 Mariam T. Heggi
Mariam T. Heggi Hanzada T. Nour El-Din
Hanzada T. Nour El-Din Dina I. Morsy
Dina I. Morsy Noha I. Abdelaziz3
Noha I. Abdelaziz3 Ahmed S. Attia
Ahmed S. Attia- 1Clinical Pharmacy Undergraduate Program, Faculty of Pharmacy, Cairo University, Cairo, Egypt
- 2Department of Microbiology and Immunology, Faculty of Pharmacy, Cairo University, Cairo, Egypt
- 3School of Pharmacy, Newgiza University, Giza, Egypt
The complement system is a fundamental part of the innate immune system that plays a key role in the battle of the human body against invading pathogens. Through its three pathways, represented by the classical, alternative, and lectin pathways, the complement system forms a tightly regulated network of soluble proteins, membrane-expressed receptors, and regulators with versatile protective and killing mechanisms. However, ingenious pathogens have developed strategies over the years to protect themselves from this complex part of the immune system. This review briefly discusses the sequence of the complement activation pathways. Then, we present a comprehensive updated overview of how the major four pathogenic groups, namely, bacteria, viruses, fungi, and parasites, control, modulate, and block the complement attacks at different steps of the complement cascade. We shed more light on the ability of those pathogens to deploy more than one mechanism to tackle the complement system in their path to establish infection within the human host.
1 The complement system: introduction
Our planet is heavily populated by microscopic organisms that are trying to evade their hosts in search of a place where they can live and prosper. Thus, the homeostasis of the host is threatened by a vast array of allergenic or toxic substances. Each pathogen has a special mechanism by which it replicates, spreads, and unintendedly alters the host’s functions. It is not surprising, therefore, that our immune system is equipped with a complex array of defensive mechanisms to oppose these alterations. At the same time, our immune system works to avoid responses that produce excessive damage to self-tissues or that might abolish beneficial commensal microbes (1).
The immune system is well-constructed upon two general systems, innate and acquired immunity. Our innate immunity functions through its soluble proteins and bioactive small molecules that are constitutively present in biological fluids, such as the complement proteins, defensins, and ficolins (1). The complement system is a highly sophisticated biological reaction system that primarily augments the opsonization, phagocytosis, and lysis of the target cells. However, it does not merely “complement” – though it upholds the name – but it forms an open border between innate and adaptive immunity, ultimately serving the purpose of maintaining homeostasis and tissue integrity (2).
Multiple functions of the complement system are carried out by effector fragments, which are inactive zymogens that are activated into proteases of 30-50 proteins, most of which are synthesized in the liver. Those proteases cleave other components successively in amplification pathways leading to exponential generation of the final effectors (3).
1.1 Complement activation
The complexity of the complement system is not solely in its composition, but it also extends to include its initiation via three pathways (Figure 1). The pathways intertwine enough to be called a maze; however, they have distinct features starting from their initiators and the pattern recognition molecules (PRMs) they recognize.
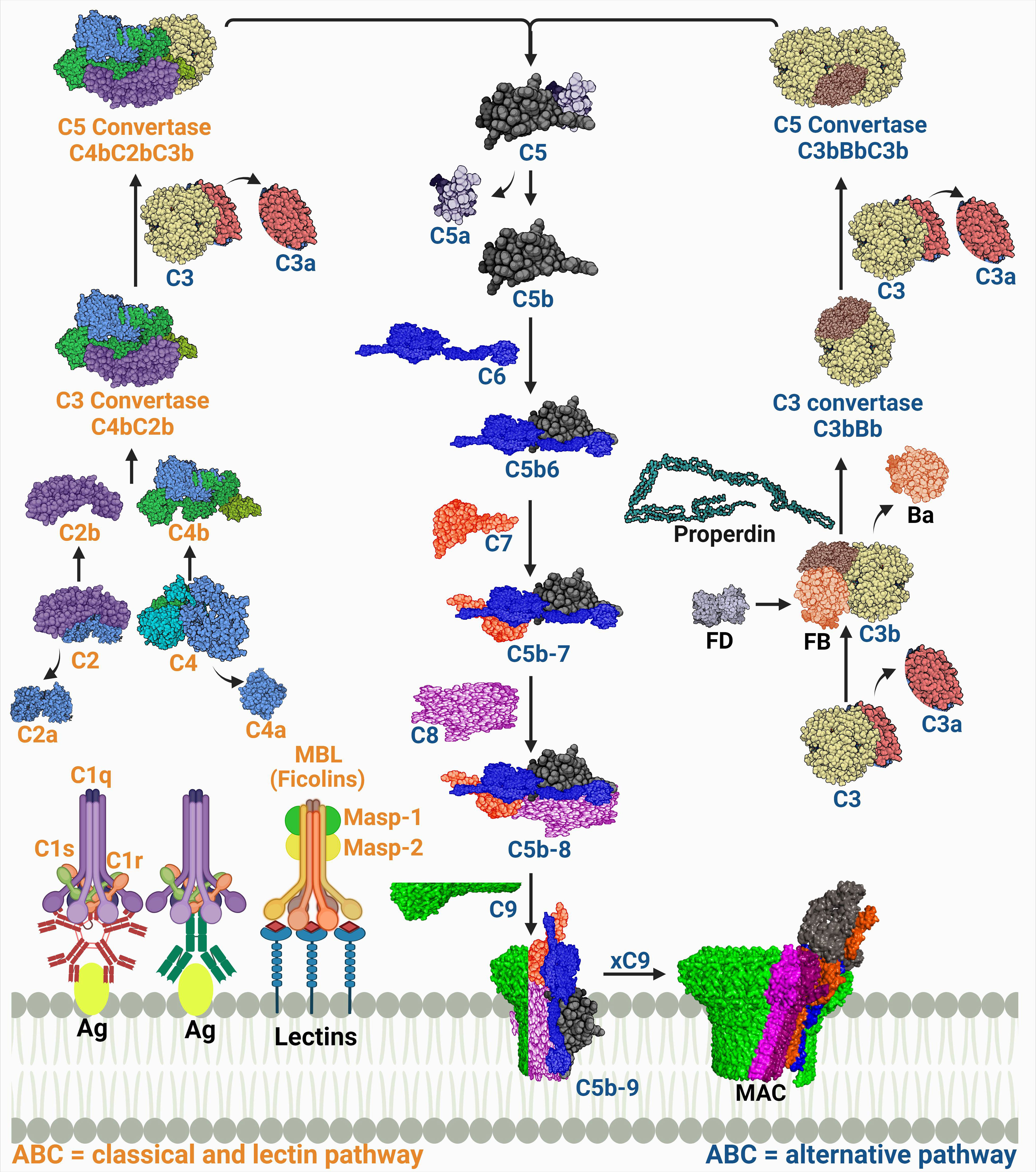
Figure 1 A schematic diagram showing the three complement activation pathways converging at a common terminal route of forming the membrane attack complex (MAC). The labels of the components of the classical and lectin pathways are colored in orange, while those of the alternative pathway are colored in dark blue. The proteins’ codes and structures were retrieved from the Protein Data Bank, while the figure was generated using BioRender.
1.1.1 Complement system initiation and generation of the C3 convertase
The PRM for the classical pathway is the C1q. It recognizes immune complexes such as antigen-bound immunoglobulin G (IgG) and IgM, thus this pathway is also known as the ‘antibody-dependent’ pathway. A conformational change then occurs allowing C1q, a hexamic collagen-like protein, to interact with C1r and C1s and form activated C1qr2s2 (4, 5). The activated C1qr2s2 subsequently cleaves C4, then C2 in a Ca2+ dependent reaction which forms the C4b and C2b complex (C4bC2b) known as the classical pathway C3 convertase. This complex is responsible for the cleavage of C3 into C3a and C3b. The latter will then be able to induce proteolytic activity and activate the common terminal pathway (6, 7). Other structures such as C-reactive protein (CRP) can also activate C1q in an antibody independent manner (8).
The C1q analog in the lectin pathway is the mannose-binding lectin (MBL). The MBL is the first known molecule to initiate this pathway and it recognizes monosaccharides exposing horizontal 3′- and 4′-OH groups as in mannose, glucose, and N-acetyl glucosamine but only when organized in a pattern fitting array (9). There are other humoral PRMs found in humans that can initiate the lectin pathway as well which are ficolins (M-, L-, and H-ficolin; also called ficolin-1, -2, and -3), and collectin 11 (CL11 or CL-K1) (10). The PRMs interact with serine proteases, known as MBL-associated serine proteases (MASPs), which are initially present as zymogens within the complexes. The complement cascade is initiated when the recognition molecules bind to their target and trigger the proteolytic step which starts with auto activation of MASP-1, which successively cleaves MASP-2. Both MASP-1 and MASP-2 cleave C2; however, C4 is cleaved only by MASP-2. Thus, both enzymes play an essential role in the subsequent assembly of the C4bC2b (CP and LP C3 convertase) (11, 12). The MASP-3 is a competitor for MASP-2 in binding to MBL. Although this competition slows LP activation, MASP-3 is considered the primary activator of the alternative pathway (AP) protease, factor D, in the human body fluids (13, 14). It is important to note that CP and LP are always in an inactive form, and they need PRMs to be activated.
On the contrary, the AP is in a constitutively activated state, referred to as the ‘tick-over’. This occurs through the spontaneous hydrolysis of a highly reactive thioester bond in C3 producing an active C3 form termed C3(H2O). This active form is able to form a complex with factor B “C3(H2O)B”. This complex is a substrate for factor D, which cleaves the B protein to form a short-lived soluble C3 convertase (C3(H2O)Bb). C3 is cleaved to give C3a, an anaphylatoxin (recruiter of inflammatory cells), and C3b that will likewise bind to factor B assembling the C3 convertase (C3bBb) and it then leads to the amplification loop (11, 14). Activated neutrophils release a protein called properdin, which stabilizes the C3bBb by binding to C3b to stop its cleavage by factors H and I. Later it was found that properdin can initiate the convertase assembly by either nonspecific covalent binding to C3b, C3bB, and/or C3bBb or by specific noncovalent binding to the target which as a result, increases the convertase assembly and C3b deposition (15). In addition, the generation of C3d from the C3 cleavage enhances the opsonization process together with iC3b and C3b (16).
All this is manifested by the presence of the C3 cleavage products, C3b and C3(H2O), in plasma in the normal physiological state. This low level of activation is the core of the complement surveillance activity. It is proposed that the AP C3 is a ‘contact-activated’ protein, which means that upon interaction with a biological or artificial interface, the C3 hydrolysis rate will be accelerated, ensuring the continuous supply of reactive C3b that can deposit on the surfaces of pathogens (17, 18).
A top view of the three pathways (Figure 1) indicates that each of them runs singularly; however, C3 is a common product of all pathways and its cleavage by the respective convertase determines the first complement effector activity. Although the three pathways are not linearly aligned, they represent a coherent network entangled on the same route to give effector functions and produce immune surveillance. The CP and the LP share the same convertase structure (C4bC2b), while the AP convertase is distinct (C3bBb) (19).
1.1.2 Generation of the C5 convertase
The classical/lectin pathway C5 convertase, C4bC2bC3b, and the alternative C5 convertase, C3bBbC3b, are both products of the binding of C3b, which is deposited from the amplification loop to the pre-assembled C3 convertases. Both C3 convertases catalyze the proteolysis of C3 into C3a and C3b and the subsequent cleavage of C5 into C5a and C5b (20). C5 cleavage is mostly managed by the AP, while properdin acts as a stabilizer for the convertase end products. However, C5 cleavage activation is not yet fully understood (21). The C3 convertase complex (C3bBb) switches the specificity of the enzyme substrate affinity from C3 to C5 cleavage into C5a and C5b (22). C5b is momentarily available to bind with C6, hence initiating the terminal pathway.
1.1.3 Assembly of the membrane attack complex
The stable complex (C5b-6) sequentially recruits proteins C7 and C8 and polymers of C9, leading to the multimeric complex assembly of the membrane attack complex (MAC). The complex penetrates the phospholipid bilayer as a result of the structural transition of its components that exposes hydrophobic regions, enabling its binding to the cell membrane (23). In order to induce a functional effect on the lipid bilayer, in which a tubular transmembrane pore is formed, 10 to 18 C9 molecules are needed (24). This pore acts as a channel, diffusing ions and small molecules, and killing the cell by osmotic instability.
However, not all cells are vulnerable to lysis by MAC. For instance, metabolically active nucleated cells can exhibit resistance and repair mechanisms against complement lysis (25). In addition, Gram-positive bacteria are naturally protected from lysis by their thick cell wall, although sub-lytic activity of the MAC can still induce signal transduction pathways (24, 26).
1.2 Complement system regulators
The undeniably important role of the complement system makes its regulation of even greater importance. Hence, a balance is maintained to ensure the effective functioning of the complement system and avoid possible autologous harm. This balance is achieved through a group of regulators and inhibitors that also keep the severity and propagation of the complement cascade on track. These regulators are either cytoplasmic or membrane-bound and they act on different and sometimes multiple levels of the cascade according to the pathway involved (27) (Figure 1). A better understanding of the role of each of these regulators is a pillar in safeguarding against pathogen evasion of the complement system (28).
1.2.1 Initiation of the C3 convertase and its regulation
The activation of the two closely connected CP and LP pathways is inhibited by the C1 inhibitor (C1INH) - a plasma serine proteinase (SERPIN1) - that irreversibly binds to, and hence inhibits, both the C1r and C1s of the CP and the MASP-1 and MASP-2 of the LP (3). Moreover, the LP is inhibited by the binding affinity of the small mannose-binding-lectin associated protein (sMAP) and MAP-1 to MBL and ficolins, as they are non-proteolytic splice products of the MASP2 and MASP1/3 genes, respectively. They possess no similar activity to MASP-1 or MASP-2 causing the inhibition of the LP (11).
The C3 convertase of the AP is regulated by factor H (FH) and its homolog factor H-Like protein 1 (FHL-1), which can either express decay accelerating activity or aid in the C3b degradation (28, 29). Also, C4 binding protein (C4BP) is a cofactor of FI that accelerates the decay of the CP and LP C3 convertase (C4bC2b), and binds C4b (8).
The membrane bound regulators, such as membrane cofactor protein (MCP or CD46) which has cofactor activity for FI and the decay accelerating factor (DAF or CD55), accelerate the decay of the C3 convertases in the three pathways. Similarly, the complement receptor 1 (CR1 or CD35) expresses decay accelerating activity and functions as a cofactor for FI-mediated cleavage of C3 convertase (3, 30). Finally, the carboxypeptidase-N inactivates the complement anaphylatoxin peptides C3a and C5a (8).
1.2.2 Regulation of the C5 convertase and MAC
Regulation of the complement system extends reaching the inhibitors of the C5 convertase. Those inhibitors include the DAF and FH that have destabilizing, and decay accelerating activity (11). The final effector molecule of the complement, the MAC, is regulated by protectin (CD59), a cell-based protein that inhibits the binding of C9 to the C5b-8 complex and hence prevents the MAC assembly (31). Additionally, soluble plasma based regulators, such as vitronectin (Vn) and clusterin, bind to C5b-7 and prevent the assembly of the MAC (28).
1.3 Complement evasion
The complex and crucial roles of the complement system in both adaptive and innate immunity have made it an integral part of the relentless rivalry between the immune system and pathogens (32). This rivalry begins with microbes trying to evade detection by the complement system, which is the very early, and somehow frontline, force of immunity to block further effector functions and reactions. There is a clear distinction between infectious microbes that are successfully recognized and eliminated, and pathogenic microbes that survive in an immunocompetent host (33). The emergence of multiple strategies to evade the complement system, impair its cascade, and prevent its effector functions is no surprise. The evasion of the complement system by a pathogen is now a battle of wits as many strategies targeting different layers of the complement system have unfolded in the past decades (34). There is a repertoire of mechanisms utilized by multiple pathogens that are well-established and understood. Known mechanisms include the recruitment and binding of complement system regulators or expression of regulator-like substances by pathogens, as well as, expression of inhibitors (35). Additionally, the secretion of proteases specifically against target complement proteins is considered a virulence mechanism (36, 37). There are common features that can be seen in the aforementioned mechanisms which are beneficial in exploiting the host defence system. The first feature is the sequence diversity of individual escape proteins (34). Also, the multiplicity of mechanisms that are utilized by a single pathogen represents another elusive approach against the complement system (38). Finally, the redundancy in the timely expression of several proteins directed to complement regulators or host defences is a common feature in multiple pathogens (33).
1.3.1 Influencing the complement activation: the beginning of the fight
As pathogens begin their survival struggle against the immune system, evasion of the complement activation, and effector functions becomes a matter of life or death. Targeting the initiation phase of the complement system is a strategy seen in multiple organisms (33). The microbial factors deployed in these mechanisms are summarized in Table 1 and Figure 2. The CP and LP have PRMs that are possible targets for microorganisms, for example, the Gram-negative bacterium Pseudomonas aeruginosa produces two proteases, Pseudomonas elastase (PaE) and Pseudomonas alkaline protease (PaAP), that cleave immunoglobulins and C1q, hence inhibiting the CP activation (47). Another mechanism is the inhibition of the C1q and binding of immunoglobulin which is seen in Neisseria meningitidis by its capsular oligosaccharides that interfere with C1q and IgG binding (46). The Streptococcus spp. have multiple evasion mechanisms including cleavage of the IgG by the IgG-protease of S. pyogenes (IdeS) and the streptococcal pyrogenic exotoxin B (SpeB), as well as the endoglycosidase (EndoS) (52, 53). Also, the capsular polysaccharides of S. pneumoniae decrease complement initiation by minimizing the binding of the complement components (54). Additionally, the streptococcal protein G (SPG) binds to the Fc portion of IgG, inhibiting its interaction with C1q (55). In the emerging zoonotic pathogen S. suis, the cell wall anchored heme-binding protein SntA was shown to bind C1q, interfering with the CP activation (56). It has also been suggested that SntA can interfere with both CP and LP through complement component consumption (56). The same pathogen secretes the IdeSsuis protease, which is highly specific to porcine IgM and hence, blocks CP activation via this immunoglobulin (57). Rickettsia spp. express the APRc, which is an Ig-binding protein that binds in a non-immune way with different immunoglobulins from different hosts, blocking the activation of the CP (48).
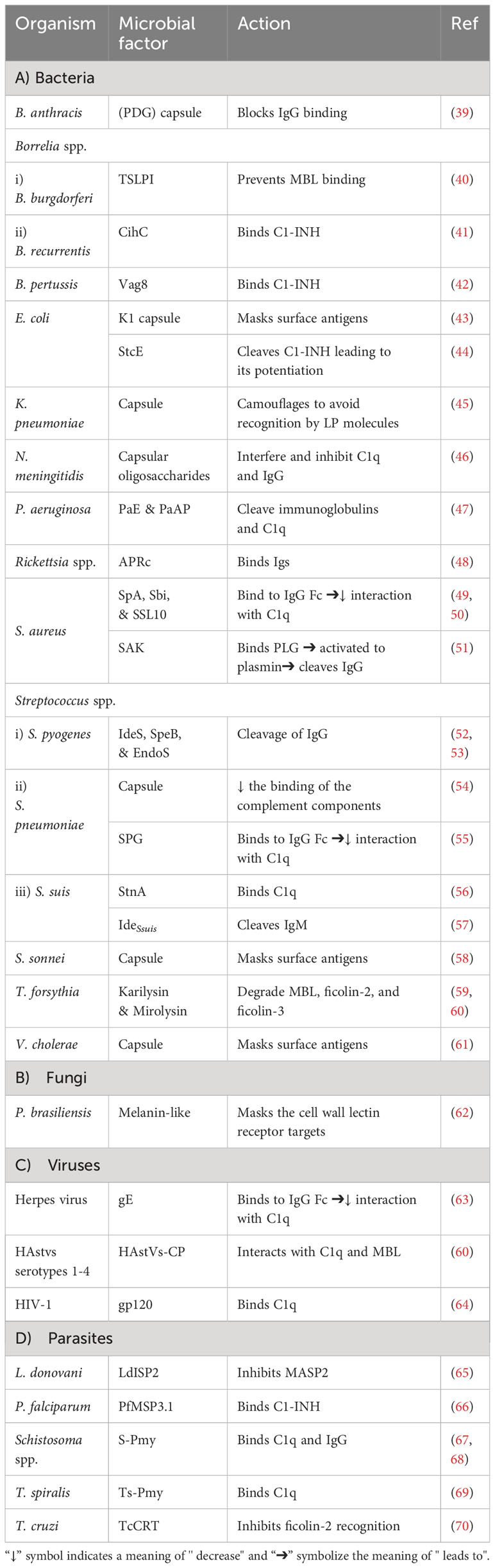
Table 1 Microbial strategies for influencing the complement activation.
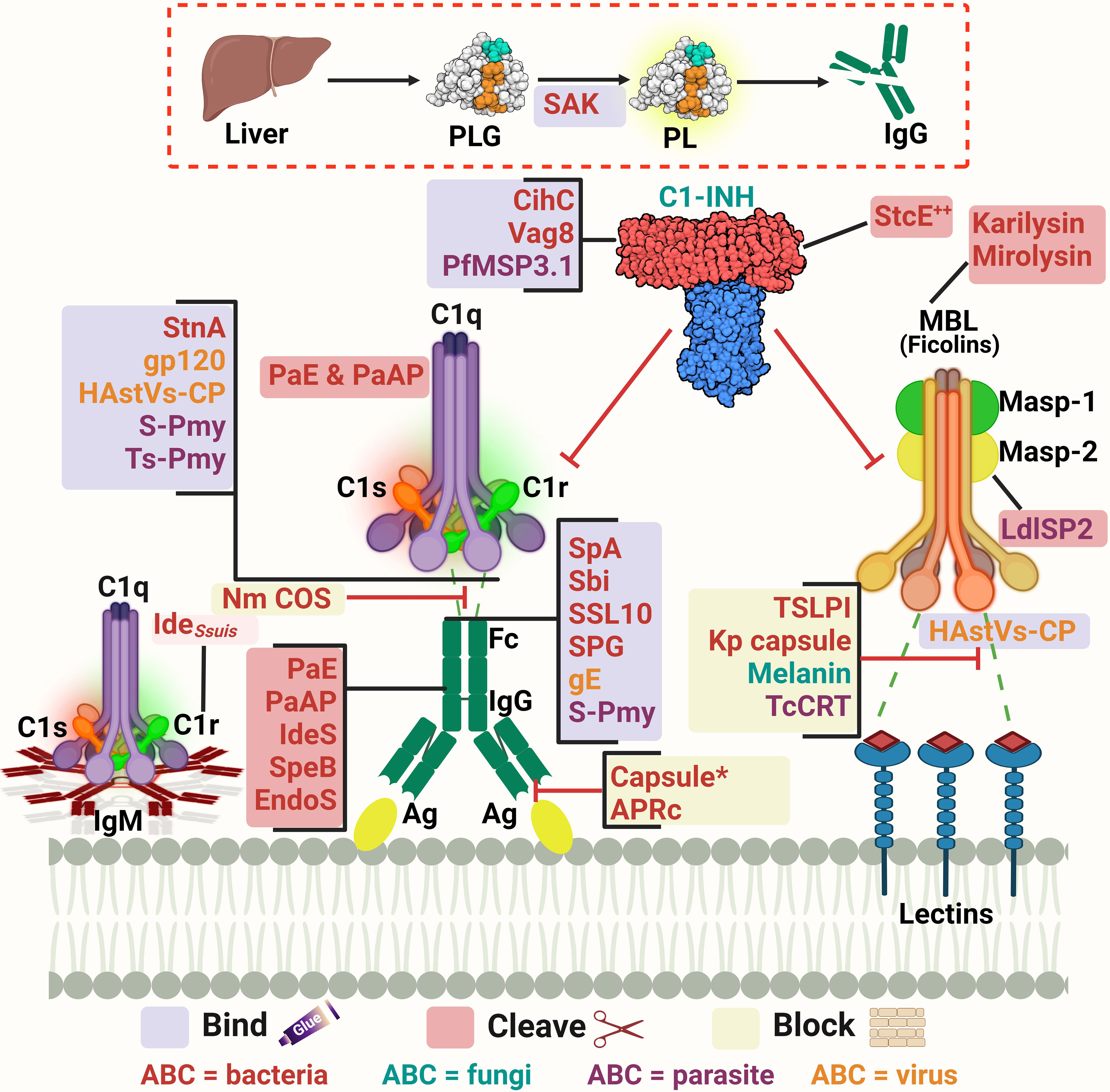
Figure 2 A schematic diagram showing microbial virulence factors involved in targeting the initiation phase of the complement system. The labels for the bacterial factors are shown in red, the fungal ones in turquoise, the parasite-related ones in purple, and finally the viral factors in orange. The mechanism by which those proteins affect the pathway is categorized into binding, shaded in purple; cleavage, which is shaded in dark pink; and blocking, which is highlighted in beige. The proteins’ codes and structures were retrieved from the Protein Data Bank, while the figure was generated using BioRender. The * indicates that the capsules of multiple pathogens perform the same function. The ++ indicates that the StcE cleavage of the C1-INH activates it rather than inhibiting it.
In the case of Staphylococcus aureus, protein A (SpA) binds to the Fc region of IgG, diminishing its interaction with C1q. There is also the staphylococcal binder of immunoglobulins (Sbi) and the staphylococcal superantigen-like protein 10 (SSL10) which inhibit the CP activation in the same way (49, 50). Moreover, S. aureus produces distinct proteolytic enzymes such as staphylokinase (SAK) which binds plasminogen (PLG) to its surface, resulting in its activation to plasmin which cleaves bound IgG and blocks the complement activation (51). On another front, the poly-γ-D-glutamate (PDG) capsule of Bacillus anthracis protects this pathogen from opsonic phagocytosis by interfering with complement activation and blocking the binding of IgG and C3b (39). The use of capsules to mask microbial surface antigens and evade complement activation and subsequent killing has been seen in other pathogens such as Escherichia coli (43), Shigella sonnei (58), and Vibrio cholerae (61).
As for viruses, the Herpes virus has several glycoproteins that target the complement system. For instance, glycoprotein E (gE) possesses Fc-receptor properties to diminish the CP-essential antibody recognition (63). The human immunodeficiency virus type 1 (HIV-1) targets the C1q as well and binds it with the envelope glycoprotein gp120 (64). While in parasites, Schistosoma spp. use the protein paramyosin (S-Pmy) which has binding affinities for both C1q and IgG (67, 68). On the other hand, the homolog of this protein in Trichinella spiralis (Ts-Pmy) inhibits the CP through binding to C1q (69).
This multitude of mechanisms that target the PRMs is not confined to the CP only but is also seen in the LP. In the case of Leishmania donovani, its inhibitor of serine peptidases 2 (LdISP2) demonstrated a potent effect on MASP2 of the LP leading to the blockage of the complement activation cascade (65). For instance, some strains of Klebsiella pneumoniae resort to camouflaging their capsular composition to avoid recognition by the LP molecules (45). The tick-borne Borrelia burgdorferi deviously benefits from the tick salivary lecithin pathway inhibitor protein (TSLPI) at the tick bite site to inhibit LP activation by preventing MBL binding (40). Also, the cleavage of the LP PRMs by proteases is seen in Tannerella forsythia, a pathogen strongly associated with periodontitis, that produces two metalloproteinases called karilysin and mirolysin that degrade the MBL ficolin-2 and ficolin-3, hence inhibiting the LP (59, 60). The parasite Trypanosoma cruzi specifically targets the LP activation by expressing the surface protein T. cruzi calreticulin (TcCRT) to inhibit the ficolin-2 mediated activation of the LP (70). The Paracoccidioides brasiliensis yeast masks the cell wall lectin receptor targets with a melanin-like pigment and hence prevents the complement-dependent phagocytosis (62). Finally, the human astroviruses (HAstVs) serotypes 1-4 are able to inhibit the complement system through their coat protein (HAstVs-CP) that interacts with both C1q and MBL, resulting in the inhibition of both the CP and LP (71).
Another mechanism adopted by some pathogens to interfere with the complement system’s initial steps is the acquisition of regulators involved in this stage, such as C1-INH. For instance, E. coli O157:H7 produces the secreted protease of C1 esterase inhibitor (StcE), which cleaves C1-INH and potentiates its inhibitory effect, thus downregulating the activation of both the CP and LP (44). B. recurrentis, the causative agent of louse-borne relapsing fever, is able to acquire the complement regulator C1-INH by the surface protein complement inhibition (CihC) (41). Likewise, Bordetella pertussis, binds the same regulator by the auto transporter protein virulence-associated gene 8 (Vag8) (42). The merozoites of the malaria causing parasite Plasmodium falciparum adopt that same strategy via its P. falciparum merozoite surface protein-3 (PfMSP3.1) (66).
1.3.2 Interfering with the intermediate stages of the complement cascade: a decisive battle
Following complement initiation, the intermediate stage of the cascade aims at the subsequent activation of the complement components through two convertases: the C3 and C5 convertases. The C3 convertase is generated as a common product for the first step of complement activation in all three pathways. However, the composition of the C3 convertase is C4bC2b in both CP and LP, while for the AP, it is C3bBb (37). This intermediate stage of the cascade is a choke point in complement evasion not solely because the three networks compile to generate those two functionally similar components, but also, as C3 cleavage gives way to two essential effector fragments, C3a and C3b (72). The mechanisms deployed by microbial pathogens to interfere with this stage are summarized in Table 2 and Figures 3, 4.
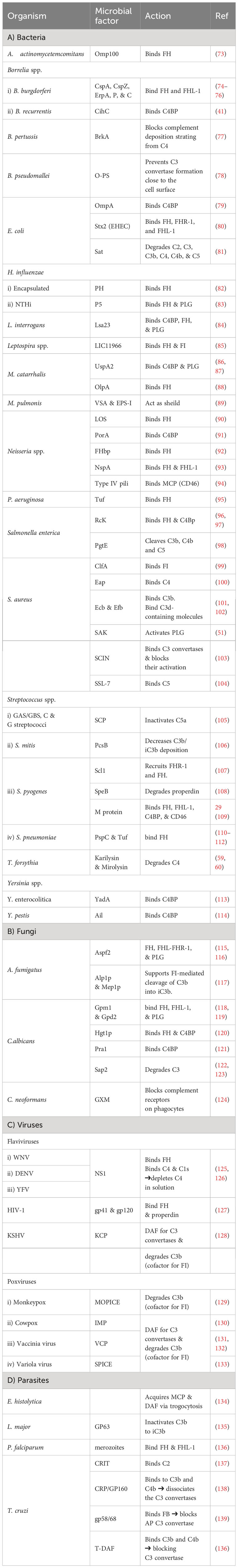
Table 2 Microbial strategies for interfering with the complement cascade intermediate stages.
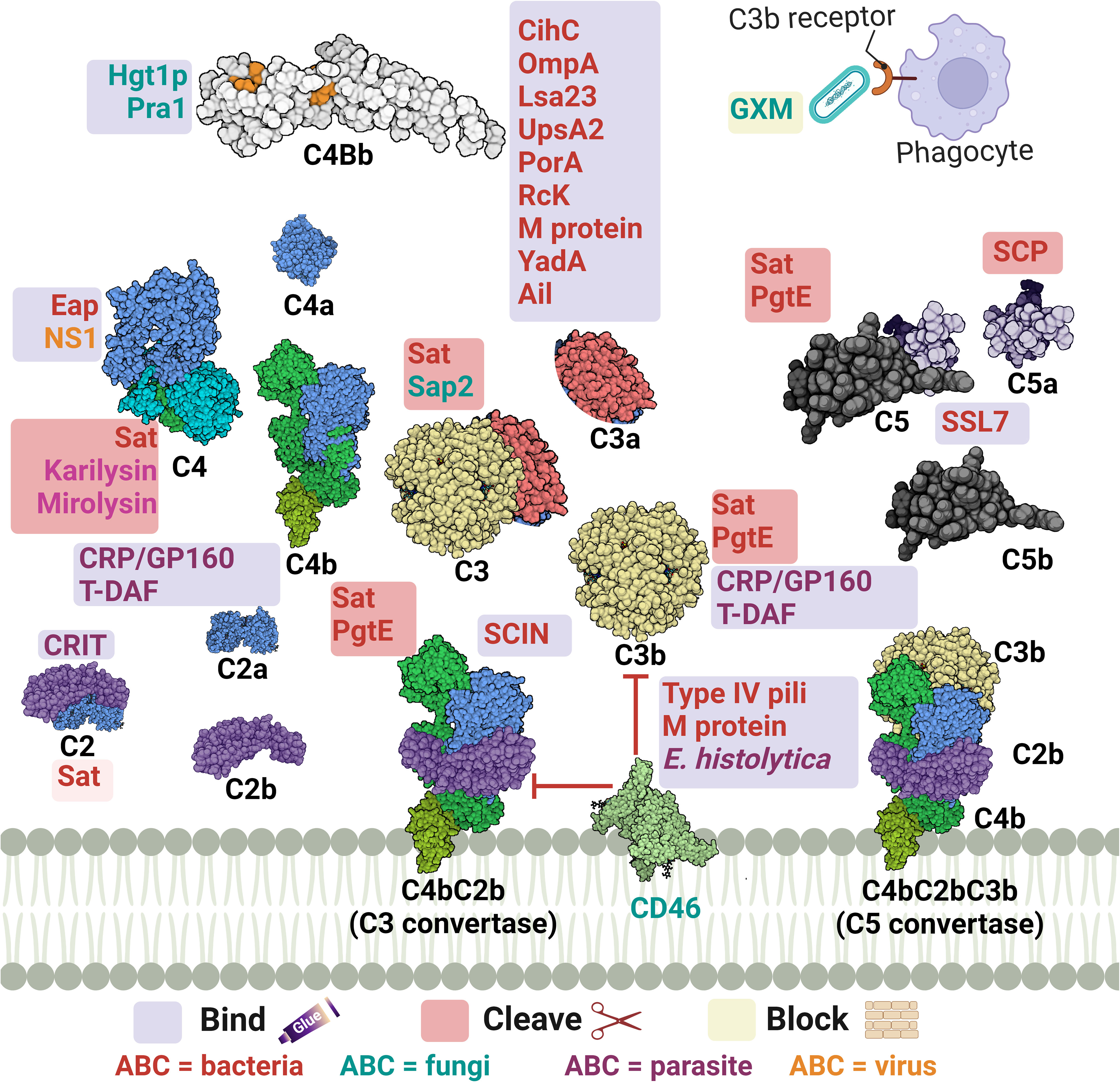
Figure 3 A schematic diagram showing microbial virulence factors involved in targeting this part of the complement cascade. The labels for the bacterial factors are shown in red, the fungal ones in turquoise, the parasite-related ones in purple, and finally the viral factors in orange. The mechanism by which those proteins affect the pathway is categorized into binding, shaded in purple; cleavage, which is shaded in dark pink; and blocking, which is highlighted in beige. The proteins’ codes and structures were retrieved from the Protein Data Bank, while the figure was generated using BioRender.
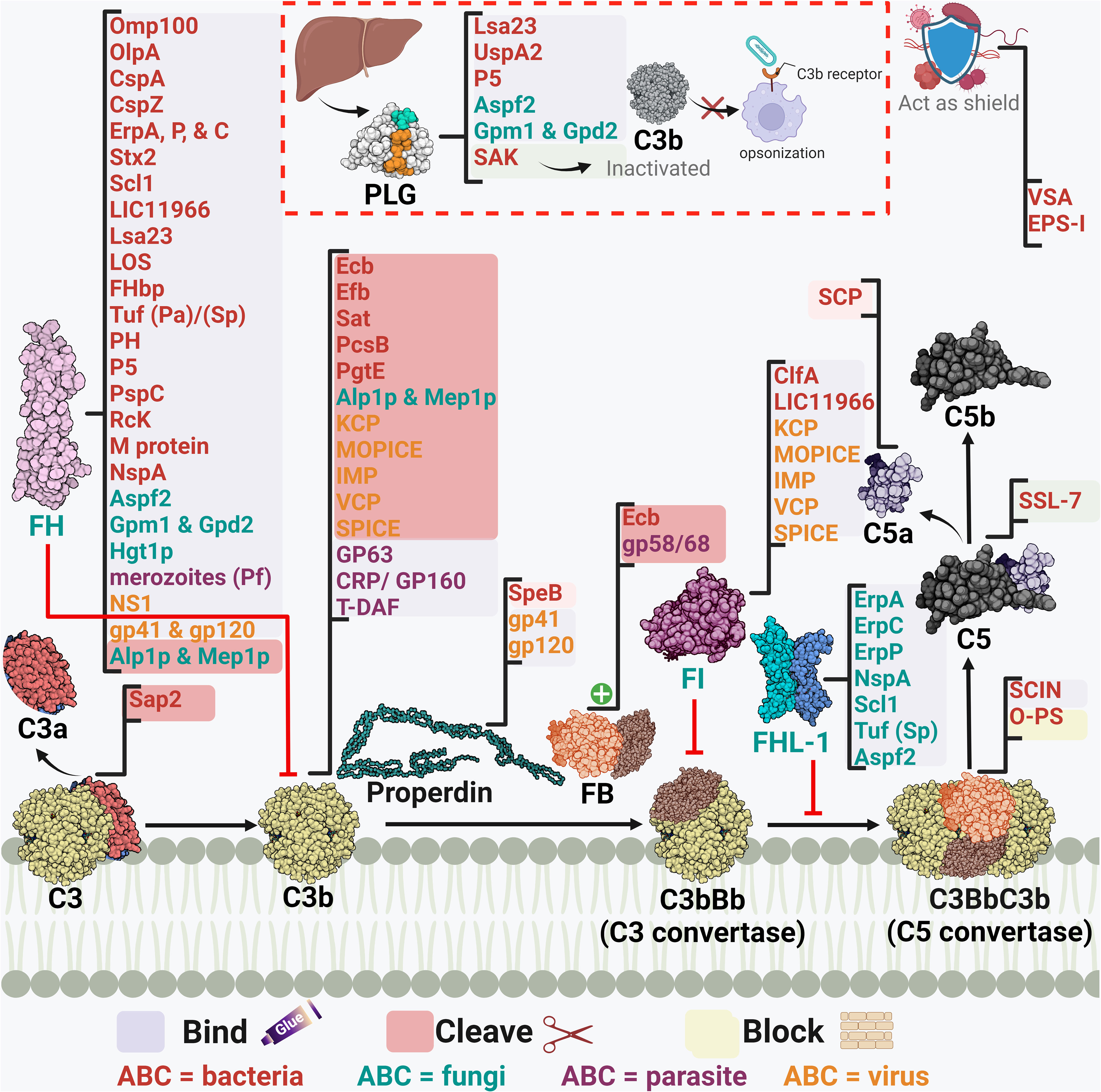
Figure 4 A schematic diagram showing microbial virulence factors involved in targeting this part of the complement cascade. The labels for the bacterial factors are shown in red, the fungal ones in turquoise, the parasite-related ones in purple, and finally the viral factors in orange. The mechanism by which those proteins affect the pathway is categorized into binding, shaded in purple; cleavage, which is shaded in dark pink; and blocking, which is highlighted in beige. The proteins’ codes and structures were retrieved from the Protein Data Bank, while the figure was generated using BioRender.
The acquisition of the hosts’ regulators is a very prominent strategy in this step as the labyrinth of multiple regulators and co-factors represents a possible getaway for microorganisms. Both C4BP and FH are complement regulators that bind to host cell surfaces, along with other FI co-factors, and are considered high-value targets for pathogens at this point (32).
Actinobacillus actinomycetemcomitans, which is associated with periodontal diseases, uses its outer membrane protein 100 (Omp100) to deposit FH at the cell surface and evade the complement (73). Neisseria spp. express lipooligosaccharides (LOS), similar to the host glycans, that enables them to recruit host regulators. Also, the sialylation of these LOS increases FH binding (90). Furthermore, the surface-exposed proteins porin A (PorA) and factor H-binding protein (FHbp) bind both C4BP (91) and FH (92), respectively enabling them to control the three pathways. Moreover, the binding of MCP (CD46) has been also demonstrated by its type IV pili (94). Similarly, P. aeruginosa binds FH by its translation elongation factor Tu (Tuf) (95). The encapsulated H. influenzae uses its protein H (PH) to bind FH while the Nontypeable H. influenzae (NTHi) uses its protein 5 (P5) to bind to the same regulator (82, 171). P5 was also shown to bind to PLG and cleave C3b following activation (83). Salmonella enterica serovars Typhimurium and Enteritidis harbour a virulence plasmid encoding the RcK outer membrane protein that recruits FH to inactivate the C3b (96). The same protein was also shown to interfere with the CP and LP via binding to C4BP (97).
Borreliaburgdorferi has several proteins on its surface that bind FH and FHL-1 such as complement regulator-acquiring surface protein (CRASP-1 or CspA) (76), complement regulator-acquiring surface protein 2 (CRASP-2 or CspZ) (75), and OspEF-related protein (Erp) types A, P, and C (74). The CihC protein of B. recurrentis that interferes with the complement cascade initiation plays another role in this stage by binding to the C4BP regulator (41).
As for S. pneumoniae, both its surface protein C (PspC) and elongation factor Tu (Tuf) can bind to FH (110–112). Moreover, the streptococcal M protein is capable of binding to 4 regulators: FH, FHL-1, C4BP, and CD46 (29, 109). Similarly, Leptospira interrogans uses its leptospiral surface adhesions 23 (Lsa23) to bind to multiple regulators including C4BP for the CP, FH for the AP, and PLG to cleave the C3b (84). In addition, the Shiga toxin 2 (Stx2) of the enterohemorrhagic E. coli (EHEC) binds to FH, FHR-1, and FHL-1 (80). The neisserial surface protein A (NspA) was shown to bind to both FH & FHL-1; however, the meningococcal NspA displayed an enhanced binding ability to these regulators over the N. gonorrhoeae homolog (93).
The binding of C4BP is seen in B. pertussis via its filamentous hemagglutinin (FHA) surface protein and another unidentified moiety; however, fha mutants are still as sensitive to killing by the complement system as their wild-type counterparts, suggesting that FHA could be playing a negligent role in the complement resistance of this pathogen (172). Moreover, C4BP is bound by multiple other pathogens, including Yersinia pestis by the attachment invasive locus (Ail) (114), Y. enterocolitica by the yersinia adhesin A (YadA) (113), E. coli by the outer membrane protein A (OmpA) (79), and Moraxella catarrhalis by the ubiquitous surface proteins A2 (UspA2) and to a lesser extent A1 (UspA1) of some M. catarrhalis strains (86). UspA2 derived from other serum-resistant M. catarrhalis strains was found to be unable to bind the C4BP regulator, suggesting that this is not the only role of UspA2 in evading killing by the complement system (153). Interestingly, the same surface protein was shown to bind PLG resulting in the degradation of both C3b and C5 (87). Also, it has been shown that the Opa-like protein A (OlpA) in some M. catarrhalis strains can bind FH and hence interferes with the AP (88). Moreover, S. aureus binds the AP regulator FI using its clumping factor A (ClfA). This binding promotes the breakdown of the surface-bound C3b into iC3b (99). Last but not least, Leptospira spp. express the LIC11966, which is an ErpY-like lipoprotein that can bind FH and FI (85).
Fungal evasion of the complement system at this stage can also happen through the recruitment of host regulators. This is seen in C. albicans which binds C4BP with its pH-regulated antigen 1 (Pra1) (121). On the other hand, both the surface protein phosphoglycerate mutase 1 (Gpm1) and the glycerol-3-phosphate dehydrogenase 2 (Gpd2) bind FH, FHL-1, and PLG (118, 119), while the high affinity glucose transporter 1 (Hgt1p) binds FH and C4BP (120). Additionally, C. albicans interferes with complement activation and complement mediated opsonization through its secreted aspartic proteases (Sap1-3), which degrade both C3b and C4b (123). Aspergillus fumigatus conidia avoids recognition by the complement system through the acquisition of some host regulators such as FH, FHL-FHR-1, and PLG with the help of its Aspf2 protein (115, 116). It is worth mentioning that other developmental stages of this fungi bind to these regulators with lower capacity, if any.
Moving to the viral side, in HIV, the acquisition of the host regulators occurs through the interaction between the host FH and properdin and the viral envelope proteins gp41 and gp120 (127). Also, the flaviviruses non-structural protein 1 (NS1) present in West Nile virus (WNV), dengue virus (DENV), and yellow fever viruses (YFV) binds FH which leads to blocking the AP (125, 126).
Another example, but this time from the perspective of parasites, is the larval stage of the cestode Echinococcus granulosus, which releases an FH binding molecule from the wall of the hydatid cyst that limits AP amplification (173, 174). Moreover, when P. falciparum merozoites are exposed to human serum they recruit FH and FHL-1 acting as a complement downregulation strategy (136). Entamoeba histolytica develops resistance to the complement by trogocytosis, a smart way of acquiring multiple membrane bound complement host regulators, that might be working collectively, via ingesting small bites of the host cells (134). Among those regulators are the MCP and DAF which are regulators of the intermediate stage of the complement cascade, in addition to CD59 which is a regulator of the late stages as will be discussed later.
Away from binding to or recruiting the host’s regulators, pathogens have adopted other approaches to interfere with the steps leading to C3 convertase formation or activation. The S. aureus pathogen possesses an arsenal of evasion proteins that target the intermediate stages of the complement cascade. Some strains use the extracellular adherence protein (Eap) to bind C4b and block the assembly of the C3 convertase, hence, targeting both the LP and CP (100). Other strains use the SAK protein which functions as a PLG activator to cleave bound C3b and block opsonization (51). They also use the Staphylococcus complement inhibitor (SCIN) which inhibits both C3 convertases by binding and stabilizing them and preventing the addition of more C3b to either of them (103). S. aureus is capable of blocking the activation and amplification loop by binding the staphylococcal extracellular fibrinogen binding protein (Efb) and its homologous extracellular complement binding protein (Ecb) to C3b and hence preventing the binding of Factor B to C3b and thus the AP C3 convertase is not formed (101, 102). The Streptococcus spp. are an equally strong foe, as the group A S. pyogenes (GAS) produce the streptococcal pyrogenic exotoxin B (SpeB) which degrades the AP regulator properdin (108). It was also reported that the overexpression of the surface immunodominant protein PcsB of S. mitis resulted in the reduction of the C3b/iC3b that are deposited on the cell surface (106). The auto-transporter of B. pertussis named Bordetella resistance to killing A (BrkA) is reported to reduce complement component deposition starting from C4, interfering with CP and LP, and moving all the way to MAC formation (77). The karilysin and mirolysin of T. forsythia exhibit efficient degradation capacity for C4 which blocks both CP and LP (59, 60). Salmonella enterica uses its outer membrane aspartate protease PgtE to cleave C3b, C4b, and C5 and it also activates the PLG, which in return increases its complement resistance. (98). Recently, the E. coli secreted autotransporter toxin (Sat) was shown to degrade multiple complement components involved in the three pathways including C2, C3, C3b, C4, C4b, and C5 (81).
In the case of fungi, some of them adopt the approach of inhibiting the cleavage of key complement proteins which in return inhibits opsonization and phagocytosis. For instance, C. albicans has multiple surface proteins, such as the C. albicans phosphoglycerate mutase1 (Gpm1p), that bind host PLG (118) and inhibit the cleavage of C3, C3b, and C5, and in return inhibits the generation of both C3b and C5 (112). C. albicans also secretes aspartic protease (Sap2) which cleaves C3 (122). On another front, other fungi adopt a different approach to degrading the key complement components, such as A. fumigatus which uses its secreted alkaline proteases (Alp1p) from the hyphal morphotype and the metalloprotease (Mep1p) from the conidial morphotype to deploy this approach. FH is cleaved by Mep1p without losing its cofactor activity. Then, FH supports FI-mediated cleavage of C3b into the inactivated form (iC3b). Thus, the fungal pathogen can be easily disseminated in the invaded host (117). A third approach, used by Cryptococcus neoformans, is by blocking the complement receptors on phagocytes and hence interfering with complement-dependent phagocytosis, which is done by the production of glucuronoxylomannan (GXM) (124).
Viruses, on the other hand, have versatile strategies for inhibiting the intermediate stages in the complement cascade. The Kaposi’s sarcoma-associated human herpes virus (KSHV) complement control protein (KCP) regulates the complement cascade by its DAF activity, which aids in accelerating the C3 convertases’ decay. At the same time, it provides cofactor activity for FI which leads to the degradation of both C4b and C3b (128). On the other hand, the NS1 protein of flaviviruses was shown to bind both C4 and C1s. This binding enhances the cleavage of C4 to C4b in solution and depletes the C4 supply, resulting in the blockage of both CP and LP (126). Poxviruses are known for their ability to evade the complement system via several proteins, the first of which to be discovered was the vaccinia virus complement control protein (VCP). Initially, studies showed that VCP binds C3b and C4b and possesses a decay accelerating activity (131). Then, its cofactor activity for FI was highlighted (132). Orthologs of the VCP are present in other poxviruses, such as the smallpox inhibitor of complement enzymes (SPICE) in the variola virus and the monkeypox inhibitor of complement enzymes (MOPICE). Those VCP orthologs are capable of inhibiting C3b and C4b with different potencies (133, 175). Yet the MOPICE functions only through its cofactor activity and lacks decay acceleration (129). Also, the cowpox virus VCP homolog, termed as the inflammation modulatory protein (IMP), possesses cofactor activity and binds C3b and C4b to inhibit the classical and alternative pathways (130).
As for parasites, they have their own tricks when it comes to inhibiting the C3 convertase. For example, T. cruzi employs surface molecules, such as complement C2 receptor inhibitor trispanning (CRIT), which binds C2 and prevents its cleavage by C1s or MASP2– blocking C3 convertase formation (137). Moreover, T. cruzi expresses the complement regulatory protein (CRP/GP160) that binds to C3b and C4b and dissociates the C3 convertases (138). In addition, the same parasite can specifically interfere with the AP C3 convertase using its glycoprotein “gp58/68” that inhibits the formation of cell-bound and fluid-phase alternative pathway C3 convertase through binding to FB (139). Also, the T-DAF binds C3b and C4b which blocks C3 convertase formation (176). On the other hand, Leishmania mainly uses the surface protease gp63 to inactivate C3b and convert it to iC3b (135).
Mycoplasma pulmonis does not interfere with the complement activation; however, it evades the complement and the phagocytosis process using a shielding strategy. The variable surface antigens (VSA) and the exopolysaccharide I (EPS-I) both contribute to shielding the pathogen against killing by the complement system and potentially other harmful molecules (89). Also, the elongated O-antigenic polysaccharide (O-PS) moieties of Burkholderia pseudomallei stretches away from the cell surface, a strategy that prevents complement-mediated killing by blocking C3 convertase formation close to the cell surface, and subsequently the membrane insertion of the TCC (78).
Efb-C and its homolog Ecb are both indispensable for the S. aureus fight against the complement system through their previously mentioned ability to prevent AP C3 convertase formation. Moreover, they possess the ability to bind C3d-containing molecules (C3, C3b, iC3b, and C3d) and they can impede all C3b-containing convertases which comprise the C5 convertases of all three complement pathways (102). Factor H-related protein-1 (FHR-1) is one of five other factor H-related (FHR) proteins that were initially identified to have complement inhibitory action by competing with FH and potentially FHL proteins. Organisms were found to recruit those proteins for their own benefit, such as special serotypes of S. pyogenes (M6 and M55) which recruit FHR-1 as well as FH by the streptococcal collagen-like protein 1 (Scl1) (107).
Stabilization of the AP C5 convertase by properdin is imperative for the cascade progression, as it is considered the main supplier of C5b to the terminal pathway (11). Stemming from this fact comes the smart strategy used by GAS to disrupt C5 convertase assembly by the cleavage of properdin through the SpeB (108). Contrary to properdin, DAF accelerates the convertases’ decay, a mechanism that is used by T. cruzi through its DAF-mimicking glycoprotein T-DAF (136).
Among the staphylococcal superantigen-like (SSL) proteins is SSL-7 which binds C5 and accordingly hinders its binding to the C5 convertase and prevents the production of C5a. Through this mechanism, S. aureus could evade complement killing by phagocytosis. Although SSL-7 was shown to form a complex with C5b and prevent MAC formation, this is unrelated to S. aureus complement evasion ability as it is already resistant to killing by MAC (104). Streptococci, including groups A, B, C, and G of human origin, produces a C5a peptidase (SCP) which specifically degrades and inactivates C5a. This complement component is considered a major human phagocyte chemotaxin. Accordingly, the SCP action delays the influx of phagocytes and aids in the pathogenesis of these pathogens (105).
Orthopoxviruses, including variola homologs, secrete soluble viral complement regulatory proteins, such as the smallpox inhibitor of complement enzymes (SPICE), which inhibit the formation of the C3/C5 convertases necessary for viral clearance (133).
1.3.3 Interference with the terminal stages: the final round
As the complement system goes through its terminal step, the microbial counter defences work to counteract the deleterious effects of the terminal complement complex (TCC) (Figure 5; Table 3). A successful microbial evasion strategy is the acquisition of the host complement regulators to block the harmful effects of this immune system component. TCC formation is regulated by multiple host regulators that microbes have already exploited to evade the lethal effect of MAC formation.
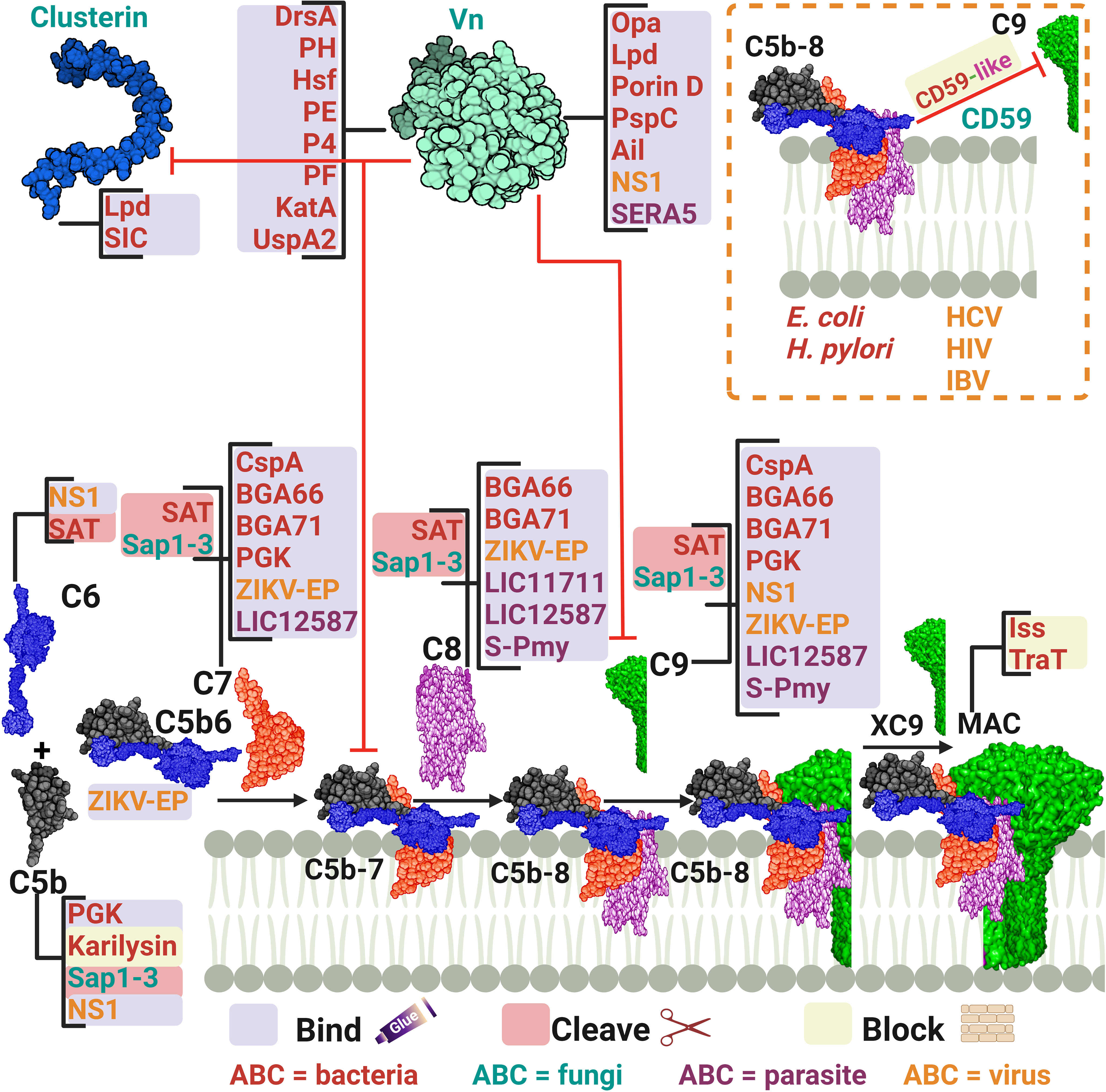
Figure 5 A schematic diagram showing microbial virulence factors involved in targeting the final stage of the complement cascade. The labels for the bacterial factors are shown in red, the fungal ones in turquoise, the parasite-related ones in purple, and finally the viral factors in orange. The mechanism by which those proteins affect the pathway is categorized into binding, shaded in purple; cleavage, which is shaded in dark pink; and blocking, which is highlighted in beige. The proteins’ codes and structures were retrieved from the Protein Data Bank, while the figure was generated using BioRender.
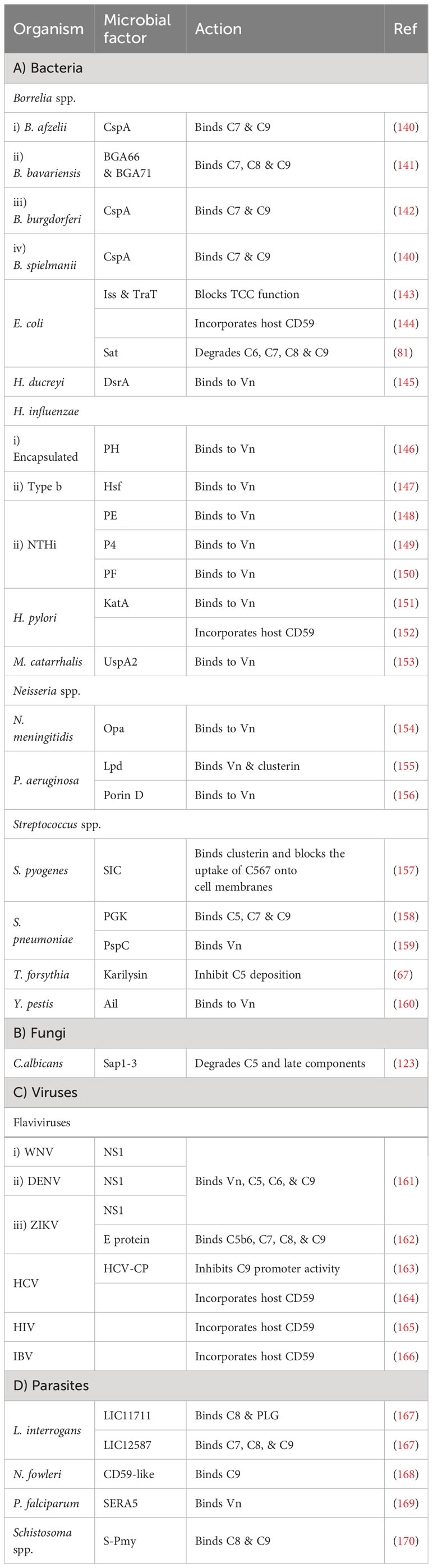
Table 3 Microbial strategies for interfering with the complement cascade late stages.
Many pathogens can bind vitronectin (Vn). They either use bound-Vn to prevent complement lysis or to “hitchhike” their way into cells. UspA2, the previously discussed protein of M. catarrhalis, was found to render complement resistance to some strains by binding Vn and hindering C9 polymerization and thus preventing MAC assembly (153). This was confirmed by testing uspA2 mutant strains which showed serum-sensitivity and by testing the wild-type strain in a Vn-depleted serum which led to a bactericidal action. Haemophilus ducreyi secretes a UspA2 homologue protein termed H. ducreyi serum resistance protein A (DsrA) which was confirmed to have a Vn binding ability (145). Likewise, H. influenzae expresses its pervasive adhesion protein E (PE) which binds Vn and hence inhibits the MAC formation, and increases the bacterial resistance among both non-typable and encapsulated H. influenzae (148). In the case of H. influenzae serotype f, protein H (PH) was shown to bind Vn to evade killing by human serum and also to better adhere to alveolar epithelial cells (146). H. influenzae serotype b, the most virulent of the encapsulated strains, utilizes its Haemophilus surface fibrils (Hsf) auto-transporter to bind both soluble and immobilized forms of Vn and enhance its serum survival (147). Moreover, two additional proteins of the non-typeable H. influenzae (NTHi) were shown to utilize the same tactic of recruiting Vn: protein 4 (P4) and protein F (PF) (149, 150). Another outer membrane adhesion protein is the opacity-associated outer membrane protein (Opa), already proven to confer resistance upon N. meningitidis, was found to bind active human Vn and hence, promoting cellular invasion (154). In P. aeruginosa, two proteins were reported to bind Vn, dihydrolipoamide dehydrogenase (Lpd) and Porin D. Lpd, a moonlighting protein offering multifunctional physiological activities independent of each other, was able to bind Vn to varying degrees among the tested strains (155). While porin D, an outer membrane protein, was shown to bind Vn via a proteomic approach (156). Both proteins effectively bound Vn and subverted TCC formation in bacterial clinical isolates. Moreover, Helicobacter pylori uses the moon-lighting approach to the phenomenon of additional, unrelated functions in often highly conserved proteins display the hydrogen peroxide-neutralizing enzyme catalase (KatA) on its surface and binds Vn to subvert the complement system (151). Also, the Y. pestis Ail protein was shown to bind functional Vn to evade TCC attack (160).
Vn acquisition is not an exclusive approach used by bacteria only as it is also found in viruses. The NS1 of the Dengue virus together with that of WNV and Zika virus (ZIKV), binds Vn directly and forms an NS1-Vn complex that inhibits C9 polymerization. This complex was identified in infected patients’ plasma, although data did not correlate its presence to disease progression (161). The serine repeat antigen 5 (SERA5) of the P. falciparum was also shown to bind Vn through its P47 N-terminal domain (169).
Gram-positive bacteria do not necessarily need to deploy evasion tactics against MAC formation owing to their possession of a hypertrophic peptidoglycan layer that shields the bacterial plasma membrane against this lytic attack (24, 26). Nevertheless, they do exploit a number of evasion strategies against MAC, something that could be explained by the recent structural analysis of the MAC which suggests activities beyond membrane penetration (24). Additionally, findings state that the whole C5b-9 is deposited on Gram-positive bacteria, yet the functional role is still not clear (177). In line with this, S. pneumoniae employs its multifunctional choline-binding protein PspC to hinder the terminal pathway by binding Vn. This was backed up by the lower levels of vitronectin binding detected in pspC mutants deficient in this protein (159).
Besides Vn, clusterin was found to be another pivotal key regulator of the TCC. P. aeruginosa Lpd was shown to bind this protein, besides binding to Vn, resulting in a reduction in C5b-9 deposition (155). For Gram-positive bacteria, the streptococcal inhibitor of complement (SIC) is an abundant protein secreted by S. pyogenes that shows comparable efficiency in binding to clusterin, although the biological relevance of this binding is doubtful (157). At the same time, it was shown that SIC can behave as the fluid phase regulator clusterin in blocking the uptake of the C5b-7 complex onto cell membranes, yet how this could benefit S. pyogenes remains debatable (157).
CD59, also known as protectin, is a glycophosphoinsitol (GPI) membrane bound human regulator of the complement that protects host cells from MAC-mediated lysis. Pathogens have acquired a tactic to incorporate the released GPI from the host cells into their membrane to protect themselves from TCC formation. For instance, it was shown that CD59 was functionally active after being inserted, in a Ca2+ dependent mechanism, in the membrane of two non-encapsulated deep rough E. coli strains (144). Moreover, H. pylori CagA-positive strains successfully incorporated the host CD59 and this binding was inversely proportional to the amount of deposited C5b-9 on the cell surface (152). Among viruses, HCV has demonstrated a similar capability of incorporating CD59 in its envelop (164). Also, the infectious bronchitis virus (IBV) adopted a similar tactic to evade the antibody-dependent complement-mediated lysis (166). In addition, HIV acquires protectin from the host cell surface to evade killing and ensure infectivity (165).
Other pathogens that do not incorporate the regulator CD59 have developed the smart tactic of expressing CD59-like proteins. For instance, the amebae Naegleria fowleri, which is responsible for primary amoebic meningoencephalitis, possess an anti-CD59 monoclonal antibody-reactive surface protein that was immunoprecipitated with C9 from human serum. At the same time, the complement sensitive non-pathogenic N. gruberi amebae lacked these reactions (168). Schistosomes have been identified to possess CD59-like protein homologs, yet their role in complement evasion remains unclear (178).
Another tactic used by microbes to interfere with the late stages of complement activation is to bind the components of the TCC, making them unavailable for membrane insertion and cell lysis. For example, the CspA of B. burgdorferi, B. afzelii, and B. spielmanii, can bind complement proteins C7 and C9, simultaneously, thus inhibiting TCC-mediated cellular destruction (140, 142). B. bavariensis has two surface proteins, termed BGA66 and BGA71, that contribute to its serum resistance by directly binding to C7, C8, and C9 and hence, preventing MAC formation (141). BGA71 can only inhibit TCC at the C7 level, while BGA66 interferes with TCC formation at different steps (141). Phosphoglycerate kinase (PGK), a pneumococcal glycolytic enzyme previously identified as a PG-binding protein of S. pneumoniae, was imputed to have an additional role as a MAC inhibitor and was found to simultaneously bind complement proteins C5, C7, and C9 (158). The Sat of E. coli, in addition to the earlier components mentioned above, are capable of degrading C6, C7, C8, and C9 (81). The karilysin of T. forsythia can inhibit C5 deposition, allowing enhanced serum survival and limiting MAC assembly (67). Moreover, the C. albicans (Sap1-3) proteases described above also degrade C5 and complement late components (123).
In viruses, the NS1 of the Dengue virus binds to complement proteins C5, C6, and C9 (161). While the Zika virus E protein binds C5b6, C7, C8, and C9 (162). Instead of depleting the TCC components, HCV suppresses the TCC by modulating complement protein synthesis through its viral core protein, which inhibits C9 promoter activity (163). L. interrogans has two surface exposed proteins, LIC11711 and LIC12587, which bind different components of the TCC, interfering with the MAC formation. LIC11711 was found to bind C8 in addition to PLG, while LIC12587 was able to bind C7, C8, and C9 (167). Finally, the paramyosin of S. mansoni, a multi-helical charged protein, can bind C1, C8, and C9, utilizing its highly charged zones (170).
E. coli strains harboring the R100 plasmid and the ColV/BM plasmid can express the outer membrane protein TraT and the increased serum survival (Iss) protein, respectively (143). Both proteins conferred a significant increase in E. coli survival in the presence of serum. However, the consumption of C6, C7, C8, and C9 was not altered when compared to non-expressing cells, indicating that it is the function of the TCC that is blocked, not its formation (143).
2 Discussion
The complement system was discovered more than 130 years ago; however, it was not until 70 years later that we realized microbes are able to evade this system with diverse strategies that have evolved over time. According to the survey conducted in this study, bacteria account for the majority of the microbial components researched, while fungal factors are the least commonly studied ones. However, in comparison with bacterial factors, viral and parasitic factors are still less studied.
Looking closer at the bacterial factors, multiple bacterial species can interfere with the complement system at the three stages of the complement cascade: early, intermediate, and late. For example, Borrelia spp., E. coli, Neisseria spp., P. aeruginosa, Streptococcus spp., and T. forsythia all have multiple virulence factors that can target the complement cascade, from blocking activation to interfering with terminal stages. S. aureus interferes with only the first two stages of the complement system; however, it has more than 10 virulence factors at its disposal, preventing the complex complement system from removing it while inside the mammalian host.
Examining the pathogens and their complement evasion tools, the spectrum of virulence factors is diverse. For instance, T. forsythia uses karilysin to interfere with the three stages of complement activation and Schistosoma spp use S-Pmy to block both activation and late stages of the complement system. On the other hand, S. aureus, although it targets only the first two stages, uses different virulence factors for each stage. Similarly, C. albicans uses seven virulence factors to target the intermediate and late stages.
When we compared the three complement stages in terms of the number of microbial factors involved, the intermediate stage was the most targeted with more than 40 reported virulence factors. This could possibly be attributed to the multiple complement components involved in this stage, especially the convertases, as well as the regulators that span across the three complement pathways. Interestingly, factor H is at the top of the list of the most targeted complement components with at least 28 virulence factors derived from the four main types of pathogens that interact with it to interfere with its role in AP activation.
It is noteworthy that most of the studies reported here have demonstrated, via various approaches, that binding of the pathogen to a complement component or regulator results in a functional role in complement evasion. For instance, this was demonstrated by generating isogenic mutants in the gene(s) encoding the proposed virulence factor and showing that this mutant is more sensitive to complement-mediated killing as compared to their wild-type (55, 56, 121, 159). More provided evidence was that the depletion of the serum from the complement regulator results in increasing susceptibility of the resistant wild-type strain to complement-mediated killing (153). Moreover, the silencing of the virulence factor or immunizing the host against it results also in an increased susceptibility to the complement (40). In addition, the proposed mechanism was supported by the ectopic expression of the virulence factor in a sensitive strain which rendered it resistant to killing by the complement system (41).
On the other hand, in some situations, proposed bacterial virulence factors exhibited good binding to the complement regulators or components, yet this was not reflected in a functional phenotype. For instance, the binding of the C4BP to B. pertussis FHA (172). Also, although the Mac/IdeS of the Group A Streptococus was shown to specifically cleave IgG interfering with the CP activation, yet knocking out the gene encoding this protease had no notable impact on multiple GAS immune evasion phenotypes (179).
Understanding the host-pathogen interactions during complement evasion is a corner stone in the treatment of several human diseases. Mimicking or inhibiting the microbial factors produced by bacteria, viruses, fungi, and parasites provides a potential platform as novel classes of antimicrobials and complement-targeting therapeutics.
3 Conclusion
The complement system plays a pivotal role in the innate immune system’s battle against invading pathogens; however, pathogens fight back in constantly evolving ways to evade this system. In this article, we have assessed more than 180 research papers to present a comprehensive overview of different mechanisms used by microorganisms to evade the complement system. We classified the pathogens’ interference with the system into three main phases: influencing the complement activation, interfering with the intermediate stages of the complement cascade, and finally, interfering with the terminal stages of membrane attack complex formation. For pathogens to achieve their goal of influencing the complement system they can mask their surface molecules, secrete proteins that inhibit or degrade complement components, recruit complement regulatory proteins, or modify their own structures to become more resistant. At each phase of the complement cascade, pathogens can deploy multiple evasion mechanisms, and use different strategies to counteract the complement system on multiple levels. Understanding how the complement system is controlled allows us to better comprehend the possible mechanisms that pathogens use to evade it. Moreover, keeping track of how pathogens are evolving in terms of their evasion strategies is crucial for developing new strategies to combat them.
Author contributions
MH: Formal analysis, Investigation, Methodology, Writing – original draft, Data curation, Visualization. HN: Data curation, Formal analysis, Writing – review & editing. DM: Data curation, Writing – original draft. NA: Data curation, Writing – original draft. AA: Writing – original draft, Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol (2010) 125:S3–23. doi: 10.1016/j.jaci.2009.12.980
2. Héja D, Kocsis A, Dobó J, Szilágyi K, Szász R, Závodszky P, et al. Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proc Natl Acad Sci (2012) 109:10498–503. doi: 10.1073/pnas.1202588109
3. Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol (2013) 33:479–92. doi: 10.1016/j.semnephrol.2013.08.001
4. Carroll MC. The role of complement and complement receptors in induction and regulation of immunity. Annu Rev Immunol (1998) 16:545–68. doi: 10.1146/annurev.immunol.16.1.545
5. Kishore U, Reid KB. C1q: structure, function, and receptors. Immunopharmacology (2000) 49:159–70. doi: 10.1016/S0162-3109(00)80301-X
6. Sim RB, Tsiftsoglou SA. Proteases of the complement system. Biochem Soc Trans (2004) 32:21–7. doi: 10.1042/bst0320021
7. Senent Y, Ajona D, González-Martín A, Pio R, Tavira B. The complement system in ovarian cancer: an underexplored old path. Cancers (2021) 13:3806. doi: 10.3390/cancers13153806
8. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11:785–97. doi: 10.1038/ni.1923
9. Kjaer TR, Thiel S, Andersen GR. Toward a structure-based comprehension of the lectin pathway of complement. Mol Immunol (2013) 56:222–31. doi: 10.1016/j.molimm.2013.05.220
10. Hansen S, Selman L, Palaniyar N, Ziegler K, Brandt J, Kliem A, et al. Collectin 11 (CL-11, CL-K1) is a MASP-1/3–associated plasma collectin with microbial-binding activity. J Immunol (2010) 185:6096–104. doi: 10.4049/jimmunol.1002185
11. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I – molecular mechanisms of activation and regulation. Front Immunol (2015) 6. doi: 10.3389/fimmu.2015.00262
12. Romano R, Giardino G, Cirillo E, Prencipe R, Pignata C. Complement system network in cell physiology and in human diseases. Int Rev Immunol (2021) 40:159–70. doi: 10.1080/08830185.2020.1833877
13. Oroszlán G, Dani R, Szilágyi A, Závodszky P, Thiel S, Gál P, et al. Extensive basal level activation of complement mannose-binding lectin-associated serine protease-3: kinetic modeling of lectin pathway activation provides possible mechanism. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.01821
14. Johnson RJ. The complement system. In: Biomaterials science. Amsterdam, Netherlands: Elsevier (2020). p. 777–90.
15. Kemper C, Atkinson JP, Hourcade DE. Properdin: emerging roles of a pattern-recognition molecule. Annu Rev Immunol (2010) 28:131–55. doi: 10.1146/annurev-immunol-030409-101250
16. Toapanta FR, Ross TM. Complement-mediated activation of the adaptive immune responses: role of C3d in linking the innate and adaptive immunity. Immunologic Res (2006) 36:197–210. doi: 10.1385/IR:36:1:197
17. Sarma JV, Ward PA. The complement system. Cell Tissue Res (2011) 343:227–35. doi: 10.1007/s00441-010-1034-0
18. Nilsson B, Nilsson Ekdahl K. The tick-over theory revisited: Is C3 a contact-activated protein? Immunobiology (2012) 217:1106–10. doi: 10.1016/j.imbio.2012.07.008
19. Pettigrew HD, Teuber SS, Gershwin ME. Clinical significance of complement deficiencies. Ann New York Acad Sci (2009) 1173:108–23. doi: 10.1111/j.1749-6632.2009.04633.x
20. Satyam A, Graef ER, Lapchak PH, Tsokos MG, Dalle Lucca JJ, Tsokos GC. Complement and coagulation cascades in trauma. Acute Med Surg (2019) 6:329–35. doi: 10.1002/ams2.426
21. Michels M, Maas RJF, Van Der Velden TJAM, Van De Kar NCAJ, Van Den Heuvel LPWJ, Volokhina EB, et al. The role of properdin in C5 convertase activity and C5b-9 formation in the complement alternative pathway. J Immunol (2021) 207:2465–72. doi: 10.4049/jimmunol.2100238
22. Rawal N, Pangburn MK. Formation of high-affinity C5 convertases of the alternative pathway of complement. J Immunol (2001) 166:2635–42. doi: 10.4049/jimmunol.166.4.2635
23. Kindt TJ, Goldsby RA, Osborne BA, Kuby J. Kuby immunology. Freeman WH, editor. New York: W.H. Freeman, New York (2003).
24. Serna M, Giles JL, Morgan BP, Bubeck D. Structural basis of complement membrane attack complex formation. Nat Commun (2016) 7:10587. doi: 10.1038/ncomms10587
25. Morgan BP. Complement membrane attack on nucleated cells: resistance, recovery and non-lethal effects. Biochem J (1989) 264:1–14. doi: 10.1042/bj2640001
26. Laarman A, Milder F, Van Strijp J, Rooijakkers S. Complement inhibition by gram-positive pathogens: molecular mechanisms and therapeutic implications. J Mol Med (Berlin Germany) (2010) 88:115–20. doi: 10.1007/s00109-009-0572-y
27. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol (2009) 9:729–40. doi: 10.1038/nri2620
28. Hovingh ES, Van Den Broek B, Jongerius I. Hijacking complement regulatory proteins for bacterial immune evasion. Front Microbiol (2016) 7. doi: 10.3389/fmicb.2016.02004
29. Horstmann RD, Sievertsen HJ, Knobloch J, Fischetti VA. Antiphagocytic activity of streptococcal M protein: selective binding of complement control protein factor H. Proc Natl Acad Sci (1988) 85:1657–61. doi: 10.1073/pnas.85.5.1657
30. Dunkelberger JR, Song W-C. Complement and its role in innate and adaptive immune responses. Cell Res (2009) 20:34–50. doi: 10.1038/cr.2009.139
31. Ninomiya H, Sims P. The human complement regulatory protein CD59 binds to the alpha-chain of C8 and to the “b” domain of C9. J Biol Chem (1992) 267:13675–80. doi: 10.1016/S0021-9258(18)42266-1
32. Bennett KM, Rooijakkers SHM, Gorham RD. Let’s tie the knot: Marriage of complement and adaptive immunity in pathogen evasion, for better or worse. Front Microbiol (2017) 8. doi: 10.3389/fmicb.2017.00089
33. Zipfel PF, Hallstrom T, Riesbeck K. Human complement control and complement evasion by pathogenic microbes-tipping the balance. Mol Immunol (2013) 56:152–60. doi: 10.1016/j.molimm.2013.05.222
34. Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol (2008) 6:132. doi: 10.1038/nrmicro1824
35. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: Role in immunity. Front Immunol (2015) 6. doi: 10.3389/fimmu.2015.00257
36. Garcia BL, Zwarthoff SA, Rooijakkers SHM, Geisbrecht BV. Novel evasion mechanisms of the classical complement pathway. J Immunol (2016) 197:2051–60. doi: 10.4049/jimmunol.1600863
37. Rosbjerg A, Genster N, Pilely K, Garred P. Evasion mechanisms used by pathogens to escape the lectin complement pathway. Front Microbiol (2017) 8:868. doi: 10.3389/fmicb.2017.00868
38. Zipfel PF, Wurzner R, Skerka C. Complement evasion of pathogens: common strategies are shared by diverse organisms. Mol Immunol (2007) 44:3850–7. doi: 10.1016/j.molimm.2007.06.149
39. Sharma S, Bhatnagar R, Gaur D. Bacillus anthracis poly-γ-d-glutamate capsule inhibits opsonic phagocytosis by impeding complement activation. Front Immunol (2020) 11:462. doi: 10.3389/fimmu.2020.00462
40. Schuijt TJ, Coumou J, Narasimhan S, Dai J, Deponte K, Wouters D, et al. A tick mannose-binding lectin inhibitor interferes with the vertebrate complement cascade to enhance transmission of the lyme disease agent. Cell Host Microbe (2011) 10:136–46. doi: 10.1016/j.chom.2011.06.010
41. Grosskinsky S, Schott M, Brenner C, Cutler SJ, Simon MM, Wallich R. Human complement regulators C4b-binding protein and C1 esterase inhibitor interact with a novel outer surface protein of Borrelia recurrentis. PloS Negl Trop Dis (2010) 4:e698. doi: 10.1371/journal.pntd.0000698
42. Marr N, Shah NR, Lee R, Kim EJ, Fernandez RC. Bordetella pertussis autotransporter Vag8 binds human C1 esterase inhibitor and confers serum resistance. PloS One (2011) 6:e20585. doi: 10.1371/journal.pone.0020585
43. Pluschke G, Mayden J, Achtman M, Levine R. Role of the capsule and the O antigen in resistance of O18: K1 Escherichia coli to complement-mediated killing. Infection Immun (1983) 42:907–13. doi: 10.1128/iai.42.3.907-913.1983
44. Lathem WW, Bergsbaken T, Welch RA. Potentiation of C1 esterase inhibitor by StcE, a metalloprotease secreted by Escherichia coli O157: H7. J Exp Med (2004) 199:1077–87. doi: 10.1084/jem.20030255
45. Sahly H, Keisari Y, Ofek I. Manno (rhamno) biose-containing capsular polysaccharides of Klebsiella pneumoniae enhance opsono-stimulation of human polymorphonuclear leukocytes. J Innate Immun (2009) 1:136–44. doi: 10.1159/000154812
46. Agarwal S, Vasudhev S, Deoliveira RB, Ram S. Inhibition of the classical pathway of complement by meningococcal capsular polysaccharides. J Immunol (2014) 193:1855–63. doi: 10.4049/jimmunol.1303177
47. Hong Y, Ghebrehiwet B. Effect of Pseudomonas aeruginosa elastase and alkaline protease on serum complement and isolated components C1q and C3. Clin Immunol Immunopathology (1992) 62:133–8. doi: 10.1016/0090-1229(92)90065-V
48. Curto P, Barro A, Almeida C, Vieira-Pires RS, Simões I. The retropepsin-type protease APRc as a novel ig-binding protein and moonlighting immune evasion factor of rickettsia. Mbio (2021) 12:e03059–03021. doi: 10.1128/mBio.03059-21
49. Atkins KL, Burman JD, Chamberlain ES, Cooper JE, Poutrel B, Bagby S, et al. S. aureus IgG-binding proteins SpA and Sbi: Host specificity and mechanisms of immune complex formation. Mol Immunol (2008) 45:1600–11. doi: 10.1016/j.molimm.2007.10.021
50. Itoh S, Hamada E, Kamoshida G, Yokoyama R, Takii T, Onozaki K, et al. Staphylococcal superantigen-like protein 10 (SSL10) binds to human immunoglobulin G (IgG) and inhibits complement activation via the classical pathway. Mol Immunol (2010) 47:932–8. doi: 10.1016/j.molimm.2009.09.027
51. Rooijakkers S, Van Wamel W, Ruyken M, Van Kessel K, Van Strijp J. Anti-opsonic properties of staphylokinase. Microbes Infection (2005) 7:476–84. doi: 10.1016/j.micinf.2004.12.014
52. Collin M, Olsén A. Effect of SpeB and EndoS from Streptococcus pyogenes on human immunoglobulins. Infection Immun (2001) 69:7187–9. doi: 10.1128/IAI.69.11.7187-7189.2001
53. Von Pawel-Rammingen U, Johansson BP, Björck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J (2002) 21:1607–15. doi: 10.1093/emboj/21.7.1607
54. Hyams C, Camberlein E, Cohen JM, Bax K, Brown JS. The streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infection Immun (2010) 78:704–15. doi: 10.1128/IAI.00881-09
55. Nitsche-Schmitz DP, Johansson HM, Sastalla I, Reissmann S, Frick I-M, Chhatwal GS. Group G streptococcal IgG binding molecules FOG and protein G have different impacts on opsonization by C1q. J Biol Chem (2007) 282:17530–6. doi: 10.1074/jbc.M702612200
56. Deng S, Xu T, Fang Q, Yu L, Zhu J, Chen L, et al. The surface-exposed protein SntA contributes to complement evasion in zoonotic Streptococcus suis. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01063
57. Rungelrath V, Weiße C, Schütze N, Müller U, Meurer M, Rohde M, et al. IgM cleavage by Streptococcus suis reduces IgM bound to the bacterial surface and is a novel complement evasion mechanism. Virulence (2018) 9:1314–37. doi: 10.1080/21505594.2018.1496778
58. Caboni M, Pedron T, Rossi O, Goulding D, Pickard D, Citiulo F, et al. An O antigen capsule modulates bacterial pathogenesis in Shigella sonnei. PloS Pathog (2015) 11:e1004749. doi: 10.1371/journal.ppat.1004749
59. Jusko M, Potempa J, Karim AY, Ksiazek M, Riesbeck K, Garred P, et al. A metalloproteinase karilysin present in the majority of Tannerella forsythia isolates inhibits all pathways of the complement system. J Immunol (2012) 188:2338–49. doi: 10.4049/jimmunol.1101240
60. Jusko M, Potempa J, Mizgalska D, Bielecka E, Ksiazek M, Riesbeck K, et al. A metalloproteinase mirolysin of Tannerella forsythia inhibits all pathways of the complement system. J Immunol (2015) 195:2231–40. doi: 10.4049/jimmunol.1402892
61. Attridge SR, Holmgren J. Vibrio cholerae O139 capsular polysaccharide confers complement resistance in the absence or presence of antibody yet presents a productive target for cell lysis: implications for detection of bactericidal antibodies. Microbial Pathogenesis (2009) 47:314–20. doi: 10.1016/j.micpath.2009.09.013
62. Da Silva MB, Marques AF, Nosanchuk JD, Casadevall A, Travassos LR, Taborda CP. Melanin in the dimorphic fungal pathogen Paracoccidioides brasiliensis: effects on phagocytosis, intracellular resistance and drug susceptibility. Microbes Infection (2006) 8:197–205. doi: 10.1016/j.micinf.2005.06.018
63. Lubinski JM, Lazear HM, Awasthi S, Wang F, Friedman HM. The herpes simplex virus 1 IgG fc receptor blocks antibody-mediated complement activation and antibody-dependent cellular cytotoxicity in vivo. J Virol (2011) 85:3239–49. doi: 10.1128/JVI.02509-10
64. Guha D, Ayyavoo V. Innate immune evasion strategies by human immunodeficiency virus type 1. ISRN AIDS (2013) 2013:954806. doi: 10.1155/2013/954806
65. Verma S, Mandal A, Ansari MY, Kumar A, Abhishek K, Ghosh AK, et al. Leishmania donovani inhibitor of serine peptidases 2 mediated inhibition of lectin pathway and upregulation of C5aR signaling promote parasite survival inside host. Front Immunol (2018) 9:63. doi: 10.3389/fimmu.2018.00063
66. Kennedy AT, Wijeyewickrema LC, Huglo A, Lin C, Pike R, Cowman AF, et al. Recruitment of human C1 esterase inhibitor controls complement activation on blood stage Plasmodium falciparum merozoites. J Immunol (2017) 198:4728–37. doi: 10.4049/jimmunol.1700067
67. Laclette J, Shoemaker C, Richter D, Arcos L, Pante N, Cohen C, et al. Paramyosin inhibits complement C1. J Immunol (1992) 148:124–8. doi: 10.4049/jimmunol.148.1.124
68. Loukas A, Jones MK, King LT, Brindley PJ, Mcmanus DP. Receptor for Fc on the surfaces of schistosomes. Infection Immun (2001) 69:3646–51. doi: 10.1128/IAI.69.6.3646-3651.2001
69. Sun R, Zhao X, Wang Z, Yang J, Zhao L, Zhan B, et al. Trichinella spiralis paramyosin binds human complement C1q and inhibits classical complement activation. PloS Negl Trop Dis (2015) 9:e0004310. doi: 10.1371/journal.pntd.0004310
70. Sosoniuk E, Vallejos G, Kenawy H, Gaboriaud C, Thielens N, Fujita T, et al. Trypanosoma cruzi calreticulin inhibits the complement lectin pathway activation by direct interaction with L-Ficolin. Mol Immunol (2014) 60:80–5. doi: 10.1016/j.molimm.2014.03.014
71. Hair PS, Gronemus JQ, Crawford KB, Salvi VP, Cunnion KM, Thielens NM, et al. Human astrovirus coat protein binds C1q and MBL and inhibits the classical and lectin pathways of complement activation. Mol Immunol (2010) 47:792–8. doi: 10.1016/j.molimm.2009.10.006
72. Ricklin D. Manipulating the mediator: modulation of the alternative complement pathway C3 convertase in health, disease and therapy. Immunobiology (2012) 217:1057–66. doi: 10.1016/j.imbio.2012.07.016
73. Asakawa R, Komatsuzawa H, Kawai T, Yamada S, Goncalves RB, Izumi S, et al. Outer membrane protein 100, a versatile virulence factor of Actinobacillus actinomycetemcomitans. Mol Microbiol (2003) 50:1125–39. doi: 10.1046/j.1365-2958.2003.03748.x
74. Alitalo A, Meri T, Lankinen H, Seppälä I, Lahdenne P, Hefty PS, et al. Complement inhibitor factor H binding to Lyme disease spirochetes is mediated by inducible expression of multiple plasmid-encoded outer surface protein E paralogs. J Immunol (2002) 169:3847–53. doi: 10.4049/jimmunol.169.7.3847
75. Hartmann K, Corvey C, Skerka C, Kirschfink M, Karas M, Brade V, et al. Functional characterization of BbCRASP-2, a distinct outer membrane protein of Borrelia burgdorferi that binds host complement regulators factor H and FHL-1. Mol Microbiol (2006) 61:1220–36. doi: 10.1111/j.1365-2958.2006.05318.x
76. Kenedy MR, Vuppala SR, Siegel C, Kraiczy P, Akins DR. CspA-mediated binding of human factor H inhibits complement deposition and confers serum resistance in Borrelia burgdorferi. Infection Immun (2009) 77:2773–82. doi: 10.1128/IAI.00318-09
77. Barnes MG, Weiss AA. BrkA protein of Bordetella pertussis inhibits the classical pathway of complement after C1 deposition. Infection Immun (2001) 69:3067–72. doi: 10.1128/IAI.69.5.3067-3072.2001
78. Deshazer D, Brett PJ, Woods DE. The type II O-antigenic polysaccharide moiety of Burkholderia pseudomallei lipopolysaccharide is required for serum resistance and virulence. Mol Microbiol (1998) 30:1081–100. doi: 10.1046/j.1365-2958.1998.01139.x
79. Prasadarao NV, Blom AM, Villoutreix BO, Linsangan LC. A novel interaction of outer membrane protein A with C4b binding protein mediates serum resistance of Escherichia coli K1. J Immunol (2002) 169:6352–60. doi: 10.4049/jimmunol.169.11.6352
80. Poolpol K, Orth-Höller D, Speth C, Zipfel PF, Skerka C, De Córdoba SR, et al. Interaction of Shiga toxin 2 with complement regulators of the factor H protein family. Mol Immunol (2014) 58:77–84. doi: 10.1016/j.molimm.2013.11.009
81. Freire CA, Silva RM, Ruiz RC, Pimenta DC, Bryant JA, Henderson IR, et al. Secreted autotransporter toxin (Sat) mediates innate immune system evasion. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.844878
82. Fleury C, Su Y-C, Hallström T, Sandblad L, Zipfel PF, Riesbeck K. Identification of a Haemophilus influenzae factor H–binding lipoprotein involved in serum resistance. J Immunol (2014) 192:5913–23. doi: 10.4049/jimmunol.1303449
83. Singh B, Al-Jubair T, Mörgelin M, Thunnissen MM, Riesbeck K. The unique structure of Haemophilus influenzae protein E reveals multiple binding sites for host factors. Infection Immun (2013) 81:801–14. doi: 10.1128/IAI.01111-12
84. Siqueira GH, Atzingen MV, De Souza GO, Vasconcellos SA, Nascimento AL. Leptospira interrogans Lsa23 protein recruits plasminogen, factor H and C4BP from normal human serum and mediates C3b and C4b degradation. Microbiology (2016) 162:295–308. doi: 10.1099/mic.0.000217
85. Ghosh KK, Prakash A, Dhara A, Hussain MS, Shrivastav P, Kumar P, et al. Role of supramolecule ErpY-like lipoprotein of Leptospira in thrombin-catalyzed fibrin clot inhibition and binding to complement factors H and I, and its diagnostic potential. Infection Immun (2019) 87:e00536–00519. doi: 10.1128/IAI.00536-19
86. Nordström T, Blom AM, Forsgren A, Riesbeck K. The emerging pathogen Moraxella catarrhalis interacts with complement inhibitor C4b binding protein through ubiquitous surface proteins A1 and A2. J Immunol (2004) 173:4598–606. doi: 10.4049/jimmunol.173.7.4598
87. Singh B, Al-Jubair T, Voraganti C, Andersson T, Mukherjee O, Su Y-C, et al. Moraxella catarrhalis binds plasminogen to evade host innate immunity. Infection Immun (2015) 83:3458–69. doi: 10.1128/IAI.00310-15
88. Bernhard S, Fleury C, Su Y-C, Zipfel PF, Koske I, Nordström T, et al. Outer membrane protein OlpA contributes to Moraxella catarrhalis serum resistance via interaction with factor H and the alternative pathway. J Infect Dis (2014) 210:1306–10. doi: 10.1093/infdis/jiu241
89. Bolland JR, Simmons WL, Daubenspeck JM, Dybvig K. Mycoplasma polysaccharide protects against complement. Microbiology (2012) 158:1867. doi: 10.1099/mic.0.058222-0
90. Ram S, Shaughnessy J, Deoliveira RB, Lewis LA, Gulati S, Rice PA. Utilizing complement evasion strategies to design complement-based antibacterial immunotherapeutics: Lessons from the pathogenic Neisseriae. Immunobiology (2016) 221:1110–23. doi: 10.1016/j.imbio.2016.05.016
91. Jarva H, Ram S, Vogel U, Blom AM, Meri S. Binding of the complement inhibitor C4bp to serogroup B Neisseria meningitidis. J Immunol (2005) 174:6299–307. doi: 10.4049/jimmunol.174.10.6299
92. Schneider MC, Exley RM, Chan H, Feavers I, Kang Y-H, Sim RB, et al. Functional significance of factor H binding to Neisseria meningitidis. J Immunol (2006) 176:7566–75. doi: 10.4049/jimmunol.176.12.7566
93. Lewis LA, Rice PA, Ram S. Role of gonococcal neisserial surface protein A (NspA) in serum resistance and comparison of its factor H binding properties with those of its meningococcal counterpart. Infection Immun (2019) 87:e00658–00618. doi: 10.1128/IAI.00658-18
94. Källström H, Liszewski MK, Atkinson JP, Jonsson AB. Membrane cofactor protein (MCP or CD46) is a cellular pilus receptor for pathogenic Neisseria. Mol Microbiol (1997) 25:639–47. doi: 10.1046/j.1365-2958.1997.4841857.x
95. Kunert A, Losse J, Gruszin C, Hühn M, Kaendler K, Mikkat S, et al. Immune evasion of the human pathogen Pseudomonas aeruginosa: Elongation factor Tuf is a factor H and plasminogen binding protein. J Immunol (2007) 179:2979–88. doi: 10.4049/jimmunol.179.5.2979
96. Ho DK, Jarva H, Meri S. Human complement factor H binds to outer membrane protein Rck of Salmonella. J Immunol (2010) 185:1763–9. doi: 10.4049/jimmunol.1001244
97. Ho DK, Tissari J, Järvinen HM, Blom AM, Meri S, Jarva H. Functional recruitment of human complement inhibitor C4B-binding protein to outer membrane protein Rck of Salmonella. PloS One (2011) 6:e27546. doi: 10.1371/journal.pone.0027546
98. Ramu P, Tanskanen R, Holmberg M, Lähteenmäki K, Korhonen TK, Meri S. The surface protease PgtE of Salmonella enterica affects complement activity by proteolytically cleaving C3b, C4b and C5. FEBS Lett (2007) 581:1716–20. doi: 10.1016/j.febslet.2007.03.049
99. Hair PS, Ward MD, Semmes OJ, Foster TJ, Cunnion KM. Staphylococcus aureus clumping factor A binds to complement regulator factor I and increases factor I cleavage of C3b. J Infect Dis (2008) 198:125–33. doi: 10.1086/588825
100. Woehl JL, Stapels DAC, Garcia BL, Ramyar KX, Keightley A, Ruyken M, et al. The extracellular adherence protein from staphylococcus aureus inhibits the classical and lectin pathways of complement by blocking formation of the C3 pro-convertase. J Immunol (Baltimore Md.: 1950) (2014) 193:6161–71. doi: 10.4049/jimmunol.1401600
101. Lee LY, Liang X, Ho O K M, Brown EL. Identification and characterization of the C3 binding domain of the Staphylococcus aureus extracellular fibrinogen-binding protein (Efb). J Biol Chem (2004) 279:50710–6. doi: 10.1074/jbc.M408570200
102. Jongerius I, Köhl J, Pandey MK, Ruyken M, Van Kessel KPM, Strijp V, et al. Staphylococcal complement evasion by various convertase-blocking molecules. J Exp Med (2007) 204:2461–71. doi: 10.1084/jem.20070818
103. Rooijakkers SH, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, et al. Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat Immunol (2005) 6:920–7. doi: 10.1038/ni1235
104. Laursen NS, Gordon N, Hermans S, Lorenz N, Jackson N, Wines B, et al. Structural basis for inhibition of complement C5 by the SSL7 protein from Staphylococcus aureus. Proc Natl Acad Sci United States America (2010) 107:3681–6. doi: 10.1073/pnas.0910565107
105. Cheng Q, Stafslien D, Purushothaman SS, Cleary P. The group B streptococcal C5a peptidase is both a specific protease and an invasin. Infection Immun (2002) 70:2408–13. doi: 10.1128/IAI.70.5.2408-2413.2002
106. Harth-Chu EN, Alves LA, Theobaldo JD, Salomão MF, Höfling JF, King WF, et al. PcsB expression diversity influences on Streptococcus mitis phenotypes associated with host persistence and virulence. Front Microbiol (2019) 10. doi: 10.3389/fmicb.2019.02567
107. Reuter M, Caswell CC, Lukomski S, Zipfel PF. Binding of the human complement regulators CFHR1 and factor H by streptococcal collagen-like protein 1 (Scl1) via their conserved C termini allows control of the complement cascade at multiple levels. J Biol Chem (2010) 285:38473–85. doi: 10.1074/jbc.M110.143727
108. Tsao N, Tsai W-H, Lin Y-S, Chuang W-J, Wang CH, Kuo C-F. Streptococcal pyrogenic exotoxin B cleaves properdin and inhibits complement-mediated opsonophagocytosis. Biochem Biophys Res Commun (2006) 339:779–84. doi: 10.1016/j.bbrc.2005.11.078
109. Giannakis E, Jokiranta TS, Ormsby RJ, Duthy TG, Male DA, Christiansen D, et al. Identification of the streptococcal M protein binding site on membrane cofactor protein (CD46). J Immunol (2002) 168:4585–92. doi: 10.4049/jimmunol.168.9.4585
110. Jarva H, Janulczyk R, Hellwage J, Zipfel PF, Bjo Rck L, Meri S. Streptococcus pneumoniae evades complement attack and opsonophagocytosis by expressing the pspC locus-encoded Hic protein that binds to short consensus repeats 8–11 of factor H. J Immunol (2002) 168:1886–94. doi: 10.4049/jimmunol.168.4.1886
111. Dave S, Carmicle S, Hammerschmidt S, Pangburn MK, Mcdaniel LS. Dual roles of PspC, a surface protein of Streptococcus pneumoniae in binding human secretory IgA and factor H. J Immunol (2004) 173:471. doi: 10.4049/jimmunol.173.1.471
112. Mohan S, Hertweck C, Dudda A, Hammerschmidt S, Skerka C, Hallström T, et al. Tuf of Streptococcus pneumoniae is a surface displayed human complement regulator binding protein. Mol Immunol (2014) 62:249–64. doi: 10.1016/j.molimm.2014.06.029
113. Kirjavainen V, Jarva H, Biedzka-Sarek M, Blom AM, Skurnik M, Meri S. Yersinia enterocolitica serum resistance proteins YadA and Ail bind the complement regulator C4b-binding protein. PloS Pathog (2008) 4:e1000140. doi: 10.1371/journal.ppat.1000140
114. Ho DK, Skurnik M, Blom AM, Meri S. Yersinia pestis Ail recruitment of C 4b-binding protein leads to factor I-mediated inactivation of covalently and noncovalently bound C 4b. Eur J Immunol (2014) 44:742–51. doi: 10.1002/eji.201343552
115. Behnsen J, Hartmann A, Schmaler J, Gehrke A, Brakhage AA, Zipfel PF. The opportunistic human pathogenic fungus Aspergillus fumigatus evades the host complement system. Infection Immun (2008) 76:820–7. doi: 10.1128/IAI.01037-07
116. Dasari P, Shopova IA, Stroe M, Wartenberg D, Martin-Dahse H, Beyersdorf N, et al. Aspf2 from Aspergillus fumigatus recruits human immune regulators for immune evasion and cell damage. Front Immunol (2018) 1635. doi: 10.3389/fimmu.2018.01635
117. Shende R, Wong SSW, Rapole S, Beau R, Ibrahim-Granet O, Monod M, et al. Aspergillus fumigatus conidial metalloprotease Mep1p cleaves host complement proteins. J Biol Chem (2018) 293:15538–55. doi: 10.1074/jbc.RA117.001476
118. Poltermann S, Kunert A, Von Der Heide M, Eck R, Hartmann A, Zipfel PF. Gpm1p is a factor H-, FHL-1-, and plasminogen-binding surface protein of Candida albicans. J Biol Chem (2007) 282:37537–44. doi: 10.1074/jbc.M707280200
119. Luo S, Hoffmann R, Skerka C, Zipfel PF. Glycerol-3-phosphate dehydrogenase 2 is a novel factor H–, factor H–like protein 1–, and plasminogen-binding surface protein of Candida albicans. J Infect Dis (2013) 207:594–603. doi: 10.1093/infdis/jis718
120. Lesiak-Markowicz I, Vogl G, Schwarzmüller T, Speth C, Lass-Flörl C, Dierich MP, et al. Candida albicans Hgt1p, a multifunctional evasion molecule: complement inhibitor, CR3 analogue, and human immunodeficiency virus–binding molecule. J Infect Dis (2011) 204:802–9. doi: 10.1093/infdis/jir455
121. Luo S, Blom AM, Rupp S, Hipler UC, Hube B, Skerka C, et al. The pH-regulated antigen 1 of Candida albicans binds the human complement inhibitor C4b-binding protein and mediates fungal complement evasion. J Biol Chem (2011) 286:8021–9. doi: 10.1074/jbc.M110.130138
122. Speth C, Rambach G, Würzner R, Lass-Flörl C. Complement and fungal pathogens: an update. Mycoses (2008) 51:477–96. doi: 10.1111/j.1439-0507.2008.01597.x
123. Gropp K, Schild L, Schindler S, Hube B, Zipfel PF, Skerka C. The yeast Candida albicans evades human complement attack by secretion of aspartic proteases. Mol Immunol (2009) 47:465–75. doi: 10.1016/j.molimm.2009.08.019
124. Dong ZM, Murphy JW. Cryptococcal polysaccharides bind to CD18 on human neutrophils. Infection Immun (1997) 65:557–63. doi: 10.1128/iai.65.2.557-563.1997
125. Chung KM, Liszewski MK, Nybakken G, Davis AE, Townsend RR, Fremont DH, et al. West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc Natl Acad Sci United States America (2006) 103:19111–6. doi: 10.1073/pnas.0605668103
126. Avirutnan P, Fuchs A, Hauhart RE, Somnuke P, Youn S, Diamond MS, et al. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J Exp Med (2010) 207:793–806. doi: 10.1084/jem.20092545
127. Stoiber H, Schneider R, Janatova J, Dierich MP. Human complement proteins C3b, C4b, factor H and properdin react with specific sites in gpl20 and gp4l, the envelope proteins of HIV1. Immunobiology (1995) 193:98–113. doi: 10.1016/S0171-2985(11)80158-0
128. Spiller OB, Mark L, Blue C, Proctor DG, Aitken J, Blom AM, et al. Dissecting the regions of virion-associated Kaposi’s sarcoma-associated herpesvirus complement control protein required for complement regulation and cell binding. J Virol (2006) 80:4068–78. doi: 10.1128/JVI.80.8.4068-4078.2006
129. Liszewski MK, Leung MK, Hauhart R, Buller RML, Bertram P, Wang X, et al. Structure and regulatory profile of the monkeypox inhibitor of complement: comparison to homologs in Vaccinia and Variola and evidence for dimer formation. J Immunol (2006) 176:3725–34. doi: 10.4049/jimmunol.176.6.3725
130. Abou-El-Hassan H, Zaraket H. Viral-derived complement inhibitors: current status and potential role in immunomodulation. Exp Biol Med (2016) 242:397–410. doi: 10.1177/1535370216675772
131. Mckenzie R, Kotwal GJ, Moss B, Hammer C, Frank M. Regulation of complement activity by vaccinia virus complement-control protein. J Infect Dis (1992) 166:1245–50. doi: 10.1093/infdis/166.6.1245
132. Sahu A, Isaacs SN, Soulika AM, Lambris JD. Interaction of vaccinia virus complement control protein with human complement proteins: factor I-mediated degradation of C3b to iC3b; inactivates the alternative complement pathway. J Immunol (1998) 160:5596–604. doi: 10.4049/jimmunol.160.11.5596
133. Rosengard AM, Liu Y, Nie Z, Jimenez R. Variola virus immune evasion design: expression of a highly efficient inhibitor of human complement. Proc Natl Acad Sci (2002) 99:8808–13. doi: 10.1073/pnas.112220499
134. Miller HW, Tam TS, Ralston KS. Entamoeba histolytica Develops Resistance to Complement Deposition and Lysis after Acquisition of Human Complement-Regulatory Proteins through Trogocytosis. Mbio (2022) 13:e03163–03121. doi: 10.1128/mbio.03163-21
135. Brittingham A, Morrison CJ, Mcmaster WR, Mcgwire BS, Chang K-P, Mosser DM. Role of the Leishmania surface protease gp63 in complement fixation, cell adhesion, and resistance to complement-mediated lysis. J Immunol (1995) 155:3102–11. doi: 10.4049/jimmunol.155.6.3102
136. Kennedy AT, Schmidt CQ, Thompson JK, Weiss GE, Taechalertpaisarn T, Gilson PR, et al. Recruitment of factor H as a novel complement evasion strategy for blood-stage Plasmodium falciparum infection. J Immunol (2016) 196:1239–48. doi: 10.4049/jimmunol.1501581
137. Inal JM, Hui K-M, Miot S, Lange S, Ramirez MI, Schneider B, et al. Complement C2 receptor inhibitor trispanning: a novel human complement inhibitory receptor. J Immunol (2005) 174:356–66. doi: 10.4049/jimmunol.174.1.356
138. Norris KA. Stable transfection of Trypanosoma cruzi epimastigotes with the trypomastigote-specific complement regulatory protein cDNA confers complement resistance. Infection Immun (1998) 66:2460–5. doi: 10.1128/IAI.66.6.2460-2465.1998
139. Fischer E, Ouaissi M, Velge P, Cornette J, Kazatchkine M. Kazatchkine MD: gp 58/68, a parasite component that contributes to the escape of the trypomastigote form of T. cruzi from damage by the human alternative complement pathway. Immunology (1988) 65:299.
140. Hammerschmidt C, Koenigs A, Siegel C, Hallström T, Skerka C, Wallich R, et al. Versatile roles of CspA orthologs in complement inactivation of serum-resistant Lyme disease spirochetes. Infection Immun (2014) 82:380–92. doi: 10.1128/IAI.01094-13
141. Hammerschmidt C, Klevenhaus Y, Koenigs A, Hallström T, Fingerle V, Skerka C, et al. BGA66 and BGA71 facilitate complement resistance of Borrelia bavariensis by inhibiting assembly of the membrane attack complex. Mol Microbiol (2016) 99:407–24. doi: 10.1111/mmi.13239
142. Hallström T, Siegel C, Mörgelin M, Kraiczy P, Skerka C, Zipfel PF. CspA from Borrelia burgdorferi inhibits the terminal complement pathway. Mbio (2013) 4:10–1128. doi: 10.1128/mBio.00481-13
143. Binns M, Mayden J, Levine R. Further characterization of complement resistance conferred on Escherichia coli by the plasmid genes traT of R100 and iss of ColV, I-K94. Infection Immun (1982) 35:654–9. doi: 10.1128/iai.35.2.654-659.1982
144. Rautemaa R, Jarvis GA, Marnila P, Meri S. Acquired resistance of Escherichia coli to complement lysis by binding of glycophosphoinositol-anchored protectin (CD59). Infection Immun (1998) 66:1928–33. doi: 10.1128/IAI.66.5.1928-1933.1998
145. Cole LE, Kawula TH, Toffer KL, Elkins C. The Haemophilus ducreyi serum resistance antigen DsrA confers attachment to human keratinocytes. Infection Immun (2002) 70:6158–65. doi: 10.1128/IAI.70.11.6158-6165.2002
146. Al-Jubair T, Mukherjee O, Oosterhuis S, Singh B, Su Y-C, Fleury C, et al. Haemophilus influenzae type F hijacks vitronectin using protein H to resist host innate immunity and adhere to pulmonary epithelial cells. J Immunol (2015) 195:5688–95. doi: 10.4049/jimmunol.1501197
147. Hallström T, Trajkovska E, Forsgren A, Riesbeck K. Haemophilus influenzae surface fibrils contribute to serum resistance by interacting with vitronectin. J Immunol (2006) 177:430–6. doi: 10.4049/jimmunol.177.1.430
148. Hallström T, Blom AM, Zipfel PF, Riesbeck K. Nontypeable Haemophilus influenzae protein E binds vitronectin and is important for serum resistance. J Immunol (2009) 183:2593–601. doi: 10.4049/jimmunol.0803226
149. Su Y-C, Mukherjee O, Singh B, Hallgren O, Westergren-Thorsson G, Hood D, et al. Haemophilus influenzae P4 interacts with extracellular matrix proteins promoting adhesion and serum resistance. J Infect Dis (2016) 213:314–23. doi: 10.1093/infdis/jiv374
150. Su YC, Jalalvand F, Mörgelin M, Blom AM, Singh B, Riesbeck K. Haemophilus influenzae acquires vitronectin via the ubiquitous Protein F to subvert host innate immunity. Mol Microbiol (2013) 87:1245–66. doi: 10.1111/mmi.12164
151. Richter C, Mukherjee O, Ermert D, Singh B, Su Y-C, Agarwal V, et al. Moonlighting of Helicobacter pylori catalase protects against complement-mediated killing by utilising the host molecule vitronectin. Sci Rep (2016) 6:1–13. doi: 10.1038/srep24391
152. Rautemaa R, Rautelin H, Puolakkainen P, Kokkola A, Karkkainen P, Meri S. Survival of Helicobacter pylori from complement lysis by binding of GPI-anchored protectin (CD59). Gastroenterology (2001) 120:470–9. doi: 10.1053/gast.2001.21197
153. Attia AS, Ram S, Rice PA, Hansen EJ. Binding of vitronectin by the Moraxella catarrhalis UspA2 protein interferes with late stages of the complement cascade. Infection Immun (2006) 74:1597–611. doi: 10.1128/IAI.74.3.1597-1611.2006
154. Sa E Cunha C, Griffiths NJ, Virji M. Neisseria meningitidis Opc invasin binds to the sulphated tyrosines of activated vitronectin to attach to and invade human brain endothelial cells. PloS Pathog (2010) 6:e1000911. doi: 10.1371/journal.ppat.1000911
155. Hallström T, Uhde M, Singh B, Skerka C, Riesbeck K, Zipfel PF. Pseudomonas aeruginosa uses dihydrolipoamide dehydrogenase (Lpd) to bind to the human terminal pathway regulators vitronectin and clusterin to inhibit terminal pathway complement attack. PloS One (2015) 10:e0137630. doi: 10.1371/journal.pone.0137630
156. Paulsson M, Singh B, Al-Jubair T, Su Y-C, Høiby N, Riesbeck K. Identification of outer membrane Porin D as a vitronectin-binding factor in cystic fibrosis clinical isolates of Pseudomonas aeruginosa. J cystic fibrosis: Off J Eur Cystic Fibrosis Soc (2015) 14:600–7. doi: 10.1016/j.jcf.2015.05.005
157. Fernie-King BA, Seilly DJ, Willers C, Würzner R, Davies A, Lachmann PJ. Streptococcal inhibitor of complement (SIC) inhibits the membrane attack complex by preventing uptake of C567 onto cell membranes. Immunology (2001) 103:390–8. doi: 10.1046/j.1365-2567.2001.01249.x
158. Blom AM, Bergmann S, Fulde M, Riesbeck K, Agarwal V. Streptococcus pneumoniae phosphoglycerate kinase is a novel complement inhibitor affecting the membrane attack complex formation. J Biol Chem (2014) 289:32499–511. doi: 10.1074/jbc.M114.610212
159. Voss S, Hallström T, Saleh M, Burchhardt G, Pribyl T, Singh B, et al. The choline-binding protein PspC of Streptococcus pneumoniae interacts with the C-terminal heparin-binding domain of vitronectin. J Biol Chem (2013) 288:15614–27. doi: 10.1074/jbc.M112.443507
160. Bartra SS, Ding Y, Fujimoto LM, Ring JG, Jain V, Ram S, et al. Yersinia pestis uses the Ail outer membrane protein to recruit vitronectin. Microbiology (2015) 161:2174–83. doi: 10.1099/mic.0.000179
161. Conde JN, Da Silva EM, Allonso D, Coelho DR, Andrade IDS, De Medeiros LN, et al. Inhibition of the membrane attack complex by Dengue Virus NS1 through interaction with vitronectin and terminal complement proteins. J Virol (2016) 90:9570–81. doi: 10.1128/JVI.00912-16
162. Malekshahi Z, Schiela B, Bernklau S, Banki Z, Würzner R, Stoiber H. Interference of the zika virus E-protein with the membrane attack complex of the complement system. Front Immunol (2020) 11:569549. doi: 10.3389/fimmu.2020.569549
163. Kim H, Meyer K, Di Bisceglie AM, Ray R. Hepatitis C virus suppresses C9 complement synthesis and impairs membrane attack complex function. J Virol (2013) 87:5858–67. doi: 10.1128/JVI.00174-13
164. Amet T, Ghabril M, Chalasani N, Byrd D, Hu N, Grantham A, et al. CD59 incorporation protects hepatitis C virus against complement-mediated destruction. Hepatology (2012) 55:354–63. doi: 10.1002/hep.24686
165. Amet T, Lan J, Shepherd N, Yang K, Byrd D, Xing Y, et al. Glycosylphosphatidylinositol anchor deficiency attenuates the production of infectious HIV-1 and renders virions sensitive to complement attack. AIDS Res Hum Retroviruses (2016) 32:1100–12. doi: 10.1089/aid.2016.0046
166. Wei Y, Ji Y, Guo H, Zhi X, Han S, Zhang Y, et al. CD59 association with infectious bronchitis virus particles protects against antibody-dependent complement-mediated lysis. J Gen Virol (2017) 98:2725. doi: 10.1099/jgv.0.000962
167. Kochi LT, Fernandes LG, Souza GO, Vasconcellos SA, Heinemann MB, Romero EC, et al. The interaction of two novel putative proteins of Leptospira interrogans with E-cadherin, plasminogen and complement components with potential role in bacterial infection. Virulence (2019) 10:734–53. doi: 10.1080/21505594.2019.1650613
168. Fritzinger AE, Toney DM, Maclean RC, Marciano-Cabral F. Identification of a Naegleria fowleri membrane protein reactive with anti-human CD59 antibody. Infection Immun (2006) 74:1189–95. doi: 10.1128/IAI.74.2.1189-1195.2006
169. Tougan T, Edula JR, Takashima E, Morita M, Shinohara M, Shinohara A, et al. Molecular camouflage of Plasmodium falciparum merozoites by binding of host vitronectin to P47 fragment of SERA5. Sci Rep (2018) 8:1–13. doi: 10.1038/s41598-018-23194-9
170. Deng J, Gold D, Loverde PT, Fishelson Z. Inhibition of the complement membrane attack complex by Schistosoma mansoni paramyosin. Infection Immun (2003) 71:6402–10. doi: 10.1128/IAI.71.11.6402-6410.2003
171. Langereis JD, De Jonge MI, Weiser JN. Binding of human factor H to outer membrane protein P5 of non-typeable H aemophilus influenzae contributes to complement resistance. Mol Microbiol (2014) 94:89–106. doi: 10.1111/mmi.12741
172. Fernandez RC, Weiss A. Serum resistance in bvg-regulated mutants of Bordetella pertussis. FEMS Microbiol Lett (1998) 163:57–63. doi: 10.1111/j.1574-6968.1998.tb13026.x
173. Ferreira AM, Breijo M, Sim RB, Dı Az A. How echinococcus granulosus deals with complement. Parasitol Today (2000) 16:168–72. doi: 10.1016/S0169-4758(99)01625-7
174. Díaz A, Casaravilla C, Allen JE, Sim RB, Ferreira AM. Understanding the laminated layer of larval Echinococcus II: immunology. Trends Parasitol (2011) 27:264–73. doi: 10.1016/j.pt.2011.01.008
175. Agrawal P, Nawadkar R, Ojha H, Kumar J, Sahu A. Complement evasion strategies of viruses: an overview. Front Microbiol (2017) 8. doi: 10.3389/fmicb.2017.01117
176. Tambourgi DV, Kipnis TL, Da Silva WD, Joiner K, Sher A, Heath S, et al. A partial cDNA clone of trypomastigote decay-accelerating factor (T-DAF), a developmentally regulated complement inhibitor of Trypanosoma cruzi, has genetic and functional similarities to the human complement inhibitor DAF. Infection Immun (1993) 61:3656–63. doi: 10.1128/iai.61.9.3656-3663.1993
177. Berends ETM, Dekkers JF, Nijland R, Kuipers A, Soppe JA, Strijp V. Distinct localization of the complement C5b-9 complex on Gram-positive bacteria. Cell Microbiol (2013) 15:1955–68. doi: 10.1111/cmi.12170
178. Acharya S, Da’dara AA, Skelly PJ. Schistosome immunomodulators. PloS Pathog (2021) 17:e1010064. doi: 10.1371/journal.ppat.1010064
Keywords: complement evasion, serum resistance, pathogens, virulence factors, regulation
Citation: Heggi MT, Nour El-Din HT, Morsy DI, Abdelaziz NI and Attia AS (2024) Microbial evasion of the complement system: a continuous and evolving story. Front. Immunol. 14:1281096. doi: 10.3389/fimmu.2023.1281096
Received: 21 August 2023; Accepted: 30 November 2023;
Published: 04 January 2024.
Edited by:
Isabelle Bekeredjian-Ding, Paul-Ehrlich-Institut (PEI), GermanyReviewed by:
Michael Kirschfink, Heidelberg University, GermanyReinhard Würzner, Innsbruck Medical University, Austria
Copyright © 2024 Heggi, Nour El-Din, Morsy, Abdelaziz and Attia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed S. Attia, YWhtZWQuYXR0aWFAcGhhcm1hLmN1LmVkdS5lZw==