 Xinzou Fan
Xinzou Fan Panyin Shu
Panyin Shu Ying Wang
Ying Wang Ning Ji
Ning Ji Dunfang Zhang
Dunfang Zhang- 1Department of Biotherapy, State Key Laboratory of Biotherapy and Cancer Center, Collaborative Innovation Center of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan, China
- 2State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, Research Unit of Oral Carcinogenesis and Management, Chinese Academy of Medical Sciences, West China Hospital of Stomatology, Sichuan University, Chengdu, Sichuan, China
Neutrophils comprise the majority of immune cells in human peripheral circulation, have potent antimicrobial activities, and are clinically significant in their abundance, heterogeneity, and subcellular localization. In the past few years, the role of neutrophils as components of the innate immune response has been studied in numerous ways, and these cells are crucial in fighting infections, autoimmune diseases, and cancer. T-helper 17 (Th17) cells that produce interleukin 17 (IL-17) are critical in fighting infections and maintaining mucosal immune homeostasis, whereas they mediate several autoimmune diseases. Neutrophils affect adaptive immune responses by interacting with adaptive immune cells. In this review, we describe the physiological roles of both Th17 cells and neutrophils and their interactions and briefly describe the pathological processes in which these two cell types participate. We provide a summary of relevant drugs targeting IL-17A and their clinical trials. Here, we highlight the interactions between Th17 cells and neutrophils in diverse pathophysiological situations.
1 Introduction
Over the past few years, our understanding of neutrophil function has considerably progressed. Neutrophils play a role in homeostatic control as part of innate immunity and can affect adaptive immunity in tissues through activation or inhibition. The concept of T-helper 17 (Th17) cells was introduced less than 20 years ago. Due to their immunomodulatory roles in autoimmune diseases and infection resistance, Th17 cells have been the subject of substantial research. Th17 cells are potent inducers of neutrophils, recruiting neutrophils to sites of inflammation via IL-17A and promoting inflammation. The mechanisms underlying Th17 cell-mediated induction of neutrophils have been studied earlier, whereas the impact of neutrophils on Th17 cells remains largely unknown. Recent studies have reported that neutrophils regulate Th17 cells. Therefore, we reviewed the current knowledge on the mechanisms by which neutrophils modulate the impact on Th17 cells.
2 Neutrophils
Neutrophils comprise the majority of myeloid leukocytes in humans, constituting 50–70% of circulating leukocytes (1). Neutrophils have a short lifespan, with a specific polymorphonuclear and defined granule content (2). Neutrophils are believed to activate and regulate innate and adaptive immunity, mainly Th17 cell-driven adaptive immune responses against infections and the subsequent activation and regulation of innate and adaptive immunity against infections (3, 4). Neutrophils interact in a complex bidirectional manner with various immune cells by expressing a wide range of cytokines and immunosuppressive and immunostimulatory molecules (5, 6). The three key ways whereby neutrophils fight pathogens are phagocytosis, degranulation, and the formation of neutrophil extracellular traps (NETs) (7). The process of neutrophil phagocytosis is receptor-mediated. The neutrophil cytoskeleton rearranges, and the plasma membrane surrounds the target organism or granules to form phagocytic vesicles. Preformed granules within neutrophils fuse rapidly with phagocytic vesicles to kill and degrade target organisms or granules (8). Neutrophil phagocytosis accompanied by degranulation. Neutrophil degranulation occurs mainly at the plasma membrane, mimicking the granule–phagosome fusion. Neutrophils perform their antimicrobial functions by releasing granule-derived soluble proteins extracellularly via degranulation to activate and deliver various antimicrobial components (9). DNA, histones, and neutrophil granule proteins form net-like structures known as NETs, produced by activated neutrophils (10). NET-binding proteins are mainly cationic bactericidal proteins, including 24 proteins such as histones, elastase, cathepsin G, myeloperoxidase (MPO), proteinase 3, neutrophil defensin, calcium-binding protein, lactoferrin, and lysozyme (11, 12). Notably, NETs can be formed in two ways, i.e., NETosis and nonlytic NETosis. In NETosis or canonical death pathway, peptidylarginine deiminase-4 converts arginine in histones to citrulline after being triggered by Ca2+ ions. The strong positive charge on histones diminishes when arginine changes to citrulline, which weakens their strong electrostatic binding to DNA in the nucleosome, leading to chromatin decompression and NET formation. Subsequently, the neutrophil nuclear membrane begins to disintegrate, and the nucleus loses its lobules, starts to swell, and undergoes granule proteolysis. Eventually, neutrophils excrete histones and granules and deconcentrate DNA to form NETs (13, 14), which, when released, kill pathogens directly or capture them and wait for further processing by phagocytes (14–16). The second is the nonlytic form of NETosis. After exposure to Staphylococcus aureus, neutrophils rapidly expel chromatin and granules to form NETs. However, this process is not accompanied by neutrophil death (7). The cytoplasm left after the neutrophils excrete chromatin and granulins can continue to perform bactericidal functions by ingesting and engulfing pathogens (17). Reactive oxygen species (ROS) are not necessary for nonlytic NETosis (18).
The antimicrobial activity of neutrophils is biotoxic. When neutrophil activation is not properly controlled, pathological damage may occur in the host. Neutrophils are associated with numerous autoimmune diseases. Phenotypic and functional neutrophil aberrations, increased levels of circulating pro-inflammatory low-density granulocytes (LDGs), increased neutrophil apoptosis, and loss of neutrophil phagocytic function are observed in patients with systemic lupus erythematosus (19). Neutrophil-forming NETs are believed to be involved in atherosclerotic thrombosis (20). They play an essential role in thrombosis by promoting the deposition of hemofibrin and the formation of hemofibrin networks (21). Activated platelets induce neutrophils to form NETs (22). Impaired directed migration and function of neutrophils during sepsis leads to harmful accumulation in the lungs and liver (23, 24). In the early stages of sepsis, platelets stimulate neutrophils to release NETs that benefit the host by wrapping and eliminating pathogens. Notably, NETs and their components can harm host tissues and endothelium during severe infections and may lead to the formation of a diffuse thrombus (25). Neutrophils play a significant role in malignancies. Neutrophils undergo ferroptosis and release immunosuppressive molecules that promote tumor cell growth in the tumor microenvironment (26). Neutrophils promote tumor cell metastasis by forming NETs and encapsulating tumors in NETs, limiting tumor killing caused by cytotoxic T cells and promoting the escape of tumor cells from the immune system (27–29).
3 Th17 cells
The first CD4 helper T lymphocytes described were Th1 and Th2 subpopulations. Th17 cells were first conceptualized in 2005 (30, 31). This subset of lymphocytes, which differentiate from naive CD4 T cells, is known for its capacity to release interleukin (IL)-17A at high levels. Th17 cells release cytokines such as IL-17F, IL-21, and IL-22, in addition to IL-17A (32, 33). Th17 differentiation is associated with retinoic acid-related orphan receptor (ROR)γt, interferon regulatory factor 4 (IRF4), basic leucine zipper ATF-like transcription factor (BATF), and aryl hydrocarbon receptor (AHR). Notably, RORγt was shown to be the first critical regulator of Th17 differentiation. It induces the expression of IL-17A and IL-17F, and lack of RORγt leads to a functional decline in Th17 (34). Moreover, IRF4 is located upstream of RORγt, and its deficiency results in the reduced capacity of naïve T cells to upregulate RORγt (35). Both Th17 and T regulatory cells (Tregs) express AHR, and Th17 cells do so relatively more highly, and the ablation of AHR significantly lowers the expression of the Th17 effector cytokine IL-22 (36). BATF is not a Th17 cell-specific transcriptional regulator and is essential for Th17 cell differentiation and related cytokine expression (37). The differentiation of Th17 in mice can be divided into three stages: (1) due to the effects of IL-6 and transforming growth factor-beta (TGF-β) on naïve T cells in mice, the activation of signal transducer and activator of transcription 3 (STAT3) causes IL-21 production; (2) IL-21 upregulates the expression of IL-17A, IL-23R, and STAT3-dependent expression of RORγt in an autocrine manner to maintain Th17 differentiation; and (3) IL-23 derived from antigen-presenting cells interacts with the IL-23 functional receptor, promoting Th17 maintenance, proliferation, and effector activities (38–41).
Th17 cells were terminally differentiated. However, Th17 cells can transdifferentiate into certain cytokines or inflammatory environments. The difference between Th1 and Th17 subsets is not as distinct as previously believed in experimental autoimmune encephalomyelitis (EAE). Notably, T cells with an in vitro bias toward Th17 cells develop a Th1-like phenotype in vivo and can produce interferon-gamma (IFN-γ) (42, 43). Th17 cells are abundant in the lamina propria of the intestinal mucosal barrier. Short-chain fatty acids promote Treg differentiation and attenuate EAE, whereas long-chain fatty acids stimulate Th17 and Th1 differentiation and exacerbate EAE (44). Bile acid metabolites regulate Th17/Treg homeostasis in the intestine (45). In autoimmune diseases, Th17 cells transdifferentiate into Tregs, secrete IL-10, and acquire regulatory and anti-inflammatory functions (46, 47). Hypoxia modulates the Th17 phenotype. Hypoxia-inducible factor 1α is required for Th17 differentiation, mediates glycolytic activity and forkhead box P3 degradation, and regulates Th17 and Treg homeostasis (48, 49). Th17 cells are heterogeneous in their phenotype, transcription, and metabolism. Th17 cells are classified into hypermetabolic and hypometabolic subsets based on their metabolism. Hypermetabolic Th17 has strong IFN-γ expression and supports transdifferentiation to Th1-like cells (50).
Th17 cells and their associated effector cytokines play crucial roles in the pathogenesis of many autoimmune diseases and mediate defense mechanisms directed against extracellular pathogenic infections. They are essential for maintaining the homeostasis of the mucosal immune microenvironment. Hosts lacking Th17 cells are highly susceptible to fungal infections. In a mouse model of systemic candidiasis in which the IL-17A/IL-17A receptor axis was essential for the host defense system in vivo, Th17 protected mice from lethal doses of the oral commensal fungus Candida albicans infection (51). Th17- and IL-17R-deficient mice exhibited severe oropharyngeal candidiasis, and the saliva of these mice showed low C. albicans-killing activity (52). In addition, Th17 cells participate in host defense against S. aureus and Citrobacter rodentium in the mucus epithelium (53). Th17 cell overexpression has a detrimental effect on the host immune response. The IL-27/IL-27R complex mediates chlamydial-protective immunity by inhibiting Th17 cell overresponse and reducing neutrophil-induced inflammation (54). Th17 cells have been associated with various autoimmune diseases. Psoriasis is an inflammatory skin disease characterized by well-defined red macules and sterile pustules. Notably, IL-17A and IL-23 are key drivers of psoriasis development. Th17 cell counts are elevated and positively linked to disease activity in the blood and skin lesions (55). Patients with psoriatic arthritis often have resident Th17 cells within the synovium that overexpress IL-17A and IL-22, exacerbating inflammation and bone reconstruction. Research using mouse models of colitis has demonstrated that inflammatory bowel disease is significantly influenced by IL-17A derived from Th17 cells. Aberrant Th17 proliferative diseases produce enormous levels of pro-inflammatory cytokines, which attract additional inflammatory cells and trigger abnormal immune responses (56). In both EAE and multiple sclerosis (MS), Th17 cells play a key pathogenic role in central nervous system (CNS) inflammation in both EAE and MS. Th17 cells are abundant in the brain and spinal cord of EAE mice, as well as in the CNS lesions, cerebrospinal fluid, and peripheral blood of patients with MS, and are positively correlated with disease activity (57–60). Mice lacking Th17 cells are resistant to EAE (61). Both asthma and chronic obstructive pulmonary disease, characterized by high mucus production, share a similar immunological pathogenesis. Notably, IL-17A regulates respiratory mucus production, and high levels of IL-17A may be linked to severe intestinal and respiratory inflammation (62, 63). Th17 cells protect the body from metabolic syndrome by regulating intestinal microbiota and fat absorption (64).
The contribution of IL-17A and Th17 cells to various diseases is the basis for their clinical targeting. Table 1 summarizes the current status of IL-17A-targeted drugs.
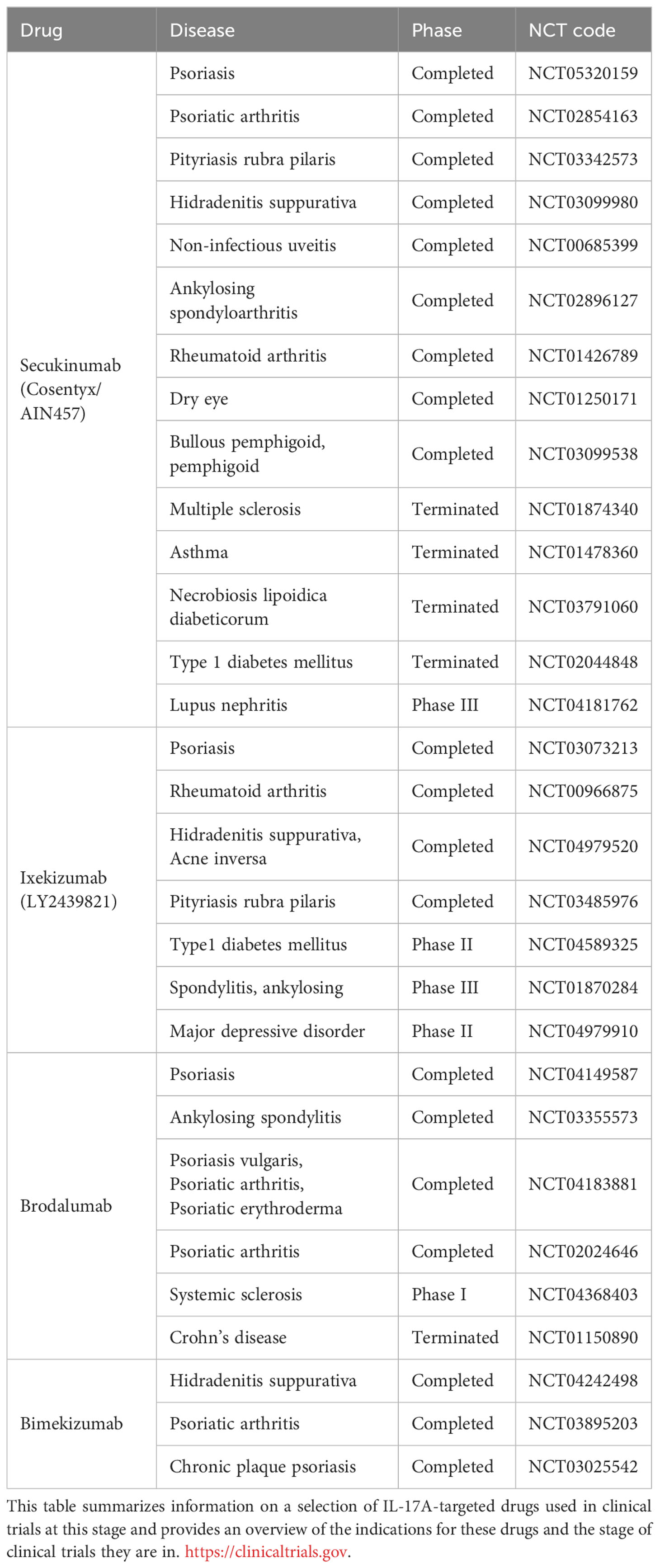
Table 1 Clinical trials using IL-17A as a target.
Secukinumab is a high-affinity, fully humanized monoclonal immunoglobulin (Ig)G1κ antibody that selectively binds to IL-17A and blocks IL-17A from binding to its receptor, thereby controlling the progression of IL-17A-associated diseases, such as psoriasis, psoriatic arthritis, ankylosing spondylitis (65–68). Secukinumab has a blocking effect and was clinically effective in a phase III clinical trial of plaque psoriasis. Psoriasis area and severity index (greater than or equal to 75) scores (PASI75) decreased by at least 75% compared to placebo and Etanercept (69). Histologically, epidermal hyperplasia reduced rapidly, and psoriasis-associated transcriptome expression was significantly reduced in genomic analysis (70). Secukinumab has been shown to be effective in controlling psoriatic arthritis. In phase III clinical trials, subcutaneous injections of 150 or 300 mg secukinumab effectively treated psoriatic arthritis. In the FUTURE trial, both 150 and 300 mg doses of secukinumab significantly improved the symptoms of psoriatic arthritis, with higher American College of Rheumatology 20% response criteria (ACR20) at 16 and 24 weeks of treatment than in patients using placebo (71–75). The proportions of patients achieving greater than or equal to 50% improvement in ACR response criteria (ACR50) and patients achieving improvements in PASI75 and PASI90 were significantly higher than in patients using placebo. Notably, ACR20 response rates at 5 years can still be as high as 70–74% (76, 77). Secukinumab improved the clinical signs and symptoms compared with those obtained with placebo use. Moreover, the improvement was sustained over a longer period than with the use of tumor necrosis factor (TNF) antagonists, making secukinumab an effective treatment modality that can be used as an alternative to TNF antagonists.
Ixekizumab is a high-affinity, recombinant humanized monoclonal IgG4κ antibody indicated for the treatment of psoriatic arthritis and moderate-to-severe plaque psoriasis by binding specifically to IL-17A and blocking IL-17A activity, reducing inflammation and achieving relief in psoriasis symptoms (78). Ixekizumab was significantly more effective than placebo in phase I and II clinical trials of psoriasis treatment. In a phase I trial, 150 mg of ixekizumab every 2 weeks resulted in maximum disease control within 6 weeks (79). Phase II trials reduced the concentration to 75 mg every 2 weeks and achieved satisfactory disease control (80). Ixekizumab is indicated to treat genital psoriasis, generalized pustular psoriasis, and psoriatic arthritis. For treating patients with genital psoriasis, 80 mg of ixekizumab was administered every 2 weeks for 12 weeks. At 12 weeks of treatment, genital pruritus scores decreased in 60% of patients using ixekizumab compared with that in patients using placebo, and 73% of patients using ixekizumab had genital pruritus scores of “0” or “1” (81). The clinical phenotype and genetic background of generalized pustular psoriasis differ from those of normal psoriasis and are uncommon in the clinic; therefore, there are not many clinical subjects of different biological agents in generalized pustular psoriasis. Promising therapeutic efficacy with limited adverse effects was observed in a German patient treated with ixekizumab for one year (82). In a psoriatic arthritis clinical trial, patients were randomized into the following four groups: 80 mg ixekizumab every 2 weeks group, 80 mg ixekizumab every 4 weeks group, 40 mg adalimumab every 2 weeks group, and a placebo group. The trial was concluded at 24 weeks, and the ACR20 rates were 62.1, 57.9, 57.4, and 30.2% for the corresponding groups, respectively (83). Ixekizumab is effective in the treatment of psoriatic arthritis.
Brodalumab is a humanized IL-17RA IgG2 monoclonal antibody. Brodalumab inhibits IL-17A downstream signaling by binding to a shared subunit of the IL-17 receptor complex and blocking multiple IL-17 family cytokines (84). Brodalumab is indicated for the treatment of moderate-to-severe plaque psoriasis. In the AMAGINE-1 phase III clinical trial, patients with plaque psoriasis were divided into the following three groups: 140 mg brodalumab every 2 weeks, 210 mg brodalumab every 2 weeks, and placebo. The PASI75 and static Physician’s Global Assessment of 0 or 1 (sPGA 0/1) ratios of these three groups were determined at 12 weeks. The PASI75 and sPGA 0/1 ratios increased significantly in subjects using Brodalumab (85). Based on the outcomes of the AMAGINE-2,3 phase III clinical trials, brodalumab (210 mg every 2 weeks) had a higher rate of PASI100 than that with ustekinumab use at 12 weeks. In addition, brodalumab provided long-term, high-level skin clearance in patients with moderate-to-severe plaque psoriasis. At 52 weeks, patients administered 210 mg brodalumab every 2 weeks rapidly achieved high levels of complete and sustained skin clearance (86). Brodalumab is effective in treating psoriatic arthritis. At 12 weeks, patients treated with 140 and 280 mg brodalumab had a higher ACR20 rate than those using placebo (87).
Bimekizumab was the first IL-17A/IL-17F dual-target inhibitor for moderate-to-severe plaque psoriasis. In the BE VIVID phase III clinical trial, patients with moderate-to-severe plaque psoriasis were divided into three groups: a 320 mg bimekizumab every 4 weeks group, a 45 or 90 mg (chosen based on body weight) ustekinumab group at weeks 0 and 4 and every 12 weeks subsequently, and a placebo group. At 16 weeks, 85% of the patients in the bimekizumab group achieved PASI90, a much higher proportion than those in the ustekinumab and placebo groups. Bimekizumab has performed well in treating moderate-to-severe plaque psoriasis, with a therapeutic efficacy superior to that obtained with ustekinumab use (88). In the BE OPTIMAL phase III trial, patients with psoriatic arthritis were divided into the following three groups: 160 mg bimekizumab every 4 weeks group, placebo group, and 40 mg adalimumab every 2 weeks group. At week 16, subjects in the placebo group were administered 160 mg of bimekizumab every 4 weeks. The proportion of patients with ACR50 was counted at week 16, and a significantly higher number of bimekizumab-treated patients than those in the placebo group achieved ACR50. Improvements in joint, skin, and imaging outcomes were relatively more pronounced in the bimekizumab-treated patients than in the placebo group (89).
4 Effects of Th17 cells on neutrophils
Th17 cells affect neutrophils via IL-17A production. Increasing the levels of granulocyte colony-stimulating factor (G-CSF), IL-17A encourages the synthesis of granulocytes and the mobilization of mature neutrophils from the bone marrow. Through peripheral blood circulation, neutrophils travel to the area of infection or inflammation and perform their effector functions (Figure 1).
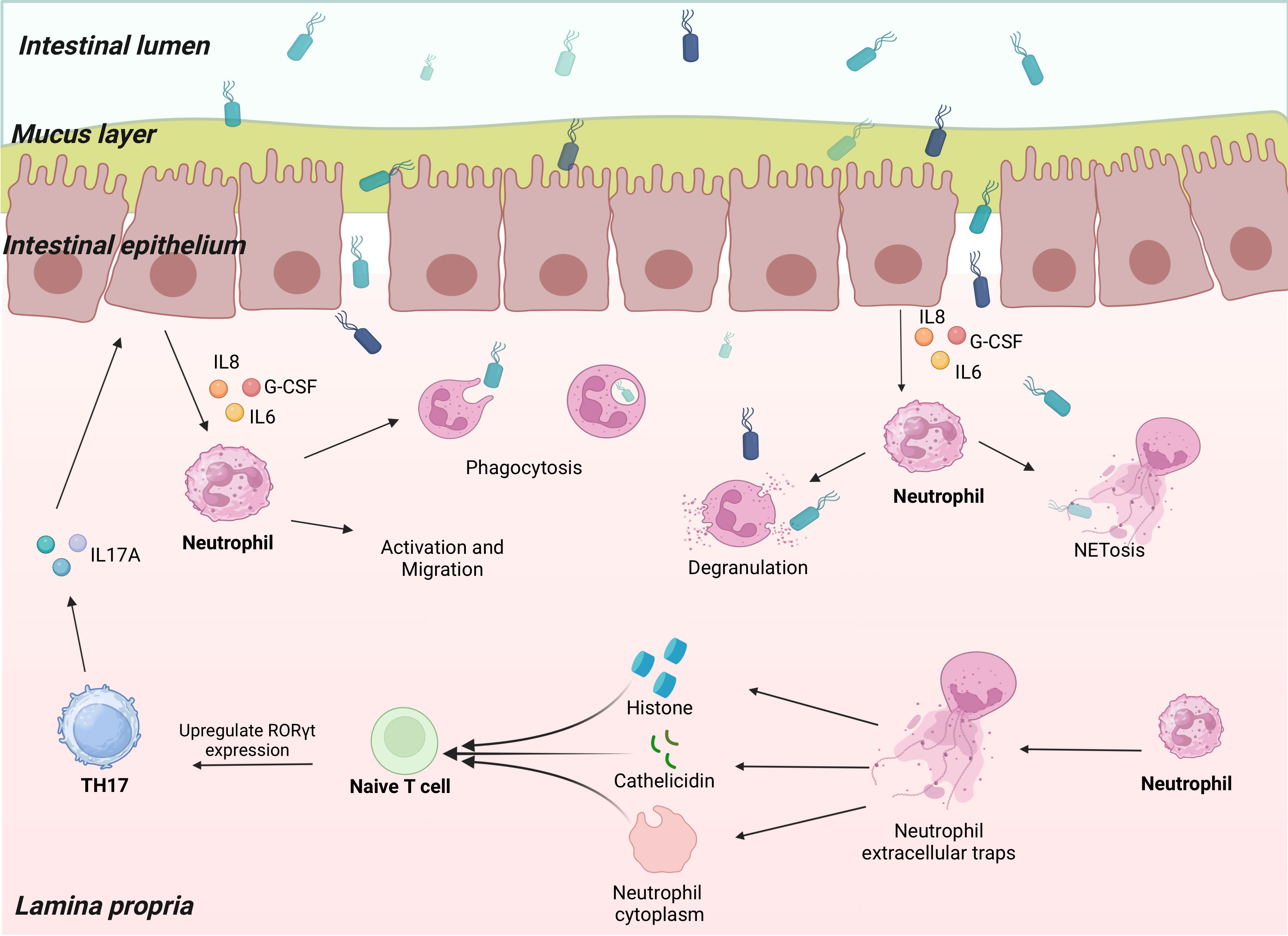
Figure 1 Neutrophils communicate with Th17 cells. Th17 cells secrete IL-17A to act on epithelial cells when the organism is infected. The epithelial cells produce a series of neutrophil chemokines that induce neutrophil activation and migration to the site of infection. Neutrophils carry out anti-infective functions through phagocytosis, degranulation, and NETosis. Through their histones, cathelicidin, and the cytoplasm remaining after NETosis, Neutrophil extracellular traps (NETs) promote naïve T cell upregulation of RORγt expression, thereby promoting Th17 differentiation.
IL-17A does not act directly on neutrophils; however, it recruits them by acting on a variety of nonimmune cells (e.g., epithelial cells, endothelial cells, and fibroblasts), causing them to release neutrophil chemokines, such as chemokine (C-X-C motif) ligand (CXCL) 1, CXCL6, and CXCL8 (90). Neutrophils in blood vessels sense these chemokines via the C-X-C motif chemokine receptor (CXCR) 1, which activates signaling downstream of vasodilator-stimulated phosphoprotein, phosphoinositide 3-kinase, and Src family kinase, mediating directional migration of neutrophils (91) (92). In addition, activated neutrophils produce matrix metalloproteinases that cleave CXCL8 family chemokines and enhance their chemotaxis (92). Overexpression of IL-17A upregulates neutrophil-targeted chemokine expression, which, in turn, increases neutrophil infiltration. Conversely, knockdown of IL-17A expression or defective IL-17A receptors results in downregulated chemokine expression and inhibition of neutrophil infiltration (91). However, IL-17A acts directly on neutrophil CXCR2 to induce neutrophil chemotaxis (93).
Moreover, IL-17A exerts a granulogenic effect (91). It intervenes in neutrophil generation by upregulating G-CSF expression, which plays a crucial role in neutrophil development. For example, G-CSF directs the differentiation of committed progenitors to myeloid lineage, promotes the proliferation of neutrophil precursors, reduces the time required for neutrophils to pass through the compartment, and promotes the release of mature neutrophils from bone marrow (94, 95). During inflammation, IL-17A, derived from Th17 cells, promotes G-CSF production, which acts on neutrophils in response to inflammation. After the inflammation subsides, neutrophils undergo apoptosis, and these apoptotic neutrophils are cleared by nearby phagocytes (such as macrophages) (94). This process is accompanied by a decrease in IL-23 levels, which, in turn, leads to a reduction in Th17-derived IL-17A production, resulting in decreased G-CSF synthesis (96, 97). Defects in G-CSF, induced by anti-IL-17A antibodies, result in elevated levels of the chemokine stromal cell-derived factor (SDF)-1α, a retention factor for neutrophils to reside in the bone marrow. Elevated levels of SDF-1α interfere with bone marrow mobilization of neutrophils, leading to a decrease in the number of circulating neutrophils (98).
The following subsections describes how Th17 cells affect neutrophils in various immune microenvironments.
4.1 Effects of Th17 cells on neutrophils in psoriasis
Characterized by epidermal proliferation and inflammation, psoriasis is a chronic relapsing inflammatory skin disease (55). The etiology of psoriasis is greatly influenced by the IL-23/IL-17A axis and, to a certain extent, reflects disease severity (99). Moreover, IL-23 activates IL-17A-producing Th17 cells and enhances their cytokine production capacity and pathogenicity. In patients with psoriatic arthritis, high levels of IL-23 and IL-17A are present in the synovium, and tissue-retained Th17 cells highly express IL-22 and IL-17A. The production of granulocyte-macrophage CSF (GM-CSF) and several chemokines, including CXCL8, is promoted in response to the IL-23/IL-17A axis (99). CXCL8 promotes neutrophil recruitment and migration, whereas GM-CSF and G-CSF replenish circulating neutrophils, which are exhausted during infiltration into the epithelium (100). In patients with psoriasis, skin lesions, serum Th17 cell counts, and IL-17A levels were positively correlated with disease activity (101). Various immune and nonimmune cells are involved in the pathogenesis of psoriasis. In addition to driving epidermal hyperproliferation and activating keratin-forming cells, IL-17A promotes the production of several cytokines and chemokines, such as the chemokine CXCL8. CXCL 8 induces neutrophil migration to lesion sites and promotes inflammation (102). Notably, IL-17A induces epithelial cells to express IL-36, contributing to neutrophil inflammatory activity and migration (103, 104). In addition, IL-17A has an apoptosis-inhibiting effect on neutrophils and promotes the production of IL-1, IL-6, and TNFα. Notably, TNFα activates neutrophils and promotes neutrophil infiltration into the epithelium, amplifies and propagates inflammation, promotes characteristic changes in psoriasis symptoms, and creates a self-sustaining inflammatory cycle (100). Neutrophils are a source of IL-17A at psoriatic lesions (99, 105). Neutrophil-derived IL-17A is a potential drug target for the treatment of psoriasis. Targeted drugs prevent neutrophils from interacting with keratin-forming cells and entering the lesion site (101). In lesions of progressive psoriasis, large aggregates of neutrophils are often observed at the epidermal site, forming neutrophilic pustules such as Kogoj spongiform pustules and Munro’s microabscess, among other typical pathologic changes (100). Neutrophils infiltrating the epidermis produce NETs that act directly on keratinocytes and are involved in the maintenance and amplification of the inflammatory environment in the skin. In addition, MPO and NETs released during neutrophil degranulation are involved in ROS generation, protein hydrolysis of inflammatory mediators, and self-antigen formation during psoriasis (106).
4.2 Effects of Th17 cells on neutrophils in asthma
Asthma is a common chronic noncommunicable respiratory disease characterized by chronic airway inflammation, hyperresponsiveness, variable reversible airflow limitation, and airway remodeling due to the prolonged course of the disease. Approximately 334 million people are affected by asthma (107). The traditional view is that dysregulation of Th2 immunity is the primary cause of asthma. However, recent studies have revealed that Th17 cell-secreted IL-17A plays a role in the development and progression of neutrophilic asthma. Patients with severe asthma have higher levels of IL-17A in their airways when their diseases are more severe (108). Th17 cells secrete cytokines, such as IL-17A, and recruit neutrophils by acting on airway epithelial cells expressing the neutrophil chemokines CXCL1 and CXCL8. Neutrophils that receive recruitment signals migrate to the airways and produce cytokines, such as IL-6, CXCL8, G-CSF, and macrophage inflammatory protein-2, which induce neutrophilic inflammation in the airway (103, 109). Most patients with asthma control their symptoms using glucocorticoid therapy. However, in patients with steroid-resistant asthma (SRA), glucocorticoid use does not adequately control the symptoms (110). Patients with SRA have large neutrophil infiltrates in the airways and high Th17/IL-17A expression (111, 112). Neutrophilic asthma exhibits significant steroid resistance and IL-17A dependence (113). Glucocorticoid receptor-β (GR-β) overexpression is linked to steroid resistance in patients with SRA. The development of steroid resistance may be facilitated by GR-β, which does not bind to corticosteroids. Notably, IL-17A induces GR-β expression in the airway epithelium of patients with asthma and generates steroid resistance in peripheral blood mononuclear cells, resulting in SRA (114). Mice overexpressing RORγt exhibit steroid-insensitive neutrophilic inflammation. Anti-IL-17A antibody, CXCR2 agonist, or anti-IL-6R antibody effectively suppress airway hyperresponsiveness and neutrophilic inflammation in RORγt-overexpressing mice (115). Airway remodeling is a crucial pathological hallmark of asthma. IL-17A, derived from Th17 cells, can activate and secrete neutrophil chemokines, such as IL-6 and CXCL8, by promoting the activation of various cells, such as epithelial cells, fibroblasts, and neutrophils, which causes neutrophils to release a variety of bioactive substances, such as neutrophil elastase, in the airways to mediate lung tissue destruction and mucus hypersecretion (112).
4.3 Effects of Th17 cells on neutrophils in other diseases
Inflammatory resorption of the bone tissue supporting the teeth is a hallmark of the chronic, progressive, and destructive disease periodontitis. According to previous studies, bacteria are the primary cause of periodontitis. Th17 cells are crucial for the progression of periodontitis and correlate with disease severity (116). Th17-derived IL-17A is involved in bone destruction in periodontitis. In the presence of IL-17A, fibroblasts and epithelial and endothelial cells of gingival tissue produce numerous inflammatory mediators, e.g., IL-6, CXCL8, IL-1β, granulocyte chemotactic protein, and GM-CSF, promoting neutrophil-mediated inflammation and causing tissue damage and alveolar bone resorption (117–121). In an experimental periodontitis model, i.e., IL-17A receptor-deficient mice infected with Pseudomonas gingivalis, neutrophil recruitment decreased at the site of periodontal lesions; however, bone loss was increased (122).
Th17 cells are present in the tumor microenvironment, and excessive inflammation caused by Th17 cells may be associated with tumorigenesis (90). Notably, IL-17A activates and recruits antitumor immune cells, such as neutrophils, macrophages, natural killer cells, and CD8+ T cells, to exert antitumor effects (123). In breast cancer, Th17 cells promote tumor development by recruiting neutrophils via IL-17A (124).
In necrotic glomerulonephritis, IL-17A deficiency leads to impaired neutrophil recruitment to the glomerulus, which, in turn, downregulates MPO and improves glomerulonephritis (125).
The Th17 cell counts are higher in the decidua than in the peripheral blood and correlate positively with neutrophil counts. Th17 cells induce protective immunity against extracellular microbes through neutrophils (126, 127).
5 Effects of neutrophils on Th17 cells
Numerous studies have documented mechanisms underlying Th17 cell-mediated induction of neutrophil chemotaxis. In recent years, as neutrophils have been studied relatively more intensively, researchers have found that they interact with Th17 cells.
The formation of NETs is a major mechanism underlying neutrophil activation to fight pathogens. DNA and histones constitute most NETs. Histones are either free or bound to DNA. Free histones are relatively more pathogenic and have pro-inflammatory effects (128–130). Large numbers of NETs and histones are present in patients with autoimmune diseases. However, the exact function of these components remains unknown (131). Wilson et al. (132) co-cultured NETs with naïve T cells under Th17-differentiation conditions and found that NETs promoted Th17 differentiation in a concentration-dependent manner. This effect is eliminated upon the application of histone inhibitors. Upregulation of RORγt expression leads to the promotion of Th17 differentiation by NET-derived histones. Specifically, histones activate toll-like receptor 2 in naïve T cells, which phosphorylates STAT3, thereby facilitating Th17 differentiation. The neutrophil antimicrobial peptide cathelicidin has potent antimicrobial activity and immunomodulatory effects and can be released from neutrophils upon degranulation, NET formation, and necrotizing death. Minns et al. (133) reported that cathelicidin derived from neutrophils released by secondary lymphoid organs in mice promotes Th17 cell differentiation in a TGF-β-dependent way by upregulating the expression of AHR and RORγt. Cathelicidin dramatically reduced Tbet expression while promoting STAT3 and Smad2/3 phosphorylation in the presence of TGF-β. Neutrophils and Th17 cells converge at sites of inflammation by producing chemokine C-C motif ligands 20/22, IL-17A, and CXCL8 (134). During severe asthma, neutrophils form nonlytic NETosis, and the remaining cytoplasm can indirectly induce Th17 cell differentiation in the lung (135). Toll-like receptor 8-dependent activated neutrophils can produce the Th17-inducing differentiation factor, IL-23, and their culture supernatant induces naïve T cells to differentiate into Th17 cells (136). In vitro, Th17 differentiation is promoted by neutrophil-derived elastase, which converts dendritic cell-derived CXCL8 into the Th17-promoted form (137). Neutrophils from patients with autoimmune diseases are defined as LDGs (19). This class of neutrophils has pro-inflammatory properties that promote the differentiation and proliferation of Th17 cells (2, 138–140). They are stimulated by IFNs and promote Th17 differentiation by upregulating the levels of costimulatory molecules and major histocompatibility complex II (141). In addition, IL-17A recruits neutrophils to lymph nodes during the Th17 response, providing additional neutrophil-derived IL-1β for Th17 differentiation (91) (Figure 1).
In addition to Th17 cells, neutrophils regulate other T cells. Neutrophils are susceptible to ferroptosis. Ferroptosis-induced oxidation of phosphatidylethanolamine and lecithin inhibits T cell proliferation (26). In sepsis, the production of macrophage-1 antigens by neutrophils inhibits T cell proliferation (142). Neutrophils can disrupt T cell function and differentiation through ROS, programmed cell death-ligand 1, and arginase overexpression to degrade arginine, which is necessary for the activation and proliferation of T cells (143–147). By expressing the suppressor surface protein CD10, neutrophils can interfere with T cell function (148). During lung infection, neutrophils inhibit T cell recruitment and activation and promote the sustained expansion of microbes in the lung (149, 150). During influenza, neutrophils promote T cell function and migration to the sites of infection (151, 152). In vitro-cultured T cells can recognize NET components through T cell receptors, lowering the threshold for T cell activation (153).
6 Conclusions
Neutrophils and Th17 cells interact closely under physiological conditions. A closed loop is formed between the two via the IL-23/IL-17A axis to maintain homeostasis in the immune microenvironment. The disruption of this regulatory loop can lead to a local imbalance in immune homeostasis, resulting in inflammatory or severe pathological reactions. Extensive research has been conducted on the effects of Th17 cells on neutrophils. However, research on how neutrophils intervene with Th17 cells in a complex immune microenvironment is still in the early stages of development. Herein, we summarize how neutrophils affect and regulate Th17 cells. The study of the relationship between neutrophils and Th17 cells has considerable potential for clinical applications because of the complex relationship between the two cells and their synergy in the immune microenvironment. Future immunotherapy may depend on targeting the different biological roles of neutrophils and their effects on T cells to develop new treatments for tumors and chronic inflammatory diseases. Th17 cells are plastic, and their role in tumors is highly dependent on the tumor microenvironment. They are recruited to the sites of malignant tumors. Exploiting the interaction of neutrophils with Th17 cells, inducing the transformation of Th17 cells in specific environments, and enhancing the Th17 antitumor response may be effective strategies for the future development of cancer immunotherapies. Neutropenia, an adverse event, has been reported in a few clinical trials of drugs targeting IL-17A, summarized in this review. Neutropenia may increase the risk of infections in patients. Reducing the incidence of neutropenia as an adverse event while targeting IL-17A and reducing the risk of infections in patients will be the focus of future studies.
Author contributions
XF: Writing – original draft. PS: Writing – original draft. YW: Writing – review & editing. NJ: Writing – review & editing. DZ: Writing – review & editing, Conceptualization, Funding acquisition.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (NO. 82171829), the Key Project of the Science and Technology Department of Sichuan Province (NO. 2022YFH0100, 2022YFS0003), the 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (NO. ZYYC21012), and Fundamental Research Funds for Central Universities (20822041E4084).
Acknowledgments
The figure in this review article was created with BioRender.com. DZ sincerely wants to commemorate Dr. Sang-A Park, who passed away suddenly in an auto accident in Bethesda, MD, USA.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer (2020) 20(9):485–503. doi: 10.1038/s41568-020-0281-y
2. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol (2019) 19(4):255–65. doi: 10.1038/s41577-019-0141-8
3. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11(8):519–31. doi: 10.1038/nri3024
4. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13(3):159–75. doi: 10.1038/nri3399
5. Jaillon S, Galdiero MR, Del Prete D, Cassatella MA, Garlanda C, Mantovani A. Neutrophils in innate and adaptive immunity. Semin Immunopathol (2013) 35(4):377–94. doi: 10.1007/s00281-013-0374-8
6. Scapini P, Cassatella MA. Social networking of human neutrophils within the immune system. Blood (2014) 124(5):710–9. doi: 10.1182/blood-2014-03-453217
7. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol (2018) 18(2):134–47. doi: 10.1038/nri.2017.105
8. Nordenfelt P, Tapper H. Phagosome dynamics during phagocytosis by neutrophils. J Leukoc Biol (2011) 90(2):271–84. doi: 10.1189/jlb.0810457
9. Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. The neutrophil. Immunity (2021) 54(7):1377–91. doi: 10.1016/j.immuni.2021.06.006
10. Hidalgo A, Libby P, Soehnlein O, Aramburu IV, Papayannopoulos V, Silvestre-Roig C. Neutrophil extracellular traps: from physiology to pathology. Cardiovasc Res (2022) 118(13):2737–53. doi: 10.1093/cvr/cvab329
11. Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood (2019) 133(20):2178–85. doi: 10.1182/blood-2018-11-844530
12. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PloS Pathog (2009) 5(10):e1000639. doi: 10.1371/journal.ppat.1000639
13. Bonaventura A, Vecchie A, Abbate A, Montecucco F. Neutrophil extracellular traps and cardiovascular diseases: an update. Cells (2020) 9(1):231. doi: 10.3390/cells9010231
14. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
15. Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol (2007) 5(8):577–82. doi: 10.1038/nrmicro1710
16. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176(2):231–41. doi: 10.1083/jcb.200606027
17. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med (2012) 18(9):1386–93. doi: 10.1038/nm.2847
18. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol (2010) 185(12):7413–25. doi: 10.4049/jimmunol.1000675
19. Silvestre-Roig C, Fridlender ZG, Glogauer M, Scapini P. Neutrophil diversity in health and disease. Trends Immunol (2019) 40(7):565–83. doi: 10.1016/j.it.2019.04.012
20. Montecucco F, Liberale L, Bonaventura A, Vecchie A, Dallegri F, Carbone F. The role of inflammation in cardiovascular outcome. Curr Atheroscler Rep (2017) 19(3):11. doi: 10.1007/s11883-017-0646-1
21. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U.S.A. (2010) 107(36):15880–5. doi: 10.1073/pnas.1005743107
22. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost (2014) 12(12):2074–88. doi: 10.1111/jth.12710
23. Qi X, Yu Y, Sun R, Huang J, Liu L, Yang Y, et al. Identification and characterization of neutrophil heterogeneity in sepsis. Crit Care (2021) 25(1):50. doi: 10.1186/s13054-021-03481-0
24. Zhu CL, Wang Y, Liu Q, Li HR, Yu CM, Li P, et al. Dysregulation of neutrophil death in sepsis. Front Immunol (2022) 13:963955. doi: 10.3389/fimmu.2022.963955
25. Zhang H, Wang Y, Qu M, Li W, Wu D, Cata JP, et al. Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis. Clin Transl Med (2023) 13(1):e1170. doi: 10.1002/ctm2.1170
26. Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature (2022) 612(7939):338–46. doi: 10.1038/s41586-022-05443-0
27. Xiao Y, Cong M, Li J, He D, Wu Q, Tian P, et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell (2021) 39(3):423–37 e7. doi: 10.1016/j.ccell.2020.12.012
28. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature (2020) 583(7814):133–8. doi: 10.1038/s41586-020-2394-6
29. Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity (2020) 52(5):856–71 e8. doi: 10.1016/j.immuni.2020.03.001
30. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6(11):1123–32. doi: 10.1038/ni1254
31. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi: 10.1038/ni1261
32. Liang SC, Long AJ, Bennett F, Whitters MJ, Karim R, Collins M, et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J Immunol (2007) 179(11):7791–9. doi: 10.4049/jimmunol.179.11.7791
33. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature (2007) 445(7128):648–51. doi: 10.1038/nature05505
34. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126(6):1121–33. doi: 10.1016/j.cell.2006.07.035
35. Brustle A, Heink S, Huber M, Rosenplanter C, Stadelmann C, Yu P, et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol (2007) 8(9):958–66. doi: 10.1038/ni1500
36. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature (2008) 453(7191):106–9. doi: 10.1038/nature06881
37. Schraml BU, Hildner K, Ise W, Lee WL, Smith WA, Solomon B, et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature (2009) 460(7253):405–9. doi: 10.1038/nature08114
38. Long D, Chen Y, Wu H, Zhao M, Lu Q. Corrigendum to "Clinical significance and immunobiology of IL-21 in autoimmunity"[J. Autoimmun. 99C (2019) p1-14]. J Autoimmun (2020) 111:102455. doi: 10.1016/j.jaut.2020.102455
39. Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol (2023) 23(1):38–54. doi: 10.1038/s41577-022-00746-9
40. Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature (2007) 448(7152):480–3. doi: 10.1038/nature05969
41. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201(2):233–40. doi: 10.1084/jem.20041257
42. Loos J, Schmaul S, Noll TM, Paterka M, Schillner M, Loffel JT, et al. Functional characteristics of Th1, Th17, and ex-Th17 cells in EAE revealed by intravital two-photon microscopy. J Neuroinflamm (2020) 17(1):357. doi: 10.1186/s12974-020-02021-x
43. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol (2011) 12(3):255–63. doi: 10.1038/ni.1993
44. Haghikia A, Jorg S, Duscha A, Berg J, Manzel A, Waschbisch A, et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity (2015) 43(4):817–29. doi: 10.1016/j.immuni.2015.09.007
45. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature (2019) 576(7785):143–8. doi: 10.1038/s41586-019-1785-z
46. Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature (2015) 523(7559):221–5. doi: 10.1038/nature14452
47. Zhang H, Madi A, Yosef N, Chihara N, Awasthi A, Pot C, et al. An IL-27-driven transcriptional network identifies regulators of IL-10 expression across T helper cell subsets. Cell Rep (2020) 33(8):108433. doi: 10.1016/j.celrep.2020.108433
48. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
49. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
50. Karmaus PWF, Chen X, Lim SA, Herrada AA, Nguyen TM, Xu B, et al. Metabolic heterogeneity underlies reciprocal fates of T(H)17 cell stemness and plasticity. Nature (2019) 565(7737):101–5. doi: 10.1038/s41586-018-0806-7
51. Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis (2004) 190(3):624–31. doi: 10.1086/422329
52. Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med (2009) 206(2):299–311. doi: 10.1084/jem.20081463
53. Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity (2009) 30(1):108–19. doi: 10.1016/j.immuni.2008.11.009
54. Zha X, Yang S, Niu W, Tan L, Xu Y, Zeng J, et al. IL-27/IL-27R mediates protective immunity against chlamydial infection by suppressing excessive th17 responses and reducing neutrophil inflammation. J Immunol (2021) 206(9):2160–9. doi: 10.4049/jimmunol.2000957
55. Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker J. Psoriasis. Lancet (2021) 397(10281):1301–15. doi: 10.1016/S0140-6736(20)32549-6
56. Jiang P, Zheng C, Xiang Y, Malik S, Su D, Xu G, et al. The involvement of TH17 cells in the pathogenesis of IBD. Cytokine Growth Factor Rev (2023) 69:28–42. doi: 10.1016/j.cytogfr.2022.07.005
57. McGinley AM, Edwards SC, Raverdeau M, Mills KHG. Th17 cells, γδ T cells and their interplay in EAE and multiple sclerosis. J Autoimmun (2018) 21:S0896-8411(18)30007-6. doi: 10.1016/j.jaut.2018.01.001
58. Lock C, Hermans G, Pedotti R, Brendolan A, SChadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med (2002) 8(5):500–8. doi: 10.1038/nm0502-500
59. Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol (2008) 172(1):146–55. doi: 10.2353/ajpath.2008.070690
60. Durelli L, Conti L, Clerico M, Boselli D, Contessa G, Ripellino P, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol (2009) 65(5):499–509. doi: 10.1002/ana.21652
61. Liu X, Lee YS, Yu CR, Egwuagu CE. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol (2008) 180(9):6070–6. doi: 10.4049/jimmunol.180.9.6070
62. Alcorn JF, Crowe CR, Kolls JK. TH17 cells in asthma and COPD. Annu Rev Physiol (2010) 72:495–516. doi: 10.1146/annurev-physiol-021909-135926
63. Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discovery (2012) 11(10):763–76. doi: 10.1038/nrd3794
64. Kawano Y, Edwards M, Huang Y, Bilate AM, Araujo LP, Tanoue T, et al. Microbiota imbalance induced by dietary sugar disrupts immune-mediated protection from metabolic syndrome. Cell (2022) 185(19):3501–19 e20. doi: 10.1016/j.cell.2022.08.005
65. Blair HA. Secukinumab: A review in psoriatic arthritis. Drugs (2021) 81(4):483–94. doi: 10.1007/s40265-021-01476-3
66. Garnock-Jones KP. Secukinumab: A review in moderate to severe plaque psoriasis. Am J Clin Dermatol (2015) 16(4):323–30. doi: 10.1007/s40257-015-0143-7
67. Blair HA. Secukinumab: A review in ankylosing spondylitis. Drugs (2019) 79(4):433–43. doi: 10.1007/s40265-019-01075-3
68. Blair HA, Dhillon S. Secukinumab: A review in ankylosing spondylitis. Drugs (2016) 76(10):1023–30. doi: 10.1007/s40265-016-0598-8
69. Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CEM, Papp K, et al. Secukinumab in plaque psoriasis — Results of two phase 3 trials. New Engl J Med (2014) 371(4):326–38. doi: 10.1056/NEJMoa1314258
70. Ly K, Smith MP, Thibodeaux Q, Reddy V, Liao W, Bhutani T. Anti IL-17 in psoriasis. Expert Rev Clin Immunol (2019) 15(11):1185–94. doi: 10.1080/1744666x.2020.1679625
71. Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, et al. Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. New Engl J Med (2015) 373(14):1329–39. doi: 10.1056/NEJMoa1412679
72. McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (2015) 386(9999):1137–46. doi: 10.1016/s0140-6736(15)61134-5
73. Nash P, Mease PJ, McInnes IB, Rahman P, Ritchlin CT, Blanco R, et al. Efficacy and safety of secukinumab administration by autoinjector in patients with psoriatic arthritis: results from a randomized, placebo-controlled trial (FUTURE 3). Arthritis Res Ther (2018) 20(1):47. doi: 10.1186/s13075-018-1551-x
74. Kivitz AJ, Nash P, Tahir H, Everding A, Mann H, Kaszuba A, et al. Efficacy and safety of subcutaneous secukinumab 150 mg with or without loading regimen in psoriatic arthritis: results from the FUTURE 4 study. Rheumatol Ther (2019) 6(3):393–407. doi: 10.1007/s40744-019-0163-5
75. Mease P, van der Heijde D, Landewé R, Mpofu S, Rahman P, Tahir H, et al. Secukinumab improves active psoriatic arthritis symptoms and inhibits radiographic progression: primary results from the randomised, double-blind, phase III FUTURE 5 study. Ann Rheum Dis (2018) 77(6):890–897. doi: 10.1136/annrheumdis-2017-212687
76. Mease PJ, Kavanaugh A, Reimold A, Tahir H, Rech J, Hall S, et al. Secukinumab provides sustained improvements in the signs and symptoms of psoriatic arthritis: final 5-year results from the phase 3 FUTURE 1 study. ACR Open Rheumatol (2019) 2(1):18–25. doi: 10.1002/acr2.11097
77. McInnes IB, Mease PJ, Kivitz AJ, Nash P, Rahman P, Rech J, et al. Long-term efficacy and safety of secukinumab in patients with psoriatic arthritis: 5-year (end-of-study) results from the phase 3 FUTURE 2 study. Lancet Rheumatol (2020) 2(4):e227–e35. doi: 10.1016/s2665-9913(20)30036-9
78. Malakouti M, Brown GE, Wang E, Koo J, Levin EC. The role of IL-17 in psoriasis. J Dermatol Treat (2014) 26(1):41–4. doi: 10.3109/09546634.2013.879093
79. Giunta A, Ventura A, Chimenti MS, Bianchi L, Esposito M. Spotlight on ixekizumab for the treatment of moderate-to-severe plaque psoriasis: design, development, and use in therapy. Drug Design Dev Ther (2017) 11:1643–51. doi: 10.2147/dddt.S92128
80. Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, Edson-Heredia E, et al. Anti–interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. New Engl J Med (2012) 366(13):1190–9. doi: 10.1056/NEJMoa1109997
81. Ryan C, Menter A, Guenther L, Blauvelt A, Bissonnette R, Meeuwis K, et al. Efficacy and safety of ixekizumab in a randomized, double-blinded, placebo-controlled phase IIIb study of patients with moderate-to-severe genital psoriasis. Br J Dermatol (2018) 179(4):844–52. doi: 10.1111/bjd.16736
82. Wilsmann-Theis D, Schnell LM, Ralser-Isselstein V, Bieber T, Schön MP, Hüffmeier U, et al. Successful treatment with interleukin-17A antagonists of generalized pustular psoriasis in patients without IL36RN mutations. J Dermatol (2018) 45(7):850–4. doi: 10.1111/1346-8138.14318
83. Mease PJ, van der Heijde D, Ritchlin CT, Okada M, Cuchacovich RS, Shuler CL, et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis (2017) 76(1):79–87. doi: 10.1136/annrheumdis-2016-209709
84. Galluzzo M, D'Adamio S, Massaro A, Piccolo A, Bianchi L, Talamonti M. Spotlight on brodalumab in the treatment of plaque psoriasis: the evidence to date. Clinical Cosmetic Investigation Dermatol (2019) 12:311–21. doi: 10.2147/ccid.S165605
85. Papp KA, Reich K, Paul C, Blauvelt A, Baran W, Bolduc C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol (2016) 175(2):273–86. doi: 10.1111/bjd.14493
86. Lebwohl M, Strober B, Menter A, Gordon K, Weglowska J, Puig L, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. New Engl J Med (2015) 373(14):1318–28. doi: 10.1056/NEJMoa1503824
87. Mease PJ, Genovese MC, Greenwald MW, Ritchlin CT, Beaulieu AD, Deodhar A, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. New Engl J Med (2014) 370(24):2295–306. doi: 10.1056/NEJMoa1315231
88. Reich K, Papp KA, Blauvelt A, Langley RG, Armstrong A, Warren RB, et al. Bimekizumab versus ustekinumab for the treatment of moderate to severe plaque psoriasis (BE VIVID): efficacy and safety from a 52-week, multicentre, double-blind, active comparator and placebo controlled phase 3 trial. Lancet (2021) 397(10273):487–98. doi: 10.1016/s0140-6736(21)00125-2
89. McInnes IB, Asahina A, Coates LC, Landewé R, Merola JF, Ritchlin CT, et al. Bimekizumab in patients with psoriatic arthritis, naive to biologic treatment: a randomised, double-blind, placebo-controlled, phase 3 trial (BE OPTIMAL). Lancet (2023) 401(10370):25–37. doi: 10.1016/s0140-6736(22)02302-9
90. Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol (2018) 15(5):458–69. doi: 10.1038/s41423-018-0004-4
91. Witowski J, Ksiązek K, Warnecke C, Kuźlan M, Korybalska K, Tayama H, et al. Role of mesothelial cell-derived granulocyte colony-stimulating factor in interleukin-17-induced neutrophil accumulation in the peritoneum. Kidney Int (2007) 71(6):514–25. doi: 10.1038/sj.ki.5002082
92. de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol (2016) 16(6):378–91. doi: 10.1038/nri.2016.49
93. Wu L, Awaji M, Saxena S, Varney ML, Sharma B, Singh RK. IL-17–CXC chemokine receptor 2 axis facilitates breast cancer progression by up-regulating neutrophil recruitment. Am J Pathol (2020) 190(1):222–33. doi: 10.1016/j.ajpath.2019.09.016
94. Liew PX, Kubes P. The neutrophil's role during health and disease. Physiol Rev (2019) 99(2):1223–48. doi: 10.1152/physrev.00012.2018
95. Lord BI, Bronchud MH, Owens S, Chang J, Howell A, Souza L, et al. The kinetics of human granulopoiesis following treatment with granulocyte colony-stimulating factor in vivo. Proc Natl Acad Sci (1989) 86(23):9499–503. doi: 10.1073/pnas.86.23.9499
96. Ley K, Smith E, Stark MA. Neutrophil-regulatory tn lymphocytes. Immunol Res (2006) 34(3):229–42. doi: 10.1385/ir:34:3:229
97. Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity (2005) 22(3):285–94. doi: 10.1016/j.immuni.2005.01.011
98. Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology (2008) 125(3):281–8. doi: 10.1111/j.1365-2567.2008.02950.x
99. Blauvelt A, Chiricozzi A. The immunologic role of IL-17 in psoriasis and psoriatic arthritis pathogenesis. Clin Rev Allergy Immunol (2018) 55(3):379–90. doi: 10.1007/s12016-018-8702-3
100. Katayama H. Development of psoriasis by continuous neutrophil infiltration into the epidermis. Exp Dermatol (2018) 27(10):1084–91. doi: 10.1111/exd.13746
101. Reich K, Papp KA, Matheson RT, Tu JH, Bissonnette R, Bourcier M, et al. Evidence that a neutrophil-keratinocyte crosstalk is an early target of IL-17A inhibition in psoriasis. Exp Dermatol (2015) 24(7):529–35. doi: 10.1111/exd.12710
102. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol (2017) 140(3):645–53. doi: 10.1016/j.jaci.2017.07.004
103. Henry CM, Sullivan GP, Clancy DM, Afonina IS, Kulms D, Martin SJ. Neutrophil-derived proteases escalate inflammation through activation of IL-36 family cytokines. Cell Rep (2016) 14(4):708–22. doi: 10.1016/j.celrep.2015.12.072
104. Tortola L, Rosenwald E, Abel B, Blumberg H, Schafer M, Coyle AJ, et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest (2012) 122(11):3965–76. doi: 10.1172/JCI63451
105. Dyring-Andersen B, Honore TV, Madelung A, Bzorek M, Simonsen S, Clemmensen SN, et al. Interleukin (IL)-17A and IL-22-producing neutrophils in psoriatic skin. Br J Dermatol (2017) 177(6):e321–e2. doi: 10.1111/bjd.15533
106. Singh TP, Zhang HH, Borek I, Wolf P, Hedrick MN, Singh SP, et al. Monocyte-derived inflammatory Langerhans cells and dermal dendritic cells mediate psoriasis-like inflammation. Nat Commun (2016) 7:13581 doi: 10.1038/ncomms13581
107. Papi A, Brightling C, Pedersen SE, Reddel HK. Asthma. Lancet (2018) 391(10122):783–800. doi: 10.1016/S0140-6736(17)33311-1
108. Ricciardolo FLM, Sorbello V, Folino A, Gallo F, Massaglia GM, Favata G, et al. Identification of IL-17F/frequent exacerbator endotype in asthma. J Allergy Clin Immunol (2017) 140(2):395–406. doi: 10.1016/j.jaci.2016.10.034
109. Roussel L, Houle F, Chan C, Yao Y, Berube J, Olivenstein R, et al. IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J Immunol (2010) 184(8):4531–7. doi: 10.4049/jimmunol.0903162
110. Bush A, Saglani S. Management of severe asthma in children. Lancet (2010) 376(9743):814–25. doi: 10.1016/S0140-6736(10)61054-9
111. Wadhwa R, Dua K, Adcock IM, Horvat JC, Kim RY, Hansbro PM. Cellular mechanisms underlying steroid-resistant asthma. Eur Respir Rev (2019) 28(153):190096. doi: 10.1183/16000617.0096-2019
112. Chesne J, Braza F, Mahay G, Brouard S, Aronica M, Magnan A. IL-17 in severe asthma. Where do we stand? Am J Respir Crit Care Med (2014) 190(10):1094–101. doi: 10.1164/rccm.201405-0859PP
113. Suzuki Y, Maazi H, Sankaranarayanan I, Lam J, Khoo B, Soroosh P, et al. Lack of autophagy induces steroid-resistant airway inflammation. J Allergy Clin Immunol (2016) 137(5):1382–9 e9. doi: 10.1016/j.jaci.2015.09.033
114. Ramakrishnan RK, Al Heialy S, Hamid Q. Role of IL-17 in asthma pathogenesis and its implications for the clinic. Expert Rev Respir Med (2019) 13(11):1057–68. doi: 10.1080/17476348.2019.1666002
115. Ano S, Morishima Y, Ishii Y, Yoh K, Yageta Y, Ohtsuka S, et al. Transcription factors GATA-3 and RORgammat are important for determining the phenotype of allergic airway inflammation in a murine model of asthma. J Immunol (2013) 190(3):1056–65. doi: 10.4049/jimmunol.1202386
116. Cheng WC, Hughes FJ, Taams LS. The presence, function and regulation of IL-17 and Th17 cells in periodontitis. J Clin Periodontol (2014) 41(6):541–9. doi: 10.1111/jcpe.12238
117. Hosokawa Y, Hosokawa I, Ozaki K, Nakanishi T, Nakae H, Matsuo T. Catechins inhibit CCL20 production in IL-17A-stimulated human gingival fibroblasts. Cell Physiol Biochem (2009) 24(5-6):391–6. doi: 10.1159/000257431
118. Konermann A, Beyer M, Deschner J, Allam JP, Novak N, Winter J, et al. Human periodontal ligament cells facilitate leukocyte recruitment and are influenced in their immunomodulatory function by Th17 cytokine release. Cell Immunol (2012) 272(2):137–43. doi: 10.1016/j.cellimm.2011.10.020
119. Takahashi N, Okui T, Tabeta K, Yamazaki K. Effect of interleukin-17 on the expression of chemokines in gingival epithelial cells. Eur J Oral Sci (2011) 119(5):339–44. doi: 10.1111/j.1600-0722.2011.00842.x
120. Takahashi K, Azuma T, Motohira H, Kinane DF, Kitetsu S. The potential role of interleukin-17 in the immunopathology of periodontal disease. J Clin Periodontol (2005) 32(4):369–74. doi: 10.1111/j.1600-051X.2005.00676.x
121. Tipton DA, Cho S, Zacharia N, Dabbous MK. Inhibition of interleukin-17-stimulated interleukin-6 and -8 production by cranberry components in human gingival fibroblasts and epithelial cells. J Periodontal Res (2013) 48(5):638–46. doi: 10.1111/jre.12050
122. Yu JJ, Ruddy MJ, Wong GC, Sfintescu C, Baker PJ, Smith JB, et al. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood (2007) 109(9):3794–802. doi: 10.1182/blood-2005-09-010116
123. Renaude E, Kroemer M, Loyon R, Binda D, Borg C, Guittaut M, et al. The fate of th17 cells is shaped by epigenetic modifications and remodeled by the tumor microenvironment. Int J Mol Sci (2020) 21(5):1653. doi: 10.3390/ijms21051673
124. Benevides L, da Fonseca DM, Donate PB, Tiezzi DG, De Carvalho DD, de Andrade JM, et al. IL17 promotes mammary tumor progression by changing the behavior of tumor cells and eliciting tumorigenic neutrophils recruitment. Cancer Res (2015) 75(18):3788–99. doi: 10.1158/0008-5472.CAN-15-0054
125. Gan PY, Steinmetz OM, Tan DS, O'Sullivan KM, Ooi JD, Iwakura Y, et al. Th17 cells promote autoimmune anti-myeloperoxidase glomerulonephritis. J Am Soc Nephrol (2010) 21(6):925–31. doi: 10.1681/ASN.2009070763
126. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol (2010) 10(7):479–89. doi: 10.1038/nri2800
127. Wang W, Sung N, Gilman-Sachs A, Kwak-Kim J. T helper (Th) cell profiles in pregnancy and recurrent pregnancy losses: th1/th2/th9/th17/th22/tfh cells. Front Immunol (2020) 11:2025. doi: 10.3389/fimmu.2020.02025
128. Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med (2013) 187(2):160–9. doi: 10.1164/rccm.201206-1037OC
129. Kutcher ME, Xu J, Vilardi RF, Ho C, Esmon CT, Cohen MJ. Extracellular histone release in response to traumatic injury: implications for a compensatory role of activated protein C. J Trauma Acute Care Surg (2012) 73(6):1389–94. doi: 10.1097/TA.0b013e318270d595
130. Tsourouktsoglou TD, Warnatsch A, Ioannou M, Hoving D, Wang Q, Papayannopoulos V. Histones, DNA, and citrullination promote neutrophil extracellular trap inflammation by regulating the localization and activation of TLR4. Cell Rep (2020) 31(5):107602. doi: 10.1016/j.celrep.2020.107602
131. Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol (2013) 33(8):2032–40. doi: 10.1161/ATVBAHA.113.301627
132. Wilson AS, Randall KL, Pettitt JA, Ellyard JI, Blumenthal A, Enders A, et al. Neutrophil extracellular traps and their histones promote Th17 cell differentiation directly via TLR2. Nat Commun (2022) 13(1):528. doi: 10.1038/s41467-022-28172-4
133. Minns D, Smith KJ, Alessandrini V, Hardisty G, Melrose L, Jackson-Jones L, et al. The neutrophil antimicrobial peptide cathelicidin promotes Th17 differentiation. Nat Commun (2021) 12(1):1285. doi: 10.1038/s41467-021-21533-5
134. Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, Costantini C, et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood (2010) 115(2):335–43. doi: 10.1182/blood-2009-04-216085
135. Krishnamoorthy N, Douda DN, Bruggemann TR, Ricklefs I, Duvall MG, Abdulnour RE, et al. Neutrophil cytoplasts induce T(H)17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci Immunol (2018) 3(26):eaao4747. doi: 10.1126/sciimmunol.aao4747
136. Tamassia N, Arruda-Silva F, Wright HL, Moots RJ, Gardiman E, Bianchetto-Aguilera F, et al. Human neutrophils activated via TLR8 promote Th17 polarization through IL-23. J Leukoc Biol (2019) 105(6):1155–65. doi: 10.1002/JLB.MA0818-308R
137. Souwer Y, Groot Kormelink T, Taanman-Kueter EW, Muller FJ, van Capel TMM, Varga DV, et al. Human T(H)17 cell development requires processing of dendritic cell-derived CXCL8 by neutrophil elastase. J Allergy Clin Immunol (2018) 141(6):2286–9 e5. doi: 10.1016/j.jaci.2018.01.003
138. Wu H, Zhen Y, Ma Z, Li H, Yu J, Xu ZG, et al. Arginase-1-dependent promotion of TH17 differentiation and disease progression by MDSCs in systemic lupus erythematosus. Sci Transl Med (2016) 8(331):331ra40. doi: 10.1126/scitranslmed.aae0482
139. Carmona-Rivera C, Kaplan MJ. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin Immunopathol (2013) 35(4):455–63. doi: 10.1007/s00281-013-0375-7
140. Chatfield SM, Thieblemont N, Witko-Sarsat V. Expanding neutrophil horizons: new concepts in inflammation. J Innate Immun (2018) 10(5-6):422–31. doi: 10.1159/000493101
141. Abi Abdallah DS, Egan CE, Butcher BA, Denkers EY. Mouse neutrophils are professional antigen-presenting cells programmed to instruct Th1 and Th17 T-cell differentiation. Int Immunol (2011) 23(5):317–26. doi: 10.1093/intimm/dxr007
142. Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest (2012) 122(1):327–36. doi: 10.1172/JCI57990
143. Aarts CEM, Hiemstra IH, Beguin EP, Hoogendijk AJ, Bouchmal S, van Houdt M, et al. Activated neutrophils exert myeloid-derived suppressor cell activity damaging T cells beyond repair. Blood Adv (2019) 3(22):3562–74. doi: 10.1182/bloodadvances.2019031609
144. Langereis JD, Pickkers P, de Kleijn S, Gerretsen J, de Jonge MI, Kox M. Spleen-derived IFN-gamma induces generation of PD-L1(+)-suppressive neutrophils during endotoxemia. J Leukoc Biol (2017) 102(6):1401–9. doi: 10.1189/jlb.3A0217-051RR
145. Feldmeyer N, Wabnitz G, Leicht S, Luckner-Minden C, Schiller M, Franz T, et al. Arginine deficiency leads to impaired cofilin dephosphorylation in activated human T lymphocytes. Int Immunol (2012) 24(5):303–13. doi: 10.1093/intimm/dxs004
146. Yachimovich-Cohen N, Even-Ram S, Shufaro Y, Rachmilewitz J, Reubinoff B. Human embryonic stem cells suppress T cell responses via arginase I-dependent mechanism. J Immunol (2010) 184(3):1300–8. doi: 10.4049/jimmunol.0804261
147. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109(4):1568–73. doi: 10.1182/blood-2006-06-031856
148. Marini O, Costa S, Bevilacqua D, Calzetti F, Tamassia N, Spina C, et al. Mature CD10(+) and immature CD10(-) neutrophils present in G-CSF-treated donors display opposite effects on T cells. Blood (2017) 129(10):1343–56. doi: 10.1182/blood-2016-04-713206
149. Ingersoll SA, Laval J, Forrest OA, Preininger M, Brown MR, Arafat D, et al. Mature cystic fibrosis airway neutrophils suppress T cell function: evidence for a role of arginase 1 but not programmed death-ligand 1. J Immunol (2015) 194(11):5520–8. doi: 10.4049/jimmunol.1500312
150. Kragh KN, Alhede M, Jensen PO, Moser C, Scheike T, Jacobsen CS, et al. Polymorphonuclear leukocytes restrict growth of Pseudomonas aeruginosa in the lungs of cystic fibrosis patients. Infect Immun (2014) 82(11):4477–86. doi: 10.1128/IAI.01969-14
151. Hufford MM, Richardson G, Zhou H, Manicassamy B, Garcia-Sastre A, Enelow RI, et al. Influenza-infected neutrophils within the infected lungs act as antigen presenting cells for anti-viral CD8(+) T cells. PloS One (2012) 7(10):e46581. doi: 10.1371/journal.pone.0046581
152. Lim K, Hyun YM, Lambert-Emo K, Capece T, Bae S, Miller R, et al. Neutrophil trails guide influenza-specific CD8(+) T cells in the airways. Science (2015) 349(6252):aaa4352. doi: 10.1126/science.aaa4352
Keywords: neutrophils, T-helper 17 cells, autoimmune diseases, interleukin-17, neutrophil extracellular traps (NETs)
Citation: Fan X, Shu P, Wang Y, Ji N and Zhang D (2023) Interactions between neutrophils and T-helper 17 cells. Front. Immunol. 14:1279837. doi: 10.3389/fimmu.2023.1279837
Received: 18 August 2023; Accepted: 05 October 2023;
Published: 18 October 2023.
Edited by:
Peng Deng, UCLA School of Dentistry, United StatesReviewed by:
Bin Zhao, Central South University, ChinaJin Koo Kim, University of California, Los Angeles, United States
Luciana Cavalheiro Marti, Albert Einstein Israelite Hospital, Brazil
Copyright © 2023 Fan, Shu, Wang, Ji and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ning Ji, amluaW5nXzEwMjNAMTI2LmNvbQ==; Dunfang Zhang, aXpkZkAxNjMuY29t
†These authors have contributed equally to this work