
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 11 January 2024
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1287857
This article is part of the Research TopicCrosstalk between Peripheral and Local Immune Response in the Pathophysiology of Stroke and Neurodegeneration Diseases, Volume IIView all 10 articles
 Zhaoyuan Gong†
Zhaoyuan Gong† Jing Guo†
Jing Guo† Bin Liu
Bin Liu Yang Guo
Yang Guo Cuicui Cheng
Cuicui Cheng Yin Jiang
Yin Jiang Ning Liang
Ning Liang Mingzhi Hu
Mingzhi Hu Tian Song
Tian Song Lu Yang
Lu Yang Huizhen Li
Huizhen Li Haili Zhang
Haili Zhang Xingyu Zong
Xingyu Zong Qianzi Che*
Qianzi Che* Nannan Shi*
Nannan Shi*Ischemic stroke (IS), which is the third foremost cause of disability and death worldwide, has inflammation and cell death as its main pathological features. IS can lead to neuronal cell death and release factors such as damage-related molecular patterns, stimulating the immune system to release inflammatory mediators, thereby resulting in inflammation and exacerbating brain damage. Currently, there are a limited number of treatment methods for IS, which is a fact necessitating the discovery of new treatment targets. For this review, current research on inflammation and cell death in ischemic stroke was summarized. The complex roles and pathways of the principal immune cells (microglia, astrocyte, neutrophils, T lymphocytes, and monocytes/macrophage) in the immune system after IS in inflammation are discussed. The mechanisms of immune cell interactions and the cytokines involved in these interactions are summarized. Moreover, the cell death mechanisms (pyroptosis, apoptosis, necroptosis, PANoptosis, and ferroptosis) and pathways after IS are explored. Finally, a summary is provided of the mechanism of action of natural pharmacological active ingredients in the treatment of IS. Despite significant recent progress in research on IS, there remain many challenges that need to be overcome.
Stroke is one of the major causes of death and disability worldwide (1). In the past three decades, the global incidence of stroke has increased by 70%, stroke mortality has increased by 43%, disability adjusted life spans lost due to stroke increased by 32%, and the economic burden of all countries has increased. Ischemic stroke (IS) is a severe insufficiency of the blood supply to the brain caused by thrombosis or embolism in the blood supply to the cerebral vessels in the functional area of the brain, resulting in an insufficient oxygen supply to the brain that leads to neuronal death and brain function defects (2). Ischemia induces cell death and neuroinflammation by promoting the production of pro-inflammatory mediators. At present, the number of drugs that can be used to treat IS [such as tissue plasminogen activator (tPA)] is limited, the clinical effect is poor, and the adverse reactions are substantial (3). Therefore, there is an urgent need for further research on IS to find more effective and safe therapeutic agents to prevent or treat IS.
Systemic inflammation, immune responses, and cell death play a key role in the occurrence and development of stroke. Post-ischemic inflammation of the injured brain is characterized by the infiltration of blood immune cells, as well as the interactions between resident microglia and invading blood immune cells (4). After IS, microglia (as resident brain macrophages) and astrocytes are activated in the innate immune system, releasing numerous inflammatory factors (5). Inflammatory factors attract peripheral immune cells to infiltrate the lesion area (6). The mechanisms of action of immune cells are complex, and these cells also interfere with each other, thus forming a complex inflammatory network. This further aggravates systemic inflammation, increases neuronal death and infarct volume, and leads to poor neurological outcomes (7). During ischemia, the blood supply to brain tissue is disrupted, which subsequently promotes a series of pathophysiological reactions leading to different types of cell death, including pyroptosis, apoptosis, necroptosis, ferroptosis, and PANoptosis, the lattermost of which is the crosstalk between pyroptosis, apoptosis, and necroptosis (8). These types of cell death all play roles in the pathogenesis of IS and induce inflammation (9).
This review summarizes—in relation to the immune system, inflammation, and cell death—the mechanisms and pathways involved in IS. In the section on the immune system and inflammation, the pathways and functions of immune cells, including microglia, astrocytes, neutrophils, T lymphocytes, and monocytes/macrophages, in the post-IS inflammatory response are reviewed. In the section on cell death, we review the pathways that mediate pyroptosis, apoptosis, necroptosis, ferroptosis, and PANoptosis. The mechanisms of natural compounds, including salidroside, baicalin, astragaloside IV, and curcumin, in the treatment of IS are also reviewed. Finally, there is a discussion of the potential future directions in this field.
The expression of pro-inflammatory factors marks the beginning of the development of cerebral ischemic inflammation and involves various cell types. The inflammatory response after IS includes two processes, namely early neural injury and late neural repair. Post-stroke inflammation is especially complex, and the interaction of different types of immune cells is crucial as a mediator of neuroinflammation. The immune system can be divided into innate and adaptive immune systems. The innate immune system includes microglia, neutrophils, and astrocytes. Microglia are the first responders in ischemic tissue (10). Moreover, T cells in the adaptive immune system play a role in central nervous system injury and repair (11) (shown in Table 1; Figure 1).
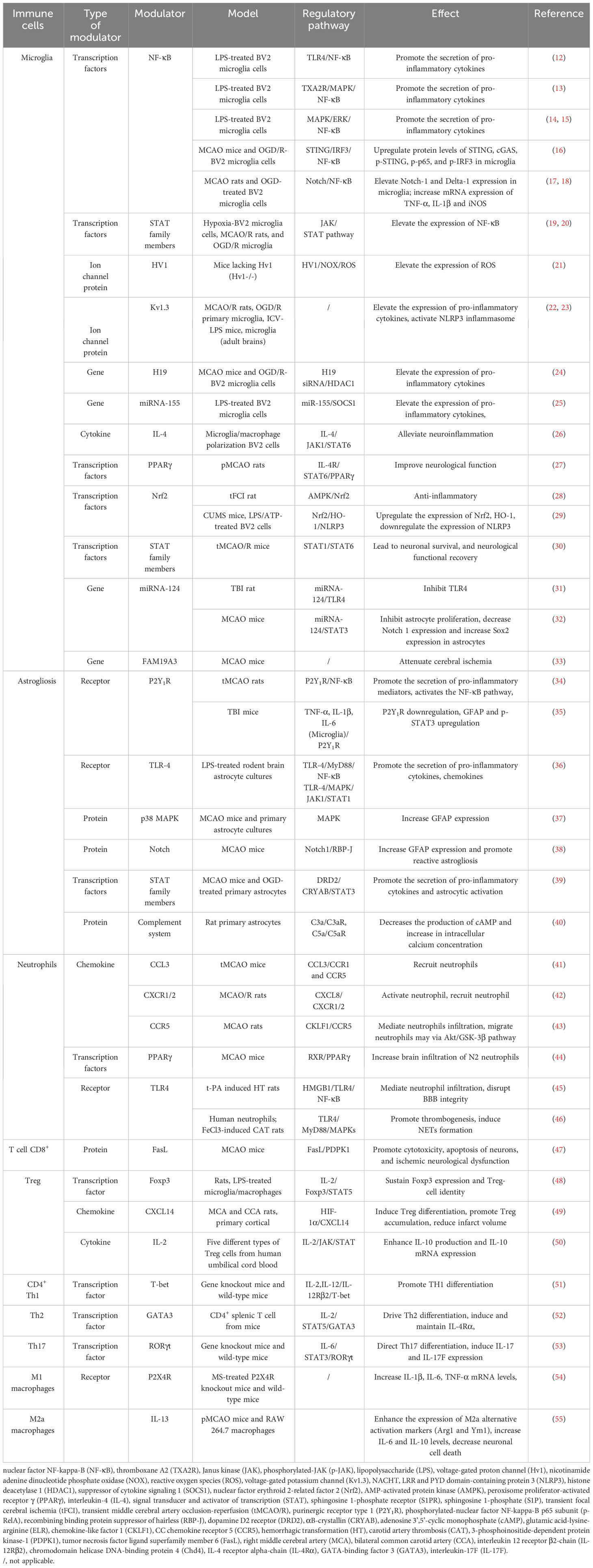
Table 1 Modulators and pathways of immune cell functions.
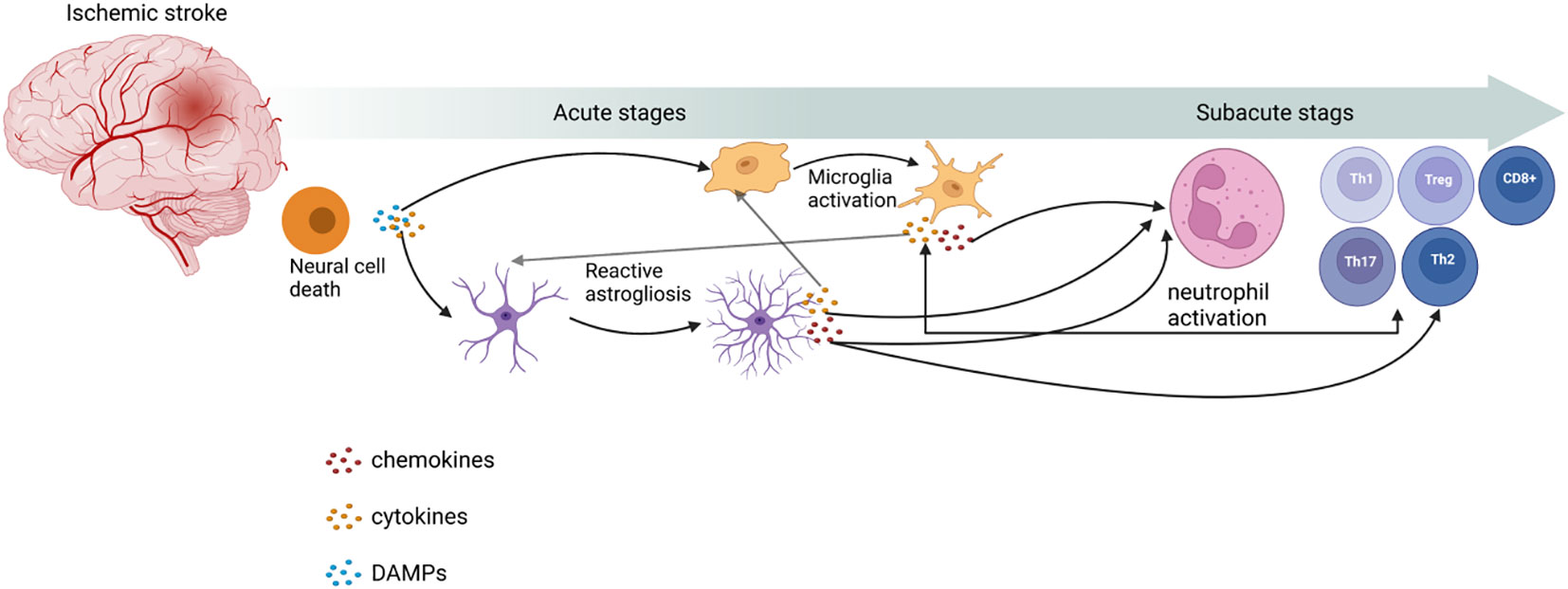
Figure 1 Role of immune cells in ischemic stroke. Post-IS, with the release of damage-associated molecular patterns (DAMPs) and cytokines from neuronal cell death, microglia activation and reactive astrogliosis occur. These processes can release cytokines and chemokines to activate neutrophils and T cells to proliferate and differentiate into different types. This image was created using BioRender.com.
Microglia, as resident CNS macrophages, play a dual role of neurotoxicity and protection in IS. When brain injury occurs, numerous DAMPs and cytokines are released, and microglia are activated with the death of nerve cells in the central area of the infarct (5); this, together with the activation of macrophages from blood-derived monocytes, constitute the innate immune response, which is the first line of defense (56). During systemic inflammation, microglia are involved in tissue damage and repair, respectively (57).
Previously, the pro-inflammatory microglia phenotype was termed M1, while the anti-inflammatory phenotype was termed M2. Since then, microglia have been demonstrated to exist in a wide range of activated states. For example, several in vivo studies failed to even find pure M1 or M2 states. In the same cell and at the same point in time, these “microglial markers” have multiple overlapping phenotypes. Therefore, a binary M1/M2 characterization is not sufficient for defining the inflammatory characteristics of microglia (58), and a systematic and careful nomenclature will greatly benefit the biological study of microglia. The current view is that moderate and precise terms should be used to properly understand the state of microglia. Using markers (genes or proteins) to identify populations of cells may be a solution, but they cannot be used as a readout of cell function (59). Wahane et al. combined transcriptomics and single-cell RNA sequencing to reveal a wide range of microglial states after spinal cord injury. The transcription profiles were diverse, with each transcription profile comprising four transcription subtypes. Furthermore, RNA-seq showed a well-defined temporal trajectory of IAM (injury-activated microglia and macrophages) gene programs over several days, including 3 days proliferation and motility, 7 days axon chemical attraction and ion channel activity, and 14 days extracellular matrix (ECM) recombination. The data also found that (i) phagocyte genes were still induced at 14 days to support the durable repair function of IAM; (ii) there was a high expression of ECM genes and nutrient factors; and (iii) the anti-inflammatory gene set signature was enriched at all stages. These three phenomena indicate that IAM has a long-lasting repair function. RNA-seq did not show significant changes in M1/M2 genes in bone marrow cells, and scRNA-seq further revealed their heterogeneous expression patterns in bone marrow subclusters. This further demonstrates the limitations of the conceptual dichotomy of pro-inflammatory and anti-inflammatory phenotypes (60).
The involvement of microglia in tissue damage reaches its peak 3–5 days after ischemic injury. At this stage, microglia play a harmful role, mainly by destroying the blood brain barrier (BBB), aggravating brain edema, and promoting neuronal apoptosis by producing and secreting many inflammatory mediators (61). The release of pro-inflammatory cytokines leads to secondary brain injury, while microglia have been shown to exhibit repair functions for nearly 14 days after injury (62). Microglia play a primarily protective role that promotes the regression of inflammation by secreting IL-4, IL-10, and transforming growth factor (TGF)-β, thereby indirectly preventing inflammation-induced damage to the blood brain barrier (BBB). The regulatory pathways of microglial cell polarization can be divided into four categories: 1) transcription factors; 2) receptors; 3) ion channels; and 4) gene modulators (shown in Table 1). Moreover, microglial phagocytosis is a double-edged sword in immune inflammation and stroke recovery. Microglia have been shown to exhibit phagocytosis, and their ability to clean neuronal debris reduces brain damage after stroke (5). Microglia invade the ischemic site of stroke earlier than macrophages, and they are the main phagocytes for the first three days after stroke (63). After ischemia, microglia infiltrate the injured brain tissue, engulfing living and dead neurons, myelin debris, apoptotic cell debris, endothelial cells, and leukocytes. During pathological cases, microglial phagocytosis can be initiated by specific “eat me” signals on specific cell types and their corresponding receptors (64).
As shown in Table 1, many pathways and mediators have been confirmed to regulate the activation of microglia. For example, NF-κB and IL-4 as regulators promote activation of microglia, respectively, and are the most extensively studied and clearly researched mediators. Some conflicting results have been found in studies on the regulatory effects of TLR and STAT, and further research is necessary to clarify their roles (65). Most of these signaling pathways overlap to varying degrees and do not appear to work independently, but rather synergistically, resulting in an inflammatory maelstrom (66). Many studies have shown that targeting microglia can effectively treat IS (67). Further research on microglial activation signal pathways after IS will help identify effective drugs that inhibit microglial activation and prevent neuroinflammation mediated by microglial activity. Therefore, in future studies, it is crucial to identify appropriate targeted intervention drugs according to the role of microglia at different times.
Astrocytes, the most abundant neuroglial cells in the brain, are essential housekeeping cells that maintain the central nervous system. Astrocytes play, as do microglia, a dual role in the pathophysiology of IS (68). After IS, damaged cells produce and release cytokines and DAMPs to stimulate receptors of astrocytes and change their phenotype. A few minutes after IS, due to reactive astrocyte proliferation, astrocytes respond to various inflammatory factors (including TGF-α, ciliary neurotrophic factor, IL-1, IL-6, and kallikrein-related peptidase 6) released by ischemic/hypoxic cells, and are subsequently activated and reproduced (69, 70). Reactive astrogliosis occurs in the peri-infarct region, and a glial scar is formed to maintain CNS homeostasis and wall off the lesion (71). During this process, astrocytes display cellular hypertrophy, proliferation, and increased expression of intermediate proteins, including glial fibrillary acidic protein (GFAP), vimentin, and nestin (72). After experiencing reactive astrogliosis, astrocytes produce and release pro-inflammatory cytokines (such as IL-6, TNF-α, IL-1α, IL-2β, and IFN-γ), chemokines (such as CXCL1/10 and CCL2/3/5), important sources of ATP, and free radicals such as NO, superoxide, and peroxynitrites (73). Thus, the activation of microglia and infiltration of white blood cells are enhanced (74). Studies have discussed the two subtypes of reactive astrocytes as A1 and A2. The A1 subtype includes astrocytes induced by IL-1α, TNF-α, and complement component subunit 1q (C1q) secreted by activated microglia. A1 subtype astrocytes induce neuronal and oligodendrocyte death. The A2 subtype can secrete IL-2, IL-10, and TGF-β, thus accelerating the regression of inflammation (75). Additionally, the A2 subtype can play an inflammatory and neuroprotective role by secreting neurotrophic factors, neuropoietic cytokines, and growth factors (76).
The response of astrocytes to Injury is a major determinant of the outcome after stroke. The gene expression of A2 astrocytes dominates the expression of A1 astrocytes (77). The JAK/STAT3 signaling pathway was found to be an important switch controlling many molecular and functional changes in reactive astrocytes in vivo and in vitro (78). However, the roles of all these molecules and pathways need to be further validated in future research (79, 80). In addition, the complexity of the multiple roles of complement protein and receptor expression in astrocytes has only recently been studied. Further in vivo and in vitro research is needed to determine astrocyte pathways and effects to determine targeted treatment strategies.
Increased numbers of leukocytes have been found to be a marker of the inflammatory response in IS. Among various types of leukocytes, neutrophils are the first to respond to ischemic brain injury (81). An in vivo study found that neutrophils were found in leptomeninges and cerebral parenchyma 6 hours and 12 hours, respectively, after permanent middle cerebral artery occlusion (pMCAO) (82). Their recruitment reached a peak on days 1-3 and gradually decreased over time (83). Neutrophils produce extensive weblike structures of DNA (neutrophil extracellular traps, NETs) that reached their peak 3-5 days after the transient middle cerebral artery occlusion (tMCAO) (84). These NETs have been associated with inflammation (85). After cerebral ischemia, neutrophils undergo conformational changes due to the presence of many adhesion molecules, which helps them to migrate through blood vessel walls to the brain tissue. Activated microglia and astrocytes release chemokines (such as CXC and CC) to promote neutrophil activation. These chemoattractants bind to the C-C chemokine receptor 5 (CCR5) and C-X-C chemokine receptor 1 (CXCR1) on the surface of neutrophils, making neutrophils the first blood-derived immune cells to migrate to damaged brain tissue (86). Neutrophils are attracted to the ischemic region by chemokines, and then infiltrate damaged brain tissue soon after injury, which aggravates inflammation (87).
Traditionally, neutrophils have been considered the main mediators of harmful inflammatory responses in IS (88). However, a significant amount of evidence suggests that neutrophils can obtain different phenotypes. As in the case of microglia, it is believed that some neutrophil subsets show different characteristics. The response phenotype of neutrophils to the ischemic environment, and the interaction between neutrophils and endothelial adhesion molecules has shifted from protective N2 to the injurious N1 phenotype (89). In vivo and in vitro studies have shown that PPARγ and TLR4 mediate the N2 phenotype of neutrophils (90, 91). However, research on the functional changes and biomarkers of the N1/N2 phenotypes of neutrophils after IS are not sufficient. This also leads to a shortage of known pathways. Therefore, further research is needed on the role of neutrophils in the inflammatory response after IS.
T lymphocytes play an important role in the process of nerve damage and repair in the late stage of IS. In the acute phase of IS, T cells chiefly react in an antigen-independent manner and are closely related to the development of the infarct volume. After 3-7 days, the T cell response gradually transforms into antigen-dependent antigen recognition (92). Brain-derived antigens are recognized by T cell receptors (TCRs) on the surface of naïve T cells. Then, T cells migrate to the brain parenchyma through cell adhesion molecules (such as P-selectin, E-selectin, VCAM-1, and ICAM-1) and chemokines. Ultimately, adaptive immune responses exacerbate ischemia-reperfusion (I/R) injury. In vivo studies involving ischemic rats demonstrated that by day 3 after ischemia, many T cells infiltrated the peripheral areas around the lesion and surrounded the infarct center, and the number of T cells increased between days 3 and 7 (93). According to different functions, T cells have multiple types marked by CD3 expression, including CD8+ cytotoxic T lymphocytes (CTL), CD4+ T helper (Th) cells, regulatory T cells (Treg), and gamma delta (γδ) T cells (94). The different roles of different types of T cells are already known, but the specific mechanism of T cell function after IS still needs further research.
CD4+ T cells, as the main effector T cells, regulate brain inflammation by producing cytokines (95). The signals derived from T cell and co-stimulatory T cell receptors and extracellular cytokines determine the phenotype of Th cells. Cytokine signals are received through multimeric receptors and propagated largely through Janus kinase/signal transducer activator of transcription (JAK/STAT) signaling pathways (96). Th cells can be divided into Th1 and Th17 (pro-inflammatory), and Th2 and Treg (anti-inflammatory) based on their cytokine secretion profile. Th1 and Th17 cells produce IL-1, IFN-γ, IL-17, IL-22, and other cytokines. Th2 and Treg cells produce IL-4, IL-10, and TGF-β (97). Different types of CD4+ T cells have their own specific transcription factors that play a crucial role in their differentiation, maintenance, and function (98).
CD8+ T cells can play a cytotoxic role through antigen recognition of the TCR and subsequent release of granzyme and perforin, forming pores on target cells and inducing apoptosis (99). Selvaraj et al. investigated the role of CD8+ T cells in stroke by establishing a tMCAO mouse model. The results showed that CD8+ T cells had an adverse effect in the chronic phase after stroke. At 30 days, there was an increase in the number of ipsilesional CD8+ T cells, revealing its association with deterioration in mouse functional outcomes (100). In recent years, there have been studies on CD8+ T cells inducing neuronal apoptosis through the FasL/PDPK1 pathway, but their mechanism of action after IS remains unclear.
γδ T cells do not require antigen recognition to activate and are detected in infarcts 6 hours after ischemia. During the onset of IS, γδ T cells mainly secrete cytokines such as IL-17, IL-21, IL-22, and IFN-γ through receptors to protect the barrier from infection and exacerbate inflammation (101). Arunachalam et al. found that Vγ6+/CCR6+ γδ T cell subtypes are the main source of IL-17 (102). However, few studies have paid attention to the signaling pathways present in γδ T cells after activation by ligands that bind to their receptors. The low number of γδ T cells, difficulty in extraction, and lack of cell lines may be the reasons for this lack of research.
The role of monocytes and macrophages in ischemic stroke is the same as that of microglia. Post-IS, pro-inflammatory monocytes infiltrate the inflamed brain, where they differentiate into macrophages that are morphologically indistinguishable from the local microglia (103). In contrast to the rapid microglial response, macrophages are rarely detected within the first 48 hours. Their level gradually increases, with a peak during the first week after stroke (104). On day 3 after stroke, the phenotypes of monocytes were found to change from the predominantly pro-inflammatory M1 to the anti-inflammatory M2 phenotype, indicating a functional shift from an enhanced immune response to inflammation resolution (105). Transcriptomic analysis of macrophages has shown that infiltrated macrophages on day 5 after stroke promote an effervescent increase and inflammation resolution after ischemic stroke (106). M2 macrophages can be further subdivided. For example, studies have classified macrophages as M2a, M2b, and M2c (107), while other authors have also classified them into an M2d subtype (108). All four M2 macrophage subtypes acquired enhanced phagocytosis and expressed IL-10, contributing to the resolution of inflammation (61). In a mouse model of ischemic stroke, researchers found that inflammatory activity peaked at 72 hours. Microglia produce relatively high levels of reactive oxygen species and TNF, while monocytes are major IL-1β producers. Although microglia show enhanced phagocytosis activity after stroke, monocytes have a significantly higher phagocytosis capacity at 72 hours (104).
Most M2 macrophages derived from monocytes can protect the blood-brain barrier from ischemic damage through vascular remodeling, physical attachment, and regression of inflammation (109). M2a macrophages express various anti-inflammatory and neurotrophic factors, such as arginase 1 (Arg1) and insulin-like growth factor-1. M2c macrophages increase the expression of TGF-β, CD163, and sphingosine kinase. However, M2b macrophages increase the production of pro-inflammatory factors, including IL-1β, IL-6, and TNF-a, which may enhance inflammation and increase blood-brain barrier permeability early in IS (110). M2d macrophages secrete VEGF-A and TNF-α, all of which are detrimental to the blood-brain barrier integrity in IS (111).
The interaction between activated microglia and astrocyte has a critical role in the process of neuroinflammation (shown in Table 2). In the first 6 hours after cerebral ischemia, microglia are first activated by pathogens or injury through TLR4, and release inflammatory mediators (112, 113). At the same time, astrocytes dependently activate TLR2, TLR3, and TLR4 to respond (114). The “molecular signal” (IL-1, TNF-α, and C1q) released by microglia can convert astrocytes into a neurotoxic A1 phenotype. For example, Dr. Ben Barres’ lab, using single-, dual-, and triple-gene knockout mice, pioneered the discovery that activated microglia secreting IL-1α, TNF-α, and C1q together are necessary and sufficient to induce A1 astrocytes (75). These neuroinflammatory reactive astrocytes lose many of their stereotypical physiological functions and secrete one or more unknown factors with strong toxicity to neurons and oligodendrocytes (115). Tarassishin et al. showed that human astrocytes and reactive astrogliosis are highly sensitive to IL-1β but unresponsive to lipopolysaccharide (LPS) stimulation. In human astrocytes, IL-1 induced both A1 and A2 responses (116). Glucagon-like peptide-1 receptor (GLP1R) is highly expressed in microglia, and is also expressed in astrocytes and neurons at reduced levels. Some studies have found that GLP1R agonists can directly prevent microglia-mediated astrocyte transformation into the A1 neurotoxic phenotype and have neuroprotective effects (117). The interleukin-1 family member interleukin-33 (IL-33) is produced by developing astrocytes, and it mainly signals to microglia and promotes synaptic phagocytosis of microglia under physiological conditions. IL-33 also drives microglia-dependent synaptic depletion in vivo. The transcriptomes of acutely isolated microglia from IL-33−/− animals showed 483 significantly altered transcripts, including reduced expression of NF-κB targets (e.g., Tnf, Nfkbia, Nfkbiz, and Tnfaip3) (118). IL-15 is also the mediator of crosstalk between astrocytes and microglia, thus aggravating brain damage after intracerebral hemorrhage. Shi et al. established a transgenic mouse model targeting IL-15 expression in astrocytes and found that the accumulation of microglia near astrocytes in the tissue around the hematoma increased after brain injury. The expression of biomarkers in M1 microglial cells increased significantly (119).
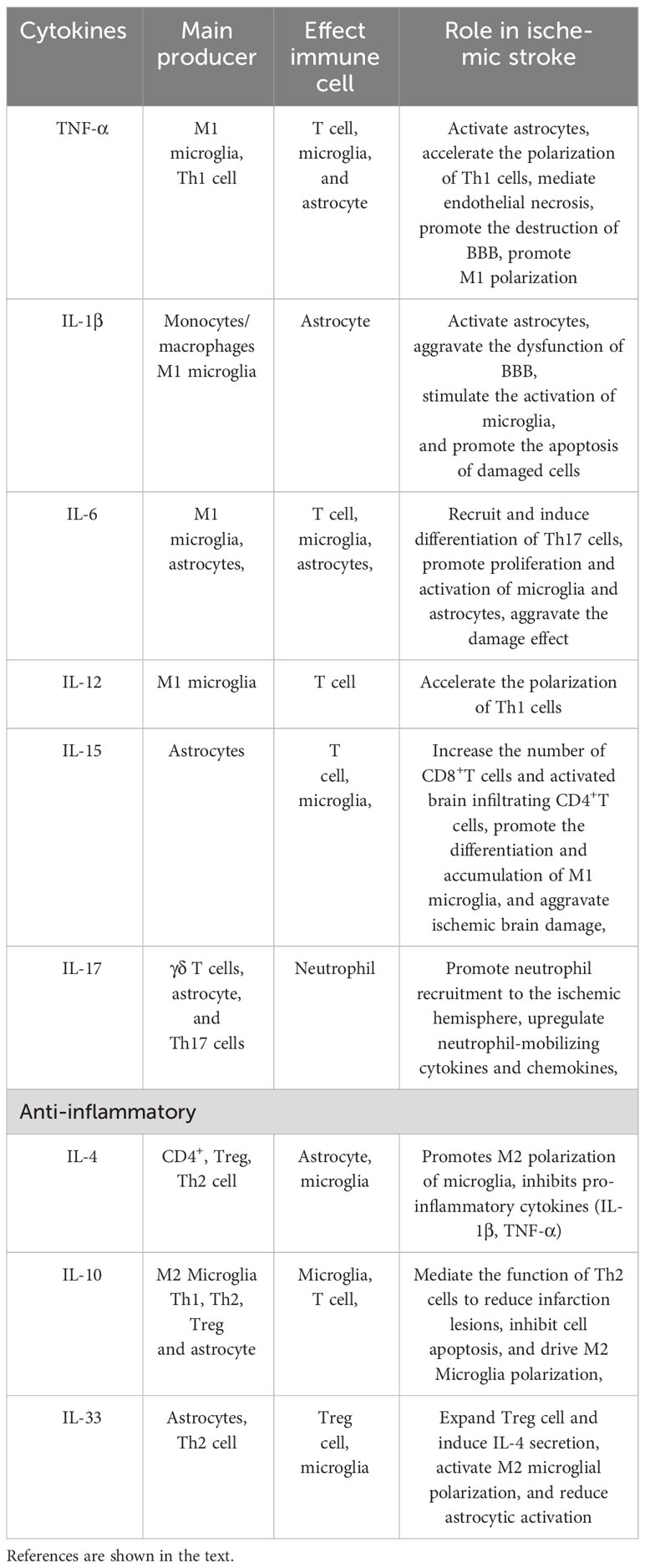
Table 2 Cytokines in mutual coordination between immune cells.
Astrocytes are the bridge between infiltrating T lymphocytes and neurons during cerebral ischemia. In vivo knockdown of interleukin-15 (IL-15) in astrocytes alleviates ischemic brain damage. Decreased levels of CD8+ T cells were also found in mice with knockdown of the IL-15 receptor α or blockade of cell-to-cell contact. Subsequent studies further confirmed the role of IL-15 from astrocytes on T cells. At the same time, a lower number of activated brain infiltrating CD4+ T cells were also found in Il15−/− mice (120). Astrocytes, γδ T cells, and Th17 cells are the main sources of interleukin-17 (IL-17) after IS. The main function of IL-17 involves coordinating local tissue inflammation by upregulating pro-inflammatory and neutrophil-mobilizing cytokines and chemokines. Kang et al. established a mouse model with specific deletions of key components of IL-17 signaling in various immune cells. It was found that astrocytes were crucial in IL-17-mediated white blood cell recruitment (121). Astrocyte-derived CXCL-1 acts as a key mediator of IL-17-initiated neutrophil chemotaxis in stroke. IL-17 secreted by γδ T cells has also been reported to attract neutrophils to the site of injury (122). Subsequently, reactive microglia engulf neutrophils in the periphery of ischemic lesions, while the local microglia loss and dystrophy occurring in the ischemic core are associated with the accumulation of neutrophils, first in perivascular spaces and later in the parenchyma (123). Following IS, central nervous system injury can trigger the release of IL-33 from astrocytes. Ito et al. found that many Treg cells accumulated in the brain of mice dependent on IL-33 after IS. The chemokines CCL1 and CCL20 drive penetration into the brain. This helps with neurological recovery in the chronic phase of ischemic brain injury (124). In the MCAO mouse model, IL-33 treatment increased the number of Treg cells in the ischemic brain. IL-33 was shown to increase the levels of anti-inflammatory cytokines in serum and brain tissue (125). IL-33 also enhanced M2 polarization marker expression in microglia. Activation of the IL-33/ST2 axis led to polarization of M2 microglia, which provided protection for ischemic neurons in an IL-10 dependent manner (126).
The crosstalk between M1 microglia and Th1/Th17 cells plays a pro-inflammatory role and contributes to brain injury. The crosstalk between M2 microglia and Th2/Treg cells plays an anti-inflammatory role and helps with brain recovery. M1 microglia secrete IL-12 and TNF-α, which induce Th1 cells, and these two types of cells work together to promote inflammation. The M1 polarization promoted by Th1 cytokines (TNF-α and IFN-γ) is associated with classic activation (127). M1 microglia secrete IL-6 and IL-23, which recruit Th17 cells and induce their differentiation (128). Th2 cells secrete IL-4 and IL-10, while Tregs secrete IL-10, further driving M2 polarization, inhibiting inflammation, and promoting tissue repair. IL-33 is suppressed in human stroke, resulting in an insufficient Th2-type response driven. In human T cells, IL-33 treatment induced IL-4 secretion while reducing astrocyte activation and increasing the number of M2 microglia (129).
After ischemia, hypoperfusion of brain tissue leads to a decrease in oxygen, ATP, and glucose, which leads to cell death over time. Ischemic tissue can be functionally divided into irreversibly injured infarcted core tissue and peripheral ischemic penumbra tissue. The infarct core is composed of dead or dying tissues and is located in the central area of the infarct area. In the penumbra, this depletion hampers cellular physiological functioning but does not induce an irreversible change. Neuronal death in IS involves a variety of cell death pathways. Apoptosis, pyroptosis, necroptosis, and PANoptosis are four key cell death pathways (Table 3; Figure 2).
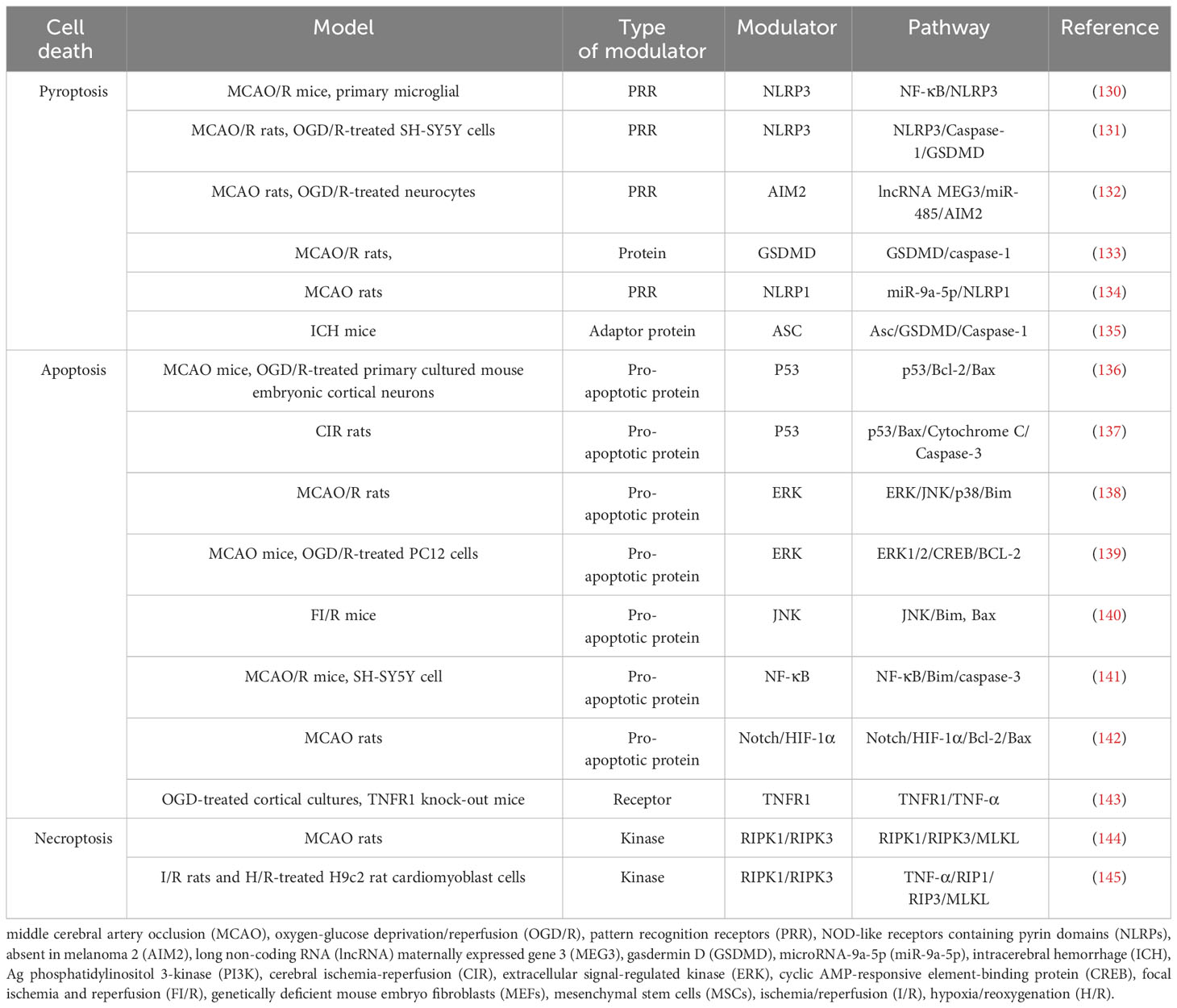
Table 3 Cell death and pathways.
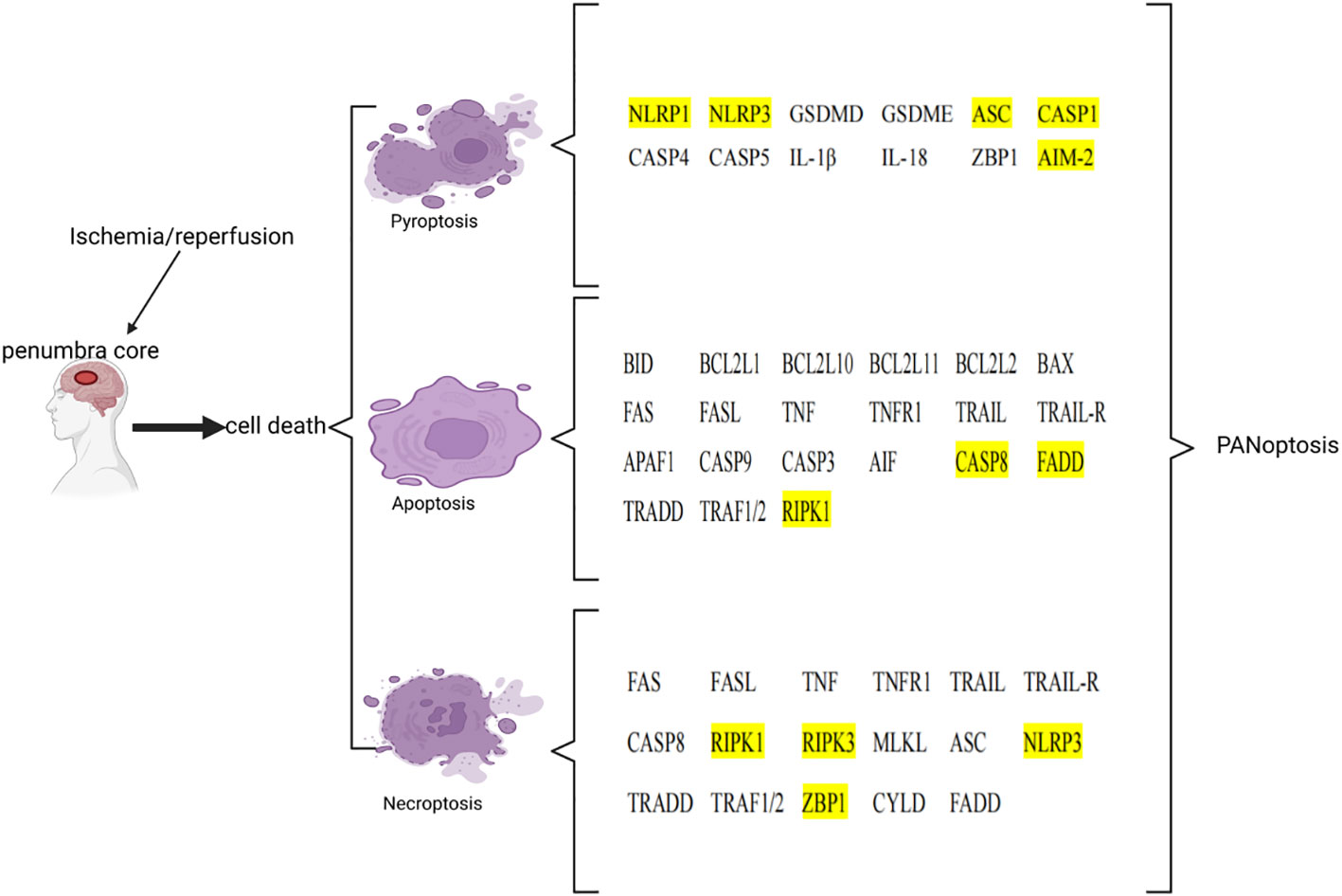
Figure 2 List of key genes involved in pyroptosis, apoptosis, and necroptosis in ischemic stroke. The genes involved in PANoptosis are marked in yellow. This image was created using BioRender.com.
Pyroptosis is a form of regulatory necrosis mediated by caspase-1 and is mainly seen in the ischemic penumbra. Pyroptosis can be divided into inflammatory and non-inflammatory pathways (146). Inflammatory pathway is the main pathway, one is the classical inflammatory pathway mediated by caspase-1, the other is the non-classical inflammatory pathway mediated by caspase-11. Inflammatory pathway of pyroptosis is an effective inducer of pro-inflammatory pathways in IS, occurring after the assembly and activation of inflammasomes (147). Inflammasomes contain pattern recognition receptors (PRRs), adapter proteins, and caspase family members (148). The adapter protein ASC has a cysteine protease recruitment domain (CARD) and a pyrin domain (PYD) (149). The structural characteristics of ASC provide support for binding of procaspase-1 to receptors. After IS, DAMPs secreted by necrotic cells in the ischemic core region are recognized by PRRs. Then, procaspase-1 autocrine signaling produces cleaved caspase-1. Cleaved caspase-1 mediates microglial pyroptosis with the release of a large number of pro-inflammatory factors (IL-1 and IL-18) that induces neuronal death (150). Caspase-1 cleaves gasdermin D (GSDMD) into N-GSDMD, which binds directly to the plasma membrane and forms pores, releasing large amounts of cytosolic content to promote inflammation. The non-classical inflammatory pathway is one in which caspase-11 mediates the “non-canonical inflammasome” to participate in IL-1 and IL-18 processing and cell death (108). Studies had shown that ccaspase-11 (mouse-derived) also cleaves GSDMD, leading to focal ptosis under LPS stimulation (151). In addition, caspase-11 has been found to promote inflammation by regulating caspase-1 expression by promoting K+ efflux (152). Non-inflammatory pyroptosis pathways are pathways in which caspase-8 is involved. It has been found that catalytic caspase-8 promotes the assembly of ASC-procaspase-1, in which caspase-8 acts as a scaffolding protein (153). In addition, caspase 8, procaspase 1, and cleaved caspase-1 were upregulated in an MCAO/R model (154). Similar to caspase-1, caspase-8 can also cleave gasdermin family proteins to induce pyroptosis. Under hypoxia conditions, nuclear transcription of GSDMC increases and caspase-8 cleaves GSDMC into N-GSDMC to induce pyroptosis after TNF-α stimulation (155). Further studies have identified a principal axis of pyroptosis extending from ROS-initiated DR6 endocytosis to caspase 8-mediated GSDMC cleavage (156).
Apoptosis is the most common form of programmed cell death in multicellular organisms that elicits no inflammatory response. It is the main mechanism of neuronal loss after IS and can be triggered either through the intrinsic or the extrinsic pathway. The intrinsic pathway is caused by DNA damage or endoplasmic reticulum stress, and the extrinsic pathway is mediated by the activation of the death receptor family members (157).
The intrinsic pathway involves a non-receptor-mediated signaling cascade (158). After IS, excitotoxicity produced through the mitochondrial pathway can mediate Ca2+ overload, leading to cell apoptosis. Glutamate binds to N-methyl-D-aspartate receptors (NMDARs), resulting in an overload of Ca2+ in neurons (159). Ca2+ activates the interaction of calpain with the Bcl-2 family proteins. Eventually, proapoptotic proteins are upregulated and mitochondrial permeability transition pores are formed (160), allowing for the release of apoptogens. The Bcl-2 protein family members (pro-apoptotic) regulate changes in the mitochondrial permeability, the release of cytochrome c, and contribute to apoptogen formation by binding with apoptotic protease activating factor-1 (Apaf-1) (161). Finally, activation of caspase leads to degradation of nuclear DNA, thus promoting cell apoptosis (162).
Following IS, the activation of immune cells during inflammation results in the release of a variety of factors (including pro-inflammatory cytokines) that trigger neuronal cell death via the extrinsic apoptotic pathways (163, 164). The extrinsic cell apoptosis pathway is triggered by the ligation of tumor necrosis factor (TNF)-family death receptors on the cell surface during external stimuli (165). After the receptor is bound, it recruits the adapter protein [Fas-associated death domain protein (FADD)] to create a death‐inducing signaling complex with procaspase-8, which activates caspase-8 (166). Caspase-8 activates the downstream effector caspase, mediating apoptosis by direct proteolytic cleavage or indirectly by catalyzing the Bcl-2 protein family members (167). Velier et al. established a pMCAO rat model and found that proteolytic processing yielding the active form of caspase-8 was active 6 hours after stroke (168).
Necroptosis, which is a lytic-programmed cell death with the ability to cause inflammation, is independent of caspase transmission. Similar to apoptosis, necroptosis is triggered by the ligation of specific death ligands to TNF-family death receptors or by pro‐caspase inhibitors (169). This process leads to de-ubiquitination of receptor interacting protein kinase 1 (RIPK1) by the de-ubiquitination enzyme CYLD (170). RIPK1 activates the kinase RIPK3 within a cytoplasmic high molecular weight complex called a necrosome. RIPK3 phosphorylates and activates the mixed lineage kinase domain-like protein (MLKL), forming the homotrimer necrosomes (171). The accumulation of necrosomes leads to increased permeability of plasma membranes and organelles. This leads to membrane damage and subsequent cell death (172, 173). The phosphorylation of MLKL and the formation of necrotic bodies are therefore considered as cellular markers of necrosis (174). After cerebral I/R injury, perivascular M1-microglia secrete TNF-α and its receptor TNFR1 on the endothelium, which serve as the main mediators triggering endothelial necroptosis (175). Necroptosis promotes the release of DAMPs, driving an inflammatory response. In vivo and in vitro studies found that RIPK3 promoted NLRP3 inflammasomes and the IL-1β inflammatory response independently of MLKL and necroptosis (176). In another study, it was found that MLKL signaling also activated NLRP3 inflammasomes and induced IL-1β secretion to promote inflammation. MLKL-induced NLRP3 inflammasome formation and IL-1β cleavage occur before cell lysis (177).
Ferroptosis refers to a new form of cell death caused by an increase of iron ion-dependent lipid peroxide (178). It is characterized by the accumulation of iron-regulated lipid peroxidation and caused by an imbalance of lipid metabolism, the depletion of glutathione (GSH), and the abnormal metabolism of iron. Excessive accumulation of iron is the key feature of ferroptosis, and most iron comes from damaged or aged red blood cells. Fe2+ produced by erythrocyte degradation can be oxidized to Fe3+, and Fe3+ binding transferrin (TF) mediates endocytosis through transferrin receptor (TFR)1 (179, 180). After the endocytosis of TF-TFR1, Fe3+ is released from TF and reduced to Fe2+ by six-transmembrane epithelial antigen of the prostate 3 (STEAP3). Finally, unbound iron is easily absorbed by neurons, resulting in intracellular iron accumulation (181). When iron is overloaded, Fe2+ generates a large number of lipid-active oxygen radicals through the Fenton reaction. Fe2+ can also participate in the synthesis of lipoxygenase and then catalyzes lipid peroxidation (182).
Lipid peroxidation is a critical process of ferroptosis (183). Ferroptosis shows obvious lipid peroxidation stress and cell membrane damage. Polyunsaturated fatty acid (PUFA)-phospholipid (PL) species are the most sensitive to peroxide because they contain highly active hydrogen atoms in their methylene bridge. In ferroptosis, acyl-CoA synthetase long-chain family member 4 (ACSL4) catalyzes fatty acids to form acyl coenzyme A and promotes fatty acid oxidation or lipid biosynthesis (184). Next, lysophosphatidylcholine acyltransferase 3 (LPCAT3) inserts the composite into the membrane phosphatidylethanolamine (PE). The ferroptotic signal is then activated (185). These lipids can be peroxidized under the catalysis of lipoxygenase (LOX) or under the induction of ROS (OH-) produced in the Fenton reaction. The resulting lipid peroxide can attack the proximal PUFA, causing a chain reaction and ferroptosis (186).
The manifestations of ferroptosis is the depletion of GSH and the inactivation of glutathione peroxidase 4 (GPX4). GSH is a tripeptide containing a sulfhydryl group, and it is composed of glutamic acid, glycine, and cysteine. It can combine with free radicals to repair cell membrane damage caused by lipid peroxide and it can clear ROS (187). GPX4 is a selenium enzyme. It can reduce oxidized lipids (L-OOH) (such as cholesterol and PL containing PUFA) to harmless lipid alcohols (L-OH) by converting GSH into oxidized glutathione (GSSG) (188). Therefore, GSH can also regulate GPX4 activity. In the process of ferroptosis, the accumulation of oxidation-reduction active iron consumes GSH reserves through the Fenton reaction, and then inhibits the activity of GPX4, leading to an overwhelming antioxidant reaction (189). The lack of GPX4 in turn leads to the accumulation of iron.
Pyroptosis and apoptosis both involve the activation of members of the caspase protease family. Studies have found that the activation of caspase-1 triggers pyroptosis and apoptosis (190). As has already been mentioned, RIPK3 and MLKL are crucial for the occurrence of necroptosis. They can also mediate the formation of NLRP3 inflammasomes and trigger pyroptosis. This widespread crosstalk between pyroptosis, apoptosis, and necroptosis led to a new form of programmed cell death called “PANoptosis”. PANoptosis is an inflammation-regulated cell death pathway. These cell death pathways are interconnected through the shared regulatory proteins called the PANoptosome. The PANoptosome is a cell death–inducing complex that is characterized by pyroptosis, apoptosis, and necroptosis molecules. It was identified as an inducer and regulator of PANoptosis. Christgen et al. found that RIPK1, RIPK3, caspase-8, NLRP3, ASC, and FADD interacted to form PANoptosomes (191). These proteins can be divided into sensors (ZBP1 and NLRP3), adapters (ASC and FADD), and catalytic effectors (RIPK1, RIPK3, caspase-1, and caspase-8) based on their functions (192). Lee et al. found that AIM2 regulated the innate immune sensors pyrin and ZBP1 to drive inflammation signal transduction and PANoptosis. The results confirmed that AIM2 mediated the assembly of multi-protein complexes, known as the AIM2 PANoptosome (193). Another study found a RIPK1 PANoptosome complex in an in vivo model of bacterial pathogen infection, which regulates all three branches of PANoptosis (194). In addition, during influenza virus infection, ZBP1 recruited RIPK3 and caspase-8 to activate ZBP1-NLRP3 inflammasomes. The formation of ZBP1-NLRP3 inflammasomes mediates PANoptosis by assembling the ZBP1 PANoptosome (195). Yan et al. confirmed the existence of PANoptosis in in vitro and in vivo models of ischemic brain injury through researching literature (196). In a following study, they demonstrated the occurrence of PANoptosis-like cell death in in vivo and in vitro models of ischemia/reperfusion injury (197). In summary, these studies indicate the presence of PANoptosis in ischemic brain injury. However, more research is needed to broaden our understanding of the basic processes of neuronal cell death and molecular targets, and to identify key molecules that regulate PANoptosis, which will lead to the development of new therapies.
The pathology of ischemic brain injury is an exceptionally complex pathological process involving a variety of cytotoxic factors and inflammatory cells in the CNS as well as in the peripheral circulatory system. Inflammation and cell death are the two main factors in IS. Inflammation and cell death, which are caused by ischemia, overlap and are interrelated. Due to the complexity of these factors and their interactions, it is very difficult to develop effective treatment methods based on the “one drug, one target” strategy, which leads to adverse outcomes in stroke treatment (198). A substantial number of studies have shown that some natural compounds (such as salidroside, baicalin, astragaloside IV, and curcumin) have protective effects on IS with few side effects (shown in Table 4).
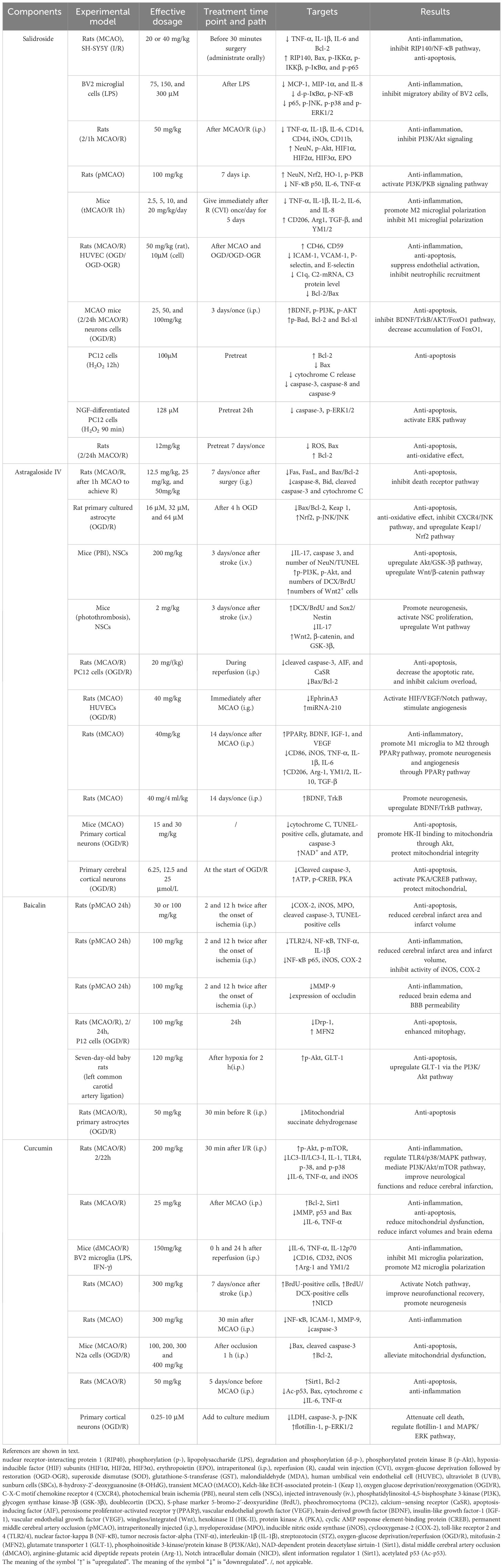
Table 4 Regulation mechanisms of natural compounds.
Salidroside (Sal) is the main bioactive component in Rhodiola rosea. In many studies of IS in vitro and in vivo in cells and animals, salidroside has demonstrated strong biological activity. Sal can significantly reduce the brain infarct size and cerebral edema by inhibiting inflammatory signaling. Sal reduces the levels of pro-inflammatory cytokines and chemokines in tissues or serum, such as TNF-α, IL-2, IL-6, IL-8, IL-1β, MCP-1, and MIP-1α (199). After cerebral ischemia, inflammatory transduction mainly depends on NF-κB, mitogen-activated protein kinases (MAPK), phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt), and phosphoinositide 3-kinase/protein kinase B (PI3K/PKB) signaling pathways. Chen et al. found Sal effectively reduced the levels of IL-6, IL-1β, and TNF-α by blocking the RIP140/NF-κB pathway (200). Sal also significantly inhibited activation of NF-κB, blocked degradation of tropomyosin-related kinase B (IκBα), and reduced p-MAPK levels (JNK, p38 and ERK1/2) (201). In a further study, it was demonstrated that Sal inhibited CD11b and inflammatory mediators through PI3K/Akt/HIF signaling. Sal significantly upregulated HIF subunits (HIF1α, HIF2α, and HIF3α) and the HIF downstream target (erythropoietin). Sal reduced CD14, CD44, and iNOS mRNA (202). Zhang et al. demonstrated that Sal reduced inflammation and brain damage through PI3K/PKB/Nrf-2/NFκB signaling transduction. Sal induced NeuN and inhibited NF-κB p50 subunit and other pro-inflammatory mediators. It prevented a significant decrease in the proportion of p-PKB/PKB in the brain (203). These studies imply that Sal may inhibit inflammatory signaling through the Nrf2, HIF, MAPK, PI3K/Akt, PI3K/PKB, and NF-κB signaling pathways. In addition, Sal acts on immune cells to recover the damage caused by IS. A recent study reported that Sal significantly inhibited the release of inflammatory factors derived from microglia. To study microglia polarization, M1 phenotypic markers (CD16, CD32, iNOS, and CD11b) and M2 phenotypic markers (CD206, Arg1, TGF-β, and YM1/2) were analyzed. The results showed that Sal promoted the transformation of microglia from the M1 phenotype to the M2 phenotype to enhance the phagocytosis of microglia. At the same time, Sal-treated M1 microglia promoted oligodendrocyte differentiation (204). Sal has been shown to effectively reduce VCAM-1, ICAM-1, P-selectin, and E-selectin, as well as neutrophil recruitment in the ischemic brain (205).
Apoptosis is one of the main mechanisms of brain injury, and Sal has been found to have significant anti apoptotic effects. Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophic factors. BDNF has a protective effect on ischemic brain injury. Zhang et al. indicated that Sal produced its anti-apoptotic effects by regulating the BDNF-mediated PI3K/Akt apoptosis pathway in a DNA-binding-dependent and -independent manners (206). Sal has been shown to inhibit the downregulation of Bcl-2, the upregulation of Bax, and the release of mitochondrial cytochrome c into the cytosol. Sal attenuated the activation of caspase-3, -8, and -9, and ultimately protected cells from apoptosis (207). Another study demonstrated that Sal induced activation of the mitogen-activated protein kinase kinase (MAPKK)/extracellular signal-related protein kinase (ERK) pathway, thereby reducing cell apoptosis (208). Shi et al. showed that Sal decreased the expression of Bax and restored the balance between pro-apoptotic and anti-apoptotic proteins (209).
These studies indicate that Sal has anti-inflammatory and anti-apoptotic effects (Figure 3). In addition, Sal also demonstrated excitotoxicity inhibition and anti-oxidant effects, and reduced damage to the BBB. Therefore, as an effective neuroprotective agent, it can be developed as a potential drug for treating stroke.
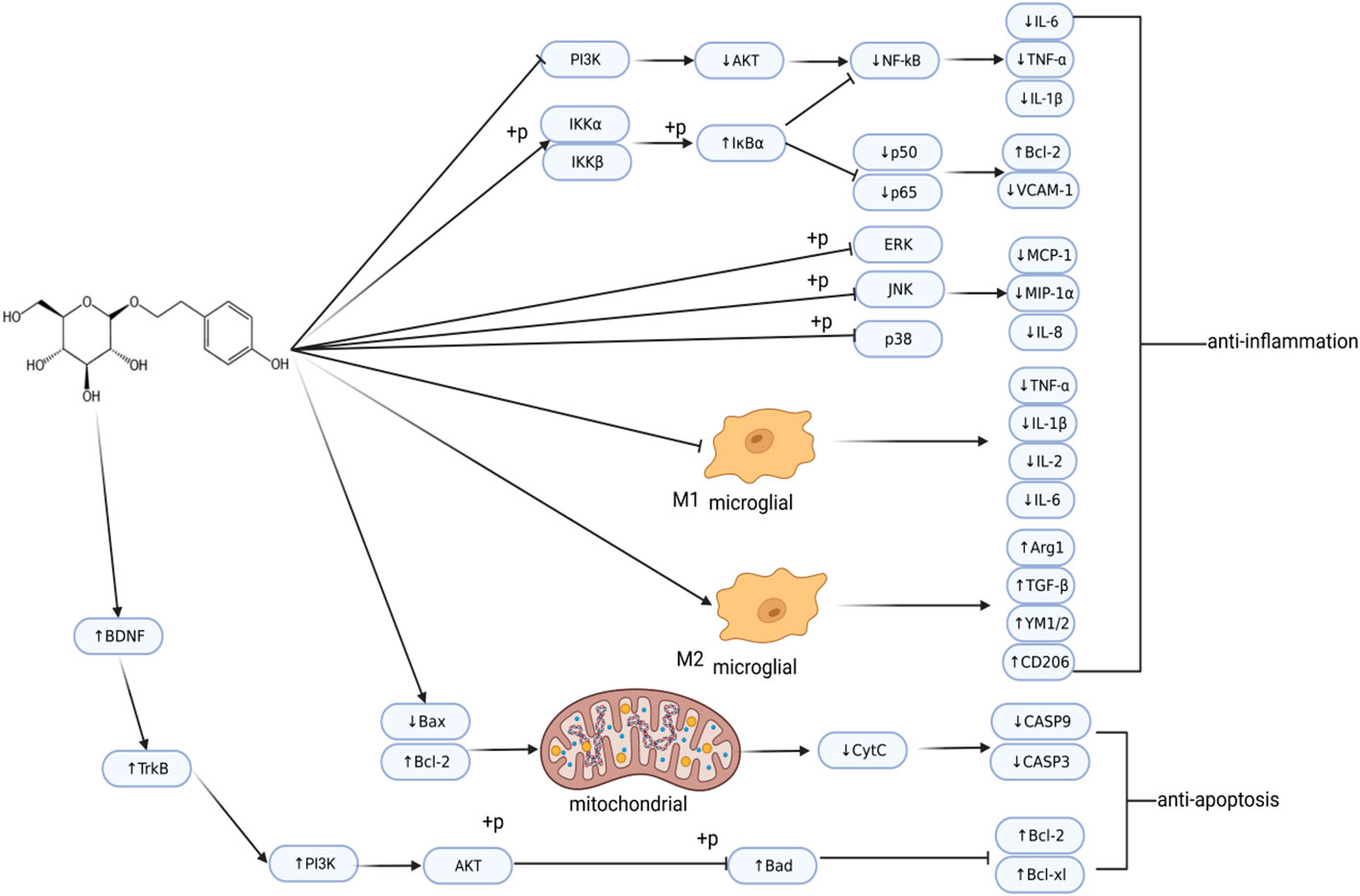
Figure 3 Regulatory network of salidroside in ischemic stroke. Post-IS, salidroside regulates inflammation and cell apoptosis. This image was created using BioRender.com.
Astragaloside IV (AS-IV) is one of the main active ingredients from Astragalus (Astragalus membranaceus (Fisch.) Bunge., Leguminosae, Huangqi in Chinese). AS-IV has been shown to significantly reduce neuronal apoptosis. AS-IV can suppress the activation of key factors in the death receptor pathway. AS-IV was found to inhibit mRNA upregulation of Fas, FasL, Caspase-8, and Bax/Bcl-2. AS-IV also inhibited the protein levels of caspase-8, Bid, cleaved caspase-3 and cytochrome C (210). AS-IV can regulate the Nrf2 signaling pathway. Yang et al. found that AS-IV induced Nrf2 through the downstream signaling pathways (MAPK pathway) to prevent cell apoptosis. AS-IV inhibited the CXCR4 receptor and downregulated the activation of the p-JNK/JNK pathway, thereby inhibiting the expression of Bax/Bcl-2 and ultimately increasing Nrf2/Keap1 signaling (211). Another study confirmed that AS-IV regulated cell apoptosis through the PI3K/Akt/GSK-3β pathway (212). The calcium-sensing receptor (CaSR) is a G-protein-coupled receptor. Its activation can increase the intracellular calcium concentration and contribute to cell apoptosis (213). AS-IV alleviated brain injury by inhibiting cell apoptosis induced by CaSR activation (214). Excitotoxicity by glutamate and mitochondrial dysfunction are common causes of cell apoptosis. AS-IV protects the integrity of mitochondria by promoting the combination of Akt and hexokinase II (HK-II). This helps to protect neurons from cell apoptosis and DNA damage (215). The PKA/CREB pathway regulates mitochondrial activity. AS-IV protects primary neurons from IS-induced apoptosis by regulating the PKA/CREB pathway and protecting mitochondrial function (216).
AS-IV also promotes the conversion of immune cells to an anti-inflammatory phenotype after IS, thereby reducing brain damage. PPARγ is a nuclear transcriptional factor that is widely expressed in microglia (217). AS-IV can promote the polarization of M1 microglia to the M2 phenotype, which plays a long-term neuroprotective role in cerebral ischemia/reperfusion injury through the PPARγ pathway (218). In addition, AS-IV also promotes angiogenesis by activating the HIF/VEGF/Notch, Wnt, and BDNF-TrkB pathways after IS, increasing cell proliferation, migration, and neovascularization (219–221).
Baicalin (BA) is a natural flavonoid compound isolated from the dried roots of Scutellaria baicalensis Georgi. BA alleviates the inflammatory reaction. BA was found to inhibit the TLR2/4 signaling pathway during cerebral ischemia, reducing expression of TLR2/4 and NF-κB in rat brain tissue. BA also attenuated the serum levels of TNF-α and IL-1β (222). In subsequent studies, Tu et al. also found that administration of BA after focal cerebral ischemia significantly reduced brain edema and BBB permeability. Overexpression of MMP-9 degraded the tight junction protein occludin, disrupting the integrity of the tight junction of the BBB (223). BA significantly downregulates the expression of MMP-9 protein and mRNA (224).
A growing body of evidence has shown the beneficial roles of BA in stroke management, such as anti-apoptosis. BA significantly inhibited neuronal apoptosis after cerebral ischemia injury in rats. Tu et al. found that BA significantly decreased MPO enzyme activity and iNOS and COX-2 mRNA expression in rat brain tissue, and significantly inhibited the expression of cleaved caspase-3 protein after IS (225). Li et al. showed that BA inhibited the expression of dynein related protein 1 (Drp-1). BA also reduced mitochondrial division and promoted the production of mitochondrial fusion protein 2 (MFN2) in an AMPK-dependent manner (226). Zhou et al. found that BA activated Akt phosphorylation and upregulated glutamate transporter 1 (GLT-1) expression through the PI3K/Akt signaling pathway. This inhibited cell apoptosis and reduced cerebral infarction volume and neuronal loss (227). BA also reduced mitochondrial succinate dehydrogenase (SDH)-mediated oxidative stress and reduced subsequent loss of glutamine synthetase (GS) (228).
Curcumin (CCM) is a compound mainly extracted from Curcuma longa. After IS, CCM can attenuate the inflammatory effect. The MAPK signaling pathway is regulated by TLR4 signaling and plays a key inflammatory role in IS. CCM alleviates inflammation of IS through the TLR4/p38/MAPK pathway. After CCM treatment, the protein levels of TLR4, p-p38, and IL-1 decreased, while the expression of IL-6, TNF-α, and iNOS increased (229). Another study demonstrated that CCM reduced inflammation by reducing levels of pro-inflammatory cytokines. Simultaneously, mitochondrial function was restored through an increase of MMP (230). In addition, CCM has a profound regulatory effect on the microglial response, promoting M2 microglia polarization and inhibiting the microglia-mediated proinflammatory response (231). However, further research is needed to confirm the involvement of curcumin and the specific mechanism of microglia phenotype regulation using stroke models. At the transcriptional level, the activation of NF-κB regulates ICAM-1, MMP-9 and caspase-3 expression (232). CCM decreased the expression of NF-κB, and subsequently attenuated the expression of the downstream mediators ICAM-1, MMP-9, and caspase-3 (233).
CCM exerts neuroprotective effects on IS and inhibits cell apoptosis. CCM reduces mitochondrial dysfunction and inhibits apoptosis by maintaining mitochondrial membrane potential and inhibiting the upregulation expression of Bax and downregulation of Bcl-2 (234). Silent information regulator 1 (Sirt1) is a class III group histone deacetylases that can protect the brain from ischemic damage (235). CCM was found to upregulate the expression of Sirt1 and Bcl-2 and downregulate the expression of acetylated p53 (Ac-p53) and Bax. Activating Sirt1 weakened cell apoptosis and promoted the neuroprotective effect of CCM (236). The MAPK signaling pathway regulates the expression of various pro-inflammatory cytokines and mediates apoptosis after ischemic injury. ERK1/2 and JNK are two of the main effectors of the MAPK signaling pathways (237). Lu et al. found that CCM reduced p-ERK1/2 and increased p-JNK protein levels. CCM also increased the level of flotilin-1 protein, thereby reducing cell death (238). CCM also improved neurofunctional recovery and promoted neurogenesis through Notch signaling after IS (239).
Therefore, based on the anti-inflammatory and anti-apoptosis effects of CCM, it may be a useful and promising neuroprotective agent against acute IS.
Immunity, inflammation, and cell death play critical roles in the occurrence and development of stroke. This review summarizes the roles and mechanisms of immune cells and cell death pathways in IS. The immune cells discussed included microglia, astrocyte, neutrophils, T lymphocytes, and monocytes/macrophages. The cell death pathways discussed included apoptosis, pyroptosis, necroptosis, PANoptosis, and ferroptosis. This review also summarized the mechanisms of natural compounds in the treatment of IS. The natural compounds discussed include salidroside, baicalin, astragaloside IV, and curcumin.
Microglia and monocytes/macrophages form the first line of defense, but are involved in damage in the early stages of ischemic stroke. Microglia can induce increased damage to the A1 neurotoxic subtype of astrocytes. Neutrophils are recruited into damaged brain tissue, which can exacerbate inflammation. Subsequently, microglia and monocytes/macrophages show anti-inflammatory and repair functions. After T cells migrate to the brain parenchyma, they differentiate into different functional types. Hence, time-defined treatments targeting different phenotypes of immune cells may provide a clear protective strategy. At the same time, the interactions between immune cells cannot be ignored. The mutual coordination between immune cells is also caused by various inflammatory mediators. After IS, peripheral immune cells and brain immune cells form a complex inflammatory network. Treatments that target only one type of immune cell may be harmful or offset the benefits of another type of immune cell, resulting in an unsatisfactory stroke prognosis. Therefore, therapeutic strategies to modulate the immune system need to be further explored to determine effective treatment measures.
Compared to PANoptosis, the key molecular pathways involved in apoptosis, pyrotosis, ferroptosis, and necroptosis are clearer. Further research is needed on the molecular basis and key pathways of PANopotosis after IS. Further research on the molecular and regulatory mechanisms of PANopotosis will have new impacts on the treatment of IS. For natural compounds in this review, mechanism research of Sal is the most extensive. All the natural compounds included in this review have therapeutic effects on inhibiting inflammation and cell apoptosis in IS. Salidroside, baicalin, astragaloside IV, and curcumin may be effective and promising candidates for the treatment of IS. However, they still have certain limitations, including whether they can show the same effect clinically as in research studies. Future research directions include 1) mechanisms for drugs to enter the central nervous system, 2) the ability to penetrate the blood-brain barrier and distribute widely in the brain, and 3) the side effects of drugs. With further research, the discovery of new drugs will lead to better treatment of IS for the benefit of public health.
NS: Conceptualization, Funding acquisition, Writing – review & editing. QC: Conceptualization, Writing – review & editing. ZG: Writing – original draft. JG: Writing – original draft. BL: Writing – review & editing. YG: Writing – review & editing. CC: Writing – review & editing. YJ: Writing – review & editing. NL: Writing – review & editing. MH: Investigation. TS: Writing – review & editing. LY: Writing – review & editing. HL: Writing – review & editing. HZ: Writing – review & editing. XZ: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the China Academy of Chinese Medical Sciences Innovation Fund (No.CI2021A00702-3), National Key Research and Development Program of China (2022YFC3501104), 2022 Qi Huang Young Scholar programme of the National Administration of Traditional Chinese Medicine (Z0841), the and the National Administration of Traditional Chinese Medicine High-Level Key Disciplines Construction Project of Traditional Chinese Medicine (zyyzdxk-2023234).
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Zhu H, Hu S, Li Y, Sun Y, Xiong X, Hu X, et al. Interleukins and ischemic stroke. Front Immunol (2022) 13:828447. doi: 10.3389/fimmu.2022.828447
2. Yu CC, Bin LL, Chen SY, Wang XF, Wang L, Du YJ. Ancient chinese herbal recipe huanglian jie du decoction for ischemic stroke: an overview of current evidence. Aging Dis (2022) 13:1733–44. doi: 10.14336/AD.2022.0311
3. Ramiro L, Simats A, García-Berrocoso T, Montaner J. Inflammatory molecules might become both biomarkers and therapeutic targets for stroke management. Ther Adv Neurol Disord (2018) 11:1–24. doi: 10.1177/1756286418789340
4. Park J, Kim JY, Kim YR, Huang M, Chang JY, Sim AY, et al. Reparative system arising from CCR2(+) monocyte conversion attenuates neuroinflammation following ischemic stroke. Transl Stroke Res (2021) 12(5):879–93. doi: 10.1007/s12975-020-00878-x
5. Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke (2012) 43:3063–70. doi: 10.1161/STROKEAHA.112.659656
6. Siniscalchi A, Iannacchero R, Anticoli S, Romana Pezzella F, De Sarro G, Gallelli L. Anti-inflammatory strategies in stroke: a potential therapeutic target. Curr Vasc Pharmacol (2015) 14:98–105. doi: 10.2174/1570161113666150923111329
7. Zhang SR, Phan TG, Sobey CG. Targeting the immune system for ischemic stroke. Trends Pharmacol Sci (2021) 42:96–105. doi: 10.1016/j.tips.2020.11.010
8. Tuo QZ, Zhang ST, Lei P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med Res Rev (2022) 42:259–305. doi: 10.1002/med.21817
9. Shu J, Yang L, Wei W, Zhang L. Identification of programmed cell death-related gene signature and associated regulatory axis in cerebral ischemia/reperfusion injury. Front Genet (2022) 13:934154. doi: 10.3389/fgene.2022.934154
10. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke (2009) 40:1849–57. doi: 10.1161/STROKEAHA.108.534503
11. Wang X, Xuan W, Zhu ZY, Li Y, Zhu H, Zhu L, et al. The evolving role of neuro-immune interaction in brain repair after cerebral ischemic stroke. CNS Neurosci Ther (2018) 24:1100–14. doi: 10.1111/cns.13077
12. Zhang J, Zheng Y, Luo Y, Du Y, Zhang X, Fu J. Curcumin inhibits LPS-induced neuroinflammation by promoting microglial M2 polarization via TREM2/ TLR4/ NF-κB pathways in BV2 cells. Mol Immunol (2019) 116:29–37. doi: 10.1016/j.molimm.2019.09.020
13. Yan A, Cai G, Xia W, Fu Y. Thromboxane A2 receptor antagonist SQ29548 suppresses the LPS-induced release of inflammatory cytokines in BV2 microglia cells via suppressing MAPK and NF-κB signaling pathways. Mol Med Rep (2017) 16:2491–6. doi: 10.3892/mmr.2017.6884
14. Qiu Z, Lu P, Wang K, Zhao X, Li Q, Wen J, et al. Dexmedetomidine inhibits neuroinflammation by altering microglial M1/M2 polarization through MAPK/ERK pathway. Neurochem Res (2020) 45:345–53. doi: 10.1007/s11064-019-02922-1
15. Chu W, Li M, Li F, Hu R, Chen Z, Lin J, et al. Immediate splenectomy down-regulates the MAPKYNF-JB signaling pathway in rat brain after severe traumatic brain injury. J Trauma Acute Care Surg (2013) 74:1446–53. doi: 10.1097/TA.0b013e31829246ad
16. Kong L, Li W, Chang E, Wang W, Shen N, Xu X, et al. mtDNA-STING axis mediates microglial polarization via IRF3/NF-κB signaling after ischemic stroke. Front Immunol (2022) 13:860977. doi: 10.3389/fimmu.2022.860977
17. Cheng M, Yang L, Dong Z, Wang M, Sun Y, Liu H, et al. Folic acid deficiency enhanced microglial immune response via the Notch1/nuclear factor kappa B p65 pathway in hippocampus following rat brain I/R injury and BV2 cells. J Cell Mol Med (2019) 23:4795–807. doi: 10.1111/jcmm.14368
18. Yao L, Kan EM, Kaur C, Dheen ST, Hao A, Lu J, et al. Notch-1 signaling regulates microglia activation via NF-κB pathway after hypoxic exposure in vivo and in vitro. PloS One (2013) 8:1–15. doi: 10.1371/journal.pone.0078439
19. Butturini E, Boriero D, Carcereri de Prati A, Mariotto S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Arch Biochem Biophys (2019) 669:22–30. doi: 10.1016/j.abb.2019.05.011
20. Ding Y, Qian J, Li H, Shen H, Li X, Kong Y, et al. Effects of SC99 on cerebral ischemia-perfusion injury in rats: Selective modulation of microglia polarization to M2 phenotype via inhibiting JAK2-STAT3 pathway. Neurosci Res (2019) 142:58–68. doi: 10.1016/j.neures.2018.05.002
21. Tian DS, Li CY, Qin C, Murugan M, Wu LJ, Liu JL. Deficiency in the voltage-gated proton channel Hv1 increases M2 polarization of microglia and attenuates brain damage from photothrombotic ischemic stroke. J Neurochem (2016) 139:96–105. doi: 10.1111/jnc.13751
22. Ma DC, Zhang NN, Zhang YN, Chen HS. Kv1.3 channel blockade alleviates cerebral ischemia/reperfusion injury by reshaping M1/M2 phenotypes and compromising the activation of NLRP3 inflammasome in microglia. Exp Neurol (2020) 332:113399. doi: 10.1016/j.expneurol.2020.113399
23. Di Lucente J, Nguyen HM, Wulff H, Jin LW, Maezawa I. The voltage-gated potassium channel Kv1.3 is required for microglial pro-inflammatory activation in vivo. Glia (2018) 66:1881–95. doi: 10.1002/glia.23457
24. Wang J, Zhao H, Fan Z, Li G, Ma Q, Tao Z, et al. Long noncoding RNA H19 promotes neuroinflammation in ischemic stroke by driving histone deacetylase 1-dependent M1 microglial polarization. Stroke (2017) 48:2211–21. doi: 10.1161/STROKEAHA.117.017387
25. Zheng X, Huang H, Liu J, Li M, Liu M, Luo T. Propofol attenuates inflammatory response in LPS-activated microglia by regulating the miR-155/SOCS1 pathway. Inflammation (2018) 41:11–9. doi: 10.1007/s10753-017-0658-6
26. He Y, Gao Y, Zhang Q, Zhou G, Cao F, Yao S. IL-4 switches microglia/macrophage M1/M2 polarization and alleviates neurological damage by modulating the JAK1/STAT6 pathway following ICH. Neuroscience (2020) 437:161–71. doi: 10.1016/j.neuroscience.2020.03.008
27. Kang R, Gamdzyk M, Luo Y, Tang H, Huang L, Lenahan C, et al. Three days delayed recanalization improved neurological function in pMCAO rats by increasing M2 microglia—Possible involvement of the IL-4R/STAT6/PPARγ Pathway. Transl Stroke Res (2023) 14:250–62. doi: 10.1007/s12975-022-01032-5
28. Wang Y, Huang Y, Xu Y, Ruan W, Wang H, Zhang Y, et al. A dual AMPK/nrf2 activator reduces brain inflammation after stroke by enhancing microglia M2 polarization. Antioxid Redox Signal (2018) 28:141–63. doi: 10.1089/ars.2017.7003
29. Tao W, Hu Y, Chen Z, Dai Y, Hu Y, Qi M. Magnolol attenuates depressive-like behaviors by polarizing microglia towards the M2 phenotype through the regulation of Nrf2/HO-1/NLRP3 signaling pathway. Phytomedicine (2021) 91:153692. doi: 10.1016/j.phymed.2021.153692
30. Lu Y, Zhou M, Li Y, Li Y, Hua Y, Fan Y. Minocycline promotes functional recovery in ischemic stroke by modulating microglia polarization through STAT1/STAT6 pathways. Biochem Pharmacol (2021) 186:114464. doi: 10.1016/j.bcp.2021.114464
31. Yang Y, Ye Y, Kong C, Su X, Zhang X, Bai W, et al. MiR-124 enriched exosomes promoted the M2 polarization of microglia and enhanced hippocampus neurogenesis after traumatic brain injury by inhibiting TLR4 pathway. Neurochem Res (2019) 44:811–28. doi: 10.1007/s11064-018-02714-z
32. Li Z, Song Y, He T, Wen R, Li Y, Chen T, et al. M2 microglial small extracellular vesicles reduce glial scar formation via the miR-124/STAT3 pathway after ischemic stroke in mice. Theranostics (2021) 11:1232–48. doi: 10.7150/thno.48761
33. Shao Y, Deng T, Zhang T, Li P, Wang Y. FAM19A3, a novel secreted protein, modulates the microglia/macrophage polarization dynamics and ameliorates cerebral ischemia. FEBS Lett (2015) 589:467–75. doi: 10.1016/j.febslet.2015.01.003
34. Kuboyama K, Harada H, Tozaki-Saitoh H, Tsuda M, Ushijima K, Inoue K. Astrocytic P2Y1 receptor is involved in the regulation of cytokine/chemokine transcription and cerebral damage in a rat model of cerebral ischemia. J Cereb Blood Flow Metab (2011) 31:1930–41. doi: 10.1038/jcbfm.2011.49
35. Shinozaki Y, Shibata K, Yoshida K, Shigetomi E, Gachet C, Ikenaka K, et al. Transformation of astrocytes to a neuroprotective phenotype by microglia via P2Y1 receptor downregulation. Cell Rep (2017) 19:1151–64. doi: 10.1016/j.celrep.2017.04.047
36. Gaire BP, Song MR, Choi JW. Sphingosine 1-phosphate receptor subtype 3 (S1P3) contributes to brain injury after transient focal cerebral ischemia via modulating microglial activation and their M1 polarization. J Neuroinflamm (2018) 15:1–14. doi: 10.1186/s12974-018-1323-1
37. Roy Choudhury G, Ryou MG, Poteet E, Wen Y, He R, Sun F, et al. Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Res (2014) 1551:45–58. doi: 10.1016/j.brainres.2014.01.013
38. Marumo T, Takagi Y, Muraki K, Hashimoto N, Miyamoto S, Tanigaki K. Notch signaling regulates nucleocytoplasmic Olig2 translocation in reactive astrocytes differentiation after ischemic stroke. Neurosci Res (2013) 75:204–9. doi: 10.1016/j.neures.2013.01.006
39. Qiu J, Yan Z, Tao K, Li Y, Li Y, Li J, et al. Sinomenine activates astrocytic dopamine D2 receptors and alleviates neuroinflammatory injury via the CRYAB/STAT3 pathway after ischemic stroke in mice. J Neuroinflamm (2016) 13:1–13. doi: 10.1186/s12974-016-0739-8
40. Sayah S, Jauneau AC, Patte C, Tonon MC, Vaudry H, Fontaine M. Two different transduction pathways are activated by C3a and C5a anaphylatoxins on astrocytes. Mol Brain Res (2003) 112:53–60. doi: 10.1016/S0169-328X(03)00046-9
41. Takami S, Minami M, Nagata I, Namura S, Satoh M. Chemokine receptor antagonist peptide, viral MIP-II, protects the brain against focal cerebral ischemia in mice. J Cereb Blood Flow Metab (2001) 21:1430–5. doi: 10.1097/00004647-200112000-00007
42. Connell BJ, Gordon JR, Saleh TM. ELR-CXC chemokine antagonism is neuroprotective in a rat model of ischemic stroke. Neurosci Lett (2015) 606:117–22. doi: 10.1016/j.neulet.2015.08.041
43. Chen C, Chu SF, Ai QD, Zhang Z, Chen NH. CKLF1/CCR5 axis is involved in neutrophils migration of rats with transient cerebral ischemia. Int Immunopharmacol (2020) 85:106577. doi: 10.1016/j.intimp.2020.106577
44. Certo M, Endo Y, Ohta K, Sakurada S, Bagetta G, Amantea D. Activation of RXR/PPARγ underlies neuroprotection by bexarotene in ischemic stroke. Pharmacol Res (2015) 102:298–307. doi: 10.1016/j.phrs.2015.10.009
45. Kong L, Ma Y, Wang Z, Liu N, Ma G, Liu C, et al. Inhibition of hypoxia inducible factor 1 by YC-1 attenuates tissue plasminogen activator induced hemorrhagic transformation by suppressing HMGB1/TLR4/NF-κB mediated neutrophil infiltration in thromboembolic stroke rats. Int Immunopharmacol (2021) 94:107507. doi: 10.1016/j.intimp.2021.107507
46. Zha C, Zhang W, Gao F, Xu J, Jia R, Cai J, et al. Anti-β2GPI/β2GPI induces neutrophil extracellular traps formation to promote thrombogenesis via the TLR4/MyD88/MAPKs axis activation. Neuropharmacology (2018) 138:140–50. doi: 10.1016/j.neuropharm.2018.06.001
47. Fan L, Zhang CJ, Zhu L, Chen J, Zhang Z, Liu P, et al. FasL-PDPK1 pathway promotes the cytotoxicity of CD8+ T cells during ischemic stroke. Transl Stroke Res (2020) 11:747–61. doi: 10.1007/s12975-019-00749-0
48. Xie L, Choudhury GR, Winters A, Yang SH, Jin K. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur J Immunol (2015) 45:180–91. doi: 10.1002/eji.201444823
49. Lee HT, Liu SP, Lin CH, Lee SW, Hsu CY, Sytwu HK, et al. A crucial role of CXCL14 for promoting regulatory T cells activation in stroke. Theranostics (2017) 7:855–75. doi: 10.7150/thno.17558
50. Tsuji-Takayama K, Suzuki M, Yamamoto M, Harashima A, Okochi A, Otani T, et al. IL-2 activation of STAT5 enhances production of IL-10 from human cytotoxic regulatory T cells, HOZOT. Exp Hematol (2008) 36:181–92. doi: 10.1016/j.exphem.2007.09.010
51. Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol (2011) 12:551–9. doi: 10.1038/ni.2030
52. Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh TY, et al. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor α-chain expression. Nat Immunol (2008) 9:1288–96. doi: 10.1038/ni.1656
53. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
54. Verma R, Cronin CG, Hudobenko J, Venna VR, McCullough LD, Liang BT. Deletion of the P2X4 receptor is neuroprotective acutely, but induces a depressive phenotype during recovery from ischemic stroke. Brain Behav Immun (2017) 66:302–12. doi: 10.1016/j.bbi.2017.07.155
55. Kolosowska N, Keuters MH, Wojciechowski S, Keksa-Goldsteine V, Laine M, Malm T, et al. Peripheral administration of IL-13 induces anti-inflammatory microglial/macrophage responses and provides neuroprotection in ischemic stroke. Neurotherapeutics (2019) 16:1304–19. doi: 10.1007/s13311-019-00761-0
56. Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, Anderson AJ. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: Evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain (2010) 133:433–47. doi: 10.1093/brain/awp322
57. Feng Y, He X, Luo S, Chen X, Long S, Liang F, et al. Chronic colitis induces meninges traffic of gut-derived T cells, unbalances M1 and M2 microglia/macrophage and increases ischemic brain injury in mice. Brain Res (Elsevier B.V.) (2019) 1707:8–17. doi: 10.1016/j.brainres.2018.11.019
58. Rosi S. A polarizing view on posttraumatic brain injury inflammatory response. Brain Circ (2016) 2:126. doi: 10.4103/2394-8108.192517
59. Paolicelli RC, Sierra A, Stevens B, Tremblay ME, Aguzzi A, Ajami B, et al. Microglia states and nomenclature: A field at its crossroads. Neuron (2022) 110:3458–83. doi: 10.1016/j.neuron.2022.10.020
60. Wahane S, Zhou X, Zhou X, Guo L, Friedl M-S, Kluge M, et al. Diversified transcriptional responses of myeloid and glial cells in spinal cord injury shaped by HDAC3 activity. Sci Adv (2021) 7:8811–37. doi: 10.1126/sciadv.abd8811
61. Qiu YM, Zhang CL, Chen AQ, Wang HL, Zhou YF, Li YN, et al. Immune cells in the BBB disruption after acute ischemic stroke: targets for immune therapy? Front Immunol (2021) 12:678744. doi: 10.3389/fimmu.2021.678744
62. Weng L, Wu Z, Zheng W, Meng H, Han L, Wang S, et al. Malibatol A enhances alternative activation of microglia by inhibiting phosphorylation of Mammalian Ste20-like kinase1 in OGD-BV-2 cells. Neurol Res (2016) 38:342–8. doi: 10.1080/01616412.2016.1174423
63. Schilling M, Besselmann M, Müller M, Strecker JK, Ringelstein EB, Kiefer R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: An investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol (2005) 196:290–7. doi: 10.1016/j.expneurol.2005.08.004
64. Jia J, Yang L, Chen Y, Zheng L, Chen Y, Xu Y, et al. The role of microglial phagocytosis in ischemic stroke. Front Immunol (2022) 12:790201. doi: 10.3389/fimmu.2021.790201
65. Lan X, Han X, Li Q, Li Q, Gao Y, Cheng T, et al. Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia. Brain Behav Immun (2017) 61:326–39. doi: 10.1016/j.bbi.2016.12.012
66. Zhao SC, Ma LS, Chu ZH, Xu H, Wu WQ, Liu F. Regulation of microglial activation in stroke. Acta Pharmacol Sin (2017) 38:445–58. doi: 10.1038/aps.2016.162
67. Lambertsen KL, Finsen B, Clausen BH. Post-stroke inflammation—target or tool for therapy? Acta Neuropathol (2019) 137:693–714. doi: 10.1007/s00401-018-1930-z
68. Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, Buckwalter MS. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia (2014) 62:1227–40. doi: 10.1002/glia.22675
69. Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, et al. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab (2008) 28:468–81. doi: 10.1038/sj.jcbfm.9600546
70. Pekna M, Siqin S, de Pablo Y, Stokowska A, Torinsson Naluai Å, Pekny M. Astrocyte responses to complement peptide C3a are highly context-dependent. Neurochem Res (2022) 48:1233–41. doi: 10.1007/s11064-022-03743-5
71. Choudhury GR, Ding S. Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol Dis (2016) 85:234–44. doi: 10.1016/j.nbd.2015.05.003
72. Liu Z, Chopp M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog Neurobiol (Elsevier Ltd.) (2016) 144:103–20. doi: 10.1016/j.pneurobio.2015.09.008
73. Hennessy E, Griffin EW, Cunningham C. Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1β and TNF-α. J Neurosci (2015) 35:8411–22. doi: 10.1523/JNEUROSCI.2745-14.2015
74. Bianco F, Pravettoni E, Colombo A, Schenk U, Möller T, Matteoli M, et al. Astrocyte-derived ATP induces vesicle shedding and IL-1β Release from microglia. J Immunol (2005) 174:7268–77. doi: 10.4049/jimmunol.174.11.7268
75. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017) 541:481–7. doi: 10.1038/nature21029
76. Linnerbauer M, Rothhammer V. Protective functions of reactive astrocytes following central nervous system insult. Front Immunol (2020) 11:573256. doi: 10.3389/fimmu.2020.573256
77. Rakers C, Schleif M, Blank N, Matušková H, Ulas T, Händler K, et al. Stroke target identification guided by astrocyte transcriptome analysis. Glia (2019) 67:619–33. doi: 10.1002/glia.23544
78. Pekna M, Pekny M. The complement system: A powerful modulator and effector of astrocyte function in the healthy and diseased central nervous system. Cells (2021) 10(7):1812. doi: 10.3390/cells10071812
79. Pekna M, Stokowska A, Pekny M. Targeting complement C3a receptor to improve outcome after ischemic brain injury. Neurochem Res (2021) 46:2626–37. doi: 10.1007/s11064-021-03419-6
80. Pekny M, Pekna M, Messing A, Steinhäuser C, Lee JM, Parpura V, et al. Astrocytes: a central element in neurological diseases. Acta Neuropathol (2016) 131:323–45. doi: 10.1007/s00401-015-1513-1
81. Ross AM, Hurn P, Perrin N, Wood L, Carlini W, Potempa K. Evidence of the peripheral inflammatory response in patients with transient ischemic attack. J Stroke Cerebrovascular Dis (2007) 16:203–7. doi: 10.1016/j.jstrokecerebrovasdis.2007.05.002
82. Perez-de-Puig I, Miró-Mur F, Ferrer-Ferrer M, Gelpi E, Pedragosa J, Justicia C, et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol (2015) 129:239–57. doi: 10.1007/s00401-014-1381-0
83. Jickling GC, Liu DZ, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting neutrophils in ischemic stroke: Translational insights from experimental studies. J Cereb Blood Flow Metab (2015) 35:888–901. doi: 10.1038/jcbfm.2015.45
84. Kang L, Yu H, Yang X, Zhu Y, Bai X, Wang R, et al. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat Commun (2020) 11(1):2488. doi: 10.1038/s41467-020-16191-y
85. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med (2014) 20:511–7. doi: 10.1038/nm.3547
86. Chen C, Chu SF, Liu DD, Zhang Z, Kong LL, Zhou X, et al. Chemokines play complex roles in cerebral ischemia. Neurochem Int (2018) 112:146–58. doi: 10.1016/j.neuint.2017.06.008
87. Kim SW, Lee H, Lee HK, Kim ID, Lee JK. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol Commun (2019) 7:94. doi: 10.1186/s40478-019-0747-x
88. Connolly ES, Winfree CJ, Springer TA, Naka Y, Liao H, Du YS, et al. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest (1996) 97:209–16. doi: 10.1172/JCI118392
89. Nourshargh S, Marelli-Berg FM. Transmigration through venular walls: A key regulator of leukocyte phenotype and function. Trends Immunol (2005) 26:157–65. doi: 10.1016/j.it.2005.01.006
90. Cuartero MI, Ballesteros I, Moraga A, Nombela F, Vivancos J, Hamilton JA, et al. N2 neutrophils, novel players in brain inflammation after stroke: Modulation by the pparγ agonist rosiglitazone. Stroke (2013) 44:3498–508. doi: 10.1161/STROKEAHA.113.002470
91. García-Culebras A, Durán-Laforet V, Peña-Martínez C, Moraga A, Ballesteros I, Cuartero MI, et al. Role of TLR4 (Toll-like receptor 4) in N1/N2 neutrophil programming after stroke. Stroke (2019) 50:2922–32. doi: 10.1161/STROKEAHA.119.025085
92. Wu F, Liu Z, Zhou L, Ye D, Zhu Y, Huang K, et al. Systemic immune responses after ischemic stroke: From the center to the periphery. Front Immunol (2022) 13:911661. doi: 10.3389/fimmu.2022.911661
93. Jander S, Kraemer M, Schroeter M, Witte OW, Stoll G. Lymphocytic infiltration and expression of intercellular adhesion molecule-1 in photochemically induced ischemia of the rat cortex. J Cereb Blood Flow Metab (1995) 15:42–51. doi: 10.1038/jcbfm.1995.5
94. Zhang D, Ren J, Luo Y, He Q, Zhao R, Chang J, et al. T cell response in ischemic stroke: from mechanisms to translational insights. Front Immunol (2021) 12:707972. doi: 10.3389/fimmu.2021.707972
95. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4+ T cell populations. Annu Rev Immunol (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
96. O’Shea JJ, Lahesmaa R, Vahedi G, Laurence A, Kanno Y. Genomic views of STAT function in CD4 + T helper cell differentiation. Nat Rev Immunol (2011) 11:239–50. doi: 10.1038/nri2958
97. Gu L, Jian Z, Stary C, Xiong X. T cells and cerebral ischemic stroke. Neurochem Res (2015) 40:1786–91. doi: 10.1007/s11064-015-1676-0
98. Li P, Spolski R, Liao W, Leonard WJ. Complex interactions of transcription factors in mediating cytokine biology in T cells. Immunol Rev (2014) 261:141–56. doi: 10.1111/imr.12199
99. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: Function, dysfunction and human pathology. Nat Rev Immunol (2015) 15:388–400. doi: 10.1038/nri3839
100. Selvaraj UM, Ujas TA, Kong X, Kumar A, Plautz EJ, Zhang S, et al. Delayed diapedesis of CD8 T cells contributes to long-term pathology after ischemic stroke in male mice. Brain Behav Immun (2021) 95:502–13. doi: 10.1016/j.bbi.2021.05.001
101. Alves de Lima K, Rustenhoven J, Da Mesquita S, Wall M, Salvador AF, Smirnov I, et al. Meningeal γδ T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat Immunol (2020) 21:1421–9. doi: 10.1038/s41590-020-0776-4
102. Arunachalam P, Ludewig P, Melich P, Arumugam TV, Gerloff C, Prinz I, et al. CCR6 (CC chemokine receptor 6) is essential for the migration of detrimental natural interleukin-17-producing γδ T cells in stroke. Stroke (2017) 48:1957–65. doi: 10.1161/STROKEAHA.117.016753
103. Thériault P, Elali A, Rivest S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res Ther (2015) 7(1):41. doi: 10.1186/s13195-015-0125-2
104. Ritzel RM, Patel AR, Grenier JM, Crapser J, Verma R, Jellison ER, et al. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflamm (2015) 12:106. doi: 10.1186/s12974-015-0329-1
105. Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J, et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci (2016) 36:4182–95. doi: 10.1523/JNEUROSCI.4317-15.2016
106. Zhang W, Zhao J, Wang R, Jiang M, Ye Q, Smith AD, et al. Macrophages reprogram after ischemic stroke and promote efferocytosis and inflammation resolution in the mouse brain. CNS Neurosci Ther (2019) 25:1329–42. doi: 10.1111/cns.13256
107. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
108. Guo H, Callaway JB, Ting JPY. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21:677–87. doi: 10.1038/nm.3893
109. ElAli A, LeBlanc NJ. The role of monocytes in ischemic stroke pathobiology: New avenues to explore. Front Aging Neurosci (2016) 8:29. doi: 10.3389/fnagi.2016.00029
110. Xue Y, Nie D, Wang LJ, Qiu HC, Ma L, Dong MX, et al. Microglial polarization: Novel therapeutic strategy against ischemic stroke. Aging Dis (2021) 12:466–79. doi: 10.14336/AD.2020.0701
111. Zeng J, Bao T, Yang K, Zhu X, Wang S, Xiang W, et al. The mechanism of microglia-mediated immune inflammation in ischemic stroke and the role of natural botanical components in regulating microglia: A review. Front Immunol (2023) 13:1047550. doi: 10.3389/fimmu.2022.1047550
112. Lambertsen KL, Clausen BH, Babcock AA, Gregersen R, Fenger C, Nielsen HH, et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci (2009) 29:1319–30. doi: 10.1523/JNEUROSCI.5505-08.2009
113. Ekdahl CT, Kokaia Z, Lindvall O. Brain inflammation and adult neurogenesis: The dual role of microglia. Neuroscience (2009) 158:1021–9. doi: 10.1016/j.neuroscience.2008.06.052
114. Holm TH, Draeby D, Owens T. Microglia are required for astroglial toll-like receptor 4 response and for optimal TLR2 and TLR3 response. Glia (2012) 60:630–8. doi: 10.1002/glia.22296
115. Guttenplan KA, Weigel MK, Adler DI, Couthouis J, Liddelow SA, Gitler AD, et al. Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat Commun (2020) 11(1):3753. doi: 10.1038/s41467-020-17514-9
116. Tarassishin L, Suh HS, Lee SC. LPS and IL-1 differentially activate mouse and human astrocytes: Role of CD14. Glia (2014) 62:999–1013. doi: 10.1002/glia.22657
117. Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med (2018) 24:931–8. doi: 10.1038/s41591-018-0051-5
118. Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science (2018) 359:1269–73. doi: 10.1126/science.aal3589
119. Shi SX, Li YJ, Shi K, Wood K, Ducruet AF, Liu Q. IL (interleukin)-15 bridges astrocyte-microglia crosstalk and exacerbates brain injury following intracerebral hemorrhage. Stroke (2020), 967–74. doi: 10.1161/STROKEAHA.119.028638
120. Lee GA, Lin TN, Chen CY, Mau SY, Huang WZ, Kao YC, et al. Interleukin 15 blockade protects the brain from cerebral ischemia-reperfusion injury. Brain Behav Immun (2018) 73:562–70. doi: 10.1016/j.bbi.2018.06.021
121. Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, et al. Astrocyte-restricted ablation of interleukin-17-induced act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity (2010) 32:414–25. doi: 10.1016/j.immuni.2010.03.004
122. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood (2012) 120:3793–802. doi: 10.1182/blood-2012-02-412726
123. Otxoa-de-Amezaga A, Miró-Mur F, Pedragosa J, Gallizioli M, Justicia C, Gaja-Capdevila N, et al. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol (2019) 137:321–41. doi: 10.1007/s00401-018-1954-4
124. Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature (2019) 565:246–50. doi: 10.1038/s41586-018-0824-5
125. Guo S, Luo Y. Brain Foxp3+ regulatory T cells can be expanded by Interleukin-33 in mouse ischemic stroke. Int Immunopharmacol (2020) 81:106027. doi: 10.1016/j.intimp.2019.106027
126. Yang Y, Liu H, Zhang H, Ye Q, Wang J, Yang B, et al. ST2/IL-33-dependent microglial response limits acute ischemic brain injury. J Neurosci (2017) 37:4692–704. doi: 10.1523/JNEUROSCI.3233-16.2017
127. Guerrero AR, Uchida K, Nakajima H, Watanabe S, Nakamura M, Okada S, et al. Blockade of interleukin-6 effects on cytokine profiles and macrophage activation after spinal cord injury in mice. Neuroprotection Regeneration Spinal Cord (2014) 9784431545:203–12. doi: 10.1007/978-4-431-54502-6_17
128. Wang S, Zhang H, Xu Y. Crosstalk between microglia and T cells contributes to brain damage and recovery after ischemic stroke. Neurol Res (2016) 38:495–503. doi: 10.1080/01616412.2016.1188473
129. Korhonen P, Kanninen KM, Lehtonen Š, Lemarchant S, Puttonen KA, Oksanen M, et al. Immunomodulation by interleukin-33 is protective in stroke through modulation of inflammation. Brain Behav Immun (2015) 49:322–36. doi: 10.1016/j.bbi.2015.06.013
130. Ran Y, Su W, Gao F, Ding Z, Yang S, Ye L, et al. Curcumin Ameliorates White Matter Injury after Ischemic Stroke by Inhibiting Microglia/Macrophage Pyroptosis through NF-κB Suppression and NLRP3 Inflammasome Inhibition. Oxid Med Cell Longev (2021) 2021:1552127. doi: 10.1155/2021/1552127
131. Xiao L, Dai Z, Tang W, Liu C, Tang B, Sase A. Astragaloside IV alleviates cerebral ischemia-reperfusion injury through NLRP3 inflammasome-mediated pyroptosis inhibition via activating Nrf2. Oxid Med Cell Longev (2021) 2021:9925561. doi: 10.1155/2021/9925561
132. Liang J, Wang Q, Li JQ, Guo T, Yu D. Long non-coding RNA MEG3 promotes cerebral ischemia-reperfusion injury through increasing pyroptosis by targeting miR-485/AIM2 axis. Exp Neurol (2020) 325:113139. doi: 10.1016/j.expneurol.2019.113139
133. Zhang D, Qian J, Zhang P, Li H, Shen H, Li X, et al. Gasdermin D serves as a key executioner of pyroptosis in experimental cerebral ischemia and reperfusion model both in vivo and in vitro. J Neurosci Res (2019) 97:645–60. doi: 10.1002/jnr.24385
134. Yuze C, Huixue Z, Xiaoyu L, Jianjian W, Xiaoming Z, Shengnan S, et al. Overexpression of microRNA-9a-5p ameliorates NLRP1 inflammasome-mediated ischemic injury in rats following ischemic stroke. Neuroscience (2020) 444:106–17. doi: 10.1016/j.neuroscience.2020.01.008
135. Gu L, Sun M, Li R, Zhang X, Tao Y, Yuan Y, et al. Didymin suppresses microglia pyroptosis and neuroinflammation through the asc/caspase-1/GSDMD pathway following experimental intracerebral hemorrhage. Front Immunol (2022) 13:810582. doi: 10.3389/fimmu.2022.810582
136. Yan H, Huang W, Rao J, Yuan J. miR-21 regulates ischemic neuronal injury via the p53/Bcl-2/Bax signaling pathway. Aging (2021) 13:22242–55. doi: 10.18632/aging.203530
137. Xie YL, Zhang B, Jing L. MiR-125b blocks Bax/Cytochrome C/Caspase-3 apoptotic signaling pathway in rat models of cerebral ischemia-reperfusion injury by targeting p53. Neurol Res (2018) 40:828–37. doi: 10.1080/01616412.2018.1488654
138. Xing Y, Yang SD, Wang MM, Dong F, Feng YS, Zhang F. Electroacupuncture alleviated neuronal apoptosis following ischemic stroke in rats via midkine and ERK/JNK/p38 signaling pathway. J Mol Neurosci (2018) 66:26–36. doi: 10.1007/s12031-018-1142-y
139. Peng T, Li S, Liu L, Yang C, Farhan M, Chen L, et al. Artemisinin attenuated ischemic stroke induced cell apoptosis through activation of ERK1/2/CREB/BCL-2 signaling pathway in vitro and in vivo. Int J Biol Sci (2022) 18:4578–94. doi: 10.7150/ijbs.69892
140. Gao Y, Signore AP, Yin W, Cao G, Yin XM, Sun F, et al. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab (2005) 25:694–712. doi: 10.1038/sj.jcbfm.9600062
141. Arumugam TV, Cheng Y-L, Choi Y, Choi Y-H, Yang S, Yun Y-K, et al. Evidence that γ-secretase-mediated notch signaling induces neuronal cell death via the nuclear factor-κB-bcl-2-interacting mediator of cell death pathway in ischemic stroke. Mol Pharmacol (2011) 80:23–31. doi: 10.1124/mol.111.071076
142. Zhao Y, Deng B, Li Y, Zhou L, Yang L, Gou X, et al. Electroacupuncture pretreatment attenuates cerebral ischemic injury via notch pathway-mediated up-regulation of hypoxia inducible factor-1α in rats. Cell Mol Neurobiol (2015) 35:1093–103. doi: 10.1007/s10571-015-0203-9
143. Badiola N, Malagelada C, Llecha N, Hidalgo J, Comella JX, Sabriá J, et al. Activation of caspase-8 by tumour necrosis factor receptor 1 is necessary for caspase-3 activation and apoptosis in oxygen-glucose deprived cultured cortical cells. Neurobiol Dis (2009) 35:438–47. doi: 10.1016/j.nbd.2009.06.005
144. Deng XX, Li SS, Sun FY. Necrostatin-1 prevents necroptosis in brains after ischemic stroke via inhibition of RIPK1-mediated RIPK3/MLKL signaling. Aging Dis (2019) 10:807–17. doi: 10.14336/AD.2018.0728
145. Hu Y, Pan H, Peng J, He J, Tang M, Yan S, et al. Resveratrol inhibits necroptosis by mediating the TNF-α/RIP1/RIP3/MLKL pathway in myocardial hypoxia/reoxygenation injury. Acta Biochim Biophys Sin (Shanghai) (2021) 53:430–7. doi: 10.1093/abbs/gmab012
146. Long J, Sun Y, Liu S, Yang S, Chen C, Zhang Z, et al. Targeting pyroptosis as a preventive and therapeutic approach for stroke. Cell Death Discov (2023) 9(1):155. doi: 10.1038/s41420-023-01440-y
147. Dong Z, Pan K, Pan J, Peng Q, Wang Y. The possibility and molecular mechanisms of cell pyroptosis after cerebral ischemia. Neurosci Bull (2018) 34:1131–6. doi: 10.1007/s12264-018-0294-7
148. Malik A, Kanneganti TD. Inflammasome activation and assembly at a glance. J Cell Sci (2017) 130:3955–63. doi: 10.1242/jcs.207365
149. Broz P, Dixit VM. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
150. Barrington J, Lemarchand E, Allan SM. A brain in flame; do inflammasomes and pyroptosis influence stroke pathology? Brain Pathol (2017) 27:205–12. doi: 10.1111/bpa.12476
151. Barrington J, Lemarchand E, Allan SM. A brain in flame; do inflammasomes and pyroptosis influence stroke pathology? Brain Pathol (2017) 27:205–12. doi: 10.1111/bpa.12476
152. Downs KP, Nguyen H, Dorfleutner A, Stehlik C. An overview of the non-canonical inflammasome. Mol Aspects Med (2020) 76:100924. doi: 10.1016/j.mam.2020.100924
153. Newton K, Wickliffe KE, Maltzman A, Dugger DL, Reja R, Zhang Y, et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature (2019) 575:679–82. doi: 10.1038/s41586-019-1752-8
154. Liu L, Cai Y, Deng C. Identification of ANXA3 as a biomarker associated with pyroptosis in ischemic stroke. Eur J Med Res (2023) 28:596. doi: 10.1186/s40001-023-01564-y
155. Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol (2020) 22:1264–75. doi: 10.1038/s41556-020-0575-z
156. Zhang JY, Zhou B, Sun RY, Ai YL, Cheng K, Li FN, et al. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res (2021) 31:980–97. doi: 10.1038/s41422-021-00506-9
157. Davis SM, Pennypacker KR. Targeting antioxidant enzyme expression as a therapeutic strategy for ischemic stroke. Neurochem Int (2017) 107:23–32. doi: 10.1016/j.neuint.2016.12.007
158. Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, et al. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif (2012) 45:487–98. doi: 10.1111/j.1365-2184.2012.00845.x
159. Lüscher C, Malenka RC. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb Perspect Biol (2012) 4:1–15. doi: 10.1101/cshperspect.a005710
160. Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, et al. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J (2016) 35:389–401. doi: 10.15252/embj.201593384
161. Sugawara T, Fujimura M, Noshita N, Kim GW, Saito A, Hayashi T, et al. Neuronal death/survival signaling pathways in cerebral ischemia. NeuroRx (2004) 1:17–25. doi: 10.1602/neurorx.1.1.17
162. Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem (2004) 90:1281–9. doi: 10.1111/j.1471-4159.2004.02572.x
163. Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J, et al. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J Neurosci (1999) 19:3809–17. doi: 10.1523/JNEUROSCI.19-10-03809.1999
164. Samary CS, Ramos AB, Maia LA, Rocha NN, Santos CL, Magalhães RF, et al. Focal ischemic stroke leads to lung injury and reduces alveolar macrophage phagocytic capability in rats. Crit Care (2018) 22:1–11. doi: 10.1186/s13054-018-2164-0
165. Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol (2007) 35:495–516. doi: 10.1080/01926230701320337
166. Muhammad IF, Borné Y, Melander O, Orho-Melander M, Nilsson J, Söderholm M, et al. FADD (Fas-associated protein with death domain), caspase-3, and caspase-8 and incidence of ischemic stroke. Stroke (2018) 49:2224–6. doi: 10.1161/STROKEAHA.118.022063
167. Xu W, Jin W, Zhang X, Chen J, Ren C. Remote limb preconditioning generates a neuroprotective effect by modulating the extrinsic apoptotic pathway and TRAIL-receptors expression. Cell Mol Neurobiol (2017) 37:169–82. doi: 10.1007/s10571-016-0360-5
168. Velier JJ, Ellison JA, Kikly KK, Spera PA, Barone FC, Feuerstein GZ. Caspase-8 and caspase-3 are expressed by different populations of cortical neurons undergoing delayed cell death after focal stroke in the rat. J Neurosci (1999) 19:5932–41. doi: 10.1523/jneurosci.19-14-05932.1999
169. Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ (2019) 26:99–114. doi: 10.1038/s41418-018-0212-6
170. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci (2019) 20:19–33. doi: 10.1038/s41583-018-0093-1
171. Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol (2014) 35:24–32. doi: 10.1016/j.semcdb.2014.02.006
172. Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep (2014) 7:971–81. doi: 10.1016/j.celrep.2014.04.026
173. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell (2014) 54:133–46. doi: 10.1016/j.molcel.2014.03.003
174. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol (2014) 15:135–47. doi: 10.1038/nrm3737
175. Chen AQ, Fang Z, Chen XL, Yang S, Zhou YF, Mao L, et al. Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain–barrier disruption after ischemic stroke. Cell Death Dis (2019) 10(7):487. doi: 10.1038/s41419-019-1716-9
176. Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun (2015) 6:6282. doi: 10.1038/ncomms7282
177. Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Núñez G, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci USA (2017) 114:E961–9. doi: 10.1073/pnas.1613305114
178. Zhou Y, Liao J, Mei Z, Liu X, Ge J. Insight into crosstalk between ferroptosis and necroptosis: novel therapeutics in ischemic stroke. Oxid Med Cell Longev (2021) 2021:9991001. doi: 10.1155/2021/9991001
179. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol (2014) 10:9–17. doi: 10.1038/nchembio.1416
180. Muckenthaler MU, Galy B, Hentze MW. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr (2008) 28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521
181. Palmer C, Menzies SL, Roberts RL, Pavlick G, Connor JR. Changes in iron histochemistry after hypoxic-ischemic brain injury in the neonatal rat. J Neurosci Res (1999) 56:60–71. doi: 10.1002/(SICI)1097-4547(19990401)56:1<60::AID-JNR8>3.0.CO;2-A
182. Seiwert N, Heylmann D, Hasselwander S, Fahrer J. Mechanism of colorectal carcinogenesis triggered by heme iron from red meat. Biochim Biophys Acta Rev Cancer (2020) 1873(1):188334. doi: 10.1016/j.bbcan.2019.188334
183. Magtanong L, Ko PJ, Dixon SJ. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ (2016) 23:1099–109. doi: 10.1038/cdd.2016.25
184. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13:91–8. doi: 10.1038/nchembio.2239
185. Hashidate-Yoshida T, Harayama T, Hishikawa D, Morimoto R, Hamano F, Tokuoka SM, et al. Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. eLife (2015) 4:e06328. doi: 10.7554/eLife.06328.001
186. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA (2016) 113:E4966–75. doi: 10.1073/pnas.1603244113
187. Aoyama K, Nakaki T. Glutathione in cellular Redox homeostasis: Association with the excitatory amino acid carrier 1 (EAAC1). Molecules (2015) 20:8742–58. doi: 10.3390/molecules20058742
188. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
189. Núñez MT, Urrutia P, Mena N, Aguirre P, Tapia V, Salazar J. Iron toxicity in neurodegeneration. BioMetals (2012) 25:761–76. doi: 10.1007/s10534-012-9523-0
190. Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun (2019) 10(1):2091. doi: 10.1038/s41467-019-09753-2
191. Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, et al. Identification of the PANoptosome: A molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol (2020) 10:237. doi: 10.3389/fcimb.2020.00237
192. Samir P, Malireddi RKS, Kanneganti TD. The PANoptosome: A deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol (2020) 10:238. doi: 10.3389/fcimb.2020.00238
193. Lee SJ, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti TD. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature (2021) 597:415–9. doi: 10.1038/s41586-021-03875-8
194. Malireddi RKS, Kesavardhana S, Karki R, Kancharana B, Burton AR, Kanneganti T-D. RIPK1 distinctly regulates yersinia -induced inflammatory cell death, PANoptosis. Immunohorizons (2020) 4:789–96. doi: 10.4049/immunohorizons.2000097
195. Zheng M, Kanneganti TD. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol Rev (2020) 297:26–38. doi: 10.1111/imr.12909
196. Yan WT, Di YY, XM Hu, WY N, LS L, Lu S, et al. Do pyroptosis, apoptosis, and necroptosis (PANoptosis) exist in cerebral ischemia? Evidence from cell and rodent studies. Neural Regener Res (2022) 17:1761–8. doi: 10.4103/1673-5374.331539
197. Yan WT, Zhao WJ, Hu XM, Ban XX, Ning WY, Wan H, et al. PANoptosis-like cell death in ischemia/reperfusion injury of retinal neurons. Neural Regener Res (2023) 18:357–63. doi: 10.4103/1673-5374.346545
198. Zhu T, Wang L, Feng Y, Sun G, Sun X. Classical active ingredients and extracts of chinese herbal medicines: pharmacokinetics, pharmacodynamics, and molecular mechanisms for ischemic stroke. Oxid Med Cell Longev (2021) 2021:8868941. doi: 10.1155/2021/8868941
199. Fan F, Yang L, Li R, Zou X, Li N, Meng X, et al. Salidroside as a potential neuroprotective agent for ischemic stroke: a review of sources, pharmacokinetics, mechanism and safety. Biomed Pharmacother (2020) 129:110458. doi: 10.1016/j.biopha.2020.110458
200. Chen T, Ma Z, Zhu L, Jiang W, Wei T, Zhou R, et al. Suppressing receptor-interacting protein 140: a new sight for salidroside to treat cerebral ischemia. Mol Neurobiol (2016) 53:6240–50. doi: 10.1007/s12035-015-9521-7
201. Hu H, Li Z, Zhu X, Lin R, Chen L. Salidroside reduces cell mobility via NF-κB and MAPK signaling in LPS-induced BV2 microglial cells. Evid-Based Complementary Altern Med (2014) 2014:383821. doi: 10.1155/2014/383821
202. Wei Y, Hong H, Zhang X, Lai W, Wang Y, Chu K, et al. Salidroside inhibits inflammation through PI3K/akt/HIF signaling after focal cerebral ischemia in rats. Inflammation (2017) 40:1297–309. doi: 10.1007/s10753-017-0573-x
203. Zhang X, Lai W, Ying X, Xu L, Chu K, Brown J, et al. Salidroside reduces inflammation and brain injury after permanent middle cerebral artery occlusion in rats by regulating PI3K/PKB/nrf2/NFκB signaling rather than complement C3 activity. Inflammation (2019) 42:1830–42. doi: 10.1007/s10753-019-01045-7
204. Liu X, Wen S, Yan F, Liu K, Liu L, Wang L, et al. Salidroside provides neuroprotection by modulating microglial polarization after cerebral ischemia. J Neuroinflamm (2018) 15:1–11. doi: 10.1186/s12974-018-1081-0
205. Wang Y, Su Y, Lai W, Huang X, Chu K, Brown J, et al. Salidroside restores an anti-inflammatory endothelial phenotype by selectively inhibiting endothelial complement after oxidative stress. Inflammation (2020) 43:310–25. doi: 10.1007/s10753-019-01121-y
206. Zhang X, Du Q, Yang Y, Wang J, Liu Y, Zhao Z, et al. Salidroside alleviates ischemic brain injury in mice with ischemic stroke through regulating BDNK mediated PI3K/Akt pathway. Biochem Pharmacol (2018) 156:99–108. doi: 10.1016/j.bcp.2018.08.015
207. Cai L, Wang H, Li Q, Qian Y, Yao W. Salidroside inhibits H2O2-induced apoptosis in PC 12 cells by preventing cytochrome c release and inactivating of caspase cascade. Acta Biochim Biophys Sin (Shanghai) (2008) 40:796–802. doi: 10.1111/j.1745-7270.2008.00463.x
208. Yu S, Shen Y, Liu J, Ding F. Involvement of ERK1/2 pathway in neuroprotection by salidroside against hydrogen peroxide-induced apoptotic cell death. J Mol Neurosci (2010) 40:321–31. doi: 10.1007/s12031-009-9292-6
209. Shi TY, Feng SF, Xing JH, Wu YM, Li XQ, Zhang N, et al. Neuroprotective effects of Salidroside and its analogue tyrosol galactoside against focal cerebral ischemia in vivo and H 2O 2-induced neurotoxicity in vitro. Neurotox Res (2012) 21:358–67. doi: 10.1007/s12640-011-9290-7
210. Yin F, Zhou H, Fang Y, Li C, He Y, Yu L, et al. Astragaloside IV alleviates ischemia reperfusion-induced apoptosis by inhibiting the activation of key factors in death receptor pathway and mitochondrial pathway. J Ethnopharmacol (2020) 248:112319. doi: 10.1016/j.jep.2019.112319
211. Yang J, Shao C, Li W, Wan H, He Y, Yang J. Protective effects of Astragaloside IV against oxidative injury and apoptosis in cultured astrocytes by regulating Nrf2/JNK signaling. Exp Brain Res (2021) 239:1827–40. doi: 10.1007/s00221-021-06096-7
212. Sun L, Han R, Guo F, Chen H, Wang W, Chen Z, et al. Antagonistic effects of IL-17 and Astragaloside IV on cortical neurogenesis and cognitive behavior after stroke in adult mice through Akt/GSK-3β pathway. Cell Death Discov (2020) 6:74. doi: 10.1038/s41420-020-00298-8
213. Sun YH, Li YQ, Feng SL, Li BX, Pan ZW, Xu CQ, et al. Calcium-sensing receptor activation contributed to apoptosis stimulates TRPC6 channel in rat neonatal ventricular myocytes. Biochem Biophys Res Commun (2010) 394:955–61. doi: 10.1016/j.bbrc.2010.03.096
214. Du SJ, Zhang Y, Zhao YM, Dong YJ, Tang JL, Zhou XH, et al. Astragaloside IV attenuates cerebral ischemia-reperfusion injury in rats through the inhibition of calcium-sensing receptor-mediated apoptosis. Int J Mol Med (2021) 47:302–14. doi: 10.3892/ijmm.2020.4777
215. Li Y, Yang Y, Zhao Y, Zhang J, Liu B, Jiao S, et al. Astragaloside IV reduces neuronal apoptosis and parthanatos in ischemic injury by preserving mitochondrial hexokinase-II. Free Radic Biol Med (2019) 131:251–63. doi: 10.1016/j.freeradbiomed.2018.11.033
216. Xue B, Huang J, Ma B, Yang B, Chang D, Liu J. Astragaloside IV protects primary cerebral cortical neurons from oxygen and glucose deprivation/reoxygenation by activating the PKA/CREB pathway. Neuroscience (2019) 404:326–37. doi: 10.1016/j.neuroscience.2019.01.040
217. Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol (2001) 169:453–9. doi: 10.1677/joe.0.1690453
218. Li L, Gan H, Jin H, Fang Y, Yang Y, Zhang J, et al. Astragaloside IV promotes microglia/macrophages M2 polarization and enhances neurogenesis and angiogenesis through PPARγ pathway after cerebral ischemia/reperfusion injury in rats. Int Immunopharmacol (2021) 92:107335. doi: 10.1016/j.intimp.2020.107335
219. Ni GX, Liang C, Wang J, Duan CQ, Wang P, Wang YL. Astragaloside IV improves neurobehavior and promotes hippocampal neurogenesis in MCAO rats though BDNF-TrkB signaling pathway. Biomed Pharmacother (2020) 130:110353. doi: 10.1016/j.biopha.2020.110353
220. Liang C, Ni GX, Shi XL, Jia L, Wang YL. Astragaloside IV regulates the HIF/VEGF/Notch signaling pathway through miRNA-210 to promote angiogenesis after ischemic stroke. Restor Neurol Neurosci (2020) 38:271–82. doi: 10.3233/RNN-201001
221. Sun L, Zhang H, Wang W, Chen Z, Wang S, Li J, et al. Astragaloside IV exerts cognitive benefits and promotes hippocampal neurogenesis in stroke mice by downregulating interleukin-17 expression via wnt pathway. Front Pharmacol (2020) 11:421. doi: 10.3389/fphar.2020.00421
222. Tu XK, Yang WZ, Shi SS, Chen Y, Wang CH, Chen CM, et al. Baicalin inhibits TLR2/4 signaling pathway in rat brain following permanent cerebral ischemia. Inflammation (2011) 34:463–70. doi: 10.1007/s10753-010-9254-8
223. Lischper M, Beuck S, Thanabalasundaram G, Pieper C, Galla HJ. Metalloproteinase mediated occludin cleavage in the cerebral microcapillary endothelium under pathological conditions. Brain Res (2010) 1326:114–27. doi: 10.1016/j.brainres.2010.02.054
224. Tu XK, Yang WZ, Liang RS, Shi SS, Chen JP, Chen CM, et al. Effect of baicalin on matrix metalloproteinase-9 expression and blood-brain barrier permeability following focal cerebral ischemia in rats. Neurochem Res (2011) 36:2022–8. doi: 10.1007/s11064-011-0526-y
225. Tu XK, Yang WZ, Shi SS, Wang CH, Chen CM. Neuroprotective effect of baicalin in a rat model of permanent focal cerebral ischemia. Neurochem Res (2009) 34:1626–34. doi: 10.1007/s11064-009-9953-4
226. Li S, Sun X, Xu L, Sun R, Ma Z, Deng X, et al. Baicalin attenuates in vivo and in vitro hyperglycemia-exacerbated ischemia/reperfusion injury by regulating mitochondrial function in a manner dependent on AMPK. Eur J Pharmacol (2017) 815:118–26. doi: 10.1016/j.ejphar.2017.07.041
227. Zhou ZQ, Li YL, Ao ZB, Wen ZL, Chen QW, Huang ZG, et al. Baicalin protects neonatal rat brains against hypoxicischemic injury by upregulating glutamate transporter 1 via the phosphoinositide 3-kinase/protein kinase B. Neural Regener Res (2017) 12:1625–31. doi: 10.4103/1673-5374.217335
228. Song X, Gong Z, Liu K, Kou J, Liu B, Liu K. Baicalin combats glutamate excitotoxicity via protecting glutamine synthetase from ROS-induced 20S proteasomal degradation. Redox Biol (2020) 34:101559. doi: 10.1016/j.redox.2020.101559
229. Huang L, Chen C, Zhang X, Li X, Chen Z, Yang C, et al. Neuroprotective effect of curcumin against cerebral ischemia-reperfusion via mediating autophagy and inflammation. J Mol Neurosci (2018) 64:129–39. doi: 10.1007/s12031-017-1006-x
230. Zhang Y, Yan Y, Cao Y, Yang Y, Zhao Q, Jing R, et al. Potential therapeutic and protective effect of curcumin against stroke in the male albino stroke-induced model rats. Life Sci (2017) 183:45–9. doi: 10.1016/j.lfs.2017.06.023
231. Liu Z, Ran Y, Huang S, Wen S, Zhang W, Liu X, et al. Curcumin protects against ischemic stroke by titrating microglia/macrophage polarization. Front Aging Neurosci (2017) 9:233. doi: 10.3389/fnagi.2017.00233
232. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res (2011) 21:103–15. doi: 10.1038/cr.2010.178
233. Li W, Suwanwela NC, Patumraj S. Curcumin prevents Reperfusion injury following ischemic stroke in rats via inhibition of NF-κB, ICAM-1, MMP-9 and caspase-3 expression. Mol Med Rep (2017) 16:4710–20. doi: 10.3892/mmr.2017.7205
234. Xie CJ, Gu AP, Cai J, Wu Y, Chen RC. Curcumin protects neural cells against ischemic injury in N2a cells and mouse brain with ischemic stroke. Brain Behav (2018) 8:1–12. doi: 10.1002/brb3.921
235. Hattori Y, Okamoto Y, Nagatsuka K, Takahashi R, Kalaria RN, Kinoshita M, et al. SIRT1 attenuates severe ischemic damage by preserving cerebral blood flow. Neuroreport (2015) 26:113–7. doi: 10.1097/WNR.0000000000000308
236. Miao Y, Zhao S, Gao Y, Wang R, Wu Q, Wu H, et al. Curcumin pretreatment attenuates inflammation and mitochondrial dysfunction in experimental stroke: The possible role of Sirt1 signaling. Brain Res Bull (2016) 121:9–15. doi: 10.1016/j.brainresbull.2015.11.019
237. Kim EK, Choi EJ. Compromised MAPK signaling in human diseases: an update. Arch Toxicol (2015) 89:867–82. doi: 10.1007/s00204-015-1472-2
238. Lu Z, Liu Y, Shi Y, Shi X, Wang X, Xu C, et al. Curcumin protects cortical neurons against oxygen and glucose deprivation/reoxygenation injury through flotillin-1 and extracellular signal-regulated kinase1/2 pathway. Biochem Biophys Res Commun (2018) 496:515–22. doi: 10.1016/j.bbrc.2018.01.089
Keywords: ischemic stroke, inflammatory, immune cells, cell death, natural compounds
Citation: Gong Z, Guo J, Liu B, Guo Y, Cheng C, Jiang Y, Liang N, Hu M, Song T, Yang L, Li H, Zhang H, Zong X, Che Q and Shi N (2024) Mechanisms of immune response and cell death in ischemic stroke and their regulation by natural compounds. Front. Immunol. 14:1287857. doi: 10.3389/fimmu.2023.1287857
Received: 02 September 2023; Accepted: 26 December 2023;
Published: 11 January 2024.
Edited by:
Xiangsheng Zhang, Capital Medical University, ChinaCopyright © 2024 Gong, Guo, Liu, Guo, Cheng, Jiang, Liang, Hu, Song, Yang, Li, Zhang, Zong, Che and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qianzi Che, Y2hlcWlhbnppMTIzQDEyNi5jb20=; Nannan Shi, MTM4MTE4MzkxNjRAdmlwLjEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.