 Massimo Fantini
Massimo Fantini Philip Martin Arlen
Philip Martin Arlen Kwong Yok Tsang
Kwong Yok Tsang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 24 November 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1275904
This article is part of the Research Topic Bringing Function to the Forefront of Cell Therapy: How Do We Demonstrate Potency? View all 7 articles
Natural killer (NK) cells are cellular components of the innate immune system that can recognize and suppress the proliferation of cancer cells. NK cells can eliminate cancer cells through direct lysis, by secreting perforin and granzymes, or through antibody-dependent cell-mediated cytotoxicity (ADCC). ADCC involves the binding of the Fc gamma receptor IIIa (CD16), present on NK cells, to the constant region of an antibody already bound to cancer cells. Cancer cells use several mechanisms to evade antitumor activity of NK cells, including the accumulation of inhibitory cytokines, recruitment and expansion of immune suppressor cells such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), modulation of ligands for NK cells receptors. Several strategies have been developed to enhance the antitumor activity of NK cells with the goal of overcoming cancer cells resistance to NK cells. The three main strategies to engineer and boost NK cells cytotoxicity include boosting NK cells with modulatory cytokines, adoptive NK cell therapy, and the employment of engineered NK cells to enhance antibody-based immunotherapy. Although the first two strategies improved the efficacy of NK cell-based therapy, there are still some limitations, including immune-related adverse events, induction of immune-suppressive cells and further cancer resistance to NK cell killing. One strategy to overcome these issues is the combination of monoclonal antibodies (mAbs) that mediate ADCC and engineered NK cells with potentiated anti-cancer activity. The advantage of using mAbs with ADCC activity is that they can activate NK cells, but also favor the accumulation of immune effector cells to the tumor microenvironment (TME). Several clinical trials reported that combining engineered NK cells with mAbs with ADCC activity can result in a superior clinical response compared to mAbs alone. Next generation of clinical trials, employing engineered NK cells with mAbs with higher affinity for CD16 expressed on NK cells, will provide more effective and higher-quality treatments to cancer patients.
Cancer is considered one of the deadliest diseases in the world. In 2020, cancer caused 10 million cancer deaths worldwide, with an estimate of 19.3 million new cases (1). Surgery, radiotherapy and chemotherapy are still considered as principal treatment options in clinical oncology. Unfortunately, especially in the case of radiotherapy and chemotherapy, their employment can cause severe side effects that do not allow patients to complete the treatment, with a consequent failure in controlling tumor growth and metastasis (2, 3).
In the last few decades, more selective and less toxic therapies have been developed. Among them, immunotherapy represents a novel and very promising approach for the treatment of cancer patients. Immunotherapy enhances the capacity of immune system to attack and eliminate cancer cells in a more selective manner with a reduced toxicity towards normal tissues (4).
The immune system targets and counteracts the proliferation of malignant cells through the immunoediting process, that is composed of three phases: elimination, equilibrium, and escape (5).
Elimination: in this phase, normal cells are transformed into cancer cells through the process of carcinogenesis. Factors that allow transformed cells to proliferate in an uncontrolled manner during the process of carcinogenesis include upregulation of the expression of oncogenes (genes that regulate cell proliferation) as well as the downregulation of the expression or loss of function of onco-suppressor genes (genes that control cell growth) (6, 7). During the elimination phase, NK cells, macrophages, granulocytes, and cytotoxic T lymphocytes (CTLs) recognize and suppress malignant cells before cancer become clinically visible (5). For example, these immune cells can suppress tumor growth and angiogenesis by releasing interferon (IFN)-γ. NK cells and CTLs can also induce death of cancer cells by apoptosis or can directly lyse them using perforin and granzymes (5, 8, 9).
Equilibrium: in this phase, most of cancer cells are in a status of dormancy due to the pressure of the immune system, but some cancer cells can develop mechanisms to resist immune recognition, such as antigen loss, genetic and epigenetic modifications that lead to a defect in antigen presentation, or overexpression of immunosuppressive molecules (5, 9).
Escape: in this phase, cancer cells acquire a more aggressive phenotype and the ability to growth without control. These features allow cancer cells to escape from the pressure of immune system and to metastasize to different organs. For example, cancer cells can induce the accumulation of MDSCs in the TME. MDSCs can release immunosuppressive cytokines which in turn can lead to the exhaustion of CTLs. MDSCs can also induce the generation of Tregs. Tregs inhibit antitumor immune response by secreting immune suppressive cytokines (5, 9–12) and by reducing NK cells cytotoxicity in the TME (13).
Potentiate the immune system through cancer immunotherapy can prevent cancer cells to enter in the escape phase of the immunoediting. In this way, the immune system should be able to keep cancer cells permanently in the elimination and/or equilibrium phase (14).
In this review we provide an overview on main cancer immunotherapy strategies based on the stimulation of NK cells, then we describe mAbs that have received FDA approval for the treatment of human cancers with emphasis on their mechanism of action involving NK cells, and finally we focus on strategies to improve antibody-based immunotherapy through the employment of engineered NK cells.
NK cells are one of the cellular components of the innate immune system. These effector cells can target and kill virus-infected cells and cancer cells. They comprise between 5% and 20% of the mononuclear cell fraction in peripheral blood lymphocyte population in humans (15, 16). One of the peculiarities of NK cells is their capacity to kill target cells without requiring their prior sensitization and the expression of major histocompatibility complex (MHC) molecules on cancer cells (17).
In accordance with the CD56 and CD16 status, NK cells can be divided into two subsets. Around 90% of NK cells found in peripheral blood and spleen belongs to the CD56dimCD16+ subset. These NK cells can kill tumor cells directly or through ADCC, but they produce a low amount of cytokines (17, 18). Conversely, remaining NK cells in peripheral blood and NK cells residing in lymph nodes and tonsils belong to the CD56brightCD16− subset. These NK cells have poor ability to kill tumor cell targets but play an important role in modulating immune responses through the secretion of regulatory cytokines, like IFN-γ (17, 18).
NK cells exert their antitumor activity through the binding of their activating receptors, such as NKp46, NKp30, NKp44, NKG2D, and killer cell immunoglobulin-like receptors (KIRs), to NK cells ligands expressed on the surface of cancer cells (19).
One example is represented by the NKG2D/NKG2D ligand (NKG2D-L) pathway (20). NKG2D binds to its ligands expressed on cancer cells, including MICA/B and UL16-binding proteins 1–6 (ULBP1–6) (21).
After the binding with their ligands, NK cells can kill cancer cells through different mechanisms, including direct lysis mediated by perforin, direct killing through death receptor-mediated pathways (i.e. TRAIL and FasL), through the release of cytokines in the TME or through ADCC (22, 23) (Figure 1).
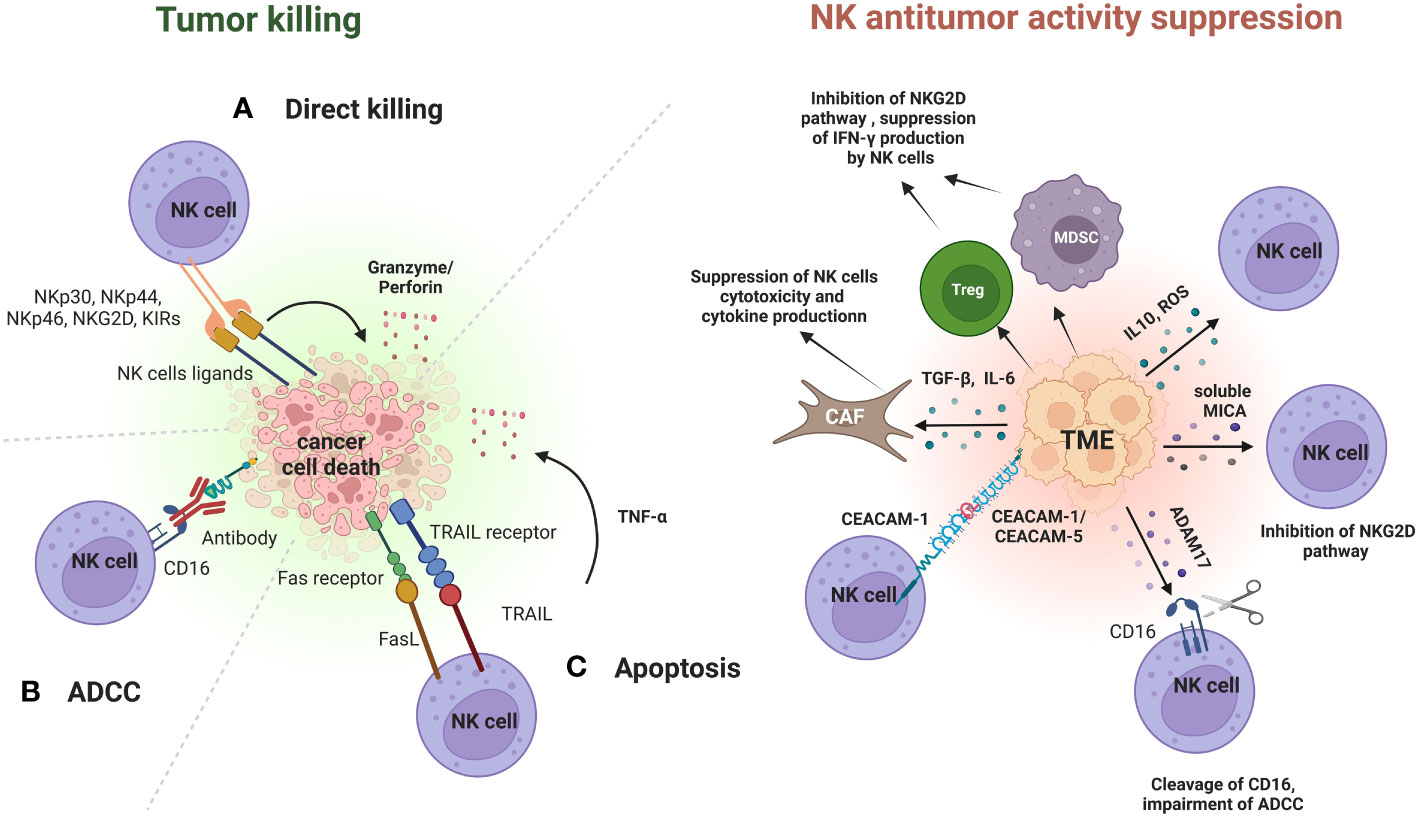
Figure 1 Role of NK cells in immunoediting and mechanisms of immune-escape by cancer cells in the TME. During the elimination phase, NK cells can recognize and suppress cancer cells growth through different mechanisms. After the binding with their ligands, NK cells can kill cancer cells through direct lysis mediated by granzyme and perforin (A), through ADCC (B) or through apoptosis mediated by TRAIL and FasL and induced by the release of cytokines (TNF-α) (C). Cancer cells in the TME have developed different strategies to inhibit NK cells antitumor activity. One of these is promotion of recruitment and expansion of Tregs and MDSCs. MDSCs and Tregs can induce NK cells exhaustion and impairment by suppressing INF-γ production, and by inhibiting NKG2D pathway. Furthermore, cancer cells can release TGF-β or IL-6 in the TME to activate cancer-associated fibroblasts (CAFs), that in turn can contribute to the suppression of NK cells cytotoxicity and cytokine production. Other mechanisms include the release of immune suppressive cytokines and of ROS within the TME. Cancer cells can also increase the expression of CEACAM-1 or CEACAM-5 on their surface. These molecules bind to CEACAM-1 expressed on NK cells and this binding can lead to the suppression of NK cells cytotoxicity. Cancer cells can also suppress the NK-mediated ADCC through the proteolytic activity of the enzyme ADAM17. ADAM17 mediates the cleavage of CD16 on NK cells, which in turn suppresses NK cells antitumor activity through ADCC. The immunosuppressive activity of cancer cells towards NK cells is also exerted through the secretion of soluble factors in the bloodstream, such as soluble MICA. Elevated serum levels of soluble MICA suppress NK cell cytotoxicity by affecting the NKG2D pathway. Figure created with BioRender.com.
Direct lysis through perforin occurs when perforin forms a pore in cell membranes of cancer cells, allowing the release of cytolytic enzymes, such as granzyme A and B, into their cytoplasm which in turn will mediate cell death. Similarly, NK cells can directly kill cancer cells through the binding of TRAIL and FasL to their receptors on cancer cells. This results in caspase activation which in turn mediates cell death by apoptosis (24, 25).
Cytokines released by NK cells have potent antitumor activity. IFN-γ can sensitize cancer cells to CD8+ T-cell killing and polarize macrophage towards M1 state with a stronger antitumoral activity. Tumor necrosis factor-α (TNF-α) can trigger caspase 8-mediated apoptosis of cancer cells and the combination of IFN-γ and TNF-α can induce senescence of cancer cells (23).
NK cells can also mediate killing of cancer cells through ADCC, a mechanism that involves the binding of the constant region (fragment crystallizable, Fc) of an antibody, including mAbs, to the Fc gamma receptor IIIa (FcƴRIIIa, CD16) present on the surface of macrophages and NK cells. This interaction allows NK cells to recognize and kill cancer cells bounded to an antibody (26).
Subjects with head and neck squamous cell carcinoma (HNSCC), oral squamous carcinoma and gastric carcinoma with high levels of NK cells infiltrating their tumors showed an increased survival and a better clinical outcome (27).
Cancer cells in the TME have developed different strategies to inhibit NK cells antitumor activity (Figure 1).
One of these is the accumulation of inhibitory cytokines, including IL-10 and TNF-α, that promote the recruitment and expansion of Tregs and MDSCs. MDSCs and Tregs can induce NK cells exhaustion and impairment by suppressing INF-γ production, and by inhibiting NKG2D pathway through membrane-bound TGF-β (27–29).
Furthermore, cancer cells can release TGF-β or IL-6 in the TME to activate cancer-associated fibroblasts (CAFs), that in turn can contribute to the suppression of NK cells cytotoxicity and cytokine production (30).
Other mechanisms include the downregulation of ligands for NK activating receptors, the overexpression of ligands for NK inhibitory receptors, the release of reactive oxygen species (ROS) within the TME (31).
Cancer cells can also increase the expression of carcinoembryonic antigen-related cell adhesion molecule (CEACAM)-1 or CEACAM-5 on their surface. These molecules bind to CEACAM-1 expressed on NK cells and this binding can lead to the suppression of NK cells cytotoxicity (32, 33).
For example, it has been observed that a direct interaction between CEACAM-1 expressed on NK cells and CEACAM-1 expressed on melanoma cells was associated with the poor prognosis of melanoma patients (32).
Cancer cells can also suppress the NK-mediated ADCC by downregulating levels of CD16 on the surface of NK cells through the enhancement of the proteolytic activity of the enzyme ADAM17. ADAM17 is a member of the metalloproteinase superfamily that mediates the cleavage of CD16. The cleavage of CD16 by ADAM17 leads to a decrease of CD16 binding avidity to antibody-coated target cells and in turn can suppress NK cells antitumor activity through ADCC, allowing cancer cells to create an immunosuppressive TME (34).
The immunosuppressive activity of cancer cells towards NK cells is also exerted far away from TME through the secretion of soluble factors in the bloodstream.
One example is represented by the impairment of NK cells antitumor activity by soluble MHC class I polypeptide–related sequence A (MICA) and soluble CEACAM-5. High serum levels of these soluble factors have been associated with cancer progression and metastasis in patients with different types of cancers (35, 36).
Elevated serum levels of soluble MICA have been proved to suppress NK cell cytotoxicity by affecting the NKG2D pathway (37). This phenomenon was associated with a poor prognosis in patients with neuroblastoma (38), hepatocellular carcinoma (39), renal cell carcinoma (40), non-small cell lung cancer (NSCLC) (41), multiple myeloma (MM) (42), pancreatic and colorectal cancer (43, 44).
NK cell-based immunotherapy represents a promising therapeutic tool for the treatment of solid and hematological tumors.
The three main strategies used to potentiate the antitumor activity of NK cells to overcome the tumor resistance at the level of the TME are boosting NK cells with modulatory cytokines, adoptive NK cell therapy, including CAR-NK cells, and the employment of engineered NK cells to enhance antibody-based immunotherapy.
There are several cytokines that are being employed to stimulate the antitumor activity of NK cells.
Major cytokines involved in the modulation of human NK cell development, survival, and activity, include interleukins, such as IL-2, IL-12, IL-15, IL-18, and IL-21 (45) (Figure 2).
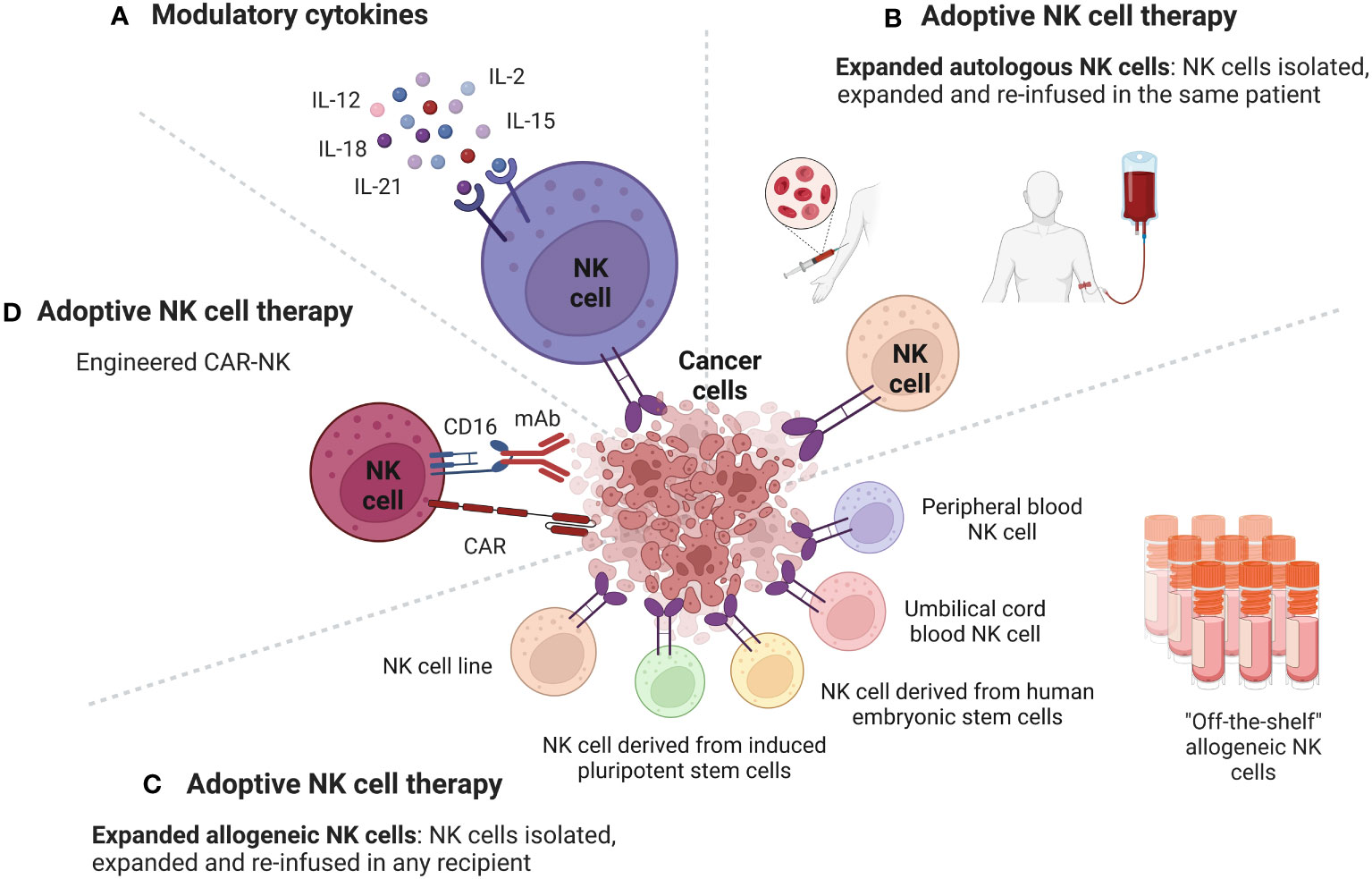
Figure 2 Strategies to potentiate the antitumor activity of NK cells to overcome the tumor resistance at the level of the TME. Cancer cells in the TME have developed several mechanisms to inhibit NK cells antitumor activity. Strategies used to potentiate the antitumor activity of NK cells to overcome the tumor resistance at the level of the TME include boosting NK cells with modulatory cytokines and adoptive NK cell therapy. (A) Cytokines, such as IL-2, IL-15, IL-12, IL-18, IL-21 can enhance the anti-cancer activity of NK cells through different mechanisms. NK cells antitumor activity can be also enhanced through the adoptive cell therapy, which consists of isolating NK cells from cellular components of immune system of a subject, engineering them ex vivo, expanding their numbers and then re-infuse them in a cancer patient. Sources of NK cells that can be used in adoptive therapy include autologous NK cells, allogeneic NK cells, and CAR-NK cells. (B) Adoptive NK cell therapy using autologous NK cells. NK cells are isolated from a cancer patient, expanded, and then re-infused in the same cancer patient. (C) Adoptive NK cell therapy using allogeneic NK cells. NK cells are isolated from a donor, expanded, and then re-infused in any recipient. The main sources for allogeneic NK cells are NK cells from peripheral blood, from umbilical cord blood, from human embryonic stem cells, from induced pluripotent stem cells and NK cell lines. (D) Adoptive NK cell therapy using engineered CAR-NK cells. CAR-NK cells can target and eliminate cancer cells with additional mechanisms beyond the CAR pathway, including ADCC. Figure created with BioRender.com.
IL-2 binds to the IL-2 receptor (IL-2R). IL-2R is composed of three subunits, IL-2Rα (CD25), IL-2Rβ (CD122), and IL-2Rγ (CD132). The binding of IL-2 and IL-2Rβ and γ subunits on NK cells activates a signal transduction that induces NK cells to proliferate and eliminate their targets (46).
Treatment with IL-2 resulted in stable clinical response in patients with metastatic melanoma, renal cancer, and advanced non-Hodgkin’s lymphoma (47).
The weaknesses of therapy with IL-2 are the dose-associated toxicity (vascular leakage favoring fluid extravasation into visceral organs) and the ability to activate and expand Tregs, which in turn can induce an impairment of antitumoral activity of NK cell (47, 48). To overcome this issue, recently, an IL-2 variant has been developed. This variant (F42K) preferentially binds to IL2Rβγ subunits, expressed on NK cells, and has a decreased capacity to bind to IL-2Rα, expressed on Tregs. It has been observed that F42K upregulates the expression of NK activation markers, enhances NK-mediated cytolytic activity and is also able to induce a reduction of Tregs in vivo (49).
A valid alternative to IL-2 to boost NK cells activity is IL-15.
IL-15 can bind to IL-2Rβ and IL-2Rγ receptor expressed on T cells, NK cells, monocytes, and neutrophils. IL-15 also binds to IL-15Rα, expressed by monocytes and dendritic cells, which is a high affinity receptor for IL-15 (50).
IL-15 has similar effects to IL-2 to NK cells, including modulation of NK cell proliferation, cytotoxicity, and cytokine production (51). The advantage of using IL-15 instead of IL-2 is that IL-15 activates Tregs less consistently compared to IL-2, does not induce a significant capillary leak, and promotes prolonged expansion and activation of NK cells (52, 53).
The first in-human clinical trial employing recombinant human IL-15 (rhIL-15) in patients with metastatic melanoma and renal cell cancer (RCC) showed that administration of IL-15 is safe and that can enhance the function and proliferation of NK cells in peripheral blood of cancer patients (53). In addition, it has been reported that CD56 bright NK cells from leukemia and MM patients stimulated with IL-15 exhibited potent antitumor responses (54).
Unfortunately, the half-life of IL-15 in vivo is very short with a limited bioactivity of IL-15 after systemic delivery. To achieve a consistent boost of NK cells IL-15 needs to be administered continuously, but constant exposure of NK cells with IL-15 can cause their exhaustion, decreased viability, and impairment of anti-tumor activity (55, 56).
One strategy to overcome these issues was the development of stable fusion proteins, comprising IL-15 and IL-15Rα. In this regard, a promising construct is N-803 (previously named ALT-803). N-803 is a complex, consisting of an IL-15 variant (IL-15N72D) bound to an IL-15 receptorα/IgG1 Fc fusion protein. Studies in vivo demonstrated that N-803 has a better stability, longer persistence in lymphoid tissues, and enhanced activity against cancer compared to IL-15 (57). N-803 can increase human NK cells antitumor activity by upregulating gene expression of NK-activating receptors and factors involved in NK cells cytotoxicity, as well as by reducing expression of NK inhibitory receptors (58).
N-803 is also able to prolong human NK cells viability in vitro (58).
Efficacy and safety of N-803 as monotherapy or in combination with conventional antitumor agents is being evaluated in several clinical trials. Two phase I clinical trials showed that N-803 was well tolerated and achieved clinical responses in patients with hematological malignancies when used as single agent (59), and in patients with advanced NSCLC when used in combination with nivolumab (60).
Another construct with promising efficacy in enhancing NK cells antitumor activity is HCW9218. HCW9218 comprises extracellular domains of the human TGF-β receptor II and a human IL-15/IL-15 receptor α complex (61). This complex showed long-lasting biological activity in vivo and enhanced proliferation, cytotoxic activity, and tumor infiltration of NK cells in a syngeneic B16F10 melanoma model (61, 62).
Another promising cytokine to boost NK cells is IL-12. IL-12 was defined as “natural killer cell stimulatory factor” (63), since can enhance antitumor NK cells activity by increasing production of IFN-γ, stimulating cytotoxicity of activated NK cells, enhancing ADCC (64).
Although IL-12 has demonstrated promising efficacy in animal models against both solid tumors and hematologic malignancies, clinical trials did not yield convincing results (64).
One of the main reasons of poor efficacy of IL-12 in cancer patients is the accumulation of immunosuppressive cells and soluble factors in the TME, which render cancer cells resistant to the antitumor activity exerted by IL-12 (65, 66). One novel approach to increase IL-12 efficacy is the employment of NHS-IL12 immunocytokine, which is composed of two IL-12 heterodimers fused to the NHS76 antibody. The first-in-human phase I trial in subjects with metastatic solid tumors showed that NHS-IL12 increased the percentage of activated and mature NK cells and tumor-infiltrating lymphocyte density, with 5 subjects having durable stable disease (SD) (67).
IL-18, a member of the IL-1 superfamily, plays a role in stimulating antitumor NK cells activity. For example, IL-18 can enhance NK cells ability to kill colorectal cancer cells via the miR-574-3p/TGF-β1 axis (68) and has been proven to enhance therapeutic effects of immune checkpoint inhibitors (ICIs) against cancer cells in animal models through accumulation of pre-mNK cells, memory-type CD8(+) T cells, and suppression of Tregs (69). Despite these promising results, the employment of recombinant IL-18 had limited activity as a single agent in patients with previously untreated metastatic melanoma (70). One of the explanations of the limited clinical activity of IL-18 is because high levels of IL-18 are associated with impairment of NK cells, suggesting that IL-18 could have a double hedge sword effect in cancer immunotherapy (71, 72).
A similar dual role in immune-regulation has been reported for IL-21. IL-21 is a member of the cytokine family comprising IL-2, IL-4, IL-7, IL-9, and IL-15 (73, 74). IL-21 can enhance antitumor NK cells activity by upregulating NKG2D receptor expression, by inducing rapid maturation of human CD34+ cell precursors towards NK cells in cooperation with IL-7 and IL-15 (73, 74), and by potentiating ADCC (75). IL-21 showed a promising activity in stimulating NK cell cytotoxicity against several types of cancers in animal models and encouraging results in humans, especially in patients with melanoma and renal cancer (76).
IL-21 can impair antitumor activities of immune cell subsets, including inhibition of dendritic cells activation and maturation, and induction of IL-10 expression in T cells and B cells. This dual activity on the immune system raised some concerns in the employment of this cytokine in cancer immunotherapy (76).
Advantages and disadvantages to stimulate NK cells through modulatory cytokines are reported in Table 1.
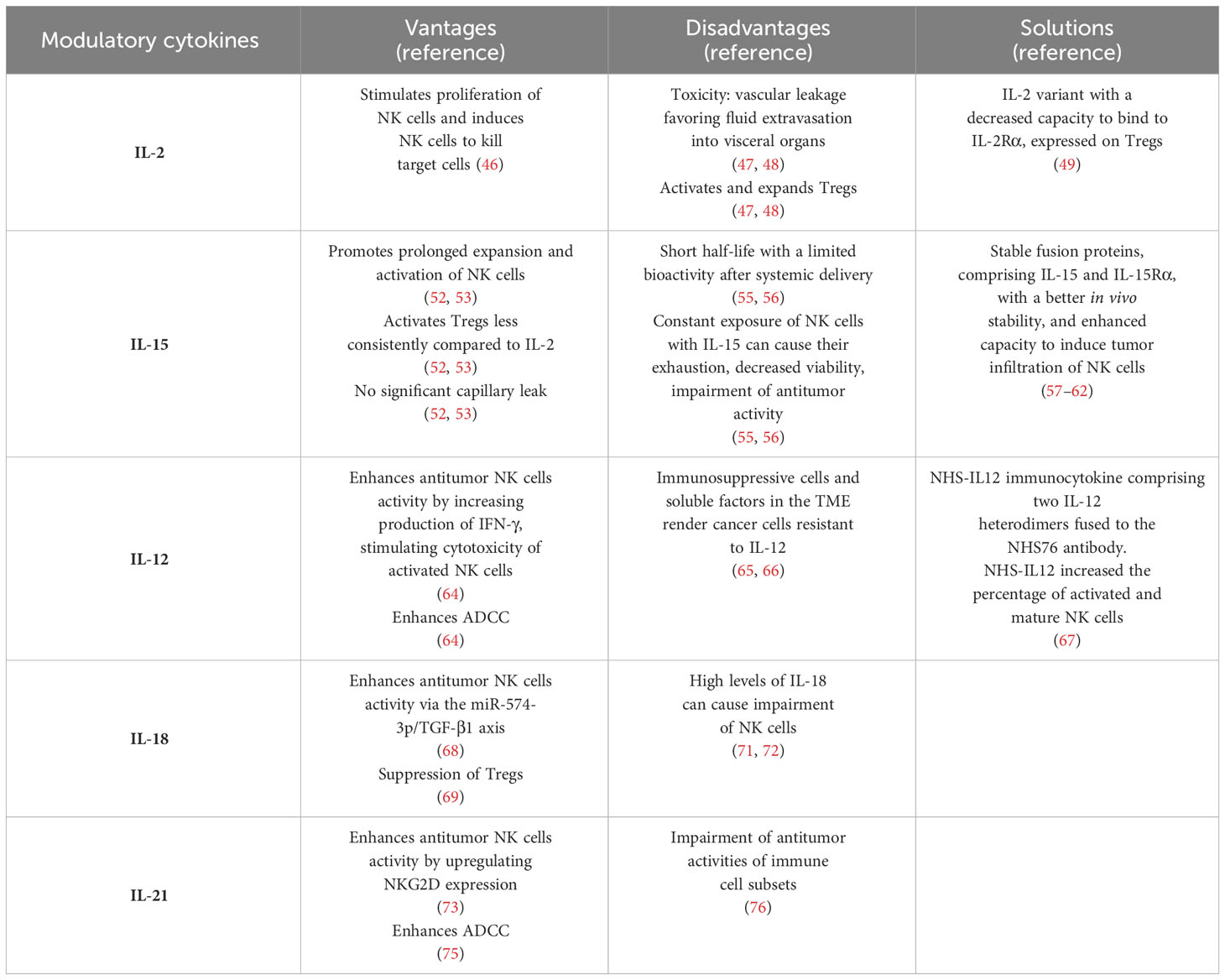
Table 1 Advantages and disadvantages to stimulate NK cells through modulatory cytokines.
Given the difficulties of achieving impressive clinical responses with manageable toxicities through the stimulation of NK cells with cytokines, a new strategy to improve NK cell-based immunotherapy was the development of engineered NK cells. Engineered NK cells are being used as adoptive NK cells and to enhance antibody-based immunotherapy.
A promising approach to enhance NK cells antitumor activity is the adoptive cell therapy, which consists of isolating NK cells from cellular components of immune system of a subject, engineering them ex-vivo, expanding their numbers and then re-infuse them in a cancer patient. Sources of NK cells employed in the adoptive NK cell therapy include autologous NK cells, allogeneic NK cells, and CAR-NK cells (Figure 2).
Adoptive NK cell therapy using autologous NK cells consists of isolating NK cells from a cancer patient, expanding them ex vivo and then re-infusing them in the same cancer patient. One of the first applications of adoptive NK cell therapy was the employment of autologous NK cells. One example is represented by NK cells from patients with lymphoma and breast cancer that were first isolated from the blood, then stimulated overnight ex vivo with IL-2 and then re-infused in the same patient (77). Despite a good stimulation of autologous NK cells, their efficacy in humans seems to be modest. In this regard, one study reported that infusion with autologous NK cells stimulated and expanded ex vivo with heat shock protein 70 (HSP70) showed SD only in one patient out of 12 patients with refractory colorectal cancer or NSCLC (78). Similarly, no clinical response was observed in patients with unresectable, locally advanced and/or metastatic digestive cancer treated with autologous NK cells expanded ex vivo with OK432, IL-2, and modified FN-CH296 induced T cells (79).
The poor clinical efficacy observed with adoptive transfer of ex vivo activated autologous NK cells may be due to the expression of MHC class I molecules on the surface of cancer cells, which bind to inhibitory receptors on autologous NK cells. This binding can suppress the activation of NK cells. Moreover, autologous NK cells isolated from cancer patients are usually exhausted or with impaired functions. In this way, is difficult to achieve effective antitumor activity (80).
The advantage of using allogeneic NK cells versus autologous NK cells is that allogeneic NK cells can be used with any recipient. This “off the shelf” source of NK cells can overcome the labor-intensive process to obtain and stabilize the one-donor, one-patient autologous NK cells.
The main sources for allogeneic NK cells are donor peripheral blood (PB), umbilical cord blood (UCB), human embryonic stem cells (hESCs), induced pluripotent stem cells (iPSCs) and NK cell lines (81, 82).
The advantage to obtain and expand allogeneic PB NK cells is that allogeneic NK cells can be isolated from peripheral blood mononuclear cells (PBMCs) by CD3 depletion and subsequent CD56 enrichment to achieve high NK cells purity (83). In addition, the off-the-shelf allogeneic PB NK cells can be re-infused to the same donor to allow further product generation and donor optimization. However, allogeneic PB NK cells present some disadvantages, such as low and/or highly variable percentage of NK cells derived from a single apheresis (especially between donors), heterogeneous population of NK cells (some express more activating receptors, some more inhibitory receptors), KIR-HLA mismatch between donor and recipient which may increase the risk of getting graft versus host disease (GVHD). In this case, to avoid immunological rejection, chemotherapy is needed before the infusion of allogeneic NK cells (82, 84).
PB NK cells can also be cryopreserved, but the process of cryopreservation can alter their cytotoxic function (85).
One strategy to overcome limitations of PB NK cells is to use UCB as source of NK cells. UCB has an abundance of highly cytotoxic CD56+ NK cells (86). NK cells derived from UCB are easy to collect, are younger and proliferate better than PB NK cells, and have less chance to trigger GVHD (87). The drawback in using UCB-derived NK cells is that they express lower levels of CD16, KIRs, IL-2R, and granzyme B as well as higher levels of inhibitory receptor NKG2A compared to PB-derived NK cells (88, 89). These features could reduce the cytotoxic activity (including mediated-ADCC) of allogeneic UCB-derived NK cells against cancer cells in vivo (88, 89). One strategy used to overcome this issue is to expand the UCB-derived NK cells in vitro using cytokines, such as IL-2, IL-12, IL-15, and IL-18 (87). Stimulation of UCB-derived NK cells with cytokines let them acquire a cytotoxicity comparable to that of PB NK cells (90).
A valid alternative to obtain a big source of allogeneic NK cells is represented by hESCs and iPSCs. The advantages of using NK cells derived from iPSC/hESC are the presence of homogenous population, the possibility to expand them on a large scale and the possibility to genetically modify them (91).
It has been proven that NK cells derived by hESC and iPSC are a homogeneous population of cells expressing CD56, KIRs, CD16, NKp44, NKp46, and NKG2D (92). This phenotype allows these NK cells to kill cancer cells similarly to PB NK cells (92). It has also been observed that hESC-derived NK cells have a more potent activity against cancer cells in vivo compared to UBC-derived NK cells (93). In addition, the possibility to modify genetically iPSCs allows to insert or delete specific genes to enhance their cytotoxicity and to have a consistent production in large scale of NK cells with identical phenotype (94).
Another strategy to have a stable and homogeneous source of allogeneic NK cells was the development of immortalized NK cell lines. The most promising NK cell line that showed antitumor activity is the NK-92 cell line.
NK-92 cell line is an immortalized IL-2-dependent human NK cell line, originated from a patient with lymphoma, that lacks ADCC activity and KIRs but can kill cancer cells through perforin and granzyme B-mediated cytolytic activity (95, 96). The limitation in the use of these cells, especially in clinic, is that they require irradiation prior to infusion into the cancer patient because of the risk they can proliferate abnormally and cause lymphoma (96).
Allogeneic NK cells are widely used in clinical trials as adoptive NK cell therapy. Allogeneic NK cells were employed to treat several types of cancers including hematological malignancies and solid tumors (82). For example, the first in human study using UBC-derived NK cells in MM patients showed that UCB-derived NK cells were well tolerated (no infusional toxicity and no GVHD) and resulted in partial response (PR) in 10 patients (97). Similarly, NK-92 cells showed safety and efficacy, with two patients that achieved a complete response (CR), in a phase I trial when used to treat refractory hematological malignancies (98). Despite promising results in blood tumors, efficacy of allogeneic NK cells in solid tumor was not so high. In a phase I study, the administration of non-HLA-matched donor PB NK cells, following a lymphodepleting chemotherapy regimen, showed SD in 2 out of 6 subjects with colorectal carcinoma up to post-infusion day 100 (99). Another phase I clinical trial reported that 8 out of 17 evaluable patients with malignant lymphoma or advanced, recurrent solid tumors, showed SD after being treated with ex vivo-expanded and highly activated NK cells (MG4101) derived from random unrelated healthy donors (100). The lower efficacy of allogeneic NK cells in solid tumors compared to blood tumors could be due to the high immune-suppressive TME of solid tumors.
Chimeric antigen receptor (CAR) technology is a relatively new strategy to enhance the potency of immune system against cancer cells. This technology was initially employed to produce CAR-T cells. CAR-T cells are generated from isolated T lymphocytes from cancer patients, that are then engineered to express CARs. These modified T lymphocytes are then expanded and reinfused into the patient to achieve a stronger antitumoral response (101).
CAR-T cells achieved promising responses against hematological malignancies, but CAR-T cell therapy failed to achieve good objective responses in patients with solid tumors. This phenomenon is due to their difficulty in infiltrating solid tumors and to the capacity of TME of solid tumors to impair the activity of CAR-T cells (101–103).
In addition, there are other limitations on the use of CAR-T cells, such as their limited expansion and short persistence, high cost and intensive manufacturing process. It is important also to report the toxicity associated with CAR-T therapy, including cytokine release syndrome (CRS), neurotoxicity, and GVHD (102, 103).
To overcome these limitations, chimeric antigen receptor-engineered NK (CAR-NK) cells have been developed.
CAR-NK cells can be generated using the same CD3ζ intracellular domain used to generate CAR-T cells or involving NK-specific activating domains, such as 2B4, DAP10 and DAP12 (104).
CAR-NK cells overcome several limitations of CAR-T cells. CAR-T cells are made from autologous T cells to avoid alloreactivity and GVHD due to a mismatch of MHC molecules between the donor and the recipient. Conversely, NK cells do not require MHC pathway to be activated. This feature reduces the risk of severe alloreactive reactions and allows to use different sources of NK cells, including peripheral and cord blood NK cells, induced pluripotent stem cells (iPSCs), and NK cell lines to generate CAR-NK cells (103, 104). In addition, CAR-NK cells can be manufactured through a less intensive process compared to CAR-T cells and infused to patients at any time (103). Moreover, CAR-NK cells release different profiles of cytokines compared to CAR-T cells upon activation, thus reducing the risk to develop CRS and neurotoxicity (105).
Another advantage of using CAR-NK cells over CAR-T cells is that NK cells can target and eliminate cancer cells with additional mechanisms beyond the CAR pathway, such as ADCC and the engagement of KIRs (104).
Recently, the safety and efficacy of CAR-NK cells against hematological malignancies have been investigated by several clinical trials. Results from these studies suggest that CAR-NK cells could be used as immunotherapy for patients with multiple myeloma, lymphoma, and leukemia (105, 106).
Unfortunately, the efficacy of CAR-NK cells to treat solid tumors is still poor. This could be due to several factors, including limited tumor infiltration, limited survival and persistence in the TME, inactivation of NK cells by the immunosuppressive TME, and low CAR transduction efficiency (103).
For example, soluble factors, such as BAG-6, galectin-9, soluble NKG2D-L, cytokines, released in the TME by cancer cells, Tregs, tumor-associated macrophages (TAM) and MDSCs can result in CAR-NK cell impairment. In addition, release of indoleamine 2, 3 dioxygenase (IDO) or ROS by CAFs, as well as hypoxia and nutrient deprivation in the TME, play a role in limiting survival, activity, and persistence of CAR-NK cells in the TME (107, 108).
To solve these limitations, a novel generation of CAR-NK cells has been developed. Most promising approach include a) modified CAR-NK cells expressing chemokine receptors to improve migration and persistence of CAR-NK cells in the TME (107, 108); b) modified CAR-NK cells targeting Tregs and CAFs in TME to reduce the release of immune-suppressive cytokines and soluble factors (108); c) engineered CAR-NK cells expressing IL-12, IL-15, and IL-18 to boost NK cells antitumor activity and prolong their survival in the TME (108, 109); d) combination of CAR-NK cells with immune checkpoint inhibitors (ICIs) to prevent NK cells exhaustion and inhibition by cancer cells (107, 108). Ongoing clinical trials are evaluating the efficacy and safety of CAR-NK cells for the treatment of different locally or advanced refractory/recurrent solid tumors, such as ovarian, endometrial, testicular, pancreatic, triple negative breast, colorectal and gastric carcinoma, but results about efficacy have not yet released (104).
Advantages and disadvantages of adoptive NK cell therapy are reported in Table 2.
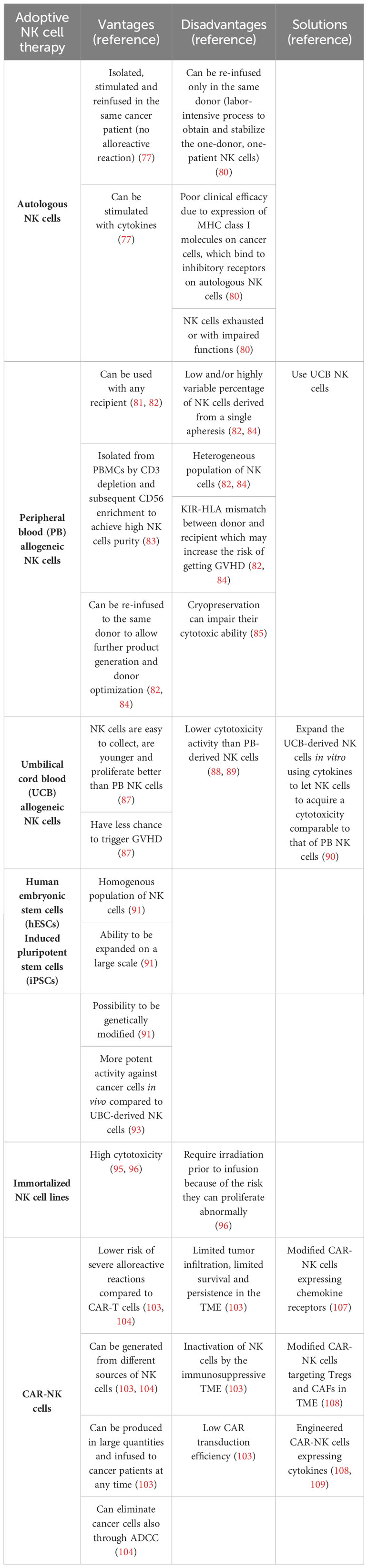
Table 2 Advantages and disadvantages of adoptive NK cell therapy.
The rationale of using antibodies for the treatment of malignant diseases was developed by Ehrlich more than a century ago (110).
Initially, antibody-based therapies employed tumor antigen allogenic or autologous polyclonal antibodies, but their use in clinic was abandoned because of their poor specificity and reproducibility.
With the development of hybridoma technology, which allowed to have tumor antigen-specific, reproducible, and immunogenic monoclonal antibodies, the employment of antibodies in the clinic became a reality (111).
Cancer cells evade immunosurveillance through several mechanisms. One of these mechanisms is the involvement of immune checkpoint pathways that allow cancer cells to inhibit antitumor immune response and to acquire a more aggressive phenotype (111–113).
During the last decades several mAbs targeting immune checkpoints pathways have been developed and approved by FDA for the treatment of human cancers.
The main pathways targeted by ICIs are cytotoxic T lymphocyte antigen-4 (CTLA-4) (112), the epidermal growth factor receptor (EGFR)-, CD20-, vascular endothelial growth factor (VEGF)-, and the programmed cell death protein-1 (PD-1)/programmed cell death protein-1 ligand (PD-L1)-mediated pathway (111).
The PD-1/PD-L1 checkpoint pathway represented one of the most targeted pathways by monoclonal antibodies used in clinic. The binding between PD-L1, over-expressed on cancer cells, and PD-1, expressed on activated T-cells, leads to cancer cells inhibiting the function of the cytotoxic T cells in the TME, and evading the pressure of the immune system. Anti–PD-1/PD-L1 mAbs are able to block the binding between PD-1 and PD-L1 to revert immunosuppressive signals by cancer cells and to enhance immune cells activity in the elimination of malignant cells (113).
Among them, nivolumab, pembrolizumab, atezolizumab, durvalumab, have demonstrated clinical efficacy against several tumor types.
Nivolumab, a fully human IgG4-blocking mAb directed against PD-1, produced objective responses ranging from 13.3 to 40% in patients with advanced melanoma (114), renal-cell (115), NSCLC (116), colorectal (117), head and neck (118), urothelial cancer (119).
The anti PD-1 mAb pembrolizumab has shown potent antitumor activity in patients with different solid tumors. Pembrolizumab provided substantial benefits in overall survival (OS), progression free survival (PFS), and overall response rate (ORR) in participants with NSCLC (120, 121), HNSCC (122), uterine (123), cervical (124), urothelial (125), Merkel-cell carcinoma (126), and melanoma (127).
A very promising clinical efficacy against PD-L1 expressing tumors has also been proved using anti-PD-L1 mAbs atezolizumab and durvalumab. For example, atezolizumab demonstrated a promising antitumor activity in patients with metastatic renal cell carcinoma, where an increased T-cell-to-regulatory T-cell ratio correlated with a better response to therapy (128, 129). In locally advanced and metastatic urothelial and in NSCLC patients, treatment with atezolizumab resulted in a significantly improved objective response rate (130–133). In addition, patients with metastatic triple-negative breast cancer with a higher PD-L1 expression showed a better clinical response than patients with a lower PD-L1 expression after treatment with atezolizumab in combination with nab-paclitaxel (134, 135). Similar results were achieved with durvalumab (136).
Although the employment of ICIs as immunotherapeutics has improved survival of cancer patients, not all patients achieved a good clinical response. This phenomenon can be due partially to tumor resistance to ICIs. Cancer cells, at level of TME, developed resistance to ICIs through reduction of expression of tumor antigens, uncontrolled activation of oncogenes, increased activity of Tregs and MDSCs (137, 138).
To overcome the resistance to ICIs, mAbs were engineered to kill cancer cells through ADCC mediated by NK cells.
The advantage of using mAbs with ADCC activity is that they can activate NK cells, but also stimulate the secretion of cytokines, which in turn facilitates the accumulation of immune effector cells to the TME (26). As already mentioned, ADCC is a mechanism that needs the binding of the Fc portion of mAbs to CD16 expressed on NK cells (26).
CD16 can have three different genotypes based on the presence of valine (V) or phenylalanine (F) at amino acid 158. Each genotype (V/V, V/F, F/F) confers to NK cells a different affinity for the Fc portion of mAbs, which in turn can result in a better or worse clinical outcome in cancer patients treated with mAbs with ADCC activity (139).
FDA approved several mAbs that can mediate ADCC. A correlation between NK cells CD16 genotype and clinical response has been observed in cancer patients treated with trastuzumab, rituximab, cetuximab, and avelumab.
Trastuzumab is a mAb which targets the human epidermal growth factor receptor 2 (HER2). HER2 is a member of the ErbB family that plays an important role in promoting oncogenic transformation and tumor growth. Overexpression of HER2 has been found in breast, ovarian, bladder, salivary gland, endometrial, pancreatic and NSCLC, and is usually linked with a poor prognosis (140).
Trastuzumab was approved by the FDA as a biological approach for the treatment of metastatic breast cancer in 1998 (141). FDA has also approved trastuzumab in combination with chemotherapy for HER2-positive metastatic cancer of the stomach or gastroesophageal junction in 2010 (142).
A pilot trial conducted in patients with operable breast tumors overexpressing HER2 demonstrated that trastuzumab was able to mediate ADCC in 15 of 18 patients (83%). Moreover, a patient which had a high ADCC activity showed a complete pathologic response, while patients unable to mount ADCC did not have a significant tumor regression after therapy. These data suggested that the response of breast cancer patients to trastuzumab may be due in part to the involvement of ADCC mechanism (143).
Different studies also showed that NK cells CD16 polymorphisms resulted in improvement of clinical outcomes in cancer patients treated with trastuzumab.
Musolino et al. demonstrated that patients with metastatic breast cancer expressing HER-2/neu and harboring the CD16 158 V/V genotype had a significantly higher trastuzumab-mediated cytotoxicity compared to other two genotypes and that patients with V/V genotype achieved a better ORR and PFS (144).
The tendency of the V/V genotype to be correlated with the objective response in metastatic HER2-positive breast cancer patients treated with trastuzumab has also been confirmed in another study by Tamura et al. (145).
Rituximab is a genetically engineered chimeric (murine-human) mAb which recognizes the B-cell-specific antigen CD20 expressed on various lymphoid malignancies, such as non-Hodgkin’s lymphomas (NHL) and B-cell chronic lymphocytic leukemia (CLL) (146, 147).
This mAb was approved by FDA for the treatment of relapsed low-grade or follicular B-cell non-Hodgkin’s lymphoma in 1997 (146), for the treatment of chronic lymphocytic leukemia in combination with fludarabine and cyclophosphamide in 2010 (148), and for the treatment of adult patients with Waldenström’s macroglobulinemia in 2018 (149).
As for trastuzumab, the CD16 158 V/V genotype resulted in an improved outcome also in patients treated with rituximab.
For example, patients with follicular lymphoma harboring CD16 V/V genotype had an improved clinical response to rituximab (150).
Similarly, Veeramani et al. suggested that the superior clinical outcome seen in patients with lymphoma harboring high affinity CD16 158 genotypes (V/F or V/V), compared to the low-affinity genotype (F/F), may be due to a quicker NK cells activation in subjects with the CD16 V/V genotype after treatment with rituximab (151).
The V/V genotype was also significantly correlated with a higher CR rate to rituximab plus cyclophosphamide/doxorubicin/vincristine/prednisone (R-CHOP) therapy in diffuse large B-cell lymphoma (DLBCL) compared with the F/F genotype (88% in V/V vs 50% in F/F) (152).
In a study conducted on patients affected by Waldenström’s macroglobulinemia, Treon et al. observed that the response trend to rituximab was significantly higher for patients with the V/V genotype compared to patients with the F/F genotype (40.0% vs 9%) (153).
Cetuximab is a chimeric immunoglobulin G1 (IgG1) which targets EGFR, a member of receptor tyrosine kinases (TK) highly expressed in several types of cancers, including breast, lung, colorectal and head and neck cancers. The binding between EGFR and its ligands activates a downstream signaling cascade that in turn promotes proliferation, differentiation, migration, and survival of cancer cells (154).
Cetuximab received FDA approval to treat patients with metastatic advanced colorectal cancer in 2004 (155). In 2011, cetuximab received FDA approval in combination with cisplatin or carboplatin and 5-fluorouracil for the first-line treatment of HNSCC (156).
In a study performed in patients with locally advanced head and neck cancer treated with cetuximab and radiotherapy, Lattanzio et al. observed that patients with elevated basal ADCC and high EGFR expression had a better chance of achieving a CR and a long OS compared to the others, suggesting a possible important role of ADCC in predicting favorable outcome (157).
Even for cetuximab it has been proved that the CD16 158 V/V genotype plays an important role in enhancing the ADCC.
In patients with metastatic colorectal cancer with KRAS mutations treated with cetuximab plus irinotecan it has been demonstrated that V/V genotype carriers had a longer PFS than F/F genotype carriers (5.5 v 3.0 months). Since patients with KRAS mutations were found to have a lower response rate and a shorter PFS than patients with non-mutated KRAS, the fact that the V/V genotype was correlated with a better PFS suggests an important role of the CD16 158 polymorfism in the ADCC activity and clinical outcome of patients treated with cetuximab (158).
Trotta et al. also demonstrated that colorectal cancer patients with the V/V genotype showed a significantly higher ADCC mediated by cetuximab and a significantly longer PFS than patients with F/F genotype (159).
Similarly, NK cells expressing the V/V genotype were found to be significantly more effective than those expressing V/F and F/F genotypes in mediating ADCC against HNSCC in presence of cetuximab, supporting the importance of the CD16 158 polymorphism in patient variability of cetuximab mediated clinical responses (160, 161).
Avelumab is a fully human IgG1 targeting PD-L1 on cancer cells.
Avelumab has been proved to mediate ADCC against a range of human cancer cells, including lung, breast, and bladder carcinomas (162–164). A preclinical study showed that subjects with the CD16 158 V/V genotype had higher capacity to kill cancer cells through ADCC mediated by avelumab than subjects with the F/F genotype (162).
Avelumab received FDA approval for the treatment of metastatic Merkel cell carcinoma (mMCC) in adults and pediatric patients aged ≥12 years and for the treatment of urothelial carcinoma in 2017 (165, 166). In 2019, avelumab received FDA approval in combination with axitinib for the treatment of patients with advanced renal cell carcinoma (167). Even if clinical studies to identify a link between CD16 polymorphism and better clinical outcome after treatment with avelumab are still ongoing, the ADCC mediated by avelumab resulted in clinical responses in several clinical trials.
In this regard, a multicenter, single-group, open-label, phase II trial showed that avelumab was associated with durable responses and was well tolerated in patients with chemotherapy-refractory metastatic Merkel cell carcinoma (168).
In a phase 3 trial, conducted in patients with advanced renal-cell carcinoma, the combination of avelumab and axitinib, a VEGF pathway inhibitor, resulted in an enhanced clinical benefit. Among patients with PD-L1-positive tumors, the ORR was 55.2% and the median PFS was 13.8 months when these agents were administered as first-line treatment (169). In a recent clinical trial, combining avelumab and cetuximab as second- or third-line treatment in patients with NSCLC, it has been proved that the clinical benefit of the combination of these two mAbs was determined by NK cell-mediated ADCC (170).
Several strategies have been developed to enhance FDA approved mAbs-mediated ADCC using engineered NK cells as effectors, including immunoligands, and adoptive NK cell therapy (Figure 3).
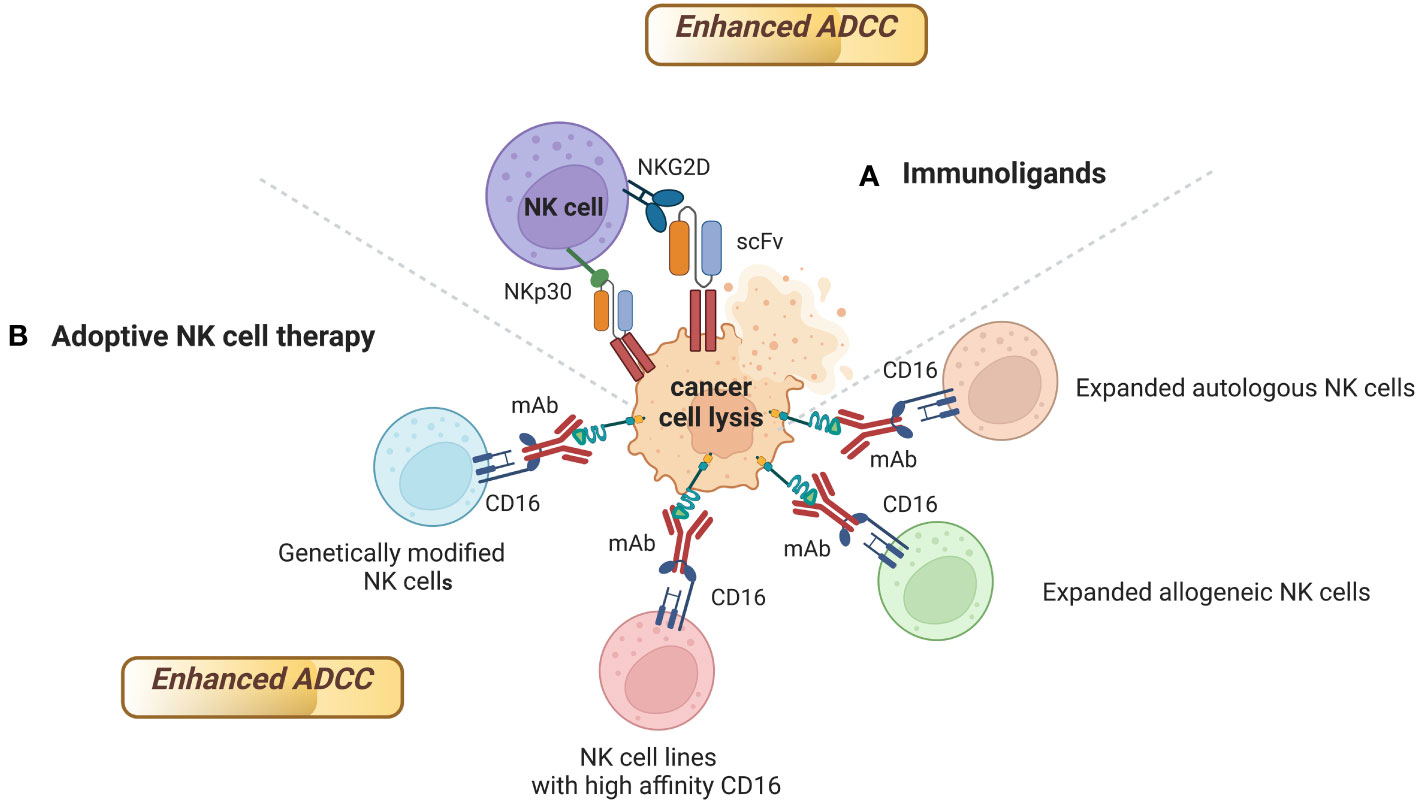
Figure 3 Strategies to enhance FDA approved mAbs-mediated ADCC using engineered NK cells as effectors. mAbs able to mediate ADCC can activate NK cells, but also stimulate the accumulation of immune effector cells to the tumor site. Engineered NK cells have been found to enhance ADCC activity of several FDA approved mAbs. Sources of engineered NK cells as effectors include (A) Immunoligands; (B) Expanded autologous or allogeneic NK cells, NK cell lines with high affinity CD16, genetically modified NK cells. Figure created with BioRender.com.
One example of the importance of immunoligands to enhance mAbs-mediated ADCC is the development of immunoligands fusing the extracellular domains of ligands of the activating NK cell receptors NKp30, NKp80, DNAM-1 and NKG2D to a single-chain fragment variable (scFv) targeting HER2 on different solid tumors (171, 172). The binding of these immunoligands to HER2 on cancer cells and to NK cell receptors enhanced ADCC mediated by trastuzumab and cetuximab against HER2 positive cancer cells (171, 172) (Table 3).
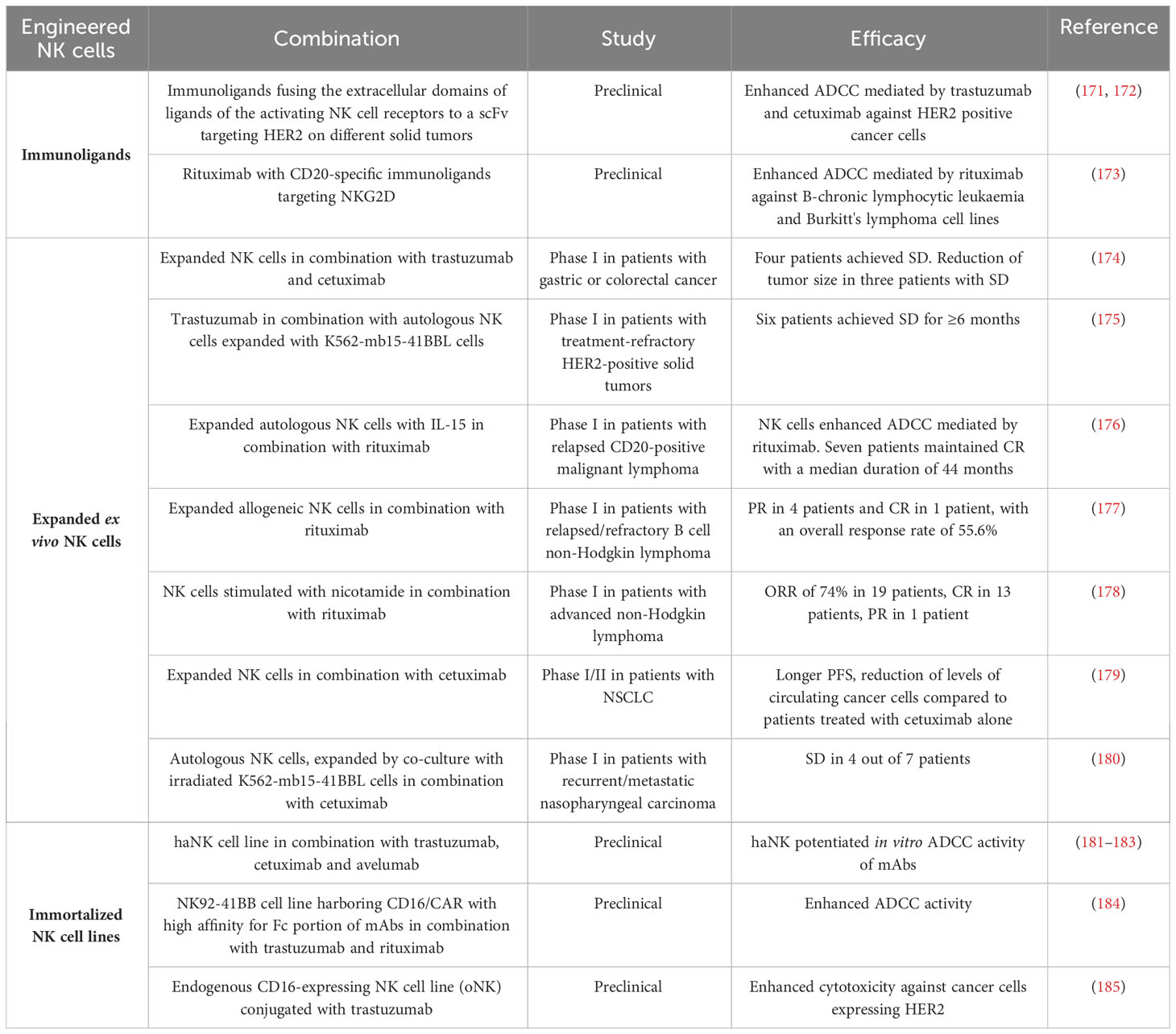
Table 3 Efficacy of engineered NK cells in enhancing FDA approved mAbs-mediated ADCC.
Similarly, it has been observed that the combination of rituximab with CD20-specific immunoligands targeting NKG2D can improve rituximab-mediated ADCC by NK cells against lymphoma (173) (Table 3).
The employment of adoptive NK cell therapy to enhance ADCC of mAbs is another promising strategy (Table 3). Sources of NK cells with a pre-clinical or clinical efficacy in enhancing ADCC of mAbs include expanded autologous or allogeneic NK cells, NK cell lines, genetically modified NK cells.
Recently, a phase I clinical trial evaluated the safety and efficacy of adoptive transfer of expanded NK cells in combination with trastuzumab and cetuximab in patients with gastric or colorectal cancer. Expanded NK cells with high expression of NKG2D and CD16 were infused in cancer patients after three days of mAbs administration. The combination was well tolerated and, among six evaluable patients, four patients achieved SD and two progressed. The treatment resulted also in a reduction of tumor size in three out of four patients with SD and this phenomenon was suggested to be linked to an enhanced whole blood IFN-γ production and reduction of Tregs (174).
Similarly, another phase I clinical trials showed that 6 of 19 patients with treatment-refractory HER2-positive solid tumors achieved SD for ≥6 months after receiving the combination of trastuzumab and autologous NK cells expanded by 10-day co-culture with K562-mb15-41BBL cells. These results support the rationale to use this combination in phase II trials (175).
A phase I study of cellular therapy for the treatment of patients with relapsed CD20-positive malignant lymphoma showed that NK cells, ex vivo expanded with IL-15 and then re-infused intravenously one day after rituximab administration, enhanced the ADCC mediated by rituximab. Seven of the nine patients maintained CR with a median duration of 44 months after NK-cell infusion. It is important to note that eight of the nine patients received chemotherapy after NK-cell infusion, rendering difficult to understand the exact contribution of the expanded NK cells to the clinical response (176). Furthermore, in a recent phase I clinical trial it has been evaluated safety and efficacy of ex vivo-expanded allogeneic NK cells (MG4101) in combination with rituximab for the treatment of relapsed/refractory B cell non-Hodgkin lymphoma. Results from the trial showed that the combined therapy was safe and resulted in PR in 4 patients and CR in 1 patient, with an ORR of 55.6% (177). In addition, two patients achieved prolonged responses and low exhaustion marker levels in T cells (177).
Similarly, a phase I clinical trial combining NK cells stimulated with NAM (GDA-201) and rituximab showed that the combination was well tolerated and yielded an ORR of 74% in 19 patients with advanced non-Hodgkin lymphoma (CR in 13 patients and PR in 1 patient) (178).
Another phase I clinical trial documented objective clinical responses in 3 out of 9 patients with liver metastasis from colorectal or pancreatic cancers after delivery of allogeneic NK cells in the liver combined with intravenous infusion of cetuximab and subcutaneous administration of IL-2 (186).
Adoptive NK cell therapy in combination with cetuximab also demonstrated to prolong survival in patients with advanced NSCLC. Liang et al. reported that patients treated with NK cell therapy + cetuximab had a significant improvement of quality of life and of the function of immune system. These features were associated with a longer PFS and a significantly reduction of levels of circulating cancer cells compared to patients treated with cetuximab alone, suggesting that the adoptive NK cell therapy can enhance the efficacy of cetuximab for the treatment of NSCLC (179).
A promising clinical response (SD in 4 out of 7 patients) was also observed in patients with nasopharyngeal carcinoma treated with a combination of autologous NK cells, expanded by co-culture with irradiated K562-mb15-41BBL cells, and cetuximab (180).
Enhancement of the ADCC activity can also be achieved using engineered NK cell lines or genetically modified NK cells (Table 3).
One example is represented by the engineered NK cell line haNK. The haNK cell line consists of the NK-92 cell line engineered to express CD16 with the high affinity (ha) genotype V/V, as well as to express IL-2. In this case, this NK cell line has the double advantage to mediate ADCC and to lyse cancer cells through an enhanced release of perforin and granzyme due to the constant expression of IL-2 (181). HaNK showed the ability to potentiate in vitro the ADCC activity of several clinical approved mAbs, including trastuzumab, cetuximab and avelumab (181–183). In addition, killing of cancer cells by irradiated haNK cells through ADCC mediated by avelumab was also shown to be similar to the killing achieved by NK cells bearing the CD16 V/V genotype (183). Another NK cell line (NK92-41BB), able to express high levels of granzyme and engineered to express chimeric genes CD16/CAR with high affinity for Fc portion of mAbs, showed a strong ADCC activity when tested in combination with trastuzumab and rituximab (184).
NK cell lines can also be generated using the Antibody-Cell Conjugation (ACC) technology. ACC provides an efficient platform to arm NK cells with antibodies targeting cancer cells to bind and kill malignant tumors. Li et al. established an NK cell line (oNK) expressing endogenous CD16 and conjugated oNK with trastuzumab and an anti-human HER2 antibody using an ACC platform. This trastuzumab-conjugated oNK, designated as ACE1702, exerted in vitro and in vivo antitumor activity against cancer cells expressing HER2, suggesting that it could be used as a novel NK cell therapy against solid tumors that express HER2 on their surface (185).
Promising results with immortalized NK cell lines provide the rationale for genetically modifying NK cells to enhance the ADCC activity of IgG1 antitumor mAbs.
Genetically modified NK cells in combination with mAbs with ADCC activity could represent a new promising strategy to achieve good clinical outcomes in several types of tumors. For example, Carlsten et al. developed genetically modified NK cells using the mRNA technology. In their study, authors demonstrated that NK cells genetically modified through electroporation with mRNA coding for CD16 158 V/V genotype potentiated NK cells cytotoxicity against lymphoma cells coated with rituximab (187). These results suggest that this approach can be utilized to improve the efficacy of adoptive NK cell-based cancer immunotherapies in combination with mAbs with ADCC activity (187).
The concept of harnessing the immune system against cancer opened the era of cancer immunotherapy for the treatment of solid and liquid cancers. The advantage of immunotherapy over chemotherapy or radiotherapy is the possibility to redirect cellular components of immune system to attack selectively cancer cells, minimizing toxicity of the treatment. Among cellular components of immune system, NK cells play a fundamental role in controlling tumor growth. NK cells can target and kill cancer cells through NK-direct killing, a mechanism that involve NK cells only, or through ADCC, a mechanism that allows NK cells to target and eliminate cancer cells via binding to an antibody already bound to a specific antigen expressed by cancer cells. Cancer cells have developed several mechanisms to evade the antitumor activity of NK cells. Immunotherapy led to the development of several strategies to enhance the antitumor activity of NK cells against cancer cells, with the goal of overcoming cancer cells resistance. In this regard, the three main strategies to engineer and boost NK cells include boosting NK cells with modulatory cytokines, adoptive NK cell therapy, and the employment of engineered NK cells to enhance antibody-based immunotherapy.
Boosting NK cells with cytokines provided the opportunity to achieve superior antitumor activity of NK cells against several types of cancers, but this strategy has the problem of systemic toxicity of cytokines, as well as the possibility to enhance proliferation of immune-suppressive cells.
Given the difficulties of achieving impressive clinical responses with manageable toxicities through the stimulation of NK cells with cytokines, NK cell-based immunotherapy was improved through the development of engineered NK cells. Engineered NK cells are being used as adoptive NK cells and to enhance antibody-based immunotherapy.
One of the first applications of adoptive NK cell therapy was the employment of autologous NK cells, which consist of isolating NK cells from a cancer patient, stimulating them ex vivo, and then reinfusing them in the same patient. Although the stimulation of autologous NK cells ex vivo can be satisfactory, their efficacy in humans seems to be modest.
The poor clinical efficacy may be due to the expression of MHC class I molecules on the surface of cancer cells, which bind to inhibitory receptors on autologous NK cells. This binding can suppress the activation of NK cells. In addition, autologous NK cells isolated from patients with malignant tumors usually are exhausted or with impaired functions (80).
To overcome this problem, the new strategy was to develop allogeneic NK cells.
The advantage of using allogeneic NK cells versus autologous NK cells is that allogeneic NK cells can be used with any recipient, thus overcoming the labor-intensive process to obtain and stabilize the one-donor, one-patient autologous NK cells (81, 82).
Allogeneic NK cells are widely used in clinical trials to treat hematological malignancies and solid tumors (82). Despite promising results in blood tumors, efficacy of allogeneic NK cells in solid tumor was modest. One reason for the lower efficacy of allogeneic NK cells in solid tumors could be due to the high immune-suppressive TME of solid tumors. A new strategy to create engineered NK cells targeting cancer cells in a more selective and aggressive manner was the development of CAR-NK cells.
The advantage of using CAR-NK cells is the fact that CAR-NK cells have a low risk of severe alloreactive reactions, can be generated by different sources of NK cells, can be produced in big quantity, and can be infused to cancer patients at any time (103).
Another advantage of using CAR-NK cells is that NK cells can target and eliminate cancer cells with additional mechanisms beyond the CAR pathway, such as ADCC and the engagement of KIRs (104).
Although CAR-NK cells achieved promising clinical responses in hematological malignancies, the efficacy of CAR-NK cells to treat solid tumors is still poor. This could be due to lack of tumor infiltration, limited persistence in the TME, inactivation of NK cells by TME (103). To solve these limitations, a novel generation of CAR-NK cells has been developed, including modified CAR-NK cells expressing chemokine receptors (107), modified CAR-NK cells targeting Tregs and CAFs in TME (108), engineered CAR-NK cells expressing IL-12, IL-15, and IL-18 (108, 109), combination of CAR-NK cells with ICIs (107, 108).
Due to the ability of cancer cells to resist to NK cells direct killing, a novel approach was to develop engineered NK cells to enhance the ADCC activity of mAbs approved for the treatment of several cancers.
Several strategies have been developed to enhance FDA approved mAbs-mediated ADCC using engineered NK cells as effectors, including immunoligands, adoptive NK cell therapy, NK cell lines, genetically modified NK cells. Several clinical trials reported that the combination of engineering NK cells with mAbs with ADCC activity can result in a superior clinical response compared to mAbs alone.
To make this combination more effective and potent, future perspectives are to further engineer both mAbs and NK cells. One strategy that is currently being implemented is to increase the affinity of mAbs to CD16 to enhance their ADCC activity.
These modifications can be done by altering the mAb Fc portion through different methodologies, such as site-directed mutagenesis, altering Fc domain glycosylation and/or taking out fucosylation of the Fc domain. In this regard, several mAbs with modified Fc portion have already shown enhanced activity compared to unmodified mAbs (188).
Some of these engineered mAbs, such as margetuximab, mogamulizumab, obinutuzumab, tafasitamab, have recently obtained approval for the treatment of several cancers (189–192).
Other strategies could be the employment of bi-specific antibodies, bi-specific killer cell engagers (BIKEs), combinations of NK cells with mAbs able to recognize and eliminate immunosuppressive cells in the TME.
For example, Pan and colleagues developed a bi-specific fusion protein (mAb04-MICA) comprising an antibody targeting vascular endothelial growth factor receptor 2 (VEGFR2) fused to a MICA α1-α2 ectodomain. Authors observed that mAb04-MICA had a better antitumor efficacy in mice bearing gastric cancer and NSCLC compared with the anti-VEGFR2 mAb ramucirumab (193).
Cancer cells can develop resistance to ADCC mediated by mAbs by inhibiting the activation and function of NK cells in the TME. One strategy to overcome this resistance is the development of BIKEs.
BiKEs concept is promoting the infiltration of NK cells to tumor site, bringing them close to cancer cells. BIKEs consist of individual heavy (VH) and light (VL) chains of the variable region of each different antibody (scFv) specific for CD16, NKG2D, NKp30 on NK cells and for tumor-associated antigens on cancer cells. In this way, NK cells can target easier cancer cells and once in close contact with them are facilitated to form cytolytic synapses that allow them to release granzymes and perforins to kill cancer cells (194). Since the antitumor activity of NK cells can also be impaired by immunosuppressive cells in the TME, the employment of mAbs able to recognize and eliminate immune-suppressive cells in the TME has the advantage of reducing NK cells impairment and exhaustion. Combining engineered NK cells with these mAbs would allow NK cells to remain in an activated status and to kill cancer cells resistant to NK cells direct killing or to ADCC mediated by mAbs.
These strategies are currently under preclinical investigation and if they achieve promising antitumor activity they could be used as rationale for the next generation of clinical trials with the scope to provide more effective and higher-quality treatments to cancer patients.
Another important point to take into consideration using NK cell therapy for treatment of cancer is the off-the-shelf applicability. NK cells can be expanded ex-vivo in large quantity using co-culture of NK cells with irradiated feeder cells, such as RPMI8866, EBV-LCL, and K562.
These feeder cells promote expansion of NK cells by presenting them activating ligands. In addition, to expand NK cells population over the time, NK cells can be periodically restimulated by feeder cells ex vivo. This expansion can be made in seed bioreactors which can allow to produce up to 1011 to 1012 cells of highly active NK cells ready to be infused to several patients at the same time. Since it is unclear if the expansion of NK cells through feeder cells can be sustained for long periods, feeder-cell free activation methods to expand NK cells are in development phase to obtain large number of stable activated NK cells with lower costs of biomanufacturing (195).
MF: Conceptualization, Data curation, Writing – original draft. PA: Funding acquisition, Visualization, Writing – review & editing. KT: Conceptualization, Data curation, Supervision, Visualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by Precision Biologics, Inc. The funder had no role in the design of the study, in the collection, analyses or interpretation of the data, in the writing of the manuscript or in the decision to publish the results.
MF, KT and PA are employees of Precision Biologics, Inc.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
2. Vilalta M, Rafat M, Graves EE. Effects of radiation on metastasis and tumor cell migration. Cell Mol Life Sci (2016) 73:2999–3007. doi: 10.1007/s00018-016-2210-5
3. Lewis A, Reed M, Walde N, Voutsadakis IA. An evaluation of the Index4 tool for chemotherapy toxicity prediction in cancer patients older than 70 years old. Sci Rep (2023) 13:1082. doi: 10.1038/s41598-023-28309-5
4. Lizée G, Overwijk WW, Radvanyi L, Gao J, Sharma P, Hwu P. Harnessing the power of the immune system to target cancer. Annu Rev Med (2013) 64:71–90. doi: 10.1146/annurev-med-112311-083918
5. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
6. Bertram JS. The molecular biology of cancer. Mol Aspects Med (2000) 21:167–223. doi: 10.1016/s0098-2997(00)00007-8
7. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
8. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol (2011) 29:235–71. doi: 10.1146/annurev-immunol-031210-101324
9. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity (2004) 21:137–48. doi: 10.1016/j.immuni.2004.07.017
10. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3:991–8. doi: 10.1038/ni1102-991
11. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science (2011) 331:1565–70. doi: 10.1126/science.1203486
12. Lakshmi Narendra B, Eshvendar Reddy K, Shantikumar S, Ramakrishna S. Immune system: a double-edged sword in cancer. Inflammation Res (2013) 62:823–34. doi: 10.1007/s00011-013-0645-9
13. Kerdiles Y, Ugolini S, Vivier E. T cell regulation of natural killer cells. J Exp Med (2013) 210:1065–8. doi: 10.1084/jem.20130960
14. Dougan M, Dranoff G. Immune therapy for cancer. Annu Rev Immunol (2009) 27:83–117. doi: 10.1146/annurev.immunol.021908.132544
15. Miller JS. The biology of natural killer cells in cancer, infection, and pregnancy. Exp Hematol (2001) 29:1157–68. doi: 10.1016/s0301-472x(01)00696-8
16. Papazahariadou M, Athanasiadis GI, Papadopoulos E, Symeonidou I, Hatzistilianou M, Castellani ML, et al. Involvement of NK cells against tumors and parasites. Int J Biol Markers (2007) 22:144–53. doi: 10.1177/172460080702200208
17. Caligiuri MA. Human natural killer cells. Blood (2008) 112:461–9. doi: 10.1182/blood-2007-09-077438
18. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol (2001) 22:633–40. doi: 10.1016/s1471-4906(01)02060-9
19. Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for Malignant diseases. Cell Mol Immunol (2013) 10:230–52. doi: 10.1038/cmi.2013.10
20. Frazao A, Rethacker L, Messaoudene M, Avril MF, Toubert A, Dulphy N, et al. NKG2D/NKG2-ligand pathway offers new opportunities in cancer treatment. Front Immunol (2019) 10:661. doi: 10.3389/fimmu.2019.00661
21. Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, et al. NKG2D and its ligands: "One for all, all for one". Front Immunol (2018) 9:476. doi: 10.3389/fimmu.2018.00476
22. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology (2007) 121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x
23. Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, et al. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol (2014) 122:91–128. doi: 10.1016/B978-0-12-800267-4.00003-1
24. Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandström N, et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J Exp Med (2019) 216:2113–127. doi: 10.1084/jem.20181454
25. Prager I, Watzl C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J Leukoc Biol (2019) 105:1319–29. doi: 10.1002/JLB.MR0718-269R
26. Seidel UJ, Schlegel P, Lang P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front Immunol (2013) 4:76. doi: 10.3389/fimmu.2013.00076
27. Baginska J, Viry E, Paggetti J, Medves S, Berchem G, Moussay E, et al. The critical role of the tumor microenvironment in shaping natural killer cell-mediated anti-tumor immunity. Front Immunol (2013) 4:490. doi: 10.3389/fimmu.2013.00490
28. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
29. Zalfa C, Paust S. Natural killer cell interactions with myeloid derived suppressor cells in the tumor microenvironment and implications for cancer immunotherapy. Front Immunol (2021) 12:633205. doi: 10.3389/fimmu.2021.633205
30. Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res (2010) 70:6945–56. doi: 10.1158/0008-5472.CAN-10-0785
31. Chretien AS, Le Roy A, Vey N, Prebet T, Blaise D, Fauriat C, et al. Cancer-induced alterations of NK-mediated target recognition: current and investigational pharmacological strategies aiming at restoring NK-mediated anti-tumor activity. Front Immunol (2014) 5:122. doi: 10.3389/fimmu.2014.00122
32. Markel G, Lieberman N, Katz G, Arnon TI, Lotem M, Drize O, et al. CD66a interactions between human melanoma and NK cells: a novel class I MHC-independent inhibitory mechanism of cytotoxicity. J Immunol (2002) 168:2803–10. doi: 10.4049/jimmunol.168.6.2803
33. Stern N, Markel G, Arnon TI, Gruda R, Wong H, Gray-Owen SD, et al. Carcinoembryonic antigen (CEA) inhibits NK killing via interaction with CEA-related cell adhesion molecule 1. J Immunol (2005) 174:6692–701. doi: 10.4049/jimmunol.174.11.6692
34. Wu J, Mishra HK, Walcheck B. Role of ADAM17 as a regulatory checkpoint of CD16A in NK cells and as a potential target for cancer immunotherapy. J Leukoc Biol (2019) 105:1297–303. doi: 10.1002/JLB.2MR1218-501R
35. Bramswig KH, Poettler M, Unseld M, Wrba F, Uhrin P, Zimmermann W, et al. Soluble carcinoembryonic antigen activates endothelial cells and tumor angiogenesis. Cancer Res (2013) 73:6584–96. doi: 10.1158/0008-5472.CAN-13-0123
36. Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICA in Malignant diseases. Int J Cancer (2006) 118:684–7. doi: 10.1002/ijc.21382
37. Luo Q, Luo W, Zhu Q, Huang H, Peng H, Liu R, et al. Tumor-derived soluble MICA obstructs the NKG2D pathway to restrain NK cytotoxicity. Aging Dis (2020) 11:118–28. doi: 10.14336/AD.2019.1017
38. Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia (2004) 6:558–68. doi: 10.1593/neo.04316
39. Li JJ, Pan K, Gu MF, Chen MS, Zhao JJ, Wang H, et al. Prognostic value of soluble MICA levels in the serum of patients with advanced hepatocellular carcinoma. Chin J Cancer (2013) 32:141–8. doi: 10.5732/cjc.012.10025
40. Qiu Y, Zhao YK, Yuan GJ, Zhu QG. Clinical significance of soluble major histocompatibility complex class I chain-related in a renal cell carcinoma patients. Asian Pac J Cancer Prev (2013) 14:5651–5. doi: 10.7314/apjcp.2013.14.10.5651
41. Wang LP, Niu H, Xia YF, Han YL, Niu P, Wang HY, et al. Prognostic significance of serum sMICA levels in non-small cell lung cancer. Eur Rev Med Pharmacol Sci (2015) 19:2226–30.
42. Zingoni A, Vulpis E, Cecere F, Amendola MG, Fuerst D, Saribekyan T, et al. MICA-129 dimorphism and soluble MICA are associated with the progression of multiple myeloma. Front Immunol (2018) 9:926. doi: 10.3389/fimmu.2018.00926
43. Onyeaghala G, Nelson HH, Thyagarajan B, Linabery AM, Panoskaltsis-Mortari A, Gross M, et al. Soluble MICA is elevated in pancreatic cancer: Results from a population based case-control study. Mol Carcinog (2017) 56:2158–64. doi: 10.1002/mc.22667
44. Cole CB, Morelli MP, Fantini M, Miettinen M, Fetsch P, Peer C, et al. First-in-human phase 1 clinical trial of anti-core 1 O-glycans targeting monoclonal antibody NEO-201 in treatment-refractory solid tumors. J Exp Clin Cancer Res (2023) 42:76. doi: 10.1186/s13046-023-02649-6
45. Romee R, Leong JW, Fehniger TA. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica (Cairo) (2014) 2014:205796. doi: 10.1155/2014/205796
46. Spolski R, Li P, Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol (2018) 18:648–59. doi: 10.1038/s41577-018-0046-y
47. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol (2014) 192:5451–8. doi: 10.4049/jimmunol.1490019
48. Vacca P, Pietra G, Tumino N, Munari E, Mingari MC, Moretta L. Exploiting human NK cells in tumor therapy. Front Immunol (2020) 10:3013. doi: 10.3389/fimmu.2019.03013
49. Sim GC, Liu C, Wang E, Liu H, Creasy C, Dai Z, et al. IL2 variant circumvents ICOS+ Regulatory T-cell expansion and promotes NK cell activation. Cancer Immunol Res (2016) 4:983–94. doi: 10.1158/2326-6066.CIR-15-0195
50. Mishra A, Sullivan L, Caligiuri MA. Molecular pathways: interleukin-15 signaling in health and in cancer. Clin Cancer Res (2014) 20:2044–50. doi: 10.1158/1078-0432.CCR-12-3603
51. Fehniger TA, Caligiuri MA. Interleukin 15: biology and relevance to human disease. Blood (2001) 97:14–32. doi: 10.1182/blood.v97.1.14
52. Waldmann TA, Lugli E, Roederer M, Perera LP, Smedley JV, Macallister RP, et al. Safety (toxicity), pharmacokinetics, immunogenicity, and impact on elements of the normal immune system of recombinant human IL-15 in rhesus macaques. Blood (2011) 117:4787–95. doi: 10.1182/blood-2010-10-311456
53. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol (2015) 33:74–82. doi: 10.1200/JCO.2014.57.3329
54. Wagner JA, Rosario M, Romee R, Berrien-Elliott MM, Schneider SE, Leong JW, et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J Clin Invest (2017) 127:4042–58. doi: 10.1172/JCI90387
55. Yang Y, Lundqvist A. Immunomodulatory effects of IL-2 and IL-15; implications for cancer immunotherapy. Cancers (Basel) (2020) 12:3586. doi: 10.3390/cancers12123586
56. Stoklasek TA, Schluns KS, Lefrançois L. Combined IL-15/IL-15Ralpha immunotherapy maximizes IL-15 activity in vivo. J Immunol (2006) 177:6072–80. doi: 10.4049/jimmunol.177.9.6072
57. Rhode PR, Egan JO, Xu W, Hong H, Webb GM, Chen X, et al. Comparison of the superagonist complex, ALT-803, to IL15 as cancer immunotherapeutics in animal models. Cancer Immunol Res (2016) 4:49–60. doi: 10.1158/2326-6066.CIR-15-0093-T
58. Fantini M, David JM, Wong HC, Annunziata CM, Arlen PM, Tsang KY. An IL-15 superagonist, ALT-803, enhances antibody-dependent cell-mediated cytotoxicity elicited by the monoclonal antibody NEO-201 against human carcinoma cells. Cancer Biother Radiopharm (2019) 34:147–59. doi: 10.1089/cbr.2018.2628
59. Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood (2018) 131:2515–27. doi: 10.1182/blood-2017-12-823757
60. Wrangle JM, Velcheti V, Patel MR, Garrett-Mayer E, Hill EG, Ravenel JG, et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomized, open-label, phase 1b trial. Lancet Oncol (2018) 19:694–704. doi: 10.1016/S1470-2045(18)30148-7
61. Liu B, Zhu X, Kong L, Wang M, Spanoudis C, Chaturvedi P, et al. Bifunctional TGF-β trap/IL-15 protein complex elicits potent NK cell and CD8+ T cell immunity against solid tumors. Mol Ther (2021) 29:2949–62. doi: 10.1016/j.ymthe.2021.06.001
62. Chaturvedi P, George V, Shrestha N, Wang M, Dee MJ, Zhu X, et al. Immunotherapeutic HCW9218 augments anti-tumor activity of chemotherapy via NK cell-mediated reduction of therapy-induced senescent cells. Mol Ther (2022) 30:1171–87. doi: 10.1016/j.ymthe.2022.01.025
63. Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, et al. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med (1989) 170:827–45. doi: 10.1084/jem.170.3.827
64. Lasek W, Zagożdżon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother (2014) 63:419–35. doi: 10.1007/s00262-014-1523-1
65. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumors. Nat Rev Immunol (2012) 12:253–68. doi: 10.1038/nri3175
66. Becker JC, Andersen MH, Schrama D, Thor Straten P. Immune-suppressive properties of the tumor microenvironment. Cancer Immunol Immunother (2013) 62:1137–48. doi: 10.1007/s00262-013-1434-6
67. Strauss J, Heery CR, Kim JW, Jochems C, Donahue RN, Montgomery AS, et al. First-in-human phase I trial of a tumor-targeted cytokine (NHS-IL12) in subjects with metastatic solid tumors. Clin Cancer Res (2019) 25:99–109. doi: 10.1158/1078-0432.CCR-18-1512
68. Li YP, Du XR, Zhang R, Yang Q. Interleukin-18 promotes the antitumor ability of natural killer cells in colorectal cancer via the miR-574-3p/TGF-β1 axis. Bioengineered (2021) 12:763–78. doi: 10.1080/21655979.2021.1880717
69. Ma Z, Li W, Yoshiya S, Xu Y, Hata M, El-Darawish Y, et al. Augmentation of immune checkpoint cancer immunotherapy with IL18. Clin Cancer Res (2016) 22:2969–80. doi: 10.1158/1078-0432.CCR-15-1655
70. Tarhini AA, Millward M, Mainwaring P, Kefford R, Logan T, Pavlick A, et al. A phase 2, randomized study of SB-485232, rhIL-18, in patients with previously untreated metastatic melanoma. Cancer (2009) 115:859–68. doi: 10.1002/cncr.24100
71. Terme M, Ullrich E, Aymeric L, Meinhardt K, Coudert JD, Desbois M, et al. Cancer-induced immunosuppression: IL-18-elicited immunoablative NK cells. Cancer Res (2012) 72:2757–67. doi: 10.1158/0008-5472.CAN-11-3379
72. Park IH, Yang HN, Lee KJ, Kim TS, Lee ES, Jung SY, et al. Tumor-derived IL-18 induces PD-1 expression on immunosuppressive NK cells in triple-negative breast cancer. Oncotarget (2017) 8:32722–30. doi: 10.18632/oncotarget.16281
73. Zhu S, Phatarpekar PV, Denman CJ, Senyukov VV, Somanchi SS, Nguyen-Jackson HT, et al. Transcription of the activating receptor NKG2D in natural killer cells is regulated by STAT3 tyrosine phosphorylation. Blood (2014) 124:403–11. doi: 10.1182/blood-2013-05-499707
74. Sivori S, Cantoni C, Parolini S, Marcenaro E, Conte R, Moretta L, et al. IL-21 induces both rapid maturation of human CD34+ cell precursors towards NK cells and acquisition of surface killer Ig-like receptors. Eur J Immunol (2003) 33:3439–47. doi: 10.1002/eji.200324533
75. Watanabe M, Kono K, Kawaguchi Y, Mizukami Y, Mimura K, Maruyama T, et al. Interleukin-21 can efficiently restore impaired antibody-dependent cell-mediated cytotoxicity in patients with esophageal squamous cell carcinoma. Br J Cancer (2010) 102:520–9. doi: 10.1038/sj.bjc.6605502
76. Croce M, Rigo V, Ferrini S. IL-21: a pleiotropic cytokine with potential applications in oncology. J Immunol Res (2015) 2015:696578. doi: 10.1155/2015/696578
77. Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR, et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplant (2003) 32:177–86. doi: 10.1038/sj.bmt.1704086
78. Krause SW, Gastpar R, Andreesen R, Gross C, Ullrich H, Thonigs G, et al. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: a clinical phase i trial. Clin Cancer Res (2004) 10:3699–707. doi: 10.1158/1078-0432.CCR-03-0683
79. Sakamoto N, Ishikawa T, Kokura S, Okayama T, Oka K, Ideno M, et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J Transl Med (2015) 13:277. doi: 10.1186/s12967-015-0632-8
80. Kang S, Gao X, Zhang L, Yang E, Li Y, Yu L. The advances and challenges of NK cell-based cancer immunotherapy. Curr Oncol (2021) 28:1077–93. doi: 10.3390/curroncol28020105
81. Berrien-Elliott MM, Jacobs MT, Fehniger TA. Allogeneic natural killer cell therapy. Blood (2023) 141:856–68. doi: 10.1182/blood.2022016200
82. Heipertz EL, Zynda ER, Stav-Noraas TE, Hungler AD, Boucher SE, Kaur N, et al. Current perspectives on "Off-the-shelf" Allogeneic NK and CAR-NK cell therapies. Front Immunol (2021) 12:732135. doi: 10.3389/fimmu.2021.732135
83. Williams SM, Sumstad D, Kadidlo D, Curtsinger J, Luo X, Miller JS, et al. Clinical-scale production of cGMP compliant CD3/CD19 cell-depleted NK cells in the evolution of NK cell immunotherapy at a single institution. Transfusion (2018) 58:1458–67. doi: 10.1111/trf.14564
84. Lizana-Vasquez GD, Torres-Lugo M, Dixon RB, Powderly JD 2nd, Warin RF. The application of autologous cancer immunotherapies in the age of memory-NK cells. Front Immunol (2023) 14:1167666. doi: 10.3389/fimmu.2023.1167666
85. Mark C, Czerwinski T, Roessner S, Mainka A, Hörsch F, Heublein L, et al. Cryopreservation impairs 3-D migration and cytotoxicity of natural killer cells. Nat Commun (2020) 11:5224. doi: 10.1038/s41467-020-19094-0
86. Condiotti R, Zakai YB, Barak V, Nagler A. Ex vivo expansion of CD56+ cytotoxic cells from human umbilical cord blood. Exp Hematol (2001) 29:104–13. doi: 10.1016/s0301-472x(00)00617-2
87. Zhao X, Cai L, Hu Y, Wang H. Cord-blood natural killer cell-based immunotherapy for cancer. Front Immunol (2020) 11:584099. doi: 10.3389/fimmu.2020.584099
88. Luevano M, Daryouzeh M, Alnabhan R, Querol S, Khakoo S, Madrigal A, et al. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum Immunol (2012) 73:248–57. doi: 10.1016/j.humimm.2011.12.015
89. Wang Y, Xu H, Zheng X, Wei H, Sun R, Tian Z. High expression of NKG2A/CD94 and low expression of granzyme B are associated with reduced cord blood NK cell activity. Cell Mol Immunol (2007) 4:377–82.
90. Verneris MR, Miller JS. The phenotypic and functional characteristics of umbilical cord blood and peripheral blood natural killer cells. Br J Haematol (2009) 147:185–91. doi: 10.1111/j.1365-2141.2009.07768.x
91. Zhu H, Kaufman DS. An improved method to produce clinical-scale natural killer cells from human pluripotent stem cells. Methods Mol Biol (2019) 2048:107–19. doi: 10.1007/978-1-4939-9728-2_12
92. Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med (2013) 2:274–83. doi: 10.5966/sctm.2012-0084
93. Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, et al. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood (2009) 113:6094–101. doi: 10.1182/blood-2008-06-165225
94. Goldenson BH, Hor P, Kaufman DS. iPSC-derived natural killer cell therapies - expansion and targeting. Front Immunol (2022) 13:841107. doi: 10.3389/fimmu.2022.841107
95. Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of Malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res (2001) 10:535–44. doi: 10.1089/15258160152509145
96. Klingemann H. The NK-92 cell line-30 years later: its impact on natural killer cell research and treatment of cancer. Cytotherapy (2023) 25:451–57. doi: 10.1016/j.jcyt.2022.12.003
97. Shah N, Li L, McCarty J, Kaur I, Yvon E, Shaim H, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol (2017) 177:457–66. doi: 10.1111/bjh.14570
98. Williams BA, Law AD, Routy B, denHollander N, Gupta V, Wang XH, et al. A phase I trial of NK-92 cells for refractory hematological Malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget (2017) 8:89256–68. doi: 10.18632/oncotarget.19204
99. Otegbeye F, Cooper B, Caimi P, Zamborsky K, Reese-Koc J, Hillian A, et al. A phase I study to determine the maximum tolerated dose of ex vivo expanded natural killer cells derived from unrelated, HLA-disparate adult donors. Transplant Cell Ther (2022) 28:250.e1–8. doi: 10.1016/j.jtct.2022.02.008
100. Yang Y, Lim O, Kim TM, Ahn YO, Choi H, Chung H, et al. Phase I study of random healthy donor-derived allogeneic natural killer cell therapy in patients with Malignant lymphoma or advanced solid tumors. Cancer Immunol Res (2016) 4:215–24. doi: 10.1158/2326-6066.CIR-15-0118
101. Fantini M, Bei R. Editorial: engineered targeted cancer immunotherapies. Front Oncol (2022) 12:953175. doi: 10.3389/fonc.2022.953175
102. Lin H, Cheng J, Mu W, Zhou J, Zhu L. Advances in universal CAR-T cell therapy. Front Immunol (2021) 12:744823. doi: 10.3389/fimmu.2021.744823
103. Shin MH, Oh E, Kim Y, Nam DH, Jeon SY, Yu JH, et al. Recent advances in CAR-based solid tumor immunotherapy. Cells (2023) 12:1606. doi: 10.3390/cells12121606
104. Pan K, Farrukh H, Chittepu VCSR, Xu H, Pan CX, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res (2022) 41:119. doi: 10.1186/s13046-022-02327-z
105. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
106. Huang R, Wen Q, Zhang X. CAR-NK cell therapy for hematological Malignancies: recent updates from ASH 2022. J Hematol Oncol (2023) 16:35. doi: 10.1186/s13045-023-01435-3
107. Włodarczyk M, Pyrzynska B. CAR-NK as a rapidly developed and efficient immunotherapeutic strategy against cancer. Cancers (Basel) (2022) 15:117. doi: 10.3390/cancers15010117
108. Valeri A, García-Ortiz A, Castellano E, Córdoba L, Maroto-Martín E, Encinas J, et al. Overcoming tumor resistance mechanisms in CAR-NK cell therapy. Front Immunol (2022) 13:953849. doi: 10.3389/fimmu.2022.953849
109. Wang X, Yang X, Yuan X, Wang W, Wang Y. Chimeric antigen receptor-engineered NK cells: new weapons of cancer immunotherapy with great potential. Exp Hematol Oncol (2022) 11:85. doi: 10.1186/s40164-022-00341-7
110. Silverstein AM. Paul Ehrlich's passion: the origins of his receptor immunology. Cell Immunol (1999) 194:213–21. doi: 10.1006/cimm.1999.1505
111. Campoli M, Ferris R, Ferrone S, Wang X. Immunotherapy of Malignant disease with tumor antigen-specific monoclonal antibodies. Clin Cancer Res (2010) 16:11–20. doi: 10.1158/1078-0432.CCR-09-2345
112. Robert C, Ghiringhelli F. What is the role of cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma? Oncologist (2009) 14:848–61. doi: 10.1634/theoncologist.2009-0028
113. Kline J, Gajewski TF. Clinical development of mAbs to block the PD1 pathway as an immunotherapy for cancer. Curr Opin Investig Drugs (2010) 11:1354–9.
114. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol (2014) 32:1020–30. doi: 10.1200/JCO.2013.53.0105
115. McDermott DF, Drake CG, Sznol M, Choueiri TK, Powderly JD, Smith DC, et al. Survival, durable response, and long-term safety in patients with previously treated advanced renal cell carcinoma receiving nivolumab. J Clin Oncol (2015) 33:2013–20. doi: 10.1200/JCO.2014.58.1041
116. Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, et al. Overall survival and long-term safety of nivolumab (Anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol (2015) 33:2004–12. doi: 10.1200/JCO.2014.58.3708
117. André T, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Nivolumab plus low-dose ipilimumab in previously treated patients with microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: 4-year follow-up from CheckMate 142. Ann Oncol (2022) 33:1052–60. doi: 10.1016/j.annonc.2022.06.008
118. Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med (2016) 375:1856–67. doi: 10.1056/NEJMoa1602252
119. Galsky MD, Saci A, Szabo PM, Han GC, Grossfeld G, Collette S, et al. Nivolumab in patients with advanced platinum-resistant urothelial carcinoma: efficacy, safety, and biomarker analyses with extended follow-up from checkMate 275. Clin Cancer Res (2020) 26:5120–28. doi: 10.1158/1078-0432.CCR-19-4162
120. Barlesi F, Garon EB, Kim DW, Felip E, Han JY, Kim JH, et al. Health-related quality of life in KEYNOTE-010: a phase II/III study of pembrolizumab versus docetaxel in patients with previously treated advanced, programmed death ligand 1-expressing NSCLC. J Thorac Oncol (2019) 14:793–801. doi: 10.1016/j.jtho.2019.01.016
121. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater. J Clin Oncol (2019) 37:537–46. doi: 10.1200/JCO.18.00149
122. Harrington KJ, Burtness B, Greil R, Soulières D, Tahara M, de Castro G Jr, et al. Pembrolizumab with or without chemotherapy in recurrent or metastatic head and neck squamous cell carcinoma: updated results of the phase III KEYNOTE-048 study. J Clin Oncol (2023) 41:790–802. doi: 10.1200/JCO.21.02508
123. Makker V, Colombo N, Herráez AC, Monk BJ, Mackay H, Santin AD, et al. Lenvatinib plus pembrolizumab in previously treated advanced endometrial cancer: updated efficacy and safety from the randomized phase III study 309/KEYNOTE-775. J Clin Oncol (2023) 41:2904–10. doi: 10.1200/JCO.22.02152
124. Colombo N, Dubot C, Lorusso D, Caceres MV, Hasegawa K, Shapira-Frommer R, et al. Pembrolizumab for persistent, recurrent, or metastatic cervical cancer. N Engl J Med (2021) 385:1856–67. doi: 10.1056/NEJMoa2112435
125. Balar AV, Castellano DE, Grivas P, Vaughn DJ, Powles T, Vuky J, et al. Efficacy and safety of pembrolizumab in metastatic urothelial carcinoma: results from KEYNOTE-045 and KEYNOTE-052 after up to 5 years of follow-up. Ann Oncol (2023) 34:289–99. doi: 10.1016/j.annonc.2022.11.012
126. Nghiem P, Bhatia S, Lipson EJ, Sharfman WH, KudChadkar RR, Brohl AS, et al. Durable tumor regression and overall survival in patients with advanced merkel cell carcinoma receiving pembrolizumab as first-line therapy. J Clin Oncol (2019) 37:693–702. doi: 10.1200/JCO.18.01896
127. Patel SP, Othus M, Chen Y, Wright GP Jr, Yost KJ, Hyngstrom JR, et al. Neoadjuvant-adjuvant or adjuvant-only pembrolizumab in advanced melanoma. N Engl J Med (2023) 388:813–23. doi: 10.1056/NEJMoa2211437
128. McDermott DF, Sosman JA, Sznol M, Massard C, Gordon MS, Hamid O, et al. Atezolizumab, an anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: long-term safety, clinical activity, and immune correlates from a phase ia study. J Clin Oncol (2016) 34:833–42. doi: 10.1200/JCO.2015.63.7421
129. Pal SK, McDermott DF, Atkins MB, Escudier B, Rini BI, Motzer RJ, et al. Patient-reported outcomes in a phase 2 study comparing atezolizumab alone or with bevacizumab vs sunitinib in previously untreated metastatic renal cell carcinoma. BJU Int (2020) 126:73–82. doi: 10.1111/bju.15058
130. van der Heijden MS, Loriot Y, Durán I, Ravaud A, Retz M, Vogelzang NJ, et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma: A long-term overall survival and safety update from the phase 3 IMvigor211 clinical trial. Eur Urol (2021) 80:7–11. doi: 10.1016/j.eururo.2021.03.024
131. Chaft JE, Oezkan F, Kris MG, Bunn PA, Wistuba II, Kwiatkowski DJ, et al. Neoadjuvant atezolizumab for resectable non-small cell lung cancer: an open-label, single-arm phase II trial. Nat Med (2022) 28:2155–61. doi: 10.1038/s41591-022-01962-5
132. Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicenter, phase 2 trial. Lancet (2016) 387:1909–20. doi: 10.1016/S0140-6736(16)00561-4
133. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet (2016) 387:1837–46. doi: 10.1016/S0140-6736(16)00587-0
134. Emens LA, Adams S, Barrios CH, Diéras V, Iwata H, Loi S, et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann Oncol (2021) 32:983–93. doi: 10.1016/j.annonc.2021.05.355
135. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med (2018) 379:2108–21. doi: 10.1056/NEJMoa1809615
136. Chen M, Jiang J, Chen J, Wang M, Lu Y, Liu L, et al. The clinical safety and efficacy of targeted PD-L1 therapy with durvalumab in solid tumors. Curr Drug Targets (2023) 24:584–98. doi: 10.2174/1389450124666230330101651
137. Teige I, Mårtensson L, Frendéus BL. Targeting the antibody checkpoints to enhance cancer immunotherapy-focus on fcγRIIB. Front Immunol (2019) 10:481. doi: 10.3389/fimmu.2019.00481
138. Bai J, Gao Z, Li X, Dong L, Han W, Nie J. Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade. Oncotarget (2017) 8:110693–707. doi: 10.18632/oncotarget.22690
139. Böttcher S, Ritgen M, Brüggemann M, Raff T, Lüschen S, Humpe A, et al. Flow cytometric assay for determination of FcgammaRIIIA-158 V/F polymorphism. J Immunol Methods (2005) 306:128–36. doi: 10.1016/j.jim.2005.08.004
140. Scholl S, Beuzeboc P, Pouillart P. Targeting HER2 in other tumor types. Ann Oncol (2001) 12 Suppl 1:S81–7. doi: 10.1093/annonc/12.suppl_1.s81
141. Brenner TL, Adams VR. First MAb approved for treatment of metastatic breast cancer. J Am Pharm Assoc (Wash) (1999) 39:236–8. doi: 10.1016/S1086-5802(16)30498-3
142. Hicks DG, Whitney-Miller C. HER2 testing in gastric and gastroesophageal junction cancers: a new therapeutic target and diagnostic challenge. Appl Immunohistochem Mol Morphol (2011) 19:506–8. doi: 10.1097/PAI.0b013e31822c3a0f
143. Varchetta S, Gibelli N, Oliviero B, Nardini E, Gennari R, Gatti G, et al. Elements related to heterogeneity of antibody-dependent cell cytotoxicity in patients under trastuzumab therapy for primary operable breast cancer overexpressing Her2. Cancer Res (2007) 67:11991–9. doi: 10.1158/0008-5472.CAN-07-2068
144. Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol (2008) 26:1789–96. doi: 10.1200/JCO.2007.14.8957
145. Tamura K, Shimizu C, Hojo T, Akashi-Tanaka S, Kinoshita T, Yonemori K, et al. FcγR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann Oncol (2011) 22:1302–7. doi: 10.1093/annonc/mdq585
146. Leget GA, Czuczman MS. Use of rituximab, the new FDA-approved antibody. Curr Opin Oncol (1998) 10:548–51. doi: 10.1097/00001622-199811000-00012
147. Plosker GL, Figgitt DP. Rituximab: a review of its use in non-Hodgkin's lymphoma and chronic lymphocytic leukemia. Drugs (2003) 63:803–43. doi: 10.2165/00003495-200363080-00005
148. Casak SJ, Lemery SJ, Shen YL, Rothmann MD, Khandelwal A, Zhao H, et al. U.S. Food and drug administration approval: rituximab in combination with fludarabine and cyclophosphamide for the treatment of patients with chronic lymphocytic leukemia. Oncologist (2011) 16:97–104. doi: 10.1634/theoncologist.2010-0306
149. Migkou M, Fotiou D, Gavriatopoulou M, Dimopoulos MA. Ibrutinib plus rituximab for the treatment of adult patients with Waldenström's macroglobulinemia: a safety evaluation. Expert Opin Drug Saf (2021) 20:987–95. doi: 10.1080/14740338.2021.1945031
150. Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol (2003) 21:3940–7. doi: 10.1200/JCO.2003.05.013
151. Veeramani S, Wang SY, Dahle C, Blackwell S, Jacobus L, Knutson T, et al. Rituximab infusion induces NK activation in lymphoma patients with the high-affinity CD16 polymorphism. Blood (2011) 118:3347–9. doi: 10.1182/blood-2011-05-351411
152. Kim DH, Jung HD, Kim JG, Lee JJ, Yang DH, Park YH, et al. FCGR3A gene polymorphisms may correlate with response to frontline R-CHOP therapy for diffuse large B-cell lymphoma. Blood (2006) 108:2720–5. doi: 10.1182/blood-2006-01-009480
153. Treon SP, Hansen M, Branagan AR, Verselis S, Emmanouilides C, Kimby E, et al. Polymorphisms in FcgammaRIIIA (CD16) receptor expression are associated with clinical response to rituximab in Waldenström's macroglobulinemia. J Clin Oncol (2005) 23:474–81. doi: 10.1200/JCO.2005.06.059
154. Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets (2012) 16:15–31. doi: 10.1517/14728222.2011.648617
156. Cohen MH, Chen H, Shord S, Fuchs C, He K, Zhao H, et al. Approval summary: Cetuximab in combination with cisplatin or carboplatin and 5-fluorouracil for the first-line treatment of patients with recurrent locoregional or metastatic squamous cell head and neck cancer. Oncologist (2013) 18:460–6. doi: 10.1634/theoncologist.2012-0458
157. Lattanzio L, Denaro N, Vivenza D, Varamo C, Strola G, Fortunato M, et al. Elevated basal antibody-dependent cell-mediated cytotoxicity (ADCC) and high epidermal growth factor receptor (EGFR) expression predict favorable outcome in patients with locally advanced head and neck cancer treated with cetuximab and radiotherapy. Cancer Immunol Immunother (2017) 66:573–79. doi: 10.1007/s00262-017-1960-8
158. Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol (2009) 27:1122–9. doi: 10.1200/JCO.2008.18.0463
159. Trotta AM, Ottaiano A, Romano C, Nasti G, Nappi A, De Divitiis C, et al. Prospective evaluation of cetuximab-mediated antibody-dependent cell cytotoxicity in metastatic colorectal cancer patients predicts treatment efficacy. Cancer Immunol Res (2016) 4:366–74. doi: 10.1158/2326-6066.CIR-15-0184
160. Taylor RJ, Chan SL, Wood A, Voskens CJ, Wolf JS, Lin W, et al. FcgammaRIIIa polymorphisms and cetuximab induced cytotoxicity in squamous cell carcinoma of the head and neck. Cancer Immunol Immunother (2009) 58:997–1006. doi: 10.1007/s00262-008-0613-3
161. López-Albaitero A, Lee SC, Morgan S, Grandis JR, Gooding WE, Ferrone S, et al. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother (2009) 58:1853–64. doi: 10.1007/s00262-009-0697-4
162. Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, et al. Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol Res (2015) 3:1148–57. doi: 10.1158/2326-6066.CIR-15-0059
163. Hicks KC, Fantini M, Donahue RN, Schwab A, Knudson KM, Tritsch SR, et al. Epigenetic priming of both tumor and NK cells augments antibody-dependent cellular cytotoxicity elicited by the anti-PD-L1 antibody avelumab against multiple carcinoma cell types. Oncoimmunology (2018) 7:e1466018. doi: 10.1080/2162402X.2018.1466018
164. Juliá EP, Amante A, Pampena MB, Mordoh J, Levy EM. Avelumab, an igG1 anti-PD-L1 immune checkpoint inhibitor, triggers NK cell-mediated cytotoxicity and cytokine production against triple negative breast cancer cells. Front Immunol (2018) 9:2140. doi: 10.3389/fimmu.2018.02140
165. Kim ES. Avelumab: first global approval. Drugs (2017) 77:929–37. doi: 10.1007/s40265-017-0749-6
166. Rodriguez-Vida A, Bellmunt J. Avelumab for the treatment of urothelial cancer. Expert Rev Anticancer Ther (2018) 18:421–29. doi: 10.1080/14737140.2018.1448271
167. Gurram S, Al Harthy M, Ball MW. The changing landscape of systemic therapy in metastatic renal cell carcinoma: an update. Discovery Med (2020) 29:191–9.
168. Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol (2016) 17:1374–85. doi: 10.1016/S1470-2045(16)30364-3
169. Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med (2019) 380:1103–115. doi: 10.1056/NEJMoa1816047
170. Della Corte CM, Fasano M, Ciaramella V, Cimmino F, Cardnell R, Gay CM, et al. Anti-tumor activity of cetuximab plus avelumab in non-small cell lung cancer patients involves innate immunity activation: findings from the CAVE-Lung trial. J Exp Clin Cancer Res (2022) 41:109. doi: 10.1186/s13046-022-02332-2
171. Peipp M, Derer S, Lohse S, Staudinger M, Klausz K, Valerius T, et al. HER2-specific immunoligands engaging NKp30 or NKp80 trigger NK-cell-mediated lysis of tumor cells and enhance antibody-dependent cell-mediated cytotoxicity. Oncotarget (2015) 6:32075–88. doi: 10.18632/oncotarget.5135
172. Kellner C, Lutz S, Oberg HH, Wesch D, Otte A, Diemer KJ, et al. Tumor cell lysis and synergistically enhanced antibody-dependent cell-mediated cytotoxicity by NKG2D engagement with a bispecific immunoligand targeting the HER2 antigen. Biol Chem (2021) 403:545–56. doi: 10.1515/hsz-2021-0229
173. Kellner C, Günther A, Humpe A, Repp R, Klausz K, Derer S, et al. Enhancing natural killer cell-mediated lysis of lymphoma cells by combining therapeutic antibodies with CD20-specific immunoligands engaging NKG2D or NKp30. Oncoimmunology (2015) 5:e1058459. doi: 10.1080/2162402X.2015.1058459
174. Ishikawa T, Okayama T, Sakamoto N, Ideno M, Oka K, Enoki T, et al. Phase I clinical trial of adoptive transfer of expanded natural killer cells in combination with IgG1 antibody in patients with gastric or colorectal cancer. Int J Cancer (2018) 142:2599–609. doi: 10.1002/ijc.31285
175. Lee SC, Shimasaki N, Lim JSJ, Wong A, Yadav K, Yong WP, et al. Phase I trial of expanded, activated autologous NK-cell infusions with trastuzumab in patients with HER2-positive cancers. Clin Cancer Res (2020) 26:4494–502. doi: 10.1158/1078-0432.CCR-20-0768
176. Tanaka J, Tanaka N, Wang YH, Mitsuhashi K, Ryuzaki M, Iizuka Y, et al. Phase I study of cellular therapy using ex vivo expanded natural killer cells from autologous peripheral blood mononuclear cells combined with rituximab-containing chemotherapy for relapsed CD20-positive Malignant lymphoma patients. Haematologica (2020) 105:e190–3. doi: 10.3324/haematol.2019.226696
177. Yoon DH, Koh Y, Jung M, Kwak JE, Shin EC, Hwang YK, et al. Phase I study: safety and efficacy of an ex vivo-expanded allogeneic natural killer cell (MG4101) with rituximab for relapsed/refractory B cell non-hodgkin lymphoma. Transplant Cell Ther (2023) 29:253.e1–9. doi: 10.1016/j.jtct.2022.12.025
178. Cichocki F, Zhang B, Wu CY, Chiu E, Day A, O'Connor RS, et al. Nicotinamide enhances natural killer cell function and yields remissions in patients with non-Hodgkin lymphoma. Sci Transl Med (2023) 15:eade3341. doi: 10.1126/scitranslmed.ade3341
179. Liang S, Lin M, Niu L, Xu K, Wang X, Liang Y, et al. Cetuximab combined with natural killer cells therapy: an alternative to chemoradiotherapy for patients with advanced non-small cell lung cancer (NSCLC). Am J Cancer Res (2018) 8:879–91.
180. Lim CM, Liou A, Poon M, Koh LP, Tan LK, Loh KS, et al. Phase I study of expanded natural killer cells in combination with cetuximab for recurrent/metastatic nasopharyngeal carcinoma. Cancer Immunol Immunother (2022) 71:2277–86. doi: 10.1007/s00262-022-03158-9
181. Jochems C, Hodge JW, Fantini M, Fujii R, Morillon YM 2nd, Greiner JW, et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget (2016) 7:86359–73. doi: 10.18632/oncotarget.13411
182. Friedman J, Padget M, Lee J, Schlom J, Hodge J, Allen C. Direct and antibody-dependent cell-mediated cytotoxicity of head and neck squamous cell carcinoma cells by high-affinity natural killer cells. Oral Oncol (2019) 90:38–44. doi: 10.1016/j.oraloncology.2019.01.017
183. Jochems C, Hodge JW, Fantini M, Tsang KY, Vandeveer AJ, Gulley JL, et al. ADCC employing an NK cell line (haNK) expressing the high affinity CD16 allele with avelumab, an anti-PD-L1 antibody. Int J Cancer (2017) 141:583–93. doi: 10.1002/ijc.30767
184. Zhao H, Zhou Z, Li G, Liu G, Lin S, Chen W, et al. An NK cell line (NK92-41BB) expressing high levels of granzyme is engineered to express the high affinity chimeric genes CD16/CAR. Cytotechnology (2021) 73:539–53. doi: 10.1007/s10616-021-00476-1
185. Li HK, Hsiao CW, Yang SH, Yang HP, Wu TS, Lee CY, et al. A novel off-the-shelf trastuzumab-armed NK cell therapy (ACE1702) using antibody-cell-conjugation technology. Cancers (Basel) (2021) 13:2724. doi: 10.3390/cancers13112724
186. Adotevi O, Godet Y, Galaine J, Lakkis Z, Idirene I, Certoux JM, et al. In situ delivery of allogeneic natural killer cell (NK) combined with Cetuximab in liver metastases of gastrointestinal carcinoma: A phase I clinical trial. Oncoimmunology (2018) 7:e1424673. doi: 10.1080/2162402X.2018.1424673
187. Carlsten M, Levy E, Karambelkar A, Li L, Reger R, Berg M, et al. Efficient mRNA-Based Genetic Engineering of Human NK Cells with High-Affinity CD16 and CCR7 Augments Rituximab-Induced ADCC against Lymphoma and Targets NK Cell Migration toward the Lymph Node-Associated Chemokine CCL19. Front Immunol (2016) 7:105. doi: 10.3389/fimmu.2016.00105
188. Baysal H, De Pauw I, Zaryouh H, Peeters M, Vermorken JB, Lardon F, et al. The right partner in crime: unlocking the potential of the anti-EGFR antibody cetuximab via combination with natural killer cell chartering immunotherapeutic strategies. Front Immunol (2021) 12:737311. doi: 10.3389/fimmu.2021.737311
189. Markham A. Margetuximab: first approval. Drugs (2021) 81:599–604. doi: 10.1007/s40265-021-01485-2
190. Ollila TA, Sahin I, Olszewski AJ. Mogamulizumab: a new tool for management of cutaneous T-cell lymphoma. Onco Targets Ther (2019) 12:1085–94. doi: 10.2147/OTT.S165615
191. Edelmann J, Gribben JG. Obinutuzumab for the treatment of indolent lymphoma. Future Oncol (2016) 12:1769–81. doi: 10.2217/fon-2016-0084
192. Davis JA, Shockley A, Glode AE. Newly approved anti-CD19 monoclonal antibodies for the treatment of relapsed or refractory diffuse large B-cell lymphoma. J Oncol Pharm Pract (2022) 28:686–90. doi: 10.1177/10781552211073575
193. Pan M, Wang F, Nan L, Yang S, Qi J, Xie J, et al. αVEGFR2-MICA fusion antibodies enhance immunotherapy effect and synergize with PD-1 blockade. Cancer Immunol Immunother (2023) 72:969–84. doi: 10.1007/s00262-022-03306-1
194. Huan T, Guan B, Li H, Tu X, Zhang C, Tang B. Principles and current clinical landscape of NK cell engaging bispecific antibody against cancer. Hum Vaccin Immunother (2023) 19:2256904. doi: 10.1080/21645515.2023.2256904
Keywords: NK cells, ADCC, modulatory cytokines, adoptive NK cell therapy, monoclonal antibody, CAR-NK
Citation: Fantini M, Arlen PM and Tsang KY (2023) Potentiation of natural killer cells to overcome cancer resistance to NK cell-based therapy and to enhance antibody-based immunotherapy. Front. Immunol. 14:1275904. doi: 10.3389/fimmu.2023.1275904
Received: 15 August 2023; Accepted: 23 October 2023;
Published: 24 November 2023.
Edited by:
Hind Rafei, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2023 Fantini, Arlen and Tsang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Massimo Fantini, bWFzc2ltby5mYW50aW5pQHByZWNpc2lvbi1iaW9sb2dpY3MuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.