 Chelsia Qiuxia Wang
Chelsia Qiuxia Wang Pei Yu Lim
Pei Yu Lim Andy Hee-Meng Tan
Andy Hee-Meng Tan- 1Immune Cell Manufacturing, Bioprocessing Technology Institute (BTI), Agency for Science, Technology and Research (A*STAR), Singapore, Singapore
- 2Food, Chemical and Biotechnology Cluster, Singapore Institute of Technology (SIT), Singapore, Singapore
Adoptive cellular immunotherapy as a new paradigm to treat cancers is exemplified by the FDA approval of six chimeric antigen receptor-T cell therapies targeting hematological malignancies in recent years. Conventional αβ T cells applied in these therapies have proven efficacy but are confined almost exclusively to autologous use. When infused into patients with mismatched human leukocyte antigen, αβ T cells recognize tissues of such patients as foreign and elicit devastating graft-versus-host disease. Therefore, one way to overcome this challenge is to use naturally allogeneic immune cell types, such as γδ T cells. γδ T cells occupy the interface between innate and adaptive immunity and possess the capacity to detect a wide variety of ligands on transformed host cells. In this article, we review the fundamental biology of γδ T cells, including their subtypes, expression of ligands, contrasting roles in and association with cancer prognosis or survival, as well as discuss the gaps in knowledge pertaining to this cell type which we currently endeavor to elucidate. In addition, we propose how to harness the unique properties of γδ T cells for cellular immunotherapy based on lessons gleaned from past clinical trials and provide an update on ongoing trials involving these cells. Lastly, we elaborate strategies that have been tested or can be explored to improve the anti-tumor activity and durability of γδ T cells in vivo.
1 Introduction to γδ T cells
Recent advances in genomic editing of cells (1–3) have propelled cellular immunotherapy as a new paradigm to treat cancers, which is rapidly gaining traction with the FDA approval since 2017 of six therapies involving T cells engineered with different chimeric antigen receptors (CARs) targeting primarily B cell malignancies [summarized in (4, 5)]. These approved therapies, and many others undergoing investigation in pre-clinical studies and clinical trials, have largely utilized conventional αβ T cells which are limited to autologous applications. If infused in a recipient patient with mismatched human leukocyte antigen (HLA), αβ T cells will recognize as foreign and attack the patient’s tissues that results in potentially life-threatening graft-versus-host disease (GvHD). One approach to circumvent the occurrence of GvHD is to use innate or innate-like immune cells such as γδ T cells, which possess characteristics rendering them appropriate for allogeneic therapy.
γδ T cells represent a small population of total leukocytes in umbilical cord blood (UCB) and peripheral blood (PB), comprising approximately 0.0045-0.035% of UCB and 0.5-5% of PB (6–9). Despite their low abundance, these cells play crucial roles in immune defense against bacterial and viral infections, as well as in immune surveillance of cancer. γδ T cells are poised to recognize intracellularly stressed cells, such as infected and tumor cells, and respond by directly eliminating such cells (10). The infected and tumor cells convey their intracellular stress to γδ T cells via a myriad of molecules. γδ T cells sense these dysfunctional cells by recognizing tumor-associated metabolic byproducts such as butyrophilins (BTNs) on tumor cells in the peripheral circulation or stress-associated proteins like MHC class I-related chain A or B (respectively MICA or MICB) upregulated on stressed cells in both PB and tissues. Engagement of these ligands by their receptors on γδ T cells activate direct killing mechanisms via granzyme B and perforin rapidly without prior exposure to pathogen- or tumor-associated antigens (Figure 1). They also stimulate secretion of effector molecules such as interferon (IFN)-γ and tumor necrosis factor (TNF)-α.
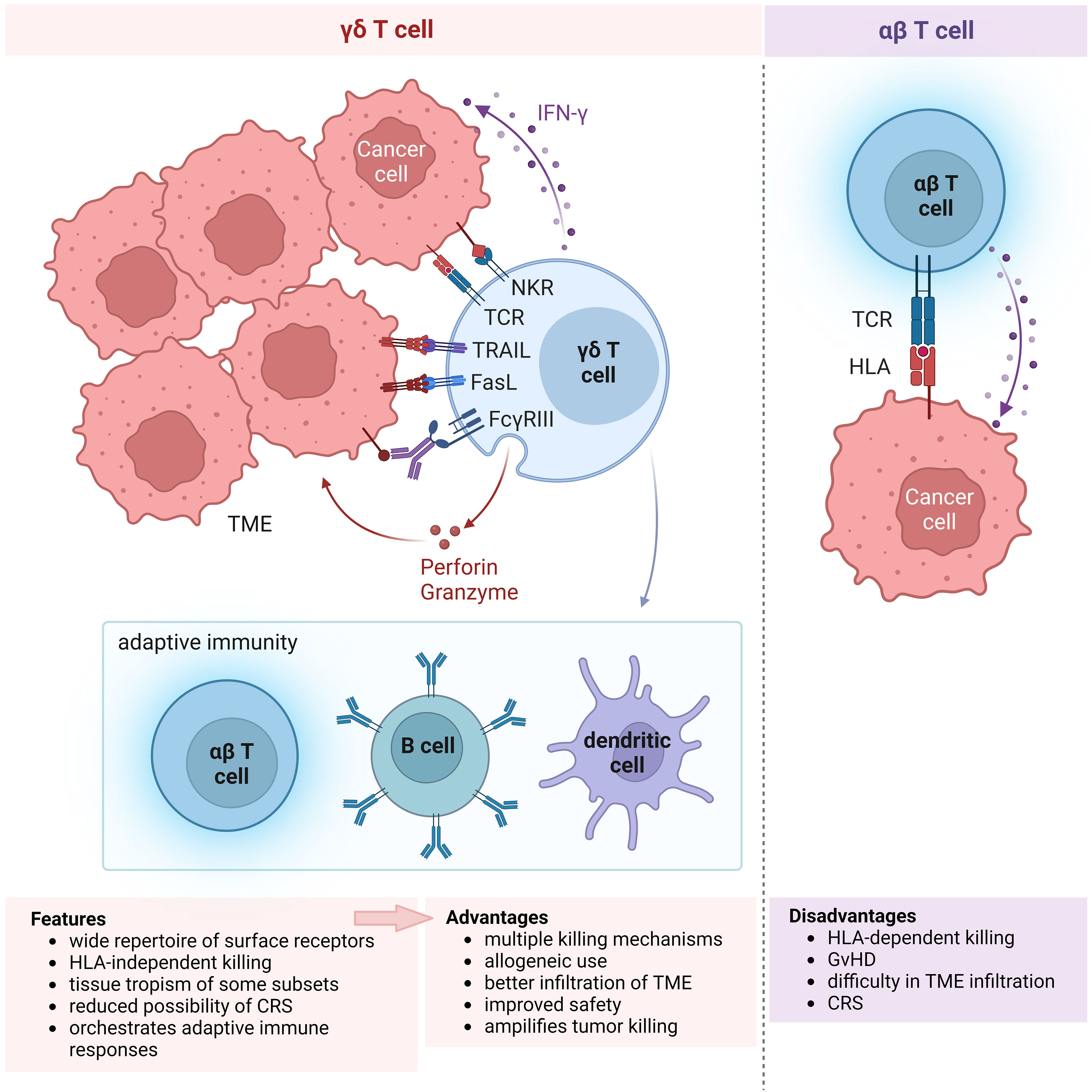
Figure 1 Features of γδ T cells propelling their choice for use in cellular immunotherapy. Schematic showing the multitude of receptors expressed on γδ T cells and their anti-tumor killing mechanisms (left), in comparison with αβ T cells (right). The wide repertoire of surface receptors confer the advantage of multiple killing mechanisms that can be elicited by γδ T cells. Moreover, their killing mechanism is independent of HLA, unlike αβ T cells, allowing for allogeneic use in cell therapy. Activation of γδ T cell killing results in engagement of the adaptive immune response, resulting in the amplification of anti-tumor cytotoxicity. Other features not illustrated in the diagram include tissue tropism of certain, e.g. Vδ1, γδ T cell subsets and the reduced possibility of CRS, presumably leading to better infiltration of TME and improved safety. All of these features of γδ T cells (bottom left) that provide advantages over αβ T cells (bottom right) for use in allogeneic immune cell therapy are summarized in the boxes below. NKR, natural killer receptor; TCR, T cell receptor; TRAIL, tumor necrosis factor (TNF)-related apoptosis-inducing ligand; FasL, Fas ligand; IFN, interferon; TME, tumor microenvironment; HLA, human leukocyte antigen; GvHD, graft-versus-host disease; CRS, cytokine release syndrome.
There are several advantages in employing γδ T cells for immunotherapy (Figure 1). Firstly, γδ T cells express a wide repertoire of cell surface receptors conferring the ability to broadly recognize a diversity of tumor ligands and thereby target multiple tumor types, unlike HLA-restricted tumor recognition by αβ T cells. This is particularly useful for tumors which have downregulated HLA class I expression to evade immune recognition by αβ T cells. Secondly, the cytotoxic function of γδ T cells is therefore activated independently of HLA which drastically reduces their chance of provoking GvHD and allows for their allogeneic use. Thirdly, γδ T cells are the major early producers of pro-inflammatory IFN-γ which triggers their anti-tumor response and orchestrates αβ T, B and dendritic cells in a cascade of adaptive immune responses that further amplify tumor killing (11). The cross-talk between γδ T cells and other immune cells are described in a recent review (12). Such interactions in the tumor microenvironment (TME) allow γδ T cells to shape its immediate environment into a tumor-suppressing one. Moreover, γδ T cell subtypes characterized by certain rearrangements of their γδ T cell receptor (TCR) intrinsically populate specific tissues, namely skin, large intestine, spleen and liver. It is thought that such tissue tropism may enhance the capacity of γδ T cell subtypes to infiltrate the TME of diverse solid tumors consisting tissues which are the physiological habitats for the respective cell subtypes. Furthermore, as engineered γδ T cells exhibit similar anti-tumor efficacy but generally secrete lower levels of cytokines compared with their similarly modified αβ counterparts, γδ T cell therapy harbors a potentially lower risk of cytokine release syndrome (CRS) (13–15).
In this review, we summarize fundamental concepts underlying the biology of γδ T cells, as well as recent developments related to their role in cancer prognosis and survival revealed by multiple lines of research evidence which will be elaborated in the following sections. We discuss the gaps in knowledge that can improve ways to harness γδ T cells for cellular immunotherapy. We also take stock of the current outlook of clinical trials relating to γδ T cell therapies that have been carried out thus far and discuss what we can learn from these trials. Lastly, we review current or propose new strategies to improve the anti-tumor efficacy of γδ T cell therapies.
2 γδ T cells: what are the gaps to be filled?
2.1 Refinement of γδ T cell subtypes and their associated ligands
Human γδ T cells can be divided into several subtypes, including Vδ1 and Vδ2 subtypes based on their expression of TCRδ chain variant, contrasting with murine γδ T cell subsets which are categorized according to their γ chain expression. While Vδ2 cells are predominantly found in blood circulation, Vδ1 cells are localized mainly in mucosal epithelial tissues. There also exist less well studied subtypes such as Vδ3 cells that reside in the liver. Regardless of their subtype based on TCRδ chain variant expression, γδ T cells can be distinguished in terms of functional potency based on their expression of cell surface receptors, including CD56 (16, 17), NKG2A (18), the SCART scavenger receptors (SCART1 and SCART2) (19), CD27 (20) and CD161 (21), signatures of which correlate with cytokine secretion and anti-tumor cytotoxicity. Interestingly, Vδ1 and Vδ2 subtypes can each be functionally differentiated by the expression of CD56. While CD56+ Vδ2 T cells have greater anti-tumor effector function compared with their CD56- counterparts, the opposite is observed of Vδ1 T cells for which positive expression of CD56 is associated with lower anti-tumor potency. It should be noted that the latter finding was based on tumor-infiltrating Vδ1 T lymphocytes derived from a single patient and hence requires further validation. Delineation of the spectrum of cytotoxic properties within each γδ T cell subset will yield added insight into the functional roles of γδ T cells (Figure 2, right, points 1 and 2).
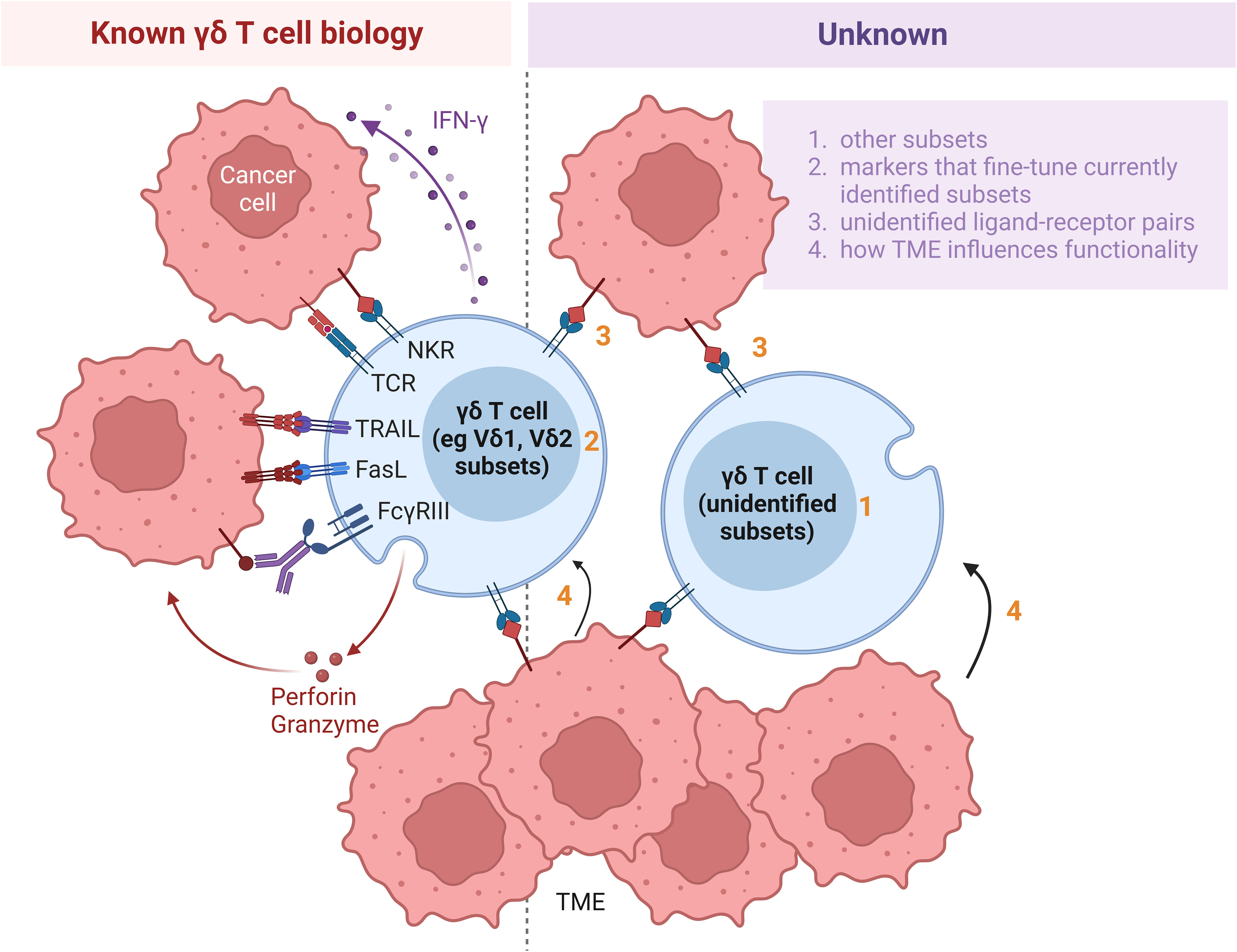
Figure 2 Hurdles impeding application of γδ T cells for cell therapy. Whilst increasing amount of information is being discovered about γδ T cell biology (left panel), much remains to be further elucidated (right panel). Firstly, various subsets, including Vδ1 and Vδ2, have been identified. However, presence of substantial Vδ1 and Vδ2 populations suggest that continuous identification of additional subsets is warranted (point 1). Secondly, within each subset, the γδ T cells can be delineated by additional markers, such as CD56, CD27 and NKG2A. Identification of such additional markers will yield added insight into the functional roles of γδ T cells (point 2). Thirdly, while several molecules have been identified as ligands to the multitude of receptors on γδ T cells, many ligands recognized by TCRγδ or other surface receptors remain currently unidentified (point 3). Furthermore, the influence of the TME on the functions of γδ T cells has been established, but the exact conditions which tune γδ T cells towards pro- versus anti-tumor subsets are not yet well defined (point 4). Research delving to uncover the unknown aspects of γδ T cell biology and their interactions with cells in the TME will improve the effectiveness of adoptive transfer of γδ T cells in immunotherapy. NKR, natural killer receptor; TCR, T cell receptor; TRAIL, tumor necrosis factor (TNF)-related apoptosis-inducing ligand; FasL, Fas ligand; IFN, interferon; TME, tumor microenvironment.
Binding of their TCR ligands activates γδ T cells to secrete IFN-γ, TNF-α and other cytotoxic effector molecules that act against tumor cells (22) (Figure 2, left). The activated γδ T cells also secrete granzyme B and perforin which aid in their cytolytic function. While many ligands remain currently unidentified (Figure 2, right, point 3), metabolites of the isoprenoid pathway, also known as phosphoantigens, or pharmacological agents that promote their accumulation have been found to efficiently activate and expand Vγ9Vδ2 T cells. Physiologically, upregulation of the mevalonate pathway in tumor cells results in the accumulation of phosphoantigens, such as isopentenyl pyrophosphate, which induce conformational changes of BTN3A1 in these cells (23). In turn, such a conformational change mediates interactions between BTN2A1 and BTN3A1 and leads to the subsequent binding of TCR Vγ9 to BTN2A1. An example of a synthetic phosphoantigen that has been assessed in clinical trials is bromohydrin pyrophosphate (BrHPP). Identification of de novo biomolecules that can preferentially stimulate other γδ T cell subsets will facilitate their ex vivo and in vivo expansion for therapeutic purpose. In addition, Vδ1 T cells can recognize ephrin receptor A2 (EphA2) (24) and MHC-related protein 1 (25), while Vδ3 T cells are activated by annexin A2 (26) on tumor cells and Vδ5 T cells bind endothelial protein C receptor (EPCR) on cytomegalovirus-infected and epithelial tumor cells (27) through their respective TCRs. Due to the tissue tropism of non-Vδ2 T cells, the ligand-receptor recognition pathways involved in the activation of non-Vδ2 T cells presumably play a more important role in γδ T cell activation in the context of solid tumors. Apart from TCR ligands, ligands induced on epithelial and tumor cells via stress or structural damage, including MICA, MICB and UL16-binding proteins (ULBPs), are recognized by NKG2D on both intraepithelial Vδ1 and circulating Vδ2 cells (14). The aforementioned ligands, among others, could be engineered in feeder cells to support ex vivo expansion of γδ T cells to attain clinically relevant numbers of γδ T cells which are estimated to be 108 to 1011 cells per infusion.
Although most γδ T cell subsets exhibit cytotoxicity against tumor cells, there exist pro-tumorigenic interleukin (IL)-17-producing (28) and PD-L1-overexpressing γδ T cells (29). These broadly termed regulatory γδ T cells (γδ Tregs) antagonize the therapeutic efficacy of cytotoxic γδ T cells and therefore suppress host immune responses (30). Interestingly, prior exposure or not to ligands during development in the murine thymus programs the effector fate of γδ T cells into respectively IFN-γ or IL-17-producing cells (31). The divergent roles of γδ T cell subsets in anti-tumor immunity have to be carefully delineated in order for their innate properties to be harnessed for immunotherapy.
2.2 Association of γδ T cells with prognosis and survival outcomes
2.2.1 γδ T cells are frequently associated with positive prognosis and survival
Notwithstanding their dual nature imprinted by thymic development, tumor-infiltrating or circulating γδ T cells are generally correlated with positive clinical outcomes or prognoses (32). Evidence from representative studies on various tumor types are described in this section (Table 1). For example, intratumoral Vδ1 T cells harvested from melanoma patients exhibited convincing anti-tumor function in vitro and when infused into patients (17). Intratumoral Vδ2 cell frequencies were found to correlate inversely with the stage of melanoma disease, with high Vδ2 frequencies observed in patients lacking cancer metastases and negligible frequencies in patients bearing advanced stage and metastatic melanomas (33). Increased intratumoral infiltration of γδ T cells was associated with overall survival benefit of gastric cancer patients (34). Moreover, Wu et al. reported that Vδ1+ cells were more abundant within triple-negative breast cancer (TNBC) vis-à-vis paired healthy tissues, especially when the cancer is in remission (44). Another study by Janssen et al. showed that the predominant population in TNBC was Vδ2- cells eliciting a proinflammatory rather than an IL-17-expressing signature (35). Interestingly, their tumor reactivity is prescribed by the diverse TCRγ and TCRδ chains and less characterized by the more “generic” anti-tumor response achieved via innate receptors such as NKG2D. Whichever the case, these findings support the observation that higher γδ T cell infiltration correlated with better survival of TNBC patients (45). Contrary to a prior study reporting the polarization of IL-17-producing Vδ1+ T cells that promote colorectal cancer (CRC) pathogenesis (28), a recent study by Meraviglia S et al. found that tumor-infiltrating γδ T cells expressing the TCRGV9-encoding gene were not the major producers of IL-17 in the CRC TME and their higher frequencies were associated with significantly longer disease-free survival rate (36). Notably, the latter study provided indirect evidence that mediators secreted by CRC cancer stem cells likely inhibited γδ T cell function in TME.
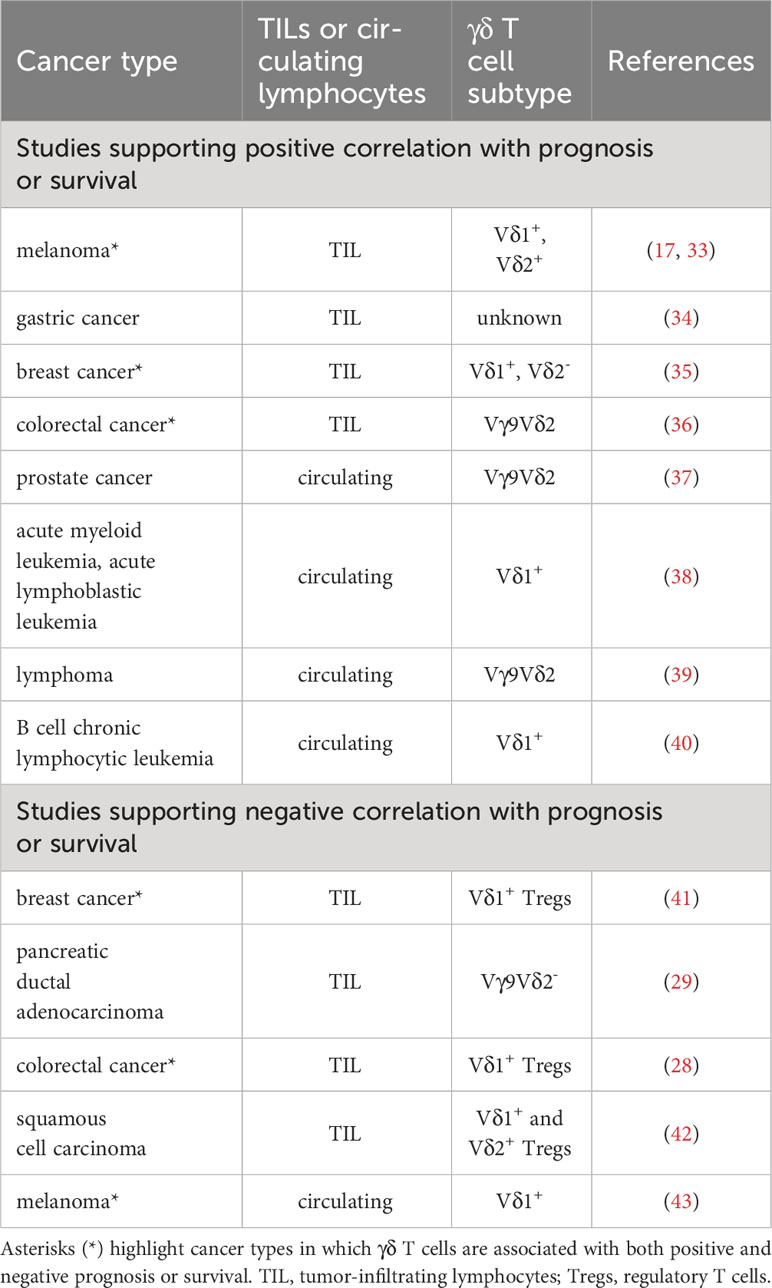
Table 1 Association of tumor-infiltrating and circulating γδ T cells with prognosis or survival of patients with different cancers.
Late-stage prostate cancer patients who were treated with zoledronate and IL-2 had superior clinical outcomes compared with zoledronate alone, as the former combination resulted in greater frequencies and more pronounced activation of peripheral γδ T cells (37). Combined zoledronate and IL-2 therapy elevated populations of γδ T cells bearing effector memory (TEM) and terminally differentiated phenotypes (TEMRA), with concomitant decrease in cell populations of naïve (Tnaïve) and central memory (TCM) phenotypes in all seven patients examined (37).
Separately, a long-term study demonstrated enhanced leukemia-free and overall survival of patients who received allogeneic hematopoietic stem cell transplantation for treatment of acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL) when their levels of donor-derived, circulating and predominantly Vδ1+ γδ T cells were high (38). γδ T cells which were responsive to proliferative stimulation by pamidronate and low-dose IL-2 contributed to effective anti-lymphoma responses in vivo while lack of γδ T cell proliferation correlated with poor objective tumor responses (39). Disease progression in patients suffering from B cell chronic lymphocytic leukemia was associated with low numbers of circulating Vδ1+ cells. Reciprocally, patients who had higher Vδ1+ cell counts maintained stable disease (40). Taken together, these studies strongly suggest that γδ T cells exert cytotoxic effects against majority of cancer types.
2.2.2 γδ T cells are occasionally associated with negative patient prognosis and survival
As earlier alluded, certain γδ T cell types are known to be tumor-promoting (Table 1) given pre-programming of different functional subsets during development and dependence on tumor context in activating selective subsets (30). For instance, Vδ1+ Tregs were found to be the dominant tumor-infiltrating lymphocyte (TIL) population in breast cancer tissues examined in 11 patients (41). These Vδ1+ Tregs, most being subsequently identified to express CD73, potently suppressed dendritic cell maturation and function, as well as cytokine secretion by CD4+ helper T cells and CD8+ effector T cells (46). The inhibitory function of Vδ1+ Tregs can be abrogated by Toll-like receptor 8 ligand engagement to enhance anti-tumor immunity (41). It was observed that γδ T cells infiltrating pancreatic ductal adenocarcinoma (PDA) overexpressed checkpoint ligands PD-L1 and Galectin-9 to directly suppress αβ T cells, hence creating an immunosuppressive TME (29). Vγ9+ cells were noticeably absent, implying that TILs were Vγ9-Vδ2- cells. The frequency of IL-17-secreting Vδ1+ γδ Tregs present in CRC positively correlated with advanced clinicopathological features of the disease (28). These pro-tumorigenic γδ Tregs were shown to promote the migration, proliferation and accumulation of myeloid-derived suppressor cells (MDSCs) via production of IL-17A, IL-8, GM-CSF and TNF-α. In both PDA and CRC, γδ TILs manifested a TEM phenotype, whereas normal healthy tissue counterparts possessed a TCM phenotype (28, 29). Furthermore, patients in the advanced stages of a type of skin cancer called squamous cell carcinoma harbored more Vδ1+ and Vδ2+ IL-17-producing γδ T cells in contrast to those in the early stages of cancer which had more IFNγ-producing cells (42). Elevated frequencies of PB Vδ1+ T cells in patients with metastatic melanoma were correlated with poorer clinical prognoses, unlike those of PB Vδ2+ counterparts which lack association (43).
While human γδ Tregs have been less studied than their murine counterparts (47), it is recognized that pro-tumorigenic γδ Tregs do not exert direct effects on tumor cells but are able to shape the TME via other cell types to become an immune suppressive one, thereby promoting oncogenic progression. Collectively, the aforementioned studies, albeit non-exhaustive, serve as a timely reminder of the opposing roles that γδ T cell subsets play in tumor immunity.
2.3 The gaps to fill for the roles of γδ T cells in anti-tumor immunity
In some studies, γδ T cells were not clearly distinguished based on subsets defined by γδ TCR usage, which could affect the interpretation of results, since the anti-tumor properties of different subsets and even populations within the same subset vary with tumor context. To account for population variations going forward, researchers should proactively include γδ T cell subset analyses in their studies. Investigating the effector phenotypes of γδ T subsets offers important insight into their recruitment patterns to tumor sites (33, 42). While identifying the wide range of ligands recognized by γδ T cells continues to pose a challenge to researchers, of greater pertinence is the choice of a specific antigen or antigens that can be used either for ex vivo activation and expansion of γδ T cells or direct administration to expand the cells in vivo. This is exemplified by the use of BrHPP or zoledronate to expand Vγ9Vδ2 T cells. Furthermore, whether γδ T cells play tumor-suppressive or promoting roles in a particular cancer type is possibly influenced by the specific TME. This can be assessed in vitro by co-incubating γδ T cells with supernatants derived from the culture of specific cancer cell types (36, 42). Whether γδ T cells are associated with good or poor prognosis for the same cancer type, such as breast cancer, CRC or melanoma (Table 1) may be dependent on the stage of cancer (42). Clearly, identification of specific molecules secreted by cancer cells in the culture supernatant that impact the “fate commitment” of γδ T cells will shed light on possible mechanisms educating the pro- or anti-tumor behavior of these cells in a given TME. Further insights into the interaction between γδ T cell biology and the TME will inform strategies of employing γδ T cells as an effective oncotherapy (Figure 2, right).
3 Harnessing γδ T cells for anti-tumor immunotherapy
3.1 Lessons learnt from past clinical trials
Several γδ T cell immunotherapy clinical trials have been carried out. In Table 2, we focus on summarizing the completed and on-going γδ T cell immunotherapy clinical trials that specifically utilized direct γδ T cell administration to provide an overview of their status, phase of trial, types of cells administered, target cancer types, and their clinical outcomes. From the accumulating number of clinical trials, we have gained invaluable insight and herein discuss the important lessons we can learn from these trials. We also put forth several strategies to advance γδ T cell immunotherapy.
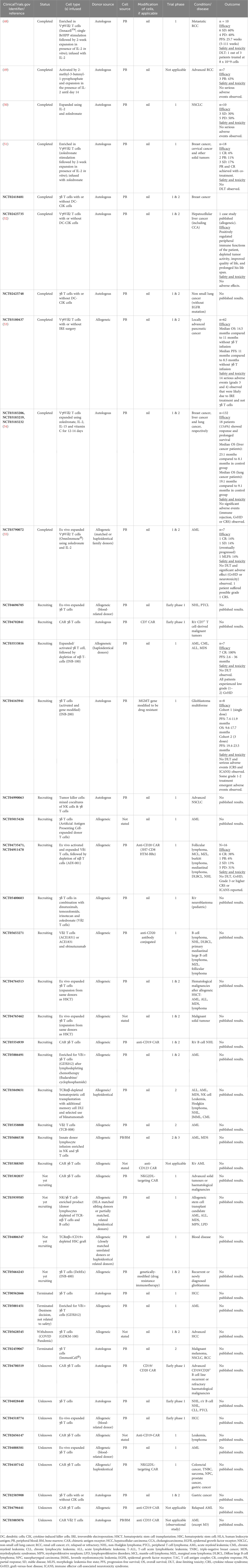
Table 2 Ongoing and past clinical trials involving direct cellular administration of unmodified and modified γδ T cells, including study outcome (if available).
Strategies to utilize γδ T cells for cancer immunotherapy are summarized in recent reviews (56–59). These include the activation or stimulation of endogenous γδ T cells via exogenous aminobisphosphonates and anti-CD3/anti-tumor antigen bispecific antibodies as well as ex vivo expansion of peripheral blood-derived γδ T cells (60). Despite their purported capability to target diverse tumor cell types, γδ T cells have performed poorly in clinical trials, yielding largely disappointing clinical outcomes exemplified by low objective tumor response rates and almost no complete responses, with the exception of IN8bio’s trial (NCT03533816) and Adicet Bio’s trial (NCT04735471) which reported 100% and 69% complete responses respectively (59) (Table 2; refer to Supplementary Table for additional fields of information). Long-term outcome data are currently limited as many of the clinical trials are still on-going and many of them are in the early phases, which focus on establishing safety profile and dose limiting toxicity. Nevertheless, γδ T cell therapy has shown to increase the overall survival and progression-free survival of patients in a limited number of studies (NCT03533816, NCT04165941, NCT03180437, NCT03183206, NCT03183219, NCT03183232) (48, 53, 54), with the longest survival outcomes observed in IN8Bio’s trial in which one patient had progression-free survival for at least 3 years. This is remarkable considering that patients treated in this trial had high-risk AML or failed multiple treatments before receiving γδ T cell therapy. While we can only speculate the reasons why these trials showed exceptional γδ T cell efficacy compared with the rest of the trials, we noted that the therapy targeted hematological malignancies for which patient outcomes are typically more favorable compared with those for solid tumors. In IN8bio’s trial, patients underwent haploidentical bone marrow (BM) transplantation followed by cyclophosphamide treatment prior to γδ T cell infusion. The regime preceding γδ T cell infusion could have synergized with the latter’s therapeutic effects. In Adicet Bio’s trial, γδ T cells were programmed with anti-CD20 CAR which likely increased their efficacy to recognize and kill B lymphoma cells. The company’s proprietary expansion process may also have enriched for the subset of cytotoxic Vδ1 T cells. Other factors to consider are discussed in the following subsections.
3.1.1 Factors affecting tumor-infiltration of γδ T cells must be considered
Even though γδ T cells are one of the major populations found in solid tumors (32), not many studies have extensively characterized their infiltration when human clinical trials are carried out. Of all the completed trials, only Nicol and colleagues reported the migration of γδ T cells after infusion. They observed that the adoptively transferred γδ T cells migrated rapidly to lungs within a few hours before travelling to the liver and spleen. However, only a small number of γδ T cells were found to traffick to tumor sites (51). More studies are needed to understand the infiltration capabilities of adoptively transferred γδ T cells in solid tumors. Knowledge on the phenotypes of γδ T cells that have successfully migrated to tumor sites will also shed light as to why patient outcomes are generally poor for solid tumors compared to hematological malignancies. One can then devise potential solutions to overcome some of the hurdles impeding solid tumor immunotherapy. For detailed discussion on tumor infiltrating γδ T cells and their clinical relevance in cancer patients, we refer readers to other review papers (61, 62). Even if γδ T cells manage to infiltrate solid tumors, another immediate hurdle that they must overcome is the hostile conditions they are subjected to within the TME.
3.1.2 The tumor microenvironment inhibits anti-tumor immune responses
γδ T cells are subjected to signals within the TME, which can drive their differentiation into different functional subsets (63). Cells in the TME comprise of immunosuppressive tumor associated macrophages, MDSCs, cancer-associated fibroblasts and tumor cells themselves, among others. These cells can secrete immunoinhibitory molecules, such as TGF-β (64), which in turn promote the pro-tumorigenic polarization of γδ T cells. In addition, γδ T cells can become exhausted and dysfunctional in the TME of certain tumors. For example, programmed cell death protein 1 (PD-1), LAG-3 and TIM3 were shown to be upregulated in γδ T cells infiltrating multiple myeloma, and together with the increased expression of the cognate ligands on tumor cells, result in their anergy (65, 66).
Cells in the TME can also directly inhibit the anti-tumor cytotoxicity of γδ T cells (67). It has been shown that PDA cells upregulate cyclooxygenase-2 (COX-2) expression in response to IFN-γ and TNF-α secreted by γδ T cells (68). COX-2 leads to an increase in PGE2 in tumor cells as a result of increased enzymatic action. As a consequence of PGE2 binding to their receptors on γδ T cells, TCR signaling is inhibited and this in turn causes the dampening of γδ T cell cytotoxic function. Elevated Cox-2 expression was also observed in breast cancer (69). Recently, it was demonstrated that IL-10 secreted by EBV-transformed lymphoblastoid B cell lines reduced the cytotoxicity of Vγ9Vδ2 T cells (70). In addition, Tregs have been shown to inhibit the proliferation of γδ T cells (71).
The anti-tumor activity of γδ T cells is also highly suppressed by tumor hypoxia in various cancers (72–74). Even if γδ T cells could infiltrate solid tumors, their cytotoxicity can be suppressed by hypoxic conditions in the TME due to apoptosis via PD-1 and reduced expression of NKG2D. In brain tumors, the use of metformin, a repurposed drug that has been shown to elicit an anti-tumor effect (75, 76), reduced hypoxia and rescued the anti-tumor effect of γδ T cells (72). In oral cancers, blockade of PD-1 or targeting hypoxia-inducible factor-1α could also help to overcome tumor hypoxia (73). On the other hand, in the case of breast cancer, cancer cells may also evade detection by γδ T cells by shedding MICA under hypoxia (74). Therefore, strategies for γδ T cells to prevail under TME conditions should be catered for specific tumor types. Taken together, more studies are required to characterize both γδ T and cancer cells, and their interactions in the TME.
3.1.3 Culture conditions during ex vivo expansion influences γδ T cell functionality
Besides understanding what happens in vivo, the process of ex vivo expansion can affect γδ T cell phenotypes and cytotoxicity. Despite the great success achieved by the two trials mentioned earlier in treating liquid tumors, we noted that the clinical outcomes in a study (NCT03790072) that also targeted liquid tumor pale in comparison, with a complete response rate of 14%. The media and/or expansion method employed could have affected the quality, quantity and ultimately the efficacy of γδ T cells produced. Xu et al. examined the effect of γδ T cells grown in the presence of different media supplements and infused into patients on the patients’ overall survival (54). Eighteen patients that were administered with Vγ9Vδ2 T cells grown in their newly formulated media supplemented with zoledronate, IL-2, IL-15 and vitamin C, were found to have better overall survival compared with patients that were administered with γδ T cells grown in media supplemented with zoledronate and IL-2. When expanded with the new formula, the authors obtained higher cell yield and observed less cell death corroborated by RNAseq results showing downregulation in expression of apoptosis-related genes. In addition, there was an increase in Vγ9Vδ2 T cells harboring terminally differentiated effector memory (CD45RA+CD27-) phenotype which were previously found to express homing receptors such as CCR5 and CXCR3 (77), and a decrease in cell populations with naïve (CD45RA+CD27+) and central memory (CD45RA-CD27+) phenotypes, although there were no significant changes in counterparts bearing effector memory (CD45RA-CD27-) phenotype. The cells also more highly expressed co-stimulatory molecules such as CD80, CD86 and MHC-II. Collectively, these data suggest that appropriate media supplements can prime γδ T cells to migrate to tumor sites and exert cytotoxic effects, thus leading to better clinical outcomes.
3.1.4 γδ T cell subtypes variably affect the clinical outcome
Another possible reason for the dismal failure is that a significant proportion of trials focused on harnessing Vγ9Vδ2 γδ T cells for therapy (Figure 3A) as they can be readily expanded ex vivo to large numbers using zoledronate and IL-2. However, Vγ9Vδ2 cells are naturally abundant in PB and do not typically home to tissues which may partially explain their limited cytotoxic capacity against solid tumors. Vδ1-enriched delta one T (DOT) cells (60), polyclonal γδ T cells comprising multiple subsets or other non-Vδ2 subsets (6) have been or could be explored as potential alternatives that demonstrate greater potency against such tumors. For example, Vγ4+ TCRs have been shown to bind butyrophilin like 3 expressed by gut epithelial cells and EPCR expressed mostly by endothelial cells (27, 78) to facilitate immunosurveillance of virus-infected and tumor cells. Such ligands are thought to mediate homing of γδ T cells to and their killing of tumors. Moreover, UCB-derived Vδ2- T cells were shown to be more cytotoxic than their Vδ2+ counterparts (8). Other tumor-associated ligands recognized by non-Vδ2 TCRs are summarized in a recent review by Dong R et al. (79) As such, the relative importance amongst the various γδ T cell subsets and which ones should be used in the application for γδ T cell therapy should be considered. In addition, incorporation of a step to specifically deplete pro-tumorigenic subsets prior infusion could improve clinical outcome.
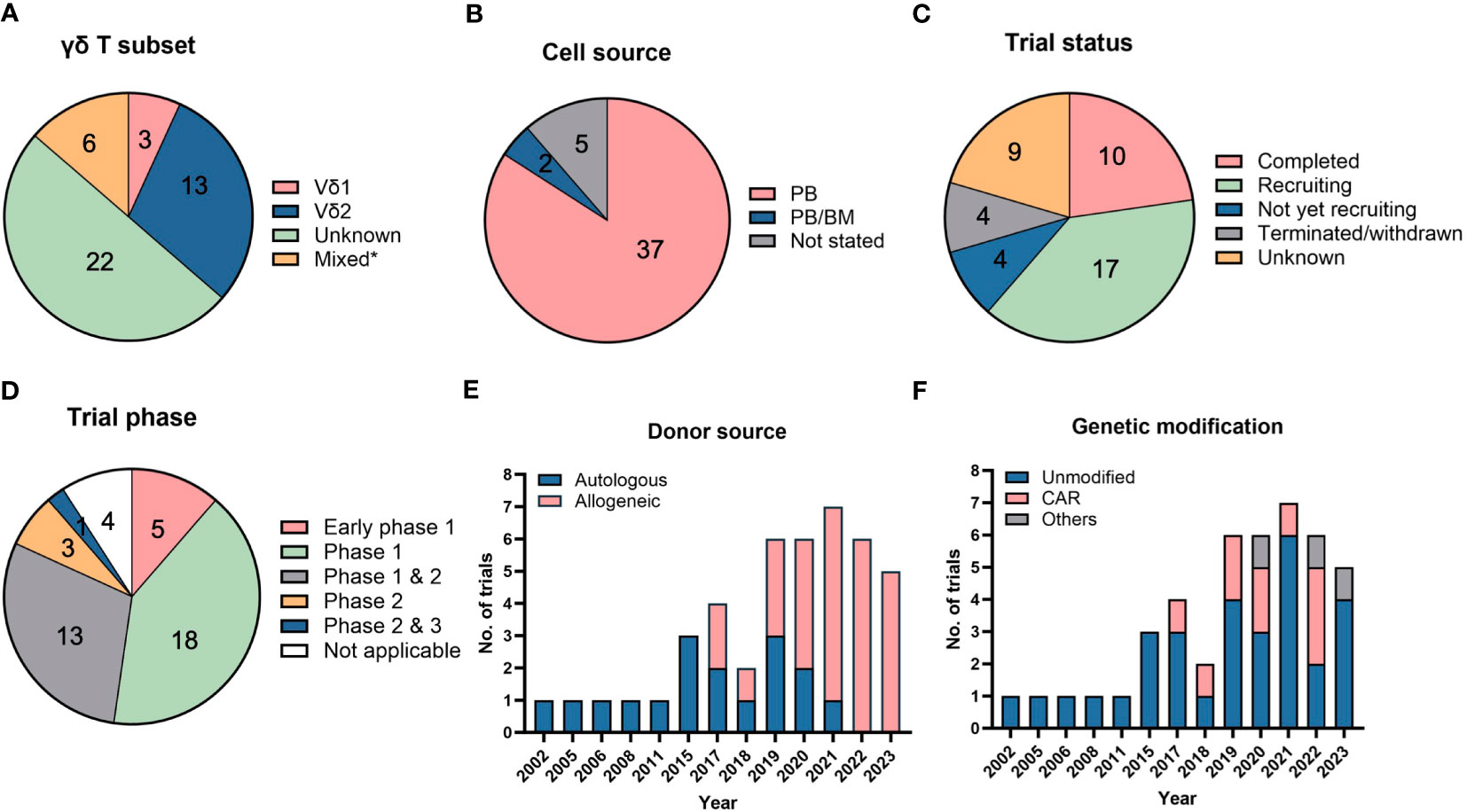
Figure 3 Clinical trials using direct cellular administration of γδ T cells. Pie charts showing the (A) γδ T subsets that were infused into patients, (B) cell sources from which γδ T cells were obtained, (C) trial status and (D) different clinical trial phases. *”Mixed” in (A) refers to the use of other cell types, namely natural killer (NK), dendritic cell-cytokine-induced killer (CIK) or CIK cells, that were infused together with γδ T cells. “Unknown” in (A) refers to trials in which details on γδ T subsets were not available. The category “not applicable” in (D) is used for trials without FDA-defined phases, according to clinicaltrials.gov, and includes observational studies. The number of trials in each category is listed within the pie charts in (A–D). Bar graphs depicting the (E) donor sources from which γδ T cells are derived and (F) types of modifications in γδ T cells. A total of 44 trials were analyzed in (A–F).
3.1.5 Cell source used for ex vivo expansion can influence the properties of γδ T cell product
Clinical trials typically rely on adult PB as a source to harvest and expand γδ T cells with a restricted TCR repertoire (Figure 3B). Expansion from UCB will generate “younger” cells equipped with a more polyclonal TCR repertoire able to recognize a broader diversity of tumor ligands but are not yet endowed with distinct homing properties characteristic of adult, tissue-resident γδ T cells (59). In-depth characterization of the functional profiles, such as cytokine secretion and varying TCR affinities towards different ligands, of polyclonal γδ T cells will be important to ascertain advantages of their therapeutic use.
3.1.6 γδ T cell therapy is safe, but its anti-tumor potency requires improvement
The clinical safety of unmodified γδ T cells has been confirmed largely by the paucity of serious adverse events following either dose escalation of aminobisphosphonates and IL-2 to stimulate their in vivo expansion in patients or ex vivo expansion and subsequent adoptive transfer to patients in multiple trials (80, 81). γδ T cell therapy is relatively safe and accompanied by low-grade adverse events such as fever, fatigue, or gastrointestinal disorder, some of which can self-resolve in a few days (49, 51). However, naturally occurring γδ T cells in most of these trials failed to promote substantial tumor regression and enforce remission, highlighting the need for targeted engineering to (1): instruct commitment of γδ T cells towards cytotoxic and not regulatory lineage and (2) restore their metabolic fitness compromised by the immunosuppressive TME.
Addressing the former challenge would entail utilizing culture conditions and specific antigens to expand and enrich γδ T cells with anti-tumor properties, i.e. cytotoxic T cells, while minimizing or depleting those with pro-tumorigenic properties, i.e. γδ Tregs. In addition, tackling the issues of T cell infiltration and immunosuppressive TME would require strategies such as rejuvenation of T cells via immune checkpoint inhibition. More details are described in section 3.3. Improving anti-tumor potency of γδ T cell therapy is a pressing issue because these relatively newer therapies will no doubt be continuously compared to currently approved CAR-T therapies for lymphoma and myeloma.
3.2 Clinical trials involving administration of unmodified or engineered γδ T cells
The strategy to engineer and expand γδ T cells in vitro followed by their in vivo infusion compared with combinatorial administration of stimulatory γδ T ligands and cytokines to selectively expand these cells in vivo likely enables more robust and precise improvement of γδ T anti-tumor efficacy. Here, we summarize in Table 2 clinical trials which implemented or are in process of implementing a regimen of ex vivo expansion followed by infusion of unmodified or CAR-modified γδ T cells into cancer patients, providing evidence that engineered γδ T cells are increasingly preferred to unmodified counterparts for tumor immunotherapy.
Out of the 44 clinical trials in Table 2, 10 are completed trials, all of which involved unmodified γδ T cells (Figure 3C). Seventeen clinical trials are currently recruiting patients. A broad range of cancers are targeted in these clinical trials with a trend towards trials targeting liquid or blood (24 trials) compared to solid malignancies (21 trials). Among blood cancers, AML is the most common cancer targeted whereas lung, liver and breast cancers are the most commonly targeted solid tumors.
Interestingly, we note that most of these trials utilized PB or BM as their cell sources and there are currently no trials utilizing γδ T cells expanded from UCB and induced pluripotent stem cells (iPSCs) (Figure 3B), although there is ongoing work in the field to generate γδ T cells from these sources (8, 82). iPSCs could potentially be an unlimited cell source for allogeneic treatment, which requires a large number of cells for scale-up. iPSCs-derived γδ T cells have been successfully generated and shown to illicit cytotoxicity effect on several cancer cell lines (83).
While majority (91%) of the clinical trials are in their early phases (Phase 1 or 2) (Figure 3D), an allogeneic treatment study using TCRαβ/CD19-depleted innate donor lymphocyte infusion has been approved for Phase 2 & 3 clinical trial in 2023 (NCT05686538) (Table 2). Besides the clinically proven advantageous safety profile of γδ T cells in not causing GvHD, another reason favoring adoption of allogeneic therapy is the difficulty in recovering and expanding sufficient numbers of autologous γδ T cells of high quality from diseased patients. This is exemplified by trials conducted by Fuda Cancer Hospital, China (NCT03183206, NCT03183219, NCT03183232) which initially planned to use patients’ own PB-derived γδ T cells but the investigators encountered challenges in expanding the cells, prompting them to source cells from allogeneic donors. Similar manufacturing obstacles in some patients were recorded by Vydra et al. when they conducted a trial using autologous γδ T cells (55). In recent years, the number of clinical trials for the allogeneic use of γδ T cells has surpassed those for autologous treatments (Figure 3E).
To further improve the specificity and efficacy of γδ T cells for targeting tumors, several strategies have been explored by others, which are discussed in Section 3.3. Equipping γδ T cells with CAR is one of the earliest and most common strategies, given the success witnessed in CAR-modified αβ T cells. Anti-CD7, anti-CD19/20, anti-CD33, and anti-CD123 CAR have been designed to target liquid cancers, while NKG2DL-targeting CAR is constructed for targeting solid tumors. Since 2017, the number of trials conducted with genetically modified γδ T cells has been increasing (Figure 3F). As of 2023, there are 10 trials that involved CAR-modified γδ T cells. Besides engineering CAR, γδ T cells have also been modified to be resistant to chemotherapy drug, temozolomide, which is useful to treat glioblastoma using combination therapy (NCT04165941, NCT05664243).
3.3 Strategies to augment anti-tumor cytotoxicity of γδ T cells
In this section, we describe how various strategies have been applied or can be adapted to improve anti-tumor efficacy of γδ T cells. Such strategies include those that were used to modify conventional αβ T cells or can complement their therapy via non-genetic engineering approaches. In vitro and pre-clinical data, where applicable, are discussed.
3.3.1 Non-genetic engineering approaches
As a first illustration, a bispecific T cell engager (BiTE) antibody construct, AMG 330, administered to leukemic patients yielded encouraging safety and anti-leukemic outcomes (84). A BiTE is a synthetic fusion protein which is designed based on linking the antibody-binding domains of two antibodies. In this example, AMG 330 simultaneously binds CD33 antigen on leukemic blasts and CD3 co-receptor on T cells, placing T cells in close proximity to CD33+ leukemic cells and ultimately mediating destruction of the latter by the former cells. In similar fashion, a bispecific tribody which recognizes Vγ9 on γδ T cells and ERBB2 (HER2/neu) on pancreatic cancer cells enhanced γδ T cell cytotoxicity against PDA in vitro and in vivo (85). Recombinant immunoligands comprising an anti-CD20 single chain variable fragment (scFv) linked to a NKG2D ligand, MICA or ULBP2, activated specific elimination of CD20+ but not CD20- lymphoma cells by ex vivo expanded Vδ1 and Vδ2 γδ T cells, a therapeutic result which could be further augmented by concurrent agonistic stimulation of the cells with BrHPP (86). Hence, exogenous application of BiTE or similarly designed molecules can be employed in combination with γδ T administration.
Another example obviating non-genetic modification involves programming γδ T cells with various combinations of cytokines to enhance their tumor killing capacity (87). Schilbach and colleagues demonstrated that the combination of IL-2, IL-12 and IL-18 synergize to significantly induce both IFN-γ and TNF-α secretion in the presence of TCR stimulus (88). The increase in TNF-α was observed even in the absence of a TCR signal. The authors also showed that in IL-2/IL-12/IL-18 stimulated γδ T cells, granzyme B and perforin protein expression was upregulated to a similar extent compared to TCR stimulation. Interestingly, the expression of FasL was increased under conditions of IL-2/IL-12/IL-18 stimulation, but not TCR stimulation. Together, these mechanisms mediate the increased anti-tumor killing capacity of cancer cells by the stimulated γδ T cells. More recently, Liu and colleagues showed that γδ T cells pre-treated with a combination of IL-12, IL-18 and IL-21 led to their enhanced inhibition of tumor growth not only in vitro, but also in vivo after adoptive transfer (89). They showed that such a pre-activation cocktail promoted the proliferation of γδ T cells and their secretion of IFN-γ and TNF-α, which can promote the anti-tumor function of endogeneous CD8+ T cells in vivo. Therefore, in the cytokine pre-treatment strategy, γδ T cells can be stimulated ex vivo by cytokine combinations to boost their anti-tumor activity. Potent cytokine-activated Vδ1+ DOT cells have been generated and appears promising for clinical use (60).
Based on newly acquired knowledge on the mechanism of Vγ9Vδ2 T cell activation, agonistic antibodies directed against BTN3A1 and BTN2A1 (90, 91) can be used to heighten sensitivity of tumor cells to γδ T cell killing and offers a promising therapeutic strategy to enhance γδ T cell cytotoxicity. An anti-BTN3A monoclonal antibody (ICT01) is currently in phase 1/2a clinical trial (NCT04243499).
Other possible strategies are targeted at overcoming the immunosuppressive effects of the TME on γδ T cells. For instance, CD137 costimulation using a recombinant CD137L protein was found to reduce the expression of IL-10 receptor, IL-10R1, thereby reducing the sensitivity of the γδ T cells to the immunosuppressive effects of endogenous IL-10 (70).
Interestingly, it was shown that acute systemic β-adrenergic receptor activation was largely responsible for the exercise-augmented mobilization, ex vivo expansion and anti-tumor activity of Vγ9Vδ2 T cells from healthy donors (92). Administration of an antagonist inhibiting both β1- and β2-adrenergic receptors abrogated these exercise-induced effects. This finding suggest that β-adrenergic receptors are potential targets to improve the potency of ex vivo expanded γδ T cells.
Novel methods of γδ T cell delivery other than the traditional intravenous infusion route could be designed to improve their efficacy in solid tumors. When CAR-T cells were delivered to tumor sites directly using biopolymer scaffolds, they were able to migrate to and kill tumor cells more effectively as compared to systemic delivery method (93). Treatment of glioblastoma has been notoriously challenging due to the difficulty in reaching the blood-brain barrier by immune cells. This could be overcome by stereotactic injection of γδ T cells directly into the brain (94, 95). These direct intratumoral delivery methods could be applied to treat solid tumors that are known to be difficult to infiltrate by immune cells.
Beyond delivering γδ T cells per se, it is noteworthy that newer strategies using cell-free extracellular vesicles, such as exosomes, confer a safety advantage over cell-based therapies and have shown promising anti-tumor efficacy. The small size (20-200 nm) of exosomes renders easy infiltration into solid tumor sites and they are resistant to the immunosuppressive TME. Such γδ T cell-derived vesicles were shown to control tumor progression of and elicit anti-tumor responses against Epstein-Barr virus-associated B-cell lymphoma, gastric carcinoma and nasopharyngeal carcinoma (96, 97). These exosomes derived from activated Vδ2 T cells were not only positive for NKG2D, which is responsible for their uptake by tumor cells, but were also positive for FasL and TRAIL, which facilitate their death-inducing properties. More recently, γδ T extracellular vesicles were used as carriers to deliver tumor-associated antigens, and the extracellular vesicles-based cancer vaccines were successful in controlling tumors in vivo (98).
3.3.2 Genetic engineering approaches
CAR-modified γδ T cells were first explored as effector cells of tumor-directed immunity in a 2004 study which demonstrated these cells efficiently recognized CAR antigen-expressing neuroblastoma and malignant B cell tumour cells as assessed by their upregulation of CD69 and secretion of IFN-α (99). Consistent with operating multiple mechanisms of cytotoxicity, CD19 CAR γδ T cells were found to exert not only CAR-directed activity against CD19+ leukemia cells but also CAR-independent activity against CD19- leukemia cells or cells which have lost expression of CD19 antigen (100), highlighting the advantage of using γδ T vis-à-vis αβ T cells. Although arming γδ T cells with CARs endows tumor specificity, CAR signaling components can be optimized to increase γδ T efficacy against hematological malignancies and solid tumors. Firstly, transduction with second-generation CARs bearing CD3ζ activation domain is known to elicit tonic signaling and exhaustion marked by PD-1 and TIM-3 upregulation in αβ and γδ T cells. Modifying the endodomain of the chimeric co-stimulatory receptors (CCRs) that replace CD3ζ with DAP10 domain in γδ T cells led to effective activation of cytotoxic responses in the presence of CCR-specific stimuli or cognate tumor cells (101). Vδ1 T cells that were genetically modified to express 4-1BB/CD3 CAR targeting the oncofetal antigen glypican-3 and a constitutively secreted form of IL-15 exhibited superior proliferation and anti-tumor activity against hepatocellular carcinoma (HCC) lines and HCC subcutaneously engrafted in immunodeficient mice compared with their non-cytokine secreting counterparts (102). Such armored CAR design which allows release of transgenic cytokine(s) of interest upon CAR signaling had previously been employed successfully in αβ T cells to counteract the inhibitory cytokine milieu of and recruit innate effector cells into the TME (103, 104). Therefore, continued innovation of CAR designs is warranted.
Beyond CAR, introduction of a tumor-specific αβ TCR and the corresponding CD4 or CD8 co-receptor for recognition of HLA-restricted tumor antigen in γδ T cells led to their pronounced cytokine secretion and cytolytic effects against leukemia (105). One obvious drawback using αβ TCR is the requirement for additional CD4 and CD8 co-receptors. The advent of CRISPR/Cas technology has opened new avenues for genetic, including TCR, modification of γδ T cells. Such targeted TCR editing enables controlled replacement of the endogenous TCR with the transgene, thereby allowing for transgene TCR to be expressed at homogeneous, physiological levels on the T cells, and consequently less functional variability compared to virus-mediated transgene integration (106). In this respect, Immatics, a clinical-stage biopharmaceutical company, has entered into a research collaboration and licensing agreement with Editas Medicine, a genome editing company, to advance off-the-shelf adoptive γδ T cell therapy platform. Reciprocally, Vγ9Vδ2 TCR was shown to effectively reprogram both CD4+ and CD8+ αβ T cells to kill a broad diversity of cancer but not normal cells, and substantially diminished but did not completely abrogate alloreactivity (107). Recently, a clinical stage immune-oncology company, Triumvira Immunologics, developed proprietary T cell Antigen Couplers (TACs) for incorporation in T cells (108). TAC consists of 3 components: a tumor antigen binding domain, a CD3 binding domain which interacts with and co-opts the native TCR and a CD4 co-receptor transmembrane and intracellular domain. When bound to its target antigen, TAC triggers the native TCR signaling cascade by recruiting downstream kinases and thereby activating T cell killing in an HLA-independent manner. TAC-modified αβ T cells are currently undergoing Phase 1 & 2 clinical trials for autologous treatment of HER2+ solid tumors (NCT04727151). HER2-targeting TAC-modified γδ T cells are similarly being developed and preclinically evaluated. TAC γδ T cells were observed to exhibit cytotoxicity against tumor xenografts that are resistant to unmodified γδ T cells (109), suggesting modifications of γδ T cells need not be restricted to CAR. However, the safety of their use requires further evaluation.
Despite the established clinical safety profile of unmodified γδ T cells, the enhanced anti-tumor efficacy achieved by modification of γδ T cells may correspondingly increase their off-tumor, on-target toxicity, resulting in undesirable side effects. To address this potential challenge, non-signaling CARs (NSCARs) lacking signaling/activation domains but retaining tumor-specific targeting capability were expressed in γδ T cells. CD5- and CD19-targeting NSCARs significantly elevated the intrinsic, HLA-independent cytotoxicity of γδ T cells against T cell and B cell ALL but expectedly did not enhance the antigen-specific cytotoxicity of αβ T cells (110). An alternative T cell therapy platform involving the concept of a synthetic agonistic receptor (SAR) originally applied in αβ can be potentially extended to γδ T cells. SAR-transduced T cells are directed by an engineered tandem scFv construct (taFv) to antigen-expressing tumor cells in a manner similar to BiTEs (111). The taFv construct comprises two scFvs, one binding the artificial antigen receptor composing an extracellular EGFRvIII domain fused to intracellular T cell-activating domains transduced in T cells and another binding a specific antigen on the surface of cancer cells, thus juxtaposing T and cancer cells. Unlike the BiTE approach which activates pan-T cells, this system specifically activates SAR-transduced T cells and is able to terminate SAR T cells via antibodies clinically approved by FDA should adverse toxicity events arise.
Strategies to improve anti-tumor cytotoxicity of γδ T cells need not be confined to improving recognition of tumor antigens. Other options include boosting the infiltration of γδ T cells into solid tumor by expressing surface proteins that can aid its migration through the extracellular matrix (ECM) surrounding the tumor whilst harnessing the diverse HLA-independent receptors of γδ T cells to target tumors, particularly those which have escaped antigen targeting. When modified to express matrix metalloprotease 14 (MMP14) enzyme that can digest the ECM, γδ T cells were able to more efficiently migrate in the tumor milieu (112). However, despite being able to kill TNBC cells effectively in vitro and showing an improved migration profile, MMP14-engineered γδ T cells could not eliminate TNBC tumors in vivo due to down regulation of γδ T cell ligands Fas, MICB and intercellular adhesion molecule 1 (ICAM-1) on breast cancer stem cells. Pre-treatment using zoledronate recovered some cancer stem cell killing by γδ T cells, suggesting that prior activation of γδ T cells may be necessary for TNBC eradication.
3.3.3 Combination therapies
In addition to the aforementioned strategies, supplementing γδ T cell therapy with immune checkpoint inhibitors, such as those targeting PD-1/PD-L1 and CTLA4 pathways (65), or novel cancer stem cell-targeting strategies may further bolster the effectiveness and durability of engineered γδ T anti-tumor responses. Rossi et al. demonstrated that γδ T cells infiltrating follicular lymphoma highly express PD-1 and anti-PD1 blockade consequently increased their cytotoxicity (113).
Another combination treatment that has shown better efficacy is the treatment of locally advanced pancreatic cancer using irreversible electroporation (IRE) with γδ T cells infusion (NCT03180437). The median overall survival of patients treated with IRE alone was 11 months but with γδ T infusions, the overall survival increased to 14.5 months, showing the potential of combination treatment in prolonging patient’s life (53).
Taking advantage of the cross-talk between γδ T cells and other immune cells in the TME, anti-tumor responses in γδ T cell therapies could be further enhanced by boosting the anti-tumor cytotoxicity mediated by other immune cells. For instance, CD137 (4-1BB) co-stimulation with recombinant human CD137L has been shown to increase NKG2D expression on NK cells, which is directly responsible for tumor cell killing (114). An added mechanism of action by these NK cells is the killing of dendritic cells which would otherwise promote inflammation and tumor growth (115).
Other plausible therapies involve targeting the tumor cells within TME, some of which have demonstrated promising preclinical results. These include the use of COX inhibitors which ameliorate the effects of the immunosuppressive TME (116) and celastrol which upregulates death receptor expression on tumor cells (117).
Similarly, patients may also develop resistance with other treatments that could be rescued by co-treatment with γδ T cells. For instance, tyrosine kinase inhibitors (TKIs) have been successful in treating various cancers such as advanced or metastatic renal cell carcinoma, non-small-cell lung cancer and HCC (118–120). However, many patients eventually develop resistance against treatment with TKIs (121). When used alone, γδ T cell therapy also showed some efficacy against these cancers (as summarized in Table 2). Therefore, these two complementary therapies potentially add to or synergize with each other in treating cancer patients (48).
4 Concluding remarks
γδ T cells are a highly promising immune subset that can be harnessed for “off-the-shelf”, allogeneic immunotherapy (Figure 1) and additionally engineered to amplify their anti-tumor efficacy. There exist several hurdles which need to be overcome in order that γδ T cells can be employed as an effective oncotherapy (Figure 2). Firstly, the challenge of translating the preclinical finds into clinical trials require extensive knowledge on γδ T cell infiltration and their plasticity within the TME. Learning how to effectively deliver γδ T cells to solid tumor sites by exploiting context-dependent mechanisms which drive γδ T cells to adopt anti- rather than pro-tumor function is absolutely crucial. Secondly, elucidating hitherto unknown ligands that activate and expand specific populations, as defined by TCR usage, of γδ T cells which play important roles in anti-tumor immunity will help to activate γδ T cell in the settings of in vivo administration or ex vivo expansion. Thirdly, culture conditions and cell source undeniably moulds the γδ T cell final product during the manufacturing process. Identifying the most optimal parameters to adopt in γδ T cell expansion ex vivo should be incorporated as part of γδ T cell therapy process development. The relative importance amongst the various γδ T cell subsets and specific depletion of pro-tumorigenic subsets prior infusion should also be considered in the application for γδ T cell therapy. Finally, innovations in modular engineering of γδ T cells and combination strategies will be crucial in improving their in vivo anti-tumor cytotoxicity and persistence to prevent tumor relapse whilst minimizing likelihood of detrimental alloreactive responses.
Author contributions
CW: Conceptualization, Data curation, Investigation, Project administration, Visualization, Writing – original draft, Writing – review & editing. PL: Data curation, Investigation, Visualization, Writing – original draft, Writing – review & editing. AT: Conceptualization, Supervision, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Medical Research Council (NMRC) Open Fund Individual Research Grant (OF-IRG, ID OFIRG21nov-0025) awarded to AH-MT and the core funds of Bioprocessing Technology Institute, Agency for Science, Technology and Research, Singapore.
Acknowledgments
We thank members of the laboratory for insightful discussions. Figures 1, 2 were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1282758/full#supplementary-material
Abbreviations
CAR, chimeric antigen receptor; HLA, human leukocyte antigen; GvHD, graft-versus-host disease; UCB, umbilical cord blood; PB, peripheral blood; BTN, butyrophilins; MICA, MHC class I-related chain A; MICB, MHC class I-related chain B; IFN, interferon; TCR, T cell receptor; TME, tumor microenvironment; CRS, cytokine release syndrome; TNF, tumor necrosis factor; BrHPP, bromohydrin pyrophosphate; EphA2, ephrin receptor A2; EPCR, endothelial protein C receptor; ULBP, UL16-binding protein; IL, interleukin; Treg, regulatory T cell; TNBC, triple-negative breast cancer; CRC, colorectal cancer; TEM, effector memory T cell; TEMRA, terminally differentiated T cell; Tnaïve, naïve T cell; TCM, central memory T cell; AML, acute myeloid leukemia; ALL, acute lymphoblastic leukemia; TIL, tumor-infiltrating lymphocyte; PDA, pancreatic ductal adenocarcinoma; MDSC, myeloid-derived suppressor cell; BM, bone marrow; PD-1, programmed cell death protein 1; COX-2, cyclooxygenase-2; DOT, Vδ1-enriched delta one T cell; iPSCs, induced pluripotent stem cells; BiTE, bispecific T cell engager; scFv, single chain variable fragment; CCR, chimeric co-stimulatory receptors; HCC, hepatocellular carcinoma; TAC, T cell Antigen Coupler; NSCAR, non-signaling CAR; SAR, synthetic agonistic receptor; taFv, tandem scFv; ECM, extracellular matrix; MMP14, matrix metalloprotease 14; IRE, irreversible electroporation; TKIs, tyrosine kinase inhibitors.
References
1. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer (2022) 21(1):1–13. doi: 10.1186/s12943-022-01559-z
2. Liu Z, Shi M, Ren Y, Xu H, Weng S, Ning W, et al. Recent advances and applications of CRISPR-Cas9 in cancer immunotherapy. Mol Cancer (2023) 22(1):1–19. doi: 10.1186/s12943-023-01738-6
3. Li C, Mei H, Hu Y. Applications and explorations of CRISPR/Cas9 in CAR T-cell therapy. Brief Funct Genomics (2020) 19(3):175–82. doi: 10.1093/bfgp/elz042
4. Wang CQ, Tan AHM. Driving CARs on the biomanufacturing road to clinical success. Cell Gene Ther Insights (2021) 7(11):1613.
5. Chen YJ, Abila B, Mostafa Kamel Y. CAR-T: what is next? Cancers (Basel) (2023) 15(3):663. doi: 10.3390/cancers15030663
6. Deniger DC, Maiti SN, Mi T, Switzer KC, Ramachandran V, Hurton LV, et al. Activating and propagating polyclonal gamma delta T cells with broad specificity for Malignancies. Clin Cancer Res (2014) 20(22):5708–19. doi: 10.1158/1078-0432.CCR-13-3451
7. Placido R, Auricchio G, Gabriele I, Galli E, Brunetti E, Colizzi V, et al. Characterization of the immune response of human cord-blood derived γδ T cells to stimulation with aminobisphosphonate compounds. Int J Immunopathol Pharmacol (2011) 24(1):101–10. doi: 10.1177/039463201102400112
8. Ng JWK, Tan KW, Guo DY, Lai JJH, Fan X, Poon Z, et al. Cord blood–derived Vδ2+ and Vδ2– T cells acquire differential cell state compositions upon in vitro expansion. Sci Adv (2023) 9(24):eadf3120. doi: 10.1126/sciadv.adf3120
9. Musha N, Yoshida Y, Sugahara S, Yamagiwa S, Koya T, Watanabe H, et al. Expansion of CD56+ NK T and γδ T cells from cord blood of human neonates. Clin Exp Immunol (2001) 113(2):220–8. doi: 10.1046/j.1365-2249.1998.00645.x
10. Cherry ABC, Gherardin NA, Sikder HI. Intracellular radar: Understanding γδ T cell immune surveillance and implications for clinical strategies in oncology. Front Oncol (2022) 12:1011081. doi: 10.3389/fonc.2022.1011081
11. Zou C, Zhao P, Xiao Z, Han X, Fu F, Fu L, et al. γδ T cells in cancer immunotherapy. Oncotarget (2016) 8(5):8900–9. doi: 10.18632/oncotarget.13051
12. Chan KF, Duarte JDG, Ostrouska S, Behren A. γδ T cells in the tumor microenvironment-interactions with other immune cells. Front Immunol (2022) 13:894315. doi: 10.3389/fimmu.2022.894315
13. Yui MA, Sharp LL, Havran WL, Rothenberg EV. Preferential activation of an IL-2 regulatory sequence transgene in TCRγδ and NKT cells: subset-specific differences in IL-2 regulation. J Immunol (2004) 172(8):4691–9. doi: 10.4049/jimmunol.172.8.4691
14. Harrer DC, Simon B, Fujii SI, Shimizu K, Uslu U, Schuler G, et al. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: A safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer (2017) 17(1):1–17. doi: 10.1186/s12885-017-3539-3
15. Wallet MA, Nishimura T, Del Casale C, Lebid A, Salantes B, Santostefano K, et al. Induced pluripotent stem cell-derived gamma delta CAR-T cells for cancer immunotherapy. Blood (2021) 138(Supplement 1):2771. doi: 10.1182/blood-2021-149095
16. Alexander AAZ, Maniar A, Cummings JS, Hebbeler AM, Schulze DH, Gastman BR, et al. Isopentenyl pyrophosphate–activated CD56+ γδ T lymphocytes display potent antitumor activity toward human squamous cell carcinoma. Clin Cancer Res (2008) 14(13):4232–40. doi: 10.1158/1078-0432.CCR-07-4912
17. Donia M, Ellebaek E, Andersen MH, Straten PT, Svane IM. Analysis of Vδ1 T cells in clinical grade melanoma-infiltrating lymphocytes. Oncoimmunology (2012) 1(8):1297. doi: 10.4161/onci.21659
18. Cazzetta V, Bruni E, Terzoli S, Carenza C, Franzese S, Piazza R, et al. NKG2A expression identifies a subset of human Vδ2 T cells exerting the highest antitumor effector functions. Cell Rep (2021) 37(3):109871. doi: 10.1016/j.celrep.2021.109871
19. Kisielow J, Kopf M, Karjalainen K. SCART scavenger receptors identify a novel subset of adult γδ T cells. J Immunol (2008) 181(3):1710–6. doi: 10.4049/jimmunol.181.3.1710
20. Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, et al. CD27 is a thymic determinant of the balance between interferon-γ- and interleukin 17–producing γδ T cell subsets. Nat Immunol (2009) 10(4):427–36. doi: 10.1038/ni.1717
21. Provine NM, Binder B, FitzPatrick MEB, Schuch A, Garner LC, Williamson KD, et al. Unique and common features of innate-like human Vδ2+ γδT cells and mucosal-associated invariant T cells. Front Immunol (2018) 9:756. doi: 10.3389/fimmu.2018.00756
22. Hayday AC. γδ T cells and the lymphoid stress-surveillance response. Immunity (2009) 31(2):184–96. doi: 10.1016/j.immuni.2009.08.006
23. Herrmann T, Karunakaran MM. Butyrophilins: γδ T cell receptor ligands, immunomodulators and more. Front Immunol (2022) 13:876493. doi: 10.3389/fimmu.2022.876493
24. Harly C, Joyce SP, Domblides C, Bachelet T, Pitard V, Mannat C, et al. Human γδ T cell sensing of AMPK-dependent metabolic tumor reprogramming through TCR recognition of EphA2. Sci Immunol (2021) 6(61):9010. doi: 10.1126/sciimmunol.aba9010
25. Le Nours J, Gherardin NA, Ramarathinam SH, Awad W, Wiede F, Gully BS, et al. A class of gd T cell receptors recognize the underside of the antigen-presenting molecule MR1. Sci (1979) (2019) 366(6472):1522–7. doi: 10.1126/science.aav3900
26. Marlin R, Pappalardo A, Kaminski H, Willcox CR, Pitard V, Netzer S, et al. Sensing of cell stress by human γδ TCR-dependent recognition of annexin A2. Proc Natl Acad Sci USA (2017) 114(12):3163–8. doi: 10.1073/pnas.1621052114
27. Willcox CR, Pitard V, Netzer S, Couzi L, Salim M, Silberzahn T, et al. Cytomegalovirus and tumor stress surveillance by binding of a human γδ T cell antigen receptor to endothelial protein C receptor. Nat Immunol (2012) 13(9):872–9. doi: 10.1038/ni.2394
28. Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity (2014) 40(5):785–800. doi: 10.1016/j.immuni.2014.03.013
29. Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, et al. γδ T cells support pancreatic oncogenesis by restraining αβ T cell activation. Cell (2016) 166(6):1485–99. doi: 10.1016/j.cell.2016.07.046
30. Wesch D, Peters C, Siegers GM. Human gamma delta T regulatory cells in cancer: Fact or fiction? Front Immunol (2014) 5:120764. doi: 10.3389/fimmu.2014.00598
31. Jensen KDC, Su X, Shin S, Li L, Youssef S, Yamasaki S, et al. Thymic selection determines γδ T cell effector fate: antigen-naive cells make interleukin-17 and antigen-experienced cells make interferon γ. Immunity (2008) 29(1):90–100. doi: 10.1016/j.immuni.2008.04.022
32. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med (2015) 21(8):938–45. doi: 10.1038/nm.3909
33. Cordova A, Toia F, la Mendola C, Orlando V, Meraviglia S, Rinaldi G, et al. Characterization of human γδ T lymphocytes infiltrating primary Malignant melanomas. PloS One (2012) 7(11):e49878. doi: 10.1371/journal.pone.0049878
34. Wang J, Lin C, Li H, Li R, Wu Y, Liu H, et al. Tumor-infiltrating γδT cells predict prognosis and adjuvant chemotherapeutic benefit in patients with gastric cancer. Oncoimmunology (2017) 6(11):e1353858. doi: 10.1080/2162402X.2017.1353858
35. Janssen A, Hidalgo JV, Beringer DX, Van Dooremalen S, Fernando F, Van Diest E, et al. γδ T-cell receptors derived from breast cancer-infiltrating T lymphocytes mediate antitumor reactivity. Cancer Immunol Res (2020) 8(4):530–43. doi: 10.1158/2326-6066.CIR-19-0513
36. Meraviglia S, Lo Presti E, Tosolini M, La Mendola C, Orlando V, Todaro M, et al. Distinctive features of tumor-infiltrating γδ T lymphocytes in human colorectal cancer. Oncoimmunology (2017) 6(10):e1347742. doi: 10.1080/2162402X.2017.1347742
37. Dieli F, Vermijlen D, Fulfaro F, Caccamo N, Meraviglia S, Cicero G, et al. Targeting human γδ T cells with zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res (2007) 67(15):7450–7. doi: 10.1158/0008-5472.CAN-07-0199
38. Godder KT, Henslee-Downey PJ, Mehta J, Park BS, Chiang KY, Abhyankar S, et al. Long term disease-free survival in acute leukemia patients recovering with increased γδ T cells after partially mismatched related donor bone marrow transplantation. Bone Marrow Transplant (2007) 39(12):751–7. doi: 10.1038/sj.bmt.1705650
39. Wilhelm M, Kunzmann V, Eckstein S, Reimer P, Weissinger F, Ruediger T, et al. γδ T cells for immune therapy of patients with lymphoid Malignancies. Blood (2003) 102(1):200–6. doi: 10.1182/blood-2002-12-3665
40. Poggi A, Venturino C, Catellani S, Clavio M, Miglino M, Gobbi M, et al. Vδ1 T lymphocytes from B-CLL patients recognize ULBP3 expressed on leukemic B cells and up-regulated by trans-retinoic acid. Cancer Res (2004) 64(24):9172–9. doi: 10.1158/0008-5472.CAN-04-2417
41. Peng G, Wang HY, Peng W, Kiniwa Y, Seo KH, Wang RF. Tumor-infiltrating γδ T cells suppress T and dendritic cell function via mechanisms controlled by a unique toll-like receptor signaling pathway. Immunity (2007) 27(2):334–48. doi: 10.1016/j.immuni.2007.05.020
42. Lo Presti E, Toia F, Oieni S, Buccheri S, Turdo A, Mangiapane LR, et al. Squamous cell tumors recruit γδ T cells producing either IL17 or IFNγ Depending on the tumor stage. Cancer Immunol Res (2017) 5(5):397–407. doi: 10.1158/2326-6066.CIR-16-0348
43. Wistuba-Hamprecht K, Di Benedetto S, Schilling B, Sucker A, SChadendorf D, Garbe C, et al. Phenotypic characterization and prognostic impact of circulating γδ and αβ T-cells in metastatic Malignant melanoma. Int J Cancer (2016) 138(3):698–704. doi: 10.1002/ijc.29818
44. Wu Y, Kyle-Cezar F, Woolf RT, Naceur-Lombardelli C, Owen J, Biswas D, et al. An innate-like Vδ1+ γδ T cell compartment in the human breast is associated with remission in triple-negative breast cancer. Sci Transl Med (2019) 11(513):eaax9364. doi: 10.1126/scitranslmed.aax9364
45. Boissière-michot F, Chabab G, Mollevi C, Guiu S, Lopez-crapez E, Ramos J, et al. Clinicopathological correlates of γδ T cell infiltration in triple-negative breast cancer. Cancers (Basel) (2021) 13(4):1–20. doi: 10.3390/cancers13040765
46. Ni C, Fang QQ, Chen WZ, Jiang JX, Jiang Z, Ye J, et al. Breast cancer-derived exosomes transmit lncRNA SNHG16 to induce CD73+γδ1 Treg cells. Signal Transduct Target Ther (2020) 5(1):1–14. doi: 10.1038/s41392-020-0129-7
47. Peters C, Kabelitz D, Wesch D. Regulatory functions of γδ T cells. Cell Mol Life Sci (2018) 75(12):2125–35. doi: 10.1007/s00018-018-2788-x
48. Bennouna J, Bompas E, Neidhardt EM, Rolland F, Philip I, Galéa C, et al. Phase-I study of Innacell gammadelta, an autologous cell-therapy product highly enriched in gamma9delta2 T lymphocytes, in combination with IL-2, in patients with metastatic renal cell carcinoma. Cancer Immunol Immunother (2008) 57(11):1599–609. doi: 10.1007/s00262-008-0491-8
49. Kobayashi H, Tanaka Y, Yagi J, Osaka Y, Nakazawa H, Uchiyama T, et al. Safety profile and anti-tumor effects of adoptive immunotherapy using gamma-delta T cells against advanced renal cell carcinoma: A pilot study. Cancer Immunol Immunother (2007) 56(4):469–76. doi: 10.1007/s00262-006-0199-6
50. Nakajima J, Murakawa T, Fukami T, Goto S, Kaneko T, Yoshida Y, et al. A phase I study of adoptive immunotherapy for recurrent non-small-cell lung cancer patients with autologous gammadelta T cells. Eur J Cardio-Thoracic Surgery (2010) 37(5):1191–7. doi: 10.1016/j.ejcts.2009.11.051
51. Nicol AJ, Tokuyama H, Mattarollo SR, Hagi T, Suzuki K, Yokokawa K, et al. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br J Cancer (2011) 105(6):778–86. doi: 10.1038/bjc.2011.293
52. Alnaggar M, Xu Y, Li J, He J, Chen J, Li M, et al. Allogenic Vγ9Vδ2 T cell as new potential immunotherapy drug for solid tumor: a case study for cholangiocarcinoma. J Immunother Cancer (2019) 7(1):36. doi: 10.1186/s40425-019-0501-8
53. Lin M, Zhang X, Liang S, Luo H, Alnaggar M, Liu A, et al. Irreversible electroporation plus allogenic Vγ9Vδ2 T cells enhances antitumor effect for locally advanced pancreatic cancer patients. Signal Transduct Target Ther (2020) 5(1):1–9. doi: 10.1038/s41392-020-00260-1
54. Xu Y, Xiang Z, Alnaggar M, Kouakanou L, Li J, He J, et al. Allogeneic Vγ9Vδ2 T-cell immunotherapy exhibits promising clinical safety and prolongs the survival of patients with late-stage lung or liver cancer. Cell Mol Immunol (2020) 18(2):427–39. doi: 10.1038/s41423-020-0515-7
55. Vydra J, Cosimo E, Lesný P, Wanless RS, Anderson J, Clark AG, et al. A phase I trial of allogeneic γδ T lymphocytes from haploidentical donors in patients with refractory or relapsed acute myeloid leukemia. Clin Lymphoma Myeloma Leuk (2023) 23(5):e232. doi: 10.1016/j.clml.2023.02.003
56. Ma L, Feng Y, Zhou Z. A close look at current γδ T-cell immunotherapy. Front Immunol (2023) 14:1140623. doi: 10.3389/fimmu.2023.1140623
57. Mensurado S, Blanco-Domínguez R, Silva-Santos B. The emerging roles of γδ T cells in cancer immunotherapy. Nat Rev Clin Oncol (2023) 20(3):178–91. doi: 10.1038/s41571-022-00722-1
58. Garber K. γδ T cells bring unconventional cancer-targeting to the clinic - again. Nat Biotechnol (2020) 38(4):389–91. doi: 10.1038/s41587-020-0487-2
59. Sebestyen Z, Prinz I, Déchanet-Merville J, Silva-Santos B, Kuball J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat Rev Drug Discov (2019) 19(3):169–84. doi: 10.1038/s41573-019-0038-z
60. Almeida AR, Correia DV, Fernandes-Platzgummer A, Da Silva CL, Da Silva MG, Anjos DR, et al. Delta one T cells for immunotherapy of chronic lymphocytic leukemia: Clinical-grade expansion/differentiation and preclinical proof of concept. Clin Cancer Res (2016) 22(23):5795–804. doi: 10.1158/1078-0432.CCR-16-0597
61. Lo Presti E, Dieli F, Fourniè JJ, Meraviglia S. Deciphering human γδ T cell response in cancer: Lessons from tumor-infiltrating γδ T cells. Immunol Rev (2020) 298(1):153–64. doi: 10.1111/imr.12904
62. Lo Presti E, Dieli F, Meraviglia S. Tumor-infiltrating γδ T lymphocytes: Pathogenic role, clinical significance, and differential programing in the tumor microenvironment. Front Immunol (2014) 5:122612. doi: 10.3389/fimmu.2014.00607
63. Lo Presti E, Pizzolato G, Corsale AM, Caccamo N, Sireci G, Dieli F, et al. γδ T cells and tumor microenvironment: from immunosurveillance to tumor evasion. Front Immunol (2018) 9:379356. doi: 10.3389/fimmu.2018.01395
64. Li MO, Wan YY, Sanjabi S, Robertson AKL, Flavell RA. Transforming growth factor-β regulation of immune responses. Annu Rev Immunol (2006) 24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737
65. Gao Z, Bai Y, Lin A, Jiang A, Zhou C, Cheng Q, et al. Gamma delta T-cell-based immune checkpoint therapy: attractive candidate for antitumor treatment. Mol Cancer (2023) 22(1):1–23. doi: 10.1186/s12943-023-01722-0
66. Castella B, Melaccio A, Foglietta M, Riganti C, Massaia M. Vγ9Vδ2 T cells as strategic weapons to improve the potency of immune checkpoint blockade and immune interventions in human myeloma. Front Oncol (2018) 8:417569. doi: 10.3389/fonc.2018.00508
67. Wesch D, Kabelitz D, Oberg HH. Tumor resistance mechanisms and their consequences on γδ T cell activation. Immunol Rev (2020) 298(1):84–98. doi: 10.1111/imr.12925
68. Gonnermann D, Oberg HH, Kellner C, Peipp M, Sebens S, Kabelitz D, et al. Resistance of cyclooxygenase-2 expressing pancreatic ductal adenocarcinoma cells against γδ T cell cytotoxicity. Oncoimmunology (2015) 4(3):1–11. doi: 10.4161/2162402X.2014.988460
69. Ristimäki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, et al. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res (2002) 62(3):632–5.
70. Pei Y, Xiang Z, Wen K, Tu CR, Wang X, Zhang Y, et al. CD137 costimulation enhances the antitumor activity of Vγ9Vδ2-T cells in IL-10-mediated immunosuppressive tumor microenvironment. Front Immunol (2022) 13:872122. doi: 10.3389/fimmu.2022.872122
71. Kunzmann V, Kimmel B, Herrmann T, Einsele H, Wilhelm M. Inhibition of phosphoantigen-mediated γδ T-cell proliferation by CD4+ CD25+ FoxP3+ regulatory T cells. Immunology (2009) 126(2):256–67. doi: 10.1111/j.1365-2567.2008.02894.x
72. Park JH, Kim HJ, Kim CW, Kim HC, Jung Y, Lee HS, et al. Tumor hypoxia represses γδ T cell-mediated antitumor immunity against brain tumors. Nat Immunol (2021) 22(3):336–46. doi: 10.1038/s41590-020-00860-7
73. Sureshbabu SK, Chaukar D, Chiplunkar SV. Hypoxia regulates the differentiation and anti-tumor effector functions of γδT cells in oral cancer. Clin Exp Immunol (2020) 201(1):40–57. doi: 10.1111/cei.13436
74. Siegers GM, Dutta I, Lai R, Postovit LM. Functional plasticity of Gamma delta T cells and breast tumor targets in hypoxia. Front Immunol (2018) 9:371670. doi: 10.3389/fimmu.2018.01367
75. Fan Y, Cheng H, Liu Y, Liu S, Lowe S, Li Y, et al. Metformin anticancer: Reverses tumor hypoxia induced by bevacizumab and reduces the expression of cancer stem cell markers CD44/CD117 in human ovarian cancer SKOV3 cells. Front Pharmacol (2022) 13:955984. doi: 10.3389/fphar.2022.955984
76. Sesen J, Dahan P, Scotland SJ, Saland E, Dang VT, Lemarié A, et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PloS One (2015) 10(4):e0123721. doi: 10.1371/journal.pone.0123721
77. Dieli F, Poccia F, Lipp M, Sireci G, Caccamo N, Di Sano C, et al. Differentiation of effector/memory Vδ2 T cells and migratory routes in lymph nodes or inflammatory sites. J Exp Med (2003) 198(3):391–7. doi: 10.1084/jem.20030235
78. Willcox CR, Vantourout P, Salim M, Zlatareva I, Melandri D, Zanardo L, et al. Butyrophilin-like 3 directly binds a human Vγ4+ T cell receptor using a modality distinct from clonally-restricted antigen. Immunity (2019) 51(5):813–825.e4. doi: 10.1016/j.immuni.2019.09.006
79. Dong R, Zhang Y, Xiao H, Zeng X. Engineering γδ T cells: recognizing and activating on their own way. Front Immunol (2022) 13:889051. doi: 10.3389/fimmu.2022.889051
80. Wilhelm M, Smetak M, Schaefer-Eckart K, Kimmel B, Birkmann J, Einsele H, et al. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J Transl Med (2014) 12(1):1–5. doi: 10.1186/1479-5876-12-45
81. Fisher JPH, Heuijerjans J, Yan M, Gustafsson K, Anderson J. γδ T cells for cancer immunotherapy: A systematic review of clinical trials. Oncoimmunology (2014) 3(1):e27572. doi: 10.4161/onci.27572
82. Zeng J, Tang SY, Wang S. Derivation of mimetic γδ T cells endowed with cancer recognition receptors from reprogrammed γδ T cell. PloS One (2019) 14(5):e0216815. doi: 10.1371/journal.pone.0216815
83. Murai N, Koyanagi-Aoi M, Terashi H, Aoi T. Re-generation of cytotoxic γδT cells with distinctive signatures from human γδT-derived iPSCs. Stem Cell Rep (2023) 18(4):853–68. doi: 10.1016/j.stemcr.2023.02.010
84. Ravandi F, Stein AS, Kantarjian HM, Walter RB, Paschka P, Jongen-Lavrencic M, et al. A phase 1 first-in-human study of AMG 330, an anti-CD33 bispecific T-cell engager (BiTE®) antibody construct, in relapsed/refractory acute myeloid leukemia (R/R AML). Blood (2018) 132(Supplement 1):25–5. doi: 10.1182/blood-2018-99-109762
85. Oberg HH, Peipp M, Kellner C, Sebens S, Krause S, Petrick D, et al. Novel bispecific antibodies increase γδ T-cell cytotoxicity against pancreatic cancer cells. Cancer Res (2014) 74(5):1349–60. doi: 10.1158/0008-5472.CAN-13-0675
86. Peipp M, Wesch D, Oberg HH, Lutz S, Muskulus A, Van De Winkel JGJ, et al. CD20-specific immunoligands engaging NKG2D enhance γδ T cell-mediated lysis of lymphoma cells. Scand J Immunol (2017) 86(4):196–206. doi: 10.1111/sji.12581
87. Song Y, Liu Y, Teo HY, Liu H. Targeting cytokine signals to enhance γδT cell-based cancer immunotherapy. Front Immunol (2022) 13:914839. doi: 10.3389/fimmu.2022.914839
88. Schilbach K, Welker C, Krickeberg N, Kaißer C, Schleicher S, Hashimoto H. In the absence of a TCR signal IL-2/IL-12/18-stimulated γδ T cells demonstrate potent anti-tumoral function through direct killing and senescence induction in cancer cells. Cancers (Basel) (2020) 12(1):130. doi: 10.3390/cancers12010130
89. Teo HY, Song Y, Yong KSM, Liu Y, Mei Y, Hanafi ZB, et al. IL12/18/21 preactivation enhances the antitumor efficacy of expanded γδT cells and overcomes resistance to anti–PD-L1 treatment. Cancer Immunol Res (2023) 11(7):978–99. doi: 10.1158/2326-6066.CIR-21-0952
90. Le Floch AC, Orlanducci F, Briantais A, Le Roy A, Chretien AS, Vey N, et al. Anti-BTN3A 20.1 agonist monoclonal antibody enhances autologous Vγ9Vδ2 T cells cytotoxicity against primary acute myeloid blasts. Blood (2019) 134(Supplement_1):5153–3. doi: 10.1182/blood-2019-125541
91. Le Floch AC, Imbert C, De Gassart A, Orlanducci F, Le Roy A, Gorvel L, et al. Targeting BTN2A1 by a unique activating mab improves Vγ9Vδ2 T cell cytotoxicity against primary acute lymphoblastic blasts. Blood (2021) 138(Supplement 1):2302. doi: 10.1182/blood-2021-152577
92. Baker FL, Bigley AB, Agha NH, Pedlar CR, O’Connor DP, Bond RA, et al. Systemic β-Adrenergic Receptor Activation Augments the ex vivo Expansion and Anti-Tumor Activity of Vγ9Vδ2 T-Cells. Front Immunol (2020) 10:497497. doi: 10.3389/fimmu.2019.03082
93. Smith TT, Moffett HF, Stephan SB, Opel CF, Dumigan AG, Jiang X, et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest (2017) 127(6):2176–91. doi: 10.1172/JCI87624
94. Chauvin C, Joalland N, Perroteau J, Jarry U, LaFrance L, Willem C, et al. NKG2D controls natural reactivity of Vγ9Vδ2 T lymphocytes against mesenchymal glioblastoma cells. Clin Cancer Res (2019) 25(23):7218–28. doi: 10.1158/1078-0432.CCR-19-0375
95. Jarry U, Chauvin C, Joalland N, Léger A, Minault S, Robard M, et al. Stereotaxic administrations of allogeneic human Vγ9Vδ2 T cells efficiently control the development of human glioblastoma brain tumors. Oncoimmunology (2016) 5(6):e1168554. doi: 10.1080/2162402X.2016.1168554
96. Wang X, Xiang Z, Liu Y, Huang C, Pei Y, Wang X, et al. Exosomes derived from Vδ2-T cells control Epstein-Barr virus-associated tumors and induce T cell antitumor immunity. Sci Transl Med (2020) 12(563):3426. doi: 10.1126/scitranslmed.aaz3426
97. Wang X, Zhang Y, Mu X, Tu CR, Chung Y, Tsao SW, et al. Exosomes derived from γδ-T cells synergize with radiotherapy and preserve antitumor activities against nasopharyngeal carcinoma in immunosuppressive microenvironment. J Immunother Cancer (2022) 10(2):e003832. doi: 10.1136/jitc-2021-003832
98. Wang X, Zhang Y, Chung Y, Tu CR, Zhang W, Mu X, et al. Tumor vaccine based on extracellular vesicles derived from γδ-T cells exerts dual antitumor activities. J Extracell Vesicles (2023) 12(9):e12360. doi: 10.1002/jev2.12360
99. Rischer M, Pscherer S, Duwe S, Vormoor J, Jürgens H, Rossig C. Human γδ T cells as mediators of chimaeric-receptor redirected anti-tumour immunity. Br J Haematol (2004) 126(4):583–92. doi: 10.1111/j.1365-2141.2004.05077.x
100. Rozenbaum M, Meir A, Aharony Y, Itzhaki O, Schachter J, Bank I, et al. Gamma-delta CAR-T cells show CAR-directed and independent activity against leukemia. Front Immunol (2020) 11:548110. doi: 10.3389/fimmu.2020.01347
101. Fisher J, Sharma R, Don DW, Barisa M, Hurtado MO, Abramowski P, et al. Engineering γδT cells limits tonic signaling associated with chimeric antigen receptors. Sci Signal (2019) 12(598):eaax1872. doi: 10.1126/scisignal.aax1872
102. Makkouk A, Yang XC, Barca T, Lucas A, Turkoz M, Wong JTS, et al. Off-the-shelf Vδ1 gamma delta T cells engineered with glypican-3 (GPC-3)-specific chimeric antigen receptor (CAR) and soluble IL-15 display robust antitumor efficacy against hepatocellular carcinoma. J Immunother Cancer (2021) 9(12):e003441. doi: 10.1136/jitc-2021-003441
103. Chmielewski M, Abken H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv Cell Gene Ther (2020) 3(3):e84. doi: 10.1002/acg2.84
104. Hawkins ER, D’Souza RR, Klampatsa A. Armored CAR T-cells: the next chapter in T-cell cancer immunotherapy. Biologics (2021) 15:95–105. doi: 10.2147/BTT.S291768
105. Van Der Veken LT, Hagedoorn RS, Van Loenen MM, Willemze R, Falkenburg JHF, Heemskerk MHM. αβ T-cell receptor engineered γδ T cells mediate effective antileukemic reactivity. Cancer Res (2006) 66(6):3331–7. doi: 10.1158/0008-5472.CAN-05-4190
106. Müller TR, Jarosch S, Hammel M, Leube J, Grassmann S, Bernard B, et al. Targeted T cell receptor gene editing provides predictable T cell product function for immunotherapy. Cell Rep Med (2021) 2(8):100374. doi: 10.1016/j.xcrm.2021.100374
107. Marcu-Malina V, Heijhuurs S, Van Buuren M, Hartkamp L, Strand S, Sebestyen Z, et al. Redirecting αβT cells against cancer cells by transfer of a broadly tumor-reactive γδT-cell receptor. Blood (2011) 118(1):50–9. doi: 10.1182/blood-2010-12-325993
108. Helsen CW, Hammill JA, Lau VWC, Mwawasi KA, Afsahi A, Bezverbnaya K, et al. The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nat Commun (2018) 9(1):1–13. doi: 10.1038/s41467-018-05395-y
109. Appelbaum J, Leung WH, Martin U, Oda K, Tampella G, Xia D, et al. 101 Engineering gamma/delta T cells with the T-Cell antigen coupler receptor effectively induces antigen-specific tumor cytotoxicity in vitro and in vivo. J Immunother Cancer (2020) 8(Suppl 3):A63.2–A64. doi: 10.1136/jitc-2020-sitc2020.0101
110. Fleischer LC, Becker SA, Ryan RE, Fedanov A, Doering CB, Spencer HT. Non-signaling chimeric antigen receptors enhance antigen-directed killing by γδ T cells in contrast to αβ T cells. Mol Ther Oncolytics (2020) 18:149–60. doi: 10.1016/j.omto.2020.06.003
111. Benmebarek MR, Cadilha BL, Herrmann M, Lesch S, Schmitt S, Stoiber S, et al. A modular and controllable T cell therapy platform for acute myeloid leukemia. Leukemia (2021) 35(8):2243–57. doi: 10.1038/s41375-020-01109-w
112. Raute K, Strietz J, Parigiani MA, Andrieux G, Thomas OS, Kistner KM, et al. Breast cancer stem cell–derived tumors escape from γδ T-cell immunosurveillance in vivo by modulating γδ T-cell ligands. Cancer Immunol Res (2023) 11(6):810. doi: 10.1158/2326-6066.CIR-22-0296
113. Rossi C, Gravelle P, Decaup E, Bordenave J, Poupot M, Tosolini M, et al. Boosting γδ T cell-mediated antibody-dependent cellular cytotoxicity by PD-1 blockade in follicular lymphoma. Oncoimmunology (2018) 8(3):1554175. doi: 10.1080/2162402X.2018.1554175
114. Maniar A, Zhang X, Lin W, Gastman BR, Pauza CD, Strome SE, et al. Human gammadelta T lymphocytes induce robust NK cell-mediated antitumor cytotoxicity through CD137 engagement. Blood (2010) 116(10):1726–33. doi: 10.1182/blood-2009-07-234211
115. Cairo C, Surendran N, Harris KM, Mazan-Mamczarz K, Sakoda Y, Diaz-Mendez F, et al. Vγ2Vδ2 T cell Costimulation Increases NK cell Killing of Monocyte-derived Dendritic Cells. Immunology (2014) 144(3):422–30. doi: 10.1111/imm.12386
116. Pu D, Yin L, Huang L, Qin C, Zhou Y, Wu Q, et al. Cyclooxygenase-2 inhibitor: A potential combination strategy with immunotherapy in cancer. Front Oncol (2021) 11:637504. doi: 10.3389/fonc.2021.637504
117. Li Z, Zhang J, Tang J, Wang R. Celastrol increases osteosarcoma cell lysis by γδ T cells through up-regulation of death receptors. Oncotarget (2016) 7(51):84388–97. doi: 10.18632/oncotarget.12756
118. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med (2007) 356(2):115–24. doi: 10.1056/NEJMoa065044
119. Sellmann L, Fenchel K, Dempke WCM. Improved overall survival following tyrosine kinase inhibitor treatment in advanced or metastatic non-small-cell lung cancer— the Holy Grail in cancer treatment? Transl Lung Cancer Res (2015) 4(3):223–7. doi: 10.21037/tlcr.2016.07.01
120. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med (2020) 382(20):1894–905. doi: 10.1056/NEJMoa1915745
Keywords: γδ T cell, Gamma/delta T cell, chimeric antigen receptor (CAR), anti-tumor immunity, cancer immunotherapy, Unconventional T cells, non-HLA-restricted T cells, cellular immunotherapy
Citation: Wang CQ, Lim PY and Tan AH-M (2024) Gamma/delta T cells as cellular vehicles for anti-tumor immunity. Front. Immunol. 14:1282758. doi: 10.3389/fimmu.2023.1282758
Received: 24 August 2023; Accepted: 26 December 2023;
Published: 11 January 2024.
Edited by:
Daniela Wesch, University of Kiel, GermanyReviewed by:
Prashant Sharma, University of Arizona, United StatesWenwei Tu, The University of Hong Kong, Hong Kong SAR, China
Copyright © 2024 Wang, Lim and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andy Hee-Meng Tan, YW5keV90YW5AYnRpLmEtc3Rhci5lZHUuc2c=; Chelsia Qiuxia Wang, Y2hlbHNpYV93YW5nQGJ0aS5hLXN0YXIuZWR1LnNn
†These authors contributed equally to this work and share first authorship
‡ORCID: Chelsia Qiuxia Wang, orcid.org/0000-0003-2471-4641
Pei Yu Lim, orcid.org/0009-0002-0834-787X
Andy Hee-Meng Tan, orcid.org/0000-0003-1558-1867