 Lihui Guo1,2
Lihui Guo1,2 Xin Su
Xin Su- 1Department of Burns and Plastic Surgery, Yanbian University Hospital, Yanji, China
- 2Department of Rheumatology and Immunology, The First Hospital of China Medical University, China Medical University, Shen Yang, China
Human Immunodeficiency Virus (HIV) has plagued human society for a long time since its discovery, causing a large number of patients to suffer and costing hundreds of millions of medical services every year. Scientists have found that HIV and antiretroviral therapy accelerate immune aging by inducing mitochondrial dysfunction, and that terminal effector memory T cells (TEMRA cells) are crucial in immune aging. This specific subset of effector memory T cells has terminally differentiated properties and exhibits high cytotoxicity and proinflammatory capacity. We therefore explored and described the interplay between exhaustion features, essential markers, functions, and signaling pathways from previous studies on HIV, antiretroviral therapy, immune senescence, and TEMRA cells. Their remarkable antiviral capacity is then highlighted by elucidating phenotypic changes in TEMRA cells during HIV infection, describing changes in TEMRA cells before, during, and after antiretroviral therapy and other drug treatments. Their critical role in complications and cytomegalovirus (CMV)-HIV superinfection is highlighted. These studies demonstrate that TEMRA cells play a key role in the antiviral response and immune senescence during HIV infection. Finally, we review current therapeutic strategies targeting TEMRA cells that may be clinically beneficial, highlight their potential role in HIV-1 vaccine development, and provide perspectives and predictions for related future applications.
1 Introduction
Human immunodeficiency virus(HIV) rapidly progresses to lethal acquired immunodeficiency syndrome (AIDS) if left untreated. However, with the advent of antiretroviral therapy, it is now possible to manage HIV infection as a chronic disease (1, 2). Despite this advancement, HIV infection disrupts the immune system’s homeostasis, leading to significant consequences (3). Long-term HIV infection-related cellular damage exposure accelerates cellular senescence, resulting in chronic inflammation and immune system failure (4, 5). Notably, inflammation has been identified as the second most influential factor, after age itself, in predicting outcomes such as survival, functional capability, and cognition (6). Additionally, antiretroviral therapy may contribute to HIV-associated inflammation and mitochondria-related aging in persons with HIV (PWH) (7–9). These factors together lead to a rise in the prevalence and morbidity of age-related comorbidities in PWH, including cancer, cardiovascular disease, metabolic illness, and neurodegenerative disorders (7, 10). Among those older than 40, the frequency of HIV infections has been rising quickly. According to a longitudinal study of an INDEPTH community in South Africa conducted in 2010, HIV prevalence in those 40 years and older was 21% and increased to 23% 5 years later (11). By 2030, the median age of PWH receiving combination antiretroviral therapy (ART) will be 56.6, up from 43.9 in 2010,thepercentage of PWH aged 50 years or older will be 73%, up from 28% in 2010. according to data from the Dutch AIDS Therapy Evaluation in the Netherlands (ATHENA) cohort (12, 13). Consequently, it is of utmost importance to further investigate the connection between HIV, antiretroviral therapy, and immunosenescence.
HIVand antiretroviral therapy accelerate immune senescence by inducing mitochondrial dysfunction (14, 15). Terminal effector memory T cells (TEMRA cells) are crucial in immune senescence. There are four subpopulations of T cells based on their expression of CD45RA and CCR7: effector memory T cells(TEM, CD45RA-/CCR7-), Naive T cells(TN, CD45RA+/CCR7+), central memory T cells(TCM, CD45RA-/CCR7+), and effector memory T cells re-expressing CD45RA(TEMRA, CD45RA+/CCR7-) (16). TEMRA cells, which are T cells that re-express CD45RA, represent terminally differentiated effector cells associated with protracted antigen exposure. Table 1 presents the primary markers of TEMRA cells, particularly senescent markers. These cells are considered hallmarks of immunosenescence and are characterized by a decline in proliferation potential but strong cytotoxicity and proinflammatory activity. They generate effective effector molecules including perforins, granzymes, IFN-γ, and TNF-α (27) (28, 29). As people age, the proportion of TEMRA cells progressively increases (30). These cells exhibit various characteristics of advanced differentiation, such as a low proliferative activity, high levels of DNA damage and the loss of telomerase activity (31–33). Telomere shortening and the activation of a senescent phenotype are brought on by the relative absence of telomerase activity in TEMRA cells (34). Senescence-associated secretory phenotype (SASP), a distinctive proinflammatory secretory program, is driven by enhanced senescence-associated -galactosidase (SA-Gal) activity. TEMRA cells thrive in an inflammatory milieu and may also exacerbate it by producing multiple proinflammatory molecules recognized as contributors to SASP (32, 35). SASP is regulated by p38 MAPK signaling and has a significant role in inflammation and organismal aging (36).CD8+TEMRA cells and CD4+TEMRA cells exhibit differences in specific senescence-related characteristics. CD8+ TEMRA cells demonstrate significant mitochondrial dysfunction primarily due to decreased mitochondrial mass, leading to compromised metabolic stability and impaired nutrient uptake (37). In contrast, CD4+TEMRA cells showcase a larger mitochondrial mass and absorb more lipids and glucose than CD8+ counterparts (38). Human T cell senescence is mostly determined by mitochondrial mass, and CD8+ TEMRA cells age more rapidly than CD4+ TEMRA cells due to their increased sensitivity to senescence (38, 39). Therefore, the research findings of TEMRA cells and related treatment methods seem to provide new ideas and solutions for the treatment of HIV.
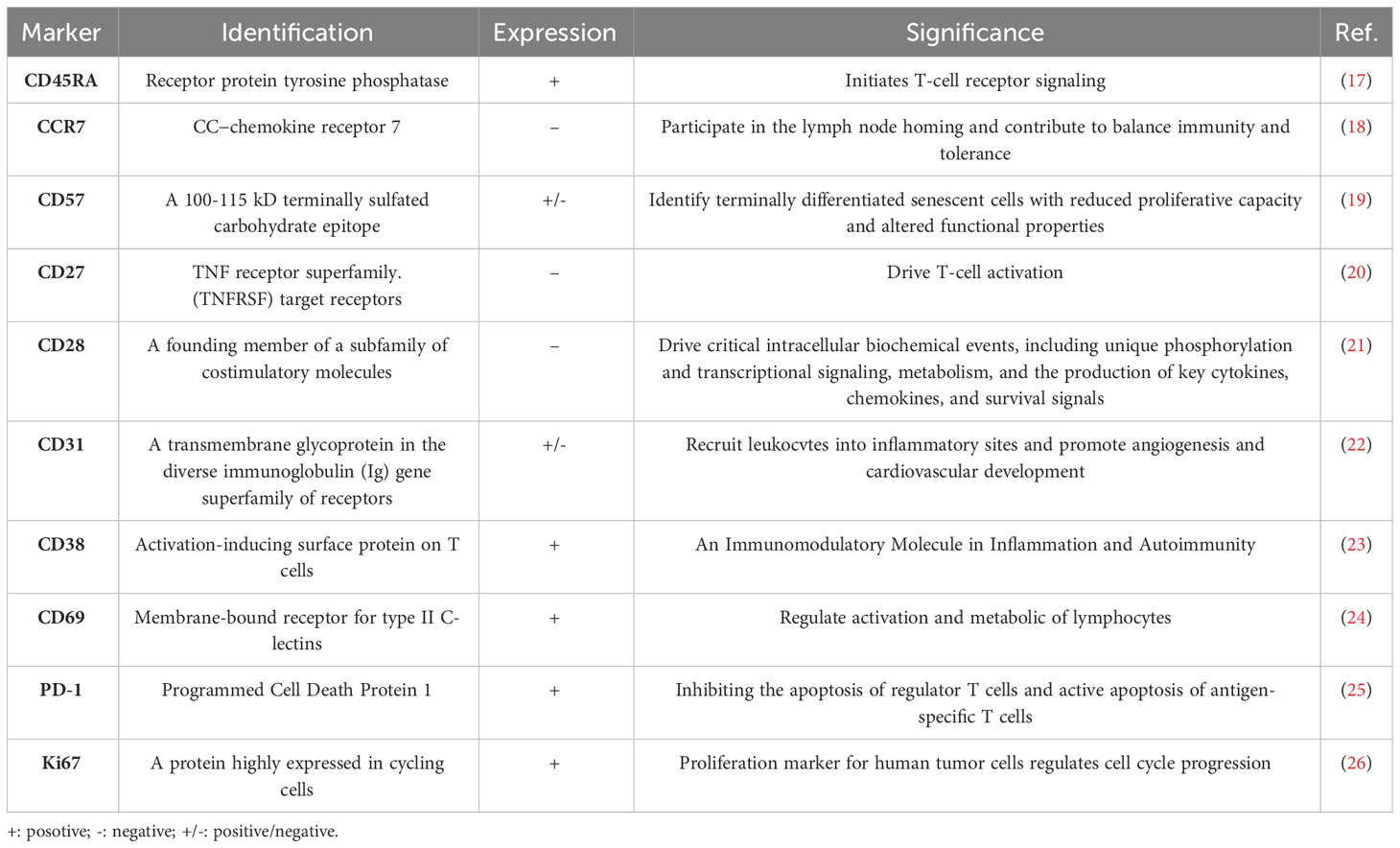
Table 1 Summary of significant markers of TEMRA cell.
Here we aim to review the correlation between HIV, antiretroviral therapy, and immune senescence, and to summarize the characteristics, function, and regulation of TEMRA cells. We comprehensively highlighted the critical role of TEMRA cells in immunosenescence under HIV infection from multiple perspectives, including their alterations, antiviral activity, changes under antiretroviral therapy and other drug therapies, age-associated complications, and cytomegalovirus (CMV)-HIV coinfection. Based on these findings, we underlined the roles of immunosenescence and TEMRA cells in treatments and vaccines for HIV.
2 Mechanism
2.1 The interaction between HIV, antiretroviral therapy, and immunosenescence
2.1.1 HIV and antiretroviral therapy cause immunosenescence and influence mitochondrial function
Immunosenescence is a dynamic and multifactorial process characterized by age-associated changes in immune responses (40, 41), which results from changes in the innate and adaptive immune systems, increasing the risk of infection, decreasing the protection provided by prior vaccines, and decreasing the response to subsequent immunizations (42, 43). Thymic involution, a significant manifestation of immunosenescence during regular aging process, reduces T cell production (44). In response to this production failure, a homeostatic process of memory T cell proliferation takes place, leading to a relative decline in T cell receptor repertoire (TCR) diversity (45).
Chronic viral infections impose a permanent stress on the immune system and reinforce immunosenescence.The enduring hyper-antigenemia such as HIV, hepatitis C virus (HCV), and CMV during the progressive decline of immune system function results in chronic inflammation, ultimately accelerating immune senescence (46, 47). The accumulation of senescent cells creates an immunosuppressive state while promoting viral replication and dissemination, ultimately exacerbating disease pathogenesis and hastening the progression to AIDS (48).
To prevent AIDS progression and the progression of viremia, PWH must be treated with antiretroviral therapy for the rest of their lives (49). The antiretroviral treatment, in particular, interferes with mitochondrial function and causes senescence in a variety of cells, despite just a brief exposure (15).
Both HIV and antiretroviral therapy cause multiple impairments to mitochondrial function, including disturbances in electron transport chain (ETC) respiration and Adenosine 5’-triphosphate (ATP) synthesis, damage to mitochondrial DNA (mtDNA), disruption of mitochondrial membrane potential (▵Ψm), and increased oxidative stress. In the setting of HIV/antiretroviral treatment, increasing oxidative stress causes more mtDNA mutations. Consequently, there is a continuous loop of mtDNA damage, decreased mitochondrial function, increased oxidative stress, and reduced ATP synthesis and cellular homeostasis (50–52). Here we demonstrate the role of TEMRA cells and immune senescence from various perspectives, which will be discussed in the following sections (Figure 1).
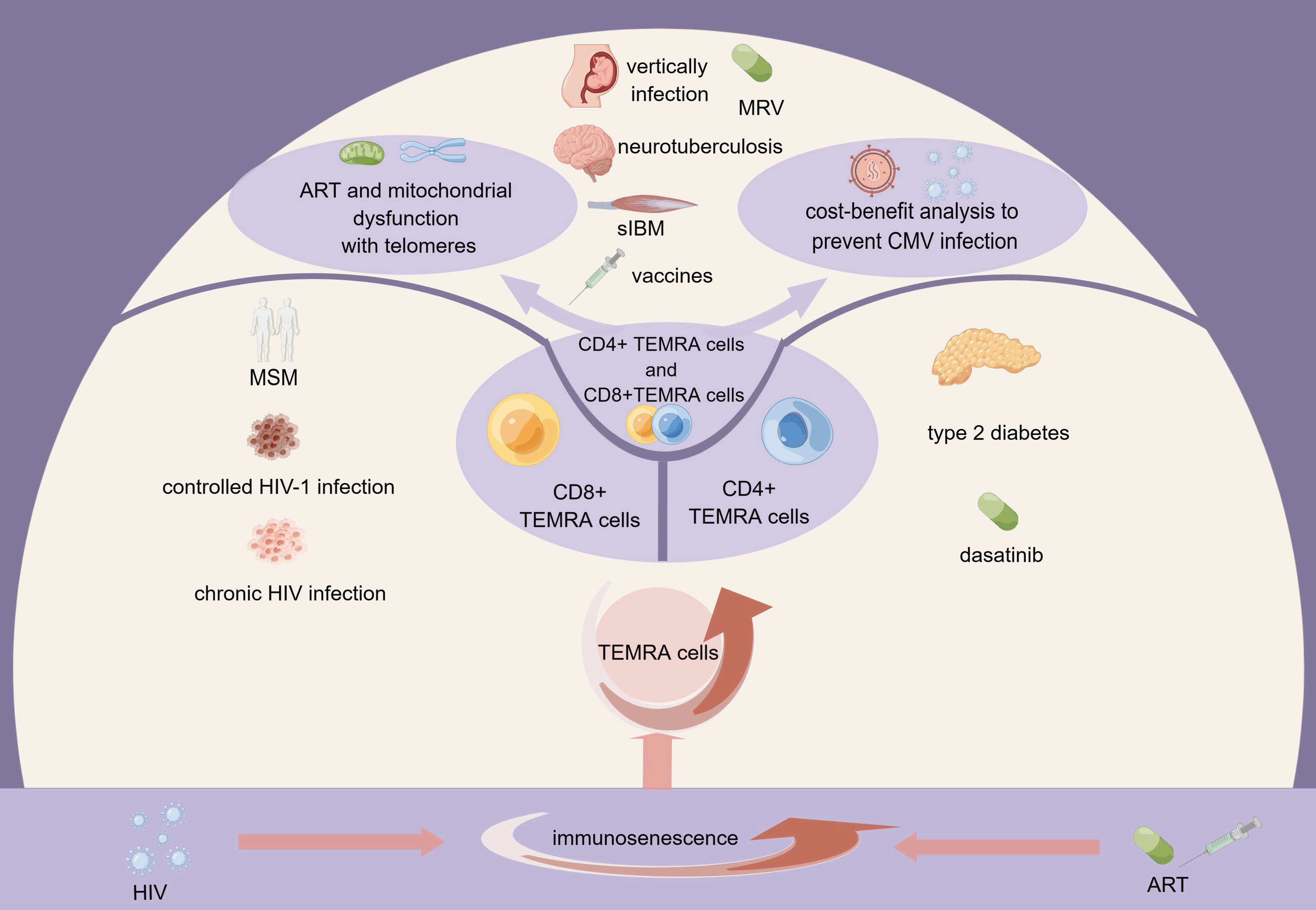
Figure 1 The role of TEMRA cells in HIV infection within the context of immunosenescence. HIV infection and antiretroviral therapy contribute to immunosenescence, resulting in the expansion of TEMRA cells. CD4+TEMRA cells, CD8+TEMRA or both have different connections with the clinical course of HIV infection, HIV infected population, drugs, and complications of HIV infection. The mitochondrial function of TEMRA cells and the cost-benefit analysis to prevent CMV infection are also noteworthy. MSM, men who have sex with men; ART, antiretroviral therapy; MRV, maraviroc; sIBM, sporadic inclusion body myositis; HIV, human immunodeficiency virus; CMV, cytomegalovirus; TEMRA cells, terminal effector memory T cells re-expressing CD45 RA.
2.1.2 CD28 and replicative senescence
CD28 has a central role in the replicative senescence program, which likely represents a characteristic end-stage state of TEMRA cells (53, 54). According to a previous study that involved individuals from various age groups, most CD8+ T cells express CD28 at birth, where the proportion of CD28- T cells gradually rises with age (55, 56).
The absence of CD28 results in upregulating p16 and p21, two vital proteins in cell cycle regulation. The proteins inhibit cyclins and cyclin-dependent kinases responsible for converting G1 to S, leading to G1 arrest and subsequent replicative senescence (57–59).In CD8+CD27− CD28− T cells, reduced phosphorylation of a serine/threonine kinase Akt (Ser473) affects the phosphorylation of human telomerase reverse transcriptase (hTERT) (60). Additionally, down-regulation of CD28 is associated with hTERT loss, resulting in reduced telomerase activity and increased telomere fragility (61).
2.2 Functions and regulations of TEMRA cells
TEMRA cells, particularly CD8+ TEMRA cells, secrete substantial quantities of cytotoxic factors like perforin and granzymes, displaying a significant level of cytotoxicity (27). Additionally, they can assemble into supramolecular attack particles to carry out cytotoxic functions (62). The morphological changes include shrinkage of cells, membrane blebbing, chromatin condensation, and nuclear fragmentation that result from perforin’s disruption of intracellular endosomal membranes (63–65).
TEMRA cells’ cytotoxic potential is regulated by transcription factors T-bet and (eomesodermin)Eomes, which produce granzyme B, granzyme H, and perforin (66). Additionally, T-bet and Eomes are regulated by mammalian target of rapamycin (mTOR), a vital cellular metabolism regulator crucial in developing CD8+ T cells (67).
TEMRA cells not only possess cytotoxic functions but also display a proinflammatory phenotype and secrete multiple inflammatory factors, such as interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), IL-1β, and IL-6 (28). While TEMRA cells initially produce abundant IFN-γ, their ability to do so gradually diminishes upon TCR stimulation. Nonetheless, IL-15 administration can restore their capacity for IFN-γ production. The exact mechanism underlying this restoration remains unknown but is believed to involve the mTOR pathway (68, 69). We summarize the biological characteristics, functions, and signaling pathways of TEMRA cells in Figure 2.
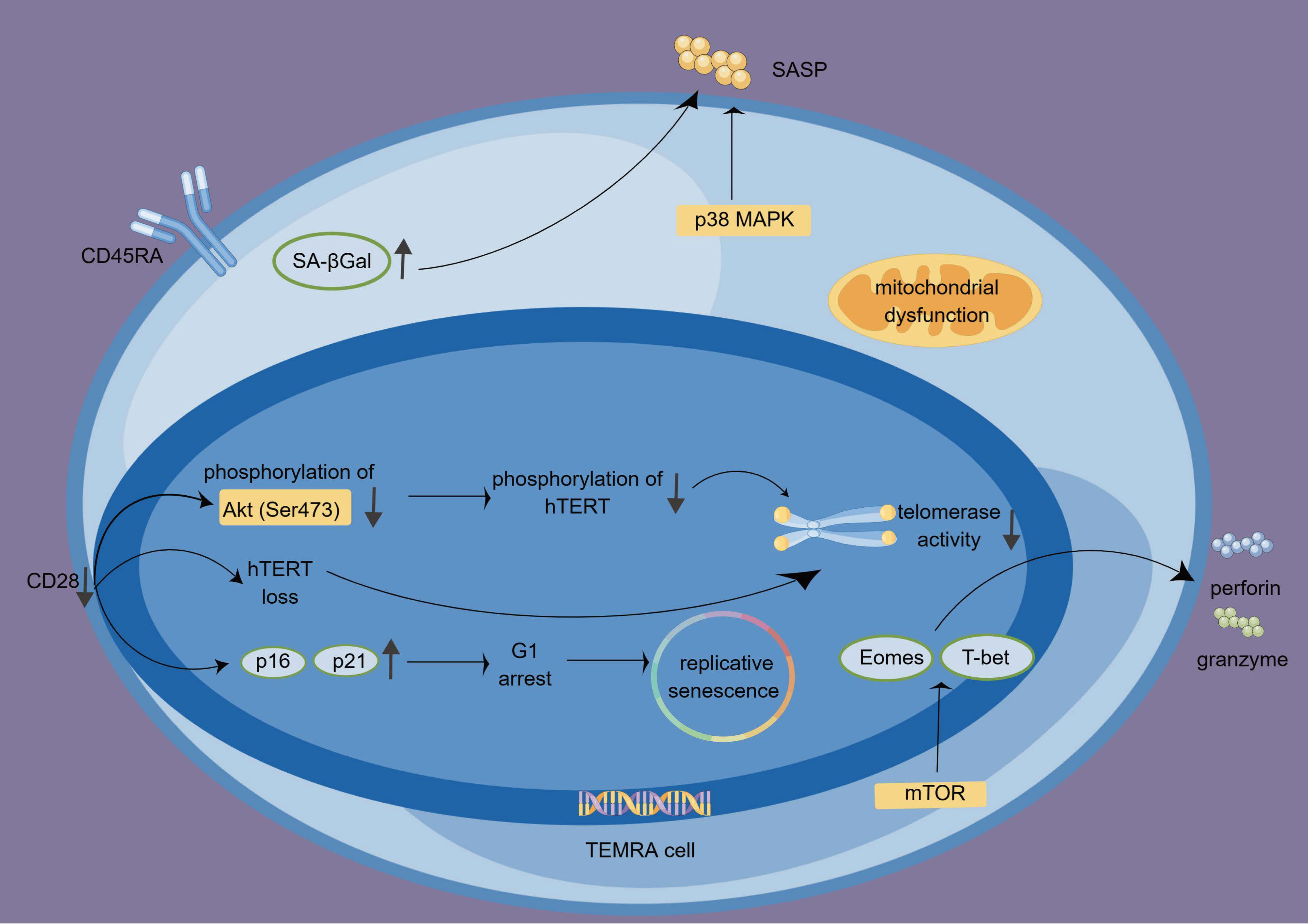
Figure 2 The features, functions, and regulations of TEMRA cells. The absence of CD28 influences the cell cycle, particularly G1 by upregulating p16 and p21, leading to replicative senescence. Additionally, the lack of CD28 contributes to the loss of hTERT and reduced telomerase activity. TEMRA cells produce granzyme and perforin and exhibit high cytotoxicity. This process is regulated by T-bet, Eomes, and mTOR signaling. P38 MAPK signaling participates in the SA-Gal-driven SASP. hTERT, human telomerase reverse transcriptase; SASP, senescence-associated secretory phenotype; TEMRA cells, terminal effector memory T cells re-expressing CD45 RA; SA-βGal, senescence-associated β-galactosidase.
2.3 Interaction of TEMRA cells and HIV
2.3.1 The alteration of TEMRA cells among PWH
2.3.1.1 Major biomarkers alteration
Several previous studies have demonstrated alterations in TEMRA cells in PWH and healthy individuals. There is an increase in the number of TEMRA cells among PWH, as well as an increase in the expression of PD-1, CD38, CD57, and Ki67 (70). Additionally, there is an increase in the number of CD4+TEMRA cells expressing PD-L1 in the viremic HIV-1+ group (71). A PD-1 molecule or PD-L1 ligand acts as a negative regulator of T-cell activity. Furthermore, CD31 expression on memory CD8+ T cells during HIV-1 infection appears to be associated with PD-1 expression on CD8+ TEMRA cells (72). CD57 has previously been proposed as a marker of proliferative history, and more recently as a marker of HIV-specific cytotoxic CD8+ T cells, and correlated with viral suppression (73). Individuals with controlled HIV-1 infection exhibit a higher likelihood of possessing antigen-specific CD8+ TEMRA cells compared to those with progressing infection (74). High expression of EOMES among CD57+ CD8+ TEMRA cells is associated with viral control during chronic HIV infection (75). Moreover, γδT cells play a crucial role in innate immunity as the primary defense against infectious diseases (76). In individuals with acute HIV and rapid progressors, memory Vδ (2) γδ T cells display a preference for the TEMRA Vδ (2) γδ T cell phenotype. The frequency of TEMRA Vδ (2) γδ T cells is positively correlated with the frequency of CD38+ T cells, indicating that HIV infection leads to an excessive activation of TEMRA Vδ (2) γδ T cells (77). Table 2 provides a summary of the significant TEMRA subsets that have experienced alterations, along with their respective characteristics and significance.
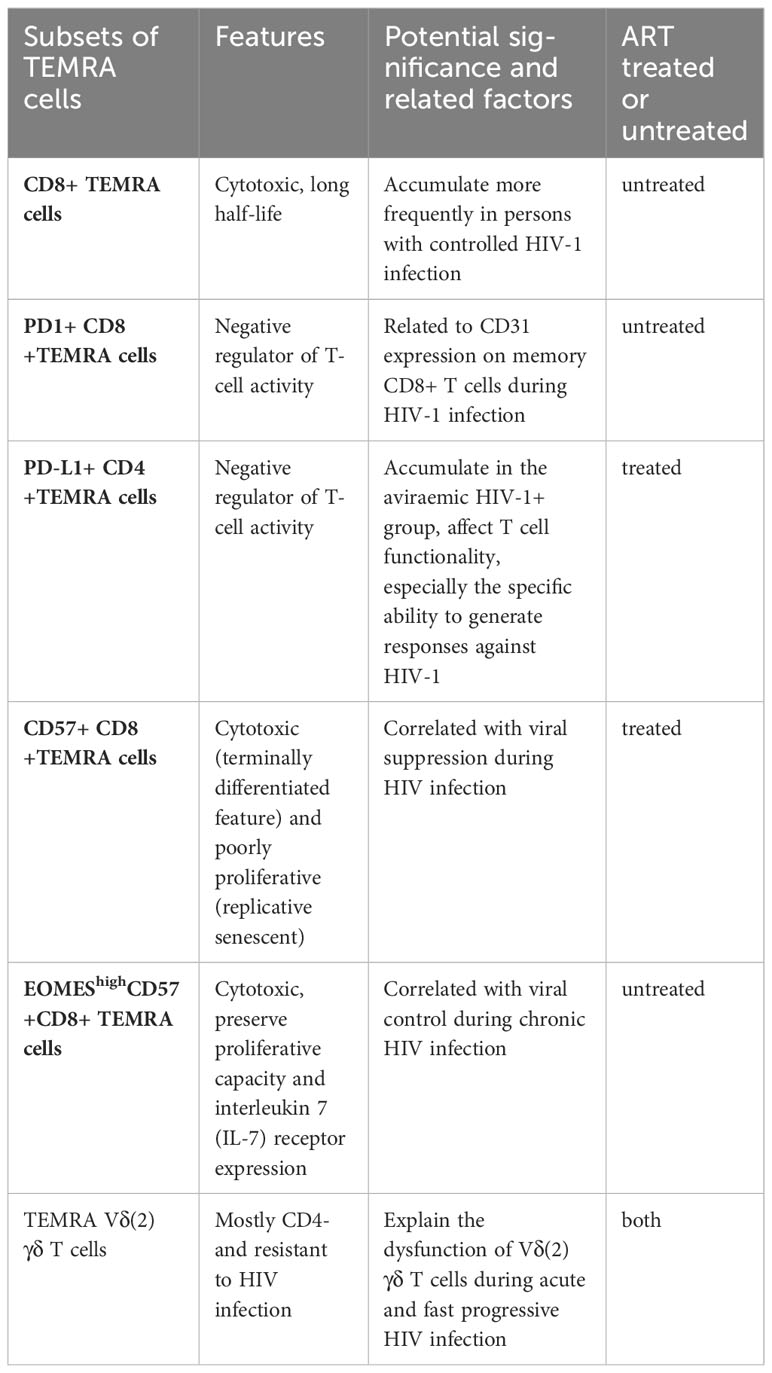
Table 2 Summary of the significant TEMRA subsets, their characteristics and significance.
2.3.1.2 The senescent and activated phenotype of significant infection populations
Male homosexuality and mother-to-child transmission of HIV pose significant global health challenges (78, 79). Additionally, PWH show a statistically significant increased frequency of CD8+ memory T cell subsets with a more activated phenotype (80). In children with vertically acquired HIV-1 infection and a detectable viral load, there is an observed increase in CD8+ TEMRA cells compared to the age-matched healthy group. These CD8+ TEMRA cells also exhibit a more senescent and activated phenotype (81).. Additionally, vertically infected children have significantly higher levels of CD4+ TEMRA cells (82). These findings suggest that HIV infection induces immune activation and drives cells toward terminal differentiation, ultimately resulting in the exhaustion and depletion of these cells.
2.3.2 Antiviral activity of TEMRA cells
CD8+ TEMRA cells have a strong antiviral effect. It has been shown that CD8+ TEMRA cells with HIV-1 specificity are associated with HIV-1 viremia control and can predict the set point of viral loads to come (83). Numerous studies have shown that CD8 T cells, rather than CD4 T cells, exhibit stronger activation in HIV infection with increased viremia (84). The use of consensus HIV-1 gag peptides before in vitro stimulation may increase CD8+ T cells’ viral suppressive capacity (VSC) in people with progressive HIV infection. Significantly, this enhanced VSC is correlated with greater levels of IFN-γ, TNF-α, and IL-10 production, CD8+ TEMRA cells also express immunological checkpoint markers, which are linked to T-cell exhaustion and the loss of T-cell effector functions in the context of sustained antigen exposure. In comparison to nonsuppressors, suppressor individuals have a greater fraction of PD1+CD160+CD8+ TEMRA cells, which may indicate the existence of more active and cytolytic cells that may inhibit viral replication (85). Despite prolonged viral suppression, HIV-1-infected patients are affected by impaired restoration of CD4+ T cells. In patients who demonstrate a good virological response to antiretroviral therapy, there is an association between insufficient CD4 T-cell numbers and altered CD8 T-cell responses, such as poor differentiation and fewer CD8+TEMRA cells, especially in response to Gag p24 stimulation in vitro (86). This impaired differentiation of CD8+ TEMRA cells reflects characteristics of progressive HIV-1 infection. Low CD4+ Tcell counts that continue to exist are expected to affect CD8+ TEM cell development, producing insufficient and defective CD8 +TEMRA cells that are specific for HIV-1.
2.3.3 TEMRA cells in PWH and the selection of HIV-resistant CD4+ T cells
Despite their remarkable resistance to CCR5 (R5)-tropic HIV-1 infection, TEMRA cells remain highly susceptible to CXCR4 (X4)-tropic HIV-1 infection (70). It helps to understand how these individuals may sustain long-term life despite chronically low CD4+ T-cell numbers because R5-tropic virus resistance in TEMRA cells begins after viral entrance but before early viral reverse transcription. In a subgroup of HIV-infected people, a substantial positive connection between the percentage of TEMRA cells and CD4+T cell counts has also been discovered (70). Based on these findings, the development of new anti-HIV-1 treatment approaches might be aided by selecting an HIV-1-resistant CD4+T cell population.
2.3.4 The changes in TEMRA cells during antiretroviral therapy
A latent viral reservoir, predominantly in CD4+ T cells, is the main reason antiretroviral therapy fails to eliminate HIV-1 in infected individuals. The majority of those treated for acute or early HIV-1 infection and HIV-1 controllers have smaller viral reservoirs. People with 50 copies of HIV-DNA per 106 peripheral blood mononuclear cells (PBMCs) were shown to have a reduced percentage of CD8+ TEMRA cells in the absence of antiretroviral treatment (87). In viraemic patients, the initiation of antiretroviral therapy result in increased proportions of CD57- TEMRA cells (88).
During antiretroviral therapy, CD127– T cells were reduced within the TEMRA subset (88). School-age children, teenagers, and young adults in the virological failure (VF) group had more CD4+ TEMRA cells than those in the low-level viremia (LLV) and virological suppression (VS) groups (89).
After antiretroviral therapy, HIV-1 DNA levels and cell associated unspliced RNA (CA usRNA) levels were negatively correlated with CD8+ CCL4-CCL5+ TEMRA cells (90). Among antiretroviral therapy responders and controllers, there was a high percentage of TEMRA cells in CD4+ T cells (91). Compared to healthy controls, viral non-controllers (VNC) had a higher absolute number of CD57+ TEMRA cells, but CD57–TEMRA cells decreased after nine months of antiretroviral therapy (88), which highlighted the importance of TEMRA cells in restraining HIV-1 virus reservoirs individuals undergoing antiretroviral therapy.
These results add to a small body of evidence that the increased cytotoxic TEMRA cells have positive antiviral effects during HIV infection and antiretroviral therapy.
3 Clinical treatment and related applications
3.1 The changes in TEMRA cells under different drugs
Maraviroc (MRV) is the first CCR5 antagonist that has been approved for the treatment of HIV infection and serves as the initial antiretroviral Medications (ARVs)targeting an endogenous chemokine receptor instead of HIV itself (92). In patients with R5 multi-resistant viruses, MRV demonstrates virological effectiveness when combined with other ARVs (93). In PWH receiving eight days of MRV monotherapy, there was a notable rise in CD8+TEMRA cells and a decrease in CD4+TEMRA cells (94). The opposite effect observed in the CD4+TEMRA cells and CD8+ TEMRA cells is likely due to the blockade of CCR5. CCR5 is a receptor expressed on the surface of CD4 +T cells and is involved in various functions, including activation, migration, and survival (95). CCR5 acts as a co-receptor for certain strains of HIV-1, allowing the virus to enter and infect CD4 T cells (96, 97). In addition to its role in viral entry, CCR5 is also involved in the activation of CD4 T cells, promoting their proliferation and cytokine production (98). Further research is needed to investigate this topic. Dasatinib, a tyrosine kinase inhibitor primarily used for the treatment of chronic myeloid leukemia (CML), has been found to have an impact on HIV-1 production in vitro. Several studies have demonstrated that dasatinib can significantly block HIV-1 production in HIV-1-infected primary CD4 +T cells (99–101). Dasatinib hinders TCR-mediated activation of CD4+ T cells and obstructs the integration and reactivation of HIV-1 provirus (101). Additionally, dasatinib can preserve its antiviral function by inhibiting the phosphorylation of SAMHD1 at T592, thus having a crucial role in limiting HIV-1 replication in CD4+ T cells (102). Dasatinib also inhibits IL-2- and IL-7-induced proliferation of CD4+ T cells (103). In PWH receiving antiretroviral therapy and dasatinib, treatment with dasatinib resulted in an average 3.3-fold decrease in the proportion of CD4+ TEMRA cells compared to those receiving solely antiretroviral therapy (104). These findings underscore the significance of CD8+ TEMRA cells in limiting the reservoir in PWH undergoing antiretroviral therapy.
3.2 TEMRA cells’ roles in complications of HIV
PWH are prone to develop various comorbidities resulting from immune deficiency, such as Type 2 diabetes, neurotuberculosis, and sporadic inclusion body myositis (sIBM). Even though PWH may live for decades on effective antiretroviral medication, this success is offset by the population’s rising burden of metabolic illnesses (105–107). A longitudinal study carried out from 2005 to 2007 revealed a correlation between an elevated presence of CD4+TEMRA cells in peripheral blood mononuclear cells (PBMC) and the onset of diabetes in PWH (108). Furthermore, subcutaneous adipose tissue (SAT) of PWH is also enriched with CD4+ TEMRA cells, exhibiting higher CD69 expression and co-expressing CD57, CX3CR1, and GPR56 associated with increased glucose intolerance. The CX3CR1 and GPR56 markers’ expression raises the possibility that TEMRA cells have antiviral selectivity. The adipose tissue of PWH could potentially serve as a significant source of inflammation in the presence of antigen stimulation (109). This collection of virus-specific cells may contribute to adipose tissue inflammation and perhaps increase the susceptibility to developing illnesses in PWH. To determine how these cells, affect adipocytes, however, further research is required. One of the most prevalent HIV-associated opportunistic illnesses of the central nervous system in India is neurotuberculosis. HIV-TB coinfection influences the frequencies and phenotypes of CD4+ TEMRA cells and CD8+ TEMRA cells. In addition, there is a higher frequency of activated CD8+ TEMRA cells than CD4+TEMRA cells (110). sIBM has been identified as a complication of HIV/AIDS since the early days of the HIV/AIDS pandemic (111, 112). The presence of TEMRA cells is a characteristic feature of sIBM in HIV+ patients, although it is not a prerequisite for the development of IBM (113).
3.3 Immunosenescence and TEMRA cells in CMV coinfection populations
In populations infected with HIV, CMV) coinfection is quite common, CMV-specific CD8+TEMRA cells typically outnumber HIV-specific T cells (114). In CMV-HIV coinfection patients, the inflated CMV epitope–specific CD4+ TEMRA could potentially contribute to the higher T cell activation (115). CMV contributes to the accelerated bone loss observed in HIV disease through the proinflammatory secretory profile of CD8+ TEMRA cells (116). Moreover, CMV contributes to cardiovascular pathologies observed in HIV. Independent associations have been observed between CMV-specific T-cell responses (including TEMRA cells) and carotid intima-media thickness (117). T-cell senescence and CMV seropositivity have been identified as predictors of cardiovascular mortality, including death from myocardial infarction and stroke (118). In CMV-positive patients experiencing myocardial ischemia and reperfusion, there is a rapid loss of CD8+ TEMRA cells, potentially due to PD-1 dependent programmed cell death (119).Therefore, CMV and HIV may collectively contribute to immunosenescence, with CMV potentially exerting a more pronounced impact than HIV (120). Clinically, age-related diseases including cardiovascular disease and bone loss may get much worse as people age, which is indicative of an expedited progression of immunosenescence in chronic HIV infection.
Chronic infection with CMV contributes to the accumulation of TEMRA cells (44). CMV infection is the most common congenital infection. Recent some case reports and a small observational study show that high-dose valacyclovir may be a safe and effective preventive measure for congenital CMV (cCMV) among women with primary CMV infection in the first trimester of pregnancy (121).While prophylactic CMV immunization during infancy may be a viable strategy, It is highly unlikely that prophylactic vaccinations against CMV and HIV-1 will be developed anytime soon. There is a link between CMV and the human immune system, demanding a careful cost-benefit analysis to prevent CMV infection (122). In addition, anti-CMV viral medications usually reduce antigenic burden only when severe immunosuppression is present, such as in organ transplant recipients or individuals in advanced stages of HIV disease (123, 124).
3.4 Role of immunosenescence and TEMRA cells in treatments and vaccines of HIV
The buildup of CD8+ TEMRA cells, which show symptoms of replicative senescence, is a hallmark of both immunosenescence and HIV infection. Despite the positive antiviral effects of TEMRA cells, reduced numbers of these cells might lessen some of the negative clinical effects that go along with them and reduce chronic inflammation caused by TEMRA cells’ release of pro-inflammatory cytokines (124). Firstly, for the homeostatic expansion of more functional cells, it may be helpful to explore physical methods to eliminate TEMRA cells from circulation or induce apoptosis in these cells (125). As an alternative to lowering the antigenic load, CD28 or telomerase gene therapy may prevent replicative senescence and improve the activity of virus specific CD8 T cells. When CMV- or HIV-specific CD8+ TEMRA cells with intact CD28 signaling molecules were reintroduced, restoring IL-2 production and proliferative response to antigens (126). Boosting telomerase activity presents a potential strategy for preventing or delaying T lymphocyte senescence. It has been discovered that hTERT promotes enhanced proliferation, telomere length stability, and sustained antiviral action in virus-specific CD8 T cells (127).
In elderly individuals, vaccine responses might be indirectly influenced by the expansion of ‘senescence’ memory cell populations. The growing proportions of these cells within the T lymphocyte pool imply a constrained “immunological space” for the naïve repertoire, potentially contributing to the diminished response of older adults to neoantigens in vaccines (125). Nonetheless, prior research has provided evidence that the modified vaccinia Ankara-based (MVA-B) vaccine elicits targeted immune responses against the vector, partly facilitated by TEMRA cells, specifically CD8+ TEMRA cells. These responses demonstrate robust polyfunctionality and persist even after administering the third dose of MVA-B (128). These findings have significant implications for HIV-1 immunology and the broader domain of HIV-1 vaccine development.
According to the current research results, we predict the strategy and applicable direction of TEMRA for HIV treatment (Box 1).
BOX 1: Future applications of TEMRA cells.
1. As combination therapy or adjuvant therapy in ART.
2. Applied to the treatment of HIV comorbidities by elimating TEMRA cells.
3. For the development of related vaccines.
4. Treatment for improving HIV drug resistance.
5. Applied to the early diagnosis of related immunodeficiency population.
4 Discussion
HIV and antiretroviral therapy contribute to the acceleration of immunosenescence by inducing mitochondrial dysfunction (129, 130).These age-associated immune responses further exacerbate the inflammatory state through the expansion of TEMRA cells, and this terminally differentiated subset displays a high cytotoxic and proinflammatory phenotype. CD28 is a critical marker for TEMRA cells, exerting influence over replicative senescence by regulating the cell cycle and hTERT. To produce effective antiviral responses, TEMRA cells are essential. They also aid in the accelerated immunological senescence seen in PWH. Therefore, we emphasize that TEMRA cells have both positive antiviral effects and negative chronic inflammatory effects during HIV infection. This expansion of TEMRA cells, specifically those expressing exhaustion markers observed in PWH, suggests the involvement of T cell-mediated immune responses in the immune senescence observed during HIV-1 infection. Besides adaptive immune cells, the inflammatory microenvironment of the innate cells may also drives TEMRA cells towards senenscence non- specifically.
TEMRA cells persist in the memory T-cell pool due to their resistance to apoptosis, gradually occupying the pool and thereby restricting the repertoire of remaining T cells (131). This review revealed that the exhaustion marker CD57 can influence the anti-apoptotic properties of TEMRA cells. CD57-TEMRA cells exhibit a longer half-life, declining until nine months after antiretroviral therapy initiation. Conversely, CD57+TEMRA cells are more vulnerable to activation-induced cell death upon antigen stimulation. The presence of HIV (either as reservoirs or with detectable viral load) may induce apoptosis in the CD57+TEMRA subset.
Furthermore, the expression of immune checkpoint markers on TEMRA cells is correlated with viral suppression. The groundwork for future research into the immunological profiles linked to viral suppression is laid by the widespread recognition of these immune checkpoint markers as signs of cellular fatigue. The expression of immune checkpoint markers has been associated with T-cell exhaustion in chronic diseases characterized by prolonged antigen exposure, resulting in a progressive decline in effector functions (132). CD4+ TEMRA cells are strongly linked to more severe immunological suppression during HIV infection, whereas CD8+ TEMRA cells exhibit robust antiviral activity. Despite our identification of the accumulation and phenotypic alterations of TEMRA cells during antiretroviral therapy and specific drug therapies, additional research is necessary to ascertain the specific subsets of TEMRA cells contributing to the suppression of viral replication. Additionally, the discrepancies in TEMRA cell markers and protein levels still require clarification. Among PWH, the diverse behaviors exhibited by TEMRA cells at various stages of differentiation may have significant implications for future treatment strategies. Specific drugs, like MRV, might have distinct effects on TEMRA cells, resulting from the blockade of the CCR5 receptor. Consequently, more extensive research is needed to examine this subject in greater detail.
We believe that these alterations in TEMRA cells, which result from comorbidities of HIV infection, are specific to certain organs. Due to the modified immune status caused by HIV infection, along with notable clinical distinctions like an earlier onset of illness and potentially accelerated disease progression, it is advisable to classify patients with varying HIV comorbidities into subgroups for future investigations. These differences in cytotoxic TEMRA cell differentiation stages may be relevant for potential novel therapeutic strategies. Long-term chronic exposure to CMV plays a significant part in accelerated immunosenescence, which is defined by replicative senescence-related traits, notably the increase of CD8+ TEMRA cells. Reactivation of CMV can also lead to severe complications. Regardless of the underlying causes for the increase of TEMRA cells in PWH, lowering the percentage of these cells may lessen many of the negative clinical outcomes and lessen the severity of age-related pathologies. Further investigation is needed to explore strategies for delaying the generation of senescent CD8+ TEMRA cells. Researchers working on vaccine development have a big difficulty because of the aging immune system. Yet, new vaccine trials to induce antibodies and T-cell immunity have already been initiated, and the results will be revealed in the upcoming years.
In conclusion, the research on TEMRA cells and the exploration of immune senescence may to provide new treatment ideas and research directions for immunodeficiency patients and PWH. The application of related combination therapy and the research and development of vaccines also have huge medical market value. We believe that with the development of technology and in-depth research in related fields, we can eventually treat and prolong the survival of PWH through various means such as drugs or vaccines.
Author contributions
LG: Writing – original draft. XL: Writing – review & editing. XS: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
All the authors acknowledge and thank their respective Institutes and Universities. Figure support was provided by Figdraw.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Egger M, May M, Chêne G, Phillips AN, Ledergerber B, Dabis F, et al. Prognosis of HIV-1-infected patients starting highly active antiretroviral therapy: a collaborative analysis of prospective studies. Lancet (2002) 360(9327):119–29. doi: 10.1016/S0140-6736(02)09411-4
2. Palella FJ Jr., Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators N Engl J Med (1998) 338(13):853–60. doi: 10.1056/NEJM199803263381301
3. Smith C, Sabin CA, Lundgren JD, Thiebaut R, Weber R, Law M, et al. Factors associated with specific causes of death amongst HIV-positive individuals in the D:A:D Study. Aids (2010) 24(10):1537–48. doi: 10.1097/QAD.0b013e32833a0918
4. Humphreys D, ElGhazaly M, Frisan T. Senescence and host-pathogen interactions. Cells (2020) 9(7):1747–64. doi: 10.3390/cells9071747
5. Baz-Martínez M, Da Silva-Álvarez S, Rodríguez E, Guerra J, El Motiam A, Vidal A, et al. Cell senescence is an antiviral defense mechanism. Sci Rep (2016) 6:37007. doi: 10.1038/srep37007
6. Arai Y, Martin-Ruiz CM, Takayama M, Abe Y, Takebayashi T, Koyasu S, et al. Inflammation, but not telomere length, predicts successful ageing at extreme old age: A longitudinal study of semi-supercentenarians. EBioMedicine (2015) 2(10):1549–58. doi: 10.1016/j.ebiom.2015.07.029
7. Van Epps P, Kalayjian RC. Human immunodeficiency virus and aging in the era of effective antiretroviral therapy. Infect Dis Clin North Am (2017) 31(4):791–810. doi: 10.1016/j.idc.2017.07.007
8. Cobos Jiménez V, Wit FW, Joerink M, Maurer I, Harskamp AM, Schouten J, et al. T-cell activation independently associates with immune senescence in HIV-infected recipients of long-term antiretroviral treatment. J Infect Dis (2016) 214(2):216–25. doi: 10.1093/infdis/jiw146
9. Sieg SF, Shive CL, Panigrahi S, Freeman ML. Probing the interface of HIV and inflammaging. Curr HIV/AIDS Rep (2021) 18(3):198–210. doi: 10.1007/s11904-021-00547-0
10. Appay V, Sauce D. Assessing immune aging in HIV-infected patients. Virulence (2017) 8(5):529–38. doi: 10.1080/21505594.2016.1195536
11. Gómez-Olivé FX, Houle B, Rosenberg M, Kabudula C, Mojola S, Rohr JK, et al. Brief report: HIV incidence among older adults in a rural South African setting: 2010-2015. J Acquir Immune Defic Syndr (2020) 85(1):18–22. doi: 10.1097/QAI.0000000000002404
12. Boender TS, Smit C, Sighem AV, Bezemer D, Ester CJ, Zaheri S, et al. AIDS Therapy Evaluation in the Netherlands (ATHENA) national observational HIV cohort: cohort profile. BMJ Open (2018) 8(9):e022516. doi: 10.1136/bmjopen-2018-022516
13. Smit M, Brinkman K, Geerlings S, Smit C, Thyagarajan K, Sighem A, et al. Future challenges for clinical care of an ageing population infected with HIV: a modelling study. Lancet Infect Dis (2015) 15(7):810–8. doi: 10.1016/S1473-3099(15)00056-0
14. Huang CY, Chiang SF, Lin TY, Chiou SH, Chow KC. HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction. PloS One (2012) 7(3):e33657. doi: 10.1371/journal.pone.0033657
15. Caron M, Auclairt M, Vissian A, Vigouroux C, Capeau J. Contribution of mitochondrial dysfunction and oxidative stress to cellular premature senescence induced by antiretroviral thymidine analogues. Antivir Ther (2008) 13(1):27–38. doi: 10.1177/135965350801300103
16. Larbi A, Fulop T. From “truly naïve” to “exhausted senescent” T cells: when markers predict functionality. Cytometry A (2014) 85(1):25–35. doi: 10.1002/cyto.a.22351
17. Rheinländer A, Schraven B, Bommhardt U. CD45 in human physiology and clinical medicine. Immunol Lett (2018) 196:22–32. doi: 10.1016/j.imlet.2018.01.009
18. Förster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol (2008) 8(5):362–71. doi: 10.1038/nri2297
19. Kared H, Martelli S, Ng TP, Pender SL, Larbi A. CD57 in human natural killer cells and T-lymphocytes. Cancer Immunol Immunother (2016) 65(4):441–52. doi: 10.1007/s00262-016-1803-z
20. Buchan SL, Rogel A, Al-Shamkhani A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood (2018) 131(1):39–48. doi: 10.1182/blood-2017-07-741025
21. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: from mechanism to therapy. Immunity (2016) 44(5):973–88. doi: 10.1016/j.immuni.2016.04.020
22. DeLisser HM, Newman PJ, Albelda SM. Molecular and functional aspects of PECAM-1/CD31. Immunol Today (1994) 15(10):490–5. doi: 10.1016/0167-5699(94)90195-3
23. Piedra-Quintero ZL, Wilson Z, Nava P, Guerau-de-Arellano M. CD38: an immunomodulatory molecule in inflammation and autoimmunity. Front Immunol (2020) 11:597959. doi: 10.3389/fimmu.2020.597959
24. Cibrián D, Sánchez-Madrid F. CD69: from activation marker to metabolic gatekeeper. Eur J Immunol (2017) 47(6):946–53. doi: 10.1002/eji.201646837
25. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res (2020) 10(3):727–42.
26. Sun X, Kaufman PD. Ki-67: more than a proliferation marker. Chromosoma (2018) 127(2):175–86. doi: 10.1007/s00412-018-0659-8
27. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
28. Ladell K, Hellerstein MK, Cesar D, Busch R, Boban D, McCune JM. Central memory CD8+ T cells appear to have a shorter lifespan and reduced abundance as a function of HIV disease progression. J Immunol (2008) 180(12):7907–18. doi: 10.4049/jimmunol.180.12.7907
29. Rodriguez RM, Suarez-Alvarez B, Lavín JL, Mosén-Ansorena D, Baragaño Raneros A, Márquez-Kisinousky L, et al. Epigenetic networks regulate the transcriptional program in memory and terminally differentiated CD8+ T cells. J Immunol (2017) 198(2):937–49. doi: 10.4049/jimmunol.1601102
30. Verma K, Ogonek J, Varanasi PR, Luther S, Bünting I, Thomay K, et al. Human CD8+ CD57- TEMRA cells: Too young to be called “old”. PloS One (2017) 12(5):e0177405. doi: 10.1371/journal.pone.0177405
31. Henson SM, Macaulay R, Riddell NE, Nunn CJ, Akbar AN. Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8(+) T-cell proliferation by distinct pathways. Eur J Immunol (2015) 45(5):1441–51. doi: 10.1002/eji.201445312
32. Callender LA, Carroll EC, Beal RWJ, Chambers ES, Nourshargh S, Akbar AN, et al. Human CD8(+) EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell (2018) 17(1):1–9. doi: 10.1111/acel.12675
33. Meyer-Olson D, Simons BC, Conrad JA, Smith RM, Barnett L, Lorey SL, et al. Clonal expansion and TCR-independent differentiation shape the HIV-specific CD8+ effector-memory T-cell repertoire in vivo. Blood (2010) 116(3):396–405. doi: 10.1182/blood-2009-11-254136
34. Coppé JP, Patil CK, Rodier F, Krtolica A, Beauséjour CM, Parrinello S, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PloS One (2010) 5(2):e9188. doi: 10.1371/journal.pone.0009188
35. Lopes-Paciencia S, Saint-Germain E, Rowell MC, Ruiz AF, Kalegari P, Ferbeyre G. The senescence-associated secretory phenotype and its regulation. Cytokine (2019) 117:15–22. doi: 10.1016/j.cyto.2019.01.013
36. Moskalev A, Stambler I, Caruso C. Innate and adaptive immunity in aging and longevity: the foundation of resilience. Aging Dis (2020) 11(6):1363–73. doi: 10.14336/AD.2020.0603
37. Henson SM, Lanna A, Riddell NE, Franzese O, Macaulay R, Griffiths SJ, et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J Clin Invest (2014) 124(9):4004–16. doi: 10.1172/JCI75051
38. Callender LA, Carroll EC, Bober EA, Akbar AN, Solito E, Henson SM. Mitochondrial mass governs the extent of human T cell senescence. Aging Cell (2020) 19(2):e13067. doi: 10.1111/acel.13067
39. Czesnikiewicz-Guzik M, Lee WW, Cui D, Hiruma Y, Lamar DL, Yang ZZ, et al. T cell subset-specific susceptibility to aging. Clin Immunol (2008) 127(1):107–18. doi: 10.1016/j.clim.2007.12.002
40. Weyand CM, Goronzy JJ. Aging of the immune system. Mechanisms and therapeutic targets. Ann Am Thorac Soc (2016) 13 Suppl 5(Suppl 5):S422–s8. doi: 10.1513/AnnalsATS.201602-095AW
41. DeVeale B, Brummel T, Seroude L. Immunity and aging: the enemy within? Aging Cell (2004) 3(4):195–208. doi: 10.1111/j.1474-9728.2004.00106.x
42. Sasaki S, Sullivan M, Narvaez CF, Holmes TH, Furman D, Zheng NY, et al. Limited efficacy of inactivated influenza vaccine in elderly individuals is associated with decreased production of vaccine-specific antibodies. J Clin Invest (2011) 121(8):3109–19. doi: 10.1172/JCI57834
43. Buffa S, Bulati M, Pellicanò M, Dunn-Walters DK, Wu YC, Candore G, et al. B cell immunosenescence: different features of naive and memory B cells in elderly. Biogerontology (2011) 12(5):473–83. doi: 10.1007/s10522-011-9353-4
44. Sauce D, Larsen M, Fastenackels S, Duperrier A, Keller M, Grubeck-Loebenstein B, et al. Evidence of premature immune aging in patients thymectomized during early childhood. J Clin Invest (2009) 119(10):3070–8. doi: 10.1172/JCI39269
45. Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol (2011) 11(4):289–95. doi: 10.1038/nri2959
46. Papagno L, Spina CA, Marchant A, Salio M, Rufer N, Little S, et al. Immune activation and CD8+ T-cell differentiation towards senescence in HIV-1 infection. PloS Biol (2004) 2(2):E20. doi: 10.1371/journal.pbio.0020020
47. Hunt PW, Martin JN, Sinclair E, Epling L, Teague J, Jacobson MA, et al. Valganciclovir reduces T cell activation in HIV-infected individuals with incomplete CD4+ T cell recovery on antiretroviral therapy. J Infect Dis (2011) 203(10):1474–83. doi: 10.1093/infdis/jir060
48. Deeks SG, Verdin E, McCune JM. Immunosenescence and HIV. Curr Opin Immunol (2012) 24(4):501–6. doi: 10.1016/j.coi.2012.05.004
49. Schank M, Zhao J, Moorman JP, Yao ZQ. The impact of HIV- and ART-induced mitochondrial dysfunction in cellular senescence and aging. Cells (2021) 10(1):174. doi: 10.3390/cells10010174
50. Graziewicz MA, Day BJ, Copeland WC. The mitochondrial DNA polymerase as a target of oxidative damage. Nucleic Acids Res (2002) 30(13):2817–24. doi: 10.1093/nar/gkf392
51. Li M, Foli Y, Liu Z, Wang G, Hu Y, Lu Q, et al. High frequency of mitochondrial DNA mutations in HIV-infected treatment-experienced individuals. HIV Med (2017) 18(1):45–55. doi: 10.1111/hiv.12390
52. Apostolova N, Gomez-Sucerquia LJ, Moran A, Alvarez A, Blas-Garcia A, Esplugues JV. Enhanced oxidative stress and increased mitochondrial mass during efavirenz-induced apoptosis in human hepatic cells. Br J Pharmacol (2010) 160(8):2069–84. doi: 10.1111/j.1476-5381.2010.00866.x
53. Saurwein-Teissl M, Lung TL, Marx F, Gschösser C, Asch E, Blasko I, et al. Lack of antibody production following immunization in old age: association with CD8(+)CD28(-) T cell clonal expansions and an imbalance in the production of Th1 and Th2 cytokines. J Immunol (2002) 168(11):5893–9. doi: 10.4049/jimmunol.168.11.5893
54. Parish ST, Wu JE, Effros RB. Sustained CD28 expression delays multiple features of replicative senescence in human CD8 T lymphocytes. J Clin Immunol (2010) 30(6):798–805. doi: 10.1007/s10875-010-9449-7
55. Jennings C, Rich K, Siegel JN, Landay A. A phenotypic study of CD8+ lymphocyte subsets in infants using three-color flow cytometry. Clin Immunol Immunopathol (1994) 71(1):8–13. doi: 10.1006/clin.1994.1044
56. Fagnoni FF, Vescovini R, Mazzola M, Bologna G, Nigro E, Lavagetto G, et al. Expansion of cytotoxic CD8+ CD28- T cells in healthy ageing people, including centenarians. Immunology (1996) 88(4):501–7. doi: 10.1046/j.1365-2567.1996.d01-689.x
57. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol (2007) 8(9):729–40. doi: 10.1038/nrm2233
58. Sharpless NE. Ink4a/Arf links senescence and aging. Exp Gerontol (2004) 39(11-12):1751–9. doi: 10.1016/j.exger.2004.06.025
59. Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Ibrahim JG, et al. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell (2009) 8(4):439–48. doi: 10.1111/j.1474-9726.2009.00489.x
60. Plunkett FJ, Franzese O, Finney HM, Fletcher JM, Belaramani LL, Salmon M, et al. The loss of telomerase activity in highly differentiated CD8+CD28-CD27- T cells is associated with decreased Akt (Ser473) phosphorylation. J Immunol (2007) 178(12):7710–9. doi: 10.4049/jimmunol.178.12.7710
61. Patrick MS, Cheng NL, Kim J, An J, Dong F, Yang Q, et al. Human T cell differentiation negatively regulates telomerase expression resulting in reduced activation-induced proliferation and survival. Front Immunol (2019) 10:1993. doi: 10.3389/fimmu.2019.01993
62. Nadler Y, González AM, Camp RL, Rimm DL, Kluger HM, Kluger Y. Growth factor receptor-bound protein-7 (Grb7) as a prognostic marker and therapeutic target in breast cancer. Ann Oncol (2010) 21(3):466–73. doi: 10.1093/annonc/mdp346
63. Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol (2008) 26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404
64. Thiery J, Keefe D, Boulant S, Boucrot E, Walch M, Martinvalet D, et al. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat Immunol (2011) 12(8):770–7. doi: 10.1038/ni.2050
65. Liu X, Lieberman J. Knocking ‘em dead: pore-forming proteins in immune defense. Annu Rev Immunol (2020) 38:455–85. doi: 10.1146/annurev-immunol-111319-023800
66. Fann M, Chiu WK, Wood WH 3rd, Levine BL, Becker KG, Weng NP. Gene expression characteristics of CD28null memory phenotype CD8+ T cells and its implication in T-cell aging. Immunol Rev (2005) 205:190–206. doi: 10.1111/j.0105-2896.2005.00262.x
67. Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity (2010) 32(1):67–78. doi: 10.1016/j.immuni.2009.10.010
68. Setoguchi R. IL-15 boosts the function and migration of human terminally differentiated CD8+ T cells by inducing a unique gene signature. Int Immunol (2016) 28(6):293–305. doi: 10.1093/intimm/dxw004
69. Setoguchi R, Matsui Y, Mouri K. mTOR signaling promotes a robust and continuous production of IFN-γ by human memory CD8+ T cells and their proliferation. Eur J Immunol (2015) 45(3):893–902. doi: 10.1002/eji.201445086
70. Oswald-Richter K, Grill SM, Leelawong M, Tseng M, Kalams SA, Hulgan T, et al. Identification of a CCR5-expressing T cell subset that is resistant to R5-tropic HIV infection. PloS Pathog (2007) 3(4):e58. doi: 10.1371/journal.ppat.0030058
71. Rosignoli G, Lim CH, Bower M, Gotch F, Imami N. Programmed death (PD)-1 molecule and its ligand PD-L1 distribution among memory CD4 and CD8 T cell subsets in human immunodeficiency virus-1-infected individuals. Clin Exp Immunol (2009) 157(1):90–7. doi: 10.1111/j.1365-2249.2009.03960.x
72. Briceño O, Peralta-Prado A, Garrido-Rodríguez D, Romero-Mora K, Chávez-Torres M, de la Barrera CA, et al. Characterization of CD31 expression in CD4+ and CD8+T cell subpopulations in chronic untreated HIV infection. Immunol Lett (2021) 235:22–31. doi: 10.1016/j.imlet.2021.04.004
73. Jensen SS, Tingstedt JL, Larsen TK, Brandt L, Gerstoft J, Kronborg G, et al. HIV-specific CD8+ T cell-mediated viral suppression correlates with the expression of CD57. J Acquir Immune Defic Syndr (2016) 71(1):8–16. doi: 10.1097/QAI.0000000000000837
74. Addo MM, Draenert R, Rathod A, Verrill CL, Davis BT, Gandhi RT, et al. Fully differentiated HIV-1 specific CD8+ T effector cells are more frequently detectable in controlled than in progressive HIV-1 infection. PloS One (2007) 2(3):e321. doi: 10.1371/journal.pone.0000321
75. Simonetta F, Hua S, Lécuroux C, Gérard S, Boufassa F, Sáez-Cirión A, et al. High eomesodermin expression among CD57+ CD8+ T cells identifies a CD8+ T cell subset associated with viral control during chronic human immunodeficiency virus infection. J Virol (2014) 88(20):11861–71. doi: 10.1128/JVI.02013-14
76. Bonneville M, O’Brien RL, Born WK. Gammadelta T cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol (2010) 10(7):467–78. doi: 10.1038/nri2781
77. Li Z, Jiao Y, Hu Y, Cui L, Chen D, Wu H, et al. Distortion of memory Vδ2 γδ T cells contributes to immune dysfunction in chronic HIV infection. Cell Mol Immunol (2015) 12(5):604–14. doi: 10.1038/cmi.2014.77
78. Fortuny C, Noguera-Julian A, Alsina L, Bellido R, Sánchez E, Muñoz-Almagro C, et al. Impact of CD4 T cell count on the outcome of planned treatment interruptions in early-treated human immunodeficiency virus-infected children. Pediatr Infect Dis J (2011) 30(5):435–8. doi: 10.1097/INF.0b013e3181ff8661
79. Nelson KM, Pantalone DW, Carey MP. Sexual health education for adolescent males who are interested in sex with males: an investigation of experiences, preferences, and needs. J Adolesc Health (2019) 64(1):36–42. doi: 10.1016/j.jadohealth.2018.07.015
80. Li L, Li XW, Ma CJ, Wang LH, Yu FT, Yang SY, et al. Accelerated aging of T-cell subsets among ART-naïve HIV-infected chinese men who have sex with men: A case-control study. Curr HIV Res (2022) 20(2):129–36. doi: 10.2174/1570162X20666220216103504
81. Díaz L, Méndez-Lagares G, Correa-Rocha R, Pacheco YM, Ferrando-Martínez S, Ruiz-Mateos E, et al. Detectable viral load aggravates immunosenescence features of CD8 T-cell subsets in vertically HIV-infected children. J Acquir Immune Defic Syndr (2012) 60(5):447–54. doi: 10.1097/QAI.0b013e318259254f
82. Sharp ER, Willberg CB, Kuebler PJ, Abadi J, Fennelly GJ, Dobroszycki J, et al. Association of differentiation state of CD4+ T cells and disease progression in HIV-1 perinatally infected children. PloS One (2012) 7(1):e29154. doi: 0.1371/journal.pone.0029154
83. Northfield JW, Loo CP, Barbour JD, Spotts G, Hecht FM, Klenerman P, et al. Human immunodeficiency virus type 1 (HIV-1)-specific CD8+ T(EMRA) cells in early infection are linked to control of HIV-1 viremia and predict the subsequent viral load set point. J Virol (2007) 81(11):5759–65. doi: 10.1128/JVI.00045-07
84. Kaplan RC, Sinclair E, Landay AL, Lurain N, Sharrett AR, Gange SJ, et al. T cell activation and senescence predict subclinical carotid artery disease in HIV-infected women. J Infect Dis (2011) 203(4):452–63. doi: 10.1093/infdis/jiq071
85. Pannus P, Adams P, Willems E, Heyndrickx L, Florence E, Rutsaert S, et al. In-vitro viral suppressive capacity correlates with immune checkpoint marker expression on peripheral CD8+ T cells in treated HIV-positive patients. Aids (2019) 33(3):387–98. doi: 10.1097/QAD.0000000000002068
86. Sachdeva N, Weinstein JE, Ashman M, Sachdeva M, Brewer TH, Gracia D, et al. Poor lymphoproliferative responses with low proportion of Gag-specific CD8 TEMRA cells in HIV-1-infected patients showing immunological and virological discordance despite prolonged suppression of plasma viremia. Viral Immunol (2010) 23(1):49–61. doi: 10.1089/vim.2009.0069
87. Gálvez C, Urrea V, Dalmau J, Jimenez M, Clotet B, Monceaux V, et al. Extremely low viral reservoir in treated chronically HIV-1-infected individuals. EBioMedicine (2020) 57:102830. doi: 10.1016/j.ebiom.2020.102830
88. Eberhard JM, Ahmad F, Hong HS, Bhatnagar N, Keudel P, Schulze Zur Wiesch J, et al. Partial recovery of senescence and differentiation disturbances in CD8(+) T cell effector-memory cells in HIV-1 infection after initiation of anti-retroviral treatment. Clin Exp Immunol (2016) 186(2):227–38. doi: 10.1111/cei.12837
89. Han J, Mu W, Zhao H, Hao Y, Song C, Zhou H, et al. HIV-1 low-level viremia affects T cell activation rather than T cell development in school-age children, adolescents, and young adults during antiretroviral therapy. Int J Infect Dis (2020) 91:210–7. doi: 10.1016/j.ijid.2019.12.001
90. Hu W, Li YJ, Zhen C, Wang YY, Huang HH, Zou J, et al. CCL5-secreting virtual memory CD8+ T cells inversely associate with viral reservoir size in HIV-1-infected individuals on antiretroviral therapy. Front Immunol (2022) 13:897569. doi: 10.3389/fimmu.2022.897569
91. Meraviglia S, Di Carlo P, Pampinella D, Guadagnino G, Presti EL, Orlando V, et al. T-cell subsets (T(CM), T(EM), T(EMRA)) and poly-functional immune response in patients with human immunodeficiency virus (HIV) infection and different T-CD4 cell response. Ann Clin Lab Sci (2019) 49(4):519–28.
92. Soriano V, Perno CF, Kaiser R, Calvez V, Gatell JM, di Perri G, et al. When and how to use maraviroc in HIV-infected patients. Aids (2009) 23(18):2377–85. doi: 10.1097/QAD.0b013e328332d32d
93. Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, et al. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med (2008) 359(14):1429–41. doi: 10.1056/NEJMoa0803152
94. Pulido I, Machmach K, Romero-Sánchez MC, Genebat M, Mendez-Lagares G, Ruiz-Mateos E, et al. T-cell changes after a short-term exposure to maraviroc in HIV-infected patients are related to antiviral activity. J Infect (2012) 64(4):417–23. doi: 10.1016/j.jinf.2011.12.017
95. Loetscher P, Uguccioni M, Bordoli L, Baggiolini M, Moser B, Chizzolini C, et al. CCR5 is characteristic of Th1 lymphocytes. Nature (1998) 391(6665):344–5. doi: 10.1038/34814
96. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature (1996) 382(6593):722–5. doi: 10.1038/382722a0
97. Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science (1996) 272(5263):872–7. doi: 10.1126/science.272.5263.872
98. Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest (1998) 101(4):746–54. doi: 10.1172/JCI1422
99. Pogliaghi M, Papagno L, Lambert S, Calin R, Calvez V, Katlama C, et al. The tyrosine kinase inhibitor Dasatinib blocks in-vitro HIV-1 production by primary CD4+ T cells from HIV-1 infected patients. Aids (2014) 28(2):278–81. doi: 10.1097/QAD.0000000000000073
100. Rodríguez-Mora S, Spivak AM, Szaniawski MA, López-Huertas MR, Alcamí J, Planelles V, et al. Tyrosine kinase inhibition: a new perspective in the fight against HIV. Curr HIV/AIDS Rep (2019) 16(5):414–22. doi: 10.1007/s11904-019-00462-5
101. Bermejo M, Ambrosioni J, Bautista G, Climent N, Mateos E, Rovira C, et al. Evaluation of resistance to HIV-1 infection ex vivo of PBMCs isolated from patients with chronic myeloid leukemia treated with different tyrosine kinase inhibitors. Biochem Pharmacol (2018) 156:248–64. doi: 10.1016/j.bcp.2018.08.031
102. Cribier A, Descours B, Valadão AL, Laguette N, Benkirane M. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep (2013) 3(4):1036–43. doi: 10.1016/j.celrep.2013.03.017
103. Coiras M, Bermejo M, Descours B, Mateos E, García-Pérez J, López-Huertas MR, et al. IL-7 induces SAMHD1 phosphorylation in CD4+ T lymphocytes, improving early steps of HIV-1 life cycle. Cell Rep (2016) 14(9):2100–7. doi: 10.1016/j.celrep.2016.02.022
104. Vigón L, Martínez-Román P, Rodríguez-Mora S, Torres M, Puertas MC, Mateos E, et al. Provirus reactivation is impaired in HIV-1 infected individuals on treatment with dasatinib and antiretroviral therapy. Biochem Pharmacol (2021) 192:114666. doi: 10.1016/j.bcp.2021.114666
105. Brown TT, Cole SR, Li X, Kingsley LA, Palella FJ, Riddler SA, et al. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med (2005) 165(10):1179–84. doi: 10.1001/archinte.165.10.1179
106. De Wit S, Sabin CA, Weber R, Worm SW, Reiss P, Cazanave C, et al. Incidence and risk factors for new-onset diabetes in HIV-infected patients: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) study. Diabetes Care (2008) 31(6):1224–9. doi: 10.2337/dc07-2013
107. Capeau J, Bouteloup V, Katlama C, Bastard JP, Guiyedi V, Salmon-Ceron D, et al. Ten-year diabetes incidence in 1046 HIV-infected patients started on a combination antiretroviral treatment. Aids (2012) 26(3):303–14. doi: 10.1097/QAD.0b013e32834e8776
108. Bailin SS, Kundu S, Wellons M, Freiberg MS, Doyle MF, Tracy RP, et al. Circulating CD4+ TEMRA and CD4+ CD28- T cells and incident diabetes among persons with and without HIV. Aids (2022) 36(4):501–11. doi: 10.1097/QAD.0000000000003137
109. Wanjalla CN, McDonnell WJ, Barnett L, Simmons JD, Furch BD, Lima MC, et al. Adipose tissue in persons with HIV is enriched for CD4(+) T effector memory and T effector memory RA(+) cells, which show higher CD69 expression and CD57, CX3CR1, GPR56 co-expression with increasing glucose intolerance. Front Immunol (2019) 10:408. doi: 10.3389/fimmu.2019.00408
110. Rao D, Venkataswamy MM, Vasanthapuram R, Satishchandra P, Desai A. Alteration of T cell phenotypes in HIV-neurotuberculosis coinfection. Cytometry B Clin Cytom (2020) 98(3):270–81. doi: 10.1002/cyto.b.21746
111. Robinson-Papp J, Simpson DM. Neuromuscular diseases associated with HIV-1 infection. Muscle Nerve (2009) 40(6):1043–53. doi: 10.1002/mus.21465
112. Catalán M, Selva-O’Callaghan A, Grau JM. Diagnosis and classification of sporadic inclusion body myositis (sIBM). Autoimmun Rev (2014) 13(4-5):363–6. doi: 10.1016/j.autrev.2014.01.016
113. Vogt S, Kleefeld F, Preusse C, Arendt G, Bieneck S, Brunn A, et al. Morphological and molecular comparison of HIV-associated and sporadic inclusion body myositis. J Neurol (2023) 270(9):4434–43. doi: 10.1007/s00415-023-11779-y
114. Lang DJ, Kovacs AA, Zaia JA, Doelkin G, Niland JC, Aledort L, et al. Seroepidemiologic studies of cytomegalovirus and Epstein-Barr virus infections in relation to human immunodeficiency virus type 1 infection in selected recipient populations. Transfusion Safety Study Group. J Acquir Immune Defic Syndr (1989) 2(6):540–9. doi: 10.1007/BF01643625
115. Abana CO, Pilkinton MA, Gaudieri S, Chopra A, McDonnell WJ, Wanjalla C, et al. Cytomegalovirus (CMV) epitope-specific CD4(+) T cells are inflated in HIV(+) CMV(+) subjects. J Immunol (2017) 199(9):3187–201. doi: 10.4049/jimmunol.1700851
116. Fausto A, Bongiovanni M, Cicconi P, Menicagli L, Ligabò EV, Melzi S, et al. Potential predictive factors of osteoporosis in HIV-positive subjects. Bone (2006) 38(6):893–7. doi: 10.1016/j.bone.2005.11.001
117. Hsue PY, Hunt PW, Sinclair E, Bredt B, Franklin A, Killian M, et al. Increased carotid intima-media thickness in HIV patients is associated with increased cytomegalovirus-specific T-cell responses. Aids (2006) 20(18):2275–83. doi: 10.1097/QAD.0b013e3280108704
118. Spyridopoulos I, Martin-Ruiz C, Hilkens C, Yadegarfar ME, Isaacs J, Jagger C, et al. CMV seropositivity and T-cell senescence predict increased cardiovascular mortality in octogenarians: results from the Newcastle 85+ study. Aging Cell (2016) 15(2):389–92. doi: 10.1111/acel.12430
119. Hoffmann J, Shmeleva EV, Boag SE, Fiser K, Bagnall A, Murali S, et al. Myocardial ischemia and reperfusion leads to transient CD8 immune deficiency and accelerated immunosenescence in CMV-seropositive patients. Circ Res (2015) 116(1):87–98. doi: 10.1161/CIRCRESAHA.116.304393
120. Pathai S, Bajillan H, Landay AL, High KP. Is HIV a model of accelerated or accentuated aging? J Gerontol A Biol Sci Med Sci (2014) 69(7):833–42. doi: 10.1093/gerona/glt168
121. Davis NL, King CC, Kourtis AP. Cytomegalovirus infection in pregnancy. Birth Defects Res (2017) 109(5):336–46. doi: 10.1002/bdra.23601
122. White DW, Suzanne Beard R, Barton ES. Immune modulation during latent herpesvirus infection. Immunol Rev (2012) 245(1):189–208. doi: 10.1111/j.1600-065X.2011.01074.x
123. Razonable RR. Oral antiviral drugs for treatment of cytomegalovirus in transplant recipients. Clin Microbiol Infect (2023) 29(9):1144–9. doi: 10.1016/j.cmi.2023.03.020
124. Effros RB. The silent war of CMV in aging and HIV infection. Mech Ageing Dev (2016) 158:46–52. doi: 10.1016/j.mad.2015.09.003
125. Effros RB. Role of T lymphocyte replicative senescence in vaccine efficacy. Vaccine (2007) 25(4):599–604. doi: 10.1016/j.vaccine.2006.08.032
126. Topp MS, Riddell SR, Akatsuka Y, Jensen MC, Blattman JN, Greenberg PD. Restoration of CD28 expression in CD28- CD8+ memory effector T cells reconstitutes antigen-induced IL-2 production. J Exp Med (2003) 198(6):947–55. doi: 10.1084/jem.20021288
127. Dagarag M, Evazyan T, Rao N, Effros RB. Genetic manipulation of telomerase in HIV-specific CD8+ T cells: enhanced antiviral functions accompany the increased proliferative potential and telomere length stabilization. J Immunol (2004) 173(10):6303–11. doi: 10.4049/jimmunol.173.10.6303
128. Gómez CE, Nájera JL, Perdiguero B, García-Arriaza J, Sorzano CO, Jiménez V, et al. The HIV/AIDS vaccine candidate MVA-B administered as a single immunogen in humans triggers robust, polyfunctional, and selective effector memory T cell responses to HIV-1 antigens. J Virol (2011) 85(21):11468–78. doi: 10.1128/JVI.05165-11
129. Onen NF, Overton ET, Seyfried W, Stumm ER, Snell M, Mondy K, et al. Aging and HIV infection: a comparison between older HIV-infected persons and the general population. HIV Clin Trials (2010) 11(2):100–9. doi: 10.1310/hct1102-100
130. Bruston F, Delbarre E, Ostlund C, Worman HJ, Buendia B, Duband-Goulet I. Loss of a DNA binding site within the tail of prelamin A contributes to altered heterochromatin anchorage by progerin. FEBS Lett (2010) 584(14):2999–3004. doi: 10.1016/j.febslet.2010.05.032
131. Gupta S, Bi R, Gollapudi S. Central memory and effector memory subsets of human CD4(+) and CD8(+) T cells display differential sensitivity to TNF-{alpha}-induced apoptosis. Ann N Y Acad Sci (2005) 1050:108–14. doi: 10.1196/annals.1313.012
Keywords: TEMRA cells, HIV, immunosenescence, antiretroviral therapy, vaccines
Citation: Guo L, Liu X and Su X (2023) The role of TEMRA cell-mediated immune senescence in the development and treatment of HIV disease. Front. Immunol. 14:1284293. doi: 10.3389/fimmu.2023.1284293
Received: 28 August 2023; Accepted: 29 September 2023;
Published: 12 October 2023.
Edited by:
Michael L. Freeman, Case Western Reserve University, United StatesReviewed by:
Celestine Wanjalla, Vanderbilt University Medical Center, United StatesCarey Shive, United States Department of Veterans Affairs, United States
Copyright © 2023 Guo, Liu and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Su, eHN1QGNtdS5lZHUuY24=
†These authors have contributed equally to this work and share last authorship