 Yanling Liang1,2
Yanling Liang1,2 Qumiao Xu
Qumiao Xu Qianqian Gao
Qianqian Gao- 1Institute of Cancer Research, Shenzhen Bay Laboratory, Shenzhen, China
- 2Human Phenome Institute, Zhangjiang Fudan International Innovation Center, Fudan University, Shanghai, China
- 3BGI Research, Hangzhou, China
- 4BGI Research, Shenzhen, China
Macrophages and neutrophils are the main components of the innate immune system and play important roles in promoting angiogenesis, extracellular matrix remodeling, cancer cell proliferation, and metastasis in the tumor microenvironment (TME). They can also be harnessed to mediate cytotoxic tumor killing effects and orchestrate effective anti-tumor immune responses with proper stimulation and modification. Therefore, macrophages and neutrophils have strong potential in cancer immunotherapy. In this review, we briefly outlined the applications of macrophages or neutrophils in adoptive cell therapies, and focused on chimeric antigen receptor (CAR)-engineered macrophages (CAR-Ms) and neutrophils (CAR-Ns). We summarized the construction strategies, the preclinical and clinical studies of CAR-Ms and CAR-Ns. In the end, we briefly discussed the limitations and challenges of CAR-Ms and CAR-Ns, as well as future research directions to extend their applications in treating solid tumors.
1 Introduction
Chimeric antigen receptor (CAR)-engineered T (CAR-T) cell therapy has demonstrated remarkable success in the treatment of hematological malignancies (1). Since the launch of the first CAR-T product Kymriah in 2017 (2), additional five CAR-T products have been approved by the Food and Drug Administration for treating B-cell lymphomas (3). The overall response rates of CAR-T cell therapy in B-cell malignancies reached 30-70%, with some case over 90% (4). Great efforts are being made to increase the efficacies of CAR-T therapy for solid tumors, which remain challenging due to many factors, such as a lack of tumor-specific surface antigens, antigen escape, insufficient infiltration of CAR-T cells into the tumor bed, dysfunctional phenotypes in the immunosuppressive microenvironment, and poor persistence (5, 6). In addition, the acute inflammatory responses induced by CAR-T treatment raised safety concerns about the risk of severe side effects, such as cytokine release syndrome (CRS) and neurotoxicity, which in some cases were lethal (7). Thus, there is a great need to develop novel therapeutical tools to extend the benefit of CAR-based immunotherapies.
Innate immune cells play important roles in responding to pathological conditions and maintaining immune homeostasis. Among them, macrophages and neutrophils are plastic immune cells with diverse functional phenotypes responsive to different environmental signals (8). Macrophages respond to specific stimuli in the tumor microenvironment (TME) and reversibly transform into anti-tumor M1 (pro-inflammatory, classically activated macrophages) or tumor-promoting M2 (anti-inflammatory, alternatively activated macrophages) phenotypes (9–11). Likewise, neutrophils in the TME have the tendency to polarize to N1 or N2 phenotypes, which can exert anti-tumor or pro-tumor functions, respectively (12). Research on targeting or modulating macrophages and neutrophils to combat tumors has attracted much attention in the field of tumor immunology (13, 14). In this review, we mainly summarized the current studies on adoptive cell therapies using macrophages or neutrophils, the advantages, and designs of CAR-engineered macrophages (CAR-Ms) and neutrophils (CAR-Ns), their preclinical and clinical progress, limitations, and the emerging research directions for the broader applications of CAR-Ms and CAR-Ns in solid tumors.
2 Adoptive cell therapies using macrophages or neutrophils
2.1 Adoptive cell therapies using macrophages
Given the key roles of macrophages in tissue repair and inflammation, macrophage-based cell therapies have been applied in various diseases. Danon et al. treated human ulcers using macrophages prepared from human peripheral blood monocytes (15). The use of activated human macrophages for the treatment of anal fissures is currently in phase 3 trials (NCT00507364). Moroni F et al. conducted the first phase I dose-increasing trial of human autologous macrophage therapy (ISRCTN 10368050) on adult patients with liver cirrhosis. All participants survived and did not undergo transplantation for one year, providing a macrophage-based treatment strategy for liver cirrhosis and other fibrotic diseases (16).
Tumor-associated macrophages (TAMs) are the most abundant immune cells in the TME with heterogenous populations, which mostly adopted M2-like phenotypes with immune-suppressive properties (17). Therefore, for adoptive cell therapies to benefit from the intrinsic superior tumor-homing and infiltrating capabilities of macrophages, effective strategies are needed to activate macrophages properly and maintain their anti-tumor characteristics in the TME. In 1988, Dumont et al. induced macrophages from human peripheral blood monocytes and used immunostimulatory compounds to activate macrophages, which showed cytotoxicity to human tumor cell lines and ovarian tumors growing on nude mice (18). The first clinical trial of adoptive immunotherapy using autologous monocyte-induced macrophages was reported in 1990 (19). The macrophage therapy was well tolerated, and some patients experienced stable disease within 6 months after the treatment, but the primary tumor did not subside (19). The reasons that macrophages failed to eliminate the tumor may be the influence of various immunosuppressive factors in the TME (20). Thus, when unmodified macrophages are transported back to the tumor site, they may be transformed into M2-like phenotypes and lead to tumor progression (21). To address these issues, De Palma et al. genetically engineered monocytes to secrete inflammatory cytokine IFN-α. Due to their high migration and tumor-homing ability, the modified monocytes aggregated at the tumor sites and induced anti-tumor immune responses within the TME (22). Macrophages engineered to express cytokines or antibodies, including IL-12 (23, 24) and bispecific T-cell engagers (25), induced strong pro-inflammatory immune responses in the TME as well. Moreover, those engineered cells may reduce the immunotoxic effects of systemic administration of cytokines or antibodies (24).
With the advantages of high biocompatibility and phagocytic ability, studies have manipulated macrophages as drug carriers, which delivered targeted therapies to diseased tissues with improved drug stability (26, 27). In addition, compared to other types of macrophages, M1-like macrophages have stronger phagocytic ability to absorb sufficient drug-loaded nanoparticles (28), thereby enhancing the suppression of tumor growth (29). Through incubating the chemotherapy drug doxorubicin (DOX) into a mouse macrophage-like cell line RAW264.7 (30), Fu et al. showed that DOX-loaded macrophages displayed stronger anti-tumor effects than DOX alone, and suppressed the lung metastasis of mouse 4T1 breast cancer (30). To delay the toxicity of DOX to primary macrophages, Pang et al. loaded M1 macrophages derived from mouse bone marrow with nanoparticles encapsulating DOX. They showed that macrophages loaded with nanoparticles were able to infiltrate effectively through the endothelial barrier into the brain tumor tissue and exhibit significant anti-glioma effects (31). However, these macrophage-based immunotherapies described above lacked the specificity to tumor.
Recently, combining the unique properties of macrophages with tumor antigen-induced activation, CAR-Ms have evolved as promising adoptive cell therapies towards solid tumors (Figure 1) (Table 1). CAR-Ms maintained the pro-inflammatory phenotypes of macrophages in the TME with potent anti-tumor effects in pre-clinical studies and clinical trials (35, 37, 38, 42, 52).
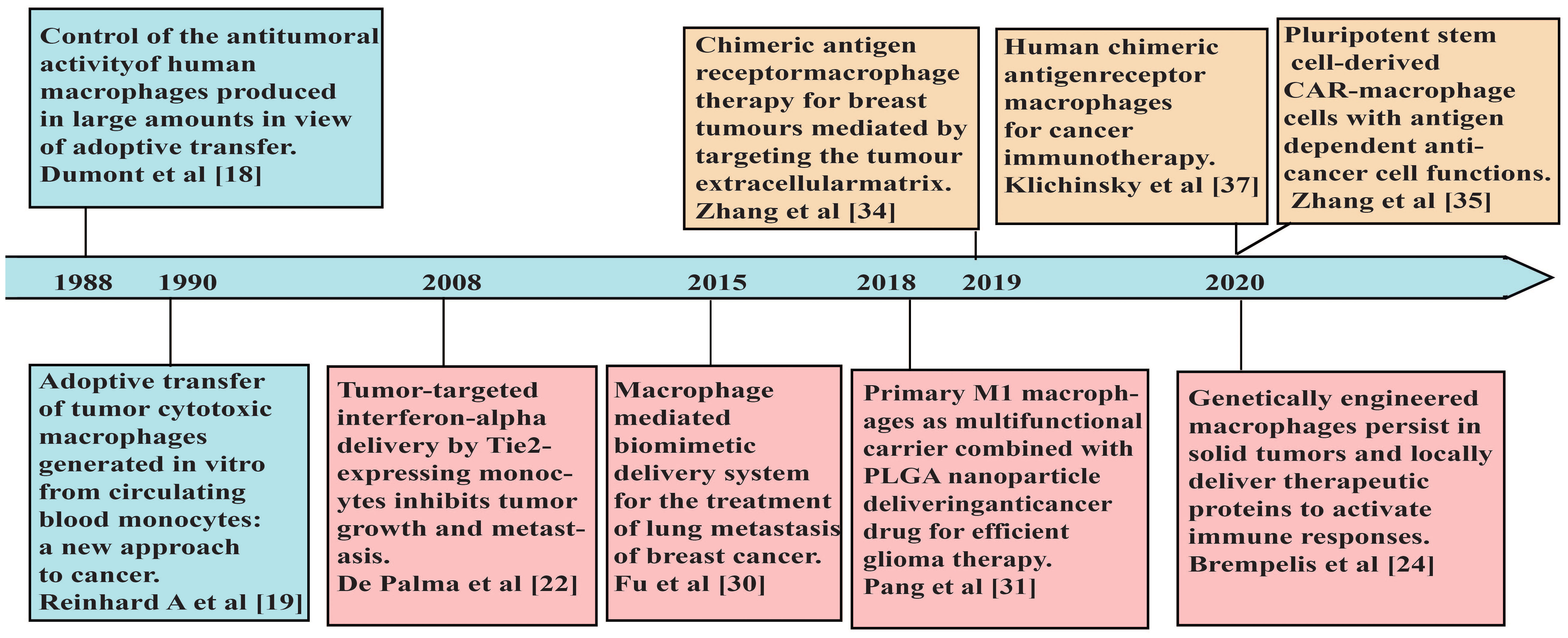
Figure 1 A brief timeline of representative studies using macrophages in adoptive cell therapies for cancers.

Table 1 Research studies on CAR-Ms.
2.2 Adoptive cell therapies using neutrophils
As the most abundant immune cells in circulation, neutrophils play central roles in the early immune responses to tissue damage or infection (53). Granulocyte infusion for the treatment of refractory neutropenia sepsis is an approved neutrophil therapy (54).
Activated neutrophils secreted various pro-inflammatory cytokines and activated T cells by antigen presentation (55). They also presented high mobility and ability to infiltrate tissues that were hard for other cells to enter, such as the blood-brain barrier (BBB) (56–58), and could be used as effective drug carriers. Xue et al. reported that neutrophils isolated from the mouse bone marrow carrying paclitaxel liposomes could penetrate the BBB and inhibit glioma recurrence in glioblastoma mouse models (59).
However, under the effects of tumor suppressive microenvironment and cytokines, neutrophils in the TME are mostly polarized to acquire N2-like phenotypes, with tumor-promoting functions through driving angiogenesis, extracellular matrix remodeling, metastasis, and immune suppression (60). Thus, cell therapy strategies using neutrophils need to consider regulating the intrinsic properties of neutrophils to effectively trigger anti-tumor responses (61). Leslie et al. suggested that infusion of immature neutrophils isolated from lipopolysaccharides (LPS)-treated mice was sufficient to reactivate the anti-tumor immunity in a hepatocellular carcinoma (HCC) mouse model (62). Akin to CAR-Ms, recent studies applied CAR engineering in neutrophils, which activated the anti-tumor functions of neutrophils, maintained the N1-like phenotypes, and drove strong proinflammatory immune responses in the TME (57, 58, 63). These studies have demonstrated promising efficacies of CAR-Ns in mouse models with glioblastoma (57, 58, 63). The exciting advance in CAR-Ns may expand the toolbox of CAR-based immunotherapies and improve the outcomes of cancer treatment.
3 Construction of CAR-Ms and CAR-Ns
3.1 CAR design
The basic principles of CAR design in the field of CAR-T cells were applicable to macrophages and neutrophils. Current studies in CAR-Ms and CAR-Ns mostly used classical CAR design consisting of an extracellular antigen recognition domain, a hinge domain, a transmembrane domain, and one or more cytoplasmic signal domains (Figure 2A). CAR-Ms or CAR-Ns were directed to well-established tumor-associated antigens, such as CD19 (64), HER2 (65), and PSMA (63) through single chain variable fragments of cognate antibodies. The intracellular activation domain from CD3ζ was also used in CAR-Ms and CAR-Ns, as the phosphorylated sites of immunoreceptor tyrosine-based activation motif (ITAM) in CD3ζ can recruit the Syk kinase which activates downstream phagocytosis and proinflammatory signaling pathways, similar to the function of Zap70 in T cells (33, 66). To further improve the phagocytosis of CAR-Ms, Liu et al. incorporated the intracellular signaling domains from FcRγ (Fc receptor γ), Megf10 (multiple EGF-like domains protein), and PI3K (phosphoinositide 3-kinases) respectively in CAR, and showed that CAR-Ms with the FcRγ intracellular domain were more powerful in phagocytizing and killing cancer cells (43). In another study, Morrissey et al. tested the signaling domains from five known mouse phagocytic receptors, including Megf10, FcRγ, Bai1 (adhesion brain-specific angiogenesis inhibitor-1), MerTK (tyrosine protein kinase Mer), and CD3ζ in CAR-Ms. Consistently, CAR-Ms with the signaling domain of FcRγ were most effective in phagocytosis, which was further promoted by adding a tandem PI3K recruitment domain (33). Similar studies in CAR-Ns need to be conducted to optimize the CAR design and enhance their functions.
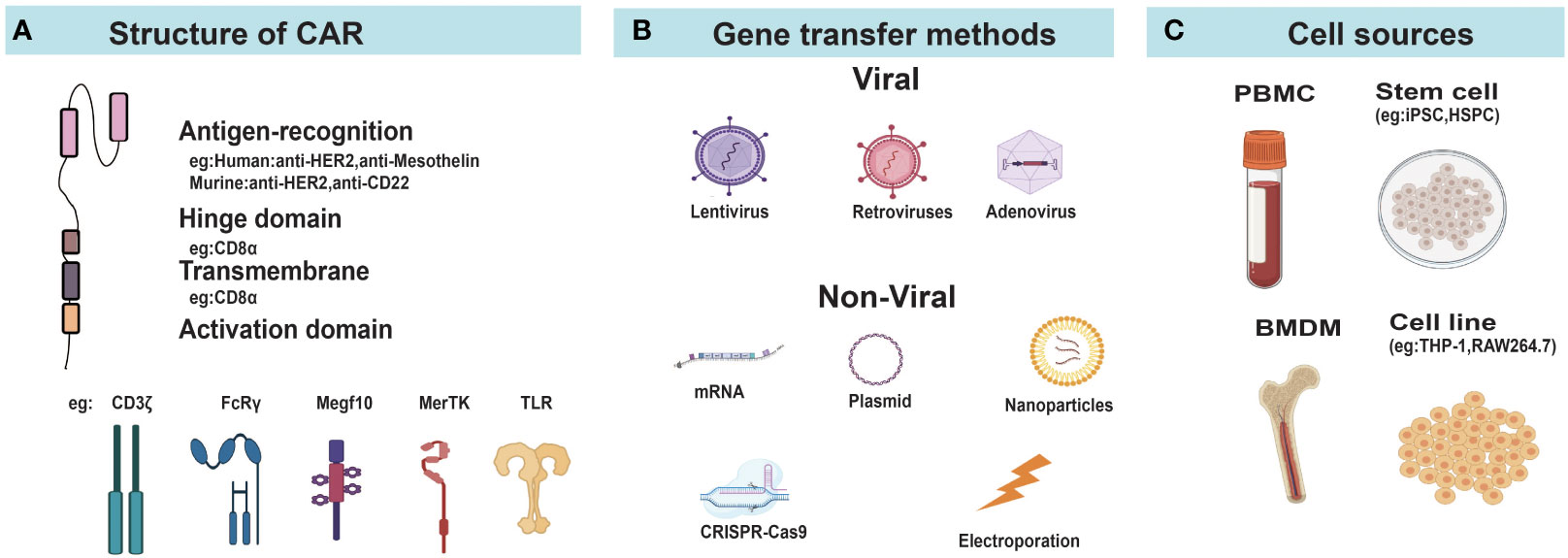
Figure 2 Construction of CAR-Ms and CAR-Ns. (A) Diagram of CAR structure. (B) Different gene transfer methods for delivering CAR genes into cells. (C) The main cellular sources for CAR-Ms and CAR-Ns. FcRγ, Fcγ receptor; MerTK, MER proto-oncogene tyrosine kinase; Megf10, Multiple EGF like domains 10; TLR, Toll-like receptors; PBMC, peripheral blood mononuclear cells; iPSC, induced pluripotent stem cells; HSPC, hematopoietic stem and progenitor cells; MSLN, mesothelin; BMDM, bone marrow derived macrophage.
3.2 Gene transfer strategies
Due to the high expression of pattern recognition receptors and non-proliferative nature of macrophages and neutrophils, they are more resistant to gene transfer compared to T cells (57, 67, 68). Different strategies based on virus, electroporation, and nanoparticles have been developed for the delivery of CAR genes to macrophages or neutrophils (Figure 2B).
3.2.1 Virus-mediated gene transfer
Lentivirus and retrovirus vectors can mediate the integration of transgenes into host genomes with high efficiencies and are commonly used for stable gene transfer to T cells. Though the primary mouse bone marrow derived macrophages (BMDMs) were infected by human immunodeficiency virus type 1 (HIV-1) lentivirus at high efficiency (64, 69), studies reported that transduction of human primary myelocytes were impeded by bone marrow specific restriction factor SAMHD1 (SAM domain and HD domain-containing protein 1) (69, 70). The virion-associated protein Vpx from HIV-2 or related simian immunodeficiency viruses was found to induce SAMHD1 degradation, which increased the infection efficacy of lentivirus to macrophages (71). Later studies used unmodified lentivirus to transduce human induced pluripotent stem cells (iPSCs) or CD34+ hematopoietic stem cells, which were then differentiated into macrophages (See section 2.3). Retroviruses (excluding the lentivirus subclass) are generally considered ineffective in transfecting non-proliferative or poorly proliferating cells (72), and the infection efficiency of wild-type retroviruses to macrophages was low (73). Introducing the nuclear localization signal sequence into the matrix protein of the C-type retrovirus spleen necrosis virus greatly increased the proportion of macrophages infected by the modified retrovirus (74). Roberts et al. reported that the retroviral vector rkat43.2, a modified variant of the retroviral transduction system rkat4 derived from moloney murine leukemia virus transduced hematopoietic stem cells to express chimeric immune receptors with over 50% efficiency. These cells were then differentiated into neutrophils with antigen-specific cytotoxicity (75). Although current studies on CAR-Ms and CAR-Ns have not utilized such modified lentivirus or retroviruses described above, they provided useful tools for future applications.
Adenovirus (AdV) belongs to the family of double-stranded DNA viruses (76). AdV vectors have the abilities to infect both dividing and nondividing cells, and are free from the risk of genomic mutations resulting from integration into the host genome (77). Gene transfer to macrophages through the common AdV serotype 5 was challenging, and the replication-incompetent chimeric AdV vector (Ad5/F35) which incorporates the knob and shaft of adenovirus serotype 35 was constructed and able to effectively infect macrophages through interacting with the cell surface protein CD46 (78, 79). In addition, Ad5/F35 activated macrophage inflammasome and provided beneficial pro-inflammatory signals, promoting the M1 polarization of CAR-Ms (37).
3.2.2 Nonviral gene transfer
With the rapid development of gene editing technologies, site-directed knock-in of CAR gene via CRISPR/Cas9 system provided a convenient strategy to perform genetic modifications (80). Zhang et al. inserted anti-GD2 CAR into the safe harbor AAVS1 gene locus of human pluripotent stem cells (hPSCs) by CRISPR/Cas9 system through electroporation followed by differentiation into macrophages with M1-like phenotypes (44). Similar strategies were applied in CAR-Ns (57, 58, 63). Chang et al. inserted the CAR construct into the AAVS1 site through CRISPR/Cas9 in human pluripotent stem cells and achieved stable and universal expression on differentiated neutrophils (57). Moreover, no insertion or deletion mutations were detected in predicted off-target sites (57). These studies suggested the applicability of CRISPR/Cas9 gene editing in generating CAR-Ms or CAR-Ns. New strategies using nanoparticles are discussed in the Section 6.
Generally, virus-mediated gene transfer strategies result in high transduction efficiency, but are limited by the sizes of exogenous genes that could be packaged and delivered (approximately 7.5 Kb, 8 Kb, and 6.5 Kb for lentivirus, retrovirus, and adenovirus, respectively). Non-viral methods can deliver larger fragments of transgenes with flexibility, and lower the manufactural time and costs (81). The properties of different gene-transfer methods are compared in Table 2.
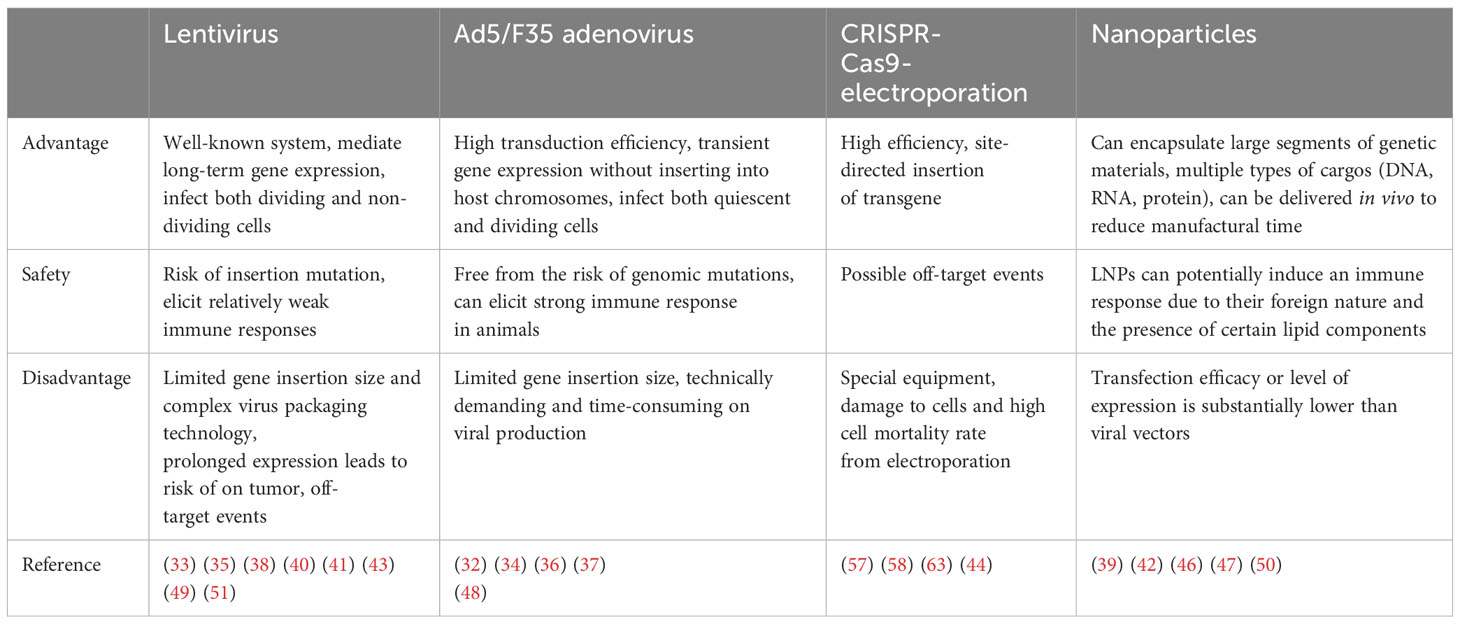
Table 2 Different gene transfer methods.
3.3 Cellular sources
Human monocytic cell line THP-1 could be induced to M1-like macrophages when cultured in the presence of phorbol myristate acetate (PMA) followed by resting and treatment by LPS and IFN-γ (82), providing a convenient resource to generate CAR-Ms for in-vitro and animal studies. Since primary macrophages in blood is scarce, peripheral blood monocytes represent the main resource for CAR-Ms used in pre-clinical and clinical studies (Figure 2C). Klichinsky et al. streamlined the process of generating CAR-Ms from peripheral blood monocytes, which differentiated CD14+ monocytes to M1-like macrophages in the presence of GM-CSF, and then transduced them by Ad5/F35 AdV vectors (37). However, it required a large quantity of monocytes for clinical treatment, raising some concerns about the number and function of circulating monocytes in cancer patients which may be affected or impaired by previous treatments, especially in advanced cancer patients (83, 84).
Stem cells provide alternative cell resources to produce CAR-Ms or CAR-Ns. They can be stably transduced to express CAR genes and then differentiated into macrophages or neutrophils. Senju et al. derived macrophages from human iPSCs in 2011 and constructed the prototype of CAR-Ms expressing only the antigen-recognition extracellular domain, lacking the intracellular signaling domains (85). Zhang et al. transduced iPSCs with lentivirus and obtained CAR-Ms with over 50-fold expansion using differentiation factors (bFGF, M-CSF, GM-CSF, etc.), which showed significant anti-cancer effects in mouse xenograft tumor models (35). Paasch et al. transduced human cord blood CD34+ hematopoietic stem and progenitor cells (HSPCs) using lentivirus to generate CAR-HSPCs, which were then differentiated into functional CAR-Ms in the presence of M-CSF/GM-CSF/IL-3 (41). Roberts et al. in 1998 first reported the generation of neutrophils with cytolysis activities from hematopoietic stem cells (75). Recently, a new chemically defined, feeder-free platform was developed, which allowed robust sequential differentiation of hematopoietic progenitor cells, myeloid progenitor cells and then CD16+ neutrophils from hPSCs using stage-specific signaling modulators in vitro (57). With the infinite self-renew capability and the potential to differentiate into diverse cell types, iPSCs represent an unlimited cell source for cell therapies (86). However, the yield and function of the final products could be largely affected by the different induction and differentiation strategies, while no widely accepted standards exist in the field of iPSCs. It is urgently needed to establish a standardized scheme to allow the production of iPSC-derived macrophages and neutrophils with high reproducibility and scalability (86).
4 Effector functions of CAR-Ms and CAR-Ns
Research studies on CAR-Ms and CAR-Ns were listed in Table 1 and Table 3. Various reports have demonstrated the multifaceted anti-tumor activities of CAR-Ms (88, 89) (Figure 3). Upon recognizing cognate antigens on tumor cells, CAR-Ms could directly clear tumors through phagocytosis. Moreover, activated CAR-Ms could secret inflammatory factors, such as IFN-γ, IL-12, and TNF-α, to promote M1 polarization and activate antigen-presenting cells (APCs) in the TME. They also upregulated the expression of major histocompatibility complex II (MHC-II), CD68, and CD80 and CD86 co-stimulatory molecules, and served as APCs to activate T cells into effector T cells to control tumor growth. Matrix proteases produced by CAR-Ms could degrade compact tumor extracellular matrix and improve the infiltration of immune cells. Furthermore, CAR-Ms could induce the epitope spreading effect and long-term immunological memory, as shown in a study that they were able to control the growth of antigen-negative tumors in the CT26 tumor model and protect the mice from tumor rechallenge (90). In addition, nitric oxide (NO) released from IFN-activated macrophages expressing inducible NO synthase (iNOS) contributed to the inflammatory death of immune-invasive tumors (91, 92).
Table 3 Research studies on CAR-Ns.
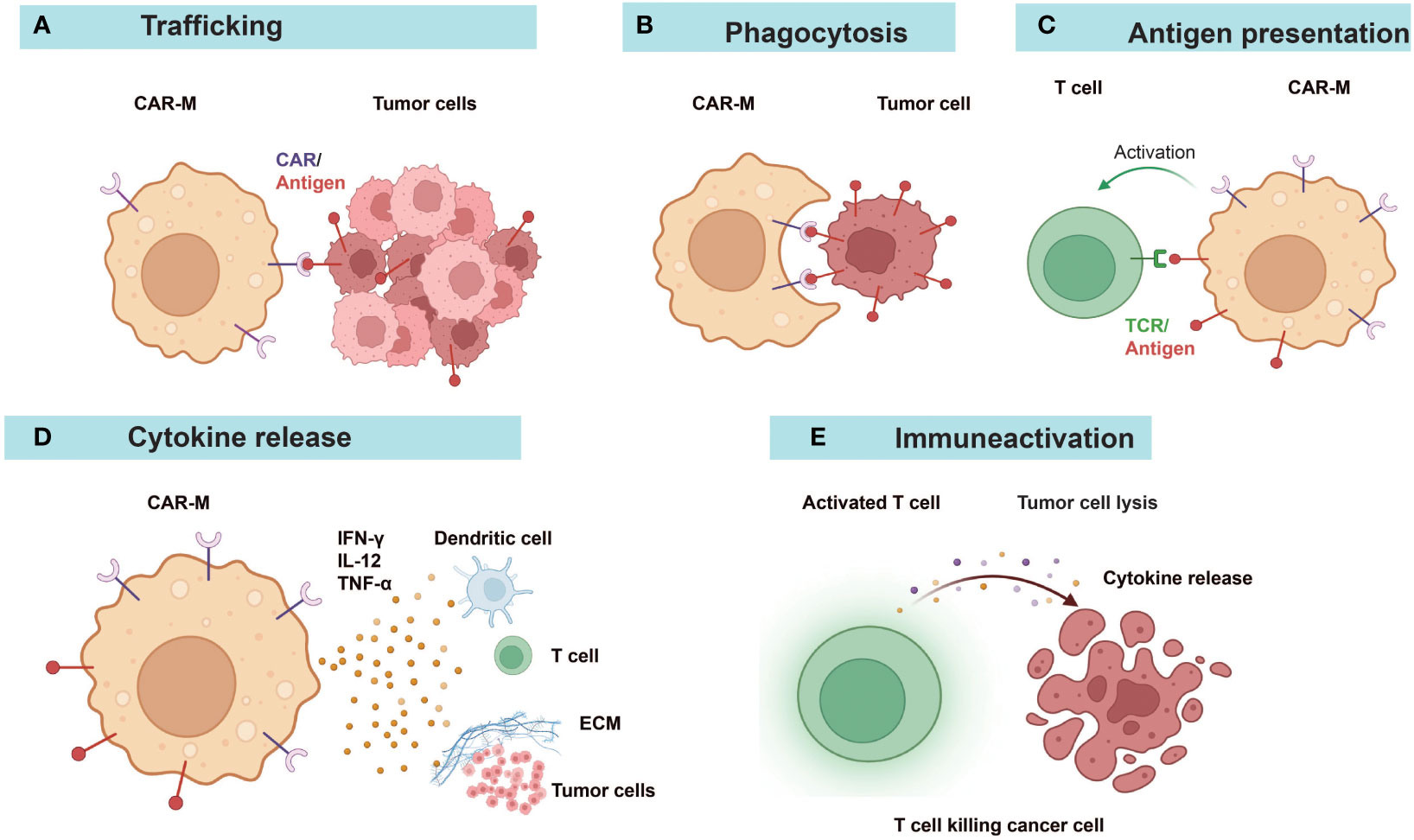
Figure 3 Diverse anti-tumor mechanisms of CAR-Ms. (A) CAR-Ms enter the TME and recognize tumor antigens. (B) Activation of CAR-Ms enhances the phagocytic activity of macrophages against tumors and (C) improves their antigen-presenting ability to T cells. (D) Secretion of pro-inflammatory cytokines alters the immune microenvironment. (E) Effector T cells activated by the functions of antigen-presentation and cytokines of CAR-Ms target and kill tumor cells. ECM, extracellular matrix.
CAR-Ns mediated tumor lysis through phagocytosis, reactive oxygen species (ROS) production, and neutrophil extracellular traps (NETs) (Figure 4). Chang et al. demonstrated that CAR constructs sustained the anti-tumor N1-like phenotype, enhanced anti-tumor and tumor-infiltrating activities of neutrophils, leading to excellent efficacy in the treatment of GBM (57). Consistent with a previous report (93), only neoplastic cells were killed while healthy cells were not attacked by activated neutrophils, indicating the good safety profile of CAR-Ns (57). Upon engagement with tumor cells, immunological synapses were formed and Syk-vav1-Erk signaling pathway was activated, which induced the phagocytic activity of neutrophils and the release of ROS (57). Through comparing CAR-Ns with wild-type neutrophils, NK cells, and CAR-engineered NK cells, the study showed that CAR-Ns significantly inhibited tumor growth and prolonged the survival of mice with orthotopic GBM xenotransplant, highlighting the potential of CAR-Ns as a novel CAR-based cell therapy (57).
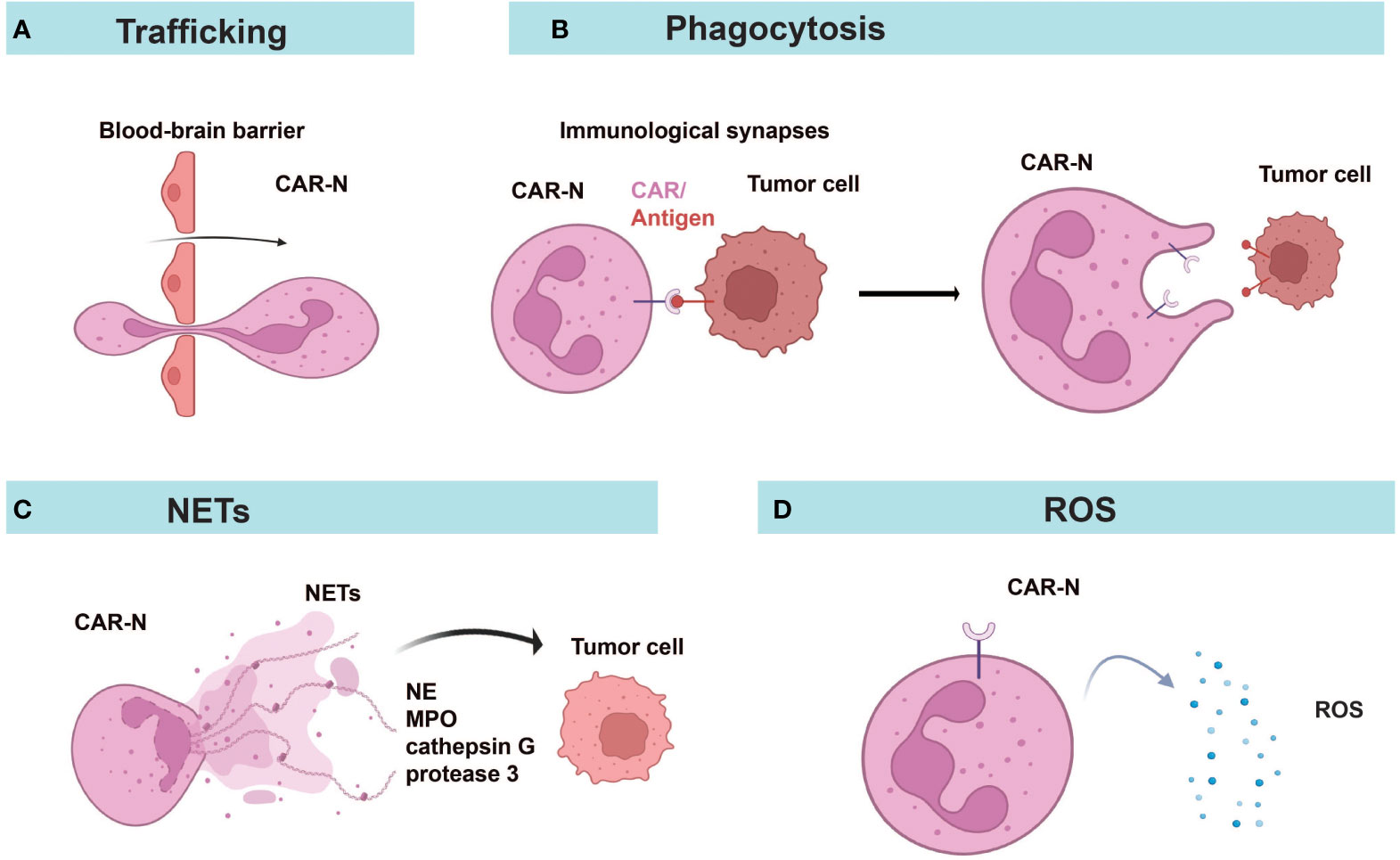
Figure 4 Diverse anti-tumor mechanisms of CAR-Ns. (A) CAR-Ns penetrate the BBB and enter the TME. (B) Immunological synapse formation and phagocytosis. (C) CAR-Ns produce NETs (neutrophil extracellular traps) and release content, such as neutrophil elastase (NE) and myeloperoxidase (MPO). (D) Secretion of reactive oxygen species (ROS) from activated CAR-Ns alters the immune microenvironment. NETs, neutrophil extracellular traps; NE, neutrophil elastase; MPO, myeloperoxidase; ROS, reactive oxygen species.
5 CAR-Ms and CAR-Ns as carriers for other therapeutic modalities
Novel strategies utilizing the tumor-homing tendency of macrophages to deliver therapeutic substances (cytokines, antibodies, chemotherapy drugs, etc.) hold the potential to further enhance the anti-tumor effects of CAR-Ms while constraining the cytotoxic effects mainly within the local tumor ecological niche and reducing the toxic effects of systemic administration (89). For example, macrophages secreting the pro-inflammatory cytokine IL-12 activated T-cell responses and prolonged survival of mice with GBM and melanoma without causing systemic toxicity (24). Macrophages releasing anti-EGFR antibodies effectively engulfed tumor cells expressing EGFR through antibody-dependent cell-mediated cytotoxicity (94). Moreover, CAR-Ms could be engineered with special control systems to release the cargo as directed. In a recent study, Liu et al. developed light-controlled CAR-Ms for drug delivery in the central nervous system (95). They showed that CAR-Ms efficiently penetrated the BBB and delivered the drug as controlled by near-infrared excitation (95).
A recent study reported that CAR-Ns carrying SiO2 nanoparticles loaded with the chemo-drug tirapazamine infiltrated to brain tumors and cleared the external tumor cells by phagocytosis (58). As neutrophils underwent apoptosis in vivo, they released the nanoparticles which resulted in effective killing of cancer cells located in the tumor core. It was estimated that 20% of nanomedicine was delivered to brain tumors through CAR-Ns, which far exceeded the conventional delivery efficiency of nanomedicine through the circulatory system (<1%) (58).
6 Clinical progress on CAR-Ms
Based on promising results in preclinical studies (37), Carisma Therapeutics initiated the first phase I multi-center clinical trial of CAR-Ms cell therapy (CT-0508) treating HER2-expressing solid tumors, including breast cancer, esophageal cancer, cholangiocarcinoma, ovarian cancer, and salivary carcinoma (NCT04660929). The updated clinical results in 2022 showed that 5 out of 9 participants achieved stable disease as the best overall response (96). CT-0508 showed no dose limiting toxicity, no severe CRS, or major organ toxicity. Myeloid Therapeutics initiated in September 2023 a phase I clinical trial of MT-302 targeting TROP2 through in vivo programming of myeloid cells using LNP encapsulating mRNA, administered to patients with advanced or metastatic epithelial tumors (NCT05969041). Another study aiming to determine the efficacy of newly developed CAR-Ms therapy using tumor organoids derived from breast cancer patients (NCT05007379) has not yet started.
7 Limitations and future directions
CAR-Ms and CAR-Ns possess unique characteristics that may help overcome some obstacles faced by CAR-T cell therapy, as they are expected to improve tumor infiltration, alleviate the immune-suppressive TME, and activate anti-tumor immune responses. Currently, they are still in the early stages as potential therapeutics with limited clinical data to evaluate their efficacies and safety profiles. Safety issues and various challenges need to be addressed to further expand their applications (Table 4). For example, activated macrophages could release large amounts of proinflammatory cytokines, such as IL-1, and IL-6, which might cause severe adverse events like CRS. Compared to the high proliferative capability of T cells, macrophages and neutrophils can hardly expand ex vivo or after infusion since they are terminally differentiated cells (97, 98). To obtain large quantities of autologous cells for treatment, especially from patients, might be a great challenge. They also have relatively short life cycles (99, 100), and probably lack persistency (101). Using iPSCs or HSPCs to generate enough functional CAR-Ms or CAR-Ns may be one of the strategies to address the problem with limited cell sources, and to achieve long-term tumor regression through repeated infusion. Besides, Dong et al. reported that M2-like peritoneal macrophages isolated from malignant ascites of gastric cancer patients could be transfected with CAR genes to prepare CAR-Ms, which exhibited M1-like polarization with anti-tumor and pro-inflammatory phenotypes, and promoted T cell proliferation (49).
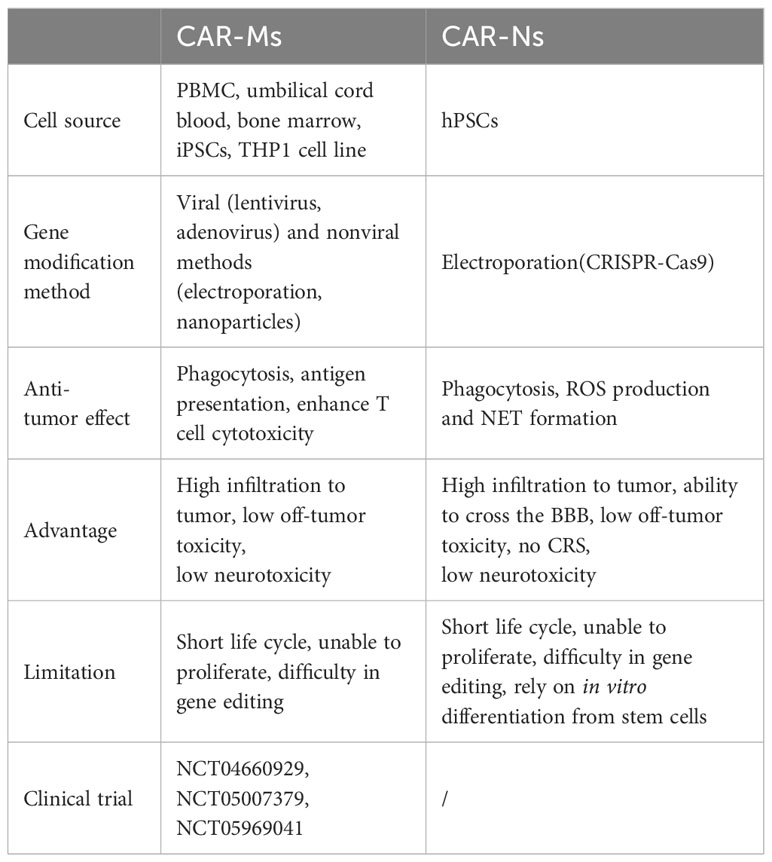
Table 4 Comparison of CAR-Ms and CAR-Ns.
7.1 In vivo production of CAR-based cells
To expedite the manufactural processes and reduce the cost, in vivo production of CAR-based therapies is under active investigations recently using nanoparticles (39, 42, 46, 50, 102). CD5-targeted lipid nanoparticles (LNPs) encapsulated with mRNAs were intravenously injected into a mouse model of heart failure, which reduced fibrosis and restored cardiac function through generating CAR-T cells targeting FAP (fibroblast activation protein) transiently in vivo. Similarly, Yang et al. delivered mRNAs encoding anti-GPC3 CAR and CD24-blocking protein in LNPs to liver macrophages, which elevated their phagocytic function and reduced tumor burden in an HCC mouse model (50). To improve the specificity of gene delivery to macrophages, Gao et al. incorporated the RP-182 peptide to the shell of nanoparticles, which activated phagocytosis and autophagy in M2-like TAMs via the mannose receptor CD206, and reprogrammed them into an antitumor M1-like phenotype (46). Plasmid DNA encoding ErbB2/Her2-specific CAR was delivered to macrophages by injecting the nanoparticles intratumorally, thereby generating CAR-Ms in situ as “living” cures which cleared the invasive tumor cells in mice with glioma (46). It is conceivable to generate CAR-Ns in vivo as studies have reported targeting neutrophils using nanoparticles to modify neutrophil function or deliver drugs (58, 103). In vivo engineering of immune cells avoided the lengthy, complex, and expensive in vitro production processes (50). Due to the stability and ease of manufacturing nanoparticles, it is possible to simplify long-term storage and reduce costs (46). Intratumoral administration of nanoparticles could directly target macrophages or neutrophils in situ and restrict the potential side effects of systemic infusion, while it is challenging in clinical applications. Experiences from intravenous injection of LNPs carrying mRNAs as vaccines might shed light on the future use of nanoparticles to generate CAR-based cells in vivo in humans (104). Due to the transient expression of exogenous genes, multiple doses are likely needed to produce potent therapeutic effects.
7.2 Functional optimization of CAR-Ms and CAR-Ns
Optimization of CAR design through assessing signaling domains and their combinations as stimulation or activation domains may further enhance the functions and reduce safety risks for CAR-Ms or CAR-Ns. For instance, various molecules are involved in functional pathways of phagocytosis, polarization, or proliferation in macrophages or neutrophils, such as the glycoprotein receptor dectin-1 (Dectin-1) (105) and MyD88 (106). The signaling domains of different receptors or signal-junction proteins are worth testing in CAR-Ms or CAR-Ns in future studies.
Various studies have implemented strategies modulating metabolic programs in T cells to enhance the functions of CAR-T cells (107). Through CRISPR screening of metabolic genes, Wang et al. found that Kelch-like ECH-associated protein 1 (KEAP1) played an important role in the pro-inflammatory activity of macrophages through inhibiting the production of itaconate. As Aconitate Decarboxylase 1 (ACOD1) is the sole enzyme to generate itaconate, ACOD1 depletion promoted pro-inflammatory activity of macrophages and enhanced the function of CAR-Ms derived from human iPSCs in solid tumors (51). Further research on the effects of metabolites is likely to discover more strategies to improve anti-tumor efficacies of CAR-Ms and CAR-Ns through metabolic reprogramming.
7.3 Combinatory therapies
Due to the limitations of single therapy in cancer treatment, combinatory therapies have become the long-term trend to increase the efficacies of CAR-based cell therapies. As noted above, CAR-Ms and CAR-Ns delivering drugs had combined anti-tumor effects of cell therapies and cytotoxicity from chemo-drugs (58, 95). Radiotherapy has been combined with CAR-T cells to avoid the tumor escape resulted from CAR-targeted antigen loss (108, 109). Pre-exposure to radiotherapy could induce immunogenic cell death and antigen release, promote HLA or CAR target expression on tumor cells, increase APC activation and immune cell infiltration, thereby reshaping the TME to favor anti-tumor immune responses (108, 110, 111). It is worth of evaluating radiotherapy combining with CAR-Ms or CAR-Ns in future studies.
Combination of different cell therapies can present synergistic anti-tumor effects. Mice treated with CAR-Ms and T cells together showed better anti-tumor responses compared to those treated with either cell therapy alone (37). This may be due to the enhanced anti-tumor activity of T cells induced by CAR-Ms. Further experiments showed that CAR-Ms cross-presented intracellular tumor-derived antigens after phagocytosis of tumor cells (112). Furthermore, another study demonstrated that CAR-Ms and CAR-T cells exhibited synergistic cytotoxicity against tumor cells in vitro (43). The inflammatory factors secreted by CAR-T cells increased the expression of co-stimulatory ligands CD86 and CD80 on CAR-Ms and enhanced the cytotoxicity of CAR-Ms by inducing M1 polarization (43).
In addition, CAR-Ms combined with antibodies, such as anti-PD-1, anti-CD47, anti-FcγRIIB and anti-Siglec-10 promoted the anti-tumor efficacies compared with CAR-Ms alone, either through enhancing the phagocytosis of macrophages or cytotoxic T cell responses (48, 113–119). Given the complexity and heterogeneity of solid tumors, it will be necessary to explore different combinatory strategies with CAR-Ms or CAR-Ns under the specific context of the TME and in clinical trials.
8 Conclusion
In conclusion, CAR-Ms and CAR-Ns have emerged as promising cell therapies with unique characteristics to combat solid cancers. Beyond cancers, they also provide novel treatment options for other diseases. For example, Li et al. utilized a surficial nanoparticle coating that locoregionally generated super bactericidal CAR-Ms to eradicate Staphylococcus aureus in mouse models, which presented a potential approach for the prevention and treatment of periprosthetic joint infection (47). Combinatory therapies with other therapeutic modalities, such as antibodies, CAR-T cells, radiotherapy, and chemotherapy are exciting directions attracting a lot of attention. Novel strategies are to be developed to further enhance the in vivo function and persistency of CAR-Ms and CAR-Ns, with effective manufactural routes to improve the yield of engineered cells and reduce the cost and time. More clinical studies are eagerly awaited to comprehensively evaluate the potency and toxicity of CAR-Ms and CAR-Ns in different diseases including solid cancers.
Author contributions
YL: Writing – original draft, Writing – review and editing. QX: Writing – original draft, Writing – review and editing. QG: Writing – original draft, Writing – review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We thank Shenzhen municipal research funding for postdoctoral fellows who stay or come to Shenzhen.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature (2023) 614(7949):635–48. doi: 10.1038/s41586-023-05707-3
2. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
3. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J (2021) 11(4):69. doi: 10.1038/s41408-021-00459-7
4. Chavez JC, Bachmeier C, Kharfan-Dabaja MA. CAR T-cell therapy for B-cell lymphomas: clinical trial results of available products. Ther Adv Hematol (2019) 10:2040620719841581. doi: 10.1177/2040620719841581
5. D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis (2018) 9(3):1–12. doi: 10.1038/s41419-018-0278-6
6. Newick K, Moon E, Albelda SM. Chimeric antigen receptor T-cell therapy for solid tumors. Mol Ther-Oncolytics (2016) 3:16006. doi: 10.1038/mto.2016.6
7. Schubert ML, Schmitt M, Wang L, Ramos CA, Jordan K, Müller-Tidow C, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol (2021) 32(1):34–48. doi: 10.1016/j.annonc.2020.10.478
8. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
9. Mishra AK, Banday S, Bharadwaj R, Ali A, Rashid R, Kulshreshtha A, et al. Macrophages as a potential immunotherapeutic target in solid cancers. Vaccines (2023) 11(1):55. doi: 10.3390/vaccines11010055
10. Bercovici N, Guérin MV, Trautmann A, Donnadieu E. The remarkable plasticity of macrophages: A chance to fight cancer. Front Immunol (2019) 10:1563. doi: 10.3389/fimmu.2019.01563
11. Christofides A, Strauss L, Yeo A, Cao C, Charest A, Boussiotis VA. The complex role of tumor-infiltrating macrophages. Nat Immunol (2022) 23(8):1148–56. doi: 10.1038/s41590-022-01267-2
12. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
13. Zhang Y, Guoqiang L, Sun M, Lu X. Targeting and exploitation of tumor-associated neutrophils to enhance immunotherapy and drug delivery for cancer treatment. Cancer Biol Med (2020) 17(1):32–43. doi: 10.20892/j.issn.2095-3941.2019.0372
14. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov (2018) 17(12):887–904. doi: 10.1038/nrd.2018.169
15. Danon D, Madjar J, Edinov E, Knyszynski A, Brill S, Diamantshtein L, et al. Treatment of human ulcers by application of macrophages prepared from a blood unit. Exp Gerontol (1997) 32(6):633–41. doi: 10.1016/S0531-5565(97)00094-6
16. Moroni F, Dwyer BJ, Graham C, Pass C, Bailey L, Ritchie L, et al. Safety profile of autologous macrophage therapy for liver cirrhosis. Nat Med (2019) 25(10):1560–5. doi: 10.1038/s41591-019-0599-8
17. Yang Q, Guo N, Zhou Y, Chen J, Wei Q, Han M. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharm Sin B (2020) 10(11):2156–70. doi: 10.1016/j.apsb.2020.04.004
18. Dumont S, Hartmann D, Poindron P, Oberling F, Faradji A, Bartholeyns J. Control of the antitumoral activity of human macrophages produced in large amounts in view of adoptive transfer. Eur J Cancer Clin Oncol (1988) 24(11):1691–8. doi: 10.1016/0277-5379(88)90069-7
19. Andreesen R, Scheibenbogen C, Brugger W, Krause S, Meerpohl H-G, Leser H-G, et al. Adoptive transfer of tumor cytotoxic macrophages generated in vitro from circulating blood monocytes: A new approach to cancer immunotherapy1. Cancer Res (1990) 50(23):7450–6.
20. Lee S, Kivimäe S, Dolor A, Szoka FC. Macrophage-based cell therapies: The long and winding road. J Control Release (2016) 240:527–40. doi: 10.1016/j.jconrel.2016.07.018
21. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. Functional differentiation. Front Immunol (2014) 5:514. doi: 10.3389/fimmu.2014.00514
22. De Palma M, Mazzieri R, Politi LS, Pucci F, Zonari E, Sitia G, et al. Tumor-targeted interferon-alpha delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell (2008) 14(4):299–311. doi: 10.1016/j.ccr.2008.09.004
23. Chinn HK, Gardell JL, Matsumoto LR, Labadie KP, Mihailovic TN, Lieberman NAP, et al. Hypoxia-inducible lentiviral gene expression in engineered human macrophages. J Immunother Cancer (2022) 10(6):e003770. doi: 10.1136/jitc-2021-003770
24. Brempelis KJ, Cowan CM, Kreuser SA, Labadie KP, Prieskorn BM, Lieberman NAP, et al. Genetically engineered macrophages persist in solid tumors and locally deliver therapeutic proteins to activate immune responses. J Immunother Cancer (2020) 8(2):e001356. doi: 10.1136/jitc-2020-001356
25. Gardell JL, Matsumoto LR, Chinn H, DeGolier KR, Kreuser SA, Prieskorn B, et al. Human macrophages engineered to secrete a bispecific T cell engager support antigen-dependent T cell responses to glioblastoma. J Immunother Cancer (2020) 8(2):e001202. doi: 10.1136/jitc-2020-001202
26. Anderson JM, McNally AK. Biocompatibility of implants: lymphocyte/macrophage interactions. Semin Immunopathol (2011) 33(3):221–33. doi: 10.1007/s00281-011-0244-1
27. Liang T, Zhang R, Liu X, Ding Q, Wu S, Li C, et al. Recent advances in macrophage-mediated drug delivery systems. Int J Nanomed (2021) 16:2703–14. doi: 10.2147/IJN.S298159
28. Papa S, Ferrari R, Paola De M, Rossi F, Mariani A, Caron I, et al. Polymeric nanoparticle system to target activated microglia/macrophages in spinal cord injury. J Control Release (2014) 174:15–26. doi: 10.1016/j.jconrel.2013.11.001
29. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
30. Fu J, Wang D, Mei D, Zhang H, Wang Z, He B, et al. Macrophage mediated biomimetic delivery system for the treatment of lung metastasis of breast cancer. J Control Release (2015) 204:11–9. doi: 10.1016/j.jconrel.2015.01.039
31. Pang L, Zhu Y, Qin J, Zhao W, Wang J. Primary M1 macrophages as multifunctional carrier combined with PLGA nanoparticle delivering anticancer drug for efficient glioma therapy. Drug Deliv (2018) 25(1):1922–31. doi: 10.1080/10717544.2018.1502839
32. Biglari A, Southgate TD, Fairbairn LJ, Gilham DE. Human monocytes expressing a CEA-specific chimeric CD64 receptor specifically target CEA-expressing tumour cells in vitro and in vivo. Gene Ther (2006) 13(7):602–10. doi: 10.1038/sj.gt.3302706
33. Morrissey MA, Williamson AP, Steinbach AM, Roberts EW, Kern N, Headley MB, et al. Chimeric antigen receptors that trigger phagocytosis. Elife (2018) 7:e36688. doi: 10.7554/eLife.36688
34. Zhang W, Liu L, Su H, Liu Q, Shen J, Dai H, et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br J Cancer (2019) 121(10):837–45. doi: 10.1038/s41416-019-0578-3
35. Zhang L, Tian L, Dai X, Yu H, Wang J, Lei A, et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J Hematol Oncol (2020) 13(1):153. doi: 10.1186/s13045-020-00983-2
36. Pierini S, Gabbasov R, Gabitova L, Ohtani Y, Klichinsky M. 132 CAR macrophages (CAR-M) elicit a systemic anti-tumor immune response and synergize with PD1 blockade in immunocompetent mouse models of HER2+ solid tumors. J Immunother Cancer (2020) 8(Suppl 3):A80–1. doi: 10.1136/jitc-2020-SITC2020.0132
37. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol (2020) 38(8):947–53. doi: 10.1038/s41587-020-0462-y
38. Niu Z, Chen G, Chang W, Sun P, Luo Z, Zhang H, et al. Chimeric antigen receptor-modified macrophages trigger systemic anti-tumour immunity. J Pathol (2021) 253(3):247–57. doi: 10.1002/path.5585
39. Kang M, Lee SH, Kwon M, Byun J, Kim D, Kim C, et al. Nanocomplex-mediated in vivo programming to chimeric antigen receptor-M1 macrophages for cancer therapy. Adv Mater (2021) 33(43):e2103258. doi: 10.1002/adma.202103258
40. Fu W, Lei C, Ma Z, Qian K, Li T, Zhao J, et al. CAR macrophages for SARS-coV-2 immunotherapy. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.669103
41. Paasch D, Meyer J, Stamopoulou A, Lenz D, Kuehle J, Kloos D, et al. Ex vivo generation of CAR macrophages from hematopoietic stem and progenitor cells for use in cancer therapy. Cells (2022) 11(6):994. doi: 10.3390/cells11060994
42. Chen C, Jing W, Chen Y, Wang G, Abdalla M, Gao L, et al. Intracavity generation of glioma stem cell-specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci Trans Med (2022) 14(656):eabn1128. doi: 10.1126/scitranslmed.abn1128
43. Liu M, Liu J, Liang Z, Dai K, Gan J, Wang Q, et al. CAR-macrophages and CAR-T cells synergistically kill tumor cells in vitro. Cells (2022) 11(22):3692. doi: 10.3390/cells11223692
44. Zhang J, Webster S, Duffin B, Bernstein MN, Steill J, Swanson S, et al. Generation of anti-GD2 CAR macrophages from human pluripotent stem cells for cancer immunotherapies. Stem Cell Rep (2023) 18(2):585–96. doi: 10.1016/j.stemcr.2022.12.012
45. Huo Y, Zhang H, Sa L, Zheng W, He Y, Lyu H, et al. M1 polarization enhances the antitumor activity of chimeric antigen receptor macrophages in solid tumors. J Trans Med (2023) 21(1):225. doi: 10.1186/s12967-023-04061-2
46. Gao L, Shi C, Yang Z, Jing W, Han M, Zhang J, et al. Convection-enhanced delivery of nanoencapsulated gene locoregionally yielding ErbB2/Her2-specific CAR-macrophages for brainstem glioma immunotherapy. J Nanobiotechnol (2023) 21(1):56. doi: 10.1186/s12951-023-01810-9
47. Li Z, Zhang S, Fu Z, Liu Y, Man Z, Shi C, et al. Surficial nano-deposition locoregionally yielding bactericidal super CAR-macrophages expedites periprosthetic osseointegration. Sci Adv (2023) 9(22):eadg3365. doi: 10.1126/sciadv.adg3365
48. Chen Y, Zhu X, Liu H, Wang C, Chen Y, Wang H, et al. The application of HER2 and CD47 CAR-macrophage in ovarian cancer. J Trans Med (2023) 21(1):654. doi: 10.1186/s12967-023-04479-8
49. Dong X, Fan J, Xie W, Wu X, Wei J, He Z, et al. Efficacy evaluation of chimeric antigen receptor-modified human peritoneal macrophages in the treatment of gastric cancer. Br J Cancer (2023) 129(3):551–62. doi: 10.1038/s41416-023-02319-6
50. Yang Z, Liu Y, Zhao K, Jing W, Gao L, Dong X, et al. Dual mRNA co-delivery for in situ generation of phagocytosis-enhanced CAR macrophages augments hepatocellular carcinoma immunotherapy. J Control Release (2023) 360:718–33. doi: 10.1016/j.jconrel.2023.07.021
51. Wang X, Su S, Zhu Y, Cheng X, Cheng C, Chen L, et al. Metabolic Reprogramming via ACOD1 depletion enhances function of human induced pluripotent stem cell-derived CAR-macrophages in solid tumors. Nat Commun (2023) 14(1):5778. doi: 10.1038/s41467-023-41470-9
52. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov (2022) 21(11):799–820. doi: 10.1038/s41573-022-00520-5
53. Rosales C. Neutrophil: A cell with many roles in inflammation or several cell types? Front Physiol (2018) 9:113. doi: 10.3389/fphys.2018.00113
54. Gea-Banacloche J. Granulocyte transfusions: A concise review for practitioners. Cytotherapy (2017) 19(11):1256–69. doi: 10.1016/j.jcyt.2017.08.012
55. Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatol (Oxford) (2010) 49(9):1618–31. doi: 10.1093/rheumatology/keq045
56. Németh T, Sperandio M, Mócsai A. Neutrophils as emerging therapeutic targets. Nat Rev Drug Discov (2020) 19(4):253–75. doi: 10.1038/s41573-019-0054-z
57. Chang Y, Syahirah R, Wang X, Jin G, Torregrosa-Allen S, Elzey BD, et al. Engineering chimeric antigen receptor neutrophils from human pluripotent stem cells for targeted cancer immunotherapy. Cell Rep (2022) 40(3):111128. doi: 10.1016/j.celrep.2022.111128
58. Chang Y, Cai X, Syahirah R, Yao Y, Xu Y, Jin G, et al. CAR-neutrophil mediated delivery of tumor-microenvironment responsive nanodrugs for glioblastoma chemo-immunotherapy. Nat Commun (2023) 14(1):2266. doi: 10.1038/s41467-023-37872-4
59. Xue J, Zhao Z, Zhang L, Xue L, Shen S, Wen Y, et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative Malignant glioma recurrence. Nat Nanotechnol (2017) 12(7):692–700. doi: 10.1038/nnano.2017.54
60. Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment. Blood (2019) 133(20):2159–67. doi: 10.1182/blood-2018-11-844548
61. Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut (2018) 67(6):1112–23. doi: 10.1136/gutjnl-2017-313738
62. Leslie J, Mackey JBG, Jamieson T, Ramon-Gil E, Drake TM, Fercoq F, et al. CXCR2 inhibition enables NASH-HCC immunotherapy. Gut (2022) 71(10):2093–106. doi: 10.1136/gutjnl-2021-326259
63. Harris JD, Chang Y, Syahirah R, Lian XL, Deng Q, Bao X. Engineered anti-prostate cancer CAR-neutrophils from human pluripotent stem cells. J Immunol Regener Med (2023) 20:100074. doi: 10.1016/j.regen.2023.100074
64. Mackensen A, Müller F, Mougiakakos D, Böltz S, Wilhelm A, Aigner M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med (2022) 28(10):2124–32. doi: 10.1038/s41591-022-02017-5
65. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol (2017) 3(8):1094–101. doi: 10.1001/jamaoncol.2017.0184
66. Bu JY, Shaw AS, Chan AC. Analysis of the interaction of ZAP-70 and syk protein-tyrosine kinases with the T-cell antigen receptor by plasmon resonance. Proc Natl Acad Sci USA (1995) 92(11):5106–10. doi: 10.1073/pnas.92.11.5106
67. Zhang X, Mosser DM. Macrophage activation by endogenous danger signals. J Pathol (2008) 214(2):161–78. doi: 10.1002/path.2284
68. Keller AA, Maeß MB, Schnoor M, Scheiding B, Lorkowski S. Transfecting macrophages. Methods Mol Biol (2018) 1784:187–95. doi: 10.1007/978-1-4939-7837-3_18
69. Laguette N, Benkirane M. How SAMHD1 changes our view of viral restriction. Trends Immunol (2012) 33(1):26–33. doi: 10.1016/j.it.2011.11.002
70. Ramezani A, Hawley RG. Overview of the HIV-1 lentiviral vector system. Curr Protoc Mol Biol (2002) 60(1):16.21.1–16.21.15. doi: 10.1002/0471142727.mb1621s60
71. Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature (2011) 474(7353):658–61. doi: 10.1038/nature10195
72. Burke B, Sumner S, Maitland N, Lewis CE. Macrophages in gene therapy: cellular delivery vehicles and in vivo targets. J Leukocyte Biol (2002) 72(3):417–28. doi: 10.1189/jlb.72.3.417
73. Jarrosson-Wuilleme L, Goujon C, Bernaud J, Rigal D, Darlix JL, Cimarelli A. Transduction of nondividing human macrophages with gammaretrovirus-derived vectors. J Virol (2006) 80(3):1152–9. doi: 10.1128/JVI.80.3.1152-1159.2006
74. Parveen Z, Krupetsky A, Engelstädter M, Cichutek K, Pomerantz RJ, Dornburg R. Spleen necrosis virus-derived C-type retroviral vectors for gene transfer to quiescent cells. Nat Biotechnol (2000) 18(6):623–9. doi: 10.1038/76458
75. Roberts MR, Cooke KS, Tran AC, Smith KA, Lin WY, Wang M, et al. Antigen-specific cytolysis by neutrophils and NK cells expressing chimeric immune receptors bearing zeta or gamma signaling domains. J Immunol (1998) 161(1):375–84. doi: 10.4049/jimmunol.161.1.375
76. Rowe WP, Huebner RJ, Gilmore LK, Parrott RH, Ward TG. Isolation of a cytopathogenic agent from human adenoids undergoing spontaneous degeneration in tissue culture. Proc Soc Exp Biol Med (1953) 84(3):570–3. doi: 10.3181/00379727-84-20714
77. Sayedahmed EE, Kumari R, Mittal SK. Current use of adenovirus vectors and their production methods. Methods Mol Biol (2019) 1937:155–75. doi: 10.1007/978-1-4939-9065-8_9
78. Yang M, Yang CS, Guo W, Tang J, Huang Q, Feng S, et al. A novel fiber chimeric conditionally replicative adenovirus-Ad5/F35 for tumor therapy. Cancer Biol Ther (2017) 18(11):833–40. doi: 10.1080/15384047.2017.1395115
79. Gabitova L, Menchel B, Gabbasov R, Pierini S, Best A, Ross K, et al. Abstract 1530: Anti-HER2 CAR monocytes demonstrate targeted anti-tumor activity and enable a single day cell manufacturing process. Cancer Res (2021) 81(13_Supplement):1530–0. doi: 10.1158/1538-7445.AM2021-1530
80. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer (2022) 21(1):78. doi: 10.1186/s12943-022-01559-z
81. Fus-Kujawa A, Prus P, Bajdak-Rusinek K, Teper P, Gawron K, Kowalczuk A, et al. An overview of methods and tools for transfection of eukaryotic cells in vitro. Front Bioeng Biotechnol (2021) 9:701031. doi: 10.3389/fbioe.2021.701031
82. Shiratori H, Feinweber C, Luckhardt S, Linke B, Resch E, Geisslinger G, et al. THP-1 and human peripheral blood mononuclear cell-derived macrophages differ in their capacity to polarize in vitro. Mol Immunol (2017) 88:58–68. doi: 10.1016/j.molimm.2017.05.027
83. Lower EE, Baughman RP. The effect of cancer and chemotherapy on monocyte function. J Clin Lab Immunol (1990) 31(3):121–5.
84. Kiss M, Caro AA, Raes G, Laoui D. Systemic reprogramming of monocytes in cancer. Front Oncol (2020) 10. doi: 10.3389/fonc.2020.01399
85. Senju S, Haruta M, Matsumura K, Matsunaga Y, Fukushima S, Ikeda T, et al. Generation of dendritic cells and macrophages from human induced pluripotent stem cells aiming at cell therapy. Gene Ther (2011) 18(9):874–83. doi: 10.1038/gt.2011.22
86. Lyadova I, Gerasimova T, Nenasheva T. Macrophages derived from human induced pluripotent stem cells: the diversity of protocols, future prospects, and outstanding questions. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.640703
87. Wang D, Starr R, Chang WC, Aguilar B, Alizadeh D, Wright SL, et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci Transl Med (2020) 12(533):eaaw2672. doi: 10.1126/scitranslmed.aaw2672
88. Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-based approaches for cancer immunotherapy. Cancer Res (2021) 81(5):1201–8. doi: 10.1158/0008-5472.CAN-20-2990
89. Sloas C, Gill S, Klichinsky M. Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.783305
90. Pierini S, Gabbasov R, Gabitova L, Ohtani Y, Shestova O, Gill S, et al. Chimeric antigen receptor macrophages (CAR-M) induce anti-tumor immunity and synergize with T cell checkpoint inhibitors in pre-clinical solid tumor models. Cancer Res (2021) 81(13_Supplement):63. doi: 10.1158/1538-7445.AM2021-63
91. Kashfi K, Kannikal J, Nath N. Macrophage reprogramming and cancer therapeutics: role of iNOS-derived NO. Cells (2021) 10(11):3194. doi: 10.3390/cells10113194
92. Kruse B, Buzzai AC, Shridhar N, Braun AD, Gellert S, Knauth K, et al. CD4(+) T cell-induced inflammatory cell death controls immune-evasive tumours. Nature (2023) 618(7967):1033–40. doi: 10.1038/s41586-023-06199-x
93. Yan J, Kloecker G, Fleming C, Bousamra M 2nd, Hansen R, Hu X, et al. Human polymorphonuclear neutrophils specifically recognize and kill cancerous cells. Oncoimmunology (2014) 3(7):e950163. doi: 10.4161/15384101.2014.950163
94. Cha EB, Shin KK, Seo J, Oh DB. Antibody-secreting macrophages generated using CpG-free plasmid eliminate tumor cells through antibody-dependent cellular phagocytosis. BMB Rep (2020) 53(8):442–7. doi: 10.5483/BMBRep.2020.53.8.024
95. Liu Y, Hu P, Zheng Z, Zhong D, Xie W, Tang Z, et al. Photoresponsive vaccine-like CAR-M system with high-efficiency central immune regulation for inflammation-related depression. Adv Mater (2022) 34(11):e2108525. doi: 10.1002/adma.202108525
96. Reiss K, Ueno N, Yuan Y, Johnson M, Dees EC, Chao J, et al. 633 A phase 1, first in human (FIH) study of autologous macrophages containing an anti-HER2 chimeric antigen receptor (CAR) in participants with HER2 overexpressing solid tumors. J Immunother Cancer (2022) 10(Suppl 2):A664–4. doi: 10.1136/jitc-2022-SITC2022.0633
97. Maalej KM, Merhi M, Inchakalody VP, Mestiri S, Alam M, Maccalli C, et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol Cancer (2023) 22(1):20. doi: 10.1186/s12943-023-01723-z
98. Kim J, Bae J-S. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflamm (2016) 2016:6058147. doi: 10.1155/2016/6058147
99. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med (2017) 214(7):1913–23. doi: 10.1084/jem.20170355
100. Lahoz-Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, et al. Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood (2016) 127(26):3431–8. doi: 10.1182/blood-2016-03-700336
101. Patel AA, Ginhoux F, Yona S. Monocytes, macrophages, dendritic cells and neutrophils: an update on lifespan kinetics in health and disease. Immunology (2021) 163(3):250–61. doi: 10.1111/imm.13320
102. Sago CD, Krupczak BR, Lokugamage MP, Gan Z, Dahlman JE. Cell subtypes within the liver microenvironment differentially interact with lipid nanoparticles. Cell Mol Bioeng (2019) 12(5):389–97. doi: 10.1007/s12195-019-00573-4
103. Völs S, Kaisar-Iluz N, Shaul ME, Ryvkin A, Ashkenazy H, Yehuda A, et al. Targeted nanoparticles modify neutrophil function in vivo. Front Immunol (2022) 13:1003871. doi: 10.3389/fimmu.2022.1003871
104. Swetha K, Kotla NG, Tunki L, Jayaraj A, Bhargava SK, Hu H, et al. Recent advances in the lipid nanoparticle-mediated delivery of mRNA vaccines. Vaccines (Basel) (2023) 11(3):658. doi: 10.3390/vaccines11030658
105. Daley D, Mani VR, Mohan N, Akkad N, Ochi A, Heindel DW, et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med (2017) 23(5):556–67. doi: 10.1038/nm.4314
106. Chen L, Zheng L, Chen P, Liang G. Myeloid differentiation primary response protein 88 (MyD88): the central hub of TLR/IL-1R signaling. J Med Chem (2020) 63(22):13316–29. doi: 10.1021/acs.jmedchem.0c00884
107. Peng JJ, Wang L, Li Z, Ku CL, Ho PC. Metabolic challenges and interventions in CAR T cell therapy. Sci Immunol (2023) 8(82):eabq3016. doi: 10.1126/sciimmunol.abq3016
108. DeSelm C, Palomba ML, Yahalom J, Hamieh M, Eyquem J, Rajasekhar VK, et al. Low-dose radiation conditioning enables CAR T cells to mitigate antigen escape. Mol Ther (2018) 26(11):2542–52. doi: 10.1016/j.ymthe.2018.09.008
109. Sugita M, Yamazaki T, Alhomoud M, Martinet J, Latouche J-B, Golden E, et al. Radiation therapy improves CAR T cell activity in acute lymphoblastic leukemia. Cell Death Dis (2023) 14(5):305. doi: 10.1038/s41419-023-05829-6
110. Liu S, Wang W, Hu S, Jia B, Tuo B, Sun H, et al. Radiotherapy remodels the tumor microenvironment for enhancing immunotherapeutic sensitivity. Cell Death Dis (2023) 14(10):679. doi: 10.1038/s41419-023-06211-2
111. Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res (2018) 78(4):1031–43. doi: 10.1158/0008-5472.CAN-17-1788
112. Villanueva MT. Macrophages get a CAR. Nat Rev Immunol (2020) 20(5):273–3. doi: 10.1038/s41577-020-0302-9
113. van der Bij GJ, GJ, Bögels M, Otten MA, Oosterling SJ, Kuppen PJ, Meijer S, et al, et al. Experimentally induced liver metastases from colorectal cancer can be prevented by mononuclear phagocyte-mediated monoclonal antibody therapy. J Hepatol (2010) 53(4):677–85. doi: 10.1016/j.jhep.2010.04.023
114. Pierini S, Gabbasov R, Gabitova L, Ohtani Y, Shestova O, Gill S, et al. Abstract 63: Chimeric antigen receptor macrophages (CAR-M) induce anti-tumor immunity and synergize with T cell checkpoint inhibitors in pre-clinical solid tumor models. Cancer Res (2021) 81(13_Supplement):63–3. doi: 10.1158/1538-7445.AM2021-63
115. Upton R, Banuelos A, Feng D, Biswas T, Kao K, McKenna K, et al. Combining CD47 blockade with trastuzumab eliminates HER2-positive breast cancer cells and overcomes trastuzumab tolerance. Proc Natl Acad Sci USA (2021) 118(29):e2026849118. doi: 10.1073/pnas.2026849118
116. Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci USA (2013) 110(27):11103–8. doi: 10.1073/pnas.1305569110
117. Roghanian A, Teige I, Kerry Mårtensson L, Cox L, Kovacek M, Ljungars A, et al. Antagonistic human fcRIIB (CD32B) antibodies have anti-tumor activity and overcome resistance to antibody therapy inVivo. Cancer Cell (2015) 27(4):473–88. doi: 10.1016/j.ccell.2015.03.005
118. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature (2019) 572(7769):392–6. doi: 10.1038/s41586-019-1456-0
Keywords: CAR macrophages, CAR neutrophils, solid tumor, cancer immunotherapy, adoptive cell therapy
Citation: Liang Y, Xu Q and Gao Q (2023) Advancing CAR-based immunotherapies in solid tumors: CAR- macrophages and neutrophils. Front. Immunol. 14:1291619. doi: 10.3389/fimmu.2023.1291619
Received: 09 September 2023; Accepted: 13 November 2023;
Published: 28 November 2023.
Edited by:
Axel Schambach, Hannover Medical School, GermanyReviewed by:
Igor Slukvin, University of Wisconsin-Madison, United StatesXiaoping Bao, Purdue University, United States
Rio Sugimura, The University of Hong Kong, Hong Kong SAR, China
Copyright © 2023 Liang, Xu and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qianqian Gao, Z2FvcXFAc3pibC5hYy5jbg==; Qumiao Xu, cXVtaWFveHVAaG90bWFpbC5jb20=