 Liuling Xiao
Liuling Xiao Miao Xian†
Miao Xian† Chuanchao Zhang
Chuanchao Zhang Qing Yi
Qing Yi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 January 2024
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1322746
This article is part of the Research Topic Current Insights in Cancer Metabolism and T Cell Based Tumor Immunity View all 5 articles
Growing evidence indicates that cellular metabolism is a critical determinant of immune cell viability and function in antitumor immunity and lipid metabolism is important for immune cell activation and adaptation to the tumor microenvironment (TME). Lipid peroxidation is a process in which oxidants attack lipid-containing carbon-carbon double bonds and is an important part of lipid metabolism. In the past decades, studies have shown that lipid peroxidation participates in signal transduction to control cell proliferation, differentiation, and cell death, which is essential for cell function execution and human health. More importantly, recent studies have shown that lipid peroxidation affects immune cell function to modulate tumor immunity and antitumor ability. In this review, we briefly overview the effect of lipid peroxidation on the adaptive and innate immune cell activation and function in TME and discuss the effectiveness and sensitivity of the antitumor ability of immune cells by regulating lipid peroxidation.
Lipid peroxidation is a complex process in which oxidants attack lipids-containing carbon-carbon double bonds, leading to oxidative degradation of lipids, formation of lipid peroxides, and subsequent cellular condition change (1). As an important aspect of lipid metabolism, lipid peroxidation is essential for the execution of cell function and human health (1–3). In recent years, accompanied by the discovery of ferroptosis that is a unique form of regulated cell death characterized by overwhelmed lipid hydroperoxides in cells (4, 5), lipid peroxidation gains increasing attention from researchers. For instance, interferon-γ (IFN-γ) secreted by activated CD8+ T cells in PD-L1 blockade therapy induced increased lipid peroxidation in tumor cells and contributed to the antitumor efficacy of immunotherapy (6). Administration of cyst(e)incase in pancreatic ductal adenocarcinoma mouse model resulted in an accumulation of lipid peroxides and consequent tumor regression (7). However, tumor develops in a complex and dynamic microenvironment that includes not only tumor cells, but also immune cells (8, 9). In TME, immune cells play important roles in tumorigenesis. They can secrete cytotoxic molecules to eliminate tumor cells, or they produce suppressive cytokines to drive chronic inflammation to stimulate cancer metastasis (8, 9). The accumulation of lipid peroxidation in TME affects both tumor and immune cells. Therefore, a comprehensive understanding of the role of lipid peroxidation in different cells in TME is essential for providing new targets and therapeutic approaches to treat cancers (10–12). Several excellent reviews have discussed the role of lipid peroxidation in tumor cells and can be referred to for details (10, 13, 14). In this review, we will focus on how lipid peroxidation mediates immune cell function and how this process affect tumor progression.
Lipid peroxidation is a metabolic process that involves the oxidative degradation of lipids and consists of three stages: initiation, propagation, and termination (1, 11, 15) (Figure 1). The mechanisms of lipid peroxidation are still not fully understood and can be catalyzed by both enzymatic and non-enzymatic formats (1, 11). For the enzymatic format, lipoxygenases (LOX) and P450 oxidoreductase have been implicated in this process (11, 15). In the initiation stage, prooxidants such as hydroxyl radicals (•OH) or peroxyl radicals (ROO•) abstract the allylic hydrogen atom from polyunsaturated fatty acyl-containing phospholipids (PUFA-PL) to form the carbon-centered phospholipid radical (PL•). The allylic hydrogen atom is usually located between two carbon-carbon bonds, and this is why lipid peroxidation is more frequently associated with polyunsaturated fatty acids (PUFA) (11, 16). In the propagation step, phospholipid radical (PL•) rapidly reacts with oxygen and experiences an arrangement to form a phospholipid peroxy radical (PLOO•), which then abstracts a hydrogen from another PUFA-PL molecule to generate a new PL• and phospholipid hydroperoxide (PLOOH). The new PL• can directly recycle to the propagation step and PLOOH can interact with cellular labile iron to generate PLOO• to continue the chain reactions (17). If glutathione peroxidase 4 (GPX4) is present, it can convert highly active PLOOH to stabilized phospholipid alcohol (PLOH) (11, 18). In the termination phase, antioxidants like vitamin E provide a hydrogen atom to the PLOO• and form PLOH (19). The propagation phase is an amplified chain reaction and leads to the production of PLOOHs. If they are not terminated by antioxidants or GPX4, these lipid hydroperoxides (PLOOHs) are unstable and can eventually lead to the formation of secondary products such as malondialdehyde (MDA), 4-hydroxynonenal (4-HNE) and other reactive aldehydes (1, 20). Although appropriate lipid peroxidation plays important roles in physiological processes including cell signaling, immune responses, and apoptosis, excessive lipid peroxidation is associated with cellular oxidative stress and participates in numerous pathological conditions, such as inflammation, neurodegenerative disorders, cardiovascular diseases, and cancers (2, 3, 14, 20).
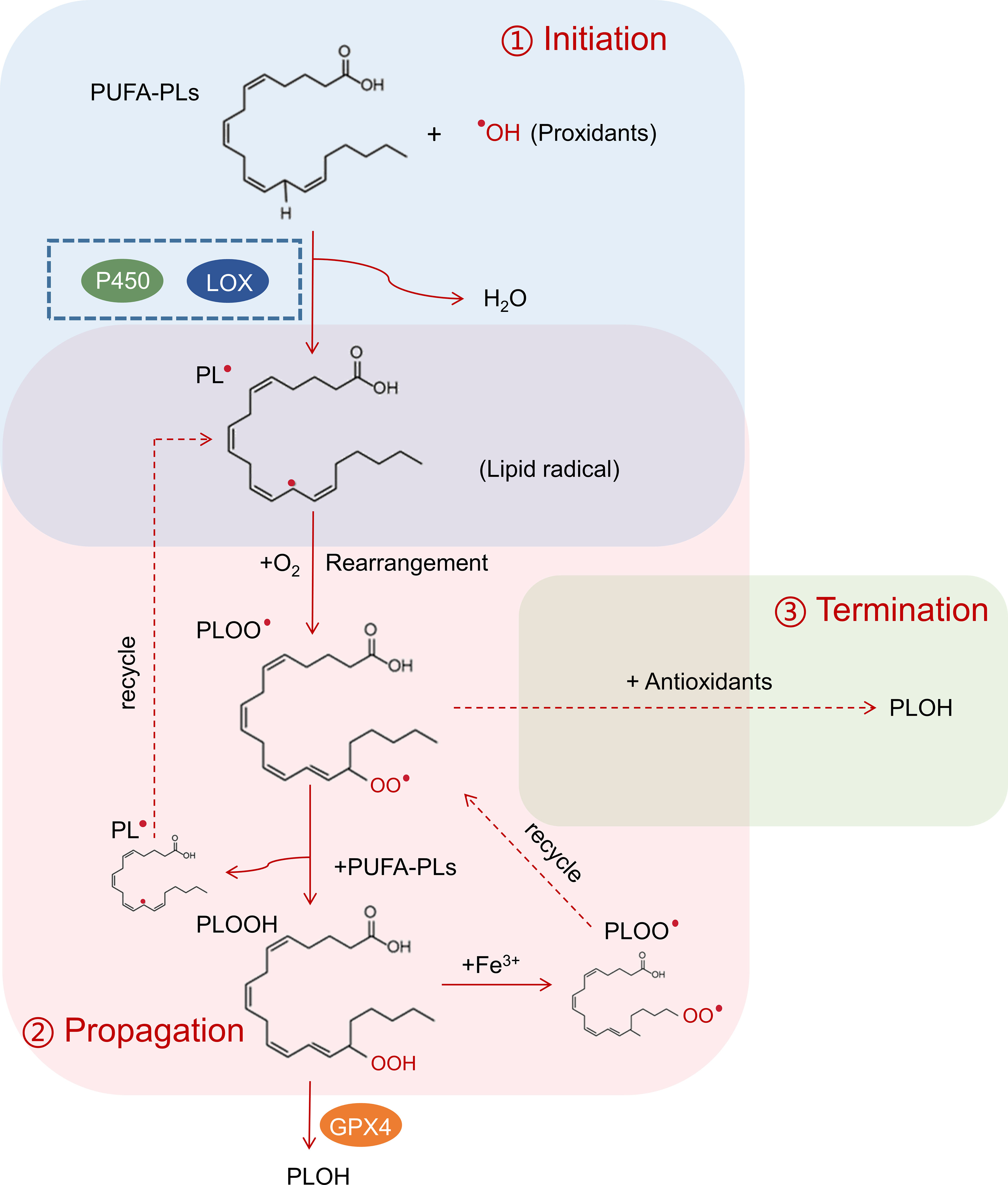
Figure 1 Process of lipid peroxidation. In the initiation process (blue box), interaction between PUFA-PLs and prooxidant hydroxyl radicals (•OH) leads to the formation of carbon-centered phospholipid radical (PL•), which can be catalyzed by lipoxygenases (LOX) and P450 oxidoreductase. In the propagation step (red box), phospholipid radical (PL•) rapidly reacts with oxygen and experiences an arrangement to form a phospholipid peroxy radical (PLOO•), which then abstracts a hydrogen from another PUFA-PL molecule to generate a new PL• and phospholipid hydroperoxide (PLOOH). The new PL• can directly recycle to the propagation step to create a chain reaction. GPX4 can convert highly active PLOOH to stabilize phospholipid alcohol (PLOH) to stop the chain reaction. In the termination phase (green box), antioxidants react with PLOO• and form PLOH.
In the context of cancer, lipid peroxidation contributes to tumor progression by promoting genomic instability, activating pro-inflammatory pathways, and inducing immune evasion (1–3). Recent years have seen significant advancements in our understanding of how lipid peroxidation affects immune cells within TME. From innate to adaptive immunity, lipid peroxidation influences the activation, polarization, and function of various immune cells as we will discuss below.
The immune system is critical for cancer initiation and progression. When a tumor develops, the host spontaneously elicits an immune response to suppress the growth of tumor cells (21). To survive and expand in vivo, tumor cells have developed various methods to avoid the recognition and attack by the immune system, which is called immune evasion (22). Therefore, interactions between tumor cells and immune cells during tumor growth are divided into three stages: elimination, equilibrium, and evasion (22). As studies on TME and cancer immunotherapy accumulate, researchers found that TME comprises a range of distinct immune cells arising from the innate and adaptive immune systems.
In the adaptive immune system, there are T cells (CD4+, CD8+ T, and NKT cells), B cells, and plasma cells in TME. CD8+ T cells are the most well-studied antitumor effect cells that can directly kill tumor cells by cytotoxic cytokines and induction of apoptosis. CD4+ T cells comprise several subsets including T helper 1 (Th1) (23), T helper 2 (Th2) (24), T helper 9 (Th9) (25, 26), T helper 17 (Th17) (27) and regulatory T (Treg) cells (28). The functions of each CD4+ T cell subset are different. Th1 and Th9 cells are antitumor effector cells, while Treg are well-known immunosuppressive cells that reduce the effectiveness of antitumor effector cells and promote tumor progression. As for NKT cells, they can exert either antitumor or immunosuppressive abilities depending on their TCR activation and immunologic context. Relative to T cells, B cells and plasma cells are less well understood and their functions on TME are being explored.
The innate immune system, including natural killer (NK) cells, tumor-associated macrophages (TAM), myeloid-derived suppressor cells (MDSC), dendritic cells (DCs), and granulocytes, acts as the body’s first line of defense and bridge to adaptive immunity (29–31). Similar to CD8+ T cells, NK cells are critical effector cells to control tumor growth in both hematologic malignancies and solid tumors. However, MDSC are well-known immunosuppressive cells in TME. Interestingly, TAM is divided into two classes, inflammatory TAM and immunosuppressive TAM, which suppress or promote tumor progression respectively. In line with TAM, DCs are a heterogeneous hematopoietic lineage with various subsets including conventional DCs (cDCs) and plasmacytoid DCs (pDCs). Although DCs are the most potent professional antigen-presenting cells, they can stimulate T cell activation to suppress tumor cell growth or become tolerant in certain TME to exhibit immunosuppressive properties. Lastly, granulocytes, including neutrophils, eosinophils, basophils, and mast cells, are complex and have distinct functions in different tumor stages.
Although there are various immune cells in TME, it has become increasingly clear that their functions are affected by metabolism conditions. Immune cells engage in a specific metabolic program to maintain their function and activity. However, highly active tumor cells can create profound changes in nutrient, oxygen concentration, and acidity in TME that affect the metabolism program in immune cells and eventually achieve immune evasion. Accumulating evidence suggests that altering immune cell metabolism is a promising approach to enhance cancer immunotherapy, and lipid peroxidation is a potential target to fulfill this goal. Therefore, we will discuss the role of lipid peroxidation in different immune cells in the following sections.
Adaptive immunity is characterized by its ability to recognize and specifically target particular antigens and plays a significant role in the body’s response to cancer. The major adaptive immune cells include T cells and B cells, and the function of adaptive immune cells is affected by lipid peroxidation (Figure 2).
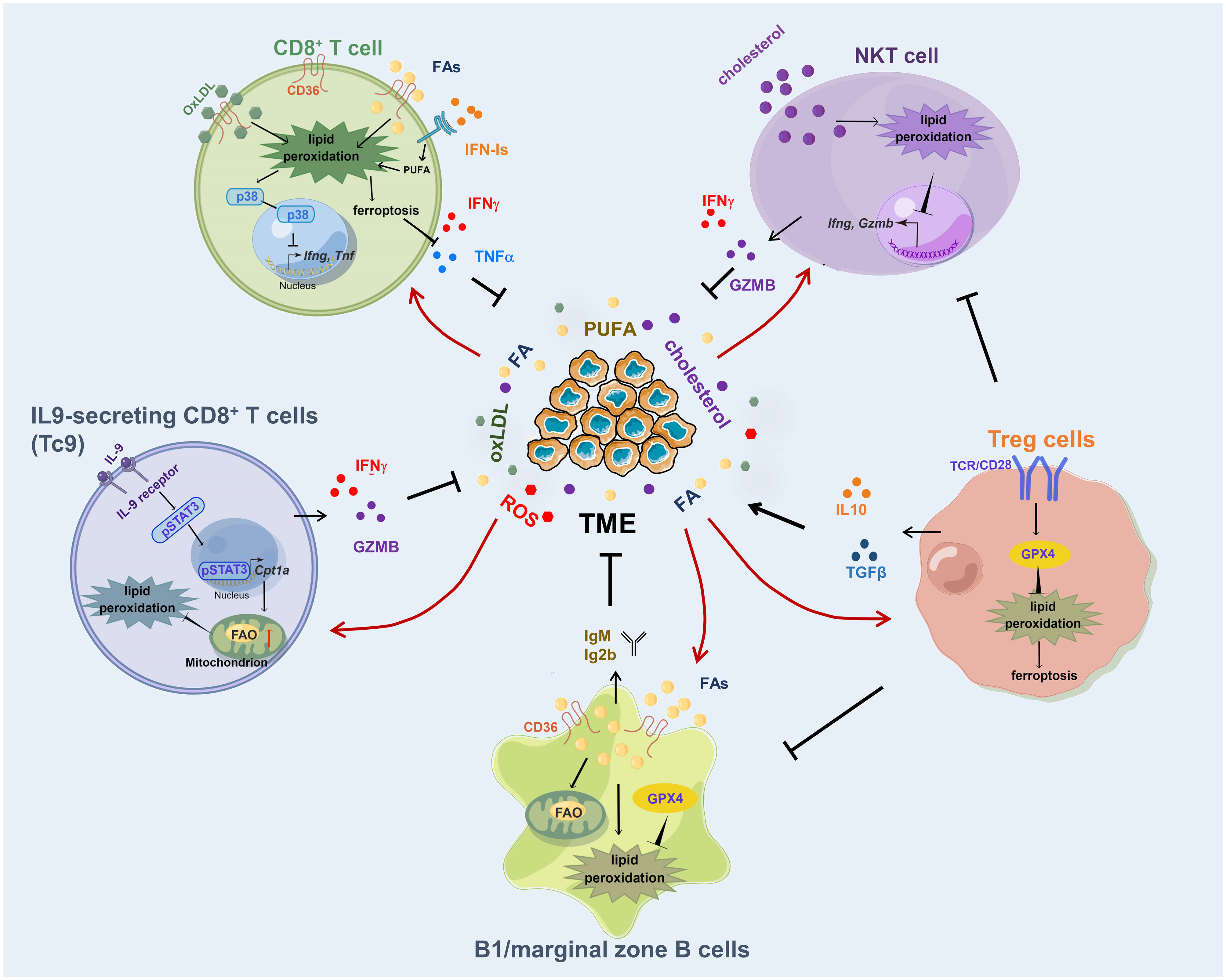
Figure 2 Lipid peroxidation in adaptive immune cells. Excessive fatty acid or OxLDL uptake through CD36 induces an increased lipid peroxidation in CD8+ T cells, resulting in ferroptosis and impaired production of cytotoxic cytokines. IL-9 activates STAT3 to upregulate fatty acid oxidation and mitochondrial activity in Tc9 cells and renders the cells with reduced lipid peroxidation and ferroptosis. GPX4 is required for Treg cells to clear lipid peroxides produced in antigen activation process. Cholesterol induces lipid peroxidation and decreases cytotoxicity of NKT cells. B1 and marginal zone B cells require GPX4 to prevent lipid peroxidation in response to increased fatty acid uptake and lipid droplet storage. FA, fatty acid; OxLDL, oxidized low-density lipoproteins.
T cells are critical components of the adaptive immune system. When naïve T cells recognize their cognate antigens by T cell receptor (TCR), they become activated and proliferative with the support of co-stimulatory signal (32). It has been reported that the lipid peroxidation products 4-HNE and MDA can impair T cell activation by the TCR signaling pathway (33, 34). Upon TCR activation, T cells would induce the production of hydrogen peroxide or superoxide anion in cellular (35) If cellular lipid peroxidation is not scavenged by GPX4 or antioxidants, rapidly accumulated membrane lipid peroxides in T cells would induce ferroptosis and suppress T cell immunity (36). In follicular helper T cells, there is an intensified lipid peroxidation, and GPX4 is the major lipid peroxidation scavenger to support follicular helper T cell survival (37).
Different from tissue-resident T cells, tumor-infiltrating T cells possess unique metabolic features (8). A recent study showed that CD8+ T cells derived from tumors displayed higher levels of lipid peroxidation than those from lymph nodes, and they were sensitive to GPX4 inhibitors-induced ferroptosis (38). Consistent with this result, other groups found that the lipid peroxidation accumulation in tumor-infiltrating CD8+ T cells was the result of excessive uptake of PUFA or oxidized lipid (39, 40). Different from PUFA, short-chain fatty acids (SCFAs) are less susceptible to lipid peroxidation and have different effects on T cells. SCFAs pentanoate and butyrate were discovered to increase the function of mTOR and inhibit class I histone deacetylase activity, which leads to elevated production of effector molecules such as CD25, IFN-γ, and TNF-α, and significantly enhances the anti-tumor activity of T cells (41). Moreover, fatty acid oxidation of certain microbiota-derived SCFAs promoted the differentiation of effector CD8+ T cells with memory potential and enhanced recall capacity (42). As the most powerful effectors in the adaptive immune system to suppress tumor progression, tumor-infiltrating CD8+ T cells utilize fatty acids as an alternative energy to maintain their function in the glucose-deprived TME (8, 43, 44). However, excessive fatty acid uptake through CD36 can induce lipid peroxidation accumulation in CD8+ T cells (45). Increased lipid peroxidation promoted CD8+ T cell ferroptosis, and impaired their cytotoxic cytokines production and antitumor ability (45). Likewise, CD36 also uptake oxidized lipid to upregulate lipid peroxidation in CD8+ T cells and activate the p38 kinase pathway to decrease the transcription of effector cytokine genes, leading to T cell exhaustion and tumor progression (40). Decreasing cellular lipid peroxidation by depleting CD36 or overexpressing GPX4 in CD8+ T cells rescued the function of T cells and achieved enhanced antitumor ability (40, 45). Besides lipids, TME is often enriched with inflammatory cytokines, and tumor-infiltrating CD8+ T cells experience persistent type-I IFN (IFN-I) signaling (46). Chronic IFN-I stimulation perturbs lipid metabolism and redox balance in exhausted CD8+ T cells, leading to aberrant lipid accumulation and elevated lipid peroxidation, which exaggerates metabolic and functional exhaustion of T cells (46).
Besides conventional CD8+ T cells, there is a specific subset of CD8+ T cells named Tc9 cells, which is generated by differentiating naïve CD8+ T cells in Th9-polarizing medium, that resists reactive oxygen species (ROS)-induced lipid peroxidation in TME (47–49). Tc9 cell-derived IL9 activated STAT3 in an autocrine manner, upregulated fatty acid oxidation (FAO) and mitochondrial activity, and rendered Tc9 cells with reduced lipid peroxidation and ferroptosis in TME, leading to longevity and enhanced antitumor activity of adoptively transferred Tc9 cells in vivo (49). This finding is consistent with previous studies that FAO was a key requirement for T cell development as modulating FAO in vivo would enhance the generation of CD8+ T memory cells (50) and increasing FAO and mitochondrial respiratory capacity by PGC-1α/PPAR complexes can support the survival of T cells (51). In addition, treating mice with the agonist of the PPAR signaling pathway has a synergistic effect with anti-PD-1 therapy (51).
Different from CD8+ T cells, studies have shown that intratumoral Treg cells were adapted to lipid-enriched and oxidative-stressed TME (39, 52, 53). In Treg cells, increased CD36 expression is necessary for mitochondrial fitness through peroxisome proliferator-activated receptor-β signaling and programs Treg cells to adapt to a lactic acid-enriched TME (39). Upon activation by antigens in TME, GPX4 was required for Treg to clear lipid peroxides produced in the activation and expansion process to support their function (54–56). When Gpx4 was deficient, excessive lipid peroxides were accumulated in Treg cells in response to TCR and co-stimulatory signals, followed by elevated production of mitochondrial superoxide and IL1β and conversion to Th17-type responses (56). Moreover, the depletion of Gpx4 in Treg cells suppressed tumor growth and enhanced antitumor immunity. However, whether and how GPX4 is highly expressed in Treg cells other than CD8+ T cells is still unknown.
NKT cells are a subset of CD1d-restricted T cells that have properties of both T cells and natural killer cells, and bridge the innate and adaptive immune systems (57). CD1d molecule is a non-classical MHC protein that presents lipid and glycolipid antigens to T cells (58). Although the number of NKT cells in peripheral blood is low, they are enriched in liver, account for 30–40% and 5–25% of liver lymphocytes in mouse and human respectively, and play important roles in suppressing the development of hepatocellular carcinoma (59). Interestingly, the antitumor ability of NKT cells was impaired by cellular lipid peroxidation accumulation (60). In obesogenic-diet-promoted hepatocellular carcinoma, excessive cholesterol production from hepatocytes impaired NKT expansion and cytotoxicity by lipid peroxide accumulation, resulting in a diminished antitumor response (60). However, the underlying mechanism of how lipid peroxidation affects the function of NKT needs further study.
Although tumor-infiltrating T cells adapted their metabolic program in lipid-enriched TME to harness lipids for energy to overcome the shortage of glucose, excessive lipid uptake, particularly oxidized lipids, can lead to the accumulation of lipid peroxidation in T cells. This accumulation has the potential to skew T cell differentiation and promote T cell dysfunction and death. Enhancing the lipid peroxidation clearance capability of effector CD8+ T cells or adoptively transferring lipid peroxidation-resistant CD8+ T cells may be promising strategies to enhance the effectiveness of cancer immunotherapy.
Although much of the immunotherapeutic strategies have been focused on T cell-mediated immunity, accumulative evidence shows that B cells also play crucial roles in modulating tumor responses (61). Similar to T cells, the function of B cells is mediated by lipid metabolism as well (62). For example, it was reported that 4-HNE, the end-product of lipid peroxidation, induced apoptosis in human leukemic B cell lines (63). In primary B1 and marginal zone B cells, they required Gpx4 to prevent lipid peroxidation in the case of increased fatty acid synthesis, lipid uptake, and lipid droplet storage (64). When Gpx4 was deficient, there was increased lipid peroxidation and impaired antibody responses in B1 and marginal zone B cells (64), which may explain why GPX4-positive patients had poor overall survival than GPX4-negative patients in B-cell lymphoma (65).
As innate immune responses are critical for controlling tumor progression, tumor cells take various actions to alter the function of innate immune cells to achieve immune evasion. In line with adaptive immune cells, the development and function of innate immune cells are modulated by cellular lipid peroxidation as well (Figure 3).
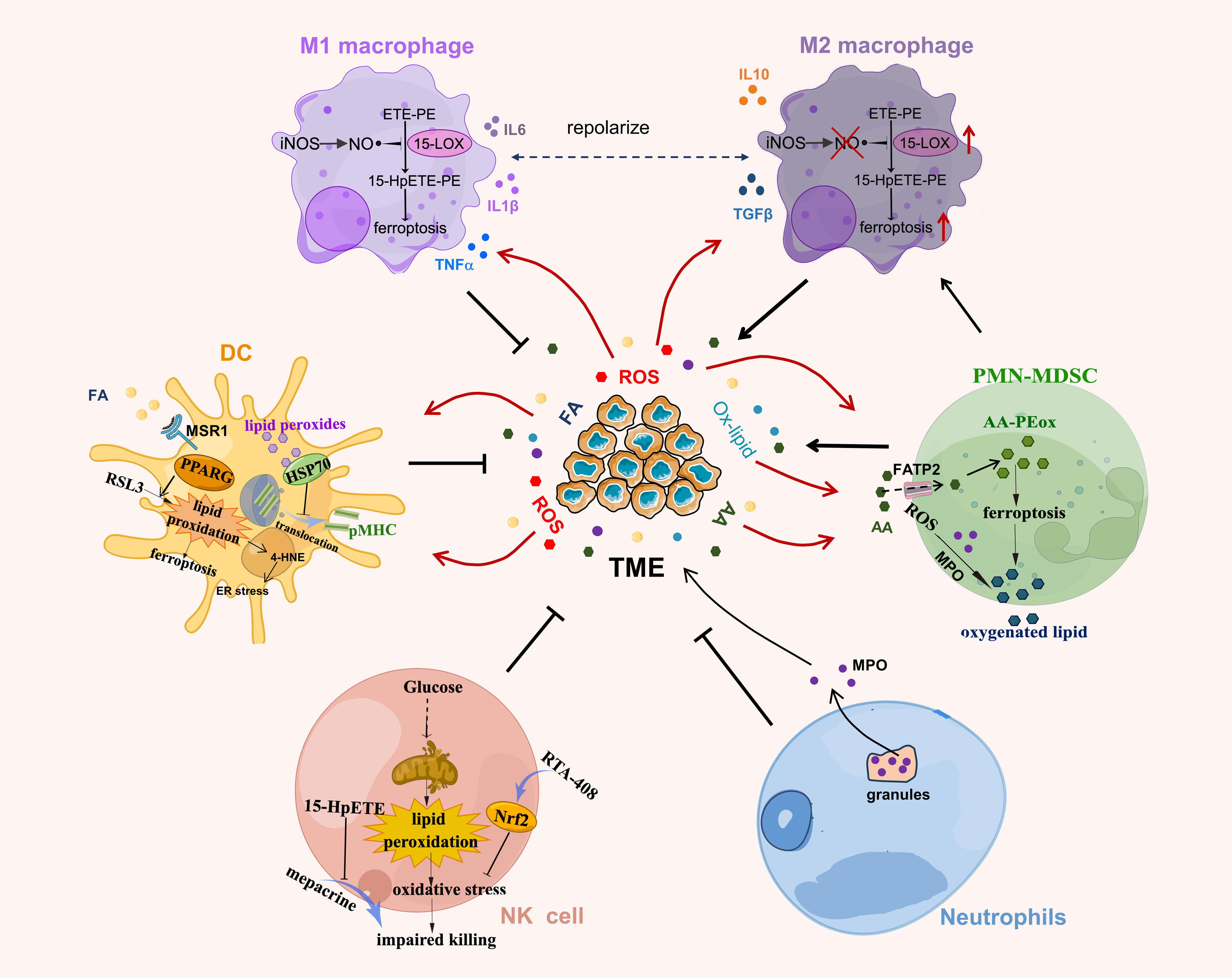
Figure 3 Lipid peroxidation in innate immune cells. NO generated by iNOS interacts with lipid peroxides to reduce the production of lipid peroxide 15-HpETE-PE that is catalyzed by 15-LOX in M1 macrophages. Lipid peroxides in DCs covalently bind to heat shock protein (HSP)70 and prevent the translocation of pMHC from late endosomes/lysosomes to cell surface. PPARγ promotes RSL3-induced lipid peroxidation and ferroptosis in DCs. The lipid peroxide 15-hydroperoxyeicosatetraenoic acid (Hp-ETE) reverses mepacrine-induced inhibition of human NK cell cytotoxicity and lipid peroxidation-associated oxidative stress induced by altered glucose metabolism induces NK cell dysfunction. Activation of antioxidant Nrf2 signaling by RTA-408 reverses this process. PMN-MDSC produce high levels of ROS and myeloperoxidase (MPO) to induce lipid peroxidation in PMN-MDSC, which spontaneously undergo ferroptosis due to increased lipid peroxides (AA-PEox) in TME. AA, arachidonic acid; Ox-lipid, oxidized lipid.
Macrophages are an important component of the immune system and can be polarized into classically activated macrophages (M1) or alternatively activated macrophages (M2) in response to microenvironmental signals (66). In general, M1 macrophages suppress tumor invasion by secreting proinflammatory cytokines whereas M2 macrophages promote tumor progression by secreting anti-inflammatory cytokines (66). Emerging evidence suggests that TAM predominantly exhibits the immunosuppressive M2-like phenotype and has increased lipid accumulation in TME by upregulating CD36 (67, 68). These changes shape them with immunosuppressive phenotype and promote the development of multiple myeloma and liver metastasis (67, 68).
As macrophages are phagocytic immune cells and usually produce ROS in digesting dying cells or microorganisms (69), it is possible that increased lipid content in macrophages induces cellular lipid peroxidation accumulation. Indeed, a recent study showed that lipid-enriched M2 macrophages had a higher level of lipid peroxide products 15-HpETE-PE than M1 macrophages (70). 15-HpETE-PE is catalyzed by the 15-lipoxygenase (15-LOX), which is encoded by Alox5 gene and can oxygenate polyunsaturated fatty acids to lipid peroxides (71). M1 macrophages had a lower expression of 15-LOX and were enriched with iNOS/NO•products. NO• generated by iNOS could interact with lipid peroxides and secondary lipid radical intermediates catalyzed by 15-LOX (70) and substitute GPX4 as an anti-ferroptotic defense, which renders M1 macrophages with high resistance to pharmacologically induced ferroptosis. Interestingly, when transferring iNOS deficient (Nos2-/-) or wild-type bone marrow cells into irradiated recipient mice followed by Lewis lung carcinoma establishment, mice reconstituted with NOS2-deficient bone marrow cells had fewer M1 and similar numbers of M2 macrophages than mice reconstituted with bone marrow from wild-type mice. These results suggest that iNOS is required for M1 macrophage survival in ROS-enriched TME. Unexpectedly, tumor burden was significantly reduced in mice with NOS2-deficient bone marrow chimers compared to the wild-type mice, suggesting that even though the number of M2 macrophages in iNOS-deficient TME is not affected, the immunosuppressive function of M2 macrophages may change. In another study, Hsu et al. reported that lipid peroxidation product 4-HNE inhibited pyroptosis and inflammasome activation of LPS stimulated macrophages (M1 macrophage) by directly disrupting the binding of NIMA-related kinases 7 to NLR Family Pyrin Domain Containing 3 (NLRP3) (72). In support of this result, Kapralov et al. showed that deletion of GPX4 inhibits the survival of M2 other than M1 macrophages (70). Those studies highlighted the role of lipid peroxidation on macrophage function and survival. As the sensitivity of M1 and M2 macrophages to lipid peroxidation products are different, targeting macrophages with lipid peroxidation inducer to eliminate M2 macrophages in TME is a potentially promising strategy to improve cancer immunotherapy efficacy, and more investigations are needed to test this hypothesis.
Different cytokines secreted by macrophages also have distinct effects on lipid peroxidation. Classic M1 macrophages mainly secrete pro-inflammatory factors like TNFα, IL1α, IL1β, IL6, IL12 and IL23. For M2 macrophages, some subtypes of M2 macrophages (M2a, M2c) produce anti-inflammatory factors such as IL10 and TGFβ, while some subtype (M2b) secrete TNFα, IL1β, IL6 and IL10 (73). Among the M1-secreted cytokines, IL6 can induce lipid peroxidation and disrupt iron homeostasis in bronchial epithelial cells, ultimately resulting in ferroptotosis (74). TNFα induces the expression of ACSL3 (75), which is required for exogenous monounsaturated fatty acids to block lipid peroxidation accumulation on the plasma membrane (76). IL1β increased lipid peroxidation in rat hippocampal tissue (77), and intraperitoneal injection of IL1β could induce accumulation of the end-products of lipid peroxidation in all structures of the brain in rats (78). As for cytokines from M2, a very recent study found that IL10 protects oligodendrocyte progenitor cells from ferroptosis by modulating the STAT3/DLK1/ACC axis to decrease the accumulation of lipid peroxidation (79). On the other hand, TGFβ, another cytokine secreted by M2 macrophages, was found to provoke redox imbalance and directly induced lipid peroxidation in hepatocellular carcinoma cells (80).
Innate immune responses are characterized by the initial influx of polymorphonuclear leukocytes, followed by monocyte-derived macrophages, ultimately leading to tissue resolution and the restoration of normal physiology (81). During this process, a crucial shift in the lipid class occurs—from pro-inflammatory lipid mediators derived from arachidonic acid to anti-inflammatory lipid mediators derived from docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA). This transition plays a pivotal role in the resolution phase (81). The acute response initiation involves changes in blood flow stimulated by prostaglandin E2 and prostaglandin I2, while leukotriene B4, produced from arachidonic acid, stimulates the recruitment of polymorphonuclear leukocytes. In later stages, the ω-3 essential fatty acids EPA and DHA are identified as substrates for the biosynthesis of potent anti-inflammatory and pro-resolving endogenous mediators within inflammatory resolving exudates (82). Notably, resolvins counterregulate the release of cytokines/chemokines, thereby suppressing tumor growth and enhancing cancer therapy (83). Given that arachidonic acid, DHA, and EPA are all PUFAs with the potential for lipid peroxidation, further investigation into the role of lipid peroxidation in the resolution process of macrophages is needed.
As professional antigen-presenting cells, DCs were reported to exhibit increased lipid accumulation in tumor-bearing mice and cancer patients (84, 85), which was caused by increased uptake of extracellular fatty acids through macrophage scavenger receptor A (MSR1) and impaired antigens presenting ability of DCs. Additionally, in the lung cancer patients, elevated intracellular lipid accumulation occurs in the form of triglycerides, and antitumor immunity of DCs is suppressed (85). Similar to CD36, MRS1 belongs to the scavenger receptor family and is critical for intracellular lipid transport and uptake (86). Moreover, proliferator-activated receptor γ (PPARγ), the master lipid metabolism regulator (87), can regulate RSL3-induced lipid peroxidation and ferroptosis in DCs. Genetic depletion of PPARγ efficiently restored the function of DCs and improved the antitumor ability of DCs vaccines (88), however, this study did not explore the underlying mechanism and another research showed that sustained PPARγ activation reduced the expression of costimulatory molecules and IL-12 in DCs, ultimately impairing their capacity to prime naïve CD4+ T cells (89). In addition, it was reported that the main composition of lipids accumulated in tumor supernatant-treated DCs was poly-unsaturated fatty acids (90). Since unsaturated fatty acids are highly susceptible to oxidation and are the main substrate of lipid peroxidation (1), DCs from tumor-bearing mice had a significantly higher level of lipid peroxides, such as oxidized linoleic acid and arachidonic acid (90).
Lipid peroxidation is a double-edged sword for the function of DCs. Proper lipid peroxide is essential for DCs maturation. Rothe et al. reported that 12/15-lipoxygenase (LOX) mediated enzymatic lipid oxidation regulates DCs activation and corresponding consecutive T cell responses (91). 12/15-LOX is encoded by Alox15 and represents the murine ortholog of human 15-LOX (92). During the DCs maturation process, the expression of 12/15-LOX was gradually downregulated and determined the threshold of DCs activation via the generation of phospholipid oxidation products. Deletion of the Alox15 in DCs accelerated maturation and aggravated T cell–dependent autoimmune responses (91). Leakage of endosomal antigens into the cytosol is a crucial step in the process of cross-presentation in DCs. Elevated production of reactive oxygen species (ROS) and lipid peroxidation induces the escape of antigens from endosomes to the cytosol, facilitating their degradation and promoting antigen presentation by DCs (93). Consistent with this study, it was observed that increased cellular lipid bodies enhance the antigen cross-presentation of human cDC2s (94), however, whether this process is related to the accumulation of lipid peroxidation remains unclear. Moreover, dysregulation of redox homeostasis induced lipid peroxidation in post-synaptic (DCs), and this was associated with increased efficiency in cross-presentation to CD8+ T cells (95).
However, overwhelming lipid peroxidation impaired the cross-presentation and antitumor ability of DCs (90). Mechanistically, oxidatively truncated lipids can covalently bind to heat shock protein 70 and prevent translocation of pMHC from late endosomes/lysosomes to the cell surface (96). In addition, lipid peroxidation byproducts 4-HNE in DCs would activate X-box binding protein 1 (XBP1) to induce ER stress, which eventually resulted in the inability of DCs to present tumor antigens and stimulate CD8+ T cell responses (97). Accordingly, DC-specific XBP1 deletion enhanced the infiltrating of effector T cells in tumors and extended host survival (97). Consistent with this result, the knockdown of XBP1 in CD8+ T cells also restored their antitumor activity in lipid laden and pro-exhausted TME (98). Therefore, blocking XBP1 or relieving ER stress to improve abnormal lipid or lipid peroxidation accumulation in immune cells may provide a novel approach to enhance the efficacy of cancer immunotherapy.
NK cells can directly kill cancer cells without prior sensitization and play a critical role in the innate immune response against tumors, however, the cytotoxicity of NK cells was impaired in immunosuppressive TME (99). In hepatic metastasis mice, restraint stress increased the content of lipid peroxidation products in liver tissue, which impaired NK cell activity (100). Although this study did not explore the mechanism of how lipid peroxidation impaired NK cell activity, Poznanski et al. recently reported that lipid peroxidation induced glucose metabolism suppression in human NK cells, resulting in NK cell dysfunction and decreased antitumor ability in a nutrient-deprived TME (101). Activation of the antioxidant Nrf2 signaling by RTA-408 can effectively decrease the expression of lipid peroxidation-associated proteins, restore glucose metabolism and cytotoxicity of NK cells, and enhance their antitumor activity (101).
In contrast, other studies reported that hydroxyl radical-mediated lipid peroxidation is required for NK cells to deliver cytotoxic factors (102). In addition, 15S-hydroperoxyeicosatetraenoic acid, the product of 15-LOX catalytic lipid peroxide, effectively reversed mepacrine-induced inhibition of human NK cell cytotoxicity against K562 cells (103). These controversial studies indicate that the function of NK cells is finely regulated by cellular lipid peroxidation and the role of lipid peroxidation on NK cells is not yet fully understood.
Neutrophils are a kind of granulocytes and are featured as lobulated nuclei and azurophilic granules, which are known to release reactive oxygen species during activation (104). About 40 years ago, Claster et al. observed that activated neutrophils could induce lipid peroxidation in human red cells, which may contribute to the pathogenesis of hemolysis-associated infectious diseases and hemoglobin can protect the cell from peroxidative damage (105). Thereafter, it was reported that neutrophil infiltration and related lipid peroxidation accumulation were associated with the process of gastric reperfusion injury (106), Crohn’s disease (107), COVID-19-associated tissue damage (108), and tumor progression (109). In a glioblastoma mouse model, neutrophils can transfer granules containing myeloperoxidase to tumor cells to induce lipid peroxide accumulation and ferroptosis in tumor cells, leading to tumor cell necrosis and glioblastoma progression (109).
Pathologically activated neutrophils are the main immunosuppressive immune cells in TME and are termed polymorphonuclear MDSC (PMN-MDSC). PMN-MDSC exclusively upregulates the expression of fatty acid transport protein 2 (FATP2) that uptake of arachidonic acid and oxidized lipid, which react with the reactive oxygen species and myeloperoxidase to induce the accumulation of lipid peroxidation in PMN-MDSC (110, 111). PMN-MDSC then transferred oxidative lipids to DCs to limit their cross-presentation ability and promote tumor progression (111). Moreover, in a recent study, Kim et al. reported that PMN-MDSC would spontaneously undergo ferroptosis due to increased lipid peroxidation in TME and release oxygenated lipids to limit T cell activity in both mouse model and humans. Inhibition of lipid peroxidation in PMN-MDSC in immunocompetent mice unexpectedly abrogated their suppressive activity and reduced tumor progression to synergize with immune checkpoint blockade to suppress tumor growth (112). MDSC upregulated the expression of system Xc- and neutral -acylsphingosine amidohydrolase (ASAH2) to avoid the excessive lipid peroxidation accumulation in the cells. This study proposed a new method of treating cancers by pharmacological induction of lipid peroxidation and ferroptosis in PMN-MDSC and revealed the complex nature of lipid peroxidation in mediating tumor immunity.
Taken together, cellular lipid peroxidation level is critical for modulating immune cell function and tumor immunity. On one hand, increased lipid peroxidation impairs cytotoxic cytokine secretion of CD8+ T cells and NKT cells, and decreases antibody production by B cells and antigen cross-presentation by DCs. On the other hand, Treg cells are adapted to lipid peroxide-enriched TME, and tumoral PMN-MDSC release oxygenated lipids to limit T cell activity to promote tumor growth. These studies demonstrate that each immune cell has different responses and sensitivity to lipid peroxidation in TME. Therefore, the complex nature of lipid peroxidation in TME must be considered when targeting lipid peroxidation to provide potential therapeutic for cancers.
Lipid peroxidation in the TME is a complex process influenced by various factors. For example, numerous studies have demonstrated that during tumor progression, the increased metabolic activity of cancer cells and infiltrating immune cells can lead to elevated ROS levels. These ROS can initiate lipid peroxidation by abstracting hydrogen atoms from unsaturated fatty acids in cell membranes. With the emergence of new methods and ongoing advancement in research, multiple factors have been discovered to be involved in the regulation of lipid peroxidation in TME.
PUFA is a type of dietary fat that contains more than one unsaturated carbon-carbon double bond in its chemical structure (113). Due to the presence of multiple double bonds in their molecular structure, PUFAs exhibit heightened susceptibility to oxidation, rendering them a primary target for lipid peroxidation (114, 115).
Many investigations have shown PUFA could induce lipid peroxidation in TME. In multiple myeloma patients, PUFA, particularly arachidonic, was notably decreased as the disease advanced through its stages (116). Subsequent in vitro experiments showed that the arachidonic acid-triggered cell death in multiple myeloma cell lines could be reversed by lipid peroxidation inhibitors (116, 117). Interestingly, arachidonic acid is also a ligand of PPARs (118) and can inhibit the growth of lung cancer cells by activating PPARγ and lipid peroxidation (119, 120). PPARγ antagonist (GW9662) was shown to decrease cellular sensitivity to lipid peroxidation-induced ferroptosis (121). However, overexpression of PPARα suppressed erastin-induced lipid peroxidation-related ferroptosis, indicating that the effect of PPAR members on lipid peroxidation may vary. Other PUFAs like DHA are also able to promote ferroptosis by increasing intracellular lipid peroxidation (122). For example, incubation of DHA elevated lipid peroxidation and paracellular permeability in caco-2 colon adenocarcinoma cells (123). Additionally, PPARγ was identified as a component activated by DHA to impair lymphoproliferative stimulation capacity of DCs (124). In breast cancer, acyl-CoA synthetase long-chain family member 4 (ACSL4) catalyzed the esterification of arachidonic acid and adrenic acid and promoted the formation of lipid peroxides (125). Integrated bioinformatics analysis further revealed that elevated levels of arachidonic acid metabolism could serve as a potential biomarker for favorable prognostic outcomes in breast cancer (126). Moreover, arachidonic acid possesses the capacity to synergize with IFN-γ derived from T cells, thereby instigating immunogenic ferroptosis within TME (127).
However, these results contradicted other studies suggesting that TME have elevated level of free fatty acid (45, 128). In these studies, researchers found that in mouse B16 melanoma and Vk*MYC multiple myeloma models, the contents of fatty acids in tumor tissues were higher than in normal tissues. Fatty acid, particularly arachidonic acid, significantly increased lipid peroxidation and ferroptosis of CD8+ T cells (45). Similarly, the accumulation of long-chain fatty acids in TME of pancreatic ductal adenocarcinoma drives lipotoxicity in tumor-infiltrating CD8+ T cells (128). Although the detection of fatty acid content is not consistent across different tumors, these findings collectively suggest that PUFAs, especially arachidonic acid, are important inducers of lipid peroxidation in the TME.
ROS encompass a group of unstable oxygen derivatives, including hydrogen peroxide (H2O2), superoxide anion (O2−), hypochlorous acid (HOCl), singlet oxygen (O2), and hydroxyl radical (•OH) (129). Numerous physiological processes within cells generate ROS, with mitochondria accounting for approximately 90% of cellular ROS production. Tumor cells, which have a heightened demand for energy to support their proliferation, have been shown to produce elevated levels of ROS compared to normal cells (130). Additionally, hypoxic TME can further stimulate the generation of mitochondrial ROS (131). The increased levels of ROS produced by tumor cells can impact immune cells within the TME. ROS interacts with membrane phospholipids, inducing lipid peroxidation. These phospholipids, rich in PUFAs, are particularly susceptible to ROS-induced damage (132). This process can initiate oxidative stress through a feedback loop triggered by fatty acid peroxidation, leading to alterations in the lipid bilayer of cell membranes and the generation of free radicals (133). Furthermore, PUFA themselves, after reacting with ROS and undergoing lipid peroxidation, can transform into reactive free radicals, thus propagating lipid peroxidation chain reactions (134).
Among the various ROS molecules, the hydroxyl radical (•OH) stands out as one of the most common and potent free radicals (135). This radical is generated through the well-known oxidation process termed the Fenton reaction, where the combination of iron (Fe2+) and hydrogen peroxide (H2O2) gives rise to hydroxyl radicals. In the Fenton reaction, iron (Fe) serves as a catalyst, especially under acidic conditions, to greatly enhance the oxidative potential of H2O2 (136). Consequently, the presence of iron in TME plays a pivotal role in promoting the production of hydroxyl radicals, which in turn can induce lipid peroxidation.
Within TME, tumor cells exhibit an overexpression of iron importers, such as DMT1 (137), and a downregulation of iron exporter ferroportin (138). This pattern reflects their heightened demand for iron, driven by increased metabolic activities associated with malignancy. In addition, M2 TAM in TME have been identified as a source of iron (139), as they exhibit an iron-releasing phenotype characterized by reduced ferritin and increased ferroportin expression (140). Consequently, in an iron-enriched TME, which fuels tumor progression and metastasis, ample opportunities arise for the generation of ROS and subsequent lipid peroxidation.
Chemotherapy drugs are a class of medications that are designed to target cancer cells to destroy or inhibit their growth (141). In recent years, accumulating evidence has shown that Several FDA-approved anti-tumor drugs can also trigger lipid peroxidation. For example, sorafenib, a protein kinase inhibitor for the treatment of kidney and liver cancer, induces lipid peroxidation and ferroptosis by suppressing the activity of system Xc− in various types of cancer cells (142–145). In addition, acyl-coA synthetase long chain family member 4 (ACSL4) and glutathione s-transferase zeta 1(GSTZ1) is required for sorafenib-induced lipid peroxidation and ferroptosis (146, 147). Another extensively utilized antitumor agent, cisplatin, has been established to increase lipid peroxide by directly depleting intracellular glutathione S-transferase (GST) and glutathione (GSH) (148–150), and exhibits a synergistic therapeutic effect when combined with erastin (151) and dihydroartemisinin (152). Poly (ADP-ribose) polymerase (PARP) inhibitor olaparib is efficacious in treating ovarian cancer (153). Recent studies showed that olaparib could promote lipid peroxidation and ferroptosis in ovarian cancer by suppressing SLC7A11, and treatment tumor-bearing mice with ferroptosis inducers sulfasalazine markedly sensitized the antitumor efficacy of olaparib (154, 155). These studies imply that chemotherapy drugs induce lipid peroxidation within tumors. Hence, patients undergoing chemotherapy treatment might experience improved responses by augmenting their regimen with additional agents that promote lipid peroxidation and ferroptosis.
Glutathione is a tripeptide molecule composed of three amino acids: cysteine, glycine, and glutamic acid (156). It plays a pivotal role in the cellular antioxidant defense system and is essential for preventing lipid peroxidation caused by free radicals and oxygen (157). During oxidative stress, GSH undergoes conversion by GSH-dependent peroxidases, leading to the formation of GSH disulfide (GSSG) through its interaction with ROS (158). With the involvement of GSH, the GPX4 transforms lipid hydroperoxides into lipid alcohols and this process effectively guards against oxidative lipid damage, lipid peroxidation, and the initiation of ferroptosis (159, 160).
Analysis of tumor samples has shown that certain types of cancer, including breast, lung, ovarian, and head and neck cancers, exhibit notably elevated levels of GSH (161). In breast tumors, GSH levels were more than twice higher than normal breast tissue, and these levels were even higher in lymph node metastases (162). In line with this, another study also reported a significant increase in the levels of GSSG (oxidized glutathione) and total glutathione in breast cancer tissues compared to adjacent cancer-free tissues (163). Similar observations were made in the cases of lung cancer, ovarian cancer, and squamous cell carcinoma (164–167). In addition, patients with elevated GSH levels in ovarian tumor tissues exhibited noticeably lower progression-free survival and overall survival rates compared to those with lower GSH levels (168). Moreover, in the case of tumors with elevated GSH levels, a corresponding decrease in lipid peroxidation was observed (169, 170), indicating that antioxidant molecules contribute to creating a microenvironment conducive to tumor cell proliferation. Nevertheless, certain types of tumors exhibit distinct scenarios, where the GSH levels within tumor tissues are notably lower than in normal tissues, as seen in brain cancers. Interestingly, this reduction of GSH levels in brain tumor tissues corresponded with an increase in lipid peroxidation, as indicated by elevated levels of thiobarbituric acid reactive substances (TBARS) (171). Furthermore, when compared to low-grade brain tumors, high-grade brain tumors exhibited a significant increase in lipid peroxidation along with a reduction in GSH (171). Collectively, these reports indicate there is an inverse correlation between lipid peroxidation and GSH level in tumor tissues, and GSH may serve as guards against lipid peroxidation to promote the development of tumors.
Small-molecule compounds are organic chemicals with low molecular weight that readily penetrate cells and influence molecular pathways by targeting vital proteins (172). In recent years, many ferroptosis inducers and inhibitors have been developed by regulating cellular lipid peroxidation across diverse tumor-related models, aiming to assess their impact on tumor cells as well as the immune cells within the tumor microenvironment.
One of the most frequently used ferroptosis inducers in scientific research is RSL3, which directly binds to GPX4 and deactivates its peroxidase activity, leading to the lethal accumulation of lipid peroxidation (173). Other compounds, such as ML162 (174), DPI7 (175), FIN56 (176), and sorafenib (143), also target GPX4 and induce the accumulation of lipid peroxidation, ultimately promoting ferroptosis. Erastin represents a different category of ferroptosis inducers. Its functional mechanism involves the inhibition of the cystine-glutamate antiporter system Xc− activity (142), which results in cells being unable to synthesize GSH. Consequently, these cells undergo excessive lipid peroxidation and ultimately succumb to ferroptosis. Sorafenib was also reported to inhibit Xc− activity (177). Though numerous studies have shown that ferroptosis inducers possess promising potential in inhibiting tumor cell growth, their potential effects on impairing the immune cells make the situation complicated (178). For example, it has been reported that CD8+ T cells have facilitated tumor ferroptosis by releasing IFN-γ, which disrupts the cystine uptake by tumor cells, triggers lipid peroxidation accumulation, and ultimately results in ferroptotic tumor cell death (6). Considering this perspective, the combination of immunotherapy with ferroptosis induction emerges as a promising strategy to effectively impair tumor growth. However, others also found that CD8+ T cells are more sensitive to ferroptosis inducers RSL3 than B16 and MC38 cancer cells (38). Intratumor CD8+ T cells underwent lipid peroxidation in lipid laden TME and became dysfunctional (40, 45). As existing lipid peroxidation/ferroptosis inducers have poor selectivity (179), targeting lipid peroxidation to enhance cancer therapy must consider the complex nature of TME.
High-throughput screening efforts have identified ferrostatin-1 and liproxstatin-1 as potent inhibitors of ferroptosis by decreasing lipid peroxidation (180–182). Cui et al. found that within the TME of gastric cancer, L-kynurenine was observed to induce lipid peroxidation and ferroptotic cell death in NK cells, which can be inhibited by ferrostatin-1 (183). In addition, treatment of immunocompetent mice with liproxstatin-1 reversed the suppressive activity of PMN-MDSCs, resulting in reduced tumor progression (112). Furthermore, when combined with immune checkpoint blockade, liproxstatin-1 displayed synergistic effects in suppressing tumor growth (112). These results showed that the use of lipid peroxidation inhibitors also holds promise in enhancing immune response in certain cancers. However, controversial results were found in other reports. In the B16 melanoma model, liproxstatin-1 treatment did not reduce tumor growth but diminished the therapeutic efficacy of immune checkpoints (6). Additionally, liproxstatin-1 administration abolished radiotherapy-induced tumor suppression in ID8 ovarian cancer (184). Therefore, as both immune cells and tumor cells respond to lipid peroxidation-induced stress within TME, cell-specific delivery of lipid peroxidation inducers or inhibitors presents a promising method to enhance cancer therapeutic outcomes.
PDT is a relatively non-invasive anticancer approach that utilizes a photosensitive drug, known as a photosensitizer, along with specific wavelength light irradiation. This combination generates ROS within the tumor tissues, thereby impeding tumor progression (185). PDT can provide the source of active oxygen for the Fenton reaction, which speeds up lipid peroxidation and improves the efficacy of PDT in antitumor therapy (186). Simultaneously, PDT triggers acute inflammation and activates both innate and adaptive immunity, inducing damage-associated molecular patterns (DAMPs) (187, 188). Considering these aspects collectively, it becomes reasonable to explore the combination of PDT with lipid peroxidation-related agents as a strategy to activate immune responses and eliminate tumor responses (189).
Nanoparticle materials have been introduced to selectively deliver lipid peroxidation inducer and combined PDT together to stimulate the immune response (186), leading to the elimination of tumor cells. Xu et al. devised a nano-platform wherein hemoglobin is conjugated with the photosensitizer chlorin e6 to construct a 2-in-1 nanoplatform with sorafenib loaded (190). This nanoplatform effectively merges oxygen-enhanced PDT, clinical chemotherapy drugs, and lipid peroxidation-induced ferroptosis to target tumor cells. Apart from sorafenib’s direct role in inducing the lethal accumulation of lipid peroxidation, PDT intensifies the ferroptosis process by recruiting immune cells that release IFN-γ, thereby sensitizing the tumor cells to ferroptosis (190). In addition, another group established a cytotoxic T-cell-inspired oncolytic system to precisely lyse cancer cells by NIR-light-controlled lipid peroxidation, which specifically induces lipid peroxidation in tumor cells and enhances T-cell-based antitumor ability (191).
Lipid peroxidation varies significantly among different types of tumors, individuals, and even in response to various therapeutic strategies. Tumor and immune cells exhibit distinct reactions in exposure to the same levels of lipid peroxidation. Therefore, nanoparticles that selectively deliver lipid peroxidation inducer to tumor cells may be a promising way to enhance cancer therapy. For example, nanoparticles co-delivering dihydroartemisinin and lipid peroxidation inducer RSL3 have been shown to efficiently induce ferroptosis in tumor cells of pancreatic ductal adenocarcinoma, melanoma, and metastatic breast tumors, and trigger T cell-based antitumor immunity (192, 193). Notably, the co-delivery system has also demonstrated a synergistic effect in inhibiting pancreatic ductal adenocarcinoma tumor growth when combined with PD-L1 blockade therapy (192). However, the safety and efficiency of this co-delivery system are still under investigation.
Although it has been demonstrated that lipid metabolism plays important roles in modulating immune cell function and targeting lipid metabolism is a novel strategy to enhance antitumor immunity, the role and clinical potential of lipid peroxidation in immune cells remain unclear. Since the discovery that therapy-resistant cancer cells exhibited sensitivity to ferroptosis (194–196), modulating cellular lipid peroxidation and induction of ferroptosis in cancer cells have been considered a potential therapeutic strategy. However, in most of these studies, the role of lipid peroxidation in the immune system was not fully considered as these studies were based on xenograft mouse tumor models. In fact, as a double-edge sword, lipid peroxidation not only affects the survival of tumor cells, but also influences the function of the immune cells (Figure 4). In this review, we summarize the effect of lipid peroxidation on the function of various immune cells in TME and emphasize the distinct sensitivities of different immune cells to tumoral lipid peroxides. These studies provide new strategies to enhance the efficacy of immunotherapy by targeting lipid peroxidation in immune cells in a cell-specific manner. For example, ex vivo pharmacologic or genetic reprogramming of CD8+ T cell lipid peroxidation prior to adoptive cellular therapy was able to enhance their effector function or longevity in vivo (40, 45, 49).
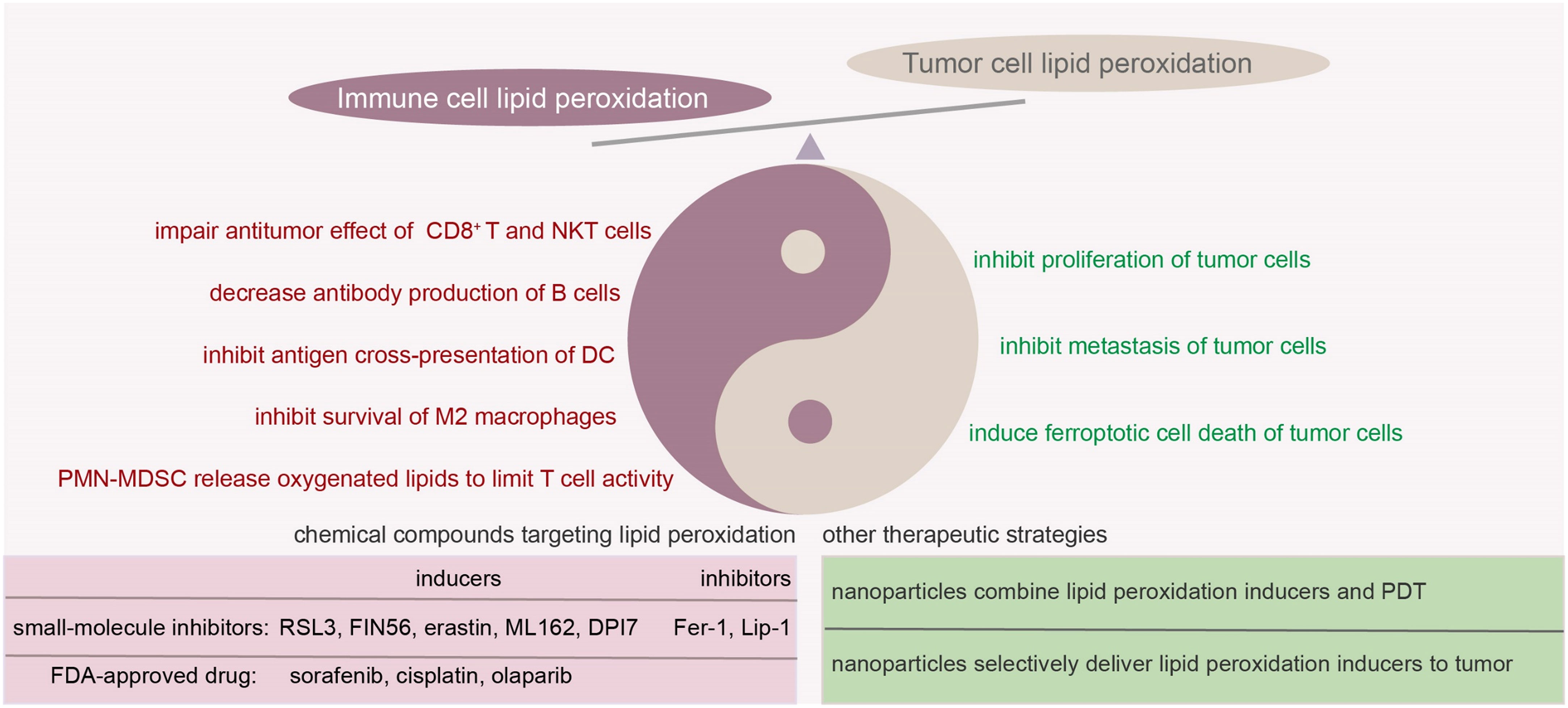
Figure 4 Double edged sword of lipid peroxidation in tumor microenvironment and potential strategies to target lipid peroxidation. In TME, tumor cells and immune cells respond differently when exposed to the same levels of lipid peroxidation. In the case of tumor cells, an increase in lipid peroxidation often aids in inhibiting tumor progression. For certain immune cells, overwhelming lipid peroxidation might potentially weaken their immune responses. Several small-molecule compounds and other therapeutical strategies were raised to target lipid peroxidation. When considering the regulation of lipid peroxidation to inhibit tumor progression, it’s essential to account for both tumor cells and immune cells. The scenarios can become quite intricate and necessitate case-by-case evaluation. Fer-1, ferrostatin-1; Lip-1, liproxstatin-1; PDT, photodynamic therapy.
Current studies have provided new insights into the molecular mechanisms of lipid peroxidation in regulating immune cell function in TME. The interaction among immune cells, tumor cells, and nutrients in TME plays an important role in the induction of lipid peroxidation in the cells, tumor development, and tumor immunity. However, the detailed underlying mechanisms driving different lipid peroxidation and sensitivity in different cell subsets await further exploration. Future work should address the specific source of lipid peroxides in TME, the key molecules in driving lipid peroxidation to ferroptosis, as well as lipid peroxidation interdependence of immune cells and cancer cells in TME. It is important to understand whether and by what mechanisms immune cells can obtain maximum antitumor ability by targeting lipid peroxidation. Undoubtably, a comprehensive understanding of lipid peroxidation in both tumor cells and immune cells within TME will provide novel and feasible therapeutic strategies for cancer.
QY: Conceptualization, Writing – review & editing. LX: Conceptualization, Writing – original draft, Writing – review & editing. MX: Writing – original draft. CZ: Writing – original draft. QG: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Startup Support from Houston Methodist Research Institute, Houston Methodist Hospital, by Cancer Prevention & Research Institute of Texas Recruitment of Established Investigator Award (RR180044), and by NCI R01s CA200539, CA239255, CA282099 and CA2855209. QY and his research group were also supported by NCI R01s (CA211073 and CA214811) and by Cancer Prevention & Research Institute of Texas High-Impact/High-Risk Research Award (RP210868).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev (2014) 2014:360438. doi: 10.1155/2014/360438
2. Barrera G, Pizzimenti S, Dianzani MU. Lipid peroxidation: control of cell proliferation, cell differentiation and cell death. Mol Aspects Med (2008) 29(1-2):1–8. doi: 10.1016/j.mam.2007.09.012
3. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun (2017) 482(3):419–25. doi: 10.1016/j.bbrc.2016.10.086
4. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
5. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab (2020) 32(6):920–37. doi: 10.1016/j.cmet.2020.10.011
6. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature (2019) 569(7755):270–4. doi: 10.1038/s41586-019-1170-y
7. Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science (2020) 368(6486):85–9. doi: 10.1126/science.aaw9872
8. Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer (2020) 20(9):516–31. doi: 10.1038/s41568-020-0273-y
9. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal (2020) 18(1):59. doi: 10.1186/s12964-020-0530-4
10. Barrera G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol (2012) 2012:137289. doi: 10.5402/2012/137289
11. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
12. Xu H, Ye D, Ren M, Zhang H, Bi F. Ferroptosis in the tumor microenvironment: perspectives for immunotherapy. Trends Mol Med (2021) 27(9):856–67. doi: 10.1016/j.molmed.2021.06.014
13. Bartolacci C, Andreani C, El-Gammal Y, Scaglioni PP. Lipid metabolism regulates oxidative stress and ferroptosis in RAS-driven cancers: A perspective on cancer progression and therapy. Front Mol Biosci (2021) 8:706650. doi: 10.3389/fmolb.2021.706650
14. Gonzalez MJ. Lipid peroxidation and tumor growth: an inverse relationship. Med Hypotheses (1992) 38(2):106–10. doi: 10.1016/0306-9877(92)90081-M
15. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13(1):81–90. doi: 10.1038/nchembio.2238
16. Hajeyah AA, Griffiths WJ, Wang Y, Finch AJ, O’Donnell VB. The biosynthesis of enzymatically oxidized lipids. Front Endocrinol (Lausanne) (2020) 11:591819. doi: 10.3389/fendo.2020.591819
17. Repetto MG, Ferrarotti NF, Boveris A. The involvement of transition metal ions on iron-dependent lipid peroxidation. Arch Toxicol (2010) 84(4):255–62. doi: 10.1007/s00204-009-0487-y
18. Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal (2018) 29(1):61–74. doi: 10.1089/ars.2017.7115
19. Cedrowski J, Litwinienko G, Baschieri A, Amorati R. Hydroperoxyl radicals (HOO(.)): vitamin E regeneration and H-bond effects on the hydrogen atom transfer. Chemistry (2016) 22(46):16441–5. doi: 10.1002/chem.201603722
20. Gentile F, Arcaro A, Pizzimenti S, Daga M, Cetrangolo GP, Dianzani C, et al. DNA damage by lipid peroxidation products: implications in cancer, inflammation and autoimmunity. AIMS Genet (2017) 4(2):103–37. doi: 10.3934/genet.2017.2.103
21. Disis ML. Immune regulation of cancer. J Clin Oncol (2010) 28(29):4531–8. doi: 10.1200/JCO.2009.27.2146
22. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3(11):991–8. doi: 10.1038/ni1102-991
23. Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity (2007) 26(5):579–91. doi: 10.1016/j.immuni.2007.03.014
24. Chang HC, Zhang S, Thieu VT, Slee RB, Bruns HA, Laribee RN, et al. PU.1 expression delineates heterogeneity in primary Th2 cells. Immunity (2005) 22(6):693–703. doi: 10.1016/j.immuni.2005.03.016
25. Lu Y, Wang Q, Xue G, Bi E, Ma X, Wang A, et al. Th9 cells represent a unique subset of CD4(+) T cells endowed with the ability to eradicate advanced tumors. Cancer Cell (2018) 33(6):1048–1060 e1047. doi: 10.1016/j.ccell.2018.05.004
26. Bi E, Ma X, Lu Y, Yang M, Wang Q, Xue G, et al. Foxo1 and Foxp1 play opposing roles in regulating the differentiation and antitumor activity of T(H)9 cells programmed by IL-7. Sci Signal (2017) 10(500). doi: 10.1126/scisignal.aak9741
27. Meisel M, Hermann-Kleiter N, Hinterleitner R, Gruber T, Wachowicz K, Pfeifhofer-Obermair C, et al. The kinase PKCalpha selectively upregulates interleukin-17A during Th17 cell immune responses. Immunity (2013) 38(1):41–52. doi: 10.1016/j.immuni.2012.09.021
28. Ramirez DE, Turk MJ. Th1-like Treg cells are dressed to suppress anti-tumor immunity. Immunity (2023) 56(7):1437–9. doi: 10.1016/j.immuni.2023.06.014
29. Hubler MJ, Kennedy AJ. Role of lipids in the metabolism and activation of immune cells. J Nutr Biochem (2016) 34:1–7. doi: 10.1016/j.jnutbio.2015.11.002
30. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14(10):1014–22. doi: 10.1038/ni.2703
31. Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol (2017) 14(12):717–34. doi: 10.1038/nrclinonc.2017.101
32. Shah K, Al-Haidari A, Sun J, Kazi JU. T cell receptor (TCR) signaling in health and disease. Signal Transduct Target Ther (2021) 6(1):412. doi: 10.1038/s41392-021-00823-w
33. Willis MS, Thiele GM, Tuma DJ, Klassen LW. T cell proliferative responses to malondialdehyde-acetaldehyde haptenated protein are scavenger receptor mediated. Int Immunopharmacol (2003) 3(10-11):1381–99. doi: 10.1016/S1567-5769(03)00136-X
34. Cambiaggi C, Dominici S, Comporti M, Pompella A. Modulation of human T lymphocyte proliferation by 4-hydroxynonenal, the bioactive product of neutrophil-dependent lipid peroxidation. Life Sci (1997) 61(8):777–85. doi: 10.1016/S0024-3205(97)00559-6
35. Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med (2002) 195(1):59–70. doi: 10.1084/jem.20010659
36. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med (2015) 212(4):555–68. doi: 10.1084/jem.20140857
37. Yao Y, Chen Z, Zhang H, Chen C, Zeng M, Yunis J, et al. Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol (2021) 22(9):1127–39. doi: 10.1038/s41590-021-00996-0
38. Drijvers JM, Gillis JE, Muijlwijk T, Nguyen TH, Gaudiano EF, Harris IS, et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8(+) T cells. Cancer Immunol Res (2021) 9(2):184–99. doi: 10.1158/2326-6066.CIR-20-0384
39. Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol (2020) 21(3):298–308. doi: 10.1038/s41590-019-0589-5
40. Xu S, Chaudhary O, Rodriguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity (2021) 54(7):1561–1577 e1567. doi: 10.1016/j.immuni.2021.05.003
41. Luu M, Riester Z, Baldrich A, Reichardt N, Yuille S, Busetti A, et al. Microbial short-chain fatty acids modulate CD8(+) T cell responses and improve adoptive immunotherapy for cancer. Nat Commun (2021) 12(1):4077. doi: 10.1038/s41467-021-24331-1
42. Bachem A, Makhlouf C, Binger KJ, de Souza DP, Tull D, Hochheiser K, et al. Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8(+) T cells. Immunity (2019) 51(2):285–297 e285. doi: 10.1016/j.immuni.2019.06.002
43. Zhang L, Romero P. Metabolic control of CD8(+) T cell fate decisions and antitumor immunity. Trends Mol Med (2018) 24(1):30–48. doi: 10.1016/j.molmed.2017.11.005
44. Wu D, Chen Y. Lipids for CD8(+) TILs: beneficial or harmful? Front Immunol (2022) 13:1020422. doi: 10.3389/fimmu.2022.1020422
45. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab (2021) 33(5):1001–1012 e1005. doi: 10.1016/j.cmet.2021.02.015
46. Chen W, Teo JMN, Yau SW, Wong MY, Lok CN, Che CM, et al. Chronic type I interferon signaling promotes lipid-peroxidation-driven terminal CD8(+) T cell exhaustion and curtails anti-PD-1 efficacy. Cell Rep (2022) 41(7):111647. doi: 10.1016/j.celrep.2022.111647
47. Visekruna A, Ritter J, Scholz T, Campos L, Guralnik A, Poncette L, et al. Tc9 cells, a new subset of CD8(+) T cells, support Th2-mediated airway inflammation. Eur J Immunol (2013) 43(3):606–18. doi: 10.1002/eji.201242825
48. Lu Y, Hong B, Li H, Zheng Y, Zhang M, Wang S, et al. Tumor-specific IL-9-producing CD8+ Tc9 cells are superior effector than type-I cytotoxic Tc1 cells for adoptive immunotherapy of cancers. Proc Natl Acad Sci U S A (2014) 111(6):2265–70. doi: 10.1073/pnas.1317431111
49. Xiao L, Ma X, Ye L, Su P, Xiong W, Bi E, et al. IL-9/STAT3/fatty acid oxidation-mediated lipid peroxidation contributes to Tc9 cell longevity and enhanced antitumor activity. J Clin Invest (2022) 132(7). doi: 10.1172/JCI153247
50. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi: 10.1038/nature08097
51. Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8(+) T cells and facilitates anti-PD-1 therapy. Cancer Immunol Res (2018) 6(11):1375–87. doi: 10.1158/2326-6066.CIR-18-0095
52. Mougiakakos D, Johansson CC, Jitschin R, Bottcher M, Kiessling R. Increased thioredoxin-1 production in human naturally occurring regulatory T cells confers enhanced tolerance to oxidative stress. Blood (2011) 117(3):857–61. doi: 10.1182/blood-2010-09-307041
53. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature (2013) 499(7459):485–90. doi: 10.1038/nature12297
54. Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, et al. Mitochondrial integrity regulated by lipid metabolism is a cell-intrinsic checkpoint for treg suppressive function. Cell Metab (2020) 31(2):422–437 e425. doi: 10.1016/j.cmet.2019.11.021
55. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
56. Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep (2021) 35(11):109235. doi: 10.1016/j.celrep.2021.109235
57. Krijgsman D, Hokland M, Kuppen PJK. The role of natural killer T cells in cancer-A phenotypical and functional approach. Front Immunol (2018) 9:367. doi: 10.3389/fimmu.2018.00367
58. Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: what’s in a name? Nat Rev Immunol (2004) 4(3):231–7. doi: 10.1038/nri1309
59. Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol (2009) 86(3):513–28. doi: 10.1189/JLB.0309135
60. Tang W, Zhou J, Yang W, Feng Y, Wu H, Mok MTS, et al. Aberrant cholesterol metabolic signaling impairs antitumor immunosurveillance through natural killer T cell dysfunction in obese liver. Cell Mol Immunol (2022) 19(7):834–47. doi: 10.1038/s41423-022-00872-3
61. Kinker GS, Vitiello GAF, Ferreira WAS, Chaves AS, Cordeiro de Lima VC, Medina TDS. B cell orchestration of anti-tumor immune responses: A matter of cell localization and communication. Front Cell Dev Biol (2021) 9:678127. doi: 10.3389/fcell.2021.678127
62. Yang F, Wan F. Lipid metabolism in tumor-associated B cells. Adv Exp Med Biol (2021) 1316:133–47. doi: 10.1007/978-981-33-6785-2_9
63. Kalinich JF, Ramakrishnan R, McClain DE, Ramakrishnan N. 4-Hydroxynonenal, an end-product of lipid peroxidation, induces apoptosis in human leukemic T- and B-cell lines. Free Radic Res (2000) 33(4):349–58. doi: 10.1080/10715760000300891
64. Muri J, Thut H, Bornkamm GW, Kopf M. B1 and marginal zone B cells but not follicular B2 cells require gpx4 to prevent lipid peroxidation and ferroptosis. Cell Rep (2019) 29(9):2731–2744 e2734. doi: 10.1016/j.celrep.2019.10.070
65. Kinowaki Y, Kurata M, Ishibashi S, Ikeda M, Tatsuzawa A, Yamamoto M, et al. Glutathione peroxidase 4 overexpression inhibits ROS-induced cell death in diffuse large B-cell lymphoma. Lab Invest (2018) 98(5):609–19. doi: 10.1038/s41374-017-0008-1
66. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
67. Yang P, Qin H, Li Y, Xiao A, Zheng E, Zeng H, et al. CD36-mediated metabolic crosstalk between tumor cells and macrophages affects liver metastasis. Nat Commun (2022) 13(1):5782. doi: 10.1038/s41467-022-33349-y
68. Su P, Wang Q, Bi E, Ma X, Liu L, Yang M, et al. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res (2020) 80(7):1438–50. doi: 10.1158/0008-5472.CAN-19-2994
69. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy (2021) 17(9):2054–81. doi: 10.1080/15548627.2020.1810918
70. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol (2020) 16(3):278–90. doi: 10.1038/s41589-019-0462-8
71. Singh NK, Rao GN. Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies. Prog Lipid Res (2019) 73:28–45. doi: 10.1016/j.plipres.2018.11.001
72. Hsu CG, Chavez CL, Zhang C, Sowden M, Yan C, Berk BC. The lipid peroxidation product 4-hydroxynonenal inhibits NLRP3 inflammasome activation and macrophage pyroptosis. Cell Death Differ (2022) 29(9):1790–803. doi: 10.1038/s41418-022-00966-5
73. Yang Y, Wang Y, Guo L, Gao W, Tang TL, Yan M. Interaction between macrophages and ferroptosis. Cell Death Dis (2022) 13(4):355. doi: 10.1038/s41419-022-04775-z
74. Han F, Li S, Yang Y, Bai Z. Interleukin-6 promotes ferroptosis in bronchial epithelial cells by inducing reactive oxygen species-dependent lipid peroxidation and disrupting iron homeostasis. Bioengineered (2021) 12(1):5279–88. doi: 10.1080/21655979.2021.1964158
75. Jung HS, Shimizu-Albergine M, Shen X, Kramer F, Shao D, Vivekanandan-Giri A, et al. TNF-alpha induces acyl-CoA synthetase 3 to promote lipid droplet formation in human endothelial cells. J Lipid Res (2020) 61(1):33–44. doi: 10.1194/jlr.RA119000256
76. Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol (2019) 26(3):420–432 e429. doi: 10.1016/j.chembiol.2018.11.016
77. Murray CA, Clements MP, Lynch MA. Interleukin-1 induces lipid peroxidation and membrane changes in rat hippocampus: An age-related study. Gerontology (1999) 45(3):136–42. doi: 10.1159/000022076
78. Pertsov SS, Koplik EV, Kalinichenko LS, Simbirtsev AS. Effect of interleukin-1beta on lipid peroxidation in emotiogenic structures of the brain in rats during acute stress. Bull Exp Biol Med (2010) 150(1):9–12. doi: 10.1007/s10517-010-1054-5
79. Wu W, Luo Z, Shen D, Lan T, Xiao Z, Liu M, et al. IL-10 protects against OPC ferroptosis by regulating lipid reactive oxygen species levels post stroke. Redox Biol (2024) 69:102982. doi: 10.1016/j.redox.2023.102982
80. Kim DH, Kim WD, Kim SK, Moon DH, Lee SJ. TGF-beta1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis (2020) 11(5):406. doi: 10.1038/s41419-020-2618-6
81. Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discovery (2004) 3(5):401–16. doi: 10.1038/nrd1383
82. Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity (2014) 40(3):315–27. doi: 10.1016/j.immuni.2014.02.009
83. Sulciner ML, Serhan CN, Gilligan MM, Mudge DK, Chang J, Gartung A, et al. Resolvins suppress tumor growth and enhance cancer therapy. J Exp Med (2018) 215(1):115–40. doi: 10.1084/jem.20170681
84. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med (2010) 16(8):880–6. doi: 10.1038/nm.2172
85. Arai R, Soda S, Okutomi T, Morita H, Ohmi F, Funakoshi T, et al. Lipid accumulation in peripheral blood dendritic cells and anticancer immunity in patients with lung cancer. J Immunol Res (2018) 2018:5708239. doi: 10.1155/2018/5708239
86. PrabhuDas MR, Baldwin CL, Bollyky PL, Bowdish DME, Drickamer K, Febbraio M, et al. A consensus definitive classification of scavenger receptors and their roles in health and disease. J Immunol (2017) 198(10):3775–89. doi: 10.4049/jimmunol.1700373
87. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med (2013) 19(5):557–66. doi: 10.1038/nm.3159
88. Han L, Bai L, Qu C, Dai E, Liu J, Kang R, et al. PPARG-mediated ferroptosis in dendritic cells limits antitumor immunity. Biochem Biophys Res Commun (2021) 576:33–9. doi: 10.1016/j.bbrc.2021.08.082
89. Klotz L, Dani I, Edenhofer F, Nolden L, Evert B, Paul B, et al. Peroxisome proliferator-activated receptor gamma control of dendritic cell function contributes to development of CD4+ T cell anergy. J Immunol (2007) 178(4):2122–31. doi: 10.4049/jimmunol.178.4.2122
90. Ramakrishnan R, Tyurin VA, Veglia F, Condamine T, Amoscato A, Mohammadyani D, et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol (2014) 192(6):2920–31. doi: 10.4049/jimmunol.1302801
91. Rothe T, Gruber F, Uderhardt S, Ipseiz N, Rossner S, Oskolkova O, et al. 12/15-Lipoxygenase-mediated enzymatic lipid oxidation regulates DC maturation and function. J Clin Invest (2015) 125(5):1944–54. doi: 10.1172/JCI78490
92. Ivanov I, Heydeck D, Hofheinz K, Roffeis J, O'Donnell VB, Kuhn H, et al. Molecular enzymology of lipoxygenases. Arch Biochem Biophys (2010) 503(2):161–74. doi: 10.1016/j.abb.2010.08.016
93. Lee W, Kingstad-Bakke B, Paulson B, Larsen A, Overmyer K, Marinaik CB, et al. Carbomer-based adjuvant elicits CD8 T-cell immunity by inducing a distinct metabolic state in cross-presenting dendritic cells. PloS Pathog (2021) 17(1):e1009168. doi: 10.1371/journal.ppat.1009168
94. Ho NI, Huis In ‘t Veld LGM, van Eck van der Sluijs J, Heuts BMH, Looman MWG, Kers-Rebel ED, et al. Saponin-based adjuvants enhance antigen cross-presentation in human CD11c(+) CD1c(+) CD5(-) CD163(+) conventional type 2 dendritic cells. J Immunother Cancer (2023) 11(8). doi: 10.1136/jitc-2023-007082
95. Calzada-Fraile D, Iborra S, Ramirez-Huesca M, Jorge I, Dotta E, Hernandez-Garcia E, et al. Immune synapse formation promotes lipid peroxidation and MHC-I upregulation in licensed dendritic cells for efficient priming of CD8(+) T cells. Nat Commun (2023) 14(1):6772. doi: 10.1038/s41467-023-42480-3
96. Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun (2017) 8(1):2122. doi: 10.1038/s41467-017-02186-9
97. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell (2015) 161(7):1527–38. doi: 10.1016/j.cell.2015.05.025
98. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8(+) T cell exhaustion in the tumor microenvironment. Cell Metab (2019) 30(1):143–156 e145. doi: 10.1016/j.cmet.2019.04.002
99. Cozar B, Greppi M, Carpentier S, Narni-Mancinelli E, Chiossone L, Vivier E. Tumor-infiltrating natural killer cells. Cancer Discovery (2021) 11(1):34–44. doi: 10.1158/2159-8290.CD-20-0655
100. Kurihara H, Koda H, Asami S, Kiso Y, Tanaka T. Contribution of the antioxidative property of astaxanthin to its protective effect on the promotion of cancer metastasis in mice treated with restraint stress. Life Sci (2002) 70(21):2509–20. doi: 10.1016/S0024-3205(02)01522-9
101. Poznanski SM, Singh K, Ritchie TM, Aguiar JA, Fan IY, Portillo AL, et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab (2021) 33(6):1205–1220 e1205. doi: 10.1016/j.cmet.2021.03.023
102. Duwe AK, Werkmeister J, Roder JC, Lauzon R, Payne U. Natural killer cell-mediated lysis involves an hydroxyl radical-dependent step. J Immunol (1985) 134(4):2637–44. doi: 10.4049/jimmunol.134.4.2637
103. Whalen MM, Doshi RN, Bader BW, Bankhurst AD. Lysophosphatidylcholine and arachidonic acid are required in the cytotoxic response of human natural killer cells to tumor target cells. Cell Physiol Biochem (1999) 9(6):297–309. doi: 10.1159/000016324
104. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med (2017) 23(3):279–87. doi: 10.1038/nm.4294
105. Claster S, Chiu DT, Quintanilha A, Lubin B. Neutrophils mediate lipid peroxidation in human red cells. Blood (1984) 64(5):1079–84. doi: 10.1182/blood.V64.5.1079.1079
106. Naito Y, Yoshikawa T, Matsuyama K, Yagi N, Arai M, Nakamura Y, et al. Neutrophils, lipid peroxidation, and nitric oxide in gastric reperfusion injury in rats. Free Radic Biol Med (1998) 24(3):494–502. doi: 10.1016/S0891-5849(97)00323-7
107. Maor I, Rainis T, Lanir A, Lavy A. Oxidative stress, inflammation and neutrophil superoxide release in patients with Crohn’s disease: distinction between active and non-active disease. Dig Dis Sci (2008) 53(8):2208–14. doi: 10.1007/s10620-007-0141-6
108. Laforge M, Elbim C, Frere C, Hemadi M, Massaad C, Nuss P, et al. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat Rev Immunol (2020) 20(9):515–6. doi: 10.1038/s41577-020-0407-1
109. Yee PP, Wei Y, Kim SY, Lu T, Chih SY, Lawson C, et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat Commun (2020) 11(1):5424. doi: 10.1038/s41467-020-19193-y
110. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature (2019) 569(7754):73–8. doi: 10.1038/s41586-019-1118-2
111. Ugolini A, Tyurin VA, Tyurina YY, Tcyganov EN, Donthireddy L, Kagan VE, et al. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight (2020) 5(15). doi: 10.1172/jci.insight.138581
112. Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature (2022) 612(7939):338–46. doi: 10.1038/s41586-022-05443-0
113. Sokola-Wysoczanska E, Wysoczanski T, Wagner J, Czyz K, Bodkowski R, Lochynski S, et al. Polyunsaturated fatty acids and their potential therapeutic role in cardiovascular system disorders-A review. Nutrients (2018) 10(10). doi: 10.3390/nu10101561
114. Rouzer CA, Marnett LJ. Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem Rev (2003) 103(6):2239–304. doi: 10.1021/cr000068x
115. Arfin S, Jha NK, Jha SK, Kesari KK, Ruokolainen J, Roychoudhury S, et al. Oxidative stress in cancer cell metabolism. Antioxid (Basel) (2021) 10(5). doi: 10.3390/antiox10050642
116. Panaroni C, Fulzele K, Soucy R, Huang C, Mukaihara K, Chattopadhyay S, et al. Polyunsaturated fatty acid (PUFA) signaling induces ferroptosis-mediated cell-death in multiple myeloma. Blood (2019) 134(Supplement_1):3108–8. doi: 10.1182/blood-2019-131906
117. Panaroni C, Fulzele K, Soucy R, Siu KT, Mukaihara K, Huang C, et al. Arachidonic acid induces ferroptosis-mediated cell-death in multiple myeloma. Blood (2018) 132(Supplement 1):4498–8. doi: 10.1182/blood-2018-99-118482
118. Beavers WN, Monteith AJ, Amarnath V, Mernaugh RL, Roberts LJ 2nd, Chazin WJ, et al. Arachidonic Acid Kills Staphylococcus aureus through a Lipid Peroxidation Mechanism. mBio (2019) 10(5). doi: 10.1128/mBio.01333-19
119. Muzio G, Trombetta A, Maggiora M, Martinasso G, Vasiliou V, Lassen N, et al. Arachidonic acid suppresses growth of human lung tumor A549 cells through down-regulation of ALDH3A1 expression. Free Radic Biol Med (2006) 40(11):1929–38. doi: 10.1016/j.freeradbiomed.2006.01.020
120. Reddy AT, Lakshmi SP, Reddy RC. PPARgamma as a novel therapeutic target in lung cancer. PPAR Res (2016) 2016:8972570. doi: 10.1155/2016/8972570
121. Venkatesh D, O’Brien NA, Zandkarimi F, Tong DR, Stokes ME, Dunn DE, et al. MDM2 and MDMX promote ferroptosis by PPARalpha-mediated lipid remodeling. Genes Dev (2020) 34(7-8):526–43. doi: 10.1101/gad.334219.119
122. Shan K, Feng N, Zhu D, Qu H, Fu G, Li J, et al. Free docosahexaenoic acid promotes ferroptotic cell death via lipoxygenase dependent and independent pathways in cancer cells. Eur J Nutr (2022) 61(8):4059–75. doi: 10.1007/s00394-022-02940-w
123. Roig-Perez S, Guardiola F, Moreto M, Ferrer R. Lipid peroxidation induced by DHA enrichment modifies paracellular permeability in Caco-2 cells: protective role of taurine. J Lipid Res (2004) 45(8):1418–28. doi: 10.1194/jlr.M300513-JLR200
124. Zapata-Gonzalez F, Rueda F, Petriz J, Domingo P, Villarroya F, Diaz-Delfin J, et al. Human dendritic cell activities are modulated by the omega-3 fatty acid, docosahexaenoic acid, mainly through PPAR(gamma):RXR heterodimers: comparison with other polyunsaturated fatty acids. J Leukoc Biol (2008) 84(4):1172–82. doi: 10.1189/jlb.1007688
125. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
126. Li W, Guo X, Chen C, Li J. The prognostic value of arachidonic acid metabolism in breast cancer by integrated bioinformatics. Lipids Health Dis (2022) 21(1):103. doi: 10.1186/s12944-022-01713-y
127. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell (2022) 40(4):365–378 e366. doi: 10.1016/j.ccell.2022.02.003
128. Manzo T, Prentice BM, Anderson KG, Raman A, Schalck A, Codreanu GS, et al. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J Exp Med (2020) 217(8). doi: 10.1084/jem.20191920
129. Yang H, Villani RM, Wang H, Simpson MJ, Roberts MS, Tang M, et al. The role of cellular reactive oxygen species in cancer chemotherapy. J Exp Clin Cancer Res (2018) 37(1):266. doi: 10.1186/s13046-018-0909-x
130. Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res (1991) 51(3):794–8.
131. Hielscher A, Gerecht S. Hypoxia and free radicals: role in tumor progression and the use of engineering-based platforms to address these relationships. Free Radic Biol Med (2015) 79:281–91. doi: 10.1016/j.freeradbiomed.2014.09.015
132. Xiao M, Zhong H, Xia L, Tao Y, Yin H. Pathophysiology of mitochondrial lipid oxidation: Role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic Biol Med (2017) 111:316–27. doi: 10.1016/j.freeradbiomed.2017.04.363
133. Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med (2020) 52(2):192–203. doi: 10.1038/s12276-020-0384-2
134. Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev (2019) 2019:5080843. doi: 10.1155/2019/5080843
135. Collin F. Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int J Mol Sci (2019) 20(10). doi: 10.3390/ijms20102407
136. Rodopulo AK. [Oxidation of tartaric acid in wine in the presence of heavy metal salts (activation of oxygen by iron)]. Izv Akad Nauk SSSR Biol (1951) 3:115–28.
137. Xue X, Ramakrishnan SK, Weisz K, Triner D, Xie L, Attili D, et al. Iron uptake via DMT1 integrates cell cycle with JAK-STAT3 signaling to promote colorectal tumorigenesis. Cell Metab (2016) 24(3):447–61. doi: 10.1016/j.cmet.2016.07.015
138. Pinnix ZK, Miller LD, Wang W, D'Agostino R Jr., Kute T, Willingham MC, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med (2010) 2(43):43ra56. doi: 10.1126/scitranslmed.3001127
139. Liang W, Ferrara N. Iron metabolism in the tumor microenvironment: contributions of innate immune cells. Front Immunol (2020) 11:626812. doi: 10.3389/fimmu.2020.626812
140. Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol (2010) 40(3):824–35. doi: 10.1002/eji.200939889
141. Debela DT, Muzazu SG, Heraro KD, Ndalama MT, Mesele BW, Haile DC, et al. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Med (2021) 9. doi: 10.1177/20503121211034366
142. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife (2014) 3:e02523. doi: 10.7554/eLife.02523
143. Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res (2014) 34(11):6417–22.
144. Zhang L, Li XM, Shi XH, Ye K, Fu XL, Wang X, et al. Sorafenib triggers ferroptosis via inhibition of HBXIP/SCD axis in hepatocellular carcinoma. Acta Pharmacol Sin (2023) 44(3):622–34. doi: 10.1038/s41401-022-00981-9
145. Li Y, Xia J, Shao F, Zhou Y, Yu J, Wu H, et al. Sorafenib induces mitochondrial dysfunction and exhibits synergistic effect with cysteine depletion by promoting HCC cells ferroptosis. Biochem Biophys Res Commun (2021) 534:877–84. doi: 10.1016/j.bbrc.2020.10.083
146. Feng J, Lu PZ, Zhu GZ, Hooi SC, Wu Y, Huang XW, et al. ACSL4 is a predictive biomarker of sorafenib sensitivity in hepatocellular carcinoma. Acta Pharmacol Sin (2021) 42(1):160–70. doi: 10.1038/s41401-020-0439-x
147. Wang Q, Bin C, Xue Q, Gao Q, Huang A, Wang K, et al. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis (2021) 12(5):426. doi: 10.1038/s41419-021-03718-4
148. Sadzuka Y, Shoji T, Takino Y. Effect of cisplatin on the activities of enzymes which protect against lipid peroxidation. Biochem Pharmacol (1992) 43(8):1872–5. doi: 10.1016/0006-2952(92)90725-X
149. Sugihara K, Nakano S, Koda M, Tanaka K, Fukuishi N, Gemba M. Stimulatory effect of cisplatin on production of lipid peroxidation in renal tissues. Jpn J Pharmacol (1987) 43(3):247–52. doi: 10.1016/S0021-5198(19)43504-X
150. Sadzuka Y, Shoji T, Takino Y. Mechanism of the increase in lipid peroxide induced by cisplatin in the kidneys of rats. Toxicol Lett (1992) 62(2-3):293–300. doi: 10.1016/0378-4274(92)90033-g
151. Cheng Q, Bao L, Li M, Chang K, Yi X. Erastin synergizes with cisplatin via ferroptosis to inhibit ovarian cancer growth in vitro and in vivo. J Obstet Gynaecol Res (2021) 47(7):2481–91. doi: 10.1111/jog.14779
152. Du J, Wang X, Li Y, Ren X, Zhou Y, Hu W, et al. DHA exhibits synergistic therapeutic efficacy with cisplatin to induce ferroptosis in pancreatic ductal adenocarcinoma via modulation of iron metabolism. Cell Death Dis (2021) 12(7):705. doi: 10.1038/s41419-021-03996-y
153. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med (2012) 366(15):1382–92. doi: 10.1056/NEJMoa1105535
154. Hong T, Lei G, Chen X, Li H, Zhang X, Wu N, et al. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol (2021) 42:101928. doi: 10.1016/j.redox.2021.101928
155. Tang S, Shen Y, Wei X, Shen Z, Lu W, Xu J. Olaparib synergizes with arsenic trioxide by promoting apoptosis and ferroptosis in platinum-resistant ovarian cancer. Cell Death Dis (2022) 13(9):826. doi: 10.1038/s41419-022-05257-y
157. Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol (2018) 217(7):2291–8. doi: 10.1083/jcb.201804161
158. Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev (2014) 94(2):329–54. doi: 10.1152/physrev.00040.2012
159. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol Med (2020) 152:175–85. doi: 10.1016/j.freeradbiomed.2020.02.027
160. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics (2019) 19(18):e1800311. doi: 10.1002/pmic.201800311
161. Gamcsik MP, Kasibhatla MS, Colvin OM. Glutathione levels in human tumors. Biomarkers (2012) 17(8):671–91. doi: 10.3109/1354750X.2012.715672
162. Perry RR, Mazetta JA, Levin M, Barranco SC. Glutathione levels and variability in breast tumors and normal tissue. Cancer (1993) 72(3):783–7. doi: 10.1002/1097-0142(19930801)72:3<783::AID-CNCR2820720325>3.0.CO;2-U
163. Yeh CC, Hou MF, Wu SH, Tsai SM, Lin SK, Hou LA, et al. A study of glutathione status in the blood and tissues of patients with breast cancer. Cell Biochem Funct (2006) 24(6):555–9. doi: 10.1002/cbf.1275
164. Zachara BA, Marchaluk-Wisniewska E, Maciag A, Peplinski J, Skokowski J, Lambrecht W. Decreased selenium concentration and glutathione peroxidase activity in blood and increase of these parameters in Malignant tissue of lung cancer patients. Lung (1997) 175(5):321–32. doi: 10.1007/PL00007578
165. Ilonen IK, Rasanen JV, Sihvo EI, Knuuttila A, Salmenkivi KM, Ahotupa MO, et al. Oxidative stress in non-small cell lung cancer: role of nicotinamide adenine dinucleotide phosphate oxidase and glutathione. Acta Oncol (2009) 48(7):1054–61. doi: 10.1080/02841860902824909
166. Abou Ghalia AH, Fouad IM. Glutathione and its metabolizing enzymes in patients with different benign and Malignant diseases. Clin Biochem (2000) 33(8):657–62. doi: 10.1016/S0009-9120(00)00181-8
167. Fiaschi AI, Cozzolino A, Ruggiero G, Giorgi G. Glutathione, ascorbic acid and antioxidant enzymes in the tumor tissue and blood of patients with oral squamous cell carcinoma. Eur Rev Med Pharmacol Sci (2005) 9(6):361–7.
168. Takenaka M, Furui T, Suzuki N, Koike T, Aoki H, Morishige K-I. Glutathione as a prognostic biomarker and a potential therapeutic target for ovarian cancer. Open J Obstetrics Gynecol (2022) 12(1):56–66. doi: 10.4236/ojog.2022.121006
169. Kolanjiappan K, Ramachandran CR, Manoharan S. Biochemical changes in tumor tissues of oral cancer patients. Clin Biochem (2003) 36(1):61–5. doi: 10.1016/S0009-9120(02)00421-6
170. Seven A, Erbil Y, Seven R, Inci F, Gulyasar T, Barutcu B, et al. Breast cancer and benign breast disease patients evaluated in relation to oxidative stress. Cancer Biochem Biophys (1998) 16(4):333–45.
171. Zengin E, Atukeren P, Kokoglu E, Gumustas MK, Zengin U. Alterations in lipid peroxidation and antioxidant status in different types of intracranial tumors within their relative peritumoral tissues. Clin Neurol Neurosurg (2009) 111(4):345–51. doi: 10.1016/j.clineuro.2008.11.008
172. Li Q, Kang C. Mechanisms of action for small molecules revealed by structural biology in drug discovery. Int J Mol Sci (2020) 21(15). doi: 10.3390/ijms21155262
173. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
174. Moosmayer D, Hilpmann A, Hoffmann J, Schnirch L, Zimmermann K, Badock V, et al. Crystal structures of the selenoprotein glutathione peroxidase 4 in its apo form and in complex with the covalently bound inhibitor ML162. Acta Crystallogr D Struct Biol (2021) 77(Pt 2):237–48. doi: 10.1107/S2059798320016125
175. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis (2020) 11(2):88. doi: 10.1038/s41419-020-2298-2
176. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol (2016) 12(7):497–503. doi: 10.1038/nchembio.2079
177. Liu MR, Zhu WT, Pei DS. System Xc(-): a key regulatory target of ferroptosis in cancer. Invest New Drugs (2021) 39(4):1123–31. doi: 10.1007/s10637-021-01070-0
178. Gong D, Chen M, Wang Y, Shi J, Hou Y. Role of ferroptosis on tumor progression and immunotherapy. Cell Death Discovery (2022) 8(1):427. doi: 10.1038/s41420-022-01218-8
179. Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol (2020) 16(5):497–506. doi: 10.1038/s41589-020-0501-5
180. Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc (2014) 136(12):4551–6. doi: 10.1021/ja411006a
181. Fan BY, Pang YL, Li WX, Zhao CX, Zhang Y, Wang X, et al. Liproxstatin-1 is an effective inhibitor of oligodendrocyte ferroptosis induced by inhibition of glutathione peroxidase 4. Neural Regener Res (2021) 16(3):561–6. doi: 10.4103/1673-5374.293157
182. Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci (2017) 3(3):232–43. doi: 10.1021/acscentsci.7b00028
183. Cui JX, Xu XH, He T, Liu JJ, Xie TY, Tian W, et al. L-kynurenine induces NK cell loss in gastric cancer microenvironment via promoting ferroptosis. J Exp Clin Cancer Res (2023) 42(1):52. doi: 10.1186/s13046-023-02629-w
184. Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discovery (2019) 9(12):1673–85. doi: 10.1158/2159-8290.CD-19-0338
185. Falk-Mahapatra R, Gollnick SO. Photodynamic therapy and immunity: an update. Photochem Photobiol (2020) 96(3):550–9. doi: 10.1111/php.13253
186. Tan L, Shen X, He Z, Lu Y. The role of photodynamic therapy in triggering cell death and facilitating antitumor immunology. Front Oncol (2022) 12:863107. doi: 10.3389/fonc.2022.863107
187. Garg AD, Nowis D, Golab J, Agostinis P. Photodynamic therapy: illuminating the road from cell death towards anti-tumour immunity. Apoptosis (2010) 15(9):1050–71. doi: 10.1007/s10495-010-0479-7
188. Castano AP, Mroz P, Hamblin MR. Photodynamic therapy and anti-tumour immunity. Nat Rev Cancer (2006) 6(7):535–45. doi: 10.1038/nrc1894
189. Mishchenko TA, Balalaeva IV, Vedunova MV, Krysko DV. Ferroptosis and photodynamic therapy synergism: enhancing anticancer treatment. Trends Cancer (2021) 7(6):484–7. doi: 10.1016/j.trecan.2021.01.013
190. Xu T, Ma Y, Yuan Q, Hu H, Hu X, Qian Z, et al. Enhanced ferroptosis by oxygen-boosted phototherapy based on a 2-in-1 nanoplatform of ferrous hemoglobin for tumor synergistic therapy. ACS Nano (2020) 14(3):3414–25. doi: 10.1021/acsnano.9b09426
191. Zuo Z, Yin H, Zhang Y, Xie C, Wang Q. A cytotoxic T cell inspired oncolytic nanosystem promotes lytic cell death by lipid peroxidation and elicits antitumor immune responses. Nat Commun (2023) 14(1):5456. doi: 10.1038/s41467-023-41335-1
192. Wang Y, Chen F, Zhou H, Huang L, Ye J, Liu X, et al. Redox dyshomeostasis with dual stimuli-activatable dihydroartemisinin nanoparticles to potentiate ferroptotic therapy of pancreatic cancer. Small Methods (2023) 7(5):e2200888. doi: 10.1002/smtd.202200888
193. Song R, Li T, Ye J, Sun F, Hou B, Saeed M, et al. Acidity-activatable dynamic nanoparticles boosting ferroptotic cell death for immunotherapy of cancer. Adv Mater (2021) 33(31):e2101155. doi: 10.1002/adma.202101155
194. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature (2017) 547(7664):453–7. doi: 10.1038/nature23007
195. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature (2017) 551(7679):247–50. doi: 10.1038/nature24297
Keywords: lipid peroxidation, TME, innate immune cells, adaptive immune cells, tumor immunity
Citation: Xiao L, Xian M, Zhang C, Guo Q and Yi Q (2024) Lipid peroxidation of immune cells in cancer. Front. Immunol. 14:1322746. doi: 10.3389/fimmu.2023.1322746
Received: 16 October 2023; Accepted: 19 December 2023;
Published: 08 January 2024.
Edited by:
Takashi MaruYama, National Institutes of Health (NIH), United StatesReviewed by:
Noa B. Martin-Cofreces, Princess University Hospital, SpainCopyright © 2024 Xiao, Xian, Zhang, Guo and Yi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Yi, UVlpQGhvdXN0b25tZXRob2Rpc3Qub3Jn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.