 Farooq Riaz
Farooq Riaz Ping Wei2
Ping Wei2 Fan Pan
Fan Pan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 20 October 2023
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1292238
T-cell-mediated autoimmune type 1 diabetes (T1D) is characterized by the immune-mediated destruction of pancreatic beta cells (β-cells). The increasing prevalence of T1D poses significant challenges to the healthcare system, particularly in countries with struggling economies. This review paper highlights the multifaceted roles of Peroxisome Proliferator-Activated Receptors (PPARs) in the context of T1D, shedding light on their potential as regulators of immune responses and β-cell biology. Recent research has elucidated the intricate interplay between CD4+ T cell subsets, such as Tregs and Th17, in developing autoimmune diseases like T1D. Th17 cells drive inflammation, while Tregs exert immunosuppressive functions, highlighting the delicate balance crucial for immune homeostasis. Immunotherapy has shown promise in reinstating self-tolerance and restricting the destruction of autoimmune responses, but further investigations are required to refine these therapeutic strategies. Intriguingly, PPARs, initially recognized for their role in lipid metabolism, have emerged as potent modulators of inflammation in autoimmune diseases, particularly in T1D. Although evidence suggests that PPARs affect the β-cell function, their influence on T-cell responses and their potential impact on T1D remains largely unexplored. It was noted that PPARα is involved in restricting the transcription of IL17A and enhancing the expression of Foxp3 by minimizing its proteasomal degradation. Thus, antagonizing PPARs may exert beneficial effects in regulating the differentiation of CD4+ T cells and preventing T1D. Therefore, this review advocates for comprehensive investigations to delineate the precise roles of PPARs in T1D pathogenesis, offering innovative therapeutic avenues that target both the immune system and pancreatic function. This review paper seeks to bridge the knowledge gap between PPARs, immune responses, and T1D, providing insights that may revolutionize the treatment landscape for this autoimmune disorder. Moreover, further studies involving PPAR agonists in non-obese diabetic (NOD) mice hold promise for developing novel T1D therapies.
Diabetes is regarded as the most critical and chronic medical condition that is illustrated by the increased blood glucose levels in association with impaired insulin activity due to the defective pancreatic beta cells (β-cell) function (1, 2). As per the definition and classification by the American Diabetes Association, diabetes has four distinct subtypes: (i) Type 1 diabetes (T1D) – an autoimmune disorder marked by insufficient insulin production that extinguishes β-cells; (ii) Type 2 diabetes (T2D) – associated with insulin resistance and a gradual decline in insulin levels accompanied by the loss of β-cells; (iii) Specific type of diabetes ascending from diverse underlying causes; and (iv) Gestational Diabetes Mellitus (GDM) – occurring in the later trimesters of pregnancy, without prior existence before conception (3). It has been estimated that diabetes impacts 9.3% of the overall global population (4), with 19.3% in elderly patients (5). Alarmingly, diabetes is escalating within certain nations (6–9).
Type 1 diabetes (T1D) is a chronic and progressive autoimmune illness. It is a long-lasting disease marked by the body’s incapability to generate insulin. Insulin, produced by β-cells, is a pivotal anabolic hormone that exerts distinct effects on protein, lipid, glucose, and growth (10, 11). T1D primarily occurs due to the death of β-cells in response to recurrent autoimmunity (12, 13). Consequently, T1D emerges as a systemic disorder defined by the hallmark trait of hyperglycemia (14). An array of investigations has underscored the substantial role of genetic, social, economic, and environmental factors in triggering autoimmune responses and ultimately driving the onset of T1D (15–21).
The immune mechanisms driving the autoimmune assault on β-cells have primarily been elucidated through studies conducted in T1D models in rodents (22). Ample evidence indicates that humoral immune responses are pivotal in producing autoantibodies that target the pancreatic β-cells (23, 24). These autoantibodies, including those against insulin, glutamic acid decarboxylase (GAD), islet cell antigens, insulinoma-associated antigen-2 (IA-2), and zinc transporter 8 (ZnT8), initiate an autoimmune response (23, 25–27). This misguided immune attack leads to the progressive destruction of insulin-producing β-cells, resulting in insulin deficiency and elevated blood sugar levels. Also, the presence of these autoantibodies is a hallmark of T1D and is often utilized in precision diagnosis (28, 29). Besides, this autoimmune process occurs when the immune system, primarily coordinated by T cells, initiates an abnormal assault on the insulin-producing β-cells within Langerhans’ pancreatic islets (30). Both CD4+ and CD8+ T cells play pivotal roles in T1D, as their significance in the development of T1D is supported by substantial evidence (31, 32). Meanwhile, it has been understood that specific major histocompatibility complex (MHC) class II haplotypes, and to a lesser extent, MHC class I haplotypes, are associated with an increased predisposition to the development of diabetes (32, 33).
Considering the impact of T cells and β-cells in the pathogenesis of T1D, the present therapeutic strategies have predominantly resolved on either overturning the ongoing immune assault or activating the regeneration of beta cells; however, the effectiveness of these therapies is limited (34, 35). Consequently, a persistent demand exists for approaches that simultaneously attenuate the immune response while enhancing β-cells function. The PPAR family stands out as a promising focus for addressing T1D using this approach because PPARs exhibit anti-inflammatory characteristics, influence the biology of β-cells, and control the lipid composition in the pancreas (36–39). This review aims to provide a comprehensive summary of the current understanding regarding the role of T cells in the pathogenesis of autoimmune T1D and how PPARs play essential roles in mediating the immune responses within the pathophysiological context of T1D. By elucidating the connections between T cell-mediated autoimmune T1D and the modulatory functions of PPARs, this review underscores the potential attractiveness of PPARs as targets for therapeutic interventions in T1D management.
The concept of a connection between the immune system and T1D was first introduced in 1973 when researchers discovered a clear connection between HLA antigens and insulin-dependent diabetes mellitus and distinguished it from insulin-independent diabetes (40). Subsequent genome-wide association investigations have established this link and revealed that HLA genes subsidize up to 50% of the genetic susceptibility to T1D, particularly the HLA class II loci. This finding strongly advocates that the selective exhibition of specific autoantigen peptides exerts a critical part in the pathogenesis of T1D (41–43). In the meantime, several meta-analyses have brought to light non-HLA high-risk genetic variations within specific genes, including but not limited to IL2RA, CTLA4, PTPN22, and INS-VNTR (variable number of tandem repeats) (44–47). Studies examining the longitudinal levels of plasma oxylipins and their connection to the risk of T1D in at-risk children showed that higher levels of certain oxylipins related to linoleic acid and alpha-linolenic acid were associated with a reduced risk of T1D (48, 49). These oxylipins, which have pro-resolving and pro-inflammatory properties, may reflect resilience to environmental triggers (50). Conversely, oxylipins related to arachidonic acid (ARA) were linked to an increased risk of T1D, possibly indicating inflammation after the onset of islet autoimmunity (48). Meanwhile, the SNP rs143070873 was strongly linked to the LA-related oxylipin 9-HODE, and rs6444933 (linked with CLDN11) was associated with the LA-related oxylipin 13 S-HODE. Additionally, a locus between LOC100131146 and MIR1302-7, rs10118380 and an intronic variant in TRPM3 were connected to the ARA-related oxylipin 11-HETE, highlighting their involvement in inflammatory signaling and oxylipin production (51). These genetic variations have been associated with a reduced ability to maintain both peripheral and central immune tolerance toward self-antigens and heightened T-cell stimulation and proliferation. These observations underscore the significant role that T cells play in the intricate process of T1D.
The phenotype of human T1D is recapitulated in the non-obese diabetic (NOD) mouse (52). These NOD mice comprehensively enhanced our knowledge of the T1D pathogenesis. Research conducted in NOD mice has elucidated that the development of T1D depends on the involvement of CD8+ and CD4+ T cells (53). The involvement of T cells in the progression of T1D has been shown in Figure 1. Notably, T1D can be transferred solely to immunocompromised syngeneic recipients when splenic T cells, CD8+ and CD4+ T cells, are transferred from a donor NOD mice (53). Conversely, detecting islet-specific autoreactive CD8+ and CD4+ T cells in insulitis lesions, pancreatic draining lymph nodes, and peripheral blood has furnished compelling findings subsidizing the autoimmune nature of T1D (54–58). This presence of autoreactive T cells points towards an impaired immune response, where central immune tolerance weakening towards self-antigens results in the loss of their immune reactivity for foreign proteins, and this weakening is believed to play a role in the insulin-producing cells for the immune attack directed at self-antigens (59–61).
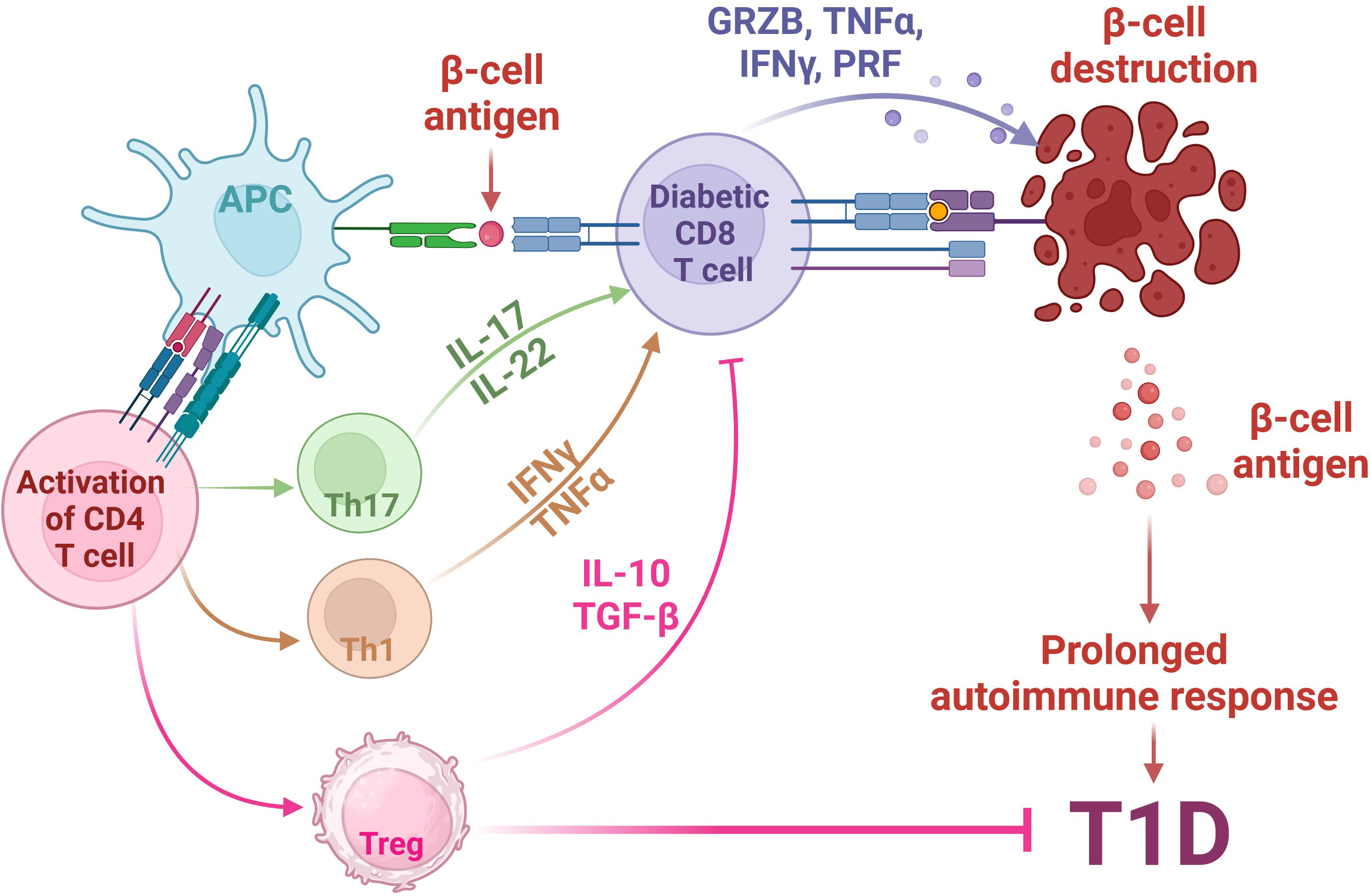
Figure 1 Involvement of T cells in the pathogenesis of autoimmune T1D. The pathogenesis of Type 1 Diabetes (T1D) involves a complex interplay between genetic susceptibility and environmental factors. In individuals predisposed to T1D, environmental triggers lead to stress in pancreatic β-cells, releasing β-cell antigens. These antigens are processed by Antigen-Presenting Cells (APCs) and presented via HLA class II MHC molecules to naïve CD4+ T cells. Activated CD4+ T cells shift towards (i) Th1 phenotype, releasing cytokines such as TNFα and INFγ; (ii) Th17 phenotype, releasing inflammatory cytokines such as IL17 and IL-22; and (iii) Immunosuppressive regulatory T cells (Tregs), which secret anti-inflammatory IL-10 and TGFβ. The activation of Th1 and Th17 activates auto-reactive cytotoxic CD8+ T cells, which then migrate to pancreatic islets, releasing cytotoxic agents, such as granzyme B (GRZB), perforin (PRF), TNFα and INFγ, ultimately leading to the destruction of β cells, a central event in T1D pathogenesis. Meanwhile, the release of anti-inflammatory IL-10 and TGFβ molecules from Tregs inhibits the auto-reactive CD8+ T cell activity, leading to the inhibition of T1D. Created with BioRender.com.
The elevated population of autoreactive CD8+ T cells (which are abundant for particular resident memory cells) in the pancreas of individuals with T1D suggests a differential peripheral regulation and/or activation in patients with T1D (62) as islet autoreactive T cells exhibit different functions than T cells that guard us from infections and cancer. For instance, it was reported that β-cell-specific CD8+ T cells extinguish the insulin-secreting β-cells, predominantly due to the intrinsic events of β-cells (57, 63–65). It was reviewed that CD8+ T cells can diminish the pancreatic β-cells via MHC class I-regulated cytotoxic mechanisms (66). Even though auto-reactive cells are also found in individuals without autoimmune conditions, the elevated numbers of CD8+ T cells, especially resident memory cells, in the pancreas of T1D individuals suggest distinct immune activity in these patients (54, 67, 68).
It is noteworthy to mention that both CD8+ T cells and CD4+ T cells can secrete various effector cytokines, such as interferon-γ (IFNγ), IL (interleukin)-1β, and tumor necrosis factor-α (TNFα) (69, 70). In T1D, the release of IFNγ by CD4 and CD8 cells may destroy β-cells and islets (69). Meanwhile, IFNγ, along with other cytokines, induces the death receptor FAS (also known as CD95) levels, stimulates the production of chemokines by β-cells and elevates their vulnerability to the autoimmune T1D (70–75). The process of β-cell apoptosis can be triggered by the activation of FAS by activated CD4 T lymphocytes that express the FAS ligand (FASL) (76–78). Additionally, the chemokines produced by β-cells contribute to the recruitment of further mononuclear cells to the site, thereby intensifying the inflammatory response (79–81).
The pathogenesis of T1D is believed to commence when there is a low-level demise of β-cells, leading to the exposure of β-cell antigens. Through MHC class II molecules, these antigens are then taken up, processed, and displayed on the cell surface of antigen-presenting cells (APCs) (82, 83). CD4+ T lymphocytes in the surrounding pancreatic lymph nodes proliferate and develop into auto-reactive CD4+ effector T cells (Teffs) in response to antigen presentation and costimulation by APCs (84–86). Immune cell-derived complement proteins (C3a and C5a), which are activated locally during the contact between T cells and APCs, help further to promote Teff growth and function (87). Within the pancreatic islets, these activated Teffs release an array of cytokines, including IFNγ and IL-2, leading to the recruitment of cytotoxic macrophages and CD8+ T lymphocytes (88, 89). These cytotoxic inflammatory cells ultimately infiltrate the islets and initiate the destruction process known as “insulitis”. β-cell death ensues, partly due to direct perforin/granzyme-mediated toxicity by CD8+ T cells and partly as a result of the release of pro-inflammatory cytokines (IFNγ, TNFα, IL-1β) by CD4+ T cells and macrophages (88, 89). Additionally, chemokines released by injured β-cells promote further recruitment of mononuclear cells, and the subsequent release of additional autoantigens enables the expansion and perpetuation of the autoreactive Teff response (90, 91).
Effector T cell subsets Th17, which produces IL-17A, a prominent pro-inflammatory cytokine, and is primarily recognized for its role in attracting other immune cells to sites of inflammation (92, 93). Numerous investigations on rodent models of diabetes suggest the participation of the Th17 subsets in the development of T1D. For instance, in the NOD, a spontaneous autoimmune diabetes model, IL-17F and IL-17A levels in the islets are associated with insulitis. It was suggested that young mice at a prediabetic stage do not exhibit increased expression of IL-17F or IL-17A in the islets. However, older diabetic mice show increased IL-17F and IL-17A, which coincides with the onset of insulitis (94). Inhibiting Th17 cells has significantly reduced diabetes development effectively (95, 96). In IL-17-depleted NOD mice, there is an adjournment in the commencement of diabetes, and insulitis is reduced (97). Additionally, in streptozotocin-induced diabetes, IL-23 plays a role in promoting diabetes development, mainly when subdiabetogenic doses of streptozotocin are administered, as it leads to the expansion of Th17 cells (98). In parallel, clinical T1D patients also support the pathogenic role of Th17. For instance, CD4+ T cells from newly diagnosed T1D adolescents are reported to produce enhanced levels of IL-17 and IL-22 (99). Interestingly, there is no noticeable increase in IFNγ levels or T-bet levels in T1D patients, indicating a bias toward a Th17 response in these individuals (99). Moreover, memory CD4+ T cells from the majority of the T1D patients display elevated secretion of IL-17 and IL-22, signifying an active Th17 response in vivo (99). Similarly, alternative investigation detected enhanced IL-17-producing CD4+ T cells in newly diagnosed T1D children (100). Notably, the circulatory CD4+ T cells in T1D patients secrete IL-17 upon activation by β-cell autoantigens (101). A proinflammatory cytokine environment that encourages Th17 development could be accountable for the elevated levels of IL-17 in T1D. In fact, monocytes from individuals with T1D dynamically express considerably more IL-6 and IL-1, encouraging memory CD4+ T cells to produce IL-17 (102).
Besides Teff, the population of CD4+ Foxp3+ regulatory T cells (Tregs) is also impaired, particularly in inflamed islets (103, 104). Foxp3+ Treg cells immunosuppressive cells, which maintain immune balance and modulate self-antigen response (105). The disruption of Foxp3+ Treg homeostasis is believed to enable the favorable differentiation and proliferation of pathogenic β cell-specific Teff (106–108). Though a few studies have concluded that the population of Tregs in the blood of T1D subjects is predominantly unbothered, the analysis of the suppressor function of Tregs isolated from T1D patients shows abridged Treg activity (109–112). Meanwhile, research involving NOD mice has highlighted the significance of Treg cells in prohibiting T1D. Notably, CD28-deficient NOD mice lack Tregs and experience an accelerated disease progression [17]. Another study showed that expression of CD226 in Tregs may lead to decreased Treg immunosuppressive function. In an attempt to elucidate the effect of CD226 in Treg, they found that specific deletion or inhibition of CD226 in Treg enhances the immunosuppressive function of Tregs, which decreases the susceptibility of T1D progression in NOD mice (106). Furthermore, approaches like administering IL-2 to augment Tregs numbers are considered a potential therapeutic avenue in diabetes (113). Although the function of T cells in autoimmune diseases, especially T1D, has been studied widely, further understanding of the role of T cells in T1D pathogenesis is necessary to develop innovative drugs that could enhance the function and population of β-cells by targeting these auto-reactive T cells and elevate the population of Tregs.
PPARs, or peroxisome proliferator-activated receptors, were discovered in the 1990s for their role in peroxisome proliferation (114). These receptors belong to the ligand-responsive nuclear hormone receptor family. They include three isoforms in mammals: PPARα, PPARβ/δ, and PPARγ. They primarily regulate lipid metabolism genes, encompassing lipogenesis, lipid storage and transport, and fatty acid oxidation (FAO) (114). These ligand-activated transcription factors are essential in regulating inflammation, energy homeostasis, and addressing issues like obesity and metabolic syndrome (115–117). They form heterodimers with the nuclear receptor RXR, bind to specific DNA-response elements in target gene promoters, recruit coactivators and facilitate chromatin remodeling to initiate DNA transcription (118, 119). Several peptides targeting PPARα (e.g., clofibrate, bezafibrate, fenofibrate) and PPARγ (e.g., ciglitazone, pioglitazone, rosiglitazone, troglitazone) have been employed to mdeicate metabolic conditions like T2D and hyperlipidemia (120–122).
PPAR isoforms exhibit significant functional and structural similarity, yet their expression patterns vary considerably. For instance, PPARα is characterized by its high expression in metabolically active tissues, e.g., adipose tissue, kidney, and liver. It is enhanced during periods of fasting and plays a pivotal role in regulating various metabolic processes. These processes include facilitating fatty acid oxidation (FAO), overseeing amino acid catabolism, modulating lipoprotein metabolism, regulating gluconeogenesis, controlling ketogenesis, and managing inflammatory responses (123, 124). PPARβ/δ, on the other hand, is ubiquitously expressed throughout the body. Its primary role revolves around promoting fatty acid oxidation (FAO). Meanwhile, the activation of PPARβ/δ employs an anti-inflammatory role, leading to abridged production of proinflammatory cytokines, contributing important roles in immune regulation (125, 126). On the contrary, PPARγ demonstrates a broader tissue distribution, with expression observed in numerous organs, including kidney, adipose tissue, intestine, and liver (127, 128). Its functions encompass the modulation of fat cell differentiation, the management of lipid storage, and the facilitation of monocyte differentiation into macrophages. In essence, PPARγ serves an essential role in controlling processes related to lipid metabolism and immune response modulation (129, 130).
In the pancreas, all three PPAR isoforms—PPARα, PPARγ, and PPARδ—are expressed in pancreatic β-cells. It has been believed that PPARα regulates fatty acid oxidation, whereas its expression is influenced by glucose levels (131, 132). Notably, high glucose levels repress PPARα expression in INS-1E cells (islets β-cell line) and primary rat islets (133, 134). Moreover, the glucose-dependent increase in insulin level appears to depend on PPARα, as glucose fails to enhance insulin levels in islets PPARα knockout mice (135). PPARα directly or indirectly impacts the key genes tangled in regulating β-cell function and development. For instance, In INS-1 cells and isolated rat islets, PPAR has been found to increase Pdx-1 levels [a transcription factor important for pancreatic and beta-cell development (136–138)]. Additionally, it was observed that PPARα knockout reduced the level of insulin, MafA [a regulator of insulin secretion (139)], Nkx6.1 [a transcription factor essential for maintaining mature β-cell function (140)], GLUT2, and glucokinase (141).
Interestingly, it was explored that PPARγ directs fatty acids toward esterification (132). Additionally, research investigating the effects of PPAR stimulation or upregulation on insulin secretion and proinsulin production has produced inconsistent findings (142–146). On the one hand, some research has shown that overexpressing PPARγ in INS-1E cells impairs glucose-stimulated insulin secretion (GSIS) (142). On the other hand, several investigations have shown that activation or upregulation of PPAR promotes GSIS in isolated islets and β-cell (147–149). Recently, it has been found that polymorphisms in the IGF1 and PPARγ genes are linked to decreased estimated glomerular filtration rates in children and adolescents with T1D, elevating their susceptibility to early renal complications and impacting the immune response (150–152).
Like PPARα, PPARγ exerts a role in regulating several critical proteins intricately regulating the function and development of β-cells. Activation of PPARγ through compounds like troglitazone (a PPARγ agonist) leads to upregulating genes such as GLUT2, glucokinase, Nkx6.1, and Pdx-1 (153, 154). Furthermore, in PPARγ pancreatic knockout mice, decreased levels of Pdx-1 protein were observed in islets (155). These findings are supported by the presence of peroxisome proliferator response elements (PPRE) sequences in the promoter regions of genes such as Pdx-1 (153, 155), GLUT2 (156), and glucokinase (157).
Although PPARδ is the most prevalent isoform in the pancreas, its impacts on fatty acid oxidation (FAO) have received little attention until recently. PPARδ activation boosts FAO more than PPARα activation does. Fatty acids in the pancreas can cause lipotoxicity and GSIS, which are both long-term impacts on insulin production (149, 158). PPARβ/δ seems to play a crucial part in pancreatic development, as evidenced by studies involving pancreatic PPARβ/δ knockout mice. These mice showed a substantial increase in the mass of β-cells and pancreatic islets (159). This elevation in β-cell mass was connected to higher plasma insulin concentrations, which led to hypoglycemia and better glucose tolerance, and it raises concerns about the detrimental effects of insulin production in the adult pancreas (159) and suggests the negative role of insulin secretion in the mature pancreas. However, these observations contrast with another study, signifying a different role for PPARβ/δ. According to this study, PPARβ/δ promotes the differentiation of beta cells from stem cells by upregulating Pdx-1 (160). This inconsistency in findings underscores the complexity of PPARβ/δ’s role in pancreatic function and development, indicating that its effects may be context-dependent and influenced by various factors. Meanwhile, it is evident that polymorphisms in the promoter region of PPARβ/δ and PPARγ subsidize the genetic susceptibility to T1D and impact the disease score of autoimmunity in islets (151). The impact of PPAR isoforms on the islets suggests that PPAR may exert crucial roles in regulating the function and biology of β-cells (161–163), and further investigations are necessary to explore the part of PPARs in β-cells and T1D.
The pathogenicity of T1D includes the intricate interactions of β-cells with various immune cells, particularly T cells (164, 165). The effect of PPARs in T cell regulation and differentiation is multifaceted and characterized by isoform-specific variations. Tregs derived from PPARα knockout mice manifest impaired suppression of CD8+ and CD4+ T cells, diminished migratory capabilities, and reduced expression of several chemokine receptors (166). This phenomenon aligns with the prolonged inflammatory response observed in PPARα knockout mice upon exposure to agents like arachidonic acid (167). In mouse models, the Fenofibrate, a PPARα agonist, has been shown to elevate Foxp3+ regulatory T cells (168, 169). Similarly, our recent study emphasizes that knockout of PPARα in mice diminishes the population of Th17 cells, whereas treating T cells with fenofibrate elevates the population of Th17 (170). Mechanistically, we found that PPARα-depletion augments the activity of IKKα, which positively contributes to the transcription of IL17A by interacting with RORγ. Meanwhile, IKKα also interacts with Foxp3 for its proteasomal degradation, thus leading to an elevated population of Th17 cells. Notably, PPARα ablation augmented the IL-17+Foxp3+ double-positive cells in the brain of the EAE-induced animal model (170). These results indicate that activation of PPAR may inhibit the formation of Th17 cells and enhance the percentage of Foxp3+ Tregs, which may slow the evolution of autoimmune disorders, including T1D.
PPARα also regulates effector T cells, with heightened PPARα expression associated with augmented secretion of Th2-related cytokines. Conversely, PPARα knockout mice exhibit a greater propensity for differentiation toward a Th1 phenotype (171). The PPARα agonist WY14643 has also been observed to curtail proliferation in human T cells and enhance depletion of T cells by arresting them in the G2/S phase (172). Patients with hyperlipidemia who undergo fenofibrate treatment experience reduced levels of IFNγ and TNFα (173). These findings are corroborated by PPARα knockout mice observations, where elevated TNFα and IFNγ levels are evident (171).
The role of PPARγ in regulating the ratio of regulatory to effector T cells is now becoming clear. In PPARγ knockout mice, decreased PPARγ activity is correlated with a higher number of effector T cells, which is distinguished by increased antigen-specific proliferation and excessive IFNγ production in response to IL-12 (174). Furthermore, it has been demonstrated that PPAR inhibits RORt expression, preventing the development of Th17 cells in both humans and mice (175). In mouse models of colitis, PPARγ agonists, such as troglitazone and rosiglitazone, have been observed to alter the immune response from Th1 to Th2, resulting in reduced Th1-related transcription factors, cytokines, chemokines, and heightened expression of Th2-associated factors (176, 177). Conversely, PPARγ deficiency is associated with a diminished CD4+ Foxp3+ regulatory T cell population (178). This is underscored by identifying a specific Treg subset characterized by high PPARγ expression within visceral adipose tissue (179). PPARγ serves a central role in orchestrating these Tregs, as evidenced by the Treg formation prevention upon Treg-specific PPARγ deletion. Additionally, PPARγ activation encourages the growth of Foxp3+ regulatory T cells, whereas PPARγ depletion in Tregs increases the responses of effector T cells (174, 178, 180). Thus, the impact of PPARγ on Tregs may be context-dependent. Notably, PPAR-γ is essential in elevating the differentiation of Tregs and regulating insulin resistance. This occurs through a synergistic mechanism that reduces the expression of pro-inflammatory cytokines such as IL-6, TNFα, and IL-1β, while simultaneously enhances the anti-inflammatory cytokines like TGF-β, and IL-10 (181). In addition to the role of PPARs in the differentiation of Th1, Th2, Treg, and Th17 subsets, PPARγ likely to influence the generation of follicular helper T cells (Tfh). It was investigated that mice with CD4 cell-specific PPARγ knockout exhibit increased Tfh cell activation and a greater propensity for germinal center formation (182).
Activation of PPARβ/δ inhibits Th17 and Th1 responses while bolstering Th2 responses (183–185). In contrast, deletion of PPARβ/δ produces an opposing outcome. This discrepancy can be attributed to PPARβ/δ’s role in promoting FAO, thereby preventing the T cell proliferative burst that occurs after antigen identification as metabolism shifts from oxidative pathways to glycolysis (186–188). Although the information regarding the role of PPARs in autoimmune diseases is limited, the available data suggests that activation of PPARs, especially PPARα, may restrict the development of autoimmune diseases.
Considering the importance of herbal medicines (189, 190), numerous researches have been conducted to find herbs and natural compounds for treating T1D (191–193). In addition, a few studies also investigated various natural compounds that exhibited potency to target PPARs and have the potency to cure T1D. For instance, it has been studied that Astilbin, a flavonoid compound initially discovered for its ability to suppress effector CD4+ T cells by inhibiting their function (194), activates the ROS-dependent PPARγ pathway which leads to the suppression of effector CD4+ T cell activities through direct binding to Cytochrome P450 1B1. Consequently, It was suggested that Astilbin exerts immune-suppressive effects by downregulating the secretion of inflammatory cytokines by CD4+ T cells in the NOD mice (195). Similarly, another flavonoid, Epigallocatechin gallate, also decreases the progression of T1D by activating PPAR-γ (196–198). Curcumin, a PPAR agonist, has shown the ability to anticipate the damage of islets by exerting a protective impact on the β-cells (199, 200). A recent study exhibits that Curcumin exerts protective effects on the autoimmune T1D (201). They found that Curcumin dampens T lymphocyte responses by inhibiting proliferation and production of IFNγ, affecting the T-bet transcription factor. It also reduces NF-κB activation in NOD lymphocytes stimulated via TCR (201, 202).
In the pancreas, activation of PPAR enhances fatty acid oxidation, which can acutely potentiate GSIS. The PPARγ-agonist pioglitazone was observed to boost GSIS in db/db mice while the PPARα-agonist fenofibrate inhibited GSIS in newborn rats with active obesity (203, 204). This discrepancy may be attributed to the minimal level of PPAR-γ in INS-1E cells. In those with recently discovered T1D, pancreatic islets exhibit reduced sulfatide levels (23% of those in control participants) and decreased sphingolipid metabolism-related enzyme levels. Fenofibrate, known to activate sulfatide biosynthesis and act as an anti-inflammatory drug (205), ultimately impeded T1D in NOD mice (151). In a 19-year-old female with newly diagnosed T1D, fenofibrate medication started seven days after diagnosis disregarded the need for insulin therapy (206).
Numerous PPAR antagonists have been synthesized, although not initially developed for diabetes treatment (207). For example, a synthetic potent PPAR-α antagonist, GW6471, is primarily employed as a pharmacological tool for identifying effects that are reliant or independent of PPARs. GW9662, which has been elucidated as a PPAR-γ antagonist, facilitates the recruitment of NCOR1 nuclear receptor corepressor 1 (NCoR). Additionally, GSK3787 and GSK0660 serve as PPAR-δ antagonists for pharmacological purposes. Notably, GSK0660, when employed solely in human retinal microvascular endothelial cells, exhibits inverse agonist activity, inhibiting the TNFα-dependent level of numerous chemokines (208, 209). Similarly in the brain, agonists for PPAR-γ (rosiglitazone), PPAR-δ (GW501516), and PPAR-α (fenofibrate), as well as their respective antagonists (GW9662, GSK0660, and GW6471), collectively reduce the production of the pro-inflammatory cytokine TNFα in rat astrocytes under the influence of lipopolysaccharide (LPS) (210).
In conclusion, the cumulative prevalence of Type 1 diabetes (T1D), particularly in Asian countries, presents a significant challenge to healthcare systems due to its associated complications, treatment costs, resource limitations, and low awareness levels. To reduce this burden and mortality due to diabetes, countries must comprehend the extent of the disease and develop effective strategies. Recent research has unveiled the intricate interplay between CD4+ T cell subsets, particularly Th17 cells and Tregs, in autoimmune diseases like T1D. Th17 cells promote inflammation and immune responses, whereas Tregs exert immunosuppressive functions, striking a balance critical for immune homeostasis. The evolving understanding of the pathogenesis and etiology of T1D emphasizes the roles of both adaptive and innate immunity in driving the autoimmune response against pancreatic β-cells. Immunotherapy shows promise in regaining self-tolerance and preventing harmful autoimmune reactions, but further investigations are needed to refine these treatments.
In the realm of molecular mechanisms, PPARs have gained attention not only as regulators of lipid metabolism but also as potent modulators of inflammation and β-cell biology. While their potential in modulating T cell responses and impacting T1D remains largely unexplored, studies on PPAR agonists in NOD mice show promise. Thus, future investigations should focus on unraveling the precise roles of PPARs in T1D pathology, offering a novel treatment approach that targets both the immune system and pancreatic function. Conversely, as the utilization of medicinal plants and their derivatives has shown promise in reducing the overall prevalence of T1D by augmenting the population of Tregs and activating the PPARs, further research should prioritize the extraction of novel herbal plants or the purification of their derivatives for consideration in diabetes treatment. This immunomodulatory effect underscores the potential of herbal remedies in T1D management. Moreover, these promising herbal interventions warrant more extensive exploration through clinical trials, potentially offering novel and effective therapeutic options for individuals living with T1D.
FR: Conceptualization, Writing – original draft, Writing – review & editing. PW: Writing – original draft, Writing – review & editing. FP: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. FP’s work was supported by the National Key R&D Program of China (2021YFC2400500) and (2022YFC2403000), the National Natural Science Foundation of China (Grant 32170925), Shenzhen Science and Technology Program (KQTD20210811090115019), the Shenzhen Science and Technology Program (JCYJ2022081800807016), the startup fund of SIAT and CAS. PW’s work was supported by the Natural Science Foundation of Chongqing Grant CSTB2022NSCQ-MSX1069, the Chongqing for overseas Scholars Grant CX2022118.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chan JCN, Lim LL, Wareham NJ, Shaw JE, Orchard TJ, Zhang P, et al. The lancet commission on diabetes: using data to transform diabetes care and patient lives. Lancet (London England) (2021) 396(10267):2019–82. doi: 10.1016/s0140-6736(20)32374-6
2. Chatterjee S, Khunti K, Davies MJ. Type 2 diabetes. Lancet (London England) (2017) 389(10085):2239–51. doi: 10.1016/s0140-6736(17)30058-2
3. Association AD. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes—2021. Diabetes Care (2020) 44(Supplement_1):S15–33. doi: 10.2337/dc21-S002
4. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the international diabetes federation diabetes atlas, 9(Th) edition. Diabetes Res Clin Pract (2019) 157:107843. doi: 10.1016/j.diabres.2019.107843
5. Sinclair A, Saeedi P, Kaundal A, Karuranga S, Malanda B, Williams R. Diabetes and global ageing among 65-99-year-old adults: findings from the international diabetes federation diabetes atlas, 9(Th) edition. Diabetes Res Clin Pract (2020) 162:108078. doi: 10.1016/j.diabres.2020.108078
6. Azeem S, Khan U, Liaquat A. The increasing rate of diabetes in Pakistan: A silent killer. Ann Med Surg (2012) (2022) 79:103901. doi: 10.1016/j.amsu.2022.103901
7. Rowley WR, Bezold C, Arikan Y, Byrne E, Krohe S. Diabetes 2030: insights from yesterday, today, and future trends. Population Health Manage (2017) 20(1):6–12. doi: 10.1089/pop.2015.0181
8. Lin X, Xu Y, Pan X, Xu J, Ding Y, Sun X, et al. Global, regional, and national burden and trend of diabetes in 195 countries and territories: an analysis from 1990 to 2025. Sci Rep (2020) 10(1):14790. doi: 10.1038/s41598-020-71908-9
9. GBD 2021 Diabetes Collaborators. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the global burden of disease study 2021. Lancet (London England) (2023) 402(10397):203–34. doi: 10.1016/s0140-6736(23)01301-6
10. Møller N, Jørgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocrine Rev (2009) 30(2):152–77. doi: 10.1210/er.2008-0027
11. Huang Z, Huang L, Waters MJ, Chen C. Insulin and growth hormone balance: implications for obesity. Trends Endocrinol Metabolism: TEM (2020) 31(9):642–54. doi: 10.1016/j.tem.2020.04.005
12. Burrack AL, Martinov T, Fife BT. T cell-mediated beta cell destruction: autoimmunity and alloimmunity in the context of type 1 diabetes. Front Endocrinol (2017) 8:343. doi: 10.3389/fendo.2017.00343
13. Donath MY. Type 1 diabetes: what is the role of autoimmunity in β Cell death? J Clin Invest (2022) 132(20):e164460. doi: 10.1172/jci164460
14. Kahanovitz L, Sluss PM, Russell SJ. Type 1 diabetes - a clinical perspective. Point Care (2017) 16(1):37–40. doi: 10.1097/poc.0000000000000125
15. Redondo MJ, Steck AK, Pugliese A. Genetics of type 1 diabetes. Pediatr Diabetes (2018) 19(3):346–53. doi: 10.1111/pedi.12597
16. Pociot F, Lernmark Å. Genetic risk factors for type 1 diabetes. Lancet (London England) (2016) 387(10035):2331–9. doi: 10.1016/s0140-6736(16)30582-7
17. Steck AK, Rewers MJ. Genetics of type 1 diabetes. Clin Chem (2011) 57(2):176–85. doi: 10.1373/clinchem.2010.148221
18. Rewers M, Ludvigsson J. Environmental risk factors for type 1 diabetes. Lancet (London England) (2016) 387(10035):2340–8. doi: 10.1016/s0140-6736(16)30507-4
19. Quinn LM, Wong FS, Narendran P. Environmental determinants of type 1 diabetes: from association to proving causality. Front Immunol (2021) 12:737964. doi: 10.3389/fimmu.2021.737964
20. Hill-Briggs F, Adler NE, Berkowitz SA, Chin MH, Gary-Webb TL, Navas-Acien A, et al. Social determinants of health and diabetes: A scientific review. Diabetes Care (2020) 44(1):258–79. doi: 10.2337/dci20-0053
21. Hill KE, Gleadle JM, Pulvirenti M, McNaughton DA. The social determinants of health for people with type 1 diabetes that progress to end-stage renal disease. Health Expectations (2015) 18(6):2513–21. doi: 10.1111/hex.12220
22. Buschard K. What causes type 1 diabetes? Lessons from animal models. APMIS Supplementum (2011) 132):1–19. doi: 10.1111/j.1600-0463.2011.02765.x
23. Kawasaki E. Anti-islet autoantibodies in type 1 diabetes. Int J Mol Sci (2023) 24(12):10012. doi: 10.3390/ijms241210012
24. Peters L, Posgai A, Brusko TM. Islet-immune interactions in type 1 diabetes: the nexus of beta cell destruction. Clin Exp Immunol (2019) 198(3):326–40. doi: 10.1111/cei.13349
25. Arvan P, Pietropaolo M, Ostrov D, Rhodes CJ. Islet autoantigens: structure, function, localization, and regulation. Cold Spring Harbor Perspect Med (2012) 2(8):a007658. doi: 10.1101/cshperspect.a007658
26. Rodriguez-Calvo T, Johnson JD, Overbergh L, Dunne JL. Neoepitopes in type 1 diabetes: etiological insights, biomarkers and therapeutic targets. Front Immunol (2021) 12:667989. doi: 10.3389/fimmu.2021.667989
27. Mistry S, Gouripeddi R, Raman V, Facelli JC. Stratifying risk for onset of type 1 diabetes using islet autoantibody trajectory clustering. Diabetologia (2023) 66(3):520–34. doi: 10.1007/s00125-022-05843-x
28. So M, Speake C, Steck AK, Lundgren M, Colman PG, Palmer JP, et al. Advances in type 1 diabetes prediction using islet autoantibodies: beyond a simple count. Endocrine Rev (2021) 42(5):584–604. doi: 10.1210/endrev/bnab013
29. Michalek DA, Onengut-Gumuscu S, Repaske DR, Rich SS. Precision medicine in type 1 diabetes. J Indian Institute Sci (2023) 103(1):335–51. doi: 10.1007/s41745-023-00356-x
30. Marré ML, Piganelli JD. Environmental factors contribute to β Cell endoplasmic reticulum stress and neo-antigen formation in type 1 diabetes. Front Endocrinol (2017) 8:262. doi: 10.3389/fendo.2017.00262
31. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol (2009) 155(2):173–81. doi: 10.1111/j.1365-2249.2008.03860.x
32. Wicker LS, Clark J, Fraser HI, Garner VE, Gonzalez-Munoz A, Healy B, et al. Type 1 diabetes genes and pathways shared by humans and nod mice. J Autoimmun (2005) 25 Suppl:29–33. doi: 10.1016/j.jaut.2005.09.009
33. Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature (2010) 464(7293):1293–300. doi: 10.1038/nature08933
34. Cobo-Vuilleumier N, Lorenzo PI, Rodríguez NG, Herrera Gómez IG, Fuente-Martin E, López-Noriega L, et al. Lrh-1 agonism favours an immune-islet dialogue which protects against diabetes mellitus. Nat Commun (2018) 9(1):1488. doi: 10.1038/s41467-018-03943-0
35. Atkinson MA, Roep BO, Posgai A, Wheeler DCS, Peakman M. The challenge of modulating β-cell autoimmunity in type 1 diabetes. Lancet Diabetes Endocrinol (2019) 7(1):52–64. doi: 10.1016/s2213-8587(18)30112-8
36. Holm LJ, Mønsted M, Haupt-Jorgensen M, Buschard K. Ppars and the development of type 1 diabetes. PPAR Res (2020) 2020:6198628. doi: 10.1155/2020/6198628
37. Choi JM, Bothwell AL. The nuclear receptor ppars as important regulators of T-cell functions and autoimmune diseases. Molecules Cells (2012) 33(3):217–22. doi: 10.1007/s10059-012-2297-y
38. Hogh KL, Craig MN, Uy CE, Nygren H, Asadi A, Speck M, et al. Overexpression of pparγ Specifically in pancreatic β-cells exacerbates obesity-induced glucose intolerance, reduces β-cell mass, and alters islet lipid metabolism in male mice. Endocrinology (2014) 155(10):3843–52. doi: 10.1210/en.2014-1076
39. Yessoufou A, Wahli W. Multifaceted roles of peroxisome proliferator-activated receptors (Ppars) at the cellular and whole organism levels. Swiss Med Weekly (2010) 140:w13071. doi: 10.4414/smw.2010.13071
40. Nerup J, Platz P, Andersen OO, Christy M, Lyngsoe J, Poulsen JE, et al. Hl-a antigens and diabetes mellitus. Lancet (London England) (1974) 2(7885):864–6. doi: 10.1016/s0140-6736(74)91201-x
41. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet (2009) 41(6):703–7. doi: 10.1038/ng.381
42. van Lummel M, van Veelen PA, de Ru AH, Janssen GM, Pool J, Laban S, et al. Dendritic cells guide islet autoimmunity through a restricted and uniquely processed peptidome presented by high-risk hla-dr. J Immunol (Baltimore Md 1950) (2016) 196(8):3253–63. doi: 10.4049/jimmunol.1501282
43. van Lummel M, van Veelen PA, de Ru AH, Pool J, Nikolic T, Laban S, et al. Discovery of a selective islet peptidome presented by the highest-risk hla-dq8trans molecule. Diabetes (2016) 65(3):732–41. doi: 10.2337/db15-1031
44. Pugliese A, Zeller M, Fernandez A Jr., Zalcberg LJ, Bartlett RJ, Ricordi C, et al. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the ins vntr-iddm2 susceptibility locus for type 1 diabetes. Nat Genet (1997) 15(3):293–7. doi: 10.1038/ng0397-293
45. Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, et al. Insulin expression in human thymus is modulated by ins vntr alleles at the iddm2 locus. Nat Genet (1997) 15(3):289–92. doi: 10.1038/ng0397-289
46. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet (2004) 36(4):337–8. doi: 10.1038/ng1323
47. Vella A, Cooper JD, Lowe CE, Walker N, Nutland S, Widmer B, et al. Localization of a type 1 diabetes locus in the il2ra/cd25 region by use of tag single-nucleotide polymorphisms. Am J Hum Genet (2005) 76(5):773–9. doi: 10.1086/429843
48. Buckner T, Vanderlinden LA, DeFelice BC, Carry PM, Kechris K, Dong F, et al. The oxylipin profile is associated with development of type 1 diabetes: the diabetes autoimmunity study in the young (Daisy). Diabetologia (2021) 64(8):1785–94. doi: 10.1007/s00125-021-05457-9
49. Buckner T, Johnson RK, Vanderlinden LA, Carry PM, Romero A, Onengut-Gumuscu S, et al. An oxylipin-related nutrient pattern and risk of type 1 diabetes in the diabetes autoimmunity study in the young (Daisy). Nutrients (2023) 15(4):945. doi: 10.3390/nu15040945
50. Gabbs M, Leng S, Devassy JG, Monirujjaman M, Aukema HM. Advances in our understanding of oxylipins derived from dietary pufas. Adv Nutr (Bethesda Md) (2015) 6(5):513–40. doi: 10.3945/an.114.007732
51. Buckner T, Johnson RK, Vanderlinden LA, Carry PM, Romero A, Onengut-Gumuscu S, et al. Genome-wide analysis of oxylipins and oxylipin profiles in a pediatric population. Front Nutr (2023) 10:1040993. doi: 10.3389/fnut.2023.1040993
52. Anderson MS, Bluestone JA. The nod mouse: A model of immune dysregulation. Annu Rev Immunol (2005) 23:447–85. doi: 10.1146/annurev.immunol.23.021704.115643
53. Phillips JM, Parish NM, Raine T, Bland C, Sawyer Y, de la Peña H, et al. Type 1 diabetes development requires both cd4+ and cd8+ T cells and can be reversed by non-depleting antibodies targeting both T cell populations. Rev Diabetic Stud RDS (2009) 6(2):97–103. doi: 10.1900/rds.2009.6.97
54. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of islet-autoreactive cd8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med (2012) 209(1):51–60. doi: 10.1084/jem.20111187
55. Velthuis JH, Unger WW, van der Slik AR, Duinkerken G, Engelse M, Schaapherder AF, et al. Accumulation of autoreactive effector T cells and allo-specific regulatory T cells in the pancreas allograft of a type 1 diabetic recipient. Diabetologia (2009) 52(3):494–503. doi: 10.1007/s00125-008-1237-z
56. Michels AW, Landry LG, McDaniel KA, Yu L, Campbell-Thompson M, Kwok WW, et al. Islet-derived cd4 T cells targeting proinsulin in human autoimmune diabetes. Diabetes (2017) 66(3):722–34. doi: 10.2337/db16-1025
57. Babon JA, DeNicola ME, Blodgett DM, Crèvecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med (2016) 22(12):1482–7. doi: 10.1038/nm.4203
58. Roep BO, Arden SD, de Vries RR, Hutton JC. T-cell clones from a type-1 diabetes patient respond to insulin secretory granule proteins. Nature (1990) 345(6276):632–4. doi: 10.1038/345632a0
59. Tan S, Li Y, Xia J, Jin CH, Hu Z, Duinkerken G, et al. Type 1 diabetes induction in humanized mice. Proc Natl Acad Sci United States America (2017) 114(41):10954–9. doi: 10.1073/pnas.1710415114
60. Skowera A, Ellis RJ, Varela-Calviño R, Arif S, Huang GC, Van-Krinks C, et al. Ctls are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J Clin Invest (2008) 118(10):3390–402. doi: 10.1172/jci35449
61. Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med (2017) 23(4):501–7. doi: 10.1038/nm.4289
62. Rodriguez-Calvo T, Ekwall O, Amirian N, Zapardiel-Gonzalo J, von Herrath MG. Increased immune cell infiltration of the exocrine pancreas: A possible contribution to the pathogenesis of type 1 diabetes. Diabetes (2014) 63(11):3880–90. doi: 10.2337/db14-0549
63. Gearty SV, Dündar F, Zumbo P, Espinosa-Carrasco G, Shakiba M, Sanchez-Rivera FJ, et al. An autoimmune stem-like cd8 T cell population drives type 1 diabetes. Nature (2022) 602(7895):156–61. doi: 10.1038/s41586-021-04248-x
64. Kronenberg D, Knight RR, Estorninho M, Ellis RJ, Kester MG, de Ru A, et al. Circulating preproinsulin signal peptide-specific cd8 T cells restricted by the susceptibility molecule hla-A24 are expanded at onset of type 1 diabetes and kill β-cells. Diabetes (2012) 61(7):1752–9. doi: 10.2337/db11-1520
65. Yeo L, Pujol-Autonell I, Baptista R, Eichmann M, Kronenberg-Versteeg D, Heck S, et al. Circulating β Cell-specific cd8(+) T cells restricted by high-risk hla class I molecules show antigen experience in children with and at risk of type 1 diabetes. Clin Exp Immunol (2020) 199(3):263–77. doi: 10.1111/cei.13391
66. Bender C, Rajendran S, von Herrath MG. New insights into the role of autoreactive cd8 T cells and cytokines in human type 1 diabetes. Front Endocrinol (2020) 11:606434. doi: 10.3389/fendo.2020.606434
67. Culina S, Lalanne AI, Afonso G, Cerosaletti K, Pinto S, Sebastiani G, et al. Islet-reactive cd8(+) T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci Immunol (2018) 3(20):eaao4013. doi: 10.1126/sciimmunol.aao4013
68. Kuric E, Seiron P, Krogvold L, Edwin B, Buanes T, Hanssen KF, et al. Demonstration of tissue resident memory cd8 T cells in insulitic lesions in adult patients with recent-onset type 1 diabetes. Am J Pathol (2017) 187(3):581–8. doi: 10.1016/j.ajpath.2016.11.002
69. El-Sheikh A, Suarez-Pinzon WL, Power RF, Rabinovitch A. Both cd4(+)and cd8(+)T cells are required for ifn-gamma gene expression in pancreatic islets and autoimmune diabetes development in biobreeding rats. J Autoimmun (1999) 12(2):109–19. doi: 10.1006/jaut.1998.0264
70. Wachlin G, Augstein P, Schröder D, Kuttler B, Klöting I, Heinke P, et al. Il-1beta, ifn-gamma and tnf-alpha increase vulnerability of pancreatic beta cells to autoimmune destruction. J Autoimmun (2003) 20(4):303–12. doi: 10.1016/s0896-8411(03)00039-8
71. Nigi L, Brusco N, Grieco GE, Licata G, Krogvold L, Marselli L, et al. Pancreatic alpha-cells contribute together with beta-cells to cxcl10 expression in type 1 diabetes. Front Endocrinol (2020) 11:630. doi: 10.3389/fendo.2020.00630
72. Igoillo-Esteve M, Marselli L, Cunha DA, Ladrière L, Ortis F, Grieco FA, et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics ccl2 expression by beta cells in type 2 diabetes. Diabetologia (2010) 53(7):1395–405. doi: 10.1007/s00125-010-1707-y
73. Martin AP, Grisotto MG, Canasto-Chibuque C, Kunkel SL, Bromberg JS, Furtado GC, et al. Islet expression of M3 uncovers a key role for chemokines in the development and recruitment of diabetogenic cells in nod mice. Diabetes (2008) 57(2):387–94. doi: 10.2337/db07-1309
74. Chen MC, Proost P, Gysemans C, Mathieu C, Eizirik DL. Monocyte chemoattractant protein-1 is expressed in pancreatic islets from prediabetic nod mice and in interleukin-1 beta-exposed human and rat islet cells. Diabetologia (2001) 44(3):325–32. doi: 10.1007/s001250051622
75. Allred MG, Chimenti MS, Ciecko AE, Chen YG, Lieberman SM. Characterization of type I interferon-associated chemokines and cytokines in lacrimal glands of nonobese diabetic mice. Int J Mol Sci (2021) 22(7):3767. doi: 10.3390/ijms22073767
76. Loweth AC, Williams GT, James RF, Scarpello JH, Morgan NG. Human islets of langerhans express fas ligand and undergo apoptosis in response to interleukin-1beta and fas ligation. Diabetes (1998) 47(5):727–32. doi: 10.2337/diabetes.47.5.727
77. Angstetra E, Graham KL, Emmett S, Dudek NL, Darwiche R, Ayala-Perez R, et al. In vivo effects of cytokines on pancreatic beta-cells in models of type I diabetes dependent on cd4(+) T lymphocytes. Immunol Cell Biol (2009) 87(2):178–85. doi: 10.1038/icb.2008.81
78. Darwiche R, Chong MM, Santamaria P, Thomas HE, Kay TW. Fas is detectable on beta cells in accelerated, but not spontaneous, diabetes in nonobese diabetic mice. J Immunol (Baltimore Md 1950) (2003) 170(12):6292–7. doi: 10.4049/jimmunol.170.12.6292
79. Cosentino C, Regazzi R. Crosstalk between macrophages and pancreatic β-cells in islet development, homeostasis and disease. Int J Mol Sci (2021) 22(4):1765. doi: 10.3390/ijms22041765
80. Wilcox NS, Rui J, Hebrok M, Herold KC. Life and death of β Cells in type 1 diabetes: A comprehensive review. J Autoimmun (2016) 71:51–8. doi: 10.1016/j.jaut.2016.02.001
81. Nackiewicz D, Dan M, Speck M, Chow SZ, Chen YC, Pospisilik JA, et al. Islet macrophages shift to a reparative state following pancreatic beta-cell death and are a major source of islet insulin-like growth factor-1. iScience (2020) 23(1):100775. doi: 10.1016/j.isci.2019.100775
82. Calderon B, Suri A, Miller MJ, Unanue ER. Dendritic cells in islets of langerhans constitutively present beta cell-derived peptides bound to their class ii mhc molecules. Proc Natl Acad Sci United States America (2008) 105(16):6121–6. doi: 10.1073/pnas.0801973105
83. Boldison J, Wong FS. Immune and pancreatic β Cell interactions in type 1 diabetes. Trends Endocrinol Metabolism: TEM (2016) 27(12):856–67. doi: 10.1016/j.tem.2016.08.007
84. Ireland JM, Unanue ER. Processing of proteins in autophagy vesicles of antigen-presenting cells generates citrullinated peptides recognized by the immune system. Autophagy (2012) 8(3):429–30. doi: 10.4161/auto.19261
85. Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Müller M, et al. Autophagy promotes mhc class ii presentation of peptides from intracellular source proteins. Proc Natl Acad Sci United States America (2005) 102(22):7922–7. doi: 10.1073/pnas.0501190102
86. Formby B, Hosszufalusi N, Chan E, Miller N, Teruya M, Takei S, et al. Quantitative and functional analyses of spleen and in situ islet immune cells before and after diabetes onset in the nod mouse. Autoimmunity (1992) 12(2):95–102. doi: 10.3109/08916939209150315
87. Cravedi P, Leventhal J, Lakhani P, Ward SC, Donovan MJ, Heeger PS. Immune cell-derived C3a and C5a costimulate human T cell alloimmunity. Am J Transplant (2013) 13(10):2530–9. doi: 10.1111/ajt.12405
88. Cantor J, Haskins K. Recruitment and activation of macrophages by pathogenic cd4 T cells in type 1 diabetes: evidence for involvement of ccr8 and ccl1. J Immunol (Baltimore Md 1950) (2007) 179(9):5760–7. doi: 10.4049/jimmunol.179.9.5760
89. Espinosa-Carrasco G, Le Saout C, Fontanaud P, Stratmann T, Mollard P, Schaeffer M, et al. Cd4(+) T helper cells play a key role in maintaining diabetogenic cd8(+) T cell function in the pancreas. Front Immunol (2017) 8:2001. doi: 10.3389/fimmu.2017.02001
90. Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol (2009) 5(4):219–26. doi: 10.1038/nrendo.2009.21
91. Li Y, Sun F, Yue TT, Wang FX, Yang CL, Luo JH, et al. Revisiting the antigen-presenting function of β Cells in T1d pathogenesis. Front Immunol (2021) 12:690783. doi: 10.3389/fimmu.2021.690783
92. Kuwabara T, Ishikawa F, Kondo M, Kakiuchi T. The role of il-17 and related cytokines in inflammatory autoimmune diseases. Mediat Inflamm (2017) 2017:3908061. doi: 10.1155/2017/3908061
93. Weaver CT, Hatton RD, Mangan PR, Harrington LE. Il-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol (2007) 25:821–52. doi: 10.1146/annurev.immunol.25.022106.141557
94. Martin-Orozco N, Chung Y, Chang SH, Wang YH, Dong C. Th17 Cells Promote Pancreatic Inflammation but Only Induce Diabetes Efficiently in Lymphopenic Hosts after Conversion into Th1 Cells. Eur J Immunol (2009) 39(1):216–24. doi: 10.1002/eji.200838475
95. Jain R, Tartar DM, Gregg RK, Divekar RD, Bell JJ, Lee HH, et al. Innocuous ifngamma induced by adjuvant-free antigen restores normoglycemia in nod mice through inhibition of il-17 production. J Exp Med (2008) 205(1):207–18. doi: 10.1084/jem.20071878
96. Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, et al. Inhibition of th17 cells regulates autoimmune diabetes in nod mice. Diabetes (2009) 58(6):1302–11. doi: 10.2337/db08-1113
97. Kuriya G, Uchida T, Akazawa S, Kobayashi M, Nakamura K, Satoh T, et al. Double deficiency in il-17 and ifn-Γ Signalling significantly suppresses the development of diabetes in the nod mouse. Diabetologia (2013) 56(8):1773–80. doi: 10.1007/s00125-013-2935-8
98. Mensah-Brown EP, Shahin A, Al-Shamisi M, Wei X, Lukic ML. Il-23 leads to diabetes induction after subdiabetogenic treatment with multiple low doses of streptozotocin. Eur J Immunol (2006) 36(1):216–23. doi: 10.1002/eji.200535325
99. Honkanen J, Nieminen JK, Gao R, Luopajarvi K, Salo HM, Ilonen J, et al. Il-17 immunity in human type 1 diabetes. J Immunol (Baltimore Md 1950) (2010) 185(3):1959–67. doi: 10.4049/jimmunol.1000788
100. Marwaha AK, Crome SQ, Panagiotopoulos C, Berg KB, Qin H, Ouyang Q, et al. Cutting edge: increased il-17-secreting T cells in children with new-onset type 1 diabetes. J Immunol (Baltimore Md 1950) (2010) 185(7):3814–8. doi: 10.4049/jimmunol.1001860
101. Arif S, Moore F, Marks K, Bouckenooghe T, Dayan CM, Planas R, et al. Peripheral and islet interleukin-17 pathway activation characterizes human autoimmune diabetes and promotes cytokine-mediated β-cell death. Diabetes (2011) 60(8):2112–9. doi: 10.2337/db10-1643
102. Bradshaw EM, Raddassi K, Elyaman W, Orban T, Gottlieb PA, Kent SC, et al. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing th17 cells. J Immunol (Baltimore Md 1950) (2009) 183(7):4432–9. doi: 10.4049/jimmunol.0900576
103. Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity (2008) 28(5):687–97. doi: 10.1016/j.immuni.2008.03.016
104. Xufré C, Costa M, Roura-Mir C, Codina-Busqueta E, Usero L, Pizarro E, et al. Low frequency of gitr+ T cells in ex vivo and in vitro expanded treg cells from type 1 diabetic patients. Int Immunol (2013) 25(10):563–74. doi: 10.1093/intimm/dxt020
105. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of cd4+Cd25+ Regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
106. Thirawatananond P, Brown ME, Sachs LK, Arnoletti JM, Yeh WI, Posgai AL, et al. Treg-specific cd226 deletion reduces diabetes incidence in nod mice by improving regulatory T cell stability. Diabetes (2023) 25:db230307. doi: 10.2337/db23-0307
107. Starosz A, Jamiołkowska-Sztabkowska M, Głowińska-Olszewska B, Moniuszko M, Bossowski A, Grubczak K. Immunological balance between treg and th17 lymphocytes as a key element of type 1 diabetes progression in children. Front Immunol (2022) 13:958430. doi: 10.3389/fimmu.2022.958430
108. Mhanna V, Fourcade G, Barennes P, Quiniou V, Pham HP, Ritvo PG, et al. Impaired activated/memory regulatory T cell clonal expansion instigates diabetes in nod mice. Diabetes (2021) 70(4):976–85. doi: 10.2337/db20-0896
109. Brusko T, Wasserfall C, McGrail K, Schatz R, Viener HL, Schatz D, et al. No alterations in the frequency of foxp3+ Regulatory T-cells in type 1 diabetes. Diabetes (2007) 56(3):604–12. doi: 10.2337/db06-1248
110. Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA, Atkinson MA. Functional defects and the influence of age on the frequency of cd4+ Cd25+ T-cells in type 1 diabetes. Diabetes (2005) 54(5):1407–14. doi: 10.2337/diabetes.54.5.1407
111. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in cd4(+)Cd25(+) T-cells from patients with type 1 diabetes. Diabetes (2005) 54(1):92–9. doi: 10.2337/diabetes.54.1.92
112. Haseda F, Imagawa A, Murase-Mishiba Y, Terasaki J, Hanafusa T. Cd4+ Cd45ra- Foxp3high activated regulatory T cells are functionally impaired and related to residual insulin-secreting capacity in patients with type 1 diabetes. Clin Exp Immunol (2013) 173(2):207–16. doi: 10.1111/cei.12116
113. Marcovecchio ML, Wicker LS, Dunger DB, Dutton SJ, Kopijasz S, Scudder C, et al. Interleukin-2 therapy of autoimmunity in diabetes (Itad): A phase 2, multicentre, double-blind, randomized, placebo-controlled trial. Wellcome Open Res (2020) 5:49. doi: 10.12688/wellcomeopenres.15697.1
114. Tyagi S, Gupta P, Saini AS, Kaushal C, Sharma S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J Advanced Pharm Technol Res (2011) 2(4):236–40. doi: 10.4103/2231-4040.90879
115. Wang YX. Ppars: diverse regulators in energy metabolism and metabolic diseases. Cell Res (2010) 20(2):124–37. doi: 10.1038/cr.2010.13
116. Stienstra R, Duval C, Müller M, Kersten S. Ppars, obesity, and inflammation. PPAR Res (2007) 2007:95974. doi: 10.1155/2007/95974
117. Kersten S, Desvergne B, Wahli W. Roles of ppars in health and disease. Nature (2000) 405(6785):421–4. doi: 10.1038/35013000
118. Bocher V, Pineda-Torra I, Fruchart JC, Staels B. Ppars: transcription factors controlling lipid and lipoprotein metabolism. Ann New York Acad Sci (2002) 967:7–18. doi: 10.1111/j.1749-6632.2002.tb04258.x
119. Smith SA. Peroxisomal proliferate-activated receptors and the regulation of lipid oxidation and adipogenesis. Biochem Soc Trans (1997) 25(4):1242–8. doi: 10.1042/bst0251242
120. Gross B, Staels B. Ppar agonists: multimodal drugs for the treatment of type-2 diabetes. Best Pract Res Clin Endocrinol Metab (2007) 21(4):687–710. doi: 10.1016/j.beem.2007.09.004
121. Botta M, Audano M, Sahebkar A, Sirtori CR, Mitro N, Ruscica M. Ppar agonists and metabolic syndrome: an established role? Int J Mol Sci (2018) 19(4):1197. doi: 10.3390/ijms19041197
122. Azhar S. Peroxisome proliferator-activated receptors, metabolic syndrome and cardiovascular disease. Future Cardiol (2010) 6(5):657–91. doi: 10.2217/fca.10.86
123. Mandard S, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci CMLS (2004) 61(4):393–416. doi: 10.1007/s00018-003-3216-3
124. Lin Y, Wang Y, Li P-f. Pparα: an emerging target of metabolic syndrome, neurodegenerative and cardiovascular diseases. Front Endocrinol (2022) 13:1074911. doi: 10.3389/fendo.2022.1074911
125. Zingarelli B, Piraino G, Hake PW, O'Connor M, Denenberg A, Fan H, et al. Peroxisome proliferator-activated receptor Δ Regulates inflammation via nf-κb signaling in polymicrobial sepsis. Am J Pathol (2010) 177(4):1834–47. doi: 10.2353/ajpath.2010.091010
126. Luquet S, Lopez-Soriano J, Holst D, Gaudel C, Jehl-Pietri C, Fredenrich A, et al. Roles of peroxisome proliferator-activated receptor delta (Ppardelta) in the control of fatty acid catabolism. A new target for the treatment of metabolic syndrome. Biochimie (2004) 86(11):833–7. doi: 10.1016/j.biochi.2004.09.024
127. Fajas L, Auboeuf D, Raspé E, Schoonjans K, Lefebvre AM, Saladin R, et al. The organization, promoter analysis, and expression of the human ppargamma gene. J Biol Chem (1997) 272(30):18779–89. doi: 10.1074/jbc.272.30.18779
128. Ricote M, Huang JT, Welch JS, Glass CK. The peroxisome proliferator-activated receptor (Pparγ) as a regulator of monocyte/macrophage function. J Leukocyte Biol (1999) 66(5):733–9. doi: 10.1002/jlb.66.5.733
129. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific ppargamma controls alternative activation and improves insulin resistance. Nature (2007) 447(7148):1116–20. doi: 10.1038/nature05894
130. Willson TM, Brown PJ, Sternbach DD, Henke BR. The ppars: from orphan receptors to drug discovery. J Medicinal Chem (2000) 43(4):527–50. doi: 10.1021/jm990554g
131. Gremlich S, Nolan C, Roduit R, Burcelin R, Peyot ML, Delghingaro-Augusto V, et al. Pancreatic islet adaptation to fasting is dependent on peroxisome proliferator-activated receptor alpha transcriptional up-regulation of fatty acid oxidation. Endocrinology (2005) 146(1):375–82. doi: 10.1210/en.2004-0667
132. Wu L, Parhofer KG. Diabetic dyslipidemia. Metabolism: Clin Exp (2014) 63(12):1469–79. doi: 10.1016/j.metabol.2014.08.010
133. Roduit R, Morin J, Massé F, Segall L, Roche E, Newgard CB, et al. Glucose down-regulates the expression of the peroxisome proliferator-activated receptor-alpha gene in the pancreatic beta -cell. J Biol Chem (2000) 275(46):35799–806. doi: 10.1074/jbc.M006001200
134. Ravnskjaer K, Boergesen M, Dalgaard LT, Mandrup S. Glucose-induced repression of pparalpha gene expression in pancreatic beta-cells involves pp2a activation and ampk inactivation. J Mol Endocrinol (2006) 36(2):289–99. doi: 10.1677/jme.1.01965
135. Bihan H, Rouault C, Reach G, Poitout V, Staels B, Guerre-Millo M. Pancreatic islet response to hyperglycemia is dependent on peroxisome proliferator-activated receptor alpha (Pparalpha). FEBS Lett (2005) 579(11):2284–8. doi: 10.1016/j.febslet.2005.03.020
136. Babu DA, Deering TG. Mirmira RG. A feat of metabolic proportions: pdx1 orchestrates islet development and function in the maintenance of glucose homeostasis. Mol Genet Metab (2007) 92(1-2):43–55. doi: 10.1016/j.ymgme.2007.06.008
137. Guo H, Sun S, Zhang X, Zhang XJ, Gao L, Zhao JJ. Ampk enhances the expression of pancreatic duodenal homeobox-1 via pparalpha, but not ppargamma, in rat insulinoma cell line ins-1. Acta Pharmacol Sin (2010) 31(8):963–9. doi: 10.1038/aps.2010.78
138. Sun Y, Zhang L, Gu HF, Han W, Ren M, Wang F, et al. Peroxisome proliferator-activated receptor-alpha regulates the expression of pancreatic/duodenal homeobox-1 in rat insulinoma (Ins-1) cells and ameliorates glucose-induced insulin secretion impaired by palmitate. Endocrinology (2008) 149(2):662–71. doi: 10.1210/en.2007-1275
139. Zhu Y, Liu Q, Zhou Z, Ikeda Y. Pdx1, neurogenin-3, and mafa: critical transcription regulators for beta cell development and regeneration. Stem Cell Res Ther (2017) 8(1):240. doi: 10.1186/s13287-017-0694-z
140. Taylor BL, Liu FF, Sander M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep (2013) 4(6):1262–75. doi: 10.1016/j.celrep.2013.08.010
141. Yessoufou A, Atègbo JM, Attakpa E, Hichami A, Moutairou K, Dramane KL, et al. Peroxisome proliferator-activated receptor-alpha modulates insulin gene transcription factors and inflammation in adipose tissues in mice. Mol Cell Biochem (2009) 323(1-2):101–11. doi: 10.1007/s11010-008-9968-1
142. Ravnskjaer K, Boergesen M, Rubi B, Larsen JK, Nielsen T, Fridriksson J, et al. Peroxisome proliferator-activated receptor alpha (Pparalpha) potentiates, whereas ppargamma attenuates, glucose-stimulated insulin secretion in pancreatic beta-cells. Endocrinology (2005) 146(8):3266–76. doi: 10.1210/en.2004-1430
143. Wang X, Zhou L, Shao L, Qian L, Fu X, Li G, et al. Troglitazone acutely activates amp-activated protein kinase and inhibits insulin secretion from beta cells. Life Sci (2007) 81(2):160–5. doi: 10.1016/j.lfs.2007.04.034
144. Ito E, Ozawa S, Takahashi K, Tanaka T, Katsuta H, Yamaguchi S, et al. Ppar-gamma overexpression selectively suppresses insulin secretory capacity in isolated pancreatic islets through induction of ucp-2 protein. Biochem Biophys Res Commun (2004) 324(2):810–4. doi: 10.1016/j.bbrc.2004.08.238
145. Bollheimer LC, Troll S, Landauer H, Wrede CE, Schölmerich J, Buettner R. Insulin-sparing effects of troglitazone in rat pancreatic islets. J Mol Endocrinol (2003) 31(1):61–9. doi: 10.1677/jme.0.0310061
146. Nakamichi Y, Kikuta T, Ito E, Ohara-Imaizumi M, Nishiwaki C, Ishida H, et al. Ppar-gamma overexpression suppresses glucose-induced proinsulin biosynthesis and insulin release synergistically with pioglitazone in min6 cells. Biochem Biophys Res Commun (2003) 306(4):832–6. doi: 10.1016/s0006-291x(03)01045-3
147. Yang C, Chang TJ, Chang JC, Liu MW, Tai TY, Hsu WH, et al. Rosiglitazone (Brl 49653) enhances insulin secretory response via phosphatidylinositol 3-kinase pathway. Diabetes (2001) 50(11):2598–602. doi: 10.2337/diabetes.50.11.2598
148. Kim HS, Noh JH, Hong SH, Hwang YC, Yang TY, Lee MS, et al. Rosiglitazone stimulates the release and synthesis of insulin by enhancing glut-2, glucokinase and beta2/neurod expression. Biochem Biophys Res Commun (2008) 367(3):623–9. doi: 10.1016/j.bbrc.2007.12.192
149. Santini E, Fallahi P, Ferrari SM, Masoni A, Antonelli A, Ferrannini E. Effect of ppar-gamma activation and inhibition on glucose-stimulated insulin release in ins-1e cells. Diabetes (2004) 53 Suppl 3:S79–83. doi: 10.2337/diabetes.53.suppl_3.s79
150. Zusi C, Rioda M, Maguolo A, Emiliani F, Unali I, Costantini S, et al. Igf1 and pparg polymorphisms are associated with reduced estimated glomerular filtration rate in a cohort of children and adolescents with type 1 diabetes. Acta Diabetol (2023) 60(10):1351–8. doi: 10.1007/s00592-023-02128-6
151. Holm LJ, Krogvold L, Hasselby JP, Kaur S, Claessens LA, Russell MA, et al. Abnormal islet sphingolipid metabolism in type 1 diabetes. Diabetologia (2018) 61(7):1650–61. doi: 10.1007/s00125-018-4614-2
152. García-Ricobaraza M, García-Bermúdez M, Torres-Espinola FJ, Segura Moreno MT, Bleyere MN, Díaz-Prieto LE, et al. Association study of rs1801282 pparg gene polymorphism and immune cells and cytokine levels in a spanish pregnant women cohort and their offspring. J Biomed Sci (2020) 27(1):101. doi: 10.1186/s12929-020-00694-3
153. Moibi JA, Gupta D, Jetton TL, Peshavaria M, Desai R, Leahy JL. Peroxisome proliferator-activated receptor-gamma regulates expression of pdx-1 and nkx6.1 in ins-1 cells. Diabetes (2007) 56(1):88–95. doi: 10.2337/db06-0948
154. Evans-Molina C, Robbins RD, Kono T, Tersey SA, Vestermark GL, Nunemaker CS, et al. Peroxisome proliferator-activated receptor gamma activation restores islet function in diabetic mice through reduction of endoplasmic reticulum stress and maintenance of euchromatin structure. Mol Cell Biol (2009) 29(8):2053–67. doi: 10.1128/mcb.01179-08
155. Gupta D, Jetton TL, Mortensen RM, Duan SZ, Peshavaria M, Leahy JL. In vivo and in vitro studies of a functional peroxisome proliferator-activated receptor gamma response element in the mouse pdx-1 promoter. J Biol Chem (2008) 283(47):32462–70. doi: 10.1074/jbc.M801813200
156. Kim HI, Kim JW, Kim SH, Cha JY, Kim KS, Ahn YH. Identification and functional characterization of the peroxisomal proliferator response element in rat glut2 promoter. Diabetes (2000) 49(9):1517–24. doi: 10.2337/diabetes.49.9.1517
157. Kim HI, Cha JY, Kim SY, Kim JW, Roh KJ, Seong JK, et al. Peroxisomal proliferator-activated receptor-gamma upregulates glucokinase gene expression in beta-cells. Diabetes (2002) 51(3):676–85. doi: 10.2337/diabetes.51.3.676
158. Ježek P, Jabůrek M, Holendová B, Plecitá-Hlavatá L. Fatty acid-stimulated insulin secretion vs. Lipotoxicity. Molecules (Basel Switzerland) (2018) 23(6):1483. doi: 10.3390/molecules23061483
159. Iglesias J, Barg S, Vallois D, Lahiri S, Roger C, Yessoufou A, et al. Pparβ/Δ Affects pancreatic β Cell mass and insulin secretion in mice. J Clin Invest (2012) 122(11):4105–17. doi: 10.1172/jci42127
160. Li L, Li T, Zhang Y, Pan Z, Wu B, Huang X, et al. Peroxisome proliferator-activated receptorβ/Δ Activation is essential for modulating P-foxo1/foxo1 status in functional insulin-positive cell differentiation. Cell Death Dis (2015) 6(4):e1715. doi: 10.1038/cddis.2015.88
161. Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PloS Genet (2012) 8(3):e1002552. doi: 10.1371/journal.pgen.1002552
162. Ravnskjaer K, Frigerio F, Boergesen M, Nielsen T, Maechler P, Mandrup S. Ppardelta is a fatty acid sensor that enhances mitochondrial oxidation in insulin-secreting cells and protects against fatty acid-induced dysfunction. J Lipid Res (2010) 51(6):1370–9. doi: 10.1194/jlr.M001123
163. Dillon JS, Yaney GC, Zhou Y, Voilley N, Bowen S, Chipkin S, et al. Dehydroepiandrosterone sulfate and beta-cell function: enhanced glucose-induced insulin secretion and altered gene expression in rodent pancreatic beta-cells. Diabetes (2000) 49(12):2012–20. doi: 10.2337/diabetes.49.12.2012
164. Agarwal G, Patel M. Review on monoclonal antibodies (Mabs) as a therapeutic approach for type 1 diabetes. Curr Diabetes Rev (2023) 20:e310823220578. doi: 10.2174/1573399820666230831153249
165. Krishnamurthy B, Lacorcia M, Kay TWH, Thomas HE, Mannering SI. Monitoring immunomodulation strategies in type 1 diabetes. Front Immunol (2023) 14:1206874. doi: 10.3389/fimmu.2023.1206874
166. Hichami A, Yessoufou A, Ghiringhelli F, Salvadori F, Moutairou K, Zwetyenga N, et al. Peroxisome proliferator-activated receptor alpha deficiency impairs regulatory T cell functions: possible application in the inhibition of melanoma tumor growth in mice. Biochimie (2016) 131:1–10. doi: 10.1016/j.biochi.2016.09.001
167. Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The pparalpha-leukotriene B4 pathway to inflammation control. Nature (1996) 384(6604):39–43. doi: 10.1038/384039a0
168. Zhou Z, Liang Y, Gao Y, Kong W, Feng J, Wang X. Fenofibrate enhances the in vitro differentiation of foxp3(+) regulatory T cells in mice. PPAR Res (2012) 2012:529035. doi: 10.1155/2012/529035
169. Cheng H, Xi Y, Chi X, Wu Y, Liu G. Fenofibrate treatment of rats with experimental autoimmune myocarditis by alleviating treg/th17 disorder. Central-European J Immunol (2016) 41(1):64–70. doi: 10.5114/ceji.2016.58817
170. Wei P, Kou W, Fu J, Chen Z, Pan F. Pparα Knockout in mice increases the th17 development by facilitating the ikkα/rorγt and ikkα/foxp3 complexes. Commun Biol (2023) 6(1):721. doi: 10.1038/s42003-023-05104-6
171. Dunn SE, Ousman SS, Sobel RA, Zuniga L, Baranzini SE, Youssef S, et al. Peroxisome proliferator-activated receptor (Ppar)Alpha expression in T cells mediates gender differences in development of T cell-mediated autoimmunity. J Exp Med (2007) 204(2):321–30. doi: 10.1084/jem.20061839
172. Morse E, Selim E, Cunard R. Pparalpha ligands cause lymphocyte depletion and cell cycle block and this is associated with augmented trb3 and reduced cyclin B1 expression. Mol Immunol (2009) 46(16):3454–61. doi: 10.1016/j.molimm.2009.08.008
173. Madej A, Okopien B, Kowalski J, Zielinski M, Wysocki J, Szygula B, et al. Effects of fenofibrate on plasma cytokine concentrations in patients with atherosclerosis and hyperlipoproteinemia iib. Int J Clin Pharmacol Ther (1998) 36(6):345–9.
174. Hontecillas R, Bassaganya-Riera J. Peroxisome proliferator-activated receptor gamma is required for regulatory cd4+ T cell-mediated protection against colitis. J Immunol (Baltimore Md 1950) (2007) 178(5):2940–9. doi: 10.4049/jimmunol.178.5.2940
175. Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S, et al. The nuclear receptor ppar gamma selectively inhibits th17 differentiation in a T cell-intrinsic fashion and suppresses cns autoimmunity. J Exp Med (2009) 206(10):2079–89. doi: 10.1084/jem.20082771
176. Saubermann LJ, Nakajima A, Wada K, Zhao S, Terauchi Y, Kadowaki T, et al. Peroxisome proliferator-activated receptor gamma agonist ligands stimulate a th2 cytokine response and prevent acute colitis. Inflamm Bowel Dis (2002) 8(5):330–9. doi: 10.1097/00054725-200209000-00004
177. Celinski K, Dworzanski T, Fornal R, Korolczuk A, Madro A, Brzozowski T, et al. Comparison of anti-inflammatory properties of peroxisome proliferator-activated receptor gamma agonists rosiglitazone and troglitazone in prophylactic treatment of experimental colitis. J Physiol Pharmacol (2013) 64(5):587–95.
178. Guri AJ, Mohapatra SK, Horne WT, Hontecillas R, Bassaganya-Riera J. The role of T cell ppar gamma in mice with experimental inflammatory bowel disease. BMC Gastroenterol (2010) 10:60. doi: 10.1186/1471-230x-10-60
179. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. Ppar-Γ Is a major driver of the accumulation and phenotype of adipose tissue treg cells. Nature (2012) 486(7404):549–53. doi: 10.1038/nature11132
180. Wohlfert EA, Nichols FC, Nevius E, Clark RB. Peroxisome proliferator-activated receptor gamma (Ppargamma) and immunoregulation: enhancement of regulatory T cells through ppargamma-dependent and -independent mechanisms. J Immunol (Baltimore Md 1950) (2007) 178(7):4129–35. doi: 10.4049/jimmunol.178.7.4129
181. Meng F, Hao P, Du H. Regulatory T cells differentiation in visceral adipose tissues contributes to insulin resistance by regulating jazf-1/ppar-Γ Pathway. J Cell Mol Med (2023) 27(4):553–62. doi: 10.1111/jcmm.17680
182. Park HJ, Kim DH, Choi JY, Kim WJ, Kim JY, Senejani AG, et al. Pparγ Negatively regulates T cell activation to prevent follicular helper T cells and germinal center formation. PloS One (2014) 9(6):e99127. doi: 10.1371/journal.pone.0099127
183. Dunn SE, Bhat R, Straus DS, Sobel RA, Axtell R, Johnson A, et al. Peroxisome proliferator-activated receptor delta limits the expansion of pathogenic th cells during central nervous system autoimmunity. J Exp Med (2010) 207(8):1599–608. doi: 10.1084/jem.20091663
184. Kanakasabai S, Chearwae W, Walline CC, Iams W, Adams SM, Bright JJ. Peroxisome proliferator-activated receptor delta agonists inhibit T helper type 1 (Th1) and th17 responses in experimental allergic encephalomyelitis. Immunology (2010) 130(4):572–88. doi: 10.1111/j.1365-2567.2010.03261.x
185. Kanakasabai S, Walline CC, Chakraborty S, Bright JJ. Pparδ deficient mice develop elevated th1/th17 responses and prolonged experimental autoimmune encephalomyelitis. Brain Res (2011) 1376:101–12. doi: 10.1016/j.brainres.2010.12.059
186. Mothe-Satney I, Murdaca J, Sibille B, Rousseau AS, Squillace R, Le Menn G, et al. A role for peroxisome proliferator-activated receptor beta in T cell development. Sci Rep (2016) 6:34317. doi: 10.1038/srep34317
187. Le Menn G, Neels JG. Regulation of immune cell function by ppars and the connection with metabolic and neurodegenerative diseases. Int J Mol Sci (2018) 19(6):1575. doi: 10.3390/ijms19061575
188. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The cd28 signaling pathway regulates glucose metabolism. Immunity (2002) 16(6):769–77. doi: 10.1016/s1074-7613(02)00323-0
189. Sujatha V. Globalisation of South Asian medicines: knowledge, power, structure and sustainability. SAGE Publications SAGE India: New Delhi India (2020) p:7–30. doi: 10.1177/2393861719883063
190. Astutik S, Pretzsch J, Ndzifon Kimengsi J. Asian medicinal plants’ Production and utilization potentials: A review. Sustainability (2019) 11(19):5483. doi: 10.3390/su11195483
191. Barzkar F, Baradaran HR, Khamseh ME, Vesal Azad R, Koohpayehzadeh J, Moradi Y. Medicinal plants in the adjunctive treatment of patients with type-1 diabetes: A systematic review of randomized clinical trials. J Diabetes Metab Disord (2020) 19(2):1917–29. doi: 10.1007/s40200-020-00633-x
192. Chang CL, Chen YC, Chen HM, Yang NS, Yang WC. Natural cures for type 1 diabetes: A review of phytochemicals, biological actions, and clinical potential. Curr Med Chem (2013) 20(7):899–907. doi: 10.2174/0929867311320070006
193. Yedjou CG, Grigsby J, Mbemi A, Nelson D, Mildort B, Latinwo L, et al. The management of diabetes mellitus using medicinal plants and vitamins. Int J Mol Sci (2023) 24(10):9085. doi: 10.3390/ijms24109085
194. Wang J, Zhao Y, Xu Q. Astilbin prevents concanavalin a-induced liver injury by reducing tnf-alpha production and T lymphocytes adhesion. J Pharm Pharmacol (2004) 56(4):495–502. doi: 10.1211/0022357023033
195. Ding S, Lu G, Wang B, Xiang J, Hu C, Lin Z, et al. Astilbin activates the reactive oxidative species/pparγ Pathway to suppress effector cd4(+) T cell activities via direct binding with cytochrome P450 1b1. Front Pharmacol (2022) 13:848957. doi: 10.3389/fphar.2022.848957
196. Fu Z, Zhen W, Yuskavage J, Liu D. Epigallocatechin gallate delays the onset of type 1 diabetes in spontaneous non-obese diabetic mice. Br J Nutr (2011) 105(8):1218–25. doi: 10.1017/s0007114510004824
197. Wan C, Ouyang J, Li M, Rengasamy KRR, Liu Z. Effects of green tea polyphenol extract and epigallocatechin-3-O-gallate on diabetes mellitus and diabetic complications: recent advances. Crit Rev Food Sci Nutr (2022) 19:1–29. doi: 10.1080/10408398.2022.2157372
198. Wan C, Hu X, Li M, Rengasamy KRR, Cai Y, Liu Z. Potential protective function of green tea polyphenol egcg against high glucose-induced cardiac injury and aging. J Funct Foods (2023) 104:105506. doi: 10.1016/j.jff.2023.105506
199. Kanitkar M, Gokhale K, Galande S, Bhonde RR. Novel role of curcumin in the prevention of cytokine-induced islet death in vitro and diabetogenesis in vivo. Br J Pharmacol (2008) 155(5):702–13. doi: 10.1038/bjp.2008.311
200. Chanpoo M, Petchpiboonthai H, Panyarachun B, Anupunpisit V. Effect of curcumin in the amelioration of pancreatic islets in streptozotocin-induced diabetic mice. J Med Assoc Thailand = Chotmaihet thangphaet (2010) 93 Suppl 6:S152–9.
201. Quispe C, Herrera-Bravo J, Javed Z, Khan K, Raza S, Gulsunoglu-Konuskan Z, et al. Therapeutic applications of curcumin in diabetes: A review and perspective. BioMed Res Int (2022) 2022:1375892. doi: 10.1155/2022/1375892
202. Castro CN, Barcala Tabarrozzi AE, Winnewisser J, Gimeno ML, Antunica Noguerol M, Liberman AC, et al. Curcumin ameliorates autoimmune diabetes. Evidence in accelerated murine models of type 1 diabetes. Clin Exp Immunol (2014) 177(1):149–60. doi: 10.1111/cei.12322
203. Liu SN, Liu Q, Li LY, Huan Y, Sun SJ, Shen ZF. Long-term fenofibrate treatment impaired glucose-stimulated insulin secretion and up-regulated pancreatic nf-kappa B and inos expression in monosodium glutamate-induced obese rats: is that a latent disadvantage? J Trans Med (2011) 9:176. doi: 10.1186/1479-5876-9-176
204. Yajima K, Hirose H, Fujita H, Seto Y, Fujita H, Ukeda K, et al. Combination therapy with ppargamma and pparalpha agonists increases glucose-stimulated insulin secretion in db/db mice. Am J Physiol Endocrinol Metab (2003) 284(5):E966–71. doi: 10.1152/ajpendo.00149.2002
205. Jin L, Hua H, Ji Y, Jia Z, Peng M, Huang S. Anti-inflammatory role of fenofibrate in treating diseases. Biomolecules Biomedicine (2023) 23(3):376–91. doi: 10.17305/bb.2022.8534
206. Buschard K, Holm LJ, Feldt-Rasmussen U. Insulin independence in newly diagnosed type 1 diabetes patient following fenofibrate treatment. Case Rep Med (2020) 2020:6865190. doi: 10.1155/2020/6865190
207. Pirat C, Farce A, Lebègue N, Renault N, Furman C, Millet R, et al. Targeting peroxisome proliferator-activated receptors (Ppars): development of modulators. J Medicinal Chem (2012) 55(9):4027–61. doi: 10.1021/jm101360s
208. Savage SR, McCollum GW, Yang R, Penn JS. Rna-seq identifies a role for the pparβ/Δ Inverse agonist gsk0660 in the regulation of tnfα-induced cytokine signaling in retinal endothelial cells. Mol Vision (2015) 21:568–76.
209. Capozzi ME, Savage SR, McCollum GW, Hammer SS, Ramos CJ, Yang R, et al. The peroxisome proliferator-activated receptor-β/Δ Antagonist gsk0660 mitigates retinal cell inflammation and leukostasis. Exp eye Res (2020) 190:107885. doi: 10.1016/j.exer.2019.107885
Keywords: type-1 diabetes, peroxisome proliferator-activated receptors, autoimmunity, CD4+ T cells, Th17, regulatory T cells
Citation: Riaz F, Wei P and Pan F (2023) PPARs at the crossroads of T cell differentiation and type 1 diabetes. Front. Immunol. 14:1292238. doi: 10.3389/fimmu.2023.1292238
Received: 11 September 2023; Accepted: 11 October 2023;
Published: 20 October 2023.
Edited by:
Reiko Shinkura, The University of Tokyo, JapanReviewed by:
Bernhard Fidelis Maier, Purdue University Indianapolis, United StatesCopyright © 2023 Riaz, Wei and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fan Pan, ZmFuLnBhbkBzaWF0LmFjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.