 Joanna Mackiewicz
Joanna Mackiewicz Malwina Lisek
Malwina Lisek Tomasz Boczek
Tomasz Boczek
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 23 November 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1281882
This article is part of the Research Topic Crosstalk between Cell Death, Oxidative Stress, and Immune Regulation View all 11 articles
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by a progressive loss of cognitive functions. While the exact causes of this debilitating disorder remain elusive, numerous investigations have characterized its two core pathologies: the presence of β-amyloid plaques and tau tangles. Additionally, multiple studies of postmortem brain tissue, as well as results from AD preclinical models, have consistently demonstrated the presence of a sustained inflammatory response. As the persistent immune response is associated with neurodegeneration, it became clear that it may also exacerbate other AD pathologies, providing a link between the initial deposition of β-amyloid plaques and the later development of neurofibrillary tangles. Initially discovered in T cells, the nuclear factor of activated T-cells (NFAT) is one of the main transcription factors driving the expression of inflammatory genes and thus regulating immune responses. NFAT-dependent production of inflammatory mediators is controlled by Ca2+-dependent protein phosphatase calcineurin (CaN), which dephosphorylates NFAT and promotes its transcriptional activity. A substantial body of evidence has demonstrated that aberrant CaN/NFAT signaling is linked to several pathologies observed in AD, including neuronal apoptosis, synaptic deficits, and glia activation. In view of this, the role of NFAT isoforms in AD has been linked to disease progression at different stages, some of which are paralleled to diminished cognitive status. The use of classical inhibitors of CaN/NFAT signaling, such as tacrolimus or cyclosporine, or adeno-associated viruses to specifically inhibit astrocytic NFAT activation, has alleviated some symptoms of AD by diminishing β-amyloid neurotoxicity and neuroinflammation. In this article, we discuss the recent findings related to the contribution of CaN/NFAT signaling to the progression of AD and highlight the possible benefits of targeting this pathway in AD treatment.
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder characterized by a progressive decline in cognitive and executive functions (1). Intensive research performed over the decades has revealed the complex mechanistic underpinnings of AD, involving a combination of age-dependent brain changes along with genetic predisposition, lifestyle, and environmental factors (1). Despite relatively wide knowledge about its etiology, the cellular determinants of AD susceptibility or resilience and the key molecular changes driving disease progression are less understood.
Neuropathologically, AD-associated degeneration is linked to the deposition of amyloid-β (Aβ) fibrillary aggregates, occurring as neuritic plaques or vascular deposits, and hyperphosphorylated microtubule-associated protein tau. The latter is a major component of neurofibrillary tangles, neuropil threads, and neuritic plaque corona (2). However, the list of detected abnormalities is much longer and involves gliosis, neuroinflammation, oxidative stress, changes in neuronal plasticity, altered expression of glutamate and cholinergic receptors, Ca2+ homeostasis imbalance, and many others. The accumulation of these changes across the lifespan leads to impaired cognition and a higher risk of AD development. It is now well-accepted that many of these pathologies are a consequence of Ca2+ signaling deregulation, which is central to amyloid-evoked degeneration (3). Indeed, numerous independent biochemical, behavioral, electrophysiological, and molecular studies, which have been the subject of excellent reviews (1, 4, 5), have confirmed a link between Ca2+ deregulation and age-related memory deficits and worsening cognitive performance. Nonetheless, it is becoming apparent that Ca2+ imbalance is not restricted to neurons but also underlies the altered function of other non-neuronal cells, especially astrocytes. The relationship between astrocytic Ca2+ deregulation as a function of neurodegenerative diseases has been thoroughly discussed in comprehensive reviews (6–8). Several laboratories have provided strong evidence to imply that Ca2+-dependent protein phosphatase calcineurin (CaN) links dysfunctional Ca2+ signaling to Aβ accumulation, neuroinflammation, and synaptotoxicity (3, 9–11). The activity of CaN is controlled by spatially and temporarily restricted intracellular Ca2+ elevations, which activate specific Ca2+ signal decoding proteins (12). CaN is required for both long-term potentiation and long-term depression, synaptic processes underlying learning and memory, and its activity positively correlated with cognitive loss in AD brains (13, 14). Perhaps, one of the most important downstream effectors linking Ca2+/CaN signaling to the gene regulatory machinery is the nuclear factor of activated T-cells (NFAT). Mounting evidence, as will be discussed in the following sections, shows that CaN/NFAT drives or exacerbates the core symptoms of AD neuropathology. It is therefore critical to understand the upstream and downstream signals leading to the pathological activation of CaN/NFAT pathway to identify new therapeutic targets and develop new treatment strategies for AD. In this review, we summarize the emerging evidence that deficient CaN/NFAT could contribute to brain degeneration in AD.
In healthy adults, CaN is widely expressed throughout the brain, with its highest concentration detected in neurons and low in glial cells (15–18). Structurally, the holoenzyme of CaN comprises two main subunits: a ~ 60 kDa catalytic subunit (CNA) and a ~19 kDa regulatory subunit (CNB) containing four Ca2+-binding EF-hand motifs (12) (Figure 1).
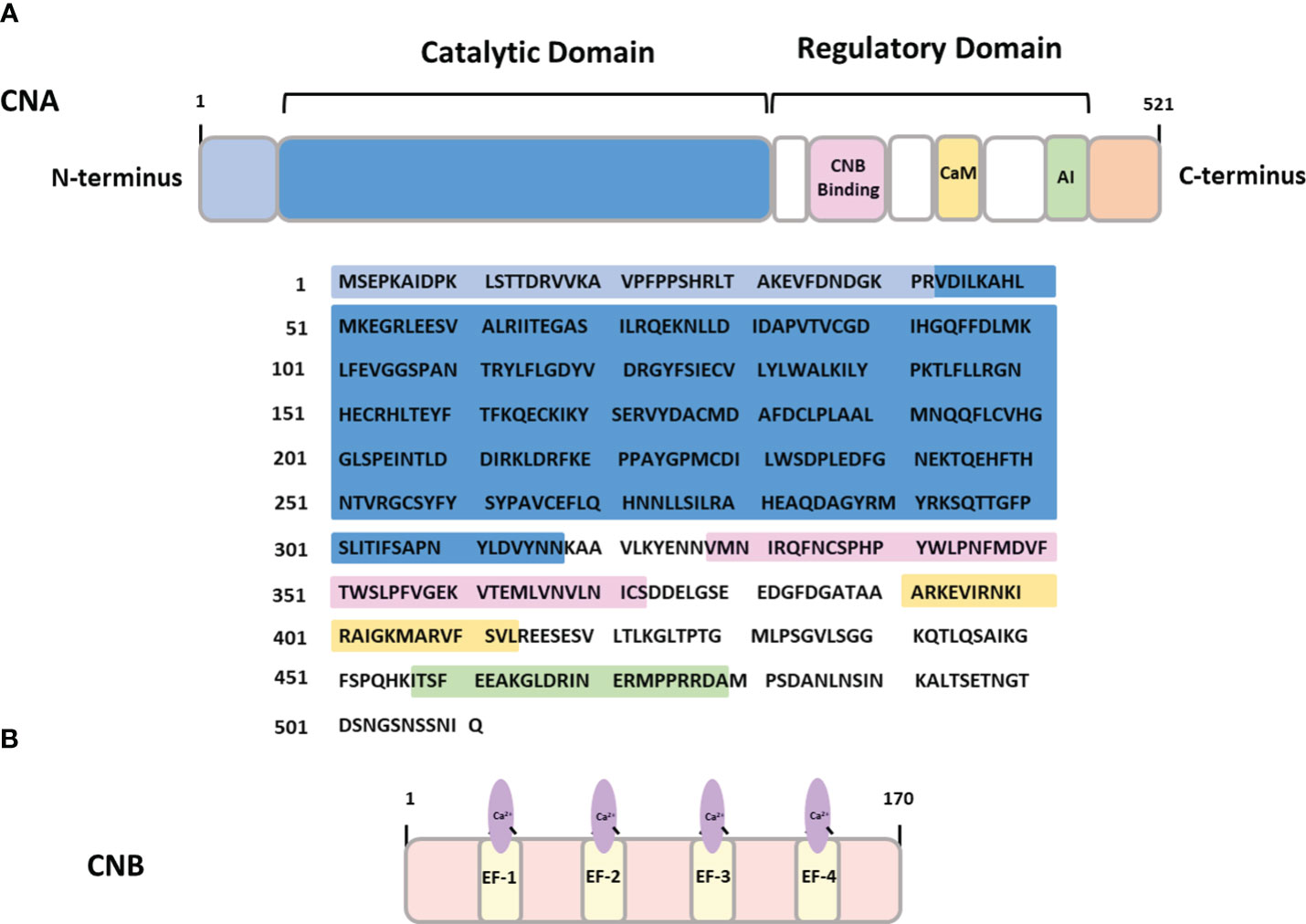
Figure 1 The structure of calcineurin (A) CNA- calcineurin subunit A containing binding sites for calcineurin subunit B (CNB binding), calmodulin binding domain (CaM) and autoinhibitory domain (AI). (B) CNB – calcineurin subunit B containing four Ca2+-binding EF hand domains.
Binding of calmodulin (CaM) to the regulatory domain of the catalytic subunit increases CaN phosphatase activity, decoding the upstream Ca2+ signals and translating them into a dephosphorylation pattern of cellular proteins. CaM itself is a small, evolutionary conserved protein expressed in all mammalian cells. It serves as a versatile Ca2+ sensor capable of responding to a wide range of Ca2+ concentrations (10-12 M - 10-16 M) due to the presence of four canonical Ca2+ binding EF hands possessing different affinities for Ca2+ (19). Upon binding Ca2+, CaM undergoes a conformational change exposing the hydrophobic surface region for Ca2+-dependent interaction with CaM-binding proteins (20). The Ca2+/CaM complex interacts with targets that mediate crucial cellular processes such as inflammation, metabolism, apoptosis, short-term and long-term memory and the immune response (21–25). Continued research indicates that CaM binds to and affects many proteins involved in the onset and progression of AD and other neurodegenerative diseases (26–28). This involvement in neurodegeneration underscores the significance of CaM in the understanding the molecular mechanisms behind these diseases.
Among many Ca2+-dependent protein phosphatases, CaN is the only one directly activated by Ca2+/CaM and is one of the most sensitive enzymes responding to Ca2+ elevations. The cooperative interaction between Ca2+, CaNB, and Ca2+/CaM allows CaN to uniquely respond to discrete Ca2+ fluctuations (29). These features make CaN particularly vulnerable to alterations in Ca2+ homeostasis observed in AD. A growing body of evidence suggests that upregulation of CaN activity is directly linked to multiple neurodegenerative insults observed in Parkinson’s disease (30), Alzheimer’s disease (31), and Huntington’s disease (32), all marked by impaired synaptic function, neuroinflammation, and neuronal loss. Moreover, abnormal activation of CaN has been observed in numerous cellular events traditionally linked to AD, including astrocyte activation, Aβ generation, neuronal apoptosis, synaptic toxicity, and behavioral deficits (14). The involvement of CaN is also supported by several studies showing altered CaN signaling in the brains of animals used to model the pathogenesis of neurodegenerative disorders (33–36).
Strong evidence for the contribution of CaN to the pathophysiology of AD in humans comes from the Taglialatela group (37). The results of a retrospective analysis of kidney transplant patients demonstrated that the administration of CaN inhbitors to prevent transplant rejection decreased the incidence of dementia and AD in this group compared to national data from the general population. The initially encouraging results were potentially limited by the small sample size. These findings have been recently replicated in a large cohort by Silva and colleagues (38), demonstrating that FK506, CsA, and sirolimus can reduce the risk of dementia compared to general population-like control. However, among the three immunosuppressive drugs, those that are capable of crossing the blood-brain barrier, like FK506, have a greater probability of reducing dementia. Since inflammatory mediators are strongly correlated with the accumulation of Aβ and induction of neuronal apoptosis, the potential role of CaN inhibitors in reducing the prevalence of dementia is not surprising. However, the study performed by Silva and colleagues delineates a new therapeutic avenue based on brain-penetrant CaN inhibitors. In addition, it has been shown that CaN is heavily involved in the regulation of gene expression (39). For upstream and downstream regulatory mechanisms of CaN, we refer to the excellent reviews published recently (12, 40–42).
Proteins belonging to the NFAT family were originally described more than three decades ago as the transcriptional activators of interleukin-2 (IL-2) in T cells (43). It is now apparent that NFAT proteins play a key role not only during T-cell activation and differentiation but also in regulating the function of several types of immune cells, including B cells, mast cells, basophils, and natural killer cells (44). NFAT family members are involved in the induction and/or coordination of the immune response by modulating the expression of a large number of immunologically important genes. These genes include cytokines such as IL-2, IL-3, IL-4, IL-5, IL-8, IL-13, granulocyte-macrophage colony-stimulating factor (GM-CSF), and tumor necrosis factor-alpha (TNF-α), as well as cell-surface receptors CD40L and CTLA-4, and the apoptotic factor FasL (45).
NFAT family members are widely distributed throughout tissues, including the brain, and can regulate distinct developmental processes (46–49). The strong correlation between NFAT expression and vertebrate development was derived from mouse genetic experiments. For instance, deletion of NFATc1 caused dramatic defects in cardiac morphogenesis (50), NFATc2 null mice displayed abnormalities in chondrogenesis (51), disruption of NFATc3 resulted in dysregulation of myogenesis (52), whereas NFATc4 deficient mice were healthy and developmentally normal. Suchting et al. (53) have discussed the developmental relationship between nerve cells and vessels, pointing out several anatomical and functional parallels. In support of this, concurrent deletion of NFATc3 and NFATc4 produced significant impairments in vascular development but also massive defects in sensory axon projection (49). NFAT signaling is recognized as an essential pathway both in the adult and developing nervous system. In the brain, NFAT-dependent gene regulation has been shown to play a critical role in neuronal survival, proliferation, and differentiation (54–56), as well as synaptogenesis, corticogenesis, and neurotransmission (57–59).
In human, the NFAT family encompasses five different members: NFAT1 [also called NFATc2 or NFATp), NFAT2 (also called NFATc1 or NFATc), NFAT3 (also called NFATc4), NFAT4 (also called NFATc3 or NFATx) and NFAT5 (also called tonicity enhancer binding protein (TonEBP) or osmotic response element-binding protein (OREBP)] that are encoded by separate genes. Moreover, the alternative splicing of each of these family members results in a different number of variants. There are eight possible variants of NFATc2 and NFATc1 in mammalian cells. The splicing of NFATc3 generates six isoforms, and up to twenty-four in the case of NFATc4 (60). On the other hand, sixteen various transcripts of NFAT5 have been identified so far (61) (Table 1). Specific NFAT isoforms appear to exhibit preferential associations with different types of neuronal cells (3, 10, 62, 63). For instance, NFATc3 is prominently expressed in astrocytes and pericytes, with comparatively lower expression in neurons. This isoform is also significantly upregulated in activated astrocytes (3). In contrast, NFATc4 has a broader distribution within neurons compared to NFATc3. NFATc1 and NFATc2, in particular, display a stronger preference for glial cells.
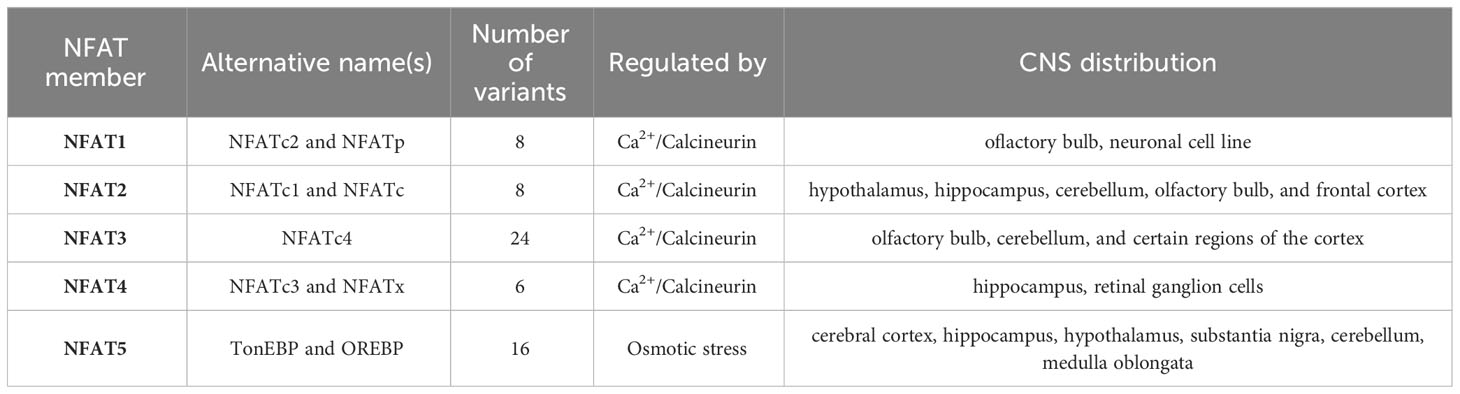
Table 1 General features of NFAT family members and their distribution in the brain.
Despite having different C- and N-terminal sequences, all family members contain a highly conserved DNA-binding domain (DBD), similar to the one in the NFκB/Rel family, and are activated, with the exception of NFAT5, by CaN (47). NFAT5 lacks an essential CaN binding site, and its activity is regulated by changes in tonicity (64). Structurally, NFAT1-4 exist as monomers and possess two tandem domains: a regulatory domain located in the N-terminus, which is known as the NFAT homology region (NHR), and a Rel homology domain (RHR), where the DBD is located (65). The NHR contains two CaN binding motifs: a Ca2+-independent PXIXIT (where X denotes any amino acid) motif in the N-terminus, and a Ca2+-dependent LXVP motif in the C-terminal portion of the NHR (65). Furthermore, the NHR possesses the nuclear localization signal (NLS) responsible for CaN-dependent nuclear translocation, extended serine-rich regions (SRR1, SRR2), and three serine-proline motifs (SP1, SP2, SP3) (65) (Figure 2). NFAT dephosphorylation is believed to expose the NLS sequence, 265-KRK-267, which is located upstream of the SP3 motif. A second potential NLS site, 682-KRK-684 is situated near the C-terminus of NFATc1 (66). It is well-documented that phosphorylation status of the regulatory domain governs DNA-binding affinity and NLS exposure. However, the mechanism of NLS activation has not been elucidated at a structural level. The most plausible hypothesis suggests that dephosphorylation initiates global changes in NFAT conformation, facilitating its transition from an inactive to an active state (67). This transition may involve alterations in interactions within NFAT itself or with binding protein partners (68–70).
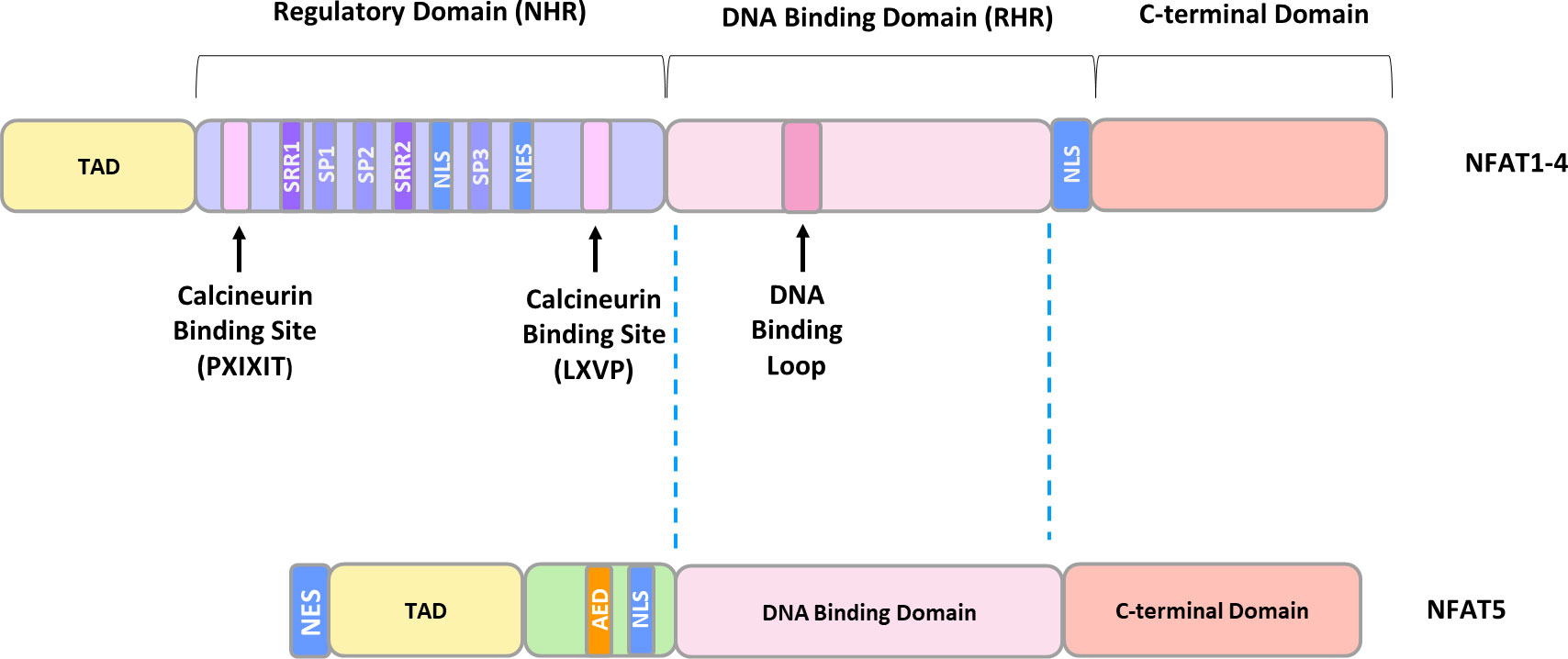
Figure 2 The general structure of NFAT proteins. TAD, transactivation domain; SRR, serine-rich region; SP, serine/proline motif; NLS, nuclear localization sequence; NES, nuclear exclusion sequence; AED, auxiliary export domain.
The NHR domain is moderately conserved and shares 22-32% sequence homology among the distinct NFAT members, whereas the Rel-homology region, consisting of approximately 270 amino acids, shares 64-72% sequence identity between various NFAT members (65, 71). In contrast, NFAT5 exists as a homodimer and possesses only a Rel DNA-binding domain structurally homologous to the Ca2+-dependent members, while the remaining 600 amino acids are entirely different (59).
Under resting conditions, the NHR domain is hyperphosphorylated within the SRR region and SP repeats, keeping the NFAT proteins in an inactive state and predominantly located in the cytoplasm (72). In response to elevated intracellular Ca2+, CaN becomes activated and dephosphorylates NFAT, leading to its rapid import into the nucleus (72). Once in the nucleus, NFATs can couple intracellular changes in Ca2+ concentration to the activation/repression of gene transcription, either acting alone or more frequently in cooperation with other transcription factors (73). Many of the NFAT transcriptional partners have not been characterized yet, but biochemical reconstructions have revealed several of them, including the cell life and death regulator activator protein 1 (AP1) and oncogenic regulators such as GATA binding protein 4 (GATA4) or myocyte enhancer factor 2 (MEF2) (46, 74). The nuclear accumulation of NFAT is counteracted by the synergistic actions of a set of protein kinases, including casein kinase 1 (CK1), glycogen synthase kinase 3 (GSK-3), and dual-specificity tyrosine-regulated kinases (DYRK) (55, 68, 75). These kinases can act in the cytosol to cause NFAT phosphorylation or in the nucleus to promote its rephosphorylation, facilitating nuclear export and transcription termination. CK1 functions as a maintenance and export kinase for NFATc2 (47). GSK-3 is classified as an export-type kinase, which phosphorylates the SP2 motif of NFATc2 and both, SP2 and SP3 motifs, of NFATc1 (47). In addition, phosphorylation of NFATc2 by GSK-3 requires previous phosphorylation by priming kinases including PKA or DYRK. Furthermore, DYRK1 functions as an export kinase which phosphorylates conserved SP3 motif in NFATc1 and NFATc2. In turn, DYRK2 can operate as a maintenance kinase, and phosphorylate the SP3 motif of NFATc1 and NFATc2 (47). Additionally, phosphorylation of SSR1 of NFATc1 by the c-Jun N-terminal kinase (JNK) or SSR1 of NFATc2 by the p38 mitogen-activated protein kinase (MAPK) has been shown to enhance cytosolic NFAT retention (69, 76, 77). However, it is not entirely clear whether kinases that phosphorylate NFAT under basal conditions also mediate its rephosphorylation in the nucleus and subsequent export back to the cytoplasm. Furthermore, NFAT transcriptional activity can be affected by different post-translational modifications, including acetylation or sumoylation, as well as phosphorylation events different from those regulating Ca2+-dependent translocation (78–80). It has been demonstrated that sumoylation plays a privileged role in extensive cellular processes and appears to be an important mediator of neuronal and synaptic function (81). In rat primary hippocampal neurons, sumoylation effectively suppressed the transcriptional activity of NFATc1, NFATc2, and NFATc3 isoforms, while in cortical neurons, only the transcriptional activity of NFATc1 and NFATc2 was affected by sumoylation (82). These findings indicate that the regulation of particular NFAT isoforms may be cell type-specific.
Regulation of subcellular localization and activity of Ca2+-dependent NFAT members is related to ligand binding to distinct cell-surface receptors (45, 46, 65, 83, 84). These receptors share a common feature: their ability to activate phosphatidylinositol-specific phospholipase C (PLC), thereby inducing Ca2+ entry through the plasma membrane. Activation of PLC triggers a cascade of events, including the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) and the release of inositol 1,4,5-triphosphate (IP3), which in turn mobilizes intracellular Ca2+ through IP3 receptors located in the membrane of the endoplasmic reticulum (ER) (85). The increase in Ca2+ concentration leads to CaN activation by binding to its CaN-B regulatory subunit or by coupling Ca2+-dependent calmodulin to CaN. Efficient CaN-dependent dephosphorylation requires a docking interaction between NFAT and CaN. The PxIxIT sequence, located at the N-terminus of the NHR domain, serves as a primary docking site for CaN. Different NFAT members have unique PxIxIT sequences with a low affinity for calcineurin (Kd = 10–30 μM), which is essential to maintain sensitivity to environmental cues and restrict constitutive activation of NFAT (86). Aramburu and colleagues replaced the PxIxIT sequence of NFATc2 with VIVIT, a higher-affinity inhibitor identified through peptide selection. This peptide binds CaN with high affinity and efficiently competes with NFAT for CaN binding, thereby inhibiting NFAT nuclear accumulation (87). In vivo studies with transgenic mice carrying a mutation in the PxIxIT sequence of NFATc2 demonstrated enhanced cytokine production by differentiated T cells and exhibited deficiencies in embryonic and hematopoietic cell maturation (86). CaN dephosphorylates 13 out of 14 serines in NFATc2, which are conserved residues in all calcineurin-dependent NFAT members (67). It has been demonstrated that dephosphorylation of all 13 serine residues in NFATc2 is required to promote its nuclear localization (67). Furthermore, mutations of twelve serines to alanine, which imitate a nearly fully dephosphorylated state of NFATc2, lead to remarkable nuclear accumulation in both, resting and CsA-treated cells.
It is not fully understood whether site-specific dephosphorylation is an organized mechanism and what the consequences are for downstream signaling in specific cell types. However, the experiments using mass spectrometric analysis indicate that the SRR1 region, which is closely adjacent to the main CaN docking site, is preferentially dephosphorylated at low CaN activity (67). NFAT mutants with restricted deletions or S → A mutations in the SRR1 motif are more susceptible to dephosphorylation in the SP repeats by CaN compared to wild-type NFATs (87, 88). CaN-regulated NFAT activation is particularly important in resting conditions when the rise in Ca2+ due to its release from intracellular stores is not sufficient for direct NFAT activation (89).
The parameters for Ca2+/CaN/NFAT activation can be regulated in various ways (Figure 3).
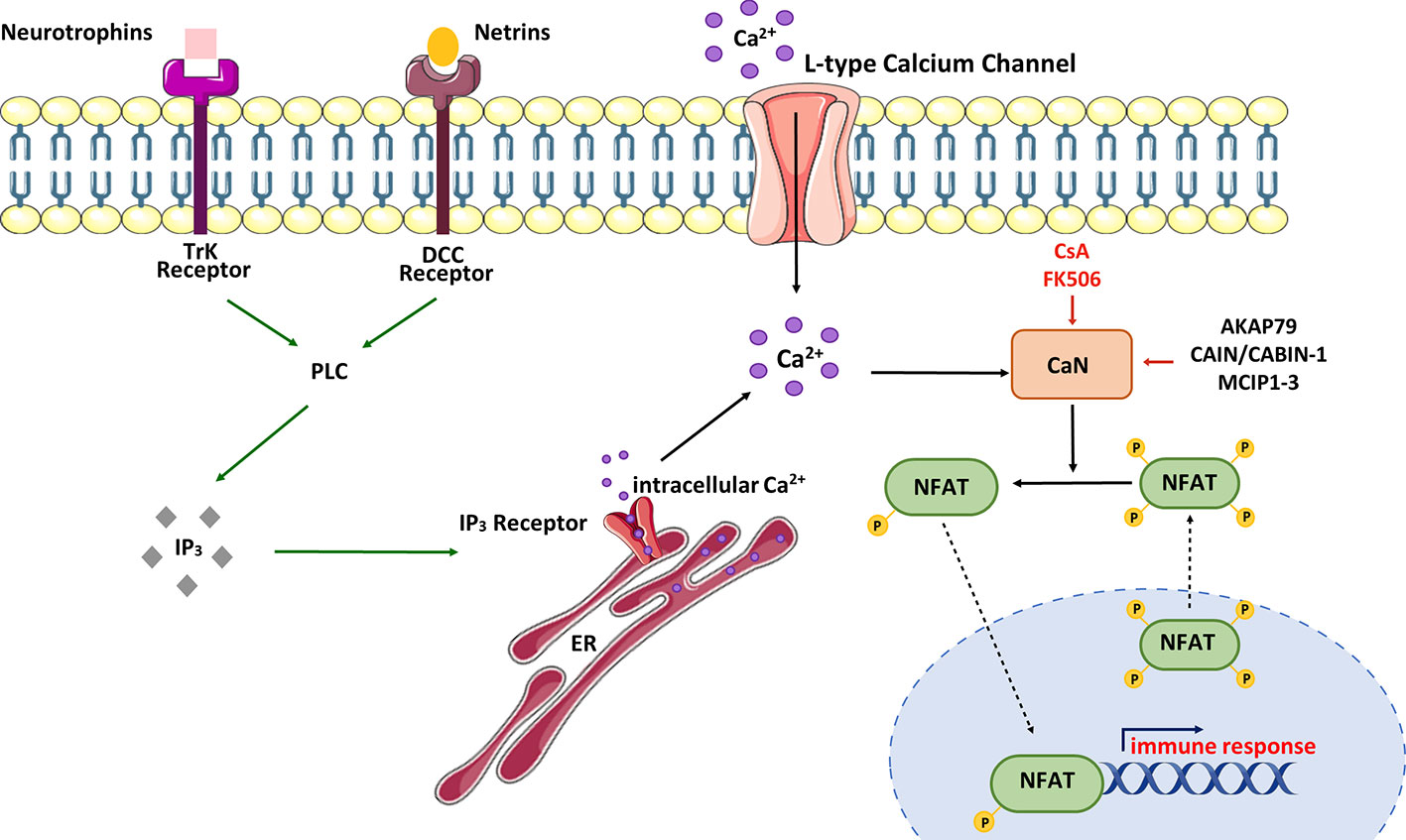
Figure 3 Schematic view of CaN/NFAT activation cycle. NFAT is activated in response to cell-surface receptors coupled to intracellular Ca2+ mobilization. Activated phospholipase C (PLC) generates inositol-1,4,5-triphosphate (IP3), which binds IP3 receptors located in the endoplasmic reticulum (ER). Stimulation of IP3 receptors produces a brief spike in Ca2+ by depleting the ER stores. Ca2+ increases promote activation of calmodulin-dependent enzymes including calcineurin (CaN). CaN dephosphorylates multiple serines in NFAT protein leading to its nuclear import and activation of NFAT-dependent transcription of genes involved in immune response. NFAT rephosphorylation by multiple kinases triggers its nuclear export and cytosolic retention. CaN activity can be modulated by immunosuppressive drugs – cyclosporine A (CsA) and tacrolimus (FK506) or CaN-interacting proteins – AKAP79 (A-kinase anchoring protein 79), MCIPs (modulatory calcineurin-interacting proteins) or CAIN/CABIN-1 (endogenous inhibitor of CaN activity). In neurons, NFAT can be selectively activated by Ca2+ influx through L-type Ca2+ channels.
In neurons, NFAT activation occurs as a result of stimulation of tyrosine kinase receptors (Trk) coupled to PLCγ (90). Neurotrophins and netrins are upstream components of this signaling pathway. For example, it has been observed that overexpression of the TrkA receptor in cortical neurons supports NFAT transcriptional activation upon nerve growth factor (NGF) treatment (91). Additionally, Groth et al. (92) demonstrated that treatment of cultured spinal neurons with NGF enhanced NFAT-dependent transcription. Brain-derived neurotrophic factor (BDNF) has also been found to increase NFAT transcriptional activity in cultured cortical neurons, and this activation was abolished by co-treatment with CaN inhibitors such as CsA or FK506 (91). To illustrate the neurotrophic significance in the NFAT signaling pathway, Graef and colleagues (91) utilized an EGFP-NFATc4 hybrid to visualize NFATc4 subcellular localization in responsive cortical neurons. The data showed that NFATc4 was imported into the nucleus, and its transcriptional activity was enhanced in response to BDNF stimulation. Moreover, cultured hippocampal pyramidal neurons treated with BDNF also exhibited increased endogenous transcriptional activity of NFATc4 (92). It is worth mentioning that inhibition of PLC activity or depletion of intracellular Ca2+ stores attenuated the ability of BDNF to activate endogenous NFAT in hippocampal neurons (92, 93). These results collectively demonstrate that NFAT transcriptional complexes appear to be downstream of neurotrophin signaling, and their activity in neuronal development is regulated through PLC-dependent mechanisms.
A growing body of evidence indicates that NFAT transcription factors can also play a pivotal role in integrating signaling pathways involved in neuronal growth driven by guidance cues during synaptogenesis. In this process, molecules such as netrins are crucial for axon guidance and control during the development of neuronal circuits (94). Supporting this, failure in the elongation of commissural axons caused by triple deletion of NFAT (c2/c3/c4) was seen in mice bearing mutation in netrin-1 or netrin DCC receptor (95, 96). Additionally, it has been demonstrated that netrin-dependent axon extension in E13 rat dorsal spinal cord explants required CaN/NFAT signaling, and stimulation with netrin-1 promoted NFAT transcriptional activity via the DCC receptor (91).
The emptying of ER Ca2+ stores is a known trigger that initiates the process of capacitative Ca2+ influx across the plasma membrane, referred to as store-operated Ca2+ entry. In neurons, Ca2+ influx may also occur via voltage- or ligand-gated Ca2+ channels, each of which is regulated in a precisely coordinated manner. It is well recognized that neuronal L-type voltage-gated Ca2+ channels (LTCC) play a crucial role in various synaptic processes underlying learning and spatial memory formation in the hippocampus and other brain areas (97, 98). The activation of LTCC by high extracellular K+ triggers the nuclear localization of NFAT in cultured hippocampal and DRG neurons (93, 99, 100). It has also been demonstrated that N-methyl-D-aspartate receptor stimulation leads to enhanced nuclear import of NFATc3 and NFATc4 in cortical neurons (101).NFAT activity can also be affected by intracellular inhibitors of CaN. Among the numerous endogenous proteins that have a pivotal role in regulating CaN activity is A-kinase anchoring protein 79 (AKAP79), which binds CaN and blocks its access to the substrates (102). It is worth mentioning that AKAP79 also binds protein kinase A (PKA), which is an NFAT kinase that prevents its nuclear import (103). Other regulators, such as CAIN/CABIN-1, can potentially inhibit CaN activity in a phosphorylation-dependent manner (104) and exert a similar effect on NFAT dephosphorylation and translocation, like the regulators of calcineurin (RCANs) or modulatory calcineurin-interacting proteins (MCIPs; MCIP1–3) (59). Studies have shown that RCANs may either positively or negatively affect the activation of CaN/NFAT signaling. Although the mechanistic explanation of how these proteins modulate CaN activity is still questionable, RCANs were found to participate in CaN biosynthesis or recycling, playing a role as chaperones (59).
Relatively insensitive to classical inhibitors of phosphatases, CaN activity can be corroborated by two widely used immunosuppressive drugs: cyclosporine A (CsA) and FK506 (tacrolimus) (105, 106). CaN/NFAT signaling may also be inhibited by other molecules that differ in chemical properties and the mechanism of action (Table 2).

Table 2 Physicochemical characteristics of CaN/NFAT inhibitors.
CsA has been employed in the treatment of organ transplant rejection since 1976 when Borel and colleagues demonstrated its immunosuppressive properties (107). The mechanism of action of CsA depends the formation of a complex with cytosolic proteins known as cyclophilins, particularly the 17 kDA cyclophilin A, which is highly abundant in T cells (108, 109). Cyclophilins, classified as immunophilins, possess peptidyl-proline-cis-trans isomerase (PPIase) activity that plays a role in ensuring proper protein folding (110). It has been established that CsA inhibits PPIase, although this effect is not associated with the mechanism of immunosuppression. The formation of a ternary complex involving CsA, cyclophilin A and CaNA inhibits calcineurin phosphatase activity and blocks the transcription of cytokine genes, including IL-2 and IL-4 (111). In addition to its impact on the CaN/NFAT pathway, CsA affects the activity of NF-κB and AP-1, transcription factors essential for the regulation of IL-2 gene expression (112, 113). CsA has been demonstrated to inhibit JNK and p38 MAPK signaling, as well as an antigen-specific Ca2+-independent response (114, 115). Several early reports have indicated both pro-apoptotic and neuroprotective effects of CsA in neuronal and neuronal-glia mixed cultures (116–121). The pharmacological normalization of CaN activity can offer a certain degree of neuroprotection (122, 123), improve synaptic function (124), and reduce glial activation (125, 126) in experimental models of brain degeneration. However, despite the undeniable benefits of CsA, its use as an immunosuppressant is limited by potential side effects, including nephrotoxicity, neurotoxicity, and hepatotoxicity (127).
Structurally, FK506 is a macrolide antibiotic with immunosuppressive properties. It forms a complex with immunophilin FK506 binding protein (FKBP12), and the FK506-FKB12 complex suppresses CaN-dependent NFAT signaling. Despite significant differences in structure and mechanism of action, CsA and FK506 surprisingly share similar biological effects (128, 129). However, FK506-induced immunosuppression is not solely due to the inhibition of the CaN/NFAT pathway but also involves other T-cell activation pathways (130). Unlike CsA, FK506 inhibits IL-2 induced IL-5 production by human CD4+ T cells and T-cell proliferation stimulated by IL-2 and IL-7 (130). Moreover, FK506, but not CsA, decreases the expression of human-affinity IL-7 receptor and is more effective in controlling IL-2 production by T cells in patients receiving kidney transplantation. These data suggest that at least some of FK506’s effects are mediated by CsA-insensitive signaling pathways (131). Regarding the immunosuppressive effects, it is evident that FK506 hinders processes further downstream in the T-cell activation cascade, beyond CaN activation. This is supported by the observed capacity of FK506 monotherapy to effectively manage steroid-resistant allograft rejection episodes, which is a critical distinction from CsA, as CsA lacks efficacy in the treatment of allograph rejection (132).
The VIVIT is a 16-mer oligopeptide (MAGPHPVIVITGPHEE) developed through an affinity- driven selection of peptides encoding mutated versions of the wild NFAT protein sequence (SPRIEIT), which is characterized by a low affinity toward the PxIxIT consensus motif (87, 133). VIVIT exhibits a remarkable 25-fold higher potency in inhibiting NFATc dephosphorylation compared to the original SPRIEIT peptide. While 10 μM CsA/cyclophilin can completely inhibit CaN activity, it remained unaffected even at a concentration of up to 100 μM VIVIT. However, the presence of 100 μM VIVIT is sufficient to disrupt the interaction between CaN and NFAT, thus preventing NFAT dephosphorylation (87). Furthermore, VIVIT demonstrates superior selectivity in inhibiting NFAT compared to CsA and FK506. VIVIT has been demonstrated to inhibit the dephosphorylation of NFATc1, NFATc2 and NFATc3, as well as the nuclear import of NFATc2. Additionally, GFP-VIVIT suppresses NFAT-mediated expression of IL-2, IL-3, IL-13, TNF-α, macrophage inflammation protein 1 (MIP-1 α), and granulocyte-macrophage colony-stimulating factor (GM-CSF) in Jurkat T cells while not affecting CsA-regulated genes such as TNF-β and lymphotoxin-β (87). To date, there have been no reported toxic side effects associated with VIVIT. Its outstanding specificity in disrupting NFAT signaling without CaN activity in a general sense, makes VIVIT highly promising for reduced toxicity compared to CsA or FK506. Similarly, its cell-permeable analog, 11-R VIVIT, has demonstrated a lack of toxicity at doses up to 10 mg/kg (134). In this analog, VIVIT was fused to the C-terminal of a poly-arginine-based protein transduction domain (11R-VIVIT, RRRRRRRRRRR-GGG-MAGPHPVIVITGPHEE).
LxVP peptides are derived from CaN-docking NFATc sequence LxVP, and they compete for binding with activated CaN (135, 136). Notably, CaN exhibits a strong affinity for the LxVP motif of NFATc1, NFATc3 and NFATc4, while its binding affinity is considerably weaker for NFATc2, the predominant isoform in activated CD4+ T cells (137). For instance, the GST-LxVPc1 peptide, consisting of 15 amino acids from human NFATc1, shows more efficient binding to CaN compared to any of the PxIxIT motifs found in NFATc1-c4. In contrast, the GST-LxVPc2 fusion peptide from NFATc2 does not bind to CaN under the same conditions. The LxVP effectively inhibits CaN activity when assessed with a phosphopeptide derived from the PKA regulatory subunit II, containing phosphoSer-95, and it increases phosphatase activity when evaluated using a nitrophenyl phosphate assay (135, 136). Therefore, similar to CsA and FK506, LxVPc1 can modify CaN’s activity through an interaction with a site distinct from the active site. When GFP-LxVPc1 fusion protein is overexpressed in HeLa cells, it effectively inhibits NFATc2 dephosphorylation and nuclear translocation upon ionophore treatment. Similarly, in Jurkat T cells, it inhibits NFATc2 dephosphorylation and the expression of luciferase under the control of the IL-2 upon PMA/ionophore stimulation (135, 136).
Dipyridamole is a medication that serves as an inhibitor of nucleoside transport and a PDE3 (phosphodiesterase 3) inhibitor. When administered chronically, it effectively prevents the formation of blood clots, and when given in high doses over a short period, it induces the dilation of blood vessels. When combined with aspirin, it is an FDA-approved treatment for secondary prevention of stroke (138). In a screening of the Prestwick Chemical Library, which contains 880 biologically active molecules, Mulero and colleagues identified dipyridamole as a compound that competes with CaNA for binding to the RCAN1 peptide (139). Functional assessments of its inhibitory effect on CaN activity revealed that dipyridamole does not interferes with phosphatase activity. Its mechanism of action appears to involve a specific interaction with NFAT substrates. In line with this, dipyridamole inhibits NFAT nuclear translocation, NFAT activity and the expression of NFAT-dependent cytokine genes in human T cells (139). It has been suggested that, beyond its antiplatelet activity, this drug may also have an immunomodulatory effect in vivo (140, 141).
The Inhibitors of NFAT-Calcineurin Association (INCAs) have been selected based on their ability to compete with the VIVIT peptide for binding to CaN. Among these, three compounds, namely INCA-1, INCA-2 and INCA-6 are capable of completely displacing VIVIT from CaN, and they achieve this at low micromolar concentrations by inducing an allosteric change in the NFAT-binding site (142). It is important to note, that the binding site of INCA1,2 and 6 is centered on Cys-266 of CaNA and does not involve PxIxIT core motif. Interestingly, particular INCAs differ in their mechanisms of action. For instance, INCA-6 is known to act on NFAT activity whereas the physiological effects of INCA2 are associated with a general inhibition of CaN activity (142). However, concentration-dependent cytotoxicity has been reported for all INCAs, suggesting potential limitation in their use in biological systems (142, 143).
A-285222, a member of bis-trifluoromethyl-pyrazole class, falls within the category of immunosuppressive agents known for their ability to inhibit NFAT activity in both human and non-human primate cells through a calcineurin-independent mechanism (144). When applied to intact T cells, A-285222 maintains NFAT in a phosphorylated state within the cytosol, preventing its nuclear accumulation, all while leaving AP-1 or NF-κB activation unaffected. Consequently, this drug effectively blocks NFAT-dependent cytokine gene transcription. It is important to note that while the drug shows potential as an immunosuppressive agent, it is associated with severe side effects, particularly neurotoxicity. Nevertheless, various cell-specific effects have been demonstrated (145–147).
Q134R is a hydroxyquinoline derivative known for its ability to penetrate the brain and inhibit both the induction of NFAT transcriptional activity and caspase-3 activity (148). Importantly, this drug does not have any impact on CaN itself, which means it avoids the immunosuppressive adverse effects associated with other agents. Preclinical studies have showed its safety, good stability, and acceptable oral bioavailability. Phase I/A trials have been successfully completed, and the drug is currently progressing into phase II trials.
Accumulating evidence suggests that NFAT activity is involved in several neuropathologies (59). In nervous tissue, abnormalities in CaN/NFAT signaling are increasingly related to a variety of pathological features associated with Alzheimer’s disease (AD), including synaptic dysfunction, glial activation, and cell death (11) The prevalence of studies, as will be discussed in subsequent sections, indicates a causative relationship between NFAT and AD, and some of the first evidence has been provided by Abdul and colleagues who demonstrated a relationship between amyloid toxicity and NFAT signaling (10).
A postmortem study of human hippocampal tissue revealed increased nuclear translocation of individual NFAT members at different stages of AD, which correlated with the severity of cognitive decline (11). NFATc2 activation is especially critical in the early stages of AD, while selective NFATc4 activation occurs later when the control of neuronal Ca2+ homeostasis and reactive oxidative species production is lost, leading to further neurodegeneration, neuronal death, and ultimately, dementia (11). Moreover, levels of several cytokines increase as NFATc2 accumulates in the nucleus (11), which is consistent with its role in neuroinflammation occurring during the early stage of AD. Excessive activation of NFATc2/c4 at different stages of AD progression may contribute to synaptic dysfunction and cognitive decline.
In agreement with this hypothesis, inhibition of CaN/NFAT signaling pathway at each stage of AD progression should prevent degeneration of neuronal processes and slow down cognitive decline. Numerous studies on animal models of AD have demonstrated that CaN inhibitors, such as tacrolimus, showed neuroprotective and/or cognitive enhancing properties and extended lifespan (123, 149–151). In addition, Abdul et al. suggested that Aβ-induced neurodegeneration is associated with selective changes in NFAT signaling (10). In line with that, over-activation of CaN, enhancing nuclear localization of NFATc2 and NFATc4, correlated with increased dementia severity in the human hippocampus, while the subcellular localization of NFATc1 was cytoplasmic (10). Increased NFATc2 activation was revealed in AD patients with mild cognitive impairments, whereas NFATc4 showed a high nuclear accumulation in patients with severe dementia and AD (10).
While these results indicate the differential contribution of NFAT isoforms to AD pathology, other reports suggest that NFAT may be an essential component of signaling pathways pharmacologically targeted in AD treatment. Lithium has been proposed as a treatment for AD and other neurodegenerative disorders (152). However, its clinical use is limited due to a high rate of associated adverse side effects, and the mechanisms underlying those effects are not fully understood. It has been demonstrated that lithium can act as a repressor of GSK-3, with Ki values for GSK-3α (~3.5 mM) and GSK-3β (~2.0 mM) that exceed therapeutic lithium serum concentrations in humans (0.5–1.2 mM) (153–155). However, lithium, like other small molecule inhibitors, is unable to selectively suppress the activity of GSK-3 isoforms, and its impact on cognitive function is lost in aged AD transgenic mice (156). For these reasons, genetic approaches have been employed to assess whether there are isoform-specific effects of GSK-3. While the majority of these studies focuses on the contribution of GSK-3β, a growing body of evidence indicates a distinct role for both isoforms in Aβ pathology (157–160). For instance, in vitro and in vivo studies have revealed that hyperphosphorylation of tau promoted by GSK-3β leads to the formation of neurofibrillary tangles, which eventually trigger neurodegenerative conditions (161–167). It has also been reported that cleavage of the amyloid precursor protein (APP) into beta-amyloid peptide is meditated by GSK-3 β (166). In addition to APP, presenilin-1 (PS1) is involved in the aggregation of the pathogenic Aβ peptide and is regulated by GSK-3β (168, 169). This is supported by the study of Ly and colleagues, which showed that specific suppression of GSK-3β, but not GSK-3α, reduced Aβ formation via NF-αB-dependent transcriptional control of β-secretase 1 (170). In contrast, Hurtado et al. (171), using AAV-delivered shRNAs and GSK-3α conditional knockout mice, suggested that GSK-3α plays a predominant role in AD pathology. However, this study was limited by the use of newborn AD model mice and putative compensatory changes in the other GSK isoform. Given that overactivated GSK-3 is hypothesized to play a central role in the pathogenesis of AD (159), the beneficial effect of lithium would stem from the normalization of GSK-3 activity and the prevention of progressive Aβ production as well as local microglial-mediated inflammatory responses.
Transgenic mice with conditional expression of dominant-negative GSK-3β showed increased neuronal apoptosis and impaired motor coordination, a phenotype that can be reversed by the expression of a dominant-negative GSK-3β form (172). It has also been hypothesized that treatment with therapeutic levels of lithium can promote neuronal loss through GSK-3β inhibition (152). These observations strongly indicate that GSK-3 activity is critical for the viability of adult neurons, and any manipulations beyond physiological GSK-3 levels may pose a potential risk of neurological toxicity. A similar effect was observed in different cell types, including Jurkat cells, differentiated immortalized hippocampal neurons (173), HL-60 promyeoloblast cells (174), human lung carcinoma A549 cells (175), human choroidal melanoma cells (176), human leukemia NB4 cells (177), K562 human erythroleukemia cells (178), human cardiomyocytes (179) and multiple myeloma (180). On the other hand, an expanding body of literature has provided substantial evidence that lithium can confer neuroprotection against various insults [for a comprehensive review, refer to (181)].
Interestingly, lithium-induced apoptosis and motor deficits were associated with elevated nuclear accumulation of NFATc4 and NFATc3, leading to increased Fas ligand (FasL) expression and its activation (152). The opposite effect was observed in mice lacking the Fas-receptor (lpr mice) or following CsA treatment. In addition, activation of the CaN/NFAT/FasL death signaling has been suggested in methamphetamine-induced neuronal apoptosis (182). The link between FasL regulation by NFAT is interesting as Fas and FasL are associated with neuronal degeneration in the AD brain and participate in Aβ-mediated cell death (183). In vivo treatment with CaN or NFAT inhibitors abolished NFAT-mediated FasL expression and attenuated neuronal apoptosis (184).
Experimental evidence suggests a possible role of NFATs in mediating the survival response. It has been demonstrated that in the presence of growth factors and neuronal activity mimicked by high extracellular KCl concentrations, both endogenous NFATc4 and exogenously expressed NFATc4 were localized to the nucleus of granule neurons (54). On the contrary, serum/K+ deprivation led to NFATc4 nuclear export, which was strongly associated with the induction of neuronal apoptosis. Treatment with the GSK-3 inhibitor lithium chloride blocked the nuclear export of NFATc4 and cell death, suggesting a correlation between NFAT localization and neuronal survival. In line with that, NFATc4 knockdown resulted in enhanced apoptosis, observed even in a rich culture medium and high K+. The expression of a constitutively active form of NFAT prevented neuronal cells from apoptosis induced by low K+ or growth factor deprivation (54).
CaN/NFAT signaling has been recognized as a target for Aβ pathology in numerous studies [for review, see (3, 11)], highlighting the importance of Aβ-mediated Ca2+ homeostasis deregulation, which creates a favorable environment for CaN/NFAT activation. A report by Wu and colleagues showed that short exposure of primary cortical neurons to Aβ oligomers led to the dynamic progression of CaN activation and resulted in morphological changes in spines and postsynaptic proteins, while longer exposure led to NFAT accumulation in the nucleus and significant spine loss (35). Similarly, the loss of spines and dendritic branching simplification evoked by Aβ treatment in primary neurons were mimicked by constitutively active NFAT (185). Transfection with VIVIT-GFP before Aβ exposure greatly improved neuronal morphology despite the persistence of Aβ in the culture medium. Spine atrophy and neuronal abnormalities seen in the vicinity of amyloid plaques in APP/PS1 mice were prevented when AAV2-VIVIT was delivered by stereotactic intracortical injections (185). This suggests that selective disruption of CaN-NFAT interaction may be protective against Aβ synaptotoxicity. These findings also illuminate a possible mechanism underlying the lack of cognitive improvement by GSK-3 inhibitors in an AD clinical trial (186). Notably, Aβ-activated GSK-3 potentiates further Aβ production, creating a positive feedback loop. Activated GSK-3 phosphorylates NFAT, inhibiting CAN/NFAT apoptotic pathway, and consequently, GSK-3 inhibitors release this inhibitory brake, promoting NFAT-mediated neurodegeneration (186). Similarly, Aβ treatment of murine hippocampal neurons caused dysregulation in intracellular Ca2+ and impaired dendritic and axonal transport of BDNF (187). Exposure of mixed neuron/astrocyte culture to Aβ stimulated CaN/NFAT activity and triggered CaN proteolytic cleavage, resulting in an overall increase in intracellular CaN activity (188). The accumulation of both CaN and NFATc4 (but not NFATc2) in the nucleus of hippocampal tissue positively correlated with a higher level of soluble Aβ, which steadily increased as the severity of dementia progressed (10).
Mounting evidence indicates that activation of CaN/NFAT signaling may be linked to enhanced generation of Aβ peptides (3, 36). To support this, Hong and colleagues (189) showed that FK506 administered to 8-month-old APP/PS1 double transgenic mice reduced Aβ accumulation in the cortex and hippocampus while increasing matrix metalloproteinase-9 (MMP-9) expression, which is known to degrade Aβ (190). Moreover, increased MMP-9 colocalized with astrocytic marker GFAP (glial fibrillary acidic protein) in FK506-treated mice indicating that drug-dependent MMP-9 up-regulation originates from activated astrocytes. In line with that, no correlation between MMP-9 immunoreactivity and the neuronal marker NeuN was observed, though FK506 increased the level of PSD-95 and synaptophysin, proteins involved in postsynaptic density formation and vesicular neurotransmitter release, respectively. Similarly, intrahippocampal injections of AAV2-VIVIT under the astrocyte-specific promoter reduced Aβ pathology, normalized spontaneous synaptic activity, prevented dendritic degeneration, and improved cognition in the 5xFAD mouse model of AD (36). Furthermore, AAV2-mediated NFAT inhibition increased glutamate transporter-1 expression in hippocampal astrocytes and reduced the number of glutamate-evoked transients (36), suggesting CaN/NFAT-dependent loss of glutamate regulatory properties underlying hyperexcitability in AD. Another study pointed out astrocytic CaN/NFATc4 signaling activated in response to Ca2+ overload as one of the key pathological mechanisms driving Aβ generation by inducing β-secretase 1 (BACE1) transcription (191) (Figure 4).
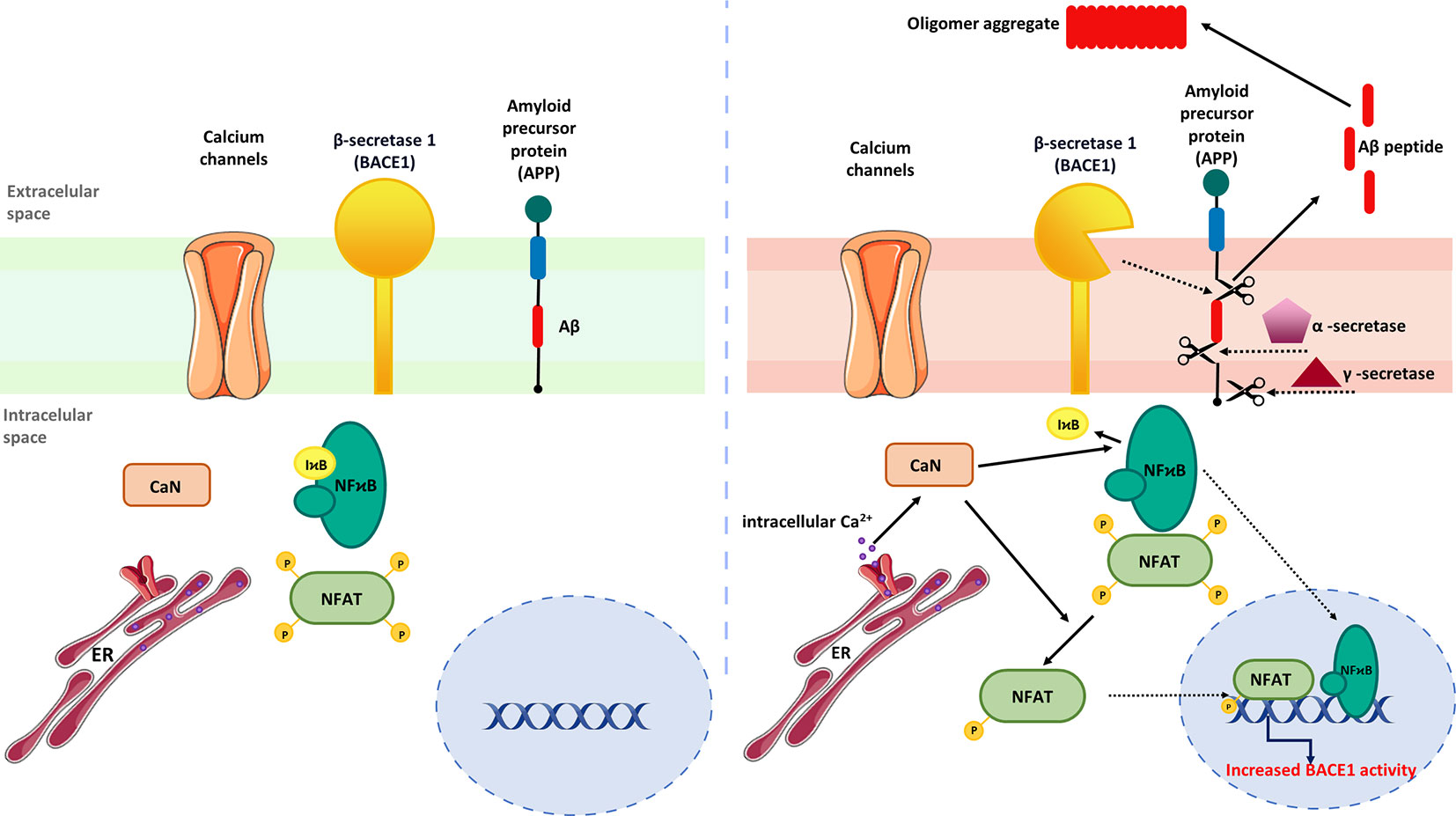
Figure 4 Regulation of astrocytic Aβ metabolism by CaN/NFAT signaling. (Left) In steady-state conditions, BACE expression is maintained at low level. (Right) Activation of astrocytes by upstream inflammatory mediators stimulates NFAT and NF-κB nuclear translocation and BACE1 upregulation. Increased BACE1 activity is associated with abnormal production of Aβ, which subsequently form amyloid plaques.
BACE1 protein and activity were found to be elevated in the AD brain, suggesting that BCAE1 up-regulation may be a phenomenon occurring early in AD or accelerating AD progression (192). Importantly, BACE1-/- mice are devoid of Aβ amyloidosis, electrophysiological dysfunctions, and cognitive deficits (192), implying targeted BACE1 inhibition to improve Aβ-mediated loss of cognitive function in humans with AD. Although not verified in AD models, Mei and coworkers demonstrated that NFATc4 directly binds the BACE1 gene promoter and regulates its expression (191). It is worth noting that the development of potent BACE1 inhibitors presents numerous challenges. One of the major hurdles is the structural similarity of BACE1 to other aspartyl proteases, a family that includes several enzymes widely expressed through the human body, such as BACE2, pepsin, renin, cathepsin D, and cathepsin E (193). Thus, it is imperative to develop effective BACE1 inhibitors without affecting other proteases and to exclude off‐target side effects (194, 195). Furthermore, another aspect of discovering successful BACE1 inhibitors is the relativity large size of the BACE1 active site (195). Moreover, another limiting factor is related to the ability of these molecules to pass through the blood-brain barrier (196). To date, despite tremendous efforts, none of the BACE1 inhibitors has succeeded in demonstrating clinical value.
Many studies now point to the contribution of neuroinflammation to the progression of neuropathological changes observed in AD. One of the signs that emerges early in the course of AD is astrocyte activation, which becomes more prominent during later stages when the amyloid and tau pathology is extensive (197). It is hypothesized that CaN/NFAT signaling serves as a critical mechanism triggering astrocyte phenotype switching and plays a pivotal role in astrogliosis and brain neuroinflammation. In astrocytes and microglia, CaN/NFAT is activated in response to a variety of neuroinflammatory mediators, including pro-inflammatory cytokines, glutamate, ATP, S100 protein, thrombin, vascular-injury factors, and Aβ. Once activated, CaN/NFAT may further potentiate the inflammatory response by driving the expression of numerous inflammatory factors, many of which are elevated in AD [reviewed in (198)]. It is well-accepted that Ca2+ homeostatic imbalance may aggravate AD through the pathological activation of neuronal networks but, in contrast to neurons, little is known about Ca2+ signaling specifically linked to glial CaN/NFAT activation. L-type voltage-dependent Ca2+ channels are likely to play a role, but the contribution of other plasma membrane transporters as well as those located in the ER cannot be ruled out (199).
Furthermore, CaN-dependent signaling may be a nodal point linking Ca2+ dyshomeostasis, Aβ accumulation, and neuroinflammation. In primary neurons, Aβ treatment stimulated a Ca2+-dependent protease calpain, which cleaves C-terminal autoinhibitory domain, producing a constitutively active truncated version of CaN (ΔCaN, 48 kDa CN-Aα) insensitive to Ca2+/CaM regulation (200) (Figure 5).
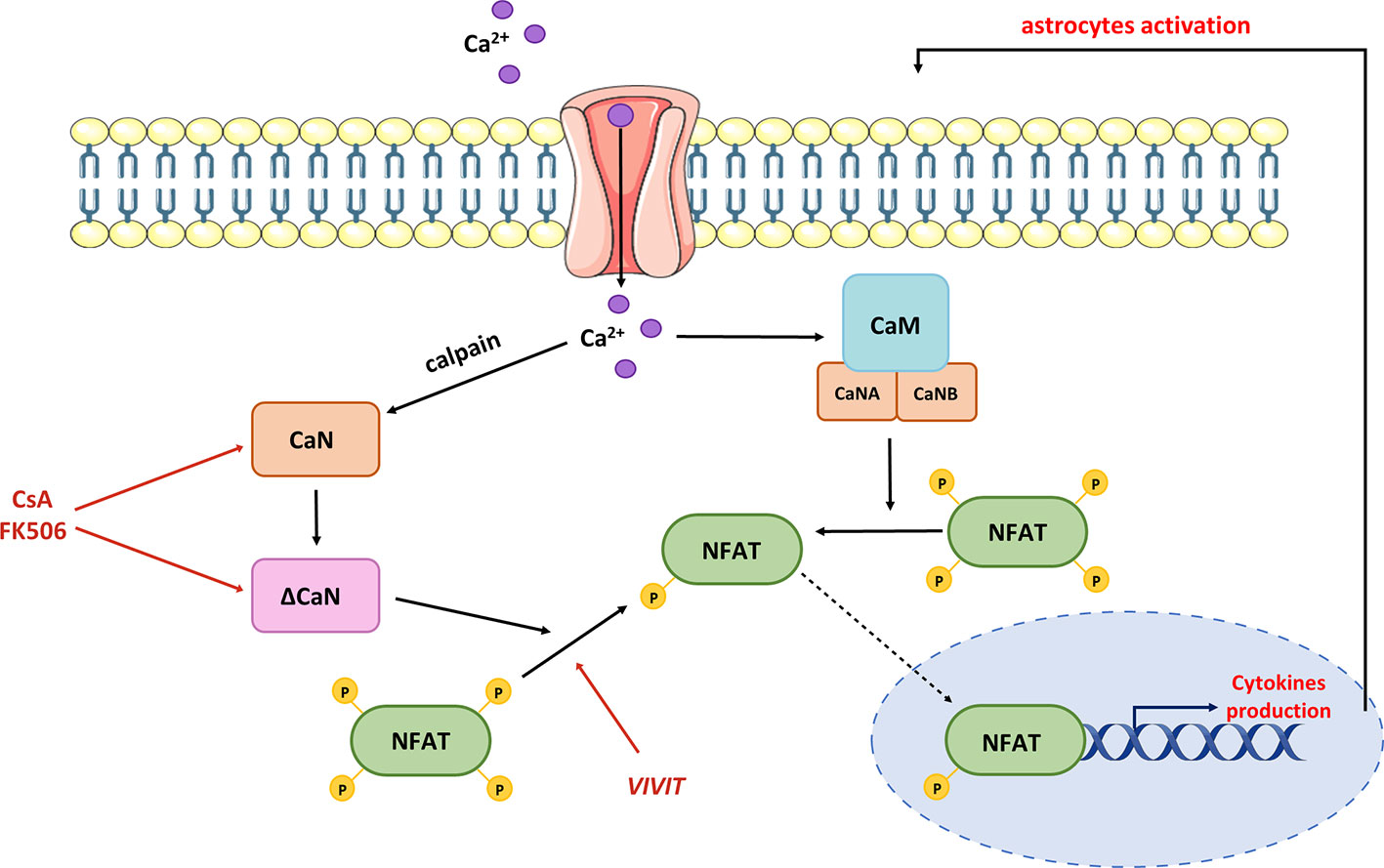
Figure 5 Contribution of hyperactive CaN/NFAT to pro-inflammatory cytokine production. Dysregulation of Ca2+ homeostasis leads to the production of truncated CaN fragment (ΔCaN), which is constitutively active. Hyperactivated CaN/NFAT sustains prolonged astrocytic activation and chronic neuroinflammation through a continuous stimulation of pro-inflammatory cytokine genes. Normalization of CaN/NFAT activity may be provided by commercial immunosuppressants such as cyclosporine A and tacrolimus (FK506), or the VIVIT peptide.
High levels of ΔCaN were detected in areas surrounding amyloid plaques in mouse and human brains (201, 202). Moreover, the levels of ΔCaN and active calpain correlated with one another in human hippocampal tissues of individuals with mild cognitive impairments when compared to age-matched controls (188). These changes were further linked to increased proteolysis of the NMDA receptor subunit 2B, which is necessary for long-term potentiation and memory formation (203, 204). This provides a mechanism by which oligomeric Aβ may contribute to brain degeneration through calpain-mediated CaN proteolysis and further overactivation of CaN/NFAT signaling. However, the molecular phenotype of ΔCaN-expressing astrocytes and their role in AD pathology have not been thoroughly studied. In primary neuronal cultures, heterologous expression of ΔCaN activated the transcription of several genes involved in immune response and morphogenesis (205). As many of these transcripts encode releasable cytokines and chemokines, CaN/NFAT signaling in activated astrocytes may contribute to cerebral vascular damage, reducing micronutrient delivery and compromising neuronal-glia interaction. The hypothesis of impaired microcirculation underlying AD development was postulated nearly 30 years ago (206) and has been widely discussed in multiple studies (207, 208). The plausible role of CaN/NFAT in AD-related vascular pathologies is further supported by recent work showing the upregulation of ΔCaN and NFAT signaling in astrocytes nearby regions of small vessel damage (209). Further evidence of aberrant astrocytic Ca2+/CaN/NFAT signaling contributing to vascular pathology associated with cognitive decline and dementia comes from the latest study of Sompol and colleagues (210). Using a diet-based model of hyperhomocysteinemia (HHcy), which recapitulates numerous features of AD, they demonstrated that VIVIT-mediated NFAT inhibition ameliorated astrocytic reactivity and improved blood flow in arterioles and capillaries. Moreover, the suppression of NFAT signaling preserved CA1 synaptic function and improved the cognitive performance of HHcy diet mice.
In addition to CaN overactivation studies, inhibition with either pharmacological drugs or targeted peptides revealed a similar relationship between CaN/NFAT and neuroinflammation. Targeting AAV2-VIVIT peptide to hippocampal astrocytes in APP/PS1 mice reduced glial activation, lowered Aβ levels, and improved cognitive and synaptic function (211). These findings align with previous reports showing high levels of CaN in activated astrocytes and its role in phenotype switching via NFAT signaling (46, 84, 205). Although the upstream signals leading to pathological activation of CaN in astrocytes are not well-characterized, heterologous expression of a constitutively active form of CaN upregulated genes linked to the activated phenotype, as well as numerous inflammatory-related genes (3). Astrocytic CaN/NFAT signaling has also been demonstrated to mediate the neurotoxic effects of several factors implicated in AD pathogenesis, including TNFα, CCL2, Cox2, GM-CSF, IL-6, IL-1β, and other cytokines (62, 202, 212–214). For instance, IL-1β promoted CaN/NFAT activation in primary astrocytes through IL-1 receptors and L-type Ca2+ channels without affecting CaN or NFAT expression levels (212). Interestingly, activated CaN/NFAT caused robust activation of CaN/NFAT in adjacent astrocytes via paracrine signaling. This observation places CaN/NFAT in the center of a positive feedback loop controlling local production of neuroinflammatory mediators.
An interesting approach based on reprogramming macrophages to become anti-inflammatory was proposed in a study conducted by Escolano and coworkers (215). By introducing the LxVP peptide that interferes with CaN-NFAT binding (for exact mechanism of action see Pharmacological inhibitors of CaN/NFAT with potential use in AD treatment section) they demonstrated that systemic or local peptide delivery via adenoviral gene transfer attenuated the inflammatory response in vivo. Mechanistically, LxVP-mediated CaN inhibition reduces the activity of MKP-1, a dual specificity protein phosphatase, releasing p38 MAPK kinase from MKP-1 repression. In murine mouse models, p38 MAPK kinase was associated with the activation of macrophage reprogramming (216). It has been reported that high dose of FK506, 500 times higher than necessary to suppress CaN, induces short-term p38 MAP kinase activation, reaching its peak at 30 min and subsequently decreasing (215). Furthermore, the application of pharmacological, non-toxic doses of CsA or FK506 effectively inhibits CaN activity, but does not trigger p38 MAPK kinase activation (215). These results strongly suggest that the anti-inflammatory phenotype of macrophages induced by LxVP requires the involvement of p38 MAP kinase. Additionally, the VIVIT peptide also failed to induce p38 activation (215). This unique feature of LxVP distinguishes its action from the properties of other CaN/NFAT inhibitors, such as CsA or FK506. However, before testing this therapy in patients, it would be particularly interesting to unravel the relationship between anti-inflammatory M2 macrophages and macrophages induced by LxVP peptide. Other strategies controlling immune cell function, such as NFAT’s O-linked β-N-acetyl glucosamine modification (217), genetic deletion of soluble epoxide hydrolase (218), or oxygen-ozone therapy that activates immune function and suppresses inflammatory responses through up-regulation of NF-κB, NFAT, and AP-1 signaling (219), should also be given special consideration.
Targeted inhibition of CaN/NFAT signaling, either by naturally occurring compounds or synthetic drugs, may effectively reduce neuroinflammation, thus offering a promising strategy in the treatment of AD (220, 221). However, the translational potential of many CaN inhibitors is markedly limited due to severe adverse effects, including neuro-muscular and renal dysfunction or progressive lymphopenia (222, 223). Despite many advantages over peptide/protein-based drugs, small chemical inhibitors of NFAT have not been widely described. Dipyridamoles can reduce inflammation and cytotoxicity and exert a neuroprotective effect under certain conditions (141, 224). However, their use is not without side effects (225). Another class of small organic molecules, known as INCAs, was initially promising due to their ability to displace VIVIT at low micromolar concentrations, but it turned out to be cytotoxic for certain cell lines when tested. Nonetheless, when administered at non-toxic low micromolar concentrations, INCA-2 and INCA-6 prevented the induction of mRNAs encoding TNFα, interferon-γ, and macrophage inflammatory proteins MIP-1α and MIP-1β (142), as well as some pro-inflammatory cytokines and chemokines (226). Other NFAT inhibitors, such as A-285222, have shown to reduce inflammation, but the experimental data on their use in neuronal or glial cells are very limited (227). Recently, a new hydroxyquinoline derivative, Q134R, has been demonstrated to reduce glial reactivity markers and improve synaptic function, without affecting Aβ load (148). Moreover, Q134R improved cognitive performance and showed no signs of lymphopenia, suggesting its efficacy comparable to CaN inhibitors but with fewer side effects.
The role of NFAT signaling in AD pathogenesis has been demonstrated in numerous studies, but the upstream signaling leading to CaN/NFAT deregulation remains largely unresolved. Therefore, in this section we discuss how the function of receptors associated with AD affects CaN/NFAT signaling (Figure 6).
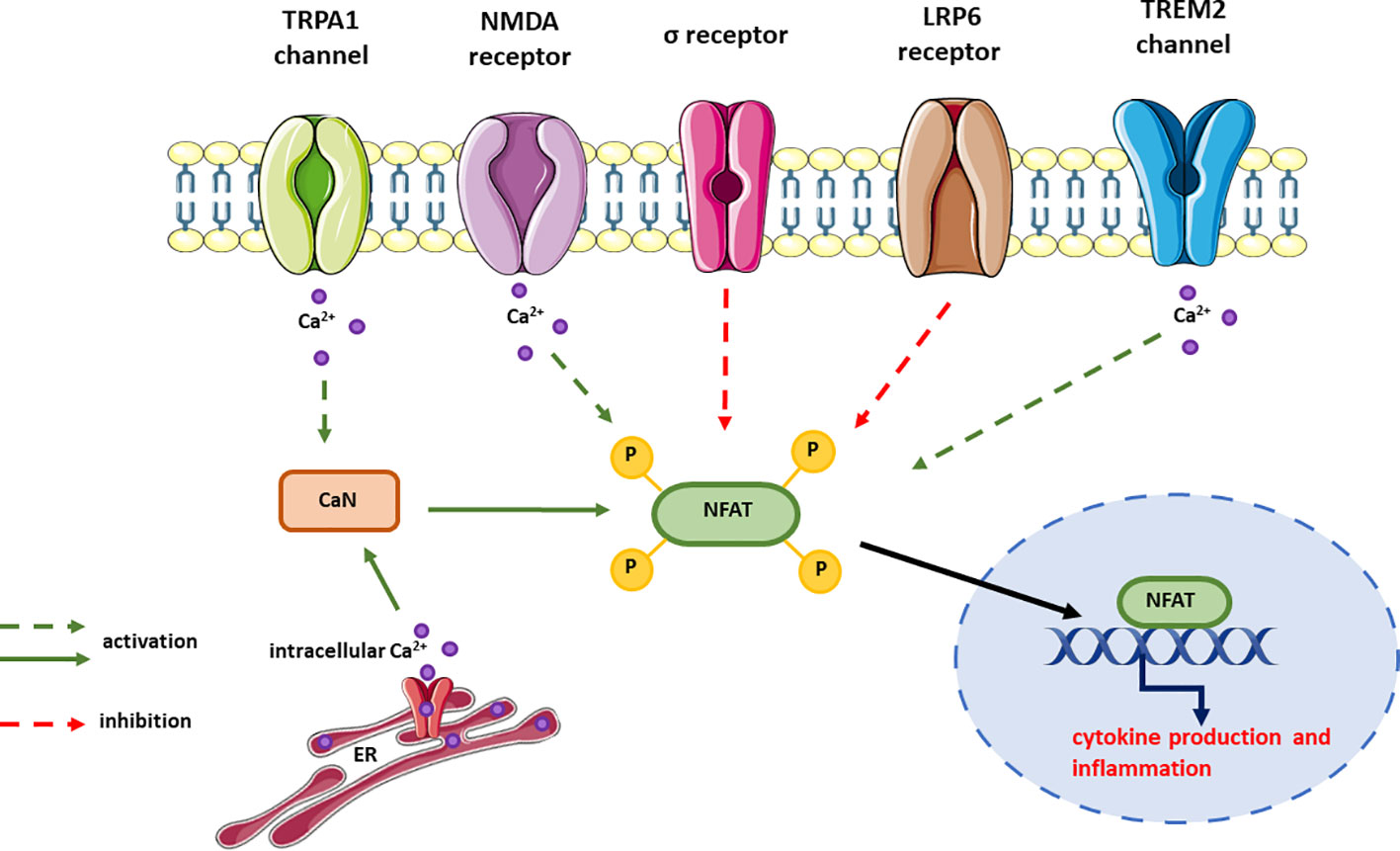
Figure 6 Receptors signaling through CaN/NFAT and their effect on NFAT activation. In most cases, the signaling pathways upstream NFAT but downstream the receptor is unknown and this is expressed as dotted line in the figure. The effect on NFAT activity has been measured either by changes in cytokine expression or release, or the activity of NFAT reporter.
As previously discussed, Aβ-induced Ca2+ overload plays a pivotal role in cytokine secretion by activating CaN and its downstream targets, including NFAT and NF-κB (198). However, the intracellular proteins that regulate Aβ-mediated Ca2+ influx and drive the inflammatory response are still largely unresolved. One of the strong candidates is the transient receptor potential ankyrin 1 (TRPA1), which is a Ca2+-permeable nonselective channel belonging to the TRP channel superfamily. It is widely expressed in the sensory neurons of the dorsal root ganglia, trigeminal ganglia, and nodose ganglia, as well as in hair cells and non-neuronal cells (228). TRPA1 responds to a variety of exogenous irritants and endogenous agonists to mediate inflammation and transmit pain (229–232). Due to these properties, TRPA1 is considered a promising target for novel analgesic and anti-inflammatory drugs. Furthermore, it is involved in temperature sensing and may play a role in the detection of infrared radiation and the avoidance of nociceptive heat (233–238).
Lee and colleagues demonstrated that TRPA1 is upregulated in the hippocampus of APP/PS1 mice (239). In this mouse model, Aβ triggers TRPA1-mediated Ca2+ accumulation and hyperexcitability of CA1 hippocampal neurons, suggesting that TRPA1 may function during the early onset of AD (240). Aβ-induced TRPA1-Ca2+ signaling has been shown to be a critical event in the activation of CaN/NFAT, leading to cytokine production and inflammation in astrocytes (239, 241). Consistent with this, genetic ablation of TRPA1 channel function in APP/PS1 mouse reduces Aβ-induced activation of CaN, decreases NFAT’s DNA-binding activity in astrocytes, and lowers the levels of pro-inflammatory cytokines IL-1β, IL-6, and IL-4 in the brain. Furthermore, loss of TRPA1 function ameliorates AD progression and improves behavioral performance (239), highlighting the importance of astrocytic TRPA1-Ca2+-CaN-NFAT signaling in the inflammatory process and AD progression.
Another channel implicated in AD pathology is the sigma receptor, a Ca2+-sensitive chaperone located at the mitochondria-associated endoplasmic reticulum (242). Upon ligand binding, the sigma receptor translocates to the plasma membrane and interacts with various ion channels and G protein-coupled receptors (243). In terms of its mechanism, it operates as more of a signaling modulator than a conventional receptor, and its function encompass the regulation of lipid metabolism, control of both voltage- and non-voltage ion channels, maintenance of intracellular Ca2+ homeostasis, modulation of electrical activity, and potentially several other roles (244). Early studies have demonstrated the loss of sigma receptor in the CA1 area of the anterior hippocampus in AD patients, which correlates with damage to CA1 cells (245). The depletion of sigma receptor in the brain is thought to manifest early in the progression of AD, primarily impacting the frontal, temporal, and occipital brain regions (246). The development of preclinical models of AD has allowed researchers to determine the correlation between receptor level and the severity of AD, as well as uncover the neuroprotective properties of sigma receptor agonists (247, 248). For instance, BD-7373 and CB-184, agonists of the sigma-2 receptor subtype, exhibit a potent inhibitory effect on NFAT and NF-κB transcription, leading to decreased expression of TNFα, IL-2, and COX-2 genes, potentially reducing brain inflammation (249). Activation of the sigma-1 receptor, on the other hand, may inhibit γ-secretase activity and reduce Aβ production (250). However, Aβ accumulation and subsequent activation of CaN/NFAT have been shown to induce sigma receptor degradation through NFAT-dependent induction of BIP protein expression, promoting E3 ligase recruitment (251).
TREM2 was initially identified and characterized in human dendritic cells derived from blood monocytes and cultured in vitro with the granulocyte macrophage colony stimulating factor and IL-4 (252). It belongs to the immunoglobulin-lectin-like receptor superfamily and plays a role in the development of the AD phenotype (253). TREM2 is predominantly expressed in tissue macrophages, where it can be found both on cell surface and within intracellular compartments. Macrophages expressing TREM2 are located in various sites, including microglia in the central nervous system, specific macrophage subset in the liver, and osteoclast in bone (253). Research has shown that alterations in TREM2 expression affect multiple functions of microglia (254). Increasing TREM2 results in elevated level of chemokine receptors, enhanced cell migration, and improved phagocytosis. Conversely, decreased TREM2 expression inhibits the engulfing of apoptotic cells while increasing the expression of pro-inflammatory cytokines (254).
Characterization of the microglial transcriptome in TREM2-deficient mice has demonstrated the essential role of TREM2 in microglial response during Aβ accumulation (255). TREM2 can be directly modulated by Aβ and other components of Aβ plaques (253). Several studies have reported that TREM2 activation increases intracellular Ca2+ level and activates the NFAT reporter system, suggesting that NFAT may transmit signals downstream of TREM2 (252, 256, 257). The precise mechanism of TREM2-dependent NFAT activation in AD is not entirely clear, but a recent study by Zhao et al. revealed that Aβ42 oligomers promote TREM2-DAP12 interaction and SYK kinase phosphorylation, which plays an important role in NFAT signaling (258). Involvement of SYK kinase activating the PLCγ-CaN-NFAT pathway was also suggested by Lessard and colleagues (259). These authors demonstrated that the loss-of-function TREM2 mutation variant R47H, which has been associated with a higher incidence of AD (260), reduced Aβ internalization and NFAT signaling, establishing a direct link between Aβ aggregates, TREM2, and NFAT transcriptional activity. Modulation of NFAT transcriptional activity may also underlie TREM2’s ability to sustain microglial response to Aβ accumulation and influence the production of inflammatory cytokines in vivo. This hypothesis is supported by the fact that apoptotic cells transduce the NFAT signal via TREM2, consistent with its function as a sensor of anionic and zwitterionic lipids presented on the surface of neurons exposed to Aβ (261, 262). The accumulation of phosphatidylserine (PS) and phosphatidylethanolamine (PE) on the neuronal cell membrane in the AD mouse brain (263) suggests that PC and PE may transduce signals via TREM2-NFAT (261, 264), potentially enhancing TREM2 activity and promoting a protective phenotype in microglia. Although it remains unclear which TREM2-regulated target genes may be responsible for the increased risk of AD, recent work with engineered TREM2 agonistic antibodies suggests that many of these genes may contain an NFAT response element (265). Activation of TREM2 by dedicated antibodies has been shown to enhance microglia-dependent clearance of Aβ plaques in 5XFAD mice, indicating their potential clinical use in AD treatment. The rationale for strategies aimed at normalizing TREM2 expression/activity in AD treatment is further supported by the latest finding of increased Trem2 expression in 5XFAD mice (266).
Imidazoline 2 receptors (I2 receptors) represent a highly heterogenous group of non-adrenergic binding sites characterized by their high-affinity binding to [3H]-idazoxan (267). Cellular distribution studies have indicated their presence on the outer mitochondrial membrane, and they have been suggested to potentially serve as novel allosteric binding sites for monoamine oxidase (MAO). Interestingly, another unrelated binding site is brain creatine kinase (268). Numerous biochemical and preclinical investigations, conducted using animal models of brain injury (269–272), suggest that ligands targeting I2 receptors exhibit neuroprotective activity, in part by lowering body temperature – an effect known to be beneficial in cerebral ischemia (273). There is also evidence to suggest that I2 receptors ligands may mitigate neurodegenerative processes, including cognitive decline, neuroinflammation, and oxidative stress (274).
Indeed, postmortem analysis of brain samples derived from AD patients revealed a higher density of imidazoline 2 receptors (I2-IRs), and several pharmacological modulators of I2-IRs activity have been successfully tested to reduce AD hallmarks (275–277). The mechanisms underlying the beneficial effects of I2-IRs modulation are complex and, so far, largely unknown. Recent reports suggest that neuroprotective mechanisms may be ligand-specific. For instance, LSL6010 alone decreased Aβ plaque formation, tau hyperphosphorylation, and the expression of microglia markers in the 5XFAD mouse model. When administered in combination with donepezil, LSL6010 additionally reduced GFAP reactivity (276). In the same AD murine model, LSL60101 (garsevil) reduced the markers of oxidative stress and decreased the expression of the pro-apoptotic FADD protein in the hippocampus (275). Using the senescence-accelerated mouse prone 8 (SAMP8) model, which is considered a late-onset AD mouse model characterized by tau hyperphosphorylation and altered APP processing (278), Vassilopoulos and colleagues demonstrated that a newly synthesized I2-IRs ligand, named B06, reduced the expression of pro-inflammatory cytokines by inhibiting CaN/NFATc1 signaling (274). Therefore, new-generation I2-IRs ligands that affect NFAT-dependent transcription hold great neuroprotective potential in AD.
LRP6 belongs to the extended low-density lipoprotein receptor family and serves as a co-receptor in the canonical Wnt signaling. LRP6 is expressed in a widespread manner across human tissues, exhibiting both weak and strong expression patterns in different tissue types (279). Because of its function in Wnt/β-catenin pathway, it is required in the regulation of cell proliferation, specification, migration during development, and it is highly expressed in different types of cancer (280). Furthermore, clinical studies and genomic analysis have confirmed that LRP6 is associated with neurodegenerative diseases including AD (281). Conditional knockout of LRP6 in mice resulted in synaptic dysfunctions, accompanied by cognitive impairments and exacerbated memory deficits in the APP/PSEN1 AD model (282). Interestingly, downregulation of LRP6 expression and LRP6-mediated Wnt signaling were observed in the human brain affected by AD compared to age-matched controls (283). Several possible mechanisms have been proposed to explain how impaired LRP6 downstream signaling may worsen AD pathology. Firstly, LRP6 directly interacts with APP, and its deficiency may promote the amyloidogenic processing of APP, leading to increased endogenous Aβ levels (283). Secondly, LRP6 loss has been shown to increase the number of hippocampal astrocytes and microglia, promoting neuroinflammation through extensive production of pro-inflammatory cytokines (283). LRP6 may also contribute to AD pathology by regulating lipid metabolism, particularly ApoE-containing lipoproteins (284, 285). However, the role of glial and astrocytic LRP6 in the context of AD has not been widely studied. Recent research by Chow and colleagues revealed that LRP6 cell surface retention serves as a bimodal switch for astrocytic fuel metabolism (286). In the absence of LRP6, Wnt signaling activates the non-canonical Ca2+‐PKC‐NFAT axis, favoring the utilization of glutamate-derived glutamine and branched chain amino acids over glucose. Increased levels of both NFATc2 and NFATc4 were observed in Wnt‐exposed GFAP‐Lrp6–/– astrocytes, but NFATc2 appears to be the isoform that regulates mitochondrial glutamine metabolism and the acquisition of a reactive phenotype. Depletion of these amino acids from the astrocyte microenvironment may impact synaptic functions and contribute to cognitive and memory deficits. Remodeling of the metabolic framework in astrocytes is an early change associated with late-onset AD (287). In the latest study, non-canonical Wnt5a/CaMKII/NFAT signaling has been shown to participate in the release of inflammatory factors and modulate the activation of microglia (288).
Physiologically, NMDARs are central to development of nervous system are involved in a numerous forms of synaptic transmission underlying learning and memory formation (289, 290). In AD, abnormal activation of NMDARs by a glutamate highly released from glial cells stimulates a massive Ca2+ influx and aberrant processing of Ca2+/CaN signaling, promoting oxidative stress, neuroinflammation and cell death (291–293). Accumulating evidence indicates that function of NMDAR is dysregulated by Aβ and the disruption in Ca2+ homeostasis and glutamatergic synaptic transmission may be related to early cognitive deficits observed in AD (294–296). There are multiply potential way by which NMDAR contributes to Aβ pathology: first, Aβ are able to bind NMDAR extracellularly suggesting direct or indirect modulation of the receptor by amyloid β oligomers (297); second, Aβ facilitates NMDAR activation, which controls Aβ production and secretion (298, 299); third, NMDAR may be important mediator in Aβ-induced neurotoxicity (300) and fourth, due to the synaptic and extrasynaptic location, NMDAR may function as a downstream target in Aβ-induced synaptic depression (301). The interaction between Aβ and NMDAR rationalizes the clinical use of memantine, a non-competitive NMDAR antagonist, in combination with acetylcholine inhibitor donepezil, to improve cognitive performance and life quality of patients with moderate to severe AD (302). The beneficial effects of memantine administration may involve reduction in neuroinflammation and overall improvement of brain function although the mechanisms of drug action are not completely understood. The participation of CaN/NFAT cannot be unequivocally excluded as other NMDAR antagonists such as MK-801 are known to abolish NMDAR-dependent NFAT activation (303, 304). Nonetheless, memantine/donepezil did not improve excessive agitation (305) and hippocampal atrophy (306) and the effectiveness of this therapy has been questioned in patients in advanced AD stages (307).
Recently, Turcu et al. developed an optimized, non-cytotoxic, memantine-like NMDAR antagonist, UB-ALT-EV, with high metabolic stability, low micromolar activity and excellent electrophysiological profile as NMDAR blocker (308). The drug rescued defective locomotion and reversed the disrupted chemotaxis behavior in C. elegans, which constitutively express human Aβ, suggesting the protection against Aβ toxicity in a manner similar to memantine. Importantly, UB-ALT-EV also enhanced short- and long-term working memory in 5XFAD mice (308). Strikingly, both UB-ALT-EV and memantine normalized CaN protein level, but only UB-ALT-EV, increased NFATc1 phosphorylation, thereby preventing its nuclear translocation (266). Further evaluation of UB-ALT-EV-exerted neuroprotection demonstrated a reduction in the production of several pro-inflammatory cytokines regulated by CaN/NFAT signaling, as well as a decrease in oxidative stress due to enhanced expression of anti-inflammatory mediators in the 5XFAD model (266). These results collectively indicate that UB-ALT-EV’s capacity to reduce gliosis arises from its modulatory effect on NMDAR-dependent Ca2+ entry and CaN/NFATc1 downstream signaling.
In mature hippocampal neurons expressing both NMDAR2A and 2B subunits, Aβ-induced hippocampal dysfunctions and ER stress were largely reversed by ifenprodil, an antagonist of NMDAR2B subunits (297). Additionally, ifenprodil prevented the depletion of ER Ca2+ content, superoxide generation, and cell death, demonstrating the involvement of NMDAR2B in Aβ neurotoxicity. Considering that NMDAR-dependent NFAT activation and NFAT-mediated transcription are disrupted in primary neurons in the presence of ifenprodil (101), its anti-inflammatory action may involve the normalization of NFAT signaling.
In the Drosophila AD model, overexpression of human amyloid precursor protein (APP) induced synaptic hyperexcitability and concurrent upregulation of Ca2+-related signaling genes, including Dmca1D (L-type Ca2+ channel), CaN, and the inositol 1,4,5-triphosphate receptor (IP3R) (309). Mechanistically, exaggerated Dmca1D expression promoted APP-dependent Ca2+ overload, leading to increased CaN activity. This, in turn, triggered NFAT-dependent transcription of IP3R. IP3R is a large-conductance cation channel located in the ER membrane, responsible for cytoplasmic Ca2+ increases that control cytoplasmic and mitochondrial processes, thereby regulating cell survival (310, 311). Aberrant Ca2+ signaling resulting from IP3R dysregulation has been implicated in several neurodegenerative diseases, including AD, and ER stress-related neuronal injury (312). For instance, IP3R has been previously shown to interact with presenilin mutants causing the familiar form of AD (FAD), leading to its gain-of-function enhancement in an Aβ-independent manner (313, 314). This gain-of-function has been suggested as a key factor behind altered IP3R-mediated Ca2+ release in sub-saturating IP3 concentrations, serving as a highly predictive diagnostic feature of AD (315). In light of this, the normalization of IP3R-dependent signaling has been found to restore normal cell function and improve memory in FAD-causing presenilin knock-in mice (316), as well as in triple-transgenic mouse models of FAD (317, 318). Furthermore, Shao et al. demonstrated that NFAT-mediated IP3R upregulation significantly contributes to synaptic downscaling machinery. The restoration of IP3R expression blocked synaptic excitability and miniature excitatory postsynaptic current (mEPSC) frequency (309). Therefore, substantial evidence supports the role of IP3R in contributing to the deregulation of Ca2+ homeostasis observed in AD. IP3R dysfunction may drive a series of pathological events leading to disease progression.
IP3R-mediated depletion of ER stores triggers the activation of SOCE (store-operated Ca2+ entry) from the extracellular milieu across the plasma membrane, resulting in a subsequent increase in cytosolic Ca2+. Altered SOCE-mediated ER store refill is one of the hallmarks of AD, as reduced SOCE has been implicated in synaptic loss and cognitive decline in genetic mouse models as well as in human AD brain samples (319). However, the molecular mechanisms by which IP3R modulates SOCE in AD are not completely understood. A recent study by Sampieri et al. (320) demonstrated that ER depletion induced by G-protein coupled receptor and phospholipase C (PLC) activation stimulates the recruitment of IP3R to the STIM1 protein, which acts as the sensor for ER calcium load. The gradual decrease in STIM1 expression in the medial frontal gyrus of pathologically confirmed AD patients has been linked with disease progression and neurodegeneration mediated by the L-type voltage-dependent Ca2+ channel (321, 322). Interestingly, STIM1 expression is sensitive to inflammation (323, 324), suggesting that changes in Ca2+ influx through SOCE channels may occur during the early stages of AD development. Using a lipopolysaccharide (LPS)-induced model of AD neuroinflammation, Sun and coworkers (325) demonstrated that SOCE-mediated activation of the PLC/CaN/NFAT pathway up-regulated NADPH oxidase and NOD-like receptor family protein 1 (NLRP1) inflammasome, both playing a pivotal role in oxidative stress and neuronal inflammation. These studies collectively indicate that NFAT activation by pathological Ca2+ signals of different origins mediates neurotoxic Aβ effects and contributes to neuronal damage.
An open question remains regarding the control mechanisms of endogenous CaN/NFAT activity. One of the candidates is the regulator of calcineurin 1 (RCAN1), a small evolutionarily conserved protein that can directly bind to and inhibit CaN activity. RCAN1 has been implicated in various forms of brain degeneration, and its increased expression has been demonstrated in the cortex of patients with AD as well as during normal brain aging (326–329). A conclusive mechanistic explanation for the role of RCAN1 in neuronal death observed in AD has not yet been presented, although several hypotheses have been proposed. Overexpression of RCAN1 is known to induce the caspase-3 mediated apoptotic pathway, which can be blocked by the antioxidant lycopene, suggesting the involvement of oxidative stress (330). In line with this, Sun and coworkers proposed an isoform-specific regulation of RCAN1 by calcium overload, activating CaN/NFAT signaling and exacerbating caspase-3 mediated death (331). According to the study by Jing and colleagues, RCAN1 overexpression disrupts mitochondrial function and promotes apoptosis through the stabilization of adenine nucleotide translocator (ANT1) mRNA and Ca2+‐dependent induction of mitochondrial permeability transition pore opening (332). This is supported by the latest study, which shows the inhibition of NFAT and NF-κB transcriptional activity by the RCAN1 RNA aptamer R1SR13, resulting in the attenuation of apoptosis in neurons (333). RCAN1 may also contribute to AD pathology by enhancing N-glycosylation in the ER, thus significantly increasing Aβ production (334). The amyloid proteins and RCAN1 appear to be mutually regulated, as Aβ enhances RCAN1 expression, and RCAN1 reciprocally induces Aβ formation and potentiates its neurotoxicity (334–337). The transcription of RCAN1 can also be activated by NF-κB, a key mediator of brain inflammation in AD (338, 339), and repressed by its own protein in a negative loop. In this regulation, the activation of CaN/NFAT signaling attenuates NF- κB activity, whereas activated NF-κB potentiates NFAT-dependent transcription.
The function of RCAN has been demonstrated to be critical during development and in healthy synapses [reviewed in (340)]. Mice deficient in rcan1/2 displayed neurological symptoms similar to CaNAβ-null mice, including enhanced locomotor activity and deficits in working memory (341). Rcan1/2 loss-of-function impaired NFAT activation, suggesting that RCAN may also allow or facilitate CaN-NFAT coupling under certain conditions. This is further supported by a study of Liu and colleagues, which demonstrates that the phosphorylation of RCAN1 at Ser94 and Ser136 by TAK1 (Transforming growth factor beta-activated kinase 1) acts as a switch between inhibitory and permissive RCAN1 functions, thereby enhancing CaN signaling and facilitating NFATc1 nuclear translocation (342). This switch requires the formation of a multimolecular complex involving TAB2 (TGFβ activated kinase 1 binding protein 2) and RCAN1, followed by the recruitment of TAK1, TAB1 and CaN. This mechanism is not observed in rcan1/2-deficient mice. As TAK1 and its binding partner TAB1 can be activated by a variety of cytokines, including IL-1β or TNFα (343), RCAN1-dependent modulation of CaN activity may have profound consequences for glial response to pro-inflammatory stimuli. Other studies also confirmed that altering RCAN1 levels either by overexpression or knock-out approaches resulted in memory deficits and impaired synaptic plasticity, both of which are frequently observed in AD patients (344, 345).
Recently, RCAN1 knockdown or overexpression has been linked to age-related deficits in rest-activity and circadian rhythms, characteristic for AD and Down syndrome (346). In the latter, perturbed CaN/NFAT signaling was associated with a higher risk of developing amyloid pathology (347). RCAN1 has also been shown to interact with Dyrk1A, which is considered a candidate protein responsible for AD in the early stages of this disease (348). This interaction allows Dyrk1A-mediated RCAN1 phosphorylation and subsequent CaN inhibition, leading to reduced NFAT activity and enhanced Tau phosphorylation (349). Besides controlling RCAN1, Dyrk1 functions upstream of the GSK-3β kinase, inhibiting NFAT (349), and may directly phosphorylate the NFAT regulatory domain, counterbalancing the effects of CaN-mediated dephosphorylation (75). These observations suggest a direct link between Dyrk1 and RCAN1 in CaN/NFAT signaling, supporting the notion that altered RCAN1/Dyrk1 expression in AD may destabilize the NFAT circuit and contribute to neuropathogenic processes. In line with that, Dyrk1 inhibition by a novel drug, KVN93, reduced neuroinflammation, improved cognitive performance, and decreased Aβ plaque deposition in 5xFAD mice (350). A promising effect toward the amelioration of phenotypic defects observed in AD was also seen with CX-4945 (silmitasertib), which has already undergone clinical trials. Originally developed as a Dyrk1-targeting drug (351), CX-4945 turned out to be a dual Dyrk-1/GSK-3β inhibitor (352) with a strong modulatory effect on AD-related CaN/NFAT signaling and Tau phosphorylation in the mouse hippocampus.
The transcription factors of the NFAT family were originally characterized for their important role in the transcription of cytokine genes and other genes critical for the immune response. The role of NFAT in the neuroinflammatory response in AD is unquestionable. Yet, the reports emerging in recent years suggest that aberrant CaN/NFAT signaling may also play a central deleterious role in brain degeneration, linking amyloid pathology, Ca2+ dysregulation, and synapse deterioration. The molecular and phenotypic changes fueled by hyperactive CaN, and cell-specific maladaptive transcriptional programs, may arise early in AD and progress with cognitive decline. These deleterious changes in transcriptional control are observed both in neurons and astrocytes and likely involve the NFAT component. The numerous studies in transgenic animal AD models showing beneficial effects of CaN and/or NFAT inhibitors are consistent with this hypothesis. Further work is needed to better characterize the upstream signals leading to CaN/NFAT overactivation in AD and dissect the actions of this pathway on different transcriptional regulatory mechanisms. Moreover, it would be desirable to unravel how these changes drive synaptic malfunction and how targeted molecular interventions may help slow down cognitive decline.
JM: Conceptualization, Resources, Software, Visualization, Writing – original draft, Writing – review & editing. ML: Software, Supervision, Visualization, Writing – original draft, Writing – review & editing. TB: Conceptualization, Funding acquisition, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Science Centre (Narodowe Centrum Nauki) grant no. UMO-2019/33/B/NZ4/00587.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. A. s. Association. 2016 Alzheimer's disease facts and figures. Alzheimers Dement (2016) 12(4):459–509. doi: 10.1016/j.jalz.2016.03.001
2. Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res Bull (2016) 126(Pt 3):238–92. doi: 10.1016/j.brainresbull.2016.08.018
4. A. s. A. C. H. Workgroup. Calcium Hypothesis of Alzheimer's disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement (2017) 13(2):178–182.e17. doi: 10.1016/j.jalz.2016.12.006
5. Pchitskaya E, Popugaeva E, Bezprozvanny I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium (2018) 70:87–94. doi: 10.1016/j.ceca.2017.06.008
6. Vardjan N, Verkhratsky A, Zorec R. Astrocytic pathological calcium homeostasis and impaired vesicle trafficking in neurodegeneration. Int J Mol Sci (2017) 18(2). doi: 10.3390/ijms18020358
7. Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev (2018) 98(1):239–389. doi: 10.1152/physrev.00042.2016
8. Sobolczyk M, Boczek T. Astrocytic calcium and cAMP in neurodegenerative diseases. Front Cell Neurosci (2022) 16:889939. doi: 10.3389/fncel.2022.889939
9. Rodríguez-Giraldo M, González-Reyes RE, Ramírez-Guerrero S, Bonilla-Trilleras CE, Guardo-Maya S, Nava-Mesa MO. Astrocytes as a therapeutic target in alzheimer's disease-comprehensive review and recent developments. Int J Mol Sci (2022) 23(21). doi: 10.3390/ijms232113630
10. Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, et al. Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci (2009) 29(41):12957–69. doi: 10.1523/JNEUROSCI.1064-09.2009
11. Abdul HM, Furman JL, Sama MA, Mathis DM, Norris CM. NFATs and alzheimer's disease. Mol Cell Pharmacol (2010) 2(1):7–14.
13. Lian Q, Ladner CJ, Magnuson D, Lee JM. Selective changes of calcineurin (protein phosphatase 2B) activity in Alzheimer's disease cerebral cortex. Exp Neurol (2001) 167(1):158–65. doi: 10.1006/exnr.2000.7534
14. Reese LC, Taglialatela G. A role for calcineurin in Alzheimer's disease. Curr Neuropharmacol (2011) 9(4):685–92. doi: 10.2174/157015911798376316
15. Goto S, Matsukado Y, Mihara Y, Inoue N, Miyamoto E. Calcineurin in human brain and its relation to extrapyramidal system. Immunohistochemical study on postmortem human brains. Acta Neuropathol (1986) 72(2):150–6. doi: 10.1007/BF00685977
16. Goto S, Matsukado Y, Mihara Y, Inoue N, Miyamoto E. The distribution of calcineurin in rat brain by light and electron microscopic immunohistochemistry and enzyme-immunoassay. Brain Res (1986) 397(1):161–72. doi: 10.1016/0006-8993(86)91381-8
17. Polli JW, Billingsley ML, Kincaid RL. Expression of the calmodulin-dependent protein phosphatase, calcineurin, in rat brain: developmental patterns and the role of nigrostriatal innervation. Brain Res Dev Brain Res (1991) 63(1-2):105–19. doi: 10.1016/0165-3806(91)90071-p
18. Kuno T, Mukai H, Ito A, Chang CD, Kishima K, Saito N, et al. Distinct cellular expression of calcineurin A alpha and A beta in rat brain. J Neurochem (1992) 58(5):1643–51. doi: 10.1111/j.1471-4159.1992.tb10036.x
19. Chin D, Means AR. Calmodulin: a prototypical calcium sensor," (in eng). Trends Cell Biol (2000) 10(8):322–8. doi: 10.1016/s0962-8924(00)01800-6
20. Zhang M, Abrams C, Wang L, Gizzi A, He L, Lin R, et al. "Structural basis for calmodulin as a dynamic calcium sensor. Structure (2012) 20(5):911–23. doi: 10.1016/j.str.2012.03.019
21. Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol (2010) 28:491–533. doi: 10.1146/annurev.immunol.021908.132550
22. Oh-hora M, Rao A. Calcium signaling in lymphocytes. Curr Opin Immunol (2008) 20(3):250–8. doi: 10.1016/j.coi.2008.04.004
23. Colbran RJ, Brown AM. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol (2004) 14(3):318–27. doi: 10.1016/j.conb.2004.05.008
24. Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature (1994) 369(6480):486–8. doi: 10.1038/369486a0
25. Hussey JW, Limpitikul WB, Dick IE. Calmodulin mutations in human disease. Channels (Austin) (2023) 17(1):2165278. doi: 10.1080/19336950.2023.2165278
26. O'Day DH, Eshak K, Myre MA. Calmodulin binding proteins and alzheimer's disease. J Alzheimers Dis (2015) 46(3):553–69. doi: 10.3233/JAD-142772
27. Berrocal M, Sepulveda MR, Vazquez-Hernandez M, Mata AM. Calmodulin antagonizes amyloid-β peptides-mediated inhibition of brain plasma membrane Ca(2+)-ATPase. Biochim Biophys Acta (2012) 1822(6):961–9. doi: 10.1016/j.bbadis.2012.02.013
28. O'Day DH. Calmodulin binding domains in critical risk proteins involved in neurodegeneration. Curr Issues Mol Biol (2022) 44(11):5802–14. doi: 10.3390/cimb44110394
29. Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev (2000) 80(4):1483–521. doi: 10.1152/physrev.2000.80.4.1483
30. Caraveo G, Auluck PK, Whitesell L, Chung CY, Baru V, Mosharov EV, et al. Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc Natl Acad Sci USA (2014) 111(34):E3544–52. doi: 10.1073/pnas.1413201111
31. Qian W, Yin X, Hu W, Shi J, Gu J, Grundke-Iqbal I, et al. Activation of protein phosphatase 2B and hyperphosphorylation of Tau in Alzheimer's disease. J Alzheimers Dis (2011) 23(4):617–27. doi: 10.3233/JAD-2010-100987
32. Pineda JR, Pardo R, Zala D, Yu H, Humbert S, Saudou F. Genetic and pharmacological inhibition of calcineurin corrects the BDNF transport defect in Huntington's disease. Mol Brain (2009) 2(33). doi: 10.1186/1756-6606-2-33
33. Overk CR, Rockenstein E, Florio J, Cheng Q, Masliah E. Differential calcium alterations in animal models of neurodegenerative disease: Reversal by FK506. Neuroscience (2015) 310:549–60. doi: 10.1016/j.neuroscience.2015.08.068
34. Mukherjee A, Morales-Scheihing D, Gonzalez-Romero D, Green K, Taglialatela G, Soto C. Calcineurin inhibition at the clinical phase of prion disease reduces neurodegeneration, improves behavioral alterations and increases animal survival. PloS Pathog (2010) 6(10):e1001138. doi: 10.1371/journal.ppat.1001138
35. Wu HY, Hudry E, Hashimoto T, Uemura K, Fan ZY, Berezovska O, et al. Distinct dendritic spine and nuclear phases of calcineurin activation after exposure to amyloid-β revealed by a novel fluorescence resonance energy transfer assay. J Neurosci (2012) 32(15):5298–309. doi: 10.1523/JNEUROSCI.0227-12.2012
36. Sompol P, Furman JL, Pleiss MM, Kraner SD, Artiushin IA, Batten SR, et al. Calcineurin/NFAT signaling in activated astrocytes drives network hyperexcitability in Aβ-bearing mice. J Neurosci (2017) 37(25):6132–48. doi: 10.1523/JNEUROSCI.0877-17.2017
37. Taglialatela G, Rastellini C, Cicalese L. Reduced incidence of dementia in solid organ transplant patients treated with calcineurin inhibitors. J Alzheimers Dis (2015) 47(2):329–33. doi: 10.3233/JAD-150065
38. Silva JD, Taglialatela G, Jupiter DC. Reduced prevalence of dementia in patients prescribed tacrolimus, sirolimus, or cyclosporine. J Alzheimers Dis (2023) 95(2):585–97. doi: 10.3233/JAD-230526
39. Im SH, Rao A. Activation and deactivation of gene expression by Ca2+/calcineurin-NFAT-mediated signaling. Mol Cells (2004) 18(1):1–9.
40. Alothaid H, Aldughaim MSK, Alamri SS, Alrahimi JSM, Al-Jadani SH. Role of calcineurin biosignaling in cell secretion and the possible regulatory mechanisms. Saudi J Biol Sci (2021) 28(1):116–24. doi: 10.1016/j.sjbs.2020.08.042
41. Ulengin-Talkish I, Cyert MS. A cellular atlas of calcineurin signaling. Biochim Biophys Acta Mol Cell Res (2023) 1870(1):119366. doi: 10.1016/j.bbamcr.2022.119366
43. Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Identification of a putative regulator of early T cell activation genes. Science (1988) 241(4862):202–5. doi: 10.1126/science.3260404
44. Kuklina EM, Shirshev SV. Role of transcription factor NFAT in the immune response. Biochem (Mosc) (2001) 66(5):467–75. doi: 10.1023/a:1010238931555
45. Kiani A, Rao A, Aramburu J. Manipulating immune responses with immunosuppressive agents that target NFAT. Immunity (2000) 12(4):359–72. doi: 10.1016/s1074-7613(00)80188-0
46. Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell (2002) 109Suppl:S67–79. doi: 10.1016/s0092-8674(02)00699-2
47. Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol (2005) 5(6):472–84. doi: 10.1038/nri1632
48. Schulz RA, Yutzey KE. Calcineurin signaling and NFAT activation in cardiovascular and skeletal muscle development. Dev Biol (2004) 266(1):1–16. doi: 10.1016/j.ydbio.2003.10.008
49. Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell (2001) 105(7):863–75. doi: 10.1016/s0092-8674(01)00396-8
50. Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, et al. The transcription factor NF-ATc is essential for cardiac valve formation. Nature (1998) 392(6672):186–90. doi: 10.1038/32426
51. Ranger AM, Gerstenfeld LC, Wang J, Kon T, Bae H, Gravallese EM, et al. The nuclear factor of activated T cells (NFAT) transcription factor NFATp (NFATc2) is a repressor of chondrogenesis. J Exp Med (2000) 191(1):9–22. doi: 10.1084/jem.191.1.9
52. Oukka M, Ho IC, de la Brousse FC, Hoey T, Grusby MJ, Glimcher LH. The transcription factor NFAT4 is involved in the generation and survival of T cells. Immunity (1998) 9(3):295–304. doi: 10.1016/s1074-7613(00)80612-3
53. Suchting S, Bicknell R, Eichmann A. Neuronal clues to vascular guidance. Exp Cell Res (2006) 312(5):668–75. doi: 10.1016/j.yexcr.2005.11.009
54. Benedito AB, Lehtinen M, Massol R, Lopes UG, Kirchhausen T, Rao A, et al. The transcription factor NFAT3 mediates neuronal survival. J Biol Chem (2005) 280(4):2818–25. doi: 10.1074/jbc.M408741200
55. Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature (2006) 441(7093):595–600. doi: 10.1038/nature04678
56. Schwartz N, Schohl A, Ruthazer ES. Neural activity regulates synaptic properties and dendritic structure in vivo through calcineurin/NFAT signaling. Neuron (2009) 62(5):655–69. doi: 10.1016/j.neuron.2009.05.007
57. de la Fuente V, Freudenthal R, Romano A. Reconsolidation or extinction: transcription factor switch in the determination of memory course after retrieval. J Neurosci (2011) 31(15):5562–73. doi: 10.1523/JNEUROSCI.6066-10.2011
58. Quadrato G, Benevento M, Alber S, Jacob C, Floriddia EM, Nguyen T, et al. Nuclear factor of activated T cells (NFATc4) is required for BDNF-dependent survival of adult-born neurons and spatial memory formation in the hippocampus. Proc Natl Acad Sci USA (2012) 109(23):E1499–508. doi: 10.1073/pnas.1202068109
59. Kipanyula MJ, Kimaro WH, Seke Etet PF. The emerging roles of the calcineurin-nuclear factor of activated T-lymphocytes pathway in nervous system functions and diseases. J Aging Res vol (2016) 2016, 5081021. doi: 10.1155/2016/5081021
60. Vihma H, Pruunsild P, Timmusk T. Alternative splicing and expression of human and mouse NFAT genes. Genomics (2008) 92(5):279–91. doi: 10.1016/j.ygeno.2008.06.011
61. Lee N, Kim D, Kim WU. Role of NFAT5 in the immune system and pathogenesis of autoimmune diseases. Front Immunol (2019) 10, 270:270. doi: 10.3389/fimmu.2019.00270
62. Manocha GD, Ghatak A, Puig KL, Kraner SD, Norris CM, Combs CK. NFATc2 modulates microglial activation in the AβPP/PS1 mouse model of alzheimer's disease. J Alzheimers Dis (2017) 58(3):775–87. doi: 10.3233/JAD-151203
63. Furman JL, Sompol P, Kraner SD, Pleiss MM, Putman EJ, Dunkerson J, et al. Blockade of astrocytic calcineurin/NFAT signaling helps to normalize hippocampal synaptic function and plasticity in a rat model of traumatic brain injury. J Neurosci (2016) 36(5):1502–15. doi: 10.1523/JNEUROSCI.1930-15.2016
64. López-Rodríguez C, Aramburu J, Jin L, Rakeman AS, Michino M, Rao A. Bridging the NFAT and NF-kappaB families: NFAT5 dimerization regulates cytokine gene transcription in response to osmotic stress. Immunity (2001) 15(1):47–58. doi: 10.1016/s1074-7613(01)00165-0
65. Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol (1997) 15:707–47. doi: 10.1146/annurev.immunol.15.1.707
66. Takeuchi K, Roehrl MH, Sun ZY, Wagner G. Structure of the calcineurin-NFAT complex: defining a T cell activation switch using solution NMR and crystal coordinates. Structure (2007) 15(5):587–97. doi: 10.1016/j.str.2007.03.015
67. Okamura H, Aramburu J, García-Rodríguez C, Viola JP, Raghavan A, Tahiliani M, et al. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol Cell (2000) 6(3):539–50. doi: 10.1016/s1097-2765(00)00053-8
68. Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science (1997) 275(5308):1930–4. doi: 10.1126/science.275.5308.1930
69. Porter CM, Havens MA, Clipstone NA. Identification of amino acid residues and protein kinases involved in the regulation of NFATc subcellular localization. J Biol Chem (2000) 275(5):3543–51. doi: 10.1074/jbc.275.5.3543
70. Neal JW, Clipstone NA. Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. J Biol Chem (2001) 276(5):3666–73. doi: 10.1074/jbc.M004888200
71. Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev (2003) 17(18):2205–32. doi: 10.1101/gad.1102703
72. Vaeth M, Feske S. NFAT control of immune function: New Frontiers for an Abiding Trooper. F1000Res (2018) 7(260). doi: 10.12688/f1000research.13426.1
73. Flanagan WM, Corthésy B, Bram RJ, Crabtree GR. Nuclear association of a T-cell transcription factor blocked by FK-506 and cyclosporin A. Nature (1991) 352(6338):803–7. doi: 10.1038/352803a0
74. Bettelli E, Dastrange M, Oukka M. Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci USA (2005) 102(14):5138–43. doi: 10.1073/pnas.0501675102
75. Gwack Y, Sharma S, Nardone J, Tanasa B, Iuga A, Srikanth S, et al. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature (2006) 441(7093):646–50. doi: 10.1038/nature04631
76. Chow CW, Rincón M, Cavanagh J, Dickens M, Davis RJ. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science (1997) 278(5343):1638–41. doi: 10.1126/science.278.5343.1638
77. Whitehurst CE, Geppert TD. MEK1 and the extracellular signal-regulated kinases are required for the stimulation of IL-2 gene transcription in T cells. J Immunol (1996) 156(3):1020–9. doi: 10.4049/jimmunol.156.3.1020
78. García-Rodríguez C, Rao A. Nuclear factor of activated T cells (NFAT)-dependent transactivation regulated by the coactivators p300/CREB-binding protein (CBP. J Exp Med (1998) 187(12):2031–6. doi: 10.1084/jem.187.12.2031
79. Yang T, Davis RJ, Chow CW. Requirement of two NFATc4 transactivation domains for CBP potentiation. J Biol Chem (2001) 276(43):39569–76. doi: 10.1074/jbc.M102961200
80. Terui Y, Saad N, Jia S, McKeon F, Yuan J. Dual role of sumoylation in the nuclear localization and transcriptional activation of NFAT1. J Biol Chem (2004) 279(27):28257–65. doi: 10.1074/jbc.M403153200
81. Henley JM, Craig TJ, Wilkinson KA. Neuronal SUMOylation: mechanisms, physiology, and roles in neuronal dysfunction. Physiol Rev (2014) 94:1249–85. doi: 10.1152/physrev.00008.2014
82. Vihma H, Luhakooder M, Pruunsild P, Timmusk T. Regulation of different human NFAT isoforms by neuronal activity. J Neurochem (2016) 137(3):394–408. doi: 10.1111/jnc.13568
83. Crabtree GR. "Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT," (in eng). Cell (1999) 96(5):611–4. doi: 10.1016/s0092-8674(00)80571-1
84. Horsley V, Pavlath GK. NFAT: ubiquitous regulator of cell differentiation and adaptation. J Cell Biol (2002) 156(5):771–4. doi: 10.1083/jcb.200111073
85. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol (2000) 1(1):11–21. doi: 10.1038/35036035
86. Müller MR, Sasaki Y, Stevanovic I, Lamperti ED, Ghosh S, Sharma S, et al. Requirement for balanced Ca/NFAT signaling in hematopoietic and embryonic development. Proc Natl Acad Sci USA (2009) 106(17):7034–9. doi: 10.1073/pnas.0813296106
87. Aramburu J, Yaffe MB, López-Rodríguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science (1999) 285(5436):2129–33. doi: 10.1126/science.285.5436.2129
88. Zhu J, Shibasaki F, Price R, Guillemot JC, Yano T, Dötsch V, et al. Intramolecular masking of nuclear import signal on NF-AT4 by casein kinase I and MEKK1. Cell (1998) 93(5):851–61. doi: 10.1016/s0092-8674(00)81445-2
89. Lewis RS. Calcium oscillations in T-cells: mechanisms and consequences for gene expression. Biochem Soc Trans (2003) 31(Pt 5):925–9. doi: 10.1042/bst0310925
90. Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol (2001) 19:497–521. doi: 10.1146/annurev.immunol.19.1.497
91. Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, et al. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell (2003) 113(5):657–70. doi: 10.1016/s0092-8674(03)00390-8
92. Groth RD, Mermelstein PG. Brain-derived neurotrophic factor activation of NFAT (nuclear factor of activated T-cells)-dependent transcription: a role for the transcription factor NFATc4 in neurotrophin-mediated gene expression. J Neurosci (2003) 23(22):8125–34. doi: 10.1523/JNEUROSCI.23-22-08125.2003
93. Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, et al. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature (1999) 401(6754):703–8. doi: 10.1038/44378
94. Nguyen T, Di Giovanni S. NFAT signaling in neural development and axon growth. Int J Dev Neurosci (2008) 26(2):141–5. doi: 10.1016/j.ijdevneu.2007.10.004
95. Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, et al. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell (1996) 87(6):1001–14. doi: 10.1016/s0092-8674(00)81795-x
96. Fazeli A, Dickinson SL, Hermiston ML, Tighe RV, Steen RG, Small CG, et al. Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature (1997) 386(6627):796–804. doi: 10.1038/386796a0
97. Langwieser N, Christel CJ, Kleppisch T, Hofmann F, Wotjak CT, Moosmang S. Homeostatic switch in hebbian plasticity and fear learning after sustained loss of Cav1.2 calcium channels. J Neurosci (2010) 30(25):8367–75. doi: 10.1523/JNEUROSCI.4164-08.2010
98. Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Müller J, et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci (2005) 25(43):9883–92. doi: 10.1523/JNEUROSCI.1531-05.2005
99. Oliveria SF, Dell'Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron (2007) 55(2):261–75. doi: 10.1016/j.neuron.2007.06.032
100. Ulrich JD, Kim MS, Houlihan PR, Shutov LP, Mohapatra DP, Strack S, et al. Distinct activation properties of the nuclear factor of activated T-cells (NFAT) isoforms NFATc3 and NFATc4 in neurons. J Biol Chem (2012) 287(45):37594–609. doi: 10.1074/jbc.M112.365197
101. Vashishta A, Habas A, Pruunsild P, Zheng JJ, Timmusk T, Hetman M. Nuclear factor of activated T-cells isoform c4 (NFATc4/NFAT3) as a mediator of antiapoptotic transcription in NMDA receptor-stimulated cortical neurons. J Neurosci (2009) 29(48):15331–40. doi: 10.1523/JNEUROSCI.4873-09.2009
102. Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science (1996) 271(5255):1589–92. doi: 10.1126/science.271.5255.1589
103. Kashishian A, Howard M, Loh C, Gallatin WM, Hoekstra MF, Lai Y. AKAP79 inhibits calcineurin through a site distinct from the immunophilin-binding region. J Biol Chem (1998) 273(42):27412–9. doi: 10.1074/jbc.273.42.27412
104. Kim MJ, Jo DG, Hong GS, Kim BJ, Lai M, Cho DH, et al. Calpain-dependent cleavage of cain/cabin1 activates calcineurin to mediate calcium-triggered cell death. Proc Natl Acad Sci USA (2002) 99(15):9870–5. doi: 10.1073/pnas.152336999
105. Tredger JM, Brown NW, Dhawan A. Calcineurin inhibitor sparing in paediatric solid organ transplantation : managing the efficacy/toxicity conundrum. Drugs (2008) 68:1385–414:no. doi: 10.2165/00003495-200868100-00004
106. Höcker B, Tönshoff B. "Calcineurin inhibitor-free immunosuppression in pediatric renal transplantation: a viable option? Paediatr Drugs (2011) 13(1):49–69. doi: 10.2165/11538530-000000000-00000
107. Borel JF, Feurer C, Gubler HU, Stähelin H. Biological effects of cyclosporin A: a new antilymphocytic agent. Agents Actions (1976) 6(4):468–75. doi: 10.1007/BF01973261
108. Handschumacher RE, Harding MW, Rice J, Drugge RJ, Speicher DW. Cyclophilin: a specific cytosolic binding protein for cyclosporin A," (in eng). Science (1984) 226(4674):544–7. doi: 10.1126/science.6238408
109. Schreiber SL. Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science (1991) 251(4991):283–7. doi: 10.1126/science.1702904
111. Matsuda S, Koyasu S. Mechanisms of action of cyclosporine. Immunopharmacology (2000) 47(2-3):119–25. doi: 10.1016/s0162-3109(00)00192-2
112. Rincón M, Flavell RA. AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO J (1994) 13(18):4370–81. doi: 10.1002/j.1460-2075.1994.tb06757.x
113. Mattila PS, Ullman KS, Fiering S, Emmel EA, McCutcheon M, Crabtree GR, et al. The actions of cyclosporin A and FK506 suggest a novel step in the activation of T lymphocytes. EMBO J (1990) 9(13):4425–33. doi: 10.1002/j.1460-2075.1990.tb07893.x
114. Metcalfe S, Alexander D, Turner J. FK506 and cyclosporin A each inhibit antigen-specific signaling in the T cell line 171 in the absence of a calcium signal. Cell Immunol (1994) 158(1):46–58. doi: 10.1006/cimm.1994.1255
115. Matsuda S, Moriguchi T, Koyasu S, Nishida E. T lymphocyte activation signals for interleukin-2 production involve activation of MKK6-p38 and MKK7-SAPK/JNK signaling pathways sensitive to cyclosporin A. J Biol Chem (1998) 273(20):12378–82. doi: 10.1074/jbc.273.20.12378
116. Kaminska B, Figiel I, Pyrzynska B, Czajkowski R, Mosieniak G. Treatment of hippocampal neurons with cyclosporin A results in calcium overload and apoptosis which are independent on NMDA receptor activation. Br J Pharmacol (2001) 133(7):997–1004. doi: 10.1038/sj.bjp.0704177
117. Canudas AM, Jordà EG, Verdaguer E, Jiménez A, Sureda FX, Rimbau V, et al. Cyclosporin A enhances colchicine-induced apoptosis in rat cerebellar granule neurons. Br J Pharmacol (2004) 141(4):661–9. doi: 10.1038/sj.bjp.0705664
118. Schnichels S, Schultheiss M, Klemm P, Blak M, Herrmann T, Melchinger M, et al. Cyclosporine A protects retinal explants against hypoxia. Int J Mol Sci (2021) 22(19). doi: 10.3390/ijms221910196
119. Chen ZR, Ma Y, Guo HH, Lu ZD, Jin QH. Therapeutic efficacy of cyclosporin A against spinal cord injury in rats with hyperglycemia. Mol Med Rep (2018) 17(3):4369–75. doi: 10.3892/mmr.2018.8422
120. Ye F, Li X, Li F, Li J, Chang W, Yuan J, et al. Cyclosporin A protects against Lead neurotoxicity through inhibiting mitochondrial permeability transition pore opening in nerve cells. Neurotoxicology (2016) 57:203–13. doi: 10.1016/j.neuro.2016.10.004
121. Schultheiss M, Schnichels S, Mlynczak T, Dipl-Ing JH, Bartz-Schmidt KU, Szurman P, et al. Cyclosporine a protects RGC-5 cells from excitotoxic cell death. J Glaucoma (2014) 23(4):219–24. doi: 10.1097/IJG.0000000000000040
122. Xiong TQ, Chen LM, Tan BH, Guo CY, Li YN, Zhang YF, et al. The effects of calcineurin inhibitor FK506 on actin cytoskeleton, neuronal survival and glial reactions after pilocarpine-induced status epilepticus in mice. Epilepsy Res (2018) 140:138–47. doi: 10.1016/j.eplepsyres.2018.01.007
123. Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron (2008) 59(2):214–25. doi: 10.1016/j.neuron.2008.06.008
124. Kim S, Violette CJ, Ziff EB. Reduction of increased calcineurin activity rescues impaired homeostatic synaptic plasticity in presenilin 1 M146V mutant, (in eng). Neurobiol Aging (2015) 36:3239–46. doi: 10.1016/j.neurobiolaging.2015.09.007
125. Shah SZA, Zhao D, Taglialatela G, Hussain T, Dong H, Sabir N, et al. Combinatory FK506 and minocycline treatment alleviates prion-induced neurodegenerative events via caspase-mediated MAPK-NRF2 pathway. Int J Mol Sci (2019) 20(5). doi: 10.3390/ijms20051144
126. Fields JA, Overk C, Adame A, Florio J, Mante M, Pineda A, et al. Neuroprotective effects of the immunomodulatory drug FK506 in a model of HIV1-gp120 neurotoxicity. J Neuroinflamm (2016) 13(1):120. doi: 10.1186/s12974-016-0585-8
127. Graham RM. Cyclosporine: mechanisms of action and toxicity. Cleve Clin J Med (1994) 61(4):308–13. doi: 10.3949/ccjm.61.4.308
128. Thomson AW, Bonham CA, Zeevi A. Mode of action of tacrolimus (FK506): molecular and cellular mechanisms. Ther Drug Monit (1995) 17(6):584–91. doi: 10.1097/00007691-199512000-00007
129. Powell JD, Zheng Y. Dissecting the mechanism of T-cell anergy with immunophilin ligands. Curr Opin Investig Drugs (2006) 7(11):1002–7.
130. Mori A, Suko M, Kaminuma O, Inoue S, Ohmura T, Hoshino A, et al. IL-2-induced IL-5 synthesis, but not proliferation, of human CD4+ T cells is suppressed by FK506. J Immunol (1997) 158(8):3659–65.
131. Rostaing L, Puyoo O, Tkaczuk J, Peres C, Rouzaud A, Cisterne JM, et al. Differences in Type 1 and Type 2 intracytoplasmic cytokines, detected by flow cytometry, according to immunosuppression (cyclosporine A vs. tacrolimus) in stable renal allograft recipients. Clin Transplant (1999) 13(5):400–9. doi: 10.1034/j.1399-0012.1999.130506.x
132. Almawi WY, Melemedjian OK. Clinical and mechanistic differences between FK506 (tacrolimus) and cyclosporin A. Nephrol Dial Transplant (2000) 15(12):1916–8. doi: 10.1093/ndt/15.12.1916
133. Aramburu J, Garcia-Cózar F, Raghavan A, Okamura H, Rao A, Hogan PG. Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol Cell (1998) 1(5):627–37. doi: 10.1016/s1097-2765(00)80063-5
134. Kuriyama M, Matsushita M, Tateishi A, Moriwaki A, Tomizawa K, Ishino K, et al. A cell-permeable NFAT inhibitor peptide prevents pressure-overload cardiac hypertrophy. Chem Biol Drug Des (2006) 67(3):238–43. doi: 10.1111/j.1747-0285.2006.00360.x
135. Martínez-Martínez S, Rodríguez A, López-Maderuelo MD, Ortega-Pérez I, Vázquez J, Redondo JM. Blockade of NFAT activation by the second calcineurin binding site. J Biol Chem (2006) 281(10):6227–35. doi: 10.1074/jbc.M513885200
136. Rodríguez A, Roy J, Martínez-Martínez S, López-Maderuelo MD, Niño-Moreno P, Ortí L, et al. A conserved docking surface on calcineurin mediates interaction with substrates and immunosuppressants. Mol Cell (2009) 33(5):616–26. doi: 10.1016/j.molcel.2009.01.030
137. Adachi S, Amasaki Y, Miyatake S, Arai N, Iwata M. Successive expression and activation of NFAT family members during thymocyte differentiation. J Biol Chem (2000) 275(19):14708–16. doi: 10.1074/jbc.275.19.14708
138. Brown DG, Wilkerson EC, Love WE. A review of traditional and novel oral anticoagulant and antiplatelet therapy for dermatologists and dermatologic surgeons. J Am Acad Dermatol (2015) 72(3):524–34. doi: 10.1016/j.jaad.2014.10.027
139. Mulero MC, Aubareda A, Orzáez M, Messeguer J, Serrano-Candelas E, Martínez-Hoyer S, et al. Inhibiting the calcineurin-NFAT (nuclear factor of activated T cells) signaling pathway with a regulator of calcineurin-derived peptide without affecting general calcineurin phosphatase activity. J Biol Chem (2009) 284(14):9394–401. doi: 10.1074/jbc.M805889200
140. Borisy AA, Elliott PJ, Hurst NW, Lee MS, Lehar J, Price ER, et al. Systematic discovery of multicomponent therapeutics. Proc Natl Acad Sci USA (2003) 100(13):7977–82. doi: 10.1073/pnas.1337088100
141. Weyrich AS, Denis MM, Kuhlmann-Eyre JR, Spencer ED, Dixon DA, Marathe GK, et al. Dipyridamole selectively inhibits inflammatory gene expression in platelet-monocyte aggregates. Circulation (2005) 111(5):633–42. doi: 10.1161/01.CIR.0000154607.90506.45
142. Kang S, Li H, Rao A, Hogan PG. Inhibition of the calcineurin-NFAT interaction by small organic molecules reflects binding at an allosteric site. J Biol Chem (2005) 280(45):37698–706. doi: 10.1074/jbc.M502247200
143. Roehrl MH, Kang S, Aramburu J, Wagner G, Rao A, Hogan PG. Selective inhibition of calcineurin-NFAT signaling by blocking protein-protein interaction with small organic molecules. Proc Natl Acad Sci USA (2004) 101(20):7554–9. doi: 10.1073/pnas.0401835101
144. Trevillyan JM, Chiou XG, Chen YW, Ballaron SJ, Sheets MP, Smith ML, et al. Potent inhibition of NFAT activation and T cell cytokine production by novel low molecular weight pyrazole compounds. J Biol Chem (2001) 276(51):48118–26. doi: 10.1074/jbc.M107919200
145. Bîrsan T, Dambrin C, Marsh KC, Jacobsen W, Djuric SW, Mollison KW, et al. Preliminary in vivo pharmacokinetic and pharmacodynamic evaluation of a novel calcineurin-independent inhibitor of NFAT. Transpl Int (2004) 17(3):145–50. doi: 10.1007/s00147-003-0676-1
146. Nilsson LM, Sun ZW, Nilsson J, Nordström I, Chen YW, Molkentin JD, et al. Novel blocker of NFAT activation inhibits IL-6 production in human myometrial arteries and reduces vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol (2007) 292(3):C1167–78. doi: 10.1152/ajpcell.00590.2005
147. Jabr RI, Wilson AJ, Riddervold MH, Jenkins AH, Perrino BA, Clapp LH. Nuclear translocation of calcineurin Abeta but not calcineurin Aalpha by platelet-derived growth factor in rat aortic smooth muscle. Am J Physiol Cell Physiol (2007) 292(6):C2213–25. doi: 10.1152/ajpcell.00139.2005
148. Sompol P, Gollihue JL, Kraner SD, Artiushin IA, Cloyd RA, Chishti EA, et al. Q134R: Small chemical compound with NFAT inhibitory properties improves behavioral performance and synapse function in mouse models of amyloid pathology. Aging Cell (2021) 20(7):e13416. doi: 10.1111/acel.13416
149. Dineley KT, Hogan D, Zhang WR, Taglialatela G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem (2007) 88(2):217–24. doi: 10.1016/j.nlm.2007.03.010
150. Taglialatela G, Hogan D, Zhang WR, Dineley KT. Intermediate- and long-term recognition memory deficits in Tg2576 mice are reversed with acute calcineurin inhibition. Behav Brain Res (2009) 200(1):95–9. doi: 10.1016/j.bbr.2008.12.034
151. Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron (2007) 53(3):337–51. doi: 10.1016/j.neuron.2007.01.010
152. Gómez-Sintes R, Lucas JJ. NFAT/Fas signaling mediates the neuronal apoptosis and motor side effects of GSK-3 inhibition in a mouse model of lithium therapy. J Clin Invest (2010) 120(7):2432–45. doi: 10.1172/JCI37873
153. Gould TD, Chen G, Manji HK. In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology (2004) 29(1):32–8. doi: 10.1038/sj.npp.1300283
154. Beaulieu JM, Caron MG. Looking at lithium: molecular moods and complex behaviour. Mol Interv (2008) 8(5):230–41. doi: 10.1124/mi.8.5.8
155. Beaulieu JM, Marion S, Rodriguiz RM, Medvedev IO, Sotnikova TD, Ghisi V, et al. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell (2008) 132(1):125–36. doi: 10.1016/j.cell.2007.11.041
156. Fiorentini A, Rosi MC, Grossi C, Luccarini I, Casamenti F. Lithium improves hippocampal neurogenesis, neuropathology and cognitive functions in APP mutant mice. PloS One (2010) 5(12):e14382. doi: 10.1371/journal.pone.0014382
157. Ma T, Tzavaras N, Tsokas P, Landau EM, Blitzer RD. Synaptic stimulation of mTOR is mediated by Wnt signaling and regulation of glycogen synthetase kinase-3. J Neurosci (2011) 31(48):17537–46. doi: 10.1523/JNEUROSCI.4761-11.2011
158. Phiel CJ, Wilson CA, Lee VM, Klein PS. "GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides," (in eng). Nature (2003) 423(6938):435–9. doi: 10.1038/nature01640
159. Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem (2008) 104(6):1433–9. doi: 10.1111/j.1471-4159.2007.05194.x
160. Kremer A, Louis JV, Jaworski T, Van Leuven F. GSK3 and alzheimer's disease: facts and fiction. Front Mol Neurosci (2011) 4:17. doi: 10.3389/fnmol.2011.00017
161. Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem (1997) 272(40):25326–32. doi: 10.1074/jbc.272.40.25326
162. Spittaels K, Van den Haute C, Van Dorpe J, Geerts H, Mercken M, Bruynseels K, et al. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem (2000) 275(52):41340–9. doi: 10.1074/jbc.M006219200
163. Leroy K, Boutajangout A, Authelet M, Woodgett JR, Anderton BH, Brion JP. The active form of glycogen synthase kinase-3beta is associated with granulovacuolar degeneration in neurons in Alzheimer's disease. Acta Neuropathol (2002) 103(2):91–9. doi: 10.1007/s004010100435
164. Sun W, Qureshi HY, Cafferty PW, Sobue K, Agarwal-Mawal A, Neufield KD, et al. Glycogen synthase kinase-3beta is complexed with tau protein in brain microtubules. J Biol Chem (2002) 277(14):11933–40. doi: 10.1074/jbc.M107182200
165. Ferrer I, BarraChina M, Puig B. Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer's disease, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol (2002) 104(6):583–91. doi: 10.1007/s00401-002-0587-8
166. Engel T, Lucas JJ, Gómez-Ramos P, Moran MA, Avila J, Hernández F. Cooexpression of FTDP-17 tau and GSK-3beta in transgenic mice induce tau polymerization and neurodegeneration. Neurobiol Aging (2006) 27(9):1258–68. doi: 10.1016/j.neurobiolaging.2005.06.010
167. Engel T, Hernández F, Avila J, Lucas JJ. Full reversal of Alzheimer's disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci (2006) 26(19):5083–90. doi: 10.1523/JNEUROSCI.0604-06.2006
168. Uemura K, Kuzuya A, Shimozono Y, Aoyagi N, Ando K, Shimohama S, et al. GSK3beta activity modifies the localization and function of presenilin 1. J Biol Chem (2007) 282(21):15823–32. doi: 10.1074/jbc.M610708200
169. Twomey C, McCarthy JV. Presenilin-1 is an unprimed glycogen synthase kinase-3beta substrate. FEBS Lett (2006) 580(17):4015–20. doi: 10.1016/j.febslet.2006.06.035
170. Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest (2013) 123(1):224–35. doi: 10.1172/JCI64516
171. Hurtado DE, Molina-Porcel L, Carroll JC, Macdonald C, Aboagye AK, Trojanowski JQ, et al. Selectively silencing GSK-3 isoforms reduces plaques and tangles in mouse models of Alzheimer's disease. J Neurosci (2012) 32(21):7392–402. doi: 10.1523/JNEUROSCI.0889-12.2012
172. Gómez-Sintes R, Hernández F, Bortolozzi A, Artigas F, Avila J, Zaratin P, et al. Neuronal apoptosis and reversible motor deficit in dominant-negative GSK-3 conditional transgenic mice. EMBO J (2007) 26(11):2743–54. doi: 10.1038/sj.emboj.7601725
173. Song L, Zhou T, Jope RS. Lithium facilitates apoptotic signaling induced by activation of the Fas death domain-containing receptor. BMC Neurosci (2004) 5, 20. doi: 10.1186/1471-2202-5-20
174. Madiehe AM, Mampuru LJ, Tyobeka EM. Induction of apoptosis in HL-60 cells by lithium. Biochem Biophys Res Commun (1995) 209(2):768–74. doi: 10.1006/bbrc.1995.1565
175. Lan Y, Liu X, Zhang R, Wang K, Wang Y, Hua ZC. Lithium enhances TRAIL-induced apoptosis in human lung carcinoma A549 cells. Biometals (2013) 26(2):241–54. doi: 10.1007/s10534-012-9607-x
176. Zhang Q, Li H, Zhao X, Zhang H. LiCl induces apoptosis via CHOP/NOXA/Mcl-1 axis in human choroidal melanoma cells. Cancer Cell Int (2021) 21(1):96. doi: 10.1186/s12935-021-01778-2
177. Li L, Song H, Zhong L, Yang R, Yang XQ, Jiang KL, et al. Lithium chloride promotes apoptosis in human leukemia NB4 cells by inhibiting glycogen synthase kinase-3 beta. Int J Med Sci (2015) 12(10):805–10. doi: 10.7150/ijms.12429
178. Tang HR, He Q, Wang FC. [Mechanism of lithium chloride-induced proliferation inhibition and apoptosis of K562 leukemic cells]. Zhongguo Shi Yan Xue Ye Xue Za Zhi (2005) 13(6):979–82.
179. Hantson P. Mechanisms of toxic cardiomyopathy. Clin Toxicol (Phila) (2019) 57(1):1–9. doi: 10.1080/15563650.2018.1497172
180. Yao R, Sun X, Xie Y, Liu L, Han D, Yao Y, et al. Lithium chloride inhibits cell survival, overcomes drug resistance, and triggers apoptosis in multiple myeloma via activation of the Wnt/β-catenin pathway. Am J Transl Res (2018) 10(8):2610–8.
181. Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol (2001) 65(4):391–426. doi: 10.1016/s0301-0082(01)00011-9
182. Jayanthi S, Deng X, Ladenheim B, McCoy MT, Cluster A, Cai NS, et al. Calcineurin/NFAT-induced up-regulation of the Fas ligand/Fas death pathway is involved in methamphetamine-induced neuronal apoptosis. Proc Natl Acad Sci USA (2005) 102(3):868–73. doi: 10.1073/pnas.0404990102
183. Su JH, Anderson AJ, Cribbs DH, Tu C, Tong L, Kesslack P, et al. Fas and Fas ligand are associated with neuritic degeneration in the AD brain and participate in beta-amyloid-induced neuronal death. Neurobiol Dis (2003) 12(3):182–93. doi: 10.1016/s0969-9961(02)00019-0
184. Luoma JI, Zirpel L. Deafferentation-induced activation of NFAT (nuclear factor of activated T-cells) in cochlear nucleus neurons during a developmental critical period: a role for NFATc4-dependent apoptosis in the CNS. J Neurosci (2008) 28(12):3159–69. doi: 10.1523/JNEUROSCI.5227-07.2008
185. Hudry E, Wu HY, Arbel-Ornath M, Hashimoto T, Matsouaka R, Fan Z, et al. Inhibition of the NFAT pathway alleviates amyloid β neurotoxicity in a mouse model of Alzheimer's disease. J Neurosci (2012) 32(9):3176–92. doi: 10.1523/JNEUROSCI.6439-11.2012
186. Ayton S, Lei P. The Aβ-induced NFAT apoptotic pathway is also activated by GSK-3 inhibition: implications for Alzheimer therapeutics. J Neurosci (2012) 32(28):9454–6. doi: 10.1523/JNEUROSCI.2143-12.2012
187. Gan KJ, Silverman MA. Dendritic and axonal mechanisms of Ca2+ elevation impair BDNF transport in Aβ oligomer-treated hippocampal neurons. Mol Biol Cell (2015) 26(6):1058–71. doi: 10.1091/mbc.E14-12-1612
188. Mohmmad Abdul H, Baig I, Levine H, Guttmann RP, Norris CM. Proteolysis of calcineurin is increased in human hippocampus during mild cognitive impairment and is stimulated by oligomeric Abeta in primary cell culture. Aging Cell (2011) 10(1):103–13. doi: 10.1111/j.1474-9726.2010.00645.x
189. Hong HS, Hwang JY, Son SM, Kim YH, Moon M, Inhee MJ. FK506 reduces amyloid plaque burden and induces MMP-9 in AβPP/PS1 double transgenic mice. J Alzheimers Dis (2010) 22(1):97–105. doi: 10.3233/JAD-2010-100261
190. Yan P, Hu X, Song H, Yin K, Bateman RJ, Cirrito JR, et al. Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J Biol Chem (2006) 281(34):24566–74. doi: 10.1074/jbc.M602440200
191. Mei Z, Yan P, Tan X, Zheng S, Situ B. Transcriptional regulation of BACE1 by NFAT3 leads to enhanced amyloidogenic processing. Neurochem Res (2015) 40(4):829–36. doi: 10.1007/s11064-015-1533-1
192. Vassar R, Kovacs DM, Yan R, Wong PC. The beta-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J Neurosci (2009) 29(41):12787–94. doi: 10.1523/JNEUROSCI.3657-09.2009
193. Al-Tel TH, Semreen MH, Al-Qawasmeh RA, Schmidt MF, El-Awadi R, Ardah M, et al. Design, synthesis, and qualitative structure-activity evaluations of novel β-secretase inhibitors as potential Alzheimer's drug leads. J Med Chem (2011) 54(24):8373–85. doi: 10.1021/jm201181f
194. Coimbra JRM, Marques DFF, Baptista SJ, Pereira CMF, Moreira PI, Dinis TCP, et al. Highlights in BACE1 inhibitors for alzheimer's disease treatment. Front Chem (2018) 6:178. doi: 10.3389/fchem.2018.00178
195. Citron M. Emerging Alzheimer's disease therapies: inhibition of beta-secretase. Neurobiol Aging (2002) 23(6):1017–22. doi: 10.1016/s0197-4580(02)00122-7
196. Yuan J, Venkatraman S, Zheng Y, McKeever BM, Dillard LW, Singh SB. Structure-based design of β-site APP cleaving enzyme 1 (BACE1) inhibitors for the treatment of Alzheimer's disease. J Med Chem (2013) 56(11):4156–80. doi: 10.1021/jm301659n
197. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement (N Y) (2018) 4:575–90. doi: 10.1016/j.trci.2018.06.014
198. Furman JL, Norris CM. Calcineurin and glial signaling: neuroinflammation and beyond. J Neuroinflamm (2014) 11:158. doi: 10.1186/s12974-014-0158-7
199. Guan PP, Cao LL, Yang Y, Wang P. Calcium ions aggravate alzheimer's disease through the aberrant activation of neuronal networks, leading to synaptic and cognitive deficits. Front Mol Neurosci (2021) 14:757515. doi: 10.3389/fnmol.2021.757515
200. Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, et al. Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem (2004) 279(6):4929–40. doi: 10.1074/jbc.M309767200
201. Pleiss MM, Sompol P, Kraner SD, Abdul HM, Furman JL, Guttmann RP, et al. Calcineurin proteolysis in astrocytes: Implications for impaired synaptic function. Biochim Biophys Acta (2016) 1862(9):1521–32. doi: 10.1016/j.bbadis.2016.05.007
202. Watanabe K, Uemura K, Asada M, Maesako M, Akiyama H, Shimohama S, et al. "The participation of insulin-like growth factor-binding protein 3 released by astrocytes in the pathology of Alzheimer's disease," (in eng). Mol Brain (2015) 8(1):82. doi: 10.1186/s13041-015-0174-2
203. Cui Y, Jin J, Zhang X, Xu H, Yang L, Du D, et al. "Forebrain NR2B overexpression facilitating the prefrontal cortex long-term potentiation and enhancing working memory function in mice," (in eng). PloS One (2011) 6(5):e20312. doi: 10.1371/journal.pone.0020312
204. Plattner F, Hernández A, Kistler TM, Pozo K, Zhong P, Yuen EY, et al. "Memory enhancement by targeting Cdk5 regulation of NR2B," (in eng). Neuron (2014) 81(5):1070–83. doi: 10.1016/j.neuron.2014.01.022
205. Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, et al. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer's models. J Neurosci (2005) 25(18):4649–58. doi: 10.1523/JNEUROSCI.0365-05.2005
206. de la Torre JC. Impaired brain microcirculation may trigger Alzheimer's disease. Neurosci Biobehav Rev (1994) 18(3):397–401. doi: 10.1016/0149-7634(94)90052-3
207. Kurz C, Walker L, Rauchmann BS, Perneczky R. Dysfunction of the blood-brain barrier in Alzheimer's disease: Evidence from human studies," (in eng). Neuropathol Appl Neurobiol (2022) 48:no. doi: 10.1111/nan.12782
208. Badimon A, Torrente D, Norris EH. "Vascular dysfunction in alzheimer's disease: alterations in the plasma contact and fibrinolytic systems," (in eng). Int J Mol Sci (2023) 24(8). doi: 10.3390/ijms24087046
209. Kraner SD, Norris CM. Astrocyte activation and the calcineurin/NFAT pathway in cerebrovascular disease. Front Aging Neurosci (2018) 10:287. doi: 10.3389/fnagi.2018.00287
210. Sompol P, Gollihue JL, Weiss BE, Lin RL, Case SL, Kraner SD, et al. Targeting astrocyte signaling alleviates cerebrovascular and synaptic function deficits in a diet-based mouse model of small cerebral vessel disease. J Neurosci (2023) 43(10):1797–813. doi: 10.1523/JNEUROSCI.1333-22.2023
211. Furman JL, Sama DM, Gant JC, Beckett TL, Murphy MP, Bachstetter AD, et al. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer's disease. J Neurosci (2012) 32(46):16129–40. doi: 10.1523/JNEUROSCI.2323-12.2012
212. Sama MA, Mathis DM, Furman JL, Abdul HM, Artiushin IA, Kraner SD, et al. Interleukin-1beta-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J Biol Chem (2008) 283(32):21953–64. doi: 10.1074/jbc.M800148200
213. Canellada A, Ramirez BG, Minami T, Redondo JM, Cano E. Calcium/calcineurin signaling in primary cortical astrocyte cultures: Rcan1-4 and cyclooxygenase-2 as NFAT target genes. Glia (2008) 56(7):709–22. doi: 10.1002/glia.20647
214. Nagamoto-Combs K, Combs CK. Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells). J Neurosci (2010) 30(28):9641–6. doi: 10.1523/JNEUROSCI.0828-10.2010
215. Escolano A, Martínez-Martínez S, Alfranca A, Urso K, Izquierdo HM, Delgado M, et al. Specific calcineurin targeting in macrophages confers resistance to inflammation via MKP-1 and p38. EMBO J (2014) 33(10):1117–33. doi: 10.1002/embj.201386369
216. Schultze JL. Precision attack on calcineurin in macrophages: a new anti-inflammatory weapon. EMBO J (2014) 33(10):1087–8. doi: 10.1002/embj.201488178
217. de Jesus T, Shukla S, Ramakrishnan P. Too sweet to resist: Control of immune cell function by O-GlcNAcylation. Cell Immunol (2018) 333:85–92. doi: 10.1016/j.cellimm.2018.05.010
218. Lee HT, Lee KI, Chen CH, Lee TS. Genetic deletion of soluble epoxide hydrolase delays the progression of Alzheimer's disease. J Neuroinflamm (2019) 16(1):267. doi: 10.1186/s12974-019-1635-9
219. Scassellati C, Ciani M, Galoforo AC, Zanardini R, Bonvicini C, Geroldi C. Molecular mechanisms in cognitive frailty: potential therapeutic targets for oxygen-ozone treatment. Mech Ageing Dev (2020) 186:111210. doi: 10.1016/j.mad.2020.111210
220. Rojanathammanee L, Puig KL, Combs CK. Pomegranate polyphenols and extract inhibit nuclear factor of activated T-cell activity and microglial activation in vitro transgenic mouse model alzheimer disease. J Nutr (2013) 143(5):597–605. doi: 10.3945/jn.112.169516
221. Rojanathammanee L, Floden AM, Manocha GD, Combs CK. Attenuation of microglial activation in a mouse model of Alzheimer's disease via NFAT inhibition. J Neuroinflamm (2015) 12:42. doi: 10.1186/s12974-015-0255-2
222. Khalil MAM, Khalil MAU, Khan TFT, Tan J. Drug-induced hematological cytopenia in kidney transplantation and the challenges it poses for kidney transplant physicians. J Transplant (2018) 2018:9429265. doi: 10.1155/2018/9429265
223. Farouk SS, Rein JL. The many faces of calcineurin inhibitor toxicity-what the FK? Adv Chronic Kidney Dis (2020) 27(1):56–66. doi: 10.1053/j.ackd.2019.08.006
224. Farinelli SE, Greene LA, Friedman WJ. Neuroprotective actions of dipyridamole on cultured CNS neurons. J Neurosci (1998) 18(14):5112–23. doi: 10.1523/JNEUROSCI.18-14-05112.1998
225. Lobner D, Choi DW. Dipyridamole increases oxygen-glucose deprivation-induced injury in cortical cell culture. Stroke (1994) 25(10):2085–9;discussion 2089-90. doi: 10.1161/01.str.25.10.2085
226. Giblin MJ, Smith TE, Winkler G, Pendergrass HA, Kim MJ, Capozzi ME, et al. Nuclear factor of activated T-cells (NFAT) regulation of IL-1β-induced retinal vascular inflammation. Biochim Biophys Acta Mol Basis Dis (2021) 1867(12):166238. doi: 10.1016/j.bbadis.2021.166238
227. Kitamura N, Kaminuma O. Isoform-selective NFAT inhibitor: potential usefulness and development. Int J Mol Sci (2021) 22(5). doi: 10.3390/ijms22052725
228. Wang Z, Ye D, Ye J, Wang M, Liu J, Jiang H, et al. The TRPA1 channel in the cardiovascular system: promising features and challenges. Front Pharmacol (2019) 10:1253. doi: 10.3389/fphar.2019.01253
229. Takahashi N, Mori Y. TRP channels as sensors and signal integrators of redox status changes. Front Pharmacol (2011) 2:58. doi: 10.3389/fphar.2011.00058
230. Talavera K, Startek JB, Alvarez-Collazo J, Boonen B, Alpizar YA, Sanchez A, et al. Mammalian transient receptor potential TRPA1 channels: from structure to disease. Physiol Rev (2020) 100(2):725–803. doi: 10.1152/physrev.00005.2019
231. Nassini R, Materazzi S, Benemei S, Geppetti P. The TRPA1 channel in inflammatory and neuropathic pain and migraine. Rev Physiol Biochem Pharmacol (2014) 167:1–43. doi: 10.1007/112_2014_18
232. Wang X, Li Y, Wei H, Yang Z, Luo R, Gao Y, et al. Molecular architecture and gating mechanisms of the Drosophila TRPA1 channel. Cell Discovery (2023) 9(1):36. doi: 10.1038/s41421-023-00527-1
233. Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell (2003) 112(6):819–29. doi: 10.1016/s0092-8674(03)00158-2
234. Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, et al. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron (2004) 41(6):849–57. doi: 10.1016/s0896-6273(04)00150-3
235. Hamada F, Rosenzweig M, Kang K, Pulver SR, Ghezzi A, Jegla TJ, et al. An internal thermal sensor controlling temperature preference in Drosophila. Nature (2008) 454(7201):217–20. doi: 10.1038/nature07001
236. Saito S, Banzawa N, Fukuta N, Saito CT, Takahashi K, Imagawa T, et al. Heat and noxious chemical sensor, chicken TRPA1, as a target of bird repellents and identification of its structural determinants by multispecies functional comparison. Mol Biol Evol (2014) 31(3):708–22. doi: 10.1093/molbev/msu001
237. Gracheva EO, Ingolia NT, Kelly YM, Cordero-Morales JF, Hollopeter G, Chesler AT, et al. Molecular basis of infrared detection by snakes. Nature (2010) 464(7291):1006–11. doi: 10.1038/nature08943
238. Kang K, Panzano VC, Chang EC, Ni L, Dainis AM, Jenkins AM, et al. Modulation of TRPA1 thermal sensitivity enables sensory discrimination in Drosophila. Nature (2011) 481(7379):76–80. doi: 10.1038/nature10715
239. Lee KI, Lee HT, Lin HC, Tsay HJ, Tsai FC, Shyue SK, et al. Role of transient receptor potential ankyrin 1 channels in Alzheimer's disease. J Neuroinflamm (2016) 13(1):92. doi: 10.1186/s12974-016-0557-z
240. Bosson A, Paumier A, Boisseau S, Jacquier-Sarlin M, Buisson A, Albrieux M. TRPA1 channels promote astrocytic Ca. Mol Neurodegener (2017) 12(1):53. doi: 10.1186/s13024-017-0194-8
241. Fernandez AM, Fernandez S, Carrero P, Garcia-Garcia M, Torres-Aleman I. Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J Neurosci (2007) 27(33):8745–56. doi: 10.1523/JNEUROSCI.1002-07.2007
242. Shi M, Chen F, Chen Z, Yang W, Yue S, Zhang J, et al. Sigma-1 receptor: A potential therapeutic target for traumatic brain injury. Front Cell Neurosci (2021) 15:685201. doi: 10.3389/fncel.2021.685201
243. Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell (2007) 131(3):596–610. doi: 10.1016/j.cell.2007.08.036
244. Pergolizzi J, Varrassi G, Coleman M, Breve F, Christo DK, Christo PJ, et al. The sigma enigma: A narrative review of sigma receptors. Cureus (2023) 15(3):e35756. doi: 10.7759/cureus.35756
245. Jansen KL, Faull RL, Storey P, Leslie RA. Loss of sigma binding sites in the CA1 area of the anterior hippocampus in Alzheimer's disease correlates with CA1 pyramidal cell loss. Brain Res (1993) 623(2):299–302. doi: 10.1016/0006-8993(93)91441-t
246. Mishina M, Ohyama M, Ishii K, Kitamura S, Kimura Y, Oda K, et al. Low density of sigma1 receptors in early Alzheimer's disease. Ann Nucl Med (2008) 22(3):151–6. doi: 10.1007/s12149-007-0094-z
247. Sałaciak K, Pytka K. Revisiting the sigma-1 receptor as a biological target to treat affective and cognitive disorders. Neurosci Biobehav Rev (2022) 132:1114–36. doi: 10.1016/j.neubiorev.2021.10.037
248. Ma WH, Chen AF, Xie XY, Huang YS. Sigma ligands as potent inhibitors of Aβ and AβOs in neurons and promising therapeutic agents of Alzheimer's disease. Neuropharmacology (2021) 190:108342. doi: 10.1016/j.neuropharm.2020.108342
249. Iñiguez MA, Punzón C, Nieto R, Burgueño J, Vela JM, Fresno M. Inhibitory effects of sigma-2 receptor agonists on T lymphocyte activation. Front Pharmacol (2013) 4:23. doi: 10.3389/fphar.2013.00023
250. Kim WS, Fu Y, Dobson-Stone C, Hsiao JT, Shang K, Hallupp M, et al. Effect of fluvoxamine on amyloid-β Peptide generation and memory. J Alzheimers Dis (2018) 62(4):1777–87. doi: 10.3233/JAD-171001
251. Fang M, Zhang P, Zhao Y, Jin A, Liu X. "Aβ mediates Sigma receptor degradation via CaN/NFAT pathway," (in eng). Am J Transl Res (2016) 8(8):3471–81.
252. Bouchon A, Hernández-Munain C, Cella M, Colonna M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med (2001) 194(8):1111–22. doi: 10.1084/jem.194.8.1111
253. Colonna M. The biology of TREM receptors. Nat Rev Immunol (2023) 23(9):580–94. doi: 10.1038/s41577-023-00837-1
254. Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med (2005) 201(4):647–57. doi: 10.1084/jem.20041611
255. Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer's disease. Nat Med (2020) 26(1):131–42. doi: 10.1038/s41591-019-0695-9
256. Okuzono Y, Sakuma H, Miyakawa S, Ifuku M, Lee J, Das D, et al. Reduced TREM2 activation in microglia of patients with Alzheimer's disease. FEBS Open Bio (2021) 11(11):3063–80. doi: 10.1002/2211-5463.13300
257. Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol (2006) 177(4):2051–5. doi: 10.4049/jimmunol.177.4.2051
258. Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, et al. TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron (2018) 97(5):1023–1031.e7. doi: 10.1016/j.neuron.2018.01.031
259. Lessard CB, Malnik SL, Zhou Y, Ladd TB, Cruz PE, Ran Y, et al. High-affinity interactions and signal transduction between Aβ oligomers and TREM2. EMBO Mol Med (2018) 10(11). doi: 10.15252/emmm.201809027
260. Ulland TK, Colonna M. TREM2 - a key player in microglial biology and Alzheimer disease. Nat Rev Neurol (2018) 14(11):667–75. doi: 10.1038/s41582-018-0072-1
261. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell (2015) 160(6):1061–71. doi: 10.1016/j.cell.2015.01.049
262. Shirotani K, Hori Y, Yoshizaki R, Higuchi E, Colonna M, Saito T, et al. Aminophospholipids are signal-transducing TREM2 ligands on apoptotic cells. Sci Rep (2019) 9(1):7508. doi: 10.1038/s41598-019-43535-6
263. Bader Lange ML, St Clair D, Markesbery WR, Studzinski CM, Murphy MP, Butterfield DA. Age-related loss of phospholipid asymmetry in APP(NLh)/APP(NLh) x PS-1(P264L)/PS-1(P264L) human double mutant knock-in mice: relevance to Alzheimer disease. Neurobiol Dis (2010) 38(1):104–15. doi: 10.1016/j.nbd.2010.01.004
264. Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, et al. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest (2015) 125(5):2161–70. doi: 10.1172/JCI77983
265. Zhao P, Xu Y, Fan X, Li L, Li X, Arase H, et al. Discovery and engineering of an anti-TREM2 antibody to promote amyloid plaque clearance by microglia in 5XFAD mice. MAbs (2022) 14(1):2107971. doi: 10.1080/19420862.2022.2107971
266. Companys-Alemany J, Turcu AL, Vázquez S, Pallàs M, Griñán-Ferré C. Glial cell reactivity and oxidative stress prevention in Alzheimer's disease mice model by an optimized NMDA receptor antagonist. Sci Rep (2022) 12(1):17908. doi: 10.1038/s41598-022-22963-x
267. Regunathan S, Reis DJ. Imidazoline receptors and their endogenous ligands. Annu Rev Pharmacol Toxicol (1996) 36:511–44. doi: 10.1146/annurev.pa.36.040196.002455
269. Han Z, Zhang HX, Tian JS, Zheng RY, Hou ST. "2-(-2-benzofuranyl)-2-imidazoline induces Bcl-2 expression and provides neuroprotection against transient cerebral ischemia in rats," (in eng). Brain Res (2010) 1361:pp. doi: 10.1016/j.brainres.2010.09.029
270. Han Z, Cheng ZH, Liu S, Yang JL, Xiao MJ, Zheng RY, et al. Neurovascular protection conferred by 2-BFI treatment during rat cerebral ischemia. Biochem Biophys Res Commun (2012) 424(3):544–8. doi: 10.1016/j.bbrc.2012.06.152
271. Han Z, Yang JL, Jiang SX, Hou ST, Zheng RY. Fast, non-competitive and reversible inhibition of NMDA-activated currents by 2-BFI confers neuroprotection," (in eng). PloS One (2013) 8:no. doi: 10.1371/journal.pone.0064894
272. Jiang SX, Zheng RY, Zeng JQ, Li XL, Han Z, Hou ST. "Reversible inhibition of intracellular calcium influx through NMDA receptors by imidazoline I(2) receptor antagonists. Eur J Pharmacol (2010) 629(1-3):12–9. doi: 10.1016/j.ejphar.2009.11.063
273. Craven JA, Conway EL. Effects of alpha 2-adrenoceptor antagonists and imidazoline2-receptor ligands on neuronal damage in global ischaemia in the rat. Clin Exp Pharmacol Physiol (1997) 24(2):204–7. doi: 10.1111/j.1440-1681.1997.tb01808.x
275. Rodriguez-Arévalo S, Bagán A, Griñán-Ferré C, Vasilopoulou F, Pallàs M, Brocos-Mosquera I, et al. Benzofuranyl-2-imidazoles as imidazoline I. Eur J Med Chem (2021) 222:113540. doi: 10.1016/j.ejmech.2021.113540
276. Vasilopoulou F, Rodríguez-Arévalo S, Bagán A, Escolano C, Griñán-Ferré C, Pallàs M. Disease-modifying treatment with I. Br J Pharmacol (2021) 178(15):3017–33. doi: 10.1111/bph.15478
277. García-Sevilla JA, Escribá PV, Walzer C, Bouras C, Guimón J. Imidazoline receptor proteins in brains of patients with Alzheimer's disease. Neurosci Lett (1998) 247(2-3):95–8. doi: 10.1016/s0304-3940(98)00265-1
278. Morley JE, Farr SA, Kumar VB, Armbrecht HJ. The SAMP8 mouse: a model to develop therapeutic interventions for Alzheimer's disease. Curr Pharm Des (2012) 18(8):1123–30. doi: 10.2174/138161212799315795
279. Brown SD, Twells RC, Hey PJ, Cox RD, Levy ER, Soderman AR, et al. Isolation and characterization of LRP6, a novel member of the low density lipoprotein receptor gene family. Biochem Biophys Res Commun (1998) 248(3):879–88. doi: 10.1006/bbrc.1998.9061
280. Alrefaei AF, Abu-Elmagd M. LRP6 receptor plays essential functions in development and human diseases. Genes (Basel) (2022) 13(1). doi: 10.3390/genes13010120
281. Jones ME, Büchler J, Dufor T, Palomer E, Teo S, Martin-Flores N, et al. A genetic variant of the Wnt receptor LRP6 accelerates synapse degeneration during aging and in Alzheimer's disease. Sci Adv (2023) 9(2):eabo7421. doi: 10.1126/sciadv.abo7421
282. Buechler J, Salinas PC. "Deficient wnt signaling and synaptic vulnerability in alzheimer's disease: emerging roles for the LRP6 receptor," (in eng). Front Synaptic Neurosci (2018) 10:38(38). doi: 10.3389/fnsyn.2018.00038
283. Liu CC, Tsai CW, Deak F, Rogers J, Penuliar M, Sung YM, et al. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer's disease. Neuron (2014) 84(1):63–77. doi: 10.1016/j.neuron.2014.08.048
284. Jaeger S, Pietrzik CU. Functional role of lipoprotein receptors in Alzheimer's disease. Curr Alzheimer Res (2008) 5(1):15–25. doi: 10.2174/156720508783884675
285. Zhao N, Liu CC, Qiao W, Bu G. "Apolipoprotein E, receptors, and modulation of alzheimer's disease," (in eng). Biol Psychiatry (2018) 83(4):347–57. doi: 10.1016/j.biopsych.2017.03.003
286. Chow HM, Sun JK, Hart RP, Cheng KK, Hung CHL, Lau TM, et al. Low-density lipoprotein receptor-related protein 6 cell surface availability regulates fuel metabolism in astrocytes. Adv Sci (Weinh) (2021) 8(16):e2004993. doi: 10.1002/advs.202004993
287. Sonntag KC, Ryu WI, Amirault KM, Healy RA, Siegel AJ, McPhie DL, et al. Late-onset Alzheimer's disease is associated with inherent changes in bioenergetics profiles. Sci Rep (2017) 7(1):14038. doi: 10.1038/s41598-017-14420-x
288. Lu Y, Liu M, Guo X, Wang P, Zeng F, Wang H, et al. miR-26a-5p alleviates CFA-induced chronic inflammatory hyperalgesia through Wnt5a/CaMKII/NFAT signaling in mice. CNS Neurosci Ther (2023) 29(5):1254–71. doi: 10.1111/cns.14099
289. Kodis EJ, Choi S, Swanson E, Ferreira G, Bloom GS. N-methyl-D-aspartate receptor-mediated calcium influx connects amyloid-β oligomers to ectopic neuronal cell cycle reentry in Alzheimer's disease. Alzheimers Dement (2018) 14(10):1302–12. doi: 10.1016/j.jalz.2018.05.017
290. Liu J, Chang L, Song Y, Li H, Wu Y. The role of NMDA receptors in alzheimer's disease. Front Neurosci (2019) 13:43(43). doi: 10.3389/fnins.2019.00043
291. Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol (2009) 7(1):65–74. doi: 10.2174/157015909787602823
292. Sattler R, Tymianski M. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol (2001) 24(1-3):107–29. doi: 10.1385/MN:24:1-3:107
293. Vesce S, Rossi D, Brambilla L, Volterra A. "Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation," (in eng). Int Rev Neurobiol (2007) 82(57-71). doi: 10.1016/S0074-7742(07)82003-4
294. Roselli F, Tirard M, Lu J, Hutzler P, Lamberti P, Livrea P, et al. Soluble beta-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J Neurosci (2005) 25(48):11061–70. doi: 10.1523/JNEUROSCI.3034-05.2005
295. Li Y, Chang L, Song Y, Gao X, Roselli F, Liu J, et al. Astrocytic gluN2A and gluN2B oppose the synaptotoxic effects of amyloid-β1-40 in hippocampal cells. J Alzheimers Dis (2016) 54(1):135–48. doi: 10.3233/JAD-160297
296. Parameshwaran K, Dhanasekaran M, Suppiramaniam V. Amyloid beta peptides and glutamatergic synaptic dysregulation. Exp Neurol (2008) 210(1):7–13. doi: 10.1016/j.expneurol.2007.10.008
297. Costa RO, Lacor PN, Ferreira IL, Resende R, Auberson YP, Klein WL, et al. Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-d-aspartate receptor in mature hippocampal cultures treated with amyloid-β oligomers. Aging Cell (2012) 11(5):823–33. doi: 10.1111/j.1474-9726.2012.00848.x
298. Verges DK, Restivo JL, Goebel WD, Holtzman DM, Cirrito JR. Opposing synaptic regulation of amyloid-β metabolism by NMDA receptors in vivo. J Neurosci (2011) 31(31):11328–37. doi: 10.1523/JNEUROSCI.0607-11.2011
299. Lesné S, et al. NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta production. J Neurosci (2005) 25(41):9367–77. doi: 10.1523/JNEUROSCI.0849-05.2005
300. Wang ZC, Zhao J, Li S. Dysregulation of synaptic and extrasynaptic N-methyl-D-aspartate receptors induced by amyloid-β. Neurosci Bull (2013) 29(6):752–60. doi: 10.1007/s12264-013-1383-2
301. Malinow R. New developments on the role of NMDA receptors in Alzheimer's disease. Curr Opin Neurobiol (2012) 22(3):559–63. doi: 10.1016/j.conb.2011.09.001
302. Owen RT. Memantine and donepezil: a fixed drug combination for the treatment of moderate to severe Alzheimer's dementia. Drugs Today (Barc) (2016) 52(4):239–48. doi: 10.1358/dot.2016.52.4.2479357
303. Ting SM, Zhao X, Zheng X, Aronowski J. Excitatory pathway engaging glutamate, calcineurin, and NFAT upregulates IL-4 in ischemic neurons to polarize microglia. J Cereb Blood Flow Metab (2020) 40(3):513–27. doi: 10.1177/0271678X19838189
304. Lisek M, Zylinska L, Boczek T. Ketamine and calcium signaling-A crosstalk for neuronal physiology and pathology. Int J Mol Sci (2020) 21(21). doi: 10.3390/ijms21218410
305. Fox C, Crugel M, Maidment I, Auestad BH, Coulton S, Treloar A, et al. Efficacy of memantine for agitation in Alzheimer's dementia: a randomised double-blind placebo controlled trial. PloS One (2012) 7(5):e35185. doi: 10.1371/journal.pone.0035185
306. Wilkinson D, Fox NC, Barkhof F, Phul R, Lemming O, Scheltens P. Memantine and brain atrophy in Alzheimer's disease: a 1-year randomized controlled trial. J Alzheimers Dis (2012) 29(2):459–69. doi: 10.3233/JAD-2011-111616
307. Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med (2003) 348(14):1333–41. doi: 10.1056/NEJMoa013128
308. Turcu AL, Companys-Alemany J, Phillips MB, Patel DS, Griñán-Ferré C, Loza MI, et al. Design, synthesis, and in vitro and in vivo characterization of new memantine analogs for Alzheimer's disease. Eur J Med Chem (2022) 236:114354. doi: 10.1016/j.ejmech.2022.114354
309. Shao L, Zhang Y, Hao Y, Ping Y. Upregulation of IP. Cell Rep (2022) 38(13):110594. doi: 10.1016/j.celrep.2022.110594
310. Bartok A, Weaver D, Golenár T, Nichtova Z, Katona M, Bánsághi S, et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat Commun (2019) 10(1):3726. doi: 10.1038/s41467-019-11646-3
311. Duchen MR. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch (2012) 464(1):111–21. doi: 10.1007/s00424-012-1112-0
312. Egorova PA, Bezprozvanny IB. Inositol 1,4,5-trisphosphate receptors and neurodegenerative disorders. FEBS J (2018) 285(19):3547–65. doi: 10.1111/febs.14366
313. Cheung KH, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, et al. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron (2008) 58(6):871–83. doi: 10.1016/j.neuron.2008.04.015
314. Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, et al. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer's disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal (2010) 3(114):ra22. doi: 10.1126/scisignal.2000818
315. Hirashima N, Etcheberrigaray R, Bergamaschi S, Racchi M, Battaini F, Binetti G, et al. Calcium responses in human fibroblasts: a diagnostic molecular profile for Alzheimer's disease. Neurobiol Aging (1996) 17(4):549–55. doi: 10.1016/0197-4580(96)00074-7
316. Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, et al. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med (1999) 5(1):101–6. doi: 10.1038/4789
317. Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron (2003) 39(3):409–21. doi: 10.1016/s0896-6273(03)00434-3
318. Shilling D, Müller M, Takano H, Mak DO, Abel T, Coulter DA, et al. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer's disease pathogenesis. J Neurosci (2014) 34(20):6910–23. doi: 10.1523/JNEUROSCI.5441-13.2014
319. Secondo A, Bagetta G, Amantea D. On the role of store-operated calcium entry in acute and chronic neurodegenerative diseases. Front Mol Neurosci (2018) 11:87(87). doi: 10.3389/fnmol.2018.00087
320. Sampieri A, Santoyo K, Asanov A, Vaca L. Association of the IP3R to STIM1 provides a reduced intraluminal calcium microenvironment, resulting in enhanced store-operated calcium entry. Sci Rep (2018) 8(1):13252. doi: 10.1038/s41598-018-31621-0
321. Pascual-Caro C, Berrocal M, Lopez-Guerrero AM, Alvarez-Barrientos A, Pozo-Guisado E, Gutierrez-Merino C, et al. STIM1 deficiency is linked to Alzheimer's disease and triggers cell death in SH-SY5Y cells by upregulation of L-type voltage-operated Ca. J Mol Med (Berl) (2018) 96(10):1061–79. doi: 10.1007/s00109-018-1677-y
322. Pascual-Caro C, Espinosa-Bermejo N, Pozo-Guisado E, Martin-Romero FJ. Role of STIM1 in neurodegeneration. World J Biol Chem (2018) 9(2):16–24. doi: 10.4331/wjbc.v9.i2.16
323. Meng M, Huo R, Ma N, Chang G, Shen X. β-carotene alleviates LPS-induced inflammation through regulating STIM1/ORAI1 expression in bovine mammary epithelial cells. Int Immunopharmacol (2022) 113(Pt A):109377. doi: 10.1016/j.intimp.2022.109377
324. Xue Y, Zhou S, Xie W, Meng M, Ma N, Zhang H, et al. STIM1-orai1 interaction exacerbates LPS-induced inflammation and endoplasmic reticulum stress in bovine hepatocytes through store-operated calcium entry. Genes (Basel) (2022) 13(5). doi: 10.3390/genes13050874
325. Sun Z, Li X, Yang L, Dong X, Han Y, Li Y, et al. SOCE-mediated NFAT1-NOX2-NLRP1 inflammasome involves in lipopolysaccharide-induced neuronal damage and Aβ generation. Mol Neurobiol (2022) 59(5):3183–205. doi: 10.1007/s12035-021-02717-y
326. Cook CN, Hejna MJ, Magnuson DJ, Lee JM. Expression of calcipressin1, an inhibitor of the phosphatase calcineurin, is altered with aging and Alzheimer's disease. J Alzheimers Dis (2005) 8(1):63–73. doi: 10.3233/jad-2005-8108
327. Sun X, Wu Y, Chen B, Zhang Z, Zhou W, Tong Y, et al. Regulator of calcineurin 1 (RCAN1) facilitates neuronal apoptosis through caspase-3 activation. J Biol Chem (2011) 286(11):9049–62. doi: 10.1074/jbc.M110.177519
328. Wu Y, Song W. Regulation of RCAN1 translation and its role in oxidative stress-induced apoptosis. FASEB J (2013) 27(1):208–21. doi: 10.1096/fj.12-213124
329. Wong H, Levenga J, Cain P, Rothermel B, Klann E, Hoeffer C. RCAN1 overexpression promotes age-dependent mitochondrial dysregulation related to neurodegeneration in Alzheimer's disease. Acta Neuropathol (2015) 130(6):829–43. doi: 10.1007/s00401-015-1499-8
330. Lim S, Hwang S, Yu JH, Lim JW, Kim H. Lycopene inhibits regulator of calcineurin 1-mediated apoptosis by reducing oxidative stress and down-regulating Nucling in neuronal cells. Mol Nutr Food Res (2017) 61(5). doi: 10.1002/mnfr.201600530
331. Sun X, Wu Y, Herculano B, Song W. RCAN1 overexpression exacerbates calcium overloading-induced neuronal apoptosis. PloS One (2014) 9(4):e95471. doi: 10.1371/journal.pone.0095471
332. Jiang H, Zhang C, Tang Y, Zhao J, Wang T, Liu H, et al. The regulator of calcineurin 1 increases adenine nucleotide translocator 1 and leads to mitochondrial dysfunctions. J Neurochem (2017) 140(2):307–19. doi: 10.1111/jnc.13900
333. Yun Y, Zhang Y, Zhang C, Huang L, Tan S, Wang P, et al. Regulator of calcineurin 1 is a novel RNA-binding protein to regulate neuronal apoptosis. Mol Psychiatry (2021) 26(4):1361–75. doi: 10.1038/s41380-019-0487-0
334. Wang T, Liu H, Wang Y, Liu C, Sun X. RCAN1 increases Aβ generation by promoting N-glycosylation via oligosaccharyltransferase. Curr Alzheimer Res (2014) 11(4):332–9. doi: 10.2174/1567205011666140331225855
335. Lloret A, Badia MC, Giraldo E, Ermak G, Alonso MD, Pallardó FV, et al. Amyloid-β toxicity and tau hyperphosphorylation are linked via RCAN1 in Alzheimer's disease. J Alzheimers Dis (2011) 27(4):701–9. doi: 10.3233/JAD-2011-110890
336. Wu Y, Deng Y, Zhang S, Luo Y, Cai F, Zhang Z, et al. Amyloid-β precursor protein facilitates the regulator of calcineurin 1-mediated apoptosis by downregulating proteasome subunit α type-5 and proteasome subunit β type-7. Neurobiol Aging (2015) 36(1):169–77. doi: 10.1016/j.neurobiolaging.2014.07.029
337. Lee S, Bang SM, Hong YK, Lee JH, Jeong H, Park SH, et al. The calcineurin inhibitor Sarah (Nebula) exacerbates Aβ42 phenotypes in a Drosophila model of Alzheimer's disease. Dis Model Mech (2016) 9(3):295–306. doi: 10.1242/dmm.018069
338. Ju Hwang C, Choi DY, Park MH, Hong JT. NF-κB as a key mediator of brain inflammation in alzheimer's disease. CNS Neurol Disord Drug Targets (2019) 18(1):3–10. doi: 10.2174/1871527316666170807130011
339. Zheng L, Liu H, Wang P, Song W, Sun X. Regulator of calcineurin 1 gene transcription is regulated by nuclear factor-kappaB. Curr Alzheimer Res (2014) 11(2):156–64. doi: 10.2174/1567205010666131212114907
340. Peiris H, Keating DJ. The neuronal and endocrine roles of RCAN1 in health and disease. Clin Exp Pharmacol Physiol (2018) 45(4):377–83. doi: 10.1111/1440-1681.12884
341. Sanna B, Brandt EB, Kaiser RA, Pfluger P, Witt SA, Kimball TR, et al. Modulatory calcineurin-interacting proteins 1 and 2 function as calcineurin facilitators in vivo. Proc Natl Acad Sci USA (2006) 103(19):7327–32. doi: 10.1073/pnas.0509340103
342. Liu Q, Busby JC, Molkentin JD. Interaction between TAK1-TAB1-TAB2 and RCAN1-calcineurin defines a signalling nodal control point. Nat Cell Biol (2009) 11(2):154–61. doi: 10.1038/ncb1823
343. Xu YR, Lei CQ. TAK1-TABs complex: A central signalosome in inflammatory responses. Front Immunol (2020) 11:608976. doi: 10.3389/fimmu.2020.608976
344. Dierssen M, Arqué G, McDonald J, Andreu N, Martínez-Cué C, Flórez J, et al. Behavioral characterization of a mouse model overexpressing DSCR1/ RCAN1. PloS One (2011) 6(2):e17010. doi: 10.1371/journal.pone.0017010
345. Hoeffer CA, Dey A, Sachan N, Wong H, Patterson RJ, Shelton JM, et al. The Down syndrome critical region protein RCAN1 regulates long-term potentiation and memory via inhibition of phosphatase signaling. J Neurosci (2007) 27(48):13161–72. doi: 10.1523/JNEUROSCI.3974-07.2007
346. Wong H, Buck JM, Borski C, Pafford JT, Keller BN, Milstead RA, et al. RCAN1 knockout and overexpression recapitulate an ensemble of rest-activity and circadian disruptions characteristic of Down syndrome, Alzheimer's disease, and normative aging. J Neurodev Disord (2022) 14(1):33. doi: 10.1186/s11689-022-09444-y
347. Asai M, Kinjo A, Kimura S, Mori R, Kawakubo T, Shirotani K, et al. Perturbed calcineurin-NFAT signaling is associated with the development of alzheimer's disease. Biol Pharm Bull (2016) 39(10):1646–52. doi: 10.1248/bpb.b16-00350
348. Janel N, Sarazin M, Corlier F, Corne H, de Souza LC, Hamelin L, et al. Plasma DYRK1A as a novel risk factor for Alzheimer's disease. Transl Psychiatry (2014) 4(8):e425. doi: 10.1038/tp.2014.61
349. Jung MS, Park JH, Ryu YS, Choi SH, Yoon SH, Kwen MY, et al. Regulation of RCAN1 protein activity by Dyrk1A protein-mediated phosphorylation. J Biol Chem (2011) 286(46):40401–12. doi: 10.1074/jbc.M111.253971
350. Lee HJ, Woo H, Lee HE, Jeon H, Ryu KY, Nam JH, et al. The novel DYRK1A inhibitor KVN93 regulates cognitive function, amyloid-beta pathology, and neuroinflammation. Free Radic Biol Med (2020) 160:575–95. doi: 10.1016/j.freeradbiomed.2020.08.030
351. Kim H, Lee KS, Kim AK, Choi M, Choi K, Kang M, et al. A chemical with proven clinical safety rescues Down-syndrome-related phenotypes in through DYRK1A inhibition. Dis Model Mech (2016) 9(8):839–48. doi: 10.1242/dmm.025668
Keywords: calcineurin, NFAT, Alzheimer’s disease, calcium, inflammation
Citation: Mackiewicz J, Lisek M and Boczek T (2023) Targeting CaN/NFAT in Alzheimer’s brain degeneration. Front. Immunol. 14:1281882. doi: 10.3389/fimmu.2023.1281882
Received: 23 August 2023; Accepted: 06 November 2023;
Published: 23 November 2023.
Edited by:
Chao Yang, Zhejiang Ocean University, ChinaReviewed by:
Huw Lewis, GlaxoSmithKline, United KingdomCopyright © 2023 Mackiewicz, Lisek and Boczek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomasz Boczek, dG9tYXN6LmJvY3pla0B1bWVkLmxvZHoucGw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.