
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 December 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1314123
 Jiulu Zhao1,2†
Jiulu Zhao1,2† Xi Zhang1,2†
Xi Zhang1,2† Yuan Li1,2†
Yuan Li1,2† Jizhang Yu1,2Zhang Chen1,2Yuqing Niu1,2Shuan Ran1,2
Jizhang Yu1,2Zhang Chen1,2Yuqing Niu1,2Shuan Ran1,2 Song Wang1,2
Song Wang1,2 Weicong Ye1,2Zilong Luo1,2
Weicong Ye1,2Zilong Luo1,2 Xiaohan Li1,2Yanglin Hao1,2Junjie Zong1,2Chengkun Xia3*Jiahong Xia1,2,4*
Xiaohan Li1,2Yanglin Hao1,2Junjie Zong1,2Chengkun Xia3*Jiahong Xia1,2,4* Jie Wu1,2,4*
Jie Wu1,2,4*The liver is a multifunctional organ that plays crucial roles in numerous physiological processes, such as production of bile and proteins for blood plasma, regulation of blood levels of amino acids, processing of hemoglobin, clearance of metabolic waste, maintenance of glucose, etc. Therefore, the liver is essential for the homeostasis of organisms. With the development of research on the liver, there is growing concern about its effect on immune cells of innate and adaptive immunity. For example, the liver regulates the proliferation, differentiation, and effector functions of immune cells through various secreted proteins (also known as “hepatokines”). As a result, the liver is identified as an important regulator of the immune system. Furthermore, many diseases resulting from immune disorders are thought to be related to the dysfunction of the liver, including systemic lupus erythematosus, multiple sclerosis, and heart failure. Thus, the liver plays a role in remote immune regulation and is intricately linked with systemic immunity. This review provides a comprehensive overview of the liver remote regulation of the body’s innate and adaptive immunity regarding to main areas: immune-related molecules secreted by the liver and the liver-resident cells. Additionally, we assessed the influence of the liver on various facets of systemic immune-related diseases, offering insights into the clinical application of target therapies for liver immune regulation, as well as future developmental trends.
The liver is an important, multifunctional organ that serves as a central hub for numerous physiological processes. It is involved not only in the synthesizing, transforming, and decomposing of proteins, carbohydrates, lipids, and vitamins within the body, but also participates in the transformation and detoxification of hormones, drugs, and other compounds (1). Additionally, the liver has the functions of bile secretion, phagocytosis and immune defense (1, 2). Notably, the liver comprises the largest reticuloendothelial phagocytic system in the human body. The hepatic sinusoid contains a large number of Kupffer cells, which can engulf exogenous substances, pathogenic microorganisms, and other particulate matter present in the blood (3, 4). In the event of infection-related mucosal damage, pathogenic substances within the intestine can breach the intestinal mucosal barrier, the primary defense line of the intestinal immune system, and access the capillaries and lymph vessels in the intestinal wall (5, 6). Subsequently, the mesenteric lymph nodes and the liver serve as the second line of defense for the intestinal immune system (7). Under typical conditions, the liver’s unique anatomy and cellular composition bestow it with immune defense and immune regulation functions. Serving as a pivotal defense barrier between the body’s internal milieu and the external environment, the liver contributes an array of secreted proteins (including hepatokines, plasma proteins, inflammatory factors, and complements, etc.) that critically participate in the regulation of immune response (8, 9).
Within this review, we summarize the effects of liver remote immune regulation on immune cells and immune homeostasis. Beginning with the distinctive anatomical structure and cellular components of the liver, we describe the intricate orchestration of innate and adaptive immune responses through various liver secreted proteins and intrahepatic immune cells. Moreover, we synopsize the regulatory mechanisms within the liver under diverse pathological conditions, with the aspiration that this review will furnish valuable insights towards a better understanding of hepatic immune modulation.
The adult liver typically weighs between 1 to 2.5 kg and has a V-shaped structure with a reddish-brown coloration. The majority of the liver resides within the right hypochondriac and epigastric regions, while a smaller portion extends into the left hypochondriac region, and is protected by the ribs and costal cartilage. The concave face of the liver interfaces with the abdominal viscera (10). The liver encompasses the Glisson system as well as the hepatic venous system; within the Glisson system are the portal vein and the proper hepatic artery. The liver also boasts an abundant blood supply, with its blood volume accounting for approximately 14% of the total human body. Blood flow in an adult liver range from 1500 to 2000 ml per minute and, unlike other abdominal organs, the liver receives a dual blood supply. The hepatic artery carries oxygenated blood from the heart, while the portal vein gathers venous blood rich in nutrients from the digestive tract (11).
The liver receives approximately 1/4 of cardiac output (12). The liver vasculature can be categorized into hepatic vessels and effluent hepatic vessels. Blood entering the liver runs through the hepatic artery and hepatic portal vein, constituting a dual vascular supply. The effluent hepatic vessels constitute the hepatic venous system. Approximately 1/4 of the liver blood supply is derived from the hepatic artery, which carries oxygenated blood and antibodies from the heart. Upon entering the liver, arterial blood divides into branches at various levels, eventually reaching the interlobular artery (13). The hepatic portal vein serves as a pivotal conduit for the liver, contributing approximately 3/4 of the liver’s blood supply. This vessel transports nutrient-laden blood from the gastrointestinal tract to the liver for metabolism (14). The blood, mixed with arterial blood, flows into sinusoidal vessels of the liver. As capillary-like structures, the flow rate of blood in these vessels is approximately half that of other capillary counterparts, which facilitates the detection of the specific molecules or pathogens by immune cells (15). The hepatic portal vein originates from the convergence of the splenic vein and the superior mesenteric vein. The portal vein additionally exhibits lateral anastomosis with the vena cava, though these channels are typically not open (16). The interconnectedness of these blood vessels means that liver-related pathological factors (e.g., cirrhosis) disrupting portal vein circulation can result in blood stasis, potentially leading to splenic congestion and hematoma formation. In instances where collateral circulation becomes open, as seen in esophageal and gastric varices, or in the event of rupture and hemorrhage, anastomoses between the portal vein and inferior vena cava via the rectal venous plexus may prompt rupture of the plexus and subsequent rectal bleeding. Alternatively, if the portal vein establishes anastomoses with both the superior and inferior vena cava through the periumbilical venous plexus, this can lead to portal hypertension and the resultant distension of periumbilical veins (17).
The hepatic artery serves as a vegetative vessel for the liver, delivering oxygen and nutrients essential for liver metabolic processes. It accounts for about 20% to 30% of the total liver blood flow is attributed to the hepatic artery, and its pressure surpasses that of the portal vein by 30-40 fold (18). The portal vein functions as a crucial conduit for the liver, constituting approximately 70%-80% of the liver’s blood supply. Characterized by lower pressure, it carries nutrient-rich blood from the digestive tract and pancreas. Upon traversing the sinusoidal space, this blood is assimilated by hepatocytes, subsequently undergoing processing. A portion is then released into the bloodstream for systemic use, while the remainder is provisionally stored within hepatocytes to serve potential demands (19) (Figure 1). Hepatocytes are usually divided into three regions within the hepatic lobules. Hepatocytes close to the portal vein are characterized by increased gluconeogenesis and beta oxidation, which results from toxins and microorganisms of gut origin, as well as blood rich in nutrients and oxygen. Conversely, hepatocytes close to the central venous are exposed to lower concentrations of nutrients and oxygen, which are associated with detoxification, enhanced glycolysis, and lipogenesis (20).
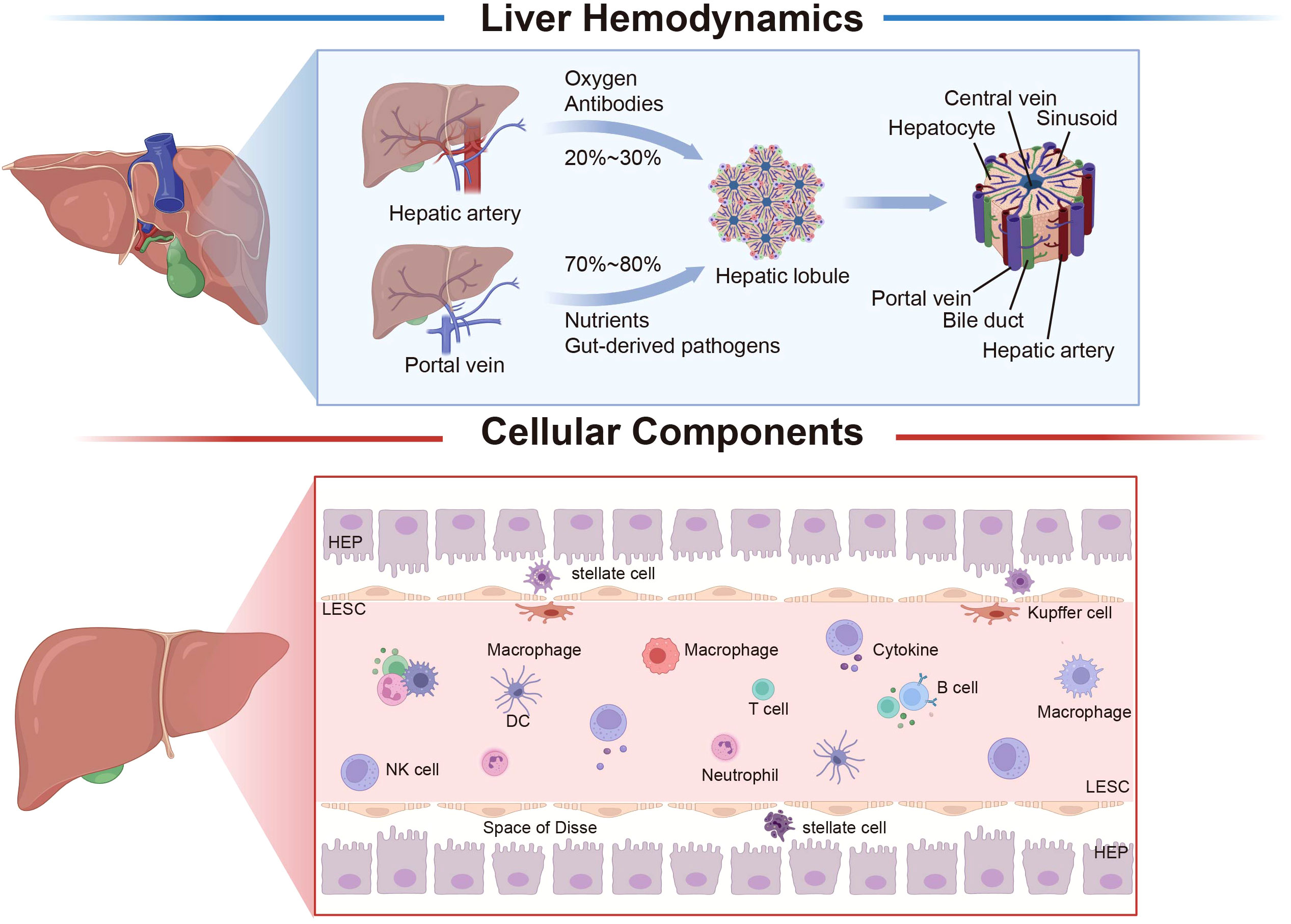
Figure 1 Liver hemodynamics and histology. The liver receives a dual blood supply from both the hepatic artery and the portal vein. Approximately 20–30% of the liver’s blood supply is derived from the hepatic artery, which primarily serves the purpose of oxygen delivery, while the remaining 70–80% originates from the portal vein, which is primarily responsible for nutrient supply. The fundamental structural and functional unit of the liver is the hepatic lobule, which is centered around the central vein, and hepatocytes radiate outward to form hepatic cord; between the hepatic cord is the hepatic sinusoid. The liver is composed of parenchymal cells (hepatocytes) and non-parenchymal cells, with liver non-parenchymal cells mainly encompassing LSEC, hepatic stellate cells, Kupffer cells, and immune cells. The liver’s various cell types interact, undergo precise regulation, and collaboratively perform specialized functions, collectively contributing to the biological functions of the liver. The space between hepatocytes in LSEC is the space of Disse, which is where hepatocytes and blood exchange substances. LSEC, liver sinusoidal endothelial cell; HEP, hepatocyte; DC, dendritic cell; NK, natural killer cell. Created with BioRender.com.
Hepatocytes constitute a vital component of the liver and perform approximately 70% of its essential functions (21)(Figure 1). Hepatocytes also create a crucial cellular barrier, which separates sinusoidal blood from tubular bile. Hepatocytes exhibit distinct tissue polarity, positioning their basement membrane towards hepatic sinusoidal endothelial cells, while their apex generates numerous bile canaliculi in direct juxtaposition with neighboring hepatocytes (22). Another hallmark of hepatocyte morphology is the creation of subendothelial positive sinusoidal spaces (Disse spaces) along both facets of the hepatocyte basal surface (23). Hepatocytes display spatial heterogeneity, with their distribution within hepatic lobules dictating their distinct functionalities (20). While liver cells in a healthy liver exhibit slow regeneration, they can undergo rapid proliferation in response to liver damage (24). Previous studies have suggested that hepatocytes exhibit a stable phenotype (25). However, recent research highlights the phenotypic plasticity of hepatocytes. For example, hepatocytes can convert into biliary-like cells in response to cholestatic injury and liver cancer (26). Moreover, the newly discovered NOTCH-YAP1/TEAD-DNMT1 axis is crucial for hepatocyte transdifferentiation (27).
LSECs constitute approximately 50% of the non-parenchymal liver cell population, making them the predominant non-parenchymal cell type in the liver (28). Hepatic sinusoidal endothelial cells form a lining along the low-shear sinusoidal capillary channels within the liver. As opposed to typical capillaries, LSECs feature window-like pore structures and the absence of a continuous basement membrane beneath the endothelium. This unique arrangement permits the formation of an unobstructed conduit between sinusoidal blood and the Disse space, facilitating the exchange of substrates between the bloodstream and liver parenchyma and regulating the movement of lipoproteins to and from hepatocytes (29) (Figure 1). LSECs stand out not only as the most permeable type of endothelial cell among mammals, but also exhibit a remarkable capacity for endocytosis. The primary receptors facilitating endocytosis include the mannose receptor, scavenger receptor, and Fcγ receptor IIb2, which enable the removal of circulating waste materials (30, 31). In a physiological context, LSECs contribute to the regulation of hepatic vascular tone, aiding in the preservation of low portal vein pressure and hepatic stellate cell quiescence. This function serves to counteract intrahepatic vasoconstriction and prevent the onset of fibrosis development (32). LSECs, as important contributors to the maintenance of liver homeostasis, previously lacked specific hallmark genes. However, a recent study identified Oit3 as marker for tracing LSECs, which is predominantly expressed in ECs of the midlobular liver. Subsequently, Oit3-CreERT2 transgenic mice were generated to investigate the complexity of LSECs in liver diseases, including sinus obstructive syndrome (SOS), providing insights into the intricate relationships between liver disease and systemic conditions (33).
Kupffer cells, an intrinsic population of liver-resident macrophages, constitute 35% of non-parenchymal liver cells and represent 90% of the total tissue macrophage population (34). These cells arise from bone marrow hematopoietic stem cells or local hematopoietic stem cells within the liver and possess a capacity for self-renewal and play a role in the clearance of microorganisms from the portal vein, thereby contributing to the maintenance of liver homeostasis (35–37). Primarily situated within the hepatic sinus, Kupffer cells are situated near sinusoidal endothelial cells, hepatic stellate cells, and natural killer cells within this sinusoidal region. They demonstrate the ability to promptly respond to intestine-originating microorganisms and their byproducts, enabling the swift execution of their functions (38, 39). In the human liver, Kupffer cells encompass distinct subsets, including classical CD14+CD16- macrophages, non-classical CD14+CD16+ macrophages, and CD16+ cells (40). Kupffer cells exhibit significant plasticity, with their phenotype and function modulated by their microenvironment, a phenomenon termed macrophage polarization (41). Classically activated and M1 macrophages, associated with pro-inflammatory responses, can be bound by lipopolysaccharides (LPS) alone or in conjunction with Th1 cytokines (e.g., IFN-γ, GM-CSF). They release pro-inflammatory cytokines such as IL-1β, IL-6, IL-12, IL-23, and TNF-α. In contrast, replace activated and M2 macrophages exhibit anti-inflammatory and immunomodulatory properties. Polarized by Th2 cytokines like IL-4 and IL-13, they then produce anti-inflammatory cytokines such as IL-10 and TGF-β (42).
HSCs, also referred to as Ito cells, reside within the Disse space and constitute approximately 5–8% of the non-parenchymal liver cell population (43). In typical conditions, HSCs remain quiescent, displaying a spindle-shaped, polygonal morphology characterized by numerous lipid droplets within the cytoplasm. These droplets are enriched with vitamin A (44). During embryonic development, HSCs originate from the mesenchymal components of the septum transversum and trace their lineage back to precursor mesothelial cells that infiltrate the liver parenchyma from the liver sac (45). Under normal circumstances, HSCs remain quiescent, but can also enter a proliferative state and subsequently differentiate into myofibroblasts upon receiving signals indicative of oxidative stress and inflammation (46). The hallmark indicator of HSC activation is the upregulation of contractile fibers α smooth muscle actin (αSMA, gene name ACTA2) (47). HSCs play a pivotal role in governing liver regeneration and are closely linked to the modulation of sinusoidal tension. They are increasingly recognized as the primary cell type influencing sinusoidal blood flow regulation, and additionally fulfill immunomodulatory function (48).
Apart from LSECs and Kupffer cells, the liver’s antigen-presenting cell population also encompasses DCs. The liver contains fewer DCs than other organs. Hepatic DCs predominantly occupy the peripheral veins and Disse space, with a minority of cells dispersed within the parenchyma (49). The activation of DCs enriched in the liver necessitates FLT3L and GM-CSF (50, 51). Liver DCs can be classified into two primary subsets: myeloid (mDCs) and plasma celloid dendritic cells (pDC) (52). Approximately one-third of hepatic CD11c myeloid dendritic cells (mDCs) exhibit CD141 expression, whereas less than 5% of circulating mDCs display this marker (53).DCs in the liver manifest an immature phenotype characterized by diminished MHC-II expression and nearly negligible levels of co-stimulatory molecules (CD40, CD80, CD86) (54).
NK cells, functioning as effector lymphocytes of the innate immune system, exhibit two distinct subpopulations within the liver. One subset consists of liver-resident NK cells (LrNK), which localize to the hepatic sinusoids and are identified as liver type 1 innate lymphocytes (ILC1). The other subset originates from circulating classical NK cells (cNK cells), resembling those found in peripheral blood and the spleen (55–58). Hepatic NK cells exhibit differences from peripheral NK cells regarding surface marker expression, cytokine profiles, and cytotoxic capacities. In mice, approximately 5–10% of hepatic lymphocytes consist of NK1.1/DX5/CD3 NK cells, whereas in humans, nearly 50% of hepatic lymphocytes are composed of NK cells characterized by CD56 and CD16 expression (59, 60).
The liver is also rich in NKT cells, which constitute a substantial portion of its cellular composition. NKT cells encompass distinct lineages, primarily categorized into type I NKT cells, referred to as invariant NKT cells (iNKT). These cells express characteristic, semi-invariant T cell receptors (TCR) comprising Vα24-Jα18. In contrast, type II NKT cells exhibit a divergent TCR repertoire (61). In mice, the liver hosts a substantial population of NK1.1+ CD3+ NKT cells, constituting up to 30–40% of hepatic lymphocytes (in comparison to 0.5–2% in peripheral blood). Of this subset, around 80% express an invariant TCR configuration. In contrast, human NKT cells are predominantly associated with type II NKT cells (62) (Figure 1).
An increasing body of research has demonstrated the liver’s role as a lymphoid tissue that contributes to immune tolerance induction, local immune responses, and the establishment of immune memory in circulating blood antigens (63, 64). The liver bridges the intestinal portal vasculature system with the systemic circulation. Within the hepatic lobule, blood travels from the portal vein triad positioned in the lobular vicinity around the portal vein to the central vein via the polarized sinusoidal network. The distinctive configuration of the hepatic lobule, the diverse assembly of constituent cells, the specialized vascular architecture, and the blood flow characteristics characterized by high volume and low flow rate collectively form a distinctive immune microenvironment within the liver and confer it with a unique immune functionality. Hepatocytes possess the ability to secrete a multitude of proteins into the bloodstream, including hepatokines, acute phase proteins, complement, and more (65). Simultaneously, the liver harbors an abundant population of immune cells engaged in immune recognition and response. These cells encompass various categories: liver-resident cells, such as Kupffer cells, LSECs, DCs, and HSCs; circulating recruiting cells include NK cells, NKT cells, neutrophils, eosinophils, and monocytes (66).
In this section, we discuss the common hepatokines with immune regulation functions. Moreover, we provide the detailed information in Table 1.
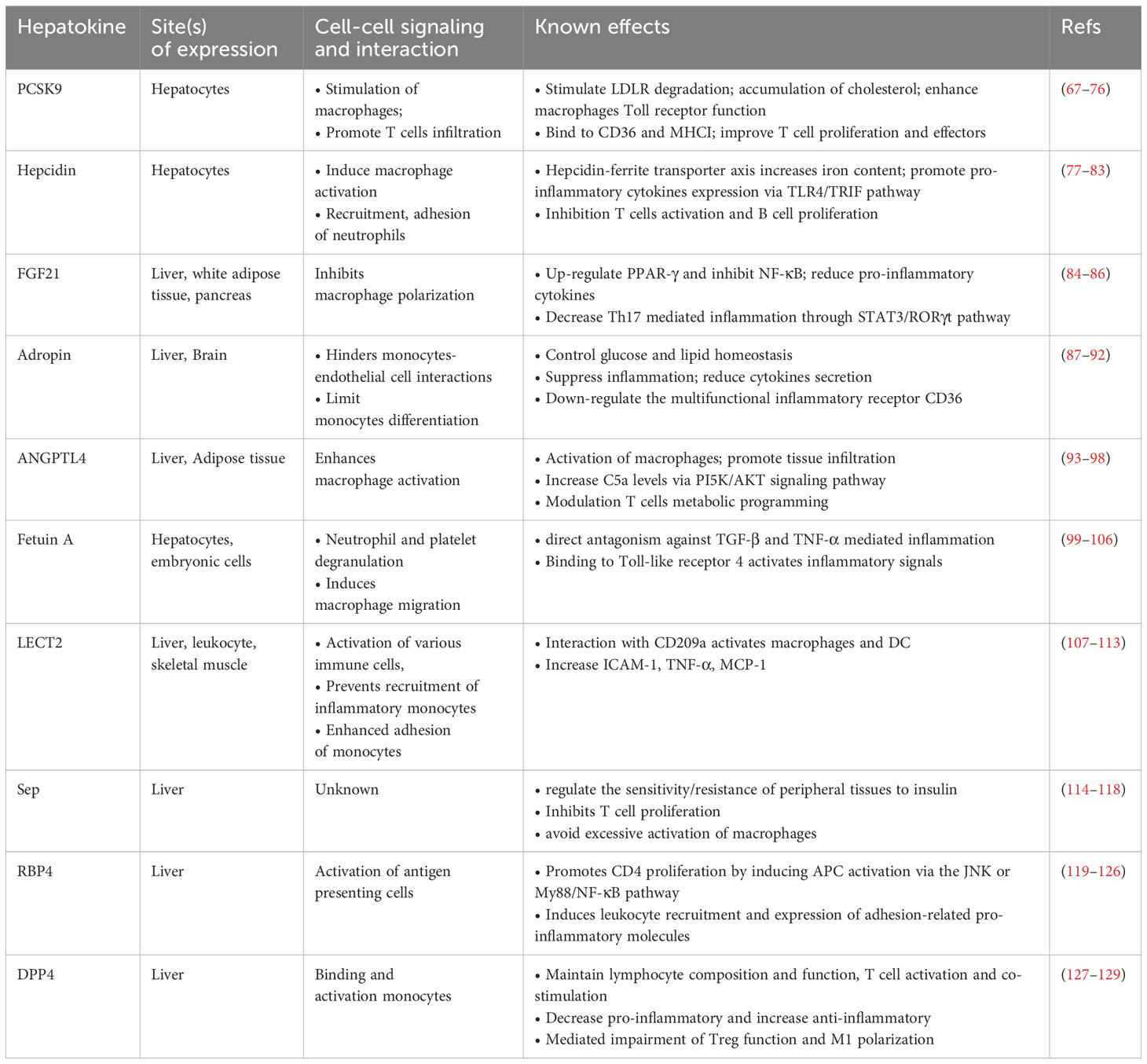
Table 1 Overview of hepatokines that affect immune cells.cc.
PCSK9, the ninth member of the proprotein convertase enzyme family, primarily localizes to the liver and plays a pivotal role in maintaining cholesterol homeostasis (130). Upon secretion into the plasma, PCSK9 binds to low-density lipoprotein receptors (LDLR) on the cell surface, leading to their subsequent degradation upon direct entry into lysosomes. This process actively modulates plasma LDL-C concentration (67) (Figure 2). Owing to its functional attributes, PCSK9 has emerged as a significant target for reducing cholesterol levels and preventing cardiovascular events (68), and an expanding body of research highlights its diverse biological functions (131). PCSK9 influences innate immunity by modulating the elimination of pathogenic LPS, a crucial component of systemic clearance and detoxification. Pharmacological inhibition of PCSK9 also has the potential to attenuate inflammatory responses and ameliorate septic shock (69, 70). Furthermore, PCSK9 can facilitate the selective buildup of cholesterol in macrophages and other immune cells. This effect is achieved through the stimulation of LDLR degradation and suppression of cholesterol reverse transport (RCT). Moreover, PCSK9 enhances lipid raft composition and bolsters Toll receptor functionality (71). Additionally, PCSK9 is capable of eliciting the synthesis of pro-inflammatory cytokines, including TNF-α and IL-6, while also facilitating the nuclear translocation of transcription factors and suppressing the generation of anti-inflammatory cytokines within macrophages (72). Aside from influencing LDLR receptors, circulating PCSK9 has the capacity to impact MHC I receptors (associated with antigen-driven immune responses) and CD36 (involved in fatty acid uptake) (132). As a target for immunotherapy, inhibiting PCSK9 can upregulate the expression of major MHC I molecules and promote the infiltration of cytotoxic T cells within tumors (73). PCSK9 hinders the recycling of MHC I to the cell surface through physical interaction with MHC I molecules, leading to their relocation and subsequent degradation within lysosomes. Inhibition of PCSK9 with small molecule compounds or monoclonal antibodies upregulates cell surface MHC I and enhances tumoral infiltrated lymphocytes (73, 74). Blocking PCSK9 improves the efficacy of anti-PD-1 therapy by promoting CD8+ T-cell infiltration, increasing inflammatory cytokines, and reducing Tregs (133). Additionally, PCSK9 modulates TCR recovery and signal transduction by suppressing LDLR expression, thereby impacting the immune response of CD8+ T cells (75). In conditions of hyperlipidemia with elevated PCSK9 levels, the increased LDLs contribute to the shift of T cells toward IL-17-producing T cells (134, 135). PCSK9 can also indirectly impact T-cell activation through oxidized LDL-induced dendritic cell maturation (136). In ankylosing spondylitis (AS), PCSK9 promotes Th1 and Th17 differentiation by activating the NF-κB pathway (137). Prior investigations by our research group demonstrated that, following heart and abdominal aorta transplantations in mice, the primary source of serum PCSK9 originated in the liver. PCSK9 influences T cell proliferation and IFN-γ production through the modulation of macrophage surface CD36 expression and the uptake of fatty acids during HTR (Figure 2) (76). In GVHD, the absence of PCSK9 can suppress the recruitment of macrophages and the expression of pro-inflammatory cytokines in aortic grafts. Additionally, PCSK9 knockout can hinder NLRP3 inflammasome activation, mitigate vascular smooth muscle cell (VSMC) migration and proliferation, and mitigate the development of allogeneic graft vascular lesions (138).
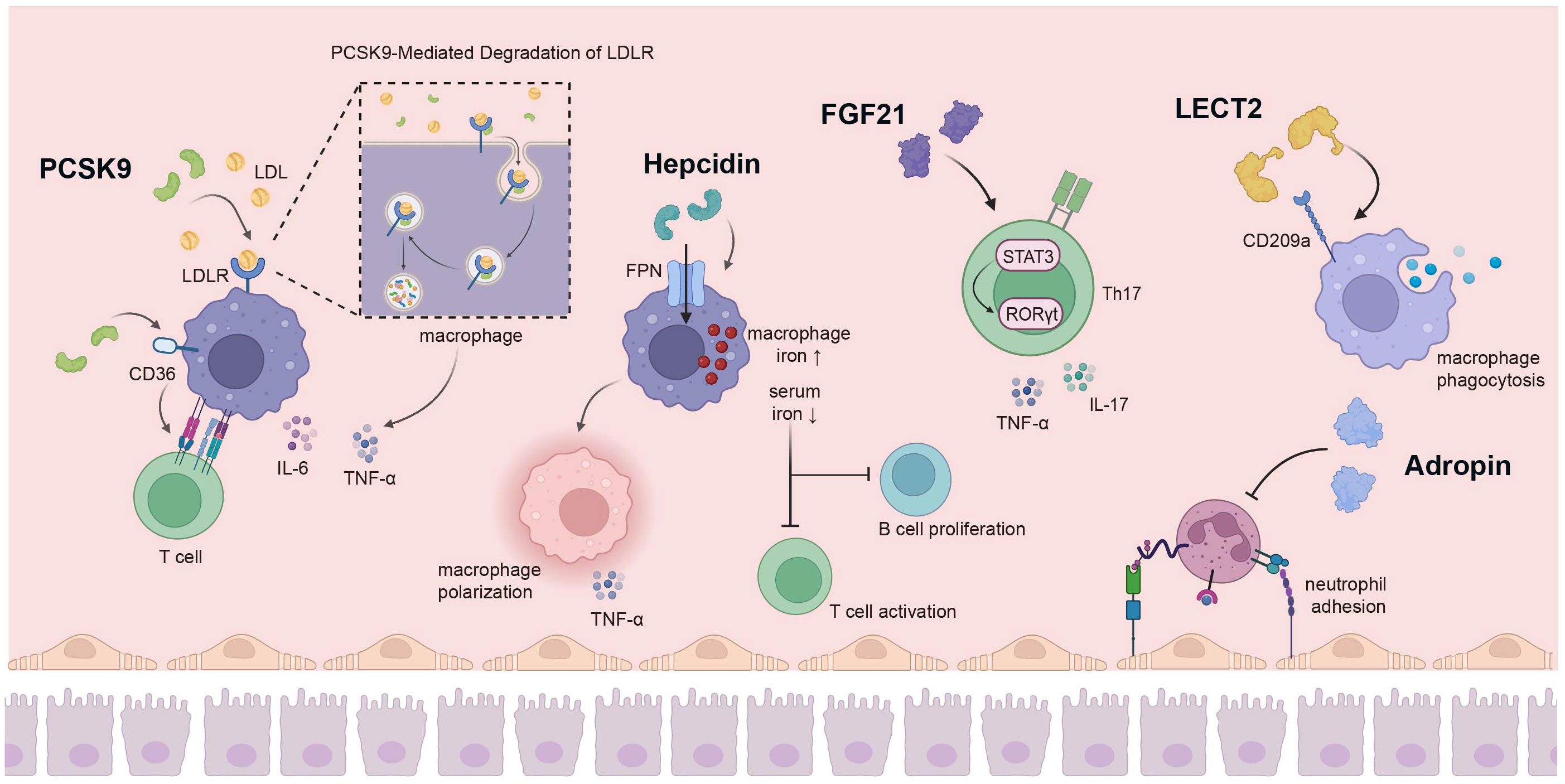
Figure 2 The liver secretes a variety of hepatokines for immune regulation. PCSK9 secreted by the liver has a variety of biological functions; it can promote macrophage cholesterol accumulation by stimulating LDLR degradation, affecting the production of pro-inflammatory cytokines. In addition, PCSK9 can act on CD36 on the surface of macrophages to affect the proliferation of T cells. Hepcidin can increase the iron level in macrophages by combining with FPN, and also promote the polarization of macrophages to pro-inflammatory direction, while low serum iron inhibits the activation of T cells and the proliferation of B cells. FGF21 regulates Th17-mediated inflammation through the STAT3/RORγt pathway. The interaction of LECT2 with CD209a can induce macrophage activation and enhance macrophage phagocytosis. Adropin hinders monocytes-endothelial cell interactions, thereby inhibiting the inflammatory response of endothelial cells and monocytes. PCSK9, Proprotein Convertase Subtilisin/Kexin 9; LDLR, low density lipoprotein receptor; FPN, ferroportin; FGF21, fibroblast growth factor 21; STAT3 signal transducer and activator of transcription 3; RORγt, retinoid-related orphan receptor-γt. Created with BioRender.com.
Hepcidin, alternatively referred to as the iron regulatory protein, is a cysteine-rich antimicrobial peptide synthesized in the liver. It plays a pivotal role in orchestrating iron homeostasis across the body, regulating plasma iron concentration, and governing overall iron content within an organism (139). Hepcidin-binding ferroportin (FPN) induces degradation, thereby modulating the release of iron from dietary sources, the recuperation of iron by macrophages, and the liberation of stored iron from hepatocytes into the bloodstream (77). The plasma concentration of hepcidin rises in response to iron accumulation and inflammation, while its levels are suppressed during active erythropoiesis (140). Iron thus serves as an indispensable micronutrient that is crucial for upholding cellular function. The stability of intracellular iron metabolism is intricately linked to the immune system’s functionality (141). In the context of infection and various inflammatory circumstances, hepcidin levels escalate under the influence of cytokines such as IL-6, IL-22, and others (142). Iron overload additionally reinforces host defenses, reshapes immune functionality, and modulates inflammatory response. The hepcidin-FPN axis contributes to augmented iron accumulation in macrophages and also governs pathogen proliferation within cells, representing a pivotal mechanism employed by the body to combat pathogenic infections (78, 79) (Figure 2). Hepcidin, derived from the liver and induced by inflammatory cytokines, reduces intestinal iron absorption and increases iron retention in macrophages. Moreover, hepcidin has the potential to prevent systemic infections of siderophilic and gram-negative bacteria by regulating iron levels (143, 144). During the early stages of infection, macrophages and neutrophils depend on hepcidin to foster the generation of pattern recognition receptor TLR4 in response to pathogens (80). The accumulation of iron within macrophages promotes their polarization towards a pro-inflammatory state; this process is coupled with the enhanced production of pro-inflammatory cytokines, including TNF-α, which is mediated by the TLR4/TRIF pathway (145). However, hepcidin-mediated hypoferremia influences leukocytogenesis, reducing the number of granulocytes but not monocytes or DCs. In addition, hypoferremia alters neutrophil effector functions and enhances mitochondrial reactive oxygen species-dependent NETosis, which is associated with chronic inflammation (146). Elevated hepcidin leads to reduced serum iron levels and can significantly compromise the immune response, influencing the reactivity of T and B cells to vaccination and infection (81) (Figure 2). Notably, earlier investigations have demonstrated that insufficient iron levels hinder the activation of T cells and the proliferation of B cells (82, 83). Mechanically, recent studies have proven that iron promotes the production of IFN-γ and IL-17A from CD4+ T cells by enhancing glucose metabolism (147, 148).
FGF21, a peptide hormone synthesized by multiple organs, governs energy equilibrium and lipid homeostasis through its interactions with FGF receptor 1 (FGFR1) and a heterodimeric complex made of β-klotho (149). The primary origin of circulating FGF21 is in the liver. It exhibits distinct metabolic functions across diverse target organs and is able to function as an autocrine, paracrine, and endocrine factor. Consequently, its biological properties are inherently complex (150, 151). FGF21 can inhibit the effect of nuclear factor NF-κB and upregulate peroxisome proliferator-activated receptor PPAR-γ on FGF receptor 1. This action mitigates the polarization of microglia and macrophages toward the M1 phenotype, consequently reducing the synthesis of pro-inflammatory cytokines (84). FGF21 induces autophagy via RACK-mediated AMPK activation and interaction with ATG5. It also further enhances cholesterol efflux and impedes the transformation of macrophages into foam cells (152). FGF21 takes on a significant anti-inflammatory role, leading to a decrease in the quantity of Th17 cells within the spleens of FGF21-treated mice. This decline is accompanied by reduced levels of IL-17, TNF-α, and IL-6, along with an increase in IL-10. FGF21 regulates Th17 mediated inflammation through the STAT3/RORγt pathway (85) (Figure 2). In the context of bacterial infection, the concentration of FGF21 in blood plasma rises in response to LPS stimulation. This elevation impacts innate immunity, leading to a decrease in the occurrence of endotoxemia and bacterial peritonitis (153). FGF21 also alleviates inflammation induced by LPS stimulation by impeding the TLR4/MYD88/NF-κB signaling pathway (86). In addition, FGF21 enhances the expression of Nrf2-ARE signaling-associated proteins, exerting an impact on inflammation and oxidative stress, as well as mitochondrial protection (154).
It has been suggested that Adropin, which is encoded by the energy homeostasis-associated gene (Enho), functions as a secreted protein expressed in both the liver and brain, acting as a determinant in regulating glucose and lipid homeostasis (87). Nevertheless, previous research findings indicate that Adropin may also exert a significant influence on inflammation, immune function, and neurological injuries (155). In mice lacking Adropin (Adropin knockout mice), elevated signals of F4/80, CD45, and MCP1 have been observed, along with substantial upregulation of TNFα and IL-6 genes (156). Adropin suppresses inflammation by diminishing levels of pro-inflammatory cytokines in tissues, including tumor necrosis factor alpha and interleukin-6 (88). Adropin also inhibits TNF-α induced adhesion of THP1 monocytes and endothelial cells. This inhibition of monocyte-endothelial cell interactions consequently restrains inflammatory responses in both endothelial and monocyte/macrophage compartments (89) (Figure 2). Furthermore, Adropin alters macrophage phenotypes towards the anti-inflammatory M2 state, as opposed to the pro-inflammatory M1 state, through the upregulation of PPAP-γ expression during differentiation from monocytes to macrophages (90). In adipose tissue, Adropin promotes the proliferation of 1T1-L2 preadipocytes by mediating ERK3/3 and AKT. Additionally, it limits the differentiation of preadipocytes into mature adipocytes, thereby curbing fat accumulation and reducing macrophage infiltration, and ultimately ameliorating inflammation (91). Moreover, deficiency in Adropin results in aberrant numbers and impaired functions of Tregs, which contributes to the development of autoimmune diseases (157). Adropin is also able to down-regulate the multifunctional inflammatory receptor CD36, which additionally provides compelling evidence for the anti-inflammatory properties of Adropin (92).
ANGPTL4 belongs to the angiopoietin-like protein family and plays a pivotal role in regulating angiogenesis, lipid metabolism, glucose metabolism, and redox reactions (158). Augmented lipid uptake is linked to the stimulation of inflammation-related genes, and Angptl4 knockout mice exhibit infiltration of neutrophils and macrophages (93). Through the SIRT1/NF-κB pathway, ANGPTL4 has the capacity to modulate the expression of inflammation-related genes induced by LPS (94). ANGPTL4 can function as a downstream target of PPARβ/δ, regulating the polarization of macrophages. ANGPTL4 amplifies macrophage activation, fosters tissue infiltration, and elevates complement component 5a (C5a) levels by activating the PI5K/AKT signaling pathway, consequently leading to hypercytokinemia (C5aR, IL-6, TNF-α, and IL-1β) (95). ANGPTL4 regulates the expression of interferon-activating gene 202B (ifi202b), impacts the monocyte-to-macrophage differentiation process, and coordinates neutrophil clearance and resolution of inflammation (96). Macrophages deficient in ANGPTL4 (-/-) adopt the M1 inflammatory phenotype due to dysregulated fatty acid metabolism, leading to substantial production of TNF-α and iNOS (97). In addition, ANGPTL4 deficiency also facilitates the immunomodulation of CD8+ T cells through metabolic reprogramming (98).
Fetuin A, a heterodimeric plasma glycoprotein primarily expressed in embryonic cells and adult hepatocytes, binds to diverse receptors and demonstrates intricate physiological and pathological roles (159). Pro-inflammatory cytokines like TNF downregulate its synthesis, leading to its classification as a negative acute phase protein (99). At higher concentrations, fetal A itself exhibits anti-inflammatory effects and effectively suppresses the generation of pro-inflammatory mediators like TNF, IL-1, and nitric oxide in bacterial endotoxin-stimulated macrophage cultures (100). By interacting with fatty acids, thyroid hormones, phosphates, and calcium ions, Fetuin A engages in diverse anti-inflammatory and inflammatory functions, in addition to facilitating neutrophil and platelet degranulation and lymphocyte stimulation (101). The anti-inflammatory effect of fetal A may stem from its direct counteraction against TGF-β and tumor necrosis factor-α (TNF-α) mediated inflammation, as well as its inhibition of pathogen-associated molecular patterns (PAMP)-triggered discharge of high-mobility hinode protein 1 (HMGBP1) by innate immune cells (102). Serving as an endogenous ligand for TLR4, Fetuin A takes on a key role in innate immunity by initiating inflammatory signals (103). Fetuin A sends chemical signals that prompt macrophage migration, shifting M2 macrophages toward the M1 phenotype and inducing the secretion of cytokines (104). Fetuin A-deficient mice have previously displayed monocyte and DC cell clustering, elevated IL-12/P40, ASC1, and IL-1 β expression, and enhanced Treg upregulation (105). Functioning as an acute-phase glycoprotein, Fetuin A regulates the generation of pro-inflammatory cytokines to uphold homeostasis amidst inflammation (106).
LECT2 is a hormone-like protein initially recognized as a neutrophil chemokine (160). Secreted by hepatocytes into the bloodstream, LECT2 serves as a multifunctional factor involved in numerous pathological conditions (161). In the bone marrow, LECT2 is capable of stimulating macrophages and modulating TNF expression through interaction with CD209a, consequently influencing HSC homeostasis (107). During bacteria-triggered sepsis, the interplay between LECT2 and CD209a can trigger macrophage activation, augmenting macrophage phagocytosis and bacterial eradication (108). Likewise, LECT2 pretreated DC cells can prompt cytokine secretion via the CD209a-JNK/P38 MAPK pathway (109). LECT2 is capable of hindering the recruitment and function of inflammatory monocytes, and its concentration is closely associated with inflammatory infiltration (110). In addition, LECT2 significantly enhances intercellular adhesion molecule-1 (ICAM-1) and the pro-inflammatory cytokine TNF-α, as well as monocyte chemotactic protein (MCP-1), via the CD209-JNK pathway, thereby reinforcing monocyte adherence to human endothelial cells (111). The expression of LECT2 has been shown to demonstrate a negative correlation with the immune infiltration of B cells, neutrophils, monocytes, cancer-associated fibroblasts, and myeloid DCs, while exhibiting a positive correlation with T cells, endothelial cells, and hematopoietic stem cells (112). LECT2-deficient (LECT2-/-) mice have exhibited a notably elevated proportion of liver NKT cells, which might contribute to the development of hepatitis (113). LECT2 also curtails the advancement of hepatocellular carcinoma (HCC) by interacting with iNKT cells, thereby obstructing β-catenin-induced inflammation (162) (Figure 2).
SeP, a secreted protein derived from the liver, functions to modulate the sensitivity or resistance of peripheral tissues to insulin (114). The essential trace element selenium (Se), integrated into Sep in the form of selenocysteine, contributes to diverse metabolic disorders linked to oxidative stress (163). The metabolism and regulation of selenium have significant implications for the physiological system, particularly the immune system (164, 165). In particular, deficiency in Se results in excessive generation of T cell oxidants, leading to the suppression of T cell proliferation upon stimulation of the TCR (115). Mice without SeP in T cells have been found to exhibit diminished numbers of mature and functional T cells within lymphoid tissue, as well as compromised T cell-dependent antibody responses (116). Sep also plays a crucial role in the activity of pro-inflammatory macrophages, and the suppression of SeP results in higher levels of inflammatory cytokine in tissues (117). SeP control oxidative bursts and cytokine production, enhance phagocytosis and killing, regulate inflammatory responses, and mitigate toxic damage arising from excessive macrophage activation (118).
RBP4, a member of the lipid carrier protein family, serves as the primary transporter of the hydrophobic molecule retinol (vitamin A) in circulation (166). RBP4 is produced in the liver and released by hepatocytes upon loading retinol and binding to the thyroxine transporter (TTR) (167). RBP4 participates in various biological activities as a vitamin A carrier, including cell proliferation, differentiation, immune regulation, bile secretion, and glucose and lipid metabolism (119, 120). Through the JNK pathway, RBP4 can activate APCs in vivo, leading to the proliferation of pro-inflammatory CD4+T cells and Th1 polarization (121). In instances of insulin resistance, RBP4 can trigger pro-inflammatory cytokine activation in macrophages via the c-Jun N-terminal protein kinase and TLR4 pathways (122). RBP4 activates innate immunity activation, leading to adaptive immunity induction. In mice with RBP4 overexpression, RBP4-induced macrophage antigen presentation and ensuing T cell activation are mediated by the MyD88 pathway, as well as downstream mitogen-activated protein kinases and NF-κB pathways (123). Furthermore, RBP4 can decrease IL-1β levels through the TLR3/MD1 receptor complex and TLR2, which activate NLRP3 inflammasomes in a glucose-dependent manner (124). Within an inflammatory microenvironment, RBP4 also enhances NOX1 and NF-κB activation, facilitates ROS accumulation, prompts M1-like polarization of Kupffer cells, and culminates in the excessive generation of inflammatory cytokines, such as TNF-α (125). Increased RBP4 levels can directly lead to endothelial inflammation, recruit leukocytes, and induce the expression of adhesion-related pro-inflammatory molecules, such as VACM-1, ICAM-1, E-selectin, and IL-6 (126).
DPP4 is a dipeptidyl protease that can exist as either a cell membrane protein or a soluble plasma protein and is synthesized and secreted by the liver. Concentrations of DPP4 are associated with body mass index and insulin resistance. Moreover, DPP4 can collaborate with plasma factor Xa to enhance the inflammatory responses of adipose tissue macrophages (ATM). Increased expression of DPP4 or external administration of DPP4 leads to decreased levels of pro-inflammatory IL-1β, IL-6, and IL-13, and augments the synthesis of the anti-inflammatory IL-10. In contrast, the DPP4 inhibitors sitagliptin and vildagliptin elevate the production of pro-inflammatory cytokines (127). DPP4 assumes a crucial function within the immune system, wherein it plays a vital role in upholding the composition and function of lymphocytes, as well as facilitating T cell activation and co-stimulation (128). DPP4 binds to the IGF2R receptor on the surface of Treg cells, triggering the activation of PKA/SP1 signaling. Consequently, this process impedes the degradation of IP3R2 and fosters the creation of mitochondria-associated ER membranes, leading to mitochondrial calcium overload in Tregs. As a result, DPP4 mediates the impairment of Treg functionality and the polarization of M1 microglia (129).
Bile acids are steroidal molecules formed by cholesterol oxidation in the liver, as well as signaling molecules and metabolic integrators. They activate the nuclear farnesol X receptor (FXR) and the membrane G protein-coupled receptor 5 (TGR5, also known as G protein-coupled bile acid receptor 1) in order to regulate glucose, lipid, and energy metabolism (168–170). Due to the intricate nature of bile acid signaling, its influence on the immune response is of paramount importance (171). The bile acid metabolite 3-oxocaryocholic acid (3-oxoLCA) regulates the accumulation of CXCR6 in hepatic NKT cells by suppressing the expression of the chemokine CXCL16 by RORγt receptor (172). Activation of the TGR5 bile acid receptor induces PKA kinase activity, which results in the ubiquitination of NLRP3, effectively inhibiting NLRP3 inflammasome-mediated lipopolysaccharide-induced systemic inflammation (173). Additionally, bilirubin, a product of heme metabolism in the liver, exhibits potent immunomodulatory effects, with high levels of bilirubin being able to induce apoptosis in immune cells (174). Within the context of Th17 cells, bilirubin modulates immune response by augmenting the downstream effects of AHR and by enhancing CD39 mediated in vitro enzymatic activity (175). Bilirubin can additionally influence cholesterol synthesis, reshaping the immune system and resulting in reduced NK cells while also promoting the expansion of DCs and MDSC populations (176) (Figure 3).
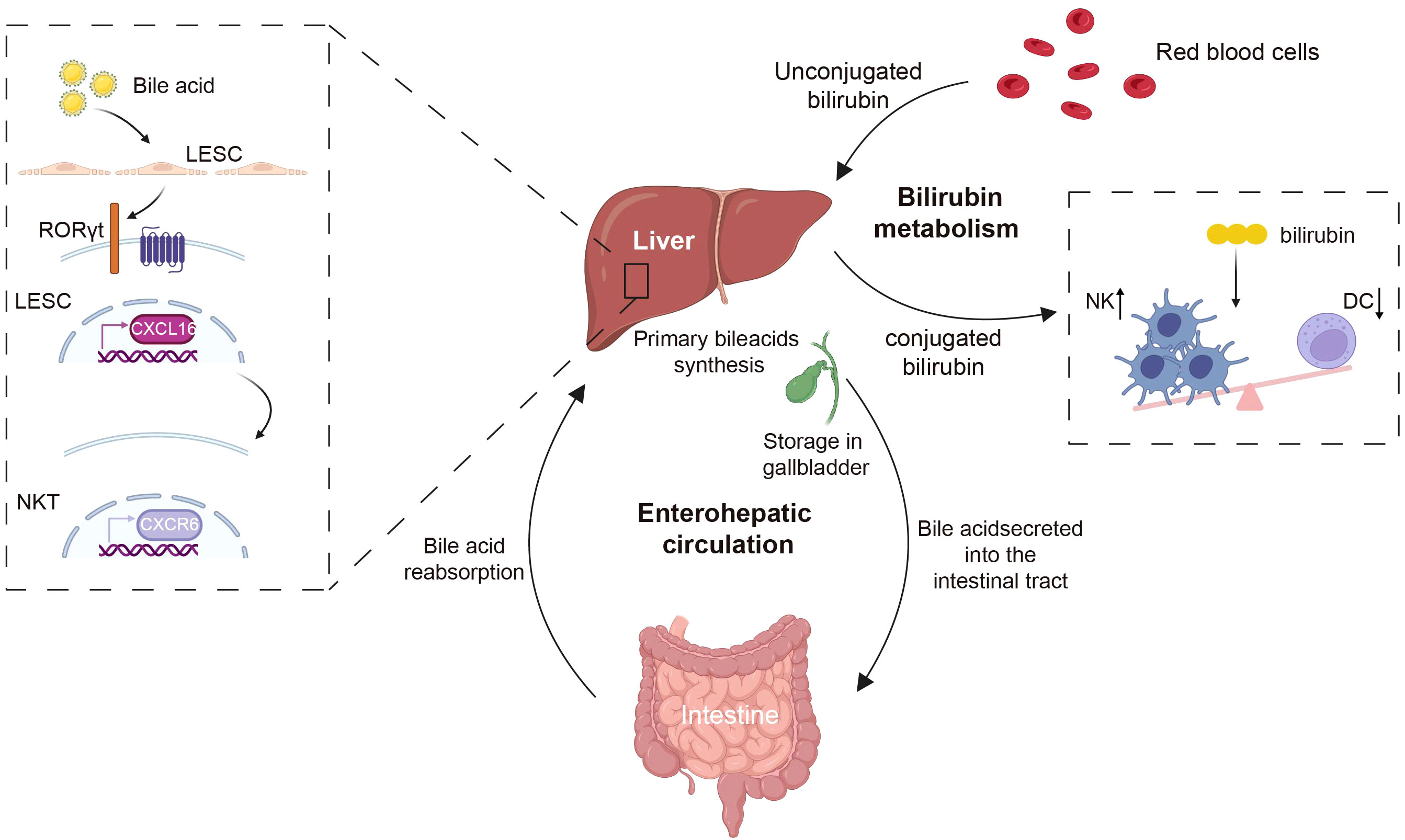
Figure 3 The liver metabolites of bile acids and bilirubin regulate immune cells. Bile acids, which are produced by the liver as cholesterol metabolites, are excreted into the intestine via the gallbladder, and are reabsorbed back to the liver through enterohepatic circulation. Bile acids can regulate the accumulation of CXCR6 in NKT cells by modulating the expression level of the chemokine CXCL16 on LSECs. Bilirubin, the byproduct of red blood cells, following interactions with hepatocytes, has the capacity to modulate the immune system by decreasing the population of NK cells and increasing DCs. CXCR6, CXC chemokine receptor 6; CXCL16, CXC chemokine ligand 16; NKT, natural killer T; NK, natural killer; DC, dendritic cell. Created with BioRender.com.
The complement system, which acts in concert with the liver, functions as an immune complex that interconnects liver homeostasis with immune responses and various effector systems (177). Activation pathways of the complement system are categorized into three types: classical, alternative, and lectin. The culmination of all complement activation pathways is the creation of a membrane attack complex (MAC), a potent cell activator and a key driver of inflammation (178, 179). The complement system offers a range of crucial protective effects; it serves not only as the primary defense against microbial intrusion, but also contributes to diverse immune and inflammatory functions while orchestrating homeostasis (180). Additional components of the complement system apart from MA are also active. C3b can coat pathogen surfaces, enabling recognition by neutrophil complement receptor CR1 and mediating pathogen uptake, degradation, and induction of NETosis (181). Complement-derived C5a also plays a role in the recruitment of neutrophils. Upon contact with the vascular endothelium, the MAC continues to activate neutrophil extracellular traps (182). In macrophages, MAC can be internalized into endosomes, activating the inflammasome and triggering IL-1β release during inflammation (183). In addition to MAC-induced IL-1β release, C5a-C5aR1 signaling potentiates the activation of the NLRP3 inflammasome and the release of IL-1β in mouse macrophages. This was also true in human monocytes (184). Circulating C1q contributes to immune differentiation by containing monocyte specialization into dedicated antigen-presenting cells. Furthermore, it curbs the production of pro-inflammatory cytokines in innate immune cells, enhances phagocytic mechanisms in macrophages, and modulates CD8+ T cell mitochondrial metabolism to curtail autoantigen responses (185, 186). The complement system can also directly exert an impact on T-cell function. C3a and C3b translocate to the cell membrane and interact with costimulatory CD46, which leads to the metabolic reprogramming of T cells (187). By binding to G protein-coupled receptors on APCs and T cells, the anaphylatoxins C3a and C5a provide costimulation and survival signals to naïve CD4+ T cells via the PI3K-AKT pathway (188, 189). C3 and C3a have been shown to have significant roles in fostering Th2 response, while C3a additionally propels innate lymphocyte (ILC2)-mediated inflammatory reactions, eliciting IL-13 and granulocyte-macrophage colony-stimulating factors, while constraining IL-10 generation (190). Complement factor H (CFH) and complement factor I (CFI) serve as pivotal plasma regulators of the complement response. Factor H (FH) serves as a fluid-phase complement regulatory protein, preventing the hyperactivation and excessive expansion of the complement system. The negative regulator of the complement system, FH, forms complexes with nucleosomes to promote the monocyte phagocytosis-induced release of anti-inflammatory cytokines (191). Complement factor H-associated protein 1 (FHR-1), a member of the complement factor H-associated proteins (FHRs), also assumes a significant role in innate immunity by impeding complement activation through the inhibition of C5 convertase. Furthermore, FHR-1 stimulates the release of inflammatory cytokines from monocytes, exhibiting a complement-independent mechanism. The paradoxical nature of its effects confers upon it a distinct role within the innate immunity process (192) (Figure 4).
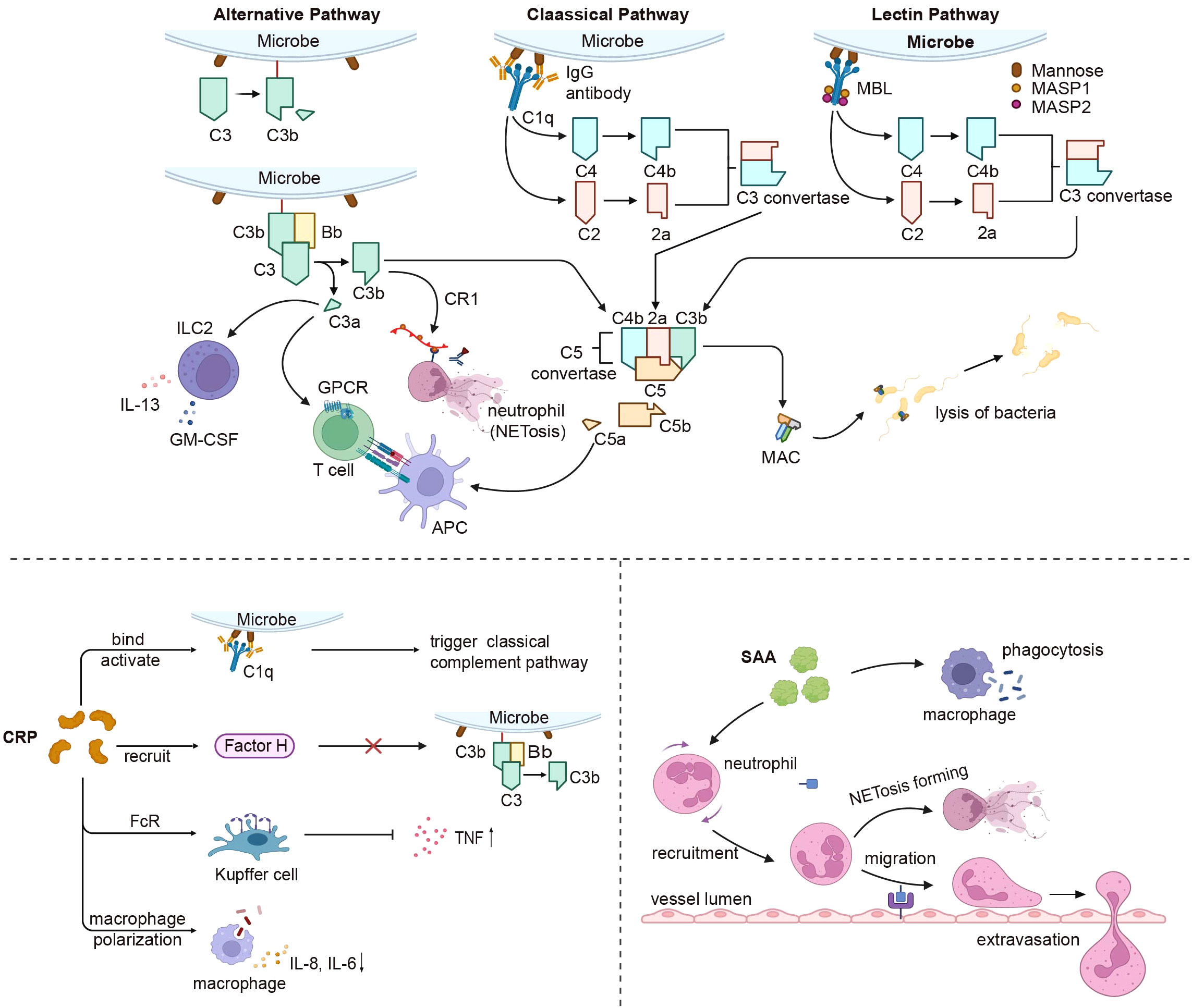
Figure 4 Liver derived innate immune proteins’ impact on the immune system. The complement system connects the liver and immune response after activation by the classical, alternative, and lectin pathways. All complement activation pathways eventually form MAC, which resists microbial invasion. C3a and C5a affect GPCR signaling in APC and T cells as anaphylactic toxins, while C3a also influences ILC2-mediated inflammatory responses. C3b can be recognized by CR1 to stimulate neutrophil NETosis. CRP triggers the classical complement pathway by binding and activating C1q, recruits factor H to prevent alternative pathway activation, and interacts with FcR on Kupffer cells, reducing TNF production and effects on macrophage polarization. SAA primarily increases neutrophil respiratory burst and migration, and also enhances macrophage phagocytosis. MAC, membrane attack complex; GPCR, G-protein-coupled receptors; APC, antigen presenting cell; ILC2, innate lymphoid cells; CR1, complement C3b/C4b Receptor 1; TNF, tumor necrosis factor; SAA, serum amyloid A. Created with BioRender.com.
CRP is an acute-phase protein present in minimal amounts in healthy individuals; however, it is swiftly synthesized and released into the bloodstream during bacterial infections and tissue damage (193). Previous studies suggested that CRP exists in at least two conformationally distinct forms, namely pentameric CRP (pCRP) and monomeric CRP (mCRP), which bind to distinct receptors and exhibit unique functions, respectively (194). CRP initiates the classical complement pathway by binding to and activating C1q, thereby promoting the assembly of the C3 convertase complex and mediating phagocytosis. Furthermore, CRP recruits factor H, which hinders the activation of C3b through alternative pathways (195, 196). It contains phosphocholine (PC), which efficiently mitigates ATP-induced monocyte inflammasome activation and averts inflammatory responses (196, 197). Furthermore, CRP engages with Fc receptors on phagocytes, functioning as an opsonin, to decrease TNF production while enhancing the phagocytic activity of Kupffer cells (198). CRP influences macrophage polarization; following CRP treatment, M2 macrophages transition to the M1 phenotype, accompanied by elevated secretion of IL-8, IL-6, and TNF in macrophages (199, 200). Increased CRP levels could potentially activate circulating monocytes by enhancing their chemotaxis response to MCP-1 (201, 202). CRP also exhibits anti-inflammatory effects through the upregulation of monocyte liver X receptor (LXR) α and activator receptor expression, as well as the downregulation of α2-macroglobulin expression (203). CRP induces the release of IL-8 by neutrophils via peroxynitrite-mediated activation of nuclear factor-κB and activator-1 (204). In the absence of an acute response, CRP serves a vital biological function by acting as a tonic inhibitor of the adaptive immune system, thereby maintaining peripheral T cell tolerance by inhibiting the maturation of DCs (205) (Figure 4).
Recent advances emphasize that pCRP exhibits both proinflammatory and anti-inflammatory properties, whereas mCRP is exclusively proinflammatory (206). The interaction of pCRP with FcγRIIa demonstrates a protective effect against autoimmune diseases by reducing the type I interferon response triggered by immune complexes (207). Both pCRP and mCRP can induce thrombus formation, activate monocytes, platelets, and neutrophils, enhance the adhesion of neutrophils and monocytes to endothelial cells, and promote the formation of neutrophil-platelet and platelet-monocyte aggregates. This process involves the production of the proinflammatory cytokines IL-1β, IL-6, and NETosis, which may result in excessive, insoluble inflammation and increased tissue damage (208). In the context of neutrophilic inflammatory responses to influenza A virus, CRP can bind to histone H4, leading to a significant inhibition of neutrophil H2O2 production, calcium influx, degranulation, and prevention of neutrophil membrane permeabilization (209). Although CRP is widely employed as a clinical marker of inflammation, it’s in vivo function and role in health and disease are still largely unestablished. Two recent lines of evidence form the basis for an improved model design, given the basic and conserved functional phenotypes of endogenous CRP in mice and rats. CRP gene knockout animals should be employed to investigate the in vivo role of human CRP. Simultaneously, the function of the human CRP is contingent upon its origin, conformation, and localization. Therefore, tissue-specific expression and conformation-locked mutants are also crucial in in vivo studies (210).
SAA functions as an innate immunomodulator against gram-negative bacteria, including Escherichia coli and Pseudomonas aeruginosa (211, 212). The opsonic effects of SAA mainly involve heightened neutrophil respiratory bursts, improved phagocytosis in monocyte-derived macrophages, and elevated production of TNF-α and IL-10 (213, 214). SAA performs biological functions via G protein-coupled receptor (GPCR) and formyl peptide receptor (FRP3) signaling, which leads to FRP3 activation, subsequently promoting migration of neutrophils and monocytes (215, 216). SAA fragments function alongside CCL3 to induce monocyte migration and interact with CXCL8 to facilitate neutrophil morphological alterations and chemotaxis (217). SAA induces strong endogenous stimulation of granulocyte colony-stimulating factor (G-CSF) production, which is facilitated by TLR2-mediated mechanisms (218). SAA signaling also triggers macrophages to secrete IL-1β via NLRP3 inflammasome activation, prompts dendritic cell maturation, and leads to the generation of IL-1, IL-6, PGE2, and IL-23, ultimately promoting CD4+ T cell secretion of IL-17A (219). As an acute-phase protein, SAA can impact insulin resistance, hepatic lipid accumulation, and liver injury by activating NF-κB signaling through its binding with TLR4 (220). Moreover, SAA participates in the crosstalk between hepatocytes and hepatic stellate cells, inducing inflammation, proliferation, and apoptosis in HSCs (221). Recent studies have discovered that SAA binds retinol and mediates the trafficking of B and T cells, as well as IgA production after bacterial infection (222, 223). In some cases, SAA may exhibit an anti-inflammatory effect (224) (Figure 4).
Apart from generating acute-phase reactive proteins, the liver also secretes chemokines that attract immune cells to sites of injury or infection in order to initiate a response (66). MCP-1 and its receptor, chemokine (C-C motif) receptor 2 (CCR2), play a critical role in recruiting and activating monocytes and macrophages at sites of tissue injury. They also modulate adhesion molecules and pro-inflammatory cytokines, such as TNFα, IL-1β, and IL-6 (225, 226). Additionally, hepatocytes secrete CXCL1 to assist in safeguarding the host against bacterial infections (227).
Blood coagulation, commonly known simply as coagulation, refers to the transformation of blood from a liquid state to a gel state, and is an important part of hemostasis. The coagulation process involves the sequential activation of a cascade of coagulation factors through enzymatic hydrolysis. This culminates in the production of thrombin, leading to the formation of a fibrin clot (228, 229). Recent years have seen mounting evidence indicating the involvement of coagulation factors, namely thrombin and fibrinogen, in tissue repair and inflammatory responses (230). Furthermore, the coagulation system is recognized as an integral part of innate immunity, and coagulation factors, as well as plasmin, have emerged as crucial mediators of inflammation (231) (Figure 5).
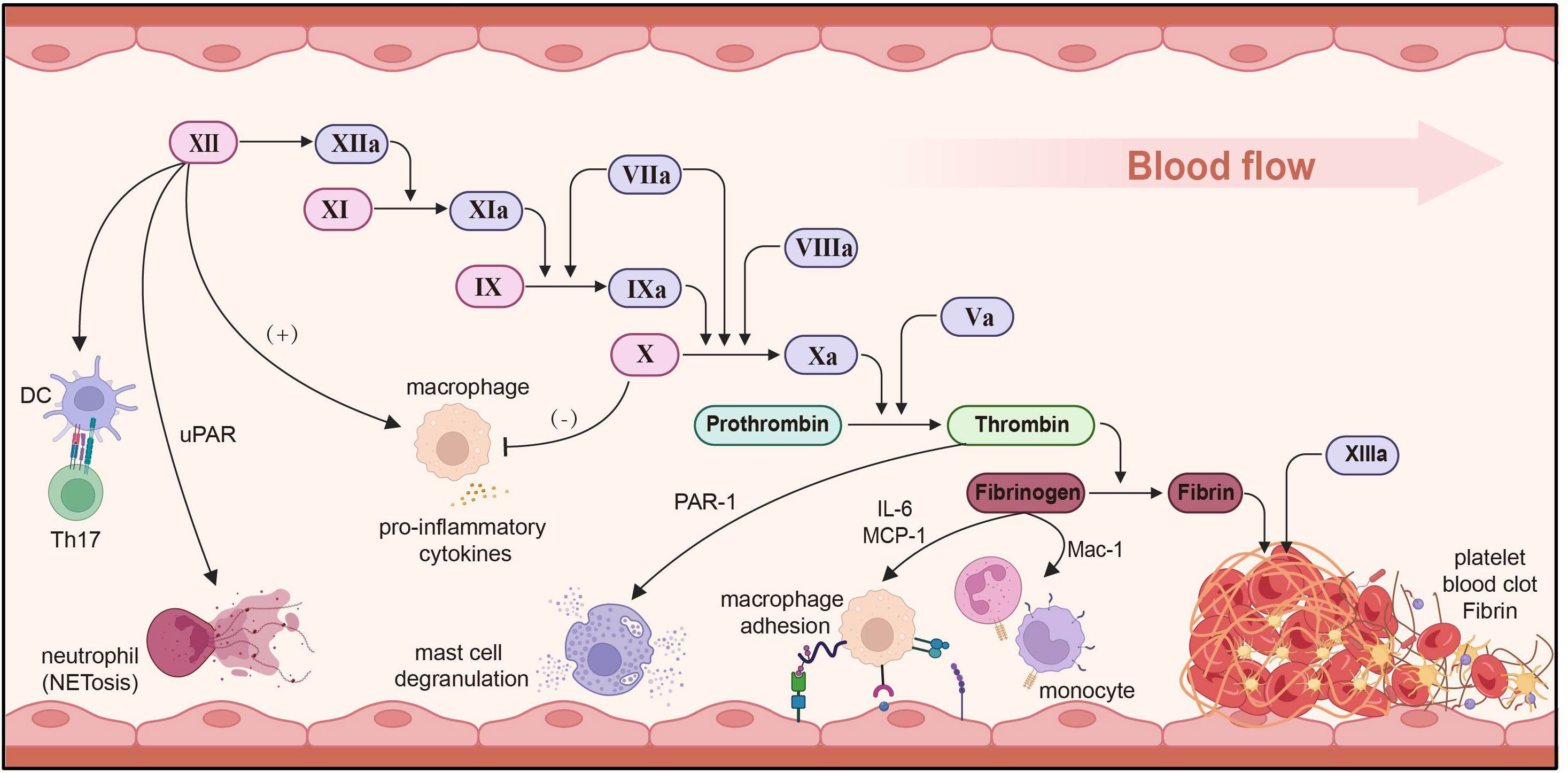
Figure 5 Immune regulation of the coagulation system. Factor XII stimulates DCs to induce Th17 production, promotes neutrophil adhesion and NET through uPAR, and induces macrophages to secrete pro-inflammatory cytokines. FXa inhibits and reduces macrophage secretion and reduces inflammation. Thrombin promotes mast cell degranulation via PAR-1. Fibrinogen secretes IL-6/MCP-1 and regulates macrophage adhesion. uPAR, urokinase-type plasminogen activator receptor; MCP-1, monocyte chemoattractant protein-1; DC, dendritic cell. Created with BioRender.com.
Evidence demonstrates that interplay among coagulation factors not only contributes to hemostasis, but also significantly impacts the progression of inflammatory diseases. Factor XII, a soluble enzyme synthesized in the liver and alternatively known as the contact factor or Hageman factor, primarily serves to trigger the activation of factor XI, plasminogen, and prekallikrein. Factor XI is a key component of the intrinsic coagulation pathway, and its activation is impeded in the absence or blockade of factor II, which impairs neutrophils’ capacity to surround bacteria (232). Aside from its thrombotic function, factor FXII also fosters inflammation by activating the bradykinin release system. It can also serve as a pro-inflammatory cytokine, provoking macrophage pro-inflammatory cytokine responses. Furthermore, it triggers CD4+ T cell antigens to produce specific IFN-γ, and factor II prompts DCs to induce Th17 cells production via a uPAR-dependent mechanism (233–235). FXII signaling promotes neutrophil adhesion, migration, and the release of neutrophil extracellular traps through urokinase plasminogen activator receptor-mediated phosphorylation, thereby promoting NETosis (236). factor FXa can also induce the release of IL-8 and monocytes chemotaxis protein by activating protease activation of receptor 8 (237). Inhibiting factor FXa diminishes macrophage accumulation and curtails the secretion of TNF-α, COX-1, and iNOS, thereby mitigating the inflammatory response (238). Thrombin elicits vasodilation and mast cell degranulation through PAR-1 activation, while also instigating the production of cytokines/chemokines IL-6 and MCP-1, while also promoting macrophage adhesion via fibrin (ogen) (239).
Fibrinogen genes are expressed almost exclusively in hepatocytes and can be stimulated during acute phase inflammation through the control of proximal promoter activity (240). In the initial stages of the innate immune response, fibrinogen bind to the bacterial surface, swiftly isolating and counteracting invading pathogens (241). Fibrinogen can invoke antibacterial effects by activating the complement system, and interacts with MBL to initiate the lectin-complement pathway (242, 243). Plasminogen and fibrinogen can also act as neutrophil surface integrin α (M) and β (2) ligands, impeding apoptosis through the activation of AKT and ERK1/2 (244). The cured fibrin matrix activates monocytes/macrophages and neutrophils through Mac-1, inciting a pro-inflammatory response (245). Fibrino-like protein 1 (FGL1) has been identified as a prominent functional ligand for LAG-3, operating independently of MHC-II. It also inhibits antigen-specific T cell activation and emerges as a potential target for the next immune checkpoint (246, 247).
The liver harbors a significant population of immune cells engaged in immune surveillance and response. These cells fall into two main categories: liver-resident cells and circulating immune cells recruited from the bloodstream. These immune components serve a multifaceted role, aiding the liver in fending off pathogenic incursions, facilitating regeneration following injury, and supporting detoxification processes, while also instigating adaptive immune responses (248) (Figure 6).
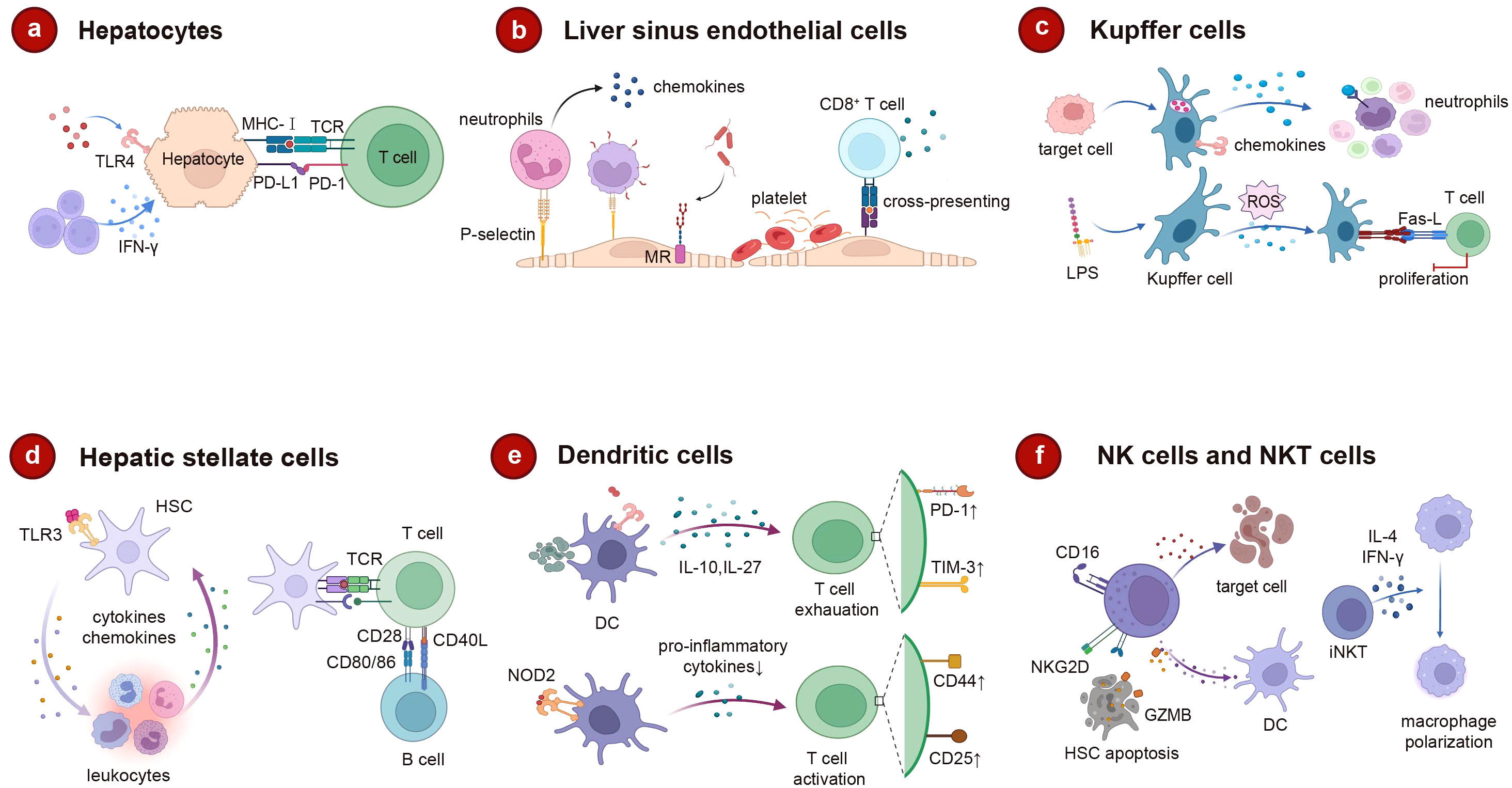
Figure 6 The role of intrahepatic cells in liver immune regulation. (A) Hepatocytes trigger adaptive immunity through MHC-I after receiving surface receptor signals. (B) The unique structure of LSEC promotes lymphocyte migration and platelet adhesion. As an endothelial cell with clearance effect, MR can mediate the rapid clearance of pathogens, and LSEC can cross-commission foreign antigens to CD8+ to induce immune tolerance. (C) Kupffer cells clear apoptosis target cells, promote neutrophil attachment, and also derive ROS to regulate apoptosis under the induction of LPS. (D) HSC not only interacts with leucocytes in a bidirectional manner, but also affects T and B cell activation as an antigen-presenting cell. (E) DC promotes T cell exhaustion through IL-10, IL-2. In turn, when NOD2 binds to its ligand, DC regulates T cell proliferation. (F) After activation, NK cells can kill target cells directly or by secreting pro-inflammatory cytokines. Increased degranulation of activated NK cells promotes HSC apoptosis through NKG2D. Activated iNKT can rapidly release IL-4 and IFN-γ mediated macrophage polarization acceleration. MHC-I, major histocompatibility complex (MHC) class I; LSEC, liver sinus endothelial cell; ROS, reactive oxygen species; LPS, lipopolysaccharide; NOD2, nucleotide-binding oligomerization domain 2; DC, dendritic cell; HSC, hepatic stellate cell; NK, natural killer; iNKT, invariant natural killer T; IFN-γ, interferon-gamma. Created with BioRender.com.
Roughly 80% of the liver’s composition is comprised of hepatocytes. While their primary responsibilities encompass material metabolism, protein synthesis, and toxin neutralization, hepatocytes also possess the capability to fulfill immune functions (249). Hepatocytes perform the synthesis of the majority of acute phase proteins and complements, serving as the foremost guardians against pathogens. Furthermore, their immune effects extend beyond this role, as hepatocytes possess the ability to adhere to the surfaces of specific microorganisms, thereby augmenting the detection of pathogens (250). Hepatocytes also possess intrinsic immune receptors that identify pathogen associated molecular pattern (PAMPs), thus instigating innate immune responses within hepatocytes. These receptors encompass cell surface receptors (e.g., TLR4), endosomal receptors (e.g., TLR3), and cytoplasmic receptors [e.g., stimulators of the IFN gene (STING)], components of the retinoic acid-inducing gene-1 (RIG-1) pathway, and members of the nucleotide-binding oligomerization domain (NOD) family) (251). Additionally, hepatocytes can also trigger an adaptive immune response. Hepatocytes possess the capacity to express MHC I molecules alongside antigen presentation-related molecules. Under inflammatory circumstances, specific hepatocytes may also be prompted to express MHC II molecules, which localize to the basolateral surface of hepatocytes. Consequently, they engage with lymphocytes and foster their activation (252, 253). Hepatic cells lack the expression of co-stimulatory molecules CD80 and CD86, making them incapable of engendering enduring activation and viability of T cells (254). Hepatocytes can also express PD-L1 in the presence of type I and type II IFN, and PD-L1 expression on hepatocytes induces apoptosis of T cells (Figure 6A).
LSECs are a type of endothelial cell with a scavenging role and are responsible for eliminating soluble macromolecular waste from tissues through high-affinity endocytic receptors, such as mannose receptors (MR) and clearance receptors (SR) (255). Additionally, endocytic receptors on LSECs play a crucial role in viral infections, facilitating the swift elimination of bloodborne viruses (256). The presentation of exogenous antigens internalized by LSECs can directly influence the modulation of adaptive immune responses (257). Functioning as the principal antigen-presenting cells within the liver, LSECs promote hepatic immune tolerance and executes anti-inflammatory functions (258, 259). LSECs display an extensive array of pattern recognition receptors (PRRs), including TLR-3, 4, 7, 9, while concurrently expressing MHC molecules and co-stimulatory molecules (260). LSECs also have the capacity to present soluble antigens to naïve CD4+ T cells, thereby inducing regulatory phenotypes. In cirrhotic patients and mouse models of liver fibrosis, LESCs are stimulated by liver damage, which promotes the immunoproteasome LMP7 levels in LESCs and the capacity of MHCII antigen presentation to CD4+ T cells (261). Additionally, LSECs are capable of cross-presenting soluble foreign antigens via MHC I molecules to CD8+ T cells, leading to the induction of immune tolerance (262, 263). The LSEC-induced T-cell activation is influenced by the antigen load and local inflammatory factors. High antigen concentrations can overcome PD-1-mediated tolerogenic responses, leading to T-cell differentiation into effector T cells (258). Circulating CD4+ cells engage in repeated interactions with hepatic sinusoidal endothelial cells, thereby suppressing the secretion of inflammatory cytokines by Th1 cells and Th17 cells (264). The distinctive structure of LSECs further facilitates the migration of lymphocytes and ensures their optimal localization (265). Compared to other types of endothelial cells, the adhesion molecular expression profile of LSECs is unique, including ICAM-1, VCAM-1, VAP-1, and stabilin-1, which is crucial for leukocyte recruitment (266). Lymphocyte recruitment involves an adhesion cascade occurring within the hepatic sinuses, influenced by a low-shear environment and intercellular communication between parenchymal and non-parenchymal cells (267, 268). Integrins located on the surface of LSECs adhere to platelets and release chemokines that facilitate the recruitment of neutrophils and lymphocytes (269). In a pro-inflammatory phenotype, dysfunctional LSECs fail to maintain Kupffer cell homeostasis, leading to the release of inflammatory mediators (270). With the stimulation of mechanical stretch, LSECs release CXCL1 to promote the recruitment of sinusoidal neutrophils and to facilitate the formation of NETosis and microthrombi (271). Injured LSECs or cancer-activated LSECs (cLSECs) enhance the proliferation of Treg cells through TGF-β, which can inhibit effector T-cell function and lead to cancer progression (272) (Figure 6B).
Kupffer cells, resident macrophages within the liver, play an important role in both antigen presentation and the response to tissue damage (273). Kupffer cells are central to the clearance of circulating antigens, and the presence of the complement receptor CRIg on their surface serves as a crucial element of the innate immune system. The absence of CRIg impairs the efficient clearance of circulating pathogen (274). Kupffer cells generate adhesion molecules that induce interactions with neutrophils, leading to the adherence of neutrophils to Kupffer cells and thereby assisting in the clearance of pathogens (275). Furthermore, Kupffer cells also possess the ability to eliminate activated and apoptotic cells from the bloodstream (276). LPS stimulates the production of reactive ROS by Kupffer cells, initiating the Fas-L transcription program that regulates apoptosis in T cells (277). Both Kupffer cells and LSECs contribute to the recruitment of neutrophils through the activation of TLR and CD44/HA mechanisms (278). Additionally, antigen presentation by Kupffer cells has the capacity to induce immune tolerance, leading to CD4+ T cell arrest and promoting the secretion of IL-10, which in turn triggers the expansion and activation of Tregs (279, 280). The chemokines generated by Kupffer cells are responsible for chemically attracting monocytes, T cells, NK cells, and DCs, thereby promoting the adhesion of T cells to endothelial cells (281). Kupffer cells have the capability to activate iNKT cells via the CD1 pathway, and this interaction is prevalent throughout the liver (282) (Figure 6C).
Quiescent HSCs express TLR3, which triggers the transcription and secretion of functional interferons as well as numerous other cytokines and chemokines. Upon activation into myofibroblasts, HSCs rapidly lose their ability to produce IFNγ (283). Stellate cells engage in bidirectional interactions with immune cells, whereby they not only respond to leukocyte regulation but also influence leukocyte chemotaxis and adhesion, thereby contributing to the modulation of leukocyte activation (284). As antigen-presenting cells, HSCs express MHC I and MHC II molecules, along with lipid-presenting CD1B and CD1C molecules, CD86, CD40, and other co-stimulatory molecules (285, 286). When exposed to pro-inflammatory cytokines like IFNγ, HSCs experience a notable upregulation of CD80, while the activation of CD40 results in elevated IL-8 secretion in HSCs and an increased release of monocyte chemotactic protein-1 (287, 288). Activated HSCs exhibit the expression of the negative costimulatory factor PD-L1 and engage with T cells through B7-H1-mediated apoptosis (289). HSCs may also counteract B cell activity using a similar mechanism (290). Increased CD54 expression on HSCs further contributes to the attenuation of T cell activation (291) (Figure 6D).
DCs are highly immunogenic APCs that excel in capturing, processing, and presenting antigens to T cells (292). However, hepatic DCs exhibit greater tolerance compared to conventional DCs. They tend to produce IL-10 and IL-27 upon LPS stimulation, resulting in a subdued T cell response mediated by IL-27 (293, 294). IL-10 secretion by DCs can modulate the balance between Th1 and Th2 cells, augment the population of IL-4 producing Th2 cells, and enhance the generation of CD4+CD25+Foxp3+ Tregs (295). Furthermore, liver-resident DCs can directly lead to T cell depletion, which encompasses T cell anergy (296). DCs are classified into classical type 1 DC (cDC1), cDC2, and plasmacytoid DC (pDC) cells, of which cDC2 cells predominate in the liver and display a tolerant nature, whereas cDC1 cells engage in antigen presentation toward T cells (297, 298). The CD103+cDC1 subtype of DCs acts as a protective variant that impacts pro-inflammatory and anti-inflammatory balance and reduces local inflammation (299). Upon binding of NOD2 on the DC surface with its ligand, it interferes with the signaling pathways of TLR4 and TLR9 in pDC. This disruption leads to reduced secretion of pro-inflammatory cytokines, such as IL-2, IL-6, TNF-α, and IFN-γ, while concurrently inducing an upregulation of B7-H1. This modulation of pDCs activity alters the regulatory influence on T cell proliferation (300) (Figure 6E).
NK cells and NKT cells are significant cellular elements within the liver microenvironment of the innate immune system. They orchestrate both cytotoxic and cytokine-mediated responses, thereby exerting a pivotal influence on the configuration of adaptive immunity (301, 302). Activation of NK cells hinges on the perturbation of the equilibrium between surface inhibitory receptors and stimulatory receptors, such as NKG2D/NKG2A (303). The NKT cell presence is essential for the activation of NK cells, contributing to the generation of IFN-γ and IL-4, which aid in facilitating NK cell activation (304). NK cells can execute direct killing of infected target cells or elicit the secretion of pro-inflammatory cytokine IFN-γ upon activation, thereby exerting cytotoxic effects (305, 306). NK cells with adequate IFN-γ levels can positively modulate CD8+ T cells through IFN-γ secretion, while, as the principal generators of IL-10, NK cells also contribute to the regulation of T cell activation (306, 307). The proliferation of Tregs can be additionally facilitated by NK cells through the generation of inhibitory factors like TGF-β and IL-2, which dampen dendritic cell activation. Moreover, the activated degranulation of NK cells augments and fosters hepatic stellate cell (HSC) apoptosis through a mechanism that is reliant on TRAIL and NKG2D (308). Unlike the killing mechanism of NK cells involving TRAIL and granzyme B, NKT cells predominantly operate through the release of pro-inflammatory cytokines and FasL (309). Activated iNKT cells can rapidly release IL-4 and expedite macrophage polarization through IFN-γ mediation (310). Furthermore, iNKT cells foster neutrophil infiltration via the IL-4/STAT6 pathway, whereas IFN-γ/STAT1 accelerates neutrophil apoptosis (311) (Figure 6F).
Advancements in liver immunology research have yielded a more profound comprehension of hepatically secreted proteins and the involvement of both parenchymal and non-parenchymal liver cells in immune responses. This progress not only contributes to unraveling the etiology of hepatitis, cirrhosis, and other hepatic disorders, but also offers novel insights into diverse, systemic immunity-linked conditions (Figure 7). Furthermore, it establishes a solid groundwork for the exploration and formulation of innovative therapeutics and strategies targeting liver-associated immune diseases. We summarize the detail information in Table 2.
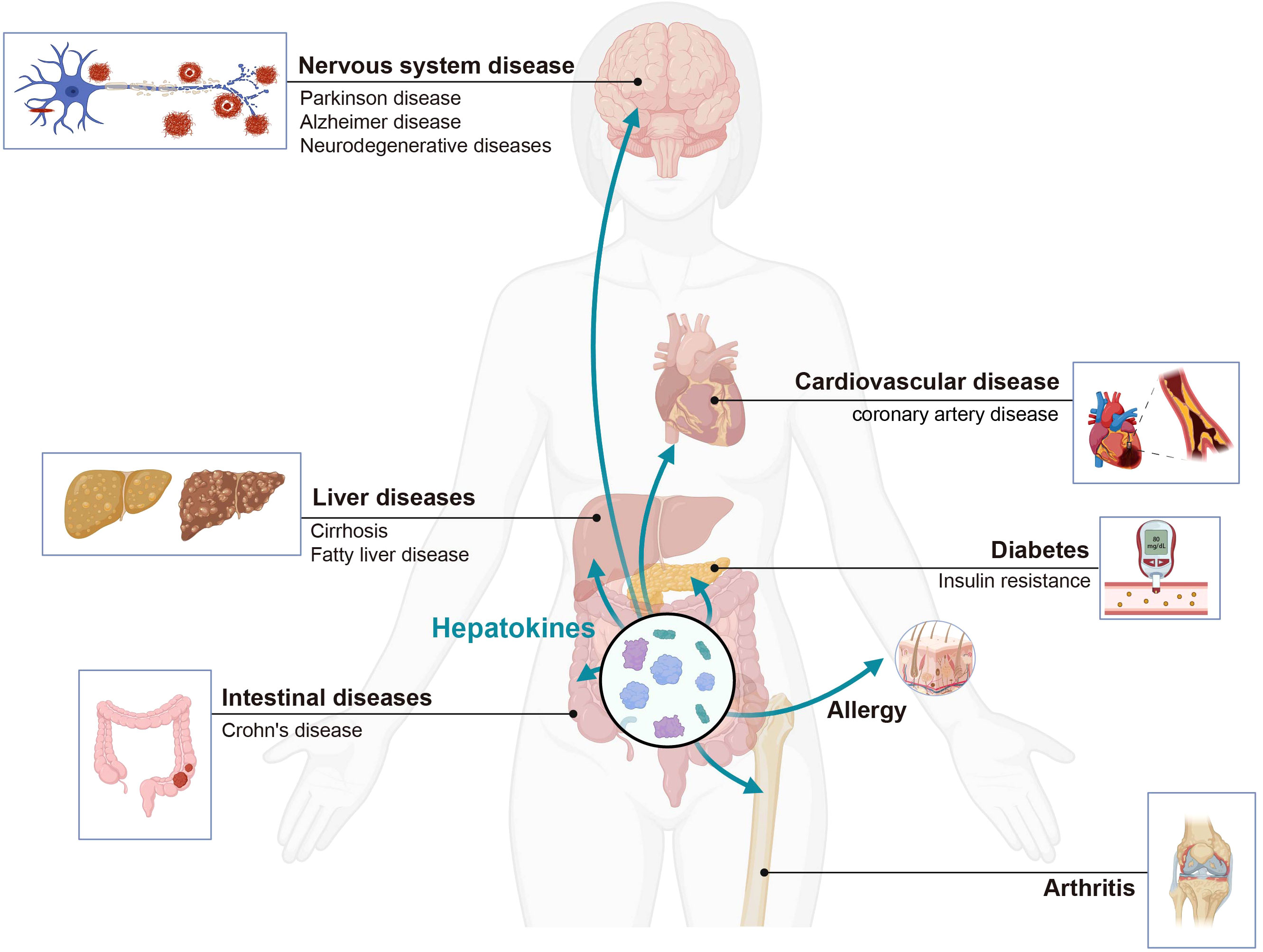
Figure 7 The liver secretes hepatokines effectively to regulate distant organs under different physiological and pathological conditions. Hepatokines affect the immune homeostasis of organs, and can lead to the development of various systemic diseases. Created with BioRender.com.
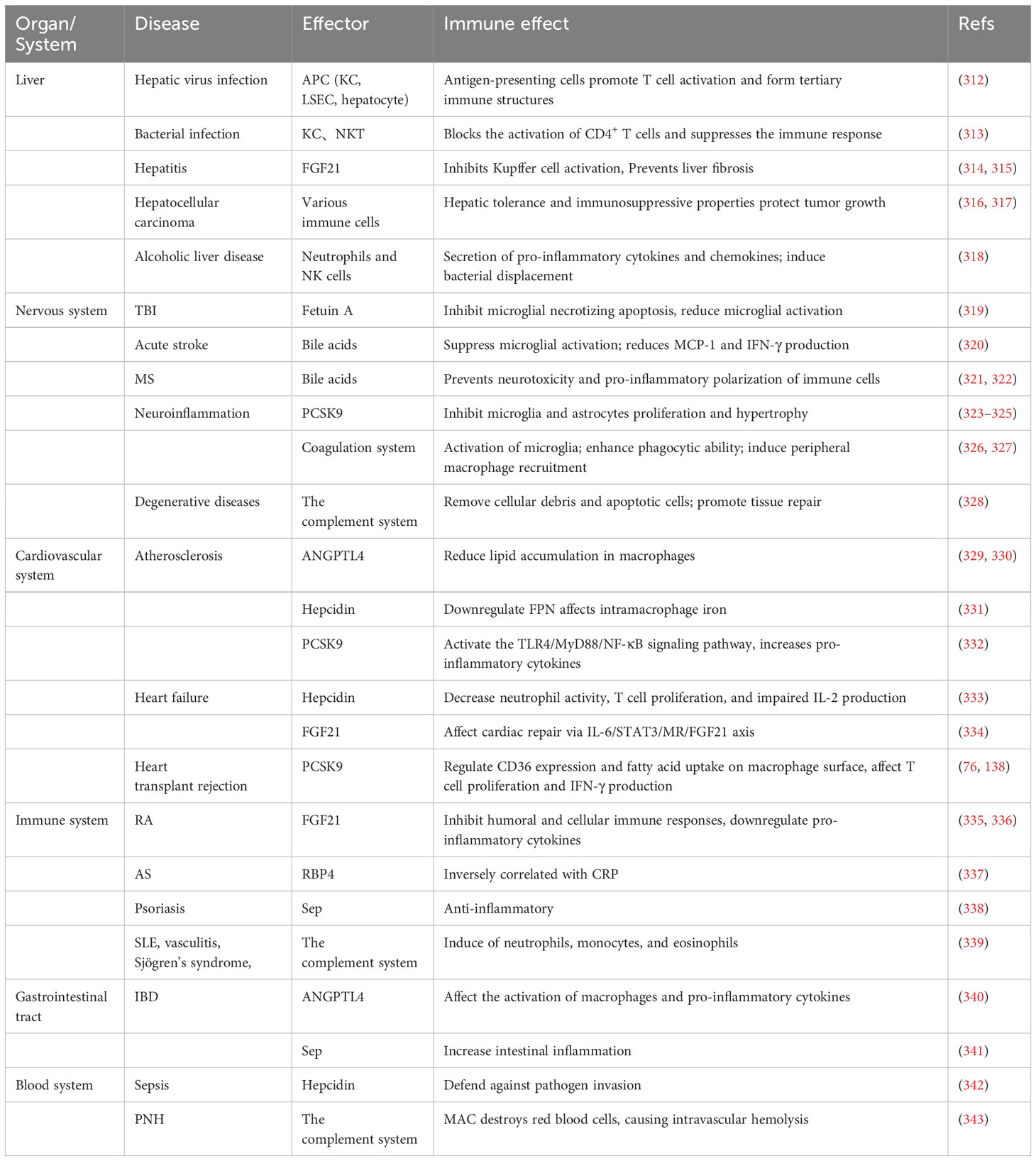
Table 2 The impact of liver remote immune regulation on various system diseases and effective molecules.
The liver serves as an immune-tolerant organ and is proficient in upholding immune tolerance toward both self-propagated and foreign antigens entering via the portal vein. Simultaneously, it is primed to mount immune responses against pathogens and is capable of accommodating liver allogeneic grafts (9). Liver-resident macrophages are equipped to engulf particles (>200 nm) through phagocytosis, while LSECs facilitate the clearance of macromolecules (<200 nm) and colloids via receptor-mediated endocytosis (344). Additionally, KCs and LSECs present accumulated antigens to the lymphocytes, instigating T cell immune tolerance through the expression of diminished levels of MHC II and co-stimulatory molecules (345). During acute liver injury, KCs generate pro-inflammatory cytokines, such as IL-1, IL-6, TNF-α, and chemokines (346). Upon viral infection of the liver, numerous antigen-presenting cells, including KCs, LSECs, and hepatocytes, swiftly become activated. This activation initiates the triggering of specific T cell activation and expansion, ultimately culminating in the development of tertiary immune structures within the liver. In the absence of pro-inflammatory cues, these tertiary immune structures cease their regulatory function over T cells, resulting in the breakdown of CTL response. Nevertheless, under suitable signal strength, these structures can lead to persistent viral infections (312). Conversely, cytokines released by KCs in response to bacterial infection can inhibit the activation of CD4+ T cells, culminating in the suppression of the immune response (313). In the early phase of microbial-induced liver inflammation, type I NKT exhibit pro-inflammatory behavior, while type II NKT cells exert inhibitory effects on NKT-mediated liver injury (347). FGF21 suppresses Kupffer cell activation, reduces monocyte infiltration, and diminishes lipid-associated macrophages amidst liver inflammation, thus exerting a preventative effect against liver fibrosis (314, 315). Additionally, the liver serves as a frequent site for malignancy within the body, with HCC displaying the highest incidence among primary tumors. Typically arising as a consequence of chronic liver inflammation, these inflammatory processes synergistically instigate tumorigenesis. Upon the initiation of tumorigenesis, the liver’s tolerance and immunosuppressive mechanisms offer a protective environment for tumor growth and epithelial-mesenchymal transition. Simultaneously, the tumor microenvironment exerts additional immune suppression, ultimately driving irreversible alterations (316, 317). Numerous liver disease patterns do not stem from trauma but rather result from unhealthy lifestyles, including conditions such as alcoholic liver disease (ALD), fatty liver, and drug-induced liver injury. The release of pro-inflammatory cytokines and chemokines by neutrophils and NK cells incites the adaptive immune response, contributing to disease development. Conversely, M2 macrophages and Tregs seem to play a role in safeguarding the liver against harm (348, 349). During alcohol-induced liver damage, alcohol consumption can potentially heighten intestinal permeability, subsequently resulting in bacterial translocation and escalated LPS levels. Beyond their direct impact on liver cells and immune cells, TLRs also possess the ability to stimulate innate immunity, thereby instigating hepatic lipid droplet deterioration and ultimately contributing to liver fibrosis (318).
Furthermore, recent studies have reported that innate immune proteins derived from the liver also contribute to liver-related diseases. In acetaminophen-induced liver injury (AILI), CRP is considered a crucial checkpoint that protects against acute liver injury by preventing excessive complement activation (350). Clinical studies on acute liver injury in COVID-19 have revealed a correlation between CRP and liver damage (351). The role of complement in nonalcoholic fatty liver disease (NFLD) and ALD is complex. For instance, C3, factor B, and factor D activate the alternative pathway, leading to the generation of anaphylatoxins C3a and C5a, which induce insulin resistance and disrupt lipid metabolism in the liver (352). C1q contributes to liver injury by activating the classical complement, whereas factor D protects against ethanol-induced inflammation and promotes hepatic healing and recovery (353, 354). Several experimental models have demonstrated that complement inhibition is beneficial for liver injury (including IRI), liver transplantation, and acute liver failure. For example, anti-C5 therapy after liver transplantation can inhibit antibody-mediated rejection, improve long-term animal survival, and reduce biliary injury and liver fibrosis. Interestingly, studies have shown that liver regeneration is dependent on complement. Complement C3 in the proliferative response could be independent of the C3a-C3aR interaction; instead, C3a and C5a appear to act through crosstalk with the local formation of cytokine networks, particularly IL-6 and TNF (177, 355). In addition to complement and CRP, SAA also participates in liver-related diseases. Stereo-seq and scRNA-seq have shown that hepatocytes secrete SAA, facilitating tumor invasion through the recruitment of macrophages that promote M2 polarization (356). Consistent with previous research on pancreatic and colorectal cancers, overexpression of SAA by hepatocytes forms a prometastatic niche in the liver (357). In previous studies, SAA has been reported to exacerbate fatty liver inflammation by promoting intrahepatic platelet aggregation during NAFLD (358). Patients with ethanol-induced liver injury, cirrhosis, and HCC exhibit increased serum levels of SAA and CRP, suggesting an association of these liver-derived innate immune proteins with liver-related diseases (359).
The nervous system stands out as the most intricate and meticulously structured bodily system, governing sensory, behavioral, autonomic, and psychiatric functions across the body. Conversely, the body possesses the ability to influence the host’s brain function and behavior via the “brain-gut axis”. Notably, a mounting body of evidence underscores the liver’s potential to impact the brain, instigating particular behaviors and modulating disease advancement (360). Within the context of inflammatory responses triggered by traumatic brain injury (TBI), Fetuin A—an acute-phase glycoprotein originating from the liver—has been found to hinder microglial necrotic apoptosis, thereby mitigating microglial activation (319). Furthermore, Fetuin A holds potential as a viable biomarker for assessing inflammation in the context of multiple sclerosis (MS) (361, 362). Bile acids, the metabolic byproducts of cholesterol, exhibit associations with immune responses linked to Alzheimer’s disease (AD) and cognitive decline (363). During acute stroke, bile acids exhibit the potential to attenuate glial activation and microglial chemotaxis, suppress MCP-1 and IFN-γ production, and mitigate inflammatory reactions within the central nervous system (CNS) (320). Empirical studies have shown that the administration of tauroursodeoxycholic acid (TUDCA) one hour following ischemia resulting from middle cerebral artery occlusion inhibited perturbations in mitochondrial membranes, extended cell viability, and mitigated both immediate and enduring harm linked to acute stroke (364). In the context of multiple sclerosis (MS), bile acids counteract neurotoxicity and pro-inflammatory polarization in immune cells and glial cells, operating in a dose-dependent manner. Consequently, they lead to diminished TNF-α and IL-1β production (321, 322). The nervous system also stands out as among the most lipid-rich tissues within the human body. Furthermore, PCSK9, functioning as a circulating inhibitor of LDLR, assumes a significant role not only within the CNS but also within the peripheral nervous system (PNS). PCSK9 possesses the capacity to influence immune cell activation within the brain, induce alterations in brain inflammatory responses, and emerge as a novel therapeutic target for addressing brain inflammation (323, 324). Administering PCSK9 inhibitors via intravenous injection can diminish the expression of NF-κB, ameliorate the proliferation and hypertrophy of microglia and astrocytes, and impede neuroinflammation (325). In the EAE model, deficiency of factor FXII was demonstrated to diminish disease-related inflammation concomitantly with heightened Th17-driven production of IL-17A. Namely, factor FXII modifies dendritic cell cytokine profiles to induce effector T cell differentiation, thereby contributing to its role in EAE (365). Simultaneously, thrombin activity within the coagulation system markedly escalates during EAE, demonstrating correlation with the intensity of microglial activation, demyelination, and axonal damage (326). Fibrin deposition is linked to microglial activation in neuroinflammation, and fibrinogen has the capacity to activate microglia, augment their phagocytic capacity, trigger recruitment of peripheral macrophages, and activate Th1 cells (327). These findings imply the feasibility of directing interventions toward the coagulation and fibrinolytic systems for potential anti-inflammatory therapy. The complement system stands as a vital element within the ancestral innate immune response, particularly in degenerative conditions like AD, Huntington’s disease, and Peake’s disease. Several complement components manifest a neuroprotective impact, engaging with complement cell surface receptors, facilitating the elimination of cellular debris and apoptotic cells, and fostering tissue repair via C3a (328).
Atherosclerotic plaques are characterized by the accumulation of lipids within macrophages, which culminate in the formation of foam cells. This progression subsequently triggers apoptosis and necrosis of cells within the plaque. Hepatocyte-specific secretion of ANGPTL4 regulates inflammation in atherosclerosis and reduces lipid accumulation in macrophages (329, 330). Hepcidin activates inflammatory response through TLR4 and transcription factor NF-κB. Additionally, it modulates intramacrophage iron efflux by downregulating FPN, thus exerting an impact on atherosclerosis development (331). Patients with atherosclerosis exhibit notably elevated PCSK9 expression in their plasma compared to healthy individuals. Beyond lipid-regulating function, PCSK9 plays a pivotal role in the unfavorable prognosis of this ailment. It not only triggers inflammatory responses, but also fosters thrombotic events and cellular demise. PCSK9 can trigger activation of the TLR4/MyD88/NF-κB signaling cascade, exert influence on the compositional makeup of macrophages, monocytes, and T cells, and augment the release of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α), thereby instigating tissue damage (332). Inhibitors of PCSK9 can regulate inflammatory cell infiltration by regulating macrophage polarization in vivo and in vitro, resulting in diminished infarct area in cases of acute myocardial infarction (AMI) and enhanced cardiac function (366). Recent investigations have unveiled the influence of PCSK9 on the localized inflammatory response of grafts, underscoring its potential as a therapeutic target to mitigate rejection (76, 138). Coagulation factor XI influences the cleavage of the extracellular matrix bone morphogenetic protein 7 (BMP7) within the heart. Furthermore, it exerts inhibitory effects on the inflammatory response in cases of heart failure, contributing to liver-heart cross-talk (367). Heart failure is closely related to inflammatory states; IL-6 in the inflammatory response is the main trigger for elevated Hepcidin, and iron deficiency due to Hepcidin can also perpetuate and amplify inflammation, including decreased neutrophil activity, defective T cell proliferation, and impaired IL-2 production (333). Moreover, the liver possesses the capacity to influence cardiac repair post-myocardial infarction through the IL-6/STAT3/MR/FGF21 axis (368). FGF21 is an influence not only on adipocytes and renal cells but also plays a role in regulating high blood pressure. Through its promotion of angiotensin-converting enzyme 2 (ACE2) induction, angiotensin II undergoes conversion to angiotensin- (1–7), resulting in the inhibition of hypertension and the reversal of vascular damage. However, inhibiting angiotensin- (1–7) via drug intervention may diminish the safeguarding impact of FGF21 against vascular dysfunction (334). Thrombosis stands out as the most feared complication within cardiovascular disease. The interplay between inflammation and thrombosis orchestrates a sequential activation of platelets and coagulation, culminating in the onset of thrombotic ailments. The crux of immunothrombosis centers on the reciprocal activation of platelets and neutrophils and the liberation of soluble mediators (CCL5, CXCL4, P-selectin, C5a), ultimately increasing vulnerability to ischemic stroke, myocardial infarction, and venous thromboembolism (369).
FGF21 has demonstrated the capacity to mitigate arthritis severity through the suppression of humoral and cellular immune responses, as well as by dampening the expression of pro-inflammatory cytokines in the cases of rheumatoid arthritis (RA) (335, 336). Abnormal expression of circulating RBP4 has been observed in ankylosing spondylitis (AS), exhibiting a negative correlation with C-reactive protein. This suggests that RBP4 could potentially serve as a biomarker for AS (337). In individuals afflicted with severe psoriasis, Sep levels display significant elevation, which subsequently recedes post-treatment. Given its involvement in modifying anti-inflammatory immune responses, Sep may emerge as a novel indicator for predicting the emergence of inflammatory and metabolic complications in psoriasis (338). Excessive activation of the complement system stands as a paramount contributor to tissue damage in autoimmune disorders. Furthermore, the deficiency of specific complement components can precipitate autoimmune conditions, including systemic lupus erythematosus, vasculitis, Sjögren’s syndrome, and antiphospholipid syndrome (370). Acquired deficiency of C1q is also prevalent among individuals suffering from systemic lupus erythematosus (SLE) (371). Inadequate activation of the complement system culminates in the liberation of pro-inflammatory mediators (C3a and C5a), thereby instigating the recruitment of neutrophils, monocytes, and eosinophils. Additionally, the formation of the MAC triggers inflammation along with cell necrosis or apoptosis, thereby exacerbating tissue damage (339). A Mendelian randomization study showed that the immunomodulatory effects of PCSK9 could have an effect on autoimmune diseases, and that PCSK9 inhibitors significantly reduced the risk of SLE but increased the risk of asthma and Crohn’s disease (372).
ANGPTL4 can modulate intestinal inflammation by impacting macrophage activation and pro-inflammatory cytokine expression (340). Numerous studies have established a connection between selenium (Se) levels and intestinal disease, wherein reduced Se levels were associated with heightened intestinal inflammation and elevated risk of intestinal tumors (341). Overexpression of LECT2 can enhance RIG-I dependent IFN-γ production, bolstering the innate immune response. Targeting LECT2 could potentially hold therapeutic significance in the context of infectious diseases and cancer (373). In cases of sepsis and infectious diseases, elevated Hepcidin levels reflect the activated state of immune cells and correlate with disease severity (342). Over the course of infection, Hepcidin prompts the reduction of extracellular iron levels, which serves as a protective mechanism, inhibiting iron intrusion by pathogens. Conversely, Hepcidin can also encourage the sequestration of iron within macrophages, thereby diminishing the immune response against intracellular infections (144). Furthermore, Hepcidin has notably been linked to inflammatory anemia and inflammatory bowel disease (IBD) (374, 375). The latest evidence suggests that DPP4 inhibitors can reduce the pro-inflammatory and profibrotic responses (including CD8+ T cells, macrophages and neutrophils) activated by ANGII in the kidneys, delay kidney damage, and reduce the incidence of proteinuria (376). Paroxysmal nocturnal hemoglobinuria (PNH) is a polyclonal hematologic disorder closely associated with complement reactions. The lack of CD55 and CD59 on the cell surface of the hematopoietic group greatly reduces the complement regulation ability of red blood cells, and after encountering infection, complement is strongly activated, resulting in red blood cells being destroyed by MAC and causing intravascular hemolysis. The prognosis of PNH has been altered after the use of C5 inhibition, and complement inhibitors have also opened up new prospects for the treatment of hematologic diseases (Table 2) (343).
As emphasized in this review, the liver, the largest physical organ in the body, serves not only as a metabolic hub but also as a crucial “immune organ.” Within the liver, a diverse array of immune cells and secreted immune molecules converge to establish a complex immune microenvironment, collectively contributing to liver immune regulation. Traditionally, the immunological perspective depicted the liver as a relatively autonomous local immune system influenced by the broader immune network. However, recent advancements in liver immunity research have unveiled the intricate interplay between the liver’s immune function and the body’s overall immune system.
In various pathological and physiological scenarios, the liver orchestrates immune responses within itself and remote organs by releasing hepatokines, innate immune proteins, coagulation factors, and other products into the bloodstream. These immune molecules wield diverse mechanisms for modulating the recognition and elimination of circulating antigens by the innate immune system, thus influencing their capacity to present antigens and initiate adaptive immune responses. Furthermore, immune molecules originating from the liver can directly govern the activation and function of adaptive immune cells. The specific modes of secretion for these signals and their role in sustaining organ function through tightly regulated interactions remain largely unexplored and necessitate further investigation. Simultaneously, intrinsic liver cells and immune cells within the liver capitalize on its distinct circulatory network to detect antigenic constituents coursing through systemic blood circulation and those originating from the gastrointestinal tract, thus engendering immune responses. The liver’s physiological architecture and cellular makeup provide the foundation for temporal and spatial immune tolerance mechanisms. As understanding of the processes of antigen capture, presentation, and recognition within the liver deepens, the clinical application of manipulating liver functions to foster immune tolerance holds increasing promising. Nonetheless, deeper exploration of the intricate interplay among liver immune cells during functional execution and assessment of the strategies of upholding liver immune homeostasis merit additional research.
Evidently, gaining a comprehensive understanding of the intricate, local and distant regulatory mechanisms governing liver immunity could enhance insight into liver function. This knowledge may subsequently uncover novel molecular targets for immune modulation, which could prove to be advantageous for diagnosing and treating immune-related disorders. Moreover, this review lays the groundwork for the exploration and advancement of innovative pharmaceuticals and strategies.
JW: Funding acquisition, Resources, Writing – original draft, Writing – review & editing. JZh: Writing – review & editing. XZ: Writing – review & editing. YL: Project administration, Validation, Writing – review & editing. JY: Project administration, Validation, Writing – review & editing. ZC: Software, Supervision, Writing – review & editing. YN: Methodology, Supervision, Writing – review & editing. SR: Methodology, Supervision, Writing – review & editing. SW: Supervision, Validation, Writing – review & editing. WY: Supervision, Writing – review & editing. ZL: Validation, Visualization, Writing – review & editing. XL: Project administration, Validation, Writing – review & editing. YH: Project administration, Validation, Writing – review & editing. JZo: Supervision, Validation, Writing – review & editing. CX: Writing – review & editing. JX: Funding acquisition, Resources, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The figures in this review were created with BioRender.com. This review work was supported by the National Natural Science Foundation of China (82071803, 82271811, 81730015, 82241217) and Fundamental Research Funds for the Central Universities (2021GCRC037, YCJJ20230232 to SR).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol (2017) 27(21):R1147–r51. doi: 10.1016/j.cub.2017.09.019
2. Robinson MW, Harmon C, O'Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol (2016) 13(3):267–76. doi: 10.1038/cmi.2016.3
3. Li W, Chang N, Li L. Heterogeneity and function of kupffer cells in liver injury. Front Immunol (2022) 13:940867. doi: 10.3389/fimmu.2022.940867
4. Sheth K, Bankey P. The liver as an immune organ. Curr Opin Crit Care (2001) 7(2):99–104. doi: 10.1097/00075198-200104000-00008
5. Wood NJ. Liver: the liver as a firewall–clearance of commensal bacteria that have escaped from the gut. Nat Rev Gastroenterol Hepatol (2014) 11(7):391. doi: 10.1038/nrgastro.2014.90
6. Lumsden AB, Henderson JM, Kutner MH. Endotoxin levels measured by a chromogenic assay in portal, hepatic and peripheral venous blood in patients with cirrhosis. Hepatology. (1988) 8(2):232–6. doi: 10.1002/hep.1840080207
7. Balmer ML, Slack E, de Gottardi A, Lawson MA, Hapfelmeier S, Miele L, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med (2014) 6(237):237ra66. doi: 10.1126/scitranslmed.3008618
8. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. (2006) 43(2 Suppl 1):S54–62. doi: 10.1002/hep.21060
9. Kubes P, Jenne C. Immune responses in the liver. Annu Rev Immunol (2018) 36:247–77. doi: 10.1146/annurev-immunol-051116-052415
10. Abdel-Misih SR, Bloomston M. Liver anatomy. Surg Clin North Am (2010) 90(4):643–53. doi: 10.1016/j.suc.2010.04.017
11. Skandalakis JE, Skandalakis LJ, Skandalakis PN, Mirilas P. Hepatic surgical anatomy. Surg Clin North Am (2004) 84(2):413–35,viii. doi: 10.1016/j.suc.2003.12.002
12. Juza RM, Pauli EM. Clinical and surgical anatomy of the liver: a review for clinicians. Clin Anat. (2014) 27(5):764–9. doi: 10.1002/ca.22350
13. Sibulesky L. Normal liver anatomy. Clin Liver Dis (Hoboken) (2013) 2(Suppl 1):S1–s3. doi: 10.1002/cld.124
14. Cheng ML, Nakib D, Perciani CT, MacParland SA. The immune niche of the liver. Clin Sci (Lond). (2021) 135(20):2445–66. doi: 10.1042/CS20190654
15. Oda M, Yokomori H, Han JY. Regulatory mechanisms of hepatic microcirculatory hemodynamics: hepatic arterial system. Clin Hemorheol Microcirc (2006) 34(1-2):11–26.
16. Corness JA, McHugh K, Roebuck DJ, Taylor AM. The portal vein in children: radiological review of congenital anomalies and acquired abnormalities. Pediatr Radiol (2006) 36(2):87–96,quiz 170-1. doi: 10.1007/s00247-005-0010-4
17. Maruyama H, Shiina S. Collaterals in portal hypertension: anatomy and clinical relevance. Quant Imaging Med Surg (2021) 11(8):3867–81. doi: 10.21037/qims-20-1328
18. Lautt WW. Colloquium series on integrated systems physiology: from molecule to function to disease. In: Hepatic Circulation: Physiology and Pathophysiology. San Rafael (CA: Morgan & Claypool Life Sciences (2009). Copyright © 2010 by Morgan & Claypool Life Sciences.
19. Carneiro C, Brito J, Bilreiro C, Barros M, Bahia C, Santiago I, et al. All about portal vein: a pictorial display to anatomy, variants and physiopathology. Insights Imaging. (2019) 10(1):38. doi: 10.1186/s13244-019-0716-8
20. Ben-Moshe S, Itzkovitz S. Spatial heterogeneity in the mammalian liver. Nat Rev Gastroenterol Hepatol (2019) 16(7):395–410. doi: 10.1038/s41575-019-0134-x
21. Si-Tayeb K, Lemaigre FP, Duncan SA. Organogenesis and development of the liver. Dev Cell (2010) 18(2):175–89. doi: 10.1016/j.devcel.2010.01.011
22. Gissen P, Arias IM. Structural and functional hepatocyte polarity and liver disease. J Hepatol (2015) 63(4):1023–37. doi: 10.1016/j.jhep.2015.06.015
23. Treyer A, Müsch A. Hepatocyte polarity. Compr Physiol (2013) 3(1):243–87. doi: 10.1002/cphy.c120009
24. Michalopoulos GK, Bhushan B. Liver regeneration: biological and pathological mechanisms and implications. Nat Rev Gastroenterol Hepatol (2021) 18(1):40–55. doi: 10.1038/s41575-020-0342-4
25. Li W, Li L, Hui L. Cell plasticity in liver regeneration. Trends Cell Biol (2020) 30(4):329–38. doi: 10.1016/j.tcb.2020.01.007
26. Nejak-Bowen K. If it looks like a duct and acts like a duct: on the role of reprogrammed hepatocytes in cholangiopathies. Gene Expr. (2020) 20(1):19–23. doi: 10.3727/105221619X15664105014956
27. Gadd VL, Aleksieva N, Forbes SJ. Epithelial plasticity during liver injury and regeneration. Cell Stem Cell (2020) 27(4):557–73. doi: 10.1016/j.stem.2020.08.016
28. Brunt EM, Gouw AS, Hubscher SG, Tiniakos DG, Bedossa P, Burt AD, et al. Pathology of the liver sinusoids. Histopathology. (2014) 64(7):907–20. doi: 10.1111/his.12364
29. Sørensen KK, Simon-Santamaria J, McCuskey RS, Smedsrød B. 3s Liver Sinusoidal Endothelial Cells. Liver Sinusoidal Endothelial Cells. Compr Physiol (2015) 5(4):1751–74. doi: 10.1002/cphy.c140078.
30. DeLeve LD. Liver sinusoidal endothelial cells and liver regeneration. J Clin Invest. (2013) 123(5):1861–6. doi: 10.1172/JCI66025
31. Sørensen KK, McCourt P, Berg T, Crossley C, Le Couteur D, Wake K, et al. The scavenger endothelial cell: a new player in homeostasis and immunity. Am J Physiol Regul Integr Comp Physiol (2012) 303(12):R1217–30. doi: 10.1152/ajpregu.00686.2011
32. Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, et al. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol (2017) 66(1):212–27. doi: 10.1016/j.jhep.2016.07.009
33. Li ZW, Ruan B, Yang PJ, Liu JJ, Song P, Duan JL, et al. Oit3, a promising hallmark gene for targeting liver sinusoidal endothelial cells. Signal Transduct Target Ther (2023) 8(1):344. doi: 10.1038/s41392-023-01621-2
34. Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int (2006) 26(10):1175–86. doi: 10.1111/j.1478-3231.2006.01342.x
35. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun (2016) 7:10321. doi: 10.1038/ncomms10321
36. Dou L, Shi X, He X, Gao Y. Macrophage phenotype and function in liver disorder. Front Immunol (2019) 10:3112. doi: 10.3389/fimmu.2019.03112
37. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518(7540):547–51. doi: 10.1038/nature13989
38. Gola A, Dorrington MG, Speranza E, Sala C, Shih RM, Radtke AJ, et al. Commensal-driven immune zonation of the liver promotes host defence. Nature. (2021) 589(7840):131–6. doi: 10.1038/s41586-020-2977-2
39. Fahrner R, Dondorf F, Ardelt M, Settmacher U, Rauchfuss F. Role of NK, NKT cells and macrophages in liver transplantation. World J Gastroenterol (2016) 22(27):6135–44. doi: 10.3748/wjg.v22.i27.6135
40. Liaskou E, Zimmermann HW, Li KK, Oo YH, Suresh S, Stamataki Z, et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology. (2013) 57(1):385–98. doi: 10.1002/hep.26016
41. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol (2013) 3(2):785–97. doi: 10.1002/cphy.c120026
42. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233(9):6425–40. doi: 10.1002/jcp.26429
43. Reynaert H, Thompson MG, Thomas T, Geerts A. Hepatic stellate cells: role in microcirculation and pathophysiology of portal hypertension. Gut. (2002) 50(4):571–81. doi: 10.1136/gut.50.4.571
44. Xuan Y, Chen S, Ding X, Wang L, Li S, Yang G, et al. Tetrahydropalmatine attenuates liver fibrosis by suppressing endoplasmic reticulum stress in hepatic stellate cells. Chin Med J (Engl) (2021) 135(5):628–30. doi: 10.1097/CM9.0000000000001883
45. Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol (2013) 3(4):1473–92. doi: 10.1002/cphy.c120035
46. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev (2008) 88(1):125–72. doi: 10.1152/physrev.00013.2007
47. Kamm DR, McCommis KS. Hepatic stellate cells in physiology and pathology. J Physiol (2022) 600(8):1825–37. doi: 10.1113/JP281061
48. Hellerbrand C. Hepatic stellate cells–the pericytes in the liver. Pflugers Arch (2013) 465(6):775–8. doi: 10.1007/s00424-012-1209-5
49. Woo J, Lu L, Rao AS, Li Y, Subbotin V, Starzl TE, et al. Isolation, phenotype, and allostimulatory activity of mouse liver dendritic cells. Transplantation. (1994) 58(4):484–91. doi: 10.1097/00007890-199408270-00015
50. Steptoe RJ, Fu F, Li W, Drakes ML, Lu L, Demetris AJ, et al. Augmentation of dendritic cells in murine organ donors by Flt3 ligand alters the balance between transplant tolerance and immunity. J Immunol (1997) 159(11):5483–91. doi: 10.4049/jimmunol.159.11.5483
51. Tokita D, Sumpter TL, Raimondi G, Zahorchak AF, Wang Z, Nakao A, et al. Poor allostimulatory function of liver plasmacytoid DC is associated with pro-apoptotic activity, dependent on regulatory T cells. J Hepatol (2008) 49(6):1008–18. doi: 10.1016/j.jhep.2008.07.028
52. Steptoe RJ, Patel RK, Subbotin VM, Thomson AW. Comparative analysis of dendritic cell density and total number in commonly transplanted organs: morphometric estimation in normal mice. Transpl Immunol (2000) 8(1):49–56. doi: 10.1016/S0966-3274(00)00010-1
53. Kelly A, Fahey R, Fletcher JM, Keogh C, Carroll AG, Siddachari R, et al. CD141+ myeloid dendritic cells are enriched in healthy human liver. J Hepatol (2014) 60(1):135–42. doi: 10.1016/j.jhep.2013.08.007
54. Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol (2012) 13(9):888–99. doi: 10.1038/ni.2370
55. Peng H, Jiang X, Chen Y, Sojka DK, Wei H, Gao X, et al. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Invest. (2013) 123(4):1444–56. doi: 10.1172/JCI66381
56. Sojka DK, Plougastel-Douglas B, Yang L, Pak-Wittel MA, Artyomov MN, Ivanova Y, et al. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife. (2014) 3:e01659. doi: 10.7554/eLife.01659
57. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi: 10.1038/ni1582
58. Takeda K, Cretney E, Hayakawa Y, Ota T, Akiba H, Ogasawara K, et al. TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood. (2005) 105(5):2082–9. doi: 10.1182/blood-2004-08-3262
59. Mikulak J, Bruni E, Oriolo F, Di Vito C, Mavilio D. Hepatic natural killer cells: organ-specific sentinels of liver immune homeostasis and physiopathology. Front Immunol (2019) 10:946. doi: 10.3389/fimmu.2019.00946
60. Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. (2012) 36(1):55–67. doi: 10.1016/j.immuni.2011.11.016
61. Berzins SP, Smyth MJ, Baxter AG. Presumed guilty: natural killer T cell defects and human disease. Nat Rev Immunol (2011) 11(2):131–42. doi: 10.1038/nri2904
62. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol (2007) 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711
63. Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol (2009) 27:147–63. doi: 10.1146/annurev.immunol.021908.132629
64. Doherty DG. Immunity, tolerance and autoimmunity in the liver: A comprehensive review. J Autoimmun (2016) 66:60–75. doi: 10.1016/j.jaut.2015.08.020
65. Gao B, Jeong WI, Tian Z. Liver: An organ with predominant innate immunity. Hepatology. (2008) 47(2):729–36. doi: 10.1002/hep.22034
66. Zhou Z, Xu MJ, Gao B. Hepatocytes: a key cell type for innate immunity. Cell Mol Immunol (2016) 13(3):301–15. doi: 10.1038/cmi.2015.97
67. Seidah NG, Awan Z, Chrétien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res (2014) 114(6):1022–36. doi: 10.1161/CIRCRESAHA.114.301621
68. Stoekenbroek RM, Lambert G, Cariou B, Hovingh GK. Inhibiting PCSK9 - biology beyond LDL control. Nat Rev Endocrinol (2018) 15(1):52–62. doi: 10.1038/s41574-018-0110-5
69. Walley KR, Thain KR, Russell JA, Reilly MP, Meyer NJ, Ferguson JF, et al. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci Transl Med (2014) 6(258):258ra143. doi: 10.1126/scitranslmed.3008782
70. Liu X, Suo R, Chan CZY, Liu T, Tse G, Li G. The immune functions of PCSK9: Local and systemic perspectives. J Cell Physiol (2019) 234(11):19180–8. doi: 10.1002/jcp.28612
71. Paciullo F, Fallarino F, Bianconi V, Mannarino MR, Sahebkar A, Pirro M. PCSK9 at the crossroad of cholesterol metabolism and immune function during infections. J Cell Physiol (2017) 232(9):2330–8. doi: 10.1002/jcp.25767
72. Basiak M, Kosowski M, Hachula M, Okopien B. Impact of PCSK9 inhibition on proinflammatory cytokines and matrix metalloproteinases release in patients with mixed hyperlipidemia and vulnerable atherosclerotic plaque. Pharm (Basel) (2022) 15(7):802. doi: 10.3390/ph15070802
73. Liu X, Bao X, Hu M, Chang H, Jiao M, Cheng J, et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. (2020) 588(7839):693–8. doi: 10.1038/s41586-020-2911-7
74. Almeida CR, Ferreira BH, Duarte IF. Targeting PCSK9: a promising adjuvant strategy in cancer immunotherapy. Signal Transduct Target Ther (2021) 6(1):111. doi: 10.1038/s41392-021-00530-6
75. Yuan J, Cai T, Zheng X, Ren Y, Qi J, Lu X, et al. Potentiating CD8(+) T cell antitumor activity by inhibiting PCSK9 to promote LDLR-mediated TCR recycling and signaling. Protein Cell (2021) 12(4):240–60. doi: 10.1007/s13238-021-00821-2
76. Zhang X, Xu H, Yu J, Cui J, Chen Z, Li Y, et al. Immune regulation of the liver through the PCSK9/CD36 pathway during heart transplant rejection. Circulation (2023). 148(4):336–53. doi: 10.1161/CIRCULATIONAHA.123.062788
77. Wang CY, Babitt JL. Liver iron sensing and body iron homeostasis. Blood. (2019) 133(1):18–29. doi: 10.1182/blood-2018-06-815894
78. Kim DK, Jeong JH, Lee JM, Kim KS, Park SH, Kim YD, et al. Inverse agonist of estrogen-related receptor γ controls Salmonella typhimurium infection by modulating host iron homeostasis. Nat Med (2014) 20(4):419–24. doi: 10.1038/nm.3483
79. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. (2004) 306(5704):2090–3. doi: 10.1126/science.1104742
80. Peyssonnaux C, Zinkernagel AS, Datta V, Lauth X, Johnson RS, Nizet V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood. (2006) 107(9):3727–32. doi: 10.1182/blood-2005-06-2259
81. Frost JN, Tan TK, Abbas M, Wideman SK, Bonadonna M, Stoffel NU, et al. Hepcidin-mediated hypoferremia disrupts immune responses to vaccination and infection. Med. (2021) 2(2):164–79.e12. doi: 10.1016/j.medj.2020.10.004
82. Teh MR, Frost JN, Armitage AE, Drakesmith H. Analysis of iron and iron-interacting protein dynamics during T-cell activation. Front Immunol (2021) 12:714613. doi: 10.3389/fimmu.2021.714613
83. Jiang Y, Li C, Wu Q, An P, Huang L, Wang J, et al. Iron-dependent histone 3 lysine 9 demethylation controls B cell proliferation and humoral immune responses. Nat Commun (2019) 10(1):2935. doi: 10.1038/s41467-019-11002-5
84. Wang D, Liu F, Zhu L, Lin P, Han F, Wang X, et al. FGF21 alleviates neuroinflammation following ischemic stroke by modulating the temporal and spatial dynamics of microglia/macrophages. J Neuroinflammation. (2020) 17(1):257. doi: 10.1186/s12974-020-01921-2
85. Li SM, Yu YH, Li L, Wang WF, Li DS. Treatment of CIA mice with FGF21 down-regulates TH17-IL-17 axis. Inflammation. (2016) 39(1):309–19. doi: 10.1007/s10753-015-0251-9
86. Gao J, Liu Q, Li J, Hu C, Zhao W, Ma W, et al. Fibroblast Growth Factor 21 dependent TLR4/MYD88/NF-κB signaling activation is involved in lipopolysaccharide-induced acute lung injury. Int Immunopharmacol. (2020) 80:106219. doi: 10.1016/j.intimp.2020.106219
87. Kumar KG, Trevaskis JL, Lam DD, Sutton GM, Koza RA, Chouljenko VN, et al. Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab (2008) 8(6):468–81. doi: 10.1016/j.cmet.2008.10.011
88. Jasaszwili M, Billert M, Strowski MZ, Nowak KW, Skrzypski M. Adropin as A fat-burning hormone with multiple functions-review of a decade of research. Molecules. (2020) 25(3):549. doi: 10.3390/molecules25030549
89. Ali II, D'Souza C, Singh J, Adeghate E. Adropin's role in energy homeostasis and metabolic disorders. Int J Mol Sci (2022) 23(15):8318. doi: 10.3390/ijms23158318
90. Sato K, Yamashita T, Shirai R, Shibata K, Okano T, Yamaguchi M, et al. Adropin contributes to anti-atherosclerosis by suppressing monocyte-endothelial cell adhesion and smooth muscle cell proliferation. Int J Mol Sci (2018) 19(5):1293. doi: 10.3390/ijms19051293
91. Jasaszwili M, Wojciechowicz T, Billert M, Strowski MZ, Nowak KW, Skrzypski M. Effects of adropin on proliferation and differentiation of 3T3-L1 cells and rat primary preadipocytes. Mol Cell Endocrinol (2019) 496:110532. doi: 10.1016/j.mce.2019.110532
92. Zhang S, Chen Q, Lin X, Chen M, Liu Q. A review of adropin as the medium of dialogue between energy regulation and immune regulation. Oxid Med Cell Longev (2020) 2020:3947806. doi: 10.1155/2020/3947806
93. Lichtenstein L, Mattijssen F, de Wit NJ, Georgiadi A, Hooiveld GJ, van der Meer R, et al. Angptl4 protects against severe proinflammatory effects of saturated fat by inhibiting fatty acid uptake into mesenteric lymph node macrophages. Cell Metab (2010) 12(6):580–92. doi: 10.1016/j.cmet.2010.11.002
94. Guo L, Li S, Zhao Y, Qian P, Ji F, Qian L, et al. Silencing angiopoietin-like protein 4 (ANGPTL4) protects against lipopolysaccharide-induced acute lung injury via regulating SIRT1 /NF-kB pathway. J Cell Physiol (2015) 230(10):2390–402. doi: 10.1002/jcp.24969
95. Jung KH, Son MK, Yan HH, Fang Z, Kim J, Kim SJ, et al. ANGPTL4 exacerbates pancreatitis by augmenting acinar cell injury through upregulation of C5a. EMBO Mol Med (2020) 12(8):e11222. doi: 10.15252/emmm.201911222
96. Wee WKJ, Low ZS, Ooi CK, Henategala BP, Lim ZGR, Yip YS, et al. Single-cell analysis of skin immune cells reveals an Angptl4-ifi20b axis that regulates monocyte differentiation during wound healing. Cell Death Dis (2022) 13(2):180. doi: 10.1038/s41419-022-04638-7
97. Ding S, Wu D, Lu Q, Qian L, Ding Y, Liu G, et al. Angiopoietin-like 4 deficiency upregulates macrophage function through the dysregulation of cell-intrinsic fatty acid metabolism. Am J Cancer Res (2020) 10(2):595–609.
98. Ding S, Lin Z, Zhang X, Jia X, Li H, Fu Y, et al. Deficiency of angiopoietin-like 4 enhances CD8(+) T cell bioactivity via metabolic reprogramming for impairing tumour progression. Immunology (2023) 170(1):28–46. doi: 10.1111/imm.13650
99. Wang H, Li W, Zhu S, Li J, D'Amore J, Ward MF, et al. Peripheral administration of fetuin-A attenuates early cerebral ischemic injury in rats. J Cereb Blood Flow Metab (2010) 30(3):493–504. doi: 10.1038/jcbfm.2009.247
100. Dziegielewska KM, Andersen NA, Saunders NR. Modification of macrophage response to lipopolysaccharide by fetuin. Immunol Lett (1998) 60(1):31–5. doi: 10.1016/S0165-2478(97)00126-0
101. Chekol Abebe E, Tilahun Muche Z, Behaile TMA, Mengie Ayele T, Mekonnen Agidew M, Teshome Azezew M, et al. The structure, biosynthesis, and biological roles of fetuin-A: A review. Front Cell Dev Biol (2022) 10:945287. doi: 10.3389/fcell.2022.945287
102. Li W, Zhu S, Li J, Huang Y, Zhou R, Fan X, et al. A hepatic protein, fetuin-A, occupies a protective role in lethal systemic inflammation. PloS One (2011) 6(2):e16945. doi: 10.1371/journal.pone.0016945
103. Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med (2012) 18(8):1279–85. doi: 10.1038/nm.2851
104. Mukhopadhyay S, Bhattacharya S. Plasma fetuin-A triggers inflammatory changes in macrophages and adipocytes by acting as an adaptor protein between NEFA and TLR-4. Diabetologia. (2016) 59(4):859–60. doi: 10.1007/s00125-016-3866-y
105. Harris VK, Bell L, Langan RA, Tuddenham J, Landy M, Sadiq SA. Fetuin-A deficiency protects mice from Experimental Autoimmune Encephalomyelitis (EAE) and correlates with altered innate immune response. PloS One (2017) 12(4):e0175575. doi: 10.1371/journal.pone.0175575
106. Mahjabeen F, Yu JJ, Chambers JP, Gupta R, Arulanandam BP. Influence of fetuin-A on chlamydia muridarum pulmonary infection. Int J Microbiol (2022) 2022:6082140. doi: 10.1155/2022/6082140
107. Lu XJ, Chen Q, Rong YJ, Yang GJ, Li CH, Xu NY, et al. LECT2 drives haematopoietic stem cell expansion and mobilization via regulating the macrophages and osteolineage cells. Nat Commun (2016) 7:12719. doi: 10.1038/ncomms12719
108. Lu XJ, Chen J, Yu CH, Shi YH, He YQ, Zhang RC, et al. LECT2 protects mice against bacterial sepsis by activating macrophages via the CD209a receptor. J Exp Med (2013) 210(1):5–13. doi: 10.1084/jem.20121466
109. Zhang X, Sun K, Tang C, Cen L, Li S, Zhu W, et al. LECT2 modulates dendritic cell function after Helicobacter pylori infection via the CD209a receptor. J Gastroenterol Hepatol (2023) 38(4):625–33. doi: 10.1111/jgh.16138
110. L'Hermitte A, Pham S, Cadoux M, Couchy G, Caruso S, Anson M, et al. Lect2 controls inflammatory monocytes to constrain the growth and progression of hepatocellular carcinoma. Hepatology. (2019) 69(1):160–78. doi: 10.1002/hep.30140
111. Hwang HJ, Jung TW, Hong HC, Seo JA, Kim SG, Kim NH, et al. LECT2 induces atherosclerotic inflammatory reaction via CD209 receptor-mediated JNK phosphorylation in human endothelial cells. Metabolism. (2015) 64(9):1175–82. doi: 10.1016/j.metabol.2015.06.001
112. Qin J, Sun W, Zhang H, Wu Z, Shen J, Wang W, et al. Prognostic value of LECT2 and relevance to immune infiltration in hepatocellular carcinoma. Front Genet (2022) 13:951077. doi: 10.3389/fgene.2022.951077
113. Saito T, Okumura A, Watanabe H, Asano M, Ishida-Okawara A, Sakagami J, et al. Increase in hepatic NKT cells in leukocyte cell-derived chemotaxin 2-deficient mice contributes to severe concanavalin A-induced hepatitis. J Immunol (2004) 173(1):579–85. doi: 10.4049/jimmunol.173.1.579
114. Misu H, Takamura T, Takayama H, Hayashi H, Matsuzawa-Nagata N, Kurita S, et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab (2010) 12(5):483–95. doi: 10.1016/j.cmet.2010.09.015
115. Shrimali RK, Irons RD, Carlson BA, Sano Y, Gladyshev VN, Park JM, et al. Selenoproteins mediate T cell immunity through an antioxidant mechanism. J Biol Chem (2008) 283(29):20181–5. doi: 10.1074/jbc.M802559200
116. Carlson BA, Yoo MH, Shrimali RK, Irons R, Gladyshev VN, Hatfield DL, et al. Role of selenium-containing proteins in T-cell and macrophage function. Proc Nutr Soc (2010) 69(3):300–10. doi: 10.1017/S002966511000176X
117. Barrett CW, Short SP, Williams CS. Selenoproteins and oxidative stress-induced inflammatory tumorigenesis in the gut. Cell Mol Life Sci (2017) 74(4):607–16. doi: 10.1007/s00018-016-2339-2
118. Campo-Sabariz J, García-Vara A, Moral-Anter D, Briens M, Hachemi MA, Pinloche E, et al. Hydroxy-selenomethionine, an organic selenium source, increases selenoprotein expression and positively modulates the inflammatory response of LPS-stimulated macrophages. Antioxidants (Basel) (2022) 11(10):1876. doi: 10.3390/antiox11101876
119. Saeed A, Dullaart RPF, Schreuder T, Blokzijl H, Faber KN. Disturbed vitamin A metabolism in non-alcoholic fatty liver disease (NAFLD). Nutrients. (2017) 10(1):29. doi: 10.3390/nu10010029
120. Li M, Wang Z, Zhu L, Shui Y, Zhang S, Guo W. Down-regulation of RBP4 indicates a poor prognosis and correlates with immune cell infiltration in hepatocellular carcinoma. Biosci Rep (2021) 41(4):BSR20210328. doi: 10.1042/BSR20210328
121. Moraes-Vieira PM, Yore MM, Dwyer PM, Syed I, Aryal P, Kahn BB. RBP4 activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab (2014) 19(3):512–26. doi: 10.1016/j.cmet.2014.01.018
122. Norseen J, Hosooka T, Hammarstedt A, Yore MM, Kant S, Aryal P, et al. Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and toll-like receptor 4-dependent and retinol-independent mechanism. Mol Cell Biol (2012) 32(10):2010–9. doi: 10.1128/MCB.06193-11
123. Moraes-Vieira PM, Castoldi A, Aryal P, Wellenstein K, Peroni OD, Kahn BB. Antigen presentation and T-cell activation are critical for RBP4-induced insulin resistance. Diabetes. (2016) 65(5):1317–27. doi: 10.2337/db15-1696
124. Moraes-Vieira PM, Yore MM, Sontheimer-Phelps A, Castoldi A, Norseen J, Aryal P, et al. Retinol binding protein 4 primes the NLRP3 inflammasome by signaling through Toll-like receptors 2 and 4. Proc Natl Acad Sci U S A. (2020) 117(49):31309–18. doi: 10.1073/pnas.2013877117
125. Yao JM, Ying HZ, Zhang HH, Qiu FS, Wu JQ, Yu CH. Exosomal RBP4 potentiated hepatic lipid accumulation and inflammation in high-fat-diet-fed mice by promoting M1 polarization of Kupffer cells. Free Radic Biol Med (2023) 195:58–73. doi: 10.1016/j.freeradbiomed.2022.12.085
126. Farjo KM, Farjo RA, Halsey S, Moiseyev G, Ma JX. Retinol-binding protein 4 induces inflammation in human endothelial cells by an NADPH oxidase- and nuclear factor kappa B-dependent and retinol-independent mechanism. Mol Cell Biol (2012) 32(24):5103–15. doi: 10.1128/MCB.00820-12
127. Han CK, Lee WF, Hsu CJ, Huang YL, Lin CY, Tsai CH, et al. DPP4 reduces proinflammatory cytokine production in human rheumatoid arthritis synovial fibroblasts. J Cell Physiol (2021) 236(12):8060–9. doi: 10.1002/jcp.30494
128. Klemann C, Wagner L, Stephan M, von Hörsten S. Cut to the chase: a review of CD26/dipeptidyl peptidase-4's (DPP4) entanglement in the immune system. Clin Exp Immunol (2016) 185(1):1–21. doi: 10.1111/cei.12781
129. Hui Y, Xu Z, Li J, Kuang L, Zhong Y, Tang Y, et al. Nonenzymatic function of DPP4 promotes diabetes-associated cognitive dysfunction through IGF-2R/PKA/SP1/ERp29/IP3R2 pathway-mediated impairment of Treg function and M1 microglia polarization. Metabolism. (2023) 138:155340. doi: 10.1016/j.metabol.2022.155340
130. Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet (2003) 34(2):154–6. doi: 10.1038/ng1161
131. Seidah NG, Prat A. The multifaceted biology of PCSK9. Endocr Rev (2022) 43(3):558–82. doi: 10.1210/endrev/bnab035
132. Seidah NG, Garçon D. Expanding biology of PCSK9: roles in atherosclerosis and beyond. Curr Atheroscler Rep (2022) 24(10):821–30. doi: 10.1007/s11883-022-01057-z
133. Wang R, Liu H, He P, An D, Guo X, Zhang X, et al. Inhibition of PCSK9 enhances the antitumor effect of PD-1 inhibitor in colorectal cancer by promoting the infiltration of CD8(+) T cells and the exclusion of Treg cells. Front Immunol (2022) 13:947756. doi: 10.3389/fimmu.2022.947756
134. Kim YU, Kee P, Danila D, Teng BB. A critical role of PCSK9 in mediating IL-17-producing T cell responses in hyperlipidemia. Immune Netw (2019) 19(6):e41. doi: 10.4110/in.2019.19.e41
135. Proto JD, Doran AC, Subramanian M, Wang H, Zhang M, Sozen E, et al. Hypercholesterolemia induces T cell expansion in humanized immune mice. J Clin Invest. (2018) 128(6):2370–5. doi: 10.1172/JCI97785
136. Liu A, Frostegård J. PCSK9 plays a novel immunological role in oxidized LDL-induced dendritic cell maturation and activation of T cells from human blood and atherosclerotic plaque. J Intern Med (2018). doi: 10.1016/j.atherosclerosis.2018.06.113
137. Cai J, Jiang Y, Chen F, Wu S, Ren H, Wang P, et al. PCSK9 promotes T helper 1 and T helper 17 cell differentiation by activating the nuclear factor-κB pathway in ankylosing spondylitis. Immun Inflammation Dis (2023) 11(5):e870. doi: 10.1002/iid3.870
138. Zou Y, Chen Z, Zhang X, Yu J, Xu H, Cui J, et al. Targeting PCSK9 ameliorates graft vascular disease in mice by inhibiting NLRP3 inflammasome activation in vascular smooth muscle cells. Front Immunol (2022) 13:894789. doi: 10.3389/fimmu.2022.894789
139. Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem (2001) 276(11):7806–10. doi: 10.1074/jbc.M008922200
140. Nemeth E, Ganz T. Hepcidin and iron in health and disease. Annu Rev Med (2023) 74:261–77. doi: 10.1146/annurev-med-043021-032816
141. Nairz M, Weiss G. Iron in infection and immunity. Mol Aspects Med (2020) 75:100864. doi: 10.1016/j.mam.2020.100864
142. Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. (2004) 113(9):1271–6. doi: 10.1172/JCI200420945
143. Casu C, Nemeth E, Rivella S. Hepcidin agonists as therapeutic tools. Blood. (2018) 131(16):1790–4. doi: 10.1182/blood-2017-11-737411
144. Michels K, Nemeth E, Ganz T, Mehrad B. Hepcidin and host defense against infectious diseases. PloS Pathog (2015) 11(8):e1004998. doi: 10.1371/journal.ppat.1004998
145. Wang L, Harrington L, Trebicka E, Shi HN, Kagan JC, Hong CC, et al. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J Clin Invest. (2009) 119(11):3322–8. doi: 10.1172/JCI39939
146. Frost JN, Wideman SK, Preston AE, Teh MR, Ai Z, Wang L, et al. Plasma iron controls neutrophil production and function. Sci Adv (2022) 8(40):eabq5384. doi: 10.1126/sciadv.abq5384
147. Lai Y, Zhao S, Chen B, Huang Y, Guo C, Li M, et al. Iron controls T helper cell pathogenicity by promoting glucose metabolism in autoimmune myopathy. Clin Transl Med (2022) 12(8):e999. doi: 10.1002/ctm2.999
148. Voss K, Sewell AE, Krystofiak ES, Gibson-Corley KN, Young AC, Basham JH, et al. Elevated transferrin receptor impairs T cell metabolism and function in systemic lupus erythematosus. Sci Immunol (2023) 8(79):eabq0178. doi: 10.1126/sciimmunol.abq0178
149. Markan KR, Naber MC, Ameka MK, Anderegg MD, Mangelsdorf DJ, Kliewer SA, et al. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes. (2014) 63(12):4057–63. doi: 10.2337/db14-0595
150. Fisher FM, Maratos-Flier E. Understanding the physiology of FGF21. Annu Rev Physiol (2016) 78:223–41. doi: 10.1146/annurev-physiol-021115-105339
151. Geng L, Lam KSL, Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat Rev Endocrinol (2020) 16(11):654–67. doi: 10.1038/s41574-020-0386-0
152. Xiaolong L, Dongmin G, Liu M, Zuo W, Huijun H, Qiufen T, et al. FGF21 induces autophagy-mediated cholesterol efflux to inhibit atherogenesis via RACK1 up-regulation. J Cell Mol Med (2020) 24(9):4992–5006. doi: 10.1111/jcmm.15118
153. Huen SC, Wang A, Feola K, Desrouleaux R, Luan HH, Hogg R, et al. Hepatic FGF21 preserves thermoregulation and cardiovascular function during bacterial inflammation. J Exp Med (2021) 218(10):e20202151. doi: 10.1084/jem.20202151
154. Conte M, Martucci M, Chiariello A, Franceschi C, Salvioli S. Mitochondria, immunosenescence and inflammaging: a role for mitokines? Semin Immunopathol (2020) 42(5):607–17. doi: 10.1007/s00281-020-00813-0
155. Algul S, Ozcelik O. Evaluating the energy regulatory hormones of nesfatin-1, irisin, adropin and preptin in multiple sclerosis. Mult Scler Relat Disord (2022) 68:104221. doi: 10.1016/j.msard.2022.104221
156. Chen X, Xue H, Fang W, Chen K, Chen S, Yang W, et al. Adropin protects against liver injury in nonalcoholic steatohepatitis via the Nrf2 mediated antioxidant capacity. Redox Biol (2019) 21:101068. doi: 10.1016/j.redox.2018.101068
157. Gao F, Fang J, Chen F, Wang C, Chen S, Zhang S, et al. Enho mutations causing low adropin: A possible pathomechanism of MPO-ANCA associated lung injury. EBioMedicine. (2016) 9:324–35. doi: 10.1016/j.ebiom.2016.05.036
158. Zhan W, Tian W, Zhang W, Tian H, Sun T. ANGPTL4 attenuates palmitic acid-induced endothelial cell injury by increasing autophagy. Cell Signal (2022) 98:110410. doi: 10.1016/j.cellsig.2022.110410
159. Icer MA, Yıldıran H. Effects of nutritional status on serum fetuin-A level. Crit Rev Food Sci Nutr (2020) 60(11):1938–46. doi: 10.1080/10408398.2019.1631751
160. Yamagoe S, Yamakawa Y, Matsuo Y, Minowada J, Mizuno S, Suzuki K. Purification and primary amino acid sequence of a novel neutrophil chemotactic factor LECT2. Immunol Lett (1996) 52(1):9–13. doi: 10.1016/0165-2478(96)02572-2
161. Xie Y, Fan KW, Guan SX, Hu Y, Gao Y, Zhou WJ. LECT2: A pleiotropic and promising hepatokine, from bench to bedside. J Cell Mol Med (2022) 26(13):3598–607. doi: 10.1111/jcmm.17407
162. Anson M, Crain-Denoyelle AM, Baud V, Chereau F, Gougelet A, Terris B, et al. Oncogenic β-catenin triggers an inflammatory response that determines the aggressiveness of hepatocellular carcinoma in mice. J Clin Invest. (2012) 122(2):586–99. doi: 10.1172/JCI43937
163. Saito Y. Selenoprotein P as a significant regulator of pancreatic β cell function. J Biochem (2020) 167(2):119–24. doi: 10.1093/jb/mvz061
164. Avery JC, Hoffmann PR. Selenium, selenoproteins, and immunity. Nutrients. (2018) 10(9):1203. doi: 10.3390/nu10091203
165. Razaghi A, Poorebrahim M, Sarhan D, Björnstedt M. Selenium stimulates the antitumour immunity: Insights to future research. Eur J Cancer. (2021) 155:256–67. doi: 10.1016/j.ejca.2021.07.013
166. Steinhoff JS, Lass A, Schupp M. Biological functions of RBP4 and its relevance for human diseases. Front Physiol (2021) 12:659977. doi: 10.3389/fphys.2021.659977
167. Steinhoff JS, Lass A, Schupp M. Retinoid homeostasis and beyond: how retinol binding protein 4 contributes to health and disease. Nutrients. (2022) 14(6):1236. doi: 10.3390/nu14061236
168. Fuchs C, Claudel T, Trauner M. Bile acid-mediated control of liver triglycerides. Semin Liver Dis (2013) 33(4):330–42. doi: 10.1055/s-0033-1358520
169. Chiang JYL, Ferrell JM. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am J Physiol Gastrointest Liver Physiol (2020) 318(3):G554–g73. doi: 10.1152/ajpgi.00223.2019
170. Perino A, Demagny H, Velazquez-Villegas L, Schoonjans K. Molecular physiology of bile acid signaling in health, disease, and aging. Physiol Rev (2021) 101(2):683–731. doi: 10.1152/physrev.00049.2019
171. Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol (2018) 15(2):111–28. doi: 10.1038/nrgastro.2017.119
172. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science (2018) 360(6391):eaan5931. doi: 10.1126/science.aan5931
173. Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. (2016) 45(4):802–16. doi: 10.1016/j.immuni.2016.09.008
174. Liu Y, Li P, Lu J, Xiong W, Oger J, Tetzlaff W, et al. Bilirubin possesses powerful immunomodulatory activity and suppresses experimental autoimmune encephalomyelitis. J Immunol (2008) 181(3):1887–97. doi: 10.4049/jimmunol.181.3.1887
175. Longhi MS, Vuerich M, Kalbasi A, Kenison JE, Yeste A, Csizmadia E, et al. Bilirubin suppresses Th17 immunity in colitis by upregulating CD39. JCI Insight (2017) 2(9):e92791. doi: 10.1172/jci.insight.92791
176. Wen G, Yao L, Hao Y, Wang J, Liu J. Bilirubin ameliorates murine atherosclerosis through inhibiting cholesterol synthesis and reshaping the immune system. J Transl Med (2022) 20(1):1. doi: 10.1186/s12967-021-03207-4
177. Thorgersen EB, Barratt-Due A, Haugaa H, Harboe M, Pischke SE, Nilsson PH, et al. The role of complement in liver injury, regeneration, and transplantation. Hepatology. (2019) 70(2):725–36. doi: 10.1002/hep.30508
178. Holers VM. Complement and its receptors: new insights into human disease. Annu Rev Immunol (2014) 32:433–59. doi: 10.1146/annurev-immunol-032713-120154
179. Anliker-Ort M, Dingemanse J, van den Anker J, Kaufmann P. Treatment of rare inflammatory kidney diseases: drugs targeting the terminal complement pathway. Front Immunol (2020) 11:599417. doi: 10.3389/fimmu.2020.599417
180. Bekassy Z, Lopatko Fagerström I, Bader M, Karpman D. Crosstalk between the renin-angiotensin, complement and kallikrein-kinin systems in inflammation. Nat Rev Immunol (2022) 22(7):411–28. doi: 10.1038/s41577-021-00634-8
181. Palmer LJ, Damgaard C, Holmstrup P, Nielsen CH. Influence of complement on neutrophil extracellular trap release induced by bacteria. J Periodontal Res (2016) 51(1):70–6. doi: 10.1111/jre.12284
182. Liu X, Wang Y, Bauer AT, Kirschfink M, Ding P, Gebhardt C, et al. Neutrophils activated by membrane attack complexes increase the permeability of melanoma blood vessels. Proc Natl Acad Sci U S A. (2022) 119(33):e2122716119. doi: 10.1073/pnas.2122716119
183. Diaz-Del-Olmo I, Worboys J, Martin-Sanchez F, Gritsenko A, Ambrose AR, Tannahill GM, et al. Internalization of the membrane attack complex triggers NLRP3 inflammasome activation and IL-1β Secretion in human macrophages. Front Immunol (2021) 12:720655. doi: 10.3389/fimmu.2021.720655
184. Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol (2017) 18(12):1288–98. doi: 10.1038/ni.3858
185. Ling GS, Crawford G, Buang N, Bartok I, Tian K, Thielens NM, et al. C1q restrains autoimmunity and viral infection by regulating CD8(+) T cell metabolism. Science. (2018) 360(6388):558–63. doi: 10.1126/science.aao4555
186. Mastellos DC, Hajishengallis G, Lambris JD. A guide to complement biology, pathology and therapeutic opportunity. Nat Rev Immunol (2023). doi: 10.1038/s41577-023-00926-1
187. West EE, Kolev M, Kemper C. Complement and the regulation of T cell responses. Annu Rev Immunol (2018) 36:309–38. doi: 10.1146/annurev-immunol-042617-053245
188. West EE, Kemper C. Complosome - the intracellular complement system. Nat Rev Nephrol. (2023) 19(7):426–39. doi: 10.1038/s41581-023-00704-1
189. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. (2013) 39(6):1143–57. doi: 10.1016/j.immuni.2013.10.018
190. Gour N, Smole U, Yong HM, Lewkowich IP, Yao N, Singh A, et al. C3a is required for ILC2 function in allergic airway inflammation. Mucosal Immunol (2018) 11(6):1653–62. doi: 10.1038/s41385-018-0064-x
191. Martin M, Leffler J, Smoląg KI, Mytych J, Björk A, Chaves LD, et al. Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ (2016) 23(5):903–11. doi: 10.1038/cdd.2015.164
192. Li X, Zong J, Si S. Complement Factor H related protein 1 and immune inflammatory disorders. Mol Immunol (2022) 145:43–9. doi: 10.1016/j.molimm.2022.03.117
193. Potempa LA, Rajab IM, Olson ME, Hart PC. C-reactive protein and cancer: interpreting the differential bioactivities of its pentameric and monomeric, modified isoforms. Front Immunol (2021) 12:744129. doi: 10.3389/fimmu.2021.744129
194. Ruiz-Fernández C, Gonzalez-Rodríguez M, Francisco V, Rajab IM, Gómez R, Conde J, et al. Monomeric C reactive protein (mCRP) regulates inflammatory responses in human and mouse chondrocytes. Lab Invest. (2021) 101(12):1550–60. doi: 10.1038/s41374-021-00584-8
195. Mortensen RF. C-reactive protein, inflammation, and innate immunity. Immunol Res (2001) 24(2):163–76. doi: 10.1385/IR:24:2:163
196. Son M. Understanding the contextual functions of C1q and LAIR-1 and their applications. Exp Mol Med (2022) 54(5):567–72. doi: 10.1038/s12276-022-00774-4
197. Richter K, Sagawe S, Hecker A, Küllmar M, Askevold I, Damm J, et al. C-reactive protein stimulates nicotinic acetylcholine receptors to control ATP-mediated monocytic inflammasome activation. Front Immunol (2018) 9:1604. doi: 10.3389/fimmu.2018.01604
198. Su L, Li N, Tang H, Lou Z, Chong X, Zhang C, et al. Kupffer cell-derived TNF-α promotes hepatocytes to produce CXCL1 and mobilize neutrophils in response to necrotic cells. Cell Death Dis (2018) 9(3):323. doi: 10.1038/s41419-018-0377-4
199. Devaraj S, Jialal I. C-reactive protein polarizes human macrophages to an M1 phenotype and inhibits transformation to the M2 phenotype. Arterioscler Thromb Vasc Biol (2011) 31(6):1397–402. doi: 10.1161/ATVBAHA.111.225508
200. Sproston NR, Ashworth JJ. Role of C-reactive protein at sites of inflammation and infection. Front Immunol (2018) 9:754. doi: 10.3389/fimmu.2018.00754
201. Eisenhardt SU, Habersberger J, Murphy A, Chen YC, Woollard KJ, Bassler N, et al. Dissociation of pentameric to monomeric C-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ Res (2009) 105(2):128–37. doi: 10.1161/CIRCRESAHA.108.190611
202. Mouliou DS. C-reactive protein: pathophysiology, diagnosis, false test results and a novel diagnostic algorithm for clinicians. Diseases (2023) 11(4):132. doi: 10.3390/diseases11040132
203. Hanriot D, Bello G, Ropars A, Seguin-Devaux C, Poitevin G, Grosjean S, et al. C-reactive protein induces pro- and anti-inflammatory effects, including activation of the liver X receptor alpha, on human monocytes. Thromb Haemost. (2008) 99(3):558–69. doi: 10.1160/TH07-06-0410
204. Khreiss T, József L, Potempa LA, Filep JG. Loss of pentameric symmetry in C-reactive protein induces interleukin-8 secretion through peroxynitrite signaling in human neutrophils. Circ Res (2005) 97(7):690–7. doi: 10.1161/01.RES.0000183881.11739.CB
205. Jimenez RV, Wright TT, Jones NR, Wu J, Gibson AW, Szalai AJ. C-reactive protein impairs dendritic cell development, maturation, and function: implications for peripheral tolerance. Front Immunol (2018) 9:372. doi: 10.3389/fimmu.2018.00372
206. Luan YY, Yao YM. The clinical significance and potential role of C-reactive protein in chronic inflammatory and neurodegenerative diseases. Front Immunol (2018) 9:1302. doi: 10.3389/fimmu.2018.01302
207. Svanberg C, Enocsson H, Govender M, Martinsson K, Potempa LA, Rajab IM, et al. Conformational state of C-reactive protein is critical for reducing immune complex-triggered type I interferon response: Implications for pathogenic mechanisms in autoimmune diseases imprinted by type I interferon gene dysregulation. J Autoimmun (2023) 135:102998. doi: 10.1016/j.jaut.2023.102998
208. Rizo-Téllez SA, Sekheri M, Filep JG. C-reactive protein: a target for therapy to reduce inflammation. Front Immunol (2023) 14:1237729. doi: 10.3389/fimmu.2023.1237729
209. Hsieh IN, White M, Hoeksema M, Deluna X, Hartshorn K. Histone H4 potentiates neutrophil inflammatory responses to influenza A virus: Down-modulation by H4 binding to C-reactive protein and Surfactant protein D. PloS One (2021) 16(2):e0247605. doi: 10.1371/journal.pone.0247605
210. Ji SR, Zhang SH, Chang Y, Li HY, Wang MY, Lv JM, et al. C-reactive protein: the most familiar stranger. J Immunol (2023) 210(6):699–707. doi: 10.4049/jimmunol.2200831
211. Hari-Dass R, Shah C, Meyer DJ, Raynes JG. Serum amyloid A protein binds to outer membrane protein A of gram-negative bacteria. J Biol Chem (2005) 280(19):18562–7. doi: 10.1074/jbc.M500490200
212. Chen R, Chen Q, Zheng J, Zeng Z, Chen M, Li L, et al. Serum amyloid protein A in inflammatory bowel disease: from bench to bedside. Cell Death Discovery (2023) 9(1):154. doi: 10.1038/s41420-023-01455-5
213. Shah C, Hari-Dass R, Raynes JG. Serum amyloid A is an innate immune opsonin for Gram-negative bacteria. Blood. (2006) 108(5):1751–7. doi: 10.1182/blood-2005-11-011932
214. Mantovani A, Garlanda C. Humoral innate immunity and acute-phase proteins. N Engl J Med (2023) 388(5):439–52. doi: 10.1056/NEJMra2206346
215. Abouelasrar Salama S, Gouwy M, Van Damme J, Struyf S. Acute-serum amyloid A and A-SAA-derived peptides as formyl peptide receptor (FPR) 2 ligands. Front Endocrinol (Lausanne). (2023) 14:1119227. doi: 10.3389/fendo.2023.1119227
216. Lee HY, Kim SD, Shim JW, Lee SY, Lee H, Cho KH, et al. Serum amyloid A induces CCL2 production via formyl peptide receptor-like 1-mediated signaling in human monocytes. J Immunol (2008) 181(6):4332–9. doi: 10.4049/jimmunol.181.6.4332
217. De Buck M, Gouwy M, Berghmans N, Opdenakker G, Proost P, Struyf S, et al. COOH-terminal SAA1 peptides fail to induce chemokines but synergize with CXCL8 and CCL3 to recruit leukocytes via FPR2. Blood. (2018) 131(4):439–49. doi: 10.1182/blood-2017-06-788554
218. Abouelasrar Salama S, De Bondt M, De Buck M, Berghmans N, Proost P, Oliveira VLS, et al. Serum amyloid A1 (SAA1) revisited: restricted leukocyte-activating properties of homogeneous SAA1. Front Immunol (2020) 11:843. doi: 10.3389/fimmu.2020.00843
219. Ather JL, Ckless K, Martin R, Foley KL, Suratt BT, Boyson JE, et al. Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J Immunol (2011) 187(1):64–73. doi: 10.4049/jimmunol.1100500
220. Jiang B, Wang D, Hu Y, Li W, Liu F, Zhu X, et al. Serum amyloid A1 exacerbates hepatic steatosis via TLR4-mediated NF-κB signaling pathway. Mol Metab (2022) 59:101462. doi: 10.1016/j.molmet.2022.101462
221. Siegmund SV, Schlosser M, Schildberg FA, Seki E, De Minicis S, UChinami H, et al. Serum amyloid A induces inflammation, proliferation and cell death in activated hepatic stellate cells. PloS One (2016) 11(3):e0150893. doi: 10.1371/journal.pone.0150893
222. Bang YJ, Hu Z, Li Y, Gattu S, Ruhn KA, Raj P, et al. Serum amyloid A delivers retinol to intestinal myeloid cells to promote adaptive immunity. Science (2021) 373(6561):eabf9232. doi: 10.1126/science.abf9232
223. Hu Z, Bang YJ, Ruhn KA, Hooper LV. Molecular basis for retinol binding by serum amyloid A during infection. Proc Natl Acad Sci U S A. (2019) 116(38):19077–82. doi: 10.1073/pnas.1910713116
224. Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Müller M, et al. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med (2010) 207(7):1453–64. doi: 10.1084/jem.20091474
225. Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology. (2011) 54(6):2185–97. doi: 10.1002/hep.24599
226. Obstfeld AE, Sugaru E, Thearle M, Francisco AM, Gayet C, Ginsberg HN, et al. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes. (2010) 59(4):916–25. doi: 10.2337/db09-1403
227. Kaltenmeier C, Wang R, Popp B, Geller D, Tohme S, Yazdani HO. Role of immuno-inflammatory signals in liver ischemia-reperfusion injury. Cells. (2022) 11(14):2222. doi: 10.3390/cells11142222
228. Lippi G, Favaloro EJ, Franchini M, Guidi GC. Milestones and perspectives in coagulation and hemostasis. Semin Thromb Hemost. (2009) 35(1):9–22. doi: 10.1055/s-0029-1214144
229. Gailani D, Broze GJ Jr. Factor XI activation in a revised model of blood coagulation. Science (1991) 253(5022):909–12. doi: 10.1126/science.1652157
230. Shrivastava S, McVey JH, Dorling A. The interface between coagulation and immunity. Am J Transplant. (2007) 7(3):499–506. doi: 10.1111/j.1600-6143.2006.01653.x
231. Schuliga M. The inflammatory actions of coagulant and fibrinolytic proteases in disease. Mediators Inflamm (2015) 2015:437695. doi: 10.1155/2015/437695
232. Stroo I, Zeerleder S, Ding C, Luken BM, Roelofs J, de Boer OJ, et al. Coagulation factor XI improves host defence during murine pneumonia-derived sepsis independent of factor XII activation. Thromb Haemost. (2017) 117(8):1601–14. doi: 10.1160/TH16-12-0920
233. Nickel KF, Long AT, Fuchs TA, Butler LM, Renné T. Factor XII as a therapeutic target in thromboembolic and inflammatory diseases. Arterioscler Thromb Vasc Biol (2017) 37(1):13–20. doi: 10.1161/ATVBAHA.116.308595
234. Vorlova S, Koch M, Manthey HD, Cochain C, Busch M, Chaudhari SM, et al. Coagulation factor XII induces pro-inflammatory cytokine responses in macrophages and promotes atherosclerosis in mice. Thromb Haemost. (2017) 117(1):176–87. doi: 10.1160/TH16-06-0466
235. Renné T, Stavrou EX. Roles of factor XII in innate immunity. Front Immunol (2019) 10:2011. doi: 10.3389/fimmu.2019.02011
236. Stavrou EX, Fang C, Bane KL, Long AT, Naudin C, Kucukal E, et al. Factor XII and uPAR upregulate neutrophil functions to influence wound healing. J Clin Invest. (2018) 128(3):944–59. doi: 10.1172/JCI92880
237. Busch G, Seitz I, Steppich B, Hess S, Eckl R, Schömig A, et al. Coagulation factor Xa stimulates interleukin-8 release in endothelial cells and mononuclear leukocytes: implications in acute myocardial infarction. Arterioscler Thromb Vasc Biol (2005) 25(2):461–6. doi: 10.1161/01.ATV.0000151279.35780.2d
238. Paraboschi EM, Khera AV, Merlini PA, Gigante L, Peyvandi F, Chaffin M, et al. Rare variants lowering the levels of coagulation factor X are protective against ischemic heart disease. Haematologica. (2020) 105(7):e365–e9. doi: 10.3324/haematol.2019.237750
239. Szaba FM, Smiley ST. Roles for thrombin and fibrin(ogen) in cytokine/chemokine production and macrophage adhesion in vivo. Blood (2002) 99(3):1053–9. doi: 10.1182/blood.V99.3.1053
240. Fish RJ, Neerman-Arbez M. Fibrinogen gene regulation. Thromb Haemost. (2012) 108(3):419–26. doi: 10.1160/TH12-04-0273
241. Påhlman LI, Mörgelin M, Kasetty G, Olin AI, Schmidtchen A, Herwald H. Antimicrobial activity of fibrinogen and fibrinogen-derived peptides–a novel link between coagulation and innate immunity. Thromb Haemost. (2013) 109(5):930–9. doi: 10.1160/TH12-10-0739
242. Endo Y, Nakazawa N, Iwaki D, Takahashi M, Matsushita M, Fujita T. Interactions of ficolin and mannose-binding lectin with fibrinogen/fibrin augment the lectin complement pathway. J Innate Immun (2010) 2(1):33–42. doi: 10.1159/000227805
243. Hoppe B. Fibrinogen and factor XIII at the intersection of coagulation, fibrinolysis and inflammation. Thromb Haemost. (2014) 112(4):649–58. doi: 10.1160/TH14-01-0085
244. Pluskota E, Soloviev DA, Szpak D, Weber C, Plow EF. Neutrophil apoptosis: selective regulation by different ligands of integrin alphaMbeta2. J Immunol (2008) 181(5):3609–19. doi: 10.4049/jimmunol.181.5.3609
245. Hulshof AM, Hemker HC, Spronk HMH, Henskens YMC, Ten Cate H. Thrombin-fibrin(ogen) interactions, host defense and risk of thrombosis. Int J Mol Sci (2021) 22(5):2590. doi: 10.3390/ijms22052590
246. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell (2019) 176(1-2):334–47.e12. doi: 10.1016/j.cell.2018.11.010
247. Qian W, Zhao M, Wang R, Li H. Fibrinogen-like protein 1 (FGL1): the next immune checkpoint target. J Hematol Oncol (2021) 14(1):147. doi: 10.1186/s13045-021-01161-8
248. Ahmed O, Robinson MW, O'Farrelly C. Inflammatory processes in the liver: divergent roles in homeostasis and pathology. Cell Mol Immunol (2021) 18(6):1375–86. doi: 10.1038/s41423-021-00639-2
249. Cai J, Zhang XJ, Li H. The role of innate immune cells in nonalcoholic steatohepatitis. Hepatology. (2019) 70(3):1026–37. doi: 10.1002/hep.30506
250. Zhao J, Liu J, Pang X, Zhang X, Wang S, Wu D. Rosiglitazone attenuates angiotensin II-induced C-reactive protein expression in hepatocytes via inhibiting AT1/ROS/MAPK signal pathway. Int Immunopharmacol. (2016) 31:178–85. doi: 10.1016/j.intimp.2015.12.026
251. Kong F, You H, Zheng K, Tang R, Zheng C. The crosstalk between pattern-recognition receptor signaling and calcium signaling. Int J Biol Macromol. (2021) 192:745–56. doi: 10.1016/j.ijbiomac.2021.10.014
252. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol (2013) 14(10):996–1006. doi: 10.1038/ni.2691
253. Jiang Y, Que W, Zhu P, Li XK. The role of diverse liver cells in liver transplantation tolerance. Front Immunol (2020) 11:1203. doi: 10.3389/fimmu.2020.01203
254. Mühlbauer M, Fleck M, Schütz C, Weiss T, Froh M, Blank C, et al. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J Hepatol (2006) 45(4):520–8. doi: 10.1016/j.jhep.2006.05.007
255. Elvevold K, Simon-Santamaria J, Hasvold H, McCourt P, Smedsrød B, Sørensen KK. Liver sinusoidal endothelial cells depend on mannose receptor-mediated recruitment of lysosomal enzymes for normal degradation capacity. Hepatology. (2008) 48(6):2007–15. doi: 10.1002/hep.22527
256. Ganesan LP, Mohanty S, Kim J, Clark KR, Robinson JM, Anderson CL. Rapid and efficient clearance of blood-borne virus by liver sinusoidal endothelium. PloS Pathog (2011) 7(9):e1002281. doi: 10.1371/journal.ppat.1002281
257. Knolle PA, Limmer A. Control of immune responses by savenger liver endothelial cells. Swiss Med Wkly (2003) 133(37-38):501–6. doi: 10.4414/smw.2003.10261
258. Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol (2018) 15(9):555–67. doi: 10.1038/s41575-018-0020-y
259. Xu X, Jin R, Li M, Wang K, Zhang S, Hao J, et al. Liver sinusoidal endothelial cells induce tolerance of autoreactive CD4+ recent thymic emigrants. Sci Rep (2016) 6:19861. doi: 10.1038/srep19861
260. Wu J, Meng Z, Jiang M, Zhang E, Trippler M, Broering R, et al. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology. (2010) 129(3):363–74. doi: 10.1111/j.1365-2567.2009.03179.x
261. Zhang Y, Yang X, Bi T, Wu X, Wang L, Ren Y, et al. Targeted inhibition of the immunoproteasome blocks endothelial MHC class II antigen presentation to CD4(+) T cells in chronic liver injury. Int Immunopharmacol. (2022) 107:108639. doi: 10.1016/j.intimp.2022.108639
262. Knolle PA, Schmitt E, Jin S, Germann T, Duchmann R, Hegenbarth S, et al. Induction of cytokine production in naive CD4(+) T cells by antigen-presenting murine liver sinusoidal endothelial cells but failure to induce differentiation toward Th1 cells. Gastroenterology. (1999) 116(6):1428–40. doi: 10.1016/S0016-5085(99)70508-1
263. Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med (2000) 6(12):1348–54. doi: 10.1038/82161
264. Carambia A, Frenzel C, Bruns OT, Schwinge D, Reimer R, Hohenberg H, et al. Inhibition of inflammatory CD4 T cell activity by murine liver sinusoidal endothelial cells. J Hepatol (2013) 58(1):112–8. doi: 10.1016/j.jhep.2012.09.008
265. Patten DA, Wilson GK, Bailey D, Shaw RK, Jalkanen S, Salmi M, et al. Human liver sinusoidal endothelial cells promote intracellular crawling of lymphocytes during recruitment: A new step in migration. Hepatology. (2017) 65(1):294–309. doi: 10.1002/hep.28879
266. Furuta K, Guo Q, Hirsova P, Ibrahim SH. Emerging roles of liver sinusoidal endothelial cells in nonalcoholic steatohepatitis. Biol (Basel) (2020) 9(11):395. doi: 10.3390/biology9110395
267. Wong J, Johnston B, Lee SS, Bullard DC, Smith CW, Beaudet AL, et al. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J Clin Invest. (1997) 99(11):2782–90. doi: 10.1172/JCI119468
268. Edwards S, Lalor PF, Nash GB, Rainger GE, Adams DH. Lymphocyte traffic through sinusoidal endothelial cells is regulated by hepatocytes. Hepatology. (2005) 41(3):451–9. doi: 10.1002/hep.20585
269. Lalor PF, Herbert J, Bicknell R, Adams DH. Hepatic sinusoidal endothelium avidly binds platelets in an integrin-dependent manner, leading to platelet and endothelial activation and leukocyte recruitment. Am J Physiol Gastrointest Liver Physiol (2013) 304(5):G469–78. doi: 10.1152/ajpgi.00407.2012
270. Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol (2019) 70(6):1278–91. doi: 10.1016/j.jhep.2019.02.012
271. Hilscher MB, Sehrawat T, Arab JP, Zeng Z, Gao J, Liu M, et al. Mechanical stretch increases expression of CXCL1 in liver sinusoidal endothelial cells to recruit neutrophils, generate sinusoidal microthombi, and promote portal hypertension. Gastroenterology. (2019) 157(1):193–209.e9. doi: 10.1053/j.gastro.2019.03.013
272. Yang M, Zhang C. The role of liver sinusoidal endothelial cells in cancer liver metastasis. Am J Cancer Res (2021) 11(5):1845–60.
273. Bennett H, Troutman TD, Sakai M, Glass CK. Epigenetic regulation of kupffer cell function in health and disease. Front Immunol (2020) 11:609618. doi: 10.3389/fimmu.2020.609618
274. Helmy KY, Katschke KJ Jr., Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. (2006) 124(5):915–27. doi: 10.1016/j.cell.2005.12.039
275. Gregory SH, Cousens LP, van Rooijen N, Döpp EA, Carlos TM, Wing EJ. Complementary adhesion molecules promote neutrophil-Kupffer cell interaction and the elimination of bacteria taken up by the liver. J Immunol (2002) 168(1):308–15. doi: 10.4049/jimmunol.168.1.308
276. Shi J, Gilbert GE, Kokubo Y, Ohashi T. Role of the liver in regulating numbers of circulating neutrophils. Blood. (2001) 98(4):1226–30. doi: 10.1182/blood.V98.4.1226
277. Uchikura K, Wada T, Hoshino S, Nagakawa Y, Aiko T, Bulkley GB, et al. Lipopolysaccharides induced increases in Fas ligand expression by Kupffer cells via mechanisms dependent on reactive oxygen species. Am J Physiol Gastrointest Liver Physiol (2004) 287(3):G620–6. doi: 10.1152/ajpgi.00314.2003
278. McDonald B, Jenne CN, Zhuo L, Kimata K, Kubes P. Kupffer cells and activation of endothelial TLR4 coordinate neutrophil adhesion within liver sinusoids during endotoxemia. Am J Physiol Gastrointest Liver Physiol (2013) 305(11):G797–806. doi: 10.1152/ajpgi.00058.2013
279. Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. (2015) 62(1):279–91. doi: 10.1002/hep.27793
280. Liu J, Yu Q, Wu W, Huang X, Broering R, Werner M, et al. TLR2 stimulation strengthens intrahepatic myeloid-derived cell-mediated T cell tolerance through inducing kupffer cell expansion and IL-10 production. J Immunol (2018) 200(7):2341–51. doi: 10.4049/jimmunol.1700540
281. Dai S, Liu F, Qin Z, Zhang J, Chen J, Ding WX, et al. Kupffer cells promote T-cell hepatitis by producing CXCL10 and limiting liver sinusoidal endothelial cell permeability. Theranostics. (2020) 10(16):7163–77. doi: 10.7150/thno.44960
282. Lee WY, Moriarty TJ, Wong CH, Zhou H, Strieter RM, van Rooijen N, et al. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat Immunol (2010) 11(4):295–302. doi: 10.1038/ni.1855
283. Wilson CL, Mann J, Walsh M, Perrugoria MJ, Oakley F, Wright MC, et al. Quiescent hepatic stellate cells functionally contribute to the hepatic innate immune response via TLR3. PloS One (2014) 9(1):e83391. doi: 10.1371/journal.pone.0083391
284. Maher JJ. Interactions between hepatic stellate cells and the immune system. Semin Liver Dis (2001) 21(3):417–26. doi: 10.1055/s-2001-17555
285. Ichikawa S, Mucida D, Tyznik AJ, Kronenberg M, Cheroutre H. Hepatic stellate cells function as regulatory bystanders. J Immunol (2011) 186(10):5549–55. doi: 10.4049/jimmunol.1003917
286. Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA, et al. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity. (2007) 26(1):117–29. doi: 10.1016/j.immuni.2006.11.011
287. Marra F, Valente AJ, Pinzani M, Abboud HE. Cultured human liver fat-storing cells produce monocyte chemotactic protein-1. Regulation by proinflammatory cytokines. J Clin Invest. (1993) 92(4):1674–80. doi: 10.1172/JCI116753
288. Schwabe RF, Schnabl B, Kweon YO, Brenner DA. CD40 activates NF-kappa B and c-Jun N-terminal kinase and enhances chemokine secretion on activated human hepatic stellate cells. J Immunol (2001) 166(11):6812–9. doi: 10.4049/jimmunol.166.11.6812
289. Yu MC, Chen CH, Liang X, Wang L, Gandhi CR, Fung JJ, et al. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology. (2004) 40(6):1312–21. doi: 10.1002/hep.20488
290. Li Y, Lu L, Qian S, Fung JJ, Lin F. Hepatic stellate cells directly inhibit B cells via programmed death-ligand 1. J Immunol (2016) 196(4):1617–25. doi: 10.4049/jimmunol.1501737
291. Schildberg FA, Wojtalla A, Siegmund SV, Endl E, Diehl L, Abdullah Z, et al. Murine hepatic stellate cells veto CD8 T cell activation by a CD54-dependent mechanism. Hepatology. (2011) 54(1):262–72. doi: 10.1002/hep.24352
292. Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol (2007) 7(7):543–55. doi: 10.1038/nri2103
293. Chen Y, Jiang G, Yang HR, Gu X, Wang L, Hsieh CC, et al. Distinct response of liver myeloid dendritic cells to endotoxin is mediated by IL-27. J Hepatol (2009) 51(3):510–9. doi: 10.1016/j.jhep.2009.04.026
294. Pillarisetty VG, Shah AB, Miller G, Bleier JI, DeMatteo RP. Liver dendritic cells are less immunogenic than spleen dendritic cells because of differences in subtype composition. J Immunol (2004) 172(2):1009–17. doi: 10.4049/jimmunol.172.2.1009
295. Bamboat ZM, Stableford JA, Plitas G, Burt BM, Nguyen HM, Welles AP, et al. Human liver dendritic cells promote T cell hyporesponsiveness. J Immunol (2009) 182(4):1901–11. doi: 10.4049/jimmunol.0803404
296. Goubier A, Dubois B, Gheit H, Joubert G, Villard-Truc F, Asselin-Paturel C, et al. Plasmacytoid dendritic cells mediate oral tolerance. Immunity. (2008) 29(3):464–75. doi: 10.1016/j.immuni.2008.06.017
297. O'Keeffe M, Hochrein H, Vremec D, Caminschi I, Miller JL, Anders EM, et al. Mouse plasmacytoid cells: long-lived cells, heterogeneous in surface phenotype and function, that differentiate into CD8(+) dendritic cells only after microbial stimulus. J Exp Med (2002) 196(10):1307–19. doi: 10.1084/jem.20021031
298. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. (2017) 127(1):55–64. doi: 10.1172/JCI88881
299. Heier EC, Meier A, Julich-Haertel H, Djudjaj S, Rau M, Tschernig T, et al. Murine CD103(+) dendritic cells protect against steatosis progression towards steatohepatitis. J Hepatol (2017) 66(6):1241–50. doi: 10.1016/j.jhep.2017.01.008
300. Castellaneta A, Sumpter TL, Chen L, Tokita D, Thomson AW. NOD2 ligation subverts IFN-alpha production by liver plasmacytoid dendritic cells and inhibits their T cell allostimulatory activity via B7-H1 up-regulation. J Immunol (2009) 183(11):6922–32. doi: 10.4049/jimmunol.0900582
301. Subleski JJ, Hall VL, Back TC, Ortaldo JR, Wiltrout RH. Enhanced antitumor response by divergent modulation of natural killer and natural killer T cells in the liver. Cancer Res (2006) 66(22):11005–12. doi: 10.1158/0008-5472.CAN-06-0811
302. Sun C, Sun H, Zhang C, Tian Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol Immunol (2015) 12(3):292–302. doi: 10.1038/cmi.2014.91
303. Sun H, Sun C, Tian Z, Xiao W. NK cells in immunotolerant organs. Cell Mol Immunol (2013) 10(3):202–12. doi: 10.1038/cmi.2013.9
304. Chen Y, Wei H, Sun R, Dong Z, Zhang J, Tian Z. Increased susceptibility to liver injury in hepatitis B virus transgenic mice involves NKG2D-ligand interaction and natural killer cells. Hepatology. (2007) 46(3):706–15. doi: 10.1002/hep.21872
305. Liaskou E, Wilson DV, Oo YH. Innate immune cells in liver inflammation. Mediators Inflamm (2012) 2012:949157. doi: 10.1155/2012/949157
306. Ali AK, Komal AK, Almutairi SM, Lee SH. Natural killer cell-derived IL-10 prevents liver damage during sustained murine cytomegalovirus infection. Front Immunol (2019) 10:2688. doi: 10.3389/fimmu.2019.02688
307. Zheng M, Sun R, Wei H, Tian Z. NK cells help induce anti-hepatitis B virus CD8+ T cell immunity in mice. J Immunol (2016) 196(10):4122–31. doi: 10.4049/jimmunol.1500846
308. Glässner A, Eisenhardt M, Krämer B, Körner C, Coenen M, Sauerbruch T, et al. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab Invest. (2012) 92(7):967–77. doi: 10.1038/labinvest.2012.54
309. Horst AK, Neumann K, Diehl L, Tiegs G. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell Mol Immunol (2016) 13(3):277–92. doi: 10.1038/cmi.2015.112
310. Goto T, Ito Y, Satoh M, Nakamoto S, Nishizawa N, Hosono K, et al. Activation of iNKT cells facilitates liver repair after hepatic ischemia reperfusion injury through acceleration of macrophage polarization. Front Immunol (2021) 12:754106. doi: 10.3389/fimmu.2021.754106
311. Wang H, Feng D, Park O, Yin S, Gao B. Invariant NKT cell activation induces neutrophil accumulation and hepatitis: opposite regulation by IL-4 and IFN-γ. Hepatology. (2013) 58(4):1474–85. doi: 10.1002/hep.26471
312. Huang LR, Wohlleber D, Reisinger F, Jenne CN, Cheng RL, Abdullah Z, et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8(+) T cells and successful immunotherapy against chronic viral liver infection. Nat Immunol (2013) 14(6):574–83. doi: 10.1038/ni.2573
313. Knolle PA, Uhrig A, Hegenbarth S, Löser E, Schmitt E, Gerken G, et al. IL-10 down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells through decreased antigen uptake via the mannose receptor and lowered surface expression of accessory molecules. Clin Exp Immunol (1998) 114(3):427–33. doi: 10.1046/j.1365-2249.1998.00713.x
314. Liu C, Schönke M, Spoorenberg B, Lambooij JM, van der Zande HJP, Zhou E, et al. FGF21 protects against hepatic lipotoxicity and macrophage activation to attenuate fibrogenesis in nonalcoholic steatohepatitis. Elife (2023) 12:e83075. doi: 10.7554/eLife.83075.sa2
315. Tacke F, Puengel T, Loomba R, Friedman SL. An integrated view of anti-inflammatory and antifibrotic targets for the treatment of NASH. J Hepatol (2023) 79(2):552–66. doi: 10.1016/j.jhep.2023.03.038
316. Brown ZJ, Tsilimigras DI, Ruff SM, Mohseni A, Kamel IR, Cloyd JM, et al. Management of hepatocellular carcinoma: A review. JAMA Surg (2023) 158(4):410–20. doi: 10.1001/jamasurg.2022.7989
317. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. (2007) 132(7):2557–76. doi: 10.1053/j.gastro.2007.04.061
318. Dunn W, Shah VH. Pathogenesis of alcoholic liver disease. Clin Liver Dis (2016) 20(3):445–56. doi: 10.1016/j.cld.2016.02.004
319. Zhao P, Wei Y, Sun G, Xu L, Wang T, Tian Y, et al. Fetuin-A alleviates neuroinflammation against traumatic brain injury-induced microglial necroptosis by regulating Nrf-2/HO-1 pathway. J Neuroinflammation. (2022) 19(1):269. doi: 10.1186/s12974-022-02633-5
320. Yanguas-Casás N, Barreda-Manso MA, Nieto-Sampedro M, Romero-Ramírez L. Tauroursodeoxycholic acid reduces glial cell activation in an animal model of acute neuroinflammation. J Neuroinflammation. (2014) 11:50. doi: 10.1186/1742-2094-11-50
321. Bhargava P, Smith MD, Mische L, Harrington E, Fitzgerald KC, Martin K, et al. Bile acid metabolism is altered in multiple sclerosis and supplementation ameliorates neuroinflammation. J Clin Invest. (2020) 130(7):3467–82. doi: 10.1172/JCI129401
322. Huang F, Pariante CM, Borsini A. From dried bear bile to molecular investigation: A systematic review of the effect of bile acids on cell apoptosis, oxidative stress and inflammation in the brain, across pre-clinical models of neurological, neurodegenerative and neuropsychiatric disorders. Brain Behav Immun (2022) 99:132–46. doi: 10.1016/j.bbi.2021.09.021
323. Lee JS, O'Connell EM, Pacher P, Lohoff FW. PCSK9 and the gut-liver-brain axis: A novel therapeutic target for immune regulation in alcohol use disorder. J Clin Med (2021) 10(8):1758. doi: 10.3390/jcm10081758
324. Jaafar AK, Techer R, Chemello K, Lambert G, Bourane S. PCSK9 and the nervous system: a no-brainer? J Lipid Res (2023) 64(9):100426. doi: 10.1016/j.jlr.2023.100426
325. Apaijai N, Moisescu DM, Palee S, McSweeney CM, Saiyasit N, Maneechote C, et al. Pretreatment with PCSK9 inhibitor protects the brain against cardiac ischemia/reperfusion injury through a reduction of neuronal inflammation and amyloid beta aggregation. J Am Heart Assoc (2019) 8(2):e010838. doi: 10.1161/JAHA.118.010838
326. Davalos D, Baeten KM, Whitney MA, Mullins ES, Friedman B, Olson ES, et al. Early detection of thrombin activity in neuroinflammatory disease. Ann Neurol (2014) 75(2):303–8. doi: 10.1002/ana.24078
327. Ryu JK, Petersen MA, Murray SG, Baeten KM, Meyer-Franke A, Chan JP, et al. Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat Commun (2015) 6:8164. doi: 10.1038/ncomms9164
328. Francis K, van Beek J, Canova C, Neal JW, Gasque P. Innate immunity and brain inflammation: the key role of complement. Expert Rev Mol Med (2003) 5(15):1–19. doi: 10.1017/S1462399403006252
329. Singh AK, Chaube B, Zhang X, Sun J, Citrin KM, Canfrán-Duque A, et al. Hepatocyte-specific suppression of ANGPTL4 improves obesity-associated diabetes and mitigates atherosclerosis in mice. J Clin Invest (2021) 131(17):e140989. doi: 10.1172/JCI140989
330. Aryal B, Rotllan N, Araldi E, Ramírez CM, He S, Chousterman BG, et al. ANGPTL4 deficiency in haematopoietic cells promotes monocyte expansion and atherosclerosis progression. Nat Commun (2016) 7:12313. doi: 10.1038/ncomms12313
331. Wunderer F, Traeger L, Sigurslid HH, Meybohm P, Bloch DB, Malhotra R. The role of hepcidin and iron homeostasis in atherosclerosis. Pharmacol Res (2020) 153:104664. doi: 10.1016/j.phrs.2020.104664
332. Ma M, Hou C, Liu J. Effect of PCSK9 on atherosclerotic cardiovascular diseases and its mechanisms: Focus on immune regulation. Front Cardiovasc Med (2023) 10:1148486. doi: 10.3389/fcvm.2023.1148486
333. Alnuwaysir RIS, Hoes MF, van Veldhuisen DJ, van der Meer P, Grote Beverborg N. Iron deficiency in heart failure: mechanisms and pathophysiology. J Clin Med (2021) 11(1):125. doi: 10.3390/jcm11010125
334. Pan X, Shao Y, Wu F, Wang Y, Xiong R, Zheng J, et al. FGF21 prevents angiotensin II-induced hypertension and vascular dysfunction by activation of ACE2/angiotensin-(1-7) axis in mice. Cell Metab (2018) 27(6):1323–37.e5. doi: 10.1016/j.cmet.2018.04.002
335. Yu Y, Li S, Liu Y, Tian G, Yuan Q, Bai F, et al. Fibroblast growth factor 21 (FGF21) ameliorates collagen-induced arthritis through modulating oxidative stress and suppressing nuclear factor-kappa B pathway. Int Immunopharmacol. (2015) 25(1):74–82. doi: 10.1016/j.intimp.2015.01.005
336. Yu D, Ye X, Che R, Wu Q, Qi J, Song L, et al. FGF21 exerts comparable pharmacological efficacy with Adalimumab in ameliorating collagen-induced rheumatoid arthritis by regulating systematic inflammatory response. BioMed Pharmacother. (2017) 89:751–60. doi: 10.1016/j.biopha.2017.02.059
337. Cui JK, Yu JZ, Xu H, Zou YQ, Zhang H, Chen SS, et al. Autophagy-lysosome inhibitor chloroquine prevents CTLA-4 degradation of T cells and attenuates acute rejection in murine skin and heart transplantation. #N/A. (2020) 10(18):8051–60. doi: 10.7150/thno.43507
338. Baran A, Nowowiejska J, Krahel JA, Kaminski TW, Maciaszek M, Flisiak I. Higher serum selenoprotein P level as a novel inductor of metabolic complications in psoriasis. Int J Mol Sci (2020) 21(13):4594. doi: 10.3390/ijms21134594
339. Ballanti E, Perricone C, Greco E, Ballanti M, Di Muzio G, Chimenti MS, et al. Complement and autoimmunity. Immunol Res (2013) 56(2-3):477–91. doi: 10.1007/s12026-013-8422-y
340. Oteng AB, Bhattacharya A, Brodesser S, Qi L, Tan NS, Kersten S. Feeding Angptl4(-/-) mice trans fat promotes foam cell formation in mesenteric lymph nodes without leading to ascites. J Lipid Res (2017) 58(6):1100–13. doi: 10.1194/jlr.M074278
341. Short SP, Pilat JM, Williams CS. Roles for selenium and selenoprotein P in the development, progression, and prevention of intestinal disease. Free Radic Biol Med (2018) 127:26–35. doi: 10.1016/j.freeradbiomed.2018.05.066
342. Hortová-Kohoutková M, Skotáková M, Onyango IG, Slezáková M, Panovský R, Opatřil L, et al. Hepcidin and ferritin levels as markers of immune cell activation during septic shock, severe COVID-19 and sterile inflammation. Front Immunol (2023) 14:1110540. doi: 10.3389/fimmu.2023.1110540
343. Duval A, Frémeaux-Bacchi V. Complement biology for hematologists. Am J Hematol (2023) 98Suppl 4:S5–s19. doi: 10.1002/ajh.26855
344. Bhandari S, Larsen AK, McCourt P, Smedsrød B, Sørensen KK. The scavenger function of liver sinusoidal endothelial cells in health and disease. Front Physiol (2021) 12:757469. doi: 10.3389/fphys.2021.757469
345. Gottwick C, Carambia A, Herkel J. Harnessing the liver to induce antigen-specific immune tolerance. Semin Immunopathol (2022) 44(4):475–84. doi: 10.1007/s00281-022-00942-8
346. Gregory SH, Wing EJ. Neutrophil-Kupffer cell interaction: a critical component of host defenses to systemic bacterial infections. J Leukoc Biol (2002) 72(2):239–48. doi: 10.1189/jlb.72.2.239
347. Kumar V. NKT-cell subsets: promoters and protectors in inflammatory liver disease. J Hepatol (2013) 59(3):618–20. doi: 10.1016/j.jhep.2013.02.032
348. Byun JS, Yi HS. Hepatic immune microenvironment in alcoholic and nonalcoholic liver disease. BioMed Res Int (2017) 2017:6862439. doi: 10.1155/2017/6862439
349. Sebode M, Schulz L, Lohse AW. "Autoimmune(-like)" Drug and herb induced liver injury: new insights into molecular pathogenesis. Int J Mol Sci (2017) 18(9):1954. doi: 10.3390/ijms18091954
350. Li HY, Tang ZM, Wang Z, Lv JM, Liu XL, Liang YL, et al. C-reactive protein protects against acetaminophen-induced liver injury by preventing complement overactivation. Cell Mol Gastroenterol Hepatol (2022) 13(1):289–307. doi: 10.1016/j.jcmgh.2021.09.003
351. Effenberger M, Grander C, Grabherr F, Griesmacher A, Ploner T, Hartig F, et al. Systemic inflammation as fuel for acute liver injury in COVID-19. Dig Liver Dis (2021) 53(2):158–65. doi: 10.1016/j.dld.2020.08.004
352. Guo Z, Fan X, Yao J, Tomlinson S, Yuan G, He S. The role of complement in nonalcoholic fatty liver disease. Front Immunol (2022) 13:1017467. doi: 10.3389/fimmu.2022.1017467
353. Zhou Y, Yuan G, Zhong F, He S. Roles of the complement system in alcohol-induced liver disease. Clin Mol Hepatol (2020) 26(4):677–85. doi: 10.3350/cmh.2020.0094
354. Wang H, Mehal W, Nagy LE, Rotman Y. Immunological mechanisms and therapeutic targets of fatty liver diseases. Cell Mol Immunol (2021) 18(1):73–91. doi: 10.1038/s41423-020-00579-3
355. Tajima T, Hata K, Kusakabe J, Miyauchi H, Badshah JS, Kageyama S, et al. Anti-complement 5 antibody ameliorates antibody-mediated rejection after liver transplantation in rats. Front Immunol (2023) 14:1186653. doi: 10.3389/fimmu.2023.1186653
356. Wu L, Yan J, Bai Y, Chen F, Zou X, Xu J, et al. An invasive zone in human liver cancer identified by Stereo-seq promotes hepatocyte-tumor cell crosstalk, local immunosuppression and tumor progression. Cell Res (2023) 33(8):585–603. doi: 10.1038/s41422-023-00831-1
357. Lee JW, Stone ML, Porrett PM, Thomas SK, Komar CA, Li JH, et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature. (2019) 567(7747):249–52. doi: 10.1038/s41586-019-1004-y
358. Li D, Xie P, Zhao S, Zhao J, Yao Y, Zhao Y, et al. Hepatocytes derived increased SAA1 promotes intrahepatic platelet aggregation and aggravates liver inflammation in NAFLD. Biochem Biophys Res Commun (2021) 555:54–60. doi: 10.1016/j.bbrc.2021.02.124
359. Wang H, Zhou H, Zhang Q, Poulsen KL, Taylor V, McMullen MR, et al. Inhibition of IRAK4 kinase activity improves ethanol-induced liver injury in mice. J Hepatol (2020) 73(6):1470–81. doi: 10.1016/j.jhep.2020.07.016
360. Endle H, Horta G, Stutz B, Muthuraman M, Tegeder I, Schreiber Y, et al. AgRP neurons control feeding behaviour at cortical synapses via peripherally derived lysophospholipids. Nat Metab (2022) 4(6):683–92. doi: 10.1038/s42255-022-00589-7
361. Harris VK, Donelan N, Yan QJ, Clark K, Touray A, Rammal M, et al. Cerebrospinal fluid fetuin-A is a biomarker of active multiple sclerosis. Mult Scler. (2013) 19(11):1462–72. doi: 10.1177/1352458513477923
362. Heinen MC, Babler A, Weis J, Elsas J, Nolte K, Kipp M, et al. Fetuin-A protein distribution in mature inflamed and ischemic brain tissue. PloS One (2018) 13(11):e0206597. doi: 10.1371/journal.pone.0206597
363. MahmoudianDehkordi S, Arnold M, Nho K, Ahmad S, Jia W, Xie G, et al. Altered bile acid profile associates with cognitive impairment in Alzheimer's disease-An emerging role for gut microbiome. Alzheimers Dement. (2019) 15(1):76–92. doi: 10.1016/j.jalz.2018.07.217
364. Rodrigues CM, Spellman SR, Solá S, Grande AW, Linehan-Stieers C, Low WC, et al. Neuroprotection by a bile acid in an acute stroke model in the rat. J Cereb Blood Flow Metab (2002) 22(4):463–71. doi: 10.1097/00004647-200204000-00010
365. Göbel K, Pankratz S, Asaridou CM, Herrmann AM, Bittner S, Merker M, et al. Blood coagulation factor XII drives adaptive immunity during neuroinflammation via CD87-mediated modulation of dendritic cells. Nat Commun (2016) 7:11626. doi: 10.1038/ncomms11626
366. Wang F, Li M, Zhang A, Li H, Jiang C, Guo J. PCSK9 modulates macrophage polarization-mediated ventricular remodeling after myocardial infarction. J Immunol Res (2022) 2022:7685796. doi: 10.1155/2022/7685796
367. Cao Y, Wang Y, Zhou Z, Pan C, Jiang L, Zhou Z, et al. Liver-heart cross-talk mediated by coagulation factor XI protects against heart failure. Science. (2022) 377(6613):1399–406. doi: 10.1126/science.abn0910
368. Sun JY, Du LJ, Shi XR, Zhang YY, Liu Y, Wang YL, et al. An IL-6/STAT3/MR/FGF21 axis mediates heart-liver cross-talk after myocardial infarction. Sci Adv (2023) 9(14):eade4110. doi: 10.1126/sciadv.ade4110
369. Stark K, Massberg S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol (2021) 18(9):666–82. doi: 10.1038/s41569-021-00552-1
370. Vignesh P, Rawat A, Sharma M, Singh S. Complement in autoimmune diseases. Clin Chim Acta (2017) 465:123–30. doi: 10.1016/j.cca.2016.12.017
371. Coss SL, Zhou D, Chua GT, Aziz RA, Hoffman RP, Wu YL, et al. The complement system and human autoimmune diseases. J Autoimmun (2023) 137:102979. doi: 10.1016/j.jaut.2022.102979
372. Xie W, Li J, Du H, Xia J. Causal relationship between PCSK9 inhibitor and autoimmune diseases: a drug target Mendelian randomization study. Arthritis Res Ther (2023) 25(1):148. doi: 10.1186/s13075-023-03122-7
373. Shirasaki T, Yamagoe S, Shimakami T, Murai K, Imamura R, Ishii KA, et al. Leukocyte cell-derived chemotaxin 2 is an antiviral regulator acting through the proto-oncogene MET. Nat Commun (2022) 13(1):3176. doi: 10.1038/s41467-022-30879-3
374. Wang CY, Babitt JL. Hepcidin regulation in the anemia of inflammation. Curr Opin Hematol (2016) 23(3):189–97. doi: 10.1097/MOH.0000000000000236
375. Dudkowiak R, Neubauer K, Poniewierka E. Hepcidin and its role in inflammatory bowel disease. Adv Clin Exp Med (2013) 22(4):585–91.
Keywords: liver, organ communication, hepatokine, immunity, immune homeostasis, immune cells, immune related diseases
Citation: Zhao J, Zhang X, Li Y, Yu J, Chen Z, Niu Y, Ran S, Wang S, Ye W, Luo Z, Li X, Hao Y, Zong J, Xia C, Xia J and Wu J (2023) Interorgan communication with the liver: novel mechanisms and therapeutic targets. Front. Immunol. 14:1314123. doi: 10.3389/fimmu.2023.1314123
Received: 11 October 2023; Accepted: 28 November 2023;
Published: 12 December 2023.
Edited by:
Uday Kishore, United Arab Emirates University, United Arab EmiratesReviewed by:
Shang-Rong Ji, Lanzhou University, ChinaCopyright © 2023 Zhao, Zhang, Li, Yu, Chen, Niu, Ran, Wang, Ye, Luo, Li, Hao, Zong, Xia, Xia and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Wu, d3VqaWU0MjZAaHVzdC5lZHUuY24=; Jiahong Xia, amlhaG9uZy54aWFAbWFpbC5odXN0LmVkdS5jbg==; Chengkun Xia, MzE2OTU0NTBAcXEuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.