 Annabelle Small
Annabelle Small Katie Lowe1
Katie Lowe1 Mihir D. Wechalekar
Mihir D. Wechalekar- 1Department of Rheumatology, College of Medicine and Public Health, Flinders University, Adelaide, SA, Australia
- 2Department of Rheumatology, Flinders Medical Centre, Adelaide, SA, Australia
Rheumatoid arthritis (RA) is one of the most prevalent autoimmune inflammatory conditions, and while the mechanisms driving pathogenesis are yet to be completely elucidated, self-reactive T cells and immune checkpoint pathways have a clear role. In this review, we provide an overview of the importance of checkpoint pathways in the T cell response and describe the involvement of these in RA development and progression. We discuss the relationship between immune checkpoint therapy in cancer and autoimmune adverse events, draw parallels with the involvement of immune checkpoints in RA pathobiology, summarise emerging research into some of the lesser-known pathways, and the potential of targeting checkpoint-related pathways in future treatment approaches to RA management.
1 Introduction
Rheumatoid arthritis (RA) is a chronic, systemic, inflammatory autoimmune disease that affects up to 1% of the population (1). Inflammation predominantly targets the synovial tissue (ST; the joint lining), leading to pain, swelling and irreversible joint deformities. Disease complications are not confined to joints, and include systemic involvement (pulmonary, cardiac, haematological, amongst others). Despite treatment advances, a majority of patients do not attain remission, and sustained remission occurs in as few as 15% of patients (2). Although the eventual inflammatory milieu in the RA ST is diverse, checkpoint, or co-stimulatory pathways controlling immune cell activation are critical in engendering, maintaining, and perpetuating the inflammatory response, driven by chronic autoimmune antigenic drive. In parallel with the rise of immune checkpoint inhibitors (ICIs) that have transformed oncologic therapeutics, has been the greater recognition of checkpoint pathways in RA pathogenesis.
In this review, we provide an overview of immune checkpoints at various stages of immune cell development and describe the involvement of these in the development and progression of RA. We discuss the involvement of immune checkpoints in RA pathobiology, the relationship between immune checkpoint therapy in cancer and autoimmune adverse events, and the potential to manipulate these pathways as a future treatment strategy.
2 Immune checkpoints: dictators of the T cell response
T cell activation is a tightly regulated, intricate, and specific process requiring multiple initiating signals. The first is provided through the interaction between a peptide antigen presented by an antigen presenting cell (APC), and a T cell expressing a cognate T cell receptor (TCR). The T cell then requires a secondary signal, referred to as co-stimulation, and the proteins that provide this are called co-stimulatory, or checkpoint molecules. In helper T cells, this is provided through the B7/CD28 co-stimulation axis (3). CD28 expressed on the T cell binds to either CD80 (B7.1) or CD86 (B7.2) on the APC, and together with TCR signalling, provides survival and activation signalling to initiate T cell proliferation (Figure 1). The extent of co-stimulation controls the extent of the initial T cell proliferative response (4), and further signalling provided through specific cytokines dictates cell lineage (5).
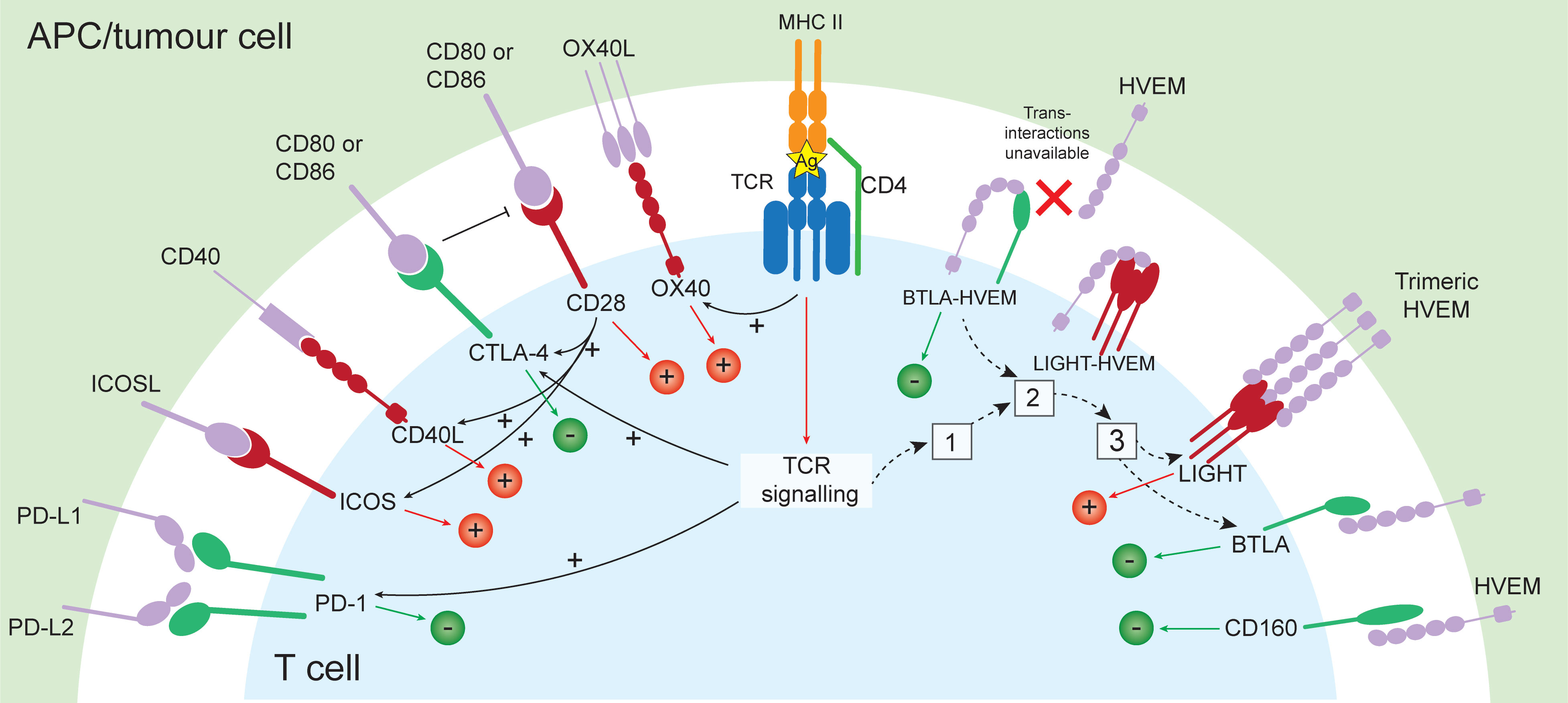
Figure 1 Co-signalling pathways with either stimulatory (+) or inhibitory function (-), and the relationship between their expression. Following presentation of antigen by APC or tumour cell and recognition by a T cell expressing a cognate TCR and co-stimulation provided through CD28 binding CD80/CD86, stimulatory (OX40, CD40L, ICOS, LIGHT) and inhibitory checkpoints are induced (CTLA-4, PD-1). In the resting state, BTLA and HVEM form a cis- complex which exerts constitutive inhibitory signalling to the T cell. Following T cell activation, LIGHT is rapidly and transiently induced (1), and LIGHT disrupts BTLA-HVEM complexes, sequestering HVEM from BTLA. This results in internalisation of HVEM (2), and leaves surface LIGHT and BTLA available for trans- interactions with HVEM (3), capable of inducing stimulatory or inhibitory signals, respectively. Ag, antigen.
Checkpoint molecule expression is largely controlled by the tissue microenvironment, and is influenced by alterations in homeostasis (6). These molecules can exert stimulatory or inhibitory signals in either direction of the interaction (i.e., in the APC and in the T cell) and cumulatively dictate the threshold of T cell activation, allowing for finetuning of the immune response (7, 8). Inhibitory checkpoints, including cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed death protein 1 (PD-1) are upregulated during cell activation, and are important for limiting the response duration, aiding resolution, and maintaining self-tolerance (7). The importance of immune checkpoints in restricting the immune response is exemplified by disturbances in these pathways. Patients with inherited CTLA-4 deficiencies present with a range of clinical features including recurrent respiratory tract infections and increased risk of autoimmunity (9), while inherited PD-1 deficiency has been linked with early-onset autoimmunity and severe tuberculosis (10). Mice deficient in CTLA-4 experience lethal lymphoproliferative disorders (11), and those deficient in PD-1 develop accelerated autoimmunity and lethal pathology in response to acute infection (12–14).
3 The T cell response in RA
The importance of the T cell response in RA has long been appreciated (15–18) and evidence suggests T cell-driven autoimmunity may occur years prior to clinical onset (19, 20). Distinct changes have been observed in the proportions of T follicular helper (Tfh) and CD19+ B cells in the lymph nodes of patients at risk of developing RA (individuals with rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA) but asymptomatic for arthritis) (21), and altered proportions of naïve T cells and T regulatory cells (Treg) have been observed in the peripheral blood (22). In lymph nodes, stromal cells from at-risk patients had distinct hypomethylated sites in genes associated with antigen processing and presentation compared to healthy controls, suggesting altered antigen presentation pathways prior to clinically identifiable disease (23), implicating stromal-lymphoid cell interactions in early RA. Finally, it is well established that RF and ACPA can often be detected in the serum for years in individuals who progress to develop inflammatory arthritis (20, 24).
In clinically identifiable early and established RA, T cells accumulate in the ST sublining layer, where they’re found in association with activated macrophages and B cells (16), and often cluster in aggregates consisting of T cells, B cells, and dendritic cells (DC) (17). While there is enormous variability in synovitis in RA (17, 25), evidence suggests a T helper 1 (Th1) bias in the ST (17), and the demonstration of T cell involvement across all stages of disease suggests that T cell targeting may be efficacious therapeutically broadly throughout disease progression.
Aligning with the importance of the T cell response in RA progression, CTLA-4 fusion protein (CTLA-4-Ig; Abatacept), a soluble protein that binds CD80/CD86 expressed by activated APCs and outcompetes CD28 expressed by T cells at the beginning of the immune response, was approved for use in RA in 2005. Abatacept therefore inhibits circulating and synovial T cell activation by inhibiting CTLA-4:CD28 interactions, and has shown remarkable efficacy in disease management (26), reducing progression of undifferentiated arthritis to RA, and reduction of radiological progression and levels of anti-CCP antibodies (27). Early outcomes from two ongoing trials further suggest Abatacept may also prevent RA development in at-risk healthy individuals (28–30).
4 Inflammatory arthritis as an adverse event of checkpoint therapy in cancer
In cancer, tumour cells can exploit checkpoint pathways controlling the adaptive immune response to neoantigens, inducing tolerance and immune evasion (31, 32). ICIs targeting co-inhibitory receptors in the tumour microenvironment have had remarkable success by unleashing the anti-tumour function of T cells. However, ICI usage is complicated by the occurrence of immune-related adverse events (irAEs) which can present with broad manifestations varying by ICI type (33). Additionally, while combinations of ICIs have shown improved oncologic therapeutic response compared to monotherapies (34), the proportion of patients who developed treatment-related severe irAEs were markedly higher with combination therapy (55%) compared with those in the nivolumab- (16.3%) or ipilimumab-only groups (27.3%), demonstrating that combination therapy may dramatically increase irAE risk (34).
Rheumatological complications occur in ~5-10% of patients treated with ICIs (35), and can manifest in the form of joint inflammation indistinguishable from RA (36), which can persist after therapy cessation (37) and exacerbate existing RA symptoms (38). Additionally, inflammatory arthritis is the most common irAE reported in trials of ICIs (39), an occurrence that has offered a serendipitous window into RA pathogenesis.
5 The role of co-stimulatory pathways in RA
5.1 CTLA-4/CD28/CD80/CD86 pathway
The most well-studied checkpoint pathway in RA is the CTLA-4/CD28/CD80/CD86 pathway. CTLA-4 is the first co-inhibitory molecule to be upregulated following lymphocyte activation (Figure 1) and is critical for controlling initial naïve T cell activation in lymph nodes (40, 41). CTLA-4 is important for Treg function, and binds CD80 with approximately 20 times the affinity of CD28 (42), enabling it to potently inhibit T cell activation. Following binding to CD80/CD86, CTLA-4 can transendocytose its ligands from the APC which are then degraded by the T cell, further impairing co-stimulation through CD28 (43–45).
The most compelling evidence of the role of the CTLA-4/CD28 pathway in the pathogenesis of RA is demonstrated by the efficacy of Abatacept in treating RA. Accordingly, polymorphisms in CTLA4 have been associated with both the development and a decreased risk of RA (46, 47). In RA, serum concentrations of soluble CTLA-4 and CD28 were higher than in healthy controls and serum CTLA-4 correlated positively with disease activity (DAS28) (48), while CTLA-4 expression on RA Tregs was lower compared with healthy controls (49). However, despite the clear role of the CTLA-4/CD28 pathway in the RA immune response, many patients do not respond adequately to Abatacept, indicating disease biology driven by alternative inflammatory pathways (possibly including other checkpoint mechanisms) and highlight the critical unmet need for the development of individualised targeted therapy in RA. Further research into the potential of targeting other co-stimulatory pathways may provide these new approaches, and our knowledge of the role of these pathways in RA is summarised below.
5.2 The PD-1 pathway
Inhibitory signalling through PD-1 regulates activated T cells in later stages of activation (Figure 1) primarily in peripheral tissues, and is associated with chronic antigen exposure (12, 40). PD-1 signalling results in immunoreceptor tyrosine-based switch motif phosphorylation and recruits the tyrosine phosphatase SHP-2, inhibiting cell proliferation and cytokine production (50). In the chronically inflamed RA synovium, in line with the relationship between PD-1/PD-L1 inhibitors and the development of irAEs, expression of PD-1 correlated with synovial inflammation (51), and PD-1 expressing T cells were enriched in the early and established RA ST and periphery (52, 53). These were T peripheral helper (Tph) cells, which contribute to pathogenesis by high production of CXCL13 and interleukin (IL)-21, enabling them to provide B cell help outside of germinal centres (53, 54). Tph cells express high levels of MAF and BATF, and unlike Tfh cells, low levels of BCL6 (53). Additionally, accumulation of PD-1+ activated B cells characterised by increased CD80, CD86, IL-1β and GM-CSF have been demonstrated in the RA ST and SF (55).
Elevated soluble PD-1 (sPD-1) produced by alternative splicing has been demonstrated in the RA periphery (56), and sPD-1 levels positively correlated with RF titres (57) and levels of TNF in the synovial fluid (58). Meanwhile, the PD-1 ligand PD-L1 was conspicuously absent from the ST (56), and sPD-1 has been demonstrated to bind PD-L1 and PD-L2 (59). Therefore, the findings of elevated PD-1+ T cells in the synovium along with elevated sPD-1 and undetectable PD-L1 suggest that the PD-1 pathway is dysregulated in RA, and this lack of PD-1-mediated inhibitory signalling may result in chronic T cell activation, thus contributing to pathogenesis (58, 60).
The evidence suggesting PD-1 pathway dysregulation in RA and the identification of pathogenic PD-1-expressing cells, particularly in early RA, suggests the therapeutic potential of targeting this pathway. In a phase IIa trial, Peresolimab, an anti-PD-1 agonistic antibody, induced a significant reduction in DAS28-CRP by week 12 (61), while another PD-1 agonist Rosnilimab, is awaiting entry into phase II trials in the near future (62). However, given the success of PD-1 inhibition in cancer, longer phase III trials will be required to fully assess the safety profiles of these therapies with regards to oncologic risk.
Finally, recent studies have highlighted the interplay between the PD-1 and CTLA-4/CD28 pathways. On APCs, PD-L1 can form a complex in cis with CD80 (63). This duplex prevents PD-1:PD-L1 binding, inhibiting PD-1 function. Approaches to disrupt this complex using anti-CD80 antibody alleviated autoimmune symptoms (including experimental arthritis) in mice (64), and therefore further study into how the interplay between these pathways contributes to RA pathogenesis is warranted to determine whether similar approaches may be of therapeutic benefit.
5.3 CD40-CD40L pathway
CD40 ligand (CD40L) is a stimulatory checkpoint upregulated on T cells following activation. CD40-CD40L signalling promotes antibody production and class-switching in B cells (65), induces proinflammatory cytokines, chemokines, and adhesion molecules, and can regulate other immune checkpoints (66). Disruption of CD40-CD40L caused by inherited loss-of-function mutations in CD40 or CD40L lead to the X-linked hyper-IgM syndrome, severely compromising humoral immunity (67). Conversely, gain-of-function mutations in CD40 are associated with RA risk and patients homozygous for these alleles express increased B cell CD40 (68). Additionally, in undifferentiated arthritis, and early and established RA, expression of CD40 and CD40LG were increased compared to osteoarthritis and healthy controls, while expanded CD8+CD40L+ ST T cells clones from RA patients expressed CD40L (69), further suggesting a role for increased local CD40- CD40L signalling in the RA ST (70). Finally, supporting the association between increased CD40-CD40L signalling and RA, a recent genome-wide protein quantitative trait locus (pQTL) study demonstrated association with elevated serum CD40 and RA risk (71).
While disruption of CD40-CD40L in patients with homozygous mutations is associated with immunodeficiency, heterozygous carriers are unaffected, indicating that therapeutic inhibition of CD40-CD40L may be well-tolerated (68). Indeed, in a phase 1b clinical trial assessing the therapeutic benefit of systemic CD40L blockade in RA, a CD40L-binding protein VIB4920 significantly decreased disease activity and demonstrated an acceptable safety profile (72). At week 12, 41.7% of patients in the VIB4920 1500 mg group achieved a DAS28-CRP score of <2.6, compared to 6.7% in the placebo group (72). Subsequently, VIB4920 has entered phase II trials alongside anti-TNF.
5.4 ICOS
The inducible T cell co-stimulator (ICOS) is expressed by T cells following CD28-co-stimulation (Figure 1) and is structurally similar to CD28, yet differs functionally by its ability to induce IL-10 following ligation, and inability to induce IL-2 (73). Interaction with its ligand ICOSL induces phosphoinositide 3-kinase (PIK3)-dependent signalling, promoting T cell differentiation and germinal centre formation (74). ICOS signalling was shown to be required for collagen induced arthritis (75, 76), and ICOS-ICOSL blockade targeting ICOSL significantly ameliorated joint inflammation, disease progression and severity (77). In RA, patients had increased proportions of ICOS+ synovial fluid T cells compared to RA peripheral blood T cells (78).
Acaziocolcept, a dual ICOS/CD28 inhibitor trialed for therapeutic efficacy in a murine model of systemic sclerosis (79), inhibited collagen-induced arthritis more potently than Abatacept, and inhibited human T cell function more effectively than inhibition of either ICOS or CD28 alone, suggesting its potential therapeutic use in human disease (80).
5.5 OX40L
OX40 (CD134), similarly to CTLA-4, PD-1 and ICOS, is predominantly expressed by T cells after activation (81). Its ligand OX40L can be induced on APC, Langerhans cells, endothelial cells, mast cells, and NK cells, suggesting a range of functions in lymphoid and non-lymphoid pathways (82). OX40 signalling on T cells provides synergistic signalling alongside CD80-CD28 that prolongs T cell proliferation and enhances IL-2 production (83), while bidirectional signalling through OX40L induces the production of proinflammatory mediators and increased antibody production (84).
In early RA, soluble OX40L was increased and positively correlated with ACPA and RF (85). In the joints, OX40L inhibited osteoclastogenesis (84), and mice lacking OX40L showed decreased bone integrity and enhanced osteoclastogenic capacity (84). Despite this, OX40L blockade in collagen induced arthritis reduced inflammation and inhibited inflammatory cytokine production by OX40L-expressing macrophages (84). Interestingly, patients with RA have been shown to have an abundance of OX40+ Tfh cells capable of producing IL-17 (86), and OX40 was enhanced on Tfh cells within the lymph nodes and demonstrated that severity of arthritis was reduced by blockade using anti-OX40L in glucose-6-phosphate isomerase-induced arthritis.
5.6 BTLA/HVEM/CD160/LIGHT regulatory network
B- and T-lymphocyte attenuator (BTLA) is a member of the CD28 immunoglobulin super family, and is expressed broadly by T and B cells, NK cells, and DCs (87). BTLA forms an intricate regulatory network together with CD160, and LIGHT (TNFSF14), sharing the ligand HVEM (herpes virus entry mediator; TNFRSF14) (88, 89). This network can elicit inhibitory or stimulatory signals depending on the cells expressing them and whether their interactions occur in cis- or trans- (Figure 1) (88). BTLA and HVEM were present in the established RA ST (90), while in the peripheral blood, BTLA was increased on circulating CD3+ T cells while HVEM and LIGHT were decreased compared to healthy controls (91). BTLA polymorphisms have also been associated with RA risk (92). HVEM also binds glycoprotein D (gD) which competes for binding with BTLA and CD160 (88, 93); efforts into blocking the BTLA-HVEM interaction using gD fragments for future cancer therapy are ongoing (94). Reciprocal approaches to free HVEM for interaction with BTLA and CD160 may induce inhibitory signalling and provide a future therapeutic approach in RA.
6 Other pathways
Research into other inhibitory immune checkpoints including TIGIT, TIM-3, and LAG-3 is currently underway in cancer (32, 95), and are tantalising future targets in RA. Increased TIGIT expression on CD4+ RA T cells correlated with autoantibody levels and DAS28 (96), and an agonistic anti-TIGIT antibody, capable of inhibiting Tfh and Tph cells and enhancing Treg function, has recently been described (97). Tregs expressing TIGIT supressed Th1 and Th17 cell responses (98) and signalling through TIGIT on functionally defective Tregs from patients with multiple sclerosis restored suppressive function (99), highlighting the potential of TIGIT-targeting. LAG-3 is constitutively expressed on Tregs; LAG-3 positive Tregs in peripheral blood were reduced in RA patients with higher disease activity, and increased following abatacept therapy (100). Additionally, soluble LAG-3 was increased in early and established RA, correlated with ACPA/RF status and erosive progression in early RA, and decreased inflammatory cytokine release in chronic RA (101). Finally, T cell expression of TIM-3, was increased in RA patients and correlated with decreased DAS28 (102).
Leukocyte-associated immunoglobulin receptors 1 and 2 (LAIR-1 and LAIR-2, respectively) are inhibitory immune receptors for collagens that are expressed broadly across the immune compartment (103, 104). LAIR-1 engagement by collagen or the complement component C1q induces inhibitory T cell signalling (105), and in collagen-induced arthritis, administration of anti-LAIR-1 antibodies significantly attenuated disease (106, 107). In RA, T cell expression of LAIR-1 is reduced compared to healthy and osteoarthritis controls and elevated on ST monocytes and macrophages (108). LAIR-2 is a secreted homolog capable of inhibiting LAIR-1-mediated inhibitory signalling, and is elevated in the RA synovial fluid (109), suggesting dysregulated LAIR-1 action in RA. Together, these suggest that manipulating inhibitory LAIR-1 signalling may offer a future therapeutic approach for RA.
VSIG4 (complement receptor immunoglobulin; CRIg) is a B7-family related protein capable of modulating T cells through interaction with a yet to be identified receptor and has additional roles in immunosuppression (110, 111). VSIG4 binds the complement fragment C3b, inhibiting the alternative complement pathway (112), and approaches utilising this function have shown efficacy in rodent models of RA (113, 114). Administration of soluble VSIG4 reduced disease severity and prevented bone erosion in murine arthritis models (114), and administration of the dual complement inhibitor CRIg-CD59 alleviated symptoms in an adjuvant-induced arthritis rat model (113). In humans, VSIG4 is expressed by tissue-resident regulatory macrophages in the healthy and RA ST from patients in remission (115), and importantly, polymorphisms in VSIG4 have been associated with RA severity (116).
Finally, the signalling lymphocytic activation molecule (SLAM) family has gained recent attention in RA. SLAMF6 was increased in osteoarthritis and early RA ST (117), and in circulating early RA PD-1+CD4+ cells (54) and inhibition of SLAMF6 on CD4+PD-1+ peripheral cells from established RA patients decreased IgG production and plasmablast differentiation (53).
7 Future directions
With their undeniable role in the RA autoimmune milieu, the potential of targeting immune checkpoint pathways with novel approaches is an area of immense interest in RA research. These include modulation utilising small molecules including non-coding microRNAs, and their use in downregulating inhibitory checkpoint pathways are currently under investigation in cancer (118, 119). These are attractive because of their ability to target not only a single gene, but entire pathways (120). Similar strategies capable of inducing these pathways may therefore be efficacious in alleviating autoimmune inflammation. Indeed, MEG3, a long non-coding RNA can modulate TIGIT expression on CD4+ T cells from aplastic anaemia patients (121).
The co-expression of checkpoint molecules on individual cells suggests the approach of targeting multiple checkpoints simultaneously, and there has been significant recent interest in targeting dual targets by bi-specific antibodies. CTLA-4/OX40 (ATOR-1015) (122), and PD-L1/CTLA-4 (KN046) bi-specifics are currently in trial for use in cancer (123). In RA, bi-specific approaches have the potential advantage of targeting checkpoint molecules specifically in the inflammatory niche. For instance, a bi-specific targeting TNF and synovial-specific domain scFv-A7, shows remarkable tissue and disease specificity for the microvascular compartment of the human arthritic ST (124). Interestingly, fewer irAEs appear to be observed following the use of bi-specific antibodies in cancer therapy compared to combination therapy, likely as a result of their site-specific nature (125).
Finally, while the adverse events associated with the use of ICIs in cancer therapy are reasonably well documented, the current lack of agonistic antibodies targeted to checkpoint pathways in human trials makes it difficult to remark on the potential risks associated with the use of checkpoint agonists for the treatment of chronic diseases such as RA. In initial trials, severe adverse events were documented while testing an anti-CD28 agonist in healthy control patients (126), while early trials of Peresolimab in RA do not suggest the development of severe adverse events (61), and in cancer, agonistic antibodies targeting OX40 showed acceptable safety profiles (127). However longer, and larger trials are required to fully assess safety profiles and effect on oncologic risk (61).
8 Concluding remarks
The role of immune checkpoint molecules in RA is a growing area of research, and the elucidation of how these pathways influence the breaching of self-tolerance and drive RA pathogenesis will undoubtedly be critical for selecting the most appropriate therapy for individual patients. However, much remains to be explored, from deciphering the complex interactions between immune cells to fine-tuning the delicate equilibrium of immune activation and suppression. As this field progresses, agonistic immune checkpoint-based therapies may emerge as valuable therapeutic tools for treating RA and may offer new avenues for improved patient care and quality of life.
Author contributions
AS: Conceptualization, Writing – original draft, Writing – review & editing. KL: Writing – original draft, Writing – review & editing. MW: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We acknowledge that AS was supported by funding from an Ideas Grant from the National Health and Medical Research Council (NHMRC) of Australia (Grant Number APP2004839).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Finckh A, Gilbert B, Hodkinson B, Bae S-C, Thomas R, Deane KD, et al. Global epidemiology of rheumatoid arthritis. Nat Rev Rheumatol (2022) 18(10):591–602. doi: 10.1038/s41584-022-00827-y
2. Prince FHM, Bykerk VP, Shadick NA, Lu B, Cui J, Frits M, et al. Sustained rheumatoid arthritis remission is uncommon in clinical practice. Arthritis Res Ther (2012) 14(2):R68. doi: 10.1186/ar3785
3. Sharpe AH, Freeman GJ. The B7–CD28 superfamily. Nat Rev Immunol (2002) 2(2):116–26. doi: 10.1038/nri727
4. Heinzel S, Marchingo JM, Horton MB, Hodgkin PD. The regulation of lymphocyte activation and proliferation. Curr Opin Immunol (2018) 51:32–8. doi: 10.1016/j.coi.2018.01.002
5. Magee CN, Boenisch O, Najafian N. The role of costimulatory molecules in directing the functional differentiation of alloreactive T helper cells. Am J Transplant (2012) 12(10):2588–600. doi: 10.1111/j.1600-6143.2012.04180.x
6. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi: 10.1038/nri3405
7. Sharpe AH. Introduction to checkpoint inhibitors and cancer immunotherapy. Immunol Rev (2017) 276(1):5–8. doi: 10.1111/imr.12531
8. Beier KC, Kallinich T, Hamelmann E. Master switches of T-cell activation and differentiation. Eur Respir J (2007) 29(4):804. doi: 10.1183/09031936.00094506
9. Verma N, Burns SO, Walker LSK, Sansom DM. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin Exp Immunol (2017) 190(1):1–7. doi: 10.1111/cei.12997
10. Ogishi M, Yang R, Aytekin C, Langlais D, Bourgey M, Khan T, et al. Inherited PD-1 deficiency underlies tuberculosis and autoimmunity in a child. Nat Med (2021) 27(9):1646–54. doi: 10.1038/s41591-021-01388-5
11. Khattri R, Auger JA, Griffin MD, Sharpe AH, Bluestone JA. Lymphoproliferative disorder in CTLA-4 knockout mice is characterized by CD28-regulated activation of Th2 responses. J Immunol (1999) 162(10):5784–91. doi: 10.4049/jimmunol.162.10.5784
12. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
13. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11(2):141–51. doi: 10.1016/S1074-7613(00)80089-8
14. Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med (2012) 209(13):2485–99. doi: 10.1084/jem.20121015
15. Weyand CM, Bryl E, Goronzy JJ. The role of T cells in rheumatoid arthritis. Arch Immunol Ther Exp (Warsz) (2000) 48(5):429–35.
16. Smith MD, J, Highton J, Palmer DG, Rozenbilds M, Roberts-Thomson PJ. Immunohistochemical analysis of synovial membranes from inflammatory and non-inflammatory arthritides: scarcity of CD5 positive B cells and IL2 receptor bearing T cells. Pathology (1992) 24(1):19–26. doi: 10.3109/00313029209063615
17. Cope AP. T cells in rheumatoid arthritis. Arthritis Res Ther (2008) 10(1):S1. doi: 10.1186/ar2412
18. Fournier C. Where do T cells stand in rheumatoid arthritis? Joint Bone Spine (2005) 72(6):527–32. doi: 10.1016/j.jbspin.2004.12.012
19. Hähnlein JS, Nadafi R, Jong T, Ramwadhdoebe TH, Semmelink JF, Maijer KI, et al. Impaired lymph node stromal cell function during the earliest phases of rheumatoid arthritis. Arthritis Res Ther (2018) 20(1):35. doi: 10.1186/s13075-018-1529-8
20. Tracy A, Buckley CD, Raza K. Pre-symptomatic autoimmunity in rheumatoid arthritis: when does the disease start? Semin Immunopathol (2017) 39(4):423–35. doi: 10.1007/s00281-017-0620-6
21. Anang DC, Ramwadhdoebe TH, Hähnlein JS, van Kuijk B, Smits N, van Lienden KP, et al. Increased frequency of CD4+ Follicular helper T and CD8+ Follicular T cells in human lymph node biopsies during the earliest stages of rheumatoid arthritis. Cells (2022) 11(7):1104. doi: 10.3390/cells11071104
22. Hunt L, Hensor EM, Nam J, Burska AN, Parmar R, Emery P, et al. T cell subsets: an immunological biomarker to predict progression to clinical arthritis in ACPA-positive individuals. Ann Rheum Dis (2016) 75(10):1884–9. doi: 10.1136/annrheumdis-2015-207991
23. Karouzakis E, Hähnlein J, Grasso C, Semmelink JF, Tak PP, Gerlag DM, et al. Molecular characterization of human lymph node stromal cells during the earliest phases of rheumatoid arthritis. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.01863
24. Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, Koning MH, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum (2004) 50(2):380–6. doi: 10.1002/art.20018
25. Dennis G Jr., Holweg CTJ, Kummerfeld SK, Choy DF, Setiadi AF, Hackney JA, et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Ther (2014) 16(2):R90–0. doi: 10.1186/ar4555
26. Maxwell L, Singh JA. Abatacept for rheumatoid arthritis. Cochrane Database Syst Rev (2009) 2009(4):Cd007277. doi: 10.1002/14651858.CD007277.pub2
27. Emery P, Durez P, Dougados M, Legerton CW, Becker JC, Vratsanos G, et al. Impact of T-cell costimulation modulation in patients with undifferentiated inflammatory arthritis or very early rheumatoid arthritis: a clinical and imaging study of abatacept (the ADJUST trial). Ann Rheum Dis (2010) 69(3):510–6. doi: 10.1136/ard.2009.119016
28. Al-Laith M, Jasenecova M, Abraham S, Bosworth A, Bruce IN, Buckley CD, et al. Arthritis prevention in the pre-clinical phase of RA with abatacept (the APIPPRA study): a multi-centre, randomised, double-blind, parallel-group, placebo-controlled clinical trial protocol. Trials (2019) 20(1):429. doi: 10.1186/s13063-019-3403-7
29. Rech J, Kleyer A, Østergaard M, Hagen M, Valor L, Tascilar K, et al. Abatacept delays the development of RA–clinical results after 18 months from the randomized, placebo-controlled ARIAA study in RA-at risk patients. Ann Rheum Dis (2022) 81:526. doi: 10.1136/annrheumdis-2022-eular.1693
30. Cope A, Jasenecova M, Vasconcelos J, Filer A, Raza K, Qureshi S, et al. Abatacept in individuals at risk of developing rheumatoid arthritis: results from the Arthritis Prevention in the Pre-Clinical Phase of RA with Abatacept (APIPPRA) trial. Ann Rheum Dis (2023) 82:86. doi: 10.1136/annrheumdis-2023-eular.1751
31. Sadeghi Rad H, Monkman J, Warkiani ME, Ladwa R, K, Rezaei N, et al. Understanding the tumor microenvironment for effective immunotherapy. Med Res Rev (2021) 41(3):1474–98. doi: 10.1002/med.21765
32. Shiravand Y, Khodadadi F, Kashani SMA, Hosseini-Fard SR, Hosseini S, Sadeghirad H, et al. Immune checkpoint inhibitors in cancer therapy. Curr Oncol (2022) 29(5):3044–60. doi: 10.3390/curroncol29050247
33. Conroy M, Naidoo J. Immune-related adverse events and the balancing act of immunotherapy. Nat Commun (2022) 13(1):392. doi: 10.1038/s41467-022-27960-2
34. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New Engl J Med (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030
35. Abdel-Wahab N, Suarez-Almazor ME. Frequency and distribution of various rheumatic disorders associated with checkpoint inhibitor therapy. Rheumatol (Oxford) (2019) 58(Suppl 7):vii40–8. doi: 10.1093/rheumatology/kez297
36. Murray-Brown W, Wilsdon TD, Weedon H, Proudman S, Sukumaran S, Klebe S, et al. Nivolumab-induced synovitis is characterized by florid T cell infiltration and rapid resolution with synovial biopsy-guided therapy. J immunother Cancer (2020) 8(1):e000281. doi: 10.1136/jitc-2019-000281
37. Tawnie JB, Julie RB, Patrick MF, Dung L, Evan JL, Jarushka N, et al. Immune checkpoint inhibitor-induced inflammatory arthritis persists after immunotherapy cessation. Ann Rheum Dis (2020) 79(3):332. doi: 10.1136/annrheumdis-2019-216109
38. Jaberg-Bentele NF, Kunz M, Abuhammad S, Dummer R. Flare-up of rheumatoid arthritis by anti-CTLA-4 antibody but not by anti-PD1 therapy in a patient with metastatic melanoma. Case Rep Dermatol (2017) 9(1):65–8. doi: 10.1159/000454875
39. Cappelli LC, Shah AA, Bingham CO 3rd. Immune-related adverse effects of cancer immunotherapy- implications for rheumatology. Rheum Dis Clin North Am (2017) 43(1):65–78. doi: 10.1016/j.rdc.2016.09.007
40. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol (2016) 39(1):98–106. doi: 10.1097/COC.0000000000000239
41. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A new member of the immunoglobulin superfamily–CTLA-4. Nature (1987) 328(6127):267–70. doi: 10.1038/328267a0
42. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med (1991) 174(3):561–9. doi: 10.1084/jem.174.3.561
43. Kennedy A, Waters E, Rowshanravan B, Hinze C, Williams C, Janman D, et al. Differences in CD80 and CD86 transendocytosis reveal CD86 as a key target for CTLA-4 immune regulation. Nat Immunol (2022) 23(9):1365–78. doi: 10.1038/s41590-022-01289-w
44. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood (2018) 131(1):58–67. doi: 10.1182/blood-2017-06-741033
45. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332(6029):600–3. doi: 10.1126/science.1202947
46. Zhou C, Gao S, Yuan X, Shu Z, Li S, Sun X, et al. Association between CTLA-4 gene polymorphism and risk of rheumatoid arthritis: a meta-analysis. Aging (Albany NY) (2021) 13(15):19397–414. doi: 10.18632/aging.203349
47. Liu W, Yang Z, Chen Y, Yang H, Wan X, Zhou X, et al. The association between CTLA-4, CD80/86, and CD28 gene polymorphisms and rheumatoid arthritis: an original study and meta-analysis. Front Med (Lausanne) (2021) 8:598076. doi: 10.3389/fmed.2021.598076
48. Cao J, Zou L, Luo P, Chen P, Zhang L. Increased production of circulating soluble co-stimulatory molecules CTLA-4, CD28 and CD80 in patients with rheumatoid arthritis. Int Immunopharmacol (2012) 14(4):585–92. doi: 10.1016/j.intimp.2012.08.004
49. Flores-Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci U.S.A. (2008) 105(49):19396–401. doi: 10.1073/pnas.0806855105
50. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol (2013) 14(12):1212–8. doi: 10.1038/ni.2762
51. Raptopoulou AP, Bertsias G, Makrygiannakis D, Verginis P, Kritikos I, Tzardi M, et al. The programmed death 1/programmed death ligand 1 inhibitory pathway is up-regulated in rheumatoid synovium and regulates peripheral T cell responses in human and murine arthritis. Arthritis Rheum (2010) 62(7):1870–80. doi: 10.1002/art.27500
52. Murray-Brown W, Guo Y, Small A, Lowe K, Weedon H, Smith MD, et al. Differential expansion of T peripheral helper cells in early rheumatoid arthritis and osteoarthritis synovium. RMD Open (2022) 8(2). doi: 10.1136/rmdopen-2022-002563
53. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature (2017) 542(7639):110–4. doi: 10.1038/nature20810
54. Lowe K, Small A, Song Q, Hao L-Y, Murray-Brown W, Proudman S, et al. Transcriptomic profiling of programmed cell death 1 (PD-1) expressing T cells in early rheumatoid arthritis identifies a decreased CD4 + PD-1 + signature post-treatment. Sci Rep (2023) 13(1):2847. doi: 10.1038/s41598-023-29971-5
55. Floudas A, Neto N, Marzaioli V, Murray K, Moran B, Monaghan MG, et al. Pathogenic, glycolytic PD-1+ B cells accumulate in the hypoxic RA joint. JCI Insight (2020) 5(21). doi: 10.1172/jci.insight.139032
56. Guo Y, Walsh AM, Canavan M, Wechalekar MD, Cole S, Yin X, et al. Immune checkpoint inhibitor PD-1 pathway is down-regulated in synovium at various stages of rheumatoid arthritis disease progression. PloS One (2018) 13(2):e0192704. doi: 10.1371/journal.pone.0192704
57. Liu C, Jiang J, Gao L, Wang X, Hu X, Wu M, et al. Soluble PD-1 aggravates progression of collagen-induced arthritis through Th1 and Th17 pathways. Arthritis Res Ther (2015) 17:340. doi: 10.1186/s13075-015-0859-z
58. Wan B, Nie H, Liu A, Feng G, He D, Xu R, et al. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J Immunol (2006) 177(12):8844–50. doi: 10.4049/jimmunol.177.12.8844
59. Khan M, Zhao Z, Arooj S, Fu Y, Liao G. Soluble PD-1: predictive, prognostic, and therapeutic value for cancer immunotherapy. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.587460
60. Canavan M, Floudas A, Veale DJ, Fearon U. The PD-1:PD-L1 axis in inflammatory arthritis. BMC Rheumatol (2021) 5(1):1–1. doi: 10.1186/s41927-020-00171-2
61. Tuttle J, Drescher E, Simón-Campos JA, Emery P, Greenwald M, Kivitz A, et al. A phase 2 trial of peresolimab for adults with rheumatoid arthritis. N Engl J Med (2023) 388(20):1853–62. doi: 10.1056/NEJMoa2209856
62. Mullard A. PD1 agonist antibody passes first phase II trial for autoimmune disease. Nat Rev Drug Discovery (2023) 22(7):526. doi: 10.1038/d41573-023-00087-9
63. Sugiura D, Maruhashi T, Okazaki IM, Shimizu K, Maeda TK, Takemoto T, et al. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science (2019) 364(6440):558–66. doi: 10.1126/science.aav7062
64. Sugiura D, Okazaki I-M, Maeda TK, Maruhashi T, Shimizu K, Arakaki R, et al. PD-1 agonism by anti-CD80 inhibits T cell activation and alleviates autoimmunity. Nat Immunol (2022) 23(3):399–410. doi: 10.1038/s41590-021-01125-7
65. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev (2009) 229(1):152–72. doi: 10.1111/j.1600-065X.2009.00782.x
66. Román-Fernández IV, García-Chagollán MA, Cerpa-Cruz S, Jave-Suárez LF, Palafox-Sánchez CA, García-Arellano S, et al. Assessment of CD40 and CD40L expression in rheumatoid arthritis patients, association with clinical features and DAS28. Clin Exp Med (2019) 19(4):427–37. doi: 10.1007/s10238-019-00568-5
67. Danese S, Sans M, Fiocchi C. The CD40/CD40L costimulatory pathway in inflammatory bowel disease. Gut (2004) 53(7):1035. doi: 10.1136/gut.2003.026278
68. Li G, Diogo D, Wu D, Spoonamore J, Dancik V, Franke L, et al. Human genetics in rheumatoid arthritis guides a high-throughput drug screen of the CD40 signaling pathway. PloS Genet (2013) 9(5):e1003487. doi: 10.1371/journal.pgen.1003487
69. Kang YM, Zhang X, Wagner UG, Yang H, Beckenbaugh RD, Kurtin PJ, et al. CD8 T cells are required for the formation of ectopic germinal centers in rheumatoid synovitis. J Exp Med (2002) 195(10):1325–36. doi: 10.1084/jem.20011565
70. Guo Y, Walsh AM, Fearon U, Smith MD, Wechalekar MD, Yin X, et al. CD40L-dependent pathway is active at various stages of rheumatoid arthritis disease progression. J Immunol (2017) 198(11):4490–501. doi: 10.4049/jimmunol.1601988
71. Zhao JH, Stacey D, Eriksson N, Macdonald-Dunlop E, Hedman ÅK, Kalnapenkis A, et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat Immunol (2023) 24(9):1540–51. doi: 10.1038/s41590-023-01588-w
72. Karnell JL, Albulescu M, Drabic S, Wang L, Moate R, Baca M, et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci Transl Med (2019) 11(489). doi: 10.1126/scitranslmed.aar6584
73. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397(6716):263–6. doi: 10.1038/16717
74. Gigoux M, Shang J, Pak Y, Xu M, Choe J, Mak TW, et al. Inducible costimulator promotes helper T-cell differentiation through phosphoinositide 3-kinase. Proc Natl Acad Sci U.S.A. (2009) 106(48):20371–6. doi: 10.1073/pnas.0911573106
75. Panneton V, Bagherzadeh Yazdchi S, Witalis M, Chang J, Suh WK. ICOS signaling controls induction and maintenance of collagen-induced arthritis. J Immunol (2018) 200(9):3067–76. doi: 10.4049/jimmunol.1701305
76. Hamel KM, Cao Y, Olalekan SA, Finnegan A. B cell-specific expression of inducible costimulator ligand is necessary for the induction of arthritis in mice. Arthritis Rheumatol (2014) 66(1):60–7. doi: 10.1002/art.38207
77. O'Dwyer R, Kovaleva M, Zhang J, Steven J, Cummins E, Luxenberg D, et al. Anti-ICOSL new antigen receptor domains inhibit T cell proliferation and reduce the development of inflammation in the collagen-induced mouse model of rheumatoid arthritis. J Immunol Res 2018 (2018) p:4089459. doi: 10.1155/2018/4089459
78. Okamoto T, Saito S, Yamanaka H, Tomatsu T, Kamatani N, Ogiuchi H, et al. Expression and function of the co-stimulator H4/ICOS on activated T cells of patients with rheumatoid arthritis. J Rheumatol (2003) 30(6):1157–63.
79. Orvain C, Cauvet A, Prudent A, Guignabert C, Thuillet R, Ottaviani M, et al. Acazicolcept (ALPN-101), a dual ICOS/CD28 antagonist, demonstrates efficacy in systemic sclerosis preclinical mouse models. Arthritis Res Ther (2022) 24(1):13. doi: 10.1186/s13075-021-02709-2
80. Dillon SR, Evans LS, Lewis KE, Debrot S, Blair TC, Mudri S, et al. Non-redundant roles of T cell costimulation pathways in inflammatory arthritis revealed by dual blockade of ICOS and CD28 with acazicolcept (ALPN-101). Arthritis Rheumatol (2023) 75(8):1344–56. doi: 10.1002/art.42484
81. Alves Costa Silva C, Facchinetti F, Routy B, Derosa L. New pathways in immune stimulation: targeting OX40. ESMO Open (2020) 5(1):e000573. doi: 10.1136/esmoopen-2019-000573
82. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev (2009) 229(1):173–91. doi: 10.1111/j.1600-065X.2009.00766.x
83. Gramaglia I, Weinberg AD, Lemon M, Croft M. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol (1998) 161(12):6510–7. doi: 10.4049/jimmunol.161.12.6510
84. Gwyer Findlay E, Danks L, Madden J, Cavanagh MM, McNamee K, McCann F, et al. OX40L blockade is therapeutic in arthritis, despite promoting osteoclastogenesis. Proc Natl Acad Sci U.S.A. (2014) 111(6):2289–94. doi: 10.1073/pnas.1321071111
85. Laustsen JK, Rasmussen TK, Stengaard-Pedersen K, Hørslev-Petersen K, Hetland ML, Østergaard M, et al. Soluble OX40L is associated with presence of autoantibodies in early rheumatoid arthritis. Arthritis Res Ther (2014) 16(5):474. doi: 10.1186/s13075-014-0474-4
86. Kurata I, Matsumoto I, Ohyama A, Osada A, Ebe H, Kawaguchi H, et al. Potential involvement of OX40 in the regulation of autoantibody sialylation in arthritis. Ann Rheum Dis (2019) 78(11):1488–96. doi: 10.1136/annrheumdis-2019-215195
87. Ritthipichai K, Haymaker CL, Martinez M, Aschenbrenner A, Yi X, Zhang M, et al. Multifaceted role of BTLA in the control of CD8(+) T-cell fate after antigen encounter. Clin Cancer Res (2017) 23(20):6151–64. doi: 10.1158/1078-0432.CCR-16-1217
88. Rodriguez-Barbosa JI, Schneider P, Weigert A, Lee K-M, Kim T-J, Perez-Simon J-A, et al. HVEM, a cosignaling molecular switch, and its interactions with BTLA, CD160 and LIGHT. Cell Mol Immunol (2019) 16(7):679–82. doi: 10.1038/s41423-019-0241-1
89. Small A, Cole S, Wang JJ, Nagpal S, Hao LY, Wechalekar MD. Attenuation of the BTLA/HVEM regulatory network in the circulation in primary sjögren's syndrome. J Clin Med (2022) 11(3). doi: 10.3390/jcm11030535
90. Shang Y, Guo G, Cui Q, Li J, Ruan Z, Chen Y. The expression and anatomical distribution of BTLA and its ligand HVEM in rheumatoid synovium. Inflammation (2012) 35(3):1102–12. doi: 10.1007/s10753-011-9417-2
91. Yang B, Huang Z, Feng W, Wei W, Zhang J, Liao Y, et al. The expression of BTLA was increased and the expression of HVEM and LIGHT were decreased in the T cells of patients with rheumatoid arthritis [corrected]. PloS One (2016) 11(5):e0155345. doi: 10.1371/journal.pone.0155345
92. Lin SC, Kuo CC, Chan CH. Association of a BTLA gene polymorphism with the risk of rheumatoid arthritis. J BioMed Sci (2006) 13(6):853–60. doi: 10.1007/s11373-006-9113-7
93. Whitbeck JC, Peng C, Lou H, Xu R, Willis SH, Ponce de Leon M, et al. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the tumor necrosis factor receptor superfamily and a mediator of HSV entry. J Virol (1997) 71(8):6083–93. doi: 10.1128/jvi.71.8.6083-6093.1997
94. Kuncewicz K, Battin C, Węgrzyn K, Sieradzan A, Wardowska A, Sikorska E, et al. Targeting the HVEM protein using a fragment of glycoprotein D to inhibit formation of the BTLA/HVEM complex. Bioorg Chem (2022) 122:105748. doi: 10.1016/j.bioorg.2022.105748
95. Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer (2019) 18(1):155. doi: 10.1186/s12943-019-1091-2
96. Luo Q, Deng Z, Xu C, Zeng L, Ye J, Li X, et al. Elevated expression of immunoreceptor tyrosine-based inhibitory motif (TIGIT) on T lymphocytes is correlated with disease activity in rheumatoid arthritis. Med Sci Monit (2017) 23:1232–41. doi: 10.12659/MSM.902454
97. Kojima M, Suzuki K, Takeshita M, Ohyagi M, Iizuka M, Yamane H, et al. Anti-human-TIGIT agonistic antibody ameliorates autoimmune diseases by inhibiting Tfh and Tph cells and enhancing Treg cells. Commun Biol (2023) 6(1):500. doi: 10.1038/s42003-023-04874-3
98. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40(4):569–81. doi: 10.1016/j.immuni.2014.02.012
99. Lucca LE, Axisa PP, Singer ER, Nolan NM, Dominguez-Villar M, Hafler DA. TIGIT signaling restores suppressor function of Th1 Tregs. JCI Insight (2019) 4(3). doi: 10.1172/jci.insight.124427
100. Nakachi S, Sumitomo S, Tsuchida Y, Tsuchiya H, Kono M, Kato R, et al. Interleukin-10-producing LAG3(+) regulatory T cells are associated with disease activity and abatacept treatment in rheumatoid arthritis. Arthritis Res Ther (2017) 19(1):97. doi: 10.1186/s13075-017-1309-x
101. Pedersen JM, Hansen AS, Skejo C, Juul-Madsen K, Junker P, Horslev-Petersen K, et al. Lymphocyte activation gene 3 is increased and affects cytokine production in rheumatoid arthritis. Arthritis Res Ther (2023) 25(1):97. doi: 10.1186/s13075-023-03073-z
102. Li S, Peng D, He Y, Zhang H, Sun H, Shan S, et al. Expression of TIM-3 on CD4+ and CD8+ T cells in the peripheral blood and synovial fluid of rheumatoid arthritis. APMIS (2014) 122(10):899–904. doi: 10.1111/apm.12228
103. Ramos MIP, Tian L, Ruiter EJ, Song C, Paucarmayta A, Singh A, et al. Cancer immunotherapy by NC410, a LAIR-2 Fc protein blocking human LAIR-collagen interaction. eLife (2021) 10:e62927. doi: 10.7554/eLife.62927.sa2
104. Olde Nordkamp MJ, van Roon JA, Douwes M, de Ruiter T, Urbanus RT, Meyaard L. Enhanced secretion of leukocyte-associated immunoglobulin-like receptor 2 (LAIR-2) and soluble LAIR-1 in rheumatoid arthritis: LAIR-2 is a more efficient antagonist of the LAIR-1-collagen inhibitory interaction than is soluble LAIR-1. Arthritis Rheum (2011) 63(12):3749–57. doi: 10.1002/art.30612
105. Park J-E, Brand DD, Rosloniec EF, Yi A-K, Stuart JM, Kang AH, et al. Leukocyte-associated immunoglobulin-like receptor 1 inhibits T-cell signaling by decreasing protein phosphorylation in the T-cell signaling pathway. J Biol Chem (2020) 295(8):2239–47. doi: 10.1074/jbc.RA119.011150
106. Myers LK, Winstead M, Kee JD, Park JJ, Zhang S, Li W, et al. 1,25-dihydroxyvitamin D3 and 20-hydroxyvitamin D3 upregulate LAIR-1 and attenuate collagen induced arthritis. Int J Mol Sci (2021) 22(24). doi: 10.3390/ijms222413342
107. Kim S, Easterling ER, Price LC, Smith SL, Coligan JE, Park J-E, et al. The role of leukocyte-associated ig-like receptor-1 in suppressing collagen-induced arthritis. J Immunol (2017) 199(8):2692–700. doi: 10.4049/jimmunol.1700271
108. Zhang Y, Lv K, Zhang CM, Jin BQ, Zhuang R, Ding Y. The role of LAIR-1 (CD305) in T cells and monocytes/macrophages in patients with rheumatoid arthritis. Cell Immunol (2014) 287(1):46–52. doi: 10.1016/j.cellimm.2013.12.005
109. Lebbink RJ, van den Berg MC, de Ruiter T, Raynal N, van Roon JA, Lenting PJ, et al. The soluble leukocyte-associated Ig-like receptor (LAIR)-2 antagonizes the collagen/LAIR-1 inhibitory immune interaction. J Immunol (2008) 180(3):1662–9. doi: 10.4049/jimmunol.180.3.1662
110. Widyagarini A, Nishii N, Kawano Y, Zhang C, Azuma M. VSIG4/CRIg directly regulates early CD8(+) T cell activation through its counter-receptor in a narrow window. Biochem Biophys Res Commun (2022) 614:100–6. doi: 10.1016/j.bbrc.2022.04.120
111. Yuan X, Yang BH, Dong Y, Yamamura A, Fu W. CRIg, a tissue-resident macrophage specific immune checkpoint molecule, promotes immunological tolerance in NOD mice, via a dual role in effector and regulatory T cells. Elife (2017) 6. doi: 10.7554/eLife.29540
112. Small AG, Al-Baghdadi M, Quach A, Hii C, Ferrante A. Complement receptor immunoglobulin: a control point in infection and immunity, inflammation and cancer. Swiss Med Wkly (2016) 146:w14301. doi: 10.4414/smw.2016.14301
113. Tao S, Yu H, You T, Kong X, Wei X, Zheng Z, et al. A dual-targeted metal–organic framework based nanoplatform for the treatment of rheumatoid arthritis by restoring the macrophage niche. ACS Nano (2023) 17(14):13917–37. doi: 10.1021/acsnano.3c03828
114. Katschke KJ Jr., Helmy KY, Steffek M, Xi H, Yin J, Lee WP, et al. A novel inhibitor of the alternative pathway of complement reverses inflammation and bone destruction in experimental arthritis. J Exp Med (2007) 204(6):1319–25. doi: 10.1084/jem.20070432
115. Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med (2020) 26(8):1295–306. doi: 10.1038/s41591-020-0939-8
116. Antunes Andrade F, Eibofner Goeldner I, Pieczarka C, Tong van H, Sena L, Skare T, et al. Impact of VSIG4 gene polymorphisms on susceptibility and functional status of rheumatoid arthritis. Int J Immunogenet (2021) 48(3):260–5. doi: 10.1111/iji.12533
117. Walsh AM, Wechalekar MD, Guo Y, Yin X, Weedon H, Proudman SM, et al. Triple DMARD treatment in early rheumatoid arthritis modulates synovial T cell activation and plasmablast/plasma cell differentiation pathways. PloS One (2017) 12(9):e0183928. doi: 10.1371/journal.pone.0183928
118. Alotaibi F. Exosomal microRNAs in cancer: Potential biomarkers and immunotherapeutic targets for immune checkpoint molecules. Front Genet (2023) 14:1052731. doi: 10.3389/fgene.2023.1052731
119. Kmiołek T, Paradowska-Gorycka A. miRNAs as biomarkers and possible therapeutic strategies in rheumatoid arthritis. Cells (2022) 11(3). doi: 10.3390/cells11030452
120. Baumann V, Winkler J. miRNA-based therapies: strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med Chem (2014) 6(17):1967–84. doi: 10.4155/fmc.14.116
121. Wang J, Liu X, Hao C, Lu Y, Duan X, Liang R, et al. MEG3 modulates TIGIT expression and CD4 + T cell activation through absorbing miR-23a. Mol Cell Biochem (2019) 454(1-2):67–76. doi: 10.1007/s11010-018-3453-2
122. Kvarnhammar AM, Veitonmäki N, Hägerbrand K, Dahlman A, Smith KE, Fritzell S, et al. The CTLA-4 x OX40 bispecific antibody ATOR-1015 induces anti-tumor effects through tumor-directed immune activation. J ImmunoTher Cancer (2019) 7(1):103. doi: 10.1186/s40425-019-0570-8
123. Yuxiang M, Jinhui X, Yuanyuan Z, Yang Z, Yan H, Yunpeng Y, et al. Phase I trial of KN046, a novel bispecific antibody targeting PD-L1 and CTLA-4 in patients with advanced solid tumors. J ImmunoTher Cancer (2023) 11(6):e006654. doi: 10.1136/jitc-2022-006654
124. Ferrari M, Onuoha SC, Fossati-Jimack L, Nerviani A, Alves PL, Pagani S, et al. Novel bispecific antibody for synovial-specific target delivery of anti-TNF therapy in rheumatoid arthritis. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.640070
125. Mortezaee K, Majidpoor J. Reinstating immunogenicity using bispecific anti-checkpoint/agent inhibitors. BioMed Pharmacother (2023) 162:114621. doi: 10.1016/j.biopha.2023.114621
126. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med (2006) 355(10):1018–28. doi: 10.1056/NEJMoa063842
Keywords: rheumatoid arthritis, T cells, immune checkpoints, co-stimulatory pathways, CTLA-4, PD-1
Citation: Small A, Lowe K and Wechalekar MD (2023) Immune checkpoints in rheumatoid arthritis: progress and promise. Front. Immunol. 14:1285554. doi: 10.3389/fimmu.2023.1285554
Received: 30 August 2023; Accepted: 07 November 2023;
Published: 24 November 2023.
Edited by:
Yuehong Chen, Sichuan University, ChinaReviewed by:
Samuel García Pérez, Fundación Biomédica Galicia Sur, SpainCopyright © 2023 Small, Lowe and Wechalekar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mihir D. Wechalekar, TWloaXIuV2VjaGFsZWthckBzYS5nb3YuYXU=