 Zhidan Luo
Zhidan Luo Yihua Zhang3†
Yihua Zhang3† Qais Waleed Saleh
Qais Waleed Saleh Zhiming Zhu
Zhiming Zhu Martin Tepel
Martin Tepel- 1Department of Geriatrics, Chongqing General Hospital, Chongqing, China
- 2Cardiovascular and Renal Research, Department of Molecular Medicine, University of Southern Denmark, Odense, Denmark
- 3Department of Cardiology, Chongqing Fifth People’s Hospital, Chongqing, China
- 4Department of Nephrology, Odense University Hospital, Odense, Denmark
- 5Department of Hypertension and Endocrinology, Daping Hospital, Chongqing, China
Forkhead Box P3 (FOXP3) is crucial for the development and suppressive function of human regulatory T cells (Tregs). There are two predominant FOXP3 splicing isoforms in healthy humans, the full-length isoform and the isoform lacking exon 2, with different functions and regulation mechanisms. FOXP3 splicing isoforms show distinct abilities in the cofactor interaction and the nuclear translocation, resulting in different effects on the differentiation, cytokine secretion, suppressive function, linage stability, and environmental adaptation of Tregs. The balance of FOXP3 splicing isoforms is related to autoimmune diseases, inflammatory diseases, and cancers. In response to environmental challenges, FOXP3 transcription and splicing can be finely regulated by T cell antigen receptor stimulation, glycolysis, fatty acid oxidation, and reactive oxygen species, with various signaling pathways involved. Strategies targeting energy metabolism and FOXP3 splicing isoforms in Tregs may provide potential new approaches for the treatment of autoimmune diseases, inflammatory diseases, and cancers. In this review, we summarize recent discoveries about the FOXP3 splicing isoforms and address the metabolic regulation and specific functions of FOXP3 splicing isoforms in Tregs.
1 Introduction
CD4+CD25+ regulatory T cells (Tregs) are a subset of T cells that mediate the immune response against antigens and inhibit conventional T-cell activation and proliferation (1, 2). Tregs generally suppress the excessive immune response and maintain immune homeostasis. In contrast, the deficiency or deactivation of Tregs leads to autoimmune diseases or allograft rejection (1, 2).
The phenotype and function of Tregs critically depend on the expression of their lineage-defining master transcription factor Forkhead Box P3 (FOXP3) (3). Unlike in mice, alternative splicing of mRNA leads to 4 different FOXP3 protein isoforms in human Tregs (4). The full-length isoform (FOXP3FL) and a shorter isoform produced by transcripts lacking exon 2 (FOXP3ΔE2) are the predominant isoforms in healthy human natural Tregs (1) and they are the primary focus of this review. Both isoforms have previously been shown to induce differentiation of CD4+ T cells to Treg phenotypes, and their relative expression varies in some autoimmune diseases, inflammatory diseases, and cancers (5, 6). The other 2 isoforms, FOXP3 with exon 7 skipping (FOXP3ΔE7) and FOXP3 missing both exon 2 and exon 7 (FOXP3ΔE2ΔE7) have been reported with very low frequency in human natural Tregs (1, 7–11). The isoforms of FOXP3 exhibit differences in functions (12–14) and regulation mechanisms (1, 15, 16).
Metabolic reprogramming in immune cells is essential for their proper function, leading to the process called immunometabolism (17). Tregs can adapt quickly to both intrinsic and extrinsic microenvironments by metabolic reprogramming to maintain their activity (18). FOXP3 seems to be a critical link between the energy metabolism and functions of Tregs. FOXP3 can regulate energy metabolism and control the differentiation, stability, and suppressive function of Tregs (19, 20). Modulation of metabolic pathways can also affect FOXP3 transcription and splicing (16, 17), which may be important for controlling the function of Tregs both in healthy and diseased subjects.
In this review, we summarize recent discoveries about the FOXP3 splicing isoforms, especially about FOXP3FL and FOXP3ΔE2, and address the specific functions and metabolic regulation of these isoforms in Tregs.
2 Functions and balance of FOXP3 isoforms
2.1 FOXP3 gene and protein structures
FOXP3 gene is located at Xp.11.23 on the X chromosome and contains 12 exons. The first exon is noncoding while the remaining 11 are coding exons (1, 8). Although some articles name all exons, including the non-coding exon as E1, most articles refer to the coding exons as E1-11 and name the first exon as the non-coding exon, which is also the way FOXP3 exons are referred to hereafter in this article. The full-length human FOXP3 protein has 431 amino acids with a molecular weight of about 47.25 kDa (1, 8). FOXP3 has 4 main domains with different functions (Figure 1). From the N terminus, it contains a repressor domain, a zinc finger, a leucine zipper motif, and finally a forkhead DNA-binding domain (1, 8).
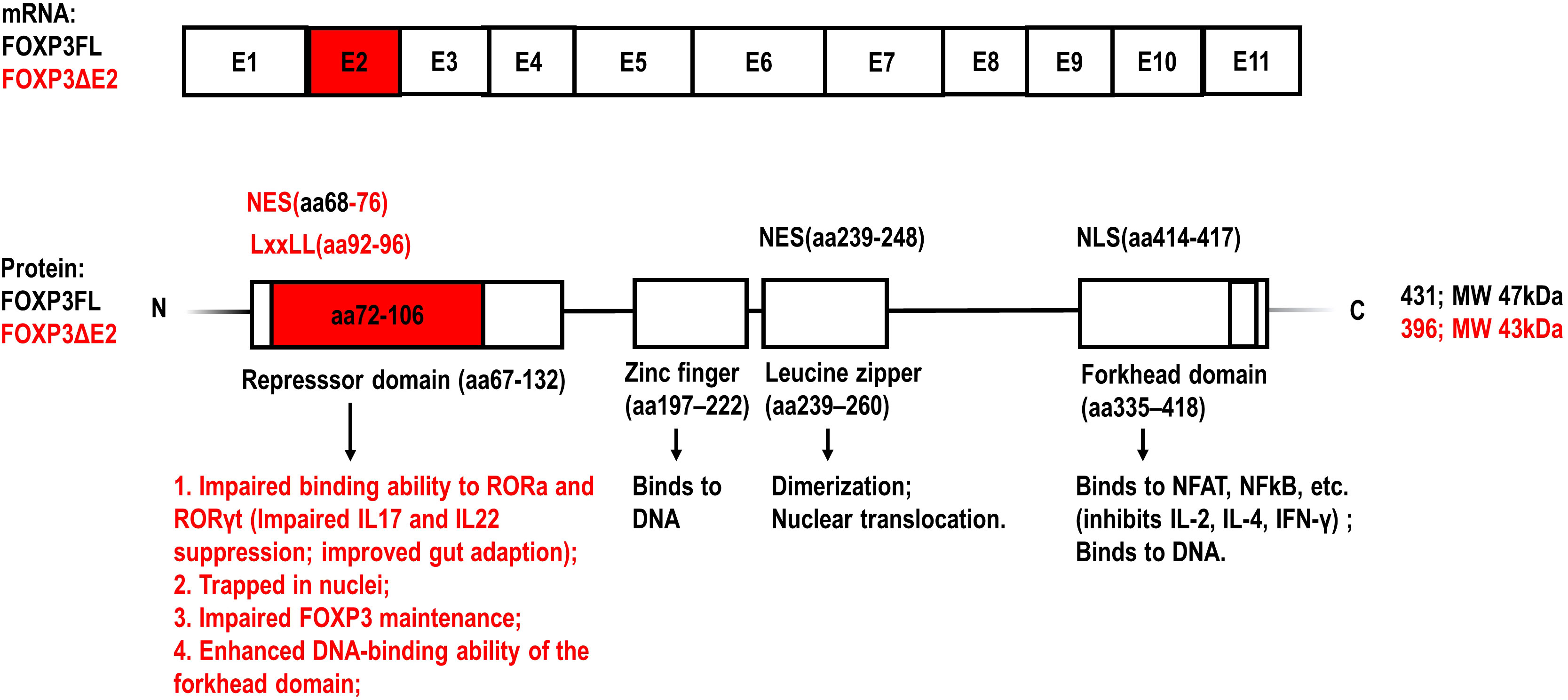
Figure 1 Schematic representation of the exons of FOXP3 mRNA and the structural domains of FOXP3 protein (1, 11, 21). The amino acid sequences encoded by FOXP3 exon 2 and the specific functions of FOXP3 isoform lacking exon2 (FOXP3ΔE2) are highlighted with red color, compared to the full-length FOXP3 isoform (FOXP3FL). NES, nuclear export signal; NLS, nuclear localization signal; LxxLL (where L is leucine and x is any amino acid) motif: a multifunctional binding sequence in transcriptional regulation.
The proline-rich repressor domain exerts a suppressive effect on target genes (22, 23). The function of the zinc-finger domain has not been elucidated clearly yet (22, 23). The leucine zipper motif enables FOXP3 dimerization, which is required for binding to the GTAAACA motif via the C-terminal forkhead domain (22, 23). The forkhead DNA-binding domain regulates the transcription of approximately 700 genes involved in a wide spectrum of inflammatory and immune responses (1). AlphaFold2 predictions and in vitro experiments demonstrate that the N-terminal domain including exon 2 of FOXP3 inhibits the DNA binding ability of the forkhead domain, which may serve as an auto-inhibitory feedback mechanism (22, 23).
Dysfunctional FOXP3 protein due to mutations in the FOXP3 gene results in the development of severe autoimmune disorders as can be observed in the “scurfy” mouse mutant and patients suffering from immune dysregulation, polyendocrinopathy, enteropathy, and X-linked syndrome (IPEX) (24, 25). Most IPEX patients die within the first 2 years of life due to severe systemic autoimmune impairments. The most common FOXP3 mutations in IPEX patients are in the FKH domain, followed by the leucine zipper domain and the repressor domain (1, 24, 26). It was reported that the IPEX patient with mutated FOXP3FL but intact FOXP3ΔE2 protein, had mild autoimmunity and impaired Treg function, although the proportion of Tregs was shown even higher than that in healthy people (22).
2.2 Different functions of FOXP3FL and FOXP3ΔE2 isoforms
Earlier in vitro studies suggested that the FOXP3FL and FOXP3ΔE2 isoforms might show similar effects on mediating Treg differentiation and function, as ectopic expression of each isoform successfully induced Treg phenotypes from CD4+ T cells (27, 28). However, recent studies indicate that FOXP3 splicing isoforms show distinct abilities in the cofactor interaction, the nuclear translocation, and the DNA-binding ability, resulting in different effects on the differentiation, cytokine secretion, suppressive function, linage stability, and environmental adaptation of Tregs (12–15, 29–31).
FOXP3 regulates Tregs through complex mechanisms, including interacting with other transcription factors to form large transcription factor complexes (21). The loss of FOXP3 exon 2 can not only impair the repressor domain function but also enhance the DNA-binding ability of the forkhead domain (22, 23), leading to different functions of FOXP3FL and FOXP3ΔE2 isoforms.
Retinoic acid receptor-related orphan receptor (ROR) α and RORγ are transcription factors belonging to the ROR family. They are expressed in many cell types including human CD4+ T cells and have an overlapping role in Th17-prone cell differentiation through the regulation of genes including IL17 and IL-22 (32, 33). Du et al. have shown that FOXP3 interacts with the activation function 2 (AF2) motif of RORα via an LxxLL motif (encoding aa 92-96 in the repressor domain) in exon 2 (Figure 1). FOXP3FL, but not FOXP3ΔE2, interacts with RORα and inhibits RORα-mediated transcriptional activation. Consequently, the mRNA expression of IL-17 and IL-22 is dramatically suppressed in Jurkat T cells expressing FOXP3FL (33).
Exon 2 of the FOXP3 gene also includes a nuclear export sequence (NES) encoding aa 68-76 in the repressor domain of the FOXP3 protein (Figure 1). Magg et al. showed that site-directed mutagenesis of NES located in exon 2 completely abolished the nuclear export of FOXP3 in human T cells (30). Hence, the transport of FOXP3ΔE2 into the cytoplasm was much slower than FOXP3FL after cellular activation and it was more likely that FOXP3ΔE2 was trapped in the nucleus, resulting in increased expression of CD25 and C-C motif chemokine receptor 4 (CCR4), reduced expression of IL-2 and IL-4, and enhanced suppressive function of FOXP3ΔE2 in vitro (30). Yet it needs to be noted that, although the increased nuclear localization of FOXP3 is usually related to enhanced transcriptional activity, the interaction with cofactors in plasma may also be limited, possibly leading to more complex results in certain conditions.
Sato et al. used human CD4+ T cells in which the endogenous FOXP3 gene was disrupted, followed by lentivirus-mediated transfer of FOXP3FL and FOXP3ΔE2 to dissect the role of each isoform and their combination (32) (Table 1). They found that co-expression of FOXP3FL and FOXP3ΔE2 induced higher FOXP3 protein expression when compared to the transfer of FOXP3FL alone or FOXP3ΔE2 alone (31) (Table 1). The transfer of FOXP3FL alone, FOXP3ΔE2 alone, and simultaneous expression of both isoforms led to comparable reductions of cytokines interleukin (IL)-2, IL-4, and Interferon-γ. In contrast, the co-expression of FOXP3FL and FOXP3ΔE2 caused a more pronounced reduction of IL-22 and IL-17A secretion compared to each isoform alone (31). Moreover, glycoprotein A repetitions predominant (GARP), which attaches the immunosuppressive cytokine transforming growth factor-beta (TGF-β) to the cell membrane, was mainly associated with the expression of the FOXP3ΔE2 isoform (31) (Table 1). Since these findings cannot be fully explained by the FOXP3 isoform-specific ROR interaction and nuclear translocation, there may be more complex mechanisms underlying the effects of FOXP3 isoforms on cytokine expression. An interaction between FOXP3FL and FOXP3ΔE2 may occur, contributing to the optimal FOXP3 protein expression and Treg phenotype. In addition, the different methods used in these studies may interfere with the comparative analysis. The supernatant cytokines detected by ELISA in the study of Sato et al. can differ from the intracellular mRNA expression in the studies of Du et al. and Magg et al. Post-transcriptional modifications should be taken into consideration.
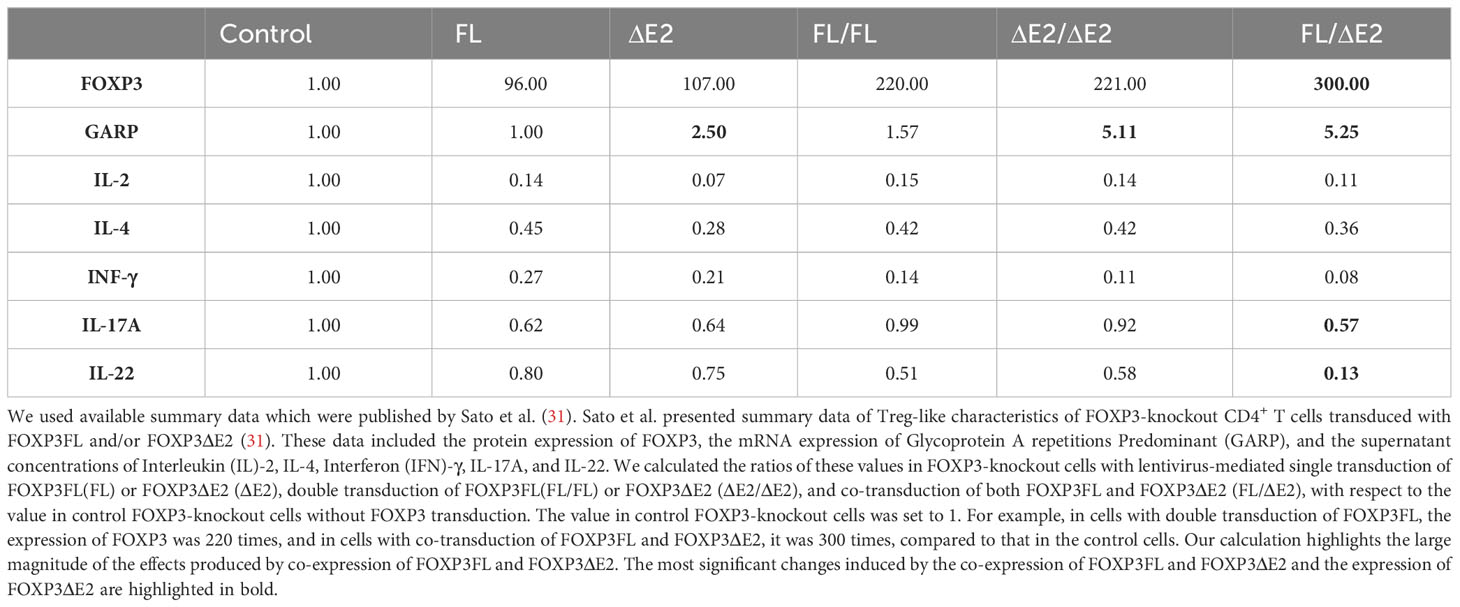
Table 1 Effects of FOXP3FL, FOXP3ΔE2, and the co-expression of both isoforms on the total expression of FOXP3 and GARP and the secretion of critical cytokines in human FOXP3-knockout CD4+ T cells.
Recent studies strongly indicate that FOXP3 exon 2 controls Treg stability and autoimmunity. There are very few cells that exclusively express the FOXP3ΔE2 isoform in the intestine-resident CD4+ cells of healthy humans (29). In contrast, in various malignant tumor tissues, the expression of FOXP3ΔE2 far exceeds that of FOXP3FL (8) and FOXP3FL is even undetectable in some cancer cells (6, 34). It suggests that the FOXP3FL isoform may be mandatory for normal Tregs. Recently, Du et al. reported that deletion of Foxp3 exon 2 in mice did not impact thymocyte development, but resulted in systemic autoimmune disease (12). Although FOXP3ΔE2 Tregs have comparable suppressive ability to FOXP3 FL in vitro, FOXP3ΔE2 Tregs in vivo exhibited intrinsic defects in the expression of phenotypic molecules including CD25, FOXP3, and Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Purified FOXP3ΔE2 Tregs lost FOXP3 expression and were sufficient to induce systemic autoimmunity after being transduced to genetic immune-deficient mice (12). The crucial role of FOXP3FL in maintaining FOXP3 expression and mediating Treg stability and immune homeostasis is further elucidated with findings by Seitz et al. (14). They analyzed subsets of Tregs from two IPEX patients and a healthy carrier and found that FOXP3FL controlled a distinct genetic program, involving the identified FOXP3 regulators DNA-binding inhibitor 3 (DI3), B-cell lymphoma 6 (BCL6), and eukaryotic translation initiation factor 4E (eIF4E) (14). These FOXP3 regulators uphold Treg cell lineage stability, while they appear nonessential for Treg cell activation (35–37).
Moreover, FOXP3FL and FOXP3ΔE2 differ in the environmental adaptation of Tregs (22), which plays a fundamental role in modulating the local gut environment, aiding mucosal tolerance, and enforcing commensalism (38, 39). Tregs can be divided into two major subsets based on their expression of additional transcriptive factors. The first subset expresses RORγ induced by commensal bacteria, which is also a key regulator for Th17 cells (38, 40). The second subset expresses Helios, which is often considered a marker for natural Tregs generated from the thymus (38, 40). Recently, Gu et al. generated Foxp3 exon 2-deficient mice by CRISPR-Cas9-based genome editing. They found that FOXP3ΔE2-bearing natural Tregs in the peripheral lymphoid organ were less sensitive to T cell antigen receptor (TCR) stimulation, due to the enhanced binding of FOXP3ΔE2 to the basic leucine zipper ATF-like transcription factor (BATF) promoter (22). In contrast, among peripherally induced RORγ+ Tregs in the colon, FOXP3ΔE2-expressing Tregs exhibited enhanced immune suppressive function over the wildtype FOXP3FL-expressing Tregs, due to the impaired FOXP3-RORγ interaction and the enhanced DNA-binding ability of the forkhead domain (22). It indicates that FOXP3ΔE2 has distinct effects on natural Tregs and peripherally induced Tregs and is beneficial for the adaptation of Tregs to the gut environment.
2.3 Changes of FOXP3FL and FOXP3ΔE2 isoforms in autoimmune and inflammatory diseases
Alternative splicing is a crucial post-transcriptional mechanism that enables reprogramming of gene expression profiles and the expansion of transcriptomic and proteomic diversity in eukaryotic organisms (41). Human FOXP3 alternative splicing exists in both physiological and pathophysiological states (8, 42). FOXP3FL and FOXP3ΔE2 are usually co-expressed in healthy human lymphocytes, but their ratio may change according to the transcriptional environment (1). Several studies have investigated the changes of FOXP3 isoforms in human autoimmune diseases and inflammatory diseases with somewhat inconsistent findings (Table 2).
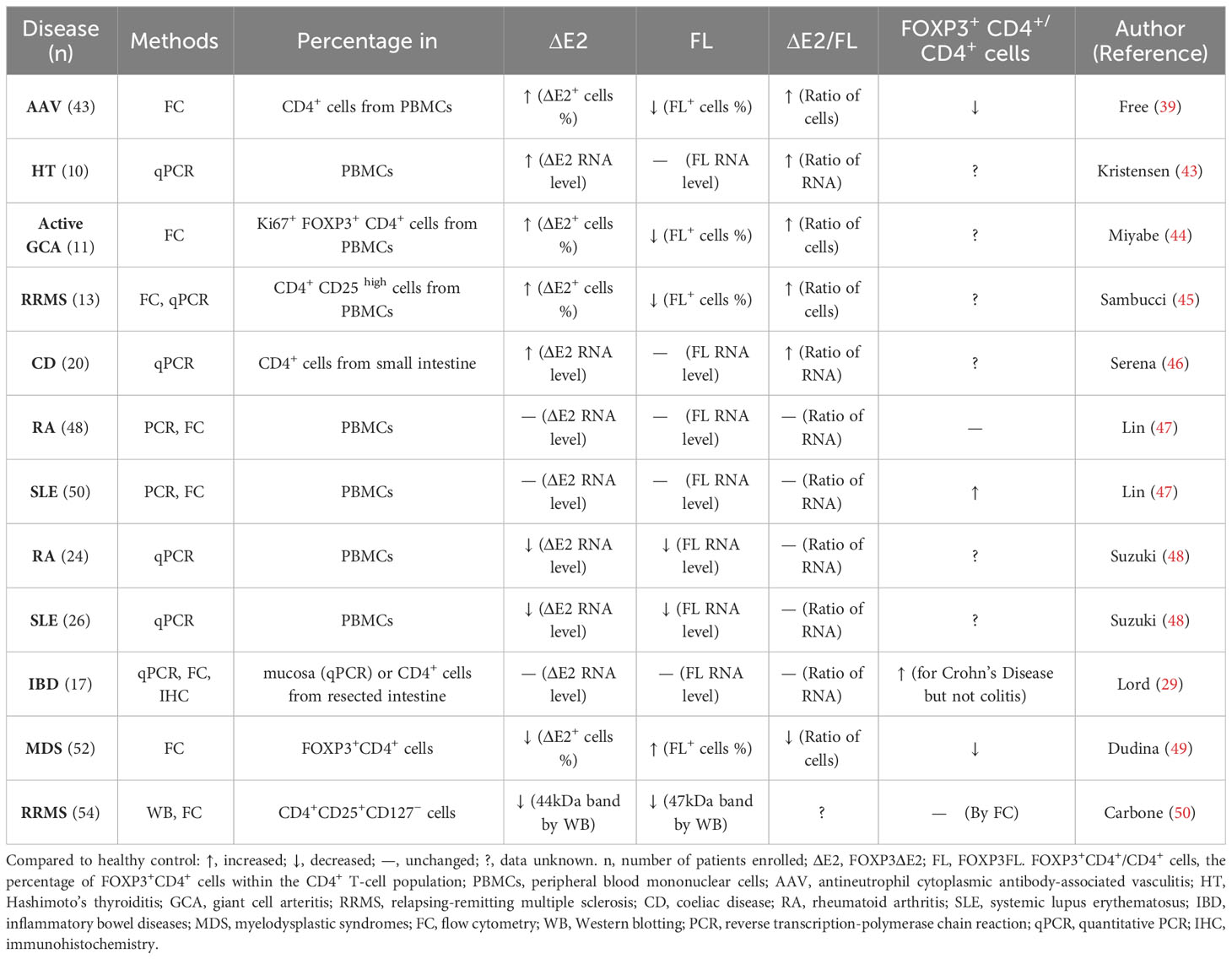
Table 2 Changes of FOXP3FL and FOXP3ΔE2 isoforms and their ratio in autoimmune diseases.
Patients with antineutrophil cytoplasmic antibody-associated vasculitis (AAV) (39), Hashimoto’s thyroiditis (HT) (43), giant cell arteritis (GCA) (44), relapsing-remitting multiple sclerosis (RRMS) (45) and coeliac disease (CD) (46) showed upregulated FOXP3ΔE2 isoforms in peripheral blood mononuclear cells (PBMCs) compared to control subjects. On the other hand, patients with rheumatoid arthritis (RA) (47), systemic lupus erythematosus (SLE) (47, 48), inflammatory bowel diseases (IBD) (29), myelodysplastic syndromes (MDS) (49) and RRMS (50) showed lower or normal FOXP3ΔE2 isoform levels. The inconsistent results shown in the studies investigating the same disease might be partly due to the differences in the disease stages and the detection methods.
Joly et al. determined the impact of alternative splicing of FOXP3 transcripts on atherosclerotic plaque stability in patients who underwent carotid endarterectomy (5). Real-time polymerase chain reaction (PCR) in a cohort of 150 patients indicated that higher plaque stability was associated with increased FOXP3ΔE2 isoform expression in the plaque (5). However, the FOXP3 isoform expression in peripheral blood mononuclear cells was not associated with plaque stability (5). Saleh et al. found that lower levels of total FOXP3 mRNA in PBMCs of kidney transplant recipients are associated with prolonged duration of inflammatory responses (51). Bruzzaniti et al. analyzed circulating peripheral Tregs in chronic obstructive pulmonary disease (COPD) subjects at different stages by staining cells with 2 specific FoxP3 antibodies: one that recognizes all splicing variants of FOXP3 and the other specific for FOXP3FL. Cytofluorimetric analysis revealed that both FOXP3FL and total FOXP3 frequencies increased at the early stage of COPD and decreased at the later exacerbated stage with severe inflammation (15). It indicated that the FOXP3FL isoform and the total FOXP3 level tend to correlate negatively with inflammation severity in disease progression.
There are some methodological concerns, about whether FOXP3 isoforms are causally associated with certain diseases: First, FOXP3 isoforms change during different stages of disease development, as shown in the studies on COPD and multiple sclerosis (7, 15, 45, 46). Second, FOXP3 splicing is organ-specific, which may result in differences between circulating T cells and tissue-resident T cells (5, 52). Third, studies using only PCR can’t show the changes in the percentage of FOXP3+ cells and the mean expression level per cell like the flow cytometry technique (12), especially when the total FOXP3 level remains unchanged in a group of CD4+ cells. Fourth, when flow cytometry is used to detect cell percentages, the difference in gating conditions may lead to distinct results, as is shown in Miyabe’s study (44). In this case, it’s better to analyze the absolute counting numbers of cell subsets rather than only percentages.
2.4 FOXP3 isoforms in cancer
Tregs are recruited to the tumor microenvironment and facilitate tumor cells to escape immune surveillance (53). Tumor-infiltrating Tregs can comprise up to 50% of intratumoral CD4+ T cells, exhibiting a more proliferative and immunosuppressive phenotype (54). High infiltration of Tregs correlates with poor prognosis in solid cancers including NSCLC, ovarian cancer, melanoma, and gastric cancer (53, 55). Thus, targeting tumor-infiltrating Tregs has become a promising anti-tumor strategy. Treg-targeting monoclonal antibodies against CD25, CTLA-4, Pd-1, and CCR4 are used to induce antitumor immunity (53). FOXP3 is a potential specific target for Treg suppression. FOXP3 in tumor-infiltrating CD4+ T cells is generally associated with an intrinsic capacity to suppress tumor immunity (53, 55). Therapeutic blocking of Tregs-specific FOXP3 reduced breast cancer growth in animal models (6). The prognostic value of CD4+FOXP3+ T cells has been confirmed in many cancers, but contradictory results have also been found in some cancers. Weed et al. demonstrated that, in patients with oral squamous cell carcinoma, the nuclear localization of FOXP3, rather than the overall expression of FOXP3, was associated with tumor recurrence within 3 years (56). As FOXP3 isoforms lacking exon 2 or exon 7 are more likely to be trapped in nuclei (30), the relationship between Treg FOXP3 isoforms and cancer prognosis needs further research.
Interestingly, FOXP3 is also expressed in various cancer cells and plays a complex role in tumor development. The functional roles of tumour-FOXP3 are inconsistent and even reversed. In breast cancer, gastric cancer, prostate cancer, and HCC, tumor FOXP3 acts as a tumor suppressor that inhibits the expression of multiple oncogenes. In contrast, tumor FOXP3 has been identified as a biomarker associated with malignant prognosis in pancreatic cancer, non-small cell lung cancer (NSCLC), thyroid cancer, and melanoma (34). The different roles of FOXP3 in cancer cells may be partly related to the alternative splicing which seems to produce more short isoforms than in normal cells. Jia et al. analyzed TCGA RNA-seq data from 9171 primary tumor tissues across 32 tumor types. Four FOXP3 isoforms including FOXP3FL, FOXP3ΔE2, FOXP3ΔE7, and an uncharacterized isoform were identified. FOXP3ΔE2 is dominant in most cancers except for acute myeloid leukemia (8). FOXP3ΔE2 over-expression in bladder cancer mediates cisplatin chemotherapy resistance (57). Besides, FOXP3ΔE2ΔE3, an isoform that doesn’t exist in Tregs, was identified in hepatocellular carcinoma cells and showed less inhibitory effect on tumor growth compared to FOXP3FL (58). By observing 10 malignant breast cancer cell lines, Zuo et al. found that none of the 10 cancer cell lines expressed FOXP3FL transcripts and 3 of them expressed FOXP3ΔE2ΔE3. The high incidence of somatic mutations contributed to the absence of FOXP3 or the abnormal expression of short FOXP3 splicing isoforms in breast cancer cells, leading to the impairment in HER2 oncogene suppression (59).
3 Metabolic regulation of FOXP3 transcription and splicing
3.1 Mechanisms controlling FOXP3 transcription and splicing during Tregs induction
Sustained FOXP3 expression, along with continuous TCR stimulation and CD28 co-stimulation, is necessary for Treg function (60, 61). In humans, anti-CD3/anti-CD28 co-stimulation of naïve CD4+ T cells can’t produce strong immunosuppressive capacity, although FOXP3 can be induced transiently (62). Signal transducer and activator of transcription 5 (STAT5) induced by IL‐2 signaling is required for maintaining FOXP3 expression (63). Furthermore, the TCR-driven Foxp3+ Tregs from naïve CD4+ T cells still require production and/or activation of TGF-β in serum-containing culture medium or in T cells (64). These essential signaling pathways are shown in Figure 2.
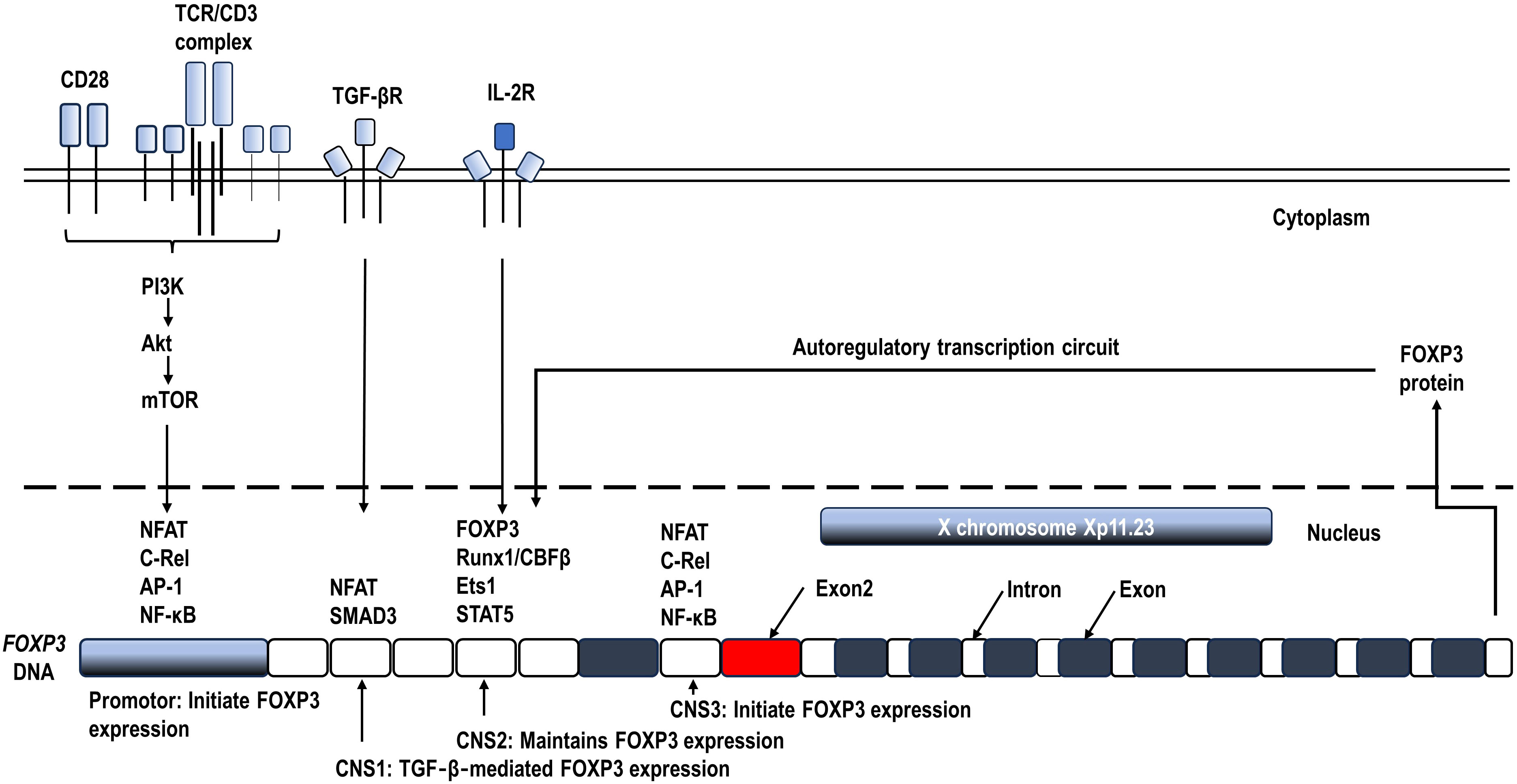
Figure 2 Schematic diagram of the mechanisms controlling FOXP3 transcription. Continuous T cell antigen receptor (TCR) stimulation and CD28 co-stimulation, along with IL‐2 signaling are required for inducing and maintaining FOXP3 expression (60, 62, 63). The FOXP3 gene locus contains 3 conserved non-coding sequences (CNS), which recruit transcription factor complexes to regulate FOXP3 transcription during Tregs activation and differentiation (1, 21).
A series of key transcription factors have been identified to form large complexes binding to the conserved non-coding sequences (CNS) of FOXP3 DNA and finely control FOXP3 transcription (21, 65) (Figure 2). To initiate FOXP3 transcription, transcription factors mainly including the nuclear factor of activated T cells (NFAT), nuclear factor kappa B (NF-κB), and activator protein 1 (AP-1) form an enhancing complex across the promotor and CNS3, integrating TCR and CD28 costimulatory signaling pathways in Treg precursors (66, 67). In addition, CNS1 enhances TCR‐ and TGF‐β‐induced FOXP3 expression by binding NFAT and Smad3 (1, 21, 68). Prolonged CD28 signals can also inhibit FOXP3 transcription by activating Akt/the mammalian target of rapamycin (mTOR) and thus inhibiting FOXO1 and FOXO3 transcription factors which are positive regulators of FOXP3 expression (69). Once expressed, along with continuous TCR and CD28 co-stimulation, FOXP3 augments and stabilizes its own transcription by forming an autoregulatory transcription circuit, which likely involves the complex of FOXP3, Runx1/CBFβ, Ets1, and STAT5 binding to CNS2 (63, 70). The accessibility of CNS2 is ensured by the demethylation of its CpG islands, known as the Treg cell-specific demethylated region (TSDR), which serves as an indicator of the stability of FOXP3 expression and Treg phenotype (71, 72). Ozay et al. found that demethylated CpGs overlapped with the STAT5 binding site, consistent with a requirement for STAT5 binding for the maintenance of FOXP3 expression (73). These key transcription factors and CNS locus can additionally recruit other cofactors to further enhance or suppress FOXP3 transcription and splicing. The cofactors related to energy metabolism including glycolysis and oxidative phosphorylation will be reviewed in the next section.
The above mechanistic studies mainly focus on the regulation of the transcription process from FOXP3 DNA to pre-mRNA, which may indirectly affect the subsequent alternative splicing. Whereas, studies on the direct regulation of FOXP3 splicing are relatively rare. This posttranscriptional modification process relies on the identification of splice sites by ribonucleoproteins and the mature mRNA generation by spliceosomes. This step is regulated by exonic or intronic enhancer/silencer auxiliary elements (74). DNA methylation has been shown to influence alternative splicing decisions by either promoting exon inclusion via recruitment of the methyl-CpG-binding protein MeCP2 or promoting alternative exon skipping via increasing RNA polymerase II (RNAPII) elongation (75). Protein kinase C θ (PKCθ), a molecular regulator of TCR downstream signaling, has been shown to phosphorylate splicing factors and inhibit FOXP3 demethylation through its modulation of two key components of RNA processing, heterogeneous nuclear ribonucleoprotein L (hnRNPL) and protein-l-isoaspartate O-methyltransferase-1 (PCMT1) (73). Minato Hirano et al. showed that the splicing of FOXP3 is strongly dependent on the RNA helicase DExD/H-Box Polypeptide 39B (DDX39B) (76).
The dynamic changes of FOXP3 splicing isoforms have been identified during the ex vivo Tregs induction. Blinova et al. reported that anti-CD3/anti-CD28 co-stimulation combined with IL-2 and TGF-β produced high numbers of mature ex vivo Tregs over 7 days of cultivation from initial CD4+ T cells (7). The proportions of FOXP3 splicing isoforms demonstrated complex and dynamic expression profiles. FOXP3FL was repressed from about 40% of the total FOXP3 isoforms at the initial to about 10% after 3-5 days’ stimulation and significantly increased to more than 90% at days 7 and 9. FOXP3ΔE2 remained at about 40% of the total FOXP3 isoforms in the first 5 days and was detected at minor levels in mature Tregs on days 7 and 9. The expression of FOXP3ΔE7 increased up to 21.6% at days 3 and 5 but became almost undetectable in mature Tregs. A similar expression pattern was observed for FOXP3ΔE2ΔE7 (7). However, the molecular mechanisms underlying the distinct changes of FOXP3 isoforms need further research.
3.2 Metabolic regulation of FOXP3 transcription and splicing by glycolysis and fatty acid oxidation
Compared to T effector cells (Teffs), Tregs at a quiescent state express low surface levels of glucose transporter 1 (GLUT1), have more activated AMP-activated protein kinase (AMPK), are rich in mitochondria, and mainly depend on lipid oxidation (20, 77). This indicates that Tregs may mainly use fatty acid oxidation for energy production. This metabolic preference in Tregs is reported to be regulated by FOXP3, which inhibits the expression of GLUT1 (20), glycolytic enzymes, and the master regulator c-Myc (19). Inhibition of glucose uptake and oxidation in inflammatory mice models promotes Tregs rather than Teffs development both in vitro and in vivo (78). Deletion of Glut1 in mice doesn’t affect the suppressive function of natural Tregs, or the in vitro Tregs induction (79). Genetically modified mice models also show that enhanced glycolysis in Tregs can decrease FOXP3 expression and disrupt their lineage stability and suppressive function (19, 80, 81). On the other hand, the treatment with etomoxir, a blocker of lipid oxidation through inhibition of carnitine palmitoyltransferase-1 (CPT1), decreases Tregs differentiation and function (77). Whereas elevated lipid oxidation by exogenous fatty acid addition or mTOR inhibitor rapamycin (82) results in modestly increased FOXP3 expression and Tregs differentiation (77, 83).
The above findings indicate that fatty acid oxidation is required for the differentiation and function maintenance of Tregs, while glycolysis is not necessary or even harmful to FOXP3 expression and functional Tregs differentiation in mice. However, it is becoming evident that this is not a fixed paradigm. Recent studies showed that activated Tregs in both mice and humans could engage in glycolytic metabolism ex vivo (84, 85). Kishore et al. showed that glycolysis initiated by the CD28-PI3K-mTORC2-mediated pathway led to glucokinase (GCK) induction, which interacted with actin filaments and promoted Tregs migration (86). Tanimine et al. observed that the glucose uptake inhibitor 2-deoxy-D-glucose (2-DG) at the onset of activation (the first 3 days) significantly decreased FOXP3 induction and cell proliferation, while at later stages of activation had little or no effect on either human thymically-derived or ex vivo-induced Tregs (87). These discoveries indicate that glycolysis is indispensable for Tregs in the initial activation, proliferation, and migration. Whereas, in the subsequent differentiation, Tregs exhibit more oxidative metabolism dependent on lipids and pyruvate and become less dependent on glucose (19, 87, 88).
Furthermore, research on FOXP3 isoform level indicates that glycolysis specifically promotes the expression of FOXP3 exon 2 (16) which mainly contributes to the stability and the suppressive function of Tregs (12). De Rosa et al. found that glycolysis promoted FOXP3FL isoform expression through the glycolytic enzyme enolase-1, leading to enhanced induction and suppressive function of Tregs in vitro (16) (Figure 3). They induced CD4+CD25+ Tregs from human CD4+CD25- cells by modest anti-CD3/anti-CD28 co-stimulation for 72 hours in the absence or presence of metabolic regulators during the first 36 hours. Tregs generated in the presence of 2-DG resulted in the nuclear translocation of enolase-1, which can bind to the promoter and CNS 2 regions of FOXP3 DNA and hinder the exon 2 expression, leading to significant FOXP3FL reduction. Enolase-1 siRNA reverted the effects of 2-DG treatment (16). Whereas Tregs generated in the presence of the fatty acid oxidation inhibitor etomoxir demonstrated slightly higher expression of both FOXP3 splicing isoforms through enhancing IL-2/STAT5 signaling (16). The FOXP3-exon 2-related suppressive activity of Tregs was impaired in human autoimmune diseases, including multiple sclerosis and type 1 diabetes, and was associated with impaired glycolysis and signaling via IL-2 (16). This study reveals that glycolysis is required for the expression of FOXP3FL that contains exon 2 during the initial induction of Tregs from human CD4+CD25- cells and unravels the underlying mechanism. However, this study’s inducing condition (weak TCR stimulation in the absence of TGF-β or IL-2) and duration (72 hours) differ from many other studies (7, 12, 87–89), which may not fully reflect the in vivo induction of Tregs. Despite the flaws, the necessity of glycolysis in FOXP3 exon 2 expression and normal function of Tregs was confirmed in later studies on freshly isolated human Tregs. Bruzzaniti et al. found that leptin overproduction in patients with severe COPD inhibited glycolysis and FOXP3FL expression, and reduced the generation and function of Tregs isolated from the blood of COPD patients (15). Adriawan et al. identified the lower expression of glycolytic enzymes, such as phosphofructokinase and enolase 1, and downregulation of FOXP3 and CD25 as well as the reduced TCR-induced calcium influx as correlates of Tregs dysfunction in patients with Giant cell arteritis (GCA). They also observed that glycolysis inhibition in healthy Tregs led to higher frequencies of FOXP3ΔE2-expressing Tregs and inhibited CD25 upregulation after 18 hours of ex vivo TCR stimulation. Like ex vivo GCA Tregs, isolated FOXP3ΔE2-expressing Tregs expressed less CD25 than FOXP3FL-expressing Tregs (90).
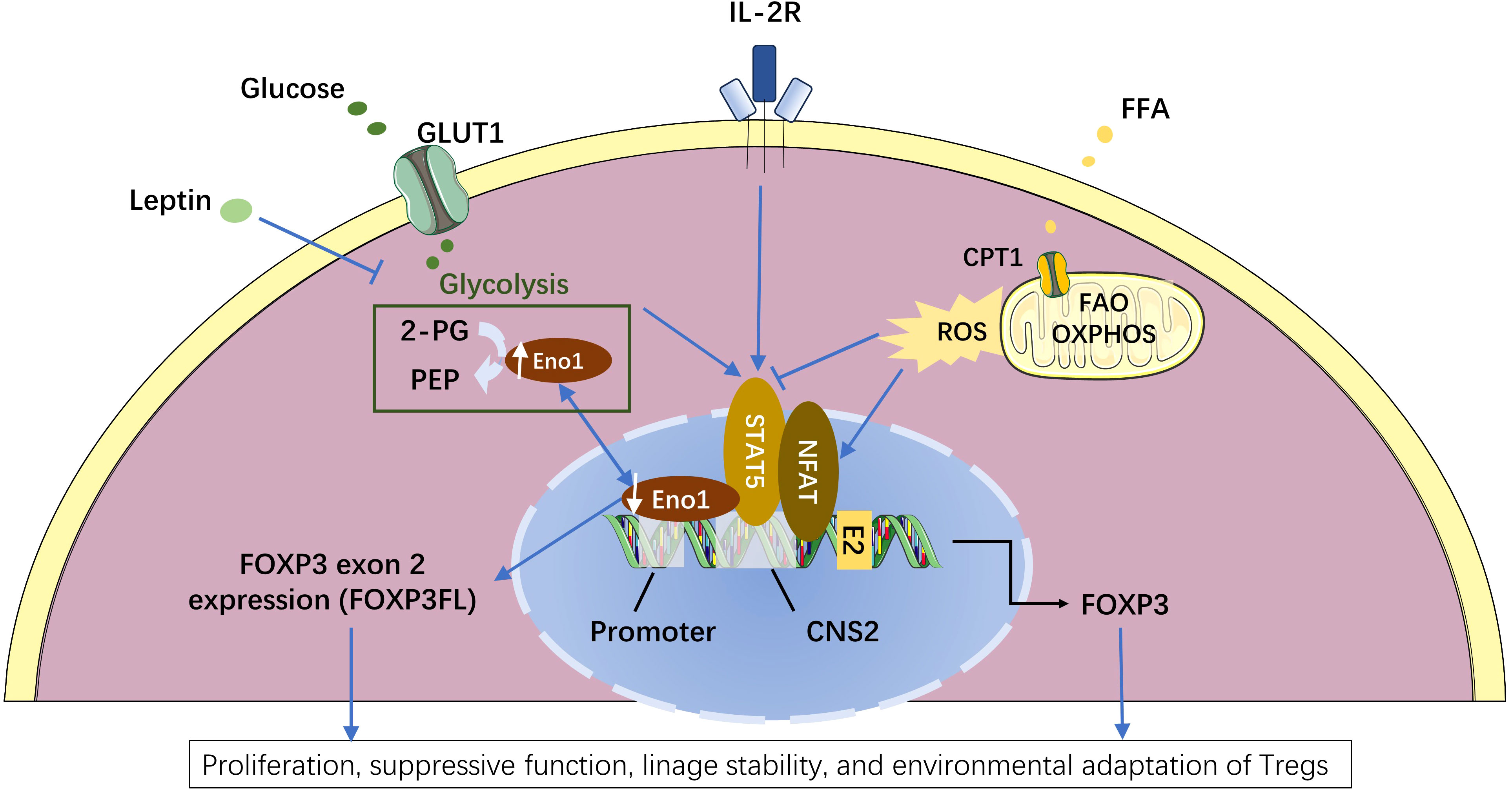
Figure 3 Metabolic regulation of FOXP3 splicing isoforms in the induced regulatory T cells (Tregs). Enolase-1 (Eno1), a glycolysis enzyme responsible for converting 2-phosphoglycerate (2-PG) to phosphoenolpyruvate (PEP), can also bind to the promoter and CNS2 locus of FOXP3 DNA and hinder the exon 2 expression (16). Enhanced glycolysis leads to more Eno1 in the cytoplasm and less nuclear Eno1, and consequently reduced the binding of Eno1 to the promoter and CNS2 locus of FOXP3. The release of this Eno1-FOXP3 binding specially promotes the expression of FOXP3 isoform containing exon 2 (FOXPFL) (16). While leptin can down-regulate FOXP3FL expression by inhibiting glycolysis (15). Compared to FOXP3 isoform lacking exon 2 (FOXP3ΔE2), FOXP3FL has stronger capacities in the proliferation, suppressive function, and lineage stability of Tregs (12, 16). Enhanced fatty acid oxidation (FAO) produces more mitochondrial reactive oxygen species (ROS) that can facilitate NFAT binding to the CNS2 enhancer of the FOXP3 locus (81–83), leading to a slight increase in the total FOXP3 transcription (16). Besides, glycolysis can promote while fatty acid oxidation can inhibit IL2-induced STAT5 signaling which is critical for FOXP3 maintenance (16, 63). The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
Therefore, it’s clear that glycolysis and fatty acid oxidation have respectively different effects on the expression of FOXP3 splicing isoforms and in the different activation stages of Tregs. Procaccini et al. illustrated a proteomic and metabolic comparison between human Tregs and CD4+CD25- conventional T (Tconv) cells in both freshly isolated and in vitro culture conditions. They found that freshly isolated human Tregs were highly glycolytic as compared to Tconv cells. The in vitro proliferation, FOXP3 expression, and suppressive activity of Tregs, induced by leptin neutralization, required both glycolysis and fatty acid oxidation, whereas those of Tconv cells require only glycolysis (85). Zhu et al. used liquid chromatography and tandem mass spectrometry (LC-MS/MS) to analyze the metabolic profiles of freshly isolated spleen Tregs in mice with sepsis with or without treatment with 2-DG or etomoxir. It was found that in severe infection, activated Tregs depend on glycolysis and fatty acid oxidation, and inhibition of metabolic pathways by either 2-DG or etomoxir reduced FOXP3 expression and increased Treg apoptosis (91). These studies confirm that both glycolysis and fatty acid oxidation are required in vivo for activated functional Tregs in both mice and humans (85, 91, 92). It may be helpful to further clarify the respective percentages of glycolysis and fatty acid oxidation in the total energy production in Tregs at different activation stages.
3.3 Metabolic regulation of FOXP3 transcription by mitochondria and reactive oxygen species
Both natural and induced Tregs have high mitochondrial mass and excessive reactive oxygen species (ROS) production (93). In activated conditions, Tregs have continuously higher ROS production compared to Teffs (19). ROS are highly reactive byproducts of normal oxygen metabolism and are involved in cellular signal transduction (94, 95), playing critical roles in human chronic inflammatory diseases (96, 97). ROS from mitochondrial oxidative phosphorylation (OXPHOS) activates NFAT in the nucleus, which then binds to the CNS2 enhancer of the FOXP3 gene and stimulates its expression (98–100) (Figure 3). In turn, FOXP3 can alter T cell metabolism and increase OXPHOS to adapt to the environment (19, 101). It has been demonstrated that Tregs exhibit reduced sensitivity to oxidative stress-induced cell death and maintain their suppressive function in a high ROS environment (102). These studies indicate that ROS generated from OXPHOS can enhance FOXP3 expression and promote the functional survival of activated Tregs.
However, excessive ROS can serve as a key mediator for promoting Treg instability in autoimmune diseases like antineutrophil cytoplasmic antibody-associated vasculitis (103) and impairing Tregs/Teffs homeostasis in cardiovascular diseases (104, 105). NADPH oxidase 2 (Nox2) is a ROS-generating enzyme that has been identified in CD4+CD25+FoxP3+ Tregs (104). Nox2-deficient Tregs produced less ROS, had increased nuclear levels of FOXP3 and NF-κB activation, and were more suppressive than wildtype Tregs (104). Compared to the wildtype, mice with CD4-targeted Nox2 deficiency had significantly higher infiltration of Tregs in the heart and minor Angiotensin II-dependent cardiovascular damage (104).
Besides ROS generation, mitochondria play a critical role in the switch of energy substances. Enhanced mitochondrial function benefits fatty acid oxidation and aerobic metabolism (77, 106). The mitochondrial transcription factor A (Tfam), which is essential for mitochondrial respiration, is also indispensable for the maintenance of Treg suppressive capacity. The genetic deletion of Tfam in Tregs impaired both their proliferation and function by enhancing DNA methylation in the TSDR of the FOXP3 locus (107).
3.4 Critical metabolic signaling pathways in the regulation of FOXP3 expression
Various signaling pathways are involved in the immunometabolism regulation of Tregs. PI3K/mTOR signaling serves as a critical link between the activating stimuli and the metabolic regulators in Tregs. Signals through the TCR, CD28, or IL-2 receptor activate the PI3K/Akt/mTOR cascade (108), which is critical for FOXP3 expression (109). mTOR activity is finely tuned by various metabolic signals (110). In response to ATP depletion, AMPK activation (111) promotes fatty acid oxidation while inhibiting mTOR-mediated glycolysis (77). The hypoxia-inducible transcription factor 1α (HIF-1α), a vital element in adaptation to varying oxygen states (112), is also activated in response to TCR activation and mTOR-mediated glycolysis (113–115). Short-chain fatty acids (SCFA) in the gut are also known to inhibit mTOR (116). Butyrate, one type of SCFA generated from gut microbiota, is reported to regulate the balance between FOXP3 isoforms in human gut-resident Tregs (46), but whether this effect of butyrate is mediated by the mTOR signaling pathway needs further verification. Besides, dietary nutrients and specific gut microbiota can also activate another transcriptional factor, aryl hydrocarbon receptor (AhR), which promotes FOXP3 expression and Treg development (117–119). Yu Y et al. revealed that the dietary intake of a moderate dose of glucose (6% w/v) induced Tregs in guts and alleviated colitis development in mice, and the mechanism might be related to glucose-induced AhR activation (119). Generally, the above-mentioned metabolic signaling pathways are closely related to both glycolysis and FOXP3 expression, but their specific roles in regulating FOXP3 alternative splicing need further research.
4 Concluding remarks and future perspectives
FOXP3 is crucial for the functional maintenance of human Tregs. The predominant FOXP3 splicing isoforms, FOXP3FL and FOXP3ΔE2, differ in functions and metabolic regulation mechanisms. The balance of FOXP3 splicing isoforms is related to autoimmune diseases, inflammatory diseases, and cancers. In response to environmental challenges, FOXP3 transcription and splicing can be finely regulated by TCR stimulation, glycolysis, fatty acid oxidation, and mitochondrial capacity. Targeting energy metabolism and FOXP3 splicing isoforms may provide new therapeutic approaches for Treg-related diseases.
Modulating Treg function has emerged as a promising approach to either upregulate or downregulate suppressive activity in the context of autoimmunity or cancer, respectively. Ex vivo expanded antigen-specific Tregs may be injected into patients with autoimmune diseases to restore Treg function (120). The stability of ex vivo expanded Tregs is one of the limitations associated with Tregs therapy (121). Proper approaches affecting glycolysis may help to balance FOXP3 isoforms, promote FOXP3 stability, and optimize Treg quality and quantity. For anti-cancer therapy, FOXP3 is a potential new target for specific Treg suppression; however, the nuclear FOXP3 is considered inaccessible by traditional therapeutics. The next-generation antisense oligonucleotides and TCR mimic antibodies targeting FOXP3-derived peptides have been developed to improve the nuclear FOXP3 accessibility in mouse models (53). Therapeutics using small molecules and splice-switching oligonucleotides may modulate alternative RNA splicing process and generate cancer-specific transcripts leading to altered functions (122, 123). However, currently no therapeutics have been described for the alternative splicing of FOXP3. Furthermore, in vivo application of metabolic regulating medicines can lead to complex effects because many types of cells can be involved. Thus, it is still a great challenge to specifically target the critical metabolic enzymes related to FOXP3 isoforms in Tregs.
The field of FOXP3 splicing isoform research is still emerging. Further studies would fully elucidate the roles of FOXP3 splicing isoforms in the development of diseases and the metabolic mechanisms regulating FOXP3 splicing. As regulation of FOXP3 expression may occur at various levels including transcriptional, RNA splicing, and post-transcriptional levels (71), methods like flow cytometry, Western blot, and immunohistochemistry using isoform-specific antibodies in combination with quantitative PCR may be required to fully demonstrate the changes in FOXP3 expression and distribution. Furthermore, Tregs from different tissues or at different disease stages, and as well as the different ex vivo treatment procedures of Tregs, may produce distinct results in FOXP3 expression and Treg function, and thus, these methods should be properly addressed and compared.
Author contributions
ZL: Funding acquisition, Writing – original draft, Writing – review & editing. YZ: Writing – original draft, Writing – review & editing. QS: Writing – review & editing. JZ: Writing – review & editing. ZZ: Funding acquisition, Writing – review & editing. MT: Conceptualization, Investigation, Project administration, Supervision, Writing – review & editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China and Chongqing Clinical Research Center for Geriatric Diseases (No. 81770801 and No. 81920108010).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mailer RKW. Alternative splicing of FOXP3-virtue and vice. Front Immunol (2018) 9:530. doi: 10.3389/fimmu.2018.00530
2. Sakaguchi S, Yamaguchi T, Nomura T and Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
3. Hori S, Murakami R. The adaptability of regulatory T cells and Foxp3. Int Immunol (2021) 33(12):803–7. doi: 10.1093/intimm/dxab045
4. Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol (2006) 24:209–26. doi: 10.1146/annurev.immunol.24.021605.090547
5. Joly AL, Seitz C, Liu S, Kuznetsov NV, Gertow K, Westerberg LS, et al. Alternative splicing of FOXP3 controls regulatory T cell effector functions and is associated with human atherosclerotic plaque stability. Circ Res (2018) 122(10):1385–94. doi: 10.1161/CIRCRESAHA.117.312340
6. Malla R, Adem M and Chakraborty A. Complexity and diversity of FOXP3 isoforms: Novel insights into the regulation of the immune response in metastatic breast cancer. Int Immunopharmacol (2023) 118:110015. doi: 10.1016/j.intimp.2023.110015
7. Blinova VG, Novachly NS, Gippius SN, Hilal A, Gladilina YA, Eliseeva DD, et al. Phenotypical and functional characteristics of human regulatory T cells during ex vivo maturation from CD4+ T lymphocytes. Appl Sci (2021) 11(13):5776–93. doi: 10.3390/app11135776
8. Jia H, Qi H, Gong Z, Yang S, Ren J, Liu Y, et al. The expression of FOXP3 and its role in human cancers. Biochim Biophys Acta Rev Cancer (2019) 1871(1):170–8. doi: 10.1016/j.bbcan.2018.12.004
9. Kaur G, Goodall JC. Jarvis LB and Hill Gaston JS. Characterisation of Foxp3 splice variants in human CD4+ and CD8+ T cells–identification of Foxp3Delta7 in human regulatory T cells. Mol Immunol (2010) 48(1-3):321–32. doi: 10.1016/j.molimm.2010.07.008
10. Mailer RK, Falk K and Rotzschke O. Absence of leucine zipper in the natural FOXP3Delta2Delta7 isoform does not affect dimerization but abrogates suppressive capacity. PLoS One (2009) 4(7):e6104. doi: 10.1371/journal.pone.0006104
11. Smith EL, Finney HM, Nesbitt AM, Ramsdell F and Robinson MK. Splice variants of human FOXP3 are functional inhibitors of human CD4+ T-cell activation. Immunology (2006) 119(2):203–11. doi: 10.1111/j.1365-2567.2006.02425.x
12. Du J, Wang Q, Yang S, Chen S, Fu Y, Spath S, et al. FOXP3 exon 2 controls T(reg) stability and autoimmunity. Sci Immunol (2022) 7(72):eabo5407. doi: 10.1126/sciimmunol.abo5407
13. Joly AL, Liu S, Dahlberg CI, Mailer RK, Westerberg LS, Andersson J. Foxp3 lacking exons 2 and 7 is unable to confer suppressive ability to regulatory T cells in vivo. J Autoimmun (2015) 63:23–30. doi: 10.1016/j.jaut.2015.06.009
14. Seitz C, Joly AL, Fang F, Frith K, Gray P and Andersson J. The FOXP3 full-length isoform controls the lineage-stability of CD4(+)FOXP3(+) regulatory T cells. Clin Immunol (2022) 237:108957. doi: 10.1016/j.clim.2022.108957
15. Bruzzaniti S, Bocchino M, Santopaolo M, Cali G, Stanziola AA, D'amato M, et al. An immunometabolic pathomechanism for chronic obstructive pulmonary disease. Proc Natl Acad Sci U.S.A. (2019) 116(31):15625–34. doi: 10.1073/pnas.1906303116
16. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol (2015) 16(11):1174–84. doi: 10.1038/ni.3269
17. Zeng H, Chi H. Metabolic control of regulatory T cell development and function. Trends Immunol (2015) 36(1):3–12. doi: 10.1016/j.it.2014.08.003
18. Sharabi A, Tsokos GC. T cell metabolism: new insights in systemic lupus erythematosus pathogenesis and therapy. Nat Rev Rheumatol (2020) 16(2):100–12. doi: 10.1038/s41584-019-0356-x
19. Angelin A, Gil-De-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab (2017) 25(6):1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
20. Kempkes RWM, Joosten I, Koenen H and He X. Metabolic pathways involved in regulatory T cell functionality. Front Immunol (2019) 10:2839. doi: 10.3389/fimmu.2019.02839
21. Ono M. Control of regulatory T-cell differentiation and function by T-cell receptor signalling and Foxp3 transcription factor complexes. Immunology (2020) 160(1):24–37. doi: 10.1111/imm.13178
22. Gu Q, Zhao X, Guo J, Jin Q, Wang T, Xu W, et al. The splicing isoform Foxp3Delta2 differentially regulates tTreg and pTreg homeostasis. Cell Rep (2023) 42(8):112877. doi: 10.1016/j.celrep.2023.112877
23. Koh KP, Sundrud MS and Rao A. Domain requirements and sequence specificity of DNA binding for the forkhead transcription factor FOXP3. PLoS One (2009) 4(12):e8109. doi: 10.1371/journal.pone.0008109
24. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
25. Mailer RK. IPEX as a consequence of alternatively spliced FOXP3. Front Pediatr (2020) 8:594375. doi: 10.3389/fped.2020.594375
26. Huang Q, Liu X, Zhang Y, Huang J, Li D and Li B. Molecular feature and therapeutic perspectives of immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. J Genet Genomics (2020) 47(1):17–26. doi: 10.1016/j.jgg.2019.11.011
27. Aarts-Riemens T, Emmelot ME, Verdonck LF and Mutis T. Forced overexpression of either of the two common human Foxp3 isoforms can induce regulatory T cells from CD4(+)CD25(-) cells. Eur J Immunol (2008) 38(5):1381–90. doi: 10.1002/eji.200737590
28. Allan SE, Passerini L, Bacchetta R, Crellin N, Dai M, Orban PC, et al. The role of 2 FOXP3 isoforms in the generation of human CD4+ Tregs. J Clin Invest (2005) 115(11):3276–84. doi: 10.1172/JCI24685
29. Lord JD, Valliant-Saunders K, Hahn H, Thirlby RC and Ziegler SF. Paradoxically increased FOXP3+ T cells in IBD do not preferentially express the isoform of FOXP3 lacking exon 2. Dig Dis Sci (2012) 57(11):2846–55. doi: 10.1007/s10620-012-2292-3
30. Magg T, Mannert J, Ellwart JW, Schmid I and Albert MH. Subcellular localization of FOXP3 in human regulatory and nonregulatory T cells. Eur J Immunol (2012) 42(6):1627–38. doi: 10.1002/eji.201141838
31. Sato Y, Liu J, Lee E, Perriman R, Roncarolo MG, Bacchetta R. Co-expression of FOXP3FL and FOXP3Delta2 isoforms is required for optimal treg-like cell phenotypes and suppressive function. Front Immunol (2021) 12:752394. doi: 10.3389/fimmu.2021.752394
32. Castro G, Liu X, Ngo K, De Leon-Tabaldo A, Zhao S, Luna-Roman R, et al. RORgammat and RORalpha signature genes in human Th17 cells. PLoS One (2017) 12(8):e0181868. doi: 10.1371/journal.pone.0181868
33. Du J, Huang C, Zhou B and Ziegler SF. Isoform-specific inhibition of ROR alpha-mediated transcriptional activation by human FOXP3. J Immunol (2008) 180(7):4785–92. doi: 10.4049/jimmunol.180.7.4785
34. Wang J, Gong R, Zhao C, Lei K, Sun X and Ren H. Human FOXP3 and tumour microenvironment. Immunology (2023) 168(2):248–55. doi: 10.1111/imm.13520
35. Koh B, Ulrich BJ, Nelson AS, Panangipalli G, Kharwadkar R, Wu W, et al. Bcl6 and Blimp1 reciprocally regulate ST2(+) Treg-cell development in the context of allergic airway inflammation. J Allergy Clin Immunol (2020) 146(5):1121–36 e9. doi: 10.1016/j.jaci.2020.03.002
36. Liu C, Zeng X, Yu S, Ren L, Sun X, Long Y, et al. Up-regulated DNA-binding inhibitor Id3 promotes differentiation of regulatory T cell to influence antiviral immunity in chronic hepatitis B virus infection. Life Sci (2021) 285:119991. doi: 10.1016/j.lfs.2021.119991
37. Volta V, Perez-Baos S, de la Parra C, Katsara O, Ernlund A, Dornbaum S, et al. A DAP5/eIF3d alternate mRNA translation mechanism promotes differentiation and immune suppression by human regulatory T cells. Nat Commun (2021) 12(1):6979. doi: 10.1038/s41467-021-27087-w
38. Pratama A, Schnell A, Mathis D and Benoist C. Developmental and cellular age direct conversion of CD4+ T cells into RORgamma+ or Helios+ colon Treg cells. J Exp Med (2020) 217(1):e20190428. doi: 10.1084/jem.20190428
39. Free ME, Bunch DO, Mcgregor JA, Jones BE, Berg EA, Hogan SL, et al. Patients with antineutrophil cytoplasmic antibody-associated vasculitis have defective Treg cell function exacerbated by the presence of a suppression-resistant effector cell population. Arthritis Rheum (2013) 65(7):1922–33. doi: 10.1002/art.37959
40. Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science (2015) 349(6251):989–93. doi: 10.1126/science.aac4263
41. Wright CJ, Smith CWJ and Jiggins CD. Alternative splicing as a source of phenotypic diversity. Nat Rev Genet (2022) 23(11):697–710. doi: 10.1038/s41576-022-00514-4
42. Andersson J, Libby P and Hansson GK. Adaptive immunity and atherosclerosis. Clin Immunol (2010) 134(1):33–46. doi: 10.1016/j.clim.2009.07.002
43. Kristensen B, Hegedus L, Madsen HO, Smith TJ and Nielsen CH. Altered balance between self-reactive T helper (Th)17 cells and Th10 cells and between full-length forkhead box protein 3 (FoxP3) and FoxP3 splice variants in Hashimoto's thyroiditis. Clin Exp Immunol (2015) 180(1):58–69. doi: 10.1111/cei.12557
44. Miyabe C, Miyabe Y, Strle K, Kim ND, Stone JH, Luster AD, et al. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann Rheum Dis (2017) 76(5):898–905. doi: 10.1136/annrheumdis-2016-210070
45. Sambucci M, Gargano F, De Rosa V, De Bardi M, Picozza M, Placido R, et al. FoxP3 isoforms and PD-1 expression by T regulatory cells in multiple sclerosis. Sci Rep (2018) 8(1):3674. doi: 10.1038/s41598-018-21861-5
46. Serena G, Yan S, Camhi S, Patel S, Lima RS, Sapone A, et al. Proinflammatory cytokine interferon-gamma and microbiome-derived metabolites dictate epigenetic switch between forkhead box protein 3 isoforms in coeliac disease. Clin Exp Immunol (2017) 187(3):490–506. doi: 10.1111/cei.12911
47. Lin SC, Chen KH, Lin CH, Kuo CC, Ling QD and Chan CH. The quantitative analysis of peripheral blood FOXP3-expressing T cells in systemic lupus erythematosus and rheumatoid arthritis patients. Eur J Clin Invest (2007) 37(12):987–96. doi: 10.1111/j.1365-2362.2007.01882.x
48. Suzuki K, Setoyama Y, Yoshimoto K, Tsuzaka K, Abe T and Takeuchi T. Decreased mRNA expression of two FOXP3 isoforms in peripheral blood mononuclear cells from patients with rheumatoid arthritis and systemic lupus erythematosus. Int J Immunopathol Pharmacol (2011) 24(1):7–14. doi: 10.1177/039463201102400102
49. Dudina GA, Donetskova AD, Litvina MM, Mitin AN, Mitina TA and Polyakov SA. Regulatory T cells and profile of FOXP3 isoforms expression in peripheral blood of patients with myelodysplastic syndromes. Adv Hematol (2018) 2018:8487403. doi: 10.1155/2018/8487403
50. Carbone F, De Rosa V, Carrieri PB, Montella S, Bruzzese D, Porcellini A, et al. Regulatory T cell proliferative potential is impaired in human autoimmune disease. Nat Med (2014) 20(1):69–74. doi: 10.1038/nm.3411
51. Saleh QW, Mohammadnejad A, Tepel M. Lower levels of FOXP3 are associated with prolonged inflammatory responses in kidney transplant recipients. Front Immunol (2023) 14:1252857. doi: 10.3389/fimmu.2023.1252857
52. Georgiev P, Charbonnier LM, Chatila TA. Regulatory T cells: The many faces of foxp3. J Clin Immunol (2019) 39(7):623–40. doi: 10.1007/s10875-019-00684-7
53. Dees S, Ganesan R, Singh S and Grewal IS. Regulatory T cell targeting in cancer: Emerging strategies in immunotherapy. Eur J Immunol (2021) 51(2):280–91. doi: 10.1002/eji.202048992
54. Togashi Y, Shitara K and Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol (2019) 16(6):356–71. doi: 10.1038/s41571-019-0175-7
55. Qiu Y, Ke S, Chen J, Qin Z, Zhang W, Yuan Y, et al. FOXP3+ regulatory T cells and the immune escape in solid tumours. Front Immunol (2022) 13:982986. doi: 10.3389/fimmu.2022.982986
56. Weed DT, Walker G, de la Fuente AC, Nazarian R, Vella JL, Gomez-Fernandez CR, et al. FOXP3 subcellular localization predicts recurrence in oral squamous cell carcinoma. PLoS One (2013) 8(8):e71908. doi: 10.1371/journal.pone.0071908
57. Zhang H, Prado K, Zhang KX, Peek EM, Lee J, Wang X, et al. Biased expression of the FOXP3Delta3 isoform in aggressive bladder cancer mediates differentiation and cisplatin chemotherapy resistance. Clin Cancer Res (2016) 22(21):5349–61. doi: 10.1158/1078-0432.CCR-15-2581
58. Gong Z, Jia H, Yu J, Liu Y, Ren J, Yang S, et al. Nuclear FOXP3 inhibits tumor growth and induced apoptosis in hepatocellular carcinoma by targeting c-Myc. Oncogenesis (2020) 9(10):97. doi: 10.1038/s41389-020-00283-x
59. Zuo T, Wang L, Morrison C, Chang X, Zhang H, Li W, et al. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/erbB2 oncogene. Cell (2021) 184(26):6378. doi: 10.1016/j.cell.2021.11.030
60. Levine AG, Arvey A, Jin W and Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol (2014) 15(11):1070–8. doi: 10.1038/ni.3004
61. Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS and Turka LA. An obligate cell-intrinsic function for CD28 in Tregs. J Clin Invest (2013) 123(2):580–93. doi: 10.1172/JCI65013
62. Tran DQ, Ramsey H and Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood (2007) 110(8):2983–90. doi: 10.1182/blood-2007-06-094656
63. Chen Q, Kim YC, Laurence A, Punkosdy GA and Shevach EM. IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo. J Immunol (2011) 186(11):6329–37. doi: 10.4049/jimmunol.1100061
64. Chen W. TGF-beta regulation of T cells. Annu Rev Immunol (2023) 41:483–512. doi: 10.1146/annurev-immunol-101921-045939
65. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K and Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature (2010) 463(7282):808–12. doi: 10.1038/nature08750
66. Mantel PY, Ouaked N, Ruckert B, Karagiannidis C, Welz R, Blaser K, et al. Molecular mechanisms underlying FOXP3 induction in human T cells. J Immunol (2006) 176(6):3593–602. doi: 10.4049/jimmunol.176.6.3593
67. Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, et al. An NF-kappaB transcription-factor-dependent lineage-specific transcriptional program promotes regulatory T cell identity and function. Immunity (2017) 47(3):450–65.e5. doi: 10.1016/j.immuni.2017.08.010
68. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI and Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol (2008) 9(2):194–202. doi: 10.1038/ni1549
69. Ouyang W, Beckett O, Ma Q, Paik JH, Depinho RA and Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol (2010) 11(7):618–27. doi: 10.1038/ni.1884
70. Bending D, Paduraru A, Ducker CB, Prieto Martin P, Crompton T and Ono M. A temporally dynamic Foxp3 autoregulatory transcriptional circuit controls the effector Treg programme. EMBO J (2018) 37(16):e99013. doi: 10.15252/embj.201899013
71. Colamatteo A, Carbone F, Bruzzaniti S, Galgani M, Fusco C, Maniscalco GT, et al. Molecular mechanisms controlling foxp3 expression in health and autoimmunity: From epigenetic to post-translational regulation. Front Immunol (2019) 10:3136. doi: 10.3389/fimmu.2019.03136
72. Minskaia E, Saraiva BC, Soares MMV, Azevedo RI, Ribeiro RM, Kumar SD, et al. Molecular markers distinguishing T cell subtypes with TSDR strand-bias methylation. Front Immunol (2018) 9:2540. doi: 10.3389/fimmu.2018.02540
73. Ozay EI, Shanthalingam S, Torres JA, Osborne BA, Tew GN and Minter LM. Protein Kinase C Theta Modulates PCMT1 through hnRNPL to Regulate FOXP3 Stability in Regulatory T Cells. Mol Ther (2020) 28(10):2220–36. doi: 10.1016/j.ymthe.2020.06.012
74. Havens MA, Duelli DM and Hastings ML. Targeting RNA splicing for disease therapy. Wiley Interdiscip Rev RNA (2013) 4(3):247–66. doi: 10.1002/wrna.1158
75. Nieto Moreno N, Giono LE, Cambindo Botto AE, Munoz MJ and Kornblihtt AR. Chromatin, DNA structure and alternative splicing. FEBS Lett (2015) 589(22):3370–8. doi: 10.1016/j.febslet.2015.08.002
76. Hirano M, Galarza-Munoz G, Nagasawa C, Schott G, Wang L, Antonia AL, et al. The RNA helicase DDX39B activates FOXP3 RNA splicing to control T regulatory cell fate. Elife (2023) 12:e76927. doi: 10.7554/eLife.76927
77. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, Maciver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
78. Michalek RD, Gerriets VA, Nichols AG, Inoue M, Kazmin D, Chang CY, et al. Estrogen-related receptor-alpha is a metabolic regulator of effector T-cell activation and differentiation. Proc Natl Acad Sci U.S.A. (2011) 108(45):18348–53. doi: 10.1073/pnas.1108856108
79. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi: 10.1016/j.cmet.2014.05.004
80. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-like receptor signaling balance T(reg) cell anabolic metabolism for suppression. Nat Immunol (2016) 17(12):1459–66. doi: 10.1038/ni.3577
81. Huynh A, Dupage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol (2015) 16(2):188–96. doi: 10.1038/ni.3077
82. Brown NF, Stefanovic-Racic M, Sipula IJ and Perdomo G. The mammalian target of rapamycin regulates lipid metabolism in primary cultures of rat hepatocytes. Metabolism (2007) 56(11):1500–7. doi: 10.1016/j.metabol.2007.06.016
83. Haxhinasto S, Mathis D and Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med (2008) 205(3):565–74. doi: 10.1084/jem.20071477
84. Priyadharshini B, Loschi M, Newton RH, Zhang JW, Finn KK, Gerriets VA, et al. Cutting edge: TGF-beta and phosphatidylinositol 3-kinase signals modulate distinct metabolism of regulatory T cell subsets. J Immunol (2018) 201(8):2215–9. doi: 10.4049/jimmunol.1800311
85. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity (2016) 44(3):712. doi: 10.1016/j.immuni.2016.02.022
86. Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D, et al. Regulatory T cell migration is dependent on glucokinase-mediated glycolysis. Immunity (2017) 47(5):875–89.e10. doi: 10.1016/j.immuni.2017.10.017
87. Tanimine N, Germana SK, Fan M, Hippen K, Blazar BR, Markmann JF, et al. Differential effects of 2-deoxy-D-glucose on in vitro expanded human regulatory T cell subsets. PLoS One (2019) 14(6):e0217761. doi: 10.1371/journal.pone.0217761
88. Chen X, Li S, Long D, Shan J and Li Y. Rapamycin facilitates differentiation of regulatory T cells via enhancement of oxidative phosphorylation. Cell Immunol (2021) 365:104378. doi: 10.1016/j.cellimm.2021.104378
89. Alissafi T, Kalafati L, Lazari M, Filia A, Kloukina I, Manifava M, et al. Mitochondrial oxidative damage underlies regulatory T cell defects in autoimmunity. Cell Metab (2020) 32(4):591–604.e7. doi: 10.1016/j.cmet.2020.07.001
90. Adriawan IR, Atschekzei F, Dittrich-Breiholz O, Garantziotis P, Hirsch S, Risser LM, et al. Novel aspects of regulatory T cell dysfunction as a therapeutic target in giant cell arteritis. Ann Rheum Dis (2022) 81(1):124–31. doi: 10.1136/annrheumdis-2021-220955
91. Zhu X, Ji W, Guo S, Zhu D, Yang Y and Liu X. Glycolytic and lipid oxidative metabolic programs are essential for freshly-isolated regulatory T cells in mice with sepsis. RSC Adv (2020) 10(35):21000–8. doi: 10.1039/d0ra01947j
92. Cluxton D, Petrasca A, Moran B and Fletcher JM. Differential regulation of human treg and th17 cells by fatty acid synthesis and glycolysis. Front Immunol (2019) 10:115. doi: 10.3389/fimmu.2019.00115
93. Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, et al. Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J (2015) 29(6):2315–26. doi: 10.1096/fj.14-268409
94. Orie NN, Zidek W and Tepel M. Increased intracellular generation of reactive oxygen species in mononuclear leukocytes from patients with diabetes mellitus type 2. Exp Clin Endocrinol Diabetes (2000) 108(3):175–80. doi: 10.1055/s-2000-7740
95. Wuensch T, Thilo F, Krueger K, Scholze A, Ristow M and Tepel M. High glucose-induced oxidative stress increases transient receptor potential channel expression in human monocytes. Diabetes (2010) 59(4):844–9. doi: 10.2337/db09-1100
96. Aranda-Rivera AK, Srivastava A, Cruz-Gregorio A, Pedraza-Chaverri J, Mulay SR and Scholze A. Involvement of inflammasome components in kidney disease. Antioxidants (Basel) (2022) 11(2):246. doi: 10.3390/antiox11020246
97. Rasmussen M, Hansen KH and Scholze A. Nrf2 protein serum concentration in human CKD shows a biphasic behavior. Antioxidants (Basel) (2023) 12(4):932. doi: 10.3390/antiox12040932
98. Chavez MD, Tse HM. Targeting mitochondrial-derived reactive oxygen species in T cell-mediated autoimmune diseases. Front Immunol (2021) 12:703972. doi: 10.3389/fimmu.2021.703972
99. Li X, Liang Y, Leblanc M, Benner C and Zheng Y. Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell (2014) 158(4):734–48. doi: 10.1016/j.cell.2014.07.030
100. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38(2):225–36. doi: 10.1016/j.immuni.2012.10.020
101. Yan Y, Huang L, Liu Y, Yi M, Chu Q, Jiao D, et al. Metabolic profiles of regulatory T cells and their adaptations to the tumor microenvironment: implications for antitumor immunity. J Hematol Oncol (2022) 15(1):104. doi: 10.1186/s13045-022-01322-3
102. Mougiakakos D, Johansson CC, Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood (2009) 113(15):3542–5. doi: 10.1182/blood-2008-09-181040
103. Shimojima Y, Kishida D, Ichikawa T, Takamatsu R, Nomura S and Sekijima Y. Oxidative stress promotes instability of regulatory T cells in antineutrophil cytoplasmic antibody-associated vasculitis. Front Immunol (2021) 12:789740. doi: 10.3389/fimmu.2021.789740
104. Emmerson A, Trevelin SC, Mongue-Din H, Becker PD, Ortiz C, Smyth LA, et al. Nox2 in regulatory T cells promotes angiotensin II-induced cardiovascular remodeling. J Clin Invest (2018) 128(7):3088–101. doi: 10.1172/JCI97490
105. Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, et al. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol (2016) 13(3):167–79. doi: 10.1038/nrcardio.2015.169
106. Luo Z, Ma L, Zhao Z, He H, Yang D, Feng X, et al. TRPV1 activation improves exercise endurance and energy metabolism through PGC-1alpha upregulation in mice. Cell Res (2012) 22(3):551–64. doi: 10.1038/cr.2011.205
107. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature (2019) 565(7740):495–9. doi: 10.1038/s41586-018-0846-z
108. Fan MY, Turka LA. Immunometabolism and PI(3)K signaling as a link between IL-2, foxp3 expression, and suppressor function in regulatory T cells. Front Immunol (2018) 9:69. doi: 10.3389/fimmu.2018.00069
109. Chapman NM, Zeng H, Nguyen TM, Wang Y, Vogel P, Dhungana Y, et al. mTOR coordinates transcriptional programs and mitochondrial metabolism of activated T(reg) subsets to protect tissue homeostasis. Nat Commun (2018) 9(1):2095. doi: 10.1038/s41467-018-04392-5
110. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 169(2):361–71. doi: 10.1016/j.cell.2017.03.035
111. He C, Li Q, Cui Y, Gao P, Shu W, Zhou Q, et al. Recurrent moderate hypoglycemia accelerates the progression of Alzheimer's disease through impairment of the TRPC6/GLUT3 pathway. JCI Insight (2022) 7(5):e154595. doi: 10.1172/jci.insight.154595
112. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
113. Ao A, Wang H, Kamarajugadda S and Lu J. Involvement of estrogen-related receptors in transcriptional response to hypoxia and growth of solid tumors. Proc Natl Acad Sci U.S.A. (2008) 105(22):7821–6. doi: 10.1073/pnas.0711677105
114. Dupage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity (2015) 42(2):227–38. doi: 10.1016/j.immuni.2015.01.007
115. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
116. Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol (2015) 8(1):80–93. doi: 10.1038/mi.2014.44
117. Oh-Oka K, Kojima Y, Uchida K, Yoda K, Ishimaru K, Nakajima S, et al. Induction of colonic regulatory T cells by mesalamine by activating the aryl hydrocarbon receptor. Cell Mol Gastroenterol Hepatol (2017) 4(1):135–51. doi: 10.1016/j.jcmgh.2017.03.010
118. Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity (2012) 36(1):92–104. doi: 10.1016/j.immuni.2011.11.011
119. Yu Y, Yang W, Yu T, Zhao X, Zhou Z, Yu Y, et al. Glucose promotes regulatory T cell differentiation to maintain intestinal homeostasis. iScience (2022) 25(9):105004. doi: 10.1016/j.isci.2022.105004
120. Mukhatayev Z, Ostapchuk YO, Fang D and Le Poole IC. Engineered antigen-specific regulatory T cells for autoimmune skin conditions. Autoimmun Rev (2021) 20(3):102761. doi: 10.1016/j.autrev.2021.102761
121. Goswami TK, Singh M, Dhawan M, Mitra S, Emran TB, Rabaan AA, et al. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders - Advances and challenges. Hum Vaccin Immunother (2022) 18(1):2035117. doi: 10.1080/21645515.2022.2035117
122. Desterro J, Bak-Gordon P, Carmo-Fonseca M. Targeting mRNA processing as an anticancer strategy. Nat Rev Drug Discovery (2020) 19(2):112–29. doi: 10.1038/s41573-019-0042-3
Keywords: forkhead box P3, splicing isoforms, metabolic regulation, regulatory T cells, suppressive function, glycolysis, fatty acid oxidation, autoimmune diseases
Citation: Luo Z, Zhang Y, Saleh QW, Zhang J, Zhu Z and Tepel M (2023) Metabolic regulation of forkhead box P3 alternative splicing isoforms and their impact on health and disease. Front. Immunol. 14:1278560. doi: 10.3389/fimmu.2023.1278560
Received: 16 August 2023; Accepted: 25 September 2023;
Published: 06 October 2023.
Edited by:
Mehdi Benamar, Harvard Medical School, United StatesReviewed by:
Manoussa Ethel Fanny, Xilio Therapeutics, Inc., United StatesQian Chen, Harvard Medical School, United States
Giuseppe Matarese, University of Naples Federico II, Italy
Copyright © 2023 Luo, Zhang, Saleh, Zhang, Zhu and Tepel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Tepel, bXRlcGVsQGhlYWx0aC5zZHUuZGs=
†These authors have contributed equally to this work and share first authorship