 Roy A. Mariuzza
Roy A. Mariuzza Daichao Wu
Daichao Wu Brian G. Pierce
Brian G. Pierce
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 17 November 2023
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1303304
This article is part of the Research TopicHarnessing TCR - Peptide/MHC binding Specificity to Tackle DiseaseView all 9 articles
Adoptive cell therapy (ACT) with tumor-specific T cells has been shown to mediate durable cancer regression. Tumor-specific T cells are also the basis of other therapies, notably cancer vaccines. The main target of tumor-specific T cells are neoantigens resulting from mutations in self-antigens over the course of malignant transformation. The detection of neoantigens presents a major challenge to T cells because of their high structural similarity to self-antigens, and the need to avoid autoimmunity. How different a neoantigen must be from its wild-type parent for it to induce a T cell response is poorly understood. Here we review recent structural and biophysical studies of T cell receptor (TCR) recognition of shared cancer neoantigens derived from oncogenes, including p53R175H, KRASG12D, KRASG12V, HHATp8F, and PIK3CAH1047L. These studies have revealed that, in some cases, the oncogenic mutation improves antigen presentation by strengthening peptide–MHC binding. In other cases, the mutation is detected by direct interactions with TCR, or by energetically driven or other indirect strategies not requiring direct TCR contacts with the mutation. We also review antibodies designed to recognize peptide–MHC on cell surfaces (TCR-mimic antibodies) as an alternative to TCRs for targeting cancer neoantigens. Finally, we review recent computational advances in this area, including efforts to predict neoepitope immunogenicity and how these efforts may be advanced by structural information on peptide–MHC binding and peptide–MHC recognition by TCRs.
Adaptive cell therapy (ACT) with tumor-specific T cells has been demonstrated to mediate durable cancer regression in patients with metastatic melanoma, breast, cervix, colon, and bile duct cancers (1–6). The therapeutic effect of these ex vivo-expanded tumor-infiltrating lymphocytes (TILs) is principally mediated by CD8+ cytotoxic T cells (7), with an additional contribution from CD4+ T cell (5). The principal target of tumor-specific T cells are neoantigens resulting from non-synonymous somatic mutations in self-antigens during malignant transformation (6, 8). The identification of neoantigens in individual patients, and of the T cells that recognize them, has been greatly accelerated by recent technical advances in high-throughput T cell-based assays and mass spectrometry (9).
A daunting challenge in the development of broadly useful neoantigen-based ACT is the unique neoantigen repertoire of individual cancer patients (private neoantigens) (8). There exist few common neoantigens among patients, even for patients with similar cancers, that can be targeted therapeutically (public neoantigens). For instance, an analysis of patients with gastrointestinal cancers found that nearly all (99%) of neoantigenic determinants (neoepitopes) targeted by neoantigen-reactive TILs were private (10). Nevertheless, a few public cancer neoantigens have been discovered (11–16). Of particular interest are neoantigens derived from oncogenes such as TP53 and KRAS that bear driver mutations. This is because driver mutations are tumor-specific, essential for cancer cell fitness and proliferation, and likely to be present in every cell within the cancer (17).
The detection of neoantigens represents a major challenge to T cells because of their high similarity to wild-type self-peptides. Exactly how different a neoantigen must be from its wild-type parent for it to overcome self-tolerance and induce a T cell response is poorly understood. With the goal of understanding T cell recognition of cancer neoantigens at the molecular level, a number of crystal structures have been recently reported of TCRs bound to various cancer neoantigens and MHC class I or class II molecules (Table 1) (18–27). These neoantigens include p53R175H (20, 21), KRASG12D (22, 23, 27), KRASG12V (24), HHATp8F (25), PIK3CAH1047L (26), and TPIT28I (18, 19). Structures have also been determined for TCRs bound to epitopes from the tumor-associated antigens NY-ESO-1, MART-1, MAGEA4, and Melan A bound to HLA-A2 (28–31). However, it is important to note these are not neoantigens but rather unmutated self-antigens that are selectively expressed or overexpressed in certain types of cancer.
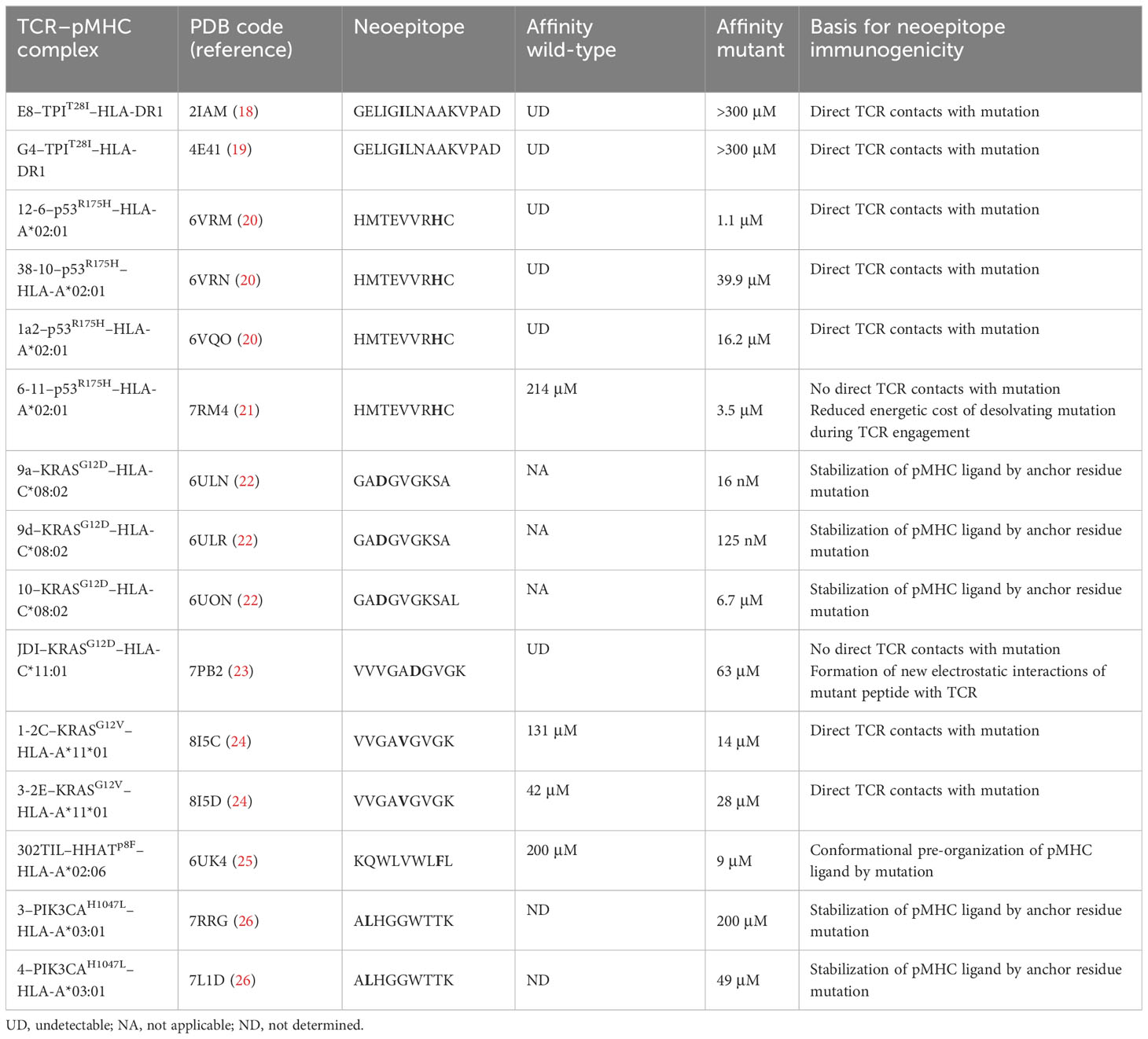
Table 1 Structures of TCR–pMHC complexes involving cancer neoantigens.
Collectively, structural studies of mutated self-antigens have provided insights into the multiple mechanisms TCRs employ to detect cancer neoantigens and into how mutations confer immunogenicity to normally cryptic self-peptides. In some cases, the mutation strengthens peptide–MHC binding, improving the presentation of neoepitopes against which the immune system is not tolerant. In other cases, the mutation does not affect peptide–MHC binding or antigen presentation, yet generates a peptide that is sufficiently different physically from its wild-type parent to be immunogenic. Detection of such peptides by T cells may occur via direct interactions between TCR and the oncogenic mutation, or may involve energetically driven or other indirect strategies not requiring direct contacts.
Here we review structural and biophysical studies of TCR recognition of cancer neoantigens with a focus on how T cells distinguish mutant from wild-type epitopes and how neoepitope-specific TCRs may be employed for ACT. We also review work on designing antibodies that mimic TCR recognition of pMHC on cell surfaces as an alternative approach to immunotherapeutic targeting of cancer neoantigens. Finally, we review recent computational efforts to predict neoepitope immunogenicity and how some of these efforts have utilized structural information on peptide binding to MHC and pMHC recognition by TCRs.
The first structural studies of TCR recognition of a cancer neoantigen involved two tumor-specific TCRs (E8 and G4) isolated from TILs of a melanoma patient in complex with the MHC class II molecule HLA-DR1 and a mutated self-peptide derived from the glycolytic enzyme triosephosphate isomerase (TPI) (Table 1) (18, 19). A natural mutation in TPI resulted in replacement of an isoleucine residue for threonine in a neoepitope corresponding to residues 23–37 of TPIT28I (GELIGILNAAKVPAD; mutant amino acid in bold) (32). This substitution resulted in >100,000-fold improved recognition of mutant TPIT28I relative to wild-type TPI, thereby revealing this neoepitope to T cells (18, 32). However, the TPIT28I neoantigen, unlike neoantigens such as p53R175H and KRASG12V that are derived from mutant oncogenes, was only expressed in a single melanoma patient. This limitation precludes wide use of TPIT28I-specific or similar TCRs in ACT.
Surprisingly, TCRs E8 and G4 displayed very low affinities for TPIT28I–HLA-DR1, as measured by surface plasmon resonance (SPR), with dissociation constants (KDs) exceeding 300 μM (Table 1) (18). However, as measured by two-dimensional (2D) mechanical-based adhesion assays, the 2D affinity of TCR E8 for TPIT28I–HLA-DR1 was comparable to other TCR–pMHC class II interactions (33). Importantly, 2D measurements are made in situ at cell–cell junctions, whereas three-dimensional (3D) measurements from SPR use soluble proteins isolated from their cellular contexts. As such, 2D parameters correspond to biology much better than their 3D counterparts (34–37). No TCR binding to wild-type TPI–HLA-DR1 was detected, in agreement with functional assays showing greatly diminished T cell activation by the wild-type peptide (18).
Replacement of isoleucine by threonine at position P3 of TPIT28I did not alter peptide affinity for HLA-DR1 or affect the conformation of the peptide in the MHC binding groove (38). Rather, structural changes in the TCR–pMHC interface are primarily responsible for improved T cell recognition of TPIT28I. TCRs E8 and G4 utilize the same Vα region (TRAV13-1) but have different CDR3α sequences and different Vβ regions (TRBV6-6 and TRBV5-8, respectively). Crystal structures of E8 and G4 bound to TPIT28I–HLA-DR1 showed that both TCRs dock on pMHC in the canonical diagonal orientation, with the Vα domain over the β1 helix of HLA-DR1 and the Vβ domain over the α1 helix (Figures 1A, B) (18, 19). However, E8 and G4 recognize TPIT28I in markedly different ways. In the E8–TPIT28I–HLA-DR1 complex, the CDR1 and CDR3 loops of both Vα and Vβ make similar contributions to contacts with TPIT28I (Figure 1C). By contrast, in the G4–TPIT28I–HLA-DR1 complex, peptide recognition is mediated almost completely by CDR3α and CDR3β (Figure 1D).
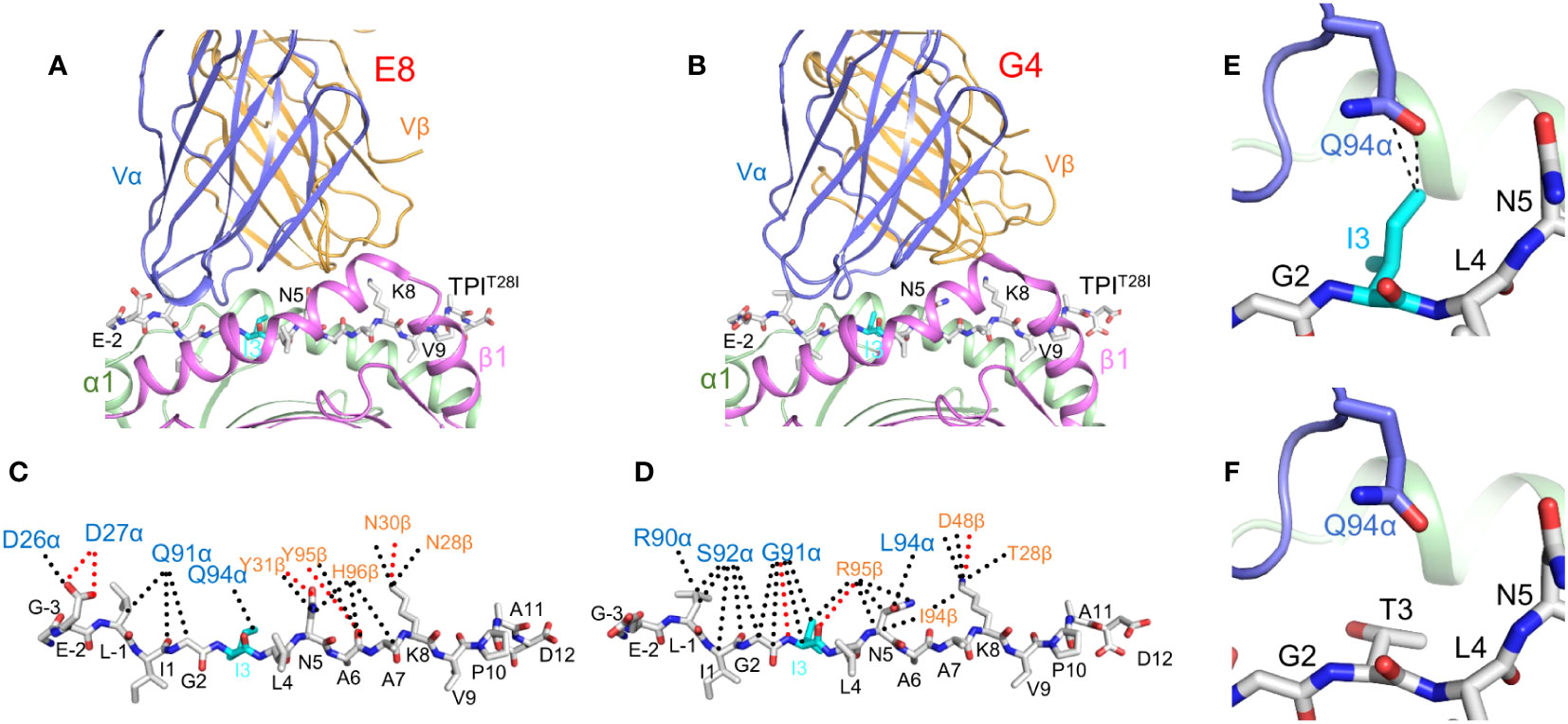
Figure 1 TCR recognition of the HLA-DR1-restricted TPIT28I cancer neoantigen. (A) Interactin of TCR E8 with TPIT28I and HLA-DR1 (ribbon diagram) (PDB accession code 2IAM) (18). TCR α chain, blue; TCR β chain, gold; MHC α chain, green; MHC β chain, magenta. The peptide is in ball-and-stick format, with carbon atoms in gray, nitrogen atoms in blue, and oxygen atoms in red. (B) Interaction of TCR G4 with TPIT28I and HLA-DR1 (4E42) (19). (C) Interactions between TCR E8 and TPIT28I. Peptide residues are identified by a one-letter amino acid designation, followed by position number. Hydrogen bonds are red dotted lines and van der Waals contacts are black dotted lines. For clarity, not all van der Waals contacts are shown. The mutant P3 Ile residue is cyan. (D) Interactions between TCR G4 and TPIT28I. (E) Van der Waals contacts (black dotted lines) between TCR E8 and mutant P3 Ile residue. (F) Absence of contacts between E8 and wild-type P3 Thr residue (2IAN) (18).
In the E8–TPIT28I–HLA-DR1 complex, CDR3α and CDR3β form a dome-shaped pocket that accommodates two TPIT28I residues, P3 Ile (the mutant amino acid) and P5 Asn. The δ1 methyl group of P3 Ile projects from the TPIT28I–HLA-DR1 surface towards CDR3α Gln94, with which it forms multiple van der Waals contacts (Figure 1E). Replacement of P3 Ile by threonine (the wild-type amino acid) results in loss of these contacts, a reduction in shape complementarity, and a decrease in buried surface at the mutation site, where P3 Ile occupies a mainly hydrophobic pocket on TCR E8 (Figure 1F) (18). In the G4–TPIT28I–HLA-DR1 complex, the main chain of P3 Ile makes three hydrogen bonds with CDR3α Gly91 and CDR3β Arg (19). In the structure of wild-type TPI–HLA-DR1 (38), P3 Thr, unlike P3 Ile, is completely buried against the HLA-DR1 α chain. This renders P3 Thr inaccessible to TCR G4, which results in a strong preference for isoleucine over threonine at P3 in T cell activation assays (18).
TP53 (tumor protein 53) was the first tumor suppressor gene to be discovered (39). It is inactivated in the large majority of human cancers (40) and is the most frequently mutated gene across all cancers (41). Driver mutations in TP53 cause most of the key features of cancer cells, notably genomic instability, proliferation, and metastasis (42, 43). A high percentage of TP53 mutations are located at positions R175, G245, R248, R273, and R282. These mutations cluster in the central DNA-binding domain of p53 and affect DNA binding (41). They are attractive targets for immunotherapy because they are associated with tumor progression and confer a growth advantage to cancer cells.
The immunogenicity of p53 mutations was demonstrated by the detection in cancer patients of T cell responses against several shared p53 neoantigens, mainly R175H and R248W (16, 44). The R175H driver mutation is the most frequent mutation in TP53. It is also the most common mutation in any tumor suppressor gene (45). Several TCRs (12-6, 38-10, 1a2, and 6-11) have been isolated from patients with epithelial cancers that recognize a neoepitope corresponding to residues 168–176 of p53R175H (HMTEVVRHC; mutant amino acid in bold) (16, 44). These TCRs are restricted by HLA-A*02:01, which is the most common MHC class I allele in the U.S. and Chinese populations (46). In a clinical trial, a breast cancer patient infused with autologous peripheral blood lymphocytes transduced with an HLA-A*02-restricted TCR (6–11) specific for p53R175H experienced ~55% tumor regression that lasted 6 months (47).
As measured by SPR, TCRs 12-6, 38-10, and 1a2 exhibited exquisite specificity for p53R175H–HLA-A2, with KDs ranging from 1 μM to 40 μM and no detectable binding to wild-type p53–HLA-A2 (Table 1). These results agree with functional assays showing that T cells transduced with these TCRs could be triggered by very low (subnanomolar) concentrations of mutant p53R175H peptide but not by wild-type p53 peptide (16, 44). By contrast, TCR 6-11 was not as highly specific for p53R175H–HLA-A2 as TCRs 12-6, 38-11 or 1a2, since its affinity for p53R175H–HLA-A2 (KD = 3.5 μM) was only ~60-fold higher than for p53–HLA-A2 (KD = 214 μM) (Table 1) (21). TCR affinities for the p53R175H neoantigen are comparable to those of TCRs specific for viral or other foreign antigens (KD = 1–50 μM) but substantially higher than the affinities of autoimmune TCRs specific for unmutated self-antigens (KD > 100 μM) (48). These affinity characteristics also apply to MHC class I-restricted TCRs that recognize other neoantigens (Table 1).
Structures of the wild-type p53–HLA-A2 and mutant p53R175H–HLA-A2 complexes were determined in order to understand how the conservative arginine-to-histidine mutation in p53, which replaces one positively charged residue by another, renders p53R175H immunogenic (Figure 2A) (20). In both complexes, the side chains of anchor residues P2 Met and P9 Cys are situated in the peptide-binding groove of HLA-A2, while the side chains of P1 His, P4 Glu, P7 Arg, and P8 Arg/His extend up from the groove. Comparison of the p53–HLA-A2 and p53R175H–HLA-A2 complexes showed that structural differences that revealed the p53R175H peptide to T cells (16, 44) are confined to the mutation site at P8 (Figure 2A) (20).
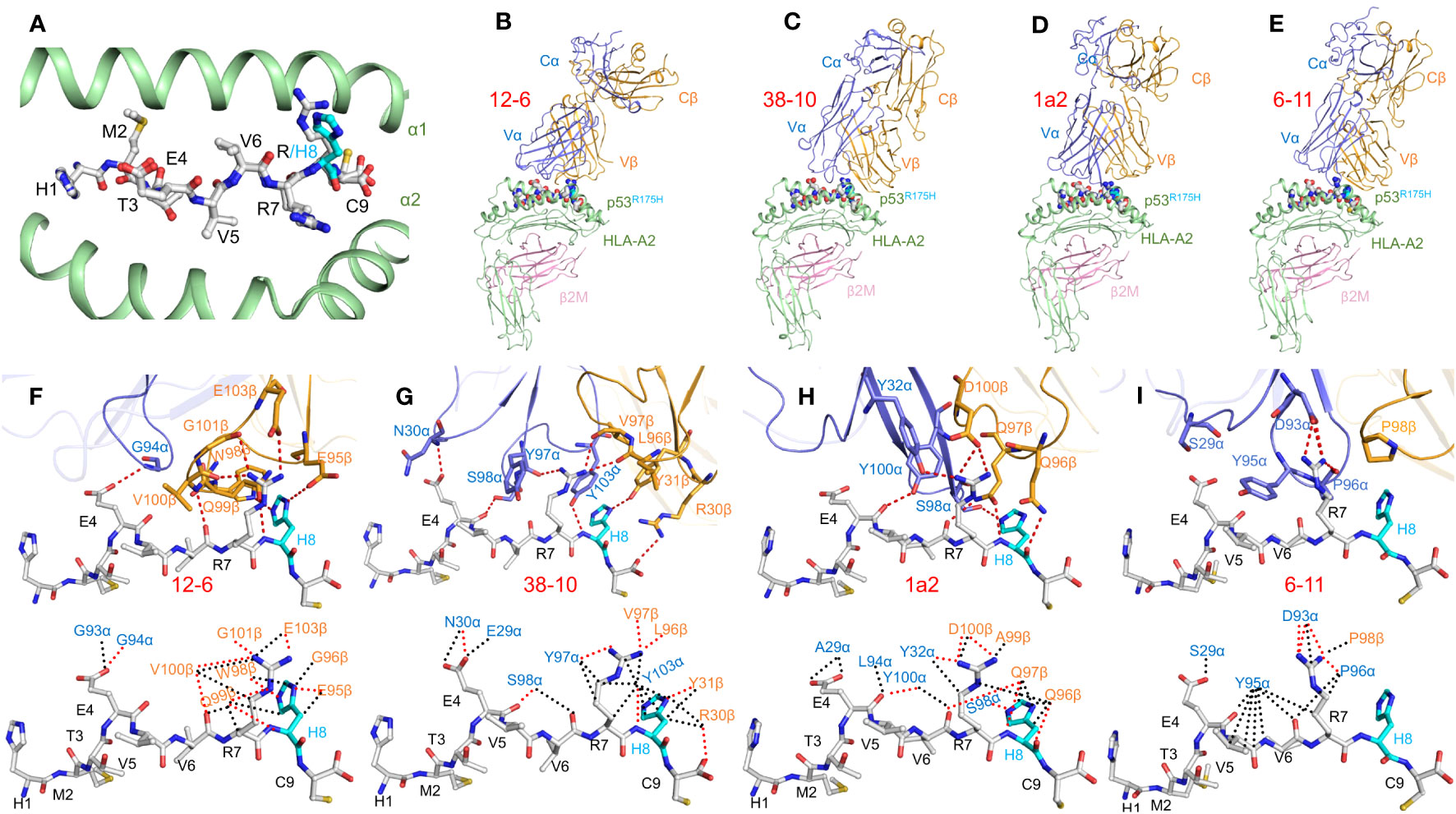
Figure 2 TCR recognition of the HLA-A2-restricted p53R175H neoepitope. (A) Conformation of wild-type and mutant p53 peptides bound to HLA-A2. Top view of the superposed p53–HLA-A2 and p53R175H–HLA-A2 complexes (6VR1 and 6VR5) (20). Carbon atoms of wild-type and mutant p53 peptides are gray; nitrogen atoms are blue; oxygen atoms are red; sulfur atoms are orange. The mutant P8 His residue is cyan. HLA-A2 is green. (B–E) Side view of the 12-6–p53R175H–HLA-A2, 38-10–p53R175H–HLA-A2, 1a2–p53R175H–HLA-A2, and 6-11–p53R175H–HLA-A2 complexes (6VRM, 6VRN, 6VQO, and 7RM4) (20, 21). TCR α chain, blue; TCR β chain, gold; HLA-A2 heavy chain, green; β2-microglobulin (β2m), pink. (F–I) (upper panels) Interactions between TCRs 12-6, 38-10, 1a2, and 6-11 and the p53R175H peptide. The side chains of contacting residues are shown in stick representation. The mutant P8 His residue is cyan. (lower panels) Comparison of interactions between 12-6, 38-10, 1a2, and 6-11 and the p53R175H peptide. Hydrogen bonds are red dotted lines and van der Waals contacts are black dotted lines.
In order to understand how TCRs 12-6, 38-10, 1a2, and 6-11 discriminate between wild-type and mutant p53 epitopes, structures were determined of their corresponding complexes with p53R175H–HLA-A2 (20, 21) (Figures 2B–E). These TCRs utilize unrelated Vα and Vβ gene segments. TCRs 12-6, 38-10, and 1a2, but not 6-11, are displaced towards the C-terminus of the p53R175H peptide. Importantly, this is the site of the driver mutation at P8 (Figures 2F–I). As a consequence, ~80% of contacts between TCRs 12-6, 38-10, and 1a2 and the p53R175H peptide involves C-terminal residues P7 Arg and P8 His, in contrast to most TCRs, including 6-11, which typically target the central portion of peptides (P4–P6) (Figure 2I) (21). In each case, the imidazole ring of P8 His is tightly sandwiched between the HLA-A2 α1 helix and the TCR CDR3 loops (Figures 2F–H). TCRs 12-6, 38-10, and 1a2 discriminate between mutant and wild-type p53 by focusing on the R175H mutation at P8 and minimizing interactions with the central and N-terminal portions of p53R175H, which are structurally identical in the wild-type peptide (20). The dramatic loss of affinity for wild-type p53 is mainly due to disruption side chain–side chain hydrogen bond interactions involving P8 His upon replacing this residue by arginine.
In sharp contrast to TCRs 12-6, 38-10, and 1a2, TCR 6-11 makes no direct contacts with the R175H mutation (Figure 2I), but is nevertheless able to distinguish mutant from wild-type p53 (21). Since the peptide residues that do contact 6-11 are highly superimposable in the structures of mutant p53R175H–HLA-A2 and unbound wild-type p53–HLA-A2 (Figure 2A), the mechanism underlying discrimination is not obvious. However, structure-based in silico mutagenesis revealed that the 60-fold loss in 6-11 binding affinity for wild-type p53 compared to p53R175H is attributable to the greater energetic cost of desolvating R175 in the wild-type p53 peptide than H175 in the mutant during complex formation (21). This indirect strategy for neoantigen recognition by 6-11 is fundamentally different from the direct strategies used by most other TCRs and emphasizes the multiple mechanisms T cells may employ to recognize tumor but not normal cells.
Mutations in the KRAS oncogene occur in ~15% of all cancers, with especially high frequencies in colorectal and pancreatic cancers (49). KRAS encodes a small GTPase that alternates between an inactive GDP-bound state and an active GTP-bound state which regulates cell survival, growth, and differentiation (50). A hotspot for oncogenic mutations in the KRAS protein is glycine at position 12, with G12D, G12V, and G12C the most common mutations (51). These driver mutations impair GTPase hydrolytic activity and lock KRAS in the active state, leading to constitutive oncogenic signaling (52). Crystal structures have been determined of TCRs bound to KRASG12D (22, 23) and KRASG12V (24) neoepitopes presented by HLA-C*08:02 and HLA-A*11:01 MHC class I molecules, respectively (Table 1).
In a landmark clinical study of ACT, a patient with metastatic colorectal cancer was treated with four different CD8+ T cell clones that targeted a KRASG12D neoepitope in the context of HLA-C*08:02 (2). All metastases that retained HLA-C*08:02 expression underwent regression. TCRs from these four clones (9a, 9b, 9c, and 9d) recognized a nonamer of KRASG12D (GADGVGKSA) bound to HLA-C*08:02 (2), while a fifth TCR (TCR10) was decamer-specific (GADGVGKSAL) (27). In marked contrast to wild-type TPI and p53 peptides, which bound tightly to MHC (18, 20), wild-type nonamer and decamer KRAS peptides did not, as indicated by the failure of these peptides, unlike their mutant counterparts, to stabilize HLA-C expression on TAP-deficient cells (22). This result suggested that TCR tumor specificity arose from preferential KRASG12D presentation by HLA-C rather than differential TCR recognition of KRASG12D over wild-type KRAS.
Crystal structures of HLA-C*08:02 in complex with KRASG12D nonamer and decamer peptides revealed that, in both cases, P3 Asp (the mutant residue) makes a salt bridge with Arg156 on the HLA-C α2 helix (Figure 3A) (22). This salt bridge cannot form with P3 Gly, which probably explains the instability of wild-type KRAS–HLA-C complexes and their inability to activate T cells. The nonamer and decamer KRASG12D peptides are anchored to HLA-C via similar interactions at the N- and C-termini, but adopt different conformations at the center between P5 Val and P8 Ser due to a bulge at P7 Lys in the decamer (Figure 3B).
Figure 3 Presentation of KRASG12D neoepitopes by HLA-C*08:02. (A) KRASG12D nonamer and decamer peptides (gray and yellow, respectively) form a salt bridge (red solid lines) with Arg156 of the HLA-C α2 helix (green) through the mutant P3 Asp anchor residue (6ULI and 6ULK) (22). Wild-type P3 Gly cannot make this salt bridge. (B) Structures KRASG12D nonamer and decamer peptides bound to HLA-C*08:02. (C) Interactions of P7 Lys of KRASG12D nonamer with CDR2β Tyr48 and Glu49 of TCR9a (orange) (6ULN) (22). (D) Interactions of CDR3α Gln98 of TCR9a (blue) with P5 Val of KRASG12D nonamer and Gln155 and Arg156 of the HLA-C α2 helix.
Structures of TCR9a and TCR9d (both Vα4/Vβ5) bound to KRASG12D nonamer and HLA-C showed that, in both complexes, P7 Lys forms a hydrogen bond with CDR2β Tyr48 and a salt bridge with CDR2β Glu49 (Figure 3C). In addition, CDR3α Gln98 makes hydrogen bonds with the carbonyl group of P5 Val and with Gln155 and Arg156 of the HLA-C α2 helix (Figure 3D). The different conformations of nonamer and decamer KRASG12D peptides (Figure 3B) result in distinct interactions with nonamer- and decamer-specific TCRs. For example, while both nonamer-specific TCR9a and decamer-specific TCR10 form salt bridge interactions with P7 Lys, TCR10 uses CDR3β Asp95 rather than CDR2β Glu49.
As measured by SPR, the affinities (KDs) of KRASG12D-specific TCRs ranged from a typical 6 μM (TCR10) to an exceptionally high 16 nM (TCR9a) (Table 1) (22). In the case of treating a colorectal cancer patient with ex vivo expanded TILs specific for KRASG12D presented by HLA-C*08:02, the transferred T cells expressed four TCRs, TCR9a, 9b, 9c, and 10 (2). Surprisingly, T cells bearing TCR9a, the highest-affinity receptor, were undetectable 40 days post-transfer. Indeed, an inverse correlation was observed between TCR affinity and in vivo persistence, with T cells expressing TCR10, the lowest-affinity receptor, maintained in the periphery the longest (9 months post-transfer) (2). A possible explanation for this counterintuitive result is that higher-affinity T cells engage their cognate antigen more effectively, leading to increased activation-induced cell death (AICD), in which activation through the TCR results in apoptosis rather than proliferation (17, 53). This clinical study, while limited to a single patient, suggests that TCRs with affinities in the low micromolar range may be most efficacious for ACT.
Poole et al. (23) isolated a TCR (JDI) from the peripheral blood mononuclear cells (PBMC) of a healthy donor that recognizes a KRASG12D decamer peptide (VVVGADGVGK) presented by HLA-A*11:01. This peptide partially overlaps, but is distinct from, the KRASG12D peptides presented by HLA-C*08:02 discussed above. TCR JDI bound mutant KRASG12D–HLA-A*11 with KD = 63 μM, with no measurable affinity for ubiquitously expressed wild-type KRAS–HLA-A*11 (Table 1), in agreement with functional assays using T cells transduced with JDI (23). With the aim of maximizing the anti-tumor activity of TCR JDI for possible immunotherapeutic applications, phage display was used to engineer a variant (JDIa41b1) with a 106-fold affinity improvement (KD = 0.7 pM) over the parental TCR that retained the ability to distinguish KRASG12D from wild-type KRAS, although JDIa41b1 did acquire measurable affinity for KRAS–HLA-A*11 (KD = 3 μM).
A major concern with engineered high-affinity TCRs is the risk of cross-reactivity (54), which may result in adverse clinical events (55). In a striking case, an affinity-enhanced TCR targeting the MAGE-A3 melanoma antigen unexpectedly cross-reacted with an epitope from the muscle protein titin, resulting in cardiovascular toxicity and death in two patients who received cells transduced with the modified TCR (56). To address this concern for affinity-enhanced JDIa41b1, the TCR was panned against a phage-displayed peptide–HLA-A*11 library encoding >106 variants to generate a peptide specificity profile (23). This profile was then used to identify bona fide self-peptides that might act as structural mimics of the neoepitope. However, none of these self-peptides were recognized by TCR JDIa41b1, easing concerns about potentially deleterious cross-reactivity. In addition, the affinity-enhanced TCR, fused to an anti-CD3 single-chain Fv fragment, mediated selective killing of cancer cells expressing KRASG12D (23).
As discussed above, poor presentation of wild-type KRAS by HLA-C*08:02 is likely the mechanism underpinning TCR specificity for KRASG12D (22). By contrast, the KRAS and KRASG12D peptides bound equally well to HLA-A*11:01, ruling out TCR selectivity based on peptide presentation. Crystal structures of TCRs JDI and JDIa41b1 bound to KRASG12D–HLA-A*11, and of JDIa41b1 bound to wild-type KRAS–HLA-A*11, have provided insights into the molecular basis for TCR selectivity for KRASG12D (Figures 4A, B) (23). These structures showed no significant differences in TCR interactions with KRASG12D versus wild-type KRAS. However, structures of KRASG12D–HLA-A*11 and KRAS–HLA-A*11 without a bound TCR revealed that both mutant and wild-type KRAS peptides underwent an induced fit conformational change upon TCR engagement (Figure 4C), with P6 Asp in the KRASG12D–HLA-A*11 complex forming multiple stabilizing interactions with the HLA-A*11 F pocket (Figure 4D). Thermodynamic analysis and molecular dynamics simulations indicate that tighter TCR binding to KRASG12D–HLA-A*11 compared to KRAS–HLA-A*11 is driven by a greater net formation of new electrostatic interactions with TCR by the mutant peptide, at the cost of a greater order-disorder transition (23). Thus, as in the case of p53R175H recognition by TCR 6-11 (21), TCRs can use indirect energetically driven strategies for preferential neoantigen binding.
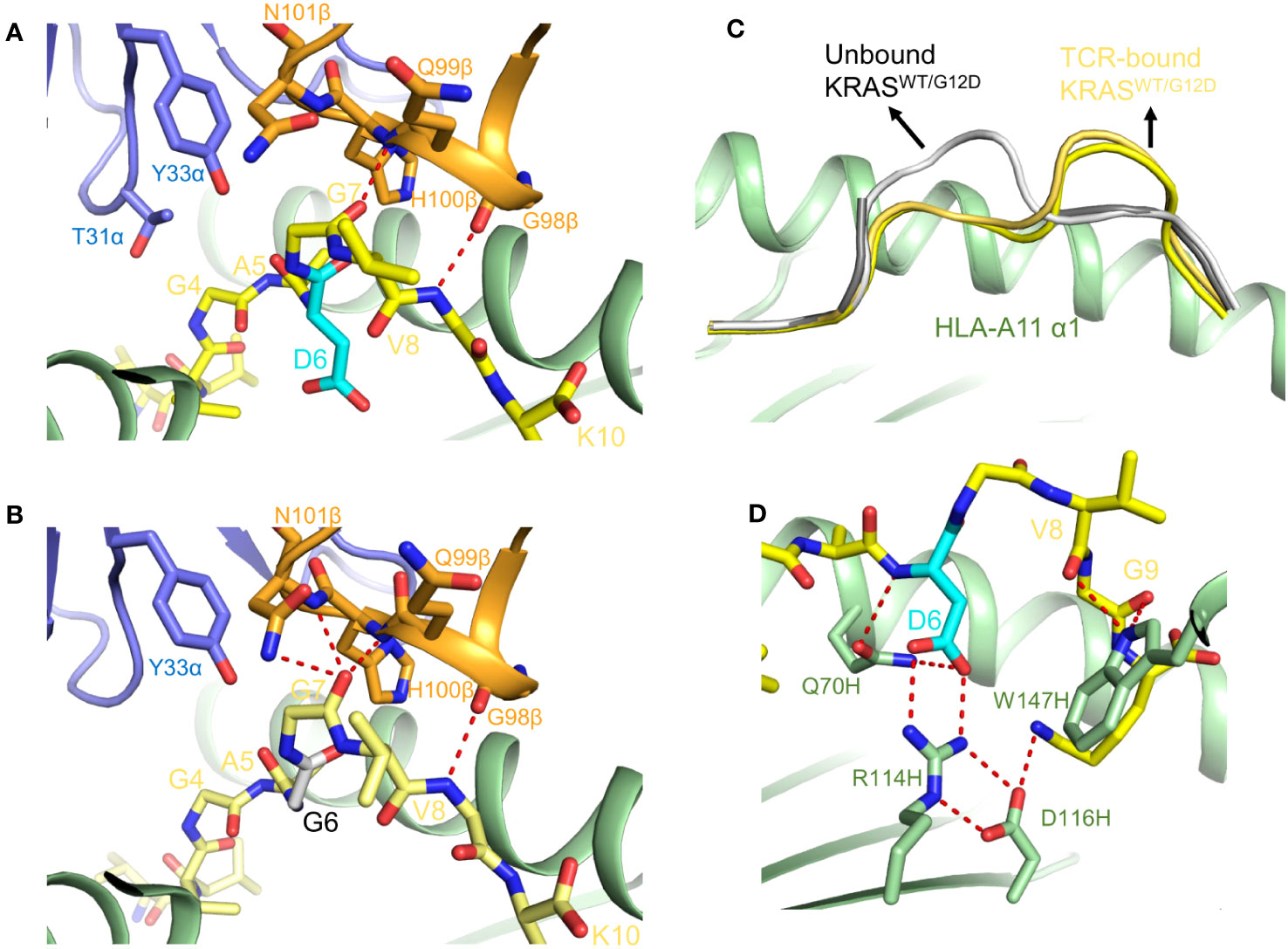
Figure 4 TCR recognition of the HLA-A*11:01-restricted KRASG12D neoepitope. (A) Interactions between TCR JDI and the KRASG12D peptide (7PB2) (23). The mutant P6 Asp residue is cyan. (B) Interactions between TCR JDI and wild-type KRAS peptide (7OW5) (23). (C) Conformational changes in mutant and wild-type KRAS peptides upon binding TCR JDI. Superposition of unbound KRASG12D–HLA-A*11 and KRAS–HLA-A*11 ligands shows that the peptides adopt an open conformation (7OW4 and 7OW3) (23). Superposition of TCR-bound KRASG12D–HLA-A*11 and KRAS–HLA-A*11 ligands shows that the peptides adopt a closed conformation. (D) Interaction network between P6 Asp and HLA F pocket residues in the TCR JDI–KRASG12D–HLA-A*11 complex.
Besides KRASG12D, another frequent oncogenic mutation in KRAS is replacement of glycine at position 12 by valine (KRASG12V) (51). Lu et al. isolated TCRs (1-2C and 3-2E) specific for a KRASG12V nonamer (VVGAVGVGK) by immunizing HLA-A*11:01 transgenic mice with this peptide (Table 1) (24). As measured by SPR, TCRs 1-2C and 3-2E bound mutant KRASG12V–HLA-A*11 with KDs of 14 μM and 28 μM, respectively, compared to 131 μM and 42 μM, respectively, for binding to wild-type KRAS–HLA-A*11.
Structures of 1-2C and 3-2E bound to KRASG12V–HLA-A*11, and of KRAS–HLA-A*11 in free form, provided insight into how these TCRs discriminate between wild-type and mutant KRAS (24). The glycine-to-valine mutation in the neoepitope is located at P5 at the center of the peptide. Residues P4 and P5 of KRASG12V are shifted significantly downward towards the peptide-binding groove of HLA-A*11 compared to their positions in wild-type KRAS, thereby avoiding steric clashes with TCRs 1-2C and 3-2E (Figure 5A). Moreover, both TCRs target the P5 Val driver mutation, albeit through completely different sets of interactions (Figures 5B, C). Therefore, specific TCR recognition of KRASG12V depends not only on its distinct conformation compared to the wild-type peptide, but also on extensive direct contacts with the mutant P5 Val residue, as observed for TCR recognition of p53R175H (20, 21).
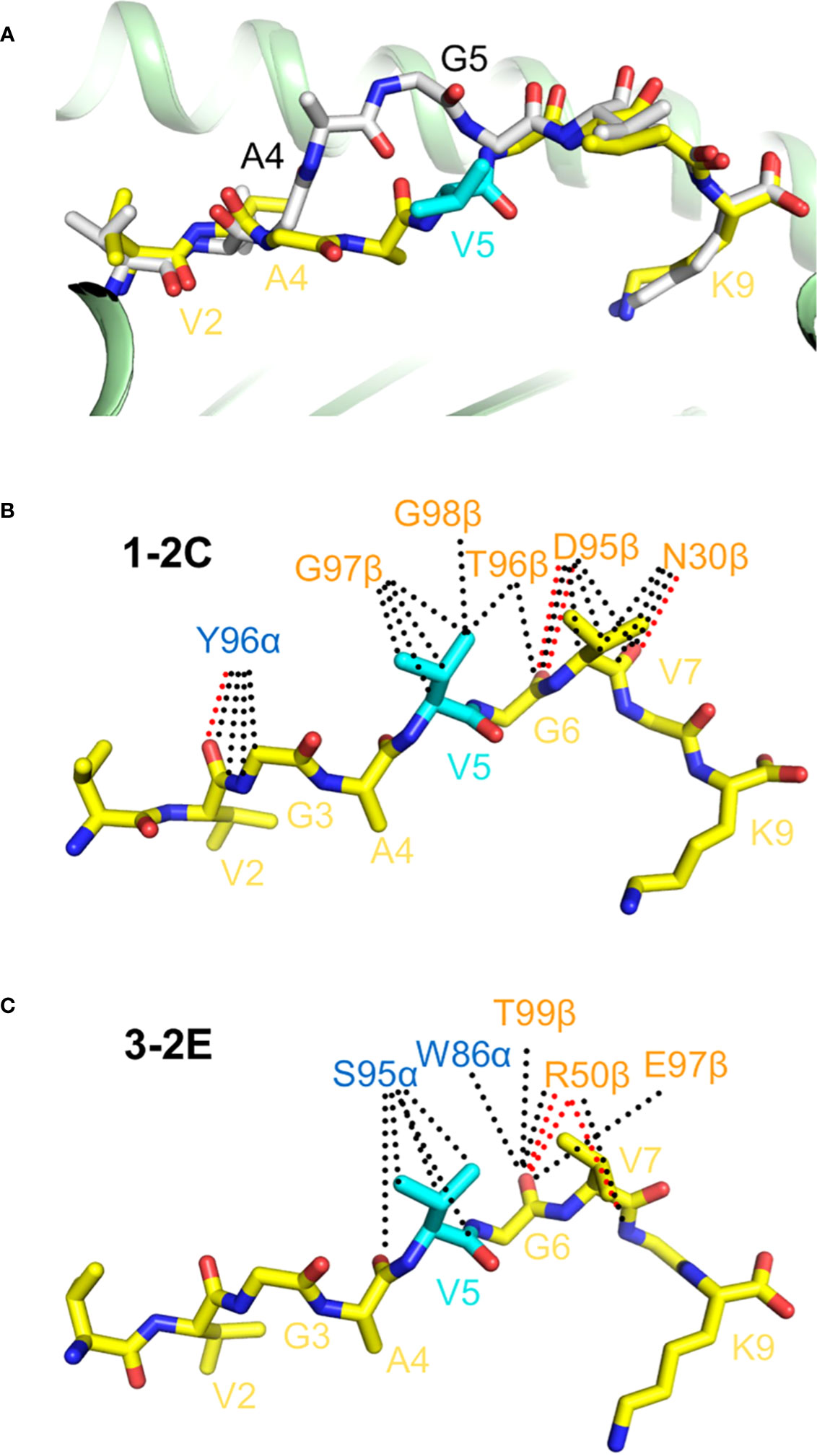
Figure 5 TCR recognition of the HLA-A*11:01-restricted KRASG12V neoepitope. (A) Superposition of wild-type KRAS–HLA-A*11:01 and mutant KRASG12V–HLA-A*11:01 structures (8I5E and 8I5C) (24). Wild-type KRAS peptide is gray; mutant KRASG12V peptide is yellow. The mutant P5 Val residue is cyan. (B) Interactions between TCR 1-C2 and the KRASG12V peptide. Hydrogen bonds are red dotted lines and van der Waals contacts are black dotted lines. (C) Interactions between TCR 3-2E and the KRASG12V peptide.
The HHATp8F neoepitope was first identified in ovarian cancer patients and derives from the hedgehog acyltransferase (HHAT) oncogene (57). It is restricted by HLA-A*02:06 and incorporates a leucine-to-phenylalanine substitution at P8 (KQWLVWLFL). Patient-derived CD8+ TILs strongly recognized HHATp8F but not wild-type HHAT. Although these peptides bound equally well to HLA-A*02:06, as measured by differential scanning fluorimetry, crystal structures of the wild-type HHAT–HLA-A*02 and mutant HHATp8F–HLA-A*02 complexes showed that the peptides differ most in the orientation of the side chain of P6 Trp, which is two residues away from the leucine-to-phenylalanine mutation at P8 (Figure 6A) (25). A 120° rotation of the P6 Trp side chain is induced by P8 Phe, which would clash with the P6 Trp side chain if it maintained the conformation seen in wild-type HHAT.

Figure 6 Pre-organization of the HHATp8F neoepitope by an immunogenic mutation. (A) Superposition of unbound wild-type HHAT–HLA-A*02 and mutant HHATp8F–HLA-A*02 structures (6UJQ and 6UJO) (25). The mutant P8 Phe residue is cyan. The conformations of the mutant and wild-type peptides are similar except for the orientation of the P6 Trp side chain. (B) Comparison of HHATp8F peptide conformations in unbound versus TCR-bound HHATp8F–HLA-A*02 structures (6UK4) (25). The mutant peptide in the TCR 302TIL–HHATp8F–HLA-A*02 complex adopts the same conformation as in unbound HHATp8F–HLA-A*02. (C) Superposition of the peptides in the neoepitope and wild-type TCR–pMHC complexes shows they have identical conformations (6UK2) (25). (D) The orientation of the P6 Trp side chain in unbound HHAT–HLA-A*02 differs from that in the TCR 302TIL–HHAT–HLA-A*02 complex.
TCR 302TIL, isolated from TILs of an ovarian cancer patient, bound HHATp8F–HLA-A*02 with KD = 9 μM compared to 200 μM for HHAT–HLA-A*02 (Table 1) (25). This 20-fold affinity differential can be explained by the structure of TCR 302TIL bound to HHATp8F– HLA-A*02. In this complex, the mutant peptide adopts the same conformation seen in unbound HHATp8F–HLA-A*02, with the P6 Trp side chain displaying a nearly identical orientation (Figure 6B). Notably, in the structure of TCR 302TIL bound to wild-type HHAT–HLA-A*02, P6 Trp adopts the same configuration as in the neoepitope complex (Figure 6C), which differs from that found in unbound HHAT–HLA-A*02 (Figure 6D). Therefore, the P8 Phe mutation pre-organizes the P6 Trp side chain into a conformation optimal for recognition by a neoantigen-specific TCR, in this way converting a self-epitope into an immunogenic epitope and enabling T cells to mediate killing of tumor but not normal cells.
PIK3CA encodes phosphoinositide 3-kinase α (PI3Kα), which is involved in cell proliferation, differentiation, motility, and survival, which are key cellular functions in cancer development. PIK3CA is among the most common genetically altered driver oncogenes, with the largest portion of mutations occurring at hotspot position H1047 (58, 59). Chandran et al. (26) isolated several TCRs from the T cells of a healthy donor that recognize a neoepitope corresponding to residues 1046–1054 of PIK3CAH1047L which contains a histidine-to-leucine mutation at position 1047 (ALHGGWTTK) (Table 1). These TCRs are restricted by the prevalent HLA-A*03:01 allele.
Crystal structures of PIK3CA and PIK3CAH1047L peptides bound to HLA-A*03:01 showed that the wild-type and mutant peptides adopt nearly identical conformations, which cannot explain the immunogenic potential of the PIK3CAH1047L neoepitope (Figure 7A). However, the thermal stability (melting temperature) of the mutant PIK3CAH1047L–HLA-A*03 complex (54 °C) is considerably higher than that of the wild-type PIK3CA–HLA-A*03 complex (37 °C) (26). Moreover, the half-life of the PIK3CAH1047L–HLA-A*03 complex (5.50 hours) is ~70 times longer than the half-life of the PIK3CA–HLA-A*03 complex (0.08 hours). The greater stability of the neoepitope complex is attributable to an optimal anchor residue at P2 (leucine rather than histidine) as a consequence of the mutation. As in the case of KRASG12D (22), the immunogenicity of PIK3CAH1047L arises from preferential presentation by MHC class I rather than differential TCR recognition.
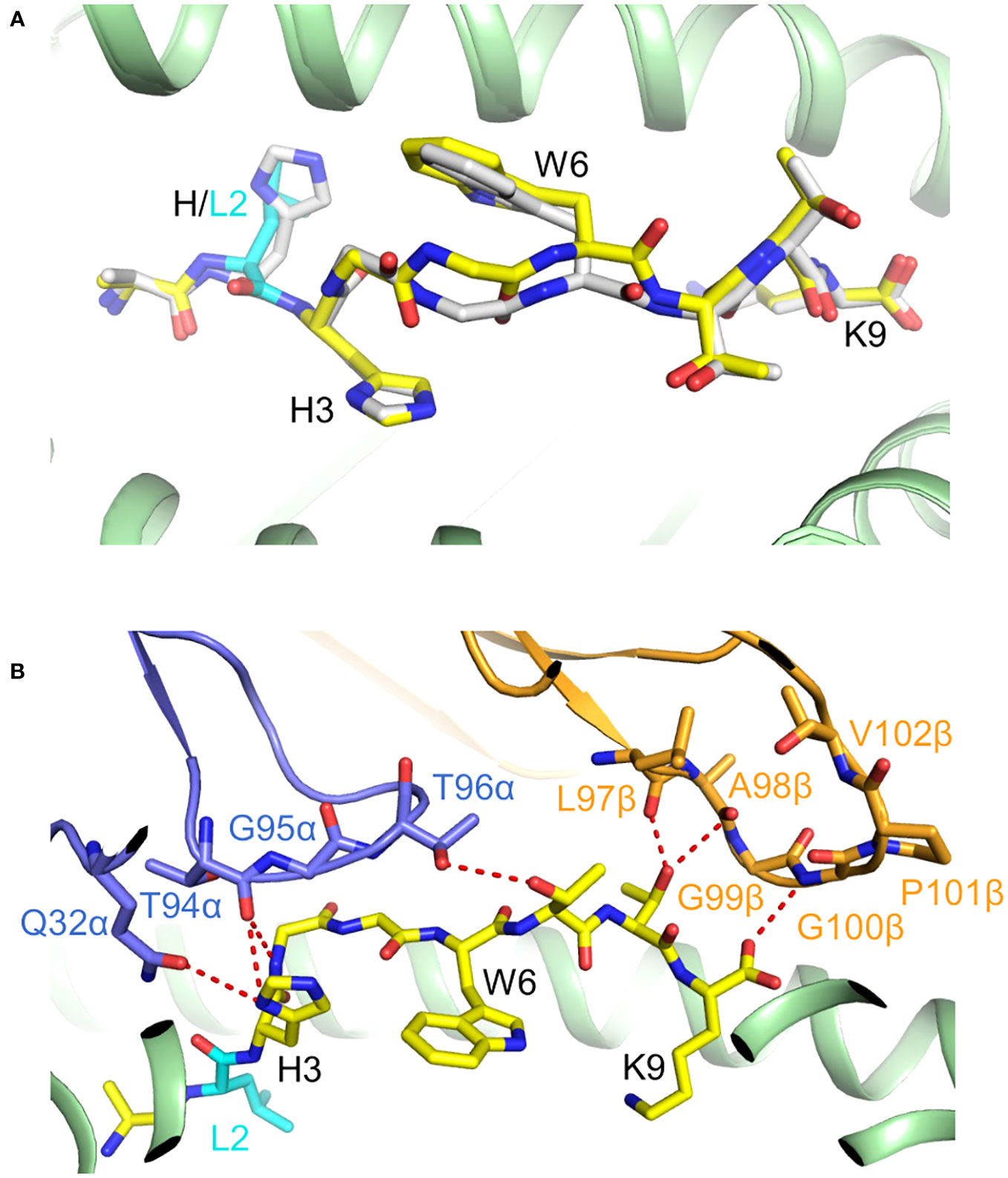
Figure 7 Presentation of PIK3CAH1047L neoepitope by HLA-C*03:01. (A) Superposition of mutant PIK3CAH1047L–HLA-A*03 and wild-type PIK3CA–HLA-A*03 complexes (7L1B and 7L1C) (26). The conformations of the mutant (yellow) and wild-type (gray) peptides are nearly identical. (B) Interactions between TCR4 and the PIK3CAH1047L peptide (7RRG) (26).
The structure of a PIK3CAH1047L-specific TCR (TCR4) bound to PIK3CAH1047L–HLA-A*03 revealed that TCR4 possesses an unusually long CDR3β loop that enables this TCR to form an extended and highly complementary interface with the neoepitope (Figure 7B). Importantly, adoptive transfer of TCR4-transduced T cells led to tumor regression in mice bearing mutant PIK3CAH1047L tumors but not wild-type PIK3CA tumors, which supports the clinical potential of TCR4 for ACT (26).
In addition to TCRs, monoclonal antibodies are also under investigation for immunotherapeutic targeting of cancer neoantigens (30, 60). These antibodies, called TCR-mimic antibodies (TCRm Abs), are designed to recognize pMHC complexes on cancer cell surfaces, similar to TCRs. They are typically isolated by screening phage or yeast libraries displaying single-chain Fv fragments with recombinant forms of the target pMHC. TCRm Abs have higher affinity than TCRs and can be readily converted to therapeutic formats such as bispecific antibodies, antibody–drug conjugates, and chimeric antigen receptors (CARs).
TCRm Abs have been described that specifically recognize several cancer neoantigen–HLA complexes, including p53R175H–HLA-A2 (61), KRASG12V–HLA-A3 (62, 63), RASQ61H–HLA-A1 (62), IDH2R140Q–HLA-B7 (64), and phosphoIRS2–HLA-A2 (65). Structural information is available for how TCRm Abs recognize three of these neoantigen–HLA complexes (Table 2) (61, 63, 64). As described below, these TCRm Abs dock on pMHC in ways that are very different from the canonical diagonal orientation of bona fide TCRs, in which Vα is positioned over the α2 helix of MHC class I and Vβ over the α1 helix. In the case of TCRs, this highly conserved docking mode is mandated by T cell signaling constraints that optimally localize CD8/Lck to CD3 in the TCR–CD3 complex (66). TCR mAbs are not subject to such signaling constraints because they are simply selected for their ability to bind pMHC targets (30, 60). This allows them to employ diverse strategies to achieve specific recognition.
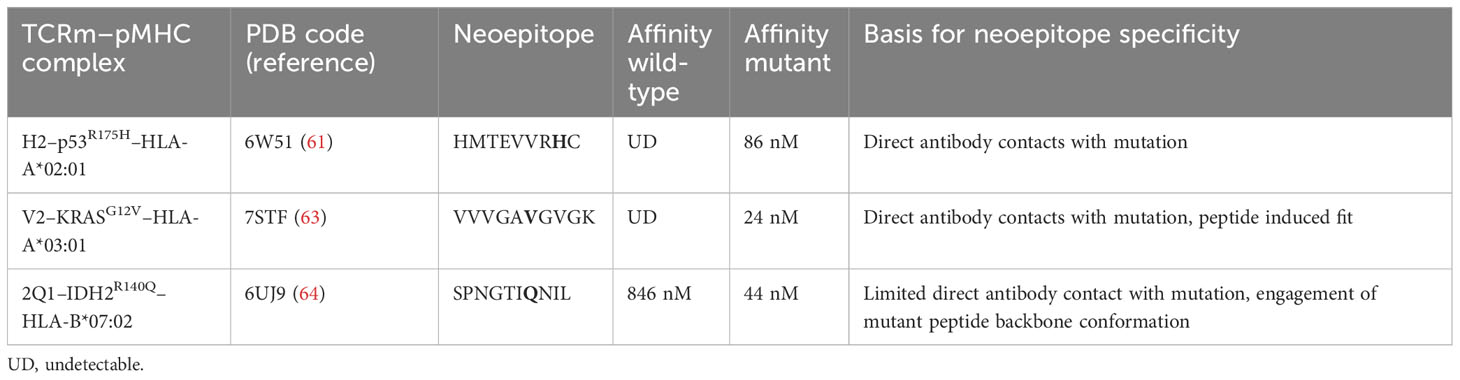
Table 2 Structures of TCR-mimic (TCRm) antibodies bound to cancer neoantigen pMHC ligands.
TCRm Ab H2 recognizes p53R175H–HLA-A2 (61), the exact same pMHC targeted by TCRs described above (20, 21). H2 bound p53R175H–HLA-A2 with nanomolar affinity but showed no detectable binding to wild-type p53–HLA-A2, thereby mimicking the exquisite neoepitope specificity of TCRs. H2 was converted to a T cell-based immunotherapeutic by fusing it to an anti-CD3 antibody (61). This bispecific antibody effectively activated T cells to lyse cancer cells presenting the p53R175H neoantigen both in vitro and in mice, despite the low density of p53R175H–HLA-A2 complexes on the cancer cell surface.
In the structure of TCRm Ab H2 bound to p53R175H–HLA-A2 (Figure 8A) (61), VL (analogous to Vα) is positioned mainly over the α1 helix of HLA-A2 and VH (analogous to Vβ) over the α2 helix (Figure 8C), which is nearly the reversed docking polarity of TCR 12-6 on p53R175H–HLA-A2 (Figures 8B, D). As a consequence, H2 and 12-6 make very different footprints on the pMHC surface (Figures 8E, F). The contributions made by individual CDRs to interactions with MHC (Figure 8G, H) and peptide (Figures 8I, J) differ radically. In the TCRm Ab H2–p53R175H–HLA-A2 complex (61), the VLCDR3 and VHCDR1–3 loops form a tight cage enveloping P7 Arg and P8 His as part of a hydrogen bonding network with VLCDR3 Tyr94 and VHCDR2 Asp54 (Figure 8K). Therefore, H2, like TCR 12-6 (Figure 2F), distinguishes mutant from wild-type p53 through direct contacts with P8 His, which is fundamentally different from the indirect strategy employed by TCR 6-11 (21).
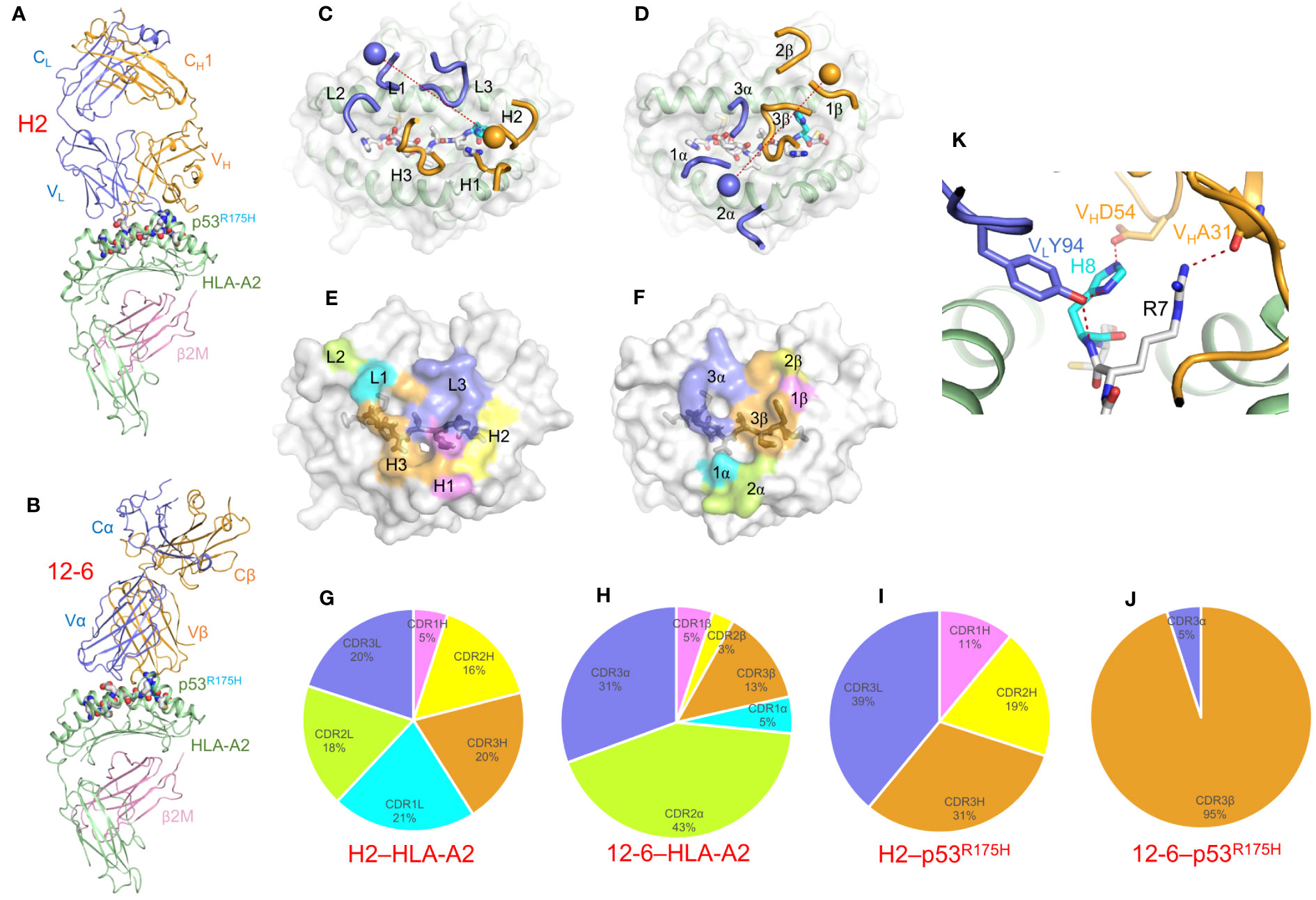
Figure 8 Recognition of p53R175H–HLA-A2 by a TCR-mimic antibody. (A) Side view of the TCRm Ab H2–p53R175H–HLA-A2 complex (6W51) (61). (B) Side view of the TCR 12-6–p53R175H–HLA-A2 complex (6VRM) (20). (C) Positions of CDR loops of TCRm Ab H2 on p53R175H–HLA-A2. VLCDRs of H2 are shown as numbered blue (L1, L2, and L3) loops; VHCDRs are shown as numbered gold (H1, H2, and H3) loops. The p53R175H peptide is drawn in gray in stick representation with the mutant P8 His residue in cyan. HLA-A2 is depicted as a light gray surface. The blue and gold spheres mark the positions of the conserved intrachain disulfide of VL and VH, respectively. The red dashed line indicates the crossing angle of TCRm Ab to pMHC. (D) Positions of CDR loops of TCR 12-6 on p53R175H–HLA-A2. CDRs of 12-6 are shown as numbered blue (CDR1α, CDR2α, and CDR3α) or gold (CDR1β, CDR2β, and CDR3β) loops. The blue and gold spheres mark the positions of the conserved intrachain disulfide of Vα and Vβ, respectively. The red dashed line indicates the crossing angle of TCR to pMHC. (E) Footprint of TCRm Ab H2 on p53R175H–HLA-A2. The areas contacted by individual CDR loops are color-coded. (F) Footprint of TCR 12-6 on p53R175H–HLA-A2. (G) Pie chart showing percentage distribution of TCRm Ab H2 contacts to HLA-A2 according to CDR. (H) Pie chart showing percentage distribution of TCR 12-6 contacts to HLA-A2 according to CDR. (I) Pie chart showing percentage distribution of TCRm Ab H2 contacts to p53R175H peptide according to CDR. (J) Pie chart showing percentage distribution of TCR 12-6 contacts to p53R175H peptide according to CDR. (K) Close-up of interactions between TCRm Ab H2 and P8 His.
Another TCRm Ab, V2, specifically recognizes KRASG12V presented by HLA-A*03:01 (62, 63), which is closely related to the pMHC targeted by TCR 1-2C (KRASG12V–HLA-A*11:01) (24). A bispecific antibody constructed by fusing V2 to an anti-CD3 antibody induced T cell activation and killing of target cancer cells expressing endogenous levels of KRASG12V neoantigen (62). In the structure of TCRm Ab V2 bound to KRASG12V–HLA-A*03 (Figure 9A) (63), VL and VH are both positioned over the α1 helix of HLA-A*03 (Figure 9C), in sharp contrast to the canonical docking topology of TCR 1-2C (Figures 9B, D). As a result, the footprints of V2 and 1-2C on the pMHC surface are very different (Figures 9E, F), as are the contributions made by individual CDRs to interactions with MHC (Figures 9G, H). However, VHCDR3 of V2, like CDR3β of 1-2C, dominates contacts with the KRASG12V peptide (Figures 9I, J). V2 engages the mutation site at P5 Val with a loose hydrophobic cage comprising VLPhe53, VHPro103, VHVal104, and VHTyr105, with the N-terminal portion of the KRASG12V peptide nearly completely untouched by V2 (Figure 9K) (63).
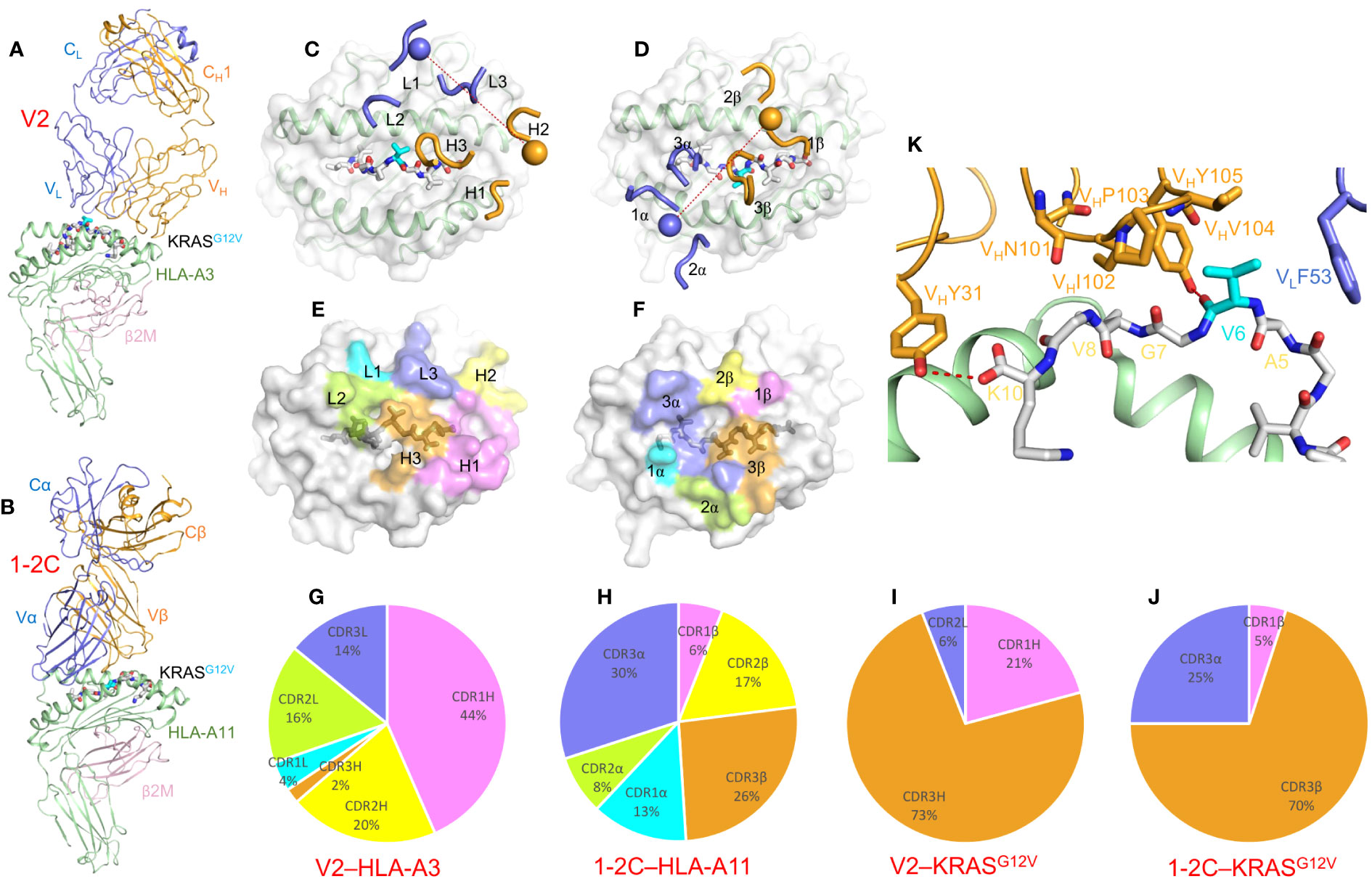
Figure 9 Recognition of KRASG12V–HLA-A*03:01 by a TCR-mimic antibody. (A) Side view of the TCRm Ab V2–KRASG12V–HLA-A*03:01 complex (7STF) (63). (B) Side view of the TCR 1-2C–KRASG12V–HLA-A*11:01 complex (8I5C) (24). (C) Positions of CDR loops of TCRm Ab V2 on KRASG12V–HLA-A*03:01. VLCDRs of V2 are shown as numbered blue (L1, L2, and L3) loops; VHCDRs are shown as numbered gold (H1, H2, and H3) loops. The KRASG12V peptide is drawn in gray in stick representation with the mutant P6 Val residue in cyan. HLA-A3 is depicted as a light gray surface. The blue and gold spheres mark the positions of the conserved intrachain disulfide of VL and VH, respectively. The red dashed line indicates the crossing angle of TCRm Ab to pMHC. (D) Positions of CDR loops of TCR 1-2C on KRASG12V–HLA-A*11:01. CDRs of 1-2C are shown as numbered blue (CDR1α, CDR2α, and CDR3α) or gold (CDR1β, CDR2β, and CDR3β) loops. The blue and gold spheres mark the positions of the conserved intrachain disulfide of Vα and Vβ, respectively. The red dashed line indicates the crossing angle of TCR to pMHC. (E) Footprint of TCRm Ab V2 on KRASG12V–HLA-A*03:01. The areas contacted by individual CDR loops are color-coded. (F) Footprint of TCR 1-2C on KRASG12V–HLA-A*11:01. (G) Pie chart showing percentage distribution of TCRm Ab V2 contacts to HLA-A*03:01 according to CDR. (H) Pie chart showing percentage distribution of TCR 1-2C contacts to HLA-A*11:01 according to CDR. (I) Pie chart showing percentage distribution of TCRm Ab V2 contacts to KRASG12V peptide according to CDR. (J) Pie chart showing percentage distribution of TCR 1-2C contacts to KRASG12V peptide according to CDR. (K) Interactions between TCRm Ab V2 and the KRASG12V peptide.
TCRm Ab 2Q1 specifically recognizes a neoantigen derived from isocitrate dehydrogenase 2 (IDH2R140Q) (SPNGTIQNIL) presented by HLA-B*07:02 (64). CAR T cells constructed from 2Q1 were cytotoxic against IDH2R140Q-bearing target cells. Structures of wild-type IDH2–HLA-B*02 and mutant IDH2R140Q–HLA-B*02 complexes showed that the peptides bound in essentially identical conformations and that P7 Arg/Gln (wild-type and mutant amino acids) is buried deep within the peptide-binding groove of the MHC molecule (64). In the structure of TCRm Ab 2Q1 bound to IDH2R140Q–HLA-B*02, the only direct interaction between P7 Gln and 2Q1 is a hydrogen bond linking P7 Gln to VHCDR3 Arg102. Elimination of this hydrogen bond by mutating VHCDR3 Arg102 to alanine completely abrogated TCRm Ab binding (64). 2Q1 docks onto pMHC in a parallel orientation rather than the canonical diagonal orientation of bona fide TCRs.
Similar to neoantigen-specific TCRm Abs (61, 63, 64), TCRm Abs specific for unmutated tumor-associated antigens such as MAGE-A1 and ESK1 also do not need to mimic TCRs completely to achieve peptide-specific recognition (67, 68). For example, a TCRm Ab specific for MAGE-A1–HLA-A1 focused on the HLA-A1 α1 helix with no contacts to N-terminal peptide residues (67). Another TCRm Ab specific for MART-1–HLA-A1 engaged pMHC with a TCR-like docking angle but its VHCDR1 and VHCDR2 loops were completely absent from MHC interactions (30). Collectively, structural studies of TCRm Ab–pMHC complexes have revealed a wide range of docking orientations that can diverge substantially from the canonical diagonal orientation of natural TCR–pMHC complexes without compromising specificity.
Accurate computational prediction of immunogenic neoantigens is of high interest for neoantigen-based vaccines and therapeutics. Neoepitope prediction faces a number of challenges, including 1) predicting whether peptides containing the somatic mutation of interest are actually generated during antigen processing, 2) predicting whether these peptides come into contact with MHC molecules in the MHC class I or class II antigen presentation pathway, 3) predicting which peptides bind MHC with sufficient affinity to be presented on the cell surface, and 4) predicting whether the displayed neoepitope–MHC complexes can be recognized by TCRs. Although much progress has been made towards meeting these challenges, at least for MHC class I-restricted neoepitopes, considerable obstacles remain.
Several recent studies have demonstrated the utility of structural information to generate neoepitope predictions or to provide key insights into predictions. In a 2019 study, Riley et al. performed modeling of candidate peptides in complex with HLA-A2, and utilized energetic features from those structural models to train a neural network predictor (69). Their method performed well relative to other methods based on their benchmarking, and while only predicting immunogenicity of nonameric peptides presented by HLA-A2 limited its practical applicability, it provided a proof of concept of such a structure-based approach, as the authors noted. A recent study that characterized neoantigen-specific TCR structural avidity utilized structural modeling in that context, finding that the amount of predicted interface contacts in template-based models of TCR–pMHC complexes was associated with higher avidity (70). Additionally, the authors generated a structure-based logistic regression model to predict avidity of TCRs without the antigen context.
Other computational neoepitope prediction studies have utilized structures to interpret and contextualize their results. One method named PRIME can predict MHC class I T cell neoepitopes based on MHC binding affinity and TCR recognition propensity (71); that algorithm performed favorably against other prediction tools, and an updated version of that algorithm (PRIME2.0) has been reported (72). In the original PRIME study, the authors found that the trained algorithm, which did not explicitly take structural information into account, was in agreement with structural features of individual TCR–pMHC complex interfaces as well as overall interface residue preferences in TCR–pMHC complex structures (71). The authors of the recently reported deep learning neoepitope prediction algorithm BigMHC reported improved performance over PRIME2.0 and a number of other predictive methods in neoepitope immunogenicity prediction (73), and they mapped the deep learning attention encodings (trained, as with the PRIME method, on sequence data) onto MHC structures to gain insights into key MHC residues.
While relatively few methods to date have directly utilized structural information in neoepitope immunogenicity prediction, with PRIME, BigMHC, and other recent machine learning-based methods (74–76) using sequence data for training, the modeling approaches noted above suggest that structural data may be helpful in prospective algorithm developments. With recent advances in deep learning-based structural modeling including AlphaFold (77), and adaptations of AlphaFold and related deep learning methods to accurately model TCRs (78), pMHCs (79), and TCR–pMHC complexes (80, 81), it is likely that such approaches would be helpful in that context. Currently determined structures of neoepitope-containing TCR–pMHC complexes (Table 1) help to illustrate the structural basis of concepts such as “agretopicity”, which is preferential mutant epitope binding by MHC (82) and is considered directly in predictive computational methods (e.g. 74, 76); additional structures of these complexes may provide new neoepitope recognition features that in turn can inform future method developments.
Structural studies have revealed that cancer neoepitopes need differ only slightly from their wild-type counterparts for them to be immunogenic in patients. However, this physiochemical similarity presents a challenge to the immune system and probably explains, at least in part, the low frequency of T cells able to recognize neoantigens with sufficient avidity to mediate efficient killing of tumor cells. Some neoepitope mutations increase peptide–MHC binding, thereby improving antigen presentation (e.g. KRASG12D and PIK3CAH1047L). Other mutations increase affinity for TCR, either through direct contacts with TCR (e.g. TPIT28I, p53R175H, and KRASG12V) or via indirect mechanisms such as conformational pre-organization of pMHC (e.g. HHATp8F). TCRm Abs use non-canonical docking topologies to recognize pMHC and provide an alternative to TCRs for immunotherapeutic targeting of cancer neoantigens. Structure-guided engineering of TCRs (23) and TCRm Abs (30, 64) provides a means for optimizing these molecules for ACT, including with CAR T cells, as well as for incorporating TCRs and TCRm Abs into alternative therapeutic formats such as bispecific agents and drug conjugates.
RM: Writing – original draft, Writing – review & editing. DW: Writing – original draft, Writing – review & editing. BP: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Institutes of Health grants GM144083 (to BP) and AI129893 (to RM) and by National Natural Science Foundation of China grants 32100985 and 32270995 (to DW).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348:62–8. doi: 10.1126/science.aaa4967
2. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
3. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
4. Stevanović S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science (2017) 356:200–5. doi: 10.1126/science.aak9510
5. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (2014) 344:641–5. doi: 10.1126/science.1251102
6. Pearlman AH, Hwang MS, Konig MF, Hsiue EH, Douglass J, DiNapoli SR, et al. Targeting public neoantigens for cancer immunotherapy. Nat Cancer (2021) 2:487–97. doi: 10.1038/s43018-021-00210-y
7. Dudley ME, Gross CA, Somerville RP, Hong Y, Schaub NP, Rosati SF, et al. Randomized selection design trial evaluating CD8+-enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. J Clin Oncol (2013) 31:2152–9. doi: 10.1200/JCO.2012.46.6441
8. Schumacher TN, Scheper W, Kvistborg P. Cancer neoantigens. Annu Rev Immunol (2019) 37:173–200. doi: 10.1146/annurev-immunol-042617-053402
9. Gopanenko AV, Kosobokova EN, Kosorukov VS. Main strategies for the identification of neoantigens. Cancers (2020) 12:2879. doi: 10.3390/cancers12102879
10. Parkhurst MR, Robbins PF, Tran E, Prickett TD, Gartner JJ, Jia L, et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discovery (2019) 9:1022–35. doi: 10.1158/2159-8290.CD-18-1494
11. Wölfel T, Hauer M, Schneider J, Serrano M, Wölfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science (1995) 269:1281–4. doi: 10.1126/science.7652577
12. Linard B, Bézieau S, Benlalam H, Labarrière N, Guilloux Y, Diez E, et al. A ras-mutated peptide targeted by CTL infiltrating a human melanoma lesion. J Immunol (2002) 168:4802–8. doi: 10.4049/jimmunol.168.9.4802
13. Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature (2014) 512:324–7. doi: 10.1038/nature13387
14. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science (2015) 350:1387–90. doi: 10.1126/science.aad1253
15. Veatch JR, Lee SM, Fitzgibbon M, Chow IT, Jesernig B, Schmitt T, et al. Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J Clin Invest (2018) 128:1563–8. doi: 10.1172/JCI98689
16. Malekzadeh P, Pasetto A, Robbins PF, Parkhurst MR, Paria BC, Jia L, et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J Clin Invest (2019) 129:1109–14. doi: 10.1172/JCI123791
17. Martinov T, Greenberg PD. Targeting driver oncogenes and other public neoantigens using T cell receptor-based cellular therapy. Annu Rev Cancer Biol (2023) 7:331–51. doi: 10.1146/annurev-cancerbio-061521-082114
18. Deng L, Langley RJ, Brown PH, Xu G, Teng L, Wang Q, et al. Structural basis for the recognition of mutant self by a tumor-specific, MHC class II-restricted T cell receptor. Nat Immunol (2007) 8:398–408. doi: 10.1038/ni1447
19. Deng L, Langley RJ, Wang Q, Topalian SL, Mariuzza RA. Structural insights into the editing of germ-line-encoded interactions between T-cell receptor and MHC class II by Vα CDR3. Proc Natl Acad Sci USA (2012) 109:14960–5. doi: 10.1073/pnas.1207186109
20. Wu D, Gallagher DT, Gowthaman R, Pierce BG, Mariuzza RA. Structural basis for oligoclonal T cell recognition of a shared p53 cancer neoantigen. Nat Commun (2020) 11:2908. doi: 10.1038/s41467-020-16755-y
21. Wu D, Gowathaman R, Pierce BG, Mariuzza RA. T cell receptors employ diverse strategies to target a p53 cancer neoantigen. J Biol Chem (2022) 298:101684. doi: 10.1016/j.jbc.2022.101684
22. Sim MJW, Lu J, Spencer M, Hopkins F, Tran E, Rosenberg SA, et al. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc Natl Acad Sci USA (2020) 117:12826–35. doi: 10.1073/pnas.1921964117
23. Poole A, Karuppiah V, Hartt A, Haidar JN, Moureau S, Dobrzycki T, et al. Therapeutic high affinity T cell receptor targeting a KRASG12D cancer neoantigen. Nat Commun (2022) 13:5333. doi: 10.1038/s41467-022-32811-1
24. Lu D, Chen Y, Jiang M, Wang J, Li Y, Ma K, et al. KRAS G12V neoantigen specific T cell receptor for adoptive T cell therapy against tumors. Nat Commun (2023) 14:6389. doi: 10.1038/s41467-023-42010-1
25. Devlin JR, Alonso JA, Ayres CM, Keller GLJ, Bobisse S, Vander Kooi CW, et al. Structural dissimilarity from self drives neoepitope escape from immune tolerance. Nat Chem Biol (2020) 16:1269–76. doi: 10.1038/s41589-020-0610-1
26. Chandran SS, Ma J, Klatt MG, Dündar F, Bandlamudi C, Razavi P, et al. Immunogenicity and therapeutic targeting of a public neoantigen derived from mutated PIK3CA. Nat Med (2022) 28:946–57. doi: 10.1038/s41591-022-01786-3
27. Sim MJW, Stotz Z, Lu J, Brennan P, Long EO, Sun PD. T cells discriminate between groups C1 and C2 HLA-C. Elife (2022) 11:e75670. doi: 10.7554/eLife.75670
28. Chen JL, Stewart-Jones G, Bossi G, Lissin NM, Wooldridge L, Choi EM, et al. Structural and kinetic basis for heightened immunogenicity of T cell vaccines. J Exp Med (2005) 201:1243–55. doi: 10.1084/jem.20042323
29. Borbulevych OY, Santhanagopolan SM, Hossain M, Baker BM. TCRs used in cancer gene therapy cross-react with MART-1/Melan-A tumor antigens via distinct mechanisms. J Immunol (2011) 187:2453–63. doi: 10.4049/jimmunol.1101268
30. Yang X, Nishimiya D, Löchte S, Jude KM, Borowska M, Savvides CS, et al. Facile repurposing of peptide–MHC-restricted antibodies for cancer immunotherapy. Nat Biotechnol (2023) 41:932–43. doi: 10.1038/s41587-022-01567-w
31. Dolton G, Rius C, Wall A, Szomolay B, Bianchi V, Galloway SAE, et al. Targeting of multiple tumor-associated antigens by individual T cell receptors during successful cancer immunotherapy. Cell (2023) 186:3333–3349.e27. doi: 10.1016/j.cell.2023.06.020
32. Pieper R, Christian RE, Gonzales MI, Nishimura MI, Gupta G, Settlage RE, et al. Biochemical identification of a mutated human melanoma antigen recognized by CD4+ T cells. J Exp Med (1999) 189:757–66. doi: 10.1084/jem.189.5.757
33. Rushdi MN, Pan V, Li K, Choi HK, Travaglino S, Hong J, et al. Cooperative binding of T cell receptor and CD4 to peptide-MHC enhances antigen sensitivity. Nat Commun (2022) 13:7055. doi: 10.1038/s41467-022-34587-w
34. Huppa JB, Axmann M, Mörtelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature (2010) 463:963–7. doi: 10.1038/nature08746
35. Adams JJ, Narayanan S, Liu B, Birnbaum ME, Kruse AC, Bowerman NA, et al. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity (2011) 35:681–93. doi: 10.1016/j.immuni.2011.09.013
36. Jiang N, Huang J, Edwards LJ, Liu B, Zhang Y, Beal CD, et al. Two-stage cooperative T cell receptor-peptide major histocompatibility complex-CD8 trimolecular interactions amplify antigen discrimination. Immunity (2011) 34:13–23. doi: 10.1016/j.immuni.2010.12.017
37. Zhu C, Chen W, Lou J, Rittase W, Li K. Mechanosensing through immunoreceptors. Nat Immunol (2019) 20:1269–78. doi: 10.1038/s41590-019-0491-1
38. Sundberg EJ, Sawicki MW, Southwood S, Andersen PS, Sette A, Mariuzza RA. Minor structural changes in a mutated human melanoma antigen correspond to dramatically enhanced stimulation of a CD4+ tumor-infiltrating lymphocyte line. J Mol Biol (2002) 319:449–61. doi: 10.1016/S0022-2836(02)00370-4
39. Garraway LA, Lander ES. Lessons from the cancer genome. Cell (2013) 153:17–37. doi: 10.1016/j.cell.2013.03.002
40. Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, et al. The evolutionary history of 2,658 cancers. Nature (2020) 578:122–8. doi: 10.1038/s41586-019-1907-7
41. Sabapathy K, Lane DP. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol (2018) 15:13–30. doi: 10.1038/nrclinonc.2017.151
42. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6:pl1. doi: 10.1126/scisignal.2004088
43. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med (2017) 23:703–13. doi: 10.1038/nm.4333
44. Lo W, Parkhurst M, Robbins PF, Tran E, Lu YC, Jia L, et al. Immunologic recognition of a shared p53 mutated neoantigen in a patient with metastatic colorectal cancer. Cancer Immunol Res (2019) 7:534–43. doi: 10.1158/2326-6066.CIR-18-0686
45. Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, et al. Toward a shared vision for cancer genomic data. N Engl J Med (2016) 375:1109–12. doi: 10.1056/NEJMp1607591
46. González-Galarza FF, Takeshita LY, Santos EJ, Kempson F, Maia MH, da Silva AL, et al. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res (2015) 43:D784–8. doi: 10.1093/nar/gku1166
47. Kim SP, Vale NR, Zacharakis N, Krishna S, Yu Z, Gasmi B, et al. Adoptive cellular therapy with autologous tumor-infiltrating lymphocytes and T-cell receptor-engineered T cells targeting common p53 neoantigens in human solid tumors. Cancer Immunol Res (2022) 10:932–46. doi: 10.1158/2326-6066.CIR-22-0040
48. Yin Y, Li Y, Mariuzza RA. Structural basis for self-recognition by autoimmune T-cell receptors. Immunol Rev (2012) 250:32–48. doi: 10.1111/imr.12002
49. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res (2012) 72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612
50. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer (2003) 3:11–22. doi: 10.1038/nrc969
51. Prior IA, Hood FE, Hartley JL. The frequency of ras mutations in cancer. Cancer Res (2020) 80:2969–74. doi: 10.1158/0008-5472.CAN-19-3682
52. Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell (2004) 5:375–87. doi: 10.1016/S1535-6108(04)00085-6
53. Presotto D, Erdes E, Duong MN, Allard M, Regamey PO, Quadroni M, et al. Fine-tuning of optimal TCR signaling in tumor-redirected CD8 T cells by distinct TCR affinity-mediated mechanisms. Front Immunol (2017) 8:1564. doi: 10.3389/fimmu.2017.01564
54. Holler PD, Chlewicki LK, Kranz DM. TCRs with high affinity for foreign pMHC show self-reactivity. Nat Immunol (2003) 4:55–62. doi: 10.1038/ni863
55. Bentzen AK, Hadrup SR. T-cell-receptor cross-recognition and strategies to select safe T-cell receptors for clinical translation. Immunooncol Technol (2019) 2:1–10. doi: 10.1016/j.iotech.2019.06.003
56. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122:863–71. doi: 10.1182/blood-2013-03-490565
57. Bobisse S, Genolet R, Roberti A, Tanyi JL, Racle J, Stevenson BJ, et al. Sensitive and frequent identification of high avidity neo-epitope specific CD8+ T cells in immunotherapy-naive ovarian cancer. Nat Commun (2018) 9:1092. doi: 10.1038/s41467-018-03301-0
58. Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell (2018) 174:1034–5. doi: 10.1016/j.cell.2018.07.034
59. Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature (2019) 575:210–6. doi: 10.1038/s41586-019-1689-y
60. Malviya M, Aretz ZEH, Molvi Z, Lee J, Pierre S, Wallisch P, et al. Challenges and solutions for therapeutic TCR-based agents. Immunol Rev (2023) 16. doi: 10.1111/imr.13233
61. Hsiue EH, Wright KM, Douglass J, Hwang MS, Mog BJ, Pearlman AH, et al. Targeting a neoantigen derived from a common TP53 mutation. Science (2021) 371(6533):eabc8697. doi: 10.1126/science.abc8697
62. Douglass J, Hsiue EH, Mog BJ, Hwang MS, DiNapoli SR, Pearlman AH, et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci Immunol (2021) 6(57):eabd5515. doi: 10.1126/sciimmunol.abd5515
63. Wright KM, DiNapoli SR, Miller MS, Aitana Azurmendi P, Zhao X, Yu Z, et al. Hydrophobic interactions dominate the recognition of a KRAS G12V neoantigen. Nat Commun (2023) 14:5063. doi: 10.1038/s41467-023-40821-w
64. Hwang MS, Miller MS, Thirawatananond P, Douglass J, Wright KM, Hsiue EH, et al. Structural engineering of chimeric antigen receptors targeting HLA-restricted neoantigens. Nat Commun (2021) 12:5271. doi: 10.1038/s41467-021-25605-4
65. Dao T, Mun SS, Molvi Z, Korontsvit T, Klatt MG, Khan AG, et al. A TCR mimic monoclonal antibody reactive with the "public" phospho-neoantigen pIRS2/HLA-A*02:01 complex. JCI Insight (2022) 7:e151624. doi: 10.1172/jci.insight.151624
66. Zareie P, Szeto C, Farenc C, Gunasinghe SD, Kolawole EM, Nguyen A, et al. Canonical T cell receptor docking on peptide-MHC is essential for T cell signaling. Science (2021) 372:eabe9124. doi: 10.1126/science.abe9124
67. Hülsmeyer M, Chames P, Hillig RC, Stanfield RL, Held G, Coulie PG, et al. A major histocompatibility complex-peptide-restricted antibody and T cell receptor molecules recognize their target by distinct binding modes: crystal structure of human leukocyte antigen (HLA)-A1-MAGE-A1 in complex with FAB-HYB3. J Biol Chem (2005) 280:2972–80. doi: 10.1074/jbc.M411323200
68. Ataie N, Xiang J, Cheng N, Brea EJ, Lu W, Scheinberg DA, et al. Structure of a TCR-mimic antibody with target predicts pharmacogenetics. J Mol Biol (2016) 428:194–205. doi: 10.1016/j.jmb.2015.12.002
69. Riley TP, Keller GLJ, Smith AR, Davancaze LM, Arbuiso AG, Devlin JR, et al. Structure based prediction of neoantigen immunogenicity. Front Immunol (2019) 10:2047. doi: 10.3389/fimmu.2019.02047
70. Schmidt J, Chiffelle J, Perez MAS, Magnin M, Bobisse S, Arnaud M, et al. Neoantigen-specific CD8 T cells with high structural avidity preferentially reside in and eliminate tumors. Nat Commun (2023) 14:3188. doi: 10.1038/s41467-023-38946-z
71. Schmidt J, Smith AR, Magnin M, Racle J, Devlin JR, Bobisse S, et al. Prediction of neo-epitope immunogenicity reveals TCR recognition determinants and provides insight into immunoediting. Cell Rep Med (2021) 2:100194. doi: 10.1016/j.xcrm.2021.100194
72. Gfeller D, Schmidt J, Croce G, Guillaume P, Bobisse S, Genolet R, et al. Improved predictions of antigen presentation and TCR recognition with MixMHCPred2.2 and Prime2.0 reveal potent SARS-CoV-2 CD8+ T-cell epitopes. Cell Syst (2023) 14:72–83. doi: 10.1016/j.cels.2022.12.002
73. Albert BA, Yang Y, Shao XM, Singh D, Smith KN, Anagnostou V, et al. Deep neural networks predict class I major histocompatibility complex epitope presentation and transfer learn neoepitope immunogenicity. Nat Mach Intell (2023) 5:861–72. doi: 10.1038/s42256-023-00694-6
74. Xia H, McMichael J, Becker-Hapak M, Onyeador OC, Buchli R, McClain E, et al. Computational prediction of MHC anchor locations guides neoantigen identification and prioritization. Sci Immunol (2023) 8:eabg2200. doi: 10.1126/sciimmunol.abg2200
75. Chu Y, Zhang Y, Wang Q, Zhang L, Wang X, Wang Y, et al. A transformer-based model to predict peptide–HLA class I binding and optimize mutated peptides for vaccine design. Nat Mach Intell (2022) 4:300–11. doi: 10.1038/s42256-022-00459-7
76. Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell (2020) 183:818–34. doi: 10.1016/j.cell.2020.09.015
77. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with Alphafold. Nature (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
78. Abanades B, Wong WK, Boyles F, Georges G, Bujotzek A, Deane CM. Immunebuilder: deep-learning models for predicting the structures of immune proteins. Commun Biol (2023) 6:575. doi: 10.1038/s42003-023-04927-7
79. Motmaen A, Dauparas J, Baek M, Abedi MH, Baker D, Bradley P. Peptide-binding specificity prediction using fine-tuned protein structure prediction networks. Proc Natl Acad Sci USA (2023) 120:e2216697120. doi: 10.1073/pnas.2216697120
80. Bradley P. Structure-based prediction of T cell receptor:peptide-MHC interactions. eLife (2023) 12:e82813. doi: 10.7554/eLife.82813
81. Yin R, Ribeiro-Filho HV, Lin V, Gowthaman R, Cheung M, Pierce BG. TCRmodel2: high-resolution modeling of T cell receptor recognition using deep learning. Nucleic Acids Res (2023) 51:W569–W76. doi: 10.1093/nar/gkad356
Keywords: cancer neoantigen, T cell, TCR, MHC, immunotherapy, immunogenicity, structure
Citation: Mariuzza RA, Wu D and Pierce BG (2023) Structural basis for T cell recognition of cancer neoantigens and implications for predicting neoepitope immunogenicity. Front. Immunol. 14:1303304. doi: 10.3389/fimmu.2023.1303304
Received: 27 September 2023; Accepted: 03 November 2023;
Published: 17 November 2023.
Edited by:
Asaf Poran, BioNTech, GermanyReviewed by:
Scheherazade Sadegh-Nasseri, Johns Hopkins University, United StatesCopyright © 2023 Mariuzza, Wu and Pierce. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roy A. Mariuzza, cm1hcml1enpAdW1kLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.