
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 08 December 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1301051
This article is part of the Research TopicCrosstalk between Peripheral and Local Immune Response in the Pathophysiology of Stroke and Neurodegeneration Diseases, Volume IIView all 10 articles
 Lingmin Zhao
Lingmin Zhao Di Ma*
Di Ma* LiJuan Wang*XingQi SuLiangShu FengLiChong Zhu
LiJuan Wang*XingQi SuLiangShu FengLiChong Zhu Ying ChenYuLei HaoXinYu Wang
Ying ChenYuLei HaoXinYu Wang JiaChun Feng
JiaChun FengAtherosclerosis is a common cardiovascular disease caused by the abnormal expression of multiple factors and genes influenced by both environmental and genetic factors. The primary manifestation of atherosclerosis is plaque formation, which occurs when inflammatory cells consume excess lipids, affecting their retention and modification within the arterial intima. This triggers endothelial cell (EC) activation, immune cell infiltration, vascular smooth muscle cell (VSMC) proliferation and migration, foam cell formation, lipid streaks, and fibrous plaque development. These processes can lead to vascular wall sclerosis, lumen stenosis, and thrombosis. Immune cells, ECs, and VSMCs in atherosclerotic plaques undergo significant metabolic changes and inflammatory responses. The interaction of cytokines and chemokines secreted by these cells leads to the onset, progression, and regression of atherosclerosis. The regulation of cell- or cytokine-based immune responses is a novel therapeutic approach for atherosclerosis. Statins are currently the primary pharmacological agents utilised for managing unstable plaques owing to their ability to enhance endothelial function, regulate VSMC proliferation and apoptosis by reducing cholesterol levels, and mitigate the expression and activity of inflammatory cytokines. In this review, we provide an overview of the metabolic changes associated with atherosclerosis, describe the effects of inflammatory responses on atherosclerotic plaques, and discuss the mechanisms through which statins contribute to plaque stabilisation. Additionally, we examine the role of statins in combination with other drugs in the management of atherosclerosis.
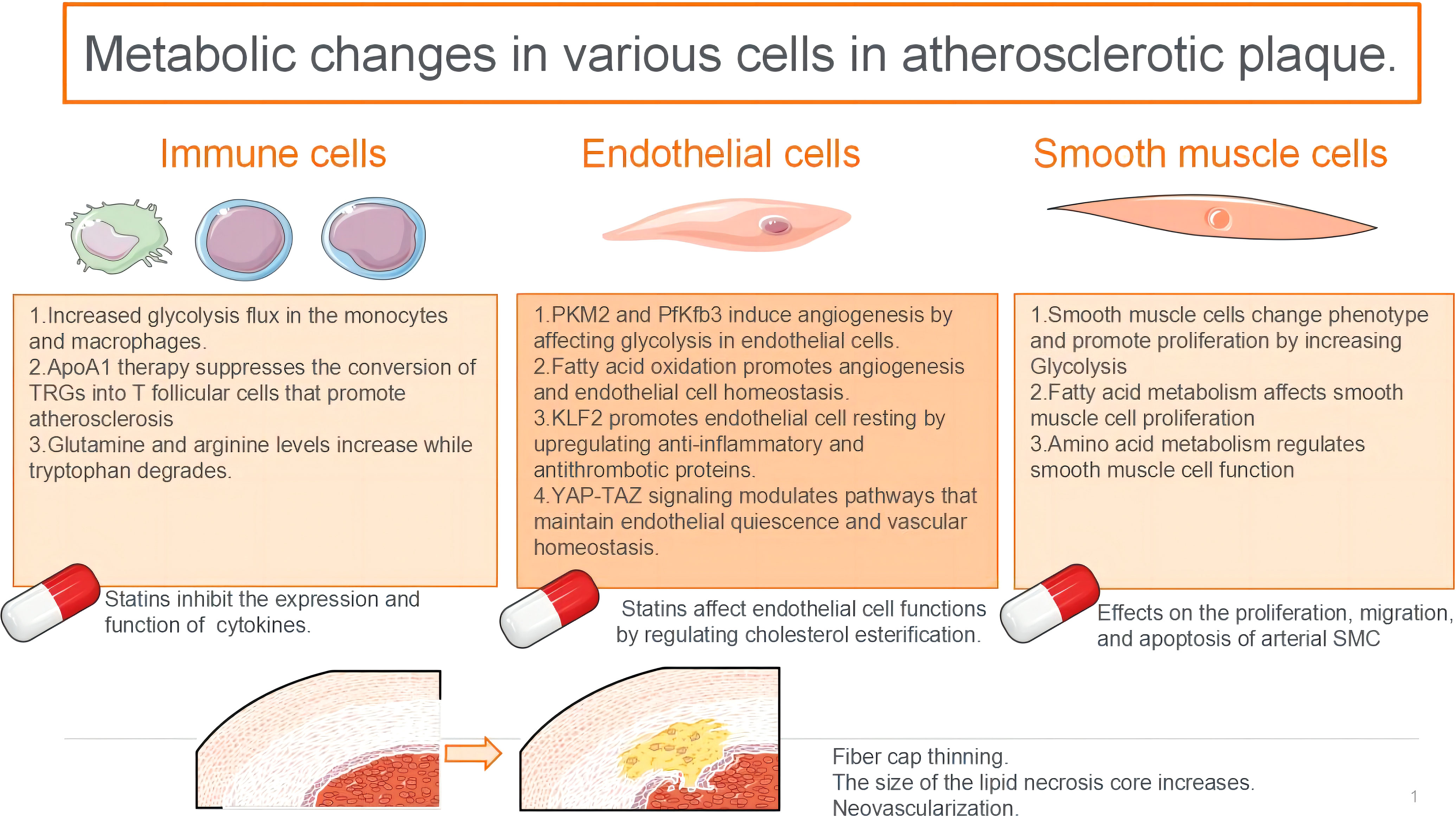
Graphical Abstract
Atherosclerosis is a common cardiovascular disease. It primarily manifests in the intima of affected arteries as lipid depositions, infiltrations of monocytes and lymphocytes, the migration and proliferation of vascular smooth muscle cells (VSMCs), and the formation of foam cells, lipid striations, and fibrous plaques, which further contributes to vascular wall sclerosis, stenosis, and thrombosis. Atherosclerosis can be asymptomatic for decades; however, when complications occur, such as the rupture of an atherosclerotic plaque, it can lead to myocardial infarction, stroke, peripheral vascular disease, and other high-fatality conditions. Treatment of these disease complications often relies on pharmaceutical interventions. In this review, we summarise the metabolic changes in some of the major cell types involved in atherosclerotic plaques and discuss the mechanisms, side effects, and progression of atherosclerotic plaques when treated with statins.
During the development of atherosclerotic lesions, the enhanced permeability of endothelial cells (ECs)facilitates the infiltration of peripheral inflammatory cells into the plaque (1). Macrophages, T cells, dendritic cells(DCs), and B cells are the most common types of immune cells found in growing arteriosclerotic plaques (2). Data from animal models show that selectively depleting or modulating the function of immune cells involved in atherosclerosis, inhibiting or blocking specific cytokines involved in inflammation and plaque development and regulating the immune cell bank and the secreted mediators in the arterial wall can impact atherosclerosis (2). Clinical trials, such as CANTOS (Canakinumab Anti-inflammatory thrombosis results study) and LoDoCo2 (low-dose colchicine for secondary prevention of stable coronary artery disease patients), also strongly suggest that immune regulation may be a relevant treatment option for human atherosclerosis (3, 4).
Metabolic regulation is closely related to the induction of immune responses. Metabolism in the microenvironment affects the proliferation and differentiation of immune cells and promotes the synthesis and secretion of immune mediators. In this section, we summarise the process of metabolic reprogramming in immune cells, particularly focusing on energy-related metabolic pathways. Moreover, we explore the potential of targeting the immune metabolism of macrophages and lymphocytes to control inflammatory responses in atherosclerotic lesions. The hypoxic environment within atherosclerotic plaques initially stimulates macrophage polarisation and enhances macrophage glycolysis.
Adenosine triphosphate (ATP) is the universal energy currency within cells. The main sources of ATP for macrophages and lymphocytes involve glucose metabolism via glycolysis and the pentose phosphate pathway (PPP). Under normal oxygen conditions, cells generate 36 ATP molecules through the citric acid cycle and oxidative phosphorylation (OXPHOS) pathway in the mitochondria. In an anaerobic environment, pyruvate is reduced as a hydrogen acceptor to lactate, resulting in a decrease in ATP production (two molecules) but an increase in ATP production rate. In the 1920s, the German scientist Warburg observed that, even in the presence of sufficient oxygen, tumour cells suppress aerobic respiration through a series of molecular mechanisms and promote efficient glycolysis reactions. This metabolic shift leads to the production of a large amount of ATP, creating a microenvironment suitable for the survival of tumour cells. This unique form of energy metabolism is known as the “Warburg effect”. In recent years, several research groups have observed that the metabolic profile of activated macrophages, induced by phagocytosis or inflammatory stimulation, undergoes reprogramming. This reprogramming involves a transition from oxidative metabolism to “Warburg metabolism” (5), enabling the rapid supply of energy required for the inflammatory process and the essential metabolic intermediates needed for biosynthesis (Figure 1).
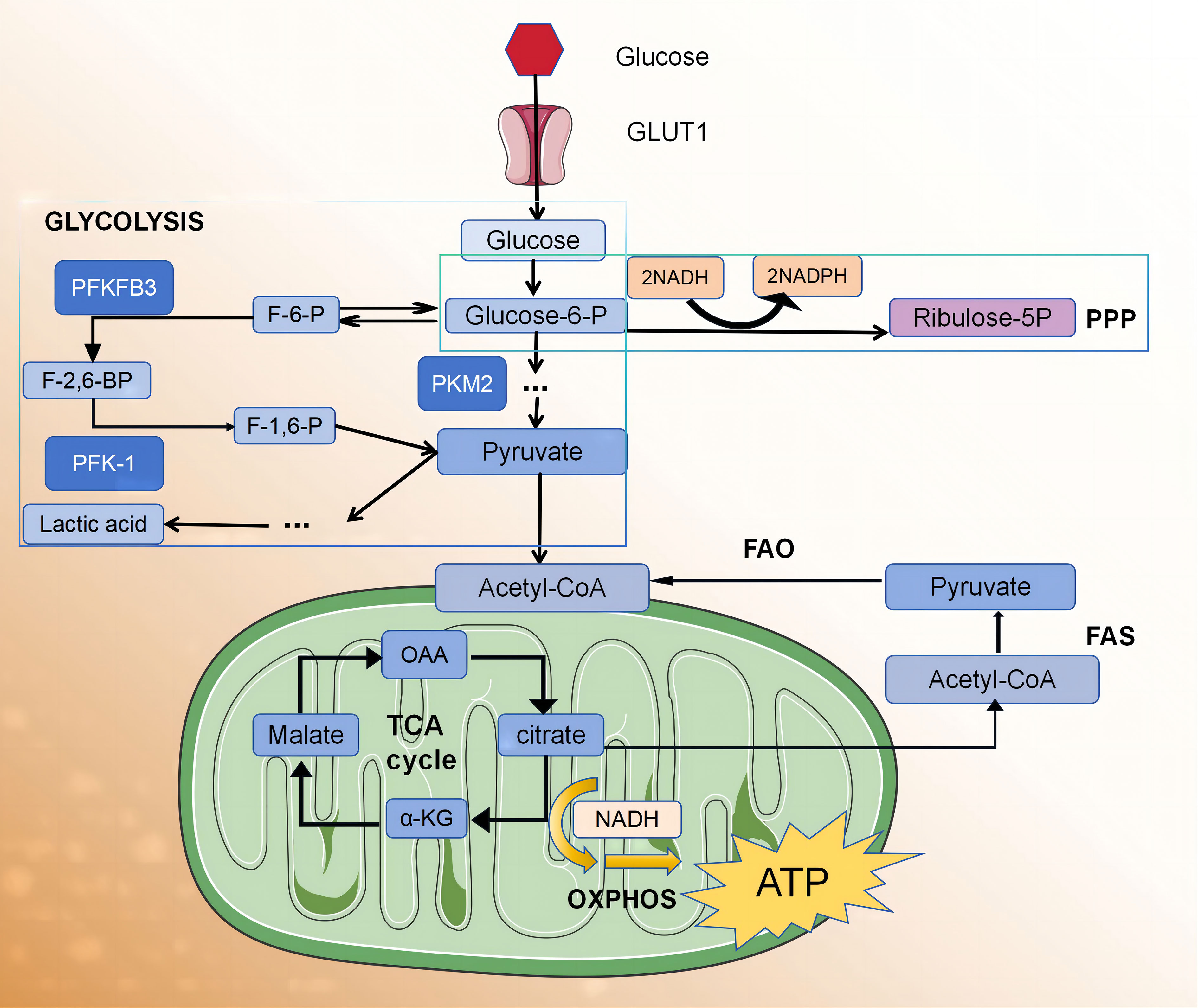
Figure 1 The glycolytic fluxes of immune cells, endothelial cells and smooth muscle cells in atherosclerotic plaque were increased.
Nonpolarized macrophages, under steady-state conditions, primarily obtain energy via the OXPHOS pathway. However, the metabolic characteristics of polarised macrophages (M1 and M2) when obtaining energy are relatively complex (6), and they exhibit distinct metabolic profiles. According to the stimulation received from the microenvironment, M1 macrophages use glycolysis and the PPP to meet their ATP requirements. Simultaneously, OXPHOS and fatty acid oxidation (FAO) in the citric acid cycle are downregulated (7–10). In contrast, M2 macrophages exhibit a metabolic profile characterised by a complete citric acid cycle and enhanced FAO and OXPHOS (11, 12).
Glycolysis serves as a fundamental mechanism for energy production within cells. The glycolytic pathway occurring in the cytoplasm facilitates the conversion of glucose into pyruvate, yielding two ATP molecules per glucose unit. It also provides metabolic intermediates required for biosynthetic pathways, including ribose, amino acid, and fatty acid synthesis. The PPP operates in conjunction with the glycolysis pathway, harnessing the energy derived from glucose 6-phosphate conversion to ribulose 5-phosphate for NADP+ reduction to nicotinamide adenine dinucleotide phosphate (NADPH). This enzymatic process generates reactive oxygen species (ROS), which exhibit pathogenic activity. Additionally, high levels of NADPH are essential for maintaining the reduced form of the antioxidant glutathione, which protects cells from oxidative stress.
The adaptation of glycolysis metabolism depends on the activation of several transcription factors, including hypoxia-inducible factor 1 (HIF1α) (13). HIF1α regulates the expression of glycolytic enzymes, glucose transporter 1 (GLUT1), and genes encoding inflammatory mediators (13–15). The upregulation of GLUT1 promotes rapid glucose uptake by M1 macrophages (10). Moreover, HIF1α supports the conversion of pyruvic acid to lactic acid by promoting the expression of lactate dehydrogenase (LDH) (16) and pyruvic acid dehydrogenase kinase (PDK) (17, 18), which are critical for restoring NAD+ levels and maintaining glycolytic flux. HIF1α regulation in macrophages predominantly occurs through two main signalling pathways, involving α expression of several genes, including toll-like receptor (TLR)/nuclear factor-κB (NF-κB) (15) and AKT/mammalian target of rapamycin (mTOR) (17–19) pathways. AKT kinase regulates macrophage polarisation in a subtype-specific manner, with AKT1 deletion promoting the M1 spectrum, while AKT2 deletion amplifies the M2 reaction (17). The other two factors that regulate glycolytic flux in M1 macrophages are 6-phosphofructose-2-kinase/fructose 2,6-diphosphatase 3 (Pfkfb3) and pyruvate kinase M2 (PKM2). Pfkfb3 catalyses the conversion of fructose-2,6-diphosphate to fructose-6-phosphate with low efficiency, thereby enhancing glycolytic flux. Moreover, M1 cells upregulate subtype PKM2, which, when overexpressed, exists in a balance between enzyme-inactive monomers or dimers and enzyme-active tetramers (20). The inactive enzymes translocate into the nucleus and bind to HIF1α, triggering the expression of HIF1α-regulated genes, whereas the enzymatically active tetramers promote glycolysis and M1 polarisation in the cytoplasm (20). In M1 macrophages, glycolysis significantly impacts macrophage functions, including phagocytosis, ROS production, and proinflammatory cytokine secretion (21). However, the role of glycolysis in M2 macrophages remains a subject of debate. Several studies have proposed that the presence of 2-deoxyglucose, an inhibitor of glycolysis, may impede M2 polarisation and impair its functionality (22, 23). There are also data indicating that M2 differentiation relies more on OXPHOS than glycolysis (24, 25). The tricarboxylic acid cycle (TCA cycle) is also complete in M2 macrophages.
Early studies using 18 fluorodeoxyglucose (18 FDG) positron emission tomography (PET) revealed elevated glucose uptake in atherosclerosis plaques of rabbits and humans compared to healthy blood vessels. Additionally, macrophage glycolysis levels, PPP activity, and TCA cycle metabolites were increased within plaques (12, 26). Notably, in vitro studies demonstrated that exposure to inflammatory factors, rather than a hypoxic environment, leads to a reduction in TCA cycle metabolism in polarized M1 macrophages, while the glycolytic metabolism level does not concurrently increase. These findings support the hypothesis that hypoxic stimulation enhances glycolysis in M1-polarized macrophages within atherosclerotic arteries (27). However, whether nonhypoxic stimulation enhances glycolysis in macrophages remains controversial (28).
Several studies have indicated that natural T cells are activated and differentiated into different subgroups after entering atherosclerotic plaques, and different T cell subgroups play distinct roles (29). For instance, a study has shown that the absence of CD8+ T cells in mice did not affect plaque size, but immunotherapy with ApoB100 P210 peptide in CD8+ ApoE-/- mice mitigated atherosclerotic lesions, suggesting that CD8+ T cells may have a different function (30). The role of CD4+ T cells in atherosclerosis is multifaceted. Static CD4+ T cells differentiate into effector T cells (Teff cell) and regulatory T cells (Treg cell) upon activation. Different Teff cell lines proliferate and differentiate rapidly depending on their microenvironment (31).
Effector CD4+ T cells in plaques primarily include Th1, Th2, and Th17 cells. Factors involved in the Th1 reaction, including tumour necrosis factor-α (TNF-α), recombinant human interferon-γ (IFN-γ), interleukin (IL)-12, and IL-18, have been proven to promote atherosclerosis through leukocyte recruitment, EC injury, and oxidative stress (32–36). However, the role of Th17 cells in atherosclerosis remains controversial. A study analysing atherosclerotic plaques in human coronary arteries showed that the cytokine IL-17 released by Th17 can synergistically increase the secretion of IL-6 with IFN-γ, promoting inflammation and atherosclerosis (37, 38). Conversely, in a study by Madhur MS et al., IL-17A did not affect atherosclerotic plaque burden in IL17AApoE-/- mice fed a high-fat diet (39, 40). IL-10 secreted by Th17 cells may prevent the recruitment of T cells and macrophages and enhance protection against atherosclerosis by promoting the transformation of macrophages from an inflammatory to an anti-inflammatory phenotype (41, 42). The role of Th2 cells in the development of atherosclerosis remains uncertain. Th2 cells secrete IL-5, IL-10, and IL-13 and activate B cells to produce antibodies, which are believed to counteract the pro-atherosclerosis effects of Th1 cells. Furthermore, a study has shown that IL-4, an autocrine growth factor of Th2 cells, did not significantly affect the development of atherosclerotic lesions in ApoE-/- or female ldlr-/- mice (43). However, another study found that IL-4 deficiency reduced the formation of atherosclerotic lesions in female ldlr-/- mice (44). Therefore, the role of IL-4 in atherosclerotic lesions remains to be further clarified. Treg cells increase plaque stability mainly by secreting transforming growth factor-β (TGF-β) and IL-10. TGF-β can inhibit the recruitment and activation of T cells and macrophages, promote the proliferation of VSMCs, and maintain atherosclerotic lesion stability (31, 45). The balance between Teff and Treg cell subsets and their respective functions significantly influences the development of atherosclerotic lesions (46). Therefore, maintaining the equilibrium of the Treg/Teff ratio and function is essential to prevent the onset of atherosclerosis and slow its progression.
As T cells transition from a quiescent state to an activated state within atherosclerotic lesions, glycolysis becomes imperative for rapid energy generation. While OXPHOS is the primary pathway for energy generation in static T cells (31, 45), activated T cells require the GLUT1 receptor to increase glucose uptake and promote the upregulation of glycolytic enzymes to catalyse glycolysis, resulting in increased pyruvic acid production (47). Subsequently, pyruvic acid can be converted into lactic acid within the cytoplasm through LDH or into acetyl coenzyme A within the mitochondria through pyruvate dehydrogenase (PDH). Additionally, the NADH and flavin adenine dinucleotide (FAD) generated during this process are primarily produced within the mitochondria and ATP is produced through OXPHOS.
Teff cells (Th1, Th2, and Th17) predominantly rely on glycolytic metabolism and glutamine catabolism for energy, whereas Treg cells primarily depend on FAO (48). This metabolic distinction between Teff and Treg subpopulations plays a pivotal role in governing the differentiation fate of CD4+ T cells and maintaining optimal immune function (46, 49). Michalek et al. (48) differentiated Th1, Th2, and Th17 cells in vitro and found increased glycolysis and GLUT1 expression in these cells. Compared to wild-type mice, GLUT1 transgenic mice showed increased glucose uptake and selective enrichment of Teff cells. Conversely, inhibiting glycolytic metabolism through treatments like 2-deoxyglucose, a hexokinase inhibitor, reduces Teff cell production and impairs immune function in vitro and in vivo (13, 48, 50, 51). These findings indicate that the Teff subpopulation undergoes increased glycolytic metabolism, a necessity for differentiation and functional specialisation.
The high activity level of PDK1 in Th17 cells leads to higher extracellular acidification and glycolysis rates than in Th1 cells (46). Furthermore, PKM2 serves as the ultimate rate-limiting enzyme in CD4+ T cell glycolysis. Hyperhomocysteinemia expedites the onset and progression of atherosclerosis by augmenting both PKM2 protein expression and activity in ApoE−/− mice (52). Therefore, targeted inhibition of the PKM2 metabolic pathway in CD4+ T cells may represent a novel strategy for treating atherosclerotic lesions.
Glycolysis promotes the activation, proliferation, and migration of Treg cells to inflammatory tissues (53–55). The expression of the Treg transcription factor, forkhead box protein 3 (FOXP3), suppresses glycolytic metabolism and enhances OXPHOS by downregulating Myc protein expression. This metabolic adaptation allows Treg cells to function effectively in a low-glucose/high-lactate environment (56). However, the anoxic conditions within plaques may decrease FOXP3 expression and impair the protective effects of Treg cells on atherosclerosis (57, 58). In summary, the induction of the glycolytic pathway plays a crucial role in modulating the differentiation balance between Treg and Teff cells. However, further research is required to elucidate how the atherosclerotic plaque microenvironment affects the metabolic and functional properties of distinct T-cell subsets (Table 1).
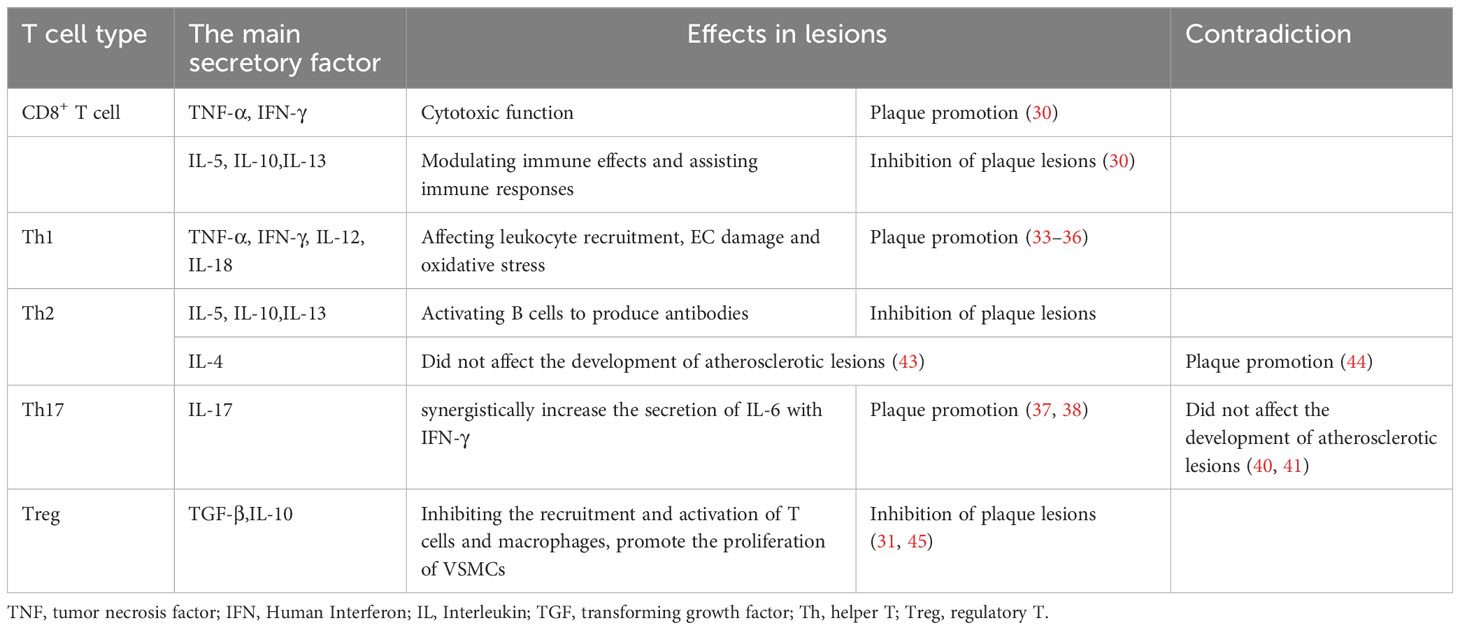
Table 1 T cell typing and function.
In atherosclerotic lesions, vascular DCs are present within the plaque and outer membrane (59). Apart from their role in lipid absorption and clearance of apoptotic cells in plaques, DCs also facilitate T cell activation and proliferation by presenting antigens derived from autologous sources. DCs secrete a diverse array of cytokines that indirectly modulate the functionality of other immune cells, thereby contributing to immune-mediated vascular wall damage and the development of atherosclerosis, including their impact on B-cell activation (60).
B cells play a significant role in the development of atherosclerosis (61); however, there is ongoing debate regarding their specific involvement in plaque formation (62). B cells can be categorised into two subtypes: B1 and B2. The former produces natural antibodies (IgM) that are believed to exert protective effects against atherosclerosis by inhibiting necrotic nucleus formation on blood vessel walls (63, 64). Conversely, B2 cells participate in adaptive immune responses and secrete cytokines IL-10 and TNF-α, which influence Treg development and potentially promote the formation of atherosclerosis (65). Studies have investigated approaches such as anti-CD20 depletion or the absence of the BAFFR receptor to mitigate damage caused by B2 cell activation and protect hypercholesterolemic mice from developing atherosclerosis (66–68).
Glycolysis plays a crucial role in the activation of B cells within atherosclerotic lesions. The B-cell receptor (BCR), an immunoglobulin located on the surface of B cells, is responsible for specific antigen recognition and binding (69). BCR regulates glucose utilisation by promoting glucose uptake and the expression of the GLUT1 transporter in B cells, leading to a rapid and sustained increase in glucose metabolism that provides essential energy and the foundation for growth. After BCR activation, there is a significant increase in glycolysis. Previous studies have demonstrated that enhanced glucose utilisation primarily involves the PI-3K signalling pathway associated with BCR activity. This pathway facilitates precise regulation of glucose utilisation within B lymphocytes through the PI-3K/AKT signalling cascade, ensuring their capacity to meet the energy demands essential for growth-related processes (70) (Table 2).
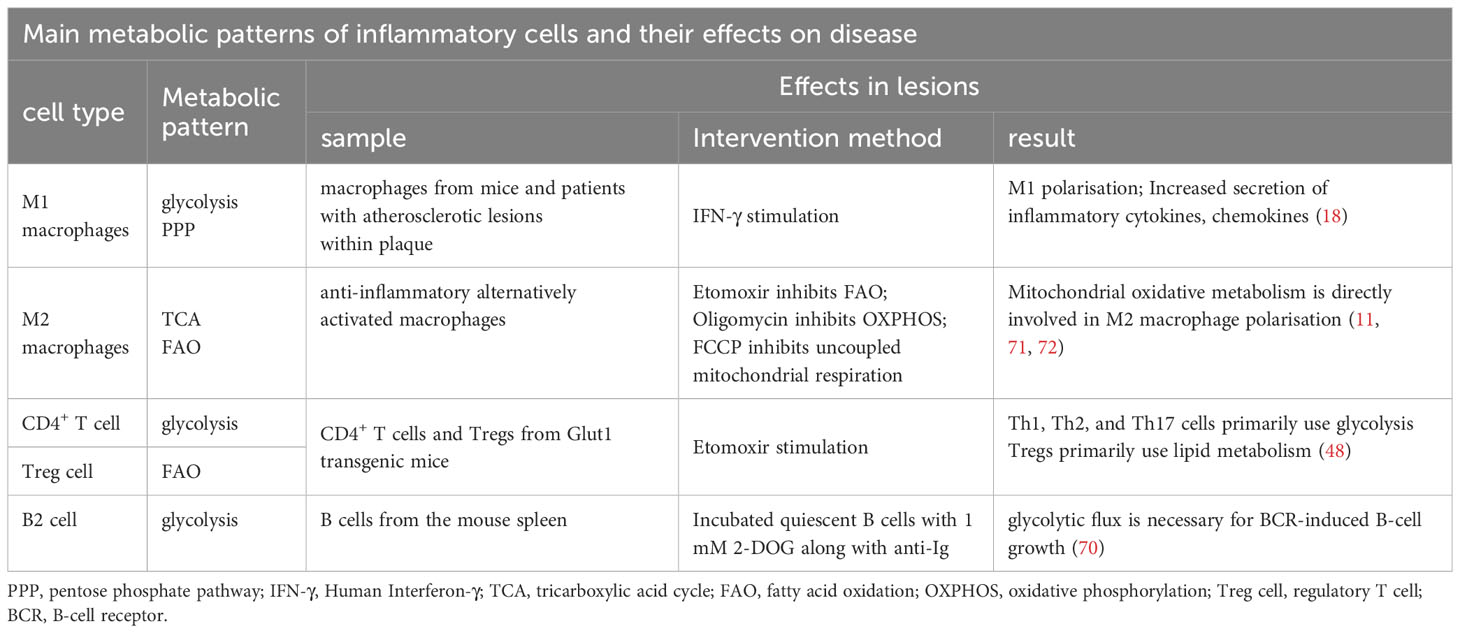
Table 2 Main metabolic patterns of inflammatory cells and their effects on disease.
During atherosclerosis, apolipoprotein A1 (ApoA1) indirectly affects T cell responses during inflammation. ApoA1 is the major protein component of high-density lipoprotein (HDL) and is produced in hepatocytes. Before being released into the plasma, it interacts with pre-HDL particles and ATP-binding cassette transporter A1 (ABCA1), acquiring phospholipids and cholesterol to form new HDL or ABCA1 (73). The formation of pre-HDL promotes the efflux of cholesterol from cells, resulting in a reduction in plaque volume. Research has shown that treating ApoE−/− mice with ApoA1 increased ABCA1 expression in Treg cells and restored cholesterol levels in these cells to normal (74). The anti-inflammatory properties of ApoA1 are also associated with changes in the lipid raft composition. Lipid rafts are microdomains on the cell membrane that are rich in sphingolipids and cholesterol, serving as enriched sites for IL-2 receptors. Lipid raft components regulate immune cell signalling and proliferation. Several studies have demonstrated that ApoA1 exerts regulatory effects on cholesterol levels, IL-2 receptor expression, and IL-6 expression in Treg cells during the progression of atherosclerosis, thereby impeding the transition from exTregs to Tfh cells and ultimately reducing atherosclerosis (73, 74).
Notably, diet-induced disruption of intracellular cholesterol metabolism is a significant factor affecting the differentiation and function of Tregs in atherosclerotic lesions. Maganto-García et al. (75) found that diet-induced hypercholesterolemia promoted the differentiation and migration of Treg cells in mouse splenic and prevented atherosclerosis. The classification of fatty acids into saturated, monounsaturated, and polyunsaturated forms is determined by the saturation level of their hydrocarbon chains. They can also be categorised as short-chain (SCFAs), medium-chain, or long-chain fatty acids (LCFAs) based on their carbon chain length. Fatty acids play a crucial role in regulating specific aspects of the body’s innate immunity and cholesterol metabolism within atherosclerotic plaques. The innate immune system relies on a diverse group of pattern-recognition receptors called toll-like receptors (TLRs) (76, 77). TLR4 is highly expressed in atherosclerosis and has multiple functions. It activates cell adhesion, enhancing the uptake of oxidized lipids by macrophages and foam cell formation (78, 79). It also influences cholesterol metabolism and its impact on atherosclerosis development (80). In contrast to saturated fatty acids, polyunsaturated acids do not activate the TLR4 signalling pathway (81–83) and instead inhibit NACHT-, leucine-rich repeat (LRR)-, and pyrin domain (PYD)-containing protein 3 (NLRP3) activity (84, 85). This is significant because NLRP3 may contribute to the pathogenesis of atherosclerosis through a signalling pathway that triggers PCSK9 secretion via IL-1β stimulation (86). LCFAs and SCFAs also play different roles in arteriosclerotic lesions. LCFAs can enhance the proliferation and differentiation of Teff cells into Th1 and Th17 cells, aggravating the progression of atherosclerosis (87). Conversely, dietary SCFAs affect Treg differentiation, which in turn improves and treats autoimmune-related diseases (87, 88).
These findings suggest the potential of using fat-free ApoA1 therapy and dietary adjustments to regulate the conversion between Treg and Tfh cells, offering potential benefits to patients with atherosclerosis and other inflammatory diseases.
Although glucose is generally considered the most important nutrient for inflammatory cells, amino acid metabolism also plays a crucial role in inflammatory cell proliferation and activation. Abnormal amino acid metabolism contributes to the occurrence and development of atherosclerotic lesions (89). Relevant studies mainly focus on monocytes and macrophages, with relatively fewer studies on lymphocytes.
Glutamine is one of the most widely studied amino acids involved in inflammation. It enters cell mitochondria through amino acid transporters and is converted to glutamic acid by the action of glutaminase. It also provides nutrients for the synthesis of other amino acids and NADPH and is an important energy source (90, 91). Glutamine, a non-essential amino acid in plasma, exerts intracellular effects on macrophage polarisation. Glutamine undergoes conversion to alpha-ketoglutaric acid (α-KG) through the enzymatic actions of glutaminase (GLS) and glutamate dehydrogenase (GDH/GLUD1). α-KG plays a crucial role in the tricarboxylic acid cycle, and its deficiency disrupts normal metabolic processes, thereby promoting the polarisation of macrophages towards the M1 phenotype and enhancing pro-inflammatory cytokine secretion. Conversely, active glutamine metabolism can stimulate macrophages to polarise towards the M2 type and secrete anti-inflammatory factors (9). Activated T cells exhibit increased glutamine uptake and metabolism, similar to cancer cells (90, 92). Depletion or deficiency of glutamine can disrupt T cell activation and proliferation (92). Rapid extracellular glutamine uptake depends on the amino acid transport weight group solute carrier family 1, member 5 (Slc1a5, also known as ASCT2). In mouse immune and autoimmune models, Slc1a5 defects impair Th1 and Th17 cell differentiation and thus affect inflammatory T cell responses (93). Under these circumstances, targeted therapy with glutamine hydrolase holds promise in preclinical models of cardio-cerebral vascular disease (94).
Leucine is an essential amino acid that affects the differentiation of Teff cells. Leucine entry into activated T cells requires the involvement of the L-leucine transporter (LAT1) (95). T cells lacking LAT1 cannot differentiate into Th1 or Th17 cells, even with appropriate polarising cytokines (96). Genetic defects in the leucine sensor sestrin 2 also limit T-cell activation and differentiation (97).
There is growing evidence that tryptophan metabolism is also involved in inflammatory responses. Overexpression of aminophenamide 2,3 dioxygenase 1 (IDO1) during inflammation can drive tryptophan consumption, produce bioactive metabolites, and control the interaction between general control non-derepressible 2 and specific receptors, thereby shifting cytokine production toward an anti-inflammatory phenotype. Conversely, the ablation of IDO1 promotes the pro-inflammatory effect of immune cells. The induction of IDO1 is associated with protection against atherosclerosis and increased plaque stability (98, 99). Other studies suggest that in addition to IDO1, other enzymes involved in tryptophan degradation, such as kynurenine 3-hydroxylase, are also involved in inflammation regulation, potentially increasing the instability of atherosclerotic plaques (100).
Arginine metabolism and its byproduct, nitric oxide (NO), play crucial roles in the early stages of atherosclerosis (101), such as involvement in immunity and affect the phenotypic polarisation of macrophages (102). Under inflammatory conditions, macrophages exhibit overexpression of inducible nitric oxide synthase (iNOS), thereby promoting arginine metabolism and subsequent NO production. Elevated NO levels can impede the repolarisation process from M1 to M2 by interfering with the electron transport chain (103). Additionally, arginine inhibits T-cell proliferation and impairs their ability to migrate to related chemokines, possibly contributing to its protective effect against atherosclerosis (104) (Table 3).
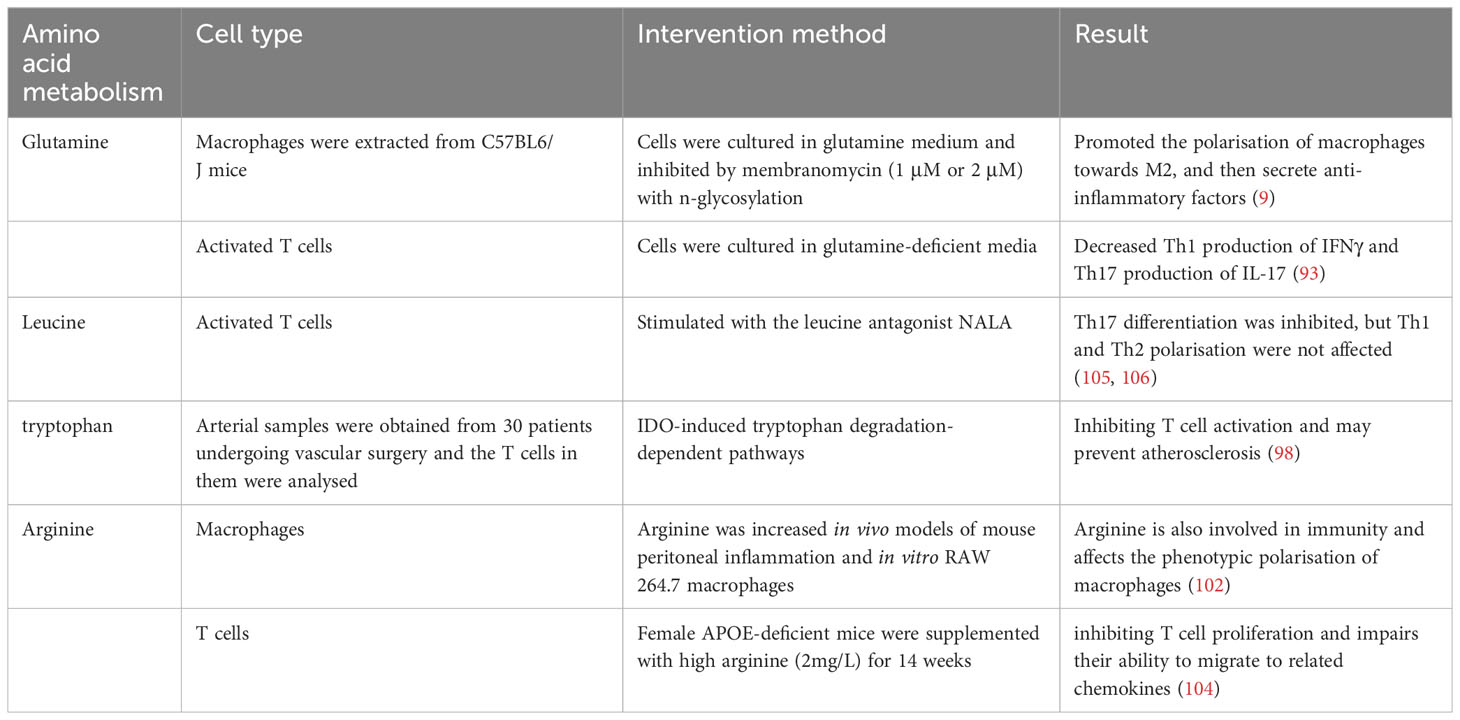
Table 3 Amino acid metabolism in inflammatory cells.
Vascular ECs play a crucial role in vascular homeostasis and disease. One of the earliest events of atherosclerosis development is the activation and dysfunction of ECs in vulnerable arterial regions. As atherosclerosis progresses, it exhibits characteristics such as the formation of a fibrous cap covering a lipid-rich necrotic core and the accumulation of leukocytes at the plaque’s periphery. These immune cells influence the phenotype of ECs and promote plaque instability. Although the endothelium was initially considered an inert and semi-permeable barrier between blood components and underlying endothelial tissues, it is now recognised as an organ with active metabolic functions that profoundly impact vascular homeostasis and atherosclerosis throughout life (107).
Metabolic pathways regulate angiogenesis, inflammation, and barrier function of ECs in atherosclerotic lesions. The following section outlines the changes in glycolysis and lipid metabolism observed in ECs within atherosclerotic plaques, as well as the regulation of EC homeostasis and function through Krüppel-like transcription factor 2 (KLF2) and yes-associated protein/the transcriptional coactivator with PDZ-binding motif (YAP/TAZ) signalling.
The formation of functional blood vessels and vascular plexuses, including processes like EC junction reorganisation, tip cell migration, stem cell proliferation, and phagocytosis (108–112), requires a substantial amount of energy in the form of ATP (108). Vascular sprouting relies on the differentiation of ECs into specialised subtypes, each with a specific function. Once the blood vessels are perfused, ECs transition into a quiescent state while being firmly anchored in the extracellular matrix. Blood vessels in which ECs germinate are regulated by genetic signalling and metabolic factors. ECs prefer to rely on glycolytic metabolism to minimise the production of ROS and rapidly produce ATP (95). Glycolytic enzymes and ATP are concentrated within the lamellar and filamentous pseudopods of ECs, facilitating their rapid motility (111).
Pfkfb3, the most prominent member of the Pfkfb family, plays a pivotal role in glycolysis by serving as the key enzyme (111). Conversely, the isozyme Pfkfb4 exhibits diminished kinase activity and can either stimulate or inhibit glycolysis (113). Signalling molecules known to induce germination in tip and stem cells, such as VEGF and FGF2 have been found to upregulate Pfkfb3 expression, subsequently promoting glycolysis. Furthermore, Pfkb3 also governs EC proliferation and influences their motility. In vitro and in vivo experiments have demonstrated that deactivating Pfkfb3 leads to reduced EC proliferation, impaired formation of filopodia/lamellipodia, and compromised directional migration, ultimately resulting in impaired vascular growth and branching in mice deficient in endothelial Pfkfb3 (114).
Alternatively, the Pfkfb3 blockade may reduce angiogenesis through other mechanisms. For example, in the context of tumour angiogenesis, lactic acid can activate HIF1α, upregulating vascular endothelial growth factor receptor 2 (VEGFR2) and promoting angiogenesis, or activate proangiogenic nuclear factors by inhibiting oxygen sensor PHD2 κB/IL-8 (115, 116). Reduced lactate levels after the Pfkfb3 blockade may inhibit angiogenesis through these mechanisms. Additionally, it has been proposed that Pfkfb3 may regulate cell proliferation through nuclear activity independent of glycolysis (117). However, further research is required to ascertain whether targeting Pfkfb3 can effectively reduce pathological angiogenesis.
Pyruvate kinases, which are involved in the production of pyruvate and ATP, are crucial for regulating glycolytic flux (109). PKM2, one of the PK isoenzymes located at the junctions of ECs expressing VE-cadherin, provides the material and energy required for promoting EC binding dynamics, migration, and proliferation through hyperactive glycolysis (118). Silencing PKM2 reduces ATP levels near EC junctions, affects the dynamics and internalisation of VE-cadherin at EC junctions, reduces the number of filopodia in endothelial tip cells, and ultimately disrupts EC junction remodelling, collective migration, and angiogenic germination.
ECs serve as gatekeepers for fatty acid transport. The fatty acids at atherosclerotic sites are associated with alterations in biophysical properties and membrane protein function within EC membranes. Polyunsaturated fatty acids not only serve as carbon sources for cultured ECs but are also regulated by ECs for transport to metabolically active tissues (119). Circulating fatty acids can be locally released from triglyceride-rich lipoproteins in the lumen of ECs via lipoprotein lipase-mediated lipolysis. Subsequently, they can enter ECs via passive diffusion or fatty acid transporters (120). The silencing or deletion of fatty acid-related genes can affect various EC functions, including migration capacity, vascular sprouting ability, and permeability regulation. Additionally, it can impact the activity of endothelial nitric oxide synthase (eNOS), leading to the production of excessive NO, an important vasodilator. Therefore, dysregulation of eNOS and excessive NO production can lead to pathological dysfunction and contribute to the progression of atherosclerosis (121).
Vascular ECs undergo different flow patterns depending on their location. For instance, the aortic arch, near the branching of the large ductal artery, and the tip of the coronary artery are exposed to oscillating shear stress known as disturbed flow (d-flow). In contrast, the larger curvature of the aorta and thoracic aorta experience high shear stress known as steady flow (s-flow). Exposure to d-flow increases the susceptibility of ECs to intima-media thickening and atherosclerosis (122). Among the numerous mechanosensitive transcription factors that differentially regulate vascular pathophysiology, our focus lies on KLF2 and YAP/TAZ.
KLF2 expression was down-regulated in ECs exposed to d-flow. KLF2 is the primary activator of eNOS expression and other regulatory genes in the ECs, Its ability to maintain optimal expression and activity is indispensable for preventing pathological alterations in blood vessels, including thrombosis, oxidative stress, and inflammation (123). While early atherosclerosis is associated with Lectin-type oxidized low-density lipoprotein receptor 1 (LOX-1) expression. The imbalance between the excessive production of ROS and inadequate antioxidant defences in atherosclerosis leads to profound oxidative stress and the transformation of low-density lipoproteins (LDL) into highly atherogenic oxidized LDL (ox-LDL). These ox-LDL particles are subsequently deposited subcutaneously and bind to the clearance receptor LOX-1. This leads to an increased expression of cell adhesion molecules in ECs, promoting increased adhesion and migration of inflammatory cells into the intima. Concurrently, endothelial dysfunction worsens owing to increased vasoconstrictor production, increased ROS, and depletion of endothelial nitrogen oxide production (124). The activation of KLF2 is crucial for LOX-1 expression under shear stress. Downregulating KLF2 increases LOX-1 expression while overexpressing it inhibits LOX-1 upregulation. KLF2 regulates the degradation of 3-hydroxy-3-methylglutaryl reductase (HRD1), an E3 ubiquitin ligase, by binding to its promoter. HRD1 expression in atherosclerotic ECs is significantly reduced due to ox-LDL. Conversely, overexpression of HRD1 prevents ox-LDL-induced apoptosis in ECs (125).
In summary, KLF2 affects EC function by regulating the expression of various proteins and is a promising target for the treatment of atherosclerosis and related diseases (Figure 2).
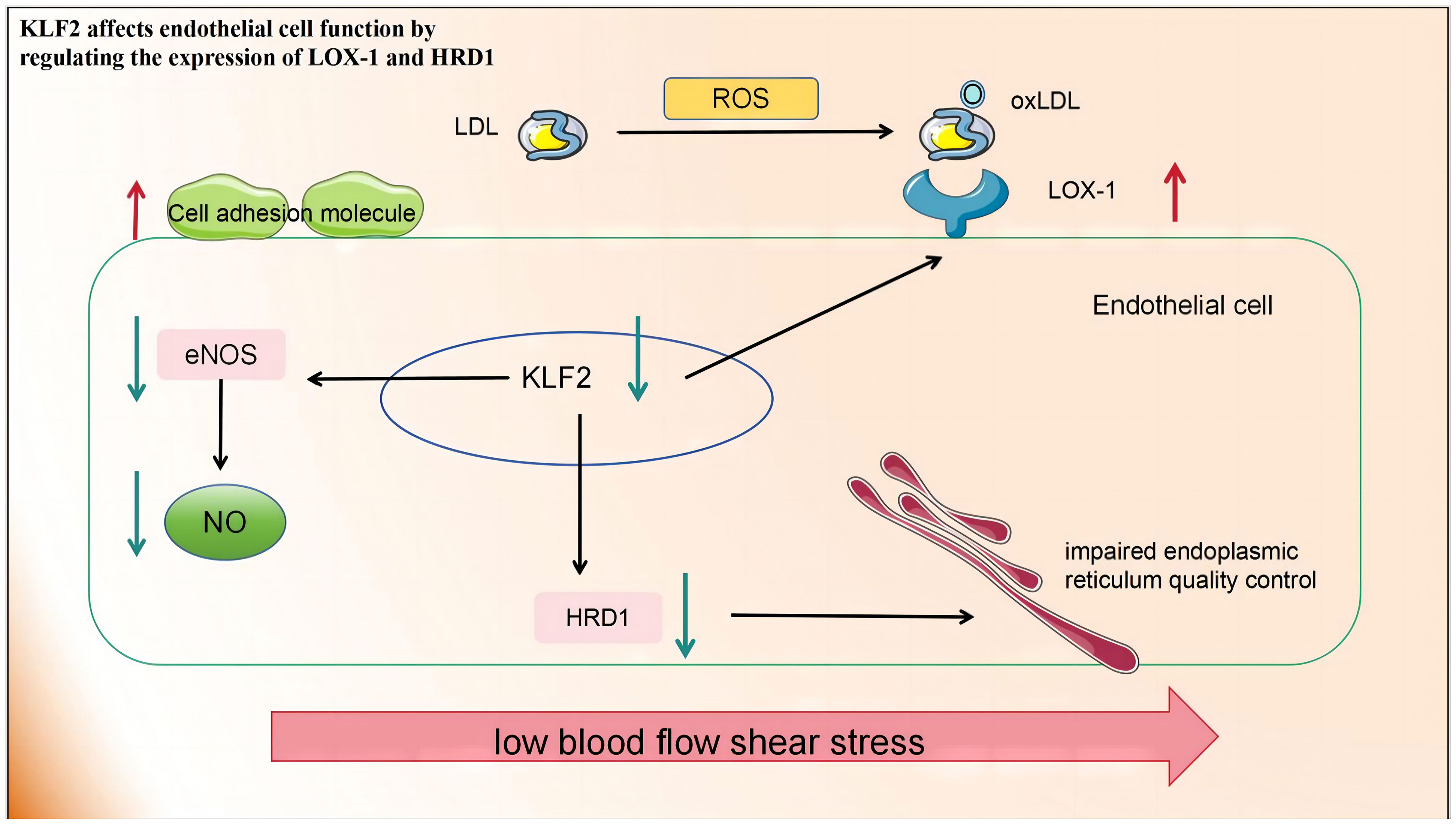
Figure 2 Low blood flow shear stress in atherosclerotic lesions leads to the downregulation of KLF2 in endothelial cells, thereby impacting endothelial cell function in the following aspects: 1) The production of eNOS and NO is diminished, consequently impairing their roles in promoting vascular relaxation and inhibiting inflammation. 2) The upregulation of LOX-1 expression on the cell membrane facilitates increased ox-LDL entry into cells, resulting in cellular damage. 3) The downregulation of HRD-1 expression involved in endoplasmic reticulum-related protein degradation pathway compromises endoplasmic reticulum quality control. KLF2, Recombinant Human Krueppel-like factor 2; eNOS, endothelial nitric oxide synthase; NO, nitric oxide; LOX-1, lectin-like oxidised LDL receptor 1; HRD-1, hydroxy-3-methylglutaryl reductase degradation.
On the contrary, YAP/TAZ was activated and translocated into the nuclei of ECs that were exposed to atherosclerotic interference in blood flow. Relevant findings were substantiated by Wang et al. through in vivo and in vitro experiments. they also found target genes such as angiogenesis inducer 61 (CYR61), connective tissue growth factor (CTGF), and ANKRD1 were upregulated. Mouse arterial surface analysis also revealed increased nuclear localisation of YAP/TAZ and elevated levels of endothelial target genes in atherosclerotic regions. In contrast, protective laminar flow inhibited YAP/TAZ activity. Knockdown of YAP/TAZ significantly reduced EC proliferation and the induction of pro-inflammatory phenotypes. Conversely, overexpression of YAP promoted EC proliferation and inflammation. Thus, inhibiting YAP/TAZ activation may be a promising therapeutic strategy for atherosclerotic protection. Notably, statins inhibit YAP/TAZ activity, thereby reducing disturbed flow-induced proliferation and inflammation (126).
In summary, the regulation of various EC functions is influenced by metabolic pathways, including those related to angiogenesis, inflammation, and barrier function. Despite advancements in understanding EC metabolism, many questions remain unanswered. A deeper comprehension of metabolic disturbances in ECs could lead to the development of novel therapeutic strategies for treating atherosclerosis (Figure 3).
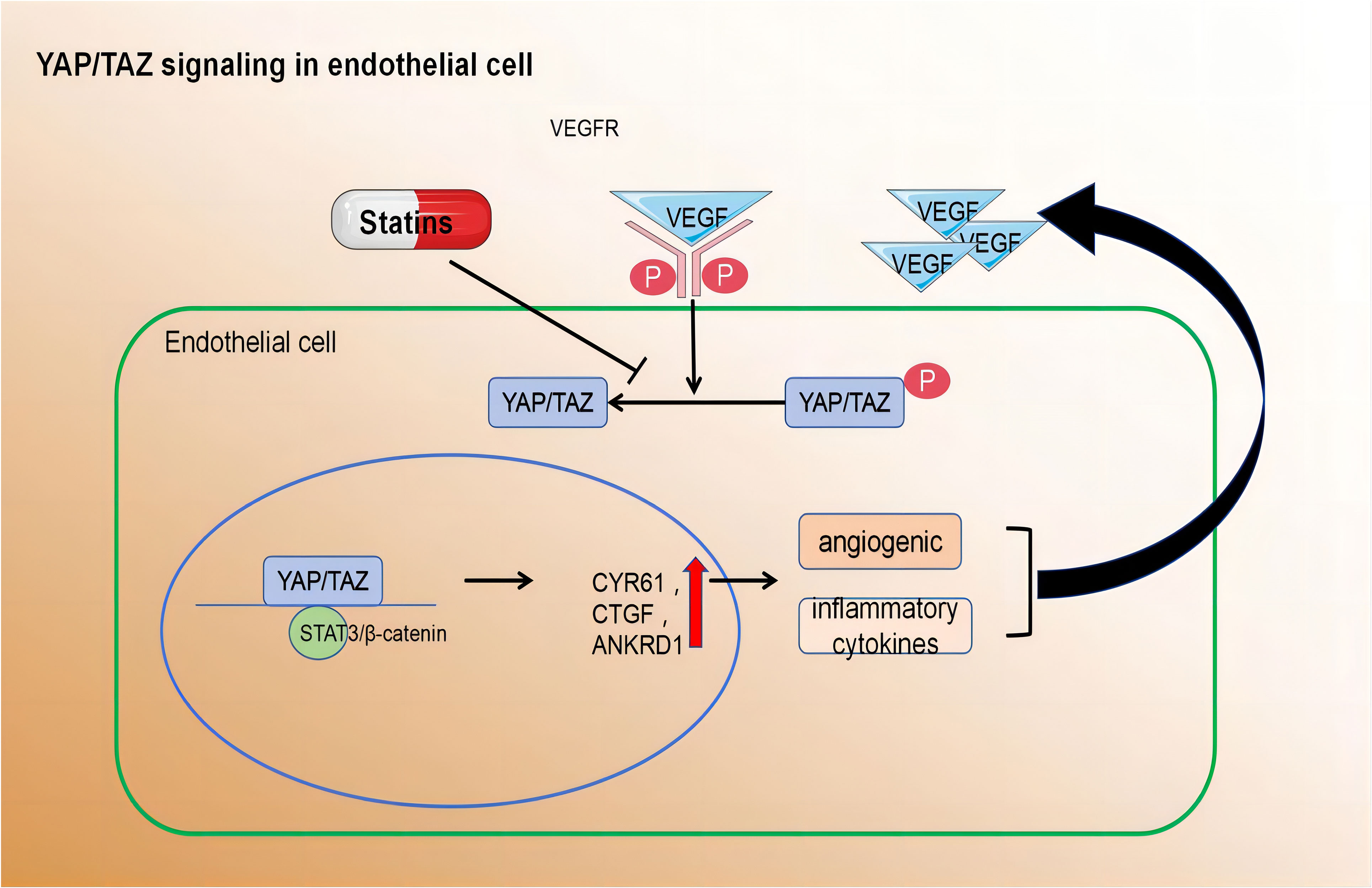
Figure 3 Pro-angiogenic growth factors, inflammatory cytokines, hypoxia, and disturbed blood flow activate YAP/TAZ leading to their translocation to the nucleus.Within the nucleus, YAP/TAZ interacts with STAT3 and β-catenin to induce transcription of downstream target genes. cysteine-rich angiogenic inducer 61 (CYR61), connective tissue growth factor (CTGF), and ankyrin repeat domain 1 (ANKRD1) Activated YAP/TAZ induce the expression of angiogenic and inflammatory cytokines.
The stability of atherosclerotic plaques depends on the thickness of the fibrous cap and the level of inflammation within it. Thinning of the vascular smooth muscle cell cap, which increases plaque rupture risk, is caused by cellular death and degradation of collagen and extracellular matrix (ECM). Vascular smooth muscle cells need to proliferate and synthesise new matrix components for effective repair. However, this process is hindered by cell death and senescence. The balance between cell proliferation, migration, death, and senescence plays a crucial role in determining the population size of vascular smooth muscle cells in atherosclerotic plaques. Understanding and regulating these processes are essential for maintaining stability during atherosclerosis development and plaque formation. Vascular smooth muscle cells are mainly found in the tunica media of arteries, exhibiting a mature “contractile” phenotype characterized by limited proliferation rates and expression of specific contractile proteins crucial for optimal vascular function (127). However, in response to vascular injury, these cells undergo a phenotypic transition from their static “contractile” state to a highly migratory and proliferative “synthetic” state. This shift significantly contributes to the development of Atherosclerosis, hypertension, and intimal hyperplasia formation (128–130). Recent research has revealed that metabolic reprogramming drives this transformation in VSMCs, involving key metabolic pathways such as glycolysis, fatty acid oxidation, and amino acid metabolism in both physiological and pathological vascular systems (130).
During the development of atherosclerotic plaques, VSMCs transition from their contractile state to a more synthetic state, involving proliferation and migration from the tunica media to the intima. This phenotypic alteration is closely linked to alterations in glucose metabolism, particularly glycolysis (131). One key driver of VSMC proliferation in atherosclerosis is the upregulation of GLUT1, a glucose transporter. This upregulation results in a substantial increase (44%) in the intracellular glucose concentration within these cells (132–134). The higher glucose levels provide ample energy and lactic acid for further VSMC proliferation. Therefore, modulating glycolysis presents a promising therapeutic avenue for treating atherosclerosis.
Inhibition of the glycosylation pathway rate-limiting enzyme PKM2 leads to a decrease in extracellular and intracellular lactate production (135). Lactate dehydrogenase A (LDHA) converts pyruvate to lactic acid, which affects the survival, proliferation, migration, and invasion of human and rat aortic SMCs (133, 134). Additionally, LDHA down-regulates adenosine monophosphate-activated protein kinase (AMPK), which is involved in the vascular injury pathway. Furthermore, pyruvate can be oxidized to form acetyl-CoA by the PDH enzyme complex in the mitochondria of SMCs, entering the TCA cycle (136). Therefore, inhibition of PDK1 weakens the TCA cycle and shifts glucose metabolism from OXPHOS to glycolytic pathways (137).
In summary, the regulation of glycolysis is a key factor influencing VSMC proliferation. Additionally, VSMCs possess functional mitochondria, which not only produce energy but also provide metabolites necessary for biomass synthesis. Targeting mitochondrial complex I activity may reduce neointimal hyperplasia by inhibiting VSMC proliferation and migration (138). Furthermore, the presence of ox-LDL induces oxidative stress and ROS production in human VSMCs. High levels of ox-LDL exacerbate VSMC apoptosis, resulting in matrix and collagen loss and the thinning of the fibrous cap (139). These intricate metabolic processes play critical roles in the development of atherosclerotic plaques and offer potential therapeutic targets for intervention.
In addition to glucose, VSMCs can also derive energy from fatty acids. FAO yields a higher amount of energy compared to glucose metabolism but requires higher oxygen levels. The presence of fatty acids can impact the utilisation of glucose and glycogen in both resting and contracting VSMCs (140). During the phenotypic transformation of VSMCs, there is a decrease in glucose oxidation and an increase in FAO. This elevation in FAO may provide VSMCs with additional energy for rapid proliferation, migration, synthesis, and secretion of the extracellular matrix (141, 142). The Randall cycle plays a pivotal role in altering the preference for utilising either glucose or fatty acids as fuel sources. This shift towards increased FAO impedes the oxidation of glucose (143). The Randall cycle has been implicated in regulating oxidative metabolism in muscle tissue, adipose tissue (143, 144), and brain energy balance (145). However, its influence on VSMC metabolism remains unclear. Exploring this aspect could present an intriguing avenue for future research.
Notably, dysfunctional FAO has been observed in plaques within the carotid artery of humans (146). This impaired FAO capacity may limit the functions of VSMCs, such as proliferation and migration, which are essential for plaque stability and vascular health.
Currently, there is a growing body of research focusing on the role of amino acid metabolism in VSMCs. Glutamine, the primary non-essential amino acid in plasma, plays a pivotal role in this process. Recent findings indicate that Slc1a5 plays a pivotal role in the efficient transportation of L-glutamine, and this transport mechanism has the potential to enhance VSMC proliferation (147). Moreover, glutamine is utilised to produce glutathione (γ-glutamyl-L-cysteinylglycine, GSH), an essential component involved in combating free radicals. Within VSMCs, both reduced GSH and its oxidized form (glutathione disulfide, GSSG) play critical roles in maintaining cellular redox balance (148). Depletion of GSH and increased DNA damage have been shown to inhibit growth and induce cell death in human VSMCs (149–151). Nitric oxide, a free radical in VSMCs, induces p53 expression and triggers programmed cell death by consuming intracellular GSH, making it a potent initiator of apoptosis (152). Additionally, it impedes mitochondrial respiration by suppressing the functions of complexes I and II; thereby affecting the relaxation of vascular smooth muscle (107). Furthermore, L-arginine effectively suppresses the proliferation and migration of VSMCs, even in the absence of NOS (153–155). Tryptophan, an essential amino acid serving as a substrate for serotonin synthesis, significantly enhances both proliferative and migratory tendencies of rat VSMCs under laboratory conditions (156). Elevated cysteine levels cause inflammation, oxidative stress, and increased proliferation and migration of VSMCs (157, 158). Notably, endogenous synthesis of hydrogen sulphide (H2S) from cysteine has protective effects on blood vessels. It inhibits NADPH oxidase, ROS production, glutathione disulfide formation, glutathione synthesis, and cysteine uptake (159, 160).
In summary, amino acid metabolism in atherosclerotic lesions affects smooth muscle proliferation and migration. These processes have profound implications for vascular health and the development of conditions such as atherosclerosis (Figure 4) (Table 4).
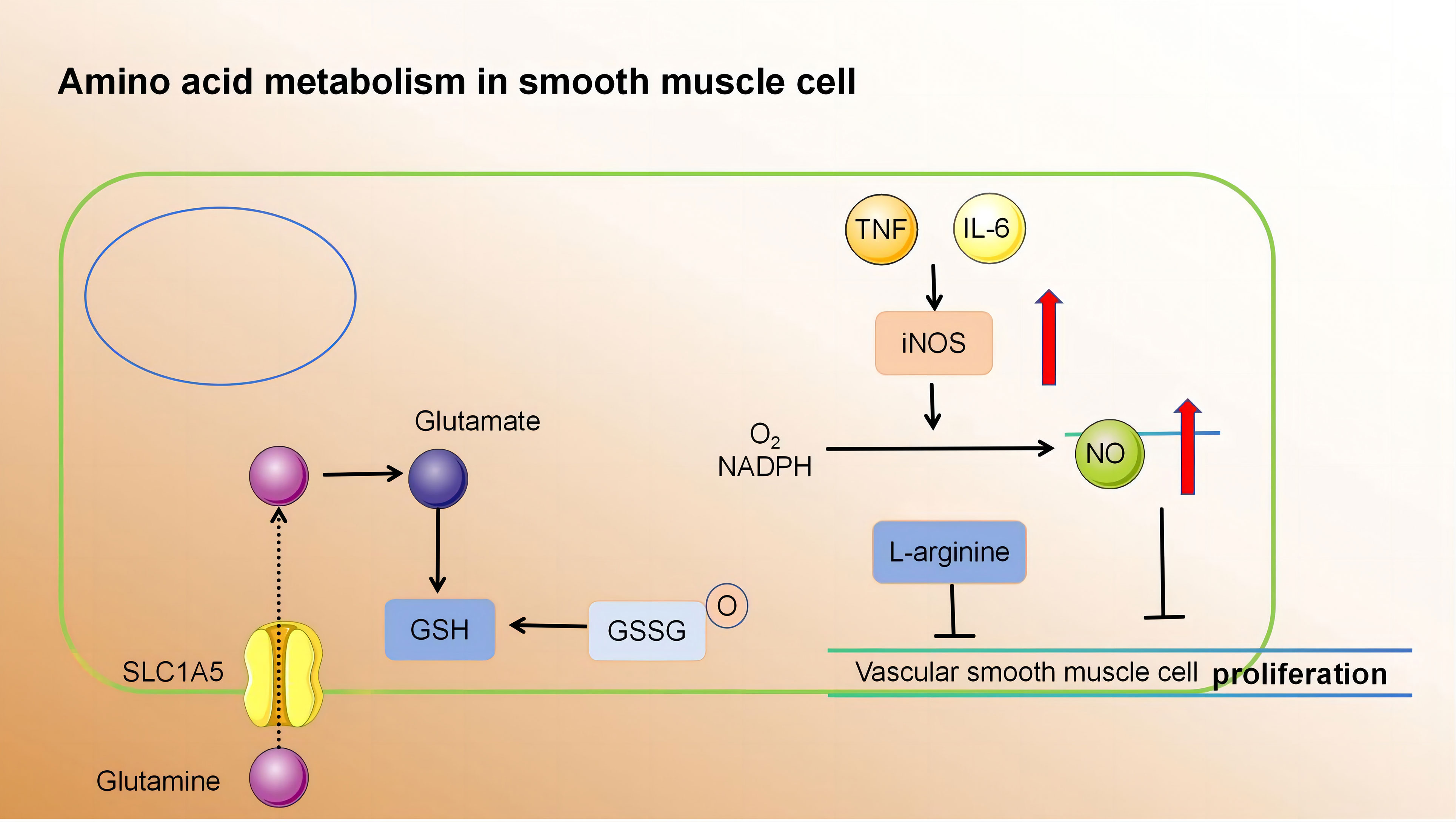
Figure 4 (1) Slc1a5 is a high-affinity L-glutamine transporter.glutamine absorption mediated by Slc1a5 can enhance the proliferation of VSMC. (2) In VSMCs, GSH and GSSG play crucial roles in cellular redox maintenance. (3) NO is a free radical. In VSMCs, NO activates programmed cell death through the consumption of intracellular GSH, thereby acting as a powerful apoptosis trigger. (4) L-arginine inhibits the proliferation and migration of VSMCs in the absence of NOS. Slc1a5, Solute Carrier Family 1, Member 5; VSMC, vascular smooth muscle cell; GSH, γ-glutamyl-L-cysteinylglycine; GSSG, Oxidized glutathione.
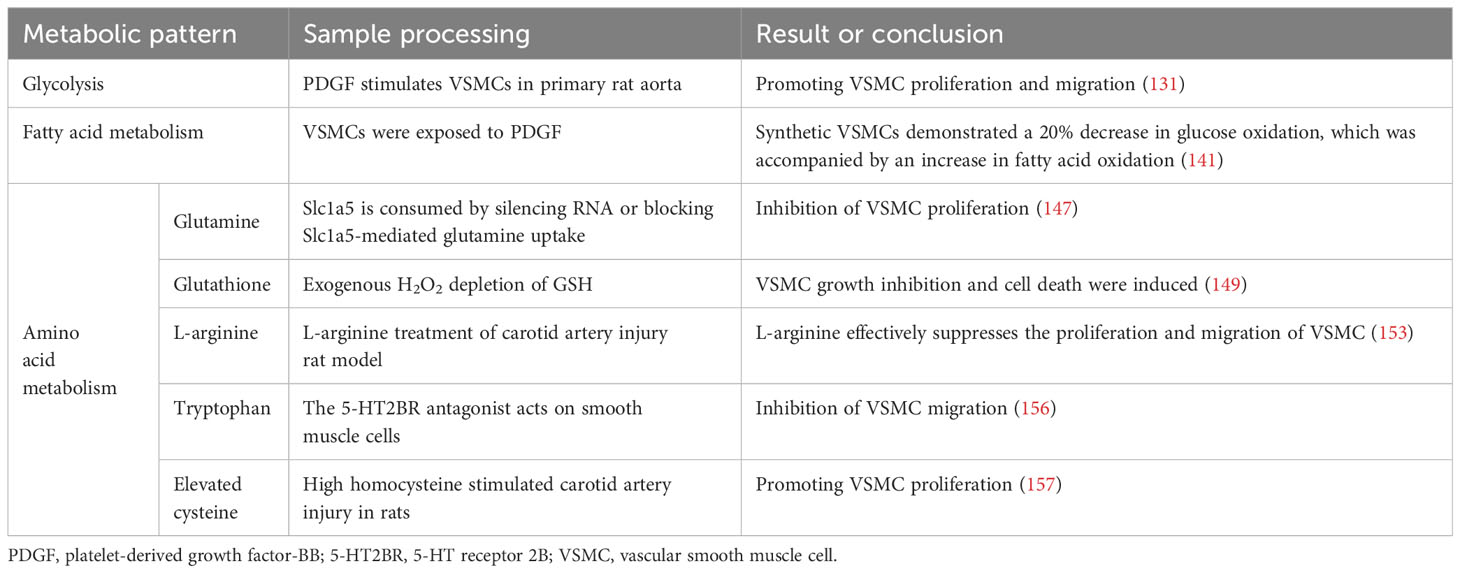
Table 4 Smooth muscle cell metabolism.
In the initial stage of plaque development, there is an accumulation and aggregation of LDL and Very Low-Density Lipoprotein (VLDL) particles beneath the endothelium. These particles undergo oxidation and enzymatic modifications, resulting in the formation of oxidized phospholipids (oxPLS). The presence of oxPLS promotes inflammation and activates ECs. Simultaneously, various adhesion molecules are expressed, leading to the recruitment of white blood cells and platelets into the endothelial lining of blood vessels (161).
Monocytes firmly adhere to ECs through the interaction between monocyte integrins and ligands on ECs. Immunohistochemical analysis of human lesions and genetic studies in mice have demonstrated the significance of monocyte integrins VLA-4 and LFA-1, as well as their respective EC ligands VCAM-1 and ICAM-1, during the early stages of atherosclerosis. Moreover, platelet aggregation on the endothelium covering atherosclerotic lesions may enhance monocyte-EC interactions by inducing NF-κB signalling, promoting the expression of adhesion molecules, and depositing platelet-derived chemical factors on activated endothelium (162).
In addition to inflammatory cells, cytokines synthesised and expressed within atherosclerotic plaques play a significant role in shaping the internal environment for these plaques. Various stimuli trigger the release of inflammatory factors, including IL-1, IL-6, IL-8, IL-12, IL-18, soluble CD40 (SCD40), and TNF. These factors exert diverse effects that contribute to increased vascular permeability caused by inflammation (SCD40), and TNF. These factors exert diverse effects that contribute to increased vascular permeability caused by inflammation (163).
Clinical research on anti-thrombotic therapy involving canakinumab has investigated the role of IL-1 in the induction of atherosclerosis. Monoclonal antibodies targeting IL-1 effectively inhibit the formation and progression of atherosclerotic plaques (164). Studies like MIRACL have demonstrated an association between stroke risk and levels of high-sensitivity C-reactive protein (hs-CRP), serum amyloid A protein (SAA), and the inflammatory marker IL-6 (165). A phase II clinical trial demonstrated the potential of ziltivekimab, an all-human monoclonal antibody targeting IL-6 ligand, to significantly reduce multiple biomarkers associated with systemic inflammation and thrombosis (166). Furthermore, elevated levels of IL-8 within atherosclerotic plaques promote the recruitment and migration of monocytes toward vascular ECs, resulting in firm adhesion (167, 168). CD40 is predominantly expressed in the pro-inflammatory M1 phenotype of macrophages (169), and plasma CD40 levels are correlated with carotid artery severity. TNF-α stimulates interstitial cells to induce the expression of various adhesion molecules and triggers the secretion of inflammatory cytokines and chemical factors, thereby enhancing the recruitment of activated white blood cells to affected areas (170). TNF-α is a pleiotropic cytokine that acts through two primary receptors, TNF-1 and TNF-2. TNF-1 mediates pro-inflammatory signals, apoptosis, and degeneration, while activation of TNF-2 by TNF-α induces anti-inflammatory and cytoprotective responses, leading to cellular proliferation, differentiation, angiogenesis, and tissue repair (171). Within plaques, there are also anti-inflammatory cytokines such as IL-10 and TGF. TGF is a multifaceted late-stage cytokine with both protective and atherogenic properties. Vascular endothelial TGF generates positive signalling cascades that inhibit inflammation, reduce vascular permeability, and slow disease progression in hyperlipidaemic mice (172). The absence of TGF-β1 results in reduced VSMC differentiation within the body, accelerated lesion formation, and increased inflammation.
In advanced atherosclerotic plaques, the fibrous cap ruptures, exposing the necrotic core to thrombotic material, which initiates platelet aggregation and subsequent clot formation. The active release of plaque-derived cytokines, proteases, and coagulation/thrombosis-related factors further promotes the progression of vulnerable plaques (173).
In unstable atherosclerotic plaques, the thickness of the fibrous cap is reduced owing to decreased collagen synthesis in SMCs and/or enhanced collagen degradation in fibroblasts. This thinning promotes the development of vulnerable plaques. Reduced abundance of VSMCs in vulnerable plaques can contribute to decreased collagen synthesis. In areas of vulnerable plaque where apoptotic cells are present, macrophages can attenuate collagen production in VSMCs without inducing cell death by secreting lower levels of TGF-β, a key stimulator of collagen synthesis in SMCs. Additionally, macrophage-derived matrix metalloproteinases (MMPs), which refer to a group of enzymes responsible for activating proteins and breaking down different types of extracellular matrix proteins, can also contribute to weakening the fibrous cap (174).
Unstable plaques are characterised by the presence of a necrotic core, which results from programmed cell death of mature macrophages and impaired phagocytic ability to engulf dying macrophages in advanced plaques (175). Early atherosclerotic lesions efficiently clear apoptotic macrophages through phagocytosis, leading to minimal compromise in cellular integrity, and limited plaque progression. Apoptotic macrophages play a crucial protective role through three essential mechanisms (1): eliminating cells before they release harmful substances into the surrounding environment; (2) inducing an anti-inflammatory response mediated by IL-10 and TGF-β; (3) enhancing cell survival by counteracting internal toxic factors. The benefits of apoptotic macrophages encompass efficient cholesterol esterification and removal, elimination of proapoptotic oxidized lipids, and activation of AKT and NF-κB signalling pathways. However, in the advanced stages of atherosclerotic lesions characterised by oxidative stress and increased inflammation, macrophage endocytosis signalling pathways are impaired. This impairment leads to secondary necrotic cell death, and elevated levels of inflammation lead to cytotoxicity (176).
The low-oxygen, inflammatory, and oxidative stress environment in atherosclerotic plaques can induce both conventional and unconventional angiogenic factors, promoting the formation of neovascularisation (177). The presence of neovascularisation in the shoulder region is typically characterised by an incomplete and immature structure, rendering it prone to leakage, which contributes to intra-plaque haemorrhage (IPH). In 1936, it was postulated that repetitive IPH is implicated in the progression of atherosclerosis and thrombosis (178). Magnetic resonance imaging (MRI) studies on human carotid atherosclerosis over the past two decades have confirmed histological observations and indicated a significant influence of IPH on plaque evolution (179). Moreover, the potential of 18F-FDG as a functional imaging technique for identifying vulnerability by analysing signals associated with plaque histology has been suggested. However, concerns regarding the accuracy and precision of PET in detecting fragile plaques remain significant. Combining 18F-FDG-PET imaging with dynamically enhanced MRI is expected to enhance the diagnosis of plaque fragility in the future (Figure 5).
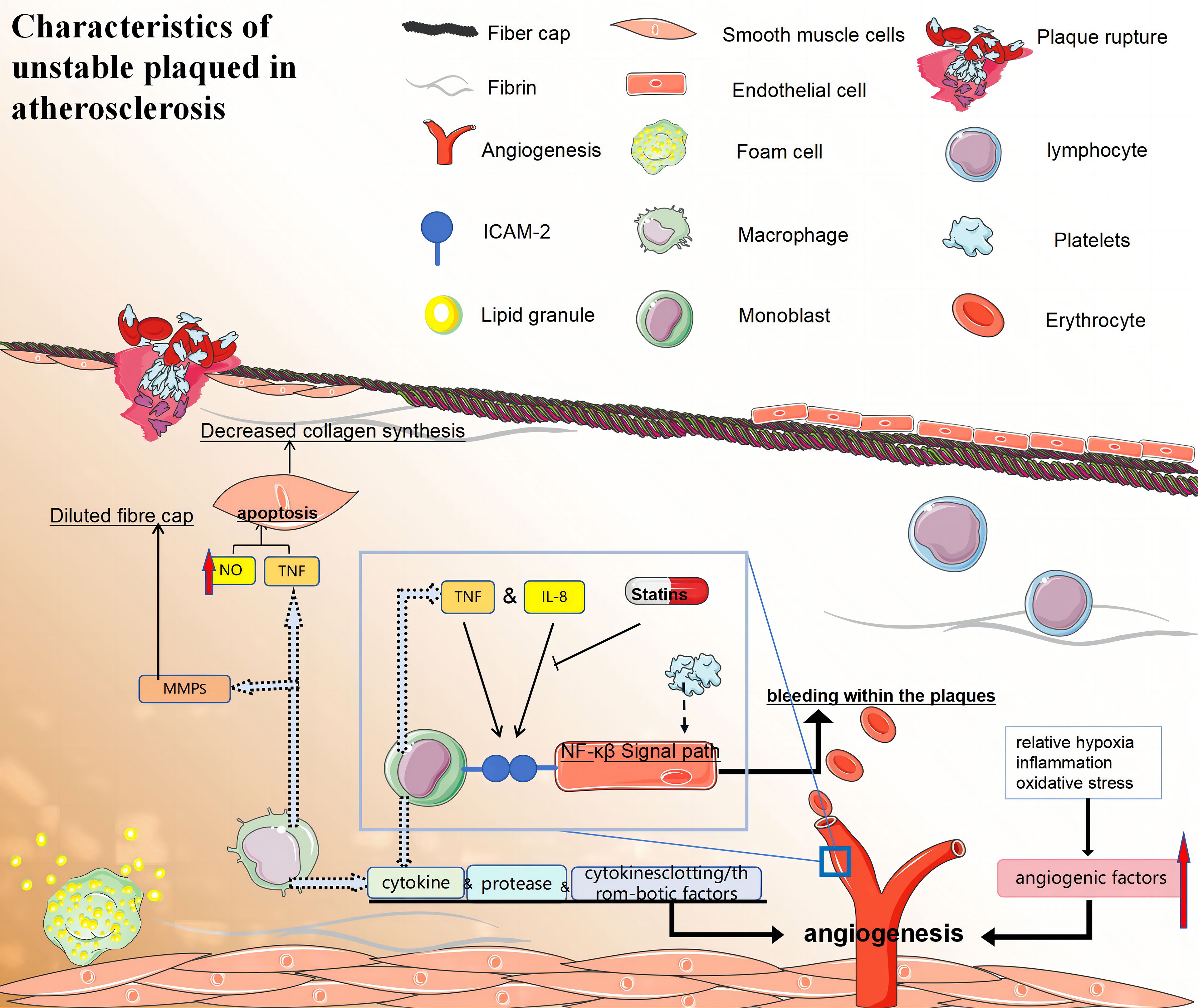
Figure 5 (1) Macrophages promote the formation of vulnerable plaques by secreting cytokines, proteases, and clotting/thrombotic factors. (2) i:The fibrous caps of late unstable atherosclerotic plaques became thinner. VSMCs may promote the formation of vulnerable plaques by reducing collagen synthesis and/or leading to collagen degradation. ii:Macrophages activate their apoptotic pathways and secrete TNF that is preapoptotic α, and that nitric oxide can trigger the apoptosis of SMC cells, which leading to a decrease in the number of VSMCs. iii:Macrophage-derived MMPS may also be involved in the dilution of fibre caps. (3) Specific local conditions (relative hypoxia, inflammation, and oxidative stress) in atherosclerotic plaques can also induce classical and non-classical angiogenic factors and promote neovascularisation. Neovascularisation increases bleeding within the plaques, which may lead to their instability and rupture. Monocytes firmly attach to endothelial cells through ICAM-1 interactions, while atherosclerotic lesions can enhance monocyte-endothelial interactions by activating NF-κB signaling. (4) Statins also exhibit anti-inflammatory properties by decreasing the expression of intercellular adhesion molecule-1 and inhibiting IL-6 secretion in monocytes and macrophages stimulated by LPS. (5) TNF:tumor necrosis factor; NF-κB:nuclear factor-κB; ICAM-1, intercellular cell adhesion molecule-1; MMPS, matrix metalloproteinase.
HMG-CoA reductase plays a crucial role in the production of cholesterol and was identified as a target enzyme for statins in the 1980s. Statins exert a wide range of effects beyond their primary function of lowering LDL cholesterol levels. Statin therapy has been associated with inflammatory responses triggered by stimulants such as oxidized LDL.
These drugs affect the function of various cells within atherosclerotic plaques through different signalling pathways. Statins enhance macrophage-mediated cholesterol esterification, increase the uptake and degradation of LDL, and promote NO synthesis in ECs. Additionally, they reduce inflammation, improve EC function, and inhibit SMC proliferation and apoptosis. These effects contribute to enhanced stability within atherosclerotic plaques, which, in turn, leads to the restoration of platelet activity and the clotting cascade. Therefore, statins are the most effective drugs for reducing lipid levels and mortality rates in patients with coronary issues. They significantly decrease the incidence of atherosclerosis in both primary and secondary prevention settings. Commonly prescribed statins include lovastatin, pravastatin, fluvastatin, simvastatin, pitavastatin, rosuvastatin, and atorvastatin.
Statins exert their pharmacological effects through a multi-faceted approach. Primarily, they act on hepatocytes and inhibit the activity of HMG-CoA reductase, a critical enzyme responsible for the synthesis of mevalonate, a precursor to cholesterol. This leads to a decrease in cellular cholesterol levels. Moreover, statins promote the removal of sterol regulatory element-binding proteins (SREBPs) from the endoplasmic reticulum by inducing protease activity. SREBPs are responsible for increasing LDL receptor expression when they translocate to the cell nucleus. The decreased cholesterol levels in liver cells lead to an increase in the number of LDL receptors on their membranes, which facilitates the efficient clearance of LDL cholesterol particles from the bloodstream. Recent research suggests that statins may also enhance the expression of PCSK9, an enzyme involved in LDL receptor breakdown, potentially affecting their effectiveness in reducing LDL-C levels and preventing coronary heart disease risk (180).
Several mechanisms underlie the antioxidant properties of statins (1): Statins reduce cholesterol levels, leading to decreased lipoprotein cholesterol and oxidative substrate levels (181). (2) They inhibit superoxide production in macrophages, thereby reducing cellular oxygen production. Additionally, statins inhibit the isopentenylation process of the p21 Rac protein in ECs, thereby impeding the formation of superoxide anions (182). This preserves the functionality of the endogenous antioxidant system, counteracting LDL oxidation (183). (3) By binding with phospholipids on lipoprotein surfaces, statins obstruct the penetration of free radicals generated during oxidative stress into the core region of lipoproteins. (4) Metabolites resulting from statin usage exhibit potent antioxidant capabilities that effectively protect against lipoprotein oxidation.
Ox-LDL promotes the activation of the NLRP3 inflammasome and its receptor pathway in plaques. The first signal is triggered by pattern recognition receptors such as TLR, which activates the NF-κB pathway, leading to the transcription of NLRP3 and other pro-inflammatory cytokines. The second signal involves the oligomerisation of activated NLRP3, ultimately forming the inflammasome, which activates pro-inflammatory cytokines. Statins exert a pleiotropic effect on the NLRP3 complex. They cause a reduction in TLR agonists and inhibit the TLR4/MyD88/NF-κB pathway. Additionally, statins inhibit the NLRP3 inflammatory response via the LOX-1/NF-κB pathway (184).
The development of endothelial dysfunction due to elevated cholesterol levels is an early event in the progression of atherosclerosis. Hindered by hypercholesterolemia, the capacity of ECs to generate NO, a pivotal regulator of anti-atherosclerotic functions, is compromised. Research has demonstrated that statins effectively enhance endothelial function by reducing cholesterol levels. In 2002, Simionescu et al. (185) discovered that simvastatin alleviated the intracellular effects of LDL and restored endothelium-dependent relaxation, possibly due to increased NO synthesis. In 2018, Geng et al. (186) demonstrated that rosuvastatin protects endothelial cells in an in vitro model of human umbilical vein endothelial cells induced by ox-LDL, through its antioxidant function and up-regulation of the expression of eNOS, an endothelial protective factor. These findings confirm the advantageous impact of statins on promoting eNOS expression and preventing LDL-induced suppression of eNOS expression (187).
Furthermore, ox-LDL induces ECs to generate adhesion molecules and selective proteins, thereby facilitating immune cell infiltration into the intima. Statin therapy inhibits EC adhesion and permeability while reducing white blood cell migration, ultimately mitigating the inflammatory response within plaques (188).
Cytokines released by macrophages and lymphocytes influence endothelial function and promote SMC proliferation, collagen degradation, and thrombosis. Statins act by inhibiting the expression and activity of these cytokines in the pathogenesis of atherosclerosis. In hypercholesterolemic rabbits, atorvastatin has been demonstrated to reduce the presence of macrophages, monocyte chemoattractant protein-1 (MCP-1), and nuclear factor NF-κB activation within the intima layer (189). Moreover, statins exhibit anti-inflammatory effects on monocytes and macrophages by downregulating intercellular adhesion molecule-1 expression induced by LPS and suppressing IL-6 secretion. The presence of crystalline cholesterol is widely acknowledged in the scientific community to play a significant role in arterial inflammation. This inflammatory response occurs due to the activation of NLRP3 (or cryopyrin) inflammasomes by cholesterol, subsequently triggering caspase-1 and leading to the release of IL-1 family cytokines (190). Additionally, studies have demonstrated that statins can effectively reduce hs-CRP levels and potentially mitigate adverse events in patients, even in the absence of evident hypercholesterolemia (191).
In atherosclerotic lesions, the proliferation, migration, and invasion of SMCs into the subendothelial layer can induce intimal hyperplasia, while the secretion of collagen fibres by SMCs can influence the thickness of fibrous caps within plaques. In 2000, Bellosta et al. (192) conducted a study using both in vitro and in vivo models. The findings suggest that fluvastatin, simvastatin, lovastatin, and atorvastatin exhibit dose-dependent inhibition of SMC migration and proliferation. Chandrasekar et al. (193) demonstrated that the pro-atherosclerotic cytokine IL-18 stimulates SMC migration in an MMP9-dependent manner, and atorvastatin inhibits this process. Zhou et al. (194) established a diabetic mouse model by utilising ApoE-/- mice and administered atorvastatin treatment. Subsequently, they quantified SMCs and collagen composition, revealing that atorvastatin effectively reduced the number of SMCs while promoting collagen fibre synthesis. Moreover, it resulted in diminished atherosclerotic plaque area and enhanced arterial plaque stability through modulation of the RAGE pathway. However, Palomino-Morales et al. (195) demonstrated that statins effectively attenuated the activity of RhoA in SMCs, consequently leading to a reduction in collagen expression. The differences among these findings may be attributed to the different animal models used. By 2021, Jo et al. (196) conducted experiments to elucidate the mechanism underlying the inhibitory effect of statins on SMC apoptosis. Upon stimulation by ox-LDL in atherosclerotic lesions, platelet-derived growth factors induce the proliferation and migration of VSMCs. Prolonged stimulation ultimately leads to VSMC apoptosis. Statins exert their inhibitory effects by suppressing p38 activation through autophagy, thereby attenuating intracellular ROS levels and preventing apoptosis.
In summary, statins can promote the stability of atherosclerotic plaques by inhibiting the proliferation, migration, and apoptosis of SMCs and by affecting the collagen secreted by SMCs.
Hyperlipidaemia and atherosclerotic plaque formation are associated with increased platelet activation and blood hypercoagulability. Elevated LDL levels promote an increase in thromboxane A2 production, which augments platelet responsiveness. Statin treatment leads to a reduction in collagen- and fibrinogen-induced platelet aggregation and thromboxane production. In a clinical trial, Barale et al. (197) assessed the impact of simvastatin treatment on platelet aggregation response and inflammatory cytokine expression in patients with hypercholesterolemia for a duration of 2 months. The findings demonstrated that in addition to ameliorating lipid distribution, simvastatin treatment also attenuated platelet aggregation rate and reduced circulating levels of pro-inflammatory factors, endothelial markers, and platelet markers. These results indicate that statins effectively reduce lipids, inhibit platelet activation, and improve inflammation levels and EC dysfunction within atherosclerotic plaques associated with primary hypercholesterolemia (Table 5).
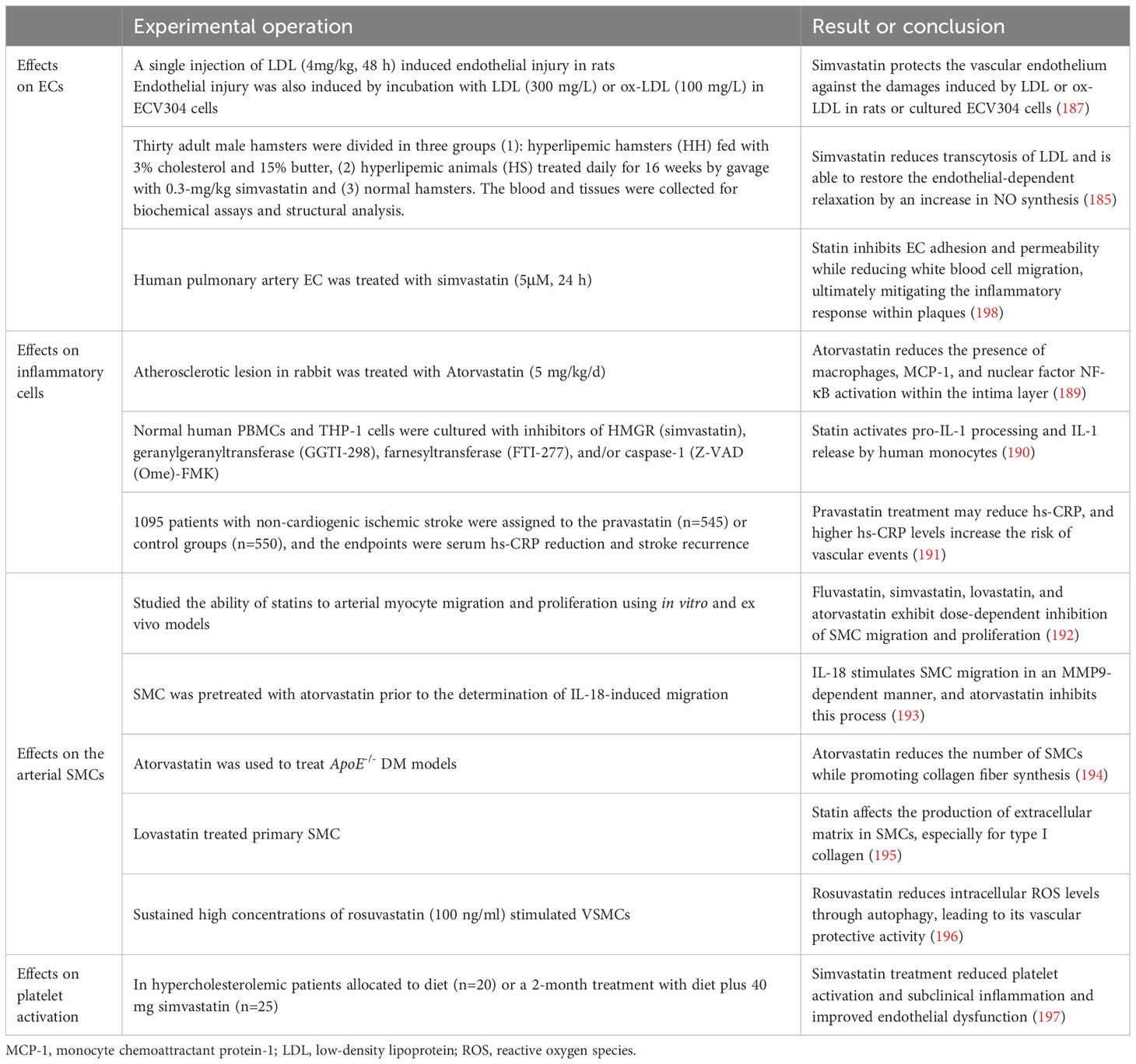
Table 5 Effects of statins on intracellular cell functions in plaques.
Statins demonstrate favourable drug tolerance but are commonly associated with adverse reactions such as hepatotoxicity and myopathy. Hanai et al. (199) have proposed a direct explanation for statin-related muscle toxicity, suggesting that these drugs can induce the expression of the Atrogin1 gene in skeletal muscles, leading to cytotoxic effects combined with interference in muscle differentiation processes like insulin induction. Moreover, some studies, including the final assessment of the JUPITER trial, have raised concerns about an elevated incidence of diabetes as a potential risk associated with the use of statins (200). Additionally, reports of potential toxic effects such as proteinuria and haematuria have also emerged (201).
Statins are powerful lipid-lowering drugs that can reduce the incidence and mortality of atherosclerosis, and they have been widely used to prevent primary and secondary cardiovascular diseases for more than 25 years. Interestingly, while statins treat atherosclerotic lesions by regulating lipid metabolism, they also regulate the function of various cells in the plaque and the secretion and expression of certain cytokines, thereby influencing the inflammatory response and plaque stability. However, since there may be other factors contributing to atherosclerotic lesions, such as hypertension and diabetes, and statins have some side effects including myopathy, the effect of statin monotherapy may be limited, and patient compliance may be low. Therefore, in clinical practice, statins are often used in combination with other lipid-lowering, antihypertensive, hypoglycaemic, and anti-inflammatory drugs.
The future of reducing atherosclerotic disease lies in combining statins with other medications. For example, inhibition of the regulatory protein PSCK9 effectively reduces plasma LDL levels, making it an ideal complement to statins (202). Experts propose that the combination of rosuvastatin and ezetimibe is safe and effective for treating hypercholesterolemia or hyperlipidaemia, regardless of diabetes or cardiovascular disease status. The fixed combination of 40 mg rosuvastatin/10 mg ezetimibe has been approved and evaluated (203). Statins can also be combined with fibrates, niacin, and omega-3 fatty acids to lower triglycerides in atherosclerosis development. However, extensive clinical studies are needed to assess the impact on cardiovascular outcomes and risk reduction for patients with hypertriglyceridemia. Hypertension is the primary risk factor for intravascular atherosclerosis, wherein the combination therapy of antihypertensive and statin treatment exhibits superior efficacy compared to monotherapy with antihypertensive agents alone in hypertensive patients without complications (204). The synergistic effect of lipid-lowering and inflammation-suppressing therapies is evident. Dicarboxylic acid, commonly prescribed as an agent for lowering blood sugar levels, not only regulates macrophage function in atherosclerosis but also suppresses inflammatory responses. By combining dimethylformic acid with statins, inflammation can effectively be inhibited while simultaneously reducing blood sugar levels and lipids, thereby enhancing the therapeutic potential for treating atherosclerosis (204, 205). Furthermore, ongoing clinical trials are investigating alternative anti-inflammatory agents targeting the CRP/IL-6/IL-1 axis such as low-dose methylidene and colchicine (206). When combined with aggressive LDL-C therapy, this approach may become the standard treatment for most patients with atherosclerosis. Aspirin and statins are well-established treatments for both atherosclerosis and coronary heart disease due to their cohesive properties and effective reduction of inflammation.
This review provides a comprehensive overview of the metabolic regulation in immune, endothelial, and smooth muscle cells and their potential contributions to the pathogenesis of atherosclerosis. Cells exposed to hypoxic conditions undergo distinct alterations in energy metabolism, including augmented glycolysis, impaired fatty acid synthesis, and abnormal amino acid metabolism. Inflammatory processes and lipid accumulation within atherosclerotic plaques are intricately linked to cellular proliferation, migration, senescence, and apoptosis. Different inflammatory responses within plaques affect plaque stability.
Statins and their combinations play an important role in the treatment of atherosclerotic lesions. In this review, the main pharmacological mechanisms of statins and their effects on the function of various cells in atherosclerotic plaques were briefly discussed. Considering the limitations and potential adverse reactions of statins in the treatment of atherosclerotic lesions, a comprehensive regimen is required, which combines statins with other lipid-lowering, antihypertensive, hypoglycaemic, antiplatelet aggregating, and anti-inflammatory drugs.
Although previous animal experiments, in vitro experiments, and clinical data analyses have provided extensive research on specific metabolic pathways, products, and the mechanisms and applications of lipid-lowering drugs, our understanding of drugs regulating inflammatory responses and influencing the pathological and physiological processes of atherosclerotic plaques remains limited. The continuous accumulation of clinical research data, utilisation of proteomics analysis, and the integration of advanced technologies will aid in the development of a more robust theoretical framework. This will enhance our understanding of angiogenesis, inflammatory changes, lipid stability, barrier function, atherosclerosis metabolic mechanisms, and treatment-related knowledge. These efforts may lead to the identification of new drug targets for treating atherosclerosis and related diseases, the development of more promising treatment strategies, and the reduction of unnecessary side effects.
LMZ: Writing – original draft, Writing – review & editing. DM: Conceptualization, Supervision, Validation, Writing – review & editing. LW: Conceptualization, Writing – original draft, Writing – review & editing. XS: Writing – original draft. LF: Writing – original draft. LCZ: Writing – original draft. YC: Writing – original draft. YH: Writing – original draft. XW: Writing – original draft. JF: Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We are highly thankful to Bethune First Hospital of Jilin University, Professor Ms. Wang, and Professor Ms. Ma for guiding us to finalise this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Schulz B, Pruessmeyer J, Maretzky T, Ludwig A, Blobel CP, Saftig P, et al. Adam10 regulates endothelial permeability and T-cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res (2008) 102(10):1192–201. doi: 10.1161/circresaha.107.169805
2. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol (2011) 12(3):204–12. doi: 10.1038/ni.2001
3. Capodanno D, Angiolillo DJ. Canakinumab for secondary prevention of atherosclerotic disease. Expert Opin Biol Ther (2018) 18(2):215–20. doi: 10.1080/14712598.2018.1420776
4. Nidorf SM, Thompson PL. Why colchicine should be considered for secondary prevention of atherosclerosis: an overview. Clin Ther (2019) 41(1):41–8. doi: 10.1016/j.clinthera.2018.11.016
5. Kondoh H, Lleonart ME, Bernard D, Gil J. Protection from oxidative stress by enhanced glycolysis; a possible mechanism of cellular immortalization. Histol Histopathol (2007) 22(1):85–90. doi: 10.14670/hh-22.85
6. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
7. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(Lps+) vs. Classically and M2(Lps-) vs. Alternatively activated macrophages. Front Immunol (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
8. Funk JL, Feingold KR, Moser AH, Grunfeld C. Lipopolysaccharide stimulation of raw 264.7 macrophages induces lipid accumulation and foam cell formation. Atherosclerosis (1993) 98(1):67–82. doi: 10.1016/0021-9150(93)90224-i
9. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity (2015) 42(3):419–30. doi: 10.1016/j.immuni.2015.02.005
10. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (Glut1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem (2014) 289(11):7884–96. doi: 10.1074/jbc.M113.522037
11. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and Pgc-1beta attenuate macrophage-mediated inflammation. Cell Metab (2006) 4(1):13–24. doi: 10.1016/j.cmet.2006.05.011
12. Yamashita A, Zhao Y, Matsuura Y, Yamasaki K, Moriguchi-Goto S, Sugita C, et al. Increased metabolite levels of glycolysis and pentose phosphate pathway in rabbit atherosclerotic arteries and hypoxic macrophage. PLoS One (2014) 9(1):e86426. doi: 10.1371/journal.pone.0086426
13. Wang T, Liu H, Lian G, Zhang SY, Wang X, Jiang C. Hif1α-induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediators Inflamm (2017) 2017:9029327. doi: 10.1155/2017/9029327
14. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. Nf-kappab links innate immunity to the hypoxic response through transcriptional regulation of Hif-1alpha. Nature (2008) 453(7196):807–11. doi: 10.1038/nature06905
15. van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by Nf-Kappab. Biochem J (2008) 412(3):477–84. doi: 10.1042/bj20080476
16. Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia response elements in the aldolase a, enolase 1, and lactate dehydrogenase a gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem (1996) 271(51):32529–37. doi: 10.1074/jbc.271.51.32529
17. Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci USA (2012) 109(24):9517–22. doi: 10.1073/pnas.1119038109
18. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. Mtor- and Hif-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345(6204):1250684. doi: 10.1126/science.1250684
19. Joshi S, Singh AR, Zulcic M, Durden DL. A macrophage-dominant Pi3k isoform controls hypoxia-induced Hif1α and Hif2α Stability and tumor growth, angiogenesis, and metastasis. Mol Cancer Res (2014) 12(10):1520–31. doi: 10.1158/1541-7786.Mcr-13-0682
20. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1α Activity and Il-1β Induction and is a critical determinant of the Warburg effect in Lps-activated macrophages. Cell Metab (2015) 21(1):65–80. doi: 10.1016/j.cmet.2014.12.005
21. Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, et al. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMedicine (2018) 30:303–16. doi: 10.1016/j.ebiom.2018.02.009
22. Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic reprogramming mediated by the mtorc2-Irf4 signaling axis is essential for macrophage alternative activation. Immunity (2016) 45(4):817–30. doi: 10.1016/j.immuni.2016.09.016
23. Tan Z, Xie N, Cui H, Moellering DR, Abraham E, Thannickal VJ, et al. Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. J Immunol (2015) 194(12):6082–9. doi: 10.4049/jimmunol.1402469
24. Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab (2018) 28(3):463–75.e4. doi: 10.1016/j.cmet.2018.08.012
25. Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol (2014) 15(9):846–55. doi: 10.1038/ni.2956
26. Pedersen SF, Græbe M, Hag AM, Højgaard L, Sillesen H, Kjær A. (18)F-Fdg imaging of human atherosclerotic carotid plaques reflects gene expression of the key hypoxia marker Hif-1α. Am J Nucl Med Mol Imaging (2013) 3(5):384–92.
27. Folco EJ, Sheikine Y, Rocha VZ, Christen T, Shvartz E, Sukhova GK, et al. Hypoxia but not inflammation augments glucose uptake in human macrophages: implications for imaging atherosclerosis with 18fluorine-labeled 2-deoxy-D-glucose positron emission tomography. J Am Coll Cardiol (2011) 58(6):603–14. doi: 10.1016/j.jacc.2011.03.044
28. Riksen NP, Stienstra R. Metabolism of innate immune cells: impact on atherosclerosis. Curr Opin Lipidol (2018) 29(5):359–67. doi: 10.1097/mol.0000000000000539
29. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol (2020) 17(7):387–401. doi: 10.1038/s41569-020-0352-5
30. Schäfer S, Zernecke A. Cd8(+) T cells in atherosclerosis. Cells (2020) 10(1):37. doi: 10.3390/cells10010037
31. Yang J, Chen Y, Li X, Qin H, Bao J, Wang C, et al. Complex interplay between metabolism and Cd4(+) T-cell activation, differentiation, and function: A novel perspective for atherosclerosis immunotherapy. Cardiovasc Drugs Ther (2023). doi: 10.1007/s10557-023-07466-9
32. Mallat Z, Ait-Oufella H, Tedgui A. The role of adaptive T cell immunity in atherosclerosis. J Lipid Res (2009) 50 Suppl(Suppl):S364–9. doi: 10.1194/jlr.R800092-JLR200
33. McKellar GE, McCarey DW, Sattar N, McInnes IB. Role for Tnf in atherosclerosis? Lessons from autoimmune disease. Nat Rev Cardiol (2009) 6(6):410–7. doi: 10.1038/nrcardio.2009.57
34. Buono C, Binder CJ, Stavrakis G, Witztum JL, Glimcher LH, Lichtman AH. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci USA (2005) 102(5):1596–601. doi: 10.1073/pnas.0409015102
35. Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C. Ifn-gamma potentiates atherosclerosis in Apoe knock-out mice. J Clin Invest (1997) 99(11):2752–61. doi: 10.1172/jci119465
36. Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E-/- mice. Am J Pathol (2000) 157(6):1819–24. doi: 10.1016/s0002-9440(10)64820-1
37. McGeachy MJ, Cua DJ, Gaffen SL. The Il-17 family of cytokines in health and disease. Immunity (2019) 50(4):892–906. doi: 10.1016/j.immuni.2019.03.021
38. Usui F, Kimura H, Ohshiro T, Tatsumi K, Kawashima A, Nishiyama A, et al. Interleukin-17 deficiency reduced vascular inflammation and development of atherosclerosis in western diet-induced apoe-deficient mice. Biochem Biophys Res Commun (2012) 420(1):72–7. doi: 10.1016/j.bbrc.2012.02.117
39. Madhur MS, Funt SA, Li L, Vinh A, Chen W, Lob HE, et al. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol (2011) 31(7):1565–72. doi: 10.1161/atvbaha.111.227629
40. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. Tgf-Beta and Il-6 drive the production of Il-17 and Il-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol (2007) 8(12):1390–7. doi: 10.1038/ni1539
41. Ou HX, Guo BB, Liu Q, Li YK, Yang Z, Feng WJ, et al. Regulatory T cells as a new therapeutic target for atherosclerosis. Acta Pharmacol Sin (2018) 39(8):1249–58. doi: 10.1038/aps.2017.140
42. Lin J, Li M, Wang Z, He S, Ma X, Li D. The role of Cd4+Cd25+ Regulatory T cells in macrophage-derived foam-cell formation. J Lipid Res (2010) 51(5):1208–17. doi: 10.1194/jlr.D000497
43. King VL, Cassis LA, Daugherty A. Interleukin-4 does not influence development of hypercholesterolemia or angiotensin ii-induced atherosclerotic lesions in mice. Am J Pathol (2007) 171(6):2040–7. doi: 10.2353/ajpath.2007.060857
44. King VL, Szilvassy SJ, Daugherty A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female Ldl receptor-/- mice. Arterioscler Thromb Vasc Biol (2002) 22(3):456–61. doi: 10.1161/hq0302.104905
45. Grönberg C, Nilsson J, Wigren M. Recent advances on Cd4(+) T cells in atherosclerosis and its implications for therapy. Eur J Pharmacol (2017) 816:58–66. doi: 10.1016/j.ejphar.2017.04.029
46. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and Pdhk1 control Cd4+ T cell subsets and inflammation. J Clin Invest (2015) 125(1):194–207. doi: 10.1172/jci76012
47. Oishi Y, Manabe I. Integrated regulation of the cellular metabolism and function of immune cells in adipose tissue. Clin Exp Pharmacol Physiol (2016) 43(3):294–303. doi: 10.1111/1440-1681.12539
48. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory Cd4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
49. Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol (2016) 28(5):514–24. doi: 10.1016/j.smim.2016.10.009
50. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for Cd4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi: 10.1016/j.cmet.2014.05.004
51. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. Hif1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of Th17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
52. Lü S, Deng J, Liu H, Liu B, Yang J, Miao Y, et al. Pkm2-dependent metabolic reprogramming in Cd4(+) T cells is crucial for hyperhomocysteinemia-accelerated atherosclerosis. J Mol Med (Berl) (2018) 96(6):585–600. doi: 10.1007/s00109-018-1645-6
53. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of foxp3 exon 2 splicing variants. Nat Immunol (2015) 16(11):1174–84. doi: 10.1038/ni.3269
54. Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D, et al. Regulatory T cell migration is dependent on glucokinase-mediated glycolysis. Immunity (2017) 47(5):875–89.e10. doi: 10.1016/j.immuni.2017.10.017
55. Zeng H, Chi H. Metabolic control of regulatory T cell development and function. Trends Immunol (2015) 36(1):3–12. doi: 10.1016/j.it.2014.08.003
56. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab (2017) 25(6):1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
57. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and toll-like receptor signaling balance T(Reg) cell anabolic metabolism for suppression. Nat Immunol (2016) 17(12):1459–66. doi: 10.1038/ni.3577
58. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(Reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
59. Cheong C, Choi JH. Dendritic cells and regulatory T cells in atherosclerosis. Mol Cells (2012) 34(4):341–7. doi: 10.1007/s10059-012-0128-9
60. Zhao Y, Zhang J, Zhang W, Xu Y. A myriad of roles of dendritic cells in atherosclerosis. Clin Exp Immunol (2021) 206(1):12–27. doi: 10.1111/cei.13634
61. Ma SD, Mussbacher M, Galkina EV. Functional role of B cells in atherosclerosis. Cells (2021) 10(2):270. doi: 10.3390/cells10020270
62. Aubry MC, Riehle DL, Edwards WD, Maradit-Kremers H, Roger VL, Sebo TJ, et al. B-lymphocytes in plaque and adventitia of coronary arteries in two patients with rheumatoid arthritis and coronary atherosclerosis: preliminary observations. Cardiovasc Pathol (2004) 13(4):233–6. doi: 10.1016/j.carpath.2004.02.005
63. Libby P, Hansson GK. Taming immune and inflammatory responses to treat atherosclerosis. J Am Coll Cardiol (2018) 71(2):173–6. doi: 10.1016/j.jacc.2017.10.081
64. Perry HM, Bender TP, McNamara CA. B cell subsets in atherosclerosis. Front Immunol (2012) 3:373. doi: 10.3389/fimmu.2012.00373
65. Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell Rna-sequencing and mass cytometry. Circ Res (2018) 122(12):1675–88. doi: 10.1161/circresaha.117.312513
66. Ketelhuth DF, Hansson GK. Adaptive response of T and B cells in atherosclerosis. Circ Res (2016) 118(4):668–78. doi: 10.1161/circresaha.115.306427
67. Sage AP, Tsiantoulas D, Baker L, Harrison J, Masters L, Murphy D, et al. Baff receptor deficiency reduces the development of atherosclerosis in mice–brief report. Arterioscler Thromb Vasc Biol (2012) 32(7):1573–6. doi: 10.1161/atvbaha.111.244731
68. Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K, et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol (2010) 185(7):4410–9. doi: 10.4049/jimmunol.1000033
69. Deng J, Lü S, Liu H, Liu B, Jiang C, Xu Q, et al. Homocysteine activates B cells via regulating Pkm2-dependent metabolic reprogramming. J Immunol (2017) 198(1):170–83. doi: 10.4049/jimmunol.1600613
70. Doughty CA, Bleiman BF, Wagner DJ, Dufort FJ, Mataraza JM, Roberts MF, et al. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood (2006) 107(11):4458–65. doi: 10.1182/blood-2005-12-4788
71. Djouadi F, Brandt JM, Weinheimer CJ, Leone TC, Gonzalez FJ, Kelly DP. The role of the peroxisome proliferator-activated receptor alpha (Ppar Alpha) in the control of cardiac lipid metabolism. Prostaglandins Leukot Essent Fatty Acids (1999) 60(5-6):339–43. doi: 10.1016/s0952-3278(99)80009-x
72. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator pgc-1. Cell (1999) 98(1):115–24. doi: 10.1016/s0092-8674(00)80611-x
73. Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta (2014) 1842(3):446–62. doi: 10.1016/j.bbadis.2013.05.017
74. Gaddis DE, Padgett LE, Wu R, McSkimming C, Romines V, Taylor AM, et al. Apolipoprotein ai prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat Commun (2018) 9(1):1095. doi: 10.1038/s41467-018-03493-5
75. Maganto-García E, Tarrio ML, Grabie N, Bu DX, Lichtman AH. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation (2011) 124(2):185–95. doi: 10.1161/circulationaha.110.006411
76. Janeway CA Jr., Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359
77. Kawai T, Akira S. Pathogen recognition with toll-like receptors. Curr Opin Immunol (2005) 17(4):338–44. doi: 10.1016/j.coi.2005.02.007
78. Roshan MH, Tambo A, Pace NP. The role of Tlr2, Tlr4, and Tlr9 in the pathogenesis of atherosclerosis. Int J Inflam (2016) 2016:1532832. doi: 10.1155/2016/1532832
79. Li H, Sun B. Toll-like receptor 4 in atherosclerosis. J Cell Mol Med (2007) 11(1):88–95. doi: 10.1111/j.1582-4934.2007.00011.x
80. Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, et al. Crosstalk between Lxr and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell (2003) 12(4):805–16. doi: 10.1016/s1097-2765(03)00384-8
81. Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, et al. Cutting edge: toll-like receptor 4 (Tlr4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for Tlr4 as the Lps gene product. J Immunol (1999) 162(7):3749–52.
82. Rocha DM, Caldas AP, Oliveira LL, Bressan J, Hermsdorff HH. Saturated fatty acids trigger Tlr4-mediated inflammatory response. Atherosclerosis (2016) 244:211–5. doi: 10.1016/j.atherosclerosis.2015.11.015
83. Rogero MM, Calder PC. Obesity, inflammation, toll-like receptor 4 and fatty acids. Nutrients (2018) 10(4):432. doi: 10.3390/nu10040432
84. Pavillard LE, Marín-Aguilar F, Bullon P, Cordero MD. Cardiovascular diseases, Nlrp3 inflammasome, and western dietary patterns. Pharmacol Res (2018) 131:44–50. doi: 10.1016/j.phrs.2018.03.018
85. Ralston JC, Lyons CL, Kennedy EB, Kirwan AM, Roche HM. Fatty acids and Nlrp3 inflammasome-mediated inflammation in metabolic tissues. Annu Rev Nutr (2017) 37:77–102. doi: 10.1146/annurev-nutr-071816-064836
86. Karasawa T, Takahashi M. Role of Nlrp3 inflammasomes in atherosclerosis. J Atheroscler Thromb (2017) 24(5):443–51. doi: 10.5551/jat.RV17001
87. Haghikia A, Jörg S, Duscha A, Berg J, Manzel A, Waschbisch A, et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity (2016) 44(4):951–3. doi: 10.1016/j.immuni.2016.04.006
88. Bhutia YD, Ganapathy V. Short, but smart: Scfas train T cells in the gut to fight autoimmunity in the brain. Immunity (2015) 43(4):629–31. doi: 10.1016/j.immuni.2015.09.014
89. Zaric BL, Radovanovic JN, Gluvic Z, Stewart AJ, Essack M, Motwalli O, et al. Atherosclerosis linked to aberrant amino acid metabolism and immunosuppressive amino acid catabolizing enzymes. Front Immunol (2020) 11:551758. doi: 10.3389/fimmu.2020.551758
90. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by Erk/Mapk during T lymphocyte activation. J Immunol (2010) 185(2):1037–44. doi: 10.4049/jimmunol.0903586
91. Chen J, Zhang S, Wu J, Wu S, Xu G, Wei D. Essential role of nonessential amino acid glutamine in atherosclerotic cardiovascular disease. DNA Cell Biol (2020) 39(1):8–15. doi: 10.1089/dna.2019.5034
92. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter Asct2 facilitation of glutamine uptake and Mtorc1 kinase activation. Immunity (2014) 40(5):692–705. doi: 10.1016/j.immuni.2014.04.007
93. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell (2018) 175(7):1780–95.e19. doi: 10.1016/j.cell.2018.10.001
94. Durante W. The emerging role of L-glutamine in cardiovascular health and disease. Nutrients (2019) 11(9):2092. doi: 10.3390/nu11092092
95. Hayashi K, Jutabha P, Endou H, Sagara H, Anzai N. Lat1 is a critical transporter of essential amino acids for immune reactions in activated human T cells. J Immunol (2013) 191(8):4080–5. doi: 10.4049/jimmunol.1300923
96. Ren W, Liu G, Yin J, Tan B, Wu G, Bazer FW, et al. Amino-acid transporters in T-cell activation and differentiation. Cell Death Dis (2017) 8(3):e2655. doi: 10.1038/cddis.2016.222
97. Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, et al. Sestrin2 is a leucine sensor for the mtorc1 pathway. Science (2016) 351(6268):43–8. doi: 10.1126/science.aab2674
98. Niinisalo P, Oksala N, Levula M, Pelto-Huikko M, Järvinen O, Salenius JP, et al. Activation of indoleamine 2,3-dioxygenase-induced tryptophan degradation in advanced atherosclerotic plaques: tampere vascular study. Ann Med (2010) 42(1):55–63. doi: 10.3109/07853890903321559
99. Liu G, Chen S, Zhong J, Teng K, Yin Y. Crosstalk between tryptophan metabolism and cardiovascular disease, mechanisms, and therapeutic implications. Oxid Med Cell Longev (2017) 2017:1602074. doi: 10.1155/2017/1602074
100. Song P, Ramprasath T, Wang H, Zou MH. Abnormal kynurenine pathway of tryptophan catabolism in cardiovascular diseases. Cell Mol Life Sci (2017) 74(16):2899–916. doi: 10.1007/s00018-017-2504-2
101. Tentolouris C, Tousoulis D, Goumas G, Stefanadis C, Davies G. Toutouzas P. L-arginine in coronary atherosclerosis. Int J Cardiol (2000) 75(2-3):123–8. doi: 10.1016/s0167-5273(00)00320-x
102. Pekarova M, Kubala L, Martiskova H, Papezikova I, Kralova S, Baldus S, et al. The unique role of dietary L-arginine in the acceleration of peritoneal macrophage sensitivity to bacterial endotoxin. Immunol Res (2013) 56(1):73–84. doi: 10.1007/s12026-012-8379-2
103. Pekarova M, Lojek A. The crucial role of L-arginine in macrophage activation: what you need to know about it. Life Sci (2015) 137:44–8. doi: 10.1016/j.lfs.2015.07.012
104. Nitz K, Lacy M, Bianchini M, Wichapong K, Kücükgöze IA, Bonfiglio CA, et al. The amino acid homoarginine inhibits atherogenesis by modulating T-cell function. Circ Res (2022) 131(8):701–12. doi: 10.1161/circresaha.122.321094
105. Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol (2009) 183(10):6095–101. doi: 10.4049/jimmunol.0803510
106. Ananieva EA, Powell JD, Hutson SM. Leucine metabolism in T cell activation: Mtor signaling and beyond. Adv Nutr (2016) 7(4):798s–805s. doi: 10.3945/an.115.011221
107. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res (2015) 116(7):1231–44. doi: 10.1161/circresaha.116.302855
108. Cantelmo AR, Brajic A, Carmeliet P. Endothelial metabolism driving angiogenesis: emerging concepts and principles. Cancer J (2015) 21(4):244–9. doi: 10.1097/ppo.0000000000000133
109. Eelen G, Cruys B, Welti J, De Bock K, Carmeliet P. Control of vessel sprouting by genetic and metabolic determinants. Trends Endocrinol Metab (2013) 24(12):589–96. doi: 10.1016/j.tem.2013.08.006
110. Stapor P, Wang X, Goveia J, Moens S, Carmeliet P. Angiogenesis revisited - role and therapeutic potential of targeting endothelial metabolism. J Cell Sci (2014) 127(Pt 20):4331–41. doi: 10.1242/jcs.153908
111. Teuwen LA, Draoui N, Dubois C, Carmeliet P. Endothelial cell metabolism: an update anno 2017. Curr Opin Hematol (2017) 24(3):240–7. doi: 10.1097/moh.0000000000000335
112. Vandekeere S, Dewerchin M, Carmeliet P. Angiogenesis revisited: an overlooked role of endothelial cell metabolism in vessel sprouting. Microcirculation (2015) 22(7):509–17. doi: 10.1111/micc.12229
113. Minchenko OH, Tsuchihara K, Minchenko DO, Bikfalvi A, Esumi H. Mechanisms of regulation of Pfkfb expression in pancreatic and gastric cancer cells. World J Gastroenterol (2014) 20(38):13705–17. doi: 10.3748/wjg.v20.i38.13705
114. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of Pfkfb3-driven glycolysis in vessel sprouting. Cell (2013) 154(3):651–63. doi: 10.1016/j.cell.2013.06.037
115. Chan DA, Kawahara TL, Sutphin PD, Chang HY, Chi JT, Giaccia AJ. Tumor vasculature is regulated by phd2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell (2009) 15(6):527–38. doi: 10.1016/j.ccr.2009.04.010
116. Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter Mct1 supports an Nf-Kb/Il-8 pathway that drives tumor angiogenesis. Cancer Res (2011) 71(7):2550–60. doi: 10.1158/0008-5472.Can-10-2828
117. Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, et al. Nuclear targeting of 6-phosphofructo-2-kinase (Pfkfb3) increases proliferation via cyclin-dependent kinases. J Biol Chem (2009) 284(36):24223–32. doi: 10.1074/jbc.M109.016816
118. Jiang H, Zou Y, Zhao J, Li X, Yang S, Zhou X, et al. Pyruvate kinase M2 mediates glycolysis in the lymphatic endothelial cells and promotes the progression of lymphatic malformations. Am J Pathol (2021) 191(1):204–15. doi: 10.1016/j.ajpath.2020.10.003
119. Mehrotra D, Wu J, Papangeli I, Chun HJ. Endothelium as a gatekeeper of fatty acid transport. Trends Endocrinol Metab (2014) 25(2):99–106. doi: 10.1016/j.tem.2013.11.001
120. Kuo A, Lee MY, Sessa WC. Lipid droplet biogenesis and function in the endothelium. Circ Res (2017) 120(8):1289–97. doi: 10.1161/circresaha.116.310498
121. Wei X, Schneider JG, Shenouda SM, Lee A, Towler DA, Chakravarthy MV, et al. De novo lipogenesis maintains vascular homeostasis through endothelial nitric-oxide synthase (Enos) palmitoylation. J Biol Chem (2011) 286(4):2933–45. doi: 10.1074/jbc.M110.193037
122. Meghwani H, Berk BC. Mst1 kinase-Cx43-Yap/Taz pathway mediates disturbed flow endothelial dysfunction. Circ Res (2022) 131(9):765–7. doi: 10.1161/circresaha.122.321921
123. Daiber A, Xia N, Steven S, Oelze M, Hanf A, Kröller-Schön S, et al. New therapeutic implications of endothelial nitric oxide synthase (Enos) function/dysfunction in cardiovascular disease. Int J Mol Sci (2019) 20(1):187. doi: 10.3390/ijms20010187
124. Kattoor AJ, Goel A, Mehta JL. Lox-1: regulation, signaling and its role in atherosclerosis. Antioxidants (Basel) (2019) 8(7):218. doi: 10.3390/antiox8070218
125. Njock MS, Cheng HS, Dang LT, Nazari-Jahantigh M, Lau AC, Boudreau E, et al. Endothelial cells suppress monocyte activation through secretion of extracellular vesicles containing antiinflammatory micrornas. Blood (2015) 125(20):3202–12. doi: 10.1182/blood-2014-11-611046
126. Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, et al. Flow-dependent Yap/Taz activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci USA (2016) 113(41):11525–30. doi: 10.1073/pnas.1613121113
127. Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev (1995) 75(3):487–517. doi: 10.1152/physrev.1995.75.3.487
128. Campbell GR, Campbell JH. Smooth muscle phenotypic changes in arterial wall homeostasis: implications for the pathogenesis of atherosclerosis. Exp Mol Pathol (1985) 42(2):139–62. doi: 10.1016/0014-4800(85)90023-1
129. Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg (2007) 45 Suppl:A:A25–32. doi: 10.1016/j.jvs.2007.03.001
130. Shi J, Yang Y, Cheng A, Xu G, He F. Metabolism of vascular smooth muscle cells in vascular diseases. Am J Physiol Heart Circ Physiol (2020) 319(3):H613–h31. doi: 10.1152/ajpheart.00220.2020
131. Heiss EH, Schachner D, Donati M, Grojer CS, Dirsch VM. Increased aerobic glycolysis is important for the motility of activated vsmc and inhibited by indirubin-3'-monoxime. Vascul Pharmacol (2016) 83:47–56. doi: 10.1016/j.vph.2016.05.002
132. Chen S, Chen H, Yu C, Lu R, Song T, Wang X, et al. Mir-638 repressed vascular smooth muscle cell glycolysis by targeting Ldha. Open Med (Wars) (2019) 14:663–72. doi: 10.1515/med-2019-0077
133. Kim JH, Bae KH, Byun JK, Lee S, Kim JG, Lee IK, et al. Lactate dehydrogenase-a is indispensable for vascular smooth muscle cell proliferation and migration. Biochem Biophys Res Commun (2017) 492(1):41–7. doi: 10.1016/j.bbrc.2017.08.041
134. Yang L, Gao L, Nickel T, Yang J, Zhou J, Gilbertsen A, et al. Lactate promotes synthetic phenotype in vascular smooth muscle cells. Circ Res (2017) 121(11):1251–62. doi: 10.1161/circresaha.117.311819
135. Zhou Q, Xu J, Liu M, He L, Zhang K, Yang Y, et al. Warburg effect is involved in apelin-13-induced human aortic vascular smooth muscle cells proliferation. J Cell Physiol (2019) 234(9):14413–21. doi: 10.1002/jcp.28218
136. Martínez-Reyes I, Chandel NS. Mitochondrial Tca cycle metabolites control physiology and disease. Nat Commun (2020) 11(1):102. doi: 10.1038/s41467-019-13668-3
137. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. Hif-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3(3):177–85. doi: 10.1016/j.cmet.2006.02.002
138. Yin J, Xia W, Wu M, Zhang Y, Huang S, Zhang A, et al. Inhibition of mitochondrial complex I activity attenuates neointimal hyperplasia by inhibiting smooth muscle cell proliferation and migration. Chem Biol Interact (2019) 304:73–82. doi: 10.1016/j.cbi.2019.03.002
139. Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, et al. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med (2006) 12(9):1075–80. doi: 10.1038/nm1459
140. Barron JT, Kopp SJ, Tow J, Parrillo JE. Fatty acid, Tricarboxylic acid cycle metabolites, and energy metabolism in vascular smooth muscle. Am J Physiol (1994) 267(2 Pt 2):H764–9. doi: 10.1152/ajpheart.1994.267.2.H764
141. Salabei JK, Hill BG. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol (2013) 1(1):542–51. doi: 10.1016/j.redox.2013.10.011
142. Scheede-Bergdahl C, Bergdahl A. Adaptation of mitochondrial expression and Atp production in dedifferentiating vascular smooth muscle cells. Can J Physiol Pharmacol (2017) 95(12):1473–9. doi: 10.1139/cjpp-2017-0227
143. Sugden MC. In appreciation of sir Philip Randle: the glucose-fatty acid cycle. Br J Nutr (2007) 97(5):809–13. doi: 10.1017/s0007114507659054
144. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet (1963) 1(7285):785–9. doi: 10.1016/s0140-6736(63)91500-9
145. Heininger K. The cerebral glucose-fatty acid cycle: evolutionary roots, regulation, and (Patho)Physiological importance. Int Rev Neurobiol (2002) 51:103–58. doi: 10.1016/s0074-7742(02)51004-7
146. Tomas L, Edsfeldt A, Mollet IG, Perisic Matic L, Prehn C, Adamski J, et al. Altered metabolism distinguishes high-risk from stable carotid atherosclerotic plaques. Eur Heart J (2018) 39(24):2301–10. doi: 10.1093/eurheartj/ehy124
147. Osman I, He X, Liu J, Dong K, Wen T, Zhang F, et al. Tead1 (Tea domain transcription factor 1) promotes smooth muscle cell proliferation through upregulating slc1a5 (Solute carrier family 1 member 5)-mediated glutamine uptake. Circ Res (2019) 124(9):1309–22. doi: 10.1161/circresaha.118.314187
148. Gupte SA, Wolin MS. Oxidant and redox signaling in vascular oxygen sensing: implications for systemic and pulmonary hypertension. Antioxid Redox Signal (2008) 10(6):1137–52. doi: 10.1089/ars.2007.1995
149. Park WH. Exogenous H2o2 induces growth inhibition and cell death of human pulmonary artery smooth muscle cells via glutathione depletion. Mol Med Rep (2016) 14(1):936–42. doi: 10.3892/mmr.2016.5307
150. Park WH, Han BR, Park HK, Kim SZ. Arsenic trioxide induces growth inhibition and death in human pulmonary artery smooth muscle cells accompanied by mitochondrial O2•- increase and Gsh depletion. Environ Toxicol (2018) 33(8):833–40. doi: 10.1002/tox.22569
151. Powell LA, Nally SM, McMaster D, Catherwood MA, Trimble ER. Restoration of glutathione levels in vascular smooth muscle cells exposed to high glucose conditions. Free Radic Biol Med (2001) 31(10):1149–55. doi: 10.1016/s0891-5849(01)00648-7
152. Zhao Z, Francis CE, Welch G, Loscalzo J, Ravid K. Reduced glutathione prevents nitric oxide-induced apoptosis in vascular smooth muscle cells. Biochim Biophys Acta (1997) 1359(2):143–52. doi: 10.1016/s0167-4889(97)00093-1
153. Holm AM, Andersen CB, Haunsø S, Hansen PR. Effects of L-arginine on vascular smooth muscle cell proliferation and apoptosis after balloon injury. Scand Cardiovasc J (2000) 34(1):28–32. doi: 10.1080/14017430050142369
154. Dubey RK, Jackson EK, Lüscher TF. Nitric oxide inhibits angiotensin ii-induced migration of rat aortic smooth muscle cell. Role of cyclic-nucleotides and angiotensin1 receptors. J Clin Invest (1995) 96(1):141–9. doi: 10.1172/jci118014
155. Kanno Y, Into T, Lowenstein CJ, Matsushita K. Nitric oxide regulates vascular calcification by interfering with Tgf- signalling. Cardiovasc Res (2008) 77(1):221–30. doi: 10.1093/cvr/cvm049
156. Liu Y, Wang Z, Li J, Ban Y, Mao G, Zhang M, et al. Inhibition of 5-hydroxytryptamine receptor 2b reduced vascular restenosis and mitigated the B-arrestin2-mammalian target of rapamycin/P70s6k pathway. J Am Heart Assoc (2018) 7(3):e006810. doi: 10.1161/jaha.117.006810
157. Chen C, Surowiec SM, Morsy AH, Ma M. Intraperitoneal infusion of homocysteine increases intimal hyperplasia in balloon-injured rat carotid arteries. Atherosclerosis (2002) 160(1):103–14. doi: 10.1016/s0021-9150(01)00573-1
158. Fan J, Zheng F, Li S, Cui C, Jiang S, Zhang J, et al. Hydrogen sulfide lowers hyperhomocysteinemia dependent on cystathionine Γ Lyase S-sulfhydration in apoe-knockout atherosclerotic mice. Br J Pharmacol (2019) 176(17):3180–92. doi: 10.1111/bph.14719
159. Hart JL. Role of sulfur-containing gaseous substances in the cardiovascular system. Front Biosci (Elite Ed) (2011) 3(2):736–49. doi: 10.2741/e282
160. Jenkins TA, Nguyen JC, Hart JL. Decreased vascular H2s production is associated with vascular oxidative stress in rats fed a high-fat western diet. Naunyn Schmiedebergs Arch Pharmacol (2016) 389(7):783–90. doi: 10.1007/s00210-016-1244-4
161. Halvorsen B, Otterdal K, Dahl TB, Skjelland M, Gullestad L, Øie E, et al. Atherosclerotic plaque stability–what determines the fate of a plaque? Prog Cardiovasc Dis (2008) 51(3):183–94. doi: 10.1016/j.pcad.2008.09.001
162. Renier G, Mamputu JC, Serri O. Benefits of gliclazide in the atherosclerotic process: decrease in monocyte adhesion to endothelial cells. Metabolism (2003) 52(8 Suppl 1):13–8. doi: 10.1016/s0026-0495(03)00212-9
163. Wautier JL, Wautier MP. Vascular permeability in diseases. Int J Mol Sci (2022) 23(7):3645. doi: 10.3390/ijms23073645
164. Carné X. [Canakinumab, a monoclonal antibody against Il-1β, with potential utility in different inflammatory processes]. Med Clin (Barc) (2011) 136 Suppl 1:34–7. doi: 10.1016/s0025-7753(11)70007-0
165. Athyros VG, Kakafika AI, Tziomalos K, Papageorgiou AA, Karagiannis A. Statins for the prevention of first or recurrent stroke. Curr Vasc Pharmacol (2008) 6(2):124–33. doi: 10.2174/157016108783955365
166. Ridker PM, Rane M. Interleukin-6 signaling and anti-interleukin-6 therapeutics in cardiovascular disease. Circ Res (2021) 128(11):1728–46. doi: 10.1161/circresaha.121.319077
167. Krishnaswamy G, Kelley J, Yerra L, Smith JK, Chi DS. Human endothelium as a source of multifunctional cytokines: molecular regulation and possible role in human disease. J Interferon Cytokine Res (1999) 19(2):91–104. doi: 10.1089/107999099314234
168. Rot A, Hub E, Middleton J, Pons F, Rabeck C, Thierer K, et al. Some aspects of il-8 pathophysiology. Iii: chemokine interaction with endothelial cells. J Leukoc Biol (1996) 59(1):39–44. doi: 10.1002/jlb.59.1.39
169. Strohm L, Ubbens H, Münzel T, Daiber A, Daub S. Role of cd40(L)-Traf signaling in inflammation and resolution-a double-edged sword. Front Pharmacol (2022) 13:995061. doi: 10.3389/fphar.2022.995061
170. Rodríguez G, Mago N, Rosa F. [Role of inflammation in atherogenesis]. Invest Clin (2009) 50(1):109–29.
171. Munjal A, Khandia R. Atherosclerosis: orchestrating cells and biomolecules involved in its activation and inhibition. Adv Protein Chem Struct Biol (2020) 120:85–122. doi: 10.1016/bs.apcsb.2019.11.002
172. Alvandi Z, Bischoff J. Endothelial-mesenchymal transition in cardiovascular disease. Arterioscler Thromb Vasc Biol (2021) 41(9):2357–69. doi: 10.1161/atvbaha.121.313788
173. Ikeda U. Inflammation and coronary artery disease. Curr Vasc Pharmacol (2003) 1(1):65–70. doi: 10.2174/1570161033386727
174. Newby AC. Metalloproteinase production from macrophages - a perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp Physiol (2016) 101(11):1327–37. doi: 10.1113/ep085567
175. Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, et al. Cd47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature (2016) 536(7614):86–90. doi: 10.1038/nature18935
176. Linton MF, Babaev VR, Huang J, Linton EF, Tao H, Yancey PG. Macrophage apoptosis and efferocytosis in the pathogenesis of atherosclerosis. Circ J (2016) 80(11):2259–68. doi: 10.1253/circj.CJ-16-0924
177. Michel JB, Thaunat O, Houard X, Meilhac O, Caligiuri G, Nicoletti A. Topological determinants and consequences of adventitial responses to arterial wall injury. Arterioscler Thromb Vasc Biol (2007) 27(6):1259–68. doi: 10.1161/atvbaha.106.137851
178. Michel JB, Martin-Ventura JL, Nicoletti A, Ho-Tin-Noé B. Pathology of human plaque vulnerability: mechanisms and consequences of intraplaque haemorrhages. Atherosclerosis (2014) 234(2):311–9. doi: 10.1016/j.atherosclerosis.2014.03.020
179. Takaya N, Yuan C, Chu B, Saam T, Polissar NL, Jarvik GP, et al. Presence of intraplaque hemorrhage stimulates progression of carotid atherosclerotic plaques: A high-resolution magnetic resonance imaging study. Circulation (2005) 111(21):2768–75. doi: 10.1161/circulationaha.104.504167
180. Taylor BA, Thompson PD. Statins and their effect on Pcsk9-impact and clinical relevance. Curr Atheroscler Rep (2016) 18(8):46. doi: 10.1007/s11883-016-0604-3
181. Lennernäs H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet (2003) 42(13):1141–60. doi: 10.2165/00003088-200342130-00005
182. Galan Moya EM, Le Guelte A, Gavard J. Paking up to the endothelium. Cell Signal (2009) 21(12):1727–37. doi: 10.1016/j.cellsig.2009.08.006
183. Saini HK, Xu YJ, Arneja AS, Tappia PS, Dhalla NS. Pharmacological basis of different targets for the treatment of atherosclerosis. J Cell Mol Med (2005) 9(4):818–39. doi: 10.1111/j.1582-4934.2005.tb00382.x
184. Koushki K, Shahbaz SK, Mashayekhi K, Sadeghi M, Zayeri ZD, Taba MY, et al. Anti-inflammatory action of statins in cardiovascular disease: the role of inflammasome and toll-like receptor pathways. Clin Rev Allergy Immunol (2021) 60(2):175–99. doi: 10.1007/s12016-020-08791-9
185. Simionescu M, Stancu C, Costache G, Sima A. Endothelial cell response to hyperlipemia. Activation-dysfunction-injury, the protective role of simvastatin. Vascul Pharmacol (2002) 38(5):275–82. doi: 10.1016/s1537-1891(02)00252-5
186. Geng J, Xu H, Yu X, Xu G, Cao H, Lin G, et al. Rosuvastatin protects against oxidized low−Density lipoprotein−Induced endothelial cell injury of atherosclerosis. In vitro. Mol Med Rep (2019) 19(1):432–40. doi: 10.3892/mmr.2018.9666
187. Jiang JL, Jiang DJ, Tang YH, Li NS, Deng HW, Li YJ. Effect of simvastatin on endothelium-dependent vaso-relaxation and endogenous nitric oxide synthase inhibitor. Acta Pharmacol Sin (2004) 25(7):893–901.
188. Kimura T, Sato K, Tomura H, Okajima F. Cross-talk between exogenous statins and endogenous high-density lipoprotein in anti-inflammatory and anti-atherogenic actions. Endocr Metab Immune Disord Drug Targets (2010) 10(1):8–15. doi: 10.2174/187153010790827939
189. Bustos C, Hernández-Presa MA, Ortego M, Tuñón J, Ortega L, Pérez F, et al. Hmg-coa reductase inhibition by atorvastatin reduces neointimal inflammation in a rabbit model of atherosclerosis. J Am Coll Cardiol (1998) 32(7):2057–64. doi: 10.1016/s0735-1097(98)00487-2
190. Massonnet B, Normand S, Moschitz R, Delwail A, Favot L, Garcia M, et al. Pharmacological inhibitors of the mevalonate pathway activate pro-Il-1 processing and Il-1 release by human monocytes. Eur Cytokine Netw (2009) 20(3):112–20. doi: 10.1684/ecn.2009.0162
191. Kitagawa K, Hosomi N, Nagai Y, Kagimura T, Ohtsuki T, Origasa H, et al. Reduction in high-sensitivity C-reactive protein levels in patients with ischemic stroke by statin treatment: Hs-Crp sub-study in J-stars. J Atheroscler Thromb (2017) 24(10):1039–47. doi: 10.5551/jat.39354
192. Bellosta S, Ferri N, Arnaboldi L, Bernini F, Paoletti R, Corsini A. Pleiotropic effects of statins in atherosclerosis and diabetes. Diabetes Care (2000) 23 Suppl 2:B72–8.
193. Chandrasekar B, Mummidi S, Mahimainathan L, Patel DN, Bailey SR, Imam SZ, et al. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on Nf-Kappab- and Ap-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J Biol Chem (2006) 281(22):15099–109. doi: 10.1074/jbc.M600200200
194. Zhou F, Tan Y, Chen XH, Wu FL, Yang DJ, Zhang XW, et al. Atorvastatin improves plaque stability in diabetic atherosclerosis through the rage pathway. Eur Rev Med Pharmacol Sci (2018) 22(4):1142–9. doi: 10.26355/eurrev_201802_14403
195. Palomino-Morales R, Perales S, Torres C, Linares A, Alejandre MJ. Effect of Hmg-Coa reductase inhibition on vascular smooth muscle cells extracellular matrix production: role of Rhoa. Curr Vasc Pharmacol (2016) 14(4):345–52. doi: 10.2174/1570161114666160229115553
196. Jo JH, Park HS, Lee DH, Han JH, Heo KS, Myung CS. Rosuvastatin inhibits the apoptosis of platelet-derived growth factor-stimulated vascular smooth muscle cells by inhibiting P38 via autophagy. J Pharmacol Exp Ther (2021) 378(1):10–9. doi: 10.1124/jpet.121.000539
197. Barale C, Frascaroli C, Senkeev R, Cavalot F, Russo I. Simvastatin effects on inflammation and platelet activation markers in hypercholesterolemia. BioMed Res Int (2018) 2018:6508709. doi: 10.1155/2018/6508709
198. Chen W, Gard JMC, Epshtein Y, Camp SM, Garcia JGN, Jacobson JR, et al. Integrin Beta 4e promotes endothelial phenotypic changes and attenuates lung endothelial cell inflammatory responses. Front Physiol (2022) 13:769325. doi: 10.3389/fphys.2022.769325
199. Hanai J, Cao P, Tanksale P, Imamura S, Koshimizu E, Zhao J, et al. The muscle-specific ubiquitin ligase atrogin-1/Mafbx mediates statin-induced muscle toxicity. J Clin Invest (2007) 117(12):3940–51. doi: 10.1172/jci32741
200. Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the Jupiter trial. Lancet (2012) 380(9841):565–71. doi: 10.1016/s0140-6736(12)61190-8
201. Su X, Zhang L, Lv J, Wang J, Hou W, Xie X, et al. Effect of Statins on Kidney Disease Outcomes: A systematic review and Meta-Analysis. Am J Kidney Dis (2016) 67(6):881–92. doi: 10.1053/j.ajkd.2016.01.016
202. Castilla-Guerra L, Del Carmen Fernandez-Moreno M, Colmenero-Camacho MA. Statins in stroke prevention: present and future. Curr Pharm Des (2016) 22(30):4638–44. doi: 10.2174/1381612822666160510125229
203. Boutari C, Karagiannis A, Athyros VG. Rosuvastatin and ezetimibe for the treatment of dyslipidemia and hypercholesterolemia. Expert Rev Cardiovasc Ther (2021) 19(7):575–80. doi: 10.1080/14779072.2021.1940959
204. Wang Y, Kuang ZM, Feng SJ, Jiang L, Chen QX, Ji XY, et al. Combined antihypertensive and statin therapy for the prevention of cardiovascular events in patients with hypertension without complications: protocol for a systematic review and meta-analysis. BMJ Open (2018) 8(5):e019719. doi: 10.1136/bmjopen-2017-019719
205. Feng X, Chen W, Ni X, Little PJ, Xu S, Tang L, et al. Metformin, macrophage dysfunction and atherosclerosis. Front Immunol (2021) 12:682853. doi: 10.3389/fimmu.2021.682853
Keywords: atherosclerosis plaque, cell metabolism, inflammatory response, statins, anti-inflammatory drug, cardiovascular and cerebrovascular diseases
Citation: Zhao L, Ma D, Wang L, Su X, Feng L, Zhu L, Chen Y, Hao Y, Wang X and Feng J (2023) Metabolic changes with the occurrence of atherosclerotic plaques and the effects of statins. Front. Immunol. 14:1301051. doi: 10.3389/fimmu.2023.1301051
Received: 24 September 2023; Accepted: 23 November 2023;
Published: 08 December 2023.
Edited by:
Xiangsheng Zhang, Capital Medical University, ChinaReviewed by:
Shuo Yuan, Yanbian University, ChinaCopyright © 2023 Zhao, Ma, Wang, Su, Feng, Zhu, Chen, Hao, Wang and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Di Ma, bWFkaTIwMTdAamx1LmVkdS5jbg==; LiJuan Wang, d2FuZ2xqNjZAamx1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.