
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 18 January 2024
Sec. B Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1298665
This article is part of the Research Topic B cells and the roles of B cell depleting therapies in autoimmune skin diseases: novel findings, challenges, and future perspective View all 5 articles
 Kazuhiro Komura1,2*
Kazuhiro Komura1,2* Koichi Yanaba3Jean-David Bouaziz4
Koichi Yanaba3Jean-David Bouaziz4 Ayumi Yoshizaki5
Ayumi Yoshizaki5 Minoru Hasegawa6
Minoru Hasegawa6 John Varga2
John Varga2 Kazuhiko Takehara7
Kazuhiko Takehara7 Takashi Matsushita7
Takashi Matsushita7Systemic sclerosis (SSc) is a rare and heterogeneous disease with no relevant environmental trigger or significant responsible gene. It has been and will continue to be difficult to identify large enough patients to conduct classic population-based epidemiologic exposure/non-exposure studies with adequate power to ascertain environmental and genetic risk factors for these entities. The complexity of pathogenesis and heterogeneity are likely to require personalized/precision medicine for SSc. Since several potential drugs are currently available for specific patients if not whole SSc, classification of SSc seems to form the foundation for a better therapeutic strategy. To date, SSc has been classified based on the extent/severity of the affected area as well as some disease markers, including the autoantibody profile. However, such an analysis should also lead to improvements in the design of appropriately stratified clinical trials to determine the effects and prediction of targeted therapies. An approach based on drug response preclinically conducted using patients’ own fibroblasts in vitro, can provide a precise disease marker/therapeutic selection for clinical practice. Because scleroderma dermal fibroblasts have a persistent hyper-productive phenotype occurring not only in person, but also in cell culture conditions. Thus, an accumulating approach based on disease markers ensures progression and de-escalation to re-establish a better life with a personally optimized drug environment after the onset of SSc.
Systemic sclerosis (SSc) or scleroderma is characterized by immune dysregulation, obliterative microvasculopathy, and fibrosis, which are significant causes of mortality occurring in the whole body (1–3). Fibrosis is caused by the excessive deposition of collagen and other extracellular matrix components produced by activated fibroblasts and α-smooth muscle actin-positive myofibroblasts. In vitro, scleroderma patient derived skin fibroblast synthesizes more collagen than fibroblasts from healthy humans (4–6). This in vitro evidence has suggested the role of epigenetics in the pathogenesis of SSc (7–11). Exogenous circumstances promote persistent cellular fibrotic phenotypes in fibroblasts, going over the endogenous predisposition in SSc patients (12–18).
Epigenetics describe heritable changes in gene expression that is independent on DNA sequence. Recent discoveries have linked to targeting DNA methylation, chromosome remodeling, and RNA turnover. Although epigenetic mechanisms help to protect cells from parasitic elements, this defense can complicate genetic manipulation. Essential to normal development, epigenetic controls are misdirected to human diseases (19). Certain environmental imprinting settings can be transmitted to future generations, although the role of these changes in the transmission of disease to humans remains unclear. The best-studied epigenetic modification of DNA is methylation of DNA by methyltransferases on cytosine residues of paired CG sequences and acetylation of lysine residues on histones by histone acetylases (20). For example, Oxidative DNA damage-mediated histone deacetylation activates the Wnt signaling pathway and fibrotic processes in SSc (21). Improved miR-150, one of miRNAs in SSc fibroblasts, normalized activated TGF beta signals and fibrosis (22). These epigenetic changes can explain why scleroderma dermal fibroblasts have a persistent hyper-productive phenotype expressed not only in person but also in cell culture conditions.
The clinical course of SSc varies substantially, and the pathogenesis and degree of fibroblast production depends on the individual burden of environmental exposure to pathogens and gene susceptibility (Figure 1). The response to antifibrotic drugs is also heterogeneous among patients, resulting in a lack of FDA-approved antifibrotic drugs for SSc from randomly controlled clinical trials with large number of patients (23–28).Traditional studies identifying anti-fibrotic drugs were conducted in optimized culture microenvironment and these in vitro studies have proposed several anti-fibrotic drugs, including Imatinib mesylate, c-Abl tyrosine kinase, and a line of PPARγ activators (23–28). However, genetics, epigenetics and environmental/exogenous affects phenotypic features of cell cultures and organism variation (29–31). In schematic pathological pathways of SSc, various endogenous factors establish heterogeneous phenotypic expression in cooperation with exogenous triggers. The pathogenetic spiral vectors may be narrowed weekly during the establishment of the phenotypic characteristics (Figures 1B, C). Within this motif, narrow portion could be a treatment target. However, existing anti-fibrotics are efficacious only in subpopulations of SSc patients, which were not sufficient to show differences in large-scale placebo-controlled studies in SSc (32, 33). This means some pathological vectors bypass the blocking by treatments.
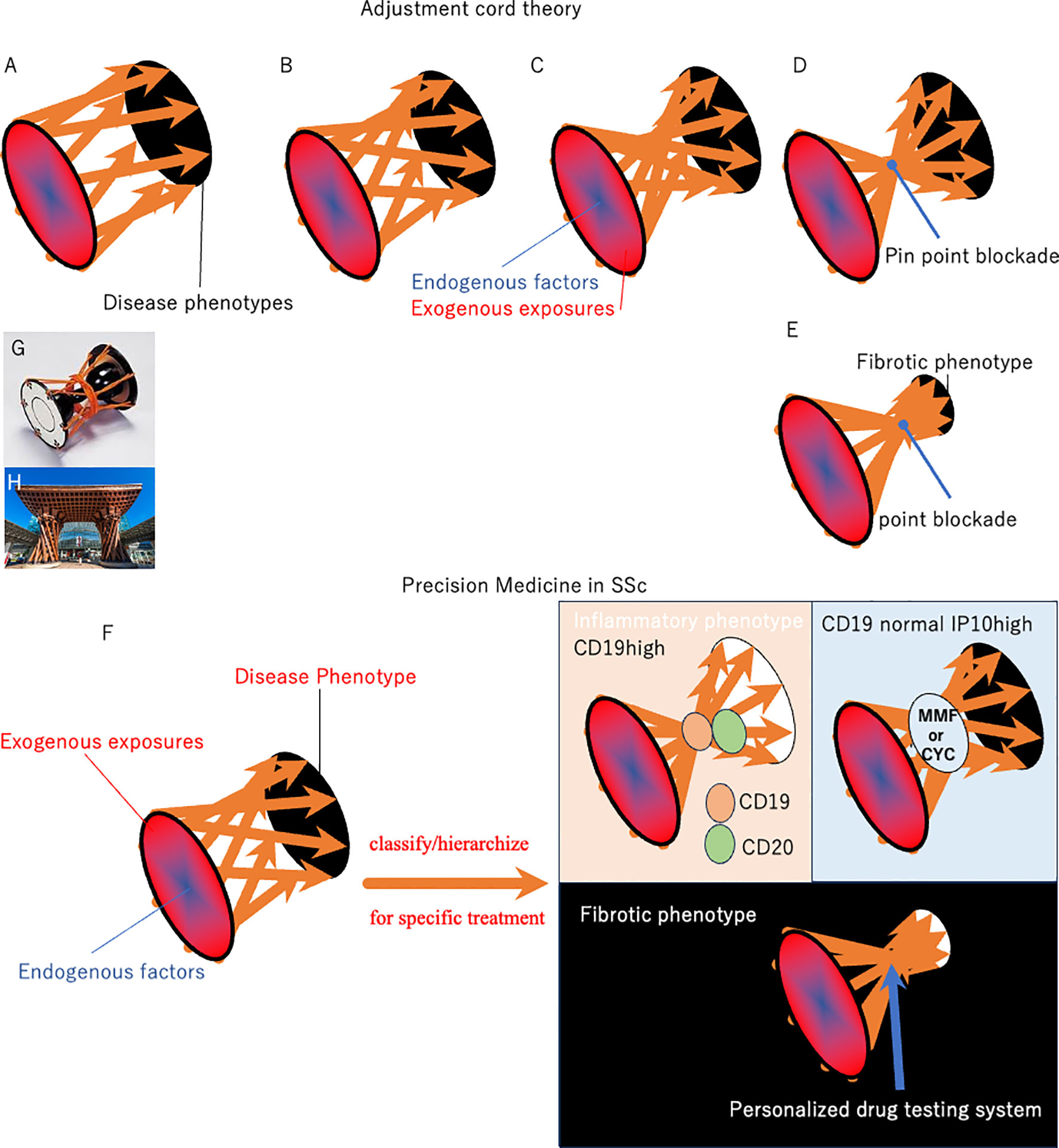
Figure 1 Schematic of pathological pathways in SSc. Various endogenous factors establish heterogeneous phenotypic expression in SSc in cooperation with exogenous triggers (A). The pathogenetic stream may be narrowed weakly during the establishment of the phenotypic characteristics (panels (B, C). The results from clinical practice indicate that antifibrotic drugs, including IM, Tocilizumab, are effective for specific SSc patients, although these were not validated in randomized trials in patients with SSc. This indicates that narrow-target therapy partially abrogates wide and complex pathways to SSc, while others bypass the blocking point, resulting in continuous development of SSc phenotypes. Considering the pathological pathway, the current approach using existing biomarkers and blocking therapy does not reach the focus point, resulting in incomplete efficacy of single-molecule blockers in SSc (panel (B). In contrast, the pathogenesis of psoriasis and atopic dermatitis consists of umbrella-like upstream, umbrella-like downstream, and focus point: IL-23 and JAK1 are near the focus point in the pathogenic motif of psoriasis and atopic dermatitis, respectively (panel (D). Alternatively, focusing on the final fibrotic process is empowered to change patient mortality or quality of life, since the final step for phenotypic fibrosis seems to be crucial for mortality and quality of life in most patients with SSc (panel (E). Biomarker studies may classify/hierarchize patients with SSc to select an efficient patient-entry design in clinical trials (panel (F). Further characterization of the key genomic and immune pathways involved will help identify novel disease mechanisms and therapeutic targets for precision medicine initiatives in SSc. The Tsuzumi column is the origin of the motif used in the pathological pathway described in this paper (panel (G). Tsuzumi, a traditional Japanese drum played in Nou, closely correlated with the traditional culture of Kanazawa City in Japan. Many citizens were initially surprised at the modern entrance when they appeared in front of Kanazawa station in 2005 (panel (H). The station’s wooden hand-drum-shaped Tsuzumi pillar and glass umbrella-shaped Motenashi Dome were controversial, because they are novel and new shapes staying in front of this traditional castle town: Kanazawa is one of the best-preserved Japanese cities after the WWII bombings. However, the station has become popular with tourists because of its beautiful sleek design. Tsuzumi changed the beat by adjusting the cord placed at the center. We hypothesize “Tsuzumi adjustment cord theory” hoping that many clinical trials tighten the center cord and discover the hub/focus point of SSc safely. Efficacy/safety and paradoxical reactions during targeting therapy trial is key findings for tightening the code to make two umbrellas and a focus point from Tuzumi pillar.
Until now most medical treatments were engineered for the “average patient.” As a result of this “one-size-fits-all” approach based on the recommendation from randomized controlled trials, treatments can be very successful for some patients, but not for others with heterogeneous diseases. Most immunological skin diseases, including SSc, do not have a standard therapy, since the biological pathophysiology and efficacy of the therapeutic intervention are highly variable between individual patients. Nonetheless, a sophisticated strategy improved therapeutic efficacy in Psoriasis, one of heterogenous immunological skin diseases. IL23 or IL17 blockade clears over 50% psoriasis patients if it continues (34–36). This indicates that IL23/IL17 aggregate complex pathological vectors in psoriasis (Figure 1D).
Precision medicine is an emerging approach for disease treatment and prevention that considers individual variability in genes and the environment for each person. Although some advances in precision medicine have been made, the practice is not currently in use for most diseases. Precision medicine can provide medical professionals with specific treatments for the correct patient group at the appropriate time (37, 38).
Excessive exposure to exogenous triggers causes disease when it exceeds the threshold limited by endogenous susceptibility. At that time, there is an opportunity to mitigate the crisis by optimally regulating external and internal exposure through circumstances and treatment. Appropriate disease markers early in disease pathogenesis are needed to correct these circumstances and provide clues for correct treatments choice. Thus, identifying appropriate disease markers/indicators for patients is an emerging issue.
Recent advances in -omics technologies over the past several years and the explosive growth in the use of genome-wide gene expression studies to analyze molecular information from individual patients have revolutionized the field of precision medicine (39). The conventional patient management approach for symptom-centered diagnosis and treatment is limited by its focus on a few late or terminal disease manifestations and by its disregard for the underlying pathogenic mechanisms. Previously, the contribution of an individual’s genetic constitution to the development of these symptoms was not routinely considered, and an emphasis on diagnostic criteria for complex human disorders enhanced these diagnostic and therapeutic limitations. It is possible to handle big data on transcription products (including coding and non-coding RNAs) and their corresponding translated protein molecules in large cohorts of patients, rendering integrative analysis of individual genetic variations (37). Such investigations will result in improvements in the precise diagnosis, in addition to a greater understanding of the complex pathogenic mechanisms responsible for comorbidities in the future (40).
With respect to clinical practice in SSc, the majority of SSc patients carry disease-specific autoantibodies, including antibodies against topoisomerase I, centromere, RNA polymerase I/III, Th/To, U3 RNP, U11/U12, Ku, human upstream-binding factor, U1RNP, and PM-Scl (27, 37, 41, 42). Since autoantibodies are disease-specific, the autoantibody profile has been progressively investigated to determine the subtype, diffuse cutaneous SSc (dcSSc) or limited cutaneous SSc (lcSSc), and predict the disease course (43, 44). Nailfold capillaroscopic microvascular patterns in SSc patients was associated with clinical phenotype and distinctive immune cell abnormalities, which might be linked to stratify individual immune cell target therapies (45). Additionally, empowered -omics studies in SSc have substantial potential to make prognostications and prospect responsiveness to antifibrotic and/or immune target therapies (46–51). In these studies, molecular signatures identified fibroproliferative, inflammatory, and other subset of SSc. Fibroproliferative subset will be linked to antifibrotic drug therapy, whereas inflammatory subset indicates immunosuppressive treatments. This is a major progression for treatment of SSc. However, a portion of this movement is possible to progress toward efficient/preferable gene alterations during the already developed therapies for SSc. Here is a substantial risk of enhancement of unnecessary stratification, or gene-editing therapies in stem/germline cells (52). “Mechanical” decision making might recommend “engineering” persistent genetical risk for SSc beyond the current ethics concern. Thus, further progress in this field requires a modern/future-level consensus.
There are many promising inputs from approximately 550 trials of SSc listed on ClinicalTrials.gov (53). Major part of these trials have used, or are using, new biologic agents or kinase inhibitors such as abatacept, tocilizumab, rituximab, inebilizumab, imatinib, dasatinib, fasudil, nintedanib, lysophosphatidic acid receptor 1 blockers, and fresolimumab, an anti-transforming growth factor (TGF) antibody (54). The results of these trials had a major impact on understanding of the pathogenesis of SSc. In this section, we focus on immunosuppressive B cell depletion therapy and antifibrotic Imatinib therapies, which show some progression of a precision medicine approach for skin sclerosis of SSc.
Recently, several studies on the role of B cells in autoimmune diseases have been published. B cells contribute to immune responses not only through antibody production but also through antigen presentation, regulating T-cell activation and production of various cytokines. Therefore, B cell-targeted clinical trials for autoimmune disorders are increasing (55). Most patients with SSc have disease-specific autoantibodies, hypergammaglobulinemia, and abnormality in B cell (56, 57). Anti-CD20 monoclonal antibodies deplete most B lymphocytes and abnormal B-cell lymphomas via antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC)-dependent mechanisms (58–60). In a murine model of SSc, B-cell depletion with an anti-CD20 monoclonal antibody ameliorated the development of skin sclerosis and autoantibody production (61). Rituximab, a chimeric monoclonal antibody against CD20, was efficacious for both skin sclerosis and ILD in SSc in case reports (62, 63), open-label trials (64–66) and small-scale RCTs (67, 68). In a case–control study of 25 patients with severe SSc (MRSS >16), rituximab treatment significantly improved skin sclerosis (mean ± SEM MRSS, from 26.6 ± 1.4 to 20.3 ± 1.8; P = 0.0001) after 6 months (range, 5–9) (66). Additionally, nine SSc patients with ILD and rituximab preserved %FVC (mean ± SEM %FVC, from 60.6 ± 2.4 to 61.3 ± 4.1; P = 0.5) (66). Consistently, rituximab significantly reduced MRSS while retaining pulmonary function at 12 months in an open-label, uncontrolled study of 20 patients with SSc (64). Therefore, B-cell depletion therapy may be beneficial for patients with SSc with severe skin sclerosis and ILD (67, 69). In addition, drugs for other B cell targets, such as CD19, are also under investigation because these efficiently improve autoantibody reactions in autoimmune disorders (70–73). Interestingly, multiple CD19-target approaches are participating in clinical trials in addition to anti-CD19 antibodies, since CD19 is relatively fragile on B cell surface, in contrast to stable CD20, during ADCC.
IM is a tyrosine kinase inhibitor that inhibits c-Abl, an important downstream signaling molecule of TGF-beta, and the platelet-derived growth factor signaling pathway, which stimulates collagen production (74). In a mouse model of bleomycin-induced lung fibrosis, IM was shown to prevent TGF-beta-induced fibroblast proliferation and ECM gene expression, resulting in the suppression of lung fibrosis (75). Moreover, in mouse models of SSc, IM inhibited both TGF-beta and platelet-derived growth factor signaling, leading to suppression of skin fibrosis (76, 77). Although IM may be a promising option for the treatment of SSc, based on the results of animal studies, its efficacy remains controversial in clinical trials. In a phase I/IIa, open-label, pilot trial with 20 patients with SSc, IM improved both %FVC and MRSS (1.74% and 3.9 points, respectively) at 12 months, although 12 of 20 patients completed the study mainly because of the adverse effects of IM (78). In a phase II pilot study, 30 SSc patients with ILD resistant to cyclophosphamide were treated with IM for 6 months (79); 19 of these 26 patients (73%) showed stabilized pulmonary function. In contrast, in a phase II RCT, included 28 patients with SSc, did not show significant improvement in skin sclerosis at 6 months (23). Further studies are needed to validate the safety and efficacy of IM with definitive inclusion subsets of SSc (80).
Figure 1 shows a schematic of the pathological pathways involved in SSc. Various endogenous factors in cooperation with exogenous triggers establish heterogeneous phenotype in SSc. The pathogenetic stream narrowed weekly during the establishment of phenotypic characteristics (Figure 1B). The results from clinical practice indicate that antifibrotic drugs, including IM, Tocilizumab, are effective for specific patients with SSc, although these were not validated in randomized trials in patients with SSc (81–83). This indicates that narrow-target therapy partially abrogates wide and complex pathways to SSc. Other spiral vectors bypass the blocking point and sometimes amplify/enhance the continuous development of SSc phenotypes (Figure 1B). By contrast, IL-23 target therapy is a focus point in the treatment of psoriasis (Figure 1D). Tumor necrosis factor, locating near the focus point, therapy sometimes cause paradoxical enhanced reactions in psoriasis (84). Great effort will tighten and clear the center of the motif in SSc, as well. Figures 1A–D directed-clearance looks like tightening the center cord of the motif “Tuzumi” (Figure 1G). We named this as adjustment cord theory.
Biomarker studies focusing on autoantibody profiles and other candidates may classify/hierarchize patients with SSc to select an efficient patient-entry design in clinical trials (Figure 1F). Further characterization of the key genomic and immune pathways involved will help to identify novel disease mechanisms and therapeutic target patients for precision medicine initiatives in SSc. For example, a higher IFN-inducible protein (IP10) level in SSc-ILD can predict the response to MMF or CYC (85, 86). CD19-positive B cell counts were associated with anti-CD20 Ab effectiveness (87) These initiated precision medicine for SSc (Figure 1F).
Alternatively, focusing on the final fibrotic process is empowered to change mortality or quality of life, since the final step for phenotypic fibrosis seems to be crucial for mortality and quality of life in most patients with SSc (Figure 1E). Fibrosis is caused by the overproduction of collagen and other ECM components by activated fibroblasts and α-smooth muscle actin positive myofibroblasts (88). Multiple physiological mechanisms regulate fibroblast activation to prevent excessive tissue remodeling and fibrosis (89). These complex reactions intensify cellular collagen overproduction, which has a long history of focus as a critical therapeutic target for SSc. Accordingly, patients belonging to the fibroproliferative intrinsic subset may respond to the antifibrotic drug IM (49). A better understanding of this stratification and precision medicine will lead to safer and more effective development of new antifibrotic drugs for SSc. However, this approach using a narrow target reagent still carries a substantial risk of adverse/paradox enhancement reactions, since it is impossible to find a focus point in SSc (Figure 1E).
SSc is a rare and diverse condition. The relevant environmental associations and the genes responsible for SSc are still unknown. Nonetheless, several potential drugs are currently available for specific patients, if not all SSc. Therefore, we propose a drug selection system using in vitro fibroblast culture (90). To select the potential drugs for a patient, we treated SSc skin fibroblast cell lines using four candidate substances/drugs affecting SSc related pathophysiologic processes.
Six SSc skin fibroblast cell lines were treated with four low molecular antifibrotic compounds, including bardoxolone (Nrf2, a key transcription factor that regulates antioxidant activity, inducer: CDDO, 5mM), imatinib (a tyrosine kinase inhibitor: IM,10mM), rosiglitazone (PPARgamma agonist: Rosi,10mM), PGJ2 (PPARgamma agonist,10mM), or DMSO (control) for 24 h. The details of the clinical data of these patients are described in patient background (Table 1). The starved fibroblasts were incubated with each reagent for 24 h. Purified mRNA from whole-cell lysates (WCL) was analyzed using real-time qPCR. Results are expressed as the ratio of Type I or II collagen mRNA levels relative to the control fibroblasts (Figure 2). As observed from the graphs in Figure 2 for two patients with CDDO has similar effect for IM (SSc-1096, SSc-I). CDDO for other three patients (SSc-1067, SSc-1066, SSc-1004) were more efficient than Im in collagen inhibition, while Rosi and PGJ2 both had very little effect in all patient cell cultures. Thus, CDDO improved all patients’ collagen synthesis(Figure 2), suggesting CDDO as first line treatment. Further, these results suggest that using IM for some patients (SSc-O) could be more efficient than CDDO. IM showed limited effect on SSc-1067 and SSc-1066 collagen production (Figure 2): SSc1066 treated by Im in past however currently treated by MTX, because IM showed limited merit for SSc 1066 (Table 1). In addition, no benefit was observed from Rosi or PGJ2, which may link to negative results of clinical trials with PPARgamma agonists. We could not find any association between clinical feature and current in vitro results (Table 1 and Figure 2) concerning the effect of CDDO and IM.

Table 1 Clinical characteristics of patients with SSc.
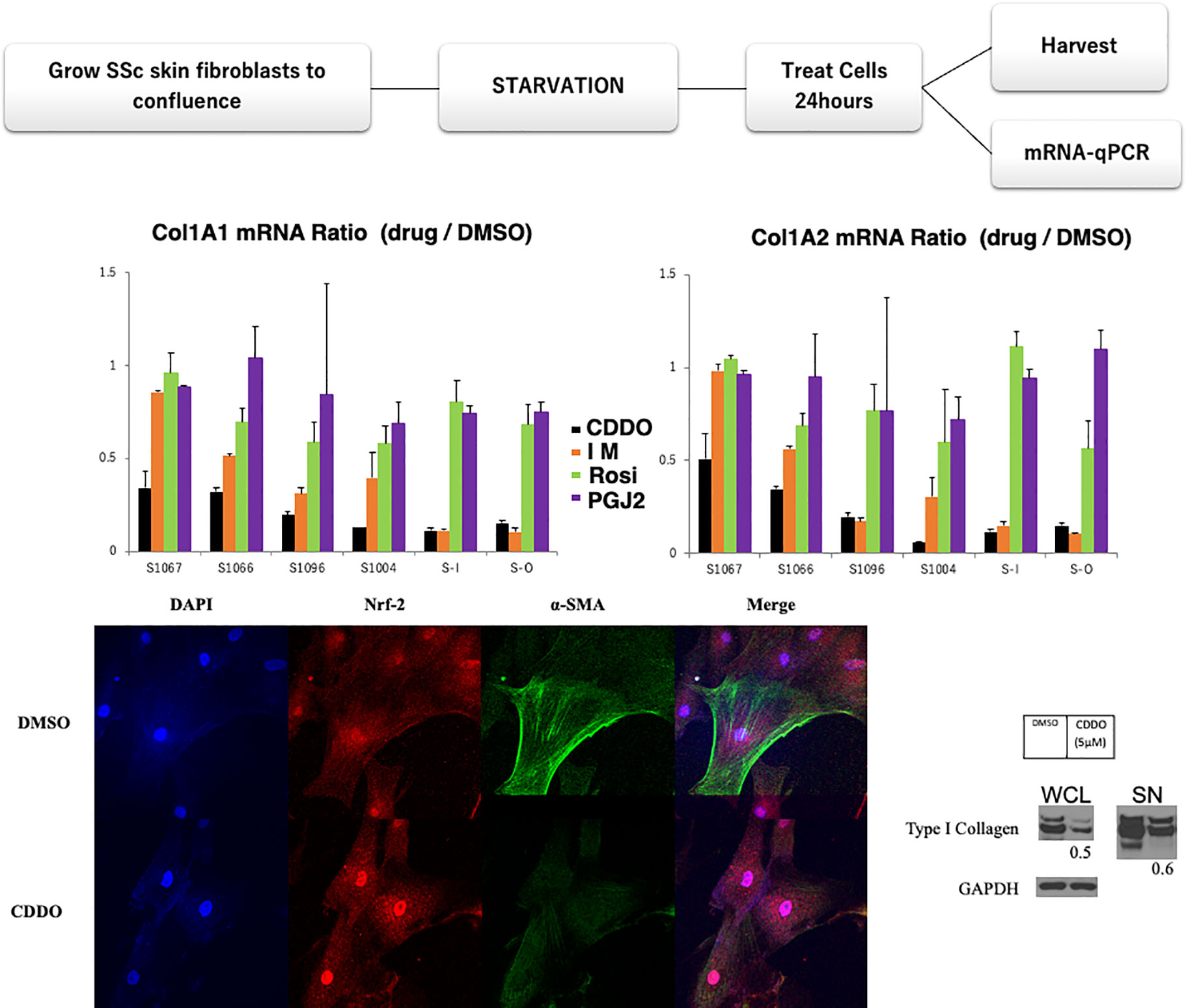
Figure 2 Personalized drug testing model in SSc. Antifibrotic drugs were selected based on the results of preclinical functional/treatment-based assessments. Six SSc skin fibroblast cell lines were treated with the anti-fibrotic agents bardoxolone (CDDO, 5µM), imatinib (IM,10µM), rosiglitazone (Rosi,10µM), PGI2 (10µM), or DMSO (as control). Starved fibroblasts were incubated with each treatment for 24 h. Purified mRNA from whole-cell lysates (WCL) was analyzed using real-time qPCR. Results are expressed as the ratio of Type I and II collagen mRNA levels relative to those from fibroblasts treated with DMSO. The anti-fibrotic drug responses were different between patients’ fibroblasts. Lower panel: CDDO treatment downregulated alpha-SMA expression in SSc skin fibroblasts with nuclear translocation of Nrf2, a key transcription factor that regulates antioxidant activity. Confluent SSc fibroblasts were incubated with DMSO (control) or 5µM CDDO for 24 h. Cells were fixed and stained with antibody for Nrf2 at 1:100 dilution (Santa Cruz) or alpha-SMA Ab (Sigma). Lower right panel: CDDO treatment downregulated type I collagen expression in SSc skin fibroblasts. Starved fibroblasts were incubated with CDDO for 24 h. WCL and culture supernatants were analyzed by Western analysis. Bands were quantitated by densitometry. Relative levels normalized with GAPDH are shown below the images. Representative images.
The anti-fibrotic effect of CDDO was confirmed by immunocytochemistry and western blotting (Figure 2 lower panels), according to the protocol described elsewhere (89). CDDO treatment downregulated alpha-SMA expression in SSc skin fibroblasts with nuclear translocation of Nrf2. This process represents an optimized environment for dermal fibroblasts to normalize collagen production. Hence, this personalized drug testing system has a potential to provide additional precision medicine for SSc. We can cultivate and investigate these tests from every patient within two months from skin biopsy. This enables physicians to know the results before starting the fundamental treatment for the patients. Further, this approach could respond the issue about other organ fibrosis, including lung. However, enrichment of the number of patients with medical outcomes and many candidate anti-fibrotic drugs will be required. In addition, other outcome indicators, including and fibronectin gene and protein expression, are also needed for this experimental design.
Classification based on drug response is likely to form the foundation for better therapeutic strategies in SSc with Fibrotic phenotype (Figure 1F). In clinical practice, we cannot clear-cut fibrotic phenotype from inflammatory phenotype, since fibrotic process is interacting with inflammatory process (91). However, personalized drug testing system could lead to improvements in the design of appropriately stratified clinical trials to determine the effects and prediction of targeted therapies.
The precision medicine in SSc is only beginning. The main hurdles to move forward include the complexity of disease, requirement for ethical consensus, and imbalance to access medical resources. The globalization of world enables to share, compare, and analyze datasets of patient for this rare disease. Thus, recent circumstances continue to develop personalized treatments and precision medicine for SSc patients, providing promising outlook.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving humans were approved by Northwestern University Institutional Review Board for Human Studies. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
KK: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. KY: Writing – review & editing, Methodology. J-DB: Methodology, Writing – review & editing. AY: Writing – review & editing. MH: Writing – review & editing. JV: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft. KT: Methodology, Writing – original draft. TM: Resources, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The authors sincerely appreciate the financial support providedby Grants-in-Aid for Young Scientists, Grants-in-Aid for Scientific Research KAKENHI-PROJECT-23791258, and the Uehara Memorial Foundation (to KK).
The authors also sincerely express their gratitude for the insights from all investigators (especially in the Northwestern scleroderma program) for preparing and discussing this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Campbell PM, LeRoy EC. Pathogenesis of systemic sclerosis: A vascular hypothesis. Semin Arthritis Rheu (1975) 4:351–68. doi: 10.1016/0049-0172(75)90017-7
2. Arnett FC, Reveille JD, Goldstein R, Pollard KM, Leaird K, Smith EA, et al. Autoantibodies to fibrillarin in systemic sclerosis (scleroderma). an immunogenetic, serologic, and clinical analysis. Arthritis Rheumatism (1996) 39:1151–60. doi: 10.1002/art.1780390712
3. Kahalefp MB, Leroy EC. Autoimmunity and vascular involvement in systemic sclerosis (SSc). Autoimmunity (2009) 31:195–214. doi: 10.3109/08916939908994064
4. LeRoy EC. Increased collagen synthesis by scleroderma skin fibroblasts in vitro a possible defect in the regulation or activation of the scleroderma fibroblast. J Clin Invest (1974) 54:880–9. doi: 10.1172/jci107827
5. Leroy EC. Connective tissue synthesis by scleroderma skin fibroblasts in cell culture. J Exp Med (1972) 135:1351–62. doi: 10.1084/jem.135.6.1351
6. Kikuchi K, Hartl CW, Smith EA, LeRoy EC, Trojanowska M. Direct demonstration of transcriptional activation of collagen gene expression in systemic sclerosis fibroblasts: Insensitivity to TGFβ1 stimulation. Biochem Bioph Res Co (1992) 187:45–50. doi: 10.1016/s0006-291x(05)81456-1
7. Lian X, Xiao R, Hu X, Kanekura T, Jiang H, Li Y, et al. DNA demethylation of CD40L in CD4+ t cells from women with systemic sclerosis: A possible explanation for female susceptibility. Arthritis Rheumatism (2012) 64:2338–45. doi: 10.1002/art.34376
8. Zhu H, Li Y, Qu S, Luo H, Zhou Y, Wang Y, et al. MicroRNA expression abnormalities in limited cutaneous scleroderma and diffuse cutaneous scleroderma. J Clin Immunol (2012) 32:514–22. doi: 10.1007/s10875-011-9647-y
9. Makino K, Jinnin M, Hirano A, Yamane K, Eto M, Kusano T, et al. The downregulation of microRNA let-7a contributes to the excessive expression of type i collagen in systemic and localized scleroderma. J Immunol (2013) 190:3905–15. doi: 10.4049/jimmunol.1200822
10. Komura K, Fujimoto M, Yanaba K, Matsushita T, Matsushita Y, Horikawa M, et al. Blockade of CD40/CD40 ligand interactions attenuates skin fibrosis and autoimmunity in the tight-skin mouse. Ann Rheumatic Dis (2008) 67:867–72. doi: 10.1136/ard.2007.073387
11. Komura K, Sato S, Hasegawa M, Fujimoto M, Takehara K. Elevated circulating CD40L concentrations in patients with systemic sclerosis. J Rheumatol (2004) 31:514–9.
12. Aslani S, Sobhani S, Gharibdoost F, Jamshidi A, Mahmoudi M. Epigenetics and pathogenesis of systemic sclerosis; the ins and outs. Hum Immunol (2018) 79:178–87. doi: 10.1016/j.humimm.2018.01.003
13. Walczyk M, Paradowska-Gorycka A, Olesinska M. Epigenetics: The future direction in systemic sclerosis. Scand J Immunol (2017) 86:427–35. doi: 10.1111/sji.12595
14. Wu H, Zhao M, Yoshimura A, Chang C, Lu Q. Critical link between epigenetics and transcription factors in the induction of autoimmunity: a comprehensive review. Clin Rev Allergy Immunol (2016) 50:333–44. doi: 10.1007/s12016-016-8534-y
15. Miao C, Xiong Y, Yu H, Zhang X, Qin M, Song T, et al. Critical roles of microRNAs in the pathogenesis of systemic sclerosis: New advances, challenges and potential directions. Int Immunopharmacol (2015) 28:626–33. doi: 10.1016/j.intimp.2015.07.042
16. Altorok N, Almeshal N, Wang Y, Kahaleh B. Epigenetics, the holy grail in the pathogenesis of systemic sclerosis. Rheumatology (2015) 54:1759–70. doi: 10.1093/rheumatology/keu155
17. Ciechomska M, van LJM, O’Reilly S. Emerging role of epigenetics in systemic sclerosis pathogenesis. Genes Immun (2014) 15:gene201444. doi: 10.1038/gene.2014.44
18. Luo Y, Wang Y, Wang Q, Xiao R, Lu Q. Systemic sclerosis: genetics and epigenetics. J Autoimmun (2013) 41:161–7. doi: 10.1016/j.jaut.2013.01.012
19. Wolffe AP, Matzke MA. Epigenetics: Regulation through repression. Science (1999) 286:481–6. doi: 10.1126/science.286.5439.481
20. Lafyatis R. Editorial: Epigenetics in systemic sclerosis. Arthritis Rheumatol (Hoboken NJ) (2016) 68:2841–4. doi: 10.1002/art.39830
21. Svegliati S, Marrone G, Pezone A, Spadoni T, Grieco A, Moroncini G, et al. Oxidative DNA damage induces the ATM-mediated transcriptional suppression of the wnt inhibitor WIF-1 in systemic sclerosis and fibrosis. Sci Signal (2014) 7:ra84. doi: 10.1126/scisignal.2004592
22. Honda N, Jinnin M, Kira-Etoh T, Makino K, Kajihara I, Makino T, et al. miR-150 down-regulation contributes to the constitutive type i collagen overexpression in scleroderma dermal fibroblasts via the induction of integrin β3. Am J Pathol (2013) 182:206–16. doi: 10.1016/j.ajpath.2012.09.023
23. Prey S, Ezzedine K, Doussau A, Grandoulier A-SS, Barcat D, Chatelus E, et al. Imatinib mesylate in scleroderma-associated diffuse skin fibrosis: a phase II multicentre randomized double-blinded controlled trial. Br J Dermatol (2012) 167:1138–44. doi: 10.1111/j.1365-2133.2012.11186.x
24. Bujor AM, Asano Y, Haines P, Lafyatis R, Trojanowska M. The c-abl tyrosine kinase controls protein kinase cδ–induced fli-1 phosphorylation in human dermal fibroblasts. Arthritis Rheumatism (2011) 63:1729–37. doi: 10.1002/art.30284
25. Soria A, Cario-Andre M, Lepreux S, Rezvani HR, Pasquet JM, Pain C, et al. The effect of imatinib (GlivecR) on scleroderma and normal dermal fibroblasts: A preclinical study. Dermatology (2008) 216:109–17. doi: 10.1159/000111507
26. Pannu J, Asano Y, Nakerakanti S, Smith E, Jablonska S, Blaszczyk M, et al. Smad1 pathway is activated in systemic sclerosis fibroblasts and is targeted by imatinib mesylate. Arthritis Rheumatism (2008) 58:2528–37. doi: 10.1002/art.23698
27. Xu GJ, Shah AA, Li MZ, Xu Q, Rosen A, Casciola-Rosen L, et al. Systematic autoantigen analysis identifies a distinct subtype of scleroderma with coincident cancer. Proc Natl Acad Sci (2016) 113:E7526–34. doi: 10.1073/pnas.1615990113
28. Wei J, Ghosh AK, Sargent JL, Komura K, Wu M, Huang Q-Q, et al. PPARγ downregulation by TGFs in fibroblast and impaired expression and function in systemic sclerosis: A novel mechanism for progressive fibrogenesis. PloS One (2010) 5:e13778. doi: 10.1371/journal.pone.0013778
29. Jimenez S, Freundlich B, Rosenbloom J. Selective inhibition of human diploid fibroblast collagen synthesis by interferons. J Clin Invest (1984) 74:1112–6. doi: 10.1172/jci111480
30. Ohta A, Uitto J. Procollagen gene expression by scleroderma fibroblasts in culture. inhibition of collagen production and reduction of proα(i) and proα1(III) collagen messenger RNA steady-state levels by retinoids. Arthritis Rheumatism (1987) 30:404–11. doi: 10.1002/art.1780300407
31. Pendergrass SA, Lemaire R, Francis IP, Mahoney JM, Lafyatis R, Whitfield ML. Intrinsic gene expression subsets of diffuse cutaneous systemic sclerosis are stable in serial skin biopsies. J Investig Dermatol (2012) 132:1363–73. doi: 10.1038/jid.2011.472
32. Lafyatis R, Mantero JC, Gordon J, Kishore N, Carns M, Dittrich H, et al. Inhibition of β-catenin signaling in the skin rescues cutaneous adipogenesis in systemic sclerosis: A randomized, double-blind, placebo-controlled trial of c-82. J Invest Dermatol (2017) 137:2473–83. doi: 10.1016/j.jid.2017.06.032
33. Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, et al. Recombinant human anti–transforming growth factor β1 antibody therapy in systemic sclerosis: A multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum (2007) 56:323–33. doi: 10.1002/art.22289
34. Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebocontrolled and ustekinumab-controlled phase 3 trials. Lancet (2018) 392:650–61. doi: 10.1016/s0140-6736(18)31713-6
35. Gordon KB, Foley P, Krueger JG, Pinter A, Reich K, Vender R, et al. Bimekizumab efficacy and safety in moderate to severe plaque psoriasis (BE READY): a multicentre, double-blind, placebo-controlled, randomised withdrawal phase 3 trial. Lancet (2021) 397:475–86. doi: 10.1016/s0140-6736(21)00126-4
36. Deodhar A, Gottlieb AB, Boehncke W-H, Dong B, Wang Y, Zhuang Y, et al. Efficacy and safety of guselkumab in patients with active psoriatic arthritis: a randomised, double-blind, placebo-controlled, phase 2 study. Lancet (2018) 391:2213–24. doi: 10.1016/s0140-6736(18)30952-8
37. Wermuth PJ, Piera-Velazquez S, Rosenbloom J, Jimenez SA. Existing and novel biomarkers for precision medicine in systemic sclerosis. Nat Rev Rheumatol (2018) 14:421–32. doi: 10.1038/s41584-018-0021-9
38. Olson MV. Precision medicine at the crossroads. Hum Genom (2017) 11:23. doi: 10.1186/s40246-017-0119-1
39. Snyder M, Weissman S, Gerstein M. Personal phenotypes to go with personal genomes. Mol Syst Biol (2009) 5:273. doi: 10.1038/msb.2009.32
40. Collins DC, Sundar R, Lim JSJ, Yap TA. Towards precision medicine in the clinic: From biomarker discovery to novel therapeutics. Trends Pharmacol Sci (2017) 38:25–40. doi: 10.1016/j.tips.2016.10.012
41. Castro SV, Jimenez SA. Biomarkers in systemic sclerosis. Biomark Med (2010) 4:133–47. doi: 10.2217/bmm.09.79
42. Steen VD, Powell DL, Medsger TA. Clinical correlations and prognosis based on serum autoantibodies in patients with systemic sclerosis. Arthritis Rheum (1988) 31:196–203. doi: 10.1002/art.1780310207
43. Smeets RL, Kersten BE, Joosten I, Kaffa C, Alkema W, Koenen HJPM, et al. Diagnostic profiles for precision medicine in systemic sclerosis; stepping forward from single biomarkers towards pathophysiological panels. Autoimmun Rev (2020) 19:102515. doi: 10.1016/j.autrev.2020.102515
44. Clark KEN, Campochiaro C, Csomor E, Taylor A, Nevin K, Galwey N, et al. Molecular basis for clinical diversity between autoantibody subsets in diffuse cutaneous systemic sclerosis. Ann Rheum Dis (2021) 80:1584–93. doi: 10.1136/annrheumdis-2021-220402
45. Kubo S, Nakayamada S, Miyazaki Y, Yoshikawa M, Yoshinari H, Satoh Y, et al. Distinctive association of peripheral immune cell phenotypes with capillaroscopic microvascular patterns in systemic sclerosis. Rheumatology (2019) 58:2273–83. doi: 10.1093/rheumatology/kez244
46. Maricq HR, Spencer-Green G, LeRoy EC. Skin capillary abnormalities as indicators of organ involvement in scleroderma (systemic sclerosis), raynaud’s syndrome and dermatomyositis. Am J Med (1976) 61:862–70. doi: 10.1016/0002-9343(76)90410-1
47. Taroni JN, Martyanov V, Mahoney JM, Whitfield ML. A functional genomic meta-analysis of clinical trials in systemic sclerosis: Toward precision medicine and combination therapy. J Invest Dermatol (2017) 137:1033–41. doi: 10.1016/j.jid.2016.12.007
48. Johnson ME, Mahoney JM, Taroni J, Sargent JL, Marmarelis E, Wu M-RR, et al. Experimentally-derived fibroblast gene signatures identify molecular pathways associated with distinct subsets of systemic sclerosis patients in three independent cohorts. PloS One (2015) 10:e0114017. doi: 10.1371/journal.pone.0114017
49. Chung L, Fiorentino DF, Benbarak MJ, Adler AS, Mariano MM, Paniagua RT, et al. Molecular framework for response to imatinib mesylate in systemic sclerosis. Arthritis Rheumatism (2009) 60:584–91. doi: 10.1002/art.24221
50. Stifano G, Sornasse T, Rice LM, Na L, Chen-Harris H, Khanna D, et al. Skin gene expression is prognostic for the trajectory of skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheumatol (Hoboken NJ) (2018) 70:912–9. doi: 10.1002/art.40455
51. Paleja B, Low AHL, Kumar P, Saidin S, Lajam A, Hazirah SN, et al. Systemic sclerosis perturbs the architecture of the immunome. Front Immunol (2020) 11:1602. doi: 10.3389/fimmu.2020.01602
52. Rosenbaum L. The future of gene editing — toward scientific and social consensus. N Engl J Med (2019) 380:971–5. doi: 10.1056/nejmms1817082
53. Baron M. Targeted therapy in systemic sclerosis. Rambam Maimonides Med J (2016) 7:e0030. doi: 10.5041/rmmj.10257
54. Campochiaro C, Allanore Y. An update on targeted therapies in systemic sclerosis based on a systematic review from the last 3 years. Arthritis Res Ther (2021) 23:155. doi: 10.1186/s13075-021-02536-5
55. Yanaba K, Bouaziz J, Matsushita T, Magro CM, St.Clair EW, Tedder TF. B-lymphocyte contributions to human autoimmune disease. Immunol Rev (2008) 223:284–99. doi: 10.1111/j.1600-065x.2008.00646.x
56. Famularo G, Giacomelli R, Alesse E, Cifone MG, Morrone S, Boirivant M, et al. Polyclonal b lymphocyte activation in progressive systemic sclerosis. J Clin Lab Immunol (1989) 29:59–63.
57. Sato S, Fujimoto M, Hasegawa M, Takehara K. Altered blood b lymphocyte homeostasis in systemic sclerosis: Expanded naive b cells and diminished but activated memory b cells. Arthritis Rheum (2004) 50:1918–27. doi: 10.1002/art.20274
58. Hamaguchi Y, Xiu Y, Komura K, Nimmerjahn F, Tedder TF. Antibody isotype-specific engagement of fcγ receptors regulates b lymphocyte depletion during CD20 immunotherapy. J Exp Med (2006) 203:743–53. doi: 10.1084/jem.20052283
59. Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, et al. The innate mononuclear phagocyte network depletes b lymphocytes through fc receptor–dependent mechanisms during AntiCD20 antibody immunotherapy. J Exp Med (2004) 199:1659–69. doi: 10.1084/jem.20040119
60. Beers SA, Chan CHT, French RR, Cragg MS, Glennie MJ. CD20 as a target for therapeutic type i and II monoclonal antibodies. Semin Hematol (2010) 47:107–14. doi: 10.1053/j.seminhematol.2010.01.001
61. Hasegawa M, Hamaguchi Y, Yanaba K, Bouaziz J-DD, Uchida J, Fujimoto M, et al. B-lymphocyte depletion reduces skin fibrosis and autoimmunity in the tight-skin mouse model for systemic sclerosis. Am J Pathol (2006) 169:954–66. doi: 10.2353/ajpath.2006.060205
62. Daoussis D, Liossis S-NC, Tsamandas AC, Kalogeropoulou C, Kazantzi A, Korfiatis P, et al. Is there a role for b-cell depletion as therapy for scleroderma? a case report and review of the literature. Semin Arthritis Rheum (2010) 40:127–36. doi: 10.1016/j.semarthrit.2009.09.003
63. Sumida H, Asano Y, Tamaki Z, Aozasa N, Taniguchi T, Takahashi T, et al. Successful experience of rituximab therapy for systemic sclerosis-associated interstitial lung disease with concomitant systemic lupus erythematosus. J Dermatol (2014) 41:418–20. doi: 10.1111/1346-8138.12461
64. Bosello SL, Luca GD, Rucco M, Berardi G, Falcione M, Danza FM, et al. Long-term efficacy of b cell depletion therapy on lung and skin involvement in diffuse systemic sclerosis. Semin Arthritis Rheum (2015) 44:428–36. doi: 10.1016/j.semarthrit.2014.09.002
65. Smith V, Piette Y, Praet JTV, Decuman S, Deschepper E, Elewaut D, et al. Two-year results of an open pilot study of a 2-treatment course with rituximab in patients with early systemic sclerosis with diffuse skin involvement. J Rheumatol (2013) 40:52–7. doi: 10.3899/jrheum.120778
66. Jordan S, Distler JHW, Maurer B, Huscher D, van LJM, Allanore Y, et al. Effects and safety of rituximab in systemic sclerosis: an analysis from the european scleroderma trial and research (EUSTAR) group. Ann Rheum Dis (2015) 74:1188. doi: 10.1136/annrheumdis-2013-204522
67. Ebata S, Yoshizaki A, Oba K, Kashiwabara K, Ueda K, Uemura Y, et al. Safety and efficacy of rituximab in systemic sclerosis (DESIRES): a double-blind, investigator-initiated, randomised, placebo-controlled trial. Lancet Rheumatol (2021) 3:e489–97. doi: 10.1016/s2665-9913(21)00107-7
68. Daoussis D, Liossis S-NC, Tsamandas AC, Kalogeropoulou C, Kazantzi A, Sirinian C, et al. Experience with rituximab in scleroderma: results from a 1-year, proof-of-principle study. Rheumatology (2010) 49:271–80. doi: 10.1093/rheumatology/kep093
69. Ebata S, Yoshizaki A, Oba K, Kashiwabara K, Ueda K, Uemura Y, et al. Safety and efficacy of rituximab in systemic sclerosis (DESIRES): open-label extension of a double-blind, investigators-initiated, randomised, placebo-controlled trial. Lancet Rheumatol (2022) 4:e546–55. doi: 10.1016/s2665-9913(22)00131-x
70. Frampton JE. Inebilizumab: First approval. Drugs (2020) 80:1259–64. doi: 10.1007/s40265-020-01370-4
71. Paul F, Marignier R, Palace J, Arrambide G, Asgari N, Bennett JL, et al. International delphi consensus on the management of AQP4-IgG+ NMOSD. Neurol Neuroimmunol Neuroinflamm (2023) 10:e200124. doi: 10.1212/nxi.0000000000200124
72. Chen D, Ireland SJ, Davis LS, Kong X, Stowe AM, Wang Y, et al. Autoreactive CD19+CD20– plasma cells contribute to disease severity of experimental autoimmune encephalomyelitis. J Immunol (2016) 196:1541–9. doi: 10.4049/jimmunol.1501376
73. Bergmann C, Muller F, Distler JHW, Gyorfi A-H, Volkl S, Aigner M, et al. Treatment of a patient with severe systemic sclerosis (SSc) using CD19-targeted CAR t cells. Ann Rheum Dis (2023) 82:1117–20. doi: 10.1136/ard-2023-223952
74. Bournia V-KK, Evangelou K, Sfikakis PP. Therapeutic inhibition of tyrosine kinases in systemic sclerosis: a review of published experience on the first 108 patients treated with imatinib. Semin Arthritis Rheumatism (2013) 42:377–90. doi: 10.1016/j.semarthrit.2012.06.001
75. Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, et al. Imatinib mesylate inhibits the profibrogenic activity of TGFbeta and prevents bleomycin-mediated lung fibrosis. J Clin Invest (2004) 114:1308–16. doi: 10.1172/jci19603
76. Distler JHH, Jungel A, Huber LC, Schulze-Horsel U, Zwerina J, Gay RE, et al. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheumatism (2007) 56:311–22. doi: 10.1002/art.22314
77. Akhmetshina A, Venalis P, Dees C, Busch N, Zwerina J, Schett G, et al. Treatment with imatinib prevents fibrosis in different preclinical models of systemic sclerosis and induces regression of established fibrosis. Arthritis Rheumatism (2009) 60:219–24. doi: 10.1002/art.24186
78. Khanna D, Saggar R, Mayes MD, Abtin F, Clements PJ, Maranian P, et al. A one-year, phase I/IIa, openlabel pilot trial of imatinib mesylate in the treatment of systemic sclerosis-associated active interstitial lung disease. Arthritis Rheumatism (2011) 63:3540–6. doi: 10.1002/art.30548
79. Fraticelli P, Gabrielli B, Pomponio G, Valentini G, Bosello S, Riboldi P, et al. Low-dose oral imatinib in the treatment of systemic sclerosis interstitial lung disease unresponsive to cyclophosphamide: a phase II pilot study. Arthritis Res Ther (2014) 16:R144. doi: 10.1186/ar4606
80. Haddon DJ, Wand HE, Jarrell JA, Spiera RF, Utz PJ, Gordon JK, et al. Proteomic analysis of sera from individuals with diffuse cutaneous systemic sclerosis reveals a multianalyte signature associated with clinical improvement during imatinib mesylate treatment. J Rheumatol (2017) 44:631–8. doi: 10.3899/jrheum.160833
81. Khanna D, Denton CP, Lin CJFJ, van LJM, TM F, ME A, et al. Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: results from the open-label period of a phase II randomised controlled trial (faSScinate). Ann Rheumatic Dis (2018) 77:212–20. doi: 10.1136/annrheumdis-2017-211682
82. Yanaba K. Strategy for treatment of fibrosis in systemic sclerosis: Present and future. J Dermatol (2016) 43:46–55. doi: 10.1111/1346-8138.13026
83. Spiera RF, Gordon JK, Mersten JN, Magro CM, Mehta M, Wildman HF, et al. Imatinib mesylate (Gleevec) in the treatment of diffuse cutaneous systemic sclerosis: results of a 1-year, phase IIa, single-arm, open-label clinical trial. Ann Rheum Dis (2011) 70:1003–9. doi: 10.1136/ard.2010.143974
84. Zakostelska ZJ, Reiss Z, Tlaskalova-Hogenova H, Rob F. Paradoxical reactions to anti-TNFα and anti-IL-17 treatment in psoriasis patients: Are skin and/or gut microbiota involved? Dermatol Ther (2023) 13:911–33. doi: 10.1007/s13555-023-00904-4
85. Assassi S, Li N, Volkmann ER, Mayes MD, Runger D, Ying J, et al. Predictive significance of serum interferon-inducible protein score for response to treatment in systemic sclerosis–related interstitial lung disease. Arthritis Rheumatol (2021) 73:1005–13. doi: 10.1002/art.41627
86. Hinchcliff M, Huang C-C, Wood TA, Mahoney JM, Martyanov V, Bhattacharyya S, et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol (2013) 133:1979–89. doi: 10.1038/jid.2013.130
87. Ebata S, Oba K, Kashiwabara K, Ueda K, Uemura Y, Watadani T, et al. Predictors of rituximab effect on modified rodnan skin score in systemic sclerosis: a machine-learning analysis of the DesiReS trial. Rheumatology (2022) 61:4364–73. doi: 10.1093/rheumatology/keac023
88. Park Y, Freedman BD, Lee EJ, Park S, Jameson JL. A dominant negative PPARγ mutant shows altered cofactor recruitment and inhibits adipogenesis in 3T3-L1 cells. Diabetologia (2003) 46:365–77. doi: 10.1007/s00125-003-1037-4
89. Wei J, Zhu H, Komura K, Lord G, Tomcik M, Wang W, et al. A synthetic PPAR-γ agonist triterpenoid ameliorates experimental fibrosis: PPAR-γ-independent suppression of fibrotic responses. Ann Rheum Dis (2014) 73:446. doi: 10.1136/annrheumdis-2012-202716
90. Komura K, Varga J. Precision medicine for patients with systemic sclerosis. J Scleroderma Related Disord (2016) 1.
Keywords: systemic sclerosis, fibrosis, precision medicine, biomarkers, therapy
Citation: Komura K, Yanaba K, Bouaziz J-D, Yoshizaki A, Hasegawa M, Varga J, Takehara K and Matsushita T (2024) Perspective to precision medicine in scleroderma. Front. Immunol. 14:1298665. doi: 10.3389/fimmu.2023.1298665
Received: 22 September 2023; Accepted: 26 December 2023;
Published: 18 January 2024.
Edited by:
Soheil Tavakolpour, Dana–Farber Cancer Institute, United StatesReviewed by:
Minghua Wu, University of Texas Health Science Center at Houston, United StatesCopyright © 2024 Komura, Yanaba, Bouaziz, Yoshizaki, Hasegawa, Varga, Takehara and Matsushita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuhiro Komura, a29tdXJha2FAbWUuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.