doi: 10.1016/j.redox.2022.102382
58. Zhou J, Xiao C, Zheng S, Wang Q, Zhu H, Zhang Y, et al. MicroRNA-214-3p aggravates ferroptosis by targeting GPX4 in cisplatin-induced acute kidney injury. Cell Stress Chaperones (2022) 27:325–36. doi: 10.1007/s12192-022-01271-3
59. Sureshbabu A, Patino E, Ma KC, Laursen K, Finkelsztein EJ, Akchurin O, et al. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight (2018) 3:e98411. doi: 10.1172/jci.insight.98411
60. Miao N, Yin F, Xie H, Wang Y, Xu Y, Shen Y, et al. The cleavage of gasdermin D by caspase-11 promotes tubular epithelial cell pyroptosis and urinary IL-18 excretion in acute kidney injury. Kidney Int (2019) 96:1105–20. doi: 10.1016/j.kint.2019.04.035
61. Li Y, Xia W, Wu M, Yin J, Wang Q, Li S, et al. Activation of GSDMD contributes to acute kidney injury induced by cisplatin. Am J Physiol Renal Physiol (2020) 318:F96–F106. doi: 10.1152/ajprenal.00351.2019
62. Xia W, Li Y, Wu M, Jin Q, Wang Q, Li S, et al. Gasdermin E deficiency attenuates acute kidney injury by inhibiting pyroptosis and inflammation. Cell Death Dis (2021) 12:139. doi: 10.1038/s41419-021-03431-2
63. Tonnus W, Maremonti F, Belavgeni A, Latk M, Kusunoki Y, Brucker A, et al. Gasdermin D-deficient mice are hypersensitive to acute kidney injury. Cell Death Dis (2022) 13:792. doi: 10.1038/s41419-022-05230-9
64. Mulay SR, Kulkarni OP, Rupanagudi KV, Migliorini A, Darisipudi MN, Vilaysane A, et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J Clin Invest (2013) 123:236–46. doi: 10.1172/JCI63679
65. Lei Y, Sehnert B, Voll RE, Jacobs-Cachá C, Soler MJ, Sanchez-Niño MD, et al. A multicenter blinded preclinical randomized controlled trial on Jak1/2 inhibition in MRL/MpJ-Fas. Kidney Int (2021) 99:1331–41. doi: 10.1016/j.kint.2021.01.024
66. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol (2015) 16:907–17. doi: 10.1038/ni.3253
67. Mázló A, Jenei V, Burai S, Molnár T, Bácsi A, Koncz G. Types of necroinflammation, the effect of cell death modalities on sterile inflammation. Cell Death Dis (2022) 13:423. doi: 10.1038/s41419-022-04883-w
68. Sarhan M, von Mässenhausen A, Hugo C, Oberbauer R, Linkermann A. Immunological consequences of kidney cell death. Cell Death Dis (2018) 9:114. doi: 10.1038/s41419-017-0057-9
69. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol (2016) 16:676–89. doi: 10.1038/nri.2016.95
70. Sato Y, Yanagita M. Immune cells and inflammation in AKI to CKD progression. Am J Physiol Renal Physiol (2018) 315:F1501–12. doi: 10.1152/ajprenal.00195.2018
71. Vallés PG, Lorenzo AG, Bocanegra V, Vallés R. Acute kidney injury: what part do toll-like receptors play? Int J Nephrol Renovasc Dis (2014) 7:241–51. doi: 10.2147/IJNRD.S37891
72. Kurts C, Panzer U, Anders HJ, Rees AJ. The immune system and kidney disease: basic concepts and clinical implications. Nat Rev Immunol (2013) 13:738–53. doi: 10.1038/nri3523
73. Molitoris BA. Therapeutic translation in acute kidney injury: the epithelial/endothelial axis. J Clin Invest (2014) 124:2355–63. doi: 10.1172/JCI72269
74. Chen H, Fang Y, Wu J, Zou Z, Zhang X, Shao J, et al. RIPK3-MLKL-mediated necroinflammation contributes to AKI progression to CKD. Cell Death Dis (2018) 9:878. doi: 10.1038/s41419-018-0936-8
75. Wang JN, Liu MM, Wang F, Wei B, Yang Q, Cai YT, et al. RIPK1 inhibitor Cpd-71 attenuates renal dysfunction in cisplatin-treated mice via attenuating necroptosis, inflammation and oxidative stress. Clin Sci (Lond) (2019) 133:1609–27. doi: 10.1042/CS20190599
76. Leng J, Zhao W, Guo J, Yu G, Zhu G, Ge J, et al. E-prostanoid 3 receptor deficiency on myeloid cells protects against ischemic acute kidney injury via breaking the auto-amplification loop of necroinflammation. Kidney Int (2023) 103:100–14. doi: 10.1016/j.kint.2022.08.019
77. Li C, Ma QY, Liu XQ, Li HD, Yu MJ, Xie SS, et al. Genetic and pharmacological inhibition of GRPR protects against acute kidney injury via attenuating renal inflammation and necroptosis. Mol Ther (2023) 31:2734–54. doi: 10.1016/j.ymthe.2023.06.016
78. Martin-Sanchez D, Guerrero-Mauvecin J, Fontecha-Barriuso M, Mendez-Barbero N, Saiz ML, Lopez-Diaz AM, et al. Bone marrow-Derived RIPK3 mediates kidney inflammation in acute kidney injury. J Am Soc Nephrol (2022) 33:357–73. doi: 10.1681/ASN.2021030383
79. Zhang S, Li R, Dong W, Yang H, Zhang L, Chen Y, et al. RIPK3 mediates renal tubular epithelial cell apoptosis in endotoxin−induced acute kidney injury. Mol Med Rep (2019) 20:1613–20. doi: 10.3892/mmr.2019.10416
80. Sanz AB, Santamaría B, Ruiz-Ortega M, Egido J, Ortiz A. Mechanisms of renal apoptosis in health and disease. J Am Soc Nephrol (2008) 19:1634–42. doi: 10.1681/ASN.2007121336
81. Lan S, Yang B, Migneault F, Turgeon J, Bourgault M, Dieudé M, et al. Caspase-3-dependent peritubular capillary dysfunction is pivotal for the transition from acute to chronic kidney disease after acute ischemia-reperfusion injury. Am J Physiol Renal Physiol (2021) 321:F335–51. doi: 10.1152/ajprenal.00690.2020
82. Rayego-Mateos S, Marquez-Expósito L, Rodrigues-Diez R, Sanz AB, Guiteras R, Doladé N, et al. : molecular mechanisms of kidney injury and repair. Int J Mol Sci (2022) 23:1542. doi: 10.3390/ijms23031542
83. Balzer MS, Doke T, Yang YW, Aldridge DL, Hu H, Mai H, et al. Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration. Nat Commun (2022) 13:4018. doi: 10.1038/s41467-022-31772-9
84. Ide S, Kobayashi Y, Ide K, Strausser SA, Abe K, Herbek S, et al. Ferroptotic stress promotes the accumulation of pro-inflammatory proximal tubular cells in maladaptive renal repair. Elife (2021) 10:e68603. doi: 10.7554/eLife.68603.sa2
85. Luan J, Fu J, Jiao C, Hao X, Feng Z, Zhu L, et al. IL-18 deficiency ameliorates the progression from AKI to CKD. Cell Death Dis (2022) 13:957. doi: 10.1038/s41419-022-05394-4
86. Srivastava A, Tomar B, Sharma P, Kumari S, Prakash S, Rath SK, et al. RIPK3-MLKL signaling activates mitochondrial CaMKII and drives intrarenal extracellular matrix production during CKD. Matrix Biol (2022) 112:72–89. doi: 10.1016/j.matbio.2022.08.005
87. Imamura M, Moon JS, Chung KP, Nakahira K, Muthukumar T, Shingarev R, et al. RIPK3 promotes kidney fibrosis via AKT-dependent ATP citrate lyase. JCI Insight (2018) 3:e94979. doi: 10.1172/jci.insight.94979
88. Landau SI, Guo X, Velazquez H, Torres R, Olson E, Garcia-Milian R, et al. Regulated necrosis and failed repair in cisplatin-induced chronic kidney disease. Kidney Int (2019) 95:797–814. doi: 10.1016/j.kint.2018.11.042
89. Wilflingseder J, Willi M, Lee HK, Olauson H, Jankowski J, Ichimura T, et al. Enhancer and super-enhancer dynamics in repair after ischemic acute kidney injury. Nat Commun (2020) 11:3383. doi: 10.1038/s41467-020-17205-5
90. Kang HM, Huang S, Reidy K, Han SH, Chinga F, Susztak K. Sox9-positive progenitor cells play a key role in renal tubule epithelial regeneration in mice. Cell Rep (2016) 14:861–71. doi: 10.1016/j.celrep.2015.12.071
91. Kumar S, Liu J, Pang P, Krautzberger AM, Reginensi A, Akiyama H, et al. Sox9 activation highlights a cellular pathway of renal repair in the acutely injured mammalian kidney. Cell Rep (2015) 12:1325–38. doi: 10.1016/j.celrep.2015.07.034
92. Wu WF, Wang JN, Li Z, Wei B, Jin J, Gao L, et al. 7-Hydroxycoumarin protects against cisplatin-induced acute kidney injury by inhibiting necroptosis and promoting Sox9-mediated tubular epithelial cell proliferation. Phytomedicine (2020) 69:153202. doi: 10.1016/j.phymed.2020.153202
93. Valentijn FA, Knoppert SN, Marquez-Exposito L, Rodrigues-Diez RR, Pissas G, Tang J, et al. Cellular communication network 2 (connective tissue growth factor) aggravates acute DNA damage and subsequent DNA damage response-senescence-fibrosis following kidney ischemia reperfusion injury. Kidney Int (2022) 102:1305–19. doi: 10.1016/j.kint.2022.06.030
94. Valentijn FA, Knoppert SN, Pissas G, Rodrigues-Diez RR, Marquez-Exposito L, Broekhuizen R, et al. CCN2 aggravates the immediate oxidative stress-DNA damage response following renal ischemia-reperfusion injury. Antioxid (Basel) (2021) 10:1542. doi: 10.3390/antiox10122020
95. Rayego-Mateos S, Marquez-Exposito L, Basantes P, Tejedor-Santamaria L, Sanz AB, Nguyen TQ, et al. CCN2 activates RIPK3, NLRP3 inflammasome, and NRF2/oxidative pathways linked to kidney inflammation. Antioxid (Basel) (2023) 12:1541. doi: 10.3390/antiox12081541
96. Marquez-Exposito L, Tejedor-Santamaria L, Valentijn FA, Tejera-Muñoz A, Rayego-Mateos S, Marchant V, et al. Oxidative stress and cellular senescence are involved in the aging kidney. Antioxid (Basel) (2022) 11:301. doi: 10.3390/antiox11020301
97. Hu Q, Lyon CJ, Fletcher JK, Tang W, Wan M, Hu TY. Extracellular vesicle activities regulating macrophage- and tissue-mediated injury and repair responses. Acta Pharm Sin B (2021) 11:1493–512. doi: 10.1016/j.apsb.2020.12.014
98. Aghajani Nargesi A, Lerman LO, Eirin A. Mesenchymal stem cell-derived extracellular vesicles for kidney repair: current status and looming challenges. Stem Cell Res Ther (2017) 8:273. doi: 10.1186/s13287-017-0727-7
99. Kosanović M, Milutinovic B, Glamočlija S, Morlans IM, Ortiz A, Bozic M. Extracellular vesicles and acute kidney injury: potential therapeutic avenue for renal repair and regeneration. Int J Mol Sci (2022) 23:3792. doi: 10.3390/ijms23073792
100. Yu Y, Chen M, Guo Q, Shen L, Liu X, Pan J, et al. Human umbilical cord mesenchymal stem cell exosome-derived miR-874-3p targeting RIPK1/PGAM5 attenuates kidney tubular epithelial cell damage. Cell Mol Biol Lett (2023) 28:12. doi: 10.1186/s11658-023-00425-0
Keywords: acute kidney injury, chronic kidney disease, cell death, fibrosis, inflammation, tissue repair
Citation: Guerrero-Mauvecin J, Villar-Gómez N, Rayego-Mateos S, Ramos AM, Ruiz-Ortega M, Ortiz A and Sanz AB (2023) Regulated necrosis role in inflammation and repair in acute kidney injury. Front. Immunol. 14:1324996. doi: 10.3389/fimmu.2023.1324996
Received: 20 October 2023; Accepted: 08 November 2023;
Published: 24 November 2023.
Edited by:
Juan Carlos Cutrin, University of Turin, ItalyReviewed by:
Nestor Ruben Lago, University of Buenos Aires, ArgentinaCopyright © 2023 Guerrero-Mauvecin, Villar-Gómez, Rayego-Mateos, Ramos, Ruiz-Ortega, Ortiz and Sanz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana B. Sanz, YXNhbnpAZmpkLmVz