
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 11 December 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1306338
This article is part of the Research Topic Case Reports in Autoimmune and Autoinflammatory Disorders View all 42 articles
 Lin-Yan Hu1,2
Lin-Yan Hu1,2 Lin Wan1,2
Lin Wan1,2 Qiu-Hong Wang1,2,3
Qiu-Hong Wang1,2,3 Xiu-Yu Shi1,2Yan Meng1,2Xiao-Fan Yang4
Xiu-Yu Shi1,2Yan Meng1,2Xiao-Fan Yang4 Guang Yang1,2,3
Guang Yang1,2,3 Li-Ping Zou1,2,3*
Li-Ping Zou1,2,3*Perforin is essentially involved in the granule-dependent killing activities of cytotoxic T lymphocytes and NK cells. Monoallelic PRF1 mutation increases the risk of autoimmune diseases, and biallelic PRF1 mutation causes familial hemophagocytic lymphohistiocytosis-2. Here, we report a case of a 12-year-old girl with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), followed by a rapidly progressive onset of hemophagocytic lymphohistiocytosis (HLH) 9 months later, alongside manifestations of demyelinating encephalopathy. Genetic sequencing revealed a heterozygous nonsense mutation in the PRF1 gene (c.984G>A; p.W328*) and a heterozygous missense mutation in the PRF1 gene (c.1349C>T; p.T450M). Eventually, she died because of no suitable allogeneic hematopoietic stem cell available in time. Our observations suggest that CIPD might represent the initial phenotype of biallelic PRF1 mutation and could serve as an early sign of subsequent HLH. A comprehensive understanding of this condition is paramount for timely diagnosis, treatment, and ultimately improved patient outcomes.
The pore-forming protein perforin (PRF1), belonging to the membrane attack complex/PRF (MACPF) protein family, is essentially involved in the granule-dependent killing activities of cytotoxic T lymphocytes (CTLs) and NK cells. Serving as a definite marker of the killing ability of immune cells, PRF1 participates in the establishment of immune homeostasis, elimination of pathogens, and tumor surveillance (1, 2). Biallelic mutations in the PRF1 gene account for up to 30% cases of familial hemophagocytic lymphohistiocytosis (FHL). Monoallelic and biallelic mutations of the PRF1 gene have also been verified to increase the risk of the development of autoimmune diseases, such as autoimmune lymphoproliferative syndrome, type 1 diabetes mellitus, and multiple sclerosis (MS). It has been shown that the frequency of missense/nonsense PRF1 variations is increased in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) patients, and patients with these variants are more likely to experience relapsing processes and axonal damage (3, 4). Previous studies have identified that three missense mutations of the PRF1 gene, c.272C>T, c.11G>A, and c.1153C>T (resulting in the p.Ala91Val, p.Arg4His, and p.Arg385Trp amino acid substitutions, respectively), and one nonsense mutation, c.1267C>T (leading to the premature stop codon p.Gln423Ter), increased the risk of developing CIDP by 4.47-fold (3). However, the relationship between PRF1 gene variants and CIDP is rarely studied and it is unknown whether patients with CIDP who carry biallelic variants in the PRF1 gene will develop FHL2.
A 12-year-old girl initially noticed weakness in her left upper extremity without any inducement. This weakness progressed to affect her left lower extremity and right limb, eventually rendering her unable to walk unassisted. She also experienced atrophy in her left limb, numbness in both hands and lower extremities, and pain in both heels. Notably, she did not report double or blurred vision, blepharoptosis, facial weakness, dysarthria, or shortness of breath. Prior to the onset of her symptoms, she had no significant medical problems. Four months after onset, she was admitted to a local hospital because of her inability to stand. Electromyography demonstrated significantly decreased motor nerve conduction velocity. Head magnetic resonance imaging (MRI), magnetic resonance angiography (MRA), and spinal MRI results revealed no abnormalities. CSF analysis showed normal white blood cells (WBC) and increased protein levels (1,295.3 mg/L). She was diagnosed with CIDP and initiated treatment with intravenous methylprednisolone (20 mg/kg*3 days) and intravenous immunoglobulin (1 g/kg*2 days) pulse therapy, followed by maintenance steroid (2 mg/kg/day), which partially improved her symptoms. However, during the process of oral steroid reduction, CIDP symptoms worsened repeatedly, promoting an increase in oral steroid dosage by her parent. Approximately 9 months after symptom onset, the girl experienced recurrent fever; at the beginning, low fever was not paid attention to, and the peak temperature gradually increased, worsening limb weakness. She was sent to a local hospital again. Blood test results indicated pancytopenia, mild abnormal liver function, slight decrease in Fib, and significantly elevated C-reactive protein, but no specific pathogen was found. As anti-infective therapy was ineffective and her condition deteriorated, she was referred to our hospital.
Upon admission, her body temperature was up to 39.8°C, and there were two to three heat peaks every day; physical examination demonstrated a pale complexion and hepatosplenomegaly. Neurological examination revealed asymmetrically reduced muscle strength in her limbs, with a Medical Research Council (MRC) score of 3/5 in the left upper limb, 4/5 in the right upper limb, and 2/5 in both lower limbs. Additionally, she exhibited apparent limb atrophy, absent tendon reflexes, impaired perception of temperature, light touch, and pinprick in the limbs, with normal cognition and cranial nerves. Blood test results indicated pancytopenia, increased serum ferritin (>2,000 ng/mL), elevated triglyceride (2.7 mmol/L), and decreased fibrinogen (1.17 g/L). Further tests, including blood culture, EBV DNA, BCG, antinuclear antibody, and anti-ENA antibody showed negative results. The morphology results of medullary cell analysis revealed hemophagocyte. CSF analysis showed normal WBC counts, increased protein levels (1,532.6 mg/L), elevated myelin basic protein (MBP) levels (4.78 nmol/L), and a positive oligoclonal band (OB). Spinal cord MRI results showed abnormal signal in the cervical vertebra and the 3rd lumbar vertebra foramina, extending beyond the spinal canal. Cervical and lumbosacral plexus MRN results revealed extensive enlargement of the brachial plexus trunk and lumbosacral plexus nerve (Figures 1A-1). Fiber track results revealed markedly thickened nerve fibers (Figures 1A-2). Fluorodeoxyglucose-positron emission tomography/computed tomography (FDG-PET/CT) results also showed an increased 18F-FDG uptake in the thickening plexus nerve, but not elsewhere. Electromyography results exhibited significantly decreased motor nerve conduction velocity (MNCV) and sensory nerve conduction velocity (SNCV) in the bilateral peroneal nerve, tibial nerve, and median nerve. It also revealed a vanished F-wave reflex in the left peroneal nerve and significantly prolonged F-wave latency in the left ulnar nerve (Table 1). Based on the patient’s history, clinical manifestations, laboratory findings, and physical examination, a diagnosis of both CIDP and HLH was confirmed.
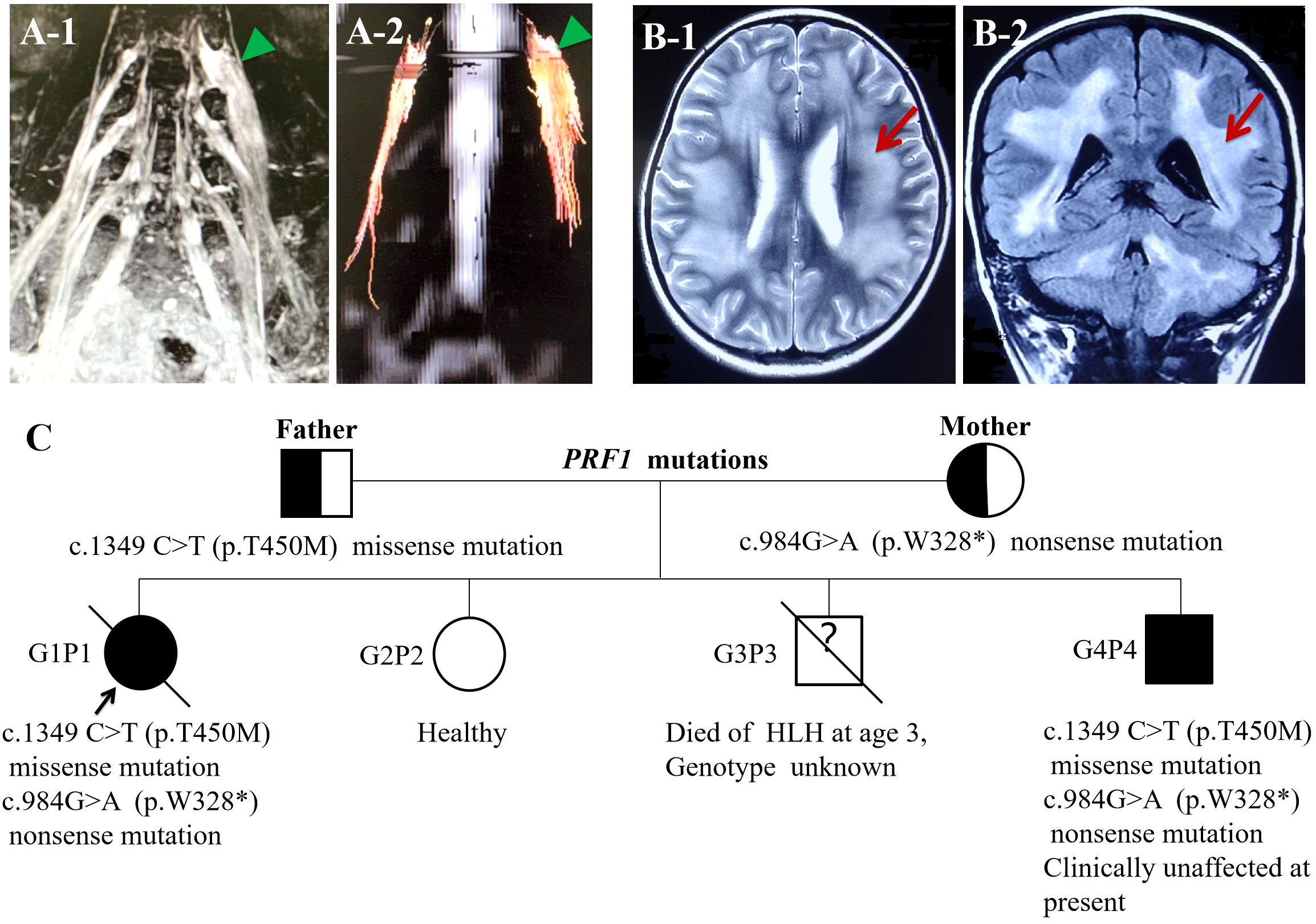
Figure 1 (A-1) Lumbosacral plexus MRN: lumbosacral plexus hypertrophy and hypersignal. (A-2) Fiber track showed markedly thickened nerve fibers. (B) Head MRI: (B-1) axial T2WI demonstrated the change of diffused, symmetrical long T2 signal in white matter of cerebral hemispheres. (B-2) Coronal FLAIR images revealed the change of generalized, symmetrical high signal in white matter of cerebral hemispheres and cerebellar hemispheres. (C) Identified complex heterozygous mutations in the PRF1 gene, c.1349 C>T (p.T450M) (paternal), and c.984>A (p.W328*) (maternal); G3P3 died of HLH at the age of 3 with an unknown genotype; G4P4 carried the same complex heterozygous mutations in the PRF1 gene and clinically unaffected.
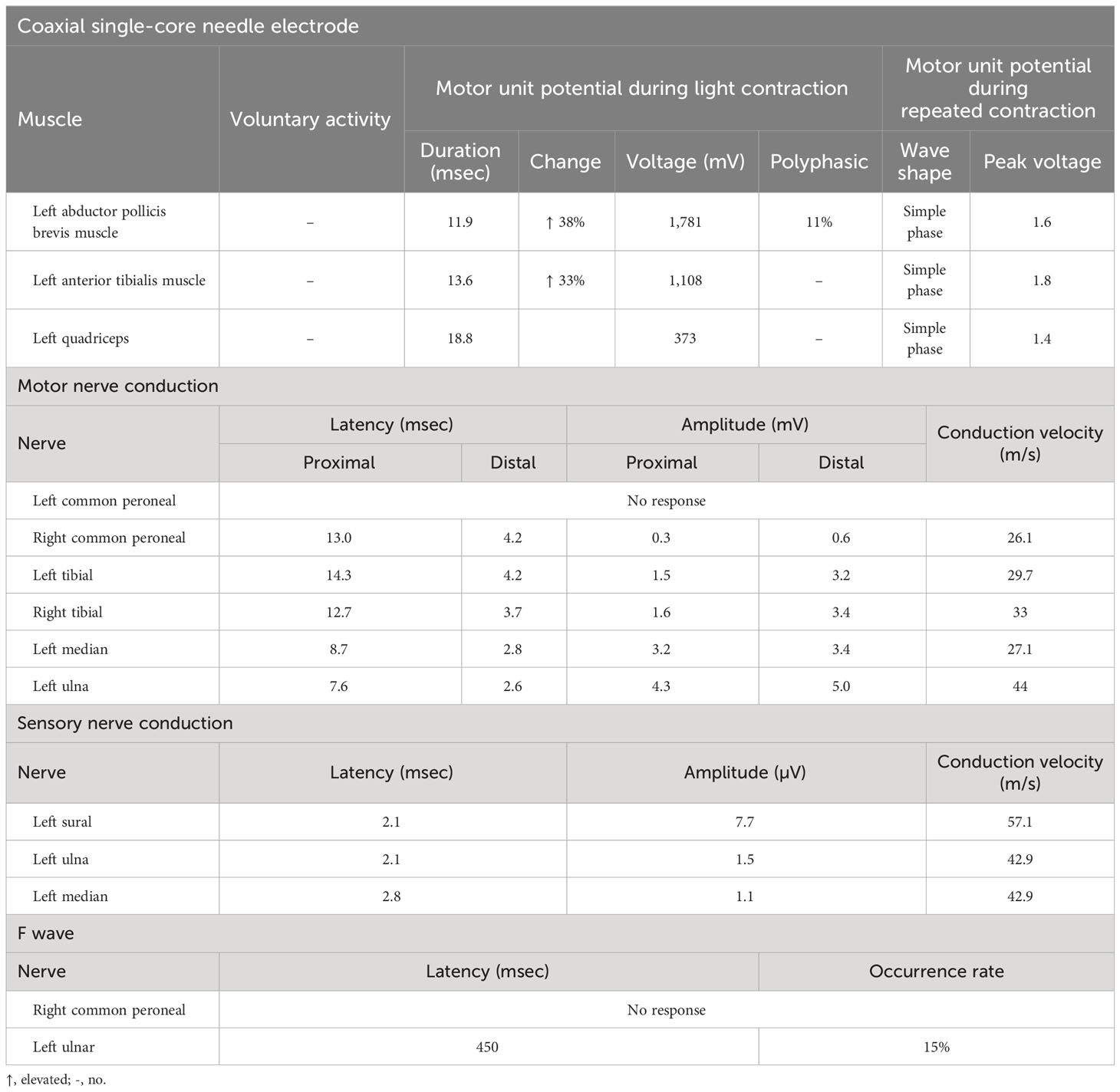
Table 1 The results of electromyography.
Regarding family history, one of her younger brothers died of HLH at the age of 3. Therefore, primary HLH was suspected, and exon sequencing revealed a heterozygous nonsense variation in the PRF1 gene (c.984G>A; p.W328*) and heterozygous missense variation in the PRF1 gene (c.1349C>T; p.T450M). The protein damage prediction results were analyzed by SIFT, PolyPhen-2, and MutationTaster. The c.1349C>T heterozygous missense variation is harmful and pathogenic, whereas c.984G>A heterozygous nonsense variation is suspected to be pathogenic. The abovementioned mutations might have caused the protein function to be affected. The mutation site c.1349C>T (p.T450M) had been reported in patients affected by FHL2 (https://www.ncbi.nlm.nih.gov/clinvar). While the pathogenicity of the variation c.984G>A has not been reported, nor has it been included in the dbSNP database. None of the abovementioned variations were polymorphic changes, which have an extremely low frequency of occurrence in the population. The compound heterozygous variants found in the PRF1 gene of the patient were inherited from their parents. According to the American College of Medical Genetics (ACMG) guidelines, the above variants might be pathogenic variants that caused the onset of the disease. She was ultimately diagnosed with FHL2. Another 3.5-year-old brother of the patient carried the same complex heterozygous mutations in the PRF1 gene but was clinically unaffected at present (Figure 1C).
During hospitalization, she developed demyelinating encephalopathy before the commencement of the HLH-2004 therapeutic regimen, as evidenced by brain MRI, which revealed generalized, symmetrical demyelinating in white matter of the cerebrum and cerebellum hemisphere (Figures 1B-1, B-2), and EEG results indicated a slow wave, although subsequent HLH-2004 protocol provided some improvement. The lack of suitable allogeneic hematopoietic stem cells (HSCs) led to disease recurrence and severe lung infections, ultimately resulting in her passing.
FHL caused by the PRF1 gene is common; however, in this case, the patient experienced symptoms of CIPD for 9 months before the onset of HLH. No direct connections between these two diseases have been reported. It is well known that perforin is important for effector functions of cytotoxic T cells and natural killer (NK) cells. Abnormal or absent perforin function because of PRF1 mutation could lead to impaired killing of target cells, uncontrolled T-cell activation, and high levels of inflammatory cytokines, in turn altering immune system activation and resulting in inflammation and risk of autoimmunity (5).
CIDP is an autoimmune-mediated demyelinating polyneuropathy with chronic progression or remission, resulting from a synergistic interaction of cellular and humoral immune responses. Pathological manifestations include multifocal demyelination of myeloid fibers, endoneuron edema, and inflammatory cell infiltration. Although the etiology and exact pathogenesis remain elusive, clonal expansion of cytotoxic T cells has been observed in the blood and peripheral nerves of patients with CIDP (6, 7). Monoallelic and biallelic pathogenic variations in the PRF1 gene have also been described in patients with CIDP (3, 8). HLH is a multisystem inflammatory disorder; biallelic PRF1 gene pathogenic mutations cause FHL-2, which is characterized by sustained overactivation and excessive proliferation of T lymphocytes and macrophages and increased cytokine levels, leading to infiltration and damage of organs including the bone marrow, liver, and spleen. Thus, we believe that both CIDP and HLH are the result of overactivation of T cells and a cytokine storm.
However, the isolated neurologic manifestations preceding HLH have long been regarded as initial manifestations of primary HLH. A list of child cases that were diagnosed with neurological diseases over the years, including CNS demyelination, MS, intracranial infection, or AIDP, which were later confirmed to have biallelic PRF1 gene pathogenic variations, is summarized in Table 2 (9–22). In a total of 24 children (including this case), from the onset of neurological symptoms to the final genetic diagnosis, the shortest time was 1 month, the longest time up to 5 years, and one-third of the patients had a brain biopsy. All patients who did not undergo hematopoietic stem cell transplantation (HSCT) died. In fact, it turns out that HLH diagnosis and initiation of treatment have always been delayed. We propose that neuropathy should not be considered a common early-onset symptom of HLH, because the diagnostic criteria for HLH do not encompass the characteristics of autoimmune-mediated neuropathy (23). When the neuropathy, such as CIDP in this case, manifests before HLH, there is a high probability that the diagnosis will be limited to neuropathy, and immunotherapy may mask early signs and symptoms of systemic involvement which can result in a missed optimal treatment window when typical HLH eventually presents (9–22). Early diagnosis is essential for better therapeutic approaches, challenging the existing viewpoint and redefining the relationship between CIDP and HLH. The term “perforinopathy” has been proposed to describe the broad-spectrum manifestations resulting from perforin deficiency caused by abnormalities in the PRF1 gene (24). As in this case, in addition to CIDP, the patient also developed leukoencephalopathy before the commencement of the HLH-2004 therapeutic regimen, and subsequent to treatment with the HLH-2004 protocol, the leukoencephalopathy gradually alleviated. Based on this progression, we attribute both CIDP and leukoencephalopathy to an immune inflammatory damage triggered by the PRF1 gene mutation. HLH represents just one extreme on the spectrum of diseases caused by PRF deficiency. A comprehensive understanding of this concept is crucial for early diagnosis, early treatment, and improved patient prognosis.

Table 2 PRF1 gene mutation loci with the nerve system as initial or isolated phenotype and characteristic analysis.
Considering the potential association between the genetic etiology and inevitable development of primary HLH, the occurrence of CIDP may serve as an early warning sign. It has been reported that patients with CIDP carrying PRF1 variations exhibit a higher incidence of relapsing forms and increased likelihood of axonal damage compared with those without these variations. The presence of PRF1 gene variations elevates the risk of developing relapsing forms by fourfold and experiencing axonal damage by 5.3-fold. In contrast, no significant difference was detected in terms of gender distribution, disease duration, INCAT disability score, response to first- and second-line treatment, development of dysautonomia, and CNS involvement (3). In our case, the child also manifested axonal injury with a recurrent course. As such, promotion of genetic analysis is recommended for CIDP patients demonstrating recurrent forms and signs of axonal damage. This allows for a window of opportunity for preparation prior to allograft HSCT, including HLA typing and suitable donor searching, thereby avoiding irreversible consequences such as death.
For the patient’s younger brother, who carries the same complex heterozygous PRF1 gene pathogenic mutations but shows no clinical signs of HLH or “perforinopathy”, the molecular analysis was sufficient for an FHL2 diagnosis. He received allogeneic HSCT before the onset of “perforinopathy” and is currently in good health.
This case highlights the potential for a neurological presentation of “perforinopathy” with CIDP as its initial manifestation. Awareness of this condition is crucial before the onset of HLH, because the disease is treatable. Certainly, for those patients with evidence of a genetic defect, allogeneic HSCT is strongly recommended, as it remains the only curative treatment for primary HLH to date.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving humans were approved by The Ethics Committee of Chinese PLA General Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
L-YH: Data curation, Formal analysis, Investigation, Writing – original draft. LW: Data curation, Formal analysis, Investigation, Writing – original draft. Q-HW: Data curation, Formal analysis, Investigation, Writing – original draft. X-YS: Supervision, Validation, Writing – review & editing. YM: Supervision, Validation, Writing – review & editing. X-FY: Validation, Writing – review & editing. GY: Conceptualization, Supervision, Validation, Writing – review & editing. L-PZ: Conceptualization, Supervision, Validation, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The 14th Five-Year National Key Research and Development Plan (2021YFC2701902); The Capital’s Funds for Health Improvement and Research (2022-1-5081); The National Natural Science Foundation of China (Nos.81071036). All funders had no role in the design and conduct of the study.
We would like to thank the patient and his parents for their cooperation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bolitho P, Voskoboinik I, Trapani JA, Smyth MJ. Apoptosis induced by the lymphocyte effector molecule perforin. Curr Opin Immunol (2007) 19(3):339–47. doi: 10.1016/j.coi.2007.04.007
2. Pipkin ME, Rao A, Lichtenheld MG. The transcriptional control of the perforin locus. Immunol Rev (2010) 235:55–72. doi: 10.1111/j.0105-2896.2010.00905.x
3. Buttini S, Cappellano G, Ripellino P, Briani C, Cocito D, Osio M, et al. Variations of the perforin gene in patients with chronic inflammatory demyelinating polyradiculoneuropathy. Genes Immun (2015) 16:99–102. doi: 10.1038/gene.2014.59
4. Sidore C, Orrù V, Cocco E, Steri M, Inshaw JR, Pitzalis M, et al. PRF1 mutation alters immune system activation, inflammation, and risk of autoimmunity. Mult Scler. (2021) 27(9):1332–40. doi: 10.1177/1352458520963937
5. Osińska I, Popko K, Demkow U. Perforin: an important player in immune response. Cent Eur J Immunol (2014) 39(1):109–15. doi: 10.5114/ceji.2014.42135
6. Schneider-Hohendorf T, Schwab N, Uçeyler N, Göbel K, Sommer C, Wiendl H. CD8+ T-cell immunity in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology (2012) 78:402–8. doi: 10.1212/WNL.0b013e318245d250
7. Mausberg AK, Dorok M, Stettner M, Müller M, Hartung HP, Dehmel T. Recovery of the T-cell repertoire in CIDP by IV immunoglobulins. Neurology (2013) 80:296–303. doi: 10.1212/WNL.0b013e31827debad
8. Iijima M, Yamamoto M, Hirayama M, Tanaka F, Katsuno M, Mori K. Clinical and electrophysiologic correlates of IVIg responsiveness in CIDP. Neurology (2005) 64:1471–5. doi: 10.1212/01.WNL.0000158680.89323.F8
9. Feldmann J, Ménasché G, Callebaut I, Minard-Colin V, Bader-Meunier B, Le Clainche L, et al. Severe and progressive encephalitis as a presenting manifestation of a novel missense perforin mutation and impaired cytolytic activity. Blood (2005) 105(7):2658–63. doi: 10.1182/blood-2004-09-3590
10. Moshous D, Feyen O, Lankisch P, Schwarz K, Schaper J, Schneider M, et al. Primary necrotizing lymphocytic central nervous system vasculitis due to perforin deficiency in a four-year-old girl. Arthritis Rheumatol (2007) 56(3):995–9. doi: 10.1002/art.22442
11. Turtzo LC, Lin DD, Hartung H, Barker PB, Arceci R, Yohay K. A neurologic presentation of familial hemophagocytic lymphohistiocytosis which mimicked septic emboli to the brain. J Child Neurol (2007) 22(7):863–8. doi: 10.1177/0883073807304203
12. Beaty AD, Weller C, Levy B, Vogler C, Ferguson WS, Bicknese A, et al. A teenage boy with late onset hemophagocytic lymphohistiocytosis with predominant neurologic disease and perforin deficiency. Pediatr Blood Cancer. (2008) 50(5):1070–2. doi: 10.1002/pbc.21438
13. Chiapparini L, Uziel G, Vallinoto C, Bruzzone MG, Rovelli A, Tricomi G, et al. Hemophagocytic lymphohistiocytosis with neurological presentation: MRI findings and a nearly miss diagnosis. Neurol Sci (2011) 32(3):473–7. doi: 10.1007/s10072-010-0467-2
14. Dias C, McDonald A, Sincan M, Rupps R, Markello T, Salvarinova R, et al. Recurrent subacute post-viral onset of ataxia associated with a PRF1 mutation. Eur J Hum Genet (2013) 21(11):1232–9. doi: 10.1038/ejhg.2013.20
15. Tesi B, Chiang SC, El-Ghoneimy D, Hussein AA, Langenskiöld C, Wali R, et al. Spectrum of atypical clinical presentations in patients with biallelic PRF1 missense mutations. Pediatr Blood Cancer. (2015) 62(12):2094–100. doi: 10.1002/pbc.25646
16. Khazal S, Polishchuk V, Soffer G, Prinzing S, Gill J, Mahadeo KM. Allogeneic hematopoietic stem cell transplantation is associated with cure and durable remission of late-onset primary isolated central nervous system hemophagocytic lymphohistiocytosis. Pediatr Transplant. (2018) 22(1):e13101. doi: 10.1111/petr.13101
17. Benson LA, Li H, Henderson LA, Solomon IH, Soldatos A, Murphy J, et al. Pediatric CNS-isolated hemophagocytic lymphohistiocytosis. Neurol Neuroimmunol Neuroinflamm. (2019) 6(3):e560. doi: 10.1212/NXI.0000000000000560
18. Feng WX, Yang XY, Li JW, Gong S, Wu Y, Zhang WH, et al. Neurologic manifestations as initial clinical presentation of familial hemophagocytic lymphohistiocytosis type2 due to PRF1 mutation in Chinese pediatric patients. Front Genet (2020) 11:126. doi: 10.3389/fgene.2020.00126
19. Caldito NG, Lorenzo J, Wang CX. Familial CNS-isolated hemophagocytic lymphohistiocytosis due to a novel PRF1 mutation triggered by SARS-CoV2. Ann Indian Acad Neurol (2022) 25(6):1170–3. doi: 10.4103/aian.aian_719_22
20. You Y, Wu W, Li B. Familial hemophagocytic phohistiocytosis induced by PRF1 mutation with neurologic manifestations as the initial clinical presentations: A case report. Med (Baltimore). (2023) 102(26):e34198. doi: 10.1097/MD.0000000000034198
21. Gupta J, Jauhari P, Kumar A, Gulati S, Chakrabarty B, Gupta AK, et al. Primary hemophagocytic lymphohistiocytosis with prolonged primary neurologic presentation. Pediatrics (2023) 151(4):e2022057848. doi: 10.1542/peds.2022-057848
22. Del Giudice E, Savoldi G, Notarangelo LD, Di Benedetto L, Manganelli F, Bruzzese E, et al. Acute inflammatory demyelinating polyradiculoneuropathy associated with perforin-deficient familial haemophagocytic lymphohistiocytosis. Acta Paediatr (2003) 92(3):398–401. doi: 10.1111/j.1651-2227.2003.tb00566.x
23. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48:124–31. doi: 10.1002/pbc.21039
Keywords: familial hemophagocytic lymphohistiocytosis, chronic inflammatory demyelinating polyradiculoneuropathy, demyelination of the central nervous system, perforin, perforinopathy
Citation: Hu L-Y, Wan L, Wang Q-H, Shi X-Y, Meng Y, Yang X-F, Yang G and Zou L-P (2023) Case Report: Chronic inflammatory demyelinating polyradiculoneuropathy rather than hemophagocytic lymphohistiocytosis—the initial phenotype of PRF1 gene mutation. Front. Immunol. 14:1306338. doi: 10.3389/fimmu.2023.1306338
Received: 03 October 2023; Accepted: 21 November 2023;
Published: 11 December 2023.
Edited by:
Mattia Bellan, University of Eastern Piedmont, ItalyReviewed by:
Wen-Xiong Chen, Guangzhou Medical University, ChinaCopyright © 2023 Hu, Wan, Wang, Shi, Meng, Yang, Yang and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Ping Zou, em91bGlwaW5nMjFAc2luYS5jb20=; em91bGlwaW5nQGhvdG1haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.