 Jason E. Hawkes
Jason E. Hawkes Sudha Visvanathan2
Sudha Visvanathan2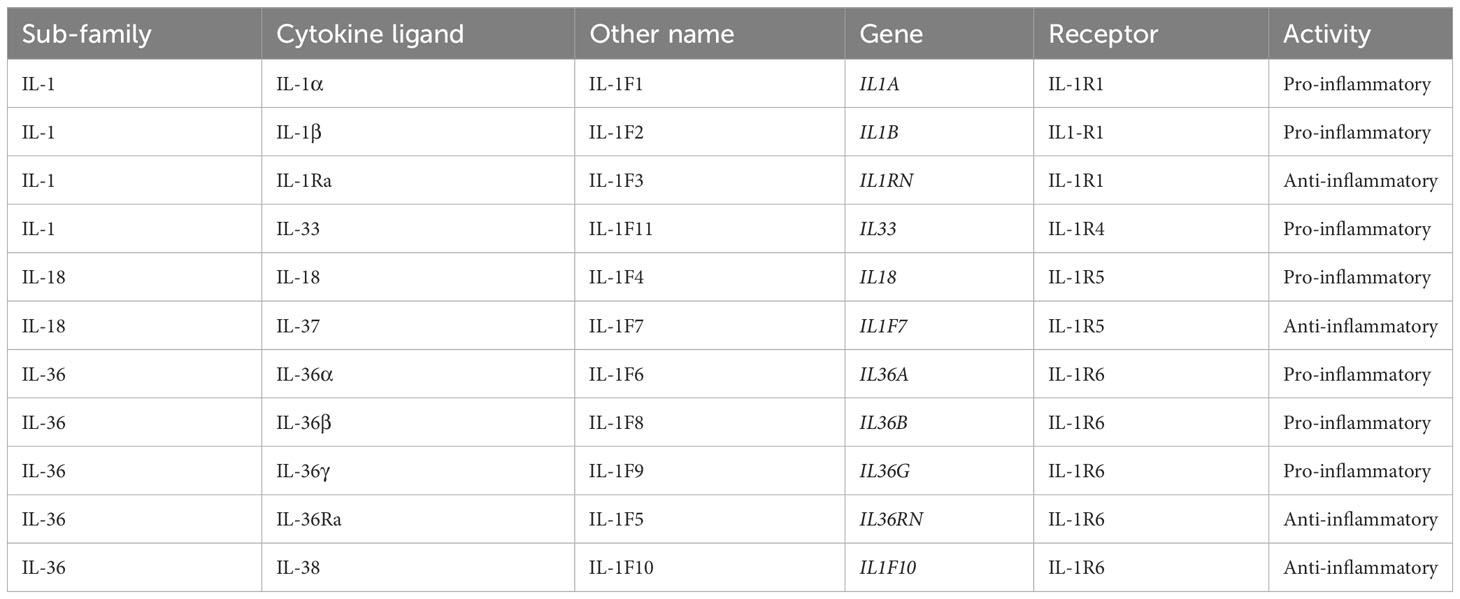

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 November 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1292941
Generalized pustular psoriasis (GPP) is a rare, chronic, inflammatory skin disorder characterized by recurrent flares associated with skin erythema, desquamation, and widespread superficial sterile pustules, which may be severe (“lakes of pus”). Systemic symptoms are often present, including malaise, fever, and skin pain. In GPP, innate immune responses are driven by abnormal activation of the interleukin (IL)-36-chemokine-neutrophil axis and excessive neutrophil infiltration. This review highlights the IL-36 pathway in the context of the IL-1 superfamily and describes how unopposed IL-36 signaling can lead to the development of GPP. Targeted inhibition of the IL-36 receptor (IL-36R) is an attractive therapeutic strategy in the treatment of GPP, including flare prevention and sustained disease control. Spesolimab is a first-in-class, humanized, monoclonal antibody that binds specifically to the IL-36R and antagonizes IL-36 signaling. Spesolimab was approved by the US Food and Drug Administration in September 2022 to treat GPP flares in adults and was subsequently approved for GPP flare treatment in other countries across the world. Anti-IL-36R therapy, such as spesolimab, can mitigate flares and address flare prevention in GPP, presumably through rebalancing IL-36 signaling and modulating the pro-inflammatory response of the downstream effectors.
Generalized pustular psoriasis (GPP) is a rare, chronic, inflammatory skin disorder, and is characterized by recurrent flares of erythema, desquamation, and the widespread eruption of superficial sterile pustules (1). In severe cases, the pustules may coalesce to form larger lesions known as “lakes of pus”. The key features of GPP are shown in Figure 1. Systemic symptoms often occur during episodic flares (1), including fever, malaise, and skin pain. Severe GPP flares often necessitate emergency or inpatient hospital care (2), due to the potential for complications, such as sepsis, heart failure, renal failure, and even death, if timely treatment is not provided (2, 3). The term “skin failure” was recently proposed to describe the potentially catastrophic endpoints of GPP (4). Estimates of GPP prevalence vary considerably in different regions of the world, ranging from approximately 2 to 120 cases per million persons (5–9). Reported mortality rates in patients with GPP are also variable, ranging from 0 to 3.3 deaths per 100 patient-years, with older studies (before 2000) stating higher rates than more recent studies (8). A 2021 study of Japanese patients with GPP who required hospitalization (N = 1516) reported a mortality rate of 4.2% (patient-year data were not available) (8, 10).
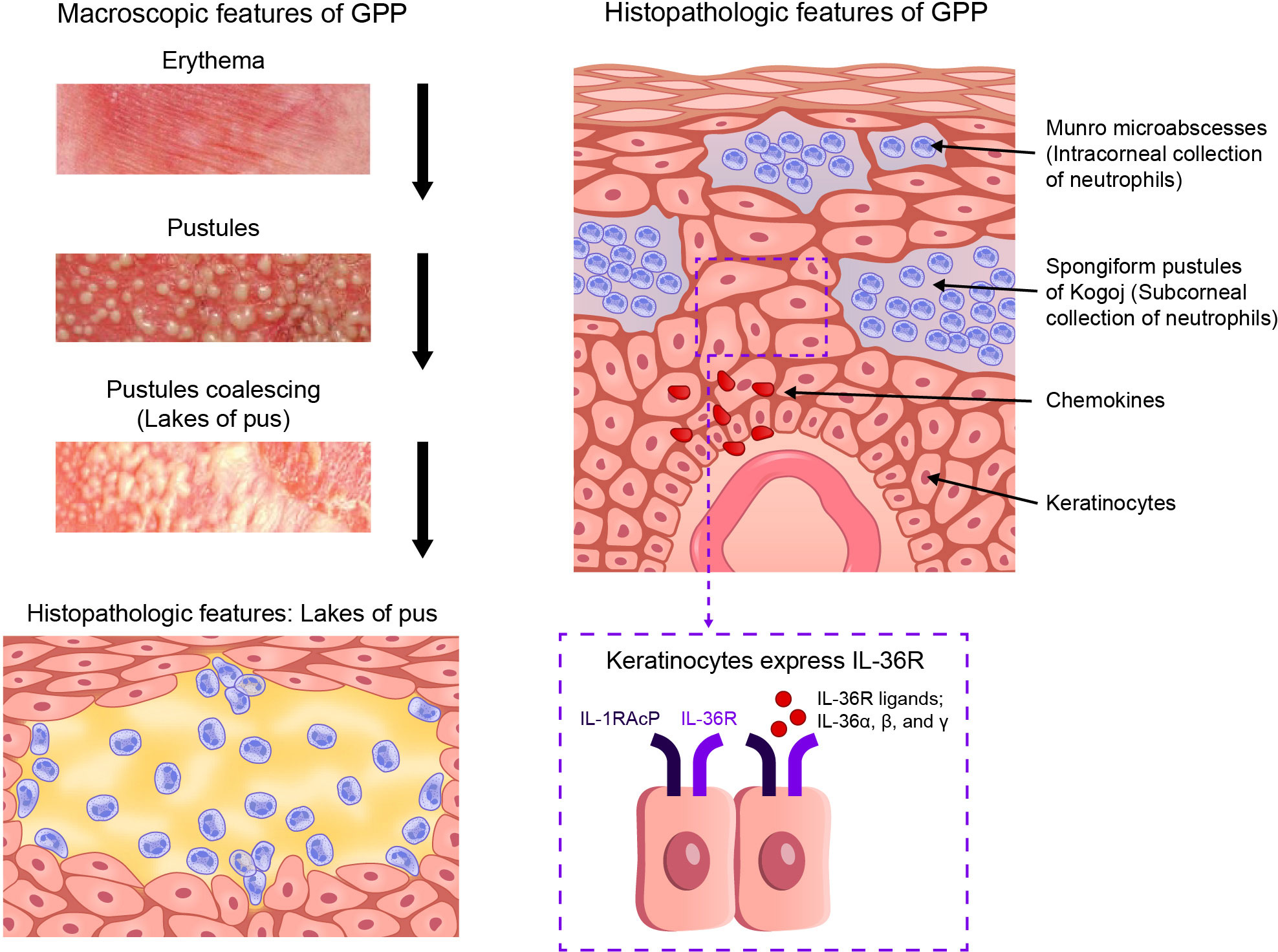
Figure 1 Key features of GPP. GPP, generalized pustular psoriasis; IL-1RAcP, interleukin-1 receptor accessory protein*; IL-36R, interleukin-36 receptor. (*Also known as IL-1R3).
The clinical course of GPP is often heterogenous, and may present as relapsing (>1 episode within weeks, months, or years) or persistent disease (episode lasting >3 months) (1). GPP flares are often precipitated by a trigger, such as infection, emotional stress, pregnancy, hypocalcemia, or exposure to sunlight. A GPP flare may also be triggered by the withdrawal of systemic corticosteroids (11), or by exposure to a range of drugs, including lithium, antimalarials, ustekinumab, and some tumor necrosis factor (TNF) antagonists (12, 13).
Recent clinical, histological, and genetic data indicate that GPP is distinct from psoriasis vulgaris (PV, also called plaque psoriasis) (1, 14–16), and warrants separate diagnosis. While GPP can manifest concurrently with PV (1, 9, 17), GPP may occur in patients with no prior history of psoriatic disease (3). Abnormal activation of the interleukin (IL)-36-chemokine-neutrophil axis, dysregulation of innate immune responses, and ensuing excessive neutrophil infiltration are implicated in the pathogenesis of GPP (18); whereas, PV is an autoimmune disease characterized predominantly by IL-23/17 signaling and self-sustaining inflammatory cycles [positive feed-forward inflammatory response (19)] that lead to disordered proliferation and abnormal differentiation of keratinocytes (20).
The purpose of this review is to provide an overview of the IL-36 pathway in the context of the IL-1 superfamily and describe how unopposed IL-36 signaling can lead to the development of GPP. We will also discuss spesolimab, a novel first-in-class humanized monoclonal antibody that binds specifically to IL-36 receptor (IL-36R) and antagonizes IL-36 signaling. Spesolimab was approved by the US Food and Drug Administration (FDA) in September 2022 for the treatment of GPP flares in adults (21, 22), and was subsequently approved for GPP flare treatment in other countries across the world.
IL-36 cytokines belong to the IL-1 superfamily of cytokines (23–25). These are important regulators of the innate immune system, manifested by inflammation (26), and are essential for skin barrier function (27). IL-1 (α and β) was discovered first and, thus, is the best characterized of the 11 members of the IL-1 cytokine superfamily (25). IL-1 cytokines include receptor agonists (IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, and IL-36γ), receptor antagonists (IL-1Ra, IL-36Ra, and IL-38), and an anti-inflammatory cytokine (IL-37) (23). Details of IL-1 family proteins and genes are presented in Table 1 (23, 28).
IL-1 cytokine members are grouped into three subfamilies (IL-1, IL-18, and IL-36) based on their consensus sequence and cognate (i.e. corresponding) receptors; IL-1 and IL-36 subfamilies share accessory protein IL-1R3 (IL-1RAcP) (26) as their co-receptor, while the IL-18 subfamily utilizes a different co-receptor (25). Binding of an agonist to its receptor causes recruitment of the co-receptor, and activation of intracellular signaling pathways that result in increased gene expression of pro-inflammatory mediators (25), as shown for IL-36 in Figure 2 (29). Formation of the receptor complex recruits intracellular adaptor proteins, including myeloid differentiation primary response 88 (Myd88), IL-1 receptor-associated kinase (IRAK), and TNF receptor-associated factor, which subsequently activate mitogen-activated protein kinase (MAPK) and nuclear factor-κB (NFκB) pathways. NFκB upregulates a broad range of pro-inflammatory gene products. IL-1 and IL-36 cytokines are negatively regulated by their receptor antagonist (IL-1Ra and IL-36Ra, respectively), via competitive binding for the receptor site (25). IL-36R is also inhibited by cytokine IL-38, which shares 40% sequence homology with IL-36Ra (and IL-1Ra) (25).
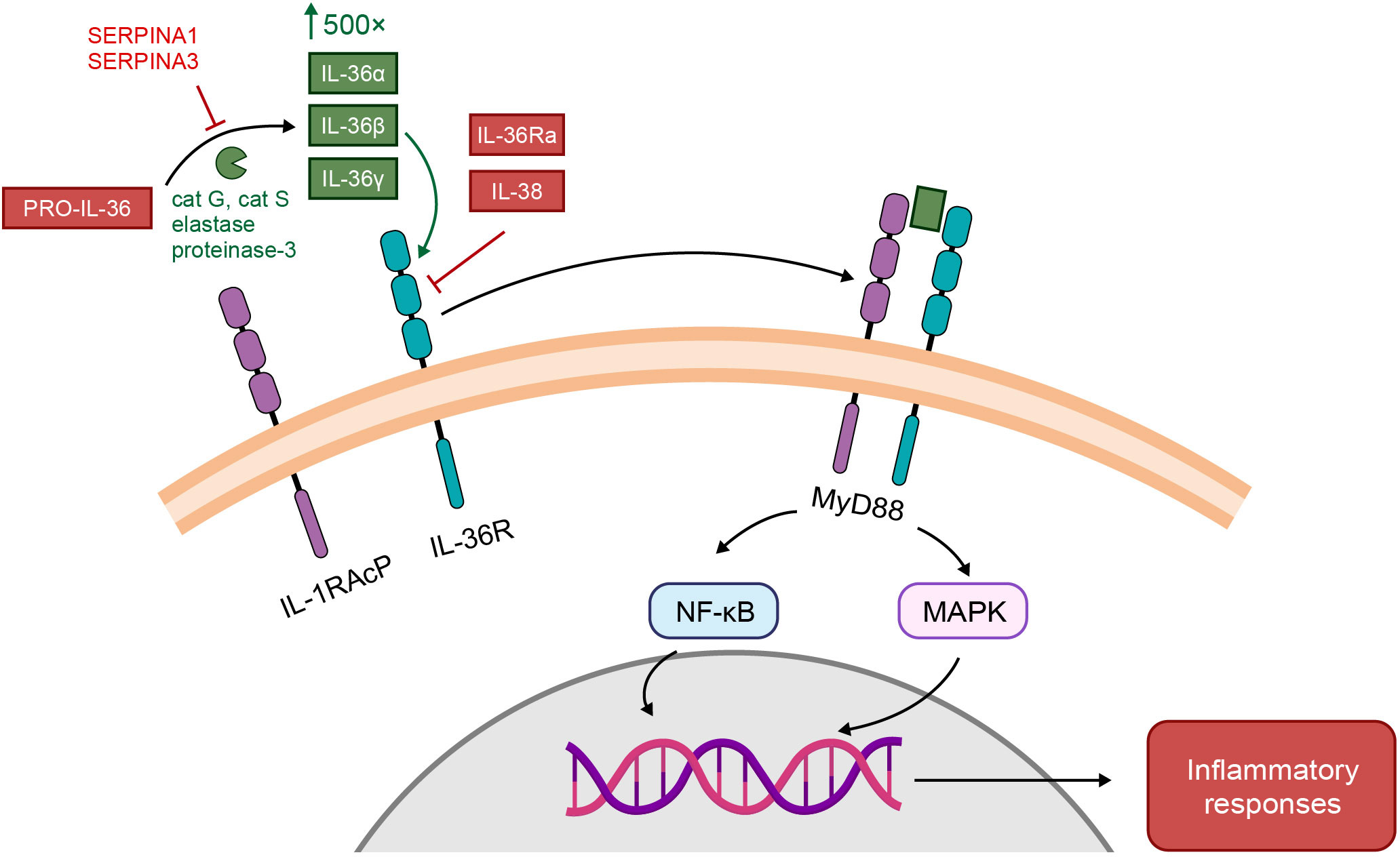
Figure 2 Receptor and signaling pathways activated by IL-1 and IL-36 (29). IL-36 activation pathway. IL-36 cytokines are secreted as low-activity precursors, pro-IL-36, which by the action of various proteases (cat G-cathepsin G; cat S-cathepsin S; elastase; proteinase-3) are cleaved into biologically active IL-36 agonists, IL-36α, IL-36β, and IL-36γ or antagonist IL-36Ra. IL-36 agonist processing increases their biologic activity by roughly 500-fold. IL-36 agonists form a binary complex with IL-36R, which recruits the IL-1 receptor accessory protein (IL-1RAcP) co-receptor. The ternary complex then binds to myeloid differentiated protein 88 (MyD88) to activate nuclear transcription factor kappa B (NF-kB) and mitogen-activated protein kinase (MAPK) signaling pathways and regulate downstream transcription of target genes and generate inflammatory responses. This pathway may be antagonized by IL-36Ra or IL-38. Alternatively, protease inhibition by SERPINA1 or SERPINA3 can prevent the generation of IL-36 agonists. From: Pathophysiology of generalized pustular psoriasis, Young KZ, Sarkar MK, Gudjonsson JE. Experimental Dermatology. 2023, Feb 13. doi: 10.1111/exd.14768. © 2023 John Wiley & Sons A/S. Reproduced with permission of John Wiley & Sons Ltd.
All IL-1 family cytokines are expressed within the skin to some extent (27). IL-36 cytokines are expressed mainly in epithelial and immune cells at barrier sites (skin, lung, and intestine) (24, 30). Receptor agonists IL-36α and IL-36β are present in healthy skin. IL-36γ is constitutively expressed at low levels by keratinocytes, and is upregulated following activation in both PV and GPP lesions (14, 31). IL-36 cytokines are released as precursors, and their activation is carried out by neutrophil-derived proteases (cathepsin G, protease 3, and elastase) (32) and cathepsin S (33).
IL-1 and IL-36 cytokines have similar actions in that their agonists bind to the cognate receptor to trigger pro-inflammatory activity and are inhibited by their respective receptor antagonist. Unopposed IL-1 or IL-36 signaling, which may be caused by loss-of-function mutations in the gene encoding the interleukin receptor antagonist (IL1RN or IL36RN, respectively), leads to autoinflammatory disease such as Deficiency of the IL-1 Receptor Antagonist (DIRA) (34, 35), and Deficiency of the IL-36 Receptor Antagonist (DITRA) (36, 37). DIRA presents at birth, or soon thereafter, with acute onset pustular dermatitis, systemic inflammation, nail dystrophy, and inflammation of the bone (sterile osteomyelitis) and periosteum; DIRA is rapidly fatal if untreated (34, 35, 38–40). DITRA usually presents in childhood with repeated and severe manifestations of GPP, including high-grade fever, but inflammation of the bone is lacking (36, 37, 40–42). DIRA and DITRA represent autosomal recessive loss-of-function mutations in genes IL1RN and IL36RN, respectively (Table 2) (34–42).
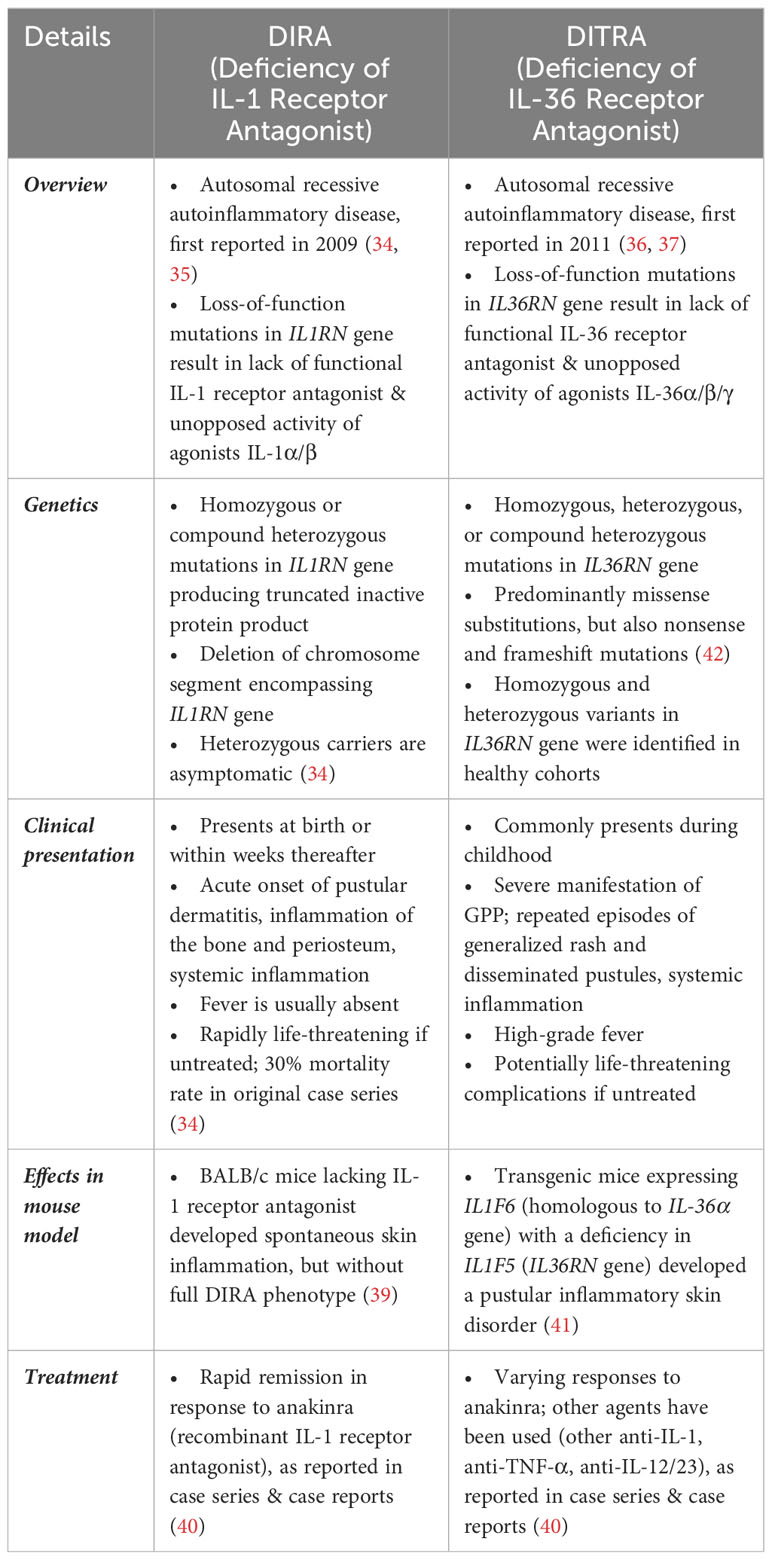
Table 2 Genetic and Clinical Features of DIRA and DITRA.
In 2011, homozygous missense mutations in the IL36RN gene were identified in nine Tunisian families in which multiple members had GPP (36). Homozygous and compound heterozygous missense IL36RN mutations were also identified in three of five unrelated individuals with GPP (without associated PV) (37). Since that discovery, multiple types of mutation have been identified in IL36RN and associated with GPP (42–44). IL36RN mutations were found far more frequently in patients with GPP alone than in those with GPP plus PV (15, 45–48), and the presence of IL36RN mutations was associated with early onset of disease (15, 44, 47). Several other genes have been identified with loss-of-function mutations that are associated with a predisposition to develop GPP; namely, CARD14 (caspase-activating recruitment domain member 14, also known as CARMA2) (49, 50), AP1S3 (adaptor protein 1 complex subunit sigma 3 (51, 52), MPO (myeloperoxidase) (53, 54), SERPINA3 (serine protease inhibitor A3) (55), and possibly SERPINA1 (56). These genes are all involved in regulating the IL-1/IL-36-chemokine-neutrophil axis (42, 57), as shown in Figure 3 (58). However, one study reported that 64% (39/61) of patients with GPP lacked any causal or disease-contributing mutations in IL36RN, CARD14, or AP1S3 genes (59). Also, none of 11 patients with a heterozygous IL36RN mutation carried a second non-coding IL36RN mutation, suggesting the presence of additional disease-causing genetic factors outside of IL36RN alone, but only 15% (3/20) of patients with an IL36RN gene mutation also carried a mutation in CARD14 or AP1S3 genes (59). Furthermore, no differences in gene expression in the profiles of patients with and without IL36RN mutations have been described to date (60). Thus, the genetic basis for GPP is not completely understood.
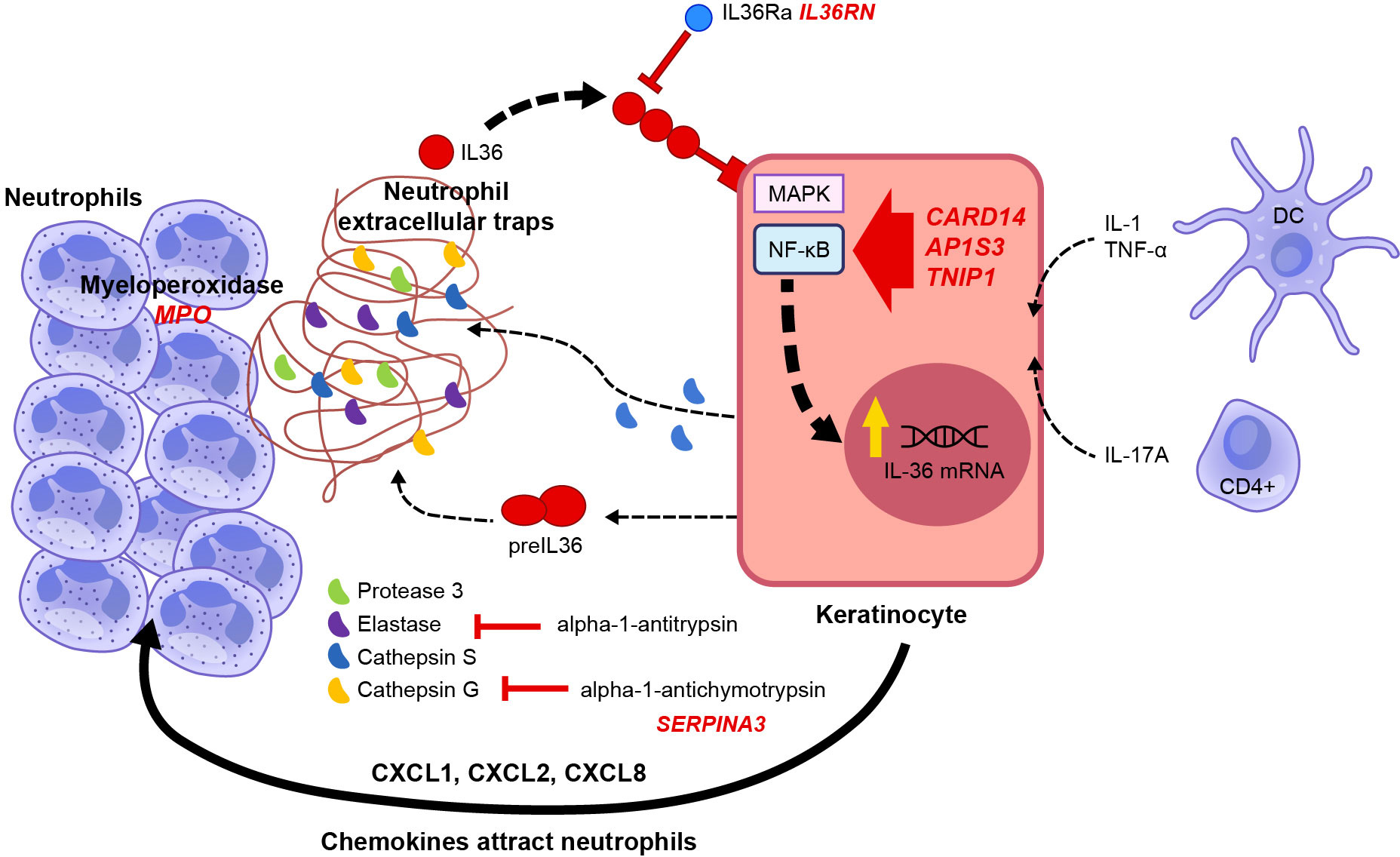
Figure 3 Genetic factors related to GPP pathogenesis (58). Schematic representation of the signal transduction pathway activated by cytokines and genes involved in IL-36 autocrine and autoinflammatory circuits. The pathogenesis of GPP is related to mutations in multiple genes, such as the human IL-1Ra gene (IL1RN), IL-36Ra (IL36RN), caspase recruitment domain-containing protein 14 (CARD14), adapter protein complex 1 subunit sigma 3 (AP1S3), TNFAIP3-interacting protein 1 (TNIP1), and the gene coding for alpha-1 antichymotrypsin, also known as serine protease inhibitor gene serpin family A member 3 (SERPINA3). IL-1, TNF, and IL-17A promote the expression of IL-36 by keratinocytes. IL-36 cytokines are released as precursors requiring enzymatical cleavage by neutrophil-derived proteases (elastase, cathepsin G, or protease 3) and keratinocyte-derived cathepsin S. Mature IL-36 cytokines have 500-fold greater biological activity than their precursors and bind to IL-36R on the keratinocyte cell surface, acting in an autocrine manner to further induce IL-36 expression. In addition, they induce the production and secretion of neutrophil chemokines CXCL1, CXCL2, CXCL6, and CXCL8 (IL-8), increasing the attraction of neutrophils to the skin. Serine protease inhibitors such as alpha-1 antitrypsin or alpha-1 antichymotrypsin (encoded by SERPINA1 and SERPINA3, respectively) can inhibit neutrophil proteases. Adapted from Iznardo et al. (25). From: Generalized Pustular Psoriasis: A Review on Clinical Characteristics, Diagnosis, and Treatment; Rivera-Díaz R, Daudén E, Carrascosa JM, Cueva P, Puig L; Dermatology and Therapy (Heidelb). 2023 Mar;13(3):673-688.
The overexpression of IL-36 in GPP and PV lesional skin in patients is similar to data obtained from mouse models of skin inflammation. Transgenic mice over-expressing IL1F6 (homologous to IL36α gene) and also bearing a deficiency in IL1F5 (IL36RN gene) developed a pustular inflammatory skin phenotype (41). Imiquimod (IMQ), an activator of Toll-like receptor-7, also induces psoriasis-like dermatitis in mice that is mediated via the pro-inflammatory IL-23/IL-17 axis (61). IL-36 receptor-deficient mice (Il36r–/–) were protected from IMQ-induced psoriasis-like dermatitis (62), whereas loss of IL-36Ra (Il36rn–/–) exacerbated disease severity (63). Additionally, mice deficient in IL-23, IL-17, or IL-22 were not as well protected from disease compared with Il36r–/– mice, indicating an additional distinct activity of IL-36 beyond induction of the IL-23/IL-17 signaling axis (63). Targeted deletion of Il36r–/– in mouse keratinocytes resulted in similar protection from IMQ-induced psoriasiform inflammation to that observed in mice with a global deficiency of Il36r–/–, demonstrating the key role of keratinocytes in IL-36-mediated effects (64). Goldstein et al. showed that Il36r signaling in keratinocytes is critical for IL-23 production and controls the recruitment of neutrophils at early treatment time points in the IMQ model (62). These data also indicate that IL-36-dervied signaling in keratinocytes may have an upstream role in the amplified “feed-forward” response by direct regulation of IL-17A expression, potentially via IL-23 induction (64). Il36a was required for the development of IMQ-induced murine psoriasis, whereas deletion of Il36a resulted in significant improvement in the skin lesions (65, 66); however, deficiency of Il36b or Il36g had no impact on reducing disease severity (66). Also, IL-36α expression was induced by IL-1α, and then acted via a feedback loop to induce IL-1α; thus, the two cytokines appear to cooperate to promote psoriasis-like dermatitis in mice (66). Additional investigation of the formation of neutrophil extracellular traps in the IMQ-mouse model demonstrated that neutrophils amplified the epidermal inflammatory responses via activation of TLR4/IL-36 cross-talk (67).
However, these findings in the IMQ-mouse model, and other murine models of psoriasis, have significant limitations due to inconsistent laboratory protocols, contradictory findings, and the inability to fully recapitulate complex, multigenic diseases such as psoriasis (68). This underscores the importance of translational human studies, and carefully designed clinical trials that allow for the collection and laboratory evaluation of tissues derived from patients with GPP. Interestingly, mice deficient in Il36rn do not exhibit any phenotypic manifestations on their skin, unlike humans with GPP. The only exception is when they are crossed with transgenic mice over-expressing Il36a or have IMQ applied to the skin. One hypothesis for this is that IL-8 is a critical cytokine for stimulating neutrophil chemotaxis in skin (only IL-8 activates both CXCR1 and CXCR2) (69). IL-8 is strongly up-regulated by IL-36 in human keratinocytes; thus, strong activation of IL-8 by IL-36 (even in the presence of an active IL-36Ra molecule in humans) may be a key driver of pustulosis. However, cases of IL36RN deficiency have even higher activation of IL-8 and the associated neutrophilic axis. Mice do not have a gene for IL-8 and depend on orthologs of IL-8 such as CXCL-1 (which is also present in GPP); thus, mice may require even higher activation of CXCL1 (as IL-8 is absent) to stimulate a neutrophil response in the skin. This threshold may require not only over-expression of IL-36, but also the absence of IL-36RN. Furthermore, mice are relatively neutropenic compared to humans; typically, only 5-10% of circulating leukocytes in mice are neutrophils, whereas >50% is more usual in humans (70). This may also suggest a higher threshold for activating a neutrophil-predominant infiltrate in mouse skin. Other murine factors may also contribute to differences in the manifestation of inflammatory skin diseases in mice, such as their predominance of gamma-delta T-lymphocytes, intrinsic differences in the immune system of various genetic strains of mice, method of disease induction (knockout models versus intralesional or topical induction), increased keratinocyte turnover, and thinner skin tissue layers (68).
IL-36 cytokines (IL-36α, IL-36β, and IL-36γ) maintain circuits that recruit and activate neutrophils, and this process plays a central role in pathogenesis of GPP (71), as shown in Figure 4. Under normal conditions, IL-36 agonist activity is balanced by a high level of IL-36Ra, while IL-36R remains inactive or signals at a low-level, and this equilibrium maintains a regulated downstream inflammatory response (72). However, if the IL-36 axis becomes hyperactivated (due to increased IL-36 agonist, or impaired IL-36Ra activity from IL36RN mutation), uncontrolled pro-inflammatory responses ensue, amplifying neutrophil chemotaxis and neutrophil-driven inflammatory responses in the skin (18). The detection of IL-36α and IL-36γ overexpression in lesional skin and peripheral blood samples from patients with GPP further supports the central role of IL-36 dysregulation in this condition (14, 73), and there is a strong association between loss-of-function mutations in IL36RN (producing faulty IL-36Ra protein) and increased susceptibility to GPP (36, 37, 46).
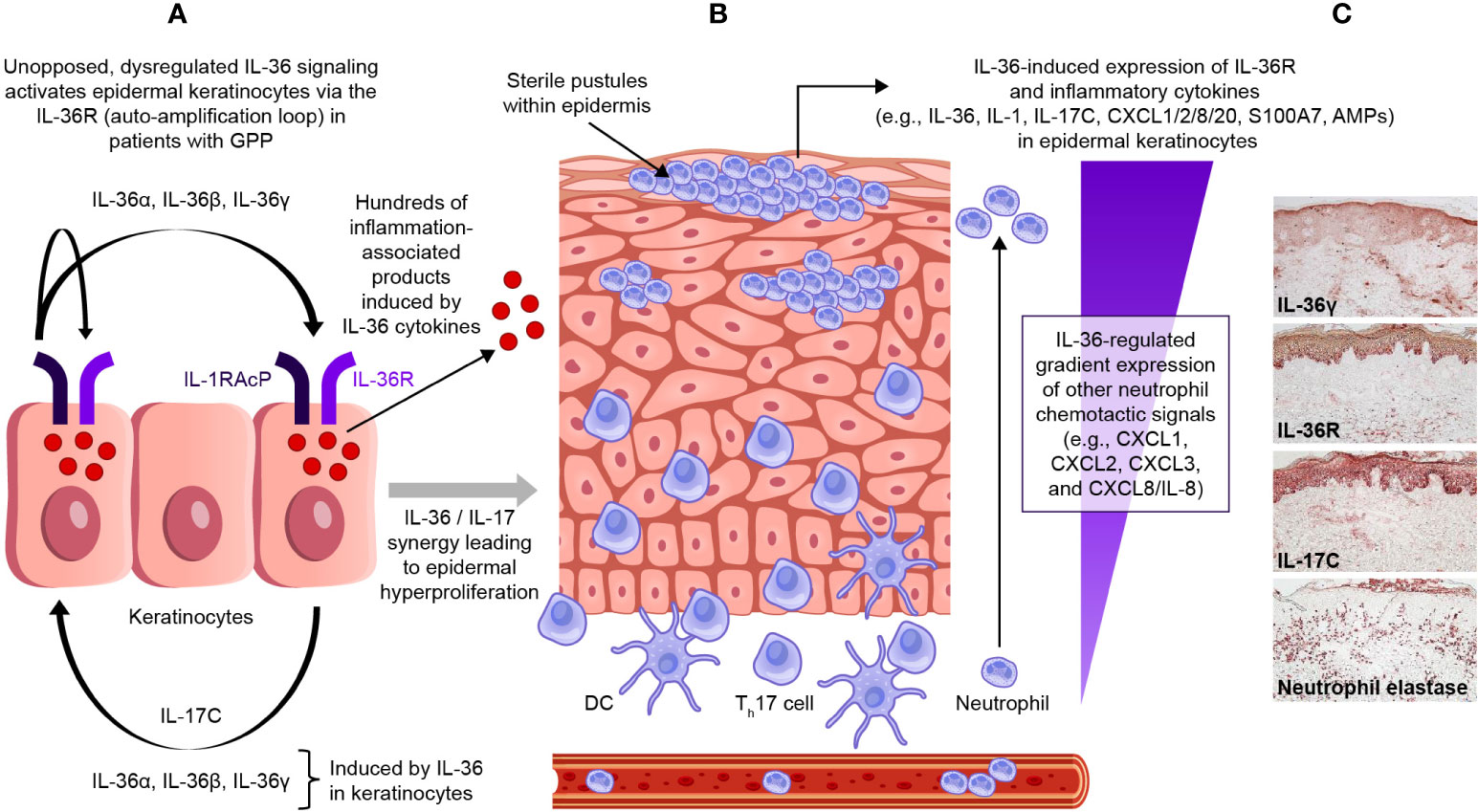
Figure 4 Dysregulated IL-36 signaling in epidermal keratinocytes in GPP. In patients with GPP, unopposed, dysregulated IL-36 signaling activates epidermal keratinocytes via the IL-36R (auto-amplification loop) (A). This induces the release of a wide range of inflammation-associated products, and leads to a gradient of IL-36-regulated neutrophil chemotactic signals (B). The expression of IL-36R is strong and dominant on epidermal keratinocytes within the viable epidermal layers (C). GPP, generalized pustular psoriasis; IL-36R, interleukin-36 receptor. [Panel C, immunohistochemical sample was kindly provided by Dr JG Krueger.].
It is likely that the IL-36 pathway plays a major role in regulating inflammatory responses at barrier epithelia, such as keratinocytes in the epidermis. From immunohistochemical (IHC) staining of human skin samples, IL-36R expression is strong and dominant on epidermal keratinocytes within the viable epidermal layers, and this pan-epidermal expression is maintained in both PV and in GPP lesions (74). In contrast, IHC staining of dermal cell types, such as fibroblasts and vascular cells, shows IL-36R expression is relatively weak in human skin (74). From in vitro experiments with cultured keratinocytes (representing a model where the agonist actions of IL-36 cytokines are relatively unopposed, due to the relative absence of IL-36Ra in the model), it is clear that IL-36 can self-induce expression of all IL-36 ligands (IL-36α, IL-36β, and IL-36γ), but also expression of other inflammatory cytokines such as TNF, IL-6, IL-8, S100A7 (psoriasin), defensins, and other anti-microbial proteins (AMPs) (75). There is also a strong synergistic interaction between IL-36 ligands and IL-17 isoforms, such as IL-17A, that are produced by activated T-cells for these gene products. The role of IL-17A in GPP disease is supported in part by case reports and small clinical trials from Japan detailing the clinical improvement and resolution of systemic symptoms in some patients with GPP treated with selective IL-17A (secukinumab and ixekizumab) and IL-17R (brodalumab) antagonists (76–80).
The breadth of the keratinocyte response to IL-36 family cytokines was only appreciated recently by whole-genome gene profiling experiments using IL-36γ and IL-17A cytokines in human keratinocytes (74). These experiments show that both IL-17A and IL-36γ induce hundreds of gene products that are only partly overlapping, while the response to these combined cytokines is up-regulation of several thousand gene products that are also highly characteristic of genes activated in PV and GPP. Included in this response are numerous chemokines that regulate neutrophil chemotaxis in skin (CXCL1, CXCL2, CXCL3, and CXCL8/IL-8), attract T-cells or inflammatory dendritic cells into skin (CXCL9 and CCL20), and induce or further amplify IL-36 synthesis/signaling in the skin (IL-17C). In addition, IL-1, IL-6, IL-19, and IL-24 are also induced, which can promote broad inflammatory responses as well as induce keratinocyte proliferation (74). Thus, the response of keratinocytes to IL-36 agonists includes increased expression of broad AMPs, the production of inflammatory cytokines and chemokines, and the broad-modification of keratinocyte gene expression programs to mimic psoriatic tissues. Figure 4C shows increased staining of IL-36γ, IL-36R, and IL-17C in the epidermis of GPP lesions, along with the influx of neutrophils into the dermis and epidermis of lesions (neutrophil elastase stain). There are extensive overlaps between gene sets induced in cultured keratinocytes by IL-36 and gene expression in GPP, but especially for CXCL chemokines that are induced by IL-36 that would be expected to regulate neutrophil trafficking (74). In addition, abnormal IL-36 activity was found to correlate with a prominent IFN-1 signature in patients with GPP and PV, and IFN-1 gene activation was also associated with extracutaneous morbidity (acute systemic flares in GPP and chronic systemic inflammation in PV) (81). However, despite the significant overlapping immune functions of IL-36 and IL-17 cytokines in the skin, the predominant role of IL-36 signaling in GPP is underscored by the variable and inconsistent clinical efficacy of off-label IL-17 antagonists for the treatment of GPP; in contrast, most patients with PV have consistent clinical responses to selective IL-17 and IL-23 inhibitors. While IL-36 could have a pathogenic role in PV, clinical studies investigating the clinical efficacy of IL-36 blockade in PV patients are lacking and are necessary to determine the exact contribution of IL-36 signaling in plaque and non-GPP subtypes.
Studies of the effects of IL-36 on inflammatory gene expression in peripheral blood mononuclear cells (PBMCs) showed that IL-36 induced expression of TNF, IL-1, IL-6, and IL-8 in normal control PBMCs, but that much higher induction of these cytokines occurred in PBMCs from a patient with a mutation in IL36RN (37). Thus, effects of amplified IL-36 signaling in GPP patients likely extends to many connective tissue and blood leukocyte cell types in which expression of IL-36R has been identified. Patients with GPP have high levels of these inflammatory cytokines in their blood prior to treatment (causing high fever, leukocytosis, and systemic inflammatory symptoms), and this may be driven by the extended release of IL-36-induced cytokines produced by various cell types under intensified IL-36 signaling in GPP.
Selective blockade of the IL-36 pathway via targeted inhibition of IL-36R is an attractive therapeutic strategy for the treatment of GPP, and other diseases involving dysregulated IL-36 signaling. Spesolimab (SPEVIGO®; Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA) is a first-in-class humanized monoclonal immunoglobulin G1 antibody that binds specifically to IL-36R and antagonizes IL-36 signaling (82). Spesolimab was approved by the US FDA in September 2022 to treat GPP flares in adults (21, 22), and regulatory approval in numerous other countries has followed (83). Key clinical trials data that supported FDA approval of spesolimab were the phase 1 proof-of-concept trial (NCT02978690; N = 7) and the phase 2 randomized, placebo-controlled trial, Effisayil™ 1 (NCT03782792; N = 53), the results of which have been published in detail (84–87). In Effisayil™ 1, spesolimab was administered as a single 900 mg dose via intravenous (IV) infusion over 90 minutes, with the option of a second 900 mg dose IV given 1 week later if symptoms persist (85). Spesolimab efficacy was assessed via the GPP Physician Global Assessment (GPPGA), and the GPP Area and Severity Index (GPPASI) (88). Biomarkers in skin and blood were evaluated (60, 89), and patient-reported outcome instruments were reported (90). In the proof-of-concept and Effisayil™ 1 trials, spesolimab treatment led to rapid (within one week) and sustained (to end of trial) clinical improvement and pustular clearance in patients with GPP flares, and was safe and well-tolerated (84, 85). Efficacy and safety were also consistent for the trial duration across prespecified subgroups in Effisayil™ 1 (91, 92). Pyrexia was observed in both spesolimab (6%) and placebo (22%) groups during Effisayil™ 1 (85), suggesting that this reported adverse event was more likely GPP-associated rather than a treatment-specific effect. Infections were the most frequent adverse reactions observed in patients treated with spesolimab (22); during the 1-week placebo-controlled period in Effisayil™ 1, infections were reported in 14% of the spesolimab group versus 6% of the placebo group (22). Effisayil™ 2 and Effisayil® ON are additional clinical trials to investigate the efficacy and safety of spesolimab in patients with a history of GPP. Effisayil™ 2 (NCT04399837; N = 123 [ (93)]) was completed in December 2022, and was published recently (94). Effisayil™ ON (NCT03886246; N = 131 [ (95)]) is an active 5-year open-label extension study, in which participants of Effisayil™ 1 and Effisayil™ 2 were recruited (96).
Pre- and post-treatment skin and blood samples were collected from participants in the phase 1 proof-of-concept trial and Effisayil® 1 trial to compare gene expression profiles in GPP lesions versus non-lesional skin, and assess molecular changes before versus after spesolimab treatment (60, 97). In lesional skin, spesolimab treatment led to significant decreases in the expression of genes associated with pro-inflammatory mediators (e.g. TNF, IL1B, IL6), neutrophil recruitment (e.g. CXCR1, CXCR2), keratinocyte-mediated inflammation and proliferation (e.g. IL20), and IL-36 ligands (IL36A, IL36B, IL36G) (60, 97), as shown in Figure 5 (60). Reductions in select serum biomarker levels were also identified; including those linked to inflammation (C-reactive protein, TNFα), neutrophilic markers (CXCL1, IL-8), innate pathways (IL-1RN, IL-6), and Th17 pathways (IL-17A, CCL20) (89). These reductions were associated with clinical improvement in GPP, as assessed by the respective primary endpoints of each trial (89). These changes in gene expression and protein biomarker data demonstrate that spesolimab treatment reverses the lesional skin molecular profile associated with GPP.
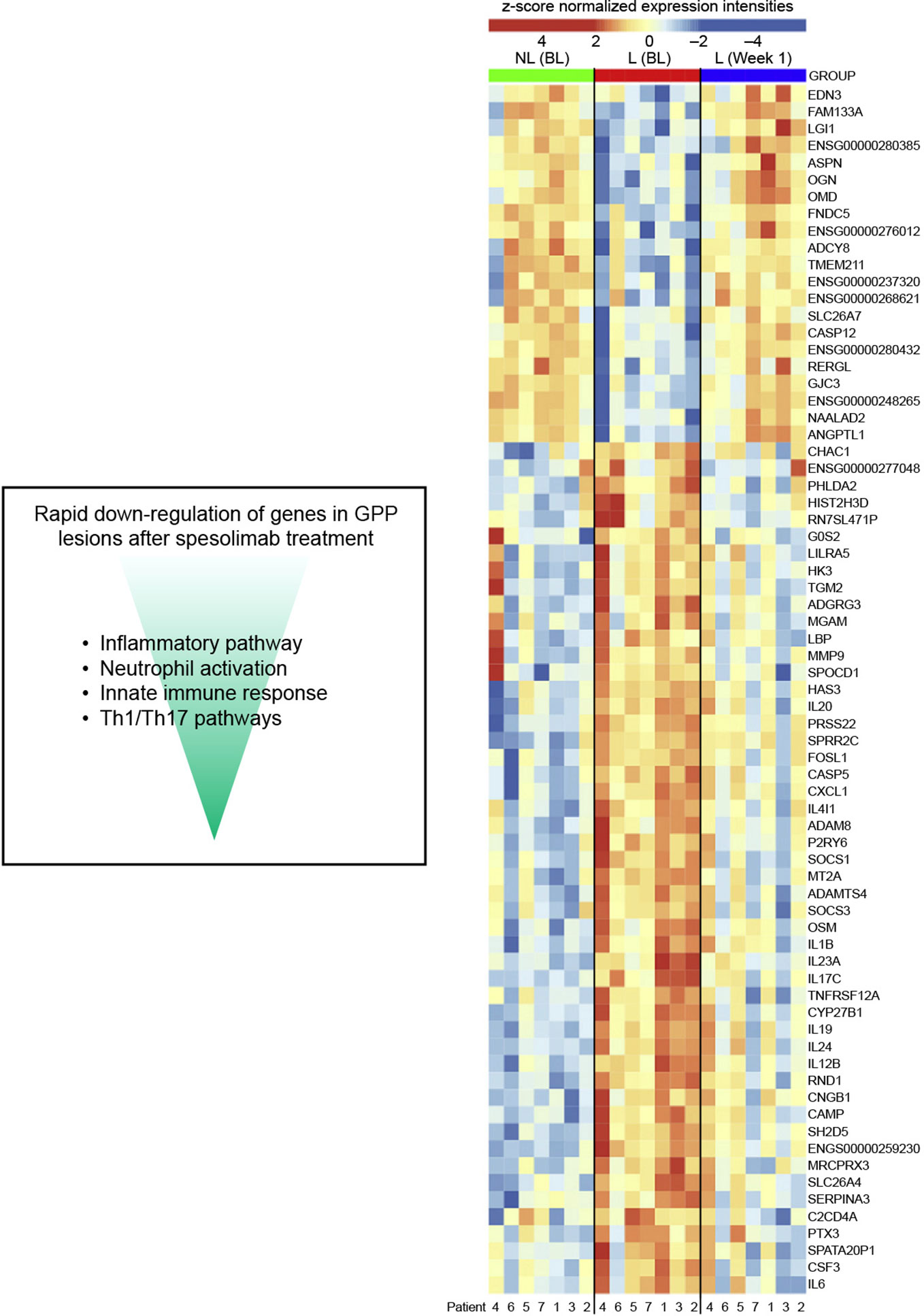
Figure 5 The effect of spesolimab on differential gene expression in GPP (60). Heatmap of 71 differentially expressed genes that are deregulated between lesional (L) and nonlesional (NL) skin at baseline (BL), and lesional skin 1 week after treatment with spesolimab compared with lesional skin at baseline (absolute log2 fold change ≥ 2, adjusted P ≤.05). GPP, generalized pustular psoriasis. From: Pustular psoriasis: Molecular pathways and effects of spesolimab in generalized pustular psoriasis; Baum P, Visvanathan S, Garcet S, Roy J, Schmid R, Bossert S, et al; J Allergy Clin Immunol. 2022;149(4):1402-12.
A second IL-36 receptor blocker, imsidolimab [AnaptysBio, Inc., San Diego, CA, USA (98)], completed a phase 2 clinical trial in patients with active moderate-to-severe GPP (GALLOP; NCT03619902; N = 8) (99, 100). Participants received imsidolimab 750 mg via IV infusion, followed by three further doses of 100 mg given subcutaneously at approximately 30-day intervals, with a 12-week follow-up. Six of the eight participants achieved the primary endpoint of improvement in Clinical Global Impression (based on the modified Japanese Dermatology Association Severity Index) at Week 4 and at Week 16 (100). A phase 3 clinical trial of imsidolimab in patients with GPP flare is now being conducted (GEMINI-1; NCT05352893; N = 45), with a long-term extension trial to follow (GEMINI-2; NCT05366855; N = 45 planned), and top-line data from GEMINI-1 are expected by the end of 2023 (101). Other investigational agents that may target the IL-36 pathway include REGN6490 (Regeneron), an antibody that blocks the IL-36 receptor, and the small molecule A-522 (AbbVie, Inc.) that antagonizes IL-36γ (102). However, phase 1 development of REGN6490 was terminated recently (103, 104).
Several other pustular skin conditions can mimic the clinical presentation of GPP (105); such as acute generalized exanthematous pustulosis (AGEP), other severe drug reactions, infections, IgA pemphigus, subcorneal pustular dermatosis (also called Sneddon-Wilkinson disease), acute psoriasis subtypes (e.g. erythrodermic and pustular disease flares), and localized forms of pustular psoriasis, such as palmoplantar pustulosis (PPP). Though IL-36 cytokines are highly expressed in AGEP lesional skin, the precise relationship between AGEP and IL-36 signaling is unclear (106–108). IL36RN mutations have been detected in a small proportion of patients with PPP, but the relationship is unclear given the lack of response in patients given an IL-36R inhibitor (15, 109, 110).
Consequently, GPP is often misdiagnosed, or may go unrecognized by clinicians who are unfamiliar with managing this disease. Patients often present in the emergency room or urgent care clinics with severe manifestations of GPP (111), which, if not treated effectively, may result in complications such as infection/sepsis, renal and liver dysfunction, or even death (112). Current biologic treatments approved for PV are often ineffective at controlling GPP; thus, GPP-specific treatment is needed to prevent and control flares. Furthermore, clinicians may be unable or unwilling to prescribe biologics that are not specifically FDA-approved for the treatment of GPP (111), and these agents are inconsistently effective for the treatment of GPP. In the CorEvitas (formerly Corrona) Psoriasis Patient Registry study, 67% of dermatologists (N = 29) reported that flare prevention was a challenge when treating patients with frequent flares (two or more episodes per year) (111). Thus, multi-level support from dermatologists, emergency physicians, and primary care providers is needed to facilitate rapid decision-making during acute illness to control skin and systemic symptoms in patients with GPP (113). Consensus guidelines for the diagnosis and management of GPP are also lacking, but are greatly needed to broaden disease understanding and improve disease management by healthcare professionals (111, 113). Importantly, the recent development of an evidence-based clinical management algorithm is now available, following a recent Delphi panel involving 21 expert dermatologists who established global consensus on the clinical course, diagnosis, treatment goals, and disease management of GPP (114).
In addition to the central role of IL-36 receptor signaling in the pathogenesis of GPP, evidence suggests that IL-36 may have a role in the pathogenesis of other diseases (115, 116); including inflammatory bowel disease (117, 118), hidradenitis suppurativa (119, 120), arthritis (121–123), systemic lupus erythematosus (124, 125), pyoderma gangrenosum (126), and Netherton syndrome (127). Phase 2 clinical trials of spesolimab in the treatment of hidradenitis suppurativa are active (NCT04876391), or recently completed (NCT04762277 (128), and a Phase 2/3 clinical trial of spesolimab in the treatment of Netherton syndrome is in progress (NCT05856526). However, a recent Phase 2b randomized placebo-controlled trial of spesolimab in patients with PPP did not meet its primary endpoint (110). Investigations are also underway into the possible role of IL-36 cytokines in fibrotic disorders (115) and malignancy (129–131).
Dysfunctional elements in the IL-36 pathway drive the clinical features and symptoms of GPP. Targeted inhibition of IL-36R (via spesolimab, imsidolimab, or other novel agents) is an attractive therapeutic strategy for the treatment of GPP. Therapies that target IL-36R, such as spesolimab, could mitigate flares, address flare prevention, and provide sustained disease control in patients with GPP, presumably through rebalancing IL-36 signaling and modulating the pro-inflammatory response of downstream effectors. However, investigation is needed to explain why patients with the same IL36RN mutations can present with differences in flare severity and frequency, and to better understand the underlying differences in the disease mechanisms in patients with GPP who do not have identifiable causal or disease-contributing mutations.
JH: Writing – original draft, Writing – review & editing. SV: Writing – original draft, Writing – review & editing. JK: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Medical writing support was funded by Boehringer Ingelheim Pharmaceuticals, Inc., (BIPI), Ridgefield, CT, USA. BIPI was given the opportunity to review the manuscript for medical and scientific accuracy, as well as intellectual property considerations.
Medical writing assistance, supported financially by Boehringer Ingelheim Pharmaceuticals, Inc. (BIPI), was provided by Debra Brocksmith, MB ChB, PhD, and Katie Crosslin, PhD, both of Envision Pharma Group.
JH serves on the medical board and scientific advisory committee of the National Psoriasis Foundation, is a councilor for the International Psoriasis Council, and has been a paid consultant for AbbVie, Arcutis, BMS, Boehringer Ingelheim, Eli Lilly and Company, Janssen, LEO, Novartis, Pfizer, Regeneron-Sanofi, Sun Pharmaceutical, UCB, VisualDx, and UpToDate. SV is an employee of Boehringer Ingelheim Pharmaceuticals, Inc. JK has received grants from and been an investigator for Boehringer Ingelheim; received personal fees from AbbVie, Baxter, Biogen Idec, Delenex Therapeutics, Kineta, Sanofi, Serono, and XenoPort; and received grants from Amgen, Bristol Myers Squibb, Dermira, Innovaderm Research, Janssen, Kadmon, Kyowa Kirin, Eli Lilly, Merck, Novartis, Parexel, and Pfizer.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Navarini AA, Burden AD, Capon F, Mrowietz U, Puig L, Koks S, et al. European consensus statement on phenotypes of pustular psoriasis. J Eur Acad Dermatol Venereol (2017) 31(11):1792–9. doi: 10.1111/jdv.14386
2. Kharawala S, Golembesky AK, Bohn RL, Esser D. The clinical, humanistic, and economic burden of generalized pustular psoriasis: a structured review. Expert Rev Clin Immunol (2020) 16(3):239–52. doi: 10.1080/1744666X.2019.1708193
3. Choon SE, Navarini AA, Pinter A. Clinical course and characteristics of generalized pustular psoriasis. Am J Clin Dermatol (2022) 23(Suppl 1):21–9. doi: 10.1007/s40257-021-00654-z
4. Koren J, Mburu S, Trigos D, Damiani G, Naldi L. Generalised pustular psoriasis: the case for rare disease and orphan designation. Br J Dermatol (2022) 187(3):411–3. doi: 10.1111/bjd.21231
5. Augey F, Renaudier P, Nicolas JF. Generalized pustular psoriasis (Zumbusch): a French epidemiological survey. Eur J Dermatol (2006) 16(6):669–73.
6. Ohkawara A, Yasuda H, Kobayashi H, Inaba Y, Ogawa H, Hashimoto I, et al. Generalized pustular psoriasis in Japan: two distinct groups formed by differences in symptoms and genetic background. Acta Derm Venereol (1996) 76(1):68–71. doi: 10.2340/00015555766871
7. Lee JY, Kang S, Park JS, Jo SJ. Prevalence of psoriasis in korea: A population-based epidemiological study using the korean national health insurance database. Ann Dermatol (2017) 29(6):761–7. doi: 10.5021/ad.2017.29.6.761
8. Prinz JC, Choon SE, Griffiths CEM, Merola JF, Morita A, Ashcroft DM, et al. Prevalence, comorbidities and mortality of generalized pustular psoriasis: A literature review. J Eur Acad Dermatol Venereol (2023) 37(2):256–73. doi: 10.1111/jdv.18720
9. Choon SE, Lai NM, Mohammad NA, Nanu NM, Tey KE, Chew SF. Clinical profile, morbidity, and outcome of adult-onset generalized pustular psoriasis: analysis of 102 cases seen in a tertiary hospital in Johor, Malaysia. Int J Dermatol (2014) 53(6):676–84. doi: 10.1111/ijd.12070
10. Miyachi H, Konishi T, Kumazawa R, Matsui H, Shimizu S, Fushimi K, et al. Treatments and outcomes of generalized pustular psoriasis: A cohort of 1516 patients in a nationwide inpatient database in Japan. J Am Acad Dermatol (2022) 86(6):1266–74. doi: 10.1016/j.jaad.2021.06.008
11. Brenner M, Molin S, Ruebsam K, Weisenseel P, Ruzicka T, Prinz JC. Generalized pustular psoriasis induced by systemic glucocorticosteroids: four cases and recommendations for treatment. Br J Dermatol (2009) 161(4):964–6. doi: 10.1111/j.1365-2133.2009.09348.x
12. Benzaquen M, Flachaire B, Rouby F, Berbis P, Guis S. Paradoxical pustular psoriasis induced by ustekinumab in a patient with Crohn’s disease-associated spondyloarthropathy. Rheumatol Int (2018) 38(7):1297–9. doi: 10.1007/s00296-018-4034-0
13. Balak DM, Hajdarbegovic E. Drug-induced psoriasis: clinical perspectives. Psoriasis (Auckl) (2017) 7:87–94. doi: 10.2147/ptt.S126727
14. Johnston A, Xing X, Wolterink L, Barnes DH, Yin Z, Reingold L, et al. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J Allergy Clin Immunol (2017) 140(1):109–20. doi: 10.1016/j.jaci.2016.08.056
15. Twelves S, Mostafa A, Dand N, Burri E, Farkas K, Wilson R, et al. Clinical and genetic differences between pustular psoriasis subtypes. J Allergy Clin Immunol Mar (2019) 143(3):1021–6. doi: 10.1016/j.jaci.2018.06.038
16. Bachelez H, Barker J, Burden AD, Navarini AA, Krueger JG. Generalized pustular psoriasis is a disease distinct from psoriasis vulgaris: evidence and expert opinion. Expert Rev Clin Immunol (2022) 18(10):1033–47. doi: 10.1080/1744666X.2022.2116003
17. Borges-Costa J, Silva R, Goncalves L, Filipe P, Soares de Almeida L, Marques Gomes M. Clinical and laboratory features in acute generalized pustular psoriasis: a retrospective study of 34 patients. Am J Clin Dermatol (2011) 12(4):271–6. doi: 10.2165/11586900-000000000-00000
18. Uppala R, Tsoi LC, Harms PW, Wang B, Billi AC, Maverakis E, et al. “Autoinflammatory psoriasis”-genetics and biology of pustular psoriasis. Cell Mol Immunol (2021) 18(2):307–17. doi: 10.1038/s41423-020-0519-3
19. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol (2017) 140(3):645–53. doi: 10.1016/j.jaci.2017.07.004
20. Grän F, Kerstan A, Serfling E, Goebeler M, Muhammad K. Current developments in the immunology of psoriasis. Yale J Biol Med (2020) 93(1):97–110.
21. Boehringer Ingelheim. FDA approves the first treatment option for generalized pustular psoriasis flares in adults (2022). Available at: https://www.boehringer-ingelheim.us/press-release/fda-approves-first-treatment-option-generalized-pustular-psoriasis-flares-adults (Accessed September 28, 2022).
22. Boehringer Ingelheim. SPEVIGO prescribing information (2022). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761244s000lbl.pdf (Accessed September 28, 2022).
23. Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity (2013) 39(6):1003–18. doi: 10.1016/j.immuni.2013.11.010
24. Iznardo H, Puig L. Exploring the role of IL-36 cytokines as a new target in psoriatic disease. Int J Mol Sci (2021) 22(9):4344. doi: 10.3390/ijms22094344
25. Iznardo H, Puig L. The interleukin-1 family cytokines in psoriasis: pathogenetic role and therapeutic perspectives. Expert Rev Clin Immunol (2021) 17(2):187–99. doi: 10.1080/1744666X.2021.1886081
26. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev (2018) 281(1):8–27. doi: 10.1111/imr.12621
27. Macleod T, Berekmeri A, Bridgewood C, Stacey M, McGonagle D, Wittmann M. The immunological impact of IL-1 family cytokines on the epidermal barrier. Front Immunol (2021) 12:808012. doi: 10.3389/fimmu.2021.808012
28. Green EA, Garrick SP, Peterson B, Berger PJ, Galinsky R, Hunt RW, et al. The role of the interleukin-1 family in complications of prematurity. Int J Mol Sci (2023) 24(3):2795. doi: 10.3390/ijms24032795
29. Young KZ, Sarkar MK, Gudjonsson JE. Pathophysiology of generalized pustular psoriasis. Exp Dermatol (2023) 32(8):1194–203. doi: 10.1111/exd.14768
30. Zhou L, Todorovic V. Interleukin-36: structure, signaling and function. Adv Exp Med Biol (2021) 21:191–210. doi: 10.1007/5584_2020_488
31. Macleod T, Ainscough JS, Hesse C, Konzok S, Braun A, Buhl AL, et al. The proinflammatory cytokine IL-36gamma is a global discriminator of harmless microbes and invasive pathogens within epithelial tissues. Cell Rep (2020) 33(11):108515. doi: 10.1016/j.celrep.2020.108515
32. Henry CM, Sullivan GP, Clancy DM, Afonina IS, Kulms D, Martin SJ. Neutrophil-derived proteases escalate inflammation through activation of IL-36 family cytokines. Cell Rep (2016) 14(4):708–22. doi: 10.1016/j.celrep.2015.12.072
33. Ainscough JS, Macleod T, McGonagle D, Brakefield R, Baron JM, Alase A, et al. Cathepsin S is the major activator of the psoriasis-associated proinflammatory cytokine IL-36gamma. Proc Natl Acad Sci U S A (2017) 114(13):E2748–57. doi: 10.1073/pnas.1620954114
34. Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med (2009) 360(23):2426–37. doi: 10.1056/NEJMoa0807865
35. Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med (2009) 360(23):2438–44. doi: 10.1056/NEJMoa0809568
36. Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med (2011) 365(7):620–8. doi: 10.1056/NEJMoa1013068
37. Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet (2011) 89(3):432–7. doi: 10.1016/j.ajhg.2011.07.022
38. Martin P, Goldstein JD, Mermoud L, Diaz-Barreiro A, Palmer G. IL-1 family antagonists in mouse and human skin inflammation. Front Immunol (2021) 12:652846. doi: 10.3389/fimmu.2021.652846
39. Shepherd J, Little MC, Nicklin MJ. Psoriasis-like cutaneous inflammation in mice lacking interleukin-1 receptor antagonist. J Invest Dermatol (2004) 122(3):665–9. doi: 10.1111/j.0022-202X.2004.22305.x
40. Gomez-Garcia F, Sanz-Cabanillas JL, Viguera-Guerra I, Isla-Tejera B, Nieto AV, Ruano J. Scoping review on use of drugs targeting interleukin 1 pathway in DIRA and DITRA. Dermatol Ther (Heidelb) (2018) 8(4):539–56. doi: 10.1007/s13555-018-0269-7
41. Blumberg H, Dinh H, Trueblood ES, Pretorius J, Kugler D, Weng N, et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med (2007) 204(11):2603–14. doi: 10.1084/jem.20070157
42. Zhou J, Luo Q, Cheng Y, Wen X, Liu J. An update on genetic basis of generalized pustular psoriasis (Review). Int J Mol Med (2021) 47(6):118. doi: 10.3892/ijmm.2021.4951
43. Setta-Kaffetzi N, Navarini AA, Patel VM, Pullabhatla V, Pink AE, Choon SE, et al. Rare pathogenic variants in IL36RN underlie a spectrum of psoriasis-associated pustular phenotypes. J Invest Dermatol (2013) 133(5):1366–9. doi: 10.1038/jid.2012.490
44. Hussain S, Berki DM, Choon SE, Burden AD, Allen MH, Arostegui JI, et al. IL36RN mutations define a severe autoinflammatory phenotype of generalized pustular psoriasis. J Allergy Clin Immunol (2015) 135(4):1067–70.e9. doi: 10.1016/j.jaci.2014.09.043
45. Capon F. IL36RN mutations in generalized pustular psoriasis: just the tip of the iceberg? J Invest Dermatol (2013) 133(11):2503–4. doi: 10.1038/jid.2013.361
46. Sugiura K, Takemoto A, Yamaguchi M, Takahashi H, Shoda Y, Mitsuma T, et al. The majority of generalized pustular psoriasis without psoriasis vulgaris is caused by deficiency of interleukin-36 receptor antagonist. J Invest Dermatol (2013) 133(11):2514–21. doi: 10.1038/jid.2013.230
47. Liu ZJ, Tian YT, Shi BY, Zhou Y, Jia XS. Association between mutation of interleukin 36 receptor antagonist and generalized pustular psoriasis: A PRISMA-compliant systematic review and meta-analysis. Med (Baltimore) (2020) 99(45):e23068. doi: 10.1097/MD.0000000000023068
48. Berki DM, Mahil SK, Burden AD, Trembath RC, Smith CH, Capon F, et al. Loss of IL36RN function does not confer susceptibility to psoriasis vulgaris. J Invest Dermatol (2014) 134(1):271–3. doi: 10.1038/jid.2013.285
49. Berki DM, Liu L, Choon SE, David Burden A, Griffiths CEM, Navarini AA, et al. Activating CARD14 mutations are associated with generalized pustular psoriasis but rarely account for familial recurrence in psoriasis vulgaris. J Invest Dermatol Dec (2015) 135(12):2964–70. doi: 10.1038/jid.2015.288
50. Jordan CT, Cao L, Roberson ED, et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am J Hum Genet (2012) 90(5):796–808. doi: 10.1016/j.ajhg.2012.03.013
51. Setta-Kaffetzi N, Simpson MA, Navarini AA, Patel VM, Lu HC, Allen MH, et al. AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am J Hum Genet (2014) 94(5):790–7. doi: 10.1016/j.ajhg.2014.04.005
52. Mahil SK, Twelves S, Farkas K, Setta-Kaffetzi N, Burden AD, Gach JE, et al. AP1S3 mutations cause skin autoinflammation by disrupting keratinocyte autophagy and up-regulating IL-36 production. J Invest Dermatol (2016) 136(11):2251–9. doi: 10.1016/j.jid.2016.06.618
53. Vergnano M, Mockenhaupt M, Benzian-Olsson N, Paulmann M, Grys K, Mahil SK, et al. Loss-of-function myeloperoxidase mutations are associated with increased neutrophil counts and pustular skin disease. Am J Hum Genet (2020) 107(3):539–43. doi: 10.1016/j.ajhg.2020.06.020
54. Haskamp S, Bruns H, Hahn M, Hoffmann M, Gregor A, Lohr S, et al. Myeloperoxidase modulates inflammation in generalized pustular psoriasis and additional rare pustular skin diseases. Am J Hum Genet (2020) 107(3):527–38. doi: 10.1016/j.ajhg.2020.07.001
55. Frey S, Sticht H, Wilsmann-Theis D, Gerschutz A, Wolf K, Lohr S, et al. Rare loss-of-function mutation in SERPINA3 in generalized pustular psoriasis. J Invest Dermatol (2020) 140(7):1451–5 e13. doi: 10.1016/j.jid.2019.11.024
56. Kantaputra P, Chaowattanapanit S, Kiratikanon S, Chaiwarith R, Choonhakarn C, Intachai W, et al. SERPINA1, generalized pustular psoriasis, and adult-onset immunodeficiency. J Dermatol (2021) 48(10):1597–601. doi: 10.1111/1346-8138.16081
57. Akiyama M. Pustular psoriasis as an autoinflammatory keratinization disease (AiKD): Genetic predisposing factors and promising therapeutic targets. J Dermatol Sci (2022) 105(1):11–7. doi: 10.1016/j.jdermsci.2021.11.009
58. Rivera-Diaz R, Dauden E, Carrascosa JM, Cueva P, Puig L. Generalized pustular psoriasis: A review on clinical characteristics, diagnosis, and treatment. Dermatol Ther (Heidelb) (2023) 13(3):673–88. doi: 10.1007/s13555-022-00881-0
59. Mössner R, Wilsmann-Theis D, Oji V, Gkogkolou P, Lohr S, Schulz P, et al. The genetic basis for most patients with pustular skin disease remains elusive. Br J Dermatol (2018) 178(3):740–8. doi: 10.1111/bjd.15867
60. Baum P, Visvanathan S, Garcet S, Roy J, Schmid R, Bossert S, et al. Pustular psoriasis: Molecular pathways and effects of spesolimab in generalized pustular psoriasis. J Allergy Clin Immunol (2022) 149(4):1402–12. doi: 10.1016/j.jaci.2021.09.035
61. van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol (2009) 182(9):5836–45. doi: 10.4049/jimmunol.0802999
62. Goldstein JD, Bassoy EY, Caruso A, Palomo J, Rodriguez E, Lemeille S, et al. IL-36 signaling in keratinocytes controls early IL-23 production in psoriasis-like dermatitis. Life Sci Alliance (2020) 3(6):e202000688. doi: 10.26508/lsa.202000688
63. Tortola L, Rosenwald E, Abel B, Blumberg H, Schafer M, Coyle AJ, et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest (2012) 122(11):3965–76. doi: 10.1172/JCI63451
64. Hernandez-Santana YE, Leon G, St Leger D, Fallon PG, Walsh PT. Keratinocyte interleukin-36 receptor expression orchestrates psoriasiform inflammation in mice. Life Sci Alliance (2020) 3(4):e201900586. doi: 10.26508/lsa.201900586
65. Campbell JJ, Ebsworth K, Ertl LS, McMahon JP, Wang Y, Yau S, et al. Efficacy of chemokine receptor inhibition in treating IL-36alpha-induced psoriasiform inflammation. J Immunol (2019) 202(6):1687–92. doi: 10.4049/jimmunol.1801519
66. Milora KA, Fu H, Dubaz O, Jensen LE. Unprocessed interleukin-36alpha regulates psoriasis-like skin inflammation in cooperation with interleukin-1. J Invest Dermatol (2015) 135(12):2992–3000. doi: 10.1038/jid.2015.289
67. Shao S, Fang H, Dang E, Xue K, Zhang J, Li B, et al. Neutrophil extracellular traps promote inflammatory responses in psoriasis via activating epidermal TLR4/IL-36R crosstalk. Front Immunol (2019) 10:746. doi: 10.3389/fimmu.2019.00746
68. Hawkes JE, Adalsteinsson JA, Gudjonsson JE, Ward NL. Research techniques made simple: murine models of human psoriasis. J Invest Dermatol (2018) 138(1):e1–8. doi: 10.1016/j.jid.2017.10.013
69. Schraufstatter IU, Chung J, Burger M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am J Physiol Lung Cell Mol Physiol (2001) 280(6):L1094–103. doi: 10.1152/ajplung.2001.280.6.L1094
70. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol (2004) 172(5):2731–8. doi: 10.4049/jimmunol.172.5.2731
71. Madonna S, Girolomoni G, Dinarello CA, Albanesi C. The significance of IL-36 hyperactivation and IL-36R targeting in psoriasis. Int J Mol Sci (2019) 20(13):3318. doi: 10.3390/ijms20133318
72. Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J Biol Chem (2011) 286(49):42594–602. doi: 10.1074/jbc.M111.267922
73. D’Erme AM, Wilsmann-Theis D, Wagenpfeil J, Holzel M, Ferring-Schmitt S, Sternberg S, et al. IL-36gamma (IL-1F9) is a biomarker for psoriasis skin lesions. J Invest Dermatol (2015) 135(4):1025–32. doi: 10.1038/jid.2014.532
74. Miura S, Garcet S, Salud-Gnilo C, Gonzalez J, Li X, Murai-Yamamura M, et al. IL-36 and IL-17A cooperatively induce a psoriasis-like gene expression response in human keratinocytes. J Invest Dermatol (2021) 141(8):2086–90. doi: 10.1016/j.jid.2021.01.019
75. Carrier Y, Ma HL, Ramon HE, Napierata L, Small C, O'Toole M, et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol (2011) 131(12):2428–37. doi: 10.1038/jid.2011.234
76. Wilsmann-Theis D, Schnell LM, Ralser-Isselstein V, Bieber T, Schon MP, Huffmeier U, et al. Successful treatment with interleukin-17A antagonists of generalized pustular psoriasis in patients without IL36RN mutations. J Dermatol (2018) 45(7):850–4. doi: 10.1111/1346-8138.14318
77. Plachouri KM, Chourdakis V, Georgiou S. The role of IL-17 and IL-17 receptor inhibitors in the management of generalized pustular psoriasis. Drugs Today (Barc) (2019) 55(9):587–93. doi: 10.1358/dot.2019.55.9.3020159
78. Imafuku S, Honma M, Okubo Y, Komine M, Ohtsuki M, Morita A, et al. Efficacy and safety of secukinumab in patients with generalized pustular psoriasis: A 52-week analysis from phase III open-label multicenter Japanese study. J Dermatol (2016) 43(9):1011–7. doi: 10.1111/1346-8138.13306
79. Saeki H, Nakagawa H, Nakajo K, Ishii T, Morisaki Y, Aoki T, et al. Efficacy and safety of ixekizumab treatment for Japanese patients with moderate to severe plaque psoriasis, erythrodermic psoriasis and generalized pustular psoriasis: Results from a 52-week, open-label, phase 3 study (UNCOVER-J). J Dermatol (2017) 44(4):355–62. doi: 10.1111/1346-8138.13622
80. Yamasaki K, Nakagawa H, Kubo Y, Ootaki K, Japanese Brodalumab Study G. Efficacy and safety of brodalumab in patients with generalized pustular psoriasis and psoriatic erythroderma: results from a 52-week, open-label study. Br J Dermatol Mar (2017) 176(3):741–51. doi: 10.1111/bjd.14702
81. Catapano M, Vergnano M, Romano M, Mahil SK, Choon SE, Burden AD, et al. IL-36 promotes systemic IFN-I responses in severe forms of psoriasis. J Invest Dermatol Apr (2020) 140(4):816–26 e3. doi: 10.1016/j.jid.2019.08.444
82. Ganesan R, Raymond EL, Mennerich D, Woska JR, Caviness G, Grimaldi C, et al. Generation and functional characterization of anti-human and anti-mouse IL-36R antagonist monoclonal antibodies. MAbs (2017) 9(7):1143–54. doi: 10.1080/19420862.2017.1353853
83. Boehringer Ingelheim. European Commission approves SPEVIGO® (spesolimab) for generalized pustular psoriasis flares (2022). Available at: https://www.boehringer-ingelheim.com/human-health/skin-diseases/gpp/european-commission-approves-spevigo-spesolimab-generalized (Accessed January 24, 2023).
84. Bachelez H, Choon SE, Marrakchi S, Burden AD, Tsai TF, Morita A, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med (2019) 380(10):981–3. doi: 10.1056/NEJMc1811317
85. Bachelez H, Choon SE, Marrakchi S, Burden AD, Tsai TF, Morita A, et al. Trial of spesolimab for generalized pustular psoriasis. N Engl J Med (2021) 385(26):2431–40. doi: 10.1056/NEJMoa2111563
86. Shao S, Wang G. Commentary on a clinical trial of spesolimab, a humanized anti-interleukin-36 receptor monoclonal antibody, in generalized pustular psoriasis. Dermatol Ther (Heidelb) (2022) 12(12):2627–35. doi: 10.1007/s13555-022-00830-x
87. Blair HA. Spesolimab: first approval. Drugs (2022) 82(17):1681–6. doi: 10.1007/s40265-022-01801-4
88. Burden AD, Choon SE, Gottlieb AB, Navarini AA, Warren RB. Clinical disease measures in generalized pustular psoriasis. Am J Clin Dermatol (2022) 23(Suppl 1):39–50. doi: 10.1007/s40257-021-00653-0
89. Farag A, Visvanathan S, Bachelez H, Morita A, Lebwohl M, Barker JN, et al. Spesolimab alters the molecular profile of lesional skin in patients with generalized pustular psoriasis with a clinical response (Abstract FC04). Br J Dermatol (Psoriasis: From Gene to Clinic 9th Int Congress; London UK; 9 to 11 December 2021) (2021) 186(1):34988979.
90. Navarini AA, Prinz JC, Morita A, Tsai TF, Viguier MA, Li L, et al. Spesolimab improves patient-reported outcomes in patients with generalized pustular psoriasis: Results from the Effisayil 1 study. J Eur Acad Dermatol Venereol (2022) 37(4):730–6. doi: 10.1111/jdv.18820
91. Morita A, Tsai TF, Yee EYW, Okubo Y, Imafuku S, Zheng M, et al. Efficacy and safety of spesolimab in Asian patients with a generalized pustular psoriasis flare: Results from the randomized, double-blind, placebo-controlled Effisayil 1 study. J Dermatol (2023) 50(2):183–94. doi: 10.1111/1346-8138.16609
92. Burden AD, Okubo Y, Zheng M. (Abstract 33007) Efficacy of spesolimab for the treatment of GPP flares across prespecified patient subgroups in the Effisayil 1 study. J Am Acad Dermatol (2022) 87(3):AB54.
93. Morita A, Choon SE, Bachelez H, Anadkat MJ, Marrakchi S, Zheng M, et al. Design of effisayil 2: A randomized, double-blind, placebo-controlled study of spesolimab in preventing flares in patients with generalized pustular psoriasis. Dermatol Ther (Heidelb) Jan (2023) 13(1):347–59. doi: 10.1007/s13555-022-00835-6
94. Morita A, Strober B, Burden AD, Choon SE, Anadkat MJ, Marrakchi S, et al. Efficacy and safety of subcutaneous spesolimab for the prevention of generalised pustular psoriasis flares (Effisayil 2): an international, multicentre, randomised, placebo-controlled trial. Lancet (2023) 402(10412):1541–51. doi: 10.1016/S0140-6736(23)01378-8
95. Boehringer Ingelheim. Effisayil™ ON: A study to test long-term treatment with spesolimab in people with generalized pustular psoriasis who took part in a previous study . Available at: https://clinicaltrials.gov/ct2/show/record/NCT03886246 (Accessed January 04, 2023).
96. Navarini AA, Bachelez H, Choon SE, Burden AD, Zheng M, Morita A, et al. (2023). Effisayil ON, an open-label, long-term extension study of spesolimab treatment in patients with generalized pustular psoriasis: interim results for flare treatment. J American Academy of Dermatology 89(3), AB44. doi: 10.1016/j.jaad.2023.07.178
97. Farag A, Visvanathan S, Bachelez H, Morita A, Lebwohl M, Barker JN, et al. Spesolimab alters the molecular profile of lesional skin in patients with generalized pustular psoriasis with a clinical response (Abstract 103). In: 4th Inflammatory Skin Disease Summit (ISDS), New York, November 3-6, 2021. Available at: https://www.isds2021.org/wp-content/uploads/2019/06/ISDS-2021_Late-Breaking-Abstracts-1.pdf.
98. AnaptysBio. (Trial protocol) A Single Arm Multiple Dose Study to Assess the Efficacy and Safety of ANB019 in Subjects with Generalized Pustular Psoriasis . Available at: https://clinicaltrials.gov/ProvidedDocs/02/NCT03619902/Prot_000.pdf (Accessed 14 April 2022).
99. AnaptysBio. AnaptysBio presents updated data from imsidolimab phase 2 GALLOP trial in generalized pustular psoriasis . Available at: https://ir.anaptysbio.com/news-releases/news-release-details/anaptysbio-presents-updated-data-imsidolimab-phase-2-gallop (Accessed October 19, 2023).
100. Warren RB, Reich A, Kaszuba A, Placek W, Griffiths CEM, Zhou J, et al. Imsidolimab, an anti-interleukin-36 receptor monoclonal antibody, for the treatment of generalized pustular psoriasis: results from the phase II GALLOP trial. Br J Dermatol (2023) 189(2):161–9. doi: 10.1093/bjd/ljad083
101. Gudjonsson JE, Randazzo B, Zhou J. Imsidolimab in the treatment of adult subjects with generalized pustular psoriasis: Design of a pivotal phase 3 clinical trial and a long-term extension study. J American Academy of Dermatology (2022) 87(3):AB70. doi: 10.1016/j.jaad.2022.06.313
102. Todorovic V, Su Z, Putman CB, Kakavas SJ, Salte KM, McDonald HA, et al. Small molecule IL-36gamma antagonist as a novel therapeutic approach for plaque psoriasis. Sci Rep (2019) 9(1):9089. doi: 10.1038/s41598-019-45626-w
103. Regeneron Pharmaceuticals Inc. Ascending dose study of the safety and tolerability of REGN6490 in healthy volunteers (NCT04616079) . Available at: https://clinicaltrials.gov/ct2/show/NCT04616079 (Accessed February 20, 2023).
104. Regeneron Pharmaceuticals Inc. Study of the safety, tolerability, and pharmacokinetics of REGN6490 in healthy Japanese adult volunteers (NCT04616105). Available at: https://clinicaltrials.gov/ct2/show/NCT04616105. (Accessed February 20, 2023).
105. Fujita H, Gooderham M, Romiti R. Diagnosis of generalized pustular psoriasis. Am J Clin Dermatol (2022) 23(Suppl 1):31–8. doi: 10.1007/s40257-021-00652-1
106. Song HS, Kim SJ, Park TI, Jang YH, Lee ES. Immunohistochemical comparison of IL-36 and the IL-23/th17 axis of generalized pustular psoriasis and acute generalized exanthematous pustulosis. Ann Dermatol (2016) 28(4):451–6. doi: 10.5021/ad.2016.28.4.451
107. Meier-Schiesser B, Feldmeyer L, Jankovic D, Mellett M, Satoh TK, Yerly D, et al. Culprit drugs induce specific IL-36 overexpression in acute generalized exanthematous pustulosis. J Invest Dermatol (2019) 139(4):848–58. doi: 10.1016/j.jid.2018.10.023
108. Stadler PC, Oschmann A, Kerl-French K, Maul JT, Oppel EM, Meier-Schiesser B, et al. Acute generalized exanthematous pustulosis: clinical characteristics, pathogenesis, and management. Dermatol (2023) 239(3):328–33. doi: 10.1159/000529218
109. Misiak-Galazka M, Zozula J, Rudnicka L. Palmoplantar pustulosis: recent advances in etiopathogenesis and emerging treatments. Am J Clin Dermatol (2020) 21(3):355–70. doi: 10.1007/s40257-020-00503-5
110. Burden AD, Bissonnette R, Navarini AA, Murakami M, Morita A, Haeufel T, et al. Spesolimab efficacy and safety in patients with moderate-to-severe palmoplantar pustulosis: A multicentre, double-blind, randomised, placebo-controlled, phase IIb, dose-finding study. Dermatol Ther (Heidelb) (2023) 13(10):2279–97. doi: 10.1007/s13555-023-01002-1
111. Strober B, Kotowsky N, Medeiros R, Mackey RH, Harrold LR, Valdecantos WC, et al. Unmet medical needs in the treatment and management of generalized pustular psoriasis flares: evidence from a survey of corrona registry dermatologists. Dermatol Ther (Heidelb) (2021) 11(2):529–41. doi: 10.1007/s13555-021-00493-0
112. Gooderham MJ, Van Voorhees AS, Lebwohl MG. An update on generalized pustular psoriasis. Expert Rev Clin Immunol (2019) 15(9):907–19. doi: 10.1080/1744666x.2019.1648209
113. Strober B, Leman J, Mockenhaupt M, Nakano de Melo J, Nassar A, Prajapati VH, et al. Unmet educational needs and clinical practice gaps in the management of generalized pustular psoriasis: global perspectives from the front line. Dermatol Ther (Heidelb) (2022) 12(2):381–93. doi: 10.1007/s13555-021-00661-2
114. Puig L, Choon SE, Gottlieb AB, Marrakchi S, Prinz JC, Romiti R, et al. Generalized pustular psoriasis: a global Delphi consensus on clinical course, diagnosis, treatment goals, and disease management. J Eur Acad Dermatol Venereol (2023) 37(4):737–52. doi: 10.1111/jdv.18851
115. Elias M, Zhao S, Le HT, Wang J, Neurath MF, Neufert C, et al. IL-36 in chronic inflammation and fibrosis - bridging the gap? J Clin Invest (2021) 131(2):e144336. doi: 10.1172/JCI144336
116. Manzanares-Meza LD, Valle-Rios R, Medina-Contreras O. Interleukin-1 receptor-like 2: one receptor, three agonists, and many implications. J Interferon Cytokine Res (2022) 42(2):49–61. doi: 10.1089/jir.2021.0173
117. Russell SE, Horan RM, Stefanska AM, Carey A, Leon G, Aguilera M, et al. IL-36alpha expression is elevated in ulcerative colitis and promotes colonic inflammation. Mucosal Immunol (2016) 9(5):1193–204. doi: 10.1038/mi.2015.134
118. Leon G, Hussey S, Walsh PT. The diverse roles of the IL-36 family in gastrointestinal inflammation and resolution. Inflammation Bowel Dis (2021) 27(3):440–50. doi: 10.1093/ibd/izaa232
119. Gudjonsson JE, Tsoi LC, Ma F, Billi AC, van Straalen KR, Vossen A, et al. Contribution of plasma cells and B cells to hidradenitis suppurativa pathogenesis. JCI Insight (2020) 5(19):e139930. doi: 10.1172/jci.insight.139930
120. Zouboulis CC, Frew JW, Giamarellos-Bourboulis EJ, Jemec GBE, Del Marmol V, Marzano AV, et al. Target molecules for future hidradenitis suppurativa treatment. Exp Dermatol (2021) 30 Suppl 1:8–17. doi: 10.1111/exd.14338
121. Boutet MA, Nerviani A, Lliso-Ribera G, Lucchesi D, Prediletto E, Ghirardi GM, et al. Interleukin-36 family dysregulation drives joint inflammation and therapy response in psoriatic arthritis. Rheumatol (Oxford) (2020) 59(4):828–38. doi: 10.1093/rheumatology/kez358
122. Frey S, Derer A, Messbacher ME, Baeten DL, Bugatti S, Montecucco C, et al. The novel cytokine interleukin-36alpha is expressed in psoriatic and rheumatoid arthritis synovium. Ann Rheum Dis (2013) 72(9):1569–74. doi: 10.1136/annrheumdis-2012-202264
123. Li T, Chubinskaya S, Esposito A, Jin X, Tagliafierro L, Loeser R, et al. TGF-beta type 2 receptor-mediated modulation of the IL-36 family can be therapeutically targeted in osteoarthritis. Sci Transl Med (2019) 11(491):eaan2585. doi: 10.1126/scitranslmed.aan2585
124. Wang XR, Xiao JP, Wang DG. Elevated levels of serum IL-36alpha in patients with systemic lupus erythematosus. BioMed Rep (2021) 15(3):76. doi: 10.3892/br.2021.1452
125. Mai SZ, Li CJ, Xie XY, Xiong H, Xu M, Zeng FQ, et al. Increased serum IL-36alpha and IL-36gamma levels in patients with systemic lupus erythematosus: Association with disease activity and arthritis. Int Immunopharmacol (2018) 58:103–8. doi: 10.1016/j.intimp.2018.03.011
126. Maronese CA, Pimentel MA, Li MM, Genovese G, Ortega-Loayza AG, Marzano AV. Pyoderma gangrenosum: an updated literature review on established and emerging pharmacological treatments. Am J Clin Dermatol (2022) 23(5):615–34. doi: 10.1007/s40257-022-00699-8
127. Barbieux C, Bonnet des Claustres M, Fahrner M, Petrova E, Tsoi LC, Gouin O, et al. Netherton syndrome subtypes share IL-17/IL-36 signature with distinct IFN-α and allergic responses. J Allergy Clin Immunol (2022) 149(4):1358–72. doi: 10.1016/j.jaci.2021.08.024
128. Alavi A, Prens EP, Kimball AB, Krueger JG, Mukhopadhyay S, Wang H, et al. Spesolimab for hidradenitis suppurativa: A proof-of-concept study. Journal of the American Academy of Dermatology (2023) 89(3):AB89. doi: 10.1016/j.jaad.2023.07.358
129. Neurath MF. IL-36 in chronic inflammation and cancer. Cytokine Growth Factor Rev (2020) 55:70–9. doi: 10.1016/j.cytogfr.2020.06.006
130. Chelvanambi M, Weinstein AM, Storkus WJ. IL-36 signaling in the tumor microenvironment. Adv Exp Med Biol (2020) 1240:95–110. doi: 10.1007/978-3-030-38315-2_8
Keywords: generalized pustular psoriasis, GPP, psoriasis, IL-36, IL-1, IL36RN, spesolimab
Citation: Hawkes JE, Visvanathan S and Krueger JG (2023) The role of the interleukin-36 axis in generalized pustular psoriasis: a review of the mechanism of action of spesolimab. Front. Immunol. 14:1292941. doi: 10.3389/fimmu.2023.1292941
Received: 12 September 2023; Accepted: 31 October 2023;
Published: 21 November 2023.
Edited by:
Toshiro Takai, Juntendo University, JapanReviewed by:
Shigetoshi Sano, Kochi Medical School Hospital, JapanCopyright © 2023 Hawkes, Visvanathan and Krueger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jason E. Hawkes, aGF3a2VzM0BnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.