 Elena Ockfen
Elena Ockfen Liza Filali
Liza Filali Diogo Pereira Fernandes
Diogo Pereira Fernandes Céline Hoffmann1
Céline Hoffmann1 Clément Thomas
Clément Thomas- 1Cytoskeleton and Cancer Progression, Department of Cancer Research, Luxembourg Institute of Health, Luxembourg, Luxembourg
- 2Faculty of Science, Technology and Medicine University of Luxembourg, Esch-sur-Alzette, Luxembourg
Cytotoxic lymphocytes (CLs), specifically cytotoxic T lymphocytes and natural killer cells, are indispensable guardians of the immune system and orchestrate the recognition and elimination of cancer cells. Upon encountering a cancer cell, CLs establish a specialized cellular junction, known as the immunological synapse that stands as a pivotal determinant for effective cell killing. Extensive research has focused on the presynaptic side of the immunological synapse and elucidated the multiple functions of the CL actin cytoskeleton in synapse formation, organization, regulatory signaling, and lytic activity. In contrast, the postsynaptic (cancer cell) counterpart has remained relatively unexplored. Nevertheless, both indirect and direct evidence has begun to illuminate the significant and profound consequences of cytoskeletal changes within cancer cells on the outcome of the lytic immunological synapse. Here, we explore the understudied role of the cancer cell actin cytoskeleton in modulating the immune response within the immunological synapse. We shed light on the intricate interplay between actin dynamics and the evasion mechanisms employed by cancer cells, thus providing potential routes for future research and envisioning therapeutic interventions targeting the postsynaptic side of the immunological synapse in the realm of cancer immunotherapy. This review article highlights the importance of actin dynamics within the immunological synapse between cytotoxic lymphocytes and cancer cells focusing on the less-explored postsynaptic side of the synapse. It presents emerging evidence that actin dynamics in cancer cells can critically influence the outcome of cytotoxic lymphocyte interactions with cancer cells.
Introduction
The anti-tumor immune response is a complex process involving a coordinated interplay between various immune cells. At the forefront of this response are cytotoxic lymphocytes (CLs), mainly natural killer (NK) cells and CD8+ cytotoxic lymphocytes (CTLs), that are essential for identifying and eliminating cancer cells. NK cells are the frontline defense against cancer cells due to their innate capability to rapidly recognize and kill target cells without prior sensitization or the need for clonal expansion (1). They play a significant role in the control of early tumors and metastasis (2, 3) while they are typically less abundant than CTLs in the tumor microenvironment (TME) of more advanced tumors (4, 5). Presently, CTLs are recognized as the most potent effectors in the anti-tumor immune response and represent the primary focus of current cancer immunotherapies involving immune-checkpoint inhibitors and genetic modification of receptors (6, 7).
NK cells have a large repertoire of surface inhibitory and activating receptors, which enable them to differentiate between healthy and diseased cells (8–11). Most host’s healthy cells express self-major histocompatibility complex class I (MHCI) molecules that act as ligands for NK cell inhibitory receptors, such as killer cell immunoglobulin-like receptors (KIRs) in humans and C-type lectin-like Ly49 receptors in mice, thus preventing NK cell activation (12–14). In contrast, cancer cells frequently downregulate MHCI molecule expression which makes them more susceptible to NK cell cytotoxicity, a process referred to as “missing-self” recognition (15). In addition, they increase the production of stress molecules recognized by activating receptors on NK cells. The combination of reduced inhibitory signals (e.g., due to low MHCI expression) and enhanced activating signal (e.g., due to the upregulation stress-induced molecules) effectively triggers the activation of NK cells. Beyond their cytotoxic function, NK cells fulfill a crucial role in modulating adaptive immune responses by releasing cytokines and chemokines and physically interacting with other immune cells, particularly dendritic cells (DCs) (16, 17). Harnessing the potent cytotoxicity and immunomodulatory functions of NK cells has emerged as a promising therapeutic strategy to treat cancer, and readers are referred to recent reviews on the subject (5, 18–20).
Unlike NK cells, CTLs need to be primed by DCs in lymph nodes, before they start to proliferate, acquire an effector phenotype, traffic to and infiltrate the tumor, and recognize and kill cancer cells (21). The specificity of CTLs for their targets is defined by their T cell receptor (TCR) and its affinity for MHCI molecules loaded with antigenic peptides. Activation of CTLs is triggered by the interaction of the TCR-CD3 complex with MCHI molecule-bound cancer-derived antigen in conjunction with co-stimulatory signals from CD8 and CD28. While being activated through different mechanisms, both NK cells and CTLs establish similar intercellular contacts with cancer cells, referred to as lytic immunological synapses (ISs), whose primary functions are to integrate signaling for activation of the killing machinery and facilitate the execution process itself (22, 23).
The prevalent and best-characterized modalities for eliminating cancer cells are the directed secretion of lytic granules containing pro-apoptotic granzymes and pore-forming perforin, as well as expression of death receptor ligands such as TNF-related apoptosis-inducing ligand (TRAIL) and Fas ligand, which can trigger caspase-dependent apoptosis in target cells expressing the cognate receptors (24–26). The lytic granule pathway is recognized as a highly efficient and rapid killing modality, while the death receptor pathway typically operates at a slower pace primarily due to complex apoptotic signaling cascades. Although these mechanisms exhibit different kinetics, they are not mutually exclusive and can work in synergy. For instance, after a few killing events, CLs can switch from a fast granzyme B-mediated killing to a slow death receptor-mediated killing, thus prolonging their killing capabilities (27).
The formation of an IS plays a crucial role in promoting the directed secretion of lytic granules toward the target cells. Following their convergence to the microtubule organizing center (MTOC) along microtubules, lytic granules undergo polarization towards the presynaptic membrane, thus ensuring their precise positioning for subsequent secretion (28). Upon reaching the secretory domain, lytic granules are secreted into the synaptic cleft, which is a narrow intercellular space. Of note, lytic granule polarization and degranulation are uncoupled events in NKs. While ICAM-1 binding to LFA-1 is sufficient to trigger polarization of granules to the IS (29, 30), additional input derived from other activating receptors is necessary to induce granule secretion (31, 32). By tightly restricting the area of secretion, the synaptic cleft not only enhances the concentration of cytotoxic molecules delivered to the target cell but also minimizes the risk of collateral damage to the surrounding healthy cells. Perforin plays a prominent role in CL-mediated cytotoxicity by creating pores in the target cell membrane, thus leading to a repair response that facilitates granzyme uptake and target cell apoptosis (33–37). Interestingly, a recent study has suggested that effective killing of cancer cells by CTLs might require sequential sublethal hits, a process referred to as “additive cytotoxicity” (38).
The prototypical architecture of the mature IS draws its origins from early investigations on T cell synapses, wherein functionalized lipid bilayers were utilized in place of an antigen presenting cell (APC), enabling high-resolution imaging (39–41). The so-called “bull’s eye” model is characterized by three concentric and functionally distinct domains and has played a pivotal role in understanding IS dynamics, encompassing variations in receptors, signaling molecules, and cytoskeletal organization (42, 43). The innermost domain, known as the central supramolecular activation domain (cSMAC), houses a significant concentration of TCRs and features a loose mesh of actin filaments (AFs) that can only be resolved using super-resolution imaging techniques (44, 45). Adjacent to the cSMAC is the peripheral SMAC (pSMAC), serving as an adhesion domain where the integrin leucocyte function-associated antigen-1 (LFA-1) accumulates and interacts with its ligand ICAM-1 on the target cell. The pSMAC’s architecture is characterized by a concentric network of contractile actin arcs assembled by formins and myosin II (46, 47). Finally, the outermost domain, referred to as the distal SMAC (dSMAC), consists of a dense and branched network of actin filaments encircling the periphery of the IS. Detailed investigations of the ISs formed between CTLs or NK cells and partner immune cells or target diseased cells have revealed a rich array of SMAC-based architectures, such as the multifocal ISs, which deviate from the monocentric topology, expanding our understanding beyond the traditional bull’s eye model (22, 48–54). While this observation may not be universally applicable to all models, an intravital microscopy-based comparative study has highlighted that, in vivo, NK cells tend to form shorter and more dynamic interactions with tumor cells, whereas CTLs establish longer-lasting contacts (55). Intriguingly, both types CLs exhibited similarly effectiveness in killing their targets, suggesting that the disparities in their interaction dynamics with target cells are likely attributable to differences in their activation mechanism and associated signaling pathways, in particular calcium influx, rather than variations in their ability to induce cell death.
Here, we begin by briefly summarizing the roles of the actin cytoskeleton at the pre-synaptic (CL) side of the IS. We then delve into a more comprehensive review of research studies that explore the organization and dynamics of the actin cytoskeleton at the post-synaptic side of the IS with a primary emphasis on cancer cells. Finally, we discuss the potential impact of actin cytoskeleton remodeling within cancer cells on the overall outcome of CL-mediated cytotoxicity.
The actin cytoskeleton at the pre-synaptic side of the CL-cancer cell IS
Extensive research spanning several decades has shed light on the diverse functions of the actin cytoskeleton and actin-generated forces at the pre-synaptic side of NK cell and CTL ISs, including initiation, organization, and maintenance of the IS; assembly of signaling complexes and patterning of receptors; activation of receptors; and controlling the exocytosis of lytic granules (22, 43, 56–59). One of the earliest and most significant events triggered by TCR engagement is the formation of a ring-shaped branched F-actin network at the cell-cell interface’s edge (dSMAC). This plays a critical role in facilitating T cell adhesion to the target cell surface (60–62). The signaling pathway involved in this process includes phosphoinositide 3-kinase (PI3K)-mediated generation of phosphatidylinositol-3,4,5-triphosphate (PIP3), which subsequently recruits DOCK2, an activator of the Rho family small GTPase Rac1. Consequently, activated Rac1 leads to the activation of WAVE2 at the plasma membrane, which in turn mediates activation of the ARP2/3 complex and actin polymerization. The organization and dynamics of this radial dendritic actin network are reminiscent of those found in the lamellipodium of migrating cells and give rise to a centripetal flow of actin (also referred to as retrograde actin flow), which is essential for assembling the TCR signaling complexes and transporting these complexes to the cSMAC where they undergo recycling through actin-dependent endocytosis (63–67). The centripetal flow of actin also directly contributes to stimulating T cell activation by providing the mechanical force required for optimal TCR signaling and promoting integrin conformational change (68–73). In turn, integrin engagement constrains the centripetal flow of actin by recruiting focal adhesion proteins such as talin and vinculin and paxillin, thus modulating the motion of signaling complexes and TCR signaling (74). A similar centripetal flow of actin driving adhesion molecules and receptors toward the IS center exists in the NK cell IS. Its velocity is modulated by the activity of ARP2/3 complex regulator Wiskott-Aldrich-Syndrome-Protein (WASp) and myosin IIA (75) and critically determines the conformation and activity of SH2 domain-containing protein tyrosine phosphatase-1 (SHP-1) - a key inhibitory regulator of the NK cell response (76–78).
The establishment of the synaptic secretory domain involves a reduction in cortical F-actin density at the IS center, which leads to the formation of a permissive network (22, 28, 44, 45, 79–82). Additionally, nanoscale actin dynamism, mediated by the ARP2/3 complex and myosin IIA, generates mechanical forces that facilitate degranulation (79, 83). In CTLs, rapid recovery of higher cortical F-actin levels follows granule exocytosis and directs termination of secretion, potentially by acting as a mechanical barrier (83, 84). Prior to their secretion into the synaptic cleft, lytic granules converge toward the MTOC and subsequently polarize along with the MTOC to the secretory domain (22, 85). The processes of granule convergence and MTOC repositioning to the IS are complex and remain not fully understood (86), with dynein, a microtubule minus-end-directed motor protein, being implicated in these processes (87, 88). Recent evidence highlights the critical role of cortical clearance of F-actin within the IS in dynein recruitment and MTOC reorientation, thus illustrating that the actin and tubulin cytoskeletons closely interact to coordinate CL polarization and exocytosis (86, 89, 90). Intriguingly, the F-actin cross-linking protein FLNa has been identified to facilitate multiple steps of NK cell cytotoxicity, including conjugation to target cells, synaptic accumulation of F-actin, and lytic granule degranulation, while limiting the production of IFN-γ and TNF-α (91). This suggests that modulation of actin dynamics contributes in steering CLs toward cytotoxicity or regulatory functions through cytokine production. Readers are referred to recent reviews for a more comprehensive description of the regulation and roles of the actin cytoskeleton at the pre-synaptic side of the IS (57, 90, 92, 93).
Actin dynamics at the post-synaptic side of the immunological synapse: lessons from lymphocyte-dendritic cell interactions
Until recently, the exploration of actin dynamics in cancer cells during their interaction with CLs and formation of the lytic IS has been limited. However, studies on other types of ISs formed by T cells and NK cells have shed light on the importance of actin dynamics in the conjugated target cells particularly dendritic cells (DCs) in regulating IS formation, organization, and activity. For a more comprehensive discussion on the actin cytoskeleton at the DC side of the IS, readers are invited to refer to a recent review that provides information beyond the scope of the present section (94). Over two decades ago, pioneering investigations unveiled the active reorganization of the actin cytoskeleton in DCs during their interaction with resting CD4+ T cells, thus leading to a robust polarization of F-actin and actin regulatory proteins, such as the actin bundling protein fascin, towards the IS (95–97). Such remodeling of the DC actin cytoskeleton is necessary for optimal cell-cell conjugation and activation of T cells.
Additional evidence of the changes in actin dynamics occurring in DCs during conjugation with T cells was provided by fluorescence recovery after photobleaching (FRAP) analysis, which revealed an overall stabilization of filamentous actin at the cell-cell interface compared to distal cortical regions (98). More recent studies show the mechanistic details underlying the role of actin cytoskeleton remodeling in DCs in the regulation of the T cell IS (99). Remarkably, depolymerization of F-actin in DCs induces the conversion of the T cell IS from a multifocal to monofocal topology (49, 99), thus supporting the involvement of the DC actin cytoskeleton in patterning the T cell IS. Similar to the T cell side of the IS, the DC side exhibits a multifocal topology that is characterized by multiple and dynamic small and larger actin patches separated by actin hypodense areas. The inhibition of the lateral movement of these patches following the genetic ablation of the Wiskott-Aldrich-Syndrome-Protein (WASP)-family verprolin-homologous protein (WAVE) regulatory complex (WRC), a critical regulator of the ARP2/3 complex, leads to prolonged cell-cell conjugation time and reduction in T cell priming efficiency (99). Such effects are primarily attributed to an overstabilization of ICAM-1 on the DC surface through the activation of ERM proteins that connect ICAM-1 and the cortical actin cytoskeleton, and the subsequent increase in synaptic ICAM-1-LFA-1 interactions and overstabilization of the IS.
As previously stated, the mechanical forces exerted by the actin cytoskeleton play critical role in modulating TCR signaling. For an in-depth understanding of this role and the underlying molecular mechanisms, readers are referred to an excellent review article (43). While the mechanical forces applied to the TCR stem from the actin-dependent pushing and pulling forces exerted by CLs on the APC surface (100, 101), they are also contingent upon the biomechanical properties of the APC - particularly the cortical stiffness, which is mostly governed by the cytoskeleton (99, 102, 103). Remarkably, DCs undergo a 2-3-fold (actin cytoskeleton-dependent) increase in cortical stiffness during their maturation, which is perceived by T cells as a co-stimulatory signal (104). As part of the underlying mechanism, cortical stiffening increases TCR complex mechanical stimulation, thus leading to enhanced downstream signaling and lowering the antigenic threshold required to activate T cells (43, 100, 101, 103–106). Of note, both the ARP2/3 complex- and formin-mediated actin polymerization contribute to the increased cortical stiffness of mature DCs (104).
Similar to T cells, NK cells physically interact with DCs via an IS. NK cells contribute to the selection of immunogenic DCs by selectively killing immature DCs. This editing process is critical for the initiation of anti-cancer immune responses by promoting successful T cell priming (107, 108). However, NK cells also engage with mature DCs within a non-lytic IS, thus leading to a reciprocal cross-talk between the two cell types, which is crucial for orchestrating and regulating the immune response against cancer. Such, non-lytic, NK-DC IS is referred to as the regulatory IS (109) and facilitates the acquisition of effector functions by naïve NK cells (110–113). In turn, DC-activated NK cells can stimulate DC maturation (114). The investigation into how mature DCs evade destruction while synapsing with NK cells has shed light on the key role of the DC actin cytoskeleton in facilitating this process (115). Throughout the conjugation process, there is a gradual relocation of the filamentous actin cytoskeleton in DCs towards the synaptic region. Such cytoskeletal polarization was associated with a blockade of NK cell cytotoxicity as indicated by the absence of MTOC and perforin polarization to the IS. Remarkably, genetic or pharmacological inhibition of actin polymerization in DCs induced the conversion of the regulatory IS into a lytic IS as indicated by NK cell MTOC and perforin polarization, as well as a significant increase in degranulation, IFN-γ production, and DC killing. As an underlying mechanism, DC-derived F-actin was shown to induce the polarization of MHCI molecules to the IS and restrict their diffusion away from the IS.
Collectively, these studies emphasize the crucial role of the synaptic actin cytoskeleton in DCs, which plays a pivotal role in modulating the organization of T cell and NK cell ISs and regulating the synaptic density of signaling molecules, thus ultimately safeguarding DCs from CL-mediated cytotoxicity. Additionally, emerging evidence suggests that the cortical stiffness of DCs, which is primarily determined by cytoskeletal dynamics, acts as a critical regulator of the mechanical response of T cells. These findings imply that specific configurations or characteristics of the cortical actin cytoskeleton of cancer cells may be indispensable for facilitating efficient CL-mediated killing. Cancer cells may exploit actin cytoskeleton remodeling to alter the morphology and activity of the lytic IS and evade immune destruction. These intriguing possibilities will be evaluated and discussed below.
Intrinsic cytoskeletal properties of cancer cells and the lytic immunological synapse
Compelling evidence suggest that mechanical forces within the lytic IS significantly influence CL cytotoxicity potential through multiple mechanisms (43, 116). As previously discussed, these forces play a crucial role as co-activating signals in both CTLs and NK cells, thus enhancing immunoreceptor affinity for ligands and promoting conformational changes in receptors and integrins, which subsequently amplify downstream signaling (117–121). Moreover, CL-originating mechanical forces have been shown to potentiate the pore-forming activity of perforin by increasing target cell membrane tension, thus enhancing target cell killing (122, 123). In turn, recent research has highlighted the significance of target cell rigidity as a pivotal factor governing the magnitude of the synaptic forces, as well as its impacts on lytic IS formation and activity (124). For instance, experiments utilizing stiff beads that were functionalized with antibodies against the NK activating receptor NKp30 and LFA-1 have proven highly effective in triggering NK cell degranulation (116). Conversely, softer beads fail to induce the NK cell’s MTOC and lytic granule polarization, and only lead to the formation of poorly stable IS.
The implications of target cell rigidity are especially pertinent to cancer because cancer cells tend to undergo softening during the metastatic cascade and the acquisition of invasive properties, primarily due to alterations in their cytoskeletal architecture (125–130). In this regard, a recent study established a compelling link between the mechanical softness of cancer cells and their ability to hinder the formation of membrane pores by perforin. The data suggested that this evasion mechanism is utilized by a subset of undifferentiated, stem cell-like, cancer cells (characterized by their intrinsic softness) to escape CTL-mediated cytotoxicity (131). The manipulation of cellular stiffness using pharmacological or genetic approaches targeting the actin cytoskeleton was proven to dictate the susceptibility of both murine cancer cell models and primary human cancer cells to perforin-mediated perforation. Most remarkably, stiffening tumor cells with the actin polymerization promoting drug jasplakinolide significantly improved antitumor T cell immunity and PD-1 blockade immunotherapy in in vivo mouse models. Softening tumor cells with the myosin II inhibitor blebbistatin had opposite effects. Another important finding of this study is the identification of MYH9 - the heavy chain of non-muscle myosin IIA that crosslinks and contracts actin filaments to produce mechanical forces (132) - as a critical molecule for perforin pore formation. A MYH9 Knockout proved sufficient to make cells softer and inhibit the pore-forming activity of perforin in several cancer cell models. Although the exact mechanism remains elusive, perforin might interact with MYH9 and thus trigger an opposite force whose strength is proportional to the amount of F-actin linked to MYH9. While this force would be sufficient to induce perforin active conformation in the case of stiff cancer cells, it would remain insufficient in the case of soft cells.
The association between the actin polymerization status of cancer cells and their susceptibility to CL-mediated lysis was previously suggested in an earlier study that focused on a variant of a non-small-cell lung carcinoma cell line exposed to in vitro CTL selection pressure (133). This particular resistant variant exhibited a substantial reduction in filamentous actin content, resulting in a consistent shift toward a more rounded cellular morphology. Transcriptomics uncovered notable dysregulation in the expression of various genes. Among those, two genes encoding cytoskeleton regulators, ephrin-A1 and scinderin, were markedly upregulated and played a pivotal role in driving alterations in cell morphology alterations and conferring resistance to CTL-mediated lysis. Although target cell perforation was not specifically evaluated in this model, it is noteworthy that the resistant cells were proficient in inducing CTL degranulation. This observation raises the possibility that these cells may exhibit resistance to pore-forming activity. Another intriguing observation was the disorganization of the IS formed between CTLs and resistant cells, characterized by a more loosely organized synaptic cleft and a reduced number of close contacts when compared to the IS formed with susceptible cells.
Another recent study has unveiled an important link between the expression levels of myocardin-related transcription factors (MRTFs), the rigidity of cancer cells and the control of tumors by CLs (134). MRTFs play a pivotal role as co-factors for the serum response factor (SRF), responding to actin polymerization by dissociating from actin monomers and relocating to the nucleus where they transcriptionally activate a range of SRF-target genes related to the actin cytoskeleton (135). MRTFs are indispensable for actin-based cell migration during physiological processes and cancer progression (136). As cancer cells disseminate via the bloodstream and infiltrate supportive perivascular niches, their ability to establish prominent metastatic lesions relies on their adhesion and spreading onto the endothelial basement membrane (137). This process critically requires MRTF activity (138, 139). Strikingly, Tello-Lafoz et al. have established that beyond their pro-metastatic function in facilitating adaptation to the metastatic niche, MRTFs paradoxically sensitize cancer cells in vivo to CL-mediated destruction, and increase the effectiveness of PD1 and CTLA4 checkpoint blockade therapies (134). Mechanistic investigations have indicated that MRTF-induced actin polymerization increases the stiffness of cancer cells, which stimulates IS formation and CL cytotoxic potential as demonstrated by increased levels of cytokine secretion and degranulation.
Further strengthening the concept that cancer cell stiffness is an actionable therapeutic point of intervention, membrane cholesterol depletion-induced stiffening of cancer cells has recently been demonstrated to enhance CTL-mediated cytotoxicity as well as the efficacy of adoptive T-cell therapy in preclinical mouse models of solid tumors (140). The vulnerability of cancer cells resulting from membrane cholesterol depletion primarily depends on actin cytoskeleton-dependent forces exerted by CTLs at the IS. It is noteworthy that, in contrast to the cytoskeleton rigidification resulting from MRTF overexpression (134), cholesterol depletion fails to induce a significant increase in CTL cytokine production or degranulation (140). This intriguing disparity underscores distinctions in underlying mechanisms among the approaches targeting the enhancement of cancer cell rigidity. Additional investigations are required to unravel and characterize these intricate mechanistic distinctions.
Actin dynamics at cancer cell side of the immunological synapse
The target cell side of the lytic IS has long been regarded as a relatively passive area and is primarily characterized by the array of molecules present on its surface. This perception has mainly evolved from the historical focus on the presynaptic cell side of the IS as well as the use of reductionists target cell models amenable to high-resolution microscopy such as glass-supported planar lipid bilayers wherein specific proteins are embedded to initiate IS formation. However, a growing body of research centered around cell-cell conjugates has underscored the dynamic nature of the cancer cell side - particularly with regards to actin dynamics and vesicular and membrane trafficking (57, 141–143).
In a recent study, we thoroughly investigated the distribution of the actin cytoskeleton within breast cancer cells upon encountering NK cells. We found that a subset of breast cancer cells responded to the attack of NK cells by exhibiting a prominent accumulation of filamentous actin within the synaptic region (144). This intriguing response somehow paralleled the cytoskeletal modifications observed in DCs during their interaction with NK cells in the context of the regulatory synapse (115, 145). Moreover, akin to DCs, the synaptic accumulation of actin filaments was associated with breast cancer cell resistance to NK cell-mediated cytotoxicity. Remarkably, such resistance could be reverted by silencing CDC42 or N-WASP, two regulators of the ARP2/3 complex, thus leading to a return of cancer cells to a susceptible phenotype (144). In a follow-up study we extended the concept of actin remodeling-mediated resistance to several chronic lymphocytic leukemia (CLL) cell lines and primary cells from CLL patients (146). Inhibition of synaptic actin accumulation using a CDC42-targeting drug - combined with an anti-HLA-G blocking antibody as a mean to increase conjugate formation - resulted in a substantial enhancement of NK cell-mediated killing of primary CLL cells.
The precise mechanism through which the polarization of cancer cells’ actin cytoskeleton to the IS leads to resistance to NK cell-mediated cytotoxicity remains unclear. The accumulation of F-actin in the postsynaptic region of the IS has been linked to diminished levels of granzyme B and a reduction in apoptosis within target cells (144, 146). However, whether this reduction stems from actin cytoskeleton-mediated resistance against perforin, the degradation of granzyme B, or inhibition of NK cell killing activity requires further investigation. Remarkably, similar to the scenario observed in the DC-NK cell inhibitory IS (115), breast cancer cells exhibiting synaptic F-actin accumulation also display increased levels of HLA-A,-B,-C and PD-L1 at the IS (144). This observation suggests that F-actin accumulation at the postsynaptic side of the IS might act as a scaffold for the clustering of NK cell inhibitory ligands, leading to potent inhibitory signal against NK cells. This remains largely speculative and will require rigorous experimental evaluation.
The mechanism responsible for initiating actin remodeling in NK cell-conjugated cancer cells remains a subject of ongoing investigation. Interestingly, K562 cells, characterized by their lack of MHCI expression and highly susceptibility to NK cell-meditated cytotoxicity have been documented as incapable of remodeling their actin cytoskeleton in response to NK cell conjugation (115). This intriguing observation raises the possibility that MHCI expression might be instrumental in driving the polarization of F-actin to the post-synaptic side of the IS. It is also unclear why discrete cells within the same cancer cell line do not uniformly exhibit the same ability to respond to NK cell attack by actin cytoskeleton remodeling (144). This variability may arise from the inherent phenotypic heterogeneity within a single cell line including factors such as the epithelial-mesenchymal transition (EMT) status of individual cells (147). This diversity can contribute to differing capacities to promptly reconfigure the actin cytoskeleton in response to NK cell IS formation. Notably, the process of EMT has been recognized as a critical driver of tumor immune evasion (148), and has been implicated in the overall proficiency of cancer cell lines to effectively polarize their actin cytoskeleton towards the IS (144).
Concluding remarks
Recent breakthroughs have prompted an increasing interest in actin dynamics at the cancer cell side of the IS and support the notion that the actin cytoskeleton of cancer cells is a double-edged sword exerting a pivotal influence on the regulation of CL cytotoxic activity. The cytoskeletal and biophysical properties of cancer cells are exploited by CLs to adjust their response to the biochemical signals present at the surface of cancer cells - a mechanism termed mechanosurveillance. Notably, the stiffening of the actomyosin cytoskeletal network, e.g., during cancer cell spread and colonization at new metastatic sites, serves as a trigger for the assembly of the IS and subsequent unleashing of lytic activity. This cytoskeleton-mediated rigidification of cancer cells amplifies force-dependent signaling in CLs, thus enhancing their proficiency in target cell recognition. Furthermore, elevated mechanical tension stands indispensable for the assembly of perforin-based transmembrane pores in cancer cells, which are pivotal for mediating cytotoxic effects.
Conversely, the softening of the actin cytoskeleton - a phenomenon often accompanying the transition of cancer cells towards an invasive and metastatic state - has emerged as a novel immune checkpoint exploited by cancer cells to dampen CL cytotoxicity. For a more comprehensive discussion on the synergistic interplay between biomechanical and biochemical cues leading to a robust antitumor immune response, readers are encouraged to refer to the recent review article “Harder, better, faster, stronger: biochemistry and biophysics in the immunosurveillance concert” (124).
Our live cell investigations into actin dynamics within cancer cells during their interaction with NK cells have revealed the remarkable ability of cancer cells to rapidly and extensively remodel their actin cytoskeleton upon engaging with NK cells, regardless of gene expression changes. This cytoskeletal remodeling is characterized by massive polarization of filamentous actin towards the IS, thus mirroring the cytoskeletal changes occurring in CLs during the IS initiation. The polarization of the cancer cell’s actin cytoskeleton provides robust protection against CL-mediated killing by efficiently reducing intracellular granzyme B levels within target cells. Nevertheless, the elucidation of the precise underlying mechanism requires further investigation. F-actin accumulation at the tumor cell side of the IS could be considered as an evasion strategy per se, possibly through inducing IS disorganization and destabilization, or as a fundamental cellular process shared across multiple synaptic evasion tactics. This remains an open question. While this remains speculative, augmenting synaptic actin dynamics could facilitate the recruitment of lysosomes and subsequent membrane repair (143), stimulate autophagy-mediated degradation of granzyme B (149), promote the synaptic recruitment of inhibitory ligands and immune checkpoint molecules (115, 144), and trigger local changes in membrane composition mediating increased resistance to perforin (145, 150). A concise summary of the primary findings and hypotheses discussed in this review article is provided in Table 1.
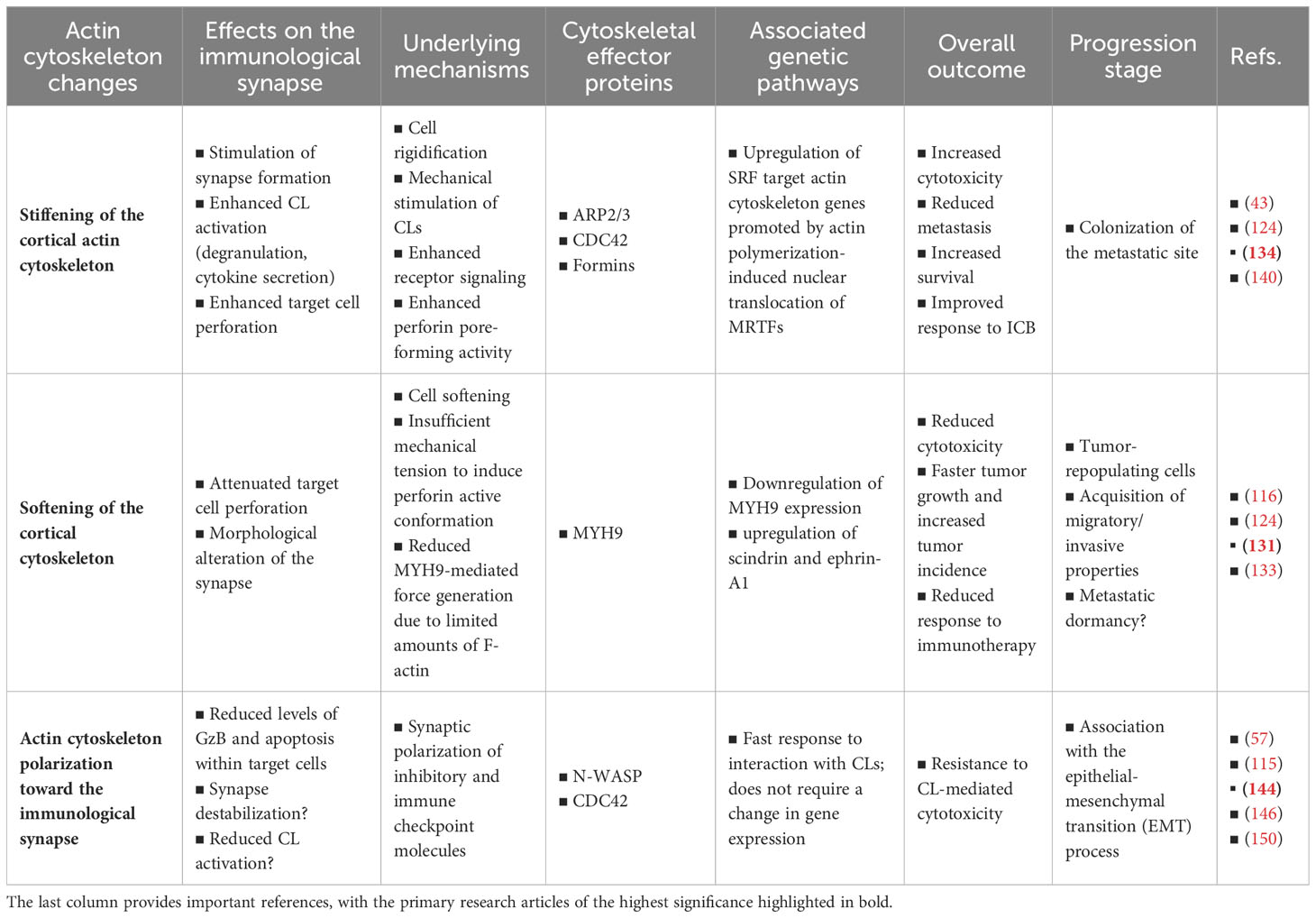
Table 1 Key findings and hypotheses regarding the impact of actin cytoskeleton remodeling within cancer cells on the lytic immunological synapse.
Innovating therapeutic approaches aiming to enhance physical contact between CLs and cancer cells or APCs and increase CL cytotoxicity against cancer cells, hold tremendous potential in cancer treatment (151). In particular, chimeric antigen receptors (CAR)-T and CAR-NK cell therapies have shown great promise – particularly in hematologic malignancies (152–160). Preclinical models and advanced intravital imaging techniques have been combined to provide real-time visualization of cancer cell elimination by CAR-CLs at the single-cell level within living organisms (161–164). Such breakthrough technologies open exciting avenues for in-depth mechanistic investigations and for translating these findings into clinical applications, such as the prospective assessment of the clinical effectiveness of CAR-based therapies. CAR synapses display several structural and functional differences compared to conventional ISs (53, 165), thus potentially limiting the effectiveness of CAR-based therapies. Recent advancements such as the incorporation of an intracellular scaffolding protein binding site, a PDZ binding domain, to the CAR of CAR-T or -NK cells, have shown improved IS formation and CL polarization, leading in turn to enhanced anti-tumor activity and prolonged survival in several in vivo tumor models including in solid tumors (166). The development of various CAR products has catalyzed the expansion of clinical trials aimed at addressing a wide spectrum of hematologic and solid malignancies (167). In the light of the present discussion, the concept of targeting the postsynaptic actin cytoskeleton, in conjunction with optimized CAR CLs, or alternative CL redirecting strategies, holds significant potential for triggering a potent anti-tumor response. The combination of CAR-based therapy with other treatment modalities, such as radiotherapy or immune checkpoint blockade, has emerged as one of the most auspicious approaches and is being currently under evaluation in clinical trials (168, 169). However, the ubiquitous expression of the actin cytoskeleton and its multiple roles in physiological processes - particularly within immune responses - pose significant challenges for the development of therapeutic approaches aimed at manipulating the organization and dynamics of the actin cytoskeleton within cancer cells. Furthermore, unlike microtubule-targeting drugs that exhibit a preference for selectively impairing rapidly dividing cancer cells and have demonstrated efficacy in treating various malignancies, actin-targeting drugs result in unacceptable levels of toxicity precluding their clinical utility. The identification of relevant molecular targets will require a thorough dissection of the cancer cell side of the IS and the development of innovative methodologies that can distinguish between the pre- and post-synaptic sides of the IS (170).
Author contributions
CT: Conceptualization, Funding acquisition, Supervision, Writing – original draft. EO: Conceptualization, Writing – original draft. LF: Supervision, Writing – review & editing. DPF: Writing – review & editing. CH: Writing – review & editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. CT, LF and CH are supported by the Luxembourg National Research Fund (FNR) and the Fondation Cancer, Luxembourg (C21/BM/15752542/SYNAPODIA and FC/2019/02/ACTIMMUNE). EO is recipient of a PhD fellowship from the Luxembourg National Research Fund, Luxembourg (PRIDE19/14254520/i2Tron). DPF is recipient of a PhD fellowship from Fonds De La Recherche Scientifique (FNRS), Belgium (Télévie 7.4594.23) and is supported by a Marian Aldred Award from Think Pink Lux, Luxembourg.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Malmberg KJ, Carlsten M, Bjorklund A, Sohlberg E, Bryceson YT, Ljunggren HG. Natural killer cell-mediated immunosurveillance of human cancer. Semin Immunol (2017) 31:20–9. doi: 10.1016/j.smim.2017.08.002
2. Raulet DH, Guerra N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nat Rev Immunol (2009) 9:568–80. doi: 10.1038/nri2604
3. Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell (2017) 32:135–54. doi: 10.1016/j.ccell.2017.06.009
4. Senovilla L, Vacchelli E, Galon J, Adjemian S, Eggermont A, Fridman WH, et al. Trial watch: Prognostic and predictive value of the immune infiltrate in cancer. Oncoimmunology (2012) 1:1323–43. doi: 10.4161/onci.22009
5. Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol (2018) 18:671–88. doi: 10.1038/s41577-018-0061-z
6. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science (2020) 367. doi: 10.1126/science.aba7365
7. Raskov H, Orhan A, Christensen JP, Gogenur I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br J Cancer (2021) 124:359–67. doi: 10.1038/s41416-020-01048-4
8. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol (2008) 9:495–502. doi: 10.1038/ni1581
9. Joncker NT, Fernandez NC, Treiner E, Vivier E, Raulet DH. NK cell responsiveness is tuned commensurate with the number of inhibitory receptors for self-MHC class I: the rheostat model. J Immunol (2009) 182:4572–80. doi: 10.4049/jimmunol.0803900
10. Vivier E, Nunes JA, Vely F. Natural killer cell signaling pathways. Science (2004) 306:1517–9. doi: 10.1126/science.1103478
11. Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood (2006) 107:159–66. doi: 10.1182/blood-2005-04-1351
12. Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity (2006) 25:331–42. doi: 10.1016/j.immuni.2006.06.013
13. Hao L, Klein J, Nei M. Heterogeneous but conserved natural killer receptor gene complexes in four major orders of mammals. Proc Natl Acad Sci USA (2006) 103:3192–7. doi: 10.1073/pnas.0511280103
14. Carrillo-Bustamante P, Kesmir C, de Boer RJ. The evolution of natural killer cell receptors. Immunogenetics (2016) 68:3–18. doi: 10.1007/s00251-015-0869-7
15. Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today (1990) 11:237–44. doi: 10.1016/0167-5699(90)90097-S
16. Gasteiger G, Rudensky AY. Interactions between innate and adaptive lymphocytes. Nat Rev Immunol (2014) 14:631–9. doi: 10.1038/nri3726
17. Ramirez-Labrada A, Pesini C, Santiago L, Hidalgo S, Calvo-Perez A, Onate C, et al. All about (NK cell-mediated) death in two acts and an unexpected encore: initiation, execution and activation of adaptive immunity. Front Immunol (2022) 13:896228. doi: 10.3389/fimmu.2022.896228
18. Huntington ND, Cursons J, Rautela J. The cancer-natural killer cell immunity cycle. Nat Rev Cancer (2020) 20:437–54. doi: 10.1038/s41568-020-0272-z
19. Bald T, Krummel MF, Smyth MJ, Barry KC. The NK cell-cancer cycle: advances and new challenges in NK cell-based immunotherapies. Nat Immunol (2020) 21:835–47. doi: 10.1038/s41590-020-0728-z
20. Laskowski TJ, Biederstadt A, Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer (2022) 22:557–75. doi: 10.1038/s41568-022-00491-0
21. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
22. Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol (2008) 8:713–25. doi: 10.1038/nri2381
23. Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity (2001) 15:751–61. doi: 10.1016/S1074-7613(01)00234-5
24. Martinez-Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res (2015) 21:5047–56. doi: 10.1158/1078-0432.CCR-15-0685
25. Prager I, Watzl C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J Leukoc Biol (2019) 105:1319–29. doi: 10.1002/JLB.MR0718-269R
26. Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol (2008) 26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404
27. Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandstrom N, et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J Exp Med (2019) 216:2113–27. doi: 10.1084/jem.20181454
28. Stinchcombe JC, Majorovits E, Bossi G, Griffiths GM. Centrosome polarization delivers secretory granules to the immunological synapse. Nature (2006) 443:462–5. doi: 10.1038/nature05071
29. Barber DF, Faure M, Long EO. LFA-1 contributes an early signal for NK cell cytotoxicity. J Immunol (2004) 173:3653–9. doi: 10.4049/jimmunol.173.6.3653
30. Mace EM, Monkley SJ, Critchley DR, Takei F. A dual role for talin in NK cell cytotoxicity: activation of LFA-1-mediated cell adhesion and polarization of NK cells. J Immunol (2009) 182:948–56. doi: 10.4049/jimmunol.182.2.948
31. Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J Exp Med (2005) 202:1001–12. doi: 10.1084/jem.20051143
32. Bryceson YT, Ljunggren HG, Long EO. Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood (2009) 114:2657–66. doi: 10.1182/blood-2009-01-201632
33. Thiery J, Keefe D, Boulant S, Boucrot E, Walch M, Martinvalet D, et al. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat Immunol (2011) 12:770–7. doi: 10.1038/ni.2050
34. Keefe D, Shi L, Feske S, Massol R, Navarro F, Kirchhausen T, et al. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity (2005) 23:249–62. doi: 10.1016/j.immuni.2005.08.001
35. Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature (1994) 369:31–7. doi: 10.1038/369031a0
36. Baran K, Dunstone M, Chia J, Ciccone A, Browne KA, Clarke CJ, et al. The molecular basis for perforin oligomerization and transmembrane pore assembly. Immunity (2009) 30:684–95. doi: 10.1016/j.immuni.2009.03.016
37. Law RH, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, Dunstone MA, et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature (2010) 468:447–51. doi: 10.1038/nature09518
38. Weigelin B, den Boer AT, Wagena E, Broen K, Dolstra H, de Boer RJ, et al. Cytotoxic T cells are able to efficiently eliminate cancer cells by additive cytotoxicity. Nat Commun (2021) 12:5217. doi: 10.1038/s41467-021-25282-3
39. Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature (1998) 395:82–6. doi: 10.1038/25764
40. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science (1999) 285:221–7. doi: 10.1126/science.285.5425.221
41. Fooksman DR, Vardhana S, Vasiliver-Shamis G, Liese J, Blair DA, Waite J, et al. Functional anatomy of T cell activation and synapse formation. Annu Rev Immunol (2010) 28:79–105. doi: 10.1146/annurev-immunol-030409-101308
42. Dieckmann NM, Frazer GL, Asano Y, Stinchcombe JC, Griffiths GM. The cytotoxic T lymphocyte immune synapse at a glance. J Cell Sci (2016) 129:2881–6. doi: 10.1242/jcs.186205
43. Blumenthal D, Burkhardt JK. Multiple actin networks coordinate mechanotransduction at the immunological synapse. J Cell Biol (2020) 219. doi: 10.1083/jcb.201911058
44. Brown AC, Oddos S, Dobbie IM, Alakoskela JM, Parton RM, Eissmann P, et al. Remodelling of cortical actin where lytic granules dock at natural killer cell immune synapses revealed by super-resolution microscopy. PloS Biol (2011) 9:e1001152. doi: 10.1371/journal.pbio.1001152
45. Rak GD, Mace EM, Banerjee PP, Svitkina T, Orange JS. Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PloS Biol (2011) 9:e1001151. doi: 10.1371/journal.pbio.1001151
46. Yi J, Wu XS, Crites T, Hammer JA 3rd. Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Mol Biol Cell (2012) 23:834–52. doi: 10.1091/mbc.E11-08-0731
47. Murugesan S, Hong J, Yi J, Li D, Beach JR, Shao L, et al. Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. J Cell Biol (2016) 215:383–99. doi: 10.1083/jcb.201603080
48. Almeida CR, Davis DM. Segregation of HLA-C from ICAM-1 at NK cell immune synapses is controlled by its cell surface density. J Immunol (2006) 177:6904–10. doi: 10.4049/jimmunol.177.10.6904
49. Brossard C, Feuillet V, Schmitt A, Randriamampita C, Romao M, Raposo G, et al. Multifocal structure of the T cell - dendritic cell synapse. Eur J Immunol (2005) 35:1741–53. doi: 10.1002/eji.200425857
50. Friedl P, den Boer AT, Gunzer M. Tuning immune responses: diversity and adaptation of the immunological synapse. Nat Rev Immunol (2005) 5:532–45. doi: 10.1038/nri1647
51. Gunzer M, Weishaupt C, Hillmer A, Basoglu Y, Friedl P, Dittmar KE, et al. A spectrum of biophysical interaction modes between T cells and different antigen-presenting cells during priming in 3-D collagen and in vivo. Blood (2004) 104:2801–9. doi: 10.1182/blood-2004-03-1193
52. Carlin LM, Eleme K, McCann FE, Davis DM. Intercellular transfer and supramolecular organization of human leukocyte antigen C at inhibitory natural killer cell immune synapses. J Exp Med (2001) 194:1507–17. doi: 10.1084/jem.194.10.1507
53. Davenport AJ, Cross RS, Watson KA, Liao Y, Shi W, Prince HM, et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc Natl Acad Sci USA (2018) 115:E2068–E76. doi: 10.1073/pnas.1716266115
54. Kumari S, Colin-York H, Irvine DJ, Fritzsche M. Not all T cell synapses are built the same way. Trends Immunol (2019) 40:977–80. doi: 10.1016/j.it.2019.09.009
55. Deguine J, Breart B, Lemaitre F, Di Santo JP, Bousso P. Intravital imaging reveals distinct dynamics for natural killer and CD8(+) T cells during tumor regression. Immunity (2010) 33:632–44. doi: 10.1016/j.immuni.2010.09.016
56. Dustin ML, Cooper JA. The immunological synapse and the actin cytoskeleton: molecular hardware for T cell signaling. Nat Immunol (2000) 1:23–9. doi: 10.1038/76877
57. Wurzer H, Hoffmann C, Al Absi A, Thomas C. Actin cytoskeleton straddling the immunological synapse between cytotoxic lymphocytes and cancer cells. Cells (2019) 8. doi: 10.3390/cells8050463
58. Roy NH, Burkhardt JK. The actin cytoskeleton: A mechanical intermediate for signal integration at the immunological synapse. Front Cell Dev Biol (2018) 6:116. doi: 10.3389/fcell.2018.00116
59. Ritter AT, Angus KL, Griffiths GM. The role of the cytoskeleton at the immunological synapse. Immunol Rev (2013) 256:107–17. doi: 10.1111/imr.12117
60. Barda-Saad M, Braiman A, Titerence R, Bunnell SC, Barr VA, Samelson LE. Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat Immunol (2005) 6:80–9. doi: 10.1038/ni1143
61. Nolz JC, Gomez TS, Zhu P, Li S, Medeiros RB, Shimizu Y, et al. The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation. Curr Biol (2006) 16:24–34. doi: 10.1016/j.cub.2005.11.036
62. Le Floc’h A, Tanaka Y, Bantilan NS, Voisinne G, Altan-Bonnet G, Fukui Y, et al. Annular PIP3 accumulation controls actin architecture and modulates cytotoxicity at the immunological synapse. J Exp Med (2013) 210:2721–37. doi: 10.1084/jem.20131324
63. Campi G, Varma R, Dustin ML. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J Exp Med (2005) 202:1031–6. doi: 10.1084/jem.20051182
64. Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity (2006) 25:117–27. doi: 10.1016/j.immuni.2006.04.010
65. McGavin MK, Badour K, Hardy LA, Kubiseski TJ, Zhang J, Siminovitch KA. The intersectin 2 adaptor links Wiskott Aldrich Syndrome protein (WASp)-mediated actin polymerization to T cell antigen receptor endocytosis. J Exp Med (2001) 194:1777–87. doi: 10.1084/jem.194.12.1777
66. Lee KH, Dinner AR, Tu C, Campi G, Raychaudhuri S, Varma R, et al. The immunological synapse balances T cell receptor signaling and degradation. Science (2003) 302:1218–22. doi: 10.1126/science.1086507
67. Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc Natl Acad Sci USA (2007) 104:20296–301. doi: 10.1073/pnas.0710258105
68. Springer TA, Dustin ML. Integrin inside-out signaling and the immunological synapse. Curr Opin Cell Biol (2012) 24:107–15. doi: 10.1016/j.ceb.2011.10.004
69. Ilani T, Vasiliver-Shamis G, Vardhana S, Bretscher A, Dustin ML. T cell antigen receptor signaling and immunological synapse stability require myosin IIA. Nat Immunol (2009) 10:531–9. doi: 10.1038/ni.1723
70. Das DK, Feng Y, Mallis RJ, Li X, Keskin DB, Hussey RE, et al. Force-dependent transition in the T-cell receptor beta-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc Natl Acad Sci USA (2015) 112:1517–22. doi: 10.1073/pnas.1424829112
71. Comrie WA, Babich A, Burkhardt JK. F-actin flow drives affinity maturation and spatial organization of LFA-1 at the immunological synapse. J Cell Biol (2015) 208:475–91. doi: 10.1083/jcb.201406121
72. Babich A, Li S, O’Connor RS, Milone MC, Freedman BD, Burkhardt JK. F-actin polymerization and retrograde flow drive sustained PLCgamma1 signaling during T cell activation. J Cell Biol (2012) 197:775–87. doi: 10.1083/jcb.201201018
73. Hu KH, Butte MJ. T cell activation requires force generation. J Cell Biol (2016) 213:535–42. doi: 10.1083/jcb.201511053
74. Jankowska KI, Williamson EK, Roy NH, Blumenthal D, Chandra V, Baumgart T, et al. Integrins modulate T cell receptor signaling by constraining actin flow at the immunological synapse. Front Immunol (2018) 9:25. doi: 10.3389/fimmu.2018.00025
75. Sabag B, Levy M, Kivelevitz J, Dashevsky N, Ben-Shmuel A, Puthenveetil A, et al. Actin retrograde flow regulated by the wiskott-aldrich syndrome protein drives the natural killer cell response. Cancers (Basel) (2022) 14. doi: 10.3390/cancers14153756
76. Matalon O, Ben-Shmuel A, Kivelevitz J, Sabag B, Fried S, Joseph N, et al. Actin retrograde flow controls natural killer cell response by regulating the conformation state of SHP-1. EMBO J (2018) 37. doi: 10.15252/embj.201696264
77. Matalon O, Fried S, Ben-Shmuel A, Pauker MH, Joseph N, Keizer D, et al. Dephosphorylation of the adaptor LAT and phospholipase C-gamma by SHP-1 inhibits natural killer cell cytotoxicity. Sci Signal (2016) 9:ra54. doi: 10.1126/scisignal.aad6182
78. Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, Long EO. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol Cell Biol (2003) 23:6291–9. doi: 10.1128/MCB.23.17.6291-6299.2003
79. Carisey AF, Mace EM, Saeed MB, Davis DM, Orange JS. Nanoscale dynamism of actin enables secretory function in cytolytic cells. Curr Biol (2018) 28:489–502.e9. doi: 10.1016/j.cub.2017.12.044
80. Ritter AT, Asano Y, Stinchcombe JC, Dieckmann NM, Chen BC, Gawden-Bone C, et al. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity (2015) 42:864–76. doi: 10.1016/j.immuni.2015.04.013
81. Mace EM, Orange JS. Dual channel STED nanoscopy of lytic granules on actin filaments in natural killer cells. Commun Integr Biol (2012) 5:184–6. doi: 10.4161/cib.18818
82. Mace EM, Orange JS. Lytic immune synapse function requires filamentous actin deconstruction by Coronin 1A. Proc Natl Acad Sci USA (2014) 111:6708–13. doi: 10.1073/pnas.1314975111
83. Hammer JA. Immunology: is actin at the lytic synapse a friend or a foe? Curr Biol (2018) 28:R155–R7. doi: 10.1016/j.cub.2018.01.013
84. Ritter AT, Kapnick SM, Murugesan S, Schwartzberg PL, Griffiths GM, Lippincott-Schwartz J. Cortical actin recovery at the immunological synapse leads to termination of lytic granule secretion in cytotoxic T lymphocytes. Proc Natl Acad Sci U.S.A. (2017) 114:E6585–E94. doi: 10.1073/pnas.1710751114
85. Mentlik AN, Sanborn KB, Holzbaur EL, Orange JS. Rapid lytic granule convergence to the MTOC in natural killer cells is dependent on dynein but not cytolytic commitment. Mol Biol Cell (2010) 21:2241–56. doi: 10.1091/mbc.e09-11-0930
86. Cassioli C, Baldari CT. Lymphocyte polarization during immune synapse assembly: centrosomal actin joins the game. Front Immunol (2022) 13:830835. doi: 10.3389/fimmu.2022.830835
87. Yi J, Wu X, Chung AH, Chen JK, Kapoor TM, Hammer JA. Centrosome repositioning in T cells is biphasic and driven by microtubule end-on capture-shrinkage. J Cell Biol (2013) 202:779–92. doi: 10.1083/jcb.201301004
88. Martin-Cofreces NB, Robles-Valero J, Cabrero JR, Mittelbrunn M, Gordon-Alonso M, Sung CH, et al. MTOC translocation modulates IS formation and controls sustained T cell signaling. J Cell Biol (2008) 182:951–62. doi: 10.1083/jcb.200801014
89. Sanchez E, Liu X, Huse M. Actin clearance promotes polarized dynein accumulation at the immunological synapse. PloS One (2019) 14:e0210377. doi: 10.1371/journal.pone.0210377
90. Calvo V, Izquierdo M. Role of actin cytoskeleton reorganization in polarized secretory traffic at the immunological synapse. Front Cell Dev Biol (2021) 9:629097. doi: 10.3389/fcell.2021.629097
91. Kim N, Yi E, Kwon SJ, Park HJ, Kwon HJ, Kim HS. Filamin A Is Required for NK Cell Cytotoxicity at the Expense of Cytokine Production via Synaptic Filamentous Actin Modulation. Front Immunol (2021) 12:792334. doi: 10.3389/fimmu.2021.792334
92. Dupre L, Boztug K, Pfajfer L. Actin dynamics at the T cell synapse as revealed by immune-related actinopathies. Front Cell Dev Biol (2021) 9:665519. doi: 10.3389/fcell.2021.665519
93. Ben-Shmuel A, Sabag B, Biber G, Barda-Saad M. The role of the cytoskeleton in regulating the natural killer cell immune response in health and disease: from signaling dynamics to function. Front Cell Dev Biol (2021) 9:609532. doi: 10.3389/fcell.2021.609532
94. Rodriguez-Fernandez JL, Criado-Garcia O. The actin cytoskeleton at the immunological synapse of dendritic cells. Front Cell Dev Biol (2021) 9:679500. doi: 10.3389/fcell.2021.679500
95. Al-Alwan MM, Rowden G, Lee TD, West KA. The dendritic cell cytoskeleton is critical for the formation of the immunological synapse. J Immunol (2001) 166:1452–6. doi: 10.4049/jimmunol.166.3.1452
96. Al-Alwan MM, Liwski RS, Haeryfar SM, Baldridge WH, Hoskin DW, Rowden G, et al. Cutting edge: dendritic cell actin cytoskeletal polarization during immunological synapse formation is highly antigen-dependent. J Immunol (2003) 171:4479–83. doi: 10.4049/jimmunol.171.9.4479
97. Benvenuti F, Hugues S, Walmsley M, Ruf S, Fetler L, Popoff M, et al. Requirement of Rac1 and Rac2 expression by mature dendritic cells for T cell priming. Science (2004) 305:1150–3. doi: 10.1126/science.1099159
98. Malinova D, Fritzsche M, Nowosad CR, Armer H, Munro PM, Blundell MP, et al. WASp-dependent actin cytoskeleton stability at the dendritic cell immunological synapse is required for extensive, functional T cell contacts. J Leukoc Biol (2016) 99:699–710. doi: 10.1189/jlb.2A0215-050RR
99. Leithner A, Altenburger LM, Hauschild R, Assen FP, Rottner K, Stradal TEB, et al. Dendritic cell actin dynamics control contact duration and priming efficiency at the immunological synapse. J Cell Biol (2021) 220. doi: 10.1083/jcb.202006081
100. Hui KL, Balagopalan L, Samelson LE, Upadhyaya A. Cytoskeletal forces during signaling activation in Jurkat T-cells. Mol Biol Cell (2015) 26:685–95. doi: 10.1091/mbc.E14-03-0830
101. Bashour KT, Gondarenko A, Chen H, Shen K, Liu X, Huse M, et al. CD28 and CD3 have complementary roles in T-cell traction forces. Proc Natl Acad Sci U.S.A. (2014) 111:2241–6. doi: 10.1073/pnas.1315606111
102. Saitakis M, Dogniaux S, Goudot C, Bufi N, Asnacios S, Maurin M, et al. Different TCR-induced T lymphocyte responses are potentiated by stiffness with variable sensitivity. Elife (2017) 6. doi: 10.7554/eLife.23190
103. Judokusumo E, Tabdanov E, Kumari S, Dustin ML, Kam LC. Mechanosensing in T lymphocyte activation. Biophys J (2012) 102:L5–7. doi: 10.1016/j.bpj.2011.12.011
104. Blumenthal D, Chandra V, Avery L, Burkhardt JK. Mouse T cell priming is enhanced by maturation-dependent stiffening of the dendritic cell cortex. Elife (2020) 9. doi: 10.7554/eLife.55995
105. Li YC, Chen BM, Wu PC, Cheng TL, Kao LS, Tao MH, et al. Cutting Edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J Immunol (2010) 184:5959–63. doi: 10.4049/jimmunol.0900775
106. Pryshchep S, Zarnitsyna VI, Hong J, Evavold BD, Zhu C. Accumulation of serial forces on TCR and CD8 frequently applied by agonist antigenic peptides embedded in MHC molecules triggers calcium in T cells. J Immunol (2014) 193:68–76. doi: 10.4049/jimmunol.1303436
107. Morandi B, Mortara L, Chiossone L, Accolla RS, Mingari MC, Moretta L, et al. Dendritic cell editing by activated natural killer cells results in a more protective cancer-specific immune response. PloS One (2012) 7:e39170. doi: 10.1371/journal.pone.0039170
108. Ferlazzo G, Moretta L. Dendritic cell editing by natural killer cells. Crit Rev Oncog (2014) 19:67–75. doi: 10.1615/CritRevOncog.2014010827
109. Mace EM, Orange JS. Multiple distinct NK-cell synapses. Blood (2011) 118:6475–6. doi: 10.1182/blood-2011-10-381392
110. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity (2007) 26:503–17. doi: 10.1016/j.immuni.2007.03.006
111. Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, et al. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med (1999) 5:405–11. doi: 10.1038/7403
112. Brilot F, Strowig T, Roberts SM, Arrey F, Munz C. NK cell survival mediated through the regulatory synapse with human DCs requires IL-15Ralpha. J Clin Invest (2007) 117:3316–29. doi: 10.1172/JCI31751
113. Borg C, Jalil A, Laderach D, Maruyama K, Wakasugi H, Charrier S, et al. NK cell activation by dendritic cells (DCs) requires the formation of a synapse leading to IL-12 polarization in DCs. Blood (2004) 104:3267–75. doi: 10.1182/blood-2004-01-0380
114. Vitale M, Della Chiesa M, Carlomagno S, Pende D, Arico M, Moretta L, et al. NK-dependent DC maturation is mediated by TNFalpha and IFNgamma released upon engagement of the NKp30 triggering receptor. Blood (2005) 106:566–71. doi: 10.1182/blood-2004-10-4035
115. Barreira da Silva R, Graf C, Munz C. Cytoskeletal stabilization of inhibitory interactions in immunologic synapses of mature human dendritic cells with natural killer cells. Blood (2011) 118:6487–98. doi: 10.1182/blood-2011-07-366328
116. Friedman D, Simmonds P, Hale A, Bere L, Hodson NW, White MRH, et al. Natural killer cell immune synapse formation and cytotoxicity are controlled by tension of the target interface. J Cell Sci (2021) 134. doi: 10.1242/jcs.258570
117. Huse M. Mechanical forces in the immune system. Nat Rev Immunol (2017) 17:679–90. doi: 10.1038/nri.2017.74
118. Zhu C, Chen W, Lou J, Rittase W, Li K. Mechanosensing through immunoreceptors. Nat Immunol (2019) 20:1269–78. doi: 10.1038/s41590-019-0491-1
119. Gonzalez C, Chames P, Kerfelec B, Baty D, Robert P, Limozin L. Nanobody-CD16 catch bond reveals NK cell mechanosensitivity. Biophys J (2019) 116:1516–26. doi: 10.1016/j.bpj.2019.03.012
120. Liu B, Chen W, Evavold BD, Zhu C. Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell (2014) 157:357–68. doi: 10.1016/j.cell.2014.02.053
121. Lee MS, Glassman CR, Deshpande NR, Badgandi HB, Parrish HL, Uttamapinant C, et al. A mechanical switch couples T cell receptor triggering to the cytoplasmic juxtamembrane regions of CD3zetazeta. Immunity (2015) 43:227–39. doi: 10.1016/j.immuni.2015.06.018
122. Basu R, Whitlock BM, Husson J, Le Floc’h A, Jin W, Oyler-Yaniv A, et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell (2016) 165:100–10. doi: 10.1016/j.cell.2016.01.021
123. Tamzalit F, Wang MS, Jin W, Tello-Lafoz M, Boyko V, Heddleston JM, et al. Interfacial actin protrusions mechanically enhance killing by cytotoxic T cells. Sci Immunol (2019) 4. doi: 10.1126/sciimmunol.aav5445
124. Tello-Lafoz M, de Jesus MM, Huse M. Harder, better, faster, stronger: biochemistry and biophysics in the immunosurveillance concert. Trends Immunol (2022) 43:96–105. doi: 10.1016/j.it.2021.12.003
125. Swaminathan V, Mythreye K, O’Brien ET, Berchuck A, Blobe GC, Superfine R. Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Cancer Res (2011) 71:5075–80. doi: 10.1158/0008-5472.CAN-11-0247
126. Xu W, Mezencev R, Kim B, Wang L, McDonald J, Sulchek T. Cell stiffness is a biomarker of the metastatic potential of ovarian cancer cells. PloS One (2012) 7:e46609. doi: 10.1371/journal.pone.0046609
127. Lekka M, Laidler P, Gil D, Lekki J, Stachura Z, Hrynkiewicz AZ. Elasticity of normal and cancerous human bladder cells studied by scanning force microscopy. Eur Biophys J (1999) 28:312–6. doi: 10.1007/s002490050213
128. Guck J, Schinkinger S, Lincoln B, Wottawah F, Ebert S, Romeyke M, et al. Optical deformability as an inherent cell marker for testing Malignant transformation and metastatic competence. Biophys J (2005) 88:3689–98. doi: 10.1529/biophysj.104.045476
129. Plodinec M, Loparic M, Monnier CA, Obermann EC, Zanetti-Dallenbach R, Oertle P, et al. The nanomechanical signature of breast cancer. Nat Nanotechnol (2012) 7:757–65. doi: 10.1038/nnano.2012.167
130. Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science (1993) 260:1124–7. doi: 10.1126/science.7684161
131. Liu Y, Zhang T, Zhang H, Li J, Zhou N, Fiskesund R, et al. Cell softness prevents cytolytic T-cell killing of tumor-repopulating cells. Cancer Res (2021) 81:476–88. doi: 10.1158/0008-5472.CAN-20-2569
132. Pecci A, Ma X, Savoia A, Adelstein RS. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene (2018) 664:152–67. doi: 10.1016/j.gene.2018.04.048
133. Abouzahr S, Bismuth G, Gaudin C, Caroll O, Van Endert P, Jalil A, et al. Identification of target actin content and polymerization status as a mechanism of tumor resistance after cytolytic T lymphocyte pressure. Proc Natl Acad Sci USA (2006) 103:1428–33. doi: 10.1073/pnas.0510454103
134. Tello-Lafoz M, Srpan K, Sanchez EE, Hu J, Remsik J, Romin Y, et al. Cytotoxic lymphocytes target characteristic biophysical vulnerabilities in cancer. Immunity (2021) 54:1037–54 e7. doi: 10.1016/j.immuni.2021.02.020
135. Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell (2003) 113:329–42. doi: 10.1016/S0092-8674(03)00278-2
136. Gau D, Roy P. SRF’ing and SAP’ing - the role of MRTF proteins in cell migration. J Cell Sci (2018) 131. doi: 10.1242/jcs.218222
137. Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature (2016) 529:298–306. doi: 10.1038/nature17038
138. Er EE, Valiente M, Ganesh K, Zou Y, Agrawal S, Hu J, et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat Cell Biol (2018) 20:966–78. doi: 10.1038/s41556-018-0138-8
139. Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol (2009) 11:257–68. doi: 10.1038/ncb1833
140. Lei K, Kurum A, Kaynak M, Bonati L, Han Y, Cencen V, et al. Cancer-cell stiffening via cholesterol depletion enhances adoptive T-cell immunotherapy. Nat BioMed Eng (2021) 5:1411–25. doi: 10.1038/s41551-021-00826-6
141. Biolato AM, Filali L, Wurzer H, Hoffmann C, Gargiulo E, Valitutti S, et al. Actin remodeling and vesicular trafficking at the tumor cell side of the immunological synapse direct evasion from cytotoxic lymphocytes. Int Rev Cell Mol Biol (2020) 356:99–130. doi: 10.1016/bs.ircmb.2020.07.001
142. McKenzie B, Khazen R, Valitutti S. Greek fire, poison arrows, and scorpion bombs: how tumor cells defend against the siege weapons of cytotoxic T lymphocytes. Front Immunol (2022) 13:894306. doi: 10.3389/fimmu.2022.894306
143. Filali L, Puissegur MP, Cortacero K, Cussat-Blanc S, Khazen R, Van Acker N, et al. Ultrarapid lytic granule release from CTLs activates Ca(2+)-dependent synaptic resistance pathways in melanoma cells. Sci Adv (2022) 8:eabk3234. doi: 10.1126/sciadv.abk3234
144. Al Absi A, Wurzer H, Guerin C, Hoffmann C, Moreau F, Mao X, et al. Actin cytoskeleton remodeling drives breast cancer cell escape from natural killer-mediated cytotoxicity. Cancer Res (2018) 78:5631–43. doi: 10.1158/0008-5472.CAN-18-0441
145. Wurzer H, Al Absi A, Hoffmann C, Thomas C. Do tumor cells escape from natural killer cell cytotoxicity by mimicking dendritic cells? Oncotarget (2019) 10:2419–20. doi: 10.18632/oncotarget.26815
146. Wurzer H, Filali L, Hoffmann C, Krecke M, Biolato AM, Mastio J, et al. Intrinsic resistance of chronic lymphocytic leukemia cells to NK cell-mediated lysis can be overcome in vitro by pharmacological inhibition of cdc42-induced actin cytoskeleton remodeling. Front Immunol (2021) 12:619069. doi: 10.3389/fimmu.2021.619069
147. Brown MS, Abdollahi B, Wilkins OM, Lu H, Chakraborty P, Ognjenovic NB, et al. Phenotypic heterogeneity driven by plasticity of the intermediate EMT state governs disease progression and metastasis in breast cancer. Sci Adv (2022) 8:eabj8002. doi: 10.1126/sciadv.abj8002
148. Terry S, Savagner P, Ortiz-Cuaran S, Mahjoubi L, Saintigny P, Thiery JP, et al. New insights into the role of EMT in tumor immune escape. Mol Oncol (2017) 11:824–46. doi: 10.1002/1878-0261.12093
149. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci USA (2013) 110:17450–5. doi: 10.1073/pnas.1304790110
150. Wurzer H, Filali L, Thomas C. How natural killer cells avoid self-destruction when killing their targets. PloS Biol (2021) 19:e3001339. doi: 10.1371/journal.pbio.3001339
151. Blanco B, Compte M, Lykkemark S, Sanz L, Alvarez-Vallina L. T cell-redirecting strategies to ‘STAb’ Tumors: beyond CARs and bispecific antibodies. Trends Immunol (2019) 40:243–57. doi: 10.1016/j.it.2019.01.008
152. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
153. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood (2015) 125:4017–23. doi: 10.1182/blood-2014-12-580068
154. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
155. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest (2016) 126:2123–38. doi: 10.1172/JCI85309
156. Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic Malignancies: interpreting clinical outcomes to date. Blood (2016) 127:3312–20. doi: 10.1182/blood-2016-02-629063
157. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
158. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
159. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
160. Alonso-Camino V, Harwood SL, Alvarez-Mendez A, Alvarez-Vallina L. Efficacy and toxicity management of CAR-T-cell immunotherapy: a matter of responsiveness control or tumour-specificity? Biochem Soc Trans (2016) 44:406–11. doi: 10.1042/BST20150286
161. He X, Yin X, Wu J, Wickstrom SL, Duo Y, Du Q, et al. Visualization of human T lymphocyte-mediated eradication of cancer cells in vivo. Proc Natl Acad Sci U.S.A. (2020) 117:22910–9. doi: 10.1073/pnas.2009092117
162. Pascoal S, Salzer B, Scheuringer E, Wenninger-Weinzierl A, Sturtzel C, Holter W, et al. A preclinical embryonic zebrafish xenograft model to investigate CAR T cells in vivo. Cancers (Basel) (2020) 12. doi: 10.3390/cancers12030567
163. Mulazzani M, Frassle SP, von Mucke-Heim I, Langer S, Zhou X, Ishikawa-Ankerhold H, et al. Long-term in vivo microscopy of CAR T cell dynamics during eradication of CNS lymphoma in mice. Proc Natl Acad Sci USA (2019) 116:24275–84. doi: 10.1073/pnas.1903854116
164. Boulch M, Cazaux M, Cuffel A, Guerin MV, Garcia Z, Alonso R, et al. Tumor-intrinsic sensitivity to the pro-apoptotic effects of IFN-gamma is a major determinant of CD4(+) CAR T-cell antitumor activity. Nat Cancer (2023) 4:968–83. doi: 10.1038/s43018-023-00570-7
165. Roda-Navarro P, Alvarez-Vallina L. Understanding the spatial topology of artificial immunological synapses assembled in T cell-redirecting strategies: A major issue in cancer immunotherapy. Front Cell Dev Biol (2019) 7:370. doi: 10.3389/fcell.2019.00370
166. Chockley PJ, Ibanez-Vega J, Krenciute G, Talbot LJ, Gottschalk S. Synapse-tuned CARs enhance immune cell anti-tumor activity. Nat Biotechnol (2023). doi: 10.1038/s41587-022-01650-2
167. Ivica NA, Young CM. Tracking the CAR-T revolution: analysis of clinical trials of CAR-T and TCR-T therapies for the treatment of cancer (1997-2020). Healthcare (Basel) (2021) 9. doi: 10.20944/preprints202107.0198.v1
168. Zhong L, Li Y, Muluh TA, Wang Y. Combination of CAR−T cell therapy and radiotherapy: Opportunities and challenges in solid tumors (Review). Oncol Lett (2023) 26:281. doi: 10.3892/ol.2023.13867
169. Rossetti R, Brand H, Lima SCG, Furtado IP, Silveira RM, Fantacini DMC, et al. Combination of genetically engineered T cells and immune checkpoint blockade for the treatment of cancer. Immunother Adv (2022) 2:ltac005. doi: 10.1093/immadv/ltac005
Keywords: actin cytoskeleton, cancer, cytotoxic lymphocytes, cytotoxic T cells, immunological synapse, natural killer cells
Citation: Ockfen E, Filali L, Pereira Fernandes D, Hoffmann C and Thomas C (2023) Actin cytoskeleton remodeling at the cancer cell side of the immunological synapse: good, bad, or both? Front. Immunol. 14:1276602. doi: 10.3389/fimmu.2023.1276602
Received: 12 August 2023; Accepted: 21 September 2023;
Published: 05 October 2023.
Edited by:
Cleo Goyvaerts, Vrije University Brussels, BelgiumReviewed by:
Yina Hsing Huang, Dartmouth College, United StatesBillur Akkaya, The Ohio State University, United States
Copyright © 2023 Ockfen, Filali, Pereira Fernandes, Hoffmann and Thomas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clément Thomas, Y2xlbWVudC50aG9tYXNAbGloLmx1