 Xingyu Wang
Xingyu Wang Lan Zhou
Lan Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 25 October 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1274816
This article is part of the Research Topic Macrophages: Allies or merciless enemies in musculoskeletal systems View all articles
Skeletal muscle is essential for body physical activity, energy metabolism, and temperature maintenance. It has excellent capabilities to maintain homeostasis and to regenerate after injury, which indispensably relies on muscle stem cells, satellite cells (MuSCs). The quiescence, activation, and differentiation of MuSCs are tightly regulated in homeostatic and regenerating muscles. Among the important regulators are intramuscular macrophages, which are functionally heterogeneous with different subtypes present in a spatiotemporal manner to regulate the balance of different MuSC statuses. During chronic injury and aging, intramuscular macrophages often undergo aberrant activation, which in turn disrupts muscle homeostasis and regenerative repair. Growing evidence suggests that the aberrant activation is mainly triggered by altered muscle microenvironment. The trained immunity that affects myeloid progenitors during hematopoiesis may also contribute. Aged immune system may contribute, in part, to the aging-related sarcopenia and compromised skeletal muscle injury repair. As macrophages are actively involved in the progression of many muscle diseases, manipulating their functional activation has become a promising therapeutic approach, which requires comprehensive knowledge of the cellular and molecular mechanisms underlying the diverse activation. To this end, we discuss here the current knowledge of multifaceted role of macrophages in skeletal muscle homeostasis, injury, and repair.
Skeletal muscle accounts for about 40% of human body mass. Besides being a crucial component of locomotor system, skeletal muscle contributes to basal energy metabolism and temperature maintenance. It also serves as an important secretory tissue, producing cytokines and other peptides (1, 2). Maintaining skeletal muscle homeostasis is critical to human health and wellbeing. Skeletal muscle homeostasis can be disrupted by many factors, such as acute injury, infection, genetic defect, and aging. Depending on the nature of the inciting factor, the outcome varies. Acute skeletal muscle injury, such as those caused by trauma or myotoxin exposure, can be repaired well unless the injury is repeated or too large. On the other hand, repeated acute injuries, large volumetric muscle loss, and chronic muscle injury caused by genetic defects cannot be completely repaired, which often leads to fibro-fatty tissue replacement in injured muscle. Muscle injury repair is a highly coordinated process, involving inflammation, myogenesis, revascularization, and remodeling of extracellular matrix (ECM) (3–5).
The capability of skeletal muscle to regenerate following injury relies on myogenic stem cells, muscle satellite cells (MuSCs), which largely stay quiescent in the homeostatic skeletal muscle (6). Depletion of MuSCs completely abolishes muscle regeneration (7). Injury-induced changes in tissue microenvironment activate MuSCs to regenerate muscle fibers. This process consists of a series of tightly-regulated proliferation and differentiation events, which have been reviewed in details by others (8–10). Briefly, quiescent MuSCs respond to extracellular signals released by injured muscle and become activated. Activated MuSCs proliferate and differentiate into myoblasts, which then fuse into multinucleated myotubes that further differentiate into muscle fibers (8–10). The quiescence, activation, and differentiation of MuSCs must be balanced to maintain stem cell pool in healthy muscle and to successfully regenerate in injured muscle. This balance is tightly regulated in a spatiotemporal manner by signals from the surrounding muscle microenvironment, which consist of extracellular matrix (ECM) and many different cell types including macrophages (11).
Macrophages predominate the inflammatory response to sterile muscle injury and contribute to the outcome of injury repair. They are functionally heterogeneous, arise from multiple origins, and actively participate in many biological processes including tissue homeostasis and injury repair (12–16). Resident macrophages maintain homeostasis of steady-state tissue via surveillance of local tissue microenvironment and response to changes. In a disease state, the functions of intramuscular macrophages can vary greatly depending on their origins and tissue microenvironment. They appear to be a double-edged sword. On the one hand, they could be essential to tissue regeneration, as seen during acute injury repair. On the other hand, they could contribute to tissue pathology, as seen during chronic inflammation (15–19). Macrophage has become a central focus of research in skeletal muscle injury and repair. In this review, we will discuss the multifaceted role of macrophages in skeletal muscle homeostasis, injury, and repair. The discussion will focus firstly on the essential roles of macrophages in maintaining homeostasis of steady state muscle and in regenerative repair of injured muscle. The pathological roles of macrophages during degenerative muscle injury repair will then be discussed. Lastly, we will discuss the roles of macrophages in aged muscle in the steady state and injury state (Figure 1).
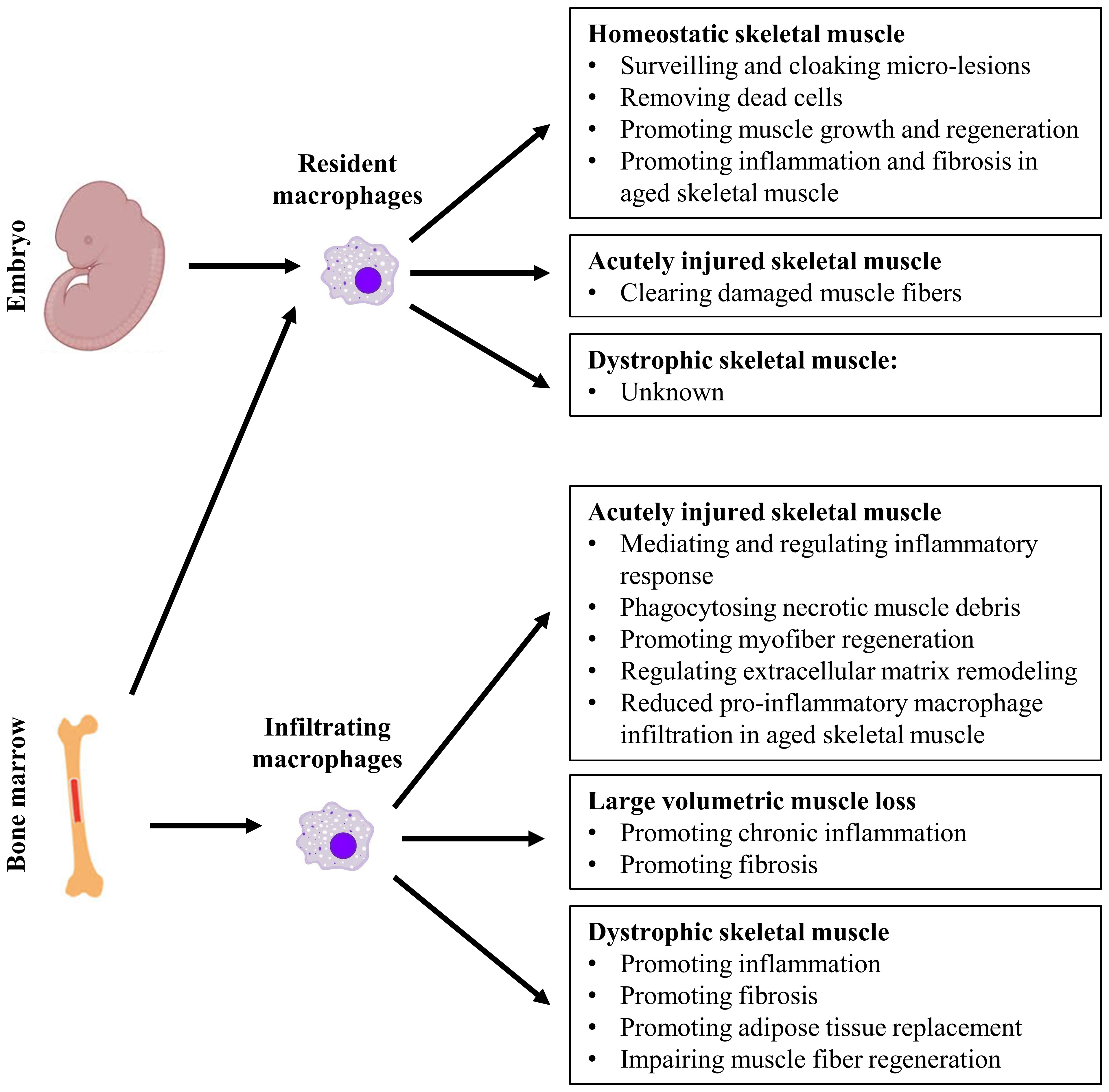
Figure 1 The multifaceted role of resident and infiltrating macrophages in homeostatic, acutely injured, and dystrophic skeletal muscles.
Tissue macrophages are classified into resident macrophages and infiltrating inflammatory macrophages. Resident macrophages are usually established during embryogenesis and are present in all adult tissues including skeletal muscle (20). Adult tissue resident macrophages originate from both embryonic and adult hematopoiesis. During embryonic hematopoiesis, tissue macrophages can be derived firstly from primitive yolk sac macrophages and secondly from fetal liver monocytes (fetal monocytes) (14, 21–28). Fetal monocytes gradually outcompete yolk sac macrophages to seed all embryonic tissues except for brain (27). Embryonic tissue resident macrophages can persist into adulthood through self-renewal. Adult hematopoiesis is originated from bone marrow hematopoietic stem cells (HSCs), which give rise to blood monocytes (adult monocytes) after birth. Adult monocytes contribute to the replenishment of resident macrophages in many tissues (29–33), including skeletal muscle (20). Different from tissue resident macrophages, infiltrating macrophages are exclusively derived from blood monocytes originated from bone marrow HSCs.
Combining single cell-based RNA sequencing (scRNAseq) and lineage-tracing technique, a recent study identified three common resident macrophage subtypes across different murine tissues, including skeletal muscle, and these functional subtypes correlate with the macrophage origins (34). The identification was based on the expression of four genes: Timd4, Lyve1, Folr2, and Ccr2. The first subtype expresses Timd4 and/or Lyve1 and/or Folr2 (TLF+), and they are self-renewal macrophages originated from yolk sac and fetal monocyte precursors. The second subtype is TLF-Ccr2+ which can be entirely replaced by blood monocytes. The third subtype is TLF-Ccr2- which expresses a high level of MHCII molecules and receives a moderate contribution from adult monocytes (34). Likewise, another study using scRNAseq analysis of human macrophages also showed that the Lyve1+ macrophages overlap strongly with fetal liver macrophages, suggesting an embryonic origin of this subtype, while the Lyve1- macrophages expressed monocytic signature genes, suggesting their monocytic origin (35). Similar resident macrophage subtypes have also been identified in skeletal muscle by other studies (20, 36). One study, combining scRNAseq and lineage-tracing, showed that adult skeletal muscle resident macrophages mostly arise from fetal monocytes and adult bone marrow HSCs, with a small percentage from primitive macrophages (20). In this study, the authors identified two macrophage subtypes: the CCR2+MHCIIhiLyve1low macrophages which are completely derived from blood monocytes, and the CCR2-MHCIIlowLyve1hi macrophages which mainly arise from embryonic origins (20). In addition, Folr2 expression is higher in CCR2- macrophages than in CCR2+ macrophages (20). Another study, using parabiosis and single-cell transcriptome analysis, identified similar muscle resident macrophage subtypes: self-renewal TIM4+Lyve1+CCR2- cells and blood replenished TIM4-Lyve1-CCR2+ cells (36). Together, these findings indicate that while the TLF expression primarily marks the embryo-derived resident macrophages, the CCR2 and MHCII expression preferentially marks the adult monocytes-derived resident macrophages in skeletal muscle. These differentially expressed markers provide useful tools for studying muscle resident macrophage subtypes of different origins. We summarize the origins, life cycles, and markers of different subpopulations of resident macrophages as well as infiltrating macrophages in Table 1.
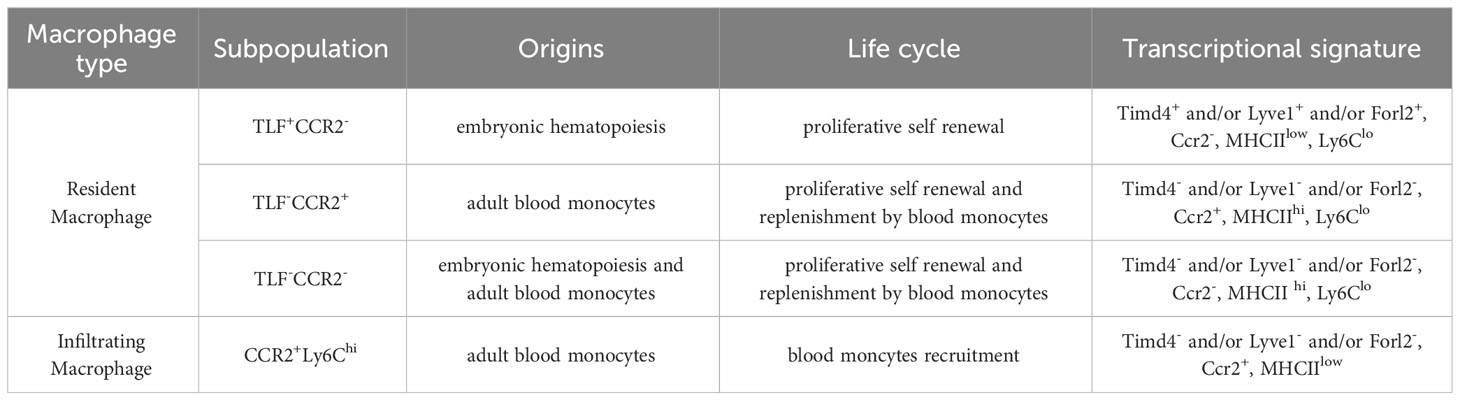
Table 1 Origins and markers of intramuscular macrophages.
Skeletal muscle resident macrophages reside in interstitial tissue as CD45+F4/80+CD64+ Ly6Clo cells, and they express muscle-specific transcription factors and display muscle-specific functions (20). Compared to the resident macrophages in other tissues, resident macrophages in skeletal muscle display a distinct transcriptome profile, which is characterized by enriched expression of genes involved in maintenance of muscle homeostasis and promotion of muscle growth and regeneration (20). Skeletal muscle resident macrophages also exhibit muscle-type specificity, as demonstrated by a much higher expression level of stress response genes by respiratory muscle resident macrophages than by limb muscle resident macrophages (20). An independent study provided additional evidence to support the homeostatic function of skeletal muscle resident macrophages (37). In multiple tissues including skeletal muscle, resident macrophages were shown to rapidly cloak tissue microlesions to prevent neutrophil chemotaxis and subsequent inflammatory tissue damage (37). This functional property of resident macrophages prevented complete death of myofibers from microlesions and maintained structural integrity of muscle in the steady state (37). Maintaining tissue homeostasis appears a ubiquitous function of resident macrophages across different tissues thanks to their ability to phagocytose dead cells and debris and to suppress immune activation (38). In addition, the transcriptome study suggests that muscle resident macrophages of different origins are specialized to exert different functions. While the CCR2+MHCIIhiLyve1low macrophages, which mainly arise from HSCs, are more antigen presenting cells, the CCR2-MHCIIlowLyve1hi macrophages, which arise from both HSCs and non-HSCs embryonic progenitors, are more active phagocytes (20). Notably, the CCR2-MHCIIlowLyve1hi subtype accounts for almost 70% of the total muscle resident macrophages (20), and the percentage is similarly to that of the TLF+ resident macrophages in heart but much higher than that of the TLF+ macrophages in other tissues (34). Cardiac tissue shares similarity to skeletal muscle. Cardiac resident macrophages are more studied and better understood than skeletal muscle resident macrophages. It was reported that LYVE1+ (TLF+) cardiac resident macrophages are localized to the areas near blood vessels (39), which might be due to the ability of LYVE1 to bind hyaluronic acid on smooth muscle cells in blood vessel walls (40). Considering both heart and skeletal muscles are highly vascularized organs, proximity to vasculature may account for a high percentage of TLF+ resident macrophages in these two tissues. Cardiac TLF+ resident macrophages are also reported to be more active in phagocytosing dead cells and tissue debris (30, 41), like the counterpart in skeletal muscle. They are also required for appropriate vascular patterning and lymphatic development (42, 43). It would interesting to explore whether muscle TLF+ resident macrophages have similar functions. On the other hand, LVVE1loMHCIIhi cardiac resident macrophages reside near nerves (39), with their functional significance not yet known in homeostatic heart. The localization and functions of skeletal muscle resident macrophage subtypes need to be further studied in the future.
A number of animal models have been used to study the roles of macrophages in acute skeletal muscle injury repair. In these models, the injury is induced by mechanical damage, contusion, freeze, myotoxin or heavy metal salt injection, or ischemia. The technical merits of different acute injury models are reviewed by Baghdadi et al. (44). Studies based on different models have revealed a similar repair process with small differences. Except for repeated injuries (45) and a large volume of muscle loss (46, 47), acute skeletal muscle injury usually repairs within 2 to 3 weeks without significant residual defects. Although the regeneration of damaged myofibers relies on the activation and differentiation of MuSCs (8–10), an adequate inflammatory response, triggered by injury and predominated by neutrophil and macrophage infiltration, is required for the complete muscle regeneration (3, 48–50). Both muscle resident macrophages and infiltrating macrophages play essential roles in acute skeletal muscle injury repair.
Studying the role of resident macrophages in muscle injury and repair has been long hampered due to the lack of specific markers to separate resident macrophages from infiltrating macrophages and the lack of effective approach to selectively target resident macrophages. The recent identification of TIM4 and Lyve1 as markers for self-renewal muscle resident macrophages helps facilitate such studies (36). By using a CSF1R inhibition and withdrawal strategy, TIM4+Lyve1+ resident macrophages can be selectively and temporally depleted from steady-state skeletal muscle (36). Using this model, one study showed that depletion of TIM4+Lyve1+ resident macrophages at the early stage of acute muscle injury repair resulted in prolonged accumulation of necrotic fibers and impaired muscle regeneration. Therefore, this subtype of self-renewal resident macrophages is required for efficient clearance of damaged fibers to promote muscle regeneration (36). Single-cell transcriptome analysis showed enriched expression of genes involved in phagocytosis and inflammatory cell chemotaxis in the TIM4+Lyve1+ resident macrophages after muscle injury, suggesting that this subtype may also play a role in initiating inflammation (36). However, the recruitment of neutrophils and inflammatory monocytes did not change after the depletion of TIM4+Lyve1+ resident macrophages, suggesting that other resident cells, including TIM4-Lyve1- resident macrophages, may compensate for this role (36). The contribution of resident macrophages to acute skeletal muscle injury repair remains largely unknown and needs to be further explored, particularly with advanced techniques such as lineage tracing and single-cell based analysis. Findings from research in cardiac resident macrophages may provide useful hints, given the similarity between these two types of tissues. Following myocardial injury, transcriptional programs of cardiac resident macrophages are induced to primarily promote phagocytosis and efferocytosis (51, 52), suggesting that cardiac resident macrophages are also required for efficient clearance of damaged tissue debris, similar to their counterpart in skeletal muscle. Self-renewing CCR2- resident cardiac macrophages resident cardiac macrophages also limit adverse remodeling (51, 53). While the CCR2+ subset promotes recruitment of neutrophils (54) and monocytes (53), the CCR2- subset inhibits monocytes recruitment (53). Whether the different subtypes of skeletal muscle resident macrophages perform similar roles following acute injury is yet to be determined.
In acutely injured skeletal muscle, infiltrating macrophages are differentiated from HSC-derived blood monocytes (55, 56), which consist of two different subsets in mice: Ly6ChiCCR2+CX3CR1lo and Ly6CloCCR2-CX3CR1hi monocytes (57). Injured skeletal muscle recruits Ly6ChiCCR2+CX3CR1lo (57) but not Ly6CloCCR2-CX3CR1hi (58) monocytes through CC chemokine receptor 2 (CCR2) chemotaxis signaling (56, 59–62). Ly6ChiCCR2+CX3CR1lo monocytes then differentiate into Ly6Chi inflammatory macrophages. In addition to blood circulation, Ly6Chi monocytes can also be deployed from spleen reservoir into inflamed tissues, including injured skeletal muscle (63, 64). After phagocytosing necrotic muscle debris, intramuscular Ly6Chi macrophages can switch into Ly6Clo macrophages (56, 65–69). This Ly6Chi-to-Ly6Clo switch, rather than the pre-existing resident macrophages, makes the major contribution to the Ly6Clo macrophage accumulation in injured muscle (56, 69, 70). The CD14hiCD16low and CD14lowCD16hi monocytes in humans are the counterparts of Ly6Chi and Ly6Clo monocytes in mice, respectively (71).
The Ly6Chi-to-Ly6Clo switch during skeletal muscle injury repair suggests major functional changes of macrophages accompanying the injury repair process. Ly6Chi and Ly6Clo macrophages have once been considered to correlate with the bipolar classification of M1 (pro-inflammatory) and M2 (anti-inflammatory, pro-regenerative, and/or pro-fibrotic) macrophages, respectively (72, 73). Accompanying the Ly6Chi-to-Ly6Clo switch, infiltrating macrophages indeed undergo pro- to anti-inflammatory functional phenotype switch, as reflected by the switches of pro- to anti-inflammatory gene expression (15, 19, 56, 65, 70, 74–77) and pro- to anti-inflammatory lipid mediator production (78, 79). However, in vivo macrophage studies indicate that this simplified bipolar paradigm of macrophage activation does not fit complex in vivo scenario (72, 80, 81). Macrophages are highly plastic cells and can rapidly switch their functional status in response to the changes of tissue microenvironment, leading to diverse macrophage subtypes. In a study using single-cell transcriptome analysis of acutely injured muscle, several macrophage subtypes were identified sequentially post injury: 1) a more pro-inflammatory subtype at day 2; 2) an Il7r+ subtype from day 3.5 to day 5; 3) a complement genes-enriched subtype from day 3.5 to day 10; 4) a subtype with enriched expression of genes of antigen presentation from day 5 to 10. Different macrophage subtypes coexist at multiple time points, with the fraction of each cluster changing over time (82). Another study found that the expression pattern of M1 and M2 genes is similar in Ly6Chi and Ly6Clo macrophages at day 3 post injury, but different between Ly6Chi macrophages at day 1 and Ly6Chi macrophages at day 3 post injury (70). Therefore, the changes of infiltrating macrophage phenotype appears to be driven by the changes in the tissue microenvironment rather than the Ly6C status (70, 82, 83).
Infiltrating macrophages are required for the complete repair of acutely injured muscle, during which macrophages of different activation status actively regulate inflammation, myogenesis, and ECM remodeling in a spatiotemporal manner. Disruption of macrophage infiltration or their spatiotemporal phenotype switch results in impaired muscle regeneration which is often accompanied by muscle fibrosis (56, 59–62, 76, 84, 85). These findings also suggest that the pre-existing resident macrophages cannot compensate the roles of infiltrating macrophages to adequately support acute muscle injury repair.
Infiltrating macrophages mediate and regulate the inflammatory response to acute muscle injury. They produce a high level of pro-inflammatory mediators at the early stage for inflammation propagation, while produce a high level of anti-inflammatory mediators at the later stage for inflammation resolution. In addition, the early infiltrated Ly6Chi macrophages are also required to clear damaged muscle debris, as blocking macrophage infiltration via CCR2 deficiency delays the clearance of necrotic muscle fibers (61). Phagocytosis of damaged tissue debris appears to drive the pro- to anti-inflammatory phenotype switch of macrophages (56, 65–69).
Infiltrating macrophages promote muscle regeneration by regulating the core process of myogenic cell activation, proliferation, differentiation, and fusion after injury. Different subtypes of macrophages appear to exert different functions. Pro-inflammatory macrophages promote myogenic cell activation and proliferation, while anti-inflammatory macrophages promote myogenic cell differentiation and fusion (56, 86–88). This is further supported by histopathological analysis of injured skeletal muscle, which shows co-localization of pro-inflammatory macrophages with proliferating satellite cells and co-localization of anti-inflammatory macrophages with differentiated myoblasts (77). Pro-inflammatory macrophages produce a high level of soluble factors known to stimulate the activation and proliferation of MuSCs, including fibronectin (89), IL-6 (90), TNF-α (91), PGE2 (92), and A Disintegrin-Like and Metalloproteinase with Thrombospondin Type 1 Motif (ADAMTS1) (93). On the other hand, anti-inflammatory macrophages produce factors that can stimulate myoblast differentiation and myofiber growth, including IL-4 (94), IGF-1 (61, 95, 96), and GDF-3 (97). The increased glutamine synthesis that accompanies the macrophage phenotype switch during acute injury repair has also been shown to boost satellite cell activation and muscle regeneration (98). Therefore, the spatiotemporal presence of macrophages of different phenotypes appears important to the proper muscle regeneration after injury. This hypothesis is further supported by the findings that targeting signaling molecules that critically involved in macrophage phenotype switch, including IGF-1 (96), Meteorin-like (Metrnl) (99), AMP-activated protein kinase-1 (AMPKα1) (65, 100), Nuclear Factor IX (Nfix) (68), CCAAT/enhancer binding protein-β (C/EBPβ) (77), and peroxisome proliferator-activated receptor-γ (PPARγ) (97), impairs muscle regeneration.
Infiltrating macrophages regulate ECM remodeling, a temporary ECM deposition and degradation process that provides an essential structural support for myogenic cell activation and differentiation during acute muscle injury repair. Besides structural support, some ECM components, such as collagen 6a (Col6a) (101) and fibronectin (89), can activate MuSCs. The primary cells that produce ECM are fibro/adipogenic progenitors (FAPs), which also support myogenic cell activation and differentiation to facilitate muscle regeneration (7, 102–104). However, impaired regenerative process can lead to aberrant FAP activation, which results in fibro-fatty tissue replacement and failure to support MuSC activation (104). Macrophages have been shown to regulate the accumulation and activation of FAPs in injured skeletal muscle. The pro-inflammatory macrophages can induce FAPs apoptosis by secreting TNF-α (105) to limit excessive FAP accumulation. On the other hand, the anti-inflammatory macrophages can enhance proliferation of fibrogenic cells through increased production pro-fibrotic factors such as TGF-β1 (106). Depleting macrophages or blocking macrophage recruitment results in not only poor muscle regeneration but also muscle fibrosis (60, 105), further supporting the importance of infiltrating macrophages in regulating FAP activity and ECM remodeling.
In summary, both resident macrophages and infiltrating macrophages are required for the complete repair of acutely injured skeletal muscle. While both resident and infiltrating macrophages clear dead cells and tissue debris, infiltrating macrophages of different activation status also actively regulate the activation, proliferation, differentiation, and growth of myogenic cells in a spatiotemporal for proper muscle regeneration (Figure 1).
Acute skeletal muscle injury usually regenerates well. However, when a critical volume of muscle is lost due to surgery or trauma, a condition known as degenerative volumetric muscle loss (VML), muscle regeneration is compromised, along with prolonged inflammatory cell infiltration and muscle fibrosis (46, 47, 107–112). VML results in pronounced disabilities ranging from atrophy and dysfunction of muscle to aggressive development of osteoarthritis (112). Comparing to the regenerative acute skeletal muscle injury, degenerative VML causes prolonged macrophage infiltration that can persist for months (108). More importantly, the macrophage phenotypes in degenerative VML appear different from those in regenerative muscle injury (110, 113).
In a well-designed study, Anderson et al. determined the threshold of critical size of muscle loss that leads to degenerative injury repair following VML (114). In mouse quadriceps, full-thickness defect of 3 mm, corresponding to 15% of total mass, is the critical threshold of un-recoverable muscle loss. Twenty-eight days post injury, quadriceps with 3-mm but not 2-mm injury showed incomplete bridging of myofibers through the defect site, persistent inflammation, fibrosis, and increased central nucleated myofibers (114). This study established an excellent mouse model for exploring cellular and molecular mechanisms underlying the pathological outcomes following VML. Single cell-based analysis exploiting this model revealed significant differences in inflammatory cell infiltration and function in the degenerative VML injury comparing to the regenerative VML injury (109, 113). While the abundance of macrophages was significantly lower in the 3-mm than in the 2-mm injured muscles, the abundance of all the other inflammatory cell types identified, including monocytes and neutrophils, was persistently increased (109). Another study of VML injury in tibias anterior muscle analyzed the spatiotemporal function of macrophages (113). By combining spatial transcriptomics and scRNAseq techniques, the authors identified an aberrantly activated scar-associated macrophage (SAM) subtype, which has been reported in other diseased tissues, including cardiac muscle (115, 116), liver (117), and lung (118). SAMs feature the expression of both pro-inflammatory and anti-inflammatory genes, as well as pro-fibrotic genes including Trem2 and Spp1. In the defect site of day 7-injured muscle, SAMs co-localize with mesenchymal-derived cells (MDCs), creating an inflammatory and pro-fibrotic milieu (113). The pro-fibrotic milieu persists 14 days post injury and leads to excessive collagen deposition (113). The study established a role for the aberrantly activated, pro-fibrotic biased macrophages in driving the pathological outcome of VML. By generating 2-mm and 3-mm VML injuries in the left and right quadriceps muscles of the same mice, respectively, leaving the size of the defect the only difference between the two injuries, Larouche et al. demonstrates that the aberrant activation of macrophages following critical VML is induced purely by the altered tissue microenvironment, which disrupts the spatiotemporal macrophage phenotype switch that normally observed during regenerative muscle injury repair (109). Therefore, improving tissue microenvironment by analyzing and targeting its molecular and cellular components may help promote injury repair.
Muscular dystrophies consist of a heterogeneous group of genetic diseases characterized by progressive muscle atrophy and weakness (119). In severe cases, such as Duchene Muscular Dystrophy (DMD), patients die prematurely from respiratory and cardiac muscle weakness (119, 120). Many muscular dystrophies are caused by mutations in the genes encoding proteins of dystrophin-glycoprotein complex (DGC) or proteins required for the proper assembly of DGC, which in turn causes fragile sarcolemma and leads to myofiber necrosis (121). These genetic defects lead to a continuous cycle of muscle degeneration and regeneration, resulting in chronic inflammation and fibro-fatty tissue replacement (122). The most studied muscular dystrophy is DMD, which is caused by the defective dystrophin gene on the X chromosome (120).
Most of the DMD studies are conducted in animal models, among which the most commonly used is mdx mice. Mdx mice display a milder phenotype compared to DMD patients (123–126). Muscle inflammation starts about 3 weeks of age, persists into 2-3 months of age, and then gradually subsides in limb muscles but not diaphragm (123–127). Correspondingly, progressive fibrosis takes place in diaphragm but not limb muscles. Along with the fibrosis development in diaphragm, respiratory function of mdx mice is impaired, which resembles human DMD patients (125, 127–129). Similar to acute injury, inflammation in mdx muscles is also predominated by macrophage infiltration (127). Recruitment of Ly6Chi monocytes by mdx muscles also depends on CCR2 (130, 131), and intramuscular Ly6Chi-to-Ly6Clo macrophage switch also occurs (130, 131). As comprehensive human study of DMD is lacking, we focus our discussion on the mouse model studies.
Macrophages in mdx leg muscles display diverse phenotypes depending on age: they appear more pro-inflammatory at 4 weeks while more pro-regenerative at 12 weeks (132). Depletion of macrophages at 4 weeks of age reduces leg muscle necrosis (133), which suggests reduced inflammation-induced muscle damage. A recent study exploiting scRNAseq analysis revealed that macrophages are highly heterogeneous in the leg muscle of 8-week-old mdx mice (134). The authors identified multiple macrophage subtypes with distinct transcriptomes, including two resident macrophage-like subtypes resembling resident macrophages in WT muscle, a pro-inflammatory subtype, an Spp1-expressing subtype reportedly to be pro-fibrotic in other tissues (135, 136), and several low-fraction subtypes (134). Importantly, the abundance of the pro-inflammatory subtype and the Spp1-expressing subtype correlated with the disease severity (134), indicating the pathogenicity of these macrophage subtypes. An independent study also indicates that Ly6Chi macrophages contribute to the fibrosis of mdx leg muscles at 8 to 10 weeks of age by producing latent TGF-β1 (137). Consistently, multiple other studies also suggest that macrophages are generally pathogenic in mdx muscles, as reducing macrophage infiltration decreases muscle damage and fibrosis while improves muscle function before 3 months of age (64, 130, 131, 138). Contrarily, there is evidence suggesting that anti-inflammatory macrophages may be beneficial to mdx (126, 127). Depleting macrophages from mdx limb muscles at 10 to 12 weeks of age compromised myogenic cell proliferation and differentiation, decreased myofiber formation, and increased fibro-fatty tissue deposition (139). Studies concerning macrophage-derived factors, including TNF-α, IL-1β, IL-6, iNOS, IL-10, IGF-1, TGF-β, and osteopontin (encoded by Spp1), provide additional evidence of the pleiotropic roles of macrophages in mdx muscles, as these factors are critically involved in inflammation, regeneration, and fibrosis of the dystrophic muscles (48, 105, 127, 132, 140–142).
Despite the fact that diaphragm but not limb muscle displays persistent inflammation and progressive fibrosis in mdx mice, the function of macrophages in the mdx diaphragm is poorly understood compared to that in the mdx limb muscles. One study showed that in 14-week-old mdx mice, Ly6Clo macrophages from diaphragm are more pro-inflammatory and pro-fibrotic than those from quadriceps (106). However, the knowledge is far from comprehensive. Future macrophage studies, particularly at a single-cell level, are required for this human DMD-resembling muscle type to provide further insight into the pathogenic roles of macrophages in muscular dystrophy.
Macrophages are highly plastic, responding and adapting to tissue microenvironment changes. During chronic inflammation, Ly6Chi monocytes/macrophages continuously infiltrate and switch to Ly6Clo macrophages in response to the constant damage of skeletal muscle. This creates a continuously altered microenvironment with mixed stimuli of both pro- and anti-inflammatory natures, disrupting the spatiotemporal presence of pro- and anti-inflammatory macrophages. Consequently, muscle regeneration is compromised. A simplified demonstration of this scenario is from a study of repeated acute skeletal muscle injury, in which muscle injury was induced twice at an interval of 4 or 10 days (45). The repeated injuries cause simultaneous presence of both pro- and anti-inflammatory macrophages in the injured muscle, as well as persistent inflammation, fibrosis, and impaired muscle regeneration (45). The altered microenvironment caused by continuous injuries in dystrophic muscle may account for the highly heterogeneous and pathogenic macrophage activation in mdx muscle (134).
Besides the asynchronous regenerative microenvironment in dystrophic muscle, a recent study revealed “trained immunity” as another important mechanism that drives the pathogenic monocyte/macrophage activation during bone marrow hematopoiesis in mdx mice (143). Trained immunity originally refers to a status of innate immune hyper-responsiveness induced by the exposure to infectious agents (144, 145). Macrophages of this status persist long after the clearance of the initial inducers, representing a type of innate immune memory. Importantly, this innate immune memory is not antigen specific and thus can promote exaggerated cytokine response to unrelated pathological challenges (144). Mechanistic studies have linked trained immunity to histone modifications (146–148) and changes in cellular metabolism (149). In addition to infections, damage-associated molecular patterns (DAMPs), released by necrotic cells and ECM breakdown during tissue injury, can also induce trained immunity (150, 151). DAMPs are recognized by pattern recognition receptors (PRRs) to trigger activation of intracellular signaling pathways that mediate the expression of inflammatory genes (152–156). PRRs, such as toll-like receptors (TLRs), have been shown to regulate trained immunity in monocytes/macrophages (143, 157). Trained immunity can be induced at the level of myeloid progenitors within bone marrow (143, 158–160), allowing its potential role in the progression of a chronic disease. Compared to wild-type (WT) mice, bone marrow-derived macrophages (BMDMs) from mdx mice showed a significantly increased basal expression of both pro- and anti-inflammatory genes. Mdx BMDMs also responded in an exaggerated fashion to heterologous inflammatory stimuli (143). This phenotypic reprogramming of BMDMs can be induced by DAMPs prepared from crushed skeletal muscle extract, and can be sustained after adoptive mdx-to-WT bone marrow transfer. TLR4 was shown to regulate the altered phenotype of mdx BMDMs through epigenetic reprogramming (143). These findings suggest that the aberrant and pathogenic macrophage activation in chronic dystrophic muscle may be contributed by the trained immunity in addition to the altered local muscle environment.
With aging, skeletal muscle suffers loss of muscle mass and concurrent accretion of fat and connective tissues, a clinical syndrome called “sarcopenia” (161, 162). Aging also impairs the capability of skeletal muscle to regenerate after injury, leading to, similar to sacopenia, the decrease in the number and size of myofibers and the deposition of adipose and fibrous tissues (163–166). It is not clear whether these two age-related defects of skeletal muscle are interconnected. They both lead to declined muscle function and compromised health condition in the elderly. Understanding the mechanisms underlying these defects is therefore critical to the development of clinical strategies to improve the function of aged skeletal muscle.
Aging impacts all tissues including immune system. Aged immune system loses its ability to mount an efficient immune response to infection or injury (167, 168). Immune cells, primarily the innate immune cells like macrophages, persistently infiltrate homeostatic tissues at a low level, and the level of several pro-inflammatory cytokines and chemokines, such as TNF-α and CCL2, increases in both tissues and circulation with aging (168, 169). These alterations lead to a hypothesis that aged immune system plays an important role in driving the aging of other tissues. This hypothesis gained a strong support from the work by Yousefzadeh et al. (170). The authors created a mouse model in which the increased burden of endogenous DNA damage was specifically induced in hematopoietic cells to cause pre-mature senescence in the immune system only. Interestingly, pre-mature senescence was also observed in non-lymphoid organs in this model, suggesting that aged immune cells can promote systemic aging. Transplantation of splenocytes from the mutant mice or aged wild-type mice into young mice induced senescence in the recipients, whereas transplantation of splenocytes from young mice into old mice suppressed senescence and tissue damage in aged recipients. In particular, the authors also showed impaired skeletal muscle regeneration following injury in the mutant mice as compared to the age-matched wild-type mice. Therefore, aged immune system plays a causal role in the aging of other tissues including skeletal muscle (170). This concept is further supported by another study showing that transplantation of young bone marrow cells into old recipients prevented sarcopenia, whereas transplantation of old bone marrow cells into young recipients reduced satellite cell number and promoted satellite cells to switch towards a fibrogenic phenotype (171).
As macrophage is the predominant immune cell type in both homeostatic and regenerating skeletal muscle, they could potentially contribute to the defects of aged skeletal muscle. Aging results in a plethora of phenotypic and functional changes in macrophages, including but not limited to the increased production of pro-inflammatory mediators such as TNF-α and IL-1β and decreased capacity of phagocytosis (170, 172). In aged mouse skeletal muscle, a recent study combining scRNAseq and flow cytometry analysis revealed significant changes in macrophage functional phenotype (173). The authors showed increased fraction of Lyve1- macrophages whereas decreased fraction of Lyve1+ macrophages in old muscle compared to young muscle, despite that the total macrophage number did not change (173). This is likely due to the continuous replenishment of embryo-derived muscle resident macrophages by blood monocytes (20). Accordingly, the expression levels of pro-inflammatory and senescence-related markers is increased in old skeletal muscle macrophages (173), suggesting an overall phenotypic shift towards a pro-inflammatory state with aging. Concurrently, the expression of Spp1 gene, which was associated with pro-fibrotic function (135, 136), was also increased in the old skeletal muscle macrophages (173). Although the authors identified multiple macrophage sub-clusters, the relative change in the fraction of these clusters between old and young muscles was not shown (173). It is thus unclear whether the phenotypic shift is due to a selective expansion of specific macrophage subtypes or a general increase in gene expression. Nevertheless, the pro-inflammatory phenotype of aged skeletal muscle macrophages may contribute to the aging of skeletal muscle, as one study showed that myeloid cell-derived TNFα promoted sarcopenia (174). In addition, CD68+CD206+ macrophages have been shown to co-localize with intramuscular adipose tissue in old human and mouse skeletal muscle (175), suggesting a potential contribution of macrophages to the aging-related adipogenic replacement of myofibers.
Growing evidence supports the notion that aging compromises the ability of skeletal muscle to regenerate following injury (163–166). As macrophages are essential for supporting muscle regeneration, the altered macrophage function may account, in part, for the aging-related regeneration defect. Indeed, decreased infiltration of pro-inflammatory macrophages in old muscles compared to young ones was reported in mouse disuse atrophy (176) and human eccentric contraction injury (177). The delivery of pro-inflammatory macrophages to the muscle that underwent disuse atrophy promoted the recovery of muscle strength in aged mice (178). Thus, the aging-related decrease in pro-inflammatory macrophage infiltration likely contributes to the compromised injury repair of aged muscle. The functional activation of infiltrating macrophages after injury also appears altered in aged muscle. One study showed that the expression of IFN‐γ responsive genes by macrophages was down‐regulated in the regenerating muscle of aged mice after injury, and IFN‐γ deficiency impaired muscle regeneration (179). By scRNAseq analysis, the authors identified an interferon‐responsive macrophage (IFNRM) subset at day 3 post injury, which was reduced in aged muscle after injury. IFNRMs specifically expressed CXCL10 which promoted MuSC proliferation. Importantly, CXCL10 treatment restored muscle regeneration in aged mice (179). Another study showed that the expression of osteopontin was significantly increased in the MuSC niche in aged skeletal muscle, which suppressed the myogenic capacity of MuSCs (180). Neutralization of osteopontin improved the regeneration of aged injured muscle (180). Notably, the Spp1 gene, which encodes osteopontin, is highly expressed by skeletal muscle macrophages in old mice (173, 180), indicating an increased pro-fibrotic activation of macrophages in aged muscle. Taken together, these findings strongly suggest that aging-related changes in infiltrating macrophages may account, in part, for the compromised regeneration of aged muscle following acute injury. Targeting these changes and rejuvenating macrophages may represent useful approaches to promote injury repair in aged muscle.
It has been increasingly recognized that macrophages can acquire highly diverse functional phenotypes and play multifaceted role in skeletal muscle homeostasis, injury, and repair. They display a high level of plasticity in response to muscle microenvironment changes. In young adult skeletal muscle, macrophages contribute to the maintenance of homeostasis in the steady state. Following acute injury, infiltrating macrophages respond to the changes in injured muscle, undergoing phenotypic switch in a spatiotemporal manner to support MuSC-mediated muscle regeneration. Resident macrophages are also required for efficient clearance of necrotic fibers to promote injury repair. In chronic conditions, such as critical VML, muscular dystrophy, and aging, macrophages undergo aberrant activation to contribute to or drive muscle pathology. The aberrant activation appears mainly triggered by altered muscle microenvironment. The trained immunity that affects myeloid progenitors during hematopoiesis may also contribute. The aged immune system may contribute, in part, to the functional changes of resident macrophages, sarcopenia, and compromised acute injury repair in aged muscle. To date, our knowledge of the origins, functions, and activation mechanisms of skeletal muscle macrophages is still limited. Future studies with state-of-the-art technologies, such as lineage tracing, single cell-based high-throughput analysis, and spatial transcriptomics, will generate new valuable insights which may lead to new therapy development.
LZ: Writing – review & editing. XW: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work was supported by the National Institute of Arthritis And Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number 1R01AR074428 (LZ). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Frontera WR, Ochala J. Skeletal muscle: a brief review of structure and function. Calcif Tissue Int (2015) 96(3):183–95. doi: 10.1007/s00223-014-9915-y
2. Iizuka K, Machida T, Hirafuji M. Skeletal muscle is an endocrine organ. J Pharmacol Sci (2014) 125(2):125–31. doi: 10.1254/jphs.14R02CP
3. Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev (2013) 93(1):23–67. doi: 10.1152/physrev.00043.2011
4. Dumont NA, Bentzinger CF, Sincennes MC, Rudnicki MA. Satellite cells and skeletal muscle regeneration. Compr Physiol (2015) 5(3):1027–59. doi: 10.1002/cphy.c140068
5. Bentzinger CF, Wang YX, Dumont NA, Rudnicki MA. Cellular dynamics in the muscle satellite cell niche. EMBO Rep (2013) 14(12):1062–72. doi: 10.1038/embor.2013.182
6. Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol (1961) 9:493–5. doi: 10.1083/jcb.9.2.493
7. Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development (2011) 138(17):3625–37. doi: 10.1242/dev.064162
8. Wang YX, Rudnicki MA. Satellite cells, the engines of muscle repair. Nat Rev Mol Cell Biol (2011) 13(2):127–33. doi: 10.1038/nrm3265
9. Dumont NA, Wang YX, Rudnicki MA. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development (2015) 142(9):1572–81. doi: 10.1242/dev.114223
10. Sousa-Victor P, Garcia-Prat L, Munoz-Canoves P. Control of satellite cell function in muscle regeneration and its disruption in ageing. Nat Rev Mol Cell Biol (2022) 23(3):204–26. doi: 10.1038/s41580-021-00421-2
11. Mashinchian O, Pisconti A, Le Moal E, Bentzinger CF. The muscle stem cell niche in health and disease. Curr Top Dev Biol (2018) 126:23–65. doi: 10.1016/bs.ctdb.2017.08.003
12. Thomas A, Wynn AC, Jeffrey W. Pollard. Macrophages biology in development, homeostasis and disease. Nature (2013) 496:445–55. doi: 10.1038/nature12034
13. Kierdorf K, Prinz M, Geissmann F, Gomez Perdiguero E. Development and function of tissue resident macrophages in mice. Semin Immunol (2015) 27(6):369–78. doi: 10.1016/j.smim.2016.03.017
14. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity (2013) 38(4):792–804. doi: 10.1016/j.immuni.2013.04.004
15. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity (2016) 44(3):450–62. doi: 10.1016/j.immuni.2016.02.015
16. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol (2016) 17(1):34–40. doi: 10.1038/ni.3324
17. McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity (2014) 41(1):36–48. doi: 10.1016/j.immuni.2014.05.010
18. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41(1):49–61. doi: 10.1016/j.immuni.2014.06.010
19. Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol (2017) 79:593–617. doi: 10.1146/annurev-physiol-022516-034356
20. Wang X, Sathe AA, Smith GR, Ruf-Zamojski F, Nair V, Lavine KJ, et al. Heterogeneous origins and functions of mouse skeletal muscle-resident macrophages. Proc Natl Acad Sci U S A (2020) 117(34):20729–40. doi: 10.1073/pnas.1915950117
21. Hoeffel G, Ginhoux F. Fetal monocytes and the origins of tissue-resident macrophages. Cell Immunol (2018) 330:5–15. doi: 10.1016/j.cellimm.2018.01.001
22. Hoeffel G, Ginhoux F. Ontogeny of tissue-resident macrophages. Front Immunol (2015) 6:486. doi: 10.3389/fimmu.2015.00486
23. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol (2014) 14(6):392–404. doi: 10.1038/nri3671
24. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature (2015) 518(7540):547–51. doi: 10.1038/nature13989
25. Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Science (2016) 353(6304):aaf4238. doi: 10.1126/science.aaf4238
26. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science (2012) 336(6077):86–90. doi: 10.1126/science.1219179
27. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity (2015) 42(4):665–78. doi: 10.1016/j.immuni.2015.03.011
28. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330(6005):841–5. doi: 10.1126/science.1194637
29. Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol (2014) 15(10):929–37. doi: 10.1038/ni.2967
30. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity (2014) 40(1):91–104. doi: 10.1016/j.immuni.2013.11.019
31. Bain CC, Hawley CA, Garner H, Scott CL, Schridde A, Steers NJ, et al. Long-lived self-renewing bone marrow-derived macrophages displace embryo-derived cells to inhabit adult serous cavities. Nat Commun (2016) 7:ncomms11852. doi: 10.1038/ncomms11852
32. Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity (2013) 39(5):925–38. doi: 10.1016/j.immuni.2013.10.004
33. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun (2016) 7:10321. doi: 10.1038/ncomms10321
34. Dick SA, Wong A, Hamidzada H, Nejat S, Nechanitzky R, Vohra S, et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci Immunol (2022) 7(67):eabf7777. doi: 10.1126/sciimmunol.abf7777
35. Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, Dunsmore G, et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity (2021) 54(8):1883–900.e5. doi: 10.1016/j.immuni.2021.07.007
36. Babaeijandaghi F, Cheng R, Kajabadi N, Soliman H, Chang CK, Smandych J, et al. Metabolic reprogramming of skeletal muscle by resident macrophages points to CSF1R inhibitors as muscular dystrophy therapeutics. Sci Transl Med (2022) 14(651):eabg7504. doi: 10.1126/scitranslmed.abg7504
37. Uderhardt S, Martins AJ, Tsang JS, Lammermann T, Germain RN. Resident macrophages cloak tissue microlesions to prevent neutrophil-driven inflammatory damage. Cell (2019) 177(3):541–55.e17. doi: 10.1016/j.cell.2019.02.028
38. Nobs SP, Kopf M. Tissue-resident macrophages: guardians of organ homeostasis. Trends Immunol (2021) 42(6):495–507. doi: 10.1016/j.it.2021.04.007
39. Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science (2019) 363(6432):aau0964. doi: 10.1126/science.aau0964
40. Lim HY, Lim SY, Tan CK, Thiam CH, Goh CC, Carbajo D, et al. Hyaluronan receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity (2018) 49(2):326–41.e7. doi: 10.1016/j.immuni.2018.06.008
41. Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, Sanchez-Diaz M, Diaz-Garcia E, Santiago DJ, et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell (2020) 183(1):94–109.e23. doi: 10.1016/j.cell.2020.08.031
42. Cahill TJ, Sun X, Ravaud C, Villa Del Campo C, Klaourakis K, Lupu IE, et al. Tissue-resident macrophages regulate lymphatic vessel growth and patterning in the developing heart. Development (2021) 148(3):dev.194563. doi: 10.1242/dev.194563
43. Leid J, Carrelha J, Boukarabila H, Epelman S, Jacobsen SE, Lavine KJ. Primitive embryonic macrophages are required for coronary development and maturation. Circ Res (2016) 118(10):1498–511. doi: 10.1161/CIRCRESAHA.115.308270
44. Baghdadi MB, Tajbakhsh S. Regulation and phylogeny of skeletal muscle regeneration. Dev Biol (2018) 433(2):200–9. doi: 10.1016/j.ydbio.2017.07.026
45. Dadgar S, Wang ZY, Johnston H, Kesari A, Nagaraju K, Chen YW, et al. Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J Cell Biol (2014) 207(1):139–58. doi: 10.1083/jcb.201402079
46. Grogan BF, Hsu JR, Skeletal Trauma Research C. Volumetric muscle loss. J Am Acad Orthop Surg (2011) 19 Suppl 1:S35–7. doi: 10.5435/00124635-201102001-00007
47. Corona BT, Rivera JC, Owens JG, Wenke JC, Rathbone CR. Volumetric muscle loss leads to permanent disability following extremity trauma. J Rehabil Res Dev (2015) 52(7):785–92. doi: 10.1682/JRRD.2014.07.0165
48. Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol Regul Integr Comp Physiol (2010) 298(5):R1173–87. doi: 10.1152/ajpregu.00735.2009
49. Tidball JG. Mechanisms of muscle injury, repair, and regeneration. Compr Physiol (2011) 1(4):2029–62. doi: 10.1002/cphy.c100092
50. Wang X, Zhou L. The many roles of macrophages in skeletal muscle injury and repair. Front Cell Dev Biol (2022) 10:952249. doi: 10.3389/fcell.2022.952249
51. Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol (2019) 20(1):29–39. doi: 10.1038/s41590-018-0272-2
52. Zaman R, Hamidzada H, Kantores C, Wong A, Dick SA, Wang Y, et al. Selective loss of resident macrophage-derived insulin-like growth factor-1 abolishes adaptive cardiac growth to stress. Immunity (2021) 54(9):2057–71.e6. doi: 10.1016/j.immuni.2021.07.006
53. Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, et al. Tissue resident CCR2- and CCR2+ Cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res (2019) 124(2):263–78. doi: 10.1161/CIRCRESAHA.118.314028
54. Li W, Hsiao HM, Higashikubo R, Saunders BT, Bharat A, Goldstein DR, et al. Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight (2016) 1(12):e87315. doi: 10.1172/jci.insight.87315
55. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol (2011) 11(11):762–74. doi: 10.1038/nri3070
56. Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med (2007) 204(5):1057–69. doi: 10.1084/jem.20070075
57. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity (2003) 19(1):71–82. doi: 10.1016/S1074-7613(03)00174-2
58. Varga T, Mounier R, Gogolak P, Poliska S, Chazaud B, Nagy L. Tissue LyC6- macrophages are generated in the absence of circulating LyC6- monocytes and Nur77 in a model of muscle regeneration. J Immunol (2013) 191(11):5695–701. doi: 10.4049/jimmunol.1301445
59. Contreras-Shannon V, Ochoa O, Reyes-Reyna SM, Sun D, Michalek JE, Kuziel WA, et al. Fat accumulation with altered inflammation and regeneration in skeletal muscle of CCR2-/- mice following ischemic injury. Am J Physiol Cell Physiol (2007) 292(2):C953–67. doi: 10.1152/ajpcell.00154.2006
60. Lu H, Huang D, Ransohoff RM, Zhou L. Acute skeletal muscle injury: CCL2 expression by both monocytes and injured muscle is required for repair. FASEB J Off Publ Fed Am Societies Exp Biol (2011) 25(10):3344–55. doi: 10.1096/fj.10-178939
61. Lu H, Huang D, Saederup N, Charo IF, Ransohoff RM, Zhou L. Macrophages recruited via CCR2 produce insulin-like growth factor-1 to repair acute skeletal muscle injury. FASEB J (2011) 25(1):358–69. doi: 10.1096/fj.10-171579
62. Sun D, Martinez CO, Ochoa O, Ruiz-Willhite L, Bonilla JR, Centonze VE, et al. Bone marrow-derived cell regulation of skeletal muscle regeneration. FASEB J (2009) 23(2):382–95. doi: 10.1096/fj.07-095901
63. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science (2009) 325(5940):612–6. doi: 10.1126/science.1175202
64. Rizzo G, Di Maggio R, Benedetti A, Morroni J, Bouche M, Lozanoska-Ochser B. Splenic Ly6Chi monocytes are critical players in dystrophic muscle injury and repair. JCI Insight (2020) 5(2):e130807. doi: 10.1172/jci.insight.130807
65. Mounier R, Theret M, Arnold L, Cuvellier S, Bultot L, Goransson O, et al. AMPKalpha1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab (2013) 18(2):251–64. doi: 10.1016/j.cmet.2013.06.017
66. Xiao YQ, Freire-de-Lima CG, Schiemann WP, Bratton DL, Vandivier RW, Henson PM. Transcriptional and translational regulation of TGF-beta production in response to apoptotic cells. J Immunol (2008) 181(5):3575–85. doi: 10.4049/jimmunol.181.5.3575
67. Johann AM, Barra V, Kuhn AM, Weigert A, von Knethen A, Brune B. Apoptotic cells induce arginase II in macrophages, thereby attenuating NO production. FASEB J (2007) 21(11):2704–12. doi: 10.1096/fj.06-7815com
68. Saclier M, Lapi M, Bonfanti C, Rossi G, Antonini S, Messina G. The transcription factor nfix requires rhoA-ROCK1 dependent phagocytosis to mediate macrophage skewing during skeletal muscle regeneration. Cells (2020) 9(3):708. doi: 10.3390/cells9030708
69. Zhang J, Qu C, Li T, Cui W, Wang X, Du J. Phagocytosis mediated by scavenger receptor class BI promotes macrophage transition during skeletal muscle regeneration. J Biol Chem (2019) 294(43):15672–85. doi: 10.1074/jbc.RA119.008795
70. Wang X, Zhao W, Ransohoff RM, Zhou L. Infiltrating macrophages are broadly activated at the early stage to support acute skeletal muscle injury repair. J Neuroimmunol (2018) 317:55–66. doi: 10.1016/j.jneuroim.2018.01.004
71. Loems Ziegler-Heitbrock PA, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJM, et al. Nomenclature of monocytes and dendritic cells in blood. Blood (2010) 116(16):e74–80. doi: 10.1182/blood-2010-02-258558
72. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep (2014) 6:13. doi: 10.12703/P6-13
73. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol (2006) 177(10):7303–11. doi: 10.4049/jimmunol.177.10.7303
74. Perdiguero E, Sousa-Victor P, Ruiz-Bonilla V, Jardi M, Caelles C, Serrano AL, et al. p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J Cell Biol (2011) 195(2):307–22. doi: 10.1083/jcb.201104053
75. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol (2005) 6(12):1191–7. doi: 10.1038/ni1276
76. Wang H, Melton DW, Porter L, Sarwar ZU, McManus LM, Shireman PK. Altered macrophage phenotype transition impairs skeletal muscle regeneration. Am J pathology (2014) 184(4):1167–84. doi: 10.1016/j.ajpath.2013.12.020
77. Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, et al. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci U S A (2009) 106(41):17475–80. doi: 10.1073/pnas.0908641106
78. Scher JU, Pillinger MH. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin Immunol (2005) 114(2):100–9. doi: 10.1016/j.clim.2004.09.008
79. Giannakis N, Sansbury BE, Patsalos A, Hays TT, Riley CO, Han X, et al. Dynamic changes to lipid mediators support transitions among macrophage subtypes during muscle regeneration. Nat Immunol (2019) 20(5):626–36. doi: 10.1038/s41590-019-0356-7
80. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
81. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci (2016) 19(8):987–91. doi: 10.1038/nn.4338
82. Oprescu SN, Yue F, Qiu J, Brito LF, Kuang S. Temporal dynamics and heterogeneity of cell populations during skeletal muscle regeneration. iScience (2020) 23(4):100993. doi: 10.1016/j.isci.2020.100993
83. Tamas Varga RM, Horvath A, Cuvellier S, Dumont F, Poliska S, Ardjoune H, et al. Highly dynamic transcriptional signature of distinct macropohage subsets during sterile inflammation, resolution, and tissue repair. J Immunol (2016) 196(11):4771–82. doi: 10.4049/jimmunol.1502490
84. Junio Dort PF, Molina T, Dumont. NA. Macrophages are key regulators of stem cells during skeletal muscle regeneration and diseases. Stem Cells Int (2019) 2019:4761427. doi: 10.1155/2019/4761427
85. Liu X, Liu Y, Zhao L, Zeng Z, Xiao W, Chen P. Macrophage depletion impairs skeletal muscle regeneration: The roles of regulatory factors for muscle regeneration. Cell Biol Int (2017) 41(3):228–38. doi: 10.1002/cbin.10705
86. Saclier M, Yacoub-Youssef H, Mackey AL, Arnold L, Ardjoune H, Magnan M, et al. Differentially activated macrophages orchestrate myogenic precursor cell fate during human skeletal muscle regeneration. Stem Cells (2013) 31(2):384–96. doi: 10.1002/stem.1288
87. Hsieh PL, Rybalko V, Baker AB, Suggs LJ, Farrar RP. Recruitment and therapeutic application of macrophages in skeletal muscles after hind limb ischemia. J Vasc Surg (2018) 67(6):1908–20 e1. doi: 10.1016/j.jvs.2017.04.070
88. Bencze M, Negroni E, Vallese D, Yacoub-Youssef H, Chaouch S, Wolff A, et al. Proinflammatory macrophages enhance the regenerative capacity of human myoblasts by modifying their kinetics of proliferation and differentiation. Mol Ther (2012) 20(11):2168–79. doi: 10.1038/mt.2012.189
89. Bentzinger CF, Wang YX, von Maltzahn J, Soleimani VD, Yin H, Rudnicki MA. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell (2013) 12(1):75–87. doi: 10.1016/j.stem.2012.09.015
90. Zhang C, Li Y, Wu Y, Wang L, Wang X, Du J. Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J Biol Chem (2013) 288(3):1489–99. doi: 10.1074/jbc.M112.419788
91. Li Y-P. TNF-α is a mitogen in skeletal muscle. Am J Physiol Cell Physiol (2002) 285:C370–C6. doi: 10.1152/ajpcell.00453.2002
92. Ho ATV, Palla AR, Blake MR, Yucel ND, Wang YX, Magnusson KEG, et al. Prostaglandin E2 is essential for efficacious skeletal muscle stem-cell function, augmenting regeneration and strength. Proc Natl Acad Sci U S A (2017) 114(26):6675–84. doi: 10.1073/pnas.1705420114
93. Du H, Shih CH, Wosczyna MN, Mueller AA, Cho J, Aggarwal A, et al. Macrophage-released ADAMTS1 promotes muscle stem cell activation. Nat Commun (2017) 8(1):669. doi: 10.1038/s41467-017-00522-7
94. Horsley V, Jansen KM, Mills ST, Pavlath GK. IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell (2003) 113(4):483–94. doi: 10.1016/S0092-8674(03)00319-2
95. Dumont N, Frenette J. Macrophages protect against muscle atrophy and promote muscle recovery in vivo and in vitro: a mechanism partly dependent on the insulin-like growth factor-1 signaling molecule. Am J pathology (2010) 176(5):2228–35. doi: 10.2353/ajpath.2010.090884
96. Tonkin J, Temmerman L, Sampson RD, Gallego-Colon E, Barberi L, Bilbao D, et al. Monocyte/macrophage-derived IGF-1 orchestrates murine skeletal muscle regeneration and modulates autocrine polarization. Mol Ther (2015) 23(7):1189–200. doi: 10.1038/mt.2015.66
97. Varga T, Mounier R, Patsalos A, Gogolak P, Peloquin M, Horvath A, et al. Macrophage PPARgamma, a lipid activated transcription factor controls the growth factor GDF3 and skeletal muscle regeneration. Immunity (2016) 45(5):1038–51. doi: 10.1016/j.immuni.2016.10.016
98. Shang M, Cappellesso F, Amorim R, Serneels J, Virga F, Eelen G, et al. Macrophage-derived glutamine boosts satellite cells and muscle regeneration. Nature (2020) 587(7835):626–31. doi: 10.1038/s41586-020-2857-9
99. Baht GS, Bareja A, Lee DE, Rao RR, Huang R, Huebner JL, et al. Meteorin-like facilitates skeletal muscle repair through a Stat3/IGF-1 mechanism. Nat Metab (2020) 2(3):278–89. doi: 10.1038/s42255-020-0184-y
100. McArthur S, Juban G, Gobbetti T, Desgeorges T, Theret M, Gondin J, et al. Annexin A1 drives macrophage skewing to accelerate muscle regeneration through AMPK activation. J Clin Invest (2020) 130(3):1156–67. doi: 10.1172/JCI124635
101. Urciuolo A, Quarta M, Morbidoni V, Gattazzo F, Molon S, Grumati P, et al. Collagen VI regulates satellite cell self-renewal and muscle regeneration. Nat Commun (2013) 4:1964. doi: 10.1038/ncomms2964
102. Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol (2010) 12(2):153–63. doi: 10.1038/ncb2015
103. Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol (2010) 12(2):143–52. doi: 10.1038/ncb2014
104. Uezumi A, Ikemoto-Uezumi M, Tsuchida K. Roles of nonmyogenic mesenchymal progenitors in pathogenesis and regeneration of skeletal muscle. Front Physiol (2014) 5:68. doi: 10.3389/fphys.2014.00068
105. Lemos DR, Babaeijandaghi F, Low M, Chang CK, Lee ST, Fiore D, et al. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat Med (2015) 21(7):786–94. doi: 10.1038/nm.3869
106. Wang X, Zhao W, Ransohoff RM, Zhou L. Identification and function of fibrocytes in skeletal muscle injury repair and muscular dystrophy. J Immunol (2016) 197(12):4750–61. doi: 10.4049/jimmunol.1601308
107. Greising SM, Rivera JC, Goldman SM, Watts A, Aguilar CA, Corona BT. Unwavering pathobiology of volumetric muscle loss injury. Sci Rep (2017) 7(1):13179. doi: 10.1038/s41598-017-13306-2
108. Aguilar CA, Greising SM, Watts A, Goldman SM, Peragallo C, Zook C, et al. Multiscale analysis of a regenerative therapy for treatment of volumetric muscle loss injury. Cell Death Discovery (2018) 4:33. doi: 10.1038/s41420-018-0027-8
109. Larouche JA, Fraczek PM, Kurpiers SJ, Yang BA, Davis C, Castor-Macias JA, et al. Neutrophil and natural killer cell imbalances prevent muscle stem cell-mediated regeneration following murine volumetric muscle loss. Proc Natl Acad Sci U S A (2022) 119(15):e2111445119. doi: 10.1073/pnas.2111445119
110. Sommerfeld SD, Cherry C, Schwab RM, Chung L, Maestas DR Jr., Laffont P, et al. Interleukin-36gamma-producing macrophages drive IL-17-mediated fibrosis. Sci Immunol (2019) 4(40):aax4783. doi: 10.1126/sciimmunol.aax4783
111. Hurtgen BJ, Ward CL, Garg K, Pollot BE, Goldman SM, McKinley TO, et al. Severe muscle trauma triggers heightened and prolonged local musculoskeletal inflammation and impairs adjacent tibia fracture healing. J Musculoskelet Neuronal Interact (2016) 16(2):122–34.
112. Garg K, Ward CL, Hurtgen BJ, Wilken JM, Stinner DJ, Wenke JC, et al. Volumetric muscle loss: persistent functional deficits beyond frank loss of tissue. J Orthop Res (2015) 33(1):40–6. doi: 10.1002/jor.22730
113. Larouche JA, Wallace EC, Spence BD, Buras E, Aguilar CA. Spatiotemporal mapping of immune and stem cell dysregulation after volumetric muscle loss. JCI Insight (2023) 8(7):162835. doi: 10.1172/jci.insight.162835
114. Anderson SE, Han WM, Srinivasa V, Mohiuddin M, Ruehle MA, Moon JY, et al. Determination of a critical size threshold for volumetric muscle loss in the mouse quadriceps. Tissue Eng Part C Methods (2019) 25(2):59–70. doi: 10.1089/ten.tec.2018.0324
115. Simoes FC, Cahill TJ, Kenyon A, Gavriouchkina D, Vieira JM, Sun X, et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat Commun (2020) 11(1):600. doi: 10.1038/s41467-019-14263-2
116. Rizzo G, Gropper J, Piollet M, Vafadarnejad E, Rizakou A, Bandi SR, et al. Dynamics of monocyte-derived macrophage diversity in experimental myocardial infarction. Cardiovasc Res (2023) 119(3):772–85. doi: 10.1093/cvr/cvac113
117. Hendrikx T, Porsch F, Kiss MG, Rajcic D, Papac-Milicevic N, Hoebinger C, et al. Soluble TREM2 levels reflect the recruitment and expansion of TREM2(+) macrophages that localize to fibrotic areas and limit NASH. J Hepatol (2022) 77(5):1373–85. doi: 10.1016/j.jhep.2022.06.004
118. Fabre T, Barron AMS, Christensen SM, Asano S, Bound K, Lech MP, et al. Identification of a broadly fibrogenic macrophage subset induced by type 3 inflammation. Sci Immunol (2023) 8(82):eadd8945. doi: 10.1126/sciimmunol.add8945
121. Rando TA. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve (2001) 24(12):1575–94. doi: 10.1002/mus.1192
122. Theret M, Saclier M, Messina G, Rossi FMV. Macrophages in skeletal muscle dystrophies, an entangled partner. J Neuromuscul Dis (2022) 9(1):1–23. doi: 10.3233/JND-210737
123. Goldspink G, Fernandes K, Williams PE, Wells DJ. Age-related changes in collagen gene expression in the muscles of mdx dystrophic and normal mice. Neuromuscul Disord (1994) 4(3):183–91. doi: 10.1016/0960-8966(94)90019-1
124. Hartel JV, Granchelli JA, Hudecki MS, Pollina CM, Gosselin LE. Impact of prednisone on TGF-beta1 and collagen in diaphragm muscle from mdx mice. Muscle Nerve (2001) 24(3):428–32. doi: 10.1002/1097-4598(200103)24:3<428::AID-MUS1018>3.0.CO;2-E
125. Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, et al. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature (1991) 352(6335):536–9. doi: 10.1038/352536a0
126. Beastrom N, Lu H, Macke A, Canan BD, Johnson EK, Penton CM, et al. mdx((5)cv) mice manifest more severe muscle dysfunction and diaphragm force deficits than do mdx Mice. Am J pathology (2011) 179(5):2464–74. doi: 10.1016/j.ajpath.2011.07.009
127. Zhou L, Porter JD, Cheng G, Gong B, Hatala DA, Merriam AP, et al. Temporal and spatial mRNA expression patterns of TGF-beta1, 2, 3 and TbetaRI, II, III in skeletal muscles of mdx mice. Neuromuscul Disord (2006) 16(1):32–8. doi: 10.1016/j.nmd.2005.09.009
128. Dupont-Versteegden EE, McCarter RJ. Differential expression of muscular dystrophy in diaphragm versus hindlimb muscles of mdx mice. Muscle Nerve (1992) 15(10):1105–10. doi: 10.1002/mus.880151008
129. Huang P, Cheng G, Lu H, Aronica M, Ransohoff RM, Zhou L. Impaired respiratory function in mdx and mdx/utrn(+/-) mice. Muscle Nerve (2011) 43(2):263–7. doi: 10.1002/mus.21848
130. Mojumdar K, Liang F, Giordano C, Lemaire C, Danialou G, Okazaki T, et al. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol Med (2014) 6(11):1476–92. doi: 10.15252/emmm.201403967
131. Zhao W, Wang X, Ransohoff RM, Zhou L. CCR2 deficiency does not provide sustained improvement of muscular dystrophy in mdx5cv mice. FASEB J (2017) 31(1):35–46. doi: 10.1096/fj.201600619r
132. Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet (2009) 18(3):482–96. doi: 10.1093/hmg/ddn376
133. Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol (2001) 155(1):123–31. doi: 10.1083/jcb.200105110
134. Saleh KK, Xi H, Switzler C, Skuratovsky E, Romero MA, Chien P, et al. Single cell sequencing maps skeletal muscle cellular diversity as disease severity increases in dystrophic mouse models. iScience (2022) 25(11):105415. doi: 10.1016/j.isci.2022.105415
135. Hoeft K, Schaefer GJL, Kim H, Schumacher D, Bleckwehl T, Long Q, et al. Platelet-instructed SPP1(+) macrophages drive myofibroblast activation in fibrosis in a CXCL4-dependent manner. Cell Rep (2023) 42(2):112131. doi: 10.1016/j.celrep.2023.112131
136. Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J (2019) 54(2):1802441. doi: 10.1183/13993003.02441-2018
137. Juban G, Saclier M, Yacoub-Youssef H, Kernou A, Arnold L, Boisson C, et al. AMPK activation regulates LTBP4-dependent TGF-beta1 secretion by pro-inflammatory macrophages and controls fibrosis in duchenne muscular dystrophy. Cell Rep (2018) 25(8):2163–76.e6. doi: 10.1016/j.celrep.2018.10.077
138. Giordano C, Mojumdar K, Liang F, Lemaire C, Li T, Richardson J, et al. Toll-like receptor 4 ablation in mdx mice reveals innate immunity as a therapeutic target in Duchenne muscular dystrophy. Hum Mol Genet (2015) 24(8):2147–62. doi: 10.1093/hmg/ddu735
139. Madaro L, Torcinaro A, De Bardi M, Contino FF, Pelizzola M, Diaferia GR, et al. Macrophages fine tune satellite cell fate in dystrophic skeletal muscle of mdx mice. PloS Genet (2019) 15(10):e1008408. doi: 10.1371/journal.pgen.1008408
140. Capote J, Kramerova I, Martinez L, Vetrone S, Barton ER, Sweeney HL, et al. Osteopontin ablation ameliorates muscular dystrophy by shifting macrophages to a pro-regenerative phenotype. J Cell Biol (2016) 213(2):275–88. doi: 10.1083/jcb.201510086
141. Ji G, Sun S, Wu H, Chen X, Cui H, Tang J, et al. Effect of AAV9-hIGF-1 on inflammatory reaction in mdx mice and its mechanism. Am J Transl Res (2020) 12(8):4488–97.
142. Villalta SA, Rinaldi C, Deng B, Liu G, Fedor B, Tidball JG. Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum Mol Genet (2011) 20(4):790–805. doi: 10.1093/hmg/ddq523
143. Bhattarai S, Li Q, Ding J, Liang F, Gusev E, Lapohos O, et al. TLR4 is a regulator of trained immunity in a murine model of Duchenne muscular dystrophy. Nat Commun (2022) 13(1):879. doi: 10.1038/s41467-022-28531-1
144. Netea MG, Schlitzer A, Placek K, Joosten LAB, Schultze JL. Innate and adaptive immune memory: an evolutionary continuum in the host's response to pathogens. Cell Host Microbe (2019) 25(1):13–26. doi: 10.1016/j.chom.2018.12.006
145. Divangahi M, Aaby P, Khader SA, Barreiro LB, Bekkering S, Chavakis T, et al. Trained immunity, tolerance, priming and differentiation: distinct immunological processes. Nat Immunol (2021) 22(1):2–6. doi: 10.1038/s41590-020-00845-6
146. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A (2012) 109(43):17537–42. doi: 10.1073/pnas.1202870109
147. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science (2014) 345(6204):1251086. doi: 10.1126/science.1251086
148. Arts RJW, Moorlag S, Novakovic B, Li Y, Wang SY, Oosting M, et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe (2018) 23(1):89–100.e5. doi: 10.1016/j.chom.2017.12.010
149. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345(6204):1250684. doi: 10.1126/science.1250684
150. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol (2014) 34(8):1731–8. doi: 10.1161/ATVBAHA.114.303887
151. Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S, et al. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature (2017) 550(7677):475–80. doi: 10.1038/nature24271
152. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol (2010) 10(12):826–37. doi: 10.1038/nri2873
153. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw (2018) 18(4):e27. doi: 10.4110/in.2018.18.e27
154. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
155. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
156. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
157. Braza MS, van Leent MMT, Lameijer M, Sanchez-Gaytan BL, Arts RJW, Perez-Medina C, et al. Inhibiting inflammation with myeloid cell-specific nanobiologics promotes organ transplant acceptance. Immunity (2018) 49(5):819–28.e6. doi: 10.1016/j.immuni.2018.09.008
158. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell (2018) 172(1-2):176–90.e19. doi: 10.1016/j.cell.2017.12.031
159. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell (2018) 172(1-2):147–61.e12. doi: 10.1016/j.cell.2017.11.034
160. Jentho E, Ruiz-Moreno C, Novakovic B, Kourtzelis I, Megchelenbrink WL, Martins R, et al. Trained innate immunity, long-lasting epigenetic modulation, and skewed myelopoiesis by heme. Proc Natl Acad Sci U.S.A. (2021) 118(42):e2102698118. doi: 10.1073/pnas.2102698118
161. Rosenberg IH. Sarcopenia: origins and clinical relevance. Clin Geriatr Med (2011) 27(3):337–9. doi: 10.1016/j.cger.2011.03.003
162. Aversa Z, Zhang X, Fielding RA, Lanza I, LeBrasseur NK. The clinical impact and biological mechanisms of skeletal muscle aging. Bone (2019) 127:26–36. doi: 10.1016/j.bone.2019.05.021
163. Gutmann E, Carlson BM. Regeneration and transplantation of muscles in old rats and between young and old rats. Life Sci (1976) 18(1):109–14. doi: 10.1016/0024-3205(76)90280-0
164. Zacks SI, Sheff MF. Age-related impeded regeneration of mouse minced anterior tibial muscle. Muscle Nerve (1982) 5(2):152–61. doi: 10.1002/mus.880050213
165. Carlson BM, Faulkner JA. Muscle transplantation between young and old rats: age of host determines recovery. Am J Physiol (1989) 256(6 Pt 1):C1262–6. doi: 10.1152/ajpcell.1989.256.6.C1262
166. Conboy IM, Conboy MJ, Smythe GM, Rando TA. Notch-mediated restoration of regenerative potential to aged muscle. Science (2003) 302(5650):1575–7. doi: 10.1126/science.1087573
167. Montecino-Rodriguez E, Berent-Maoz B, Dorshkind K. Causes, consequences, and reversal of immune system aging. J Clin Invest (2013) 123(3):958–65. doi: 10.1172/JCI64096
168. Pinti M, Appay V, Campisi J, Frasca D, Fulop T, Sauce D, et al. Aging of the immune system: Focus on inflammation and vaccination. Eur J Immunol (2016) 46(10):2286–301. doi: 10.1002/eji.201546178
169. Fagiolo U, Cossarizza A, Scala E, Fanales-Belasio E, Ortolani C, Cozzi E, et al. Increased cytokine production in mononuclear cells of healthy elderly people. Eur J Immunol (1993) 23(9):2375–8. doi: 10.1002/eji.1830230950
170. Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, et al. An aged immune system drives senescence and ageing of solid organs. Nature (2021) 594(7861):100–5. doi: 10.1038/s41586-021-03547-7
171. Wang Y, Wehling-Henricks M, Welc SS, Fisher AL, Zuo Q, Tidball JG. Aging of the immune system causes reductions in muscle stem cell populations, promotes their shift to a fibrogenic phenotype, and modulates sarcopenia. FASEB J (2019) 33(1):1415–27. doi: 10.1096/fj.201800973R
172. De Maeyer RPH, Chambers ES. The impact of ageing on monocytes and macrophages. Immunol Lett (2021) 230:1–10. doi: 10.1016/j.imlet.2020.12.003
173. Krasniewski LK, Chakraborty P, Cui CY, Mazan-Mamczarz K, Dunn C, Piao Y, et al. Single-cell analysis of skeletal muscle macrophages reveals age-associated functional subpopulations. Elife (2022) 11:e77974. doi: 10.7554/eLife.77974.sa2
174. Wang Y, Welc SS, Wehling-Henricks M, Tidball JG. Myeloid cell-derived tumor necrosis factor-alpha promotes sarcopenia and regulates muscle cell fusion with aging muscle fibers. Aging Cell (2018) 17(6):e12828. doi: 10.1111/acel.12828
175. Cui CY, Driscoll RK, Piao Y, Chia CW, Gorospe M, Ferrucci L. Skewed macrophage polarization in aging skeletal muscle. Aging Cell (2019) 18(6):e13032. doi: 10.1111/acel.13032
176. Reidy PT, McKenzie AI, Mahmassani ZS, Petrocelli JJ, Nelson DB, Lindsay CC, et al. Aging impairs mouse skeletal muscle macrophage polarization and muscle-specific abundance during recovery from disuse. Am J Physiol Endocrinol Metab (2019) 317(1):E85–98. doi: 10.1152/ajpendo.00422.2018
177. Ahmadi M, Karlsen A, Mehling J, Soendenbroe C, Mackey AL, Hyldahl RD. Aging is associated with an altered macrophage response during human skeletal muscle regeneration. Exp Gerontol (2022) 169:111974. doi: 10.1016/j.exger.2022.111974
178. Ferrara PJ, Yee EM, Petrocelli JJ, Fix DK, Hauser CT, de Hart N, et al. Macrophage immunomodulation accelerates skeletal muscle functional recovery in aged mice following disuse atrophy. J Appl Physiol (1985) (2022) 133(4):919–31. doi: 10.1152/japplphysiol.00374.2022
179. Zhang C, Cheng N, Qiao B, Zhang F, Wu J, Liu C, et al. Age-related decline of interferon-gamma responses in macrophage impairs satellite cell proliferation and regeneration. J Cachexia Sarcopenia Muscle (2020) 11(5):1291–305. doi: 10.1002/jcsm.12584
Keywords: skeletal muscle, macrophage, homeostasis, injury repair, aging
Citation: Wang X and Zhou L (2023) The multifaceted role of macrophages in homeostatic and injured skeletal muscle. Front. Immunol. 14:1274816. doi: 10.3389/fimmu.2023.1274816
Received: 08 August 2023; Accepted: 13 October 2023;
Published: 25 October 2023.
Edited by:
Mohamad Alaa Terkawi, Hokkaido University, JapanReviewed by:
Xiaoguang Liu, Guangzhou Sport University, ChinaCopyright © 2023 Wang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xingyu Wang, d2FuZ3gyMEBidS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.