
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 13 November 2023
Sec. Immunological Tolerance and Regulation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1282874
 Silvia Redondo-García1‡
Silvia Redondo-García1‡ Christopher Barritt1,2,3‡
Christopher Barritt1,2,3‡ Charys Papagregoriou1†‡
Charys Papagregoriou1†‡ Muchaala Yeboah1
Muchaala Yeboah1 Björn Frendeus1,4
Björn Frendeus1,4 Mark S. Cragg1,5
Mark S. Cragg1,5 Ali Roghanian1,5*
Ali Roghanian1,5*Human leukocyte immunoglobulin (Ig)-like receptors (LILR) are a family of 11 innate immunomodulatory receptors, primarily expressed on lymphoid and myeloid cells. LILRs are either activating (LILRA) or inhibitory (LILRB) depending on their associated signalling domains (D). With the exception of the soluble LILRA3, LILRAs mediate immune activation, while LILRB1-5 primarily inhibit immune responses and mediate tolerance. Abnormal expression and function of LILRs is associated with a range of pathologies, including immune insufficiency (infection and malignancy) and overt immune responses (autoimmunity and alloresponses), suggesting LILRs may be excellent candidates for targeted immunotherapies. This review will discuss the biology and clinical relevance of this extensive family of immune receptors and will summarise the recent developments in targeting LILRs in disease settings, such as cancer, with an update on the clinical trials investigating the therapeutic targeting of these receptors.
The human immune system is composed of a network of complex effector cells, organs and tissues, all of which are tightly regulated to maintain immune homeostasis (1). One axis of immune regulation is through the dynamic integration of signals from the myriad of leukocyte activating and inhibitory cell surface receptors (1, 2).
Inhibitory receptors have recently been in the spotlight due to the development of immune checkpoint inhibitors for cancer immunotherapy. Current immunotherapies directed against the inhibitory receptors, such as programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), have shown efficacy in various types of cancers that were previously untreatable (3). In addition, CTLA-4 Ig (abatacept) is being used to treat a number of autoimmune conditions, such as rheumatoid arthritis (RA) and type 1 diabetes (4). A variety of other cell-surface receptors are implicated in the regulation of the immune system and are potential targets for immunotherapy. One such family of receptors are the human LILRs, which play key roles in a wide range of immunological processes. Their ligation through interaction with endogenous or exogenous ligands can reprogram leukocytes and alter their functions (5, 6). Given their central roles in immunoregulation, LILRs are implicated in several pathologies. Hence, their targeting provides an attractive approach for the treatment of human disease.
This review will discuss LILR biology, immune responses mediated by each LILR, and their contribution to human health and disease. Furthermore, it will discuss the potential of targeting LILRs in treating a broad-spectrum of disorders, ranging from cancer to autoimmunity with reference to ongoing clinical trials.
LILRs are a family of immune receptors with immunomodulatory roles in innate and adaptive immunity. The LILR gene family were independently discovered by different investigators around the same time (7). LILRs were originally identified in 1997 by the Colonna laboratory (8), followed by the Cosman laboratory (9). Due to their discovery by different investigators, these genes were assigned several different names (e.g., ILT, LIR, MIR, CD85). LILR is the current standardised nomenclature for this receptor family, which was approved by the HUGO gene nomenclature committee in 2015 (10). LILRs are classified into two subfamilies: activating (LILRA) and inhibitory (LILRB).
LILRs are type 1 transmembrane glycoproteins structurally and functionally similar to killer cell Ig-like receptors (KIR) expressed on natural killer (NK) cells and some subsets of T lymphocytes (11, 12). LILR genes are located adjacent to the KIRs within the leukocyte receptor complex on chromosome 19 at 19q13.4, encoding for 11 functional genes and two pseudogenes (13, 14). The LILR gene cluster is believed to have originated from an activating founder gene, which after gene duplications gave rise to the current family and organisation (12). The LILR region consists of around 497 kb, divided into telomeric (~211 kb) and centromeric (~154 kb) regions, separated by a central region (~132 kb) (12). There are multiple polymorphisms in the receptor binding site of the LILRs (13, 15–20). In particular, LILRB3 and LILRA6 are considered as highly polymorphic and are found as different allelic variants, while LILRA3 and LILRA6 show copy number variations (13, 15–20). Interestingly, LILRA3 shows an extremely high allele frequency of deletion in the Japanese population (21). In addition, although the LILR region in humans is relatively stable, a haplotype lacking LILRA3 due to a 6.7 kb deletion exists (12).
LILRs are primarily expressed on myeloid antigen-presenting cells (APC), such as monocytes and dendritic cells (DC), but also on granulocytes, NK cells, T and B lymphocytes, hematopoietic stem cells (22, 23), and non-immune cells, such as endothelial cells and neurons (12, 19) (Figures 1, 2). LILRs are membrane-bound receptors, except for LILRA3. However, all LILRs also exist in soluble form as a result of alternative splicing (11, 19, 24). In addition, extracellular Ig-like domains of LILRB1, LILRB2, LILRB4, LILRA1, LILRA3 and LILRA5 are found in human sera or the supernatants of leukocytes (11, 25–29). These soluble LILR variants may act as decoy receptors, as demonstrated for LILRB1 (26).
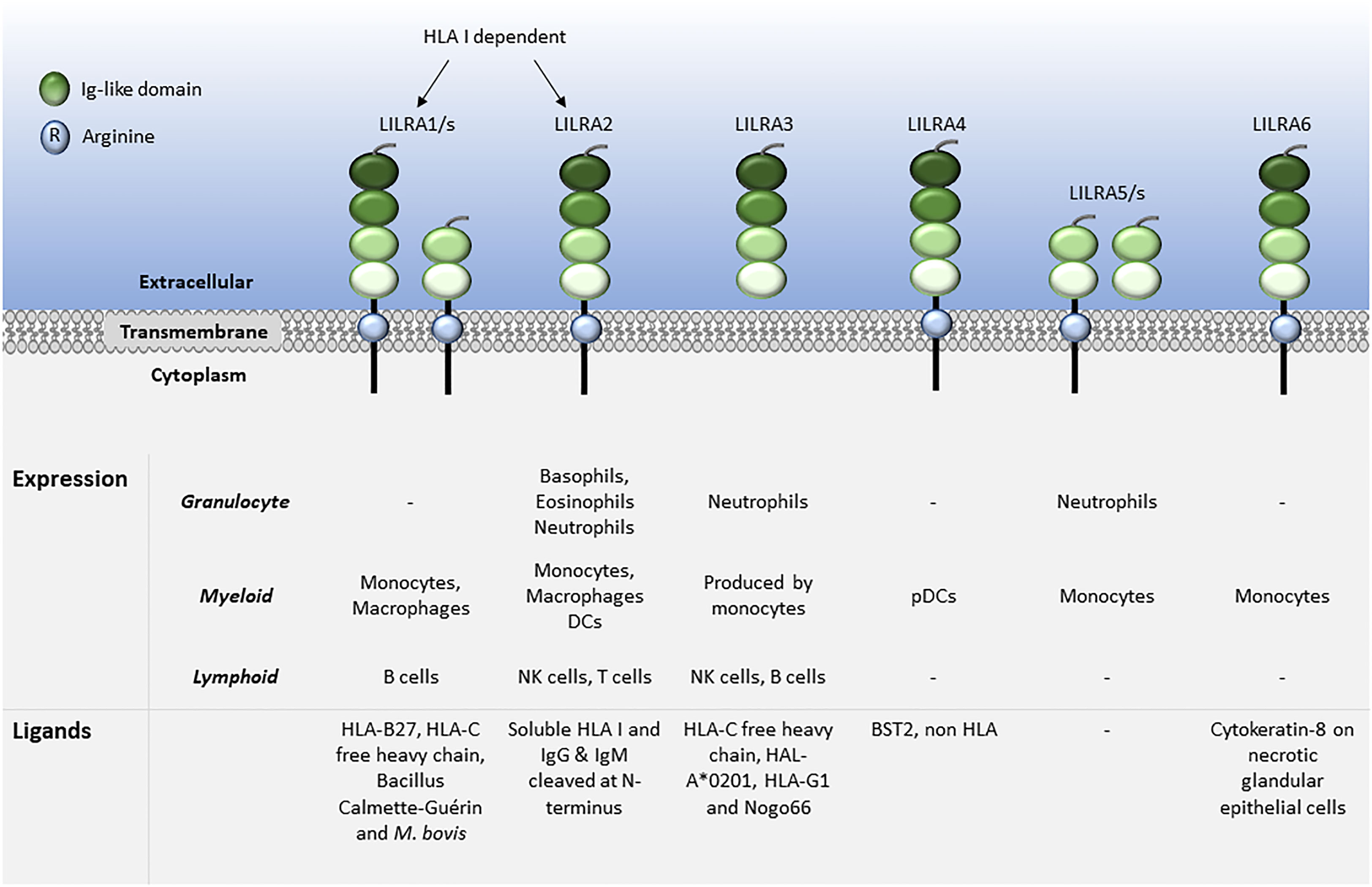
Figure 1 Leukocyte expression and ligand profiles of LILRAs. LILRAs have 2 to 4 extracellular lg-like domains, a transmembrane domain with a positively charged arginine residue and a truncated intracellular tail.
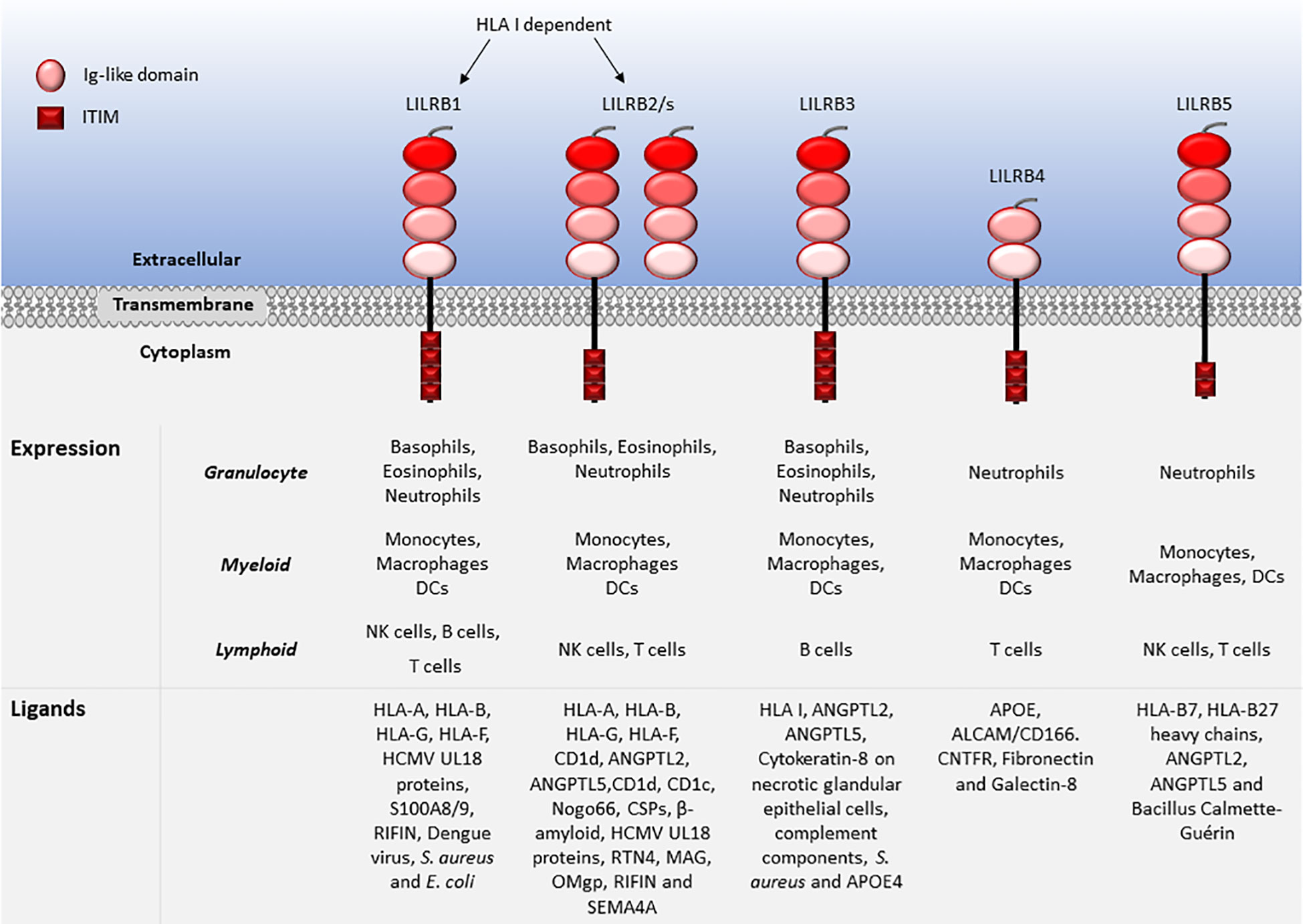
Figure 2 Leukocyte expression and ligand profiles of LILRBs. LILRBs have 2 to 4 extracellular lg-like domains and their cytoplasmic regions are composed of long ITIM-containing motifs exhibiting their inhibitory nature.
The family have 2-4 C2-type Ig-like domains in their extracellular domains. However, their different cytoplasmic tails transduce either activating or inhibitory signalling (14). Apart from LILRA3 that only exists as a soluble form, the other five activating receptors (LILRA1, LILRA2, LILRA4, LILRA5 and LILRA6) have a shorter cytoplasmic tail and a positively charged arginine residue in the transmembrane domain (Figure 1). As a result, LILRAs transduce signals through an association with immunoreceptor tyrosine-based activation motif (ITAM)-containing high affinity IgE Fc epsilon receptor type I γ chain (FcεRIγ) (30) (Figure 3). In contrast, the five inhibitory receptors (LILRB1-5) signal through their immunoreceptor tyrosine-based inhibitory motifs (ITIM) (14, 30) (Figure 2). Together, LILRs fine-tune the immune response according to relevant local stimuli. Their dysfunction is therefore associated with pathologies ranging from autoimmunity to immunosuppression.
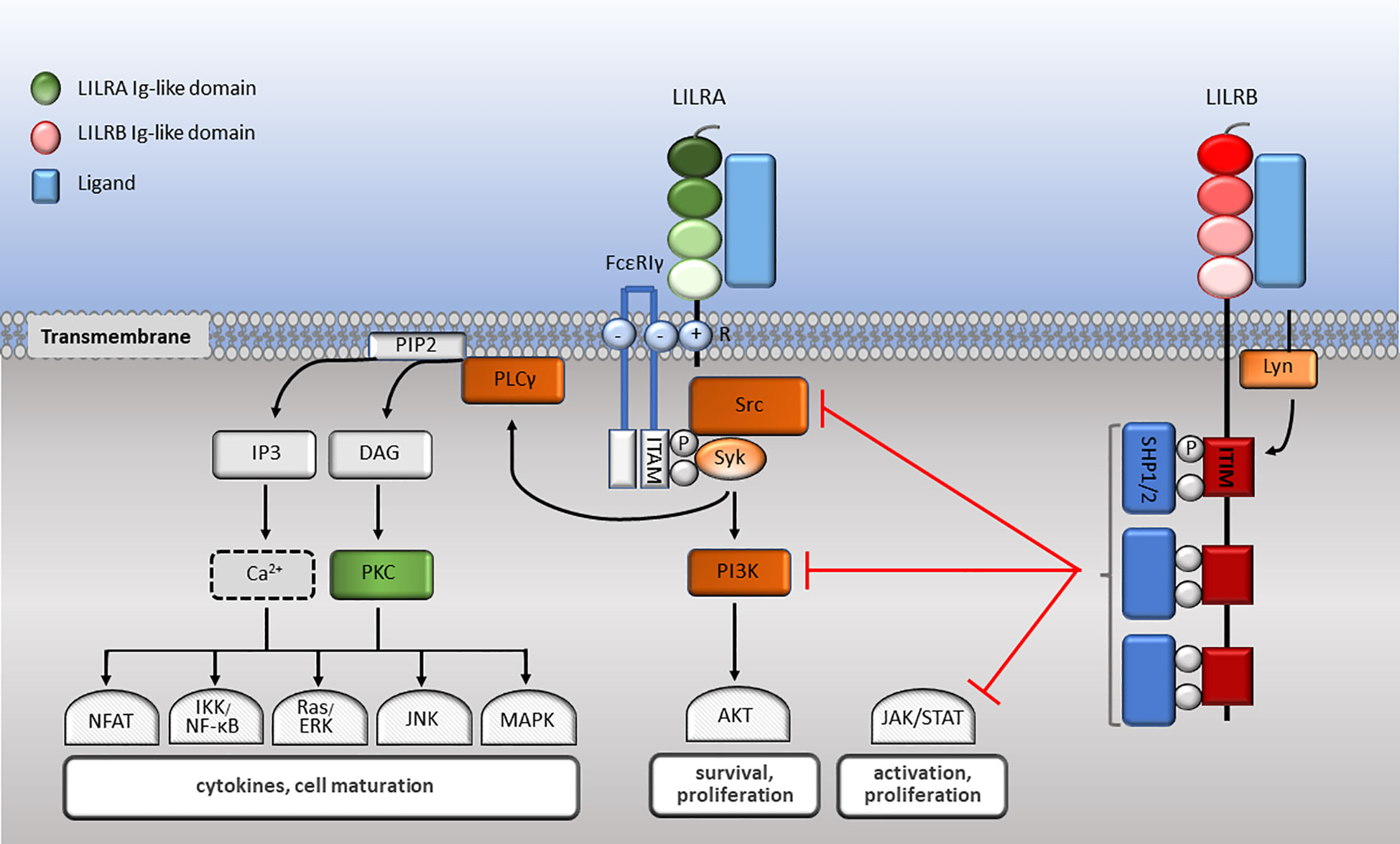
Figure 3 LILR signalling pathways. LILRA intracellular domain interacts with the dimeric FcεRIγ-chain comprised of cytoplasmic ITAM motifs. Phosphorylation of ITAM-bearing tyrosine residues by Src family kinases recruits Syk that mediates activating signalling cascades. Upon LILRB ligation, activated Lyn phosphorylates their ITIM domains, engaging phosphatases, which in turn abrogate activating signalling cascades essential for effector cell maturation and function.
The crystal structure of a number of LILRs have been resolved. A partial structure of LILRB1 (distal D1 and D2) was resolved at 2.1Å resolution, confirming an Ig-like structure for both extracellular domains. It comprises β-sheets with fused helical regions, with a similar LILRB1 folding arrangement to that of the homologous KIR molecules (31). Nam et al. resolved the two membrane-proximal domains (D3 and D4) of LILRB1, as structurally similar to D1 and D2 (32). Based on the LILRB1 crystal structure model, Willcox et al. resolved LILRB2 by homology modelling to 1.8Å resolution (33). The folding of LILRB2 was predicted to be similar to LILRB1 apart from fewer α-helical structures (33). At the ligand binding interface, limited plasticity and flexibility were reported for both receptors due to the angles between the domains and the staggered assembly of the Ig-like domains (34). Willcox and colleagues further reported the crystal structure of the extracellular D1 and D2 of LILRB1 (35). The LILRB4 ectodomain was resolved at 1.7Å, depicting two Ig-like domains, similar in structure to the other LILR members. Although, LILRB4 D2 is similar to D4 of other LILRs, it contains helices that have not been reported before for this family. Reduced interdomain contact sites were also observed at the D1-D2 interface, which was associated with an obtuse interdomain angle of 107° (36). The crystal structure of LILRA2 indicated shifts in the amino acid residues that determine binding to human leukocyte antigen (HLA), explaining why it does not bind to HLA (37). The crystal structure of LILRA5 has also been reported (38), but the structures of other LILRs have not yet been determined.
The putative murine orthologues of LILRs are the paired Ig-like receptors (PIR), which possess six Ig-like domains and, similar to LILRs, are activating (PIR-A) or inhibitory (PIR-B) (19, 39, 40). PIR-B is the human ortholog of LILRB2-3, while glycoprotein 49B1 (gp49B1) is an orthologue of LILRB4 (41). Similar to LILRs, PIR genes are located within the leukocyte receptor complex on chromosome 7 (39). Resembling its human counterparts, PIR-A associates non-covalently with the ITAM-bearing FcγR adaptor molecule to transduce signals (42), while PIR-B contains 4 ITIMs in its cytoplasmic tail and binds to mouse major histocompatibility complex class I (39). These paired receptors are expressed on B cells, DCs, monocytes, macrophages, neutrophils, eosinophils, mast cells and megakaryocytes (42–47). These similarities in genomic location, expression profiles, structure and ligand affinity have identified PIR-A/B as the murine orthologues of human LILRs (19, 39, 40, 42). However, PIRs exhibit low overall homology to human LILRBs ranging from 45% to 54% as well as a wider tissue expression and greater regulatory effects than LILRs. Consequently, knowledge of PIRs (and gp49B1) may be limited when extrapolating to LILR biology.
LILRs were initially characterised as HLA class I (HLA I) binding molecules. Later studies demonstrated that LILRs can be classed into two groups based on their ligands. Group 1 LILRs (LILRA1, LILRA2, LILRA3, LILRB1 and LILRB2) contain highly conserved HLA I binding sites, enabling the interaction with classical and non-classical HLA I or HLA I-like proteins. In contrast, group 2 LILRs (LILRA4, LILRA5, LILRA6, LILRB3, LILRB4 and LILRB5) interact with HLA I/β2-microglobulin (β2m) independent ligands (35). In this second group, LILRB5 is an exception since it interacts with angiopoietin-like proteins (ANGPTL) but also binds to HLA I heavy chains (12). Ligand profiles and known immunoregulatory functions of LILRs are summarised in Table 1 and Figures 1, 2.
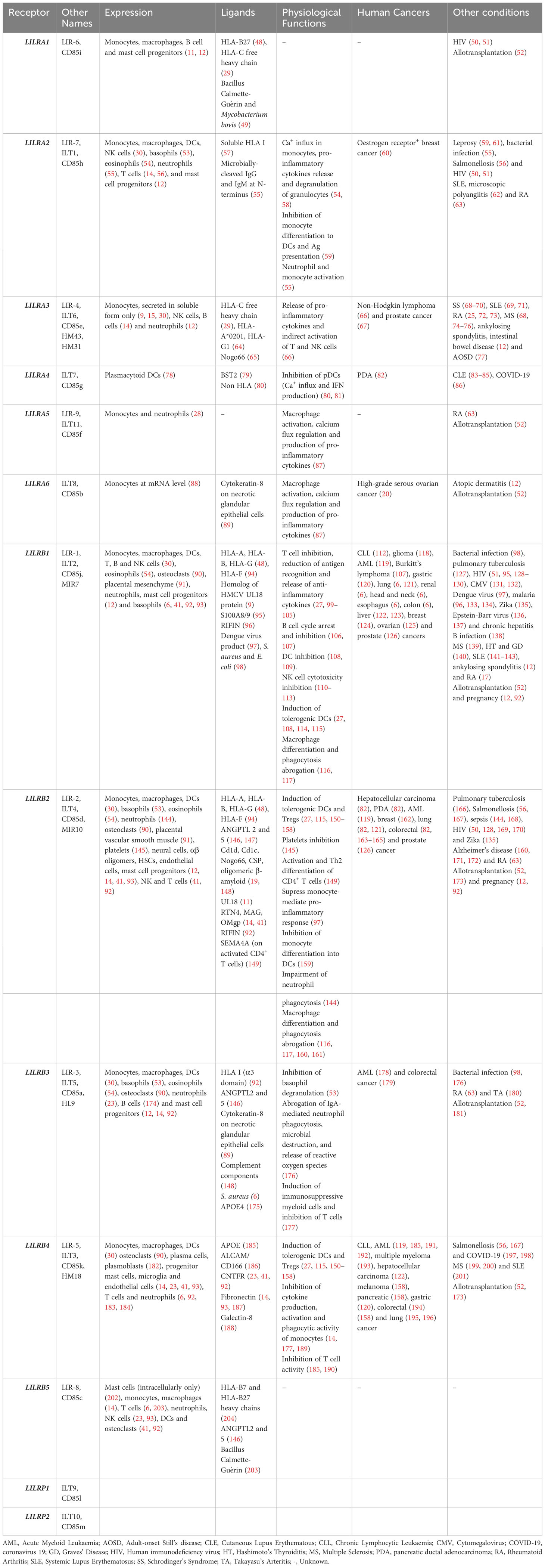
Table 1 Overview of the expression, ligands and physiological functions of LILRs in health and disease.
Structural analysis of LILRB1-HLA I interaction has revealed that LILRB1 interacts with the highly conserved α3 β2m domains of HLA I, unlike T cell receptors (TCR) which bind to α1 and α2 domains, indicating they may bind simultaneously and demonstrating that LILRB1 may have a wider number of binding partners (35, 48). The interactions between LILRBs and HLA I may provide an inhibitory balancing force preventing immune activation to self and termination of immune responses.
The highest affinity LILR ligand is the non-classical HLA I molecule HLA-G, found in several forms including in disulphide-linked dimer or β2m-free isoforms. HLA-G interacts with LILRB1 and LILRB2 with different affinities (114, 205). LILRB1 lacks the reactivity to β2m-free HLA-G or HLA-B27, while LILRB2 interacts with the β2m-free form of HLA-B27 (48). Although LILRB2 exhibits overlapping HLA I recognition to LILRB1, it dominantly recognises the hydrophobic site of HLA-G D3 (48, 205). LILRB1 was shown to bind to HLA-G with 3 fold higher affinity compared to other HLA I molecules (150). In normal physiology HLA-G is expressed on foetal placental trophoblasts, enabling the invasion of the placental decidua during implantation and facilitating maternal tolerance to the semi-allogenic foetus (206). Enhanced expression of HLA-G contributes to the pathogenesis of viral infections and cancer by downregulating immune responses (207–213). Both HLA-G expression and dimerisation upregulate expression of LILRBs and inhibit T cell activity in vitro (214, 215). Additionally, LILRB1 binds to various pathogens, including, Staphylococcus aureus (S. aureus) and Escherichia coli (E. coli), opsonised dengue virus, cytomegalovirus (CMV), calcium-binding proteins S100A8 and A9 and repetitive interspersed family of polypeptides (RIFIN) (9, 19, 23, 94–98). LILRB2 binds to ANGPTLs (similar to LILRB3 and LILRB5), HLA I-like proteins, Nogo66, complement split products (CSP), oligomeric β-amyloid, UL18, RTN4, MAG, OMgp, RIFIN and SEMA4A in activated CD4+ T cells (11, 14, 19, 41, 92, 146–149). LILRB3 is the least studied LILRB and its natural ligands have not been fully elucidated. Although, regarded as an orphan receptor, recent findings suggest that LILRB3 interacts with ANGPTL2 and 5, complement components, and cytokeratin-associated proteins exposed on necrotic tumour cells and bacteria such as S. aureus (6, 23, 89, 92, 98, 146, 148). Hence, LILRB3 engagement by ligands expressed on necrotic cancer cells or pathogens may subvert immune responses. Recently, apolipoprotein (APOE) 4 was reported as a putative LILRB3 ligand, which is recognised by the D2/D4 regions LILRB3 (175). LILRB4 has been described to bind to APOE (185), ALCAM/CD166 (186), galectin-8 (188), CNTFR (23, 41, 92) and fibronectin (93, 187), while LILRB5 binds to ANGPTLs, HLA-B7 and HLA-B27 heavy chains and Bacillus Calmette-Guérin (146, 204).
The ligands for LILRAs are less characterised and may function as an autoregulatory mechanism for cell activation. They include HLA molecules for LILRA1, LILRA2 and LILRA3 (29, 48, 57, 64). Similar to LILRB5, LILRA1 binds to Bacillus Calmette-Guérin and also to Mycobacterium bovis (49). Moreover, LILRA2 was shown to recognise IgG and IgM cleaved by proteases secreted by microorganisms such as Mycoplasma hyorhinis, Legionella (L.) pneumophila, Streptococcus pneumonia and Candida albicans. Interestingly, stimulation of primary monocytes via LILRA2 inhibited L. pneumophila growth (55). LILRA3 binds to Nogo66 (65), while LILRA4 binds to the bone marrow stromal cell antigen 2 (BST2) (79). However, there are no described ligands for LILRA5. Finally, LILRA6 is known to bind to cytokeratin 8 in necrotic glandular epithelial cells, similar to LILRB3 (89).
LILRs signal via their associated ITAMs or ITIMs (Figure 3). As described above, LILRAs possess a transmembrane domain with a positively charged arginine and their short cytoplasmic domain has no kinase or docking motifs (30, 58, 81, 87). The arginine residue in LILRA2, LILRA4 and LILRA5 associate with a charged residue on FcεRIγ (58, 81, 87). Upon receptor crosslinking, Src kinases are activated and phosphorylate the ITAM tyrosine residues, which allows the phosphorylation of Src homology 2 domain (SH2) on Syk and ZAP70 tyrosine kinases (216) (Figure 3). ITAM-mediated signalling propagates the nuclear translocation of nuclear factor (NF)-κB and nuclear factor of activated T cells (NFAT), phosphoinositide (PI) 3-kinase (PI3K) activation, which activates membrane-bound serine/threonine-specific protein kinases (AKT and BTK), as well as interacting with Ras to activate the Ras/Raf pathway. As a result, ligation of LILRAs propagates the proliferation, maturation and survival of immune cells (217, 218) (Figure 3). The signalling mechanisms of LILRA1 and LILRA6 remain to be identified, although the structural similarity with the other LILRAs suggests that they also signal though FcεRIγ.
LILRBs impose their inhibitory signalling through ITIMs (30, 190). Upon receptor binding with the ligand, the Src-family protein Lyn becomes autophosphorylated, phosphorylating ITIM tyrosine residues. In turn, SH2 containing protein tyrosine phosphatases SHP-1 and SHP-2 are recruited to the phosphorylated sites. These phosphatases proceed to negatively regulate Syk and PI3K cell signalling (30). Consequently, downstream signalling pathways such as MAPK, JNK, Ras/ERK, NFAT and NF-κB are abrogated. This leads to attenuation of cytokine secretion and effector cell maturation, survival and function (218) (Figure 3). As an example, upon co-ligation of FcγRI with LILRB1/LILRB2, FcγR-mediated PTK-dependent signalling is abrogated (54, 116). Nevertheless, how multiple ligands and activating and inhibitory LILRs act in concert to modulate immune responses need further investigation.
LILR functions have been primarily studied in terms of regulatory mechanisms exerted by LILRBs and, as such, little is known about the activating roles of LILRAs. Dividing the functions of LILRs as either activating or inhibitory, based on the presence of ITAMs or ITIMs may be too simplistic (219, 220). There have been suggestions that under certain conditions ITIM-bearing receptors can enhance leukocyte functions and ITAM-bearing receptors may inhibit the immune system (81, 217, 219–223). Based on the broad expression of LILRs across an array of immune cells and non-immune cells, their roles in controlling both innate and adaptive immunity are divided into leukocyte subsets herein, and the functional role of LILRs is discussed with regard to immune activation and tolerance (Table 1, Figure 4).
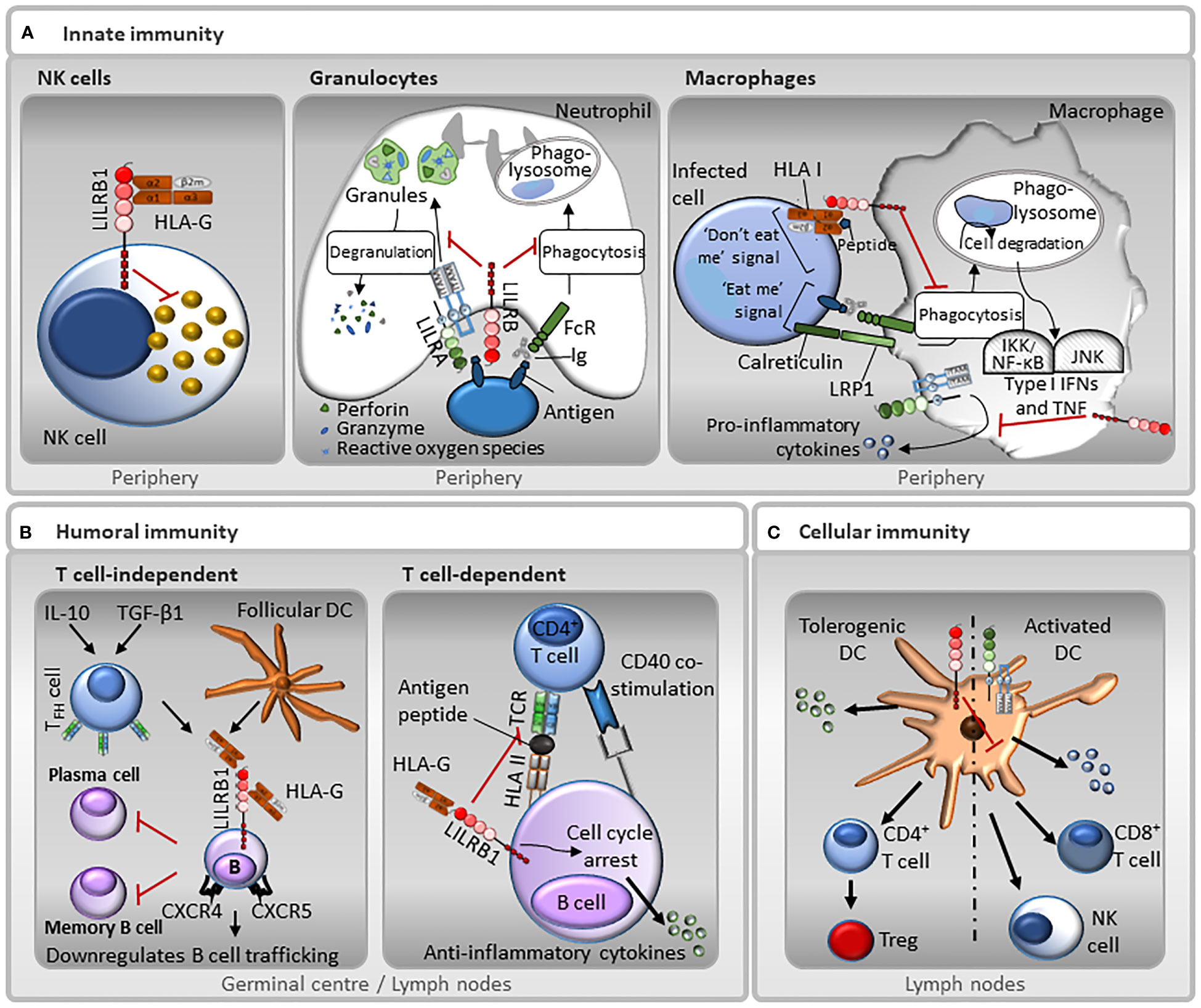
Figure 4 Representative functions of LILRs in innate and adaptive immunity. (A) Innate immunity: LILRB1 is expressed on NK cells, and therefore may be involved in missing self, whereby the receptor recognises HLA I molecules on target cells, and those that do not express HLA I are destroyed. In neutrophils, LILRAs, such as LILRA2 and LILRA5, stimulate degranulation and pro-inflammatory cytokines release, while LILRBs, such as LILRB3, have the opposite effect and block their phagocytic activity. Moreover, the paired-receptors LILRA6 and LILRB3 recognise bacterially-infected cells. Interaction between LILRBs, such as LILRB1, with HLA I abrogates FcγR-mediated phagocytic function of macrophages. In addition, LILRAs, such as LILRA3, LILRA5 and LILRA6 promote the secretion of pro-inflammatory cytokines. However, this secretion is suppressed by LILRBs. (B) Humoral immunity: LILRB1 inhibits B cell responses in a T cell-independent manner. TGF-β1 and IL-10 induce follicular dendritic cells (DC) and follicular helper T cells (TFH) to secrete HLA-G. HLA-G binds to LILRB1 on the surface of germinal centre B cells resulting in a down-regulation of chemokine receptors CXCR4 and CXCR5 and inhibiting B cell trafficking. This interaction also inhibits differentiation into antibody-secreting plasma cells and memory B cells. Moreover, LILRB1 regulates B cell responses in a T cell-dependent manner. B cells can present antigen to T cells. LILRB1-HLA-G interaction can prevent antigen presentation and inhibits B cell proliferation by causing cell cycle arrest in the G0/G1 phase by disrupting the mTOR pathway mediated by SHP-2. (C) Cellular immunity: Ligation of LILRBs during DC development renders DCs tolerogenic by increasing the threshold of activation. Normal DCs have low levels of LILRBs. In contrast, tolerogenic DCs that express increased levels of LILRBs, promote anti-inflammatory cytokines release, CD4+ helper T cells activation and their conversion into Tregs. Conversely, LILRAs activate DCs towards a cytotoxic phenotype, inducing the secretion of pro-inflammatory cytokines that induce NK and CD8+ T cells activation.
LILRAs are abundantly expressed on monocytes with some expression on granulocytes. LILRA crosslinking leads to cell activation resulting in calcium influx, selective cytokine release and degranulation (28, 53, 54, 58, 87). LILRA2, the most studied LILRA to date, as well as LILRA5, are implicated in stimulating degranulation and release of pro-inflammatory cytokines, such as IL-1β, IL-12 and tumour necrosis factor (TNF)-α and other factors involved in the early phases of eosinophils- and basophils-mediated inflammatory responses (54, 224). Microbially-cleaved Ig products activate primary human neutrophils via LILRA2 (55).
LILRB2, LILRB3 and LILRB5 are involved in regulating neutrophil activation and function (19, 23). LILRB2 is expressed on the surface of neutrophils as well as within the granules, inhibits their phagocytic capacity and leads to exocytosis of LILRB2 to the cell surface (144). This phenomenon of increased expression of inhibitory receptors upon activation may provide an inhibitory feedback loop (144). LILRB3 was recently found to be highly expressed on resting neutrophils and secreted upon their activation. Prolonged ligation of LILRB3 abrogated IgA-mediated neutrophil effector functions such as phagocytosis, microbial destruction and release of reactive oxygen species, suggesting that LILRB3 could be a novel checkpoint inhibitor on neutrophils (176). Similarly, co-ligation of PIR-B with FcεRI is able to abrogate IgE-mediated mast cell activation and serotonin secretion (42). A murine homologue of LILRB4, gp49B, is also expressed in mouse neutrophils and plays a regulatory role in lipopolysaccharide (LPS)-induced adhesion and microangiopathy (183, 184).
The expression of LILRs varies on macrophages and DCs at different maturation phases. DCs and macrophages detect surface microbial molecules through their pattern-recognition receptors, such as Toll-like receptors (TLR). However, APCs are also able to adopt a tolerogenic phenotype and orchestrate immune tolerance (150–153). Although LILRAs induce immune effector function, they can be inhibitory when ligated concurrently with an unrelated activating receptor on APCs (56, 217). Upon ligation of LILRA2 on monocytes, TLR-mediated antimicrobial activity was reduced by increased production of IL-10 (61). Furthermore, activation of LILRA2 on monocytes impaired their GM-CSF-mediated differentiation into immature DCs and supressed antigen (Ag) presentation and adaptive T cell response (59). In addition, LILRs can mediate cytokine secretion and affect the expression of co-stimulatory receptors on professional APCs. Accordingly, ligation of LILRA3 on monocytes and B cells increases the secretion of pro-inflammatory cytokines and indirectly induces proliferation of NK cells and CD8+ T cells (66). Similarly, LILRA2 ligation on monocytes is able to regulate TLR4 (56). Interestingly, both LILRA2 and LILRA3 inhibit LPS-mediated secretion of TNF-α by monocytes (63, 210). In addition, while treatment of monocytes with IL-10 and interferon (IFN)-γ increases secretion of soluble LILRA3, TNF-α reduces its expression (25, 225). Although less studied, LILRA4 inhibits the secretion of inflammatory cytokines by plasmacytoid DCs (pDC) (80). Furthermore, crosslinking of LILRA5 and LILRA6 on monocytes induces tyrosine kinase phosphorylation, which in turn mediates calcium flux and secretion of pro‐inflammatory cytokines (IL‐1β, TNF‐α, IL‐6), suggesting a potential role in inflammation. However, their underlying functions alongside LILRA1 remain unknown (87).
LILRBs can detect soluble factors including CSPs in response to microbial infections through classical lectin or alternative pathways of complement activation. Moreover, interaction of LILRB2 and C4d can suppress monocyte-mediated pro-inflammatory responses (97) and promote endocytosis of C4d (148). Tolerogenic APCs are unable to activate T cells, and they alternatively induce Ag-specific regulatory T (Treg) cells (153). Ligation of LILRB1 on monocytes during differentiation into DCs renders them tolerogenic (tDC), which in turn become resistant to LPS stimulation and unable to activate autologous T cells (27, 108, 114, 115). It also leads to increased expression of the NF-κB inhibitor ABIN1, key in maintaining functional DCs (226). Moreover, LILRB1 signalling inhibits DC activation mediated by OSCAR, which activates DCs via the FcRγ chain (108, 109). Banchereau et al. (227) showed that human Langerhans cells which do not express LILRBs were able to efficiently prime cytotoxic CD8+ T cells, whereas LILRB1- and LILRB2-expressing dermal CD14+ DCs were less efficient at priming cytotoxic T cells. Blockade of LILRB1/LILRB2 on dermal DCs enhanced T cell cytotoxicity (227). Similarly, tDCs exhibit high expression of LILRB2 and LILRB4, playing an essential role in tDC activity (27, 115, 150–153). Co-culture of T cells with an APC line transfected to express LILRB2 and LILRB4 extracellular domains demonstrated that only LILRB2 relies on its intracellular signalling to induce Tregs, whereas the extracellular Ig-like domains of LILRB4 and soluble LILRB4 were able to induce Tregs irrespective of their signalling potential (154, 155). Moreover, LILRB4 silencing in DCs promotes the release of pro-inflammatory cytokines and consequently the proliferation and migration of T cells (156). LILRB4 ligation on APCs leads to the upregulation of the co-stimulatory molecule CD86 (167), unlike LILRB2 which leads to its downregulation, indicating that LILRB2 and LILRB4 limit T cell responses via distinct mechanisms (150, 168, 169). In addition, LILRB2 and LILRB4 promote the differentiation of suppressor T cells (152, 155–158), and LILRB4 interacts with receptors on T cells and antagonises CD8+ T cells to promote the development of Tregs (154, 228). This suggests LILRB4 functions as an inducer of immune tolerance, while LILRB2 affects APC function and secondary co-stimulation via distinct mechanisms (150, 168, 169). Similarly, in PIR-B-/- mice humoral T helper (Th) 2 responses are enhanced in response to T-dependent Ags due to impaired DC maturation compared to wildtype mice (229).
LILRB2-HLA-G interaction inhibits the differentiation of monocytes to DCs and maturation via IL-6 and STAT3 signalling (159). Moreover, LILRB4-/- DCs stimulated via LPS-mediated TLR signalling exhibit increased pro-inflammatory cytokine/chemokine synthesis and secretion (156). Under physiological conditions, LILRB1 and LILRB4 are downregulated upon DC activation. This loss of inhibitory receptors may be essential for the maturation of DCs (230). Stimulation of DCs maturing in vitro with immunosuppressive agents, such as niflumic acid, IL-10, IFN-α and IFN-β, leads to the development of tDCs with increased LILRB2 and LILRB4 expression (27, 128, 151, 199, 231). Aspirin and 1,25-dihydroxyvitamin D3 (vitamin D) render DCs tolerogenic that are unable to stimulate T cells, only upon upregulation of LILRB4 (199, 232, 233).
LILRBs, therefore, act as myeloid checkpoint receptors to limit overt immune responses. LILRB1 ligation on tumour-associated macrophages (TAM) was shown to abrogate phagocytosis of HLA I+ tumours, which could be ablated with LILRB1 monoclonal antibodies (mAbs) (117). Similarly, combining LILRB2 and PD-1 blockade mediated E. coli phagocytic removal, which was associated with suppressed SHP1/2 phosphorylation, promoting pro-inflammatory macrophage activity (160). LILRB2 blocking reprograms macrophages to a more pro-inflammatory state and enhances the activation of T cells, increasing the efficiency of anti-PD-1 therapy (161). LILRB1 and LILRB2 co-ligation with FcγRI on monocytes mediates SHP-1 activation, abrogating downstream phosphorylation and intracellular calcium mobilisation (116). Ligation of LILRB3 on monocytes induces immunosuppressive myeloid cells, which inhibit T cell responses in vitro, and inhibit allograft rejection in humanised mice (177). Furthermore, LILRB4 activation results in recruitment of phosphatases that contribute to the dephosphorylation of FcγRI-activated tyrosine kinases and inhibition of FcγR-mediated phagocytosis (189).
Soluble isoforms of LILRBs (sLILRB), generated by alternative mRNA splicing, can also regulate immune responses. Jones et al. showed that sLILRB1 can compete with membrane-bound LILRB1 for binding to their natural ligands. This suggests that sLILRBs may act as decoy receptors in modulating immune effector functions (26). Similarly, recombinant sLILRB2 is able to restore the proliferation of T cells rendered inactive by tDCs (27). Notably, mature DCs treated with IL-10 abrogate shedding of sLILRB2, while increasing the expression of the surface-bound LILRB2 (27).
NK cells are cytotoxic against cells deficient for surface HLA I. Control of NK cell activation is regulated by activating and inhibitory receptors. Like KIRs, some LILRs can recognise HLA I molecules and influence NK cell effector functions. LILRB1 inhibits NK cell cytotoxicity as a result of HLA I interaction, which inhibits FcγRIIIA-dependent lysis of target cells (110). HLA-G-mediated LILRB1 ligation on NK cells inhibits activation, polarisation of lytic granules and IFN-γ production (111). LILRB1 blockade augments NK cell activation and proliferation and is associated with IL-2 production by CD4+ T cells (112). In addition, LILRB1 can regulate initial ligand recognition by abrogating the adhesion of NK cells to target cells (113).
LILRB1 is the main LILR found on T cells, however, its expression is variable among CD8+ and CD4+ T cells, as not all T cells express LILRB1 (27, 99–104). LILRB1 abrogates TCR signalling by dephosphorylating the TCR-ζ chain of ITAM domains, which in turn suppress downstream signal transduction mediated by ZAP70 and linker for activation of T cells (105). LILRB1-mediated inhibition of T cells is characterised by a reduction in Ag recognition, CD3-mediated clonal expansion, proliferation, chemotaxis, resistance to TLR stimulation and a shift in the cytokine profile in favour of anti-inflammatory cytokines (27, 99–104). As CD8+ T cells mature, they acquire cytotoxic potential with an increase in perforin within the cell; LILRB1 expression increases in parallel, possibly to protect self (136). Crosslinking of surface LILRB1 or CTLA-4 on T cells leads to inhibition of Ag-specific CD4+ T cell proliferation, and IL-13, IFN-γ and IL-2 release, as well as an increase in TGF-β and IL-10 secretion (99). Apart from mediating inhibitory signals to T cells (111, 234), both surface-bound and soluble forms of LILRB1 and LILRB2 limit activating signals by antagonising the HLA I-CD8 interactions (228). Moreover, LILRB2 is expressed in CD4+ T cells and regulates Th2 differentiation (149). Also, LILRB4 has been found expressed on T cells, and it suppresses T cell activity mediated by APOE/the intracellular domain of LILRB4/SHP-2/NF-κB/urokinase receptor/arginase-1 (ARG1) axis (185, 190). Finally, there are contradictory reports on whether T cells express LILRB5 or not, which may be due to the nature of the reagents and assay conditions used by the investigators (203, 204). Additionally, LILRA2 has been found on T cells at low levels (56), and regulates T cells indirectly by modulating the behavior of other cells, such as APCs (59).
Some LILRs are expressed by B cells but they primarily impact B cell responses by modifying APC and T cell responses (235). Transcripts of LILRA1, LILRA3 and LILRB3 are found in B cells, and LILRB4 is present in plasmablasts (11, 174, 182). However, only LILRB1 has a clear role in B cells. HLA-G binding to LILRB1 on B cells inhibits both T cell-dependent and -independent activation of naïve and memory B cells (106). Furthermore, LILRB1 interaction with HLA-G leads to B cell G0/G1 cell cycle arrest as a result of mTOR/AKT and PKC pathway modification (106). LILRB1-ligated B cells exhibit reduced Ig secretion and increased secretion of anti-inflammatory cytokines (106, 107).
Apart from the pivotal roles of LILRs in maintaining immune homeostasis, they can mediate pathogenesis during bacterial, viral and parasitic infections, as extensively reviewed elsewhere (49). Below is a summary of their key roles in infection.
Mycobacterium leprae infection mediates strong Th1 cell-mediated immune responses, to give rise to the tuberculoid form of leprae. In contrast, lepromatous leprosy infection involves higher bacterial load, dominance in Th2 cytokine secretion and strong humoral immune responses (61). LILRB3, LILRB5 and especially the activating LILRA2, are overexpressed in skin biopsies from patients with lepromatous leprosy, which is associated with inhibition of TLR-mediated microbial killing, secretion of type 2 cytokines with an increase in IL-10:IL-12 ratio (61). Genetic profiling and immune labelling of skin lesions of these two forms has revealed a substantial regulation of LILRA2 on the disseminated lepromatous leprosy lesions over the limited tuberculoid form. Pre-treatment with LILRA2 antibodies reduces TLR-mediated antimicrobial activity (59, 61). In the case of Mycobacterium tuberculosis, its major niche to persist are macrophages and myeloid-derived suppressor cells (MDSCs). Blocking LILRB2 reprograms myeloid cells to be more pro-inflammatory and enhances the killing of intracellular Mycobacterium tuberculosis(166). Moreover, patients with active pulmonary tuberculosis have a higher frequency of LILRB1+ CD56dim FcγRIIIA+ NK cells, which correlates with disease severity (127). Conversely, recent data implicates LILRA2 in pathogen sensing and activation of innate immunity against microbial pathogens via the recognition of cleaved IgM and IgG products by proteases from S. pneumonia, Legionella pneumophila, Mycoplasma hyorhinis and Candida albicans(55). Neutrophils and monocytes expressing LILRA2 are activated by these cleaved Igs, enhancing immune responses against these bacteria (55).
Infection with Salmonella typhimurium can modulate APCs, especially macrophages and DCs. Exposure of APCs to Salmonella mediates upregulation of LILRB2 and LILRB4 and downregulation of LILRA2. This tuning in the balance of the LILR family members suppresses innate immune responses by increasing the IL-10:IL-12 ratio (56, 167). Mouse fibroblast cells generated to express PIR-B, LILRB1 or LILRB3 are all able to recognise Gram positive S. aureus, while LILRB1 is also able to bind E. coli(98). LILRB3 was recently reported to inhibit neutrophil effector functions and microbial killing, whereby ligation of LILRB3 abrogated IgA-mediated phagocytic uptake, reactive oxygen species generation and microbial killing of S. capitis(176).
The key pathogenic element of sepsis is systemic inflammation. However, most patients suffer signs of severe immunosuppression and fail to address the primary bacterial infection. Immune dysregulation during sepsis is associated with increased LILRB2 expression on monocytes and organ dysfunction (168). LILRB2+ monocytes express lower levels of CD86 and have an increase in IL-10:IL-12 cytokine ratio (168). In addition, LILRB2 upregulation found on healthy donor-derived activated neutrophils is impaired in septic patients with a consequent inhibition of their phagocytic function, proposing LILRB2 as a therapeutic target to prevent neutrophil dysfunction and exacerbated inflammation (144). Additionally, the antibiotic amoxicillin binds to HLA I, increasing NK cells cytolysis due to the inhibition of LILRB1 binding (236).
PIR-A and PIR-B are able to recognise cell wall components of both Gram positive and negative bacteria (98). Wildtype mice exhibit greater mortality than PIR-B-/- mice upon S. aureus infection. Stimulation of macrophages from PIR-B-/- mice with S. aureus results in increased levels of TLR-induced inflammatory cytokines IL-6 and IL-1β, compared to wildtype macrophages (237). Moreover, PIR-B is upregulated on macrophages after LPS treatment and negatively regulates the secretion of pro-inflammatory cytokines during E. coli infection (238).
Bacterial infections result in overexpression of most of the LILRBs, modulating leukocytes to a more anti-inflammatory state and blocking their effector properties. In contrast, LILRAs have an opposite role, depending on the type of infection.
Viruses interact with LILRs to suppress antiviral responses (9). The CMV gene product UL18 binds to LILRB1 on DCs rendering them resistant to maturation signals and unable to activate naïve T cells (115, 239), potentially so that CMV-infected cells can avoid elimination (9, 240). Analysis of memory T cells from CMV patients found high LILRB1 expression, with levels appearing to increase over time (241). Additionally, lung transplant recipients with elevated levels of LILRB1 on lymphocytes are at increased likelihood of CMV infection (131). However, investigations into the role of the UL18-LILRB1 interaction on T cells has yielded contradictory results. One study found that LILRB1 on cytotoxic T cells mediates lysis of virally-infected cells expressing UL18 independently of TCR, while cells infected with human CMV defective for UL18 were not lysed (132). In contrast, others have demonstrated that UL18 protects infected cells from LILRB1+ NK cell cytolysis. This protection was abrogated if cells were infected with CMV containing an UL18 mutant. In addition, UL18 mediated the activation of LILRB1- NK cells, which can mask LILRB1+ NK cell inhibition (242). Moreover, LILRB1 is highly expressed on viral-specific CD8+ T cells in Epstein-Barr virus-infected individuals (136, 137). LILRB1 expression is elevated on viral-specific CD8+ T and NK cells and interacts with viral products to downregulate immunity (129, 136, 137). NK cell activity was impaired in patients with chronic hepatitis B. Circulating CD56dim FcγRIIIA+ NK cells had increased LILRB1 in immunotolerant patients, which positively correlated with their serum viral load. Interestingly, LILRB1+ CD56dim NK cells were reduced with antiviral therapy, and LILRB1 blockade increased their cytotoxicity (138).
The interaction between LILRs and HIV infection is emerging as an important determinant of HIV progression (50). Upon HIV infection, DC dysfunction correlates with the upregulation of LILRB1 and LILRB2 (128) and downregulation of LILRA1 and LILRA2 (50, 51). In these patients, LILRB1 is upregulated on CD8+ T and NK cells, while LILRB2 expression is increased on myelomonocytic cells due to the increase in IL-10. These monocytes are defective in Ag presentation, which in turn abrogates the antiviral T cell responses and CD4+ T cell proliferation (128). These results indicate that the presence of high IL-10 levels in the sera of HIV+ patients impede Ag presentation of APCs by increasing LILRB2 expression. More recently, LILRB2 affinity for HLA I molecules was shown to positively correlate to the viral load in the majority of untreated HIV-1 patients. DCs in this cohort of patients were shown to have impaired Ag presentation ability as a result of LILRB2 crosslinking by HLA I molecules (170). In contrast, LILRBs can also enhance APC activity to stimulate T cells from HIV-1 elite controllers (51). These DCs express elevated levels of LILRB1 and LILRB3, blockade of which diminishes the enhanced Ag presenting properties (51). This enhanced T cell stimulating ability is contrary to other in vitro studies, which demonstrate that LILRB1 reduces the capability of DCs to stimulate T cells (115, 239). The interactions between LILRs on immune cells and HLA I expressed on HIV-infected cells is important to the response against infection (50). Specific HIV escape mutations when loaded as epitopes on HLA I diminish recognition by TCRs and enhance binding to LILRB2, resulting in the development of tolerogenic myelomonocytic cells (50, 169). Moreover, HLA-G is elevated in sera, and on monocytes and T cells of HIV-infected individuals (209, 243, 244). LILRB1 has been found overexpressed on NK cells after HIV-1 infection and these LILRB1+ NK cells control virus replication in DCs (130). However, the same laboratory has demonstrated that the inflammatory protein S100A9 expressed on HIV-infected DCs interacts with LILRB1 on NK cells and reduces DC cytotoxicity despite increased TNF-α secretion (95). These discrepancies potentially relate to different virality and stage of the disease (129).
pDCs are the only cell type known to express LILRA4 and are important in innate responses to viruses and tumours, producing significant quantities of IFNs following TLR7 and TLR9 ligation (78, 79, 81, 245). Indeed, LILRA4 is used as a marker of pDC subpopulations in coronavirus-19 (COVID-19)-infected patients (246), an APC subset that is reduced in severe cases (86). The only known ligand for LILRA4 is BST2, which prevents prolonged IFN production and assures TLR response by pDCs. BST2 expression is stimulated on a variety of cells by IFN and TLR7/9 ligands and is elevated during HIV infection (79, 247). The ability of IFN to induce BST2, which in turn interacts with LILRA4 to downregulate the IFN-producing pDCs, may serve as a negative feedback loop limiting IFN production (79).
Dengue virus is able to facilitate infection of myeloid cells by using antibody opsonisation to bind to activating FcγRs (97). Crosslinking of activating FcγRs leads to the induction of type-1 IFNs though Syk signalling, responses potentially deleterious to the internalised virus. To avoid this, viral proteins co-ligate LILRB1 on myeloid cells, which recruit phosphatases to inhibit Syk, preventing productive signalling. However, the ligand of LILRB1 on dengue virus remains unknown (97). In COVID-19 patients, LILRB4 expression is linked to disease severity and associates with a strong expansion of MDSCs and poor T cell responses, increasing immunosuppression (197, 198). Similarly, polymorphisms in LILRB1 and HLA-G are linked to higher risk of Zika virus transmission from mother to foetus, while certain polymorphisms in LILRB2 have a protective function (135).
Interestingly, the D3-4 region of PIR-B has been recently described to bind reovirus, allowing infection and producing serotype-dependent neuropathogenesis in infected mice (248).
Infection with the parasite Plasmodium falciparum, which develops into malaria, is associated with inflammatory cytokine production. LILRB1 has been shown to be upregulated on apoptotic B cells in the peripheral blood of patients with severe malaria compared to healthy controls. These early apoptotic LILRB1+ CD19+ B cells contribute to the inflammatory cytokine storm and impairment of immune memory (249). RIFINs, which are the causative targets of the malarial parasite, act as ligands for inhibitory receptors. A recent study proposed that LILRB1-binding RIFINs mimic the binding interface of the natural ligands of LILRB1 at the immunological synapse of NK cells, which suppresses NK cell cytotoxicity (96). LILRB2 also binds to RIFIN expressed on Plasmodium falciparum-infected erythrocytes, proposing it to produce a similar immune evasion to LILRB1 (133, 134). Additionally, infection with Toxoplasma gondii during pregnancy provokes a downregulation of LILRB4, switching macrophages and decidual MDSCs to a more pro-inflammatory state, contributing to adverse outcomes during pregnancy (250). In contrast, there is an upregulation of LILRB2 in non-classical monocytes of infants born to placental malaria mothers, enhancing susceptibility to the disease (251).
The immunomodulatory capacity of LILRs has been associated with autoimmune diseases and neurodegenerative disorders (Table 1). However, the functions of LILRs in these settings have not been fully elucidated.
Hashimoto’s thyroiditis (HT) and Graves’ disease patients express elevated levels of LILRB1 on peripheral CD4+, CD8+ and NK cells as well as thyroid tissue (HT patients). However, stimulation of these cells in vitro in the presence of a LILRB1 mAb has revealed that the receptor has an attenuated and defective ability to inhibit T cell proliferation. This reduced activity was mediated by IL-10 and contributed to poor control of inflammation in autoimmune disease (140).,
Two studies looking at a western European population found an association between the deletion of LILRA3 and an increased risk of multiple sclerosis (MS), whereas a study of a Polish population found that LILRA3 deletion was associated with later onset of MS (68, 74–76). In patients with MS, abundant expression of HLA-G and LILRB1 in areas of activated microglia, central nervous system (CNS) phagocytic cells, and periplaque tissues indicates that LILRB1-HLA-G interaction can regulate immune homeostasis of the CNS (139). Furthermore, LILRB4 is downregulated on monocytes during MS relapse (200). MS patients treated with IFN-β and vitamin D3 exhibit DC tolerance, in a LILRB4-dependent manner (199, 200). In addition, in the experimental autoimmune encephalopathy (EAE) mouse model of MS, sLILRB4 binds to immune cells and reduces the secretion of pro-inflammatory cytokines, delaying the evolution of the disease (252). Interestingly, it has been reported that glatiramer acetate (GA), a therapeutic molecule for relapsing-remitting MS, interacts with PIR-B on MDSCs and reduces pro-inflammatory responses. In addition, soluble GA competitively interacts with LILRB2 and LILRB3, modulating the alternative activation of monocytes and macrophages (253).
LILRB2 and PIR-B bind to oligomeric β-amyloid forms, which are involved in memory deficits and loss of synaptic plasticity. Interestingly, PIR-B-deficient mice do not have signs of damage caused by β-amyloid peptide or synaptic loss, implying its role in β-amyloid-induced Alzheimer’s disease (171, 172). Hence, many efforts have been made to improve synapsis elimination by disrupting LILRB2-β-amyloid interactions, for instance, with structure-guided small molecule inhibitors that physically impede the binding (254).
Examination of peripheral blood mononuclear cells (PBMC) from systemic lupus erythematosus (SLE) patients has revealed reduced expression of LILRB1 on CD4+ and CD8+ T cells, B cells and DCs, with LILRB1 on these cells demonstrating a diminished inhibitory function compared to healthy donors (141, 142). Moreover, LILRs possess high levels of polymorphisms that have been implicated with different autoimmune disorders, including SLE. A splice-site single nucleotide polymorphism (SNP) in LILRA2 gives rise to novel isoforms expressed on monocytes and is associated with higher susceptibility to SLE and microscopic polyangiitis (62). Furthermore, high expression and functionality of LILRA3 are associated with higher susceptibility to SLE and an increased disease activity and severity when induced in CD14+ monocytes (69, 71). Specific SNPs within LILRB4 observed in SLE patients are associated with its decreased surface expression on DCs, further correlating with increased serum type I IFNs and TNF-α (201). These results suggest that LILRBs have a potential role in the pathogenesis of SLE. LILRA4, BST2 and type I IFNs are orchestrated in a loop that regulates pDCs activation (24, 79). The release of autoantigens from dying keratinocytes induces neutrophil extracellular traps (NET) that promote the activation of LILRA4-expressing pDCs. This persistent activation drives the release of type I IFNs, provoking cutaneous lupus erythematous (CLE). Type I IFNs are increased in CLE patients (83) and CLE is known as a type I IFN disease (84, 85). Hence, LILRA4 has been studied as a specific target for some autoimmune disorders (255).
Genotyping studies suggest that patients homozygous for LILRA3 deficiency exhibit higher susceptibility to Sjogren’s syndrome (SS) (68–70). LILRA3 shares close homology with LILRA2, LILRB1 and LILRB2, so it may bind to their ligands either agonistically or antagonistically, possibly accounting for the contrary associations with LILRA3 in inflammatory diseases (25, 200). A risk allele (rs103294) in LILRA3 is involved in the deletion of the gene, and the epistasis of LILRA3 and HLA-B*52 might play an important role in Takayasu’s arteritis (TA), possibly by over activating NK cells (256). However, deeper analyses are needed to confirm an actual correlation. Additionally, genome-wide association studies of TA patients have identified a SNP which is associated with reduced LILRB3 expression as a susceptibility allele (180).
Neutrophil activation with high degree of NET formation is associated with the pathogenesis of adult-onset Still’s disease (AOSD). In a recent study, LILRA3 was reported to act as a novel genetic risk factor for AOSD, with elevated plasma LILRA3 levels in AOSD patients. NET formation was enhanced in neutrophils from AOSD patients upon LILRA3 stimulation (77).
Aberrant expression of LILRs has been associated with several arthritis syndromes. LILRA2, LILRA3, LILRA5, LILRB2, and LILRB3 are found at elevated levels in the sera and synovial fluid of RA patients, correlating with disease severity (25, 63, 72, 73, 87, 257). Significantly lower numbers of LILRA2+, LILRB2+ and LILRB3+ inflammatory cells were detected in RA patients who responded to anti-rheumatic therapy compared to healthy controls, as a result of the partial blocking of LILRA2-mediated secretion of TNF-α (63, 87, 257). Additionally, LILRA3 promotes pro-inflammatory responses in fibroblast-like synoviocytes, promoting their activation, migration and invasion in vitro (73). Anti-rheumatic drugs downregulate synovial expression of LILRB2, LILRB3 and LILRA2 in responding patients. However, this is not replicated in vitro, suggesting that the drugs do not act directly to impact LILR expression (257). While LILRA2 and LILRA5 are expressed highly in patients treated with methotrexate, LILRB2 is elevated in patients treated with prednisone (anti-inflammatory) (63). The potential relevance of these receptors in rheumatic inflammation is underlined by the ability of LILRB1, LILRB2 and LILRA2 to engage with HLA-B27, a haplotype associated with several inflammatory diseases. LILRB2 has been implicated in the pathogenesis of spondylarthritis, since LILRB2 can recognise several HLA-B27 isoforms and regulate innate and adaptive inflammatory responses (48). Moreover, LILRB1 binds to sHLA-G in RA patients protecting them against inflammation. However, this binding is not seen in advanced RA patients with long-term chronic inflammation, which impedes the immunosuppression and reduction of inflammation mediated by LILRB1 (258). Due to the high polymorphic nature of LILRs, different alleles can confer susceptibility to RA. A haplotype of LILRB1 that leads to reduced surface expression of the receptor is associated with high susceptibility to RA in HLA-DRB1 shared epitope-negative patients, possibly because of insufficient inhibitory signalling in their leukocytes (17).
APCs derived both from the recipient and donor are able to present Ags to T cells, and play a key role in transplant immunity (259). Consequently, alloreactive T cells are stimulated, which result in allogeneic graft rejection. Elevated levels of circulating T suppressor cells, tolerogenic APCs and HLA-G augment immunosuppression and are associated with a more favourable allotransplant acceptance (153, 260, 261). As receptors for HLA-G, LILRBs can be considered as therapeutic targets for medicating transplantation tolerance. In an organ transplantation setting, LILRB-mediated inhibition of T cells induced immune tolerance to allow allograft acceptance (249). LILRB1, LILRB2 and LILRB4 play fundamental roles in the immunosuppression cascade (Figure 4). Rejection-free heart, liver and kidney transplant recipients all possess alloantigen-specific CD8+ T suppressor cells (153, 260, 261). These T cells are able to induce LILRB2 and LILRB4 expression on donor DCs and monocytes and abrogate the expression of CD80/CD86 co-stimulatory molecules and alloreactive CD4+ Th cell proliferation (153, 260, 261). T suppressor cell-mediated tolerance extends to non-professional APCs including donor endothelial cells to confer tolerance of APCs (262, 263). In addition, LILRB1 was found highly expressed in circulating non-classical and intermediate monocytes of kidney transplant recipients. Interestingly, myeloid cells from kidney biopsies showed an upregulation of LILRB1, LILRB2 and LILRB3 after antibody-mediated rejection (ABMR), whereas circulating non-classical monocytes specifically had higher levels of LILRB3 and LILRB4 after ABMR (52).
Transplanted human pancreatic islet cells are tolerated by PBMC-engrafted NOD/SCID mice when treated with sLILRB4. This graft acceptance is associated with expansion of CD8+ T suppressor cells and diminished Th reactivity against graft HLA alloantigens (158, 264). The immunosuppression induced by the drug rapamycin is associated with increased LILRB2 and LILRB4 expression on DCs and a related increase in Tregs, T suppressor cells and serum HLA-G (173). Moreover, interaction of LILRB1 and LILRB2 with soluble and membrane-bound HLA-G from transplant patient sera augments Tregs and MDSCs and reduces T cell proliferation, enhancing the survival of skin allograft (215, 265–273). Similar findings were demonstrated in animal studies, where PIR-B was shown to enhance allotransplant acceptance. UVB-irradiated DCs that were unable to stimulate CD4+ T cells induced tolerance in heart transplant recipient rats, characterised by T suppressor cells and upregulation of PIR-B on APCs. Re-transplantation of PIR-B+ APC heart allografts to a second recipient failed to elicit rejection, indicating that these PIR-B+ APCs are responsible for tolerance (274). In addition, LILRB2+ DCs in LILRB2 transgenic mice induced tolerance against skin allografts when treated with HLA-G microbeads, via STAT3 and IDO activation and T cell suppression (273, 275, 276). HLA-G treatment of LILRB1 transgenic mice that previously received allogenic skin grafts resulted in expansion of MDSCs, which was associated with prolonged allograft survival (271).
Graft-versus Host Disease (GVHD) is the foremost impediment of allogeneic hematopoietic stem cell transplantation (HSCT), which in turn is associated with rejection of the allograft. PIR-B-deficient mice that received allogeneic T cells exhibited aggravated GVHD compared to wildtype mice as a result of the stimulation of PIR-B-deficient DCs (277). Similarly, acute GVHD was abrogated in mice that received PIR-B-transfected DCs, which were deficient in CD80/CD86 co-stimulatory molecules (278). A clinical study reported that 5.4% of patients that received HSCT, but not solid organ, had LILRB3-reactive antibodies directed against LILRB3+ DCs. These patients also expressed LILRB3 on leukemic cells, proposing LILRB3 as a GVH and graft-versus-leukaemia target (181). Moreover, our group demonstrated that mAb-mediated ligation of LILRB3 in humanised mice induces tolerance, allowing the engraftment of allogenic lymphoma cells (177). Collectively, these studies demonstrate that LILRBs are key regulators of immune tolerance and allograft acceptance and present an exciting therapeutic opportunity. Contrary to LILRBs, few studies have analysed the role of LILRAs in allotransplantation. A recent study showed that LILRA1 was highly expressed in circulating FcγRIIIA+ CD14- non-classical monocytes after kidney transplantation. Additionally, LILRA5 and LILRA6 were found overexpressed in circulating non-classical monocytes after ABMR and LILRA5 was highly expressed in myeloid cells from kidney tissues (52).
Pregnancy can be considered a type of allotransplant. Although the mechanisms that prevent foetal rejection by the maternal immune system remain incompletely known, LILRs have been implicated. HLA-G is expressed in trophoblasts during pregnancy, hence its interaction with LILRBs is considered essential (92). During pregnancy LILRB1 ligation inhibits the cytotoxicity of NK cells, while LILRB2 promotes M2 macrophage polarisation and MDSC suppressive activity (12, 92). Moreover, LILRB2 promotes DC tolerance and MDSC activation by binding to sHLA-G, and both LILRB1 and LILRB2 regulate B and T cell functions to maintain pregnancy. Considering all of this and their relevant role in placental vascular remodelling and foetal development, LILRBs are being considered as biomarkers of recurrent implantation failure (273, 279).
In addition to expression on immune cells and their dysregulation within the tumour microenvironment (TME), LILRs may also be present on cancer cells, to support tumourigenesis and suppress anti-tumour immunity. Hence, LILRs may be exploited as potential targets in cancer immunotherapy. It is now appreciated that LILRs may play central roles in a number of hallmarks of cancer: immune-evasion, inflammation, tumour cell proliferation and metastasis (6, 41).
The implication of LILRAs in cancer has not been fully addressed. In oestrogen receptor-positive breast cancer patients, LILRA2 gene expression correlated with tumour shrinkage (60), and in pancreatic ductal adenocarcinoma (PDA) higher transcript levels were associated with relapse-free survival (82). In the case of LILRA3, its ligation on monocytes was proposed to stimulate CD8+ T cells and NK cells in vitro, suggesting LILRA3 may be immunostimulatory (66). In addition, genetic deletion of LILRA3 leads to predisposition to non-Hodgkin’s lymphoma (66), while its presence is more common in prostate cancer patients of Chinese Han origin than in healthy controls (67). Notably, LILRA4 ligation can inhibit IFN-α and TNF-α production from pDCs. pDC infiltration in human tumours has been associated with poor prognosis, linked to impaired ability to produce the tumouricidal IFN-α (80, 280). More recently BST2 has been identified as a ligand for LILRA4, which is also expressed on several human cancers and downregulates IFN-α production, implying a mechanism through which tumours interact with LILRA4 to suppress immunity (79, 245). Additionally, a study looking at PDA patients found that higher LILRA4 expression is associated with better overall survival (OS) (82). Finally, a genome-wide association study showed that duplications at LILRA6 were associated with high-grade serous ovarian cancer susceptibility (20).
In contrast to LILRAs, there is compelling evidence that LILRBs are implicated in tumourigenesis, as well as tumour immune-evasion and progression. Examination of human cancer cell lines and tumour specimens has highlighted three main mechanisms. Firstly, aberrant LILRB expression occurs in several human cancers but not healthy adjacent tissues, with the expression of LILRBs and HLA-G found to correlate with poorly differentiated, more advanced or aggressive cancers in most cases (80, 120, 121, 124, 206–208, 210, 211, 213, 281–283). Secondly, IL-10 contributes to the LILRB : HLA-G axis of immunosuppression, as it upregulates LILRB and HLA-G (27, 98, 114, 115, 128, 143, 151, 282). Thirdly, recent advances have implicated LILRB signalling and expression directly with tumour progression and worse therapeutic response (119, 284). Directly or indirectly, many important LILRB functions involve modulation of myeloid cells (Table 1, Figure 5). Since the role of LILRBs in cancer have recently been reviewed elsewhere (6, 41, 93, 249), a brief summary is outlined here.
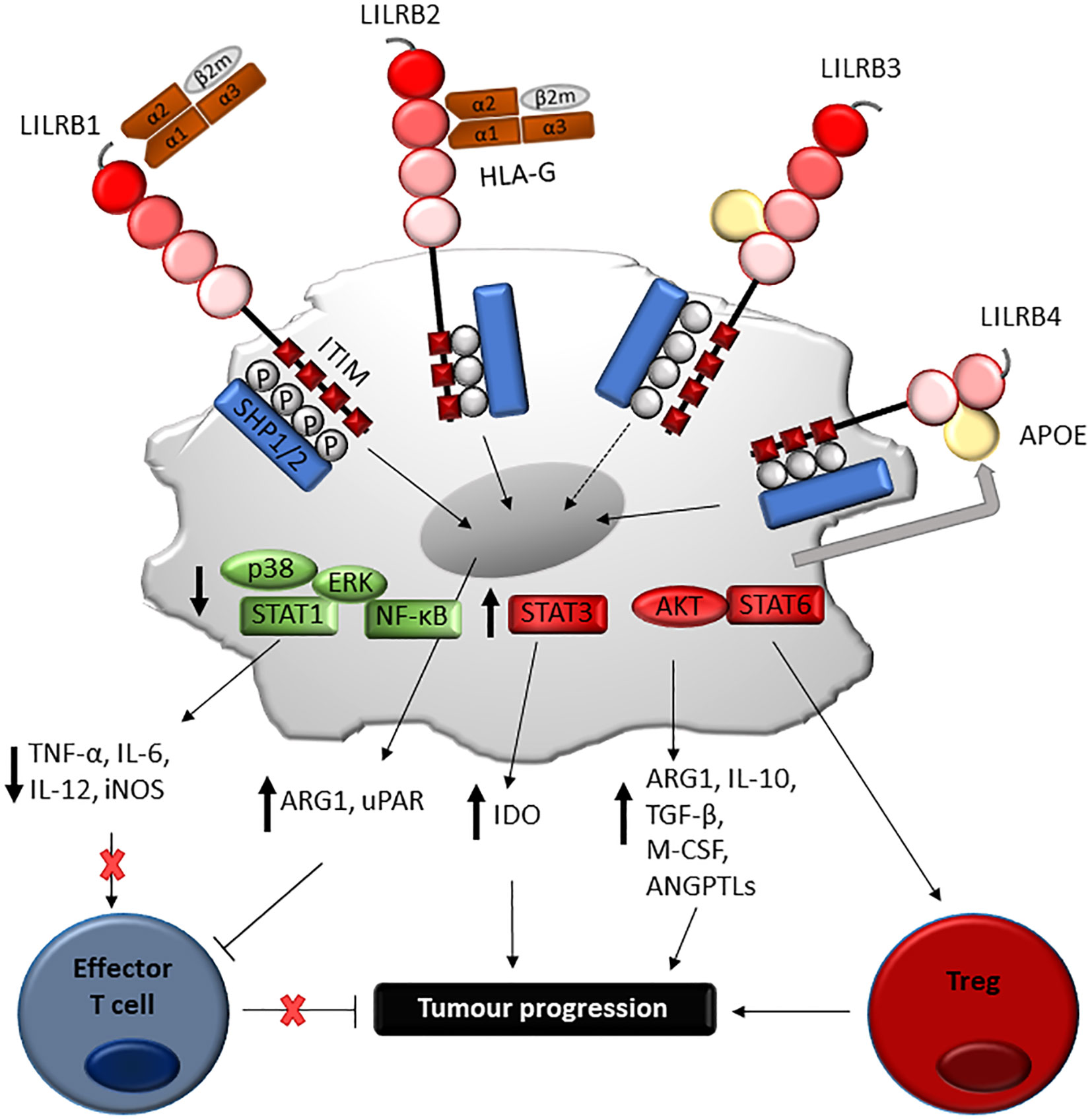
Figure 5 Proposed mechanism of LILRB-mediated immune-evasion and tumour progression via myeloid cells. Engagement of LILRB1 and LILRB2 with HLA-G on myeloid-derived suppressor cells (MDSC) activates STAT6 and STAT3-mediated cascades, which in turn induces ARG1 and IDO production responsible for T cell suppression. LILRB4 ligation by APOE on monocytic AML cells mediates SHP-2 inhibitory signalling, which in turn positively regulate the NF-κB pathway. This leads to ARG1 production and urokinase-type plasminogen activator receptor (uPAR), responsible for T cell suppression and support of leukaemia migration. Although further studies are needed, LILRB3 ligation can mediate similar processes, such as induction of amphiregulin.
LILRB1 is found on a variety of cancers including breast and prostate cancers, hepatocellular carcinoma, B cell lymphoma, acute myeloid leukaemia (AML), acute lymphoblastic leukaemia (ALL) and gastric cancer cell lines (110, 120, 124, 126). Moreover, a pan-cancer genomics analysis showed that LILRB1 was highly mutated in various cancers (119). In addition to expression on tumour cells, higher levels of LILRB1 have been demonstrated on the peripheral blood of non-small cell lung cancer (NSCLC), renal, head and neck, oesophagus and colon cancer patients than healthy individuals (6). Histological analysis of breast cancer biopsies revealed LILRB1 expression on CD68+ macrophages and CD8+ T cells (124). There is strong evidence that LILRB1 mediates cancer immune-evasion. Additionally, LILRB1 has been recently defined as a prognosis marker in ovarian cancer; high expression correlating with immunosuppression and its levels on immune cells associating with the clinical subtype and stage, resistance to platinum treatment and PD-1/PD-L1 mAb therapy (125). LILRB1 and LILRB4 are also expressed on human primary gastric cancer specimens compared to healthy tissue, with high expression correlating with advanced disease (120). Expression of LILRB1 on gastric cancer cell lines induces resistance to NK cell cytotoxicity (120, 283). Interestingly, LILRB1 gene and protein levels correlate with a shorter progression-free survival and poor clinical outcome in high but not operated intermediate-risk prostate cancer patients, indicating its correlation with tumour grade (126). Similarly, LILRB1 was found in the highest grade glioma patients, and correlated with M2 macrophage markers, proliferation, migration and invasion of glioma cells, lack of response to immunotherapy and poor prognosis (118). Furthermore, blocking of LILRB1 combined with rituximab and anti-CD47 enhanced antibody-dependent cellular phagocytosis (ADCP) of chronic lymphocytic leukaemia (CLL) cells (285). LILRB1 expression has been associated with poor AML survival, adverse prognostic impact, the inhibition of genes related to immune activation and dysfunctional CD8+ T cells (119). Expectedly, LILRB1 ligation by HLA-G on tumour cells induces tumour immune-evasion (206–208, 210, 211, 213, 281, 282).
LILRB2 is expressed on several types of cancers, including colon, breast, pancreas, lung, hepatocellular and prostate cancers and leukaemia (82, 126). In prostate cancer, LILRB2 together with LILRB3 and LILRB5 expression have been associated with reduced recurrence-free survival in intermediate but not high-risk patients (126). Furthermore, its overexpression in hepatocellular carcinoma, colon and NSCLC is associated with poor prognosis (82). Interestingly, in colorectal cancer it has been recently described that tumour-derived LILRB2 promotes tumour growth by increasing angiogenesis, and its blocking sensitises tumours to bevacizumab (anti-VEGF-A) treatment (163). As such, LILRB2 binding to HLA-G is associated with advanced stage and poor OS due to the increase in proliferation, migration and invasion of colorectal cancer cells (164). Moreover, in clear cell renal cell carcinoma LILRB2 increases the infiltration of macrophages, which have pro-angiogenic functions, and induces VEGF-C production (165). Additionally, LILRB2 is found on stromal macrophages, fibroblasts and plasma cells within the TME of primary breast cancer patients (162). Expression of LILRB2 on tumours correlates with higher levels of IL-10. Elevated levels of IL-10 in LILRB2+ breast cancer tissue positively correlates with advanced disease and lymph node metastasis, as well as reduction in tumour-infiltrating lymphocytes (TIL) (162). Moreover, LILRB2+ tissues in NSCLC have reduced numbers of TILs compared to LILRB2- tissues (121). In addition, LILRB2 is upregulated in NSCLC patients, inducing M2 macrophage polarisation and impairing T cell function, whose inhibition reverses its immunosuppressive role (286). Moreover, LILRB2 expression is associated with adverse prognostic impact in AML patients and lower OS (119). The ANGPTL2-LILRB2 interaction contributes to metastasis of pancreatic and lung cancers, correlating with poor survival. Oncogene mutations important in the carcinogenesis of PDA lead to the overexpression of LILRB2 and secretion of ANGPTL2 in pancreatic cancer lesions (284, 287).
LILRB3 has not yet been extensively studied with respect to tumour immune-evasion and development. LILRB3 has been found to be expressed in leukaemia and a few solid cancers, such as hepatocellular and colorectal cancers, and its expression is associated with a poor OS (119, 122, 179, 288). Perna et al., identified LILRB3 as being overexpressed on primary human AML samples and leukemic stem cells, while absent on healthy HSCs (178). Moreover, LILRB3 expression is linked to adverse prognostic impact in AML patients, with the highest LILRB3 expression found in M5 monocytic AML subtype, which correlates with worse OS. LILRB3 activates TRAF2 in AML cells, but not healthy monocytes, promoting NF-κB signalling and inhibiting anti-tumoural T cell activity (119, 288, 289). Interestingly, ectopic expression of LILRB3 on colorectal cancer cells associates with lower TILs and its high expression within the TME correlates with worse OS (179). LILRB3 as well as LILRA6 (> 90% extracellular homology) have been found to interact with cytokeratin-associated proteins on necrotic glandular epithelial cells, which may enhance tumour immune-evasion (89).
LILRB4 is expressed on a number of cancers, including AML, multiple myeloma, gastric cancer, melanoma, colorectal, pancreatic, hepatocellular, NSCLC and ovarian cancers (119, 158, 158, 185, 191–196, 290). LILRB4 has been associated with tumour immune-evasion with lower expression correlating with higher sensitivity to killing by NK cells in gastric cancer (120). LILRB4 is highly expressed on MDSCs of patients with NSCLC, correlating with poor OS due to the immunosuppressive environment and the enhanced migration, invasion and pro-angiogenic ability of NSCLC cells by binding to APOE (195, 196). In addition, LILRB4 expression on tumour-infiltrating cells and particularly MDSCs correlates with postoperative recurrence and shorter OS and relapse-free survival (290). Interestingly, its blockade prevented leukaemia metastasis and enhanced immunotherapy (185). Moreover, LILRB4 has been found on TAMs in several cancer types, and its blocking enhances the infiltration of anti-tumour immune cells due to the increased secretion of IL-1β and inducible nitric oxide synthase (184). sLILRB4 has been associated with immunosuppression and is found elevated in the sera of cancer patients (melanoma, colorectal and pancreatic), raising the possibility that it contributes to tumour escape (158). Humanised-SCID mice transplanted with several different allogenic tumour cell lines developed tumours when injected simultaneously with sLILRB4, unlike tumour cells that were injected alone (158). TILs found in these tumours were anergic with no tumour cell necrosis observed (158). T cells isolated from lymph nodes of the sLILRB4-treated mice failed to elicit T cell proliferation in a mixed lymphocyte reaction (MLR), a phenomena also observed when a MLR was conducted using human sera from cancer patients (158). The addition of a LILRB4 mAb or depletion of sLILRB4 increased T cell reactivity, demonstrating that sLILRB4 in the sera of cancer patients inhibits T cell proliferation (158). Deng et al. reported a potential mechanism for LILRB4-mediated AML progression (185). LILRB4 expression was shown to be restricted to monocytic AML cells, with ligation by APOE recruiting SHP-2 to the phosphorylated ITIM, leading to regulation of the NF-κB pathway and T cell suppression (185).
LILRB5 is also expressed in different tumours but its functions remain unclear (119, 122, 203). LILRB5 mRNA has been detected in NK cells (203), with NK cells from hepatocellular cancer patients blood expressing higher levels of LILRB5 than those from healthy donors. The same was observed in TAMs compared to healthy tissue (122). Moreover, and opposite to other members of the family, LILRB5 was associated with a favourable outcome in AML patients (119).
The mouse homolog PIR-B inhibits CD8+ T cell infiltration and promotes M2 macrophage polarisation (179). PIR-B-/- MDSCs exhibit an M1-like phenotype upon entry into the periphery and result in reduced suppressive function associated with impaired Treg activity, and accelerated lung tumour growth and metastases (291). In addition, deficiency in PIR-B results in increased differentiation of AML cells, indicating that PIR-B maintains AML cell stemness and promotes leukaemia development by arresting transformed cells in an undifferentiated state (146).
Recent success in the use of immune checkpoint blockade, including pembrolizumab, nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4), has paved the way for the development of novel immune checkpoint inhibitors (292). Apart from their potential use to predict immunotherapy responses, the powerful immunomodulatory capacity of LILRs supports this family of receptors as potential therapeutic targets (119). Several immunomodulatory approaches have been proposed to target LILR family members. In particular, antibodies can exert potent immunomodulatory functions with the ability to either activate (agonistic) or block (antagonistic) the activity of the respective targets (249).
Targeting LILRBs could achieve allotransplantations and prevent autoimmunity. Work by Suciu-Foca and colleagues has highlighted the Ag-specific immune tolerance mediated by LILRB2+ LILRB4+ tDCs and proposed them as therapeutic targets for allotransplantation, while avoiding the side effects of indiscriminate immunosuppression (293). Ex vivo expansion of tDCs with T suppressor cells may enable the transfer of donor-specific tolerance to mediate transplant tolerance. Moreover, treatment with sLILRB4 has the potential to dampen over-active immunity but may lack specificity (293). In addition, treatment with recombinant human LILRB4-extracellular domain-Fc fusion-protein has been shown to induce DC tolerance, reducing the progression of the disease, while blocking it exacerbated SLE (187). Alternatively, agonistic LILRB mAbs that block immune effector functions can be used to treat autoimmune syndromes that involve exacerbated immune activation. We recently demonstrated the potential of an agonistic LILRB3 mAb to reprogram myeloid cells (177). LILRB3 mAb treatment induced tolerance in vivo and enabled successful engraftment of allogeneic tumour cells in a humanised mouse model. This immunosuppressive efficacy may be exploited as a therapy for transplantation and autoimmunity (177). Another study showed that GA acts as a ligand for PIR-B, LILRB2 and LILRB3 on MDSCs, whose activation promotes Th2 immunity and the release of cytokines that suppress autoimmunity (253).
LILRAs can also be used as immune modulators. In CLE, pDCs expressing LILRA4 are essential in the immunopathology of the disease. For that reason, blocking LILRA4 can improve CLE patients’ outcome. In this regard, an anti-LILRA4 (clone VIB7734) that can deplete pDCs, reduced type I IFN release and disease severity in the skin (255). In fact, this mAb was evaluated in two Phase 1 clinical trials in patients with different type I IFN-mediated autoimmune diseases, including CLE. However, the primary endpoint in the Phase 2 trial was not met. Nevertheless, a Phase 2 clinical trial in SLE was recently completed. Additionally, the same mAb has been investigated in a Phase 1 clinical trial to treat and prevent acute lung injury in patients with COVID-19 (Table 2).
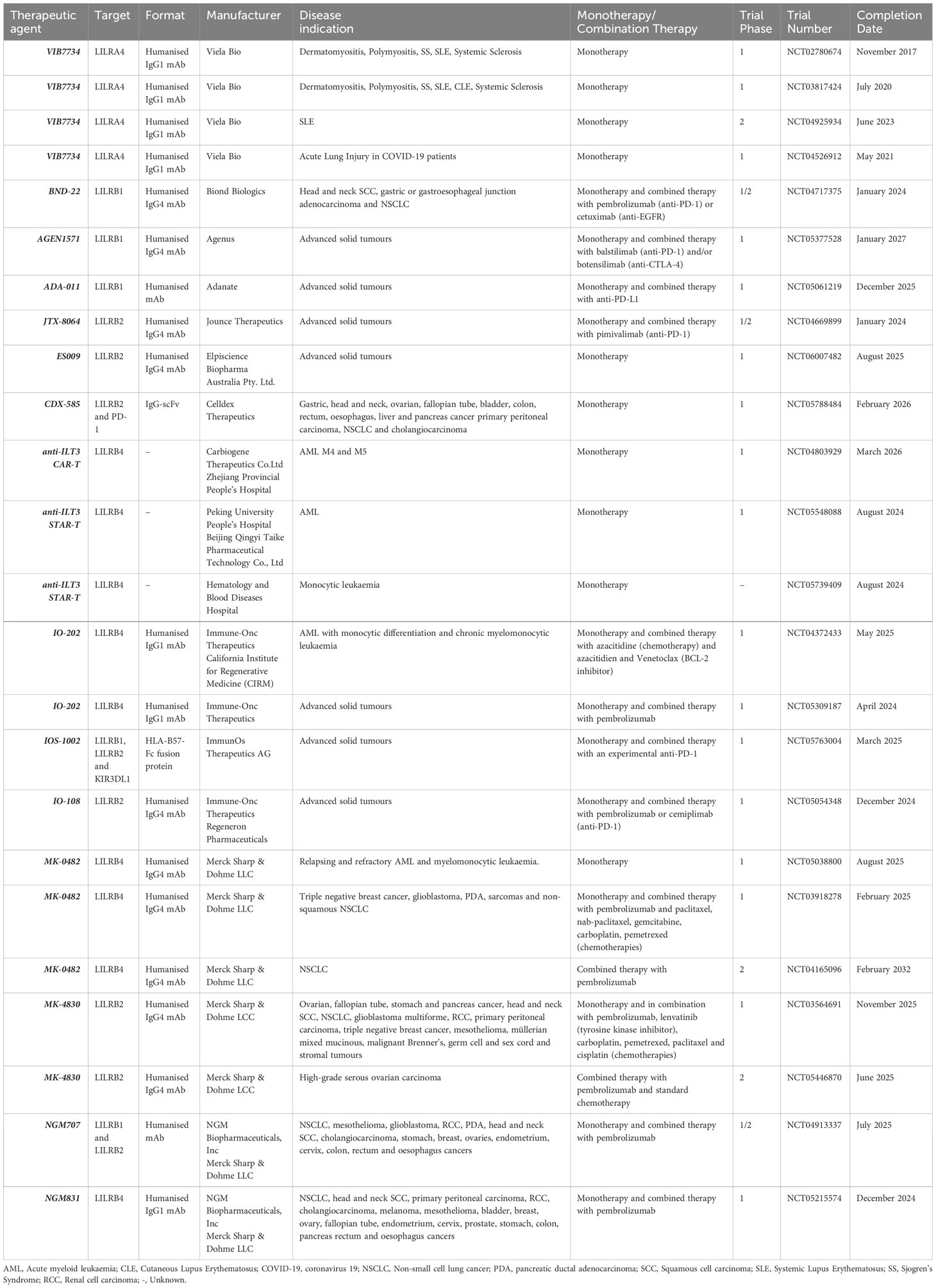
Table 2 A comprehensive list of clinical trials investigating the therapeutic targeting of LILRs to date.
Immunomodulatory approaches targeting LILRs may also provide durable therapy for cancer. Blockade of LILRBs may simultaneously inhibit tumour progression and promote anti-tumour immune responses. Indeed, several studies have addressed this therapeutic potential, especially by targeting LILRB1, LILRB2 and LILRB4 (93, 119). Furthermore, combination with other classes of immunotherapies, such as anti-PD-1, are being investigated in the clinic (Table 2). Mandel et al. developed BND-22, a first-in-class LILRB1 blocking antibody to treat murine and humanised mouse tumour models by increasing the activity of NK and T cells and the phagocytic potential of macrophages (294). A Phase 1/2 clinical trial is currently evaluating the safety, tolerability and anti-tumour effect of BND-22/SAR444881 in advanced solid tumours (unresectable or metastatic disease). Another Phase 1 clinical trial is underway to examine the potency of AGEN1571, a novel LILRB1 mAb, as monotherapy or combined with anti-PD-1 or anti-CTLA-4 in advanced solid tumours. Interestingly, AGEN1571 can polarise macrophages towards a pro-inflammatory phenotype and enhances the activity of CD8+ T, NK and NKT cells in preclinical models (295). Additionally, patient recruitment is underway for a Phase 1 clinical trial involving another LILRB1 mAb, ADA-011, as monotherapy or combined with a PD-L1 inhibitor in advanced solid tumours. Moreover, a recent preclinical study showed that dual mAb blockade of LILRB1 and PD-1 enhances CD8+ T cell activation and as a result augments the cytolytic efficacy of bispecific T cell engager (BiTE) molecules (296). Similarly, Zhang and colleagues recently developed and tested the efficacy of an antagonistic LILRB1 mAb. They specifically focused on the activity on NK cells, where LILRB1 expression is significantly upregulated in cancer patients, and demonstrated that LILRB1 blockade increases the tumouricidal activity of NK cells against several types of human solid and haematological cancers in preclinical settings (297). In addition, blockade of LILRB1 and NKG2A mediated NK cell cytotoxic killing of primary human ALL and AML blasts (110).
Blockade of LILRB2 has been demonstrated to reduce cancer cell proliferation, migration and invasion of cancer cells (284). Preclinical studies in NSCLC showed that LILRB2 blockade reprograms immunosuppressive myeloid cells and promotes antitumour immunity via SHP1/2, AKT and STAT6 inhibition, suppressing granulocytic MDSCs and Treg infiltration and improving checkpoint inhibitor efficacy (160). Moreover, LILRB2 antagonism increases inflammatory macrophages by interfering with M-CSF maturation (160). Umiker and colleagues demonstrated that the blockade of LILRB2 with JTX-8064 in different tumour types reprogrammes macrophages and DCs by inhibiting HLA I ligand binding. In addition, JTX-8064 improved the efficacy of anti-PD-1 therapy (161). JTX-8064 is currently in a Phase 1/2 clinical trial as monotherapy or in combination with the PD-1 mAb pimivalimab in advanced refractory solid malignancies with potential clinical benefits (298). Moreover, in another Phase 1 clinical trial, a human LILRB2 mAb (MK-4830) is being used as monotherapy or in combination with pembrolizumab (anti-PD-1) to treat advanced solid tumours. Initial results showed 11 objective responses to the combination and 1 to the monotherapy with durable responses in heavily pre-treated patients and no dose-limiting toxicities. Furthermore, LILRB2 blockade alleviated the myeloid-suppressive compartment, improving T cell response to pembrolizumab (299). IO-108 is another LILRB2 mAb that is in a Phase 1 clinical trial to treat solid tumours (as monotherapy or in combination with pembrolizumab) with promising results reportedly due to its activation of cytotoxic T lymphocytes and APCs, and repolarisation of macrophages (300). NGM831, an antagonistic LILRB2 antibody, is being investigated as monotherapy or in combination with pembrolizumab in advanced or metastatic solid tumours in a Phase 1 clinical trial. In preclinical studies, NGM831 modulated tDCs to a more stimulatory and responsive phenotype, stimulating allogenic T cells in combination with anti-PD-1 (301). Additionally, a recent Phase 1 clinical trial is currently recruiting patients with advanced solid tumours to evaluate the safety, tolerability and clinical activity of a new human LILRB2 mAb (ES009). Previous in vitro and ex vivo studies demonstrated that blocking LILRB2 with ES009 reprograms myeloid cells to a pro-inflammatory phenotype and enhances T cell activation (302).
Furthermore, NGM707, a mAb that recognises both LILRB1 and LILRB2 is in a Phase 1/2 clinical trial as monotherapy or combined with pembrolizumab in advanced or metastatic solid tumour malignancies. Preliminary data from the Phase 1 trial showed that NGM707 was well tolerated and developed early signs of anti-tumour activity by reprogramming myeloid cells (303, 304). Similarly, a new Phase 1 clinical trial with IOS-1002, a first-in-class molecule that targets LILRB1, LILRB2 and KIR3DL1, is being investigated in patients with advanced solid tumours. It is being evaluated as monotherapy and in combination with anti-PD-1, and preclinical data have shown that it significantly increases the anti-tumourigenic responses of macrophages, T and NK cells (305). CDX-585 is a novel tetravalent IgG-scFv bispecific antibody targeting both PD-1 and LILRB2, which is undergoing Phase 1 clinical trial in advanced malignancies. In preclinical studies, CDX-585 improved T cell activation, resulted in macrophage polarisation towards M1 and enhanced anti-tumour responses in a humanised mouse model of melanoma (306).
LILRB4 has also been widely evaluated for its therapeutic potential (307). IO-202 is being investigated in relapsed/refractory myelomonocytic and monocytic AML and relapsed/refractory chronic myelomonocytic leukaemia (308). In addition, IO-202 is undergoing clinical trials as monotherapy or in combination with pembrolizumab in solid tumours (308). Similarly, Di Meo and colleagues developed a LILRB4 BiTE that showed a high efficacy in potentiating T cell cytotoxicity against multiple myeloma cells in vitro and in vivo, and prolonged survival of tumour-bearing mice (193). MK-0482 is being tested in patients with relapsed/refractory myelomonocytic and monocytic AML and relapsed/refractory chronic myelomonocytic leukaemia. However, it is important to highlight its toxicity; myositis was observed in two patients and led to death of one of them. The same antibody is being used as monotherapy or in combination with pembrolizumab in a Phase 1 clinical trial in heavily pre-treated advanced solid tumours and in a Phase 2 clinical trial in advanced NSCLC. A novel anti-LILRB4 chimeric antigen receptor (CAR) T cell therapy recently demonstrated potent elimination of human LILRB4+ AML cells in preclinical models with no toxicity on normal CD34+ hematopoietic cells (307). An early Phase 1 clinical study is evaluating the safety and efficacy of this anti-LILRB4 CAR-T cell immunotherapy in AML patients. Similarly, LILRB4 synthetic T cell antigen receptor (STAR)-T cells have been developed and are in a Phase 1 clinical trial for the treatment of relapsed/refractory AML (309) and monocytic AML. Interestingly, Huang and colleagues developed a bispecific LILRB4 x CD3 antibody for monocytic AML with promising preclinical results (310). Moreover, a T-cell engager targeting LILRB4, NGM936, to treat AML has been developed, which induces T cell cytotoxicity against LILRB4+ cells in preclinical studies (311). Finally, an antibody-drug conjugate has been developed from a humanised anti-LILRB4, inducing cytotoxicity against LILRB4+ AML cells (312).
LILRs are emerging as important mediators of immune homeostasis, regulating the balance between tolerance and immune activation. Increasing evidence supports LILRs’ central involvement in various human pathologies, ranging from oncology to autoimmune disorders, associated with suppressed immunity or exacerbated immune activation, respectively. Our understanding of the human LILR biology and crosstalk is limited by our understanding of their ligands, with ligands for only some LILRs identified to date. The lack of direct LILR homologues in the mouse and specific reagents have impaired the study of this important family of immune receptors. This poses a major hurdle for studying LILR biology and requires the need for the development of novel mouse models, including LILRA and/or LILRB transgenic mice and knock-in/-out mice (eg, PIR- and LILR+), faithfully expressing these receptors. As such, more recent engineering advances in generating humanised mice have begun to allow the study of this complex receptor family in a more ‘physiological’ context and will undoubtedly continue to support the study of these receptors and other elusive immune receptors (177, 218, 313, 314). These models could allow us to further examine the ligand profiles, functions and therapeutic potential of the LILR family members.
In regard to their therapeutic potential, the high homology among certain LILRs must be taken into consideration. Without this, targeting the inhibitory LILRs could simultaneously mediate LILRA activation or inhibition, which may interfere with the desired outcome. The recent preclinical and clinical evidence propose LILRBs as ideal targets for immunotherapies against various pathologies including cancer. Most exciting is the emerging dual role of LILRBs in promoting carcinogenesis and immune-evasion, which propose the development of novel and highly potent immunotherapies for reducing tumour burden and immunosuppression. As such, it is anticipated that novel LILR-targeting modalities currently in clinical trials will soon make their way into the clinic.
SR-G: Writing – original draft, Writing – review & editing. CB: Writing – original draft. CP: Writing – original draft, Writing – review & editing. MY: Writing – original draft. BF: Writing – review & editing. MSC: Funding acquisition, Supervision, Writing – review & editing. AR: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We are also grateful for the funding from the Biotechnology and Biological Sciences Research Council (BBSRC), Medical Research Council (MRC), Blood Cancer UK (14043), Cancer Research UK, Wessex Medical Research and Breast Cancer Now (BCN 2022FebPR1540). The authors declare that this study also received funding from BioInvent International. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication. SR-G was supported by a BCN project grant awarded to AR. MY was supported by a BBSRC iCASE PhD studentship and CP was supported by an MRC iCASE PhD studentship awarded to AR and MSC.
We would like to thank the members of the Antibody and Vaccine Group, University of Southampton, and BioInvent International for useful discussions and support.
BF is an employee of BioInvent International. AR and MSC receive institutional support from BioInvent International. AR acts as a consultant for Epsilogen. MSC has consulted for BioInvent International, Boehringer Ingelheim, GSK, Radiant, iteos, Surrozen, Hanall and Mestag and received research funding from BioInvent International, GSK, UCB and iTeos. He is a member of the GSK Immunology Network.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Levi-Schaffer F, Mandelboim O. Inhibitory and coactivating receptors recognising the same ligand: immune homeostasis exploited by pathogens and tumours. Trends Immunol (2018) 39(2):112–22. doi: 10.1016/j.it.2017.10.001
2. Rumpret M, Drylewicz J, Ackermans LJE, Borghans JAM, Medzhitov R, Meyaard L. Functional categories of immune inhibitory receptors. Nat Rev Immunol (2020) 20(12):771–80. doi: 10.1038/s41577-020-0352-z
3. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372(26):2521–32. doi: 10.1056/NEJMoa1503093
4. Edner NM, Carlesso G, Rush JS, Walker LSK. Targeting co-stimulatory molecules in autoimmune disease. Nat Rev Drug Discov (2020) 19(12):860–83. doi: 10.1038/s41573-020-0081-9
5. Brown D, Trowsdale J, Allen R. The LILR family: modulators of innate and adaptive immune pathways in health and disease. Tissue Antigens (2004) 64(3):215–25. doi: 10.1111/j.0001-2815.2004.00290.x
6. De Louche CD, Roghanian A. Human inhibitory leukocyte Ig-like receptors: from immunotolerance to immunotherapy. JCI Insight (2022) 7(2):e151553. doi: 10.1172/jci.insight.151553
7. Hirayasu K, Arase H. Leukocyte immunoglobulin-like receptor (LILR). In: Choi S, editor. Encyclopedia of signaling molecules. Cham: Springer International Publishing (2018). p. 2854–61. doi: 10.1007/978-3-319-67199-4_101689
8. Colonna M, Samaridis J, Cella M, Angman L, Allen RL, O’Callaghan CA, et al. Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J Immunol (1998) 160(7):3096–100. doi: 10.4049/jimmunol.160.7.3096
9. Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, et al. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity (1997) 7(2):273–82. doi: 10.1016/S1074-7613(00)80529-4
10. Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. Genenames.org: the HGNC resources in 2015. Nucleic Acids Res (2015) 43(Database issue):D1079–85. doi: 10.1093/nar/gku1071
11. Borges L, Hsu ML, Fanger N, Kubin M, Cosman D. A family of human lymphoid and myeloid Ig-like receptors, some of which bind to MHC class I molecules. J Immunol (1997) 159(11):5192–6. doi: 10.4049/jimmunol.159.11.5192
12. Storm L, Bruijnesteijn J, de Groot NG, Bontrop RE. The genomic organization of the LILR region remained largely conserved throughout primate evolution: implications for health and disease. Front Immunol (2021) 12:716289. doi: 10.3389/fimmu.2021.716289
13. Wilson MJ, Torkar M, Haude A, Milne S, Jones T, Sheer D, et al. Plasticity in the organization and sequences of human KIR/ILT gene families. Proc Natl Acad Sci USA (2000) 97(9):4778–83. doi: 10.1073/pnas.080588597
14. Yang T, Qian Y, Liang X, Wu J, Zou M, Deng M. LILRB4, an immune checkpoint on myeloid cells. Blood Sci (2022) 4(2):49–56. doi: 10.1097/BS9.0000000000000109
15. Torkar M, Haude A, Milne S, Beck S, Trowsdale J, Wilson MJ. Arrangement of the ILT gene cluster: a common null allele of the ILT6 gene results from a 6.7-kbp deletion. Eur J Immunol (2000) 30(12):3655–62. doi: 10.1002/1521-4141(200012)30:12<3655::AID-IMMU3655<3.0.CO;2-Y
16. Norman P, Carey BS, Stephens HF, Vaughan R. DNA sequence variation and molecular genotyping of natural killer leukocyte immunoglobulin-like receptor, LILRA3. Immunogenetics (2003) 55(3):165–71. doi: 10.1007/s00251-003-0561-1
17. Kuroki K, Tsuchiya N, Shiroishi M, Rasubala L, Yamashita Y, Matsuta K, et al. Extensive polymorphisms of LILRB1 (ILT2, LIR1) and their association with HLA-DRB1 shared epitope negative rheumatoid arthritis. Hum Mol Genet (2005) 14(16):2469–80. doi: 10.1093/hmg/ddi247
18. López-Álvarez MR, Jones DC, Jiang W, Traherne JA, Trowsdale J. Copy number and nucleotide variation of the LILR family of myelomonocytic cell activating and inhibitory receptors. Immunogenetics (2014) 66(2):73–83. doi: 10.1007/s00251-013-0742-5
19. Lewis Marffy AL, McCarthy AJ. Leukocyte immunoglobulin-like receptors (LILRs) on human neutrophils: modulators of infection and immunity. Front Immunol (2020) 11:857. doi: 10.3389/fimmu.2020.00857
20. Reid BM, Permuth JB, Chen YA, Fridley BL, Iversen ES, Chen Z, et al. Genome-wide analysis of common copy number variation and epithelial ovarian cancer risk. Cancer Epidemiol Biomarkers Prev (2019) 28(7):1117–26. doi: 10.1158/1055-9965.EPI-18-0833
21. Hirayasu K, Ohashi J, Tanaka H, Kashiwase K, Ogawa A, Takanashi M, et al. Evidence for natural selection on leukocyte immunoglobulin-like receptors for HLA class I in Northeast Asians. Am J Hum Genet (2008) 82(5):1075–83. doi: 10.1016/j.ajhg.2008.03.012
22. Anderson KJ, Allen RL. Regulation of T-cell immunity by leucocyte immunoglobulin-like receptors: innate immune receptors for self on antigen-presenting cells. Immunology (2009) 127(1):8–17. doi: 10.1111/j.1365-2567.2009.03097.x
23. Zhang M, Yang J, Zhang J, Huang C, Liu H, Zhang P, et al. Research progress of B subfamily of leucocyte immunoglobulin-like receptors in inflammation. Int J Immunogenet (2023) 50(3):107–16. doi: 10.1111/iji.12618
24. Hirayasu K, Arase H. Functional and genetic diversity of leukocyte immunoglobulin-like receptor and implication for disease associations. J Hum Genet (2015) 60(11):703–8. doi: 10.1038/jhg.2015.64
25. An H, Chandra V, Piraino B, Borges L, Geczy C, McNeil HP, et al. Soluble LILRA3, a potential natural antiinflammatory protein, is increased in patients with rheumatoid arthritis and is tightly regulated by interleukin 10, tumor necrosis factor-α, and interferon-γ. J Rheumatol (2010) 37(8):1596–606. doi: 10.3899/jrheum.091119
26. Jones DC, Roghanian A, Brown DP, Chang C, Allen RL, Trowsdale J, et al. Alternative mRNA splicing creates transcripts encoding soluble proteins from most LILR genes. Eur J Immunol (2009) 39(11):3195–206. doi: 10.1002/eji.200839080
27. Beinhauer BG, McBride JM, Graf P, Pursch E, Bongers M, Rogy M, et al. Interleukin 10 regulates cell surface and soluble LIR-2 (CD85d) expression on dendritic cells resulting in T cell hyporesponsiveness in vitro. Eur J Immunol (2004) 34(1):74–80. doi: 10.1002/eji.200324550
28. Borges L, Kubin M, Kuhlman T. LIR9, an immunoglobulin-superfamily–activating receptor, is expressed as a transmembrane and as a secreted molecule. Blood (2002) 101(4):1484–6. doi: 10.1182/blood-2002-05-1432
29. Jones DC, Kosmoliaptsis V, Apps R, Lapaque N, Smith I, Kono A, et al. HLA class I allelic sequence and conformation regulate leukocyte Ig-like receptor binding. J Immunol (2011) 186(5):2990–7. doi: 10.4049/jimmunol.1003078
30. Colonna M, Nakajima H, Navarro F, López-Botet M. A novel family of Ig-like receptors for HLA class I molecules that modulate function of lymphoid and myeloid cells. J Leukocyte Biol (1999) 66(3):375–81. doi: 10.1002/jlb.66.3.375
31. Chapman TL, Heikema AP, West AP Jr., Bjorkman PJ. Crystal structure and ligand binding properties of the D1D2 region of the inhibitory receptor LIR-1 (ILT2). Immunity (2000) 13(5):727–36. doi: 10.1016/s1074-7613(00)00071-6
32. Nam G, Shi Y, Ryu M, Wang Q, Song H, Liu J, et al. Crystal structures of the two membrane-proximal Ig-like domains (D3D4) of LILRB1/B2: alternative models for their involvement in peptide-HLA binding. Protein Cell (2013) 4(10):761–70. doi: 10.1007/s13238-013-3908-x
33. Willcox BE, Thomas LM, Chapman TL, Heikema AP, West AP Jr., Bjorkman PJ. Crystal structure of LIR-2 (ILT4) at 1.8 A: differences from LIR-1 (ILT2) in regions implicated in the binding of the Human Cytomegalovirus class I MHC homolog UL18. BMC Struct Biol (2002) 2:6. doi: 10.1186/1472-6807-2-6
34. Wang Q, Song H, Cheng H, Qi J, Nam G, Tan S, et al. Structures of the four Ig-like domain LILRB2 and the four-domain LILRB1 and HLA-G1 complex. Cell Mol Immunol (2020) 17(9):966–75. doi: 10.1038/s41423-019-0258-5
35. Willcox BE, Thomas LM, Bjorkman PJ. Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat Immunol (2003) 4(9):913–9. doi: 10.1038/ni961
36. Cheng H, Mohammed F, Nam G, Chen Y, Qi J, Garner LI, et al. Crystal structure of leukocyte Ig-like receptor LILRB4 (ILT3/LIR-5/CD85k): a myeloid inhibitory receptor involved in immune tolerance. J Biol Chem (2011) 286(20):18013–25. doi: 10.1074/jbc.M111.221028
37. Chen Y, Gao F, Chu F, Peng H, Zong L, Liu Y, et al. Crystal structure of myeloid cell activating receptor leukocyte Ig-like receptor A2 (LILRA2/ILT1/LIR-7) domain swapped dimer: molecular basis for its non-binding to MHC complexes. J Mol Biol (2009) 386(3):841–53. doi: 10.1016/j.jmb.2009.01.006
38. Shiroishi M, Kajikawa M, Kuroki K, Ose T, Kohda D, Maenaka K. Crystal structure of the human monocyte-activating receptor, “Group 2” leukocyte Ig-like receptor A5 (LILRA5/LIR9/ILT11). J Biol Chem (2006) 281(28):19536–44. doi: 10.1074/jbc.M603076200
39. Takai T. Paired immunoglobulin-like receptors and their MHC class I recognition. Immunology (2005) 115(4):433–40. doi: 10.1111/j.1365-2567.2005.02177.x
40. Vlieg HC, Huizinga EG, Janssen BJC. Structure and flexibility of the extracellular region of the PirB receptor. J Biol Chem (2019) 294(12):4634–43. doi: 10.1074/jbc.RA118.004396
41. Deng M, Chen H, Liu X, Huang R, He Y, Yoo B, et al. Leukocyte immunoglobulin-like receptor subfamily B: therapeutic targets in cancer. Antib Ther (2021) 4(1):16–33. doi: 10.1093/abt/tbab002
42. Uehara T, Bléry M, Kang D-W, Chen C-C, Ho LH, Gartland GL, et al. Inhibition of IgE-mediated mast cell activation by the paired Ig-like receptor PIR-B. J Clin Invest (2001) 108(7):1041–50. doi: 10.1172/JCI12195
43. Kubagawa H, Chen C-C, Le Hong H, Shimada T, Gartland L, Mashburn C, et al. Biochemical nature and cellular distribution of the paired immunoglobulin-like receptors, PIR-A and PIR-B. J Exp Med (1999) 189(2):309–18. doi: 10.1084/jem.189.2.309
44. Kubagawa H, Burrows PD, Cooper MD. A novel pair of immunoglobulin-like receptors expressed by B cells and myeloid cells. Proc Natl Acad Sci USA (1997) 94(10):5261–6. doi: 10.1073/pnas.94.10.5261
45. Pereira S, Zhang H, Takai T, Lowell CA. The inhibitory receptor PIR-B negatively regulates neutrophil and macrophage integrin signaling. J Immunol (2004) 173(9):5757–65. doi: 10.4049/jimmunol.173.9.5757
46. Munitz A, McBride ML, Bernstein JS, Rothenberg ME. A dual activation and inhibition role for the paired immunoglobulin-like receptor B in eosinophils. Blood (2008) 111(12):5694–703. doi: 10.1182/blood-2007-12-126748
47. Baruch-Morgenstern NB, Shik D, Moshkovits I, Itan M, Karo-Atar D, Bouffi C, et al. Paired immunoglobulin-like receptor A is an intrinsic, self-limiting suppressor of IL-5-induced eosinophil development. Nat Immunol (2014) 15(1):36–44. doi: 10.1038/ni.2757
48. Shiroishi M, Kuroki K, Rasubala L, Tsumoto K, Kumagai I, Kurimoto E, et al. Structural basis for recognition of the nonclassical MHC molecule HLA-G by the leukocyte Ig-like receptor B2 (LILRB2/LIR2/ILT4/CD85d). Proc Natl Acad Sci USA (2006) 103(44):16412–7. doi: 10.1073/pnas.0605228103
49. Abdallah F, Coindre S, Gardet M, Meurisse F, Naji A, Suganuma N, et al. Leukocyte immunoglobulin-like receptors in regulating the immune response in infectious diseases: A window of opportunity to pathogen persistence and a sound target in therapeutics. Front Immunol (2021) 12:717998. doi: 10.3389/fimmu.2021.717998
50. Lichterfeld M, Yu XG. The emerging role of leukocyte immunoglobulin-like receptors (LILRs) in HIV-1 infection. J Leukocyte Biol (2012) 91(1):27–33. doi: 10.1189/jlb.0811442
51. Huang J, Burke PS, Cung TDH, Pereyra F, Toth I, Walker BD, et al. Leukocyte immunoglobulin-like receptors maintain unique antigen-presenting properties of circulating myeloid dendritic cells in HIV-1-infected elite controllers. J Virol (2010) 84(18):9463–71. doi: 10.1128/JVI.01009-10
52. Lamarthée B, Genet C, Cattin F, Danger R, Giral M, Brouard S, et al. Single-cell mapping of leukocyte immunoglobulin-like receptors in kidney transplant rejection. Front Transplant (2022) 1:952785. doi: 10.3389/frtra.2022.952785
53. Sloane DE, Tedla N, Awoniyi M, MacGlashan DW, Borges L, Austen KF, et al. Leukocyte immunoglobulin-like receptors: novel innate receptors for human basophil activation and inhibition. Blood (2004) 104(9):2832–9. doi: 10.1182/blood-2004-01-0268
54. Tedla N, Bandeira-Melo C, Tassinari P, Sloane DE, Samplaski M, Cosman D, et al. Activation of human eosinophils through leukocyte immunoglobulin-like receptor 7. Proc Natl Acad Sci (2003) 100(3):1174–9. doi: 10.1073/pnas.0337567100
55. Hirayasu K, Saito F, Suenaga T, Shida K, Arase N, Oikawa K, et al. Microbially cleaved immunoglobulins are sensed by the innate immune receptor LILRA2. Nat Microbiol (2016) 1:16054. doi: 10.1038/nmicrobiol.2016.54
56. Lu HK, Mitchell A, Endoh Y, Hampartzoumian T, Huynh O, Borges L, et al. LILRA2 selectively modulates LPS-mediated cytokine production and inhibits phagocytosis by monocytes. PloS One (2012) 7(3):e33478. doi: 10.1371/journal.pone.0033478
57. Ottonello L, Ghio M, Contini P, Bertolotto M, Bianchi G, Montecucco F, et al. Nonleukoreduced red blood cell transfusion induces a sustained inhibition of neutrophil chemotaxis by stimulating in vivo production of transforming growth factor-β1 by neutrophils: role of the immunoglobulinlike transcript 1, sFasL, and sHLA-I. Transfusion (2007) 47(8):1395–404. doi: 10.1111/j.1537-2995.2007.01268.x
58. Nakajima H, Samaridis J, Angman L, Colonna M. Cutting edge: human myeloid cells express an activating ILT receptor (ILT1) that associates with fc receptor γ-chain. J Immunol (1999) 162(1):5–8. doi: 10.4049/jimmunol.162.1.5
59. Lee DJ, Sieling PA, Ochoa MT, Krutzik SR, Guo B, Hernandez M, et al. LILRA2 activation inhibits dendritic cell differentiation and antigen presentation to T cells. J Immunol (2007) 179(12):8128–36. doi: 10.4049/jimmunol.179.12.8128
60. Goto-Yamaguchi L, Yamamoto-Ibusuki M, Yamamoto Y, Fujiki Y, Tomiguchi M, Sueta A, et al. Therapeutic predictors of neoadjuvant endocrine therapy response in estrogen receptor-positive breast cancer with reference to optimal gene expression profiling. Breast Cancer Res Treat (2018) 172(2):353–62. doi: 10.1007/s10549-018-4933-5
61. Bleharski JR, Li H, Meinken C, Graeber TG, Ochoa M-T, Yamamura M, et al. Use of genetic profiling in leprosy to discriminate clinical forms of the disease. Science (2003) 301(5639):1527–30. doi: 10.1126/science.1087785
62. Mamegano K, Kuroki K, Miyashita R, Kusaoi M, Kobayashi S, Matsuta K, et al. Association of LILRA2 (ILT1, LIR7) splice site polymorphism with systemic lupus erythematosus and microscopic polyangiitis. Genes Immun (2008) 9(3):214–23. doi: 10.1038/gene.2008.5
63. Tedla N, An H, Borges L, Vollmer-Conna U, Bryant K, Geczy C, et al. Expression of activating and inhibitory leukocyte immunoglobulin-like receptors in rheumatoid synovium: correlations to disease activity. Tissue Antigens (2011) 77(4):305–16. doi: 10.1111/j.1399-0039.2011.01633.x
64. Ryu M, Chen Y, Qi J, Liu J, Fan Z, Nam G, et al. LILRA3 binds both classical and non-classical HLA class I molecules but with reduced affinities compared to LILRB1/LILRB2: structural evidence. PloS One (2011) 6(4):e19245. doi: 10.1371/journal.pone.0019245
65. An H, Brettle M, Lee T, Heng B, Lim CK, Guillemin GJ, et al. Soluble LILRA3 promotes neurite outgrowth and synapses formation through a high-affinity interaction with Nogo 66. J Cell Sci (2016) 129(6):1198–209. doi: 10.1242/jcs.182006
66. Low HZ, Reuter S, Topperwien M, Dankenbrink N, Peest D, Kabalak G, et al. Association of the LILRA3 deletion with B-NHL and functional characterization of the immunostimulatory molecule. PloS One (2013) 8(12):e81360. doi: 10.1371/journal.pone.0081360
67. Xu J, Mo Z, Ye D, Wang M, Liu F, Jin G, et al. Genome-wide association study in Chinese men identifies two new prostate cancer risk loci at 9q31.2 and 19q13.4. Nat Genet (2012) 44(11):1231–5. doi: 10.1038/ng.2424
68. Koch S, Goedde R, Nigmatova V, Epplen JT, Muller N, de Seze J, et al. Association of multiple sclerosis with ILT6 deficiency. Genes Immun (2005) 6(5):445–7. doi: 10.1038/sj.gene.6364187
69. Du Y, Su Y, He J, Yang Y, Shi Y, Cui Y, et al. Impact of the leucocyte immunoglobulin-like receptor A3 (LILRA3) on susceptibility and subphenotypes of systemic lupus erythematosus and Sjögren’s syndrome. Ann Rheumatic Dis (2014) 74:2070–5. doi: 10.1136/annrheumdis-2013-204441
70. Kabalak G, Dobberstein SB, Matthias T, Reuter S, The YH, Dörner T, et al. Association of immunoglobulin-like transcript 6 deficiency with Sjögren’s syndrome. Arthritis Rheumatism (2009) 60(10):2923–5. doi: 10.1002/art.24804
71. Du Y, Sun F, Zhou M, Wu X, Sun W, Jiang Y, et al. The expression and clinical significance of different forms of LILRA3 in systemic lupus erythematosus. Clin Rheumatol (2019) 38(11):3099–107. doi: 10.1007/s10067-019-04624-z
72. Du Y, Cui Y, Liu X, Hu F, Yang Y, Wu X, et al. Contribution of functional LILRA3, but not nonfunctional LILRA3, to sex bias in susceptibility and severity of anti–citrullinated protein antibody–positive rheumatoid arthritis. Arthritis Rheumatol (2014) 66(4):822–30. doi: 10.1002/art.38308
73. Liu M, Tang Y, Du Y, Zhang J, Hu F, Zou Y, et al. Leukocyte Ig-like receptor A3 facilitates inflammation, migration and invasion of synovial tissue-derived fibroblasts via ERK/JNK activation. Rheumatology (2023). doi: 10.1093/rheumatology/kead359
74. Kabalak G, Koch S, Dobberstein B, The YH, Matthias T, Schnarr S, et al. Immunoglobulin-like transcripts as risk genes for autoimmunity. Ann N Y Acad Sci (2007) 1110:10–4. doi: 10.1196/annals.1423.002
75. Ordóñez D, Sanchez AJ, Martinez-Rodriguez JE, Cisneros E, Ramil E, Romo N, et al. Multiple sclerosis associates with LILRA3 deletion in Spanish patients. Genes Immun (2009) 10(6):579–85. doi: 10.1038/gene.2009.34
76. Wiśniewski A, Wagner M, Nowak I, Bilińska M, Pokryszko-Dragan A, Jasek M, et al. 6.7-kbp deletion in LILRA3 (ILT6) gene is associated with later onset of the multiple sclerosis in a Polish population. Hum Immunol (2013) 74(3):353–7. doi: 10.1016/j.humimm.2012.12.006
77. Wang M, Liu M, Jia J, Shi H, Teng J, Liu H, et al. Association of the leukocyte immunoglobulin-like receptor A3 gene with neutrophil activation and disease susceptibility in adult-onset still’s disease. Arthritis Rheumatol (2021) 73(6):1033–43. doi: 10.1002/art.41635
78. Cho M, Ishida K, Chen J, Ohkawa J, Chen W, Namiki S, et al. SAGE library screening reveals ILT7 as a specific plasmacytoid dendritic cell marker that regulates type I IFN production. Int Immunol (2008) 20(1):155–64. doi: 10.1093/intimm/dxm127
79. Cao W, Bover L. Signaling and ligand interaction of ILT7: receptor-mediated regulatory mechanisms for plasmacytoid dendritic cells. Immunol Rev (2010) 234(1):163–76. doi: 10.1111/j.0105-2896.2009.00867.x
80. Tsukamoto N, Okada S, Onami Y, Sasaki Y, Umezawa K, Kawakami Y. Impairment of plasmacytoid dendritic cells for IFN production by the ligand for immunoglobulin-like transcript 7 expressed on human cancer cells. Clin Cancer Res (2009) 15(18):5733–43. doi: 10.1158/1078-0432.CCR-09-0171
81. Cao W, Rosen DB, Ito T, Bover L, Bao M, Watanabe G, et al. Plasmacytoid dendritic cell–specific receptor ILT7–FcϵRIγ inhibits Toll-like receptor–induced interferon production. J Exp Med (2006) 203(6):1399–405. doi: 10.1084/jem.20052454
82. Gao Q, Mo S, Han C, Liao X, Yang C, Wang X, et al. Comprehensive analysis of LILR family genes expression and tumour-infiltrating immune cells in early-stage pancreatic ductal adenocarcinoma. IET Syst Biol (2023) 17(2):39–57. doi: 10.1049/syb2.12058
83. Goel RR, Kotenko SV, Kaplan MJ. Interferon lambda in inflammation and autoimmune rheumatic diseases. Nat Rev Rheumatol (2021) 17(6):349–62. doi: 10.1038/s41584-021-00606-1
84. Little AJ, Vesely MD. Cutaneous lupus erythematosus: current and future pathogenesis-directed therapies. Yale J Biol Med (2020) 93(1):81–95.
85. Niebel D, de Vos L, Fetter T, Bragelmann C, Wenzel J. Cutaneous lupus erythematosus: an update on pathogenesis and future therapeutic directions. Am J Clin Dermatol (2023) 24(4):521–40. doi: 10.1007/s40257-023-00774-8
86. Ren X, Wen W, Fan X, Hou W, Su B, Cai P, et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell (2021) 184(23):5838. doi: 10.1016/j.cell.2021.01.053
87. Mitchell A, Rentero C, Endoh Y, Hsu K, Gaus K, Geczy C, et al. LILRA5 is expressed by synovial tissue macrophages in rheumatoid arthritis, selectively induces pro-inflammatory cytokines and IL-10 and is regulated by TNF-α, IL-10 and IFN-γ. Eur J Immunol (2008) 38(12):3459–73. doi: 10.1002/eji.200838415
88. Bashirova AA, Apps R, Vince N, Mochalova Y, Yu XG, Carrington M. Diversity of the human LILRB3/A6 locus encoding a myeloid inhibitory and activating receptor pair. Immunogenetics (2014) 66(1):1–8. doi: 10.1007/s00251-013-0730-9
89. Jones DC, Hewitt CR, Lopez-Alvarez MR, Jahnke M, Russell AI, Radjabova V, et al. Allele-specific recognition by LILRB3 and LILRA6 of a cytokeratin 8-associated ligand on necrotic glandular epithelial cells. Oncotarget (2016) 7(13):15618–31. doi: 10.18632/oncotarget.6905
90. Mori Y, Tsuji S, Inui M, Sakamoto Y, Endo S, Ito Y, et al. Inhibitory immunoglobulin-like receptors LILRB and PIR-B negatively regulate osteoclast development. J Immunol (2008) 181(7):4742–51. doi: 10.4049/jimmunol.181.7.4742
91. McIntire RH, Sifers T, Platt JS, Ganacias KG, Langat DK, Hunt JS. Novel HLA-G-binding leukocyte immunoglobulin-like receptor (LILR) expression patterns in human placentas and umbilical cords. Placenta (2008) 29(7):631–8. doi: 10.1016/j.placenta.2008.04.007
92. Wang J, Zhao SJ, Wang LL, Lin XX, Mor G, Liao AH. Leukocyte immunoglobulin-like receptor subfamily B: A novel immune checkpoint molecule at the maternal-fetal interface. J Reprod Immunol (2023) 155:103764. doi: 10.1016/j.jri.2022.103764
93. Zhang CC. A perspective on LILRBs and LAIR1 as immune checkpoint targets for cancer treatment. Biochem Biophys Res Commun (2022) 633:64–7. doi: 10.1016/j.bbrc.2022.09.019
94. Lepin EJM, Bastin JM, Allan DSJ, Roncador G, Braud VM, Mason DY, et al. Functional characterization of HLA-F and binding of HLA-F tetramers to ILT2 and ILT4 receptors. Eur J Immunol (2000) 30(12):3552–61. doi: 10.1002/1521-4141(200012)30:12<3552::AID-IMMU3552>3.0.CO;2-L
95. Arnold V, Cummings J-S, Moreno-Nieves UY, Didier C, Gilbert A, Barré-Sinoussi F, et al. S100A9 protein is a novel ligand for the CD85j receptor and its interaction is implicated in the control of HIV-1 replication by NK cells. Retrovirology (2013) 10:122–. doi: 10.1186/1742-4690-10-122
96. Saito F, Hirayasu K, Satoh T, Wang CW, Lusingu J, Arimori T, et al. Immune evasion of Plasmodium falciparum by RIFIN via inhibitory receptors. Nature (2017) 552(7683):101–5. doi: 10.1038/nature24994
97. Chan KR, Ong EZ, Tan HC, Zhang SL-X, Zhang Q, Tang KF, et al. Leukocyte immunoglobulin-like receptor B1 is critical for antibody-dependent dengue. Proc Natl Acad Sci (2014) 111(7):2722–7. doi: 10.1073/pnas.1317454111
98. Nakayama M, Underhill DM, Petersen TW, Li B, Kitamura T, Takai T, et al. Paired ig-like receptors bind to bacteria and shape TLR-mediated cytokine production. J Immunol (2007) 178(7):4250–9. doi: 10.4049/jimmunol.178.7.4250
99. Saverino D, Merlo A, Bruno S, Pistoia V, Grossi CE, Ciccone E. Dual effect of CD85/leukocyte ig-like receptor-1/ig-like transcript 2 and CD152 (CTLA-4) on cytokine production by antigen-stimulated human T cells. J Immunol (2002) 168(1):207–15. doi: 10.4049/jimmunol.168.1.207
100. Merlo A, Saverino D, Tenca C, Grossi CE, Bruno S, Ciccone E. CD85/LIR-1/ILT2 and CD152 (Cytotoxic T lymphocyte antigen 4) inhibitory molecules down-regulate the cytolytic activity of human CD4(+) T-cell clones specific for mycobacterium tuberculosis. Infection Immun (2001) 69(10):6022–9. doi: 10.1128/IAI.69.10.6022-6029.2001
101. Xu Z, Ho S, Chang C-C, Liu Z, Li M, Vasilescu ER, et al. ILT3.Fc inhibits the production of exosomes containing inflammatory microRNA in supernatants of alloactivated T cells. Hum Immunol (2014) 75(8):756–9. doi: 10.1016/j.humimm.2014.05.006
102. Saverino D, Fabbi M, Ghiotto F, Merlo A, Bruno S, Zarcone D, et al. The CD85/LIR-1/ILT2 inhibitory receptor is expressed by all human T lymphocytes and down-regulates their functions. J Immunol (2000) 165(7):3742–55. doi: 10.4049/jimmunol.165.7.3742
103. Bahri R, Hirsch F, Josse A, Rouas-Freiss N, Bidere N, Vasquez A, et al. Soluble HLA-G inhibits cell cycle progression in human alloreactive T lymphocytes. J Immunol (2006) 176(3):1331–9. doi: 10.4049/jimmunol.176.3.1331
104. Morandi F, Ferretti E, Bocca P, Prigione I, Raffaghello L, Pistoia V. A novel mechanism of soluble HLA-G mediated immune modulation: downregulation of T cell chemokine receptor expression and impairment of chemotaxis. PloS One (2010) 5(7):e11763. doi: 10.1371/journal.pone.0011763
105. Purbhoo MA, Liu H, Oddos S, Owen DM, Neil MAA, Pageon SV, et al. Dynamics of subsynaptic vesicles and surface microclusters at the immunological synapse. Sci Signaling (2010) 3(121):ra36–r. doi: 10.1126/scisignal.2000645
106. Naji A, Menier C, Morandi F, Agaugué S, Maki G, Ferretti E, et al. Binding of HLA-G to ITIM-bearing ig-like transcript 2 receptor suppresses B cell responses. J Immunol (2014) 192(4):1536–46. doi: 10.4049/jimmunol.1300438
107. Naji A, Menier C, Maki G, Carosella ED, Rouas-Freiss N. Neoplastic B-cell growth is impaired by HLA-G/ILT2 interaction. Leukemia (2012) 26(8):1889–92. doi: 10.1038/leu.2012.62
108. Tenca C, Merlo A, Merck E, Bates EEM, Saverino D, Simone R, et al. CD85j (Leukocyte ig-like receptor-1/ig-like transcript 2) inhibits human osteoclast-associated receptor-mediated activation of human dendritic cells. J Immunol (2005) 174(11):6757–63. doi: 10.4049/jimmunol.174.11.6757
109. Cella M, Döhring C, Samaridis J, Dessing M, Brockhaus M, Lanzavecchia A, et al. A novel inhibitory receptor (ILT3) expressed on monocytes, macrophages, and dendritic cells involved in antigen processing. J Exp Med (1997) 185(10):1743–51. doi: 10.1084/jem.185.10.1743
110. Godal R, Bachanova V, Gleason M, McCullar V, Yun GH, Cooley S, et al. Natural killer cell killing of acute myelogenous leukemia and acute lymphoblastic leukemia blasts by killer cell immunoglobulin-like receptor-negative natural killer cells after NKG2A and LIR-1 blockade. Biol Blood Marrow Transplant (2010) 16(5):612–21. doi: 10.1016/j.bbmt.2010.01.019
111. Favier B, LeMaoult J, Lesport E, Carosella ED. ILT2/HLA-G interaction impairs NK-cell functions through the inhibition of the late but not the early events of the NK-cell activating synapse. FASEB J (2010) 24(3):689–99. doi: 10.1096/fj.09-135194
112. Villa-Alvarez M, Sordo-Bahamonde C, Lorenzo-Herrero S, Gonzalez-Rodriguez AP, Payer AR, Gonzalez-Garcia E, et al. Ig-like transcript 2 (ILT2) blockade and lenalidomide restore NK cell function in chronic lymphocytic leukemia. Front Immunol (2018) 9:2917. doi: 10.3389/fimmu.2018.02917
113. Forte P, Pazmany L, Matter-Reissmann UB, Stussi G, Schneider MK, Seebach JD. HLA-G inhibits rolling adhesion of activated human NK cells on porcine endothelial cells. J Immunol (2001) 167(10):6002–8. doi: 10.4049/jimmunol.167.10.6002
114. Apps R, Gardner L, Sharkey AM, Holmes N, Moffett A. A homodimeric complex of HLA-G on normal trophoblast cells modulates antigen-presenting cells via LILRB1. Eur J Immunol (2007) 37(7):1924–37. doi: 10.1002/eji.200737089
115. Young NT, Waller ECP, Patel R, Roghanian A, Austyn JM, Trowsdale J. The inhibitory receptor LILRB1 modulates the differentiation and regulatory potential of human dendritic cells. Blood (2007) 111(6):3090–6. doi: 10.1182/blood-2007-05-089771
116. Fanger NA, Cosman D, Peterson L, Braddy SC, Maliszewski CR, Borges L. The MHC class I binding proteins LIR-1 and LIR-2 inhibit Fc receptor-mediated signaling in monocytes. Eur J Immunol (1998) 28(11):3423–34. doi: 10.1002/(SICI)1521-4141(199811)28:11<3423::AID-IMMU3423>3.0.CO;2-2
117. Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol (2018) 19(1):76–84. doi: 10.1038/s41590-017-0004-z
118. Zou R, Zhong X, Liang K, Zhi C, Chen D, Xu Z, et al. Elevated LILRB1 expression predicts poor prognosis and is associated with tumor immune infiltration in patients with glioma. BMC Cancer (2023) 23(1):403. doi: 10.1186/s12885-023-10906-2
119. Xu ZJ, Zhang XL, Jin Y, Wang SS, Gu Y, Ma JC, et al. Pan-cancer analysis reveals distinct clinical, genomic, and immunological features of the LILRB immune checkpoint family in acute myeloid leukemia. Mol Ther Oncolytics (2022) 26:88–104. doi: 10.1016/j.omto.2022.05.011
120. Zhang Y, Lu N, Xue Y, Zhang M, Li Y, Si Y, et al. Expression of immunoglobulin-like transcript (ILT)2 and ILT3 in human gastric cancer and its clinical significance. Mol Med Rep (2012) 5(4):910–6. doi: 10.3892/mmr.2012.744
121. Sun Y, Liu J, Gao P, Wang Y, Liu C. Expression of ig-like transcript 4 inhibitory receptor in human non-small cell lung cancer. Chest (2008) 134(4):783–8. doi: 10.1378/chest.07-1100
122. Fan J, Li J, Han J, Zhang Y, Gu A, Song F, et al. Expression of leukocyte immunoglobulin-like receptor subfamily B expression on immune cells in hepatocellular carcinoma. Mol Immunol (2021) 136:82–97. doi: 10.1016/j.molimm.2021.05.011
123. Fan J, Wang L, Chen M, Zhang J, Li J, Song F, et al. Analysis of the expression and prognosis for leukocyte immunoglobulin-like receptor subfamily B in human liver cancer. World J Surg Oncol (2022) 20(1):92. doi: 10.1186/s12957-022-02562-w
124. Lefebvre S, Antoine M, Uzan S, McMaster M, Dausset J, Carosella ED, et al. Specific activation of the non-classical class I histocompatibility HLA-G antigen and expression of the ILT2 inhibitory receptor in human breast cancer. J Pathol (2002) 196(3):266–74. doi: 10.1002/path.1039
125. Xu X, Yin S, Wang Y, Zhu Q, Zheng G, Lu Y, et al. LILRB1+ immune cell infiltration identifies immunosuppressive microenvironment and dismal outcomes of patients with ovarian cancer. Int Immunopharmacol (2023) 119:110162. doi: 10.1016/j.intimp.2023.110162
126. Vittrant B, Bergeron A, Molina OE, Leclercq M, Legare XP, Hovington H, et al. Immune-focused multi-omics analysis of prostate cancer: leukocyte Ig-Like receptors are associated with disease progression. Oncoimmunology (2020) 9(1):1851950. doi: 10.1080/2162402X.2020.1851950
127. Wang X, Meng X, Zheng Y, Jiang J, Yang B, Liu Y, et al. Increased frequency of ILT2-expressing CD56(dim)CD16(+) NK cells correlates with disease severity of pulmonary tuberculosis. Tuberculosis (Edinb) (2014) 94(5):469–74. doi: 10.1016/j.tube.2014.03.009
128. Vlad G, Piazza F, Colovai A, Cortesini R, Della Pietra F, Suciu-Foca N, et al. Interleukin-10 induces the upregulation of the inhibitory receptor ILT4 in monocytes from HIV positive individuals. Hum Immunol (2003) 64(5):483–9. doi: 10.1016/S0198-8859(03)00040-5
129. O’Connor GM, Holmes A, Mulcahy F, Gardiner CM. Natural Killer cells from long-term non-progressor HIV patients are characterized by altered phenotype and function. Clin Immunol (2007) 124(3):277–83. doi: 10.1016/j.clim.2007.05.016
130. Scott-Algara D, Arnold V, Didier C, Kattan T, Pirozzi G, Barré-Sinoussi F, et al. The CD85j(+) NK cell subset potently controls HIV-1 replication in autologous dendritic cells. PloS One (2008) 3(4):e1975. doi: 10.1371/journal.pone.0001975
131. Berg L, Riise GC, Cosman D, Bergström T, Olofsson S, Kärre K, et al. LIR-1 expression on lymphocytes, and cytomegalovirus disease in lung-transplant recipients. Lancet (2003) 361(9363):1099–101. doi: 10.1016/S0140-6736(03)12855-3
132. Saverino D, Ghiotto F, Merlo A, Bruno S, Battini L, Occhino M, et al. Specific recognition of the viral protein UL18 by CD85j/LIR-1/ILT2 on CD8+ T cells mediates the non-MHC-restricted lysis of human cytomegalovirus-infected cells. J Immunol (2004) 172(9):5629–37. doi: 10.4049/jimmunol.172.9.5629
133. Harrison TE, Morch AM, Felce JH, Sakoguchi A, Reid AJ, Arase H, et al. Structural basis for RIFIN-mediated activation of LILRB1 in malaria. Nature (2020) 587(7833):309–12. doi: 10.1038/s41586-020-2530-3
134. Sakoguchi A, Saito F, Hirayasu K, Shida K, Matsuoka S, Itagaki S, et al. Plasmodium falciparum RIFIN is a novel ligand for inhibitory immune receptor LILRB2. Biochem Biophys Res Commun (2021) 548:167–73. doi: 10.1016/j.bbrc.2021.02.033
135. Moraes AG, Ayo CM, Elpidio LNS, Souza VH, Yamanaka AHU, Nogueira ML, et al. HLA-G, LILRB1 and LILRB2 variants in zika virus transmission from mother to child in a population from south and southeast of Brazil. Curr Issues Mol Biol (2022) 44(7):2783–93. doi: 10.3390/cimb44070191
136. Ince MN, Harnisch B, Xu Z, Lee S-K, Lange C, Moretta L, et al. Increased expression of the natural killer cell inhibitory receptor CD85j/ILT2 on antigen-specific effector CD8 T cells and its impact on CD8 T-cell function. Immunology (2004) 112(4):531–42. doi: 10.1046/j.1365-2567.2004.01907.x
137. Anfossi N, Doisne J-M, Peyrat M-A, Ugolini S, Bonnaud O, Bossy D, et al. Coordinated expression of ig-like inhibitory MHC class I receptors and acquisition of cytotoxic function in human CD8+ T cells. J Immunol (2004) 173(12):7223–9. doi: 10.4049/jimmunol.173.12.7223
138. Zhang Y, Tong S, Li S, Wang X, Ren H, Yin W. Increased ILT2 expression contributes to dysfunction of CD56(dim)CD16(+)NK cells in chronic hepatitis B virus infection. Antiviral Res (2022) 205:105385. doi: 10.1016/j.antiviral.2022.105385
139. Wiendl H, Feger U, Mittelbronn M, Jack C, Schreiner B, Stadelmann C, et al. Expression of the immune-tolerogenic major histocompatibility molecule HLA-G in multiple sclerosis: implications for CNS immunity. Brain (2005) 128(11):2689–704. doi: 10.1093/brain/awh609
140. Doníz-Padilla L, Paniagua AE, Sandoval-Correa P, Monsiváis-Urenda A, Leskela S, Marazuela M, et al. Analysis of expression and function of the inhibitory receptor ILT2 in lymphocytes from patients with autoimmune thyroid disease. Eur J Endocrinol (2011) 165(1):129–36. doi: 10.1530/EJE-11-0109
141. Monsiváis-Urenda A, Niño-Moreno P, Abud-Mendoza C, Baranda L, Layseca-Espinosa E, López-Botet M, et al. Analysis of expression and function of the inhibitory receptor ILT2 (CD85j/LILRB1/LIR-1) in peripheral blood mononuclear cells from patients with systemic lupus erythematosus (SLE). J Autoimmun (2007) 29(2–3):97–105. doi: 10.1016/j.jaut.2007.05.003
142. Monsiváis-Urenda A, Gómez-Martin D, Santana-de-Anda K, Cruz-Martínez J, Alcocer-Varela J, González-Amaro R. Defective expression and function of the ILT2/CD85j regulatory receptor in dendritic cells from patients with systemic lupus erythematosus. Hum Immunol (2013) 74(9):1088–96. doi: 10.1016/j.humimm.2013.05.006
143. Figueroa-Vega N, Galindo-Rodríguez G, Bajaña S, Portales-Pérez D, Abud-Mendoza C, Sánchez-Torres C, et al. Phenotypic analysis of IL-10-treated, monocyte-derived dendritic cells in patients with systemic lupus erythematosus. Scandinavian J Immunol (2006) 64(6):668–76. doi: 10.1111/j.1365-3083.2006.01849.x
144. Baudhuin J, Migraine J, Faivre V, Loumagne L, Lukaszewicz A-C, Payen D, et al. Exocytosis acts as a modulator of the ILT4-mediated inhibition of neutrophil functions. Proc Natl Acad Sci United States America (2013) 110(44):17957–62. doi: 10.1073/pnas.1221535110
145. Fan X, Shi P, Dai J, Lu Y, Chen X, Liu X, et al. Paired immunoglobulin-like receptor B regulates platelet activation. Blood (2014) 124(15):2421–30. doi: 10.1182/blood-2014-03-557645
146. Zheng J, Umikawa M, Cui C, Li J, Chen X, Zhang C, et al. Inhibitory receptors bind ANGPTLs and support blood stem cells and leukaemia development. Nature (2012) 485(7400):656–60. doi: 10.1038/nature11095
147. Deng M, Lu Z, Zheng J, Wan X, Chen X, Hirayasu K, et al. A motif in LILRB2 critical for Angptl2 binding and activation. Blood (2014) 124(6):924–35. doi: 10.1182/blood-2014-01-549162
148. Hofer J, Forster F, Isenman DE, Wahrmann M, Leitner J, Holzl MA, et al. Ig-like transcript 4 as a cellular receptor for soluble complement fragment C4d. FASEB J (2016) 30(4):1492–503. doi: 10.1096/fj.15-275594
149. Lu N, Li Y, Zhang Z, Xing J, Sun Y, Yao S, et al. Human Semaphorin-4A drives Th2 responses by binding to receptor ILT-4. Nat Commun (2018) 9(1):742. doi: 10.1038/s41467-018-03128-9
150. Ristich V, Liang S, Zhang W, Wu J, Horuzsko A. Tolerization of dendritic cells by HLA-G. Eur J Immunol (2005) 35(4):1133–42. doi: 10.1002/eji.200425741
151. Manavalan JS, Rossi PC, Vlad G, Piazza F, Yarilina A, Cortesini R, et al. High expression of ILT3 and ILT4 is a general feature of tolerogenic dendritic cells. Transplant Immunol (2003) 11(3–4):245–58. doi: 10.1016/S0966-3274(03)00058-3
152. Suciu-Foca N, Manavalan JS, Scotto L, Kim-Schulze S, Galluzzo S, Naiyer AJ, et al. Molecular characterization of allospecific T suppressor and tolerogenic dendritic cells: review. Int Immunopharmacol (2005) 5(1):7–11. doi: 10.1016/j.intimp.2004.09.003
153. Chang CC, Ciubotariu R, Manavalan JS, Yuan J, Colovai AI, Piazza F, et al. Tolerization of dendritic cells by TS cells: the crucial role of inhibitory receptors ILT3 and ILT4. Nat Immunol (2002) 3(3):237–43. doi: 10.1038/ni760
154. Vlad G, Liu Z, Zhang Q-Y, Cortesini R, Suciu-Foca N. Immunosuppressive activity of recombinant ILT3. Int Immunopharmacol (2006) 6(13–14):1889–94. doi: 10.1016/j.intimp.2006.07.017
155. Kim-Schulze S, Scotto L, Vlad G, Piazza F, Lin H, Liu Z, et al. Recombinant ig-like transcript 3-fc modulates T cell responses via induction of th anergy and differentiation of CD8+ T suppressor cells. J Immunol (2006) 176(5):2790–8. doi: 10.4049/jimmunol.176.5.2790
156. Chang C-C, Liu Z, Vlad G, Qin H, Qiao X, Mancini DM, et al. Ig-like transcript 3 regulates expression of proinflammatory cytokines and migration of activated T cells. J Immunol (2009) 182(9):5208–16. doi: 10.4049/jimmunol.0804048
157. Chang C-C, Vlad G, D’Agati VD, Liu Z, Zhang Q-y, Witkowski P, et al. BCL6 is required for differentiation of ig-like transcript 3-fc–induced CD8+ T suppressor cells. J Immunol (2010) 185(10):5714–22. doi: 10.4049/jimmunol.1001732
158. Suciu-Foca N, Feirt N, Zhang Q-Y, Vlad G, Liu Z, Lin H, et al. Soluble ig-like transcript 3 inhibits tumor allograft rejection in humanized SCID mice and T cell responses in cancer patients. J Immunol (2007) 178(11):7432–41. doi: 10.4049/jimmunol.178.11.7432
159. Liang S, Ristich V, Arase H, Dausset J, Carosella ED, Horuzsko A. Modulation of dendritic cell differentiation by HLA-G and ILT4 requires the IL-6—STAT3 signaling pathway. Proc Natl Acad Sci (2008) 105:8357–62. doi: 10.1073/pnas.0803341105
160. Chen HM, van der Touw W, Wang YS, Kang K, Mai S, Zhang J, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest (2018) 128(12):5647–62. doi: 10.1172/JCI97570
161. Umiker B, Hashambhoy-Ramsay Y, Smith J, Rahman T, Mueller A, Davidson R, et al. Inhibition of LILRB2 by a novel blocking antibody designed to reprogram immunosuppressive macrophages to drive T-cell activation in tumors. Mol Cancer Ther (2023) 22(4):471–84. doi: 10.1158/1535-7163.MCT-22-0351
162. Liu J, Wang L, Gao W, Li L, Cui X, Yang H, et al. Inhibitory receptor immunoglobulin-like transcript 4 was highly expressed in primary ductal and lobular breast cancer and significantly correlated with IL-10. Diagn Pathol (2014) 9:85–. doi: 10.1186/1746-1596-9-85
163. Liu J, Zhang F, He J, Wang S, Wang L, Li J, et al. Tumor-derived Immunoglobulin-like transcript 4 facilitates angiogenesis of colorectal cancer. Am J Cancer Res (2023) 13(2):419–35.
164. Cai Z, Wang L, Han Y, Gao W, Wei X, Gong R, et al. Immunoglobulin−like transcript 4 and human leukocyte antigen−G interaction promotes the progression of human colorectal cancer. Int J Oncol (2019) 54(6):1943–54. doi: 10.3892/ijo.2019.4761
165. García M, Palma MB, Verine J, Miriuka S, Inda AM, Errecalde AL, et al. The immune-checkpoint HLA-G/ILT4 is involved in the regulation of VEGF expression in clear cell renal cell carcinoma. BMC Cancer (2020) 20(1):624. doi: 10.1186/s12885-020-07113-8
166. Singh VK, Khan A, Xu Y, Mai S, Zhang L, Mishra A, et al. Antibody-mediated LILRB2-receptor antagonism induces human myeloid-derived suppressor cells to kill mycobacterium tuberculosis. Front Immunol (2022) 13:865503. doi: 10.3389/fimmu.2022.865503
167. Brown DP, Jones DC, Anderson KJ, Lapaque N, Buerki RA, Trowsdale J, et al. The inhibitory receptor LILRB4 (ILT3) modulates antigen presenting cell phenotype and, along with LILRB2 (ILT4), is upregulated in response to Salmonella infection. BMC Immunol (2009) 10:56–. doi: 10.1186/1471-2172-10-56
168. Baffari E, Fiume D, Caiazzo G, Sinistro A, Natoli S, Almerighi C, et al. Upregulation of the inhibitory receptor ILT4 in monocytes from septic patients. Hum Immunol (2013) 74(10):1244–50. doi: 10.1016/j.humimm.2013.07.012
169. Lichterfeld M, Kavanagh DG, Williams KL, Moza B, Mui SK, Miura T, et al. A viral CTL escape mutation leading to immunoglobulin-like transcript 4–mediated functional inhibition of myelomonocytic cells. J Exp Med (2007) 204(12):2813–24. doi: 10.1084/jem.20061865
170. Bashirova AA, Martin-Gayo E, Jones DC, Qi Y, Apps R, Gao X, et al. LILRB2 interaction with HLA class I correlates with control of HIV-1 infection. PloS Genet (2014) 10(3):e1004196. doi: 10.1371/journal.pgen.1004196
171. Kawaguchi Y, Matsubayashi J, Kawakami Y, Nishida R, Kurihara Y, Takei K. LOTUS suppresses amyloid beta-induced dendritic spine elimination through the blockade of amyloid beta binding to PirB. Mol Med (2022) 28(1):154. doi: 10.1186/s10020-022-00581-7
172. Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, et al. Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science (2013) 341(6152):1399–404. doi: 10.1126/science.1242077
173. Stallone G, Pontrelli P, Infante B, Gigante M, Netti GS, Ranieri E, et al. Rapamycin induces ILT3highILT4high dendritic cells promoting a new immunoregulatory pathway. Kidney Int (2014) 85(4):888–97. doi: 10.1038/ki.2013.337
174. Borges L, Cosman D. LIRs/ILTs/MIRs, inhibitory and stimulatory Ig-superfamily receptors expressed in myeloid and lymphoid cells. Cytokine Growth Factor Rev (2000) 11(3):209–17. doi: 10.1016/s1359-6101(00)00007-1
175. Zhou J, Wang Y, Huang G, Yang M, Zhu Y, Jin C, et al. LilrB3 is a putative cell surface receptor of APOE4. Cell Res (2023) 33(2):116–30. doi: 10.1038/s41422-022-00759-y
176. Zhao Y, van Woudenbergh E, Zhu J, Heck AJR, van Kessel KPM, de Haas CJC, et al. The orphan immune receptor LILRB3 modulates fc receptor-mediated functions of neutrophils. J Immunol (2020) 204(4):954–66. doi: 10.4049/jimmunol.1900852
177. Yeboah M, Papagregoriou C, Jones DC, Chan HTC, Hu G, McPartlan JS, et al. LILRB3 (ILT5) is a myeloid cell checkpoint that elicits profound immunomodulation. JCI Insight (2020) 5(18):e141593. doi: 10.1172/jci.insight.141593
178. Perna F, Berman SH, Soni RK, Mansilla-Soto J, Eyquem J, Hamieh M, et al. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell (2017) 32(4):506–19 e5. doi: 10.1016/j.ccell.2017.09.004
179. Shi W, Zhang F, Chen X, Wang S, Zhang H, Yang Z, et al. Tumor-derived immunoglobulin like transcript 5 induces suppressive immunocyte infiltration in colorectal cancer. Cancer Sci (2022) 113(6):1939–54. doi: 10.1111/cas.15360
180. Renauer PA, Saruhan-Direskeneli G, Coit P, Adler A, Aksu K, Keser G, et al. Identification of susceptibility loci in IL6, RPS9/LILRB3, and an intergenic locus on chromosome 21q22 in takayasu arteritis in a genome-wide association study. Arthritis Rheumatol (2015) 67(5):1361–8. doi: 10.1002/art.39035
181. Pfistershammer K, Lawitschka A, Klauser C, Leitner J, Weigl R, Heemskerk MH, et al. Allogeneic disparities in immunoglobulin-like transcript 5 induce potent antibody responses in hematopoietic stem cell transplant recipients. Blood (2009) 114(11):2323–32. doi: 10.1182/blood-2008-10-183814
182. Inui M, Hirota S, Hirano K, Fujii H, Sugahara-Tobinai A, Ishii T, et al. Human CD43+ B cells are closely related not only to memory B cells phenotypically but also to plasmablasts developmentally in healthy individuals. Int Immunol (2015) 27(7):345–55. doi: 10.1093/intimm/dxv009
183. Mitsune A, Yamada M, Fujino N, Numakura T, Ichikawa T, Suzuki A, et al. Upregulation of leukocyte immunoglobulin-like receptor B4 on interstitial macrophages in COPD; their possible protective role against emphysema formation. Respir Res (2021) 22(1):232. doi: 10.1186/s12931-021-01828-3
184. Sharma N, Atolagbe OT, Ge Z, Allison JP. LILRB4 suppresses immunity in solid tumors and is a potential target for immunotherapy. J Exp Med (2021) 218(7):e20201811. doi: 10.1084/jem.20201811
185. Deng M, Gui X, Kim J, Xie L, Chen W, Li Z, et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature (2018) 562(7728):605–9. doi: 10.1038/s41586-018-0615-z
186. Xu Z, Chang CC, Li M, Zhang QY, Vasilescu EM, D’Agati V, et al. ILT3.Fc-CD166 interaction induces inactivation of p70 S6 kinase and inhibits tumor cell growth. J Immunol (2018) 200(3):1207–19. doi: 10.4049/jimmunol.1700553
187. Su MT, Inui M, Wong YL, Takahashi M, Sugahara-Tobinai A, Ono K, et al. Blockade of checkpoint ILT3/LILRB4/gp49B binding to fibronectin ameliorates autoimmune disease in BXSB/Yaa mice. Int Immunol (2021) 33(8):447–58. doi: 10.1093/intimm/dxab028
188. Wang Y, Yufan S, Deng S, Song T, Wang Y, Xu J. Galectin-8 is a major ligand of LILRB4 prompting MDSC functions in the tumor microenvironment. Biorxiv (2022). doi: 10.1101/2022.07.27.501694
189. Park M, Raftery MJ, Thomas PS, Geczy CL, Bryant K, Tedla N. Leukocyte immunoglobulin-like receptor B4 regulates key signalling molecules involved in FcγRI-mediated clathrin-dependent endocytosis and phagocytosis. Sci Rep (2016) 6:35085. doi: 10.1038/srep35085
190. Li Z, Deng M, Huang F, Jin C, Sun S, Chen H, et al. LILRB4 ITIMs mediate the T cell suppression and infiltration of acute myeloid leukemia cells. Cell Mol Immunol (2020) 17(3):272–82. doi: 10.1038/s41423-019-0321-2
191. Dobrowolska H, Gill KZ, Serban G, Ivan E, Li Q, Qiao PY, et al. Expression of immune inhibitory receptor ILT3 in acute myeloid leukemia with monocytic differentiation. Cytometry Part B-Clinical Cytometry (2013) 84B(1):21–9. doi: 10.1002/cyto.b.21050
192. Atfy M, Ebian H, Elhefni A, Atteia H. The usefulness of immunoglobulin-like transcript-3 receptor expression in the diagnosis of acute myeloid leukemia with monocytic differentiation. Egyptian J Haematol (2014) 39(3):122–7. doi: 10.4103/1110-1067.148235
193. Di Meo F, Iyer A, Akama K, Cheng R, Yu C, Cesarano A, et al. A target discovery pipeline identified ILT3 as a target for immunotherapy of multiple myeloma. Cell Rep Med (2023) 4(7):101110. doi: 10.1016/j.xcrm.2023.101110
194. Liu J, Lu CX, Zhang F, Lv W, Liu C. Expression of ILT3 predicts poor prognosis and is inversely associated with infiltration of CD45RO+ T cells in patients with colorectal cancer. Pathol Res Pract (2018) 214(10):1621–5. doi: 10.1016/j.prp.2018.07.026
195. de Goeje PL, Bezemer K, Heuvers ME, Dingemans A-MC, Groen HJ, Smit EF, et al. Immunoglobulin-like transcript 3 is expressed by myeloid-derived suppressor cells and correlates with survival in patients with non-small cell lung cancer. OncoImmunology (2015) 4(7):e1014242. doi: 10.1080/2162402X.2015.1014242
196. Li J, Gao A, Zhang F, Wang S, Wang J, Wang J, et al. ILT3 promotes tumor cell motility and angiogenesis in non-small cell lung cancer. Cancer Lett (2021) 501:263–76. doi: 10.1016/j.canlet.2020.10.048
197. Tomic S, Dokic J, Stevanovic D, Ilic N, Gruden-Movsesijan A, Dinic M, et al. Reduced expression of autophagy markers and expansion of myeloid-derived suppressor cells correlate with poor T cell response in severe COVID-19 patients. Front Immunol (2021) 12:614599. doi: 10.3389/fimmu.2021.614599
198. Patel H, Ashton NJ, Dobson RJB, Andersson LM, Yilmaz A, Blennow K, et al. Proteomic blood profiling in mild, severe and critical COVID-19 patients. Sci Rep (2021) 11(1):6357. doi: 10.1038/s41598-021-85877-0
199. Waschbisch A, Sanderson N, Krumbholz M, Vlad G, Theil D, Schwab S, et al. Interferon beta and vitamin D synergize to induce immunoregulatory receptors on peripheral blood monocytes of multiple sclerosis patients. PloS One (2014) 9(12):e115488. doi: 10.1371/journal.pone.0115488
200. Jensen MA, Yanowitch RN, Reder AT, White DM, Arnason BGW. Immunoglobulin-like transcript 3, an inhibitor of T cell activation, is reduced on blood monocytes during multiple sclerosis relapses and is induced by interferon β-1b. Multiple Sclerosis (2010) 16(1):30–8. doi: 10.1177/1352458509352794
201. Jensen MA, Patterson KC, Kumar AA, Kumabe M, Franek BS, Niewold TB. Functional genetic polymorphisms in ILT3 are associated with decreased surface expression on dendritic cells and increased serum cytokines in lupus patients. Ann Rheumatic Dis (2013) 72(4):596–601. doi: 10.1136/annrheumdis-2012-202024
202. Tedla N, Lee C-W, Borges L, Geczy CL, Arm JP. Differential expression of leukocyte immunoglobulin-like receptors on cord blood-derived human mast cell progenitors and mature mast cells. J Leukocyte Biol (2008) 83(2):334–43. doi: 10.1189/jlb.0507314
203. Hogan LE, Jones DC, Allen RL. Expression of the innate immune receptor LILRB5 on monocytes is associated with mycobacteria exposure. Sci Rep (2016) 6:21780. doi: 10.1038/srep21780
204. Zhang Z, Hatano H, Shaw J, Olde Nordkamp M, Jiang G, Li D, et al. The leukocyte immunoglobulin-like receptor family member LILRB5 binds to HLA-class I heavy chains. PloS One (2015) 10(6):e0129063. doi: 10.1371/journal.pone.0129063
205. Kuroki K, Matsubara H, Kanda R, Miyashita N, Shiroishi M, Fukunaga Y, et al. Structural and functional basis for LILRB immune checkpoint receptor recognition of HLA-G isoforms. J Immunol (2019) 203(12):3386–94. doi: 10.4049/jimmunol.1900562
206. Carosella ED, Moreau P, LeMaoult J, Rouas-Freiss N. HLA-G: from biology to clinical benefits. Trends Immunol (2008) 29(3):125–32. doi: 10.1016/j.it.2007.11.005
207. Paul P, Rouas-Freiss N, Khalil-Daher I, Moreau P, Riteau B, Le Gal FA, et al. HLA-G expression in melanoma: A way for tumor cells to escape from immunosurveillance. Proc Natl Acad Sci USA (1998) 95(8):4510–5. doi: 10.1073/pnas.95.8.4510
208. Le Gal F-A, Riteau B, Sedlik C, Khalil-Daher I, Menier C, Dausset J, et al. HLA-G-mediated inhibition of antigen-specific cytotoxic T lymphocytes. Int Immunol (1999) 11(8):1351–6. doi: 10.1093/intimm/11.8.1351
209. Lozano JM, González R, Kindelán JM, Rouas-Freiss N, Caballos R, Dausset J, et al. Monocytes and T lymphocytes in HIV-1-positive patients express HLA-G molecule. AIDS (2002) 16(3):347–51. doi: 10.1097/00002030-200202150-00005
210. Nückel H, Rebmann V, Dürig J, Dührsen U, Grosse-Wilde H. HLA-G expression is associated with an unfavorable outcome and immunodeficiency in chronic lymphocytic leukemia. Blood (2005) 105(4):1694–8. doi: 10.1182/blood-2004-08-3335
211. Rouas-Freiss N, Moreau P, Ferrone S, Carosella ED. HLA-G proteins in cancer: do they provide tumor cells with an escape mechanism? Cancer Res (2005) 65(22):10139–44. doi: 10.1158/0008-5472.CAN-05-0097
212. Huang J, Burke P, Yang Y, Seiss K, Beamon J, Cung T, et al. Soluble HLA-G inhibits myeloid dendritic cell function in HIV-1 infection by interacting with leukocyte immunoglobulin-like receptor B2. J Virol (2010) 84(20):10784–91. doi: 10.1128/JVI.01292-10
213. Agaugué S, Carosella ED, Rouas-Freiss N. Role of HLA-G in tumor escape through expansion of myeloid-derived suppressor cells and cytokinic balance in favor of Th2 versus Th1/Th17. Blood (2011) 117(26):7021–31. doi: 10.1182/blood-2010-07-294389
214. LeMaoult J, Zafaranloo K, Le Danff C, Carosella ED. HLA-G up-regulates ILT2, ILT3, ILT4, and KIR2DL4 in antigen presenting cells, NK cells, and T cells. FASEB J (2005) 19:662–4. doi: 10.1096/fj.04-1617fje
215. Zhong M, Weng X, Liang Z, Lu S, Li J, Chen X, et al. Dimerization of soluble HLA-G by igG-fc fragment augments ILT2-mediated inhibition of T-cell alloresponse. Transplantation (2009) 87(1):8–15. doi: 10.1097/TP.0b013e31818b6141
216. Hamerman JA, Ni M, Killebrew JR, Chu C-L, Lowell CA. The expanding roles of ITAM adapters FcRγ and DAP12 in myeloid cells. Immunol Rev (2009) 232(1):42–58. doi: 10.1111/j.1600-065X.2009.00841.x
217. Blank U, Launay P, Benhamou M, Monteiro RC. Inhibitory ITAMs as novel regulators of immunity. Immunol Rev (2009) 232(1):59–71. doi: 10.1111/j.1600-065X.2009.00832.x
218. van der Touw W, Chen HM, Pan PY, Chen SH. LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer Immunol Immunother (2017) 66(8):1079–87. doi: 10.1007/s00262-017-2023-x
219. O’Neill LAJ. When signaling pathways collide: positive and negative regulation of toll-like receptor signal transduction. Immunity (2008) 29(1):12–20. doi: 10.1016/j.immuni.2008.06.004
220. Barrow AD, Trowsdale J. You say ITAM and I say ITIM, let’s call the whole thing off: the ambiguity of immunoreceptor signalling. Eur J Immunol (2006) 36(7):1646–53. doi: 10.1002/eji.200636195
221. Pinheiro da Silva F, Aloulou M, Skurnik D, Benhamou M, Andremont A, Velasco IT, et al. CD16 promotes Escherichia coli sepsis through an FcR[gamma] inhibitory pathway that prevents phagocytosis and facilitates inflammation. Nat Med (2007) 13(11):1368–74. doi: 10.1038/nm1665
222. da Silva FP, Aloulou M, Benhamou M, Monteiro RC. Inhibitory ITAMs: a matter of life and death. Trends Immunol (2008) 29(8):366–73. doi: 10.1016/j.it.2008.05.001
223. Tai L-H, Goulet M-L, Belanger S, Toyama-Sorimachi N, Fodil-Cornu N, Vidal SM, et al. Positive regulation of plasmacytoid dendritic cell function via Ly49Q recognition of class I MHC. J Exp Med (2008) 205(13):3187–99. doi: 10.1084/jem.20080718
224. Borges L, Kubin M, Kuhlman T. LIR9, an immunoglobulin-superfamily-activating receptor, is expressed as a transmembrane and as a secreted molecule. Blood (2003) 101(4):1484–6. doi: 10.1182/blood-2002-05-1432
225. Lee THY, Mitchell A, Liu Lau S, An H, Rajeaskariah P, Wasinger V, et al. Glycosylation in a mammalian expression system is critical for the production of functionally active leukocyte immunoglobulin-like receptor A3 protein. J Biol Chem (2013) 288(46):32873–85. doi: 10.1074/jbc.M113.478578
226. Khanolkar RC, Kalogeropoulos M, Lawrie A, Roghanian A, Vickers MA, Young NT. Leukocyte Ig-Like receptor B1 restrains dendritic cell function through increased expression of the NF-κB regulator ABIN1/TNIP1. J Leukocyte Biol (2016) 100:737–46. doi: 10.1189/jlb.1A0915-420RRR
227. Banchereau J, Zurawski S, Thompson-Snipes L, Blanck J-P, Clayton S, Munk A, et al. Immunoglobulin-like transcript receptors on human dermal CD14+ dendritic cells act as a CD8-antagonist to control cytotoxic T cell priming. Proc Natl Acad Sci (2012) 109(46):18885–90. doi: 10.1073/pnas.1205785109
228. Shiroishi M, Tsumoto K, Amano K, Shirakihara Y, Colonna M, Braud VM, et al. Human inhibitory receptors Ig-like transcript 2 (ILT2) and ILT4 compete with CD8 for MHC class I binding and bind preferentially to HLA-G. Proc Natl Acad Sci USA (2003) 100(15):8856–61. doi: 10.1073/pnas.1431057100
229. Ujike A, Takeda K, Nakamura A, Ebihara S, Akiyama K, Takai T. Impaired dendritic cell maturation and increased T(H)2 responses in PIR-B(-/-) mice. Nat Immunol (2002) 3(6):542–8. doi: 10.1038/ni801
230. Ju X-S, Hacker C, Scherer B, Redecke V, Berger T, Schuler G, et al. Immunoglobulin-like transcripts ILT2, ILT3 and ILT7 are expressed by human dendritic cells and down-regulated following activation. Gene (2004) 331(0):159–64. doi: 10.1016/j.gene.2004.02.018
231. Švajger U, Vidmar A, Jeras M. Niflumic acid renders dendritic cells tolerogenic and up-regulates inhibitory molecules ILT3 and ILT4. Int Immunopharmacol (2008) 8(7):997–1005. doi: 10.1016/j.intimp.2008.03.006
232. Penna G, Roncari A, Amuchastegui S, Daniel KC, Berti E, Colonna M, et al. Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4+Foxp3+ regulatory T cells by 1,25-dihydroxyvitamin D3. Blood (2005) 106(10):3490–7. doi: 10.1182/blood-2005-05-2044
233. Buckland M, Jago CB, Fazekasova H, Scott K, Tan PH, George AJT, et al. Aspirin-treated human DCs up-regulate ILT-3 and induce hyporesponsiveness and regulatory activity in responder T cells. Am J Transplant (2006) 6(9):2046–59. doi: 10.1111/j.1600-6143.2006.01450.x
234. Karre K. Natural killer cell recognition of missing self. Nat Immunol (2008) 9(5):477–80. doi: 10.1038/ni0508-477
235. Vlad G, Cortesini R, Suciu-Foca N. CD8+ T suppressor cells and the ILT3 master switch. Hum Immunol (2008) 69(11):681–6. doi: 10.1016/j.humimm.2008.08.286
236. Morel E, Bellón T. Amoxicillin conjugates to HLA class I molecules and interferes with signalling through the ILT2/LIR-1/CD85j inhibitory receptor. Allergy (2007) 62(2):190–6. doi: 10.1111/j.1398-9995.2006.01285.x
237. Nakayama M, Kurokawa K, Nakamura K, Lee BL, Sekimizu K, Kubagawa H, et al. Inhibitory receptor paired ig-like receptor B is exploited by staphylococcus aureus for virulence. J Immunol (2012) 189(12):5903–11. doi: 10.4049/jimmunol.1201940
238. Munitz A, Cole ET, Beichler A, Groschwitz K, Ahrens R, Steinbrecher K, et al. Paired immunoglobulin-like receptor B (PIR-B) negatively regulates macrophage activation in experimental colitis. Gastroenterology (2010) 139(2):530–41. doi: 10.1053/j.gastro.2010.04.006
239. Wagner CS, Walther-Jallow L, Buentke E, Ljunggren H-G, Achour A, Chambers BJ. Human cytomegalovirus-derived protein UL18 alters the phenotype and function of monocyte-derived dendritic cells. J Leukocyte Biol (2008) 83(1):56–63. doi: 10.1189/jlb.0307181
240. Yang Z, Bjorkman PJ. Structure of UL18, a peptide-binding viral MHC mimic, bound to a host inhibitory receptor. Proc Natl Acad Sci United States America (2008) 105(29):10095–100. doi: 10.1073/pnas.0804551105
241. Northfield J, Lucas M, Jones H, Young NT, Klenerman P. Does memory improve with age? CD85j (ILT-2//LIR-1) expression on CD8+ T cells correlates with/`memory inflation/’ in human cytomegalovirus infection. Immunol Cell Biol (2005) 83(2):182–8. doi: 10.1111/j.1440-1711.2005.01321.x
242. Prod’homme V, Griffin C, Aicheler RJ, Wang ECY, McSharry BP, Rickards CR, et al. The human cytomegalovirus MHC class I homolog UL18 inhibits LIR-1(+) but activates LIR-1(–) NK cells. J Immunol (Baltimore Md: 1950) (2007) 178(7):4473–81. doi: 10.4049/jimmunol.178.7.4473
243. Tripathi P, Agrawal S. The role of human leukocyte antigen E and G in HIV infection. AIDS (2007) 21(11):1395–404. doi: 10.1097/QAD.0b013e32810c8bbc
244. Donaghy L, Gros F, Amiot L, Mary C, Maillard A, Guiguen C, et al. Elevated levels of soluble non-classical major histocompatibility class I molecule human leucocyte antigen (HLA)-G in the blood of HIV-infected patients with or without visceral leishmaniasis. Clin Exp Immunol (2007) 147(2):236–40. doi: 10.1111/j.1365-2249.2006.03268.x
245. Cao W, Bover L, Cho M, Wen X, Hanabuchi S, Bao M, et al. Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J Exp Med (2009) 206(7):1603–14. doi: 10.1084/jem.20090547
246. Cai G, Du M, Bosse Y, Albrecht H, Qin F, Luo X, et al. SARS-coV-2 impairs dendritic cells and regulates DC-SIGN gene expression in tissues. Int J Mol Sci (2021) 22(17). doi: 10.3390/ijms22179228
247. Tavano B, Galao RP, Graham DR, Neil SJD, Aquino VN, Fuchs D, et al. Ig-like transcript 7, but not bone marrow stromal cell antigen 2 (Also known as HM1.24, tetherin, or CD317), modulates plasmacytoid dendritic cell function in primary human blood leukocytes. J Immunol (2013) 190(6):2622–30. doi: 10.4049/jimmunol.1202391
248. Shang P, Simpson JD, Taylor GM, Sutherland DM, Welsh OL, Aravamudhan P, et al. Paired immunoglobulin-like receptor B is an entry receptor for mammalian orthoreovirus. Nat Commun (2023) 14(1):2615. doi: 10.1038/s41467-023-38327-6
249. Zhang J, Mai S, Chen HM, Kang K, Li XC, Chen SH, et al. Leukocyte immunoglobulin-like receptors in human diseases: an overview of their distribution, function, and potential application for immunotherapies. J Leukoc Biol (2017) 102(2):351–60. doi: 10.1189/jlb.5MR1216-534R
250. Li Y, Guo J, Zhang H, Li Z, Ren Y, Jiang Y, et al. LILRB4 regulates the function of decidual MDSCs via the SHP-2/STAT6 pathway during Toxoplasma gondii infection. Parasites Vectors (2023) 16(1):237. doi: 10.1186/s13071-023-05856-4
251. Dechavanne C, Nouatin O, Adamou R, Edslev S, Hansen A, Meurisse F, et al. Placental malaria is associated with higher LILRB2 expression in monocyte subsets and lower anti-malarial igG antibodies during infancy. Front Immunol (2022) 13:909831. doi: 10.3389/fimmu.2022.909831
252. Xu Z, Lin CC, Ho S, Vlad G, Suciu-Foca N. Suppression of experimental autoimmune encephalomyelitis by ILT3.Fc. J Immunol (2021) 206(3):554–65. doi: 10.4049/jimmunol.2000265
253. van der Touw W, Kang K, Luan Y, Ma G, Mai S, Qin L, et al. Glatiramer acetate enhances myeloid-derived suppressor cell function via recognition of paired ig-like receptor B. J Immunol (2018) 201(6):1727–34. doi: 10.4049/jimmunol.1701450
254. Cao Q, Shin WS, Chan H, Vuong CK, Dubois B, Li B, et al. Inhibiting amyloid-beta cytotoxicity through its interaction with the cell surface receptor LilrB2 by structure-based design. Nat Chem (2018) 10(12):1213–21. doi: 10.1038/s41557-018-0147-z
255. Karnell JL, Wu Y, Mittereder N, Smith MA, Gunsior M, Yan L, et al. Depleting plasmacytoid dendritic cells reduces local type I interferon responses and disease activity in patients with cutaneous lupus. Sci Transl Med (2021) 13(595):eabf8442. doi: 10.1126/scitranslmed.abf8442
256. Terao C, Yoshifuji H, Matsumura T, Naruse TK, Ishii T, Nakaoka Y, et al. Genetic determinants and an epistasis of LILRA3 and HLA-B*52 in Takayasu arteritis. Proc Natl Acad Sci U.S.A. (2018) 115(51):13045–50. doi: 10.1073/pnas.1808850115
257. Huynh OA, Hampartzoumian T, Arm JP, Hunt J, Borges L, Ahern M, et al. Down-regulation of leucocyte immunoglobulin-like receptor expression in the synovium of rheumatoid arthritis patients after treatment with disease-modifying anti-rheumatic drugs. Rheumatology (2007) 46(5):742–51. doi: 10.1093/rheumatology/kel405
258. Veit TD, Chies JA, Switala M, Wagner B, Horn PA, Busatto M, et al. The paradox of high availability and low recognition of soluble HLA-G by LILRB1 receptor in rheumatoid arthritis patients. PloS One (2015) 10(4):e0123838. doi: 10.1371/journal.pone.0123838
259. Alegre ML, Lakkis FG, Morelli AE. Antigen presentation in transplantation. Trends Immunol (2016) 37(12):831–43. doi: 10.1016/j.it.2016.09.003
260. Ciubotariu R, Vasilescu R, Ho E, Cinti P, Cancedda C, Poli L, et al. Detection of T suppressor cells in patients with organ allografts. Hum Immunol (2001) 62(1):15–20. doi: 10.1016/S0198-8859(00)00226-3
261. Colovai AI, Mirza M, Vlad G, Wang Su, Ho E, Cortesini R, et al. Regulatory CD8+CD28– T cells in heart transplant recipients. Hum Immunol (2003) 64(1):31–7. doi: 10.1016/S0198-8859(02)00742-5
262. Cortesini R, Suciu-Foca N. ILT3+ ILT4+ Tolerogenic endothelial cells in transplantation. Transplantation (2006) 82:S30–2. doi: 10.1097/01.tp.0000231437.12890.64
263. Manavalan JS, Kim-Schulze S, Scotto L, Naiyer AJ, Vlad G, Colombo PC, et al. Alloantigen specific CD8+CD28– FOXP3+ T suppressor cells induce ILT3+ ILT4+ tolerogenic endothelial cells, inhibiting alloreactivity. Int Immunol (2004) 16(8):1055–68. doi: 10.1093/intimm/dxh107
264. Vlad G, D’Agati VD, Zhang Q-Y, Liu Z, Ho EK, Mohanakumar T, et al. Immunoglobulin-like transcript 3-fc suppresses T-cell responses to allogeneic human islet transplants in hu-NOD/SCID mice. Diabetes (2008) 57(7):1878–86. doi: 10.2337/db08-0054
265. Naji A, Durrbach A, Carosella ED, Rouas-Freiss N. Soluble HLA-G and HLA-G1 expressing antigen-presenting cells inhibit T-cell alloproliferation through ILT-2/ILT-4/fasL-mediated pathways. Hum Immunol (2007) 68(4):233–9. doi: 10.1016/j.humimm.2006.10.017
266. Qiu J, Terasaki PI, Miller J, Mizutani K, Cai J, Carosella ED. Soluble HLA-G expression and renal graft acceptance. Am J Transplant (2006) 6(9):2152–6. doi: 10.1111/j.1600-6143.2006.01417.x
267. Lila N, Amrein C, Guillemain R, Chevalier P, Latremouille C, Fabiani J-N, et al. Human leukocyte antigen-G expression after heart transplantation is associated with a reduced incidence of rejection. Circulation (2002) 105(16):1949–54. doi: 10.1161/01.CIR.0000015075.89984.46
268. Ezeakile M, Portik-Dobos V, Wu J, Horuzsko DD, Kapoor R, Jagadeesan M, et al. HLA-G dimers in the prolongation of kidney allograft survival. J Immunol Res (2014) 2014:153981. doi: 10.1155/2014/153981
269. Luque J, Torres MI, Aumente MD, Marín J, García-Jurado G, González R, et al. Soluble HLA-G in heart transplantation: their relationship to rejection episodes and immunosuppressive therapy. Hum Immunol (2006) 67(4–5):257–63. doi: 10.1016/j.humimm.2006.02.034
270. Le Rond S, Azéma C, Krawice-Radanne I, Durrbach A, Guettier C, Carosella ED, et al. Evidence to support the role of HLA-G5 in allograft acceptance through induction of immunosuppressive/regulatory T cells. J Immunol (2006) 176(5):3266–76. doi: 10.4049/jimmunol.176.5.3266
271. Zhang W, Liang S, Wu J, Horuzsko A. Human inhibitory receptor ILT2 amplifies CD11b(+)Gr1(+) myeloid-derived suppressor cells that promote long-term survival of allografts. Transplantation (2008) 86(8):1125–34. doi: 10.1097/TP.0b013e318186fccd
272. Wu J, Zhang W, Hernandez-Lopez P, Fabelo E, Parikh M, Mulloy LL, et al. Isoforms of human leukocyte antigen-G and their inhibitory receptors in human kidney allograft acceptance†. Hum Immunol (2009) 70(12):988–94. doi: 10.1016/j.humimm.2009.07.023
273. Kostlin N, Ostermeir AL, Spring B, Schwarz J, Marme A, Walter CB, et al. HLA-G promotes myeloid-derived suppressor cell accumulation and suppressive activity during human pregnancy through engagement of the receptor ILT4. Eur J Immunol (2017) 47(2):374–84. doi: 10.1002/eji.201646564
274. Liu J, Liu Z, Witkowski P, Vlad G, Manavalan JS, Scotto L, et al. Rat CD8+ FOXP3+ T suppressor cells mediate tolerance to allogeneic heart transplants, inducing PIR-B in APC and rendering the graft invulnerable to rejection. Transplant Immunol (2004) 13(4):239–47. doi: 10.1016/j.trim.2004.10.006
275. Ristich V, Zhang W, Liang S, Horuzsko A. Mechanisms of prolongation of allograft survival by HLA-G/ILT4-modified dendritic cells. Hum Immunol (2007) 68(4):264–71. doi: 10.1016/j.humimm.2006.11.008
276. Yu J, Wang Y, Yan F, Zhang P, Li H, Zhao H, et al. Noncanonical NF-kappaB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol (2014) 193(5):2574–86. doi: 10.4049/jimmunol.1400833
277. Nakamura A, Kobayashi E, Takai T. Exacerbated graft-versus-host disease in Pirb-/- mice. Nat Immunol (2004) 5(6):623–9. doi: 10.1038/ni1074
278. Zhao J, Luo Y, Wang X, Zhou H, Li Q, You Y, et al. Prevention of murine acute graft-versus-host disease by recipient-derived paired immunoglobulin-like receptor B lentivirus-transfected dendritic cells. Acta Haematol (2010) 124(3):134–40. doi: 10.1159/000315553
279. Nowak I, Wilczynska K, Wilczynski JR, Malinowski A, Radwan P, Radwan M, et al. KIR, LILRB and their ligands’ Genes as potential biomarkers in recurrent implantation failure. Arch Immunol Ther Exp (Warsz) (2017) 65(5):391–9. doi: 10.1007/s00005-017-0474-6
280. Pinto A, Rega A, Crother TR, Sorrentino R. Plasmacytoid dendritic cells and their therapeutic activity in cancer. Oncoimmunology (2012) 1(5):726–34. doi: 10.4161/onci.20171
281. Zilberman S, Schenowitz C, Agaugué S, Favier B, Riteau B, Rouzier R, et al. HLA-G1 and HLA-G5 active dimers are present in Malignant cells and effusions: The influence of the tumor microenvironment. Eur J Immunol (2012) 42(6):1599–608. doi: 10.1002/eji.201141761
282. Rouas-Freiss N, Moreau P, LeMaoult J, Carosella ED. The dual role of HLA-G in cancer. J Immunol Res (2014) 2014:10. doi: 10.1155/2014/359748
283. Wan R, Wang ZW, Li H, Peng XD, Liu GY, Ou JM, et al. Human leukocyte antigen-G inhibits the anti-tumor effect of natural killer cells via immunoglobulin-like transcript 2 in gastric cancer. Cell Physiol Biochem (2017) 44(5):1828–41. doi: 10.1159/000485819
284. Carbone C, Piro G, Fassan M, Tamburrino A, Mihaela Mina M, Zanotto M, et al. An angiopoietin-like protein 2 autocrine signaling promotes EMT during pancreatic ductal carcinogenesis. Oncotarget (2015) 6(15):13822–34. doi: 10.18632/oncotarget.2635
285. Zeller T, Lutz S, Munnich IA, Windisch R, Hilger P, Herold T, et al. Dual checkpoint blockade of CD47 and LILRB1 enhances CD20 antibody-dependent phagocytosis of lymphoma cells by macrophages. Front Immunol (2022) 13:929339. doi: 10.3389/fimmu.2022.929339
286. Chen X, Gao A, Zhang F, Yang Z, Wang S, Fang Y, et al. ILT4 inhibition prevents TAM- and dysfunctional T cell-mediated immunosuppression and enhances the efficacy of anti-PD-L1 therapy in NSCLC with EGFR activation. Theranostics (2021) 11(7):3392–416. doi: 10.7150/thno.52435
287. Liu X, Yu X, Xie J, Zhan M, Yu Z, Xie L, et al. ANGPTL2/LILRB2 signaling promotes the propagation of lung cancer cells. Oncotarget (2015) 6(25):21004–15. doi: 10.18632/oncotarget.4217
288. Wu G, Xu Y, Schultz RD, Chen H, Xie J, Deng M, et al. LILRB3 supports acute myeloid leukemia development and regulates T-cell antitumor immune responses through the TRAF2-cFLIP-NF-kappaB signaling axis. Nat Cancer (2021) 2(11):1170–84. doi: 10.1038/s43018-021-00262-0
289. Lasry A, Aifantis I. LILRB3 as a regulator of AML survival. Nat Cancer (2021) 2(11):1122–3. doi: 10.1038/s43018-021-00285-7
290. Kumata S, Notsuda H, Su M-T, Saito-Koyama R, Tanaka R, Suzuki Y, et al. Prognostic impact of LILRB4 expression on tumor-infiltrating cells in resected non-small cell lung cancer. Thorac Cancer (2023) 14(n/a):2057–68. doi: 10.1111/1759-7714.14991
291. Ma G, Pan PY, Eisenstein S, Divino CM, Lowell CA, Takai T, et al. Paired immunoglobin-like receptor-B regulates the suppressive function and fate of myeloid-derived suppressor cells. Immunity (2011) 34(3):385–95. doi: 10.1016/j.immuni.2011.02.004
292. Gubens MA, Sequist LV, Stevenson JP, Powell SF, Villaruz LC, Gadgeel SM, et al. Pembrolizumab in combination with ipilimumab as second-line or later therapy for advanced non-small-cell lung cancer: KEYNOTE-021 cohorts D and H. Lung Cancer (2019) 130:59–66. doi: 10.1016/j.lungcan.2018.12.015
293. Vlad G, Chang CC, Colovai AI, Vasilescu ER, Cortesini R, Suciu-Foca N. Membrane and soluble ILT3 are critical to the generation of T suppressor cells and induction of immunological tolerance. Int Rev Immunol (2010) 29(2):119–32. doi: 10.3109/08830180903281185
294. Mandel I, Haves Ziv D, Goldshtein I, Peretz T, Alishekevitz D, Fridman Dror A, et al. BND-22, a first-in-class humanized ILT2-blocking antibody, promotes antitumor immunity and tumor regression. J Immunother Cancer (2022) 10(9):e004859. doi: 10.1136/jitc-2022-004859
295. Udartseva O, Pupecka-Swider M, Garcia-Broncano P, Guo N, Campbell S, Klakus E, et al. Abstract 2906: AGEN1571 is a novel high-affinity ILT2 antagonist antibody that promotes adaptive and innate immune responses. Cancer Res (2022) 82(12_Supplement):2906. doi: 10.1158/1538-7445.Am2022-2906
296. Kim A, Han CJ, Driver I, Olow A, Sewell AK, Zhang Z, et al. LILRB1 blockade enhances bispecific T cell engager antibody-induced tumor cell killing by effector CD8(+) T cells. J Immunol (2019) 203(4):1076–87. doi: 10.4049/jimmunol.1801472
297. Chen H, Chen Y, Deng M, John S, Gui X, Kansagra A, et al. Antagonistic anti-LILRB1 monoclonal antibody regulates antitumor functions of natural killer cells. J Immunother Cancer (2020) 8(2):e000515. doi: 10.1136/jitc-2019-000515
298. Papadopoulos K, Li T, Lakhani N, Powderly J, George T, Teoh DGK, et al. 172P Phase I study of JTX-8064, a LILRB2 (ILT4) inhibitor, as monotherapy and combination with pimivalimab (pimi), a PD-1 inhibitor (PD-1i), in patients (pts) with advanced solid tumors. Immuno-Oncology Technol (2022) 16:28–9. doi: 10.1016/j.iotech.2022.100284
299. Siu LL, Wang D, Hilton J, Geva R, Rasco D, Perets R, et al. First-in-class anti-immunoglobulin-like transcript 4 myeloid-specific antibody MK-4830 abrogates a PD-1 resistance mechanism in patients with advanced solid tumors. Clin Cancer Res (2022) 28(1):57–70. doi: 10.1158/1078-0432.CCR-21-2160
300. Ma J-T, Liu X, Stafford R, McCutcheon K, Chen H, Huang T, et al. Abstract 601: IO-108, A fully human therapeutic antibody blocking the myeloid checkpoint LILRB2/ILT4, promotes innate and adaptive anti-cancer immunity in preclinical studies. Cancer Res (2022) 82(12_Supplement):601. doi: 10.1158/1538-7445.Am2022-601
301. Chen P, Crawley SC, Lin VY, Kapoor V, Paavola KJ, Chen H-I, et al. Abstract LB216: Pre-clinical characterization of NGM831, an ILT3 antagonist antibody for the treatment of solid tumors. Cancer Res (2022) 82(12_Supplement):LB216–LB. doi: 10.1158/1538-7445.Am2022-lb216
302. Niu X, Wang C, Zhao J, Hu J, Hu Y, Sun J, et al. 1062 ES009, a LILRB2-specific blocking antibody, reprograms myeloid cells into pro-inflammation phenotype and potentiates T cell activation. J ImmunoTher Cancer (2022) 10(Suppl 2):A1104–A. doi: 10.1136/jitc-2022-SITC2022.1062
303. Naing A, Wang JS, Sharma MR, Sommerhalder D, Gandhi L, Oh DY, et al. 174P First-in-human study of NGM707, an ILT2/ILT4 dual antagonist antibody in advanced or metastatic solid tumors: Preliminary monotherapy dose escalation data. Immuno-Oncology Technol (2022) 16:29–30. doi: 10.1016/j.iotech.2022.100286
304. Mondal K, Song C, Tian J, Ho C, Rivera LB, Huang J, et al. Abstract LB156: Preclinical evaluation of NGM707, a novel anti-ILT2/anti-ILT4 dual antagonist monoclonal antibody. Cancer Res (2021) 81(13_Supplement):LB156–LB. doi: 10.1158/1538-7445.Am2021-lb156
305. Rafiei A, Kumar A, Gualandi M, Yang C-L, Smith SR, Curran MA, et al. Poster: A stabilized HLA-B57 open format (IOS-1002) binding to LILRB1, LILRB2 and KIR3DL1 induces potent anti-tumor efficacy. Protein Antibody Eng Summit (2022).
306. Murphy MB, Vitale L, Xia S, Peng Z, O’Neill T, Lillquist J, et al. CDX-585, a bispecific antibody with dual targeting of ILT4 and PD-1 checkpoint pathways. Immuno (2023) 3(3):273–88. doi: 10.3390/immuno3030018
307. John S, Chen H, Deng M, Gui X, Wu G, Chen W, et al. A novel anti-LILRB4 CAR-T cell for the treatment of monocytic AML. Mol Ther (2018) 26(10):2487–95. doi: 10.1016/j.ymthe.2018.08.001
308. Naing A, Powderly J, Pelster M, Spira A, Schneider R, Woodard P, et al. 747 A phase 1 trial of IO-202, an antagonist antibody targeting myeloid checkpoint LILRB4 (ILT3), as monotherapy and in combination with pembrolizumab in adult patients with advanced relapsed or refractory solid tumors. J ImmunoTher Cancer (2022) 10(Suppl 2):A780–A. doi: 10.1136/jitc-2022-SITC2022.0747
309. Rui W, Lei L, Zhang Z, Wu C, Xia Y, Liu Y, et al. Abstract 3185: Development of LILRB4 biparatopic synthetic T-cell receptor and antigen receptor (STAR)-T cells for the treatment of acute myeloid leukemia (AML). Cancer Res (2023) 83(7_Supplement):3185. doi: 10.1158/1538-7445.Am2023-3185
310. Huang T, Hong K, McCutcheon K, Ma J-T, Zhu Y, Deng M, et al. Abstract LB217: A novel bispecific LILRB4/CD3 antibody with potent killing of monocytic acute myeloid leukemia cells. Cancer Res (2023) 83(8_Supplement):LB217–LB. doi: 10.1158/1538-7445.Am2023-lb217
311. Lin VY, Iyer A, Akama K, Cheng R, Yang H, Aguayo J, et al. Preclinical characterization of NGM936, a novel bispecific T cell engager targeting ILT3 for the treatment of acute myeloid leukemia with monocytic differentiation. Blood (2022) 140(Supplement 1):9063–4. doi: 10.1182/blood-2022-160209
312. Anami Y, Deng M, Gui X, Yamaguchi A, Yamazaki CM, Zhang N, et al. LILRB4-targeting antibody–drug conjugates for the treatment of acute myeloid leukemia. Mol Cancer Ther (2020) 19(11):2330–9. doi: 10.1158/1535-7163.Mct-20-0407
313. Chuprin J, Buettner H, Seedhom MO, Greiner DL, Keck JG, Ishikawa F, et al. Humanized mouse models for immuno-oncology research. Nat Rev Clin Oncol (2023) 20(3):192–206. doi: 10.1038/s41571-022-00721-2
Keywords: LILR, immune tolerance, cancer, autoimmunity, infection, immunomodulation, immunotherapy
Citation: Redondo-García S, Barritt C, Papagregoriou C, Yeboah M, Frendeus B, Cragg MS and Roghanian A (2023) Human leukocyte immunoglobulin-like receptors in health and disease. Front. Immunol. 14:1282874. doi: 10.3389/fimmu.2023.1282874
Received: 25 August 2023; Accepted: 20 September 2023;
Published: 13 November 2023.
Edited by:
Petr O. Ilyinskii, Selecta Biosciences, United StatesReviewed by:
Kouyuki Hirayasu, Kanazawa University, JapanCopyright © 2023 Redondo-García, Barritt, Papagregoriou, Yeboah, Frendeus, Cragg and Roghanian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali Roghanian, QS5Sb2doYW5pYW5Ac290b24uYWMudWs=; YXJvZ2hhbmlAbWl0LmVkdQ==
†Present address: Charys Papagregoriou, The Centre for the Study of Haematological and other Malignancies, Nicosia, Cyprus
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.