
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 December 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1291990
This article is part of the Research Topic Challenges Associated with Identifying Preclinical Animal Models for the Development of Immune-based Therapies View all 17 articles
 Erik P. Karmele1
Erik P. Karmele1 Ana Laura Moldoveanu2Irem Kaymak2
Ana Laura Moldoveanu2Irem Kaymak2 Bat-Erdene Jugder3Rebecca L. Ursin3
Bat-Erdene Jugder3Rebecca L. Ursin3 Kyle J. Bednar1
Kyle J. Bednar1 Daniele Corridoni2*
Daniele Corridoni2* Tatiana Ort1*
Tatiana Ort1*Inflammatory bowel disease (IBD) is an umbrella term for two conditions (Crohn’s Disease and Ulcerative Colitis) that is characterized by chronic inflammation of the gastrointestinal tract. The use of pre-clinical animal models has been invaluable for the understanding of potential disease mechanisms. However, despite promising results of numerous therapeutics in mouse colitis models, many of these therapies did not show clinical benefits in patients with IBD. Single cell RNA-sequencing (scRNA-seq) has recently revolutionized our understanding of complex interactions between the immune system, stromal cells, and epithelial cells by mapping novel cell subpopulations and their remodeling during disease. This technology has not been widely applied to pre-clinical models of IBD. ScRNA-seq profiling of murine models may provide an opportunity to increase the translatability into the clinic, and to choose the most appropriate model to test hypotheses and novel therapeutics. In this review, we have summarized some of the key findings at the single cell transcriptomic level in IBD, how specific signatures have been functionally validated in vivo, and highlighted the similarities and differences between scRNA-seq findings in human IBD and experimental mouse models. In each section of this review, we highlight the importance of utilizing this technology to find the most suitable or translational models of IBD based on the cellular therapeutic target.
Inflammatory bowel disease (IBD), consisting of ulcerative colitis (UC) and Crohn’s disease (CD), is a chronic relapsing and remitting inflammatory mediated disorder of the lining of the gastrointestinal tract, resulting in clinical symptoms (1). Changes and functional alterations of immune cells and non-hematopoietic cells (including epithelial and stromal cells) are hallmarks of the onset and sustained inflammation associated with IBD (2, 3). Much of what we know about human IBD pathogenesis is attributed to the use of numerous pre-clinical models of IBD. These models include chemically-induced, spontaneous, immune cell-induced, and microbiota-dependent colitis models. These models have been extensively reviewed (4, 5) and the most common pre-clinical IBD models are summarized in Table 1. While numerous pre-clinical models of IBD exist, no single model fully recapitulates the complexity observed in human IBD. Thus, there remains a need to further validate these pre-clinical models of IBD and identify mucosal cell subpopulations with their associated disease-relevant phenotype which may overlay with human IBD. This understanding will potentially increase their translatability into the clinic and help define the best model to test novel therapeutic targets and modalities.
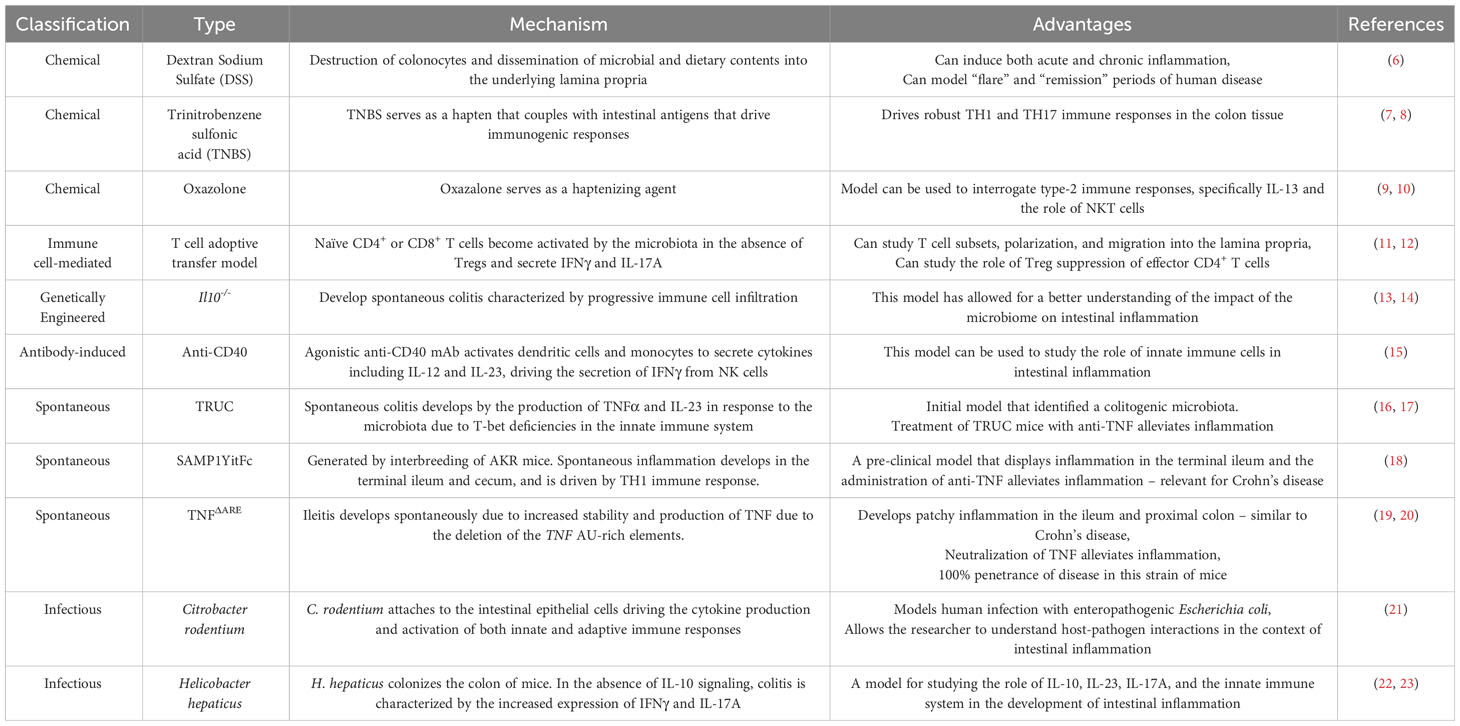
Table 1 Summary of the most commonly used pre-clinical models of IBD.
Multimodal single-cell technologies, such as single cell RNA-sequencing (scRNA-seq), have allowed for in-depth analysis and a deeper understanding of tissue architecture and function at individual-cell high-resolution (24). ScRNA-seq has been instrumental to our understanding of immunology and autoimmune diseases, where complex cellular heterogeneity and interactions between immune cells, stromal cells, and epithelial cells are paramount for disease progression. These technologies have enabled an improved understanding of human IBD by identifying novel or rare cell types that were previously undiscovered and how their abundance or function changes during intestinal inflammation may be a driver of disease (25). While scRNA-seq technology has been applied to numerous UC and CD cohorts to better understand the complex biology between immune cells and non-hematopoietic cells, this technology has only begun to be applied to pre-clinical models of IBD for comparative studies with the human condition.
While studies have enriched specific cell populations from pre-clinical models of IBD and characterized these cells by scRNA-seq (26–28), there are gaps in the knowledge of the entire makeup of the inflamed tissue. In this review, we have summarized the key findings from a longitudinal scRNA-seq study that analyzed immune cells, stromal cells, and epithelial cells. Furthermore, we highlight the similarities and differences between scRNA-seq in murine models compared to human IBD. Finally, we emphasize the need for utilizing scRNA-seq technology to find suitable translational models based on the therapeutic target.
While there are other studies utilizing scRNA-seq in enriched cell populations from pre-clinical models, highlighted below is one study that has applied scRNA-seq technology to comprehensively describe the landscape of the epithelial cell, stromal cell, and immune cell populations during the onset and resolution of intestinal inflammation (29). Ho et al. subjected wild-type mice to 1.5% dextran sodium sulfate (DSS) drinking water for 6 days, then placed mice on regular drinking water for the remainder of the study. Colon tissue was collected from mice on days 0, 3, 6, 9, 12, and 15 in biological triplicates. ScRNA-seq was performed on viable cells and 14,624 cells were profiled. ScRNA-seq analysis identified 15 clusters including: mononuclear phagocytes (Il1b, C1qa, and C1qb), epithelial: enteroendocrine cells (Scgn and Pcsk1n), epithelial: absorptive and secretory cells (Muc2, Spink4, Lypd8, and Elf3), endothelial cells (Pecam1 and Flt1), lymphatic cells (Lyve1), stromal cells (Col1a1, Pdgfra, and Spon2), two myofibroblast clusters (Acta2 and Myh11), interstitial cells of Cajal (Ano1 and Kit), enteric glial cells (S100b), T cells (Cd3d and Cd3g), plasma cells (Igha and Mzb1), plasmacytoid dendritic cells (Siglech and Ccr9), B cells (Cd19 and Cd22), and granulocytes (Cd14, S100a8, and S100a9) (29).
To assess changes in the cellular composition, the numbers of cells in every cluster were counted at each sampling time point. Epithelial cells, stromal cells, myofibroblasts, interstitial cells of Cajal, and enteric glial cells were reduced in number with inflammation and did not repopulate during the resolution phase. In contrast, the numbers of granulocytes and B cells increased with inflammation and did not return to homeostatic numbers measured at steady state. Mononuclear phagocytes peaked in number 6 days after DSS administration and returned to baseline at the end of the study after the removal of DSS drinking water. Lastly, plasma cells decreased at days 6 and 9, but recovered in total numbers by the end of the resolution phase on drinking water at day 12. Furthermore, differentially expressed gene (DEG) analysis demonstrated phenotypic changes in mononuclear phagocytes and stromal cell clusters across all time points. The authors selected 96 “IBD risk genes” to assess how the DEGs identified in their longitudinal scRNA-seq study correlated with human IBD, 79 of the selected 96 human IBD risk genes were expressed at one or more time points and in one or more of the dysregulated cell clusters, suggesting that a dynamic expression pattern is under regulation during intestinal inflammation (29). One example is Ifng, which was found to be expressed during the time points associated with inflammation within the T cell cluster (29).
While scRNA-seq has been seldom applied to pre-clinical models of IBD, scRNA-seq has been more readily applied to multiple CD and UC patient cohorts to identify novel cell types, cell-cell interactions, and to generate new hypotheses of IBD pathogenesis. We will discuss in the next section the differences that have been found in human IBD using scRNA-seq in the epithelial cell, stromal cell and immune cell compartments compared to the limited murine studies including but not limited to the study described above.
The lining of the gastrointestinal tract (GI) is composed of diverse populations of intestinal epithelial cell lineages with specialized functions. These epithelial cells are derived from a common stem-cell precursor, which expresses LGR5 (30). Intestinal epithelial cells contribute to intestinal homeostasis by maintaining barrier function and secreting mediators in response to the microbiota and underlying immune cells in the lamina propria.
The first scRNA-seq study analyzing human colonic epithelial cells from healthy volunteers or patients with UC, sampled from clinically inflamed and noninflamed mucosa, was published by Parikh et al. in 2019 (31). Visualization of scRNA-seq data identified 10 clusters of intestinal epithelial cells including undifferentiated cells, absorptive colonocytes, goblet cells, and unique enteroendocrine cell populations including L-cells, enterochromaffin cells, and precursor-like cells, and 5 clusters of stem cells. In UC, two additional clusters were identified representing inflammation-associated goblet cells and intraepithelial immune cells. Not only did this analysis provide a comprehensive landscape of the epithelial cells that are present in the healthy colon, but also identified their dysregulation in inflamed UC. One of these subsets were “BEST4/OTOP2” colonocytes. BEST4 encodes a calcium-sensitive chloride channel, while OTOP2 encodes a proton-conducting ion channel. These findings highlighted the power of scRNA-seq technology in identifying novel cell populations which could contribute to intestinal homeostasis and/or inflammation. Another prominent cell type linked to disease pathogenesis and barrier function are goblet cells. Goblet cells are mucus-secreting epithelial cells that are critical for the maintenance of barrier function and prevention of bacterial translocation. In IBD both their function and numbers become dysregulated (32). ScRNA-seq analysis of healthy human and UC colonic tissue revealed a distinct cluster of inflammation-associated goblet cells that had decreased expression of WFDC2 (31), a gene encoding an anti-protease enzyme (33). To explore the significance of the loss of WFDC2 expression in goblet cells in intestinal homeostasis, Wfdc2+/- mice were generated. These mice had abnormalities in colonic epithelial intercellular tight junction proteins, mild-to-modest epithelial cell hyperplasia, immune cell infiltration into the tissue, and proximity of gram-negative and gram-positive bacteria to the colonic epithelium compared to littermate controls (31). Although these mice were not subjected to an induced model of colitis, this study demonstrates how findings from human scRNA-seq samples can be translated in an in vivo setting of gastrointestinal tract dysregulation.
These novel populations become critically important in IBD as breakdown in intestinal barrier function is a hallmark phenotype. There has been great interest in underpinning the mechanisms and contributions of epithelial subsets in this loss of barrier function and to target barrier regeneration with novel therapeutics. In other studies, the authors have performed scRNA-seq analysis on healthy and CD or UC cohorts to assess the transcriptomic changes in epithelial cell subsets. One common finding amongst these scRNA-seq analyses is a depletion of BEST4+/OTOP2+ colonocytes in UC and CD compared to healthy controls (31, 34, 35). While BEST4-expressing colonocytes are present in the human GI tract, this cell population is absent in the murine gut (36).
The primary function of microfold (M) cells is the transport of antigens from the lumen of the intestine to gut-associated lymphoid tissues such as Peyer’s patches (37). ScRNA-seq uncovered an expansion of M cells in UC patients compared to healthy controls (34). Furthermore, these cells expressed elevated levels of CCL20 and CCL23, important chemokines driving immune cell recruitment into the inflamed tissue. The expansion of M cells, detected by immunohistochemistry, held true in a DSS colitis model and treatment with an anti-TNF antibody abrogated the expansion of M cells in the inflamed tissue in mice, suggesting the pro-inflammatory milieu in IBD can impact the differentiation of intestinal epithelial cells during disease (38).
In addition to aiding in our understanding of cell populations in barrier breakdown at the single cell level, scRNA-seq has contributed to identifying novel biological pathways in epithelial cells. Gasdermin-D (GSDM) plays a pivotal role in driving cell death through the pore-forming activity of the protein’s N-terminus and has recently been discovered to be required for IL-1β release (39, 40). ScRNA-seq of colonic tissue between healthy and UC patients revealed that secretory and absorptive progenitors, as well as colonocytes, from UC patients express elevated levels of GSDM (31, 41). This finding highlights a new mechanism by which GSDM-mediated IL-1β release by intestinal epithelial cells could sustain intestinal inflammation. In yet another example of how scRNA-seq translated into an in vivo model, Gsdmd+/- mice are protected from DSS-induced colitis as indicated by a decreased infiltration of neutrophils, macrophages, and T cells, and decreased expression of inflammatory cytokines and chemokines (41). This study reiterates the power of scRNA-seq in identifying pathways in previously underappreciated cell populations that are critical for inflammation.
In the longitudinal DSS study described above by Ho et al. (29), upon administration of DSS, enteroendocrine and both absorptive and secretory epithelia cell clusters were reduced. Thus, applying scRNA-seq to additional pre-clinical IBD models would be advantageous to find a suitable model to translate scRNA-seq findings in epithelial cells in human IBD.
Stromal cells are part of the mesenchymal compartment of the intestine, and represent a distinctive heterogeneous population comprised of non-hematopoietic, non-epithelial cell types (42). Mesenchymal cells regulate homeostasis by promoting extracellular matrix (ECM) turnover through the secretion of ECM factors, such as collagen and glycoproteins, self-renewal of the intestinal epithelial barrier through WNT/β-catenin signaling and colon regeneration through production of prostaglandins (43). However, in the context of inflammation these pathways are subverted into inflammatory processes. During intestinal inflammation, dysregulation of Wnt signaling leads to impaired wound healing characterized by the activation of fibroblasts and myofibroblasts, the accumulation of collagen-rich ECM, culminating in fibrosis. Additionally, TGF-β1 signaling in fibroblasts leads to their transition to an activated phenotype, marked by an increase in ECM production, leading to fibrosis. ScRNA-seq has played a significant role in elucidating the heterogeneous function of stromal cells and highlighted novel ‘cross-talks’ with other cells in the intestinal microenvironment. In the first report using unbiased single-cell profiling of over 16,500 human colonic mesenchymal cells from healthy and treatment-naïve UC patients, Kinchen and colleagues have shown how the colonic mesenchyme compartment remodels in the context of IBD (44). Specifically, the authors identified key stromal cell populations involved in immune homeostasis in intestinal inflammation. They identified a distinct cluster found near the epithelial crypts marked by the expression of SOX6, F3 (CD142) and WTN genes, which was important in mediating epithelial cell self-renewal in physiological conditions. Interestingly, scRNA-seq of stromal cells from the DSS colitis model revealed similarities between human and mouse stromal cell clusters, with SOX6 being highly conserved between the two (44).
ScRNA-seq of UC colonic mesenchyme revealed changes in gene expression and cell composition within the stromal compartment (44). Single-cell analysis identified 12 distinct clusters, with a significant expansion of a cluster marked by genes involved in response to TNF and leukocyte migration. Highly ranked markers included lymphocyte trafficking chemokines, such as CCL19 and CCL21; T cell co-stimulatory TNF-superfamily ligand (TNFSF14/LIGHT) and MHC II invariant chain (CD74); CD24; IL-33 and Lysyl oxidases. In UC, the enrichment of this cluster was associated with a decrease in the SOX6 expressing cluster in inflamed tissues, further highlighting the heterogeneous nature of the stromal-driven responses. Thus, this study revealed a divergent response in the context of disease, marked by an increase in pro-inflammatory fibroblasts and a reduction in the stromal subsets associated with epithelial self-renewal and resolution of disease. The role of stromal cells in IBD was further validated in the DSS model of colitis (44). ScRNA-seq analysis revealed high expression of Lox and LoxI1 in the mesenchymal populations. Lastly, the authors investigated whether blockade of this pathway could ameliorate DSS. The Lox family of lysyl oxidases generate oxidative stress responses, disturbing the redox balance within the tissues, which in turn mediate recruitment of inflammatory mediators, thus sustaining IBD pathogenicity. Indeed, administration of Lox/LoxI1 inhibitor, β-aminopropionitrile, improved the clinical score of mice receiving DSS drinking water (44).
Kong et al. profiled 720,633 cells from the terminal ileum and colon of CD and non-IBD donors by scRNA-seq (45). The authors identified CHMP1A, TBX3, and RNF168 as disease-associated genes which are affiliated with myofibroblast activation (45). To validate the roles of these three genes in fibrosis development, the authors generated and pooled siRNA oligos to knockdown CHMP1A, TBX3, and RNF168 in normal human intestinal fibroblasts. Knockdown of CHMP1A, TBX3, and RNF168 significantly impaired TGF-β-driven collagen gene expression (45).
To gain a better understanding of the complexity of the fibroblast cellular networks and how they underly stricture formation in CD, Mukherjee and colleagues have generated the first scRNA-seq atlas of strictured bowel from CD patients (46). Specifically, scRNA-seq was performed on CD resections containing non-involved, inflamed non-strictured and strictured segments, as well as non-IBD bowel segments. ScRNA-seq analysis revealed significant differences in the cellular composition between non-inflamed, inflamed and strictured segments. Comparison between non-strictured and strictured segments revealed significant changes in lymphocytes, smooth muscle cells, endothelial cells, fibroblasts and myeloid cells within the epithelial and lamina propria fractions. Additionally, the strictures were highly heterogenous with regards to cell composition, with 19 distinct cell types being identified, further clustered into fibroblasts, smooth muscle and endothelial populations (46).
Further analysis of the fibroblast compartment revealed six fibroblast populations enriched in CD strictures as opposed to non-strictured segments (46). These fibroblasts were characterized by an inflammatory phenotype, marked by the expression of pro-fibrotic markers involved in TGF-β signaling, macrophage differentiation and ECM production. However, the predominant cluster in these strictures were the MMP+/WNT5A+ fibroblasts, which were previously found to be enriched in the mucosa of patients with UC resistant to anti-TNF therapy (46). Among the populations within the stromal niche, CXCL14+ fibroblasts were the main contributors to transcriptional changes in strictures. These fibroblasts were associated with increased ECM production and CXCL14 displayed pro-fibrotic functions (46).
More importantly, the authors identified Cadherin11 (CDH11) as the only cell surface receptor co-expressed in both MMP+/WNT5A+ and CXCL14+ fibroblasts. Interestingly, CDH11 expression was shown to increase in CD strictures. The therapeutic potential of targeting CDH11 was shown in a DSS-induced fibrosis model of colitis in Cdh11 knockout mice (46). In this model, mice are subjected to two cycles of DSS drinking water followed by a recovery period with normal drinking water. Despite no differences in inflammation being observed in Cdh11 knockout mice compared to wild-type controls, Cdh11-knockout mice were protected from fibrosis development. Similarly, administration of an anti-Cdh11 mAb ameliorated DSS-driven fibrosis. In a longitudinal scRNA-seq analysis in the DSS model, Ho et al. observed that stromal cell clusters had strong influences on other clusters, highlighting their potential role in intestinal inflammation (29). Looking closely at these interactions, the authors identified relevant genes during the recovery phase, with the highest expression observed in Serpina3n, a serine protease inhibitor. To validate these observations, the authors examined the progression of DSS colitis in a Serpina3n knockout mouse. Although the disease progression was similar to wild-type mice, the knockout mice recovered much faster. Furthermore, the authors examined the mechanisms by which Serpina3n ameliorated DSS-induced intestinal inflammation. Using scRNA-seq on colon treated with Serpina3n protein, they observed that genes associated with ECM organization and cytokine mediated signaling pathways were enriched in stromal cells. Additionally, there was a decrease in inflammatory interactions and in the number of neutrophils, warranting the therapeutic potential of Serpina3n to accelerate recovery from intestinal inflammation. Overall, scRNA-seq has been crucial in characterizing the heterogeneity of the stromal population, as well as their interactions with the immune compartment in IBD. Alternatively, applying this technology to preclinical models of IBD could help uncover important mechanisms underlying resistance to current therapies. To address this, West and colleagues used transcriptomic approaches to uncover relevant genes associated with anti-TNF resistance. Oncostatin M (OSM) is expressed by stromal cells and was identified as a potential biomarker associated with anti-TNF failure (47). This observation was validated in a mouse model of IBD driven by oral Helicobacter hepaticus infection and anti-IL-10R blocking antibody, which mirrors the T cell-dependent pathology driving resistance to anti-TNF therapy. Using this model, treatment with an oncostatin receptor-Fc significantly attenuated colitis (47). In a more recent study, bulk and single-cell transcriptomics have been applied to inflamed tissue from three IBD patients’ cohorts undergoing surgical resection (48). The authors observed similar gene signatures from therapy non-responders, marked by genes indicating high neutrophil infiltration and activation of fibroblasts at sites of deep ulceration. Further analysis revealed important neutrophil-attractant signatures in fibroblasts, marked by an increase in IL-1R signaling, suggesting that this pathway is important in the inflammatory fibroblast/neutrophil recruitment in patients that are nonresponsive to current therapy (48).
In active IBD, macrophages secrete pro-inflammatory cytokines and react to commensal bacteria, with CD exhibiting aggregates of macrophages and other inflammatory cells (granulomas) associated with higher resection risk (49). IBD susceptibility involves various genetic loci linked to monocyte and macrophage function, including NOD2, ATG16L1, and IRGM (50–52). In 2019, two scRNA-seq studies explored intestinal macrophages in CD and UC. One scRNA-seq analysis of 11 ileal CD patients revealed resident and inflammatory macrophage subtypes. The resident subtype expressed C1Qs, CSF1R, MAFB, and MRC1, while inflammatory macrophages showed elevated levels of chemokines (CXCL2, CXCL3, CXCL8) and cytokines (IL23, IL6, IL1B, IL1A, TNF, OSM) Notably, pro-inflammatory interactions between non-epithelial stromal cells and macrophage-derived oncostatin-M (OSM) were found within the inflamed intestine, linked to anti-TNF-resistant CD (53). The second scRNA-seq study conducted on 18 UC patients revealed the presence of an inflammatory monocyte cluster with prominent levels of OSM expression that were associated with disease progression and resistance to anti-TNF treatment. A macrophage cluster was also identified that closely interacts with B cells and T cells. This macrophage cluster exhibited an enrichment of genes linked to IBD susceptibility, such as GPR65, ADCY7, PTGER4, PTPRC, and SH2B3 (34). A very recent scRNA-seq study provided a therapeutic map for CD and UC after anti-TNF therapy in a longitudinal manner (54). Importantly, the presence of a granuloma signature, characterized by C1Qhigh IL1Blow resident macrophage cells, was specifically associated with non-responders in CD. Moreover, genes associated with an inflammatory monocyte profile (S100A9, S100A12, FTH1, IL1RN) showed higher levels in non-responders but were reduced in responders. The latest study utilizing scRNA-seq and spatial analysis (CosMx Spatial Molecular Imaging) found that resident macrophages were present in both healthy and active IBD patients (55). Notably, inflammatory macrophages displayed patient-specific adaptation, characterized by an alternative activation pattern featuring the expression of EGFR ligands, NRG1, HBEGF, CLEC10A, and ASGR1, differing from the conventional M1 signature. The study also suggested a potential role of interactions between M2 or inflammation-dependent associated macrophages (representing the predominant inflammation-dependent macrophage state) and inflammatory fibroblasts in contributing to disease pathophysiology.
Through scRNA-seq studies in mice (56–61), we have gained insights into the diverse inflammatory roles of gut macrophages. In the DSS-induced colitis study, the abundance of Il1b+ C1qa+ C1qb+ macrophages increased in diseased animals, peaking post DSS administration, then returning to baseline during water administration (29). The mouse single-cell findings align with how intestinal cues influence transcription factors shaping macrophage subsets. The recent single-cell investigation using the Helicobacter hepaticus-induced colitis model, highlights interferon regulatory factor 5 (IRF5)’s role in mucosal macrophage differentiation. Irf5 knockout hindered CD11c+ mucosal macrophage formation, unveiling IRF5’s contribution to pro-inflammatory CD11c+F4/80+ macrophages during inflammation, driving colitis development (60). IRF5’s link to inflammatory macrophages and genetic risk factors for UC and CD underlines these mouse findings’ translational relevance to human IBD (62).
Dendritic cells (DCs) are antigen presenting cells that bridge both innate and adaptive immune responses. In the intestine, DCs continuously sample antigens by extending processes through the epithelial cells and glycocalyx layers into the lumen, or by remaining in the lamina propria or Peyer’s patches to process antigens that may have translocated (63). While multiple scRNA-seq studies in humans have begun to showcase the importance of different DC subsets in driving IBD pathogenesis, scRNA-seq studies in mouse models of IBD lag, specifically in the inclusion of diverse DC subsets, such as plasmacytoid DCs (pDCs), conventional DCs (cDCs), and monocyte-derived DCs (moDCs) (34, 45, 53, 54, 64). Corbin et al. used Helicobacter hepaticus-induced colitis coupled with an anti-IL-10R mAb in Irf5-/- mice and found the frequencies of DC populations during ongoing inflammation remained unchanged compared to steady-state (60). ScRNA-seq analysis on mononuclear phagocytes taken from the inflamed colonic lamina propria of CX3CR1+ wild-type and Irf5-/- mice. Within the four clusters of DCs, all lacked Cd64 expression, but included Flt3, Cd11c, and the DC-specific MHCII genes H2-DMb2 and H2-Oa. DC2-like cells were identified by Sirpa, Kmo, Cd209a, and Cd7 expression. The strong expression profile of Pu.1 but low Flt3 expression within these cells from these clusters suggested some may be moDCs. The other two clusters were likely representative of cDC1s (termed “Xcr1 DC” and expressing Xcr1high Irf8high Sirpalow) and a smaller group of Ccr7+ migratory DCs. Ho et al. also identified by scRNA-seq that pDCs decrease in number shortly after DSS exposure, however, the numbers stabilize over the resolution phase (29). Overall, this experiment showed that the heterogeneity of DCs could be identified and classified in the inflamed colonic tissue from these mice using scRNA-seq. The scarcity of murine single-cell studies necessitates further scRNA-seq in IBD models for a better understanding of these cells’ pivotal roles.
CD4+ T cell function in the intestine can be broadly categorized as effector (pro-inflammatory) or regulatory (anti-inflammatory). Regulatory T cells (Tregs) are CD4+ T cells that are critical for tolerance and dampening excessive immune responses against dietary and microbial antigens, an integral mechanism for maintaining intestinal homeostasis (65). In IBD, this balance of anti-inflammatory Treg function is lost and exuberant pro-inflammatory T cell responses against the commensal microbes drives and sustains chronic inflammation. In the paragraphs below, we will mainly highlight Treg and TH17 biology (63).
ScRNA-seq studies have been instrumental in advancing our understanding of the complex cellular heterogeneity and gene expression patterns of intestinal Treg cells in UC and CD. ScRNA-seq uncovered previously unidentified FOXP3+ cell subtypes involved in IBD. In a related study, Corridoni et al. (66) utilized multi-modal scRNA-seq to examine colonic CD8+ T cells in healthy individuals and those with UC. Interestingly, they noted an elevated presence of double-positive CD4+ CD8+ FOXP3+ T cells in UC patients. Smillie et al. (34) noted heightened levels of CD8+IL-17+ T cells and Tregs in inflamed UC tissues. They observed a TNF expression shift towards Tregs during inflammation. The study proposed that TNF+ Treg cells could significantly influence IBD progression, potentially causing resistance to TNF antibodies and playing a crucial role in CD8+ T cell adaptability. In healthy tissue, Tregs represented 1% of TNF expression, but this increased to over 14% during inflammation. Interestingly, in another study by Devlin et al. 67) found increased FOXP3+/BATF+ Treg cells in inflamed UC patients. They used scRNA-seq on CD45+ hematopoietic cells from UC patients with and without ileal pouch-anal anastomosis. These Tregs were enriched in inflamed UC and pouchitis samples, suggesting a role for BATF in their recruitment during inflammation. ScRNA-seq conducted by Boland et al. (68) revealed transcriptionally distinct Tregs expressing ZEB2 in UC. Among Treg gene profiles, 288 genes differed between healthy individuals and UC patients, including SATB1, KLF2, MYC, and ITGB1, associated with Treg function. Notably, ZEB2, linked to CD8+ T cell function, exhibited high expression in UC-related Treg cells. The reduction of ZEB2 enhanced suppressive function in murine Treg cells.
Huang et al. (69) analyzed initial diagnosis colon biopsies from healthy individuals and CD/UC patients, finding enriched Treg cells (slightly more in CD) and CD8+GZMK+ effector memory T cells in both conditions. Additionally, Jaeger et al. (70) used scRNA-seq and multi-parameter flow cytometry or mass cytometry to examine CD patients’ inflamed terminal ileum tissue. Inflammatory tissue IEL showed reduced Tregs, CD8+T, γδT and TFH but elevated TH17 cells. Lamina propria (LP) analysis revealed decreased Tregs and TFH cells, while CD8+T cells and TH17 cells increased. Tregs in LP were enriched in GPX1 and GLRX, associated with oxidative stress protection via FOXP3. GPX1 expression in LP Tregs hinted at activated anti-oxidative pathways, pertinent to CD due to GPX1 gene’s CD risk connections. Martin et al. (53) examined ileal samples from 13 CD patients at the single-cell level. In their pursuit of unraveling the cellular profile linked to resistance against anti-TNF drug therapy, they observed an abundance of Tregs with elevated IL-10 levels, along with low IL-17A expression within inflamed ileal lesions. Additionally, Treg populations have been detected in CD and UC patients by Mitsialis et al. (71). They observed pro-inflammatory memory-like IL17A+ Treg subsets expanded uniquely in active UC. Among IBD-enriched memory Tregs, one group was FOXP3+, while another was FOXP3low with strong pro-inflammatory cytokine expression (IFNG+TNF+IL17A+/–), indicating altered Treg function in IBD. Additionally, they observed an unusual expansion of novel IL1B+ Tregs in peripheral active CD compared to inactive CD and active UC. Furthermore, in a recent development by Nie et al., a computational tool called “scIBD” provided an integrated assessment of scRNA-seq data associated with IBD. This platform enabled the recognition of different cell types and IBD-associated genes. The meta-analysis corroborated the greater prevalence of Tregs in both UC and CD compared to healthy tissues, consistent with prior findings (72).
The study by Miragaia et al. (73) conducted single-cell analysis to comparatively investigate human and murine colon Treg populations. In mice, three distinct Treg subpopulations were identified: non-lymphoid tissue (NLT), suppressive, and lymphoid tissue-like (LT). NLT Tregs displayed a GATA3+-subpopulation phenotype, expressing Gata3, Nrp1, Areg, Il1rl1, and Ikzf2, while suppressive Tregs resembled the peripherally derived RORγt+-subpopulation. LT-like Tregs exhibited LT-associated gene expression (Sell, Ccr7, Tcf7, Bcl2) and fewer NLT-associated genes (Klrg1, Cd44, Icos, Rora, Tnfrsf9, Itgae), signifying functional diversity. Notably, 17 human-mouse colon Treg markers overlapped, including Tnfrsf4, Lgals1, Srgn, Cxcr6, Maf, and Ikzf3, indicating conserved roles. Paralogous genes demonstrated inter-organism expression pattern substitutions (e.g., Pim1-Pim2), hinting at the evolution of cellular communication pathways. Despite interspecies disparities, the study underscores the conservation of the TNFRSF-NF-κB-pathway between mice and humans (73). Surprisingly, the scRNA-seq study mentioned earlier, which focused on the murine DSS model (29), did not identify Tregs in any of the collected time course samples. However, a very recent preprint (74) took a different approach. Using MERFISH, a method for spatially resolved single-cell transcriptome profiling, they studied spatial and cellular changes during gut inflammation initiation and recovery in a DSS model. In day 9 samples, corresponding to peak inflammation after DSS administration, mucosal remodeling was observed, along with the appearance of diverse cell populations, including Tregs. Thus, the limited investigation of murine Tregs at the single-cell level underscores the need for further scRNA-seq studies on murine IBD models to gain a deeper understanding of the roles played by this essential cell population.
Naïve CD4+ T cell polarization towards an effector phenotype requires signals from antigen-presentation, co-stimulation, and cytokines in the local tissue environment. In vitro polarization of human CD4+ T cells towards a TH17 phenotype requires TGF-β, IL-1β, IL-6, IL-23 and the expression of RAR-related orphan receptor g (RORγT) (75). Polarized TH17 cells secrete IL-17A, IL-17F, and IL-22 – cytokines which are associated with the development of inflammation. While not required for TH17 differentiation, IL-23 signaling, through the IL-23 receptor complex, promotes RORγT stability and the acquisition of a pathogenic phenotype in these cells. IL-23 is upregulated in numerous autoimmune diseases, including IBD (76). Furthermore, genome-wide association studies have identified IL23R as a genetic susceptibility locus for IBD. A key role for IL-23 in IBD pathogenesis has been highlighted by the clinical efficacy of anti-IL-23 biologics (77–79).
Jaeger et al. used scRNA-seq analysis to compare T cells from intra epithelial lymphocytes (IELs) and the lamina propria (LP) found in the terminal ileum of adults with severe CD to measure heterogeneity in T cell lineages and subsets (70). There was a marked increase in pro-inflammatory, activated CD39+CCR6+ and CD39+CD4+ TH17 cells but decreased CD8+T, γδT, TFH and Treg cells in the IEL compartment of CD patients, demonstrating that loss of balance between TH17 and Treg cells plays an important role in disease progression. Within IELs, they identified unique T cell clusters, including one with NKp30+γδT cells expressing RORγt and producing IL-26 upon NKp30 engagement. Some of these T cell subsets expressed TH17 markers including RORC, IL23R, IL22, CXCR4, and IL26. While these RORγt+ γδ T cells have been shown and described in mouse models (80, 81), Jaeger et al. identified this subset of γδ T cells in the IEL of humans. γδ T cells can constitute as much as 40% of the IELS in the intestine. These cells can have cytotoxic, immunoregulatory, and tissue repair functions within the intestinal mucosa (82). However, the role of human γδ T cells in CD remains to be fully elucidated, with multiple studies showing either a decrease or increase in number in the blood or inflamed tissue (82). Similar analysis in the LP found a biased TH17/Treg ratio in CD. The CD39+ TH17 cells that were found within the IEL may prove to be a double-edged sword, as their expression of GZMB and CCL4 could make them pathogenic and able to recruit other inflammatory cell types to increase epithelial damage (70). Conversely, their production of IL-17 and IL-26 may enhance epithelial barrier integrity and protection (70). Overall, this study showed an altered spatial distribution of T cell subsets between the two cellular compartments, with TH17s in the LP bearing more markers of quiescence (i.e., CXCR4 and CD39) than in the IEL compartment that may correlate with transmural inflammation during IBD pathogenesis (70).
An atlas of over 350,000 cells from the colonic mucosa of patients with and without UC was used to reveal 51 cell subsets, including epithelial, stromal, and immune cells (34). When reviewing T cells, several CD4+ T cell subsets showed increased expression of IL17A, showcasing an expansion of TH17 cells with pro-inflammatory phenotypes. However, the CD8+ T cell subsets showed the largest induction on IL17A. Using an in-situ analysis, the authors found CD8 co-expressed on both CD4- and CD4+ cells. These CD4+CD8+IL17+ T cells have been largely uncharacterized, however, these cells correlated with activated cytotoxic programs and genetic signatures conducive to TH17 pathogenicity and tissue damage (e.g., RBPJ and IL23R) that have been reported in mice (83). Kong et al. also showed IL23R to be an IBD risk gene in cells taken from the colon and terminal ileum of patients with CD, but they showed the greatest mean expression to be in innate lymphoid cells (ILCs) in both tissue types (45).
A recent study in mice showed that colonic immunopathology driven by Red 40, and IL-23 may not actually depend on these classical TH17 responses; as IL-17A and IL-17F blockade did not prevent colitis development in mice that conditionally expressed IL23 in CX3CR1-positive myeloid cells (84). Additionally, they sought to define this mechanism further. They used the T cell transfer colitis model, an invaluable model to dissect pathogenic and protective mechanisms of CD4+ T cells (11) and scRNA-seq to identify a new population of IFNγ-secreting CD4+ cytotoxic T lymphocytes that were induced by IL-23 and the food dye Red 40 to promote colitis pathogenesis (27). Without IFNγ, IL-23 and Red 40 did not induce progressive colitis development and prevented colonic epithelial cell death, showcasing perhaps a specialized relationship between IL-23, IL-17, IFNγ, and overall TH17 signaling in the development of IBD in mice (27).
In a longitudinal DSS mouse model, Ho et al. looked at how cell phenotypes changed during inflammation (29). Using a pseudotime analysis, significant changes were observed in T cell subsets (which expressed Cd3d and Cd3g). The IBD risk gene Ifng was expressed over the entire DSS-induced disease time course in the T cell cluster but only at day 6 in stromal cells, indicating subtle dynamic phenotypic changes in different cell types. Capturing these subtle changes over time in a human model is difficult, exemplifying how useful mouse models of IBD can be, especially when paired with scRNA-seq analysis.
CD8+ T cells are a cytotoxic T cell subset that drive cell-death through numerous pathways including perforin and granzyme release and by expressing FAS ligand (85). CD8+ T cells are typically in lower abundance in the intestine at steady state compared to CD4+ T cells (86). Despite numerous evidences that IBD pathology is associated with exuberant CD4+ T cell responses, recent scRNA-seq data revealed that CD8+ T cells also play a role in IBD pathogenesis (25, 66, 68).
T cells sorted from the IELs of healthy donors and CD patients revealed different CD8+ clusters (70). One of these clusters expressed genes associated with an effector phenotype (KLRG1, GZMB, GZMK, and IFNG). Whereas, the other CD8+ cluster highly expressed the tissue residency marker ITGAE. When these cells were analyzed further using a new clustering strategy, this highlighted the expression of IL7R, IL2 and TCF7, genes associated with a memory phenotype. The authors hypothesized that the resident memory CD8+ T cell cluster might gain cytotoxic and antimicrobial functions via S100A family members during inflammation. In the same study, scRNA-seq of CD8+ T cells in the LP identified three clusters: effector CD8+ T cells (KLRG1, EOMES, GZMB), a second group expressing CD160 and ITGA1, and a third group expressing NK cell receptors such as KLRC1, KLRD1, and ENTPD1 (70). In summary, similar and unique CD8+ T cell clusters were identified in the IEL and LP compartments of CD patients.
A second scRNA-seq analysis of colonic T cells revealed that CD8+ and γδ T cells both expressed ENTPD1 (64). This gene codes an ectonucleotidase that converts ATP and ADP to AMP. In colonic CD8+ T cells from pediatric cases of colitis, decreased ENTPD1 expression was observed in association with a defective cyclic AMP (cAMP) pathway. Mechanistically, the reduction of cAMP is believed to result in platelet aggregation and hyperinflammation. The authors then used DSS-induced acute colitis mouse model to test the effect of dipyrimodole, a phosphodiesterase (PDE) inhibitor. PDE is an enzyme the degrades and inactivates cAMP and cGMP, therefore, inhibiting PDE will increase cAMP levels in vivo. Administering mice dipyrimodole increased the colon length, prevented body weight loss, improved barrier function, and crypt structure in the DSS treated mice. As a result, they demonstrated increased expression of Entpd1 and elevated cAMP levels also influenced platelet aggregation and TNFα production, linking the findings observed in a pre-clinical model of colitis back to human IBD (64).
In yet another study, scRNA-seq of CD8+ T cells from dissociated colonic tissues of UC patients, identified tissue resident (ITGAE and DUSP4), naïve (SELL, CCR7, and TCF7) double positive CD8+CD4+ (GZMA, RORA, and CCR6) and increased numbers of IL26 expressing T cells (66). Although IL26 is not expressed in mice, human IL-26 can signal through the IL-10RB and IL-20RA heterodimeric receptor in mice (87). Deeper analysis of the IL26-expressing CD8+ T cells displayed both an ILC3 signature (RORC, CKIT and AHR), a TH17 signature, and several exhaustion markers (PDCD1, HAVCR2, and CTLA4). Interestingly, these IL26 expressing CD8+ T cells co-expressed IL23R and IL17A, which has also been shown in a recent longitudinal human single cell therapeutic atlas of IBD (54). To further assess the role of IL26 in UC, the authors subjected humanized IL26 transgenic mice to DSS-induced colitis and tested the efficacy of an anti-IL-26 monoclonal antibody. Using these tools, the authors demonstrated that IL-26 has a protective role in the acute model of colitis in mice; however, whether IL-26 has the same protective effect under chronic inflammation conditions remains to be elucidated (66).
While typically associated with protecting the host from infection, a pathogenic role for resident memory CD8+ T cells (Trm) in autoimmune diseases have become appreciated (53, 68, 88). Boland and colleagues identified four CD8+ Trm clusters, expressing high levels of CD69, ITGAE, CD101, CCR6, ITGA1 and lower levels of KLF2 and S1PR1, with unique clonal expansion of a cluster of CD8+ Trm cells in UC patients compared to healthy controls (68). This cluster also expressed transcripts encoding cytotoxic granules, metabolic regulators, and the transcription factors ZEB2 and EOMES. The authors then used two pre-clinical models of colitis to demonstrate a pathogenic role for CD8+ T cells. First, Il10 knockout mice fed piroxicam had a marked accumulation of CD8+ T cells in the colon. The administration of an anti-CD8 mAb in these mice reduced weight loss and colon pathology. Secondly, the adoptive transfer of CD8+ T cells transduced with Eomes retroviral constructs into Rag1 deficient mice significantly increased weight loss and more colonic inflammation after DSS administration (68).
Plasma cells are terminally differentiated B cells that secrete immunoglobulin (Ig), including IgM, IgE, IgA, and IgG. While IgA levels are highly abundant in the intestinal tissue and contribute to intestinal homeostasis (89), anti-microbial IgG in the gut is also observed at steady-state (90, 91). The large presence of anti-microbial IgG observed in IBD has renewed interest due to an FCGR2A variant associated with disease pathogenesis (92, 93). Thus, the role of IgG in the inflamed mucosa of IBD patients is an area of active investigation.
Huang et al. identified increases in CD138+ plasma cells in pediatric CD subjects by scRNA-seq (64). More specifically, seven B cell clusters including tissue resident B cells (CD44, CD69), CD19+ B cells, CD27high B cells, CD27low B cells, and two CD138+ plasma cell clusters expressing IGHG1 and IGHA1 respectively were identified. Interestingly, the tissue resident marker (CD103) was increased in plasmablasts. In addition, they observed the transition from IgA1 to IgG1 in the IBD subjects, in line with the study showing the impact of CXCR4+IgG+ plasma cells to the IBD pathogenesis (94). In another study scRNA-seq study by Boland et al. characterized different subsets of B cells and plasma cells (68). They observed an increase in the IgG1+ plasmablasts in UC whereas IgA2+ plasma cells were elevated in the healthy controls. All together, these scRNA-seq studies highlight the presence of B cells in the inflamed mucosa and bias towards IgG class-switching in plasma cells in human IBD. Uzzan and colleagues identified twenty clusters of B cells isolated from the LP when comparing healthy and UC patients (95). These clusters included naïve B cells (IGHD, FCER2, and CD72), memory B cells (CD27 and TNFRSF13B), atypical memory B cells (FCRL5, FCRL4, and DUSP4), germinal center-like B cells (AICDA, BCL6, and FAS) and plasma cell populations based on the expression of immunoglobulin genes. Additionally, they detected an IFNG signature specific to a naive B cell cluster expressing IGHD, FCER2, and CD72 suggesting a shift in the systemic humoral response towards a pro-inflammatory IgG B cell phenotype in UC.
Kong et al. collected tissue from CD patients and non-IBD donors from inflamed and non-inflamed regions of the terminal ileum and the colon and characterized the cellular networks by scRNA-seq (45). B cells were sub-clustered into plasma cells (SDC1, MZB1, SSR4, XBP1), B cells (BANK, MS4A1/CD30, ASDAM28, VPREB3) and germinal center B cells (LRMP, GPT2, PAG1). Plasma cells were a higher percentage of the sample in inflamed CD in the terminal ileum (45). Martin et al. described the GIMATS module, consisting of the presence of IgG-expressing plasma cells, inflammatory mononuclear phagocytes, activated T cells, and stromal cells, is expressed in a subset of ileal CD and is linked to anti-TNF therapy resistance (53).
On the other hand, in the mouse scRNA-seq study Ho et al. described decrease in the IgA producing plasma cell population at most severe time points of intestinal inflammation, whereas IgA plasma cell numbers recovered during the resolution phase (29). While differences in B cell biology have been identified between healthy and human IBD, the role of B cells has begun to be explored in pre-clinical models of IBD. In a separate study by Frede et al, they performed another longitudinal DSS-colitis study where scRNA-seq was performed on enriched B cells from the colonic LP on days 0 and 14 after DSS exposure in mice (26). While no differences in B cell cluster numbers were observed, the authors identified an expansion in an IFN-induced B cell cluster marked by Serpina3g, Serpina3f, Stat1, Tgtp2, and Zpb1 during the mucosal healing stage of disease at day 14. Subsequent experiments where B cells were depleted by administering CD19cre-iDTRHET mice diphtheria toxin during DSS administration enhanced tissue regeneration, suggesting a pathogenic role of B cells in mucosal healing (26).
The advances and application of single cell transcriptomics to human IBD samples have revolutionized our understanding of the complex and dynamic interactions between epithelial cell, stromal cell, and immune cell populations. Additionally, this technology has also been instrumental in identifying novel cell types and rare populations of cells, which were likely not detected previously. While numerous human IBD scRNA-seq studies were highlighted in this review, it is evident that the application of this powerful technology has not been applied enough to pre-clinical models of IBD to translate findings found in human disease. To our knowledge, the only pre-clinical model that applied scRNA-seq to generate a comprehensive landscape of epithelial cells, stromal cells, and immune cells was the longitudinal DSS study by Ho et al. (29). However, this study did not identify all cell types that have been identified by scRNA-seq in human studies (Figure 1). For example, a recent study combining scRNA-seq with spatial analysis (65) uncovered notable diversity within neutrophils in IBD colonic mucosa. Neutrophils were categorized into three distinct states (N1, N2, and N3) based on their unique gene expression profiles. Furthermore, novel clusters of innate lymphoid cells have been identified and contribute to intestinal inflammation (96, 97). Thus, by expanding the use of scRNA-seq to other pre-clinical models of IBD, we will be better equipped to select the most appropriate model to translate and functionally validate findings from human IBD studies.
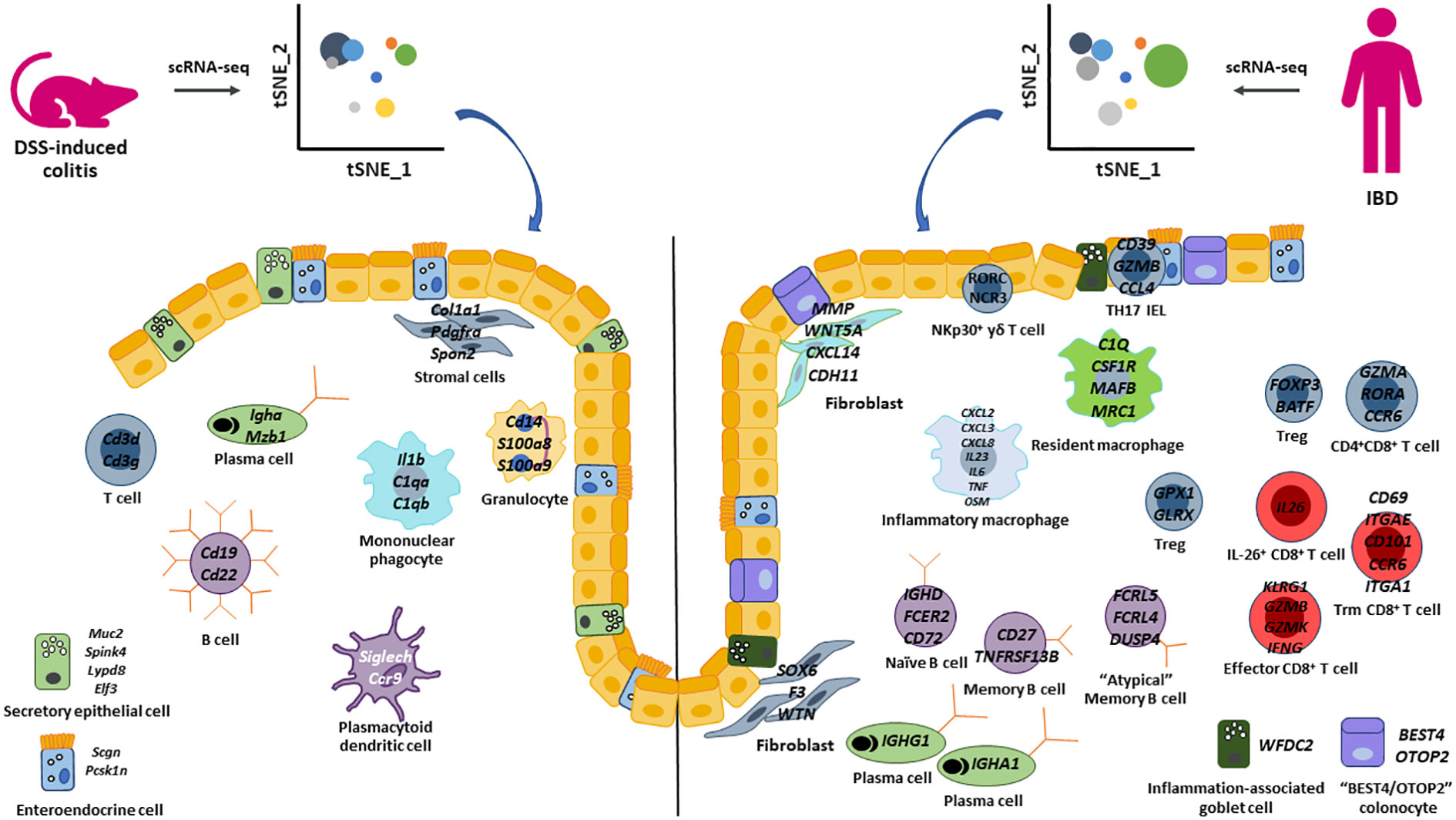
Figure 1 Summary of epithelial cell, stromal cell, and immune cell populations identified by scRNA-seq in the DSS-induced colitis model and human IBD. Highlighted key cell populations identified in this review that describe similar and different cell populations between the DSS-colitis pre-clinical model and human IBD.
Furthermore, by expanding the use of scRNA-seq to all pre-clinical models to generate comprehensive disease atlases with epithelial cells, stromal cells, and immune cells will warrant the opportunity to generate novel hypotheses for therapeutic strategies (Figure 2). Overlaying gene signatures at the single cell level from human IBD and numerous pre-clinical models would significantly increase the likelihood of selecting the most appropriate pre-clinical model to test novel first- and best-in-class therapeutics and modalities to treat and ultimately cure IBD.
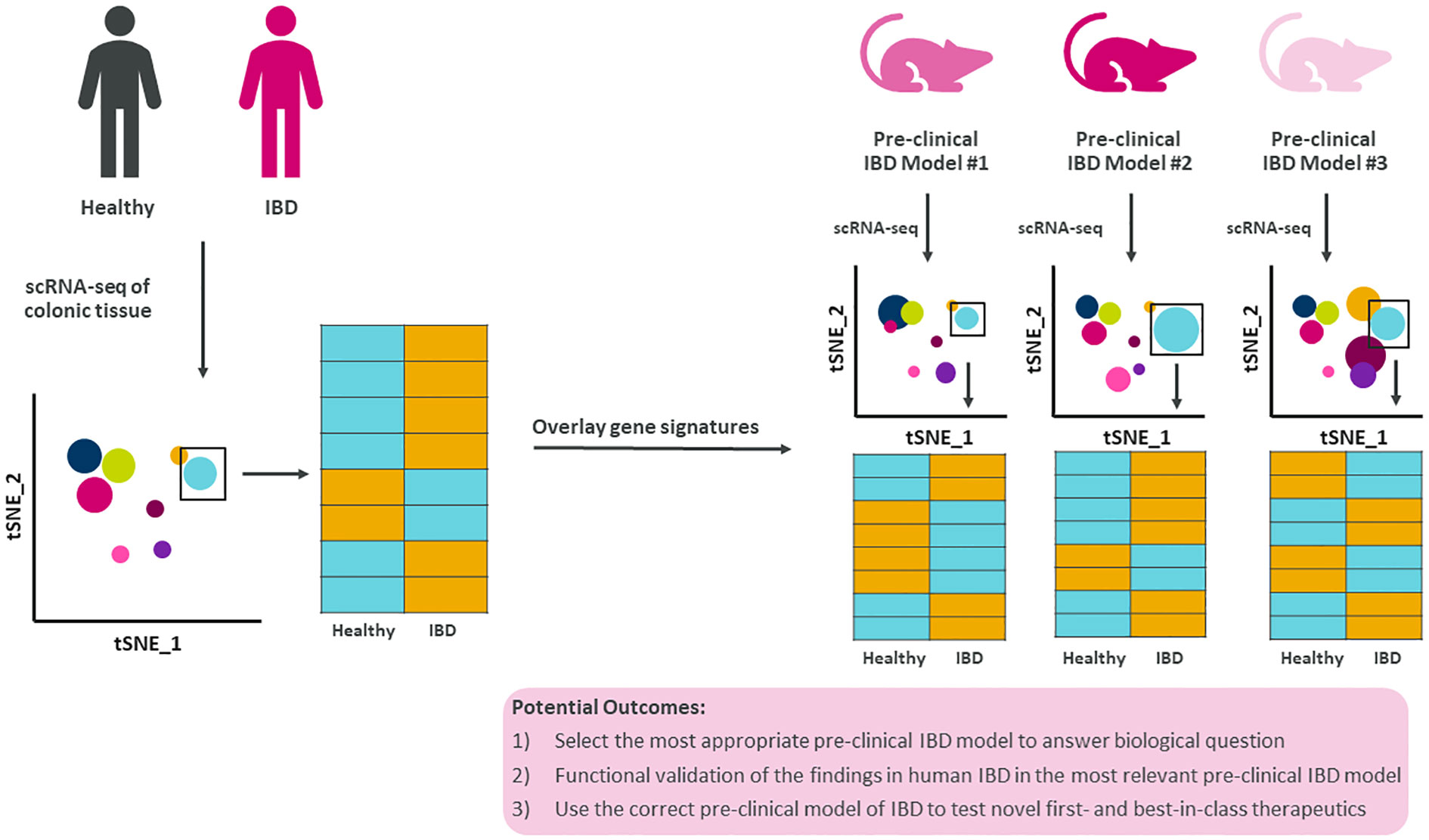
Figure 2 Summary figure of how translating single cell findings in human to pre-clinical models improves our understanding of disease. Human IBD gene signatures at the single cell level could be overlayed with single cell gene signatures from multiple pre-clinical models of IBD allowing for data-driven selection of the most appropriate pre-clinical model to test novel biology, questions, and therapeutics.
EPK: Conceptualization, Writing – original draft, Writing – review & editing. AM: Conceptualization, Writing – original draft, Writing – review & editing. IK: Conceptualization, Writing – original draft, Writing – review & editing. B-EJ: Conceptualization, Writing – original draft, Writing – review & editing. RLU: Conceptualization, Writing – original draft, Writing – review & editing. KJB: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. DC: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. TO: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We would like to thank Jessica Örnros, Julie Zacharczuk, and Bartosz Poniatowski for assistance with illustrations.
EPK, AM, IK, B-EJ, RLU, KJB, DC, TO are employed by the company AstraZeneca.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Singh S, Blanchard A, Walker JR, Graff LA, Miller N, Bernstein CN. Common symptoms and stressors among individuals with inflammatory bowel diseases. Clin Gastroenterol Hepatol (2011) 9(9):769–75. doi: 10.1016/j.cgh.2011.05.016
2. Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res (2019) 2019:7247238. doi: 10.1155/2019/7247238
3. Barnhoorn MC, Hakuno SK, Bruckner RS, Rogler G, Hawinkels L, Scharl M. Stromal cells in the pathogenesis of inflammatory bowel disease. J Crohns Colitis (2020) 14(7):995–1009. doi: 10.1093/ecco-jcc/jjaa009
4. Katsandegwaza B, Horsnell W, Smith K. Inflammatory bowel disease: A review of pre-clinical murine models of human disease. Int J Mol Sci (2022) 23(16). doi: 10.3390/ijms23169344
5. Baydi Z, Limami Y, Khalki L, Zaid N, Naya A, Mtairag EM, et al. An update of research animal models of inflammatory bowel disease. ScientificWorldJournal (2021) 2021:7479540. doi: 10.1155/2021/7479540
6. Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (Dss)-induced colitis in mice. Curr Protoc Immunol (2014) 104:15 25 1–15 25 14. doi: 10.1002/0471142735.im1525s104
7. Antoniou E, Margonis GA, Angelou A, Pikouli A, Argiri P, Karavokyros I, et al. The tnbs-induced colitis animal model: an overview. Ann Med Surg (Lond) (2016) 11:9–15. doi: 10.1016/j.amsu.2016.07.019
8. Morris GP, Beck PL, Herridge MS, Depew WT, Szewczuk MR, Wallace JL. Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology (1989) 96(3):795–803. doi: 10.1016/S0016-5085(89)80079-4
9. Boirivant M, Fuss IJ, Chu A, Strober W. Oxazolone colitis: A murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J Exp Med (1998) 188(10):1929–39. doi: 10.1084/jem.188.10.1929
10. Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a th2 colitis model resembling ulcerative colitis, is mediated by il-13-producing nk-T cells. Immunity (2002) 17(5):629–38. doi: 10.1016/s1074-7613(02)00453-3
11. Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by cd4+Cd25+ Regulatory T cells. J Immunol (2003) 170(8):3939–43. doi: 10.4049/jimmunol.170.8.3939
12. Ostanin DV, Bao J, Koboziev I, Gray L, Robinson-Jackson SA, Kosloski-Davidson M, et al. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am J Physiol Gastrointest Liver Physiol (2009) 296(2):G135–46. doi: 10.1152/ajpgi.90462.2008
13. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell (1993) 75(2):263–74. doi: 10.1016/0092-8674(93)80068-p
14. Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and cd4(+) th1-like responses. J Clin Invest (1996) 98(4):1010–20. doi: 10.1172/JCI118861
15. Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, et al. Differential activity of il-12 and il-23 in mucosal and systemic innate immune pathology. Immunity (2006) 25(2):309–18. doi: 10.1016/j.immuni.2006.05.017
16. Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell (2007) 131(1):33–45. doi: 10.1016/j.cell.2007.08.017
17. Garrett WS. Glimcher LH. T-bet-/- rag2-/- ulcerative colitis: the role of T-bet as a peacekeeper of host-commensal relationships. Cytokine (2009) 48(1-2):144–7. doi: 10.1016/j.cyto.2009.07.007
18. Pizarro TT, Pastorelli L, Bamias G, Garg RR, Reuter BK, Mercado JR, et al. Samp1/yitfc mouse strain: A spontaneous model of crohn's disease-like ileitis. Inflammation Bowel Dis (2011) 17(12):2566–84. doi: 10.1002/ibd.21638
19. Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of tnf biosynthesis in mice lacking tnf au-rich elements: implications for joint and gut-associated immunopathologies. Immunity (1999) 10(3):387–98. doi: 10.1016/s1074-7613(00)80038-2
20. Kontoyiannis D, Boulougouris G, Manoloukos M, Armaka M, Apostolaki M, Pizarro T, et al. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced crohn's-like inflammatory bowel disease. J Exp Med (2002) 196(12):1563–74. doi: 10.1084/jem.20020281
21. Higgins LM, Frankel G, Douce G, Dougan G, MacDonald TT. Citrobacter rodentium infection in mice elicits a mucosal th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect Immun (1999) 67(6):3031–9. doi: 10.1128/IAI.67.6.3031-3039.1999
22. Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, et al. Il-23 plays a key role in helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med (2006) 203(11):2485–94. doi: 10.1084/jem.20061082
23. Fox JG, Ge Z, Whary MT, Erdman SE, Horwitz BH. Helicobacter hepaticus infection in mice: models for understanding lower bowel inflammation and cancer. Mucosal Immunol (2011) 4(1):22–30. doi: 10.1038/mi.2010.61
24. Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. Mrna-seq whole-transcriptome analysis of a single cell. Nat Methods (2009) 6(5):377–82. doi: 10.1038/nmeth.1315
25. Corridoni D, Chapman T, Antanaviciute A, Satsangi J, Simmons A. Inflammatory bowel disease through the lens of single-cell rna-seq technologies. Inflammation Bowel Dis (2020) 26(11):1658–68. doi: 10.1093/ibd/izaa089
26. Frede A, Czarnewski P, Monasterio G, Tripathi KP, Bejarano DA, Ramirez Flores RO, et al. B cell expansion hinders the stroma-epithelium regenerative cross talk during mucosal healing. Immunity (2022) 55(12):2336–51.e12. doi: 10.1016/j.immuni.2022.11.002
27. Chen L, He Z, Reis BS, Gelles JD, Chipuk JE, Ting AT, et al. Ifn-gamma(+) cytotoxic cd4(+) T lymphocytes are involved in the pathogenesis of colitis induced by il-23 and the food colorant red 40. Cell Mol Immunol (2022) 19(7):777–90. doi: 10.1038/s41423-022-00864-3
28. Gurtner A, Borrelli C, Gonzalez-Perez I, Bach K, Acar IE, Nunez NG, et al. Active eosinophils regulate host defence and immune responses in colitis. Nature (2023) 615(7950):151–7. doi: 10.1038/s41586-022-05628-7
29. Ho YT, Shimbo T, Wijaya E, Kitayama T, Takaki S, Ikegami K, et al. Longitudinal single-cell transcriptomics reveals a role for serpina3n-mediated resolution of inflammation in a mouse colitis model. Cell Mol Gastroenterol Hepatol (2021) 12(2):547–66. doi: 10.1016/j.jcmgh.2021.04.004
30. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene lgr5. Nature (2007) 449(7165):1003–7. doi: 10.1038/nature06196
31. Parikh K, Antanaviciute A, Fawkner-Corbett D, Jagielowicz M, Aulicino A, Lagerholm C, et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature (2019) 567(7746):49–55. doi: 10.1038/s41586-019-0992-y
32. Surawicz CM, Haggitt RC, Husseman M, McFarland LV. Mucosal biopsy diagnosis of colitis: acute self-limited colitis and idiopathic inflammatory bowel disease. Gastroenterology (1994) 107(3):755–63. doi: 10.1016/0016-5085(94)90124-4
33. Bingle L, Cross SS, High AS, Wallace WA, Rassl D, Yuan G, et al. Wfdc2 (He4): A potential role in the innate immunity of the oral cavity and respiratory tract and the development of adenocarcinomas of the lung. Respir Res (2006) 7(1):61. doi: 10.1186/1465-9921-7-61
34. Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell (2019) 178(3):714–30 e22. doi: 10.1016/j.cell.2019.06.029
35. Maddipatla SC, Kolachala VL, Venkateswaran S, Dodd AF, Pelia RS, Geem D, et al. Assessing cellular and transcriptional diversity of ileal mucosa among treatment-naive and treated crohn's disease. Inflamm Bowel Dis (2023) 29(2):274–85. doi: 10.1093/ibd/izac201
36. Burclaff J, Bliton RJ, Breau KA, Ok MT, Gomez-Martinez I, Ranek JS, et al. A proximal-to-distal survey of healthy adult human small intestine and colon epithelium by single-cell transcriptomics. Cell Mol Gastroenterol Hepatol (2022) 13(5):1554–89. doi: 10.1016/j.jcmgh.2022.02.007
37. Lelouard H, Fallet M, de Bovis B, Meresse S, Gorvel JP. Peyer's patch dendritic cells sample antigens by extending dendrites through M cell-specific transcellular pores. Gastroenterology (2012) 142(3):592–601 e3. doi: 10.1053/j.gastro.2011.11.039
38. Bennett KM, Parnell EA, Sanscartier C, Parks S, Chen G, Nair MG, et al. Induction of colonic M cells during intestinal inflammation. Am J Pathol (2016) 186(5):1166–79. doi: 10.1016/j.ajpath.2015.12.015
39. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
40. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of gsdmd by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
41. Bulek K, Zhao J, Liao Y, Rana N, Corridoni D, Antanaviciute A, et al. Epithelial-derived gasdermin D mediates nonlytic il-1beta release during experimental colitis. J Clin Invest (2020) 130(8):4218–34. doi: 10.1172/JCI138103
42. Nowarski R, Jackson R, Flavell RA. The stromal intervention: regulation of immunity and inflammation at the epithelial-mesenchymal barrier. Cell (2017) 168(3):362–75. doi: 10.1016/j.cell.2016.11.040
43. Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell (2013) 13(4):392–402. doi: 10.1016/j.stem.2013.09.006
44. Kinchen J, Chen HH, Parikh K, Antanaviciute A, Jagielowicz M, Fawkner-Corbett D, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell (2018) 175(2):372–86 e17. doi: 10.1016/j.cell.2018.08.067
45. Kong L, Pokatayev V, Lefkovith A, Carter GT, Creasey EA, Krishna C, et al. The landscape of immune dysregulation in crohn's disease revealed through single-cell transcriptomic profiling in the ileum and colon. Immunity (2023) 56(2):444–58.e5. doi: 10.1016/j.immuni.2023.01.002
46. Mukherjee PK, Nguyen QT, Li J, Zhao S, Christensen SM, West GA, et al. Stricturing crohn's disease single-cell rna sequencing reveals fibroblast heterogeneity and intercellular interactions. Gastroenterology (2023) 165:1180–96. doi: 10.1053/j.gastro.2023.07.014
47. West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med (2017) 23(5):579–89. doi: 10.1038/nm.4307
48. Friedrich M, Pohin M, Jackson MA, Korsunsky I, Bullers SJ, Rue-Albrecht K, et al. Il-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med (2021) 27(11):1970–81. doi: 10.1038/s41591-021-01520-5
49. Heresbach D, Alexandre JL, Branger B, Bretagne JF, Cruchant E, Dabadie A, et al. Frequency and significance of granulomas in a cohort of incident cases of crohn's disease. Gut (2005) 54(2):215–22. doi: 10.1136/gut.2004.041715
50. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous snps identifies a susceptibility variant for crohn disease in atg16l1. Nat Genet (2007) 39(2):207–11. doi: 10.1038/ng1954
51. Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, et al. Sequence variants in the autophagy gene irgm and multiple other replicating loci contribute to crohn's disease susceptibility. Nat Genet (2007) 39(7):830–2. doi: 10.1038/ng2061
52. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of nod2 leucine-rich repeat variants with susceptibility to crohn's disease. Nature (2001) 411(6837):599–603. doi: 10.1038/35079107
53. Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, et al. Single-cell analysis of crohn's disease lesions identifies a pathogenic cellular module associated with resistance to anti-tnf therapy. Cell (2019) 178(6):1493–508 e20. doi: 10.1016/j.cell.2019.08.008
54. Thomas T, Rich-Griffin C, Pohin M, Friedrich M, Aschenbrenner D, Pakpoor J, et al. A longitudinal single-cell therapeutic atlas of anti-tumour necrosis factor treatment in inflammatory bowel disease. BioRvix (2023). doi: 10.1101/2023.05.05.539635%JbioRxiv
55. Garrido-Trigo A, Corraliza AM, Veny M, Dotti I, Melon-Ardanaz E, Rill A, et al. Macrophage and neutrophil heterogeneity at single-cell spatial resolution in human inflammatory bowel disease. Nat Commun (2023) 14(1):4506. doi: 10.1038/s41467-023-40156-6
56. Kang B, Alvarado LJ, Kim T, Lehmann ML, Cho H, He J, et al. Commensal microbiota drive the functional diversification of colon macrophages. Mucosal Immunol (2020) 13(2):216–29. doi: 10.1038/s41385-019-0228-3
57. Wuggenig P, Kaya B, Melhem H, Ayata CK, Swiss IBDCI, Hruz P, et al. Loss of the branched-chain amino acid transporter cd98hc alters the development of colonic macrophages in mice. Commun Biol (2020) 3(1):130. doi: 10.1038/s42003-020-0842-3
58. Scott CL, T'Jonck W, Martens L, Todorov H, Sichien D, Soen B, et al. The transcription factor zeb2 is required to maintain the tissue-specific identities of macrophages. Immunity (2018) 49(2):312–25.e5. doi: 10.1016/j.immuni.2018.07.004
59. Rohm TV, Keller L, Bosch AJT, AlAsfoor S, Baumann Z, Thomas A, et al. Targeting colonic macrophages improves glycemic control in high-fat diet-induced obesity. Commun Biol (2022) 5(1):370. doi: 10.1038/s42003-022-03305-z
60. Corbin AL, Gomez-Vazquez M, Berthold DL, Attar M, Arnold IC, Powrie FM, et al. Irf5 guides monocytes toward an inflammatory cd11c(+) macrophage phenotype and promotes intestinal inflammation. Sci Immunol (2020) 5(47). doi: 10.1126/sciimmunol.aax6085
61. De Schepper S, Verheijden S, Aguilera-Lizarraga J, Viola MF, Boesmans W, Stakenborg N, et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell (2018) 175(2):400–15 e13. doi: 10.1016/j.cell.2018.07.048
62. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. Irf5 promotes inflammatory macrophage polarization and th1-th17 responses. Nat Immunol (2011) 12(3):231–8. doi: 10.1038/ni.1990
63. Yan JB, Luo MM, Chen ZY, He BH. The function and role of the th17/treg cell balance in inflammatory bowel disease. J Immunol Res (2020) 2020:8813558. doi: 10.1155/2020/8813558
64. Huang B, Chen Z, Geng L, Wang J, Liang H, Cao Y, et al. Mucosal profiling of pediatric-onset colitis and ibd reveals common pathogenics and therapeutic pathways. Cell (2019) 179(5):1160–76 e24. doi: 10.1016/j.cell.2019.10.027
65. Ma H, Tao W, Zhu S. T lymphocytes in the intestinal mucosa: defense and tolerance. Cell Mol Immunol (2019) 16(3):216–24. doi: 10.1038/s41423-019-0208-2
66. Corridoni D, Antanaviciute A, Gupta T, Fawkner-Corbett D, Aulicino A, Jagielowicz M, et al. Single-cell atlas of colonic cd8(+) T cells in ulcerative colitis. Nat Med (2020) 26(9):1480–90. doi: 10.1038/s41591-020-1003-4
67. Devlin JC, Axelrad J, Hine AM, Chang S, Sarkar S, Lin JD, et al. Single-cell transcriptional survey of ileal-anal pouch immune cells from ulcerative colitis patients. Gastroenterology (2021) 160(5):1679–93. doi: 10.1053/j.gastro.2020.12.030
68. Boland BS, He Z, Tsai MS, Olvera JG, Omilusik KD, Duong HG, et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci Immunol (2020) 5(50). doi: 10.1126/sciimmunol.abb4432
69. Huang LJ, Mao XT, Li YY, Liu DD, Fan KQ, Liu RB, et al. Multiomics analyses reveal a critical role of selenium in controlling T cell differentiation in crohn's disease. Immunity (2021) 54(8):1728–44 e7. doi: 10.1016/j.immuni.2021.07.004
70. Jaeger N, Gamini R, Cella M, Schettini JL, Bugatti M, Zhao S, et al. Single-cell analyses of crohn's disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat Commun (2021) 12(1):1921. doi: 10.1038/s41467-021-22164-6
71. Mitsialis V, Wall S, Liu P, Ordovas-Montanes J, Parmet T, Vukovic M, et al. Single-cell analyses of colon and blood reveal distinct immune cell signatures of ulcerative colitis and crohn's disease. Gastroenterology (2020) 159(2):591–608.e10. doi: 10.1053/j.gastro.2020.04.074
72. Nie H, Lin P, Zhang Y, Wan Y, Li J, Yin C, et al. Single-cell meta-analysis of inflammatory bowel disease with scibd. Nat Comput Sci (2023) 3(6):522–31. doi: 10.1038/s43588-023-00464-9
73. Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, et al. Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity (2019) 50(2):493–504 e7. doi: 10.1016/j.immuni.2019.01.001
74. Cadinu P, Sivanathan KN, Misra A, Xu RJ, Mangani D, Yang E, et al. Charting the cellular biogeography in colitis reveals fibroblast trajectories and coordinated spatial remodeling. bioRxiv (2023). doi: 10.1101/2023.05.08.539701
75. Bhaumik S, Basu R. Cellular and molecular dynamics of th17 differentiation and its developmental plasticity in the intestinal immune response. Front Immunol (2017) 8:254. doi: 10.3389/fimmu.2017.00254
76. Abdo AIK, Tye GJ. Interleukin 23 and autoimmune diseases: current and possible future therapies. Inflammation Res (2020) 69(5):463–80. doi: 10.1007/s00011-020-01339-9
77. Feagan BG, Sandborn WJ, D'Haens G, Panes J, Kaser A, Ferrante M, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe crohn's disease: A randomised, double-blind, placebo-controlled phase 2 study. Lancet (2017) 389(10080):1699–709. doi: 10.1016/S0140-6736(17)30570-6
78. Sandborn WJ, Ferrante M, Bhandari BR, Berliba E, Feagan BG, Hibi T, et al. Efficacy and safety of mirikizumab in a randomized phase 2 study of patients with ulcerative colitis. Gastroenterology (2020) 158(3):537–49.e10. doi: 10.1053/j.gastro.2019.08.043
79. Sandborn WJ, D'Haens GR, Reinisch W, Panes J, Chan D, Gonzalez S, et al. Guselkumab for the treatment of crohn's disease: induction results from the phase 2 galaxi-1 study. Gastroenterology (2022) 162(6):1650–64.e8. doi: 10.1053/j.gastro.2022.01.047
80. Khairallah C, Chu TH, Sheridan BS. Tissue adaptations of memory and tissue-resident gamma delta T cells. Front Immunol (2018) 9:2636. doi: 10.3389/fimmu.2018.02636
81. Barros-Martins J, Schmolka N, Fontinha D, Pires de Miranda M, Simas JP, Brok I, et al. Effector gammadelta T cell differentiation relies on master but not auxiliary th cell transcription factors. J Immunol (2016) 196(9):3642–52. doi: 10.4049/jimmunol.1501921
82. Catalan-Serra I, Sandvik AK, Bruland T, Andreu-Ballester JC. Gammadelta T cells in crohn's disease: A new player in the disease pathogenesis? J Crohns Colitis (2017) 11(9):1135–45. doi: 10.1093/ecco-jcc/jjx039
83. Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, et al. Single-cell genomics unveils critical regulators of th17 cell pathogenicity. Cell (2015) 163(6):1400–12. doi: 10.1016/j.cell.2015.11.009
84. He Z, Chen L, Catalan-Dibene J, Bongers G, Faith JJ, Suebsuwong C, et al. Food colorants metabolized by commensal bacteria promote colitis in mice with dysregulated expression of interleukin-23. Cell Metab (2021) 33(7):1358–71 e5. doi: 10.1016/j.cmet.2021.04.015
85. Zhang N, Bevan MJ. Cd8(+) T cells: foot soldiers of the immune system. Immunity (2011) 35(2):161–8. doi: 10.1016/j.immuni.2011.07.010
86. Shale M, Schiering C, Powrie F. Cd4(+) T-cell subsets in intestinal inflammation. Immunol Rev (2013) 252(1):164–82. doi: 10.1111/imr.12039
87. Hor S, Pirzer H, Dumoutier L, Bauer F, Wittmann S, Sticht H, et al. The T-cell lymphokine interleukin-26 targets epithelial cells through the interleukin-20 receptor 1 and interleukin-10 receptor 2 chains. J Biol Chem (2004) 279(32):33343–51. doi: 10.1074/jbc.M405000200
88. Thomas MF, Slowikowski K, Manakongtreecheep K, Sen P, Tantivit J, Nasrallah M, et al. Altered interactions between circulating and tissue-resident cd8 T cells with the colonic mucosa define colitis associated with immune checkpoint inhibitors. BioRvix (2021). doi: 10.1101/2021.09.17.460868%JbioRxiv
89. Spencer J, Bemark M. Human intestinal B cells in inflammatory diseases. Nat Rev Gastroenterol Hepatol (2023) 20(4):254–65. doi: 10.1038/s41575-023-00755-6
90. Kamada N, Sakamoto K, Seo SU, Zeng MY, Kim YG, Cascalho M, et al. Humoral immunity in the gut selectively targets phenotypically virulent attaching-and-effacing bacteria for intraluminal elimination. Cell Host Microbe (2015) 17(5):617–27. doi: 10.1016/j.chom.2015.04.001
91. Spencer J, Sollid LM. The human intestinal B-cell response. Mucosal Immunol (2016) 9(5):1113–24. doi: 10.1038/mi.2016.59
92. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491(7422):119–24. doi: 10.1038/nature11582
93. Castro-Dopico T, Dennison TW, Ferdinand JR, Mathews RJ, Fleming A, Clift D, et al. Anti-commensal igg drives intestinal inflammation and type 17 immunity in ulcerative colitis. Immunity (2019) 50(4):1099–114.e10. doi: 10.1016/j.immuni.2019.02.006
94. Buckner CM, Moir S, Kardava L, Ho J, Santich BH, Kim LJ, et al. Cxcr4/igg-expressing plasma cells are associated with human gastrointestinal tissue inflammation. J Allergy Clin Immunol (2014) 133(6):1676–85 e5. doi: 10.1016/j.jaci.2013.10.050
95. Uzzan M, Martin JC, Mesin L, Livanos AE, Castro-Dopico T, Huang R, et al. Ulcerative colitis is characterized by a plasmablast-skewed humoral response associated with disease activity. Nat Med (2022) 28(4):766–79. doi: 10.1038/s41591-022-01680-y
96. Kokkinou E, Soini T, Pandey RV, van Acker A, Theorell J, Czarnewski P, et al. The single-cell transcriptional landscape of innate and adaptive lymphocytes in pediatric-onset colitis. Cell Rep Med (2023) 4(5):101038. doi: 10.1016/j.xcrm.2023.101038
Keywords: pre-clinical models, Inflammatory bowel disease (IBD), single cell RNA-sequencing (scRNA-seq), epithelial cell, stromal cell, immune cell
Citation: Karmele EP, Moldoveanu AL, Kaymak I, Jugder B-E, Ursin RL, Bednar KJ, Corridoni D and Ort T (2023) Single cell RNA-sequencing profiling to improve the translation between human IBD and in vivo models. Front. Immunol. 14:1291990. doi: 10.3389/fimmu.2023.1291990
Received: 10 September 2023; Accepted: 29 November 2023;
Published: 19 December 2023.
Edited by:
Silke Paust, Jackson Laboratory for Genomic Medicine, United StatesReviewed by:
Gianluca Matteoli, KU Leuven, BelgiumCopyright © 2023 Karmele, Moldoveanu, Kaymak, Jugder, Ursin, Bednar, Corridoni and Ort. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniele Corridoni, ZGFuaWVsZS5jb3JyaWRvbmlAYXN0cmF6ZW5lY2EuY29t; Tatiana Ort, VGF0aWFuYS5vcnRAYXN0cmF6ZW5lY2EuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.