 Yawen Xie
Yawen Xie Guoyu Zhao1
Guoyu Zhao1 Xianli Lei
Xianli Lei Na Cui
Na Cui Hao Wang
Hao Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 December 2023
Sec. Cytokines and Soluble Mediators in Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1297408
The mammalian target of rapamycin (mTOR), an evolutionarily highly conserved serine/threonine protein kinase, plays a prominent role in controlling gene expression, metabolism, and cell death. Programmed cell death (PCD) is indispensable for maintaining homeostasis by removing senescent, defective, or malignant cells. Necroptosis, a type of PCD, relies on the interplay between receptor-interacting serine-threonine kinases (RIPKs) and the membrane perforation by mixed lineage kinase domain-like protein (MLKL), which is distinguished from apoptosis. With the development of necroptosis-regulating mechanisms, the importance of mTOR in the complex network of intersecting signaling pathways that govern the process has become more evident. mTOR is directly responsible for the regulation of RIPKs. Autophagy is an indirect mechanism by which mTOR regulates the removal and interaction of RIPKs. Another necroptosis trigger is reactive oxygen species (ROS) produced by oxidative stress; mTOR regulates necroptosis by exploiting ROS. Considering the intricacy of the signal network, it is reasonable to assume that mTOR exerts a bifacial effect on necroptosis. However, additional research is necessary to elucidate the underlying mechanisms. In this review, we summarized the mechanisms underlying mTOR activation and necroptosis and highlighted the signaling pathway through which mTOR regulates necroptosis. The development of therapeutic targets for various diseases has been greatly advanced by the expanding knowledge of how mTOR regulates necroptosis.
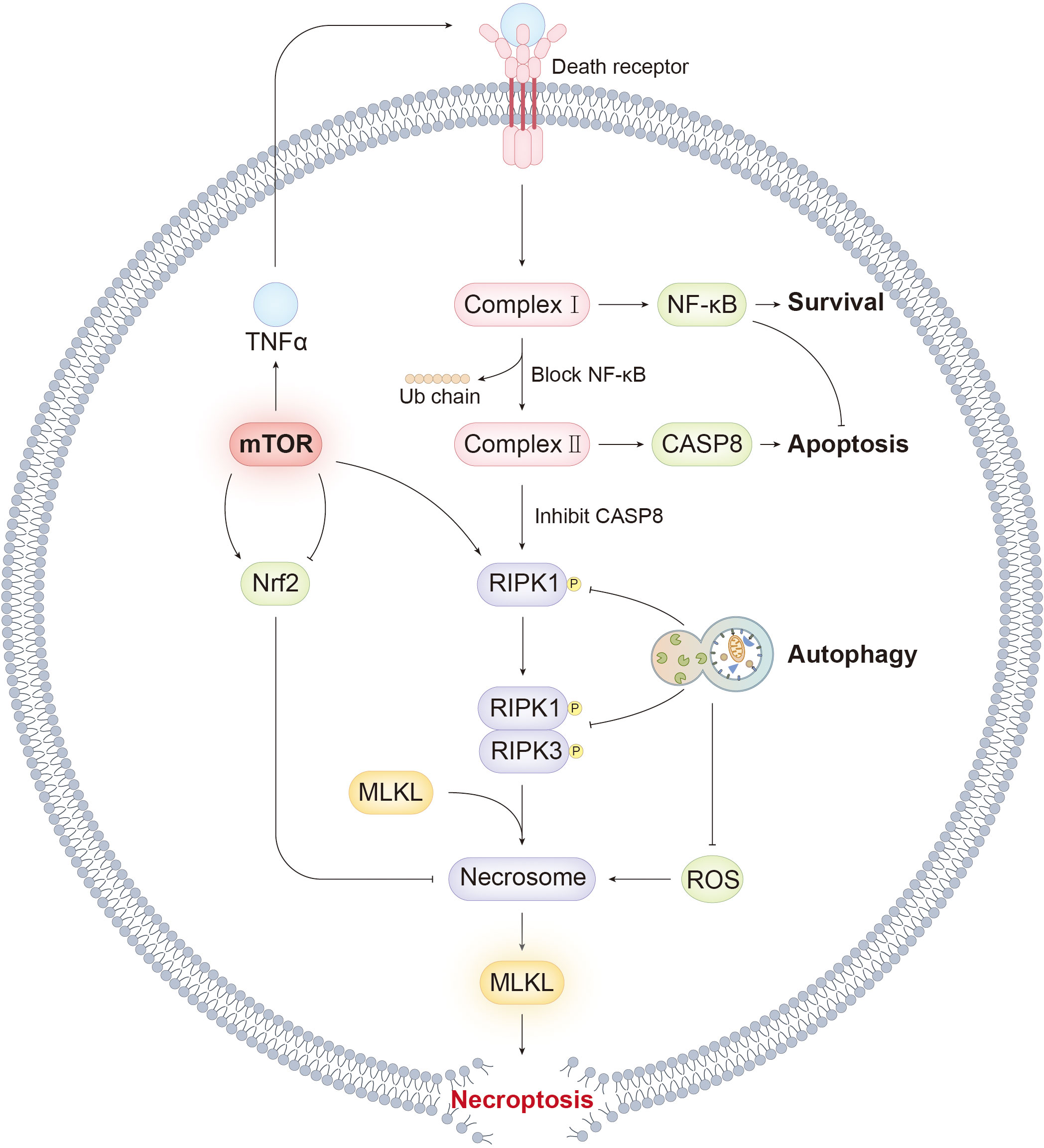
Graphical Abstract Our review summarized the molecular mechanism of necroptosis. mTOR, as a key hub for cell fate, regulates necroptosis by various way. The graphical abstract illustrates the association between mTOR and necroptosis and highlights key signaling events responsible for the transition from apoptosis and autophagy to necroptosis.
The equilibrium between cell proliferation and death is fundamental for maintaining normal physiological functions. Cell demise facilitates the removal of aging, injured, or dysplastic cells that harm the body’s homeostasis. Hence, vigilant monitoring and elimination of the pernicious cells are essential for development, resistance to various pathogens, and homeostasis maintenance (1). Apoptosis was the first discovered programmed cell death (PCD), whereas necrosis was considered for an extended time to be a typical unprogrammed cell death with no signaling pathway involved. Hirsch et al. found that inhibiting apoptosis may initiate a transition from apoptosis to necrosis, a pivotal milestone in identifying regulated necrotic cell death (2). An increasing number of types of regulated necrosis have been recognized, such as ferroptosis, pyroptosis, and others, among which necroptosis has been investigated most thoroughly (3–5). Necroptosis is characterized by the formation of a necrosome consisting of RIPKs and the activation of MLKL with similar morphological changes to necrosis, including cellular swelling, impairment of plasma membrane integrity, and release of intracellular substances, leading to an inflammatory response (6–8).
Investigating the underlying mechanisms of necroptosis regulation yields potentially effective therapeutic targets. The mammalian target of rapamycin (mTOR), an evolutionally conserved serine/threonine protein kinase, serves as a hub for catabolism and cell growth and plays an essential role in the complex signaling network consisting of numerous regulatory pathways associated with cell death (9). mTOR is involved in the assembly of two different complexes, mTORC1 and mTORC2, by collaborating with other subunits. Given the relatively limited understanding of mTORC2, this review primarily focused on the role of mTORC1. mTORC1 has been identified as an autophagy inhibitor, and increasing evidence indicates that it also regulates necroptosis precisely, thereby identifying new potential therapeutic targets for various disorders (10, 11).
Our objective is to provide a comprehensive literature on the regulatory mechanisms of mTOR in necroptosis by updating a synopsis of recent advances in this area and highlighting clinical applications in necroptosis-related diseases.
mTOR, an evolutionally conserved kinase, is pivotal in regulating cellular metabolism, growth, and death (9). It was first confirmed 30 years ago by gene screening in budding yeasts and subsequently in mammalian cells (12–16). The mTOR complex and relevant upstream and downstream pathways are discussed in this section (Figure 1).
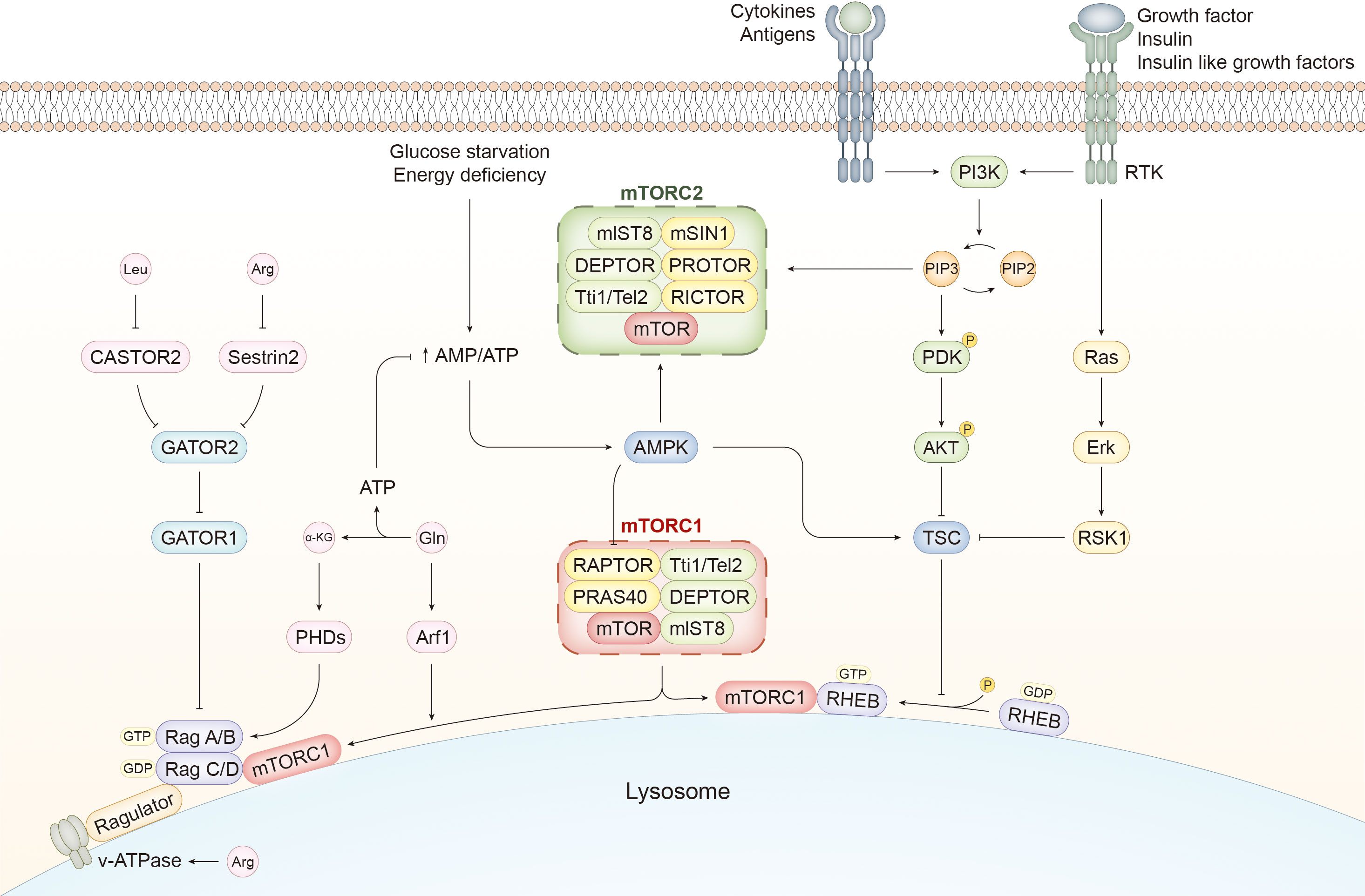
Figure 1 mTORC pathways and activation mechanisms of mTORC1. The components of mTORC1 are slightly different from those of mTORC2. RAPTOR and PRAS40 are unique to mTORC1, whereas mTORC2 contains mSIN1, PROTOR, and RICTOR. Several external stimuli, such as immune signals and growth factors, activate PI3K, which catalyzes the transversion of PIP2 to PIP3. The latter can induce the activation of PDK and AKT by phosphorylation. PIP3 can also activate mTORC2. AKT is a potent stimulator of mTORC1 activity by inhibiting TSC, a GTPase-activating protein. mTORC1 is recruited to the lysosome and activated by RHEB with GTP binding, whereas TSC facilitates the transition from GTP to GDP and suppresses RHEB-mediated mTORC1 activation. In addition to the PI3K/AKT/mTORC1 pathway, growth factors can activate the Ras/Erk/RSK1 pathway to ameliorate the negative effect of TSC on mTORC1 activity. Cellular energetic status is another determiner of mTORC1 activity. AMPK can detect the increasing ratio of AMP to ATP caused by glucose deprivation and inhibit mTORC1 directly and indirectly by phosphorylating RAPTOR and activating TSC, respectively. However, AMPK serves as an activator of mTORC2. In addition to RHEB, mTORC1 is recruited and activated by the Ragulator-Rag complex induced by amino acids. Rag A/B and Rag C/D bound to GTP and GDP, respectively, facilitate the translocation of mTORC1 to the lysosome and activate it with the assistance of Ragulator. CASTOR1 and Sestrin2 sense the alterations in the content of Leucine (Leu) and Arginine (Arg) and mediate the mTORC1-activating signals by suppressing GATOR2, which mitigates the negative effect of GATOR1 on Rag heterodimer. Changes in Arg in the lumen of lysosomes are sensed by v-ATPase, which activates mTORC1 through the Ragulator-Rag pathway. Glutamine (Gln) is converted to α-KG during glutaminolysis, which generates ATP to inhibit AMPK and activate mTORC1. α-KG regulates the nucleotide-binding state of Rag B via PHDs, and Gln enhances the translocation of mTORC1 to the lysosome via Arf1. PI3K, phosphoinositide 3 kinase; RTK, receptor tyrosine kinase; PIP3, phosphatidylinositol 3,4,5 trisphosphate; PDK, phosphoinositide-dependent kinase 1; mTOR, mammalian target of rapamycin; RAPTOR, regulatory-associated proteins of mTOR; PRAS40, 40-kDa pro-rich Akt-substrate; RICTOR, Rapamycin-insensitive companion of mTOR; mSIN1, mammalian stressed-activated map-kinase interacting protein1; PROTOR, protein observed with RICTOR; AMPK, adenosine 5′-monophosphate-activated protein kinase; TSC, tuberous sclerosis complex1; RHEB, Ras homolog enriched in brain; α-KG, α-ketoglutarate; PHD, prolyl hydroxylases; Arf1, ADP-ribosylation factor-1; Leu, leucine; Arg, arginine; Gln, glutamine.
mTOR attaches itself to different components to form mTORC1 and mTORC2, both of which share the same catalytic subunit mTOR and other elements, including mlST8, DEPTOR, and Tti1/Tel2. Regulatory-associated proteins of mTOR (RAPTOR) and the 40-kDa pro-rich Akt-substrate (PRAS40) are the kernels of mTORC1. Rapamycin-insensitive companion of mTOR (RICTOR), mammalian stressed-activated map-kinase interacting protein1 (mSIN1), and protein observed with RICTOR (PROTOR) are exclusive to mTORC2 (17).
Rapamycin, an allosteric inhibitor of mTOR, forms a complex with FK506 binding protein 12 (FKBP12), which can bind to mTOR and hinder the recruitment of the substrate, thereby blocking the interaction between the substrate and the active site of mTOR (18, 19). mTORC1 and mTORC2 have divergent susceptibilities to rapamycin because of the difference between RAPTOR and RICTOR. Rapamycin significantly suppresses mTORC1 but exhibits a relatively mild inhibitory effect on mTORC2, which may be apparent in cell lines or tissues after prolonged treatment (20–22).
Environmental stimuli, immune signals, and metabolic status are the primary upstream signals that activate mTOR (23). Various pathways are associated with mTORC1 activation, such as phosphoinositide 3-kinase (PI3K)/AKT, adenosine 5′-monophosphate-activated protein kinase (AMPK), tuberous sclerosis complex1 (TSC1)/TSC2/Ras homolog enriched in brain (RHEB), VAM6/Rag GTPase, and others (24). RHEB is a GTPase embedded in the lysosome membrane whose activity depends on the type of nucleotide it binds to. mTORC1 can be recruited to the lysosome and activated directly through GTP attachment to RHEB (25, 26).
Environmental stimuli, specifically signals from growth factors, insulin, and insulin-like growth factors, are efficient activators of mTORC1, predominantly mediated by the PI3K/AKT pathway (27). Receptor tyrosine kinases (RTKs) stimulated by growth factors activate PI3K to enable the production of phosphatidylinositol 3,4,5 trisphosphate (PIP3), which transfers the activating signals to phosphoinositide-dependent kinase 1 (PDK1) and AKT (28, 29). TSC acts as a GTPase-activating protein by promoting the conversion of GTP to GDP by irritating the GTPase activity of RHEB to alter its nucleotide binding state to the inactive mode and indirectly inhibits mTORC1 (30). In brief, TSC is suppressed by the PI3K/AKT pathway to facilitate the positive effect of RHEB on mTORC1. Furthermore, downstream of RTKs, the Ras/Erk/p90 RSK1 axis acts as both a direct and indirect activator of mTORC1 by inhibiting TSC and phosphorylating RAPTOR, respectively (31–34).
Antigens, cytokines, and various other immune signals are the main stimulators of mTORC1 activation (35, 36). For example, a toll-like receptor (TLR) is triggered by its legend and conveys the activating signals to mTORC1 through the PI3K/AKT pathway (37, 38).
Considering the critical role of mTOR in metabolism, changes in energy and nutrient abundance are expected to regulate mTORC1. Under the circumstances of glucose starvation or energy depletion, the ratio of AMP to ATP increases due to a decline in ATP, facilitating AMPK activation. AMPK can enhance the inhibitory effect of TSC on mTORC1 by phosphorylating TSC (39). Moreover, it has been shown that mTORC1 is inhibited directly by AMPK by phosphorylating RAPTOR (40).
In addition to glucose, intracellular amino acid levels activate mTORC1, which is mediated by the RAS-related GTP binding protein (RAG)/Ragulator pathway. The heterodimer consisting of RAG A/B and RAG C/D can be ligated to the lysosome with Ragulator as a bridge and transformed to its active state with GTP and GDP attached to RAG A/B and RAG C/D, respectively (41, 42). The RAG heterodimer promotes the transposition of mTORC1 to lysosomes and activates it, whereas GATOR1 suppresses mTORC1 activation by inhibiting RAG A/B (42, 43). GATOR2 can counteract the negative effect of GATOR1 on mTORC1 (43). Sestrin2, when bound to leucine, is deprived of the ability to restrain GATOR2 and mediates the effect of leucine on mTORC1 activation, which gives rise to the interaction between GATOR1 and GATOR2 to activate mTORC1 (44–46). CASTOR1 can detect intracellular arginine, mitigating its suppressive role in GATOR2 to activate mTORC1 indirectly (47). The arginine inside the lysosome activates mTORC1 by interacting with the RAG-Ragulator complex; v-ATPase assists in perceiving changes in arginine levels (48–50). Glutamine activates mTORC1 in both a RAG-dependent and -independent manner. Glutamine is decomposed during glutaminolysis, which yields α-ketoglutarate (α-KG), enabling loading of GTP to RAG B by prolyl hydroxylases (PHDs) (51). The breakdown process also contributes to ATP generation and serves as a bioenergetic resource to ameliorate AMPK-induced mTORC1 inhibition (52, 53). Increasing evidence has substantiated that mTORC1 is activated by glutamine independently of RAG through ADP-ribosylation factor-1 (Arf1), which promotes the recruitment and translocation of mTORC1 to lysosomes (51, 54, 55).
Regarding the mechanisms underlying the activation of mTORC2, it has been well established that PI3K functions upstream of mTORC2 via PIP3, which binds to mSin1 and reverses its inhibitory effect on mTOR to activate it (56). Furthermore, PI3K strengthens the interactions between mTORC2 and the ribosome, which serves as the site of mTORC2 activation (57). In contrast to mTORC1, mTORC2 is activated by AMPK with glucose withdrawal (58). However, more in-depth research on mTORC2 activation is in high demand.
As a key regulator of cellular metabolism and growth, mTORC1 can promote anabolism and inhibit catabolism by controlling the synthesis of proteins, nucleotides, and lipids (59). This part of the review focuses on regulating protein synthesis by mTORC1.
p70-S6 kinase 1 (p70-S6K1) and 4E binding protein 1 (4E-BP1) are the main substrates of mTORC1 and are pivotal to translation. mTORC1 activates p70S6K through phosphorylation, which increases gene expression and protein synthesis by activating various substrates implicated in mRNA maturation and translation. 4E-BP1 inhibits protein production by binding to eukaryotic initiation factor 4E (eIF4E) to block transcription. 4EBP phosphorylated by mTORC1 results in the loss of its affinity for eIF4E, thus reversing its negative effect on transcription and protein synthesis (60–62).
Some substrates of mTORC2 have been identified, including AKT, serum and glucocorticoid-induced protein kinase 1 (SGK1), and protein kinase C-α (PKC-α) (63–65). AKT functions upstream of NF-κB, Bad, and FOXO to regulate cell death. SGK1 and PKC-α activated by mTORC2 are associated with cytoskeleton remodeling and migration (66–68).
A necrosome composed of RIPKs is required for necroptosis, with activated MLKL serving as the executor of membrane perforation. RIPK1 comprises the N-terminal kinase, intermediate domain, and C-terminal death domain (DD). Several regulatory sites exist in the intermediate domain, such as ubiquitination sites and RIP homotypic interaction motifs (RHIM) (69, 70). RIPK3 and RIPK1 share a similar molecular structure; however, the C-terminal domain of RIPK3 does not encompass DD, which differs from RIPK1 (71). MLKL activated by RIPK3 is integral for the terminal stage of necroptosis. MLKL can form an amyloid-like oligomer that translocates to the plasma membrane and mediates membrane perforation (72, 73).
Necroptosis is initiated by binding the corresponding ligand to the death receptor. Death receptors, belonging to the TNFR (TNF receptor) superfamily, receive and transduce several extracellular signals, including FasL, TNFα, TNF-related apoptosis-inducing ligand (TRAIL), and so on, among which the TNFα-induced mechanism has been studied most thoroughly (74, 75). In addition to death receptors, pattern-recognition receptors (PRRs), such as TLR3 and TLR4, contribute to necroptosis induction by recognizing relevant pathogen-associated molecular patterns (PAMPs) (76, 77). This section provides an overview of the mechanisms of necroptosis induced by various signals (Figure 2).
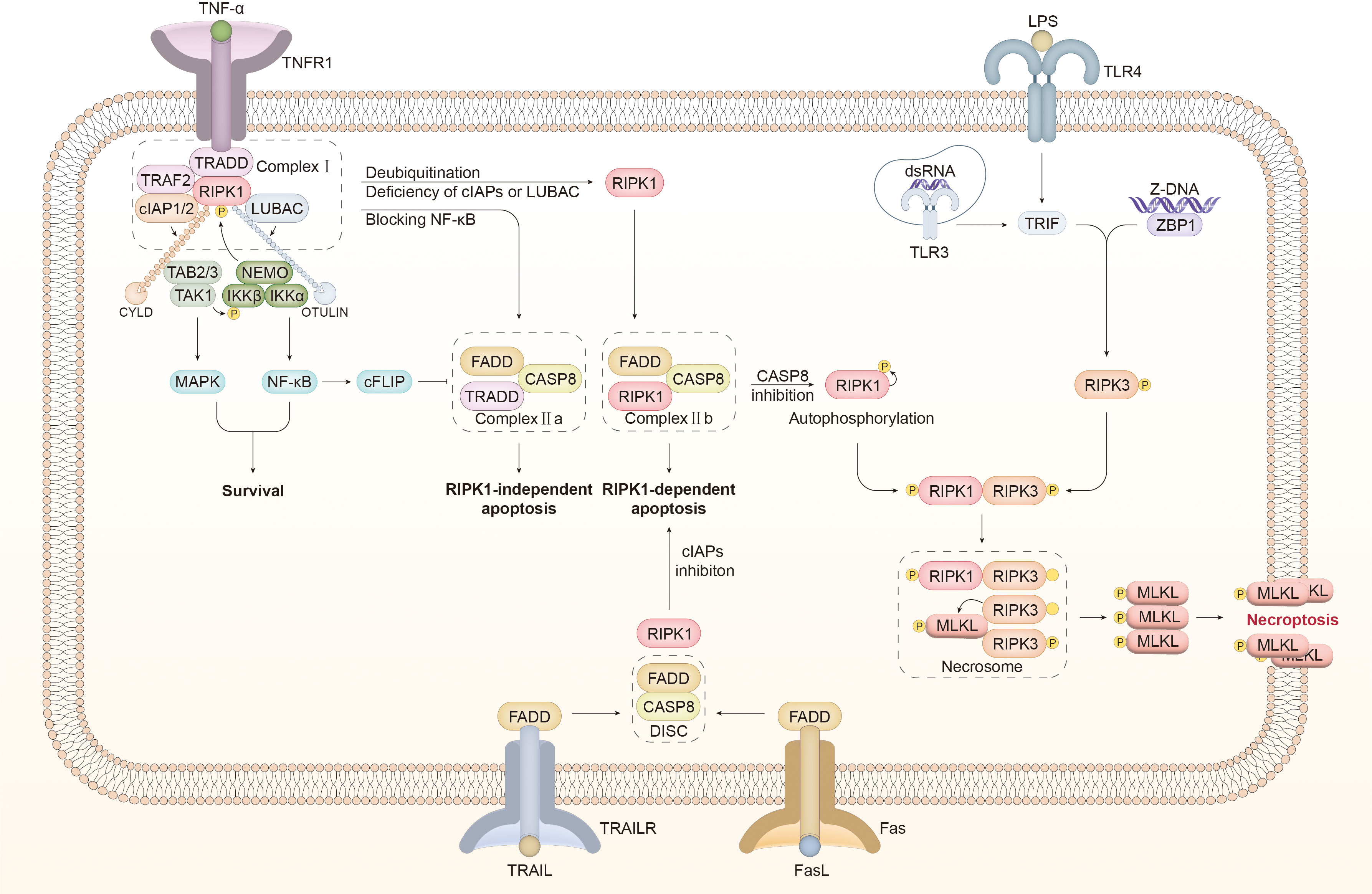
Figure 2 Molecular mechanisms of necroptosis A wide range of signals act as an inducer of necroptosis. TNFR1 bound with TNFα promotes the recruitment of TRADD to assembly complex I, which comprises TRAF2, RIPK1, cIAP1/2, and LUBAC. cIAP1/2 and LUBAC catalyze the connection of K63- and M1-linked ubiquitin chains to RIPK1, which provides a platform for the recruitment of TAK1, TAB2/3, and the IKK complex consisting of NEMO, IKKα, and IKKβ. The kinases mentioned above can inhibit the activity of RIPK1 through phosphorylation to counteract the shift from complex I to complex II. In addition, MAPK and NF-κB signaling pathways are activated downstream of the kinases mentioned above to induce survival. NF-κB facilitates the synthesis of cFLIP, which inhibits the activity of caspase 8 (CASP8). Upon blocking NF-κB, TRADD assembles with FADD and CASP8 to form complex IIa, which mediates RIPK1-independent apoptosis. In certain cases, including cIAP1/2 or LUBAC inhibition and CYLD-mediated RIPK1 deubiquitination, RIPK1 is dissociated from complex I to form complex IIb with FADD and CASP8, inducing RIPK1-dependent apoptosis. Inactivating CASP8 is a prerequisite for the detachment of RIPK1 from complex IIb. RIPK1 is supposed to undergo conformational changes through autophosphorylation and interact with RIPK3 through the RHIM domain. Phosphorylated RIPK3 is oligomerized to recruit and activate MLKL, leading to the assembly of the necrosome. The activated MLKL oligomer is translocated to the cell membrane, mediating membrane perforation and necroptosis. In addition to TNFα, the connection of FasL to Fas and TRAIL to TRAILR transfers a pro-necroptotic signal. DISC comprises FADD and CASP8 to induce apoptosis. Under cIAP1/2 inhibition, DISC associates with RIPK1 to form complex IIb, which can transfer to the necrosome with CASP8 inhibition. TLR3/4 recruits RHIM-containing TRIF in the intracellular domain after dsRNA and LPS are identified, to directly activate the RIPK3/MLKL pathway and thereby trigger necroptosis. ZBP connected to Z-DNA interacts with RIPK3 through the RHIM domain to stimulate downstream MLKL and induce necroptosis. TNF-α, tumor necrosis factor-α; TNFR1, tumor necrosis factor receptor; TRADD, TNF receptor type 1 associated death domain; TRAF2, TNF receptor associated factor 2; RIPK, receptor-interacting serine-threonine kinase; LUBAC, linear ubiquitin chain assembly complex; cIAP1/2, cellular inhibitors of apoptosis 1/2; TAK1, transforming growth factor-β-activated kinase1; TAB2, TAK1-binding protein2; IKKα/β, IKK complex constituted of IκB kinase α/β; NEMO, NF-κB essential modulator; MAPK, mitogen-activated protein kinase; CYLD, Cylindromatosis; OTULIN, OTU deubiquitinase with linear linkage specificity; cFLIP, cellular FLICE-like inhibitory protein; CASP, caspase; FADD, Fas associated death domain; DISC, death-inducing signaling complex; MLKL, mixed lineage kinase domain-like protein; TLR, toll-like receptor; TRIF, Toll/IL-1 receptor domain-containing adaptor protein inducing interferon-β; dsRNA, double-stranded RNA; LPS, lipopolysaccharide; TRAIL, TNF-related apoptosis-inducing ligand.
The binding of TNFα to TNFR1 recruits downstream molecules to DD located in the intracellular region of TNFR1, such as TNF receptor type 1 associated death domain (TRADD), TNF receptor-associated factor 2 (TRAF2), and RIPK1. Cellular inhibitors of apoptosis 1/2 (cIAP1/2) are connected to TRADD through TRAF2 (78–80). The binding of TNFR1, TRADD, RIPK1, TRAF2, cIAP1/2, and the linear ubiquitin chain assembly complex (LUBAC) promotes the formation of complex I, resulting in multiple outcomes, including survival, apoptosis, and necroptosis (81, 82).
Complex I-induced cellular survival requires the involvement of ubiquitin ligase. E3 ubiquitin ligases comprising RING E3s, HECT E3s, and RBR E3s are highly substrate-specific. The orientation of the substrate is determined by the conformational flexibility of the substate-binding subunit of E3 ubiquitin ligases (83, 84). cIAP1/2, a homodimeric RING E3 ubiquitin ligase, enables the ligation of the K63-linked ubiquitin chain to RIPK1 on Lys377 (78, 84). LUBAC, another linear ubiquitin E3 ligase, interacts with the K63-linked ubiquitin chain on cIAP1/2 to attach the M1-linked ubiquitin chain to RIPK1 (85, 86). Ubiquitination of RIPK1 catalyzed by cIAP1/2 and LUBAC is propitious for recruiting an array of kinases, including transforming growth factor-β-activated kinase1 (TAK1), TAK1-binding protein2 (TAB2), TAB3, and IKK complex constituted of IκB kinase α (IKKα), IKKβ, and NF-κB essential modulator (NEMO). TAK1 phosphorylates IKKβ to enhance the IKK complex-mediated inhibitory effect on RIPK1 by exerting its kinase activity, which blocks the assembly of complex II and relieves its cytotoxicity. Furthermore, the aforementioned kinases stimulate the NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways, which are both beneficial to cell survival (86–88).
TNFα-induced apoptosis is mediated by complexes IIa and IIb. In addition to activating NF-κB and MAPK pathways, the polyubiquitin chain also interferes with the interplay of RIPK1 and RIPK3 to suppress the transformation from complex I to complex II (89, 90). The deubiquitinating enzyme catalyzes the hydrolysis by which the ubiquitin chain is cleaved from the ubiquitinated protein (91). Cylindromatosis (CYLD), a lysine 63 deubiquitinase, boosts the elimination of the K63-linked ubiquitin chain from RIPK1 and interrupts the NF-κB pathway. OTU deubiquitinase with linear linkage specificity (OTULIN) is another deubiquitinase that exhibits a specific property to degrade the M1-linked ubiquitin chain (92, 93). OTULIN positively affects LUBAC by inhibiting its auto-ubiquitination, and OTULIN deficiency leads to TNFα-induced apoptosis and necroptosis (94, 95). Complex I is destabilized by deubiquitinated RIPK1 or the absence of cIAP1/2, thereby facilitating the transition to complex IIb (96, 97). Cellular FLICE-like inhibitory protein (cFLIP) is structurally analogous to caspase 8 but has no catalytic activity; therefore, it acts as a caspase 8 inhibitor (98). NF-κB activated by complex I can promote the production of cFLIP to inhibit apoptosis (79). Inhibiting the NF-κB pathway or protein synthesis will lead to cFLIP deficiency, which will facilitate the transformation from complex I to complex IIa, induce apoptosis via complete activation of caspase 8, and suppress necroptosis through the cleavage of RIPKs (79, 99). Complex IIa consists of FADD, TRADD, and caspase 8, whereas complex IIb consists of FADD, RIPK1, and caspase 8 (100). When cFLIP production is blocked by NF-κB inhibition, complex IIa leads to RIPK1-independent apoptosis, and RIPK1 phosphorylation on S321 catalyzed by the TAK1 fan facilitates this process. In contrast, RIPK1, with dephosphorylated S321, participates in the assembly of complex IIb (101). DD is essential for RIPK1 dimerization and activation during the transformation process from complex I to complex IIb, contributing to caspase 8 activation by RIPK1 through its kinase domain, thereby inducing RIPK1-dependent apoptosis (101, 102).
The onset of necroptosis is dependent on the formation of complex IIc. Complex II c, also known as necrosome, comprises MLKL, RIPK1, and RIPK3 (100, 103). TNFα combines with TNFR1 to initiate the assembly of complex I. RIPK1 is deubiquitinated by CYLD or is incapable of undergoing ubiquitination with pharmacological or genetic inhibition of cIAP1/2. Both of these conditions are favorable for NF-κB pathway suppression and the transformation from complex I to complex IIa and IIb, which impedes the generation of cFLIP and facilitates apoptosis, respectively. Caspase 8 inactivation or RIPK3 overexpression prevents apoptosis to promote the formation of necrosomes (103). The assembly of necrosomes requires the kinase activity of RIPK1, which is inhibited by necrostatin-1; this inhibitory effect is specific to necroptosis (104, 105). RIPK1 has multiple autophosphorylation sites, among which the N-terminal S161 site is proposed to induce a conformational alteration of RIPK1, leading to the exposure of the RHIM domain and RIPK1 activation (104, 106). Phosphorylated RIPK1 interacts with RIPK3 through the RHIM domain to activate RIPK3 and facilitate the assembly of the RIPK3 oligomer (107). Phosphorylation of Ser 232 of RIPK3 benefits the recruitment of MLKL (108). By phosphorylating MLKL at its Thr357/Ser358 site, RIPK3 facilitates the formation of MLKL oligomer and conformational changes that are essential for its translocation to the membrane and subsequent perforation, which in turn triggers necroptosis (109, 110).
Upon receiving the signal of FasL in conjunction with Fas, FADD is recruited to the intracellular domain of Fas, where it acts as an adaptor for caspase 8 to promote the assembly of death-inducing signaling complex (DISC) and induce extrinsic apoptosis (100). In the absence of cIAPs, RIPK1 is involved in the assembly of complex IIb, which can be converted into a necrosome upon caspase 8 inactivation (100, 111). TRAIL mediates caspase8-dependent apoptosis by recruiting FADD and necroptosis when cIAPs and capsase8 are inhibited, which is similar to FasL (112, 113).
Mounting evidence suggests that reactive oxygen species (ROS) are an important mediator of necroptosis (106, 114). RIPKs are regulated by ROS, which is considered an upstream pro-necroptotic molecule. ROS induces RIPK1 autophosphorylation on S161, which is essential for RIPK1 activation. Autophosphorylated activated RIPK1 is required for RIPK1-RIPK3 co-localization and subsequent necrosome assembly (106). The findings of Bin LU et al. suggested that ROS improves the expression levels of RIPK1 and RIPK3 in glioma cells (115). ROS enhances RIPK1-RIPK3 interaction and necrosome stability, thereby facilitating TNFα-induced necroptosis (115, 116). On the other hand, the production of ROS is also instigated by RIPK3 by activating vital metabolic enzymes (117, 118). Zhentao Yang et al. revealed that RIPK3 boosts aerobic respiration by targeting the pyruvate dehydrogenase complex, which accounts for the increased mitochondrial ROS (mtROS) induction in TNFα-triggered necroptosis (119). The aforementioned findings indicate the existence of a positive feedback regulation loop between RIPKs and ROS during necroptosis. Intriguingly, a recent study by Chi G. Weindel et al. further revealed that mtROS released from gasdermin D (GSDMD)-induced mitochondrial perforation is a potent driver of necroptosis through RIPK3 and that RIPK3 also enhances mtROS production, which indicates that mtROS mediates the pyroptosis-to-necroptosis switch (114).
However, there is a controversy concerning the role of mtROS in necroptosis. Sudan He et al. observed that ROS quenching exerted opposing effects on TNFα-induced necroptosis in L929 and HT-29 cells and concluded that the role of ROS in necroptosis is dependent on cell type (120). Mitochondria depletion blocked necroptosis-related mtROS production but failed to influence TNFα-induced necroptosis dependent on RIPK3, which contradicted the hypothesis that mtROS is a necroptotic trigger (121). Therefore, it is imperative to illustrate the relationship between ROS and RIPKs in necroptosis in various cells and their underlying mechanisms.
TLR is a main category of PRRs that induce necroptosis when stimulated by pathogen-associated molecular patterns (PAMPs). TLR3 and TLR4 are recognition molecules of viral dsRNA and lipopolysaccharide (LPS), respectively, functioning as a platform to recruit Toll/IL-1 receptor domain-containing adaptor protein inducing interferon-β (TRIF), which contains the RHIM domain, to interact with RIPK3 and induces necrosome-mediated necroptosis (77).
In addition to TLR, Z-DNA binding protein1 (ZBP1) is an RHIM-containing PRR capable of inducing necroptosis. Viral Z-DNA transmits an activation signal to ZBP1, which can directly activate RIPK3 and its downstream effector MLKL in an RIPK1-independent manner. It has been revealed that the mutation in the RHIM domain of RIPK1 results in increased perinatal mortality in RIPK1RHIM/RHIM mice. However, this effect could be reversed by knocking out RIPK3, MLKL, or ZBP1 (122, 123). This finding indicates that RIPK1 inhibits ZBP1-induced necroptosis through the RHIM domain, likely due to the competitive interplay between RIPK1 and ZBP1 with RIPK3.
More novel types of cell death have been discovered gradually, including apoptosis, necroptosis, pyroptosis, ferroptosis, and so on. It is pertinent to emphasize the conditions under which favor necroptosis.
Pan-caspase inhibitor Z-VAD-FMK ignited the switch from apoptosis to necroptosis (2). More initiators of necroptosis have been discovered, which partially overlap with those of extrinsic apoptosis and can be roughly categorized into two groups. One group is dominated by death receptors, including TNFR1, Fas, and TRAILR (72, 124, 125). The other one consists of the receptors which contain the RHIM domain and activate RIPK3 directly (76, 77, 126).
Activated death receptors induce the constitution of complex I (TNFα-TNFR1 and TRAIL-TRAILR) or DISC (FasL-Fas), which triggers extrinsic apoptosis and necroptosis (79). Complex I transfers a pro-survival signal through NF-κB (88). When NF-κB is blocked or RIPK1 is deubiquitinated, complex I shifts to complex II, causing extrinsic apoptosis (127, 128). Inactivating caspase-8 can tip the balance in favor of necroptosis through RIPK1-RIPK3 interaction and necrosome assembly (82, 129). In addition, some RHIM-containing factors activate RIPK3 directly. TLR3 and TLR4 recruit TRIF to activate RIPK3 through the RHIM domain. ZBP1 activated by Z-DNA is another mediator of necroptosis through direct interplay with RIPK3 (130, 131).
Membrane shrinkage and blebbing, chromatin condensation, and DNA fragmentation are typical morphological apoptotic characteristics (132). Apoptotic bodies prevent the inflammatory effect, which is different from necroptosis.
The cascade caspase activation is indispensable for apoptosis. Initiating signals of extrinsic apoptosis are shared with those of necroptosis. The crosstalk between extrinsic apoptosis and necroptosis induced by death receptors has been summarized before. Intrinsic apoptosis is caused by the imbalance of pro-apoptotic BH3-only proteins and anti-apoptotic Bcl-2 proteins, leading to mitochondrial outer membrane permeabilization (MOMP) (133, 134).
The executors of apoptosis and necroptosis are different as well. Necrosome activates and promotes MLKL oligomerization for membrane translocation and perforation (105, 109, 118). Apoptosis is reliant on caspase (135). In intrinsic apoptosis, the substances released from MOMP activate caspase-9 and subsequent caspase-3 and -7 (136). Caspase-8 activated in extrinsic apoptosis not only contributes to the caspase-3 and -7 but also triggers MOMP by cleaving BH3-only proteins (137). Caspase-3 and -7 activate the downstream DNase or undermine the electron transport chain to implement apoptosis (138, 139).
Similar to necroptosis, pyroptosis is another necrotic PCD featured by Gasdermin-mediated membrane perforation and release of proinflammatory cytokines. The inflammasome is the key for pyroptosis, rather than necrosome in necroptosis.
Pyroptosis is reliant on caspases to facilitate the cleavage of pyroptotic executors, which is different from necroptosis. PRRs activate NF-κB to promote the transcription of pyroptosis-related molecules, including NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3), pro-IL-18, and pro-IL-1β (127, 140). NLRP3 activating signals include K+ efflux, Ca2+ mobilization, lysosomal enzymes, mitochondrial DNA, and ROS (141–145). NLRP3 inflammasome cleaves GSDMD, pro-IL-18, and pro-IL-1β through caspase-1 to induce pyroptosis in the canonical inflammasome pathway (146). Caspase-4/5/11 is activated by LPS to cleave GSDMD and mediate non-canonical pyroptosis (147–149).
The cleavage product, GSDMD-N, is the pyroptosis executor by perforating membrane (150–152). Pro-IL-18 and pro-IL-1β are cleaved and released through the membrane pore (153). Despite that necroptosis and pyroptosis are both characterized by membrane perforation, the mechanisms are distinct and mediated by MLKL and GSDMD respectively.
Ferroptosis is a type of programmed necrosis driven by lipid peroxidation characterized by the involvement of iron overload. The imbalance of oxidation and anti-oxidation systems is the foundation of ferroptosis.
Firstly, the engagement of iron is an apparent difference between ferroptosis and necroptosis. Fe3+ combined with the transferrin is conjugated to the transferrin receptor and internalized to be reduced to Fe2+ (154, 155). Fe2+ can replenish the labile iron pool to participate in lipid peroxidation by iron-dependent Fenton reaction or bind to ferritin to prevent oxidative cytotoxicity (156). Secondly, the antioxidant system is another integral part of ferroptosis but seems to be less essential in necroptosis. System Xc-glutathione (GSH)-GPX4 axis is the predominant antioxidant system for inhibiting ferroptosis (157, 158). What’s more, it is suspected that lipid peroxidation leads to the ultimate cell death but the inner mechanisms have not been elucidated yet (4, 156). Compared to ferroptosis, necroptosis-mediated cellular damage is more highly MLKL-dependent and less reliant on lipid peroxidation.
Autophagy plays an integral role in the regulation of necroptosis. mTORC1 has been universally recognized as an autophagy inhibitor through various mechanisms, such as phosphorylating unc-51-like kinase 1 (ULK1), interrupting transcription factor EB (TFEB) translocation to nuclear, and interfering with lysosome reformation (159, 160). Thus, autophagy serves as a bridge for mTOR to regulate necroptosis (Figure 3).
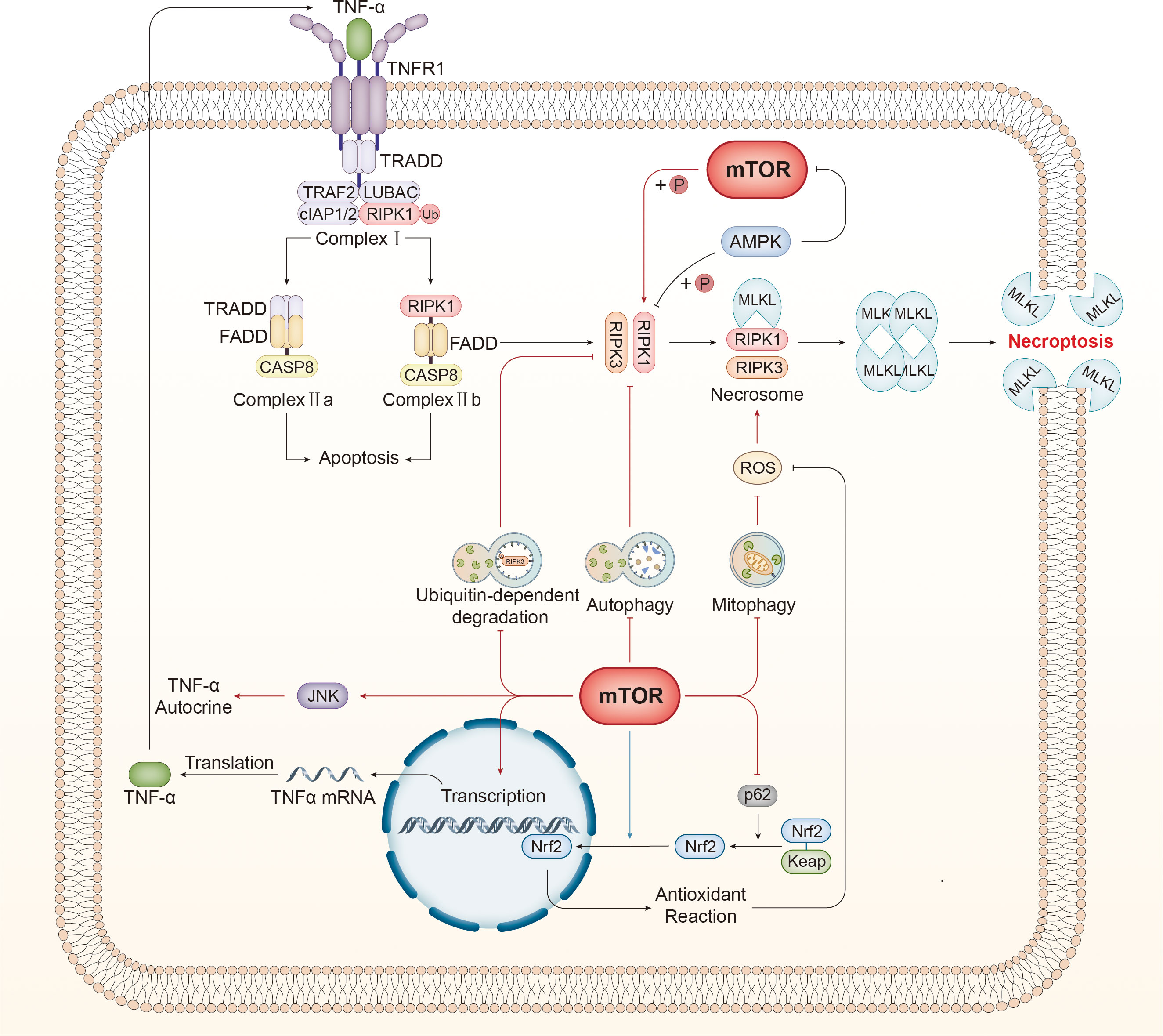
Figure 3 mTOR regulates necroptosis via various mechanisms. mTOR inhibits autophagy, which suppresses necroptosis by disturbing the interaction between RIPK1 and RIPK3. Autophagy specifically recognizes and degrades ubiquitinated RIPK3 and negatively affects necroptosis. Furthermore, damaged mitochondria are the primary source of ROS, which can activate RIPK1 directly and promote the interaction between RIPK1 and RIPK3, facilitating the assembly of necrosomes. Mitophagy restrains ROS production by eliminating injured mitochondria. Furthermore, RIPK1 can be activated by mTOR directly through phosphorylation. mTOR can elevate the level of TNRα mRNA and promote the autocrine effect of TNFα by activating the JNK pathway, both of which can boost necroptosis. Tipping the balance of the redox reaction is another approach for mTOR to regulate necroptosis. p62 assists with the release and stabilization of Nrf2 by counteracting Keap1, and the released Nrf2 translocates to the nucleus and governs the expression of genes associated with antioxidant defense to antagonize necroptosis. mTOR displays bidirectional effects on this process. On the one hand, mTOR inhibits p62 to promote necroptosis, thereby suppressing the antioxidant reaction. In contrast, mTOR promotes nuclear translocation to inhibit necroptosis. TNF-α, tumor necrosis factor-α; TNFR1, tumor necrosis factor receptor; TRADD, TNF receptor type 1 associated death domain; TRAF2, TNF receptor associated factor 2; RIPK, receptor-interacting serine-threonine kinase; LUBAC, linear ubiquitin chain assembly complex; cIAP1/2, cellular inhibitors of apoptosis 1/2; TAK1, transforming growth factor-β-activated kinase1; TAB2, TAK1-binding protein2; IKKα/β, IKK complex constituted of IκB kinase α/β; NEMO, NF-κB essential modulator; MAPK, mitogen-activated protein kinase; CYLD, Cylindromatosis; OTULIN, OTU deubiquitinase with linear linkage specificity; cFLIP, cellular FLICE-like inhibitory protein; CASP, caspase; FADD, Fas associated death domain; DISC, death-inducing signaling complex; MLKL, mixed lineage kinase domain-like protein; TLR, toll-like receptor; TRIF, Toll/IL-1 receptor domain-containing adaptor protein inducing interferon-β; dsRNA, double-stranded RNA; LPS, lipopolysaccharide; ROS, reactive oxygen species; Nrf2, nuclear factor-erythroid 2-related factor 2; Keap1, Kelch-like ECH-associated protein 1.
Autophagy disrupts the interaction between RIPK1 and RIPK3 to inhibit necroptosis. Di Ge et al. verified that 11-methoxytabersonine (11-MT) induced necroptosis in A549 and H157 cell lines, and autophagy was also initiated by activating the AMPK/mTORC1 pathway (161). 3-methyladenine (3-MA) and chloroquine (CQ), both of which inhibit autophagy, were utilized to investigate the relationship between autophagy and necroptosis in detail. The combined administration of 11-MT and autophagy inhibitors may intensify the interaction between RIPK1 and RIPK, thereby accelerating necroptosis, which confirmed that mTORC1 promoted necroptosis by downregulating autophagy (161).
Autophagy also has a prominent influence on the levels of cellular RIPKs to inhibit necroptosis. RIPK1 and MLKL are suppressed by increased autophagy resulting from inactivated PI3K/AKT/mTOR/p70S6K pathway (162). AMPK-mTOR is a potential target for treating spinal cord injury (SCI) (163). Inactive mTOR dephosphorylates TFEB to promote the expression of autophagy-related genes and elevated autophagic flux results in the decline of RIPKs and MLKL, which inhibits necroptosis and facilitates the neurofunction restoration (163). However, another study about SCI yielded the opposite conclusion that autophagy may deteriorate the necroptosis-mediated injury because rapamycin antagonizes the edaravone-induced downregulation of phosphorylated necroptotic proteins through autophagy (164). The discrepancy probably derives from different cell types because neurons are the focus in the former, while human brain microvascular endothelial cells are targeted in the latter. Therefore, the effect of autophagy on necroptosis seems to be context-dependent and cell-type specific.
mTORC1 regulates ubiquitin-dependent RIPK3 degradation through autophagy. Yadong Xie and his colleagues found that mTOR hyperactivated by the Western diet or TSC1 knockout caused substantial RIPK3 accumulation through the blockade of autophagy in intestinal epithelial cells, and p62 was involved in the autophagic degradation of RIPK3 as a specific recognizer for ubiquitinated cargo (11). TRIM11, an E3 ubiquitin ligase, catalyzes polyubiquitination of RIPK3 for autophagic degradation, which was abrogated by mTORC1 overexpression (11). Consequently, this study revealed two mechanisms by which mTOR upregulates necroptosis in an autophagy-dependent manner. Kevin Bray et al. (165) also verified that mTOR inhibits the autophagic degradation of ubiquitinated RIPK3 and interferes with TRIM11 to hinder RIPK3 ubiquitination modification to protect RIPK3 from p62-mediated autophagic degradation.
Mitophagy, another vital subtype of autophagy, has been demonstrated to be involved in necroptosis. Especially the damaged mitochondria are the main site of ROS production, which is closely associated with necroptosis. Mitophagy can eliminate injured or fragmentized mitochondria to restrain the production of ROS, thereby preventing necroptosis. Bo Xu et al. revealed that exosomes produced from Schwann cells induced AMPK-mediated mitophagy to subdue the generation of ROS and alleviate necroptosis in PC12 cells but failed to explore the role of mTORC1 in the mechanisms related to mitophagy and necroptosis despite the close relationship between AMPK and mTORC1 (166). Mitochondrial permeability transition pore opening, ROS production, and mitochondrial fission are considered mediators of necroptosis evidence by phosphorylated MLKL locating mitochondrial fractions, which can be alleviated by rapamycin through mitophagy-dependent removal of impaired mitochondria (167, 168). A recent study found that a triggering receptor expressed on myeloid cells-1 (TREM-1) induced necroptosis in macrophages by activating mTOR. The study further concluded that mTOR could elevate the expression levels of mitochondrial fission process protein 1 (MTFP) and phosphatase phosphoglycerate mutase family member 5 (PGAM5) to activate dynamin-related protein 1 (DRP1)-mediated mitochondria fission and subsequent mitophagy (169). Despite the limited evidence for this recently identified pathway, it remains a plausible mechanism by which mTOR regulates necroptosis in a mitophagy-dependent manner.
Nevertheless, there is more than one signaling pathway that connects mitophagy and necroptosis, and mitophagy may potentially serve a pro-necroptotic effect via certain mechanisms rather than an anti-necroptotic role. For instance, Sorafenib has been found to induce mitophagy through the mTORC1-TFEB pathway, and mitophagy-mediated MFN degradation contributed to the increased expression of MAM constituents and induced excessive contact between mitochondria and the ER, an intracellular calcium pool. The process mentioned above resulted in Ca2+ flow from the ER to the mitochondria, and this Ca2+ overload triggered necroptosis (170).
The key molecules that play a crucial role in necroptosis are directly regulated by mTOR (Figure 3). The kinase activity of mTORC1 is effectively utilized to phosphorylate RIPK1, activating RIPK1 and subsequent regulation of necroptosis. Abe et al. observed that rapamycin, an mTOR inhibitor, exhibited a protective effect on cardiomyocytes against necroptosis induced by TNFα/z-VAD-fmk (zVAD) (171). Subsequent investigations have indicated that mTORC1 selectively phosphorylates different RIPK1 sites (e.g., Ser166 and Ser320) and that phosphorylation of these two sites is positively and negatively correlated with RIPK1 activity, respectively (171). Rapamycin promoted the phosphorylation at Ser320 and decreased phosphorylation at Ser166, thereby impairing the activity of RIPK1. Given that Ser320 is required for the reciprocal interaction between RIPK1 and RIPK3, phosphorylation of Ser320 induced by mTOR inhibitor hindered the RIPK1-RIPK3 connection (171). Consequently, mTORC1 facilitates necroptosis by regulating RIPK1 activity and the interaction between RIPK1 and RIPK3 through phosphorylation.
As AMPK has a well-established role as a mTORC1 inhibitor, RIPK1 phosphorylation by AMPK has been increasingly recognized. Tao Zhang et al. uncovered that AMPK stimulated by glucose deprivation restricts RIPK1 activation by phosphorylating RIPK1 S415, thereby inhibiting necroptosis during short-term energy stress (172). Prolonged 2-DG treatment to mimic long-term glucose starvation leads to a decrease and an increase in p-RIPK1 at Ser415 and Ser166, respectively, which implies that AMPK mediates the conversion from cellular protection to necroptosis by regulating RIPK1 phosphorylation at different sites in the context of glucose deficiency (172). However, another study reported that activated AMPK inhibits necroptosis through the enhancement of pro-caspase 8 stability, rather than regulating the level of p-RIPK1 in acute pancreatitis mice models (173). The divergent conclusions mentioned above can be attributed to the different activators of AMPK and the cell types. Different metabolic status leads to diverse types of cell death in different cells. Consequently, it is necessary and meaningful to investigate the relationship between metabolic status and necroptosis and the connections between AMPK and RIPK1 in necroptosis.
It is widely recognized that TNFα is a crucial signal that initiates necroptosis. Consequently, mTOR also targets necroptosis to modulate it. It was demonstrated that TNFα/z-VAD-fmk (zVAD)-induced necroptosis was significantly suppressed by a PI3K/mTOR inhibitor or miRNA-mediated mTOR gene silencing (174). An increase in TNFα mRNA was detected in the cells undergoing necroptosis, suggesting that necroptosis was accompanied by a significant rise in TNFα synthesis and secretion. To elucidate the inherent relationship between mTOR and TNFα, researchers used a combination of PI3K/mTORC1 inhibitor and miRNA knockdown of mTOR or AKT. Their findings revealed that TNFα mRNA was significantly decreased in the absence of mTOR, which indicated that PI3K/AKT/mTORC1 could upregulate the synthesis of TNFα (174). c-Jun N-terminal kinase (JNK) is involved in TNFα synthesis and autocrine and is a downstream effector of mTOR to regulate necroptosis (175–177). McNamara et al. found that the level of active JNK relevant to necroptosis increased in cells where AKT/mTOR was suppressed by siRNA knockdown or inhibitors. This finding suggested that TNFα autocrine was promoted through AKT/mTOR/JNK to exacerbate the necroptosis and establish a positive feedback loop (174). In addition to TNFα, it is noteworthy that TRAIL, another necroptosis inducer, may be associated with TSC2 and mTOR. A recent study revealed that TSC2 facilitates the mTORC2-skewed profile in malignant cells to defend against the attack from cytotoxic T cells. TSC2 ablation leads to mTORC1 overactivation and increased expression of TRAIL receptor to increase the susceptibility to necroptosis, indicating the close relationship between TSC2-mTOR and TRAIL signaling and their pivotal role in necroptosis (178).
Moreover, it’s conceivable that mTORC1 is also regulated by necroptosis-related molecules. Rune Busk Damgaard et al. used hepatocyte-specific knockout of OTULIN (OtulinΔhep) mice to establish that the lack of OTULIN led to mTORC1-associated steatohepatitis, hepatic fibrosis, and cancer (179). Abnormal mTORC1 activation was indicated in OtulinΔhep mice, and the administration of the mTORC1 inhibitor Rapamycin alleviated the liver injury and pathology caused by OTULIN insufficiency, which might be associated with TSC and RHEB. However, the intrinsic mechanism by which OTULIN regulates mTORC1 and its contribution to necroptosis still requires further research.
ROS is regarded as a signal of necroptosis (180–182). Mechanically, RIPK1 autophosphorylation and activation are promoted by ROS, which facilitates the recruitment of RIPK3 and the assembly of necrosomes. Thus, oxidative stress connects mTOR to necroptosis through ROS (Figure 3).
mTOR regulates the generation of ROS. Q Liu et al. revealed that the pharmacological or genetic blockade of AKT/mTOR undermined the production of ROS without affecting the interaction between RIPK1 and RIPK3 in HT22 cells treated with TNFα/zVAD to alleviate necroptosis, indicating that AKT/mTOR functioned downstream of RIPK1 but upstream of ROS generation (183). Forkhead box subclass O (FOXO) transcription factors, which govern antioxidant defenses, were substantiated to be involved in the regulatory mechanism of AKT/mTOR regarding redox reaction and oxidative stress (183, 184). FOXO phosphorylated by AKT is deprived of pro-transcription capacity and is removed from the nucleus when the 14-3-3 protein binds to it (185). Sustained AKT/mTOR inhibition relieved the phosphorylation suppression of FOXO and facilitated antioxidant response to downregulate necroptosis (183). Since AKT has been widely acknowledged as an upstream inhibitor of FOXO, mTOR, a downstream molecule of AKT, still warrants further exploration for its relationship with FOXO and potential regulatory mechanism.
Nuclear factor-erythroid 2-related factor 2 (Nrf2) is another target of mTOR to regulate oxidative stress. Similar to FOXO, Nrf2 is another transcription factor that controls the expression of antioxidant genes. Kelch-like ECH-associated protein 1 (Keap1), a ubiquitin ligase complex, binds to Nrf2 to facilitate its degradation by ubiquitination. p62 has an affinity for Keap1 and is therefore considered a guardian of Nrf2. Keap1 is inhibited upon binding to p62, facilitating the release and stabilization of Nrf2 for translocation to the nucleus and subsequent antioxidant response (186, 187). It has been revealed that the mTOR inhibitor CCI-779 facilitated the dissociation of Nrf2 from Keap1 by upregulating p62 to enhance the antioxidant reaction but blocked the nuclear translocation of Nrf2 to prohibit antioxidant defense (165).
Intriguingly, the pro-necroptotic and anti-necroptotic roles of mTOR were well illustrated in this study (165). In contrast to the views of the majority of the studies above, this study concluded that the mTOR inhibitor-induced necroptosis through different mechanisms. However, it is not contradictory because the regulatory network between mTOR and necroptosis is complicated by the intersection of numerous signaling pathways. The positive or negative regulatory effect of mTOR on necroptosis is determined by the predominant mechanism, which may be influenced by various factors, including cell type and stimulus. Kevin Bray et al. demonstrated that the effect of mTOR inhibitors blocking the nuclear translocation of Nrf2 was more significant than inducing mitophagy to eliminate ROS and relieving Nrf2 from Keap1, which was ultimately manifested as the promotional role of mTOR inhibitors on necroptosis (165).
Necroptosis considered a proinflammatory cell death, has been authenticated to be associated with a variety of diseases. We will discuss potential implications in immune-related disorders and infectious diseases by mTOR through necroptosis regulation, such as autoimmune disease, sepsis and related complications, graft rejection, and infection (131, 188–190).
The AKT/mTOR pathway promotes TNFα-induced necroptosis by increasing TNFα production and activating JNK, both of which facilitate TNFα autocrine and deteriorate the damage caused by necroptosis (174). The finding that mTOR regulates necroptosis through TNFα implies that the blockade of the signaling pathway could be a potential direction in suppressing pathologic inflammation and consequently alleviate the inflammatory injury, which is particularly beneficial in treating acute pancreatitis, bacterial infections and systemic inflammatory response syndrome (SIRS). Zhong et al. identified a novel mechanism by which mTORC1 promoted DRP1-mediated mitochondrial fission through downstream molecules, PGAM5 and MTFP, thereby aggravating the inflammatory damage induced by necroptosis in alveolar macrophages treated with LPS, indicating a new therapeutic target for LPS-triggered acute lung injury (169).
The pathogenesis of IBDs, including Crohn’s disease and ulcerative colitis, is related to the necroptosis of intestinal epithelial cells, a process in which mTOR has been identified as a participant. Xie et al. revealed that mTOR overactivation promoted necroptosis of epithelial cells to damage the intestinal barrier and induce intestinal inflammation and inflammation-related cancer (11). Mechanically, mTOR inhibited autophagic degradation of RIPK3 by downregulating RIPK3 ubiquitination to promote necroptosis, which provided more precise therapeutic directions for IBDs (11). Moreover, the role of necroptosis in other autoimmune diseases has been validated, including autoimmune hepatitis, lupus nephritis, and autoimmune lymphoproliferative syndrome (191–194). However, investigations into the potential therapeutic value of mTOR in the aforementioned autoimmune disorders through necroptosis are lacking.
It has been demonstrated that mTOR plays a role in immune defense against pathogens by regulating necroptosis, which provides a possible target for treating infectious diseases. Macrophages infected with Candida albicans, a common fungal pathogen, undergo MLKL-mediated necroptosis considerably. mTORC1 contributes to the activation of RIPK1, RIPK3, and MLKL and specific knockout of Tsc1 in macrophage/neutrophil gives rise to the prominent increase in fungal burden and cell death, suggesting the potential of TSC1-mTORC1 in defending pathogen infections (195). Moreover, AKT/mTOR is inhibited by ursolic acid to promote autophagy and suppress the necroptosis of macrophages, enhancing the anti-infection effect against Mycobacterium tuberculosis (196). Nevertheless, further studies are required to validate the role of mTOR-regulated necroptosis in other pathogen infections, such as SARS-CoV-2.
In terms of graft rejection, substantial evidence proves the function of necroptosis in organ transplantation (190, 197–199). However, only one study observed that the cardiac allograft models survived longer with the treatment of mTOR inhibitor sirolimus, which may be associated with necroptosis (190). Therefore, more research is in demand to explore whether and how mTOR is engaged in regulating necroptosis in graft rejection.
The regulatory mechanism of mTOR on necroptosis benefits the treatment of various tumors. It has been verified that auranofin can induce necroptosis by inhibiting the PI3K/AKT/mTOR pathway to restrain the exacerbation of non-small cell lung cancer (NSCLC); however, additional research is needed to elucidate the underlying mechanism (200). In lung cancer, the combination administration of 11-MT and an autophagy inhibitor exhibited more efficient therapeutic benefits compared to 11-MT used alone. This can be attributed to the finding that the AMPK/mTOR pathway inhibited the interaction between RIPK1 and RIPK3 via autophagy, suppressing necroptosis (161). Moreover, the therapeutic value of regulating necroptosis via mTOR has also been confirmed in schwannoma. Necroptosis of schwannoma cells is induced by LiCl through the AKT-mTOR-p70S6K axis to restrain the deterioration of schwannoma (201).
Regarding urinary tumors, the role of mTOR in regulating necroptosis has been gradually realized. Jin et al. used GNE-493 to reveal a new therapeutic target for prostate cancer by blocking the PI3K-AKT-mTOR pathway and inducing ROS production which brought about oxidative damage and necroptosis in prostate cancer cells (202). In renal cell carcinoma, an mTOR inhibitor has been demonstrated to promote autophagy-mediated RIPK clearance to inhibit necroptosis, but it also exerts a positive role in necroptosis by suppressing Nrf2 nuclear translocation and consequent antioxidant defense. Therefore, autophagy inhibitors are expected to enhance the efficacy of mTOR inhibitors in treating renal cell cancer (165).
There are numerous poisonous agents. For instance, Sorafenib is a targeted drug for hepatocellular carcinoma but is restricted in its clinical applicability due to its myocardial toxicity to some extent (203, 204). mTOR can be targeted to protect the myocardium from Sorafenib, which was validated to degrade MFN2 through mTOR-regulated mitophagy, disrupting the balance of mitochondrial Ca2+ and inducing necroptosis (170). Therefore, mTOR represents a potentially effective treatment for myocardial damage induced by Sorafenib. Additionally, saturated fatty acids can trigger necroptosis in cardiomyocytes to provoke oxidative damage (205). Mingyue Zhao et al. found that the AKT/mTOR signaling pathway activated by palmitic acid elevated the levels of RIPK1 and RIPK to induce the necroptosis of cardiomyocytes, which is likely to induce myocardial hypertrophy. They concluded that mTOR inhibition could reverse necroptosis triggered by increased saturated fatty acid to treat myocardial hypertrophy (206).
Despite most studies holding the view that mTOR is indispensable in promoting necroptosis, a few studies have reached the opposite conclusion. Intake of the plastic degradation product microplastic (MP) and plastic additive Di (2-ethylhexyl) phthalate (DEHP) can elicit the toxicity of skeletal muscle. Liu et al. investigated its potential mechanisms and proposed that DEHP/MP triggered necroptosis by inhibiting the PI3K/AKT/mTOR signaling pathway and stimulating oxidative stress, which suggested that mTOR could be targeted to curtail the plastic-induced muscle toxicity (207).
An increasing number of studies have substantiated the role of necroptosis in neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Multiple Sclerosis, and amyotrophic lateral sclerosis (ALS) (208–211). However, explorations into the therapeutic utility of mTOR-regulated necroptosis in these disorders are lacking. Indirect evidence indicates the treatment potential to some extent. p62 is involved in the TNFα-induced necroptosis by interacting with RIPK1 in AD, which is aggravated by comprised autophagy through aberrant p62 accumulation (212). As a well-established autophagy inhibitor, mTOR is likely to regulate necroptosis through the autophagy-p62-RIPK1 axis and serve as a potential therapeutic target in AD. A recent study found that the activation of necroptosis by the long noncoding RNA MEG3 is associated with AD (213). The strong correlation between MEG3 and mTOR activity has been established in numerous diseases, including neuroblastoma, endometrial carcinoma, and cisplatin-induced nephrotoxicity (214–217). Thus, mTOR may mediate MEG3-induced necroptosis. Due to the lack of direct evidence, further investigations are urgently required.
In other neurological diseases, a study revealed that AKT/mTOR contributed to ROS production to exacerbate necroptosis in the mouse hippocampal neuronal cell line HT-22. However, the study did not delve deeper into the potential involvement of this mechanism in neurological diseases through in vivo experiments (183). Ying Wang et al. revealed the treatment mechanisms of LiCl in schwannoma by activating AKT/mTOR axis to induce necroptosis (201).
Since the therapeutic potentials reviewed above are shed light on by animal models or in vitro experiments, it is obligatory to keep track of the advances in the clinical trials of mTOR inhibitors with verified necroptosis-regulating roles.
A wide range of basic research has confirmed that various mTOR inhibitors have the potential to regulate necroptosis, which has been summarized before in this review. Rapamycin (also named sirolimus) has been substantiated to regulate necroptosis as a well-characterized mTOR inhibitor. For instance, the treatment of rapamycin mitigates the injury of intestinal epithelial cells by recovering the mitophagy flux to alleviate necroptosis, which is verified in neurons, macrophages, cardiomyocytes, and so on (167, 169, 218, 219). Additionally, other mTOR inhibitors exert important effects on necroptosis, such as AZD2014 (also named vistusertib), everolimus, and Torin1 (171, 220). The advances in clinical trials related to mTOR inhibitors will be discussed in the section and summarized in Table 1.
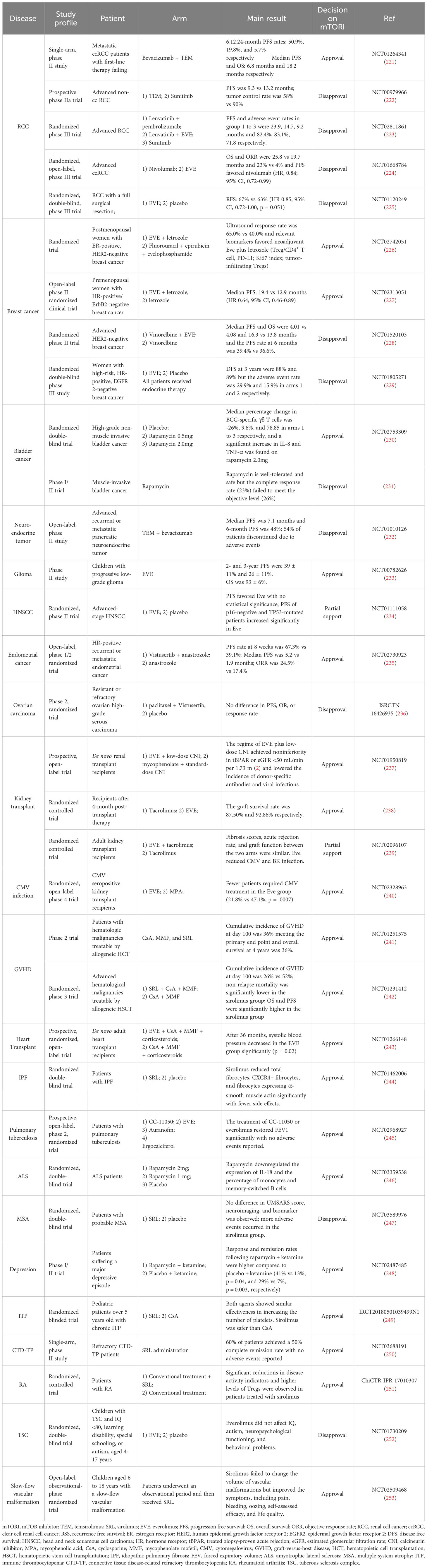
Table 1 Non-exhaustive summary of the application of mTOR inhibitors in clinical trials.
Despite preclinical experiments confirming the role of mTOR inhibitors in a variety of diseases, most clinical trials focus on their application in organ transplant as a potent immunosuppressant. In terms of graft-versus-host disease (GVHD) prophylaxis, the addition of sirolimus to standard therapy is propitious to reducing the incidence and severity of graft-versus-host disease (GVHD) and improving the life quality in kidney transplant recipients (241, 242, 254, 255). Additionally, everolimus improves the prognosis of liver or heart transplants with fewer adverse effects (256, 257). Since the use of immunosuppressant increases the susceptibility to infections, whether the administration of mTOR inhibitors influence infections has been paid attention to in organ transplant. Tedesco-Silva, H. et al. compared the efficacy of everolimus and low-dose calcineurin inhibitor with standard therapy (calcineurin inhibitor and mycophenolic acid) and found that the incidence of cytomegalovirus (CMV) infection decreased significantly in patients treated with everolimus (258). Moreover, sirolimus also prevents CMV recurrence in CMV-positive kidney transplant recipients (259). Hence, mTOR inhibitor is an effective and safe therapeutic strategy in organ transplant.
The value of mTOR inhibitors in antitumor therapy has been investigated, especially in urinary tumors. A phase II clinical trial confirmed that combining cyclophosphamide and everolimus leads to the depletion of Tregs and myeloid-derived suppressor cells, which improves the survival of metastatic renal cell cancer (RCC) patients (260). Another phase 2 study proved that the combination of lapatinib plus everolimus is effective in restricting the expansion of tumors with controllable side effects in non-clear cell RCC (261). However, the effectiveness of mTOR inhibitors in treating RCC remains controversial. A phase 3 trial exhibited a longer recurrence-free survival in patients receiving everolimus than those treated with placebo but the result failed to meet the statistical requirement and support the adjuvant treatment of everolimus for RCC (225). Other regimens display improved therapeutic efficacy over mTOR inhibitors in RCC. Lenvatinib plus pembrolizumab prolonged the progression-free and overall survival and improved health-related quality-of-life in patients with advanced RCC compared with Lenvatinib plus everolimus (223, 262). In addition to RCC, the immunomodulatory effect of mTOR inhibitors has been employed in the treatment of bladder cancer. The count of γδT cells specific to BCG and the production of cytokines are boosted by rapamycin, suggesting that rapamycin is a potential adjuvant agent of bladder cancer by enhancing the immune response to BCG to exert antitumor effect (230).
Moreover, the application of mTOR inhibitors has been investigated extensively in breast cancer and other tumors, but few researchers have reached a supportive or positive conclusion. Triple treatment composed of ribociclib, everolimus, and endocrine therapy shows a clinical benefit with manageable safety profile in advanced breast cancer (263). Everolimus plus letrozole or exemestane benefits the outcome and has lower adverse effects in postmenopausal advanced breast cancer patients with hormone receptor-positive and human epidermal growth factor receptor 2-negative (226, 264). Nonetheless, several clinical trials refute the therapeutic efficacy of everolimus in survival improvement in breast cancer (229, 265, 266). A randomized phase II trial demonstrated that the treatment of everolimus and letrozole prolonged the progression-free survival in recurrent endometrial carcinoma (267). Inhibiting PI3K/AKT by everolimus or temsirolimus is tolerable and beneficial to overall survival in glioma and glioblastoma patients (233, 268).
mTOR inhibitor is a promising therapeutic target for autoimmune diseases. Sirolimus downregulates the indicators of disease activity and elevates Tregs in patients with rheumatoid arthritis more significantly than conventional therapy (251). Over half of patients with connective tissue disease-related thrombocytopenia (CTD-TP) who received oral sirolimus administration achieved complete remission, suggesting that sirolimus is an alternative in treating CTD-TP (250). Similarly, sirolimus increases the count of platelets in pediatric chronic immune thrombocytopenia (249).
Furthermore, mTOR inhibitors can also be used as adjuvant agents to augment the efficacy of conventional drugs in treating tuberculosis. Researchers evaluated the lung function and sputum culture to compare the efficacy of different adjuvants plus standard tuberculosis treatment, concluding that everolimus is a potential adjuvant in tuberculosis therapy with safety and well-tolerance (245). In terms of respiratory diseases, the treatment of sirolimus contributes to a significant decrease in circulating fibrocytes to alleviate lung fibrosis, which implies the effect of sirolimus on treating idiopathic pulmonary fibrosis (244).
Regarding neurological disorders, mTOR is a promising therapeutic target as well. Patients of ALS receiving rapamycin underwent a decline in proinflammatory cytokines and an increase in Tregs to inhibit aberrant neuroinflammation (246).
The precise regulation of cell death is principal to sustaining equilibrium, and abnormal cell death is closely associated with various disorders. As a key molecule in cellular metabolism, growth, and death, mTOR plays an essential role in regulating the complex signaling network. There have been increasing studies that have demonstrated the vital effect of necroptosis regulated by mTOR on a wide range of diseases, such as IBD, sepsis, lung cancer, and others.
We summarized the upstream and downstream mechanisms of mTOR and the molecular mechanisms of necroptosis (Graphical Abstract). Recent advances in the mechanisms by which mTOR regulates necroptosis are also highlighted in this review. mTOR, established as a presentative autophagy inhibitor, controls the interaction of RIPKs and their removal through autophagy to regulate necroptosis in a roundabout manner. Several necroptosis-relevant molecules are directly regulated by mTOR, such as RIPKs and TNFα. mTOR also utilizes oxidative stress to control necroptosis. However, subsequent investigations have suggested that mTOR exerts a bidirectional effect on necroptosis. It is determined by numerous factors, such as the stimulus category and cell types. Distinct signaling pathways in which mTOR is involved in the regulation of necroptosis are likely to lead to diverse outcomes; the ultimate effect is contingent upon the dominant mechanism.
Investigations into the mechanisms by which mTOR regulates necroptosis have shed more light on potential therapeutic targets for an extensive array of diseases, including immune-related diseases, cancer, drug toxicity, and more. An incremental body of basic research substantiates the value of targeting mTOR in treating various disorders. Based on the fundamental findings, considerable clinical trials have attempted to apply mTOR inhibitors in therapeutic strategies for transplant rejection, tumor, autoimmune diseases, etc. and presented promising prospects.
Therefore, as a pivotal hub for regulating cell activities, mTOR exerts an essential role in controlling necroptosis through autophagy and other mechanisms. Necroptosis is under the bidirectional regulation of mTOR by virtue of complicated cellular signaling networks but the underlying mechanisms require further investigation. Although both basic experiments and clinical trials have indicated the therapeutic implications of mTOR mediated by necroptosis, substantial evidence from mechanism studies and clinical trials is in enormous demand to demonstrate its therapeutic value and ultimately fulfil clinical transformation of mTOR in various diseases such as cancer, sepsis, and immune-related disorders.
YX: Visualization, Writing – original draft, Writing – review & editing. GZ: Writing – review & editing. XL: Writing – review & editing. NC: Conceptualization, Supervision, Writing – review & editing. HW: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work was supported by National High Level Hospital Clinical Research Funding (No. 2022-PUMCH-B-126), National Key R&D Program of China 2022YFC2009803 from Ministry of Science and Technology of the People’s Republic of China, National Natural Science Foundation of China (No. 82072226), CAMS Innovation Fund for Medical Sciences (CIFMS) 2021-I2M-1-062 from Chinese Academy of Medical Sciences, and Beijing Municipal Science and Technology Commission (No. Z201100005520049).
I would like to express my sincere gratitude to all the tutors and colleagues for their selfless assistance and encouragement during the process of writing this review. Thanks for their contributions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bedoui S, Herold MJ, Strasser A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol (2020) 21(11):678–95. doi: 10.1038/s41580-020-0270-8
2. Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, et al. The apoptosis-necrosis paradoxApoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene (1997) 15(13):1573–81. doi: 10.1038/sj.onc.1201324
3. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol (2005) 1(2):112–9. doi: 10.1038/nchembio711
4. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
5. Boise LH, Collins CM. Salmonella-induced cell death: apoptosis, necrosis or programmed cell death? Trends Microbiol (2001) 9(2):64–7. doi: 10.1016/S0966-842X(00)01937-5
6. Zhang G, Wang J, Zhao Z, Xin T, Fan X, Shen Q, et al. Regulated necrosis, a proinflammatory cell death, potentially counteracts pathogenic infections. Cell Death Disease (2022) 13(7):637. doi: 10.1038/s41420-022-00861-5
7. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol (2014) 15(2):135–47. doi: 10.1038/nrm3737
8. Guo X, Chen Y, Liu Q. Necroptosis in heart disease: Molecular mechanisms and therapeutic implications. J Mol Cell Cardiol (2022) 169:74–83. doi: 10.1016/j.yjmcc.2022.05.006
9. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi: 10.1016/j.cell.2012.03.017
10. Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest (2015) 125(1):25–32. doi: 10.1172/JCI73939
11. Xie Y, Zhao Y, Shi L, Li W, Chen K, Li M, et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and cancer. J Clin Invest (2020) 130(4):2111–28. doi: 10.1172/JCI133264
12. Cafferkey R, Young PR, McLaughlin MM, Bergsma DJ, Koltin Y, Sathe GM, et al. Dominant missense mutations in a novel yeast protein related to mammalian phosphatidylinositol 3-kinase and VPS34 abrogate rapamycin cytotoxicity. Mol Cell Biol (1993) 13(10):6012–23. doi: 10.1128/mcb.13.10.6012-6023.1993
13. Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell (1993) 73(3):585–96. doi: 10.1016/0092-8674(93)90144-F
14. Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature (1994) 369(6483):756–8. doi: 10.1038/369756a0
15. Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell (1994) 78(1):35–43. doi: 10.1016/0092-8674(94)90570-3
16. Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem (1995) 270(2):815–22. doi: 10.1074/jbc.270.2.815
17. Hung CM, Garcia-Haro L, Sparks CA, Guertin DA. mTOR-dependent cell survival mechanisms. Cold Spring Harbor Perspect Biol (2012) 4(12):a008771. doi: 10.1101/cshperspect.a008771
18. Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature (2013) 497(7448):217–23. doi: 10.1038/nature12122
19. Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell (1992) 69(7):1227–36. doi: 10.1016/0092-8674(92)90643-Q
20. Kennedy BK, Lamming DW. The mechanistic target of rapamycin: the grand conducTOR of metabolism and aging. Cell Metab (2016) 23(6):990–1003. doi: 10.1016/j.cmet.2016.05.009
21. Popova NV, Jücker M. The role of mTOR signaling as a therapeutic target in cancer. Int J Mol Sci (2021) 22(4):1743. doi: 10.3390/ijms22041743
22. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell (2006) 22(2):159–68. doi: 10.1016/j.molcel.2006.03.029
23. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol (2012) 12(5):325–38. doi: 10.1038/nri3198
24. Zou Z, Tao T, Li H, Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell bioscience (2020) 10:31. doi: 10.1186/s13578-020-00396-1
25. Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, et al. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature (2017) 552(7685):368–73. doi: 10.1038/nature25023
26. Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol (2003) 5(6):566–71. doi: 10.1038/ncb996
27. Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol (2015) 25(9):545–55. doi: 10.1016/j.tcb.2015.06.002
28. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol (2002) 4(9):648–57. doi: 10.1038/ncb839
29. Wataya-Kaneda M. Mammalian target of rapamycin and tuberous sclerosis complex. J Dermatol science. (2015) 79(2):93–100. doi: 10.1016/j.jdermsci.2015.04.005
30. Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol (2002) 4(9):658–65. doi: 10.1038/ncb840
31. Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. (2005) 121(2):179–93. doi: 10.1016/j.cell.2005.02.031
32. Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA (2004) 101(37):13489–94. doi: 10.1073/pnas.0405659101
33. Carriere A, Romeo Y, Acosta-Jaquez HA, Moreau J, Bonneil E, Thibault P, et al. ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J Biol Chem (2011) 286(1):567–77. doi: 10.1074/jbc.M110.159046
34. Carrière A, Cargnello M, Julien LA, Gao H, Bonneil E, Thibault P, et al. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr biology: CB. (2008) 18(17):1269–77. doi: 10.1016/j.cub.2008.07.078
35. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol (Baltimore Md: 1950). (2017) 198(3):1006–14. doi: 10.4049/jimmunol.1601515
36. Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol (2015) 27(4):286–96. doi: 10.1016/j.smim.2015.08.001
37. Troutman TD, Hu W, Fulenchek S, Yamazaki T, Kurosaki T, Bazan JF, et al. Role for B-cell adapter for PI3K (BCAP) as a signaling adapter linking Toll-like receptors (TLRs) to serine/threonine kinases PI3K/Akt. Proc Natl Acad Sci United States America. (2012) 109(1):273–8. doi: 10.1073/pnas.1118579109
38. Troutman TD, Bazan JF, Pasare C. Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle (Georgetown Tex). (2012) 11(19):3559–67. doi: 10.4161/cc.21572
39. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. (2003) 115(5):577–90. doi: 10.1016/S0092-8674(03)00929-2
40. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell (2008) 30(2):214–26. doi: 10.1016/j.molcel.2008.03.003
41. Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. (2010) 141(2):290–303. doi: 10.1016/j.cell.2010.02.024
42. Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Sci (New York NY). (2008) 320(5882):1496–501. doi: 10.1126/science.1157535
43. Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Sci (New York NY). (2013) 340(6136):1100–6. doi: 10.1126/science.1232044
44. Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, et al. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep (2014) 9(1):1–8. doi: 10.1016/j.celrep.2014.09.014
45. Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, et al. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep (2014) 9(4):1281–91. doi: 10.1016/j.celrep.2014.10.019
46. Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Sci (New York NY). (2016) 351(6268):43–8. doi: 10.1126/science.aab2674
47. Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, et al. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell. (2016) 165(1):153–64. doi: 10.1016/j.cell.2016.02.035
48. Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, et al. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell. (2017) 171(3):642–54.e12. doi: 10.1016/j.cell.2017.09.046
49. Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Sci (New York NY). (2015) 347(6218):188–94. doi: 10.1126/science.1257132
50. Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Sci (New York NY). (2011) 334(6056):678–83. doi: 10.1126/science.1207056
51. Durán RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell (2012) 47(3):349–58. doi: 10.1016/j.molcel.2012.05.043
52. Bodineau C, Tomé M, Courtois S, Costa ASH, Sciacovelli M, Rousseau B, et al. Two parallel pathways connect glutamine metabolism and mTORC1 activity to regulate glutamoptosis. Nat Commun (2021) 12(1):4814. doi: 10.1038/s41467-021-25079-4
53. Bodineau C, Tomé M, Murdoch PDS, Durán RV. Glutamine, MTOR and autophagy: a multiconnection relationship. Autophagy. (2022) 18(11):2749–50. doi: 10.1080/15548627.2022.2062875
54. Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, et al. MetabolismDifferential regulation of mTORC1 by leucine and glutamine. Sci (New York NY). (2015) 347(6218):194–8. doi: 10.1126/science.1259472
55. Meng D, Yang Q, Wang H, Melick CH, Navlani R, Frank AR, et al. Glutamine and asparagine activate mTORC1 independently of Rag GTPases. J Biol Chem (2020) 295(10):2890–9. doi: 10.1074/jbc.AC119.011578
56. Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, et al. PtdIns(3,4,5)P3-dependent activation of the mTORC2 kinase complex. Cancer discovery. (2015) 5(11):1194–209. doi: 10.1158/2159-8290.CD-15-0460
57. Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. (2011) 144(5):757–68. doi: 10.1016/j.cell.2011.02.014
58. Kazyken D, Magnuson B, Bodur C, Acosta-Jaquez HA, Zhang D, Tong X, et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci Signaling (2019) 12(585):eaav 3249. doi: 10.1126/scisignal.aav3249
59. Tan VP, Miyamoto S. Nutrient-sensing mTORC1: Integration of metabolic and autophagic signals. J Mol Cell Cardiol (2016) 95:31–41. doi: 10.1016/j.yjmcc.2016.01.005
60. Ben-Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol (2017) 45:72–82. doi: 10.1016/j.ceb.2017.02.012
61. Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J (2012) 441(1):1–21. doi: 10.1042/BJ20110892
62. Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, et al. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev (1999) 13(11):1422–37. doi: 10.1101/gad.13.11.1422
63. García-Martínez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J (2008) 416(3):375–85. doi: 10.1042/BJ20081668
64. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Sci (New York NY). (2005) 307(5712):1098–101. doi: 10.1126/science.1106148
65. Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J (2008) 27(14):1919–31. doi: 10.1038/emboj.2008.119
66. Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol (2004) 6(11):1122–8. doi: 10.1038/ncb1183
67. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr biology: CB. (2004) 14(14):1296–302. doi: 10.1016/j.cub.2004.06.054
68. Pópulo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci (2012) 13(2):1886–918. doi: 10.3390/ijms13021886
69. Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem (2002) 277(11):9505–11. doi: 10.1074/jbc.M109488200
70. Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. (1995) 81(4):513–23. doi: 10.1016/0092-8674(95)90072-1
71. Wu XN, Yang ZH, Wang XK, Zhang Y, Wan H, Song Y, et al. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death differentiation. (2014) 21(11):1709–20. doi: 10.1038/cdd.2014.77
72. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol (2014) 16(1):55–65. doi: 10.1038/ncb2883
73. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell (2014) 54(1):133–46. doi: 10.1016/j.molcel.2014.03.003
74. Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol (Baltimore Md: 1950). (1988) 141(8):2629–34. doi: 10.4049/jimmunol.141.8.2629
75. Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. (2008) 135(7):1311–23. doi: 10.1016/j.cell.2008.10.044
76. He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci United States America. (2011) 108(50):20054–9. doi: 10.1073/pnas.1116302108
77. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. RIP3, and MLKL. J Biol Chem (2013) 288(43):31268–79. doi: 10.1074/jbc.M113.462341
78. Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci United States America. (2008) 105(33):11778–83. doi: 10.1073/pnas.0711122105
79. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. (2003) 114(2):181–90. doi: 10.1016/S0092-8674(03)00521-X
80. Csomos RA, Brady GF, Duckett CS. Enhanced cytoprotective effects of the inhibitor of apoptosis protein cellular IAP1 through stabilization with TRAF2. J Biol Chem (2009) 284(31):20531–9. doi: 10.1074/jbc.M109.029983
81. Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. (2009) 138(2):229–32. doi: 10.1016/j.cell.2009.07.006
82. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol (2010) 11(10):700–14. doi: 10.1038/nrm2970
83. Gadd MS, Bulatov E, Ciulli A. Serendipitous SAD solution for DMSO-soaked SOCS2-elonginC-elonginB crystals using covalently incorporated dimethylarsenic: insights into substrate receptor conformational flexibility in cullin RING ligases. PloS One (2015) 10(6):e0131218. doi: 10.1371/journal.pone.0131218
84. Morreale FE, Walden H. Types of ubiquitin ligases. Cell. (2016) 165(1):248–.e1. doi: 10.1016/j.cell.2016.03.003
85. Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell (2009) 36(5):831–44. doi: 10.1016/j.molcel.2009.10.013
86. Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. (2011) 471(7340):591–6. doi: 10.1038/nature09816
87. Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell (2008) 30(6):689–700. doi: 10.1016/j.molcel.2008.05.014
88. Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med (2003) 9(5):575–81. doi: 10.1038/nm849
89. de Almagro MC, Goncharov T, Newton K, Vucic D. Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination. Cell Death disease. (2015) 6(6):e1800. doi: 10.1038/cddis.2015.158
90. Wei R, Xu LW, Liu J, Li Y, Zhang P, Shan B, et al. SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination. Genes Dev (2017) 31(11):1162–76. doi: 10.1101/gad.299776.117
91. Clague MJ, Urbé S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol (2019) 20(6):338–52. doi: 10.1038/s41580-019-0099-1
92. Keusekotten K, Elliott PR, Glockner L, Fiil BK, Damgaard RB, Kulathu Y, et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell. (2013) 153(6):1312–26. doi: 10.1016/j.cell.2013.05.014
93. Rivkin E, Almeida SM, Ceccarelli DF, Juang YC, MacLean TA, Srikumar T, et al. The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature. (2013) 498(7454):318–24. doi: 10.1038/nature12296
94. Douglas T, Saleh M. Post-translational modification of OTULIN regulates ubiquitin dynamics and cell death. Cell Rep (2019) 29(11):3652–63.e5. doi: 10.1016/j.celrep.2019.11.014
95. Kupka S, Reichert M, Draber P, Walczak H. Formation and removal of poly-ubiquitin chains in the regulation of tumor necrosis factor-induced gene activation and cell death. FEBS J (2016) 283(14):2626–39. doi: 10.1111/febs.13644
96. Khan I, Yousif A, Chesnokov M, Hong L, Chefetz I. A decade of cell death studies: Breathing new life into necroptosis. Pharmacol Ther (2021) 220:107717. doi: 10.1016/j.pharmthera.2020.107717
97. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol (2017) 18(2):127–36. doi: 10.1038/nrm.2016.149
98. Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell (2011) 43(3):449–63. doi: 10.1016/j.molcel.2011.06.011
99. He MX, He YW. A role for c-FLIP(L) in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes. Cell Death differentiation. (2013) 20(2):188–97. doi: 10.1038/cdd.2012.148
100. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. (2015) 517(7534):311–20. doi: 10.1038/nature14191
101. Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun (2017) 8(1):359. doi: 10.1038/s41467-017-00406-w
102. Meng H, Liu Z, Li X, Wang H, Jin T, Wu G, et al. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc Natl Acad Sci United States America. (2018) 115(9):E2001–e9. doi: 10.1073/pnas.1722013115
103. Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev (2013) 27(15):1640–9. doi: 10.1101/gad.223321.113
104. Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol (2008) 4(5):313–21. doi: 10.1038/nchembio.83
105. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. (2009) 137(6):1112–23. doi: 10.1016/j.cell.2009.05.037
106. Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun (2017) 8:14329. doi: 10.1038/ncomms14329
107. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. (2012) 150(2):339–50. doi: 10.1016/j.cell.2012.06.019
108. Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, Wu X, et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem (2013) 288(23):16247–61. doi: 10.1074/jbc.M112.435545
109. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell (2012) 148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031
110. Petrie EJ, Sandow JJ, Jacobsen AV, Smith BJ, Griffin MDW, Lucet IS, et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat Commun (2018) 9(1):2422. doi: 10.1038/s41467-018-04714-7
111. Geserick P, Hupe M, Moulin M, Wong WW, Feoktistova M, Kellert B, et al. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J Cell Biol (2009) 187(7):1037–54. doi: 10.1083/jcb.200904158
112. von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat Rev Cancer. (2017) 17(6):352–66. doi: 10.1038/nrc.2017.28
113. Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. (2000) 12(6):611–20. doi: 10.1016/S1074-7613(00)80212-5
114. Weindel CG, Martinez EL, Zhao X, Mabry CJ, Bell SL, Vail KJ, et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell. (2022) 185(17):3214–31.e23. doi: 10.1016/j.cell.2022.06.038
115. Lu B, Gong X, Wang ZQ, Ding Y, Wang C, Luo TF, et al. Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation. Acta pharmacologica Sinica. (2017) 38(11):1543–53. doi: 10.1038/aps.2017.112
116. Schenk B, Fulda S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene. (2015) 34(47):5796–806. doi: 10.1038/onc.2015.35
117. Morgan MJ, Kim YS. Roles of RIPK3 in necroptosis, cell signaling, and disease. Exp Mol Med (2022) 54(10):1695–704. doi: 10.1038/s12276-022-00868-z
118. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Sci (New York NY). (2009) 325(5938):332–6. doi: 10.1126/science.1172308
119. Yang Z, Wang Y, Zhang Y, He X, Zhong CQ, Ni H, et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat Cell Biol (2018) 20(2):186–97. doi: 10.1038/s41556-017-0022-y
120. He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. (2009) 137(6):1100–11. doi: 10.1016/j.cell.2009.05.021
121. Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep (2013) 5(4):878–85. doi: 10.1016/j.celrep.2013.10.034
122. Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature. (2016) 540(7631):124–8. doi: 10.1038/nature20558
123. Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature. (2016) 540(7631):129–33. doi: 10.1038/nature20559
124. Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med (1998) 188(5):919–30. doi: 10.1084/jem.188.5.919
125. Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death differentiation. (2012) 19(12):2003–14. doi: 10.1038/cdd.2012.90
126. Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe (2012) 11(3):290–7. doi: 10.1016/j.chom.2012.01.016
127. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death differentiation. (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
128. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res (2019) 29(5):347–64. doi: 10.1038/s41422-019-0164-5
129. Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med (1998) 187(9):1477–85. doi: 10.1084/jem.187.9.1477
130. Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J (2017) 36(17):2529–43. doi: 10.15252/embj.201796476
131. Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. (2020) 580(7803):391–5. doi: 10.1038/s41586-020-2129-8
132. Kist M, Vucic D. Cell death pathways: intricate connections and disease implications. EMBO J (2021) 40(5):e106700. doi: 10.15252/embj.2020106700
133. Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death differentiation. (2006) 13(8):1396–402. doi: 10.1038/sj.cdd.4401963
134. Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol (2019) 20(3):175–93. doi: 10.1038/s41580-018-0089-8
135. Kumar S, Dorstyn L, Lim Y. The role of caspases as executioners of apoptosis. Biochem Soc Trans (2022) 50(1):33–45. doi: 10.1042/BST20210751
136. He S, Wang X. RIP kinases as modulators of inflammation and immunity. Nat Immunol (2018) 19(9):912–22. doi: 10.1038/s41590-018-0188-x
137. Ketelut-Carneiro N, Fitzgerald KA. Apoptosis, pyroptosis, and necroptosis-oh my! The many ways a cell can die. J Mol Biol (2022) 434(4):167378. doi: 10.1016/j.jmb.2021.167378
138. Larsen BD, Sørensen CS. The caspase-activated DNase: apoptosis and beyond. FEBS J (2017) 284(8):1160–70. doi: 10.1111/febs.13970
139. Ricci JE, Muñoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, et al. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell. (2004) 117(6):773–86. doi: 10.1016/j.cell.2004.05.008
140. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
141. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. (2018) 560(7717):198–203. doi: 10.1038/s41586-018-0372-z
142. Weber K, Schilling JD. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J Biol Chem (2014) 289(13):9158–71. doi: 10.1074/jbc.M113.531202
143. Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem (2007) 282(5):2871–9. doi: 10.1074/jbc.M608083200
144. Katsnelson MA, Rucker LG, Russo HM, Dubyak GR. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol (Baltimore Md: 1950). (2015) 194(8):3937–52. doi: 10.4049/jimmunol.1402658
145. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci United States America. (2012) 109(28):11282–7. doi: 10.1073/pnas.1117765109
146. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526(7575):660–5. doi: 10.1038/nature15514
147. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. (2015) 526(7575):666–71. doi: 10.1038/nature15541
148. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Sci (New York NY). (2013) 341(6151):1250–3. doi: 10.1126/science.1240988
149. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535(7610):153–8. doi: 10.1038/nature18629
150. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. (2016) 535(7610):111–6. doi: 10.1038/nature18590
151. Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. (2021) 591(7848):131–6. doi: 10.1038/s41586-021-03218-7
152. Kayagaki N, Stowe IB, Alegre K, Deshpande I, Wu S, Lin Z, et al. Inhibiting membrane rupture with NINJ1 antibodies limits tissue injury. Nature. (2023) 618(7967):1072–7. doi: 10.1038/s41586-023-06191-5
153. Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol (2020) 20(3):143–57. doi: 10.1038/s41577-019-0228-2
154. Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep (2020) 30(10):3411–23.e7. doi: 10.1016/j.celrep.2020.02.049
155. Gammella E, Buratti P, Cairo G, Recalcati S. The transferrin receptor: the cellular iron gate. Metallomics: integrated biometal science. (2017) 9(10):1367–75. doi: 10.1039/C7MT00143F
156. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. (2021) 17(9):2054–81. doi: 10.1080/15548627.2020.1810918
157. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
158. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death differentiation. (2016) 23(2):270–8. doi: 10.1038/cdd.2015.93
159. Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem (2009) 284(18):12297–305. doi: 10.1074/jbc.M900573200
160. Napolitano G, Esposito A, Choi H, Matarese M, Benedetti V, Di Malta C, et al. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun (2018) 9(1):3312. doi: 10.1038/s41467-018-05862-6
161. Ge D, Tao HR, Fang L, Kong XQ, Han LN, Li N, et al. 11-methoxytabersonine induces necroptosis with autophagy through AMPK/mTOR and JNK pathways in human lung cancer cells. Chem Pharm bulletin. (2020) 68(3):244–50. doi: 10.1248/cpb.c19-00851
162. Bray K, Mathew R, Lau A, Kamphorst JJ, Fan J, Chen J, et al. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PloS One (2012) 7(7):e41831. doi: 10.1371/journal.pone.0041831
163. Xu B, Zhou Z, Fang J, Wang J, Tao K, Liu J, et al. Exosomes derived from schwann cells alleviate mitochondrial dysfunction and necroptosis after spinal cord injury via AMPK signaling pathway-mediated mitophagy. Free Radical Biol Med (2023) 208:319–33. doi: 10.1016/j.freeradbiomed.2023.08.026
164. Zhong WJ, Zhang J, Duan JX, Zhang CY, Ma SC, Li YS, et al. TREM-1 triggers necroptosis of macrophages through mTOR-dependent mitochondrial fission during acute lung injury. J Trans Med (2023) 21(1):179. doi: 10.1186/s12967-023-04027-4
165. Song Z, Song H, Liu D, Yan B, Wang D, Zhang Y, et al. Overexpression of MFN2 alleviates sorafenib-induced cardiomyocyte necroptosis via the MAM-CaMKIIδ pathway in vitro and in vivo. Theranostics (2022) 12(3):1267–85. doi: 10.7150/thno.65716
166. Abe K, Yano T, Tanno M, Miki T, Kuno A, Sato T, et al. mTORC1 inhibition attenuates necroptosis through RIP1 inhibition-mediated TFEB activation. Biochim Biophys Acta Mol basis disease. (2019) 1865(12):165552. doi: 10.1016/j.bbadis.2019.165552
167. Zhang T, Xu D, Trefts E, Lv M, Inuzuka H, Song G, et al. Metabolic orchestration of cell death by AMPK-mediated phosphorylation of RIPK1. Sci (New York NY). (2023) 380(6652):1372–80. doi: 10.1126/science.abn1725
168. Wang K, Zhao A, Tayier D, Tan K, Song W, Cheng Q, et al. Activation of AMPK ameliorates acute severe pancreatitis by suppressing pancreatic acinar cell necroptosis in obese mice models. Cell Death discovery. (2023) 9(1):363. doi: 10.1038/s41421-023-00527-1
169. McNamara CR, Ahuja R, Osafo-Addo AD, Barrows D, Kettenbach A, Skidan I, et al. Akt Regulates TNFα synthesis downstream of RIP1 kinase activation during necroptosis. PloS One (2013) 8(3):e56576. doi: 10.1371/journal.pone.0056576
170. Nguyen MT, Satoh H, Favelyukis S, Babendure JL, Imamura T, Sbodio JI, et al. JNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J Biol Chem (2005) 280(42):35361–71. doi: 10.1074/jbc.M504611200
171. Zhou Z, Tao T, Ji Y, Yang H, Wang Y, Cheng C, et al. SSeCKS promotes tumor necrosis factor-alpha autocrine via activating p38 and JNK pathways in Schwann cells. Cell Mol neurobiology. (2010) 30(5):701–7. doi: 10.1007/s10571-009-9494-z
172. Christofferson DE, Li Y, Hitomi J, Zhou W, Upperman C, Zhu H, et al. A novel role for RIP1 kinase in mediating TNFα production. Cell Death disease. (2012) 3(6):e320. doi: 10.1038/cddis.2012.64
173. Damgaard RB, Jolin HE, Allison MED, Davies SE, Titheradge HL, McKenzie ANJ, et al. OTULIN protects the liver against cell death, inflammation, fibrosis, and cancer. Cell Death differentiation. (2020) 27(5):1457–74. doi: 10.1038/s41418-020-0532-1
174. Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. (2008) 135(7):1161–3. doi: 10.1016/j.cell.2008.12.004
175. Vanlangenakker N, Vanden Berghe T, Vandenabeele P. Many stimuli pull the necrotic trigger, an overview. Cell Death differentiation. (2012) 19(1):75–86. doi: 10.1038/cdd.2011.164
176. Hsu SK, Chang WT, Lin IL, Chen YF, Padalwar NB, Cheng KC, et al. The role of necroptosis in ROS-mediated cancer therapies and its promising applications. Cancers (2020) 12(8):2185. doi: 10.3390/cancers12082185
177. Liu Q, Qiu J, Liang M, Golinski J, van Leyen K, Jung JE, et al. Akt and mTOR mediate programmed necrosis in neurons. Cell Death disease. (2014) 5(2):e1084. doi: 10.1038/cddis.2014.69
178. Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. (2002) 419(6904):316–21. doi: 10.1038/nature01036
179. Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. (2005) 24(50):7410–25. doi: 10.1038/sj.onc.1209086
180. Yu C, Xiao JH. The keap1-nrf2 system: A mediator between oxidative stress and aging. Oxid Med Cell longevity. (2021) 2021:6635460. doi: 10.1155/2021/6635460
181. Silva-Islas CA, Maldonado PD. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol Res (2018) 134:92–9. doi: 10.1016/j.phrs.2018.06.013
182. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci (2019) 20(1):19–33. doi: 10.1038/s41583-018-0093-1
183. Deng B, Yang D, Wu H, Wang L, Wu R, Zhu H, et al. Ketamine inhibits TNF-α-induced cecal damage by enhancing RIP1 ubiquitination to attenuate lethal SIRS. Cell Death discovery. (2022) 8(1):72. doi: 10.1038/s41420-022-00869-x
184. Pavlosky A, Lau A, Su Y, Lian D, Huang X, Yin Z, et al. RIPK3-mediated necroptosis regulates cardiac allograft rejection. Am J transplantation: Off J Am Soc Transplant Am Soc Transplant Surgeons. (2014) 14(8):1778–90. doi: 10.1111/ajt.12779
185. Mistry P, Kaplan MJ. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin Immunol (Orlando Fla). (2017) 185:59–73. doi: 10.1016/j.clim.2016.08.010
186. Guo C, Fu R, Zhou M, Wang S, Huang Y, Hu H, et al. Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. J autoimmunity. (2019) 103:102286. doi: 10.1016/j.jaut.2019.05.014
187. Li X, Zhang Y, Wang J, Li Y, Wang Y, Shi F, et al. zVAD alleviates experimental autoimmune hepatitis in mice by increasing the sensitivity of macrophage to TNFR1-dependent necroptosis. J autoimmunity. (2022) 133:102904. doi: 10.1016/j.jaut.2022.102904
188. Zou R, Nie C, Pan S, Wang B, Hong X, Xi S, et al. Co-administration of hydrogen and metformin exerts cardioprotective effects by inhibiting pyroptosis and fibrosis in diabetic cardiomyopathy. Free Radical Biol Med (2022) 183:35–50. doi: 10.1016/j.freeradbiomed.2022.03.010
189. Li T, Xie Y, Shi L, Sun Y, Wen J, Deng Z, et al. TSC1 suppresses macrophage necroptosis for the control of infection by fungal pathogen candida albicans. ImmunoHorizons. (2021) 5(2):90–101. doi: 10.4049/immunohorizons.2000093
190. Shen J, Fu Y, Liu F, Ning B, Jiang X. Ursolic acid promotes autophagy by inhibiting akt/mTOR and TNF-α/TNFR1 signaling pathways to alleviate pyroptosis and necroptosis in mycobacterium tuberculosis-infected macrophages. Inflammation. (2023) 46(5):1749–63. doi: 10.1007/s10753-023-01839-w
191. Lau A, Wang S, Jiang J, Haig A, Pavlosky A, Linkermann A, et al. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am J transplantation: Off J Am Soc Transplant Am Soc Transplant Surgeons. (2013) 13(11):2805–18. doi: 10.1111/ajt.12447
192. Kwok C, Pavlosky A, Lian D, Jiang J, Huang X, Yin Z, et al. Necroptosis is involved in CD4+ T cell-mediated microvascular endothelial cell death and chronic cardiac allograft rejection. Transplantation. (2017) 101(9):2026–37. doi: 10.1097/TP.0000000000001578
193. Chen H, McKeen T, Chao X, Chen A, Deng F, Jaeschke H, et al. The role of MLKL in hepatic ischemia-reperfusion injury of alcoholic steatotic livers. Int J Biol Sci (2022) 18(3):1096–106. doi: 10.7150/ijbs.67533
194. Li H, Hu J, Wu S, Wang L, Cao X, Zhang X, et al. Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells. Oncotarget. (2016) 7(3):3548–58. doi: 10.18632/oncotarget.6516
195. Wang Y, Zhang Q, Wang B, Li P, Liu P. LiCl treatment induces programmed cell death of schwannoma cells through AKT- and MTOR-mediated necroptosis. Neurochemical Res (2017) 42(8):2363–71. doi: 10.1007/s11064-017-2256-2
196. Jin L, Zhang W, Yao MY, Tian Y, Xue BX, Tao W. GNE-493 inhibits prostate cancer cell growth via Akt-mTOR-dependent and -independent mechanisms. Cell Death discovery. (2022) 8(1):120. doi: 10.1038/s41420-022-00911-y
197. Gounder MM, Mahoney MR, Van Tine BA, Ravi V, Attia S, Deshpande HA, et al. Sorafenib for advanced and refractory desmoid tumors. New Engl J Med (2018) 379(25):2417–28. doi: 10.1056/NEJMoa1805052
198. Chaar M, Kamta J, Ait-Oudhia S. Mechanisms, monitoring, and management of tyrosine kinase inhibitors-associated cardiovascular toxicities. OncoTargets Ther (2018) 11:6227–37. doi: 10.2147/OTT.S170138
199. Park EJ, Lee AY, Park S, Kim JH, Cho MH. Multiple pathways are involved in palmitic acid-induced toxicity. Food Chem toxicology: an Int J published Br Ind Biol Res Assoc (2014) 67:26–34. doi: 10.1016/j.fct.2014.01.027
200. Zhao M, Lu L, Lei S, Chai H, Wu S, Tang X, et al. Inhibition of receptor interacting protein kinases attenuates cardiomyocyte hypertrophy induced by palmitic acid. Oxid Med Cell longevity. (2016) 2016:1451676. doi: 10.1155/2016/1451676
201. Liu X, Zhang Y, Sun X, Zhang W, Shi X, Xu S. Di-(2-ethyl hexyl) phthalate induced oxidative stress promotes microplastics mediated apoptosis and necroptosis in mice skeletal muscle by inhibiting PI3K/AKT/mTOR pathway. Toxicology. (2022) 474:153226. doi: 10.1016/j.tox.2022.153226
202. Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Sci (New York NY). (2016) 353(6299):603–8. doi: 10.1126/science.aaf6803
203. Iannielli A, Bido S, Folladori L, Segnali A, Cancellieri C, Maresca A, et al. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in parkinson’s disease models. Cell Rep (2018) 22(8):2066–79. doi: 10.1016/j.celrep.2018.01.089
204. Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, et al. Activation of necroptosis in multiple sclerosis. Cell Rep (2015) 10(11):1836–49. doi: 10.1016/j.celrep.2015.02.051
205. Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, et al. Necroptosis activation in Alzheimer’s disease. Nat Neurosci (2017) 20(9):1236–46. doi: 10.1038/nn.4608
206. Xu C, Wu J, Wu Y, Ren Z, Yao Y, Chen G, et al. TNF-α-dependent neuronal necroptosis regulated in Alzheimer’s disease by coordination of RIPK1-p62 complex with autophagic UVRAG. Theranostics. (2021) 11(19):9452–69. doi: 10.7150/thno.62376
207. Balusu S, Horré K, Thrupp N, Craessaerts K, Snellinx A, Serneels L, et al. MEG3 activates necroptosis in human neuron xenografts modeling Alzheimer’s disease. Sci (New York NY). (2023) 381(6663):1176–82. doi: 10.1126/science.abp9556
208. Ye M, Gao R, Chen S, Wei M, Wang J, Zhang B, et al. Downregulation of MEG3 and upregulation of EZH2 cooperatively promote neuroblastoma progression. J Cell Mol Med (2022) 26(8):2377–91. doi: 10.1111/jcmm.17258
209. Ye M, Lu H, Tang W, Jing T, Chen S, Wei M, et al. Downregulation of MEG3 promotes neuroblastoma development through FOXO1-mediated autophagy and mTOR-mediated epithelial-mesenchymal transition. Int J Biol Sci (2020) 16(15):3050–61. doi: 10.7150/ijbs.48126
210. Sun KX, Wu DD, Chen S, Zhao Y, Zong ZH. LncRNA MEG3 inhibit endometrial carcinoma tumorigenesis and progression through PI3K pathway. Apoptosis: an Int J programmed Cell death. (2017) 22(12):1543–52. doi: 10.1007/s10495-017-1426-7
211. Jing X, Han J, Zhang J, Chen Y, Yuan J, Wang J, et al. Long non-coding RNA MEG3 promotes cisplatin-induced nephrotoxicity through regulating AKT/TSC/mTOR-mediated autophagy. Int J Biol Sci (2021) 17(14):3968–80. doi: 10.7150/ijbs.58910
212. Wang S, Yang Y, Yu Z, Song M, Liu Z, Shan S, et al. Mitophagy suppresses motor neuron necroptotic mitochondrial damage and alleviates necroptosis that converges to SARM1 in acrylamide-induced dying-back neuropathy. J neurochemistry. (2023) 166(3):588–608. doi: 10.1111/jnc.15889
213. Ogasawara M, Yano T, Tanno M, Abe K, Ishikawa S, Miki T, et al. Suppression of autophagic flux contributes to cardiomyocyte death by activation of necroptotic pathways. J Mol Cell Cardiol (2017) 108:203–13. doi: 10.1016/j.yjmcc.2017.06.008
214. Xu D, Ma Y, Peng C, Gan Y, Wang Y, Chen Z, et al. Differently surface-labeled polystyrene nanoplastics at an environmentally relevant concentration induced Crohn’s ileitis-like features via triggering intestinal epithelial cell necroptosis. Environ Int (2023) 176:107968. doi: 10.1016/j.envint.2023.107968
215. Lin JX, Xu CY, Wu XM, Che L, Li TY, Mo SM, et al. Rab7a-mTORC1 signaling-mediated cholesterol trafficking from the lysosome to mitochondria ameliorates hepatic lipotoxicity induced by aflatoxin B1 exposure. Chemosphere. (2023) 320:138071. doi: 10.1016/j.chemosphere.2023.138071
216. Sandmaier BM, Kornblit B, Storer BE, Olesen G, Maris MB, Langston AA, et al. Addition of sirolimus to standard cyclosporine plus mycophenolate mofetil-based graft-versus-host disease prophylaxis for patients after unrelated non-myeloablative haemopoietic stem cell transplantation: a multicentre, randomised, phase 3 trial. Lancet Haematology. (2019) 6(8):e409–e18. doi: 10.1016/S2352-3026(19)30088-2
217. Wegener A, Andersen NS, Friis LS, Petersen SL, Schjødt I, Kornblit B, et al. Triple-drug graft-versus-host disease prophylaxis after HLA-matched unrelated donor nonmyeloablative allogenic hematopoietic stem cell transplantation. Transplant Cell Ther (2023) 29(9):575.e1–.e6. doi: 10.1016/j.jtct.2023.05.022
218. Kornblit B, Storer BE, Andersen NS, Maris MB, Chauncey TR, Petersdorf EW, et al. Sirolimus with CSP and MMF as GVHD prophylaxis for allogeneic transplantation with HLA antigen-mismatched donors. Blood. (2020) 136(13):1499–506. doi: 10.1182/blood.2020005338
219. Pidala J, Hamadani M, Dawson P, Martens M, Alousi AM, Jagasia M, et al. Randomized multicenter trial of sirolimus vs prednisone as initial therapy for standard-risk acute GVHD: the BMT CTN 1501 trial. Blood. (2020) 135(2):97–107. doi: 10.1182/blood.2019003125
220. Cillo U, Saracino L, Vitale A, Bertacco A, Salizzoni M, Lupo F, et al. Very early introduction of everolimus in de novo liver transplantation: results of a multicenter, prospective, randomized trial. Liver transplantation: Off Publ Am Assoc Study Liver Dis Int Liver Transplant Society. (2019) 25(2):242–51. doi: 10.1002/lt.25400
221. Makrakis D, Wright JL, Roudier MP, Garcia J, Vakar-Lopez F, Porter MP, et al. A phase 1/2 study of rapamycin and cisplatin/gemcitabine for treatment of patients with muscle-invasive bladder cancer. Clin genitourinary cancer. (2023) 21(2):265–72. doi: 10.1016/j.clgc.2022.12.003
222. Abuzakhm SM, Sukrithan V, Fruth B, Qin R, Strosberg J, Hobday TJ, et al. A phase II study of bevacizumab and temsirolimus in advanced extra-pancreatic neuroendocrine tumors. Endocrine-related Cancer (2023) 30(11):e220301. doi: 10.1530/ERC-22-0301
223. Tesch H, Stoetzer O, Decker T, Kurbacher CM, Marmé F, Schneeweiss A, et al. Efficacy and safety of everolimus plus exemestane in postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative locally advanced or metastatic breast cancer: Results of the single-arm, phase IIIB 4EVER trial. Int J cancer. (2019) 144(4):877–85. doi: 10.1002/ijc.31738
224. Nathan CO, Hayes DN, Karrison T, Harismendy O, Flores JM, Moore-Medlin T, et al. A randomized multi-institutional phase II trial of everolimus as adjuvant therapy in patients with locally advanced squamous cell cancer of the head and neck. Clin Cancer research: an Off J Am Assoc Cancer Res (2022) 28(23):5040–8. doi: 10.1158/1078-0432.CCR-21-4290
225. Wu W, Chen J, Deng H, Jin L, He Z, Rao N, et al. Neoadjuvant everolimus plus letrozole versus fluorouracil, epirubicin and cyclophosphamide for ER-positive, HER2-negative breast cancer: a randomized pilot trial. BMC cancer. (2021) 21(1):862. doi: 10.1186/s12885-021-08612-y
226. Slomovitz BM, Filiaci VL, Walker JL, Taub MC, Finkelstein KA, Moroney JW, et al. A randomized phase II trial of everolimus and letrozole or hormonal therapy in women with advanced, persistent or recurrent endometrial carcinoma: A GOG Foundation study. Gynecologic Oncol (2022) 164(3):481–91. doi: 10.1016/j.ygyno.2021.12.031
227. Heudel P, Frenel JS, Dalban C, Bazan F, Joly F, Arnaud A, et al. Safety and efficacy of the mTOR inhibitor, vistusertib, combined with anastrozole in patients with hormone receptor-positive recurrent or metastatic endometrial cancer: the VICTORIA multicenter, open-label, phase 1/2 randomized clinical trial. JAMA Oncol (2022) 8(7):1001–9. doi: 10.1001/jamaoncol.2022.1047
228. Banerjee S, Giannone G, Clamp AR, Ennis DP, Glasspool RM, Herbertson R, et al. Efficacy and safety of weekly paclitaxel plus vistusertib vs paclitaxel alone in patients with platinum-resistant ovarian high-grade serous carcinoma: the OCTOPUS multicenter, phase 2, randomized clinical trial. JAMA Oncol (2023) 9(5):675–82. doi: 10.1001/jamaoncol.2022.7966
229. Kaley TJ, Panageas KS, Pentsova EI, Mellinghoff IK, Nolan C, Gavrilovic I, et al. Phase I clinical trial of temsirolimus and perifosine for recurrent glioblastoma. Ann Clin Trans neurology. (2020) 7(4):429–36. doi: 10.1002/acn3.51009
230. Moore HCF, Barlow WE, Somlo G, Gralow JR, Schott AF, Hayes DF, et al. A randomized trial of fulvestrant, everolimus, and anastrozole for the front-line treatment of patients with advanced hormone receptor-positive breast cancer, SWOG S1222. Clin Cancer research: an Off J Am Assoc Cancer Res (2022) 28(4):611–7. doi: 10.1158/1078-0432.CCR-21-3131
231. Schnitzbauer AA, Filmann N, Adam R, Bachellier P, Bechstein WO, Becker T, et al. mTOR inhibition is most beneficial after liver transplantation for hepatocellular carcinoma in patients with active tumors. Ann surgery. (2020) 272(5):855–62. doi: 10.1097/SLA.0000000000004280
232. Assis BPS, Lasmar MF, Fabreti-Oliveira RA, Araujo SA, Oliveira J, Wanderley DC, et al. An open-label randomized clinical trial to evaluate the efficacy of everolimus versus tacrolimus in triple maintenance immunosuppressive therapy for kidney transplant patients. Braz J Med Biol Res = Rev Bras pesquisas medicas e biologicas. (2021) 54(4):e9369. doi: 10.1590/1414-431x20209369
233. Wallis RS, Ginindza S, Beattie T, Arjun N, Likoti M, Edward VA, et al. Adjunctive host-directed therapies for pulmonary tuberculosis: a prospective, open-label, phase 2, randomised controlled trial. Lancet Respir Med (2021) 9(8):897–908. doi: 10.1016/S2213-2600(20)30448-3
234. Taber DJ, Chokkalingam A, Su Z, Self S, Miller D, Srinivas T. Randomized controlled trial assessing the impact of everolimus and low-exposure tacrolimus on graft outcomes in kidney transplant recipients. Clin transplantation. (2019) 33(10):e13679. doi: 10.1111/ctr.13679
235. Kaminski H, Kamar N, Thaunat O, Bouvier N, Caillard S, Garrigue I, et al. Incidence of cytomegalovirus infection in seropositive kidney transplant recipients treated with everolimus: A randomized, open-label, multicenter phase 4 trial. Am J transplantation: Off J Am Soc Transplant Am Soc Transplant Surgeons. (2022) 22(5):1430–41. doi: 10.1111/ajt.16946
236. Andreassen AK, Broch K, Eiskjær H, Karason K, Gude E, Mølbak D, et al. Blood pressure in de novo heart transplant recipients treated with everolimus compared with a cyclosporine-based regimen: results from the randomized SCHEDULE trial. Transplantation. (2019) 103(4):781–8. doi: 10.1097/TP.0000000000002445
237. Palma JA, Martinez J, Millar Vernetti P, Ma T, Perez MA, Zhong J, et al. mTOR inhibition with sirolimus in multiple system atrophy: A randomized, double-blind, placebo-controlled futility trial and 1-year biomarker longitudinal analysis. Movement disorders: Off J Movement Disord Society. (2022) 37(4):778–89. doi: 10.1002/mds.28923
238. Abdallah CG, Averill LA, Gueorguieva R, Goktas S, Purohit P, Ranganathan M, et al. Modulation of the antidepressant effects of ketamine by the mTORC1 inhibitor rapamycin. Neuropsychopharmacology: Off Publ Am Coll Neuropsychopharmacol (2020) 45(6):990–7. doi: 10.1038/s41386-020-0644-9
239. Overwater IE, Rietman AB, Mous SE, Bindels-de Heus K, Rizopoulos D, Ten Hoopen LW, et al. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology. (2019) 93(2):e200–e9. doi: 10.1212/WNL.0000000000007749
240. Maruani A, Tavernier E, Boccara O, Mazereeuw-Hautier J, Leducq S, Bessis D, et al. Sirolimus (Rapamycin) for slow-flow malformations in children: the observational-phase randomized clinical PERFORMUS trial. JAMA Dermatol (2021) 157(11):1289–98. doi: 10.1001/jamadermatol.2021.3459
241. Viana LA, Cristelli MP, Basso G, Santos DW, Dantas MTC, Dreige YC, et al. Conversion to mTOR inhibitor to reduce the incidence of cytomegalovirus recurrence in kidney transplant recipients receiving preemptive treatment: A prospective, randomized trial. Transplantation. (2023) 107(8):1835–45. doi: 10.1097/TP.0000000000004559
242. Gustafsson F, Andreassen AK, Andersson B, Eiskjær H, Rådegran G, Gude E, et al. Everolimus initiation with early calcineurin inhibitor withdrawal in de novo heart transplant recipients: long-term follow-up from the randomized SCHEDULE study. Transplantation. (2020) 104(1):154–64. doi: 10.1097/TP.0000000000002702
243. Andreassen AK, Broch K, Eiskjær H, Karason K, Gude E, Mølbak D, et al. Blood Pressure in De Novo Heart Transplant Recipients Treated With Everolimus Compared With a Cyclosporine-based Regimen: Results From the Randomized SCHEDULE Trial. Transplantation. (2019) 103(4):781–8. doi: 10.1097/TP.0000000000002445
244. Fan Y, Sun T, Shao Z, Zhang Q, Ouyang Q, Tong Z, et al. Effectiveness of Adding Everolimus to the First-line Treatment of Advanced Breast Cancer in Premenopausal Women Who Experienced Disease progression while receiving selective estrogen receptor modulators: A phase 2 randomized clinical trial. JAMA Oncol (2021) 7(10):e213428. doi: 10.1001/jamaoncol.2021.3428
245. Motzer RJ, Escudier B, George S, Hammers HJ, Srinivas S, Tykodi SS, et al. Nivolumab versus everolimus in patients with advanced renal cell carcinoma: Updated results with long-term follow-up of the randomized, open-label, phase 3 CheckMate 025 trial. Cancer. (2020) 126(18):4156–67. doi: 10.1002/cncr.33033
246. Decker T, Marschner N, Muendlein A, Welt A, Hagen V, Rauh J, et al. VicTORia: a randomised phase II study to compare vinorelbine in combination with the mTOR inhibitor everolimus versus vinorelbine monotherapy for second-line chemotherapy in advanced HER2-negative breast cancer. Breast Cancer Res Treat (2019) 176(3):637–47. doi: 10.1007/s10549-019-05280-2
247. Palma JA, Martinez J, Millar Vernetti P, Ma T, Perez MA, Zhong J, et al. mTOR Inhibition with Sirolimus in Multiple System Atrophy: A Randomized, Double-Blind, Placebo-Controlled Futility Trial and 1-Year Biomarker Longitudinal Analysis. Movement Disord Off J Movement Disord Society. (2022) 37(4):778–89. doi: 10.1002/mds.28923
248. Abdallah CG, Averill LA, Gueorguieva R, Goktas S, Purohit P, Ranganathan M, et al. Modulation of the antidepressant effects of ketamine by the mTORC1 inhibitor rapamycin. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol (2020) 45(6):990–7. doi: 10.1038/s41386-020-0644-9
249. Bergmann L, Grünwald V, Maute L, Grimm MO, Weikert S, Schleicher J, et al. A randomized Phase IIa Trial with Temsirolimus versus Sunitinib in Advanced Non-Clear Cell Renal Cell Carcinoma: An Intergroup Study of the CESAR Central European Society for Anticancer Drug Research-EWIV and the Interdisciplinary Working Group on Renal Cell Cancer (IAGN) of the German Cancer Society. Oncol Res Treat (2020) 43(7-8):333–9. doi: 10.1159/000508450
250. Bamias A, Karavasilis V, Gavalas N, Tzannis K, Samantas E, Aravantinos G, et al. The combination of bevacizumab/temsirolimus after first-line anti-VEGF therapy in advanced renal-cell carcinoma: a clinical and biomarker study. Int J Clin Oncol (2019) 24(4):411–9. doi: 10.1007/s10147-018-1361-9
251. Mandrioli J, D’Amico R, Zucchi E, De Biasi S, Banchelli F, Martinelli I, et al. Randomized, double-blind, placebo-controlled trial of rapamycin in amyotrophic lateral sclerosis. Nat Commun (2023) 14(1):4970. doi: 10.1038/s41467-023-40734-8
252. Overwater IE, Rietman AB, Mous SE, Bindels-de Heus K, Rizopoulos D, Ten Hoopen LW, et al. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology. (2019) 93(2):e200–e9. doi: 10.1212/WNL.0000000000007749
253. Maruani A, Tavernier E, Boccara O, Mazereeuw-Hautier J, Leducq S, Bessis D, et al. Sirolimus (Rapamycin) for Slow-Flow Malformations in Children: The Observational-Phase Randomized Clinical PERFORMUS Trial. JAMA Dermatol (2021) 157(11):1289–98. doi: 10.1001/jamadermatol.2021.3459
254. Tedesco-Silva H, Pascual J, Viklicky O, Basic-Jukic N, Cassuto E, Kim DY, et al. Safety of everolimus with reduced calcineurin inhibitor exposure in de novo kidney transplants: an analysis from the randomized TRANSFORM study. Transplantation. (2019) 103(9):1953–63. doi: 10.1097/TP.0000000000002626
255. Huijts CM, Lougheed SM, Bodalal Z, van Herpen CM, Hamberg P, Tascilar M, et al. The effect of everolimus and low-dose cyclophosphamide on immune cell subsets in patients with metastatic renal cell carcinoma: results from a phase I clinical trial. Cancer immunology immunotherapy: CII. (2019) 68(3):503–15. doi: 10.1007/s00262-018-2288-8
256. Hutson TE, Michaelson MD, Kuzel TM, Agarwal N, Molina AM, Hsieh JJ, et al. A single-arm, multicenter, phase 2 study of lenvatinib plus everolimus in patients with advanced non-clear cell renal cell carcinoma. Eur urology. (2021) 80(2):162–70. doi: 10.1016/j.eururo.2021.03.015
257. Ryan CW, Tangen CM, Heath EI, Stein MN, Meng MV, Alva AS, et al. Adjuvant everolimus after surgery for renal cell carcinoma (EVEREST): a double-blind, placebo-controlled, randomised, phase 3 trial. Lancet (London England). (2023) 402(10407):1043–51. doi: 10.1016/S0140-6736(23)00913-3
258. Motzer R, Alekseev B, Rha SY, Porta C, Eto M, Powles T, et al. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. New Engl J Med (2021) 384(14):1289–300. doi: 10.1056/NEJMoa2035716
259. Motzer R, Porta C, Alekseev B, Rha SY, Choueiri TK, Mendez-Vidal MJ, et al. Health-related quality-of-life outcomes in patients with advanced renal cell carcinoma treated with lenvatinib plus pembrolizumab or everolimus versus sunitinib (CLEAR): a randomised, phase 3 study. Lancet Oncol (2022) 23(6):768–80. doi: 10.1016/S1470-2045(22)00212-1
260. Ji N, Mukherjee N, Reyes RM, Gelfond J, Javors M, Meeks JJ, et al. Rapamycin enhances BCG-specific γδ T cells during intravesical BCG therapy for non-muscle invasive bladder cancer: a randomized, double-blind study. J immunotherapy Cancer (2021) 9(3):e001941. doi: 10.1136/jitc-2020-001941
261. Bardia A, Modi S, Oliveira M, Cortes J, Campone M, Ma B, et al. Phase ib dose-escalation/expansion trial of ribociclib in combination with everolimus and exemestane in postmenopausal women with HR(+), HER2(-) advanced breast cancer. Clin Cancer research: an Off J Am Assoc Cancer Res (2020) 26(24):6417–28. doi: 10.1158/1078-0432.CCR-20-1068
262. Bachelot T, Cottu P, Chabaud S, Dalenc F, Allouache D, Delaloge S, et al. Everolimus added to adjuvant endocrine therapy in patients with high-risk hormone receptor-positive, human epidermal growth factor receptor 2-negative primary breast cancer. J Clin oncology: Off J Am Soc Clin Oncol (2022) 40(32):3699–708. doi: 10.1200/JCO.21.02179
263. Guarneri V, Giorgi CA, Cinieri S, Bengala C, Mariani G, Bisagni G, et al. Everolimus plus aromatase inhibitors as maintenance therapy after first-line chemotherapy: Final results of the phase III randomised MAIN-A (MAINtenance Afinitor) trial. Eur J Cancer (Oxford England: 1990). (2021) 154:21–9. doi: 10.1016/j.ejca.2021.05.008
264. Wright KD, Yao X, London WB, Kao PC, Gore L, Hunger S, et al. A POETIC Phase II study of continuous oral everolimus in recurrent, radiographically progressive pediatric low-grade glioma. Pediatr Blood cancer. (2021) 68(2):e28787. doi: 10.1002/pbc.28787
265. Wen HY, Wang J, Zhang SX, Luo J, Zhao XC, Zhang C, et al. Low-dose sirolimus immunoregulation therapy in patients with active rheumatoid arthritis: A 24-week follow-up of the randomized, open-label, parallel-controlled trial. J Immunol Res (2019) 2019:7684352. doi: 10.1155/2019/7684352
266. Wu C, Wang Q, Xu D, Li M, Zeng X. Sirolimus for patients with connective tissue disease-related refractory thrombocytopenia: a single-arm, open-label clinical trial. Rheumatol (Oxford England). (2021) 60(6):2629–34. doi: 10.1093/rheumatology/keaa645
267. Mousavi-Hasanzadeh M, Bagheri B, Mehrabi S, Eghbali A, Eghbali A. Sirolimus versus cyclosporine for the treatment of pediatric chronic immune thrombocytopenia: A randomized blinded trial. Int immunopharmacology. (2020) 88:106895. doi: 10.1016/j.intimp.2020.106895
Keywords: mTOR, necroptosis, autophagy, RIPK, ROS
Citation: Xie Y, Zhao G, Lei X, Cui N and Wang H (2023) Advances in the regulatory mechanisms of mTOR in necroptosis. Front. Immunol. 14:1297408. doi: 10.3389/fimmu.2023.1297408
Received: 20 September 2023; Accepted: 01 December 2023;
Published: 18 December 2023.
Edited by:
A. Brent Carter, University of Alabama at Birmingham, United StatesReviewed by:
Nieves Peltzer, University of Cologne, GermanyCopyright © 2023 Xie, Zhao, Lei, Cui and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Na Cui, cHVtY2hjbkAxNjMuY29t; Hao Wang, bmV3d2FuZ2hhb0Bob3RtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.