 Areya Tabatabai1†
Areya Tabatabai1† Aastha Arora1†
Aastha Arora1† Svenja Höfmann1†
Svenja Höfmann1† Maximilian Jauch1
Maximilian Jauch1 Bastian von Tresckow1
Bastian von Tresckow1 Julia Hansen2,3,4,5,6
Julia Hansen2,3,4,5,6 Ruth Flümann2,3,4,5,6Ron D. Jachimowicz2,3,4,5,6
Ruth Flümann2,3,4,5,6Ron D. Jachimowicz2,3,4,5,6 Sebastian Klein1
Sebastian Klein1 Hans Christian Reinhardt1*
Hans Christian Reinhardt1* Gero Knittel1*
Gero Knittel1*- 1Department of Hematology and Stem Cell Transplantation, University Hospital Essen, West German Cancer Center, German Cancer Consortium Partner Site Essen, Center for Molecular Biotechnology, University of Duisburg-Essen, Essen, Germany
- 2Department I of Internal Medicine, University of Cologne, Faculty of Medicine and University Hospital Cologne, Center for Integrated Oncology Aachen Bonn, Cologne, Germany
- 3Center for Molecular Medicine, University of Cologne, Cologne, Germany
- 4Cologne Excellence Cluster on Cellular Stress Response in Aging-Associated Diseases (CECAD), University of Cologne, Cologne, Germany
- 5Mildred Scheel School of Oncology Aachen Bonn Cologne Düsseldorf (MSSO ABCD), Faculty of Medicine and University Hospital of Cologne, Cologne, Germany
- 6Max Planck Institute for Biology of Ageing, Cologne, Germany
Diffuse large B cell lymphoma (DLBCL) is a genetically highly heterogeneous disease. Yet, to date, the vast majority of patients receive standardized frontline chemo-immune-therapy consisting of an anthracycline backbone. Using these regimens, approximately 65% of patients can be cured, whereas the remaining 35% of patients will face relapsed or refractory disease, which, even in the era of CAR-T cells, is difficult to treat. To systematically tackle this high medical need, it is important to design, generate and deploy suitable in vivo model systems that capture disease biology, heterogeneity and drug response. Recently published, large comprehensive genomic characterization studies, which defined molecular sub-groups of DLBCL, provide an ideal framework for the generation of autochthonous mouse models, as well as an ideal benchmark for cell line-derived or patient-derived mouse models of DLBCL. Here we discuss the current state of the art in the field of mouse modelling of human DLBCL, with a particular focus on disease biology and genetically defined molecular vulnerabilities, as well as potential targeting strategies.
Introduction
Malignant lymphomas constitute a highly diverse spectrum of diseases that originate from various distinct stages of lymphocyte development. Collectively, they rank as the sixth most common cancer entity (1–3). Lymphomas are further divided into Hodgkin- (HL) and non-Hodgkin lymphomas (NHL) (1). Among the NHLs, cases derived from B- and T cells are distinguished (1). Additionally, lymphomas can further be subcategorized based on their clinical features, classifying them as either indolent or aggressive lymphomas (1). This review exclusively focusses on aggressive B-NHL and will only discuss indolent lymphomas when these entities provide critical information relevant for our understanding of the overall disease biology. We will also omit an in-depth discussion of Burkitt lymphoma models, to allow enough space for a more detailed review of diffuse large B cell lymphoma (DLBCL) models.
The majority of aggressive lymphomas originate from the germinal center (GC) - a highly specialized immunological structure - which transiently forms upon antigen encounter of a naïve B cell in the context of a T cell-dependent adaptive immune response. This specialized anatomic structure serves to facilitate B cell receptor (BCR) affinity maturation and class switch recombination (4). In line with their GC origin, Burkitt lymphomas (BL), follicular lymphomas (FL) and diffuse large B cell lymphomas (DLBCL) are GC-experienced, as evidenced by somatic hypermutation (SHM)-mediated mutagenesis within their BCR variable regions – an irreversible marker of GC passage (5–8). The functional output of the GC reaction is the development of a population of plasma cells capable of secreting high-affinity antibodies neutralizing the initially recognized antigen, as well as the production of memory B cells, which possess the ability to rapidly differentiate into antibody-secreting plasma cells upon antigen re-exposure.
While the generation of memory B cells and plasma cells is of critical importance for efficient control of infections and thus for survival, the GC reaction, due to its unique biological features, also poses the risk of lymphoma development. GCs are established upon antigen encounter and T cell co-stimulation through which B cells are activated via B cell receptor (BCR)-, CD40- and Toll-like receptor (TLR) signaling. Altogether, this signaling input triggers NFκB activation and the subsequent expression of genes mediating B cell activation and proliferation to drive GC initiation (9, 10). The central regulator of the GC response is the transcriptional repressor BCL6. Once GCs are established upon antigen encounter, BCL6 orchestrates the GC reaction through the repression of numerous genes involved in distinct biological processes, including apoptosis, DNA damage response and genome maintenance, cell cycle checkpoint signaling, as well as plasma cell differentiation (4, 11, 12).
Particularly the transient repression of DNA damage response programs in conjunction with the ability to override cell cycle checkpoints and the attenuation of the apoptotic cell death machinery, enable dark zone B cells to establish a hyper-proliferative program. This growth acceleration is associated with an extraordinarily high risk of accumulating oncogenic aberrations. These genomic lesions can manifest as single nucleotide variants resulting from off-target Activation Induced Cytidine Deaminase (AID)-mediated deamination events, as well as structural variations through erroneous AID-mediated class-switch recombination (3, 5, 13–17). GC B cells can undergo repeated transitions between the hyperproliferative dark zone compartment and the light zone, where they compete for antigen presented on follicular dendritic cells and T cell help provided by T-follicular helper cells (5, 12, 14). BCL6 further facilitates the retention of GC B cells within the GC reaction by preventing the induction of plasma cell differentiation programs (5, 12, 14). Only upon signal integration from the BCR, CD40, BAFF and TLRs can high-affinity GC B cells accumulate a robust NFkB signal, which drives IRF4 expression leading to BCL6 silencing and subsequent BLIMP1 expression, ultimately terminating the GC program and facilitating post-GC differentiation (10, 18, 19).
In summary, the GC reaction serves a critical purpose in diversifying the BCR repertoire and generating high-affinity antibodies to eradicate infectious agents. However, this immense potential for affinity maturation comes at the cost of a significant risk of accumulating lymphoma-promoting genomic aberrations. Hence, it is perhaps not surprising that the majority of mature B-NHLs originate from GC B cells, as evidenced by these lymphomas typically displaying genomic signatures indicative of GC experience, such as hypermutated immunoglobulin variable regions or the expression of a class-switched constant region. As diffuse large B cell lymphoma is the most common lymphoma, this review will primarily focus on this entity and the associated molecularly defined subtypes. As this topic has been reviewed previously (5, 14), we primarily focus on novel alleles that have recently emerged.
Given the extraordinary architectural and biological complexity, as well as the dynamic nature of the GC, it is perhaps not surprising that sufficient in vitro models of this lymphoid structure are still lacking. Activation and expansion of naïve B cells ex vivo can be achieved by stimulation of the Toll-like receptor pathway with LPS or CpG (20). Several combinations of CD40 and BCR stimulation by activating antibodies alone or together with IL-4 and/or IL-21 treatment result in B cell expansion and generation of cells with memory B or plasma cell phenotypes (20–22). Fibroblasts stably expressing BAFF and CD40L have been used for the in vitro generation of GC B cells that then differentiate into memory B or plasma cells (23). B and T cells isolated from genetically engineered mice expressing ovalbumin-specific B cell- and T cell receptors can be employed in vitro as a model system recapitulating phagocytic antigen uptake (23). While these systems are able to mimic B cell expansion and plasma cell/memory B cell differentiation to some extent, they fail to recapitulate the cellular complexity of the GC response with TFH cells, follicular dendritic cells and B cells as the main participants. Additionally, the compartmentalization of the GC into the dark and light zone is not well-represented in cell culture models, which generates a temporal and spatial profile of stimuli provided to the B cell. Lastly, different stimulus combinations (BCR, TLR, CD40, cytokines) prompt B cells to proliferate in vitro, but potentially differ from stimulus intensities in vivo. Thus, genetically engineered mouse models (GEMMs) of GC-derived lymphomas have played, and will likely continue to play, an important role in unraveling the intricate GC biology and lymphomagenesis. However, when conceptualizing and designing proper in vivo modeling experiments, it is of utmost importance to have a robust understanding of human disease biology, as our ultimate goal is to model the human scenario. Thus, prior to discussing lymphoma GEMMs, it is appropriate to briefly review the genomic complexity of human DLBCL.
Traditionally, DLBCL has been subdivided according to transcriptome-based clustering into germinal center B cell-like (GCB) and activated B cell-like (ABC) DLBCL (4, 24, 25). This classification, rooted in the cell of origin (COO), distinguishes subtypes with distinct biology, pathogenesis and clinical response to frontline chemo-immune therapy (4, 26, 27). In addition to transcriptome-based subtyping, two independent comprehensive genomic analyses recently provided a framework for a molecular subtyping algorithm for DLBCL (2, 3). The Dana-Farber group defined 5 DLBCL clusters (3). These clusters were defined by 1) BCL6 structural variants in combination with NOTCH2 aberrations (C1), 2) Bi-allelic TP53 inactivation (TP53 mutations and 17p copy number losses) in combination with haploinsufficiencies of 9p21.13/CDKN2A and 13q14.2/RB1 (C2), 3) BCL2 mutations with concordant BCL2 structural variants in combination with EZH2-, CREBBP- and KMT2D mutations and additional activating alterations of the PI3K pathway (C3), 4) mutations in linker and core histone genes in combination with aberrations in immune evasion molecules, NFkB and RAS/JAK/STAT signaling molecules (C4) and 5) 18q gains, likely affecting BCL2 and/or MALT1 in combination with MYD88- and CD79B mutations, as well as lesions that enforce a plasma cell differentiation block (aberrations in TBL1XR1, PRDM1 and SPIB1)(C5 DLBCL). Notably, subgroups associated with a particularly high risk of treatment failure include C2, C3 and C5. In a parallel approach, the NCI group employed a supervised approach, allowing the classification of ~50% of the cases into four genetically defined DLBCL subtypes with substantial overlap with these clusters (2). This approach enabled the clustering of lymphomas with co-occurring MYD88- and CD79B mutations (MCD), BCL6 rearrangements and NOTCH2 mutations (BN2), EZH2 mutations and BCL2 rearrangements (EZB), as well as NOTCH1 mutations (N1) (2). More recent work of the same group extended this classification system by two additional clusters, defined by aneuploidy together with TP53 inactivating mutations (A53), as well as mutations in SGK1 cooccurring with TET2 lesions (ST2) (28).
While anthracycline-based first-line chemo-immune therapy regimens achieve cure rates of approximately 65% in patients with DLBCL, relapsed or refractory disease remains a major clinical challenge. Until recently, the standard of care in second-line was confined to either intensive salvage therapy followed by high-dose consolidation and autologous stem cell rescue, or a number of palliative regimens for patients deemed ineligible for intensive consolidation. However, chimeric antigen receptor (CAR) T cells are currently revolutionizing the therapeutic landscape in r/r DLBCL, and anti-CD19 CAR-T cells are now firmly established in second and third-line treatment algorithms, substantially reducing the need to deploy high-dose chemotherapy regimens (29). While these products appear to be curative in approximately 35-40% of patients, representing a significant advancement in our treatment strategies, the majority of r/r DLBCL patients treated with CAR-T cells still do not experience long-term benefit. Thus, establishing pre-clinical models that faithfully recapitulate central aspects of DLBCL subtype-specific disease biology in vivo, to serve as experimental platforms for the conceptual development of novel therapeutic approaches, remains a major goal in translational lymphoma research.
Cre alleles with relevance to DLBCL modeling
Throughout the recent years, numerous highly relevant mouse models of aggressive lymphoma have been generated, to mimic critical features of the human disease with ever increasing precision. This development has been fueled in large parts by our expanding knowledge of recurrent genomic aberrations in human lymphomas, as well as an understanding of the molecular dialogue between lymphoma cells and cellular components of their microenvironment, as well as immune surveillance mechanisms. In parallel to this enhanced understanding of human disease biology, which serves as a guiding principle in GEMM design, technology developments in gene targeting and the availability of multiple highly specific recombinase tools have further catalyzed the development of mouse models that faithfully mimic the human disease. These genome editing tools include the Cre/loxP, Flp/FRT and Dre/Rox recombinase systems and the integrase system PhiC31/attB/attP (30). Inducibility of these recombinase systems can be achieved by fusing the recombinases to a modified estrogen receptor (ER) ligand binding domain, which leads to cytoplasmic retention of the fusion protein in the absence of the tamoxifen. Only upon tamoxifen binding, the recombinase fusion protein enters the nucleus to mediate DNA recombination. Inducibility can further be achieved through recombinase fusions to the progesterone receptor, which allows activation via the progesterone analog RU-486, or through dihydrofolate reductase (DHFR) fusions, which enable trimethoprim-induced stabilization of the recombinase fusion protein (31–33). Lastly, photoactivatable recombinases have been developed, recently (31). Altogether, these tools allow researchers to mimic inducible multistep tumorigenesis through the use of dual or triple recombinase strategies. The recent development of CRISPR/Cas9 or dCas9 technology, which is also available in a conditional fashion, for instance through a Rosa26Lox-STOP-Lox.Cas9-P2A-EGFP allele (34), has further increased our ability to precisely edit the murine genome.
In the context of B-NHL modeling, a number of Cre alleles have been developed. These include Cd19Cre, where the Cre expression cassette was targeted into exon 2 of the endogenous Cd19 allele (35). Similarly, to create an inducible Cre allele, an ERT2-fused Cre cDNA was targeted into exon 2 of the Cd19 locus, yielding a Cd19CreERT2 allele (36). The Mb1Cre allele was generated by replacing exons 2 and 3 of the Cd79a gene with a codon-optimized Cre (37). The ATG Start codon of Cd79a exon 1 was deleted, leading to abolished endogenous Cd79a expression (37). Both, Mb1Cre and Cd19Cre are expressed at early B cell developmental stages, enabling Cre-mediated genome editing throughout B cell development. In contrast, the bacterial artificial chromosome (BAC) transgenic Cd21Cre allele is expressed when transitional B cells differentiate into mature long-lived peripheral B cells (38). In addition to these alleles, the Cγ1Cre and AicdaCre alleles allow recombination at the (pre-)germinal center stage of B cell development. The Cγ1Cre allele was generated by introducing Cre cDNA preceded by an internal ribosomal entry site into the 3’ region of the Cγ1 locus upstream of an internal polyadenylation site, essentially leading to co-expression of the Cγ1 germline transcript and Cre from the endogenous Cγ1 locus (39). While CSR has been regarded a hallmark of the germinal center process, recent data suggests that germline transcript expression peaks already prior to GC entry in a pre-GC B cell population that is primed for CSR before entering the germinal center (40, 41). Expression of Aicda has been detected 12h after the induction of germline transcript expression, 24 hours before the appearance of GC B cells and peaks during the GC reaction (41). The AicdaCre allele was generated by targeting Cre cDNA into exon 1 of the Aicda locus (42). Next to a constitutive AicdaCre allele, an AicdaCreERT2 allele was generated by targeting exon 2 of the Aicda locus leading the expression of an ERT2-fused Cre recombinase, which also contains the first four N-terminal amino acids of the AID protein (43).
Autochthonous mouse models of diffuse large B cell lymphoma
When generating mouse models of aggressive lymphoma, it is crucial to precisely phrase the question these models should address. Typically, the goal of generating mouse models is for these tools to serve as preclinical avatars of the human disease, which can be deployed to either understand the biology of certain genomic aberrations, or to serve as model systems for preclinical drug development. Particularly in the latter context, it is critical that the genomic context in which individual genetic aberrations are induced in the murine genome mimics the situation observed in human neoplastic lesions. In the subsequent paragraphs, we will discuss murine alleles with relevance to human lymphomagenesis and will describe allele combinations that led to the development of valuable preclinical models (Figure 1 and Table 1). We will organize our discussion according to the clustering of mutations that occurs in human DLBCL (2, 3, 28).
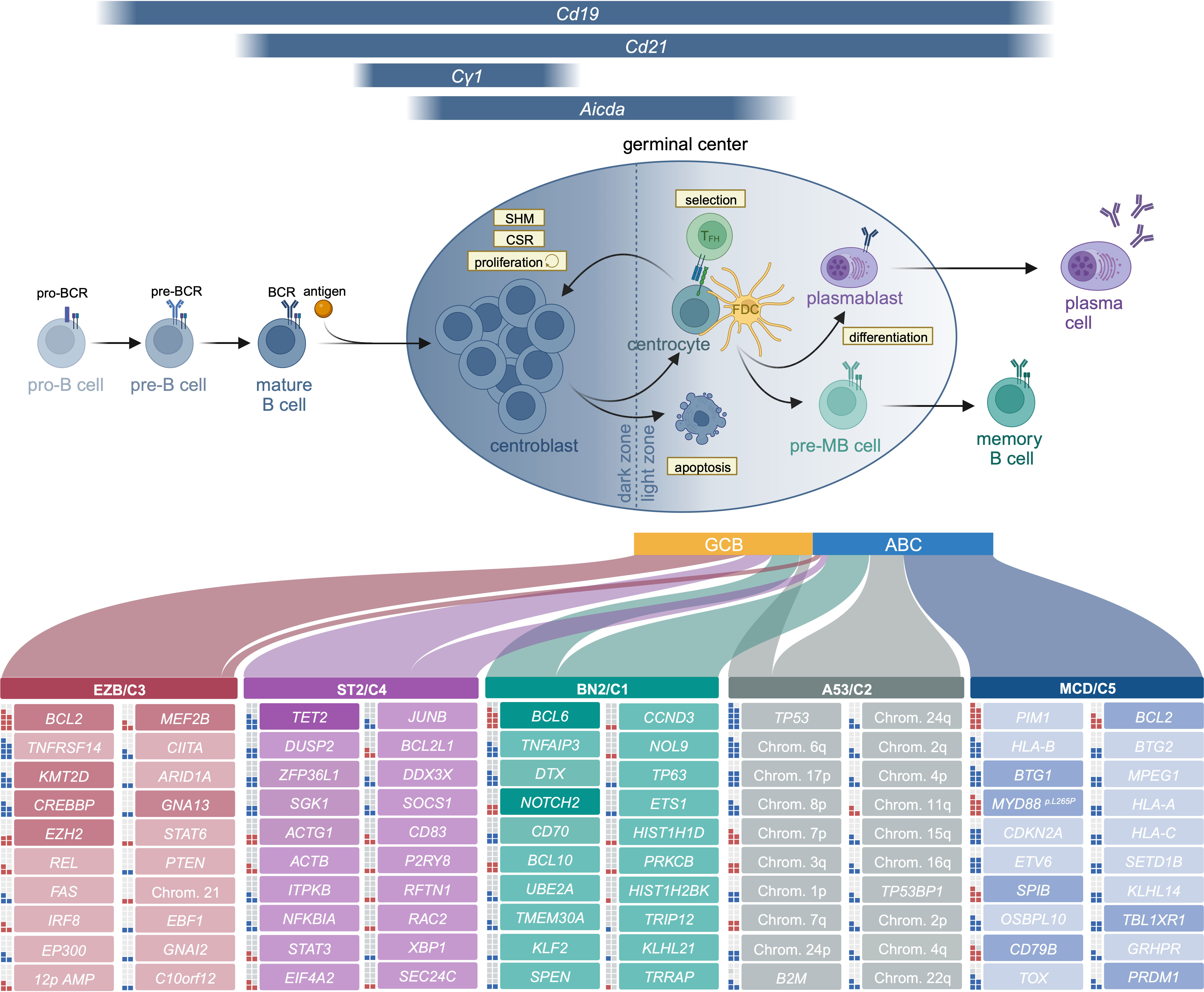
Figure 1 B cells develop in the bone marrow, where they pass through the pro- and pre-B cell stages before they enter the periphery as antigen-naïve mature B cells. Binding of antigen results in the activation of the B cell and its migration to the follicle, where the germinal center structure is formed. This structure is subdivided into two areas, the dark and the light zone. Proliferation predominantly occurs in the dark zone. Moreover, AID-driven somatic hypermutation (SHM), resulting in B cell receptor diversification, is also largely confined to the dark zone. B cells migrate to the light zone, where they compete for antigen presented by follicular dendritic cells (FDC) and CD40 stimulation provided by T follicular helper cells (TFH). Signals from T helper cells, either at the pre-GC stage or in the GC light zone promote class switch recombination (CSR). While high-affinity B cells are provided with those signals, prompting them to either recycle to the dark zone or differentiate into the memory B or plasma cell lineage, low-affinity B cells do not receive pro-survival stimuli and undergo apoptosis. While GCB DLBCL cells show features of light zone germinal center B cells, the ABC subtype more closely resembles plasmablast- or pre-MB stages. The frequency of ABC and GCB DLBCL within each genetic cluster is represented by the width of the respective link (unclassified cases are omitted for clarity of visualization but exist at different frequencies in all clusters: EZB, 9%; ST2, 22%; BN2, 42%; A53, 5%; MCD, 1%). The top 20 mutated genes for each genetic cluster are listed. Genes that have been investigated in a mouse model in the context of lymphoma are shaded with a higher color intensity. Squares to the left of each gene represent the frequency of alteration within the respective cluster (each square equals 10%, putative oncogenes are marked in red, tumor suppressors in blue). Frequencies of mutations, genetic clusters and transcriptomic subtypes are extracted from (28).
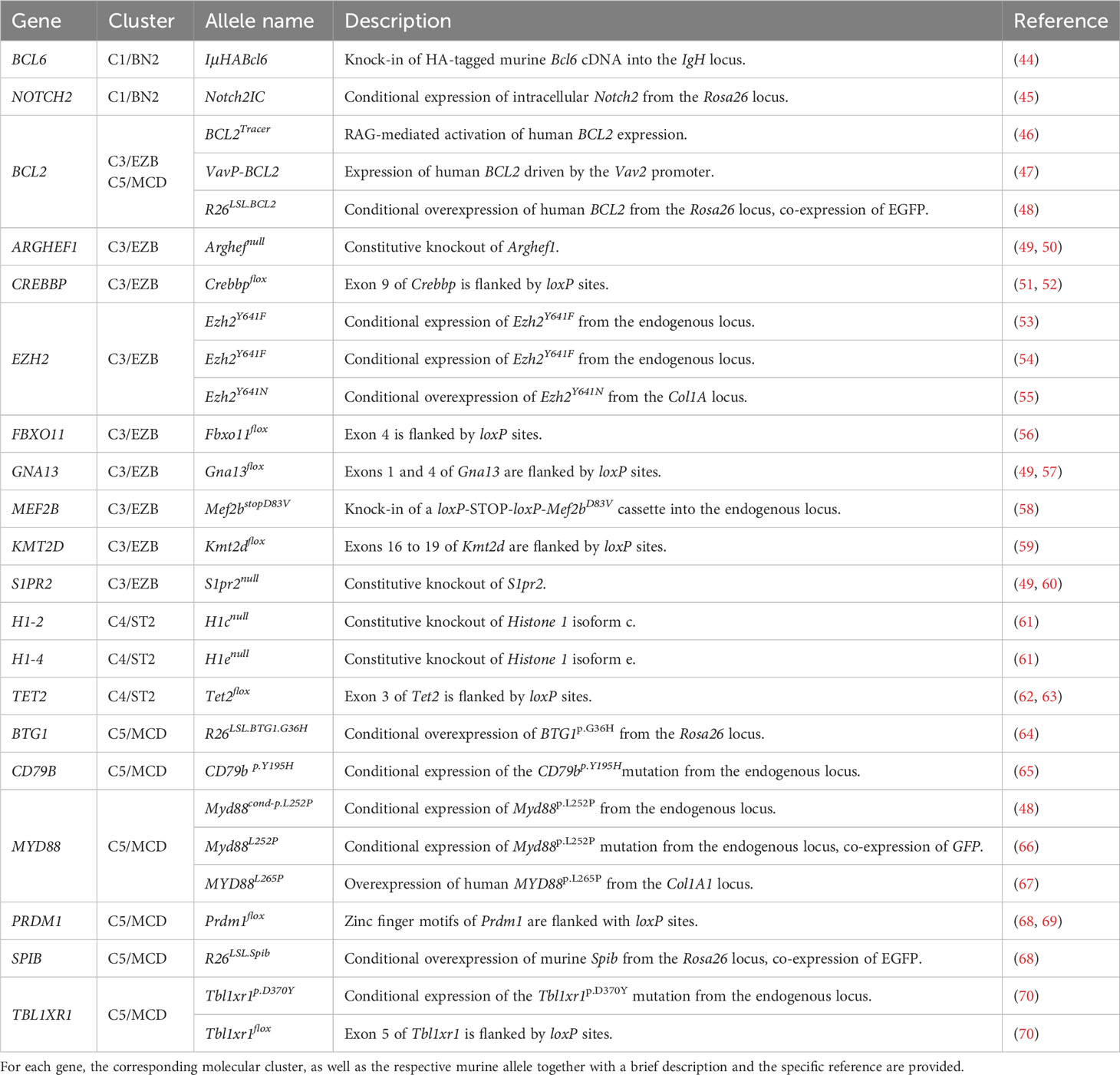
Table 1 Overview of genes that are recurrently altered in human DLBCL.
Cluster 1/BN2 modeling
Structural variants involving BCL6 are the hallmark feature of C1/BN2 DLBCL (2, 3). BCL6 itself is considered the master regulator of the GC reaction, and its continued expression prevents GC exit and terminal differentiation (11, 44, 71–73). BCL6 expression is tightly controlled through various regulatory elements in the BCL6 promoter region, including an IRF4 binding site. IRF4, in turn, acts as a transcriptional repressor involved in silencing BCL6 expression to promote GC exit (19). In lymphoma, BCL6 expression is sustained through different mechanisms. These include indirect effects, such as mutational inactivation of FBXO11 (56, 74), which is involved in mediating proteasomal degradation of BCL6, as well as mutations in CREBBP and MEF2B, which lead to enhanced and maintained BCL6 transcription (75, 76). Additionally, genetic alterations directly affecting the BCL6 gene, either though mutations within the 5’ non-coding region or via rearrangements and subsequent promoter substitution (19, 77–80). Collectively, these aberrations prevent GC exit by maintaining the BCL6-driven GC program, ultimately retaining B cells in a hyperproliferative state. In this state, BCL6-mediated blunting of the DNA damage response, for instance through repression of TP53, ATR, CHK1, CDKN1A, and others, facilitates genomic instability (4, 81–88).
In an attempt to model the human DLBCL- and FL-associated t (3, 14) (q27;q32) rearrangement, a knockin model was generated, where HA-tagged Bcl6 was targeted into the murine Igh locus, placing it under the control of the Iµ promoter (44). These IµHA.Bcl6/wt animals displayed spontaneous GC hyperplasia, with a notable predominance of dark zone over light zone GC B cells. Moreover, these mice developed spontaneous lymphomas, albeit with a long latency of >15 months (44). These lesions closely resembled central aspects of human DLBCL with respect to morphology, presence of AID-mediated somatic hypermutation, as well as non-random clonal numerical and structural cytogenetic abnormalities. These abnormalities included Myc-Igh rearrangements, in the vast majority of cases (44). It is worth mentioning that conditional alleles of a number of genes that are frequently altered in BCL6-rearranged human DLBCL, such as Tnfaip3, Notch2 and Spen, are readily available. Exploring the possibility of creating compound mutant models to investigate a potential oncogenic cooperation between these genes could be of great interest (3, 45, 89, 90).
Cluster 2/A53 modeling
Cluster 2 DLBCLs are characterized by the prevalence of bi-allelic TP53-inactivating mutations and 17p copy number losses, which frequently co-occur with copy number losses affecting 9p21.13/CDKN2A and 13q14.2/RB1, as well as 1q23.3/MCL1 copy number gains (3). Up to this point, no systematic cluster 2 DLBCL modeling has been reported. However, there are various Trp53 alleles, including conditional knockout alleles, as well as alleles that permit the conditional expression of mutant versions of Trp53 (91–93). Similarly, conditional Rb1 knockout alleles and conditional Mcl1 overexpression alleles are available, which would in principle allow cluster 2 DLBCL modeling attempts (94–97).
Cluster 3/EZB DLBCL modeling
In human DLBCL, cluster 3/EZB cases are dominated by GCB-DLBCL cases with respect to cell of origin classification (3). Molecularly, cluster 3 DLBCL is dominated by co-occurring BCL2 mutations and rearrangements that position BCL2 downstream of the IGH enhancer (3). In addition to these BCL2 aberrations, cluster 3/EZB harbors recurrent mutations in genes encoding the epigenetic regulators KMT2D, EZH2 and CREBBP, the transcription factor MEF2B and the Guanine Nucleotide-Binding Protein Subunit Alpha-13 (GNA13), as well as the F-box protein 11 (FBXO11), among others (2, 3, 28). Numerous alleles capturing these genes have been generated and we will discuss them in the following paragraphs.
The t(14;18) translocation can be detected in 85% of follicular lymphoma (FL) and 30% of GCB-DLBCL. This rearrangement, which places BCL2 under the transcriptional control of the IGH regulatory elements, occurs early during B cell development, as a result of erroneous VDJ recombination. Intriguingly, this translocation, which clearly represents a selected event during B cell transformation, by itself does not have substantial transformative capacity, as the t(14;18) is detectable in approximately 70% of healthy adults who never develop FL or GCB-DLBCL. Several distinct alleles have been generated to model the t(14;18) translocation in vivo (46, 47, 98–100). For the purpose of this review, we will focus on the three most commonly used alleles in B-NHL modeling.
The VavP-BCL2 allele was generated by juxtaposing the human BCL2 cDNA to the Vav2 promoter, which drives pan-hematopoietic expression of BCL2 at a developmental stage earlier than the time at which the t(14;18) translocation typically occurs (47, 101). Despite the non-B cell-restricted BCL2 expression in the VavP-BCL2 model, these mice develop clonal, somatically Ighv-mutated B cell lymphomas, which display a follicular growth pattern, as well as PNA and BCL6 expression and lack of post-GC markers (47). VavP-BCL2 mice were crossed with a number of relevant alleles, including Crebbpfl, Kmt2dfl, Ezh2Y641N and Ezh2Y641F to model aggressive B-NHL in vivo (see details below) (51, 102, 103). In addition, VavP-BCL2-derived hematopoietic stem cells were used as a platform to assess the effects of additional gain and loss of function genetic aberrations introduced by viral transduction prior to transplantation into irradiated recipient animals (55).
One of the major drawbacks of the VavP-BCL2 model is the ubiquitous BCL2 expression in the hematopoietic lineage. This limitation is circumvented by the introduction of the BCL2-Ig model, which was generated by classical transgenesis and expresses a BCL2 minigene under the transcriptional control of IG regulatory elements (98). This strategy limits ectopic BCL2 expression to the B cell lineage. When these transgenic animals were immunized with sheep red blood cells, they developed follicular lymphoma (40%) and plasmablastic lymphoma (20%) with a latency of more than 500 days (58).
The development of the mosaic BCL2Tracer transgenic model, in which RAG-mediated VDJ recombination flips exon 3 of a human BCL2 sequence from inverse into direct orientation allowing correct splicing and subsequent expression of BCL2 under the transcriptional control of a constitutive CMV promoter and 3’-located IGH intronic enhancer (Eµ), constitutes a further step in modeling the t(14; 18) (46). This design offers two critical advantages, as it faithfully mimics the sporadic nature of the t(14; 18) rearrangement, as well as the RAG recombinase-mediated occurrence of the recombination event at the pro-/pre- B cell developmental stage (46). It is important to note, however, that the BCL2Tracer mice did not develop spontaneous lymphomas during the reported 9 months observation period (46).
Next to BCL2 aberrations, cluster 3 DLBCLs are characterized by recurrent mutations in the epigenetic modifiers KMT2D, EZH2 and CREBBP. KMT2D constitutes the catalytic activity of the complex of proteins associated with Set1 (COMPASS), which regulates transcriptional activity through mediating mono- and di-methylation of histone 3 lysine 4 (H3K4) at enhancer and super-enhancer regions within the chromatin. In human DLBCL and FL, KMT2D aberrations are typically loss of function events, either leading to truncations or deleterious missense mutations within the SET domain, indicating that KMT2D acts as a tumor suppressor gene. A Kmt2dfl allele is available (59) and when Kmt2d was deleted during early B cell development by Cd19Cre, the majority of Cd19Cre/wt;Kmt2dfl/fl mice died from lymphoma within 338 days. The developing oligoclonal lymphomas were of pre-GC origin and had undergone VDJ recombination. However, these lymphomas displayed no signs of class switch recombination or somatic hypermutation and lacked expression of the murine GC B cell marker PNA (104). Interestingly, when crossed with Cγ1Cre, Kmt2dfl animals did not develop lymphoma within an observation period of 18 months (103). However, VavP-BCL2;Cγ1Cre/wt;Kmt2dfl/fl mice developed significantly more clonal B cell lymphomas than VavP-BCL2 controls (103). Morphologically, these lymphomas covered the entire spectrum from low to high-grade FL and DLBCL (103). These clonal lesions stained positive for B220, PAX5 and BCL6, displayed evidence of somatic hypermutation and were thus likely GC-experienced (103). These data indicate that GC-specific loss of Kmt2d cooperates with BCL2 in GC B cell-derived FL and DLBCL lymphomagenesis.
As loss of KMT2D biochemically results in decreased (H3K4) methylation and subsequently altered gene expression, it was proposed that pharmacological inhibition of the KDM5 family, which mediates H3K4me3/me2 demethylation, might, at least partially, restore H3K4 methylation in KMT2D-deficient cells, leading to the reinstated expression of KMT2D-controlled genes. Indeed, KDM5 inhibition using a series of α-ketoglutarate-competitive small molecule inhibitors, promoted increased H3K4me3 levels and had a growth-repressing effect in KMT2D-defective cell lines and xenograft lymphoma models (105). Thus, KDM5 inhibition might be a viable therapeutic strategy for the treatment of KMT2D-mutant GC-derived lymphomas. However, high intracellular α-ketoglutarate concentrations complicate the use of α-ketoglutarate-competitive small molecule inhibitors. Hence, the development of potent KDM5 PROTAC degraders is highly desirable to further develop the concept of KDM5 repression for the treatment of KMT2D-mutant lymphomas.
The methyltransferase EZH2 is part of the Polycomb Repressive Complex 2 (PRC2) and catalyzes the deposition of repressive H3K27me3 marks at cell type and context-specific chromatin regions, which in GC B cells include genes that control proliferation and cell checkpoint signaling, including Cdkn1a and Cdkn1b, as well as genes involved in driving terminal plasma cell differentiation, such as Prdm1 and Irf4 (55, 106, 107). Approximately 30% of human GCB-DLBCLs and particularly cluster 3/EZB cases harbor heterozygous EZH2 mutations, typically located within the EZH2 SET domain coding region affecting p.Y641 (2, 3, 28). These SET domain mutations appear to convey an enhanced trimethylation efficiency to mutant EZH2 (108).
Several Ezh2 alleles have been generated and their role in B cell biology and lymphomagenesis has been investigated exhaustively. Important evidence for a critical role of Ezh2 in GC B cell biology and GC formation was provided by the assessment of Cγ1Cre;Ezh2fl/fl mice, which displayed massively impaired GC B cell expansion following sheep red blood cell (SRBC) immunization-mediated GC induction. A similar lack of GC B cell expansion upon SRBC immunization was observed when C57BL/6 wildtype animals were treated with the EZH2 inhibitor GSK-503, firmly establishing a central role for EZH2 in establishing GCs (55).
To gain mechanistic insight into the biological function of the EZH2 hotspot mutations observed in human DLBCL, two additional alleles were generated, namely an Ezh2Y641F and Ezh2Y641N strain. Both alleles are conditionally Cre-inducible. Expression of the Ezh2Y641F allele is driven by the endogenous Ezh2 promoter and expression of the Ezh2Y641N allele is driven off a CAG promoter (53, 55). When crossed with Cγ1Cre, these animals did not develop overt lymphoma. However, further analyses revealed that following immunization, both Cγ1Cre;Ezh2Y641F/wt and Cγ1Cre;Ezh2Y641N/wt mice displayed GC hyperplasia and enhanced H3K27 trimethylation and subsequent silencing of EZH2 target genes, which appears to be dependent on BCL6 to ultimately form the CBX8- BCOR repressive complex (53, 55). Of note, reminiscent of the conditional Kmt2d knockout mice, when Ezh2Y641F mice were crossed with Cd19Cre animals in which recombination occurs at early B cell developmental stages, the resulting Cd19Cre/wt;Ezh2Y641F/wt mice develop DLBCL within 12 months (54). Lymphoma development in this setting was further enhanced when Trp53 was co-deleted in Cd19Cre/wt;Ezh2Y641F/wt;Trp53fl/fl mice (54).
Next to an oncogenic cooperation between Ezh2 and Bcl6 (53), HSC transplant experiments revealed an oncogenic cooperation between BCL2 and EZH2 Y641N/F 81,83. In brief, VavP-BCL2 HSCs were transduced with either empty vector, Ezh2wt or Ezh2Y641F constructs and subsequently transplanted into lethally irradiated recipient mice. These recipients were then immunized with SRBCs (monthly). Whereas the majority of VavP-BCL2;Ezh2Y641F and 20% of VavP-BCL2;Ezh2wt bone marrow chimeras developed lymphoma displaying morphological features of DLBCL with hepatosplenomegaly on day 111, none of the VavP-BCL2 chimeras displayed overt lymphoma manifestation at that stage (55). Of note, results obtained in an autochthonous setting do not support these observations made in the transplant setting. Here, Cγ1Cre;VavP-BCL2;Ezh2Y641F/wt and Cγ1Cre;VavP-BCL2;Ezh2Y641N/wt mice did not succumb to lymphoma significantly earlier than VavP-BCL2 controls (102). VavP-BCL2, Cγ1Cre;VavP-BCL2;Ezh2Y641F/wt and Cγ1Cre;VavP-BCL2;Ezh2Y641N/wt did, however, die significantly earlier than Cγ1Cre, Cγ1Cre;Ezh2Y641F/wt and Cγ1Cre;Ezh2Y641N/wt controls (102). It is important to note that these results are likely confounded by the development of glomerulonephritis and other autoimmune diseases which occur in VavP-BCL2 mice with almost 100% penetrance, resulting in a median survival of around 6 to 9 months, representing a limiting factor for employing this allele in the study of GC B cell lymphomagenesis (47, 55, 61, 64, 109).
Next to the above-described role of EZH2 gain-of-function mutations in B-NHL lymphomagenesis and chromatin remodeling, EZH2 also emerges as a potential drug target. By using the above-mentioned HSC transplant experiments, as well as the autochthonous Cγ1Cre;VavP-BCL2;Ezh2Y641F/wt and Cγ1Cre;VavP-BCL2;Ezh2Y641N/wt models, it was shown that expression of the MHCI and MHCII antigen presentation machinery was substantially reduced in Ezh2-mutant lymphoma cells, compared to wildtype controls (102). Coincidingly, Ezh2-mutant lymphomas were characterized by a drastically reduced T cell infiltration of the local lymphoma microenvironment (102). These observations suggest that mutationally enhanced EZH2 H3K27me3 activity promotes escape from lymphoma-suppressing immune surveillance through the repression of lymphoma cell autonomous MHCI/II expression. From a therapeutic point of view, it is interesting to note that pharmacological inhibition of mutant EZH2 activity with EPZ-6438 reduced H3K27me3 in DLBCL cell lines (102). Moreover, EPZ-6438 exposure significantly increased surface MHCI/II surface expression in the majority of the investigated EZH2-mutant GCB-DLBCL cell lines, whereas EZH2 wildtype cell lines did not display substantial changes in MHCI/II expression (102).
The KAT3 family histone and non-histone acetyl-transferase CREBBP is frequently affected in FL and DLBCL, either by truncating or missense mutations affecting the catalytic HAT domain (2, 3, 28, 75). In DLBCL, CREBBP mutations are enriched in C3 DLBCL (2, 3, 28). Biochemically, CREBBP, together with its paralog EP300, exerts transcriptional control through H3K18 and H3K27 enhancer and promoter acetylation. In a series of experiments using human lymphoma cell lines, RNAi-mediated depletion of Crebbp on a VavP-BCL2 background and GC-specific conditional Crebbp knockout, two independent groups recently demonstrated that CREBBP loss promotes the development of GC-derived lymphomas in vivo (51, 109). Mechanistically, CREBBP loss-of-function led to substantially reduced H3K27 acetylation at enhancers and super-enhancers, leading to subsequent transcriptional repression of a series of genes involved in the BCR-, TLR- and CD40 receptor signaling cascades, genome maintenance and cell cycle regulation, as well as genes regulating GC and plasma cell development and antigen presentation, including the MHCII complex (51, 109). A further inspection of the affected genes revealed that CREBBP-regulated enhancers are largely overlapping targets of the BCL6/SMRT/HDAC3 complexes (109). The role of HDAC3 was confirmed using GC B cells derived from Hdac3-deficient mice (109). In these experiments, Hdac3 deficiency rescued repression of the Crebbp/BCL6-regulated transcripts and suppressed Crebbp-mutant lymphomas in vitro and in vivo (109). Particularly this HDAC3 involvement is of potential therapeutic interest, as subtype-specific HDAC inhibitors are currently being developed and could potentially be used to treat CREBBP-mutant lymphomas (110–113). From a modeling point of view, it is important to note that homo- or heterozygous Crebbp-deficiency (in Cd19Cre/wt;Crebbpfl/fl or Cd19Cre/wt;Crebbpfl/wt and Cγ1Cre;Crebbpfl/fl or Cγ1Cre;Crebbpfl/wt mice) did not lead to significant lymphoma (FL/DLBCL) development (51). However, Cγ1Cre;Crebbpfl/wt;VavP-BCL2 animals displayed a significant increase in lymphoma incidence, compared to Cγ1Cre;Crebbpfl/wt controls (51). These lesions mimicked critical aspects of human FL, including clonal GC B cell origin indicated by BCL6 expression, as well as lack of IRF4 and CD138 positivity and the presence of clonal Ig rearrangements carrying mutations as evidence of somatic hypermutation (51). There were no significant survival differences between Cγ1Cre;Crebbpfl/wt;VavP-BCL2 and Cγ1Cre;Crebbpwt/wt;VavP-BCL2 animals, which is likely due to the development of glomerulonephritis and other autoimmune diseases that develop in VavP-BCL2 mice with almost 100% penetrance (47).
Next to genomic aberrations affecting epigenetic modifiers, such as EZH2, KMT2D, and CREBBP, transcription in is also rewired in GC-derived lymphomas via mutations in the gene encoding the transcription factor MEF2B. In B cells, MEF2B expression is restricted to GC B cells, where it controls the expression of a plethora of genes, including BCL6 (58, 76). Approx. 15% of DLBCL cases display protein-damaging MEF2B mutations (2, 3, 28), affecting either the N-terminal DNA-binding domain or the C-terminal protein tails, which has been shown to be the target of different post-transcriptional modifications, including phosphorylation and sumoylation (5, 76). Particularly the effect of the MEF2B p.D83V mutation was investigated in more detail (58). These experiments revealed that MEF2Bwt-driven transcription of a luciferase reporter was readily abrogated when either HDAC5, HDAC7, or CABIN1 (which is part of the HUCA complex consisting of HIRA, UBN1, CABIN1, and ASF1A) were co-expressed (58). In marked contrast, MEF2BD83V-mediated reporter expression was unaffected by HDAC5, HDAC7, or CABIN1 co-expression, indicating that this mutation mediates escape from negative regulation (58). These data were further corroborated through the generation of a Cre-inducible Mef2bD83V allele in which a loxP-STOP-loxP (LSL) cassette was inserted into the endogenous locus followed by an exon 3 harboring the p.D83V mutation (58). When these animals were crossed with Cd21Cre, the resulting Cd21Cre/wt;Mef2bD83V/wt mice displayed an increased abundance of GC B cells, as well as GC hyperplasia, compared to Mef2b wildtype controls (58). Moreover, approximately 20% of Cd21Cre/wt;Mef2bD83V/wt mice developed lymphomas (FL and DLBCL) at late time points. In a further extension of these experiments, Cd21Cre/wt;Mef2bD83V/wt;BCL2-Ig mice were generated (58). More than 90% of these animals developed GC-experienced, somatically hypermutated clonal lymphomas, in which the malignant lesions morphologically and phenotypically mimicked human FL and DLBCL (58).
While C3 DLBCL is clearly enriched for mutations in genes encoding epigenetic modifiers, transcription factors and BCL2, two additional genes with different functions are also recurrently altered in C3 DLBCL and will be discussed briefly. These are GNA13 and FBXO11. The guanine nucleotide binding protein Gα13 (encoded by GNA13) serves as a signaling molecule downstream of the G-protein-coupled receptor sphingosine-1 phosphatase receptor-2 (S1PR2) and relays signals mediating physical confinement of B cells in the GC through repression of migration and negative regulation of the AKT pathway (5, 14, 114). The Gα13 pathway is affected by inactivating mutations in approximately 20% of GCB-DLBCL, including lesions in GNA13, S1PR2, ARHGEF1 and P2RY8 (2, 3, 5, 14, 49). Moreover, S1PR2 was recently shown to act as a tumor suppressor in ABC-DLBCL and S1PR2 expression was prognostic in that setting (115). The biology of Gα13 in lymphoma was recently further dissected through the generation of two distinct B cell-specific loss-of-function models, namely a GC B cell-specific deletion in the AicdaCre;Gna13fl/fl setting (116) and a pan-B cell disruption of Gα13 expression in the Mb1Cre/wt;Gna13fl/fl setting (49). In both scenarios, animals displayed an increased abundance of GC B cells, disrupted GC anatomy with defective spatial distribution of dark zone and light zone B cells, irregular B cell migratory phenotypes and an increased somatic hypermutation activity. It is important to note that S1pr2 deficiency does not fully phenocopy Gna13 loss. While Gna13- and Arhgef1 deficiency lead to leukemic effusion of GC B cells into the lymph- and blood stream, S1pr2-deficiency does not (49). This incomplete phenocopy fueled the hypothesis that additional Gα13-coupled G-protein-coupled receptors might regulate GC confinement. Indeed, in human DLBCL and Burkitt’s lymphoma sequencing data, the orphan receptor P2RY8 was identified as recurrently mutated. And while there is no murine orthologue of human P2RY8, overexpression of P2RY8 repressed GC B cell proliferation in murine Peyer’s patches and mesenteric lymph nodes, reminiscent of what could be observed with S1pr2 overexpression. Importantly, P2RY8-mediated GC growth suppression required the presence of Gα13 and was undetectable in Gna13-deficient settings, indicating that multiple G-protein-coupled receptors signal via Gα13 to confine GC B cells to the GC. Moreover, S1pr2 constitutive knockout mice spontaneously develop clonal GC-derived DLBCL-like lymphomas displaying increased AID activity with approximately 50% penetrance, lending further support to the hypothesis that the Gα13 pathway is critically involved in lymphomagenesis (117).
FBXO11 belongs to the family of F-box proteins (118). The F-box is a protein domain consisting of approximately 40 amino acids, which primarily functions as a protein:protein interaction domain (118). It is through this F-box domain that F-box proteins interact with other components of SCF ubiquitin ligase complexes, which consist of SKP1, CUL1 and an F-box protein, such as FBXO11 (118). SCF ubiquitin ligase complexes deploy F-box proteins for the purpose of specific substrate recognition to ultimately drive the proteasomal degradation of a selected set of target proteins (119). F-box proteins are further subdivided according to the presence of additional protein domains into an FBXW family (harboring a WD40 domain), and FBXL family (containing a Leucine-rich repeat) and an FBXO family (F-box only or F-box and other domains) (119). It is through these non-F-box domains that F-box proteins confer substrate specificity to the SCF complex (119).
FBXO11-containing SCF complexes mediate the ubiquitinylation and subsequent proteasomal degradation of a number of lymphomagenesis-relevant substrates, including PRDM1 and BCL6 (5, 74, 119, 120). FBXO11 is affected by deleterious genomic aberrations in approximately 5% of human DLBCL cases, particularly in C3 DLBCL and these FBXO11 aberrations are associated with increased BCL6 expression levels (2, 3, 28, 74). The in vivo effects of Fbxo11 deletion in GC B cells was assessed in Cγ1Cre/wt;Fbxo11fl/fl and Cγ1Cre/wt;Fbxo11fl/wt mice, in which exon 4 of the Fbxo11 gene was flanked by loxP sites (56). Analyses in these animals revealed that upon immunization Fbxo11 deficiency in GC B cells leads to an increased number of GC B cells, GC hyperplasia, a shift within the GC B cell compartment towards an increased percentage of dark zone cells, compared to light zone B cells, as well as increased BCL6 expression as a function of Fbxo11 gene dose (56). The role of Fbxo11 as a tumor suppressor in lymphomagenesis was formally established in Cγ1Cre/wt;Fbxo11fl/fl and Cγ1Cre/wt;Fbxo11fl/wt mice that received 6 immunizations with SRBCs over the course of their lifespan (56). In these experiments, animals were euthanized at 17 to 18 months. At autopsy, 5% of wildtype controls, approximately 35% of Cγ1Cre/wt;Fbxo11fl/wt mice and approximately 20% of Cγ1Cre/wt;Fbxo11fl/fl animals developed various types of lymphoproliferation, including florid follicular hyperplasia, lymphoproliferative disease and DLBCL (56). Particularly in the Cγ1Cre/wt;Fbxo11fl/wt setting, DLBCL development was recorded (56).
While the above-detailed models cover a lot of ground in DLBCL modeling, it is important to note that a recent comprehensive genomic analysis revealed that human DLBCLs harbor a median of 17 (range 0-48) genetic drivers (3). A further analysis provided an additional dissection of human EZB cases into those that displayed the so-called “DHIT gene expression signature (DHIT+)” and those lacking this signature (28). The DHIT signature is derived from double hit lymphomas, which harbor combined structural variants affecting MYC and BCL2 (28). In this dataset, DHIT+ cases displayed a significantly worse overall survival, than DHIT- cases (28). Moreover, a significant co-clustering of MYC (46%) and TP53 (43%) aberrations was observed within the DHIT+ cases (28). In contrast, MYC and TP53 lesions were observed in DHIT- cases in only 4% and 15%, respectively (28). Mutations in other genes, such as GNA13, DDX3X and FOXO1 were also enriched in the DHIT+ cases, albeit less pronounced (28). These observations provide a framework for the design and generation of more advanced C3/EZB models, such as Cγ1Cre/wt;Ezh2Y641F/wt;Rosa26LSL.Bcl2/LSL.Myc;Tp53fl/fl.
Cluster 4/ST2 modelling
Cluster 4 DLBCL is dominated by highly recurrent mutations in genes encoding for the histone H1 isoforms H1B, C, D, and E. We note, however, that these H1 mutations do not exclusively occur in C4 DLBCL, but can also be detected in other clusters, such as the C5/MCD cluster. Mechanistically, these so-called linker-histones bind to nucleosomes to promote chromatin compaction, which essentially enables the folding of chromatin into higher-order structures, and subsequently allowing the further epigenetic regulation through the recruitment of additional histone modifiers (121). Lymphoma-associated histone H1 mutations, most commonly affecting H1C and H1E are typically missense mutations within the globular C-terminal domain, which impairs chromatin binding (61, 122, 123). The tumor-suppressive role of H1c and H1e was recently confirmed in vivo, using constitutive H1c-/-;H1e-/- animals (61). Deploying chromatin conformation capture analyses in sorted H1c-/-;H1e-/- and wildtype control GC B cells revealed a profound architectural remodeling of the genome characterized by numerous focal shifts in chromatin constitution from a compacted to a relaxed state (61). In these cells, chromatin decompaction was associated with increased histone H3K36 dimethylation, as well as reduced H3K27 trimethylation (61). A further analysis demonstrated that decompacted genes in H1c-/-;H1e-/- GC-B cells were enriched for iPS cell reprogramming, mesenchymal-transition states, stem cell transcription factor cistromes, as well as H2K27me3-marked genes in haematopoietic cells (61). In subsequent competitive bone marrow chimera experiments, it was shown that H1c-/-;H1e-/- GC-B cells displayed a competitive advantage within the GC, which was associated with an increased abundance of cycling light zone B cells (61). Fittingly, H1c-/-;H1e-/-;VavP-BCL2 animals showed a prominent disruption of lymph node architecture and infiltration of extranodal tissues, such as lung and liver with immunoblastic cells together with a prominent CD3-positive T cell infiltrate (61). Survival monitoring of different cohorts of experimental mice revealed that H1cwt/-;H1ewt/-;VavP-BCL2 and H1c-/-;H1e-/-;VavP-BCL2 animals displayed a significantly shorter overall survival than VavP-Bcl2 controls (61). In these experiments, a trend towards increased lethality of H1cwt/-;H1ewt/-;VavP-BCL2, compared to H1c-/-;H1e-/-;VavP-BCL2 animals was observed, possibly indicating that certain isoform specific functions, such as interactions with additional epigenetic modifiers, need to be retained for a full-blown competitive advantage of H1c and H1e-defective GC-B cells (61). This observation is also in line with the observation that H1 mutations typically occur in a heterozygous fashion in human DLBCL (2, 3, 61).
TET2 (ten-eleven translocation 2), the gene encoding the methylcytosine dioxygenase 2, which catalyzes the conversion of the modified genomic base 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC), is mutated in approximately 5-10% of human GCB-DLBCL. TET2 mutations are enriched in ST2 cluster DLBCL, which shares genomic features with C4 DLBCL (2, 3, 28, 124). TET2 mutations are associated with clonal hematopoiesis and consistently patients with TET2-mutant lymphoma typically carry the identical mutation in their hematopoietic stem cells (HSCs) (125). Whether these TET2 aberrations play a causal role in B cell lymphomagenesis in these patients or merely represent a clonal hematopoiesis scar was systematically assessed in experimental mice by conditional deletion in either HSCs (VavCre) or specifically in the B cell lineage (Cd19Cre) (62). In these models, Tet2 deficiency led to a stalled transit GC transit of B cells and subsequent preneoplastic GC hyperplasia, reduced class switch recombination and a block in terminal plasma cell differentiation (62). Of note, Tet2 disruption at the GC stage in Cg1Cre/wt;Tet2fl/flmice did not lead to an expansion of GC B cells or any other detectable shift in the relative abundance of different B cell populations, compared to Cg1Cre/wt;Tet2wt/wtanimals (62). Consistent with the impaired GC exit phenotype in Cd19Cre;Tet2fl/fland VavCre;Tet2fl/flmice, Tet2 deficiency was associated with reduced enhancer cytosine hydroxymethylation and reduced expression of genes that promote GC exit, such as Prdm1, in these settings (62). A further analysis revealed that there is substantial overlap of the enhancers and genes that are repressed in Tet2-deficient settings and CREBBP-mutant lymphomas, suggesting a similarly rewired transcriptome that is brought about by these distinct genomic lesions (62). Perhaps not surprisingly, TET2 and CREBBP mutations are mutually exclusive in human lymphomas (62).
Cluster 5/MCD modeling
Cluster 5/MCD DLBCL is dominated by ABC-DLBCL cases and is enriched for co-occurring mutations leading to activation of BCR signaling (CD79B) and the Toll-like receptor (TLR) pathway (MYD88, particularly through the highly recurrent p.L265P hotspot mutation) (2, 3, 28, 48, 65, 68, 126, 127). In addition, C5/MCD DLBCL cases almost uniformly harbor copy number gains on 18q, which, among others, includes the BCL2 locus (2, 3, 28). BCL2 gains and amplifications are a defining feature of MCD DLBCL (2, 28). C5/MCD cases are further enriched for aberrations leading to a block in plasma cell differentiation (SPIB gains and loss of function lesions affecting PRDM1 and TBL1XR1), as well as lesions mediating escape from immune surveillance (HLA-A) (2, 3, 28, 68, 128).
A number of relevant alleles mimicking recurrent aberrations in C5/MCD DLBCL have been generated in recent years and have enabled a precise modelling of this DLBCL subtype (48, 64, 65, 68–70, 127). A first step towards generation of a C5/MCD model was the development of a conditional Myd88p.L252P allele (Myd88c-p.L252P) that is expressed from the endogenous locus upon Cre-mediated recombination (48). In this model, murine Myd88p.L252P is at the orthologous position of human Myd88p.L265P 116. When this Myd88p.L252P allele was crossed with Cd19Cre, AicdaCre or Cd21Cre mice, all of the resulting animals developed splenomegaly with disrupted splenic architecture and non-clonal lymphoproliferative infiltration of the liver, as well as clonal DLBCL-like disease in a subset of cases (25%, 33% and 33%, respectively) (48).
In parallel, with the goal of mimicking BCL2 amplification in vivo, a novel conditional allele in which BCL2.IRES.GFP was targeted into the Rosa26 locus, was developed (48). This modeling strategy is rationalized by the observation that, in contrast to C3/EZB DLBCL where BCL2 is typically affected by structural variants, BCL2 is typically amplified in C5/MCD (2, 3, 28). In this model, expression of human BCL2 cDNA from the Rosa26 locus is prevented by the insertion of a loxP.STOP.loxP cassette upstream of the translation-initiating codon (48).
In a next step Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/wt mice were generated, which expired significantly earlier than Cd19Cre/wt;Myd88p.L252P/wt, Cd19Cre/wt;Rosa26BCL2.IRES.GFP/wt and Cd19Cre/wt controls (48). Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/wt animals almost uniformly succumbed to clonal lymphoma (penetrance of 83%), which displayed morphological features of DLBCL and stained positive for IRF4 and CD138, while being negative for B220 and BCL6, consistent with a plasmablastic, rather than a classical DLBCL phenotype (126).
A further refinement of the model was recently published, where the parental Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/wt model was modified to either harbor a conditional Prdm1 loss or conditional Spib overexpression from the Rosa26 locus, in order to install a robust plasma cell differentiation blockade in the resulting Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/wt;Prdm1fl/fl and Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/LSL.Spib.IRES.GFP mice (68). As expected these mice developed DLBCL-like lymphomas and displayed a significantly reduced overall survival, compared to the Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/wt parental strain (68). A more detailed molecular analysis using whole-exome sequencing, transcriptomics, flow-cytometry, as well as mass cytometry revealed that Prdm1- or Spib-altered lymphomas display molecular features of pre-memory and light-zone B cells, whereas lymphomas derived from the parental Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/wt strain were enriched for late light-zone and plasmablast-associated molecular features (48, 68, 126). Thus, in contrast to the parental strain, which rather represents a model of plasmablastic lymphoma, engineering of a B cell-specific Prdm1 deletion or Spib overexpression converts this model into a disease that faithfully resembles C5/MCD DLBCL (48, 68, 126).
In a further refinement of this model, a conditional allele that expresses the Cd79b p.Y195H mutation from the endogenous locus, mimicking the recurrent p.Y196H mutation within the CD79B immunoreceptor tyrosine-based activation motif (ITAM) found in human DLBCL cases, was generated (65). When four hallmark genetic aberrations in C5/MCD DLBCL were modelled in AicdaCre/wt;Myd88p.L252P/wt;Cd79bp.Y195H/wt;Rosa26LSL.BCL2.IRES.GFP/wt;Prdm1fl/fl mice, these animals displayed a drastically increased in GC size and number at 8 weeks of age, compared to controls (65). AicdaCre/wt;Myd88p.L252P/wt;Cd79bp.Y195H/wt;Rosa26LSL.BCL2.IRES.GFP/wt;Prdm1fl/fl animals developed splenomegaly and B cells in these animals displayed immune-phenotypes consistent with dark zone GC B cells and memory B cells (65). A further analysis, using bone marrow chimeras, revealed that 5 of 8 AicdaCre/wt;Myd88p.L252P/wt;Cd79bp.Y195H/wt;Rosa26LSL.BCL2.IRES.GFP/wt;Prdm1fl/fl animals displayed disrupted splenic architecture due to a proliferation of large atypical lymphoid cells that morphologically resembled DLBCL (65). 2 of 5 animals carrying these intra-splenic lymphomas also harbored retroperitoneal lesions involving lymph nodes or accessory splenic tissue (65). Unfortunately, there was no further analysis of lymphomagenesis in the autochthonous setting, preventing a direct comparison with previous versions of C5/MCD modeling approaches. At first glance, the use of the Cd19Cre allele, which drives recombination already at the pro-B cell stage of B cell development, in the Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/wt, Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/wt;Prdm1fl/fl and Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/LSL.Spib.IRES.GFP models could be perceived as not ideal. However, in this regard it is important to note that particularly the MYD88 p.L265P mutation is detectable at low allele frequency in CD34-positive hematopoietic stem cells and B cell precursor cells of patients with lymphoplasmacytic lymphoma/Waldenstrom´s macroglobulinemia (127). These data indicate that the MYD88 p.L265P mutation may arise early during B cell development and may require additional hits that are acquired during the GC reaction to facilitate full-blown transformation, similar to the t(14;18) rearrangements in FL, that are caused by the RAG recombinase, which is active in pre-B cells but not mature B cells. Moreover, the Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/wt, Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/wt;Prdm1fl/fl and Cd19Cre/wt;Myd88p.L252P/wt;Rosa26LSL.BCL2.IRES.GFP/LSL.Spib.IRES.GFP models display features of GC passage, including evidence of somatic hypermutation and class-switch recombination, which again suggests their GC origin (48, 68, 126). Against this background, a dual recombinase strategy enabling early Myd88 mutation and GC-specific mutation of the additional C5/MCD hallmark genes (such as Cd79b, Bcl2, Prdm1 and others) may be a desirable strategy for future modeling attempts.
An additional gene that is frequently affected by mutations in C5/MCD DLBCL is TBL1XR1, which encodes for a core component of the SMRT/NCOR1 complex, which in is recruited to chromatin by BCL6 in GC B cells (129). Biochemical experiments using a TBL1XR1 p.Y446S mutant, which frequently occurs in human DLBCL, revealed that although the interaction with the SMRT/HDAC3 complex was preserved, the only significant change in the TBL1XR1mut interactome was a robust shift away from an interaction with BCL6 towards an interaction with the transcription factor BACH2 (70), which plays a critical role in memory B cell generation (130). These data indicate that mutant TBL1XR1 may redirect the SMRT complex toward BACH2, which in turn drives a transcriptional response mediating memory B cell fate in parallel to installing a plasma cell differentiation block via maintained BACH2-mediated PRDM1 transcriptional repression (70).
To study the potential role of Tbl1xr1 in lymphomagenesis, a Tbl1xr1 allele, which allows conditional expression of the p.D370Y mutation from the endogenous locus, was generated (70). In line with transcriptional rewiring downstream of mutant Tbl1xr1, RNA-Seq analysis of GC B cells derived from these animals revealed an enrichment for ABC-DLBCL-associated gene expression signatures and NF-kB signaling, which are normally repressed in GC B by the BCL6-SMRT complex (70). Interestingly, it was also observed that GC B cells derived from Tbl1xr1 p.D370Y mutant mice displayed de-repression of Gpr183 and S1pr1, which are transcriptionally silenced in the wildtype setting (70). Of note, silencing of Gpr183 and S1pr1 is critical for containment of GC B cells in lymphoid follicles (70). Thus, de-repression of these genes may drive expansion of post-GC B cells, as well as extra-nodal accumulation of these cells. Further support for a tumor-suppressive role of TBL1XR1 stems from the analysis of immunized VavP-BCL2;Cd19Cre/wt;Tbl1xr1fl/fl animals, which were shown to develop predominantly extra-nodal lymphomas, at a time point at which no tumors were detectable in VavP-BCL2;Cd19Cre/wt controls (70). B220+ cells in the VavP-BCL2;Cd19Cre/wt;Tbl1xr1fl/fl setting largely lacked GC B cell markers and displayed a relative expansion of (pre-) memory B cell populations (70). Morphologically, tumors in VavP-BCL2;Cd19Cre/wt;Tbl1xr1fl/fl mice were largely composed of large atypical immunoblasts, mimicking human extranodal ABC-DLBCL (70). These cells infiltrated extra-nodal tissues, such as liver and kidneys, while mostly sparing the lymph nodes (70). Tumors showed somatic mutations in the JH4 intron and the Pim1 locus (a known AID off-target), indicative of GC passage (70). Similar results were obtained in VavP-BCL2;Cγ1Cre/wt;Tbl1xr1fl/fl animals (70). Altogether, these data pinpoint a role for TBL1XR1 in skewing the transcriptome towards a memory B cell phenotype and a plasma cell differentiation block, which may promote GC re-entry instead of plasma cell differentiation upon antigen recall.
While the selection of TBL1XR1 mutations in C5/MCD DLBCL help to explain their memory B cell phenotypes and a plasma cell differentiation block, an explanation for the competitive fitness advantage of these cells remained largely elusive. A recent study focusing on the role of B cell translocation gene 1 (BTG1) helped to shed light on the mechanistic basis of competition within the GC reaction (64). BTG1 mutations are detected in approximately 70% of C5/MCD DLBCL and are typically heterozygous missense aberrations, clustering at the N-terminal portion of the protein between an N-terminal hydrophobic domain and the LxxLL motif (2, 3, 28, 64). Particularly the glutamine residue in position 36 is most frequently replaced by a histidine (64). It was further shown that these N-terminal aberration can induce conformational changes of BTG1 (131). To assess the biological role of BTG1 in the GC, two alleles were generated, namely a Rosa26LSL.BTG1.G36H and Rosa26LSL.BTG1.wt allele, where expression of either the mutant or the wildtype is prevented by a loxP-STOP-loxP cassette in the absence of Cre recombinase (64). The competitive fitness of BTG1 p.Q36H-mutant B cells was assessed in an elegant in vivo experimental system, in which Cd19Cre/wt;Rosa26LSL.BTG1.G36H/wt;Cd45.1 and Cd19wt/wt;Rosa26LSL.BTG1.G36H/wt;Cd45.1 mice were crossed with a B1-8hi allele, which encodes a B cell receptor with high (4-hydroxy-3-nitrophenyl)acetyl (NP) antigen affinity in B cells with a Λ-immunoglobulin light chain that facilitates GC entry upon NP immunization (64, 132). Upon adoptive transfer of these cells into Cd45.2 wildtype recipients, and subsequent immunization with the T cell-dependent antigen NP-ovalbumin, BTG1 p.Q36H-mutant B cells displayed a progressive competitive advantage, reaching approximately 90% of all GC B cells at day 14 (64). In contrast, no competitive advantage was observed when the Rosa26LSL.BTG1.wt allele was used (64). Mechanistically, it was shown that BTG1-mutant B cells displayed a significant enrichment of gene set signatures associated with TFH cell help, light zone to dark zone recycling B cells and MYC/mTORC1 signaling, compared to controls (64). Further experiments revealed that BTG1 represses MYC on a posttranscriptional level and that this repressive function is lost in mutant BTG1 (64). The oncogenic potential of the BTG1 p.Q36H mutation was further investigated in a bone marrow chimera setting, where recipients transplanted with Cγ1Cre/wt;VavP-BCL2;Rosa26LSL.BTG1.G36H/wt HSCs developed clonal DLBCL-like lymphoma with evidence of somatic hypermutation and passed away significantly earlier than Cγ1wt/wt;VavP-BCL2;Rosa26LSL.BTG1.G36H/wt and Cγ1wt/wt;Rosa26LSL.BTG1.G36H/wt controls (64). Moreover, Cγ1Cre/wt;VavP-BCL2;Rosa26LSL.BTG1.G36H/wt transplanted animals displayed extra-nodal infiltration of malignant B cells into lungs, kidneys, and liver (64).
Next to the above-discussed genomic aberrations, C5/MCD lymphomas frequently display CDKN2A deletions, which through its gene products p16 and p19 controls the RB1 and TP53 pathways, respectively (2, 28). A number of conditional Cdkn2a alleles exist and it would be interesting to assess the role of Cdkn2a deletions on the background of the various C5/MCD models detailed above. It might be particularly interesting to assess whether Cdkn2a deficiency impacts lymphoma tropism and might promote primary CNS lymphoma development, which frequently harbors a C5/MCD genetic makeup (3, 133).
Patient-derived mouse models of aggressive lymphoma
Human cancer cell line-derived xenograft (CDX) models are heavily used for in vivo pharmacology studies. The relatively low-cost and usability of CDX models make them attractive preclinical tools, however they typically do not fully recapitulate the disease complexity (134). CDX models are often cultured 2D over many passages in serum-containing media and implanted into immuno-compromised mice, such as NOD-SCID or Nude mice for drug efficacy studies (135). With the immune system as a crucial component of the antitumor response, and as immune-checkpoint-inhibitors (ICIs) are emerging as the standard of care for several cancer indications, this type of immuno-deficient mouse model system is not ideal for representing human biology (136). Furthermore, the abysmal correlation between therapeutic efficacy shown in CDX models and efficacy in humans, calls for innovation in preclinical mouse models, as <5% of clinical-stage cancer drugs reach regulatory approval (137).
A more complex preclinical mouse model system compared to CDX models are patient-derived xenograft models (PDX). PDX models are established from implanting tumor tissue from a patient into an immuno-compromised or humanized mouse. In contrast to CDX models, PDX models are not artificially grown and selected in vitro prior to implantation and are instead serially passaged in mice (138). One powerful advantage of PDX models is that the tumors can maintain the molecular heterogeneity of the patient sample. PDX models have been shown to exhibit clonal dynamics and acquired mutations, which emulate a similar trajectory as the primary tumor, and this is maintained over serial passages. This consistent clonal evolution between primary patient samples and PDX models give them a superior advantage over CDX models for functional analyses, and are ultimately better predictors of clinical response (139, 140). However, the PDX model system ultimately comes with its own limitations. First, commercially available PDX models are substantially more costly than CDX models. Establishing and maintaining an internal PDX biobank also comes with its financial, logistic and regulatory hurdles with regards to the acquisition of primary patient material and laboratory animal welfare (141). Another disadvantage of PDX models is the lack of the tumor microenvironment and human stromal component that is inherently missing in immuno-compromised mice. The main approach to bridge the human immune system and patient-derived tumors in mice is the humanized PDX model system. Humanized PDX models are developed by implanting human CD34+ cells from the umbilical cord or human PBMCs in irradiated immuno-deficient mice followed by the implantation of a patient-derived tumors. However, difficulties with the engraftment rate and the onset of graft-versus-host disease, due to HLA-mismatches between the donor and host are several roadblocks that have slowed the progress of this model system (142, 143).
Several PDX platforms of aggressive lymphomas have been established in an attempt to faithfully recapitulate the human disease biology and enable preclinical pharmacology (144, 145). Margaret Shipp and colleagues established a cohort of LBCL PDX models by implanting primary tumors underneath the renal capsule of NOD SCID Il2rγnull (NSG) mice. In total, 9 out of 28 (32%) PDX models were successfully propagated and considered a stable model. IHC characterization of these PDX models showed consistent immunophenotyping with the diagnosed LBCLs, indicating retained morphological features of the primary tumors. Furthermore, through RNA-seq analyses, the PDX models were correctly classified as ABC- (6/9 66%) or GCB-DLBCL (2/9 22%) based on their transcriptional signatures. Further genetic characterization of these PDX models revealed frequently mutated genes and chromosomal rearrangements commonly found in primary DLBCL. Lastly, the authors could show a differential sensitivity of spleen tyrosine kinase (SYK) inhibition in ABC-DLBCL PDX models, compared to GCB-DLBCL PDX models (144).
The lab of Michael Wang at MD Anderson also established a cohort of lymphoma PDX models comprised of DLBCL, MCL, MZL, BL, and FL (145). Similar to the previously mentioned PDX cohort, these PDX models also showed similar immunophenotypes and genetic profiles compared to the primary tumors. Following the characterization of the PDX models, an in vitro drug screen was performed with compounds that were used to treat the primary tumors in the patients. The PDX models exhibited similar response or resistance to the respective compounds as shown in patients, showcasing a robust pharmacology platform that can be enabled for personalized medicine. Moreover, the group developed primary and acquired ibrutinib-resistant PDX models to potentially reveal resistance mechanisms to BTK inhibitors. Through reverse phase protein array (RPPA) assays, they found an upregulation of PI3K pathway members, as well as BCL-2 family members following ibrutinib treatment. Follow-up combination efficacy studies in the ibrutinib-resistant PDX models with ibrutinib and idelalisib, a PI3Kδ inhibitor, ultimately overcame the ibrutinib resistance and led to significant tumor growth inhibition (145).
The two aforementioned groups faithfully established PDX platforms that can be utilized for pre-clinical pharmacology and drug discovery efforts. One possible innovation of these platforms could be to establish PDX models via intravenous transplantation of the primary tumor cells instead of subcutaneous or other orthotopic methods. This could potentially represent the disease biology in a more clinically relevant way as the tumor cells may home to organs that are commonly invaded by lymphoma cells i.e. spleen and lymph nodes. Another innovative idea might be to generate a cohort of PDX models that represent the newly classified molecular subtypes of DLBCL to further develop the idea of a genetically guided personalized medicine approach of treatment (146).
Concluding remarks and perspectives
In vivo experimentation always has to be well-justified against the three Rs (reduce, refine, replace) of animal research. The passing of the FDA Modernization Act 2.0 law in December 2022 further underscores this. However, none of the currently available alternative in vitro model systems is capable of fully recapitulating the complex B cell activation process with the germinal center at its core, a structure central to DLBCL lymphomagenesis, nor do they faithfully model the complex lymphoma microenvironment. Therefore to date, mouse models remain a cornerstone for disease modelling and pre-clinical evaluation of potential drugs.
GEMMs, as a result of their immunocompetency and autochthonous lymphoma manifestation, represent the model system that most closely recapitulates the genetic and immunological complexity of human lymphoma. The presence of a complex tumor microenvironment, including immune cells, allows the investigation of compounds that modulate or might be influenced by the TME, including immune therapies (68, 126). However, tumors in GEMMs usually manifest with high variance after months of latency and their diagnosis ideally involves imaging methods (53, 68, 126, 147). This makes treatment experiments in GEMMs laborious and expensive. CDX/PDX models, isogenic transplantation systems of murine lymphoma cell lines or lymphoma organoids might provide more cost- and time-effective platforms for larger experimental setups with multiple treatment cohorts. Further, transplantable lymphoma cell lines obtained from GEMMs could be engineered to perform functional CRISPR screening in vivo, which would provide a platform to screen and test multiple therapeutic vulnerabilities in an unbiased manner.
The genesis of precision genome editing using the CRISPR/Cas9 system has enabled fast-track creation of ever-more sophisticated animal models of human lymphomas, and greatly expanded the toolkit of available preclinical models (34, 64, 148). While the generation of novel alleles has become much more efficient with these technological developments, the ex vivo editing of hematopoietic stem cells followed by transplantation into irradiated recipients provides a shortcut method to investigate the role of a gene-of-interest in lymphoma (104, 109, 149). By using Cre-dependent overexpression vectors or gRNA vectors in conjunction with a Cre-dependent Cas9 allele, the introduced modifications could manifest B cell-specifically.
Altogether, the choice of the experimental platform strongly depends on the research question at hand. For economic an animal welfare reasons, in vitro systems should be the first choice whenever appropriate. However, the complex interaction of healthy and malignant B cells with their environment requires the use of in vivo models in many situations. While these interactions are recapitulated by different transplantation-based systems to some extent (150, 151), autochthonous tumors developing in GEMMs most faithfully replicate human disease in terms of lymphomagenesis and tumor microenvironment and therefore will remain indispensable tools in lymphoma research in the foreseeable future.
Author contributions
AT: Writing – original draft, Writing – review & editing. AA: Writing – original draft, Writing – review & editing. SH: Writing – original draft, Writing – review & editing. MJ: Writing – original draft, Writing – review & editing. BvT: Writing – review & editing. JH: Writing – review & editing. RF: Writing – review & editing. RJ: Writing – review & editing. SK: Writing – original draft, Writing – review & editing. HCR: Writing – original draft, Writing – review & editing. GK: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded through the Deutsche Forschungsgemeinschaft (RE 2246/13-1, SFB 1399/1 2019, SFB 1430/1 2021, SFB 1530/1 2022 to HCR), the Else Kröner-Fresenius Stiftung (EKFS-2014-A06 to HCR, 2016_Kolleg.19 to HCR, RJ), the Deutsche Krebshilfe (70115679, 1117240 and 70113041 to HCR) and a Mildred Scheel Nachwuchszentrum Grant (Grant number 70113307), the German Ministry of Education and Research (BMBF e:Med 01ZX1303A to HCR), the consortium HiRisk-HiGain ERAPERMED 2020-090 (01KU2104), the MERCUR Foundation (IGNITE consortium, Ex-2021-0033), as well as the NRW Network consortium CANTAR (NW21-062B).
Acknowledgments
We acknowledge support by the Open Access Publication Fund of the University of Duisburg-Essen. We apologize to our colleagues for the omission of many seminal contributions to the field, and their corresponding references, owing to space constraints. We thank members of the Jachimowicz and Reinhardt laboratories for critically reading the manuscript and for providing critical comments and ideas.
Conflict of interest
HCR received consulting and lecture fees from Roche, Novartis, BMS, Abbvie, AstraZeneca, Vertex and Merck; H. received research funding from Gilead Sciences and AstraZeneca. HCR is a co-founder of CDL Therapeutics GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Campo E, Jaffe ES, Cook JR, Quintanilla-Martinez L, Swerdlow SH, Anderson KC, et al. The international consensus classification of mature lymphoid neoplasms: a report from the clinical advisory committee. Blood (2022) 140(11):1229–53. doi: 10.1182/blood.2022015851
2. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med (2018) 378(15):1396–407. doi: 10.1056/NEJMoa1801445
3. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med (2018) 24(5):679–90. doi: 10.1038/s41591-018-0016-8
4. Shaffer AL 3rd, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol (2012) 30:565–610. doi: 10.1146/annurev-immunol-020711-075027
5. Meyer SN, Koul S, Pasqualucci L. Mouse models of germinal center derived B-cell lymphomas. Front Immunol (2021) 12:710711. doi: 10.3389/fimmu.2021.710711
6. Kuppers R, Klein U, Hansmann ML, Rajewsky K. Cellular origin of human B-cell lymphomas. N Engl J Med (1999) 341(20):1520–9. doi: 10.1056/NEJM199911113412007
7. Hsu FJ, Levy R. Preferential use of the VH4 Ig gene family by diffuse large-cell lymphoma. Blood (1995) 86(8):3072–82. doi: 10.1182/blood.V86.8.3072.3072
8. Stevenson F, Sahota S, Zhu D, Ottensmeier C, Chapman C, Oscier D, et al. Insight into the origin and clonal history of B-cell tumors as revealed by analysis of immunoglobulin variable region genes. Immunol Rev (1998) 162:247–59. doi: 10.1111/j.1600-065X.1998.tb01446.x
9. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol (2015) 15(3):137–48. doi: 10.1038/nri3804
10. Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity (2013) 38(5):918–29. doi: 10.1016/j.immuni.2013.04.009
11. Basso K, Dalla-Favera R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv Immunol (2010) 105:193–210. doi: 10.1016/S0065-2776(10)05007-8
12. Hatzi K, Melnick A. Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol Med (2014) 20(6):343–52. doi: 10.1016/j.molmed.2014.03.001
13. Knittel G, Rehkamper T, Nieper P, Schmitt A, Flumann R, Reinhardt HC. DNA damage pathways and B-cell lymphomagenesis. Curr Opin Hematol (2018) 25(4):315–22. doi: 10.1097/MOH.0000000000000433
14. Mossadegh-Keller N, Brisou G, Beyou A, Nadel B, Roulland S. Human B lymphomas reveal their secrets through genetic mouse models. Front Immunol (2021) 12:683597. doi: 10.3389/fimmu.2021.683597
15. Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature (2001) 412(6844):341–6. doi: 10.1038/35085588
16. Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, et al. AID is required for c-myc/IgH chromosome translocations vivo. Cell (2004) 118(4):431–8. doi: 10.1016/j.cell.2004.08.006
17. Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell (2009) 36(4):631–41. doi: 10.1016/j.molcel.2009.11.007
18. Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol (2006) 7(7):773–82. doi: 10.1038/ni1357
19. Saito M, Gao J, Basso K, Kitagawa Y, Smith PM, Bhagat G, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell (2007) 12(3):280–92. doi: 10.1016/j.ccr.2007.08.011
20. Hawkins ED, Turner ML, Wellard CJ, Zhou JH, Dowling MR, Hodgkin PD. Quantal and graded stimulation of B lymphocytes as alternative strategies for regulating adaptive immune responses. Nat Commun (2013) 4:2406. doi: 10.1038/ncomms3406
21. Rush JS, Hodgkin PD. B cells activated via CD40 and IL-4 undergo a division burst but require continued stimulation to maintain division, survival and differentiation. Eur J Immunol (2001) 31(4):1150–9. doi: 10.1002/1521-4141(200104)31:4<1150::AID-IMMU1150>3.0.CO;2-V
22. Wortis HH, Teutsch M, Higer M, Zheng J, Parker DC. B-cell activation by crosslinking of surface IgM or ligation of CD40 involves alternative signal pathways and results in different B-cell phenotypes. Proc Natl Acad Sci USA (1995) 92(8):3348–52. doi: 10.1073/pnas.92.8.3348
23. Nojima T, Haniuda K, Moutai T, Matsudaira M, Mizokawa S, Shiratori I, et al. In-vitro derived germinal centre B cells differentially generate memory B or plasma cells. vivo. Nat Commun (2011) 2:465. doi: 10.1038/ncomms1475
24. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature (2000) 403(6769):503–11. doi: 10.1038/35000501
25. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood (2016) 127(20):2375–90. doi: 10.1182/blood-2016-01-643569
26. Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med (2008) 359(22):2313–23. doi: 10.1056/NEJMoa0802885
27. Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med (2002) 346(25):1937–47. doi: 10.1056/NEJMoa012914
28. Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell (2020) 37(4):551–568 e514. doi: 10.1016/j.ccell.2020.03.015
29. Boardman AP, Salles G. CAR T-cell therapy in large B cell lymphoma. Hematol Oncol (2023) 41 Suppl 1(Suppl 1):112–8. doi: 10.1002/hon.3153
30. Birling MC, Gofflot F, Warot X. Site-specific recombinases for manipulation of the mouse genome. Methods Mol Biol (2009) 561:245–63. doi: 10.1007/978-1-60327-019-9_16
31. Tian X, Zhou B. Strategies for site-specific recombination with high efficiency and precise spatiotemporal resolution. J Biol Chem (2021) 296:100509. doi: 10.1016/j.jbc.2021.100509
32. Whitfield J, Littlewood T, Evan GI, Soucek L. The estrogen receptor fusion system in mouse models: a reversible switch. Cold Spring Harb Protoc (2015) 2015(3):227–34. doi: 10.1101/pdb.top069815
33. Sando R 3rd, Baumgaertel K, Pieraut S, Torabi-Rander N, Wandless TJ, Mayford M, et al. Inducible control of gene expression with destabilized Cre. Nat Methods (2013) 10(11):1085–8. doi: 10.1038/nmeth.2640
34. Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell (2014) 159(2):440–55. doi: 10.1016/j.cell.2014.09.014
35. Rickert RC, Roes J, Rajewsky K, lymphocyte-specific -B. Cre-mediated mutagenesis in mice. Nucleic Acids Res (1997) 25(6):1317–8. doi: 10.1093/nar/25.6.1317
36. Yasuda T, Wirtz T, Zhang B, Wunderlich T, Schmidt-Supprian M, Sommermann T, et al. Studying Epstein-Barr virus pathologies and immune surveillance by reconstructing EBV infection in mice. Cold Spring Harb Symp Quant Biol (2013) 78:259–63. doi: 10.1101/sqb.2013.78.020222
37. Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, et al. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci USA (2006) 103(37):13789–94. doi: 10.1073/pnas.0605944103
38. Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell (2004) 117(6):787–800. doi: 10.1016/j.cell.2004.05.014
39. Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Hao Z, et al. Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc Natl Acad Sci USA (2006) 103(19):7396–401. doi: 10.1073/pnas.0602353103
40. King HW, Orban N, Riches JC, Clear AJ, Warnes G, Teichmann SA, et al. Single-cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics. Sci Immunol (2021) 6(56). doi: 10.1126/sciimmunol.abe6291
41. Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, et al. Class-switch recombination occurs infrequently in germinal centers. Immunity (2019) 51(2):337–350 e337. doi: 10.1016/j.immuni.2019.07.001
42. Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell (2008) 135(6):1028–38. doi: 10.1016/j.cell.2008.09.062
43. Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol (2009) 10(12):1292–9. doi: 10.1038/ni.1814
44. Cattoretti G, Pasqualucci L, Ballon G, Tam W, Nandula SV, Shen Q, et al. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell (2005) 7(5):445–55. doi: 10.1016/j.ccr.2005.03.037
45. Hampel F, Ehrenberg S, Hojer C, Draeseke A, Marschall-Schroter G, Kuhn R, et al. CD19-independent instruction of murine marginal zone B-cell development by constitutive Notch2 signaling. Blood (2011) 118(24):6321–31. doi: 10.1182/blood-2010-12-325944
46. Sungalee S, Mamessier E, Morgado E, Gregoire E, Brohawn PZ, Morehouse CA, et al. Germinal center reentries of BCL2-overexpressing B cells drive follicular lymphoma progression. J Clin Invest (2014) 124(12):5337–51. doi: 10.1172/JCI72415
47. Egle A, Harris AW, Bath ML, O'Reilly L, Cory S. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood (2004) 103(6):2276–83. doi: 10.1182/blood-2003-07-2469
48. Knittel G, Liedgens P, Korovkina D, Seeger JM, Al-Baldawi Y, Al-Maarri M, et al. B cell-specific conditional expression of Myd88p.L252P leads to the development of diffuse large B cell lymphoma in mice. Blood (2016) 127(22):2732–41. doi: 10.1182/blood-2015-11-684183
49. Muppidi JR, Schmitz R, Green JA, Xiao W, Larsen AB, Braun SE, et al. Loss of signalling via Galpha13 in germinal centre B-cell-derived lymphoma. Nature (2014) 516(7530):254–8. doi: 10.1038/nature13765
50. Francis SA, Shen X, Young JB, Kaul P, Lerner DJ. Rho GEF Lsc is required for normal polarization, migration, and adhesion of formyl-peptide-stimulated neutrophils. Blood (2006) 107(4):1627–35. doi: 10.1182/blood-2005-03-1164
51. Zhang J, Vlasevska S, Wells VA, Nataraj S, Holmes AB, Duval R, et al. The CREBBP acetyltransferase is a haploinsufficient tumor suppressor in B-cell lymphoma. Cancer Discov (2017) 7(3):322–37. doi: 10.1158/2159-8290.CD-16-1417
52. Kang-Decker N, Tong C, Boussouar F, Baker DJ, Xu W, Leontovich AA, et al. Loss of CBP causes T cell lymphomagenesis in synergy with p27Kip1 insufficiency. Cancer Cell (2004) 5(2):177–89. doi: 10.1016/S1535-6108(04)00022-4
53. Beguelin W, Teater M, Gearhart MD, Calvo Fernandez MT, Goldstein RL, Cardenas MG, et al. EZH2 and BCL6 cooperate to assemble CBX8-BCOR complex to repress bivalent promoters, mediate germinal center formation and lymphomagenesis. Cancer Cell (2016) 30(2):197–213. doi: 10.1016/j.ccell.2016.07.006
54. Souroullas GP, Jeck WR, Parker JS, et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med (2016) 22(6):632–40. doi: 10.1038/nm.4092
55. Beguelin W, Popovic R, Teater M, Simon JM, Liu JY, Paulk J, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell (2013) 23(5):677–92. doi: 10.1016/j.ccr.2013.04.011
56. Schneider C, Kon N, Amadori L, Shen Q, Schwartz FH, Tischler B, et al. FBXO11 inactivation leads to abnormal germinal-center formation and lymphoproliferative disease. Blood (2016) 128(5):660–6. doi: 10.1182/blood-2015-11-684357
57. Ruppel KM, Willison D, Kataoka H, Wang A, Zheng YW, Cornelissen I, et al. Essential role for Galpha13 in endothelial cells during embryonic development. Proc Natl Acad Sci USA (2005) 102(23):8281–6. doi: 10.1073/pnas.0503326102
58. Brescia P, Schneider C, Holmes AB, Shen Q, Hussein S, Pasqualucci L, et al. MEF2B instructs germinal center development and acts as an oncogene in B cell lymphomagenesis. Cancer Cell (2018) 34(3):453–465 e459. doi: 10.1016/j.ccell.2018.08.006
59. Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife (2013) 2:e01503. doi: 10.7554/eLife.01503.027
60. Kono M, Mi Y, Liu Y, Kono M, Mi . The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J Biol Chem (2004) 279(28):29367–73. doi: 10.1074/jbc.M403937200
61. Yusufova N, Kloetgen A, Teater M, Osunsade A, Camarillo JM, Chin CR, et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature (2021) 589(7841):299–305. doi: 10.1038/s41586-020-3017-y
62. Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Beguelin W, Fontan L, et al. TET2 deficiency causes germinal center hyperplasia, impairs plasma cell differentiation, and promotes B-cell lymphomagenesis. Cancer Discov (2018) 8(12):1632–53. doi: 10.1158/2159-8290.CD-18-0657
63. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell (2011) 20(1):11–24. doi: 10.1016/j.ccr.2011.06.001
64. Mlynarczyk C, Teater M, Pae J, Chin CR, Wang L, Arulraj T, et al. BTG1 mutation yields supercompetitive B cells primed for Malignant transformation. Science (2023) 379(6629):eabj7412. doi: 10.1126/science.abj7412
65. Pindzola GM, Razzaghi R, Tavory RN, Nguyen HT, Morris VM, Li M, et al. Aberrant expansion of spontaneous splenic germinal centers induced by hallmark genetic lesions of aggressive lymphoma. Blood (2022). doi: 10.1182/blood.2022015926
66. Schmidt K, Sack U, Graf R, Winkler W, Popp O, Mertins P, et al. B-cell-specific myd88 L252P expression causes a premalignant gammopathy resembling Igm MGUS. Front Immunol (2020) 11:602868. doi: 10.3389/fimmu.2020.602868
67. Sewastianik T, Guerrera ML, Adler K, Dennis PS, Wright K, Shanmugam V, et al. Human MYD88L265P is insufficient by itself to drive neoplastic transformation in mature mouse B cells. Blood Adv (2019) 3(21):3360–74. doi: 10.1182/bloodadvances.2019000588
68. Flumann R, Hansen J, Pelzer BW, Nieper P, Lohmann T, Kisis I, et al. Distinct genetically-determined origins of Myd88/Bcl2-driven aggressive lymphoma rationalize targeted therapeutic intervention strategies. Blood Cancer Discov (2022) 4(1):78–97. doi: 10.1158/2643-3230.BCD-22-0007
69. Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity (2003) 19(4):607–20. doi: 10.1016/S1074-7613(03)00267-X
70. Venturutti L, Teater M, Zhai A, Chadburn A, Babiker L, Kim D, et al. TBL1XR1 mutations drive extranodal lymphoma by inducing a pro-tumorigenic memory fate. Cell (2020) 182(2):297–316 e227. doi: 10.1016/j.cell.2020.05.049
71. Bunting KL, Melnick AM. New effector functions and regulatory mechanisms of BCL6 in normal and Malignant lymphocytes. Curr Opin Immunol (2013) 25(3):339–46. doi: 10.1016/j.coi.2013.05.003
72. Cardenas MG, Oswald E, Yu W, Xue F, MacKerell AD Jr., Melnick AM. The expanding role of the BCL6 oncoprotein as a cancer therapeutic target. Clin Cancer Res (2017) 23(4):885–93. doi: 10.1158/1078-0432.CCR-16-2071
73. Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev (2012) 247(1):172–83. doi: 10.1111/j.1600-065X.2012.01112.x
74. Duan S, Cermak L, Pagan JK, Rossi M, Martinengo C, di Celle PF, et al. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature (2012) 481(7379):90–3. doi: 10.1038/nature10688
75. Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature (2011) 471(7337):189–95. doi: 10.1038/nature09730
76. Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M, et al. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol (2013) 14(10):1084–92. doi: 10.1038/ni.2688
77. Pasqualucci L, Migliazza A, Basso K, Houldsworth J, Chaganti RS, Dalla-Favera R. Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood (2003) 101(8):2914–23. doi: 10.1182/blood-2002-11-3387
78. Pasqualucci L, Migliazza A, Fracchiolla N, William C, Neri A, Baldini L, et al. BCL-6 mutations in normal germinal center B cells: evidence of somatic hypermutation acting outside Ig loci. Proc Natl Acad Sci USA (1998) 95(20):11816–21. doi: 10.1073/pnas.95.20.11816
79. Wang X, Li Z, Naganuma A, Ye BH. Negative autoregulation of BCL-6 is bypassed by genetic alterations in diffuse large B cell lymphomas. Proc Natl Acad Sci USA (2002) 99(23):15018–23. doi: 10.1073/pnas.232581199
80. Ye BH, Lista F, Lo Coco F, Knowles DM, Offit K, Chaganti RS, et al. Alterations of a zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science (1993) 262(5134):747–50. doi: 10.1126/science.8235596
81. Cerchietti LC, Ghetu AF, Zhu X, Da Silva GF, Zhong S, Matthews M, et al. A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell (2010) 17(4):400–11. doi: 10.1016/j.ccr.2009.12.050
82. Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature (2004) 432(7017):635–9. doi: 10.1038/nature03147
83. Phan RT, Saito M, Basso K, Niu H, Dalla-Favera R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat Immunol (2005) 6(10):1054–60. doi: 10.1038/ni1245
84. Ranuncolo SM, Polo JM, Melnick A. BCL6 represses CHEK1 and suppresses DNA damage pathways in normal and Malignant B-cells. Blood Cells Mol Dis (2008) 41(1):95–9. doi: 10.1016/j.bcmd.2008.02.003
85. Ranuncolo SM, Wang L, Polo JM, Dell'Oso T, Dierov J, Gaymes TJ, et al. BCL6-mediated attenuation of DNA damage sensing triggers growth arrest and senescence through a p53-dependent pathway in a cell context-dependent manner. J Biol Chem (2008) 283(33):22565–72. doi: 10.1074/jbc.M803490200
86. Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet (2012) 28(3):128–36. doi: 10.1016/j.tig.2011.12.002
87. Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol (2009) 21(2):245–55. doi: 10.1016/j.ceb.2009.01.018
88. Reinhardt HC, Yaffe MB. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat Rev Mol Cell Biol (2013) 14(9):563–80. doi: 10.1038/nrm3640
89. Yabe D, Fukuda H, Aoki M, Yamada S, Takebayashi S, Shinkura R, et al. Generation of a conditional knockout allele for mammalian Spen protein Mint/SHARP. Genesis (2007) 45(5):300–6. doi: 10.1002/dvg.20296
90. Nakagawa MM, Thummar K, Mandelbaum J, Pasqualucci L, Rathinam CV. Lack of the ubiquitin-editing enzyme A20 results in loss of hematopoietic stem cell quiescence. J Exp Med (2015) 212(2):203–16. doi: 10.1084/jem.20132544
91. Marino S, Vooijs M, van der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev (2000) 14(8):994–1004. doi: 10.1101/gad.14.8.994
92. Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell (2004) 119(6):847–60. doi: 10.1016/j.cell.2004.11.004
93. Lee CL, Moding EJ, Huang X, Li Y, Woodlief LZ, Rodrigues RC, et al. Generation of primary tumors with Flp recombinase in FRT-flanked p53 mice. Dis Model Mech (2012) 5(3):397–402. doi: 10.1242/dmm.009084
94. Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature (2003) 424(6945):223–8. doi: 10.1038/nature01764
95. Vooijs M, van der Valk M, te Riele H, Berns A. Flp-mediated tissue-specific inactivation of the retinoblastoma tumor suppressor gene in the mouse. Oncogene (1998) 17(1):1–12. doi: 10.1038/sj.onc.1202169
96. Campbell KJ, Bath ML, Turner ML, Vandenberg CJ, Bouillet P, Metcalf D, et al. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood (2010) 116(17):3197–207. doi: 10.1182/blood-2010-04-281071
97. Zhou P, Qian L, Bieszczad CK, Noelle R, Binder M, Levy NB, et al. Mcl-1 in transgenic mice promotes survival in a spectrum of hematopoietic cell types and immortalization in the myeloid lineage. Blood (1998) 92(9):3226–39. doi: 10.1182/blood.V92.9.3226
98. McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, et al. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell (1989) 57(1):79–88. doi: 10.1016/0092-8674(89)90174-8
99. Strasser A, Harris AW, Cory S. E mu-bcl-2 transgene facilitates spontaneous transformation of early pre-B and immunoglobulin-secreting cells but not T cells. Oncogene (1993) 8(1):1–9.
100. Xiang H, Noonan EJ, Wang J, Duan H, Ma L, Michie S, et al. The immunoglobulin heavy chain gene 3' enhancers induce Bcl2 deregulation and lymphomagenesis in murine B cells. Leukemia (2011) 25(9):1484–93. doi: 10.1038/leu.2011.115
101. Ogilvy S, Metcalf D, Print CG, Bath ML, Harris AW, Adams JM. Constitutive Bcl-2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc Natl Acad Sci USA (1999) 96(26):14943–8. doi: 10.1073/pnas.96.26.14943
102. Ennishi D, Takata K, Beguelin W, Duns G, Mottok A, Farinha P, et al. Molecular and genetic characterization of MHC deficiency identifies EZH2 as therapeutic target for enhancing immune recognition. Cancer Discov (2019) 9(4):546–63. doi: 10.1158/2159-8290.CD-18-1090
103. Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med (2015) 21(10):1190–8. doi: 10.1038/nm.3940
104. Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med (2015) 21(10):1199–208. doi: 10.1038/nm.3943
105. Heward J, Konali L, D'Avola A, Close K, Yeomans A, Philpott M, et al. KDM5 inhibition offers a novel therapeutic strategy for the treatment of KMT2D mutant lymphomas. Blood (2021) 138(5):370–81. doi: 10.1182/blood.2020008743
106. Beguelin W, Rivas MA, Calvo Fernandez MT, Teater M, Purwada A, Redmond D, et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat Commun (2017) 8(1):877. doi: 10.1038/s41467-017-01029-x
107. Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest (2013) 123(12):5009–22. doi: 10.1172/JCI70626
108. McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci USA (2012) 109(8):2989–94. doi: 10.1073/pnas.1116418109
109. Jiang Y, Ortega-Molina A, Geng H, Ying HY, Hatzi K, Parsa S, et al. CREBBP inactivation promotes the development of HDAC3-dependent lymphomas. Cancer Discov (2017) 7(1):38–53. doi: 10.1158/2159-8290.CD-16-0975
110. Parveen R, Harihar D, Chatterji BP. Recent histone deacetylase inhibitors in cancer therapy. Cancer (2023) 129(21):3372–80. doi: 10.1002/cncr.34974
111. Sarkar R, Banerjee S, Amin SA, Adhikari N, Jha T. Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: A review. Eur J Med Chem (2020) 192:112171. doi: 10.1016/j.ejmech.2020.112171
112. Mondello P, Tadros S, Teater M, Fontan L, Chang AY, Jain N, et al. Selective inhibition of HDAC3 targets synthetic vulnerabilities and activates immune surveillance in lymphoma. Cancer Discov (2020) 10(3):440–59. doi: 10.1158/2159-8290.CD-19-0116
113. Wagner FF, Lundh M, Kaya T, McCarren P, Zhang YL, Chattopadhyay S, et al. An isochemogenic set of inhibitors to define the therapeutic potential of histone deacetylases in beta-cell protection. ACS Chem Biol (2016) 11(2):363–74. doi: 10.1021/acschembio.5b00640
114. Lu E, Cyster JG. G-protein coupled receptors and ligands that organize humoral immune responses. Immunol Rev (2019) 289(1):158–72. doi: 10.1111/imr.12743
115. Flori M, Schmid CA, Sumrall ET, Tzankov A, Law CW, Robinson MD, et al. The hematopoietic oncoprotein FOXP1 promotes tumor cell survival in diffuse large B-cell lymphoma by repressing S1PR2 signaling. Blood (2016) 127(11):1438–48. doi: 10.1182/blood-2015-08-662635
116. Healy JA, Nugent A, Rempel RE, Moffitt AB, Davis NS, Jiang X, et al. GNA13 loss in germinal center B cells leads to impaired apoptosis and promotes lymphoma. vivo. Blood (2016) 127(22):2723–31. doi: 10.1182/blood-2015-07-659938
117. Cattoretti G, Mandelbaum J, Lee N, Chaves AH, Mahler AM, Chadburn A, et al. Targeted disruption of the S1P2 sphingosine 1-phosphate receptor gene leads to diffuse large B-cell lymphoma formation. Cancer Res (2009) 69(22):8686–92. doi: 10.1158/0008-5472.CAN-09-1110
118. Kipreos ET, Pagano M. The F-box protein family. Genome Biol (2000) 1(5):REVIEWS3002. doi: 10.1186/gb-2000-1-5-reviews3002
119. Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol (2013) 14(6):369–81. doi: 10.1038/nrm3582
120. Horn M, Geisen C, Cermak L, Becker B, Nakamura S, Klein C, et al. DRE-1/FBXO11-dependent degradation of BLMP-1/BLIMP-1 governs C. elegans developmental timing and maturation. Dev Cell (2014) 28(6):697–710. doi: 10.1016/j.devcel.2014.01.028
121. Fyodorov DV, Zhou BR, Skoultchi AI, Bai Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol (2018) 19(3):192–206. doi: 10.1038/nrm.2017.94
122. Okosun J, Bodor C, Wang J, Araf S, Yang CY, Pan C, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet (2014) 46(2):176–81. doi: 10.1038/ng.2856
123. Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood (2014) 123(10):1487–98. doi: 10.1182/blood-2013-05-500264
124. Morin RD, Arthur SE, Hodson DJ. Molecular profiling in diffuse large B-cell lymphoma: why so many types of subtypes? Br J Haematol (2022) 196(4):814–29. doi: 10.1111/bjh.17811
125. Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell (2011) 20(1):25–38. doi: 10.1016/j.ccr.2011.06.003
126. Flumann R, Rehkamper T, Nieper P, Pfeiffer P, Holzem A, Klein S, et al. An autochthonous mouse model of myd88- and BCL2-driven diffuse large B-cell lymphoma reveals actionable molecular vulnerabilities. Blood Cancer Discov (2021) 2(1):70–91. doi: 10.1158/2643-3230.BCD-19-0059
127. Rodriguez S, Celay J, Goicoechea I, Jimenez C, Botta C, Garcia-Barchino MJ, et al. Preneoplastic somatic mutations including MYD88(L265P) in lymphoplasmacytic lymphoma. Sci Adv (2022) 8(3):eabl4644. doi: 10.1126/sciadv.abl4644
128. Venturutti L, Rivas MA, Pelzer BW, Flumann R, Hansen J, Karagiannidis I, et al. An aged/autoimmune B-cell program defines the early transformation of extranodal lymphomas. Cancer Discov (2022). doi: 10.1158/2159-8290.c.6549789.v1
129. Hatzi K, Geng H, Doane AS, Meydan C, LaRiviere R, Cardenas M, et al. Histone demethylase LSD1 is required for germinal center formation and BCL6-driven lymphomagenesis. Nat Immunol (2019) 20(1):86–96. doi: 10.1038/s41590-018-0273-1
130. Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol (2016) 17(7):861–9. doi: 10.1038/ni.3460
131. Kots E, Mlynarczyk C, Melnick A, Khelashvili G. Conformational transitions in BTG1 antiproliferative protein and their modulation by disease mutants. Biophys J (2022) 121(19):3753–64. doi: 10.1016/j.bpj.2022.04.023
132. Shih TA, Roederer M, Nussenzweig MC. Role of antigen receptor affinity in T cell-independent antibody responses. vivo. Nat Immunol (2002) 3(4):399–406. doi: 10.1038/ni776
133. Hernandez-Verdin I, Kirasic E, Wienand K, Mokhtari K, Eimer S, Loiseau H, et al. Molecular and clinical diversity in primary central nervous system lymphoma. Ann Oncol (2023) 34(2):186–99. doi: 10.1016/j.annonc.2022.11.002
134. Day CP, Merlino G, Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell (2015) 163(1):39–53. doi: 10.1016/j.cell.2015.08.068
135. Yanguas-Casas N, Pedrosa L, Fernandez-Miranda I, Sanchez-Beato M. An overview on diffuse large B-cell lymphoma models: towards a functional genomics approach. Cancers (Basel) (2021) 13(12):2893. doi: 10.3390/cancers13122893
136. Twomey JD, Zhang B. Cancer immunotherapy update: FDA-approved checkpoint inhibitors and companion diagnostics. AAPS J (2021) 23(2):39. doi: 10.1208/s12248-021-00574-0
137. Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov (2006) 5(9):741–54. doi: 10.1038/nrd2110
138. Liu Y, Wu W, Cai C, Zhang H, Shen H, Han Y. Patient-derived xenograft models in cancer therapy: technologies and applications. Signal Transduct Target Ther (2023) 8(1):160. doi: 10.1038/s41392-023-01419-2
139. Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature (2015) 518(7539):422–6. doi: 10.1038/nature13952
140. Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D, et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol (2017) 28(10):2595–605. doi: 10.1093/annonc/mdx416
141. Mattar M, McCarthy CR, Kulick AR, Qeriqi B, Guzman S, de StanChina E. Establishing and maintaining an extensive library of patient-derived xenograft models. Front Oncol (2018) 8:19. doi: 10.3389/fonc.2018.00019
142. Gitto SB, George E, Medvedev S, Simpkins F, Powell DJ Jr. Humanized patient-derived xenograft models of ovarian cancer. Methods Mol Biol (2022) 2424:255–74. doi: 10.1007/978-1-0716-1956-8_17
143. Kang Y, Armstrong AJ, Hsu DS. An autologous humanized patient-derived xenograft (PDX) model for evaluation of nivolumab immunotherapy in renal cell cancer: a case report. Stem Cell Investig (2022) 9:8. doi: 10.21037/sci-2022-029
144. Chapuy B, Cheng H, Watahiki A, Ducar MD, Tan Y, Chen L, et al. Diffuse large B-cell lymphoma patient-derived xenograft models capture the molecular and biological heterogeneity of the disease. Blood (2016) 127(18):2203–13. doi: 10.1182/blood-2015-09-672352
145. Zhang L, Nomie K, Zhang H, Bell T, Pham L, Kadri S, et al. B-cell lymphoma patient-derived xenograft models enable drug Discov and are a platform for personalized therapy. Clin Cancer Res (2017) 23(15):4212–23. doi: 10.1158/1078-0432.CCR-16-2703
146. Zhang MC, Tian S, Fu D, Wang L, Cheng S, Yi HM, et al. Genetic subtype-guided immunochemotherapy in diffuse large B cell lymphoma: The randomized GUIDANCE-01 trial. Cancer Cell (2023) 41(10):1705–16. doi: 10.1016/j.ccell.2023.09.004
147. Riabinska A, Lehrmann D, Jachimowicz RD, Knittel G, Fritz C, Schmitt A, et al. ATM activity in T cells is critical for immune surveillance of lymphoma. vivo. Leukemia (2019) 34(3):771–86. doi: 10.1038/s41375-019-0618-2
148. Ely ZA, Mathey-Andrews N, Naranjo S, Gould SI, Mercer KL, Newby GA, et al. A prime editor mouse to model a broad spectrum of somatic mutations. vivo. Nat Biotechnol (2023) 1–13. doi: 10.1038/s41587-023-01783-y
149. Hu Q, Xu T, Zhang M, Zhang H, Liu Y, Li HB, et al. Diverging regulation of Bach2 protein and RNA expression determine cell fate in early B cell response. Cell Rep (2022) 40(1):111035. doi: 10.1016/j.celrep.2022.111035
150. Kim KJ, Kanellopoulos-Langevin C, Merwin RM, Sachs DH, Asofsky R. Establishment and characterization of BALB/c lymphoma lines with B cell properties. J Immunol (1979) 122(2):549–54. doi: 10.4049/jimmunol.122.2.549
Keywords: diffuse large B cell lymphoma (DLBCL), genetically engineered (GE) animals, lymphoma, animal models, mouse models
Citation: Tabatabai A, Arora A, Höfmann S, Jauch M, von Tresckow B, Hansen J, Flümann R, Jachimowicz RD, Klein S, Reinhardt HC and Knittel G (2023) Mouse models of diffuse large B cell lymphoma. Front. Immunol. 14:1313371. doi: 10.3389/fimmu.2023.1313371
Received: 10 October 2023; Accepted: 10 November 2023;
Published: 06 December 2023.
Edited by:
Juan M. Zapata, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Anthony Uren, University of Plymouth, United KingdomKatia Basso, Columbia University, United States
Copyright © 2023 Tabatabai, Arora, Höfmann, Jauch, von Tresckow, Hansen, Flümann, Jachimowicz, Klein, Reinhardt and Knittel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans Christian Reinhardt, Y2hyaXN0aWFuLnJlaW5oYXJkdEB1ay1lc3Nlbi5kZQ==; Gero Knittel, Z2Vyby5rbml0dGVsQHVrLWVzc2VuLmRl
†These authors have contributed equally to this work