
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 21 November 2023
Sec. Microbial Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1273524
This article is part of the Research TopicInflammasomes in Human Diseases and MetabolismView all 8 articles
 Yaxuan Xing1,2
Yaxuan Xing1,2 Longmei Yan1,2,3Xiaoya Li1,2
Longmei Yan1,2,3Xiaoya Li1,2 Zhijie Xu1,2,3Xianyu Wu1,2,3Huirong Gao1,2,3Yiduo Chen1,2,3
Zhijie Xu1,2,3Xianyu Wu1,2,3Huirong Gao1,2,3Yiduo Chen1,2,3 Xiaojuan Ma1,2*
Xiaojuan Ma1,2* Jiangang Liu1,2*Jingchun Zhang1,2*
Jiangang Liu1,2*Jingchun Zhang1,2*Atrial fibrillation (AF) is a common clinical arrhythmia whose pathogenesis has not been fully elucidated, and the inflammatory response plays an important role in the development of AF. The inflammasome is an important component of innate immunity and is involved in a variety of pathophysiologic processes. The NLRP3 inflammasome is by far the best studied and validated inflammasome that recognizes multiple pathogens through pattern recognition receptors of innate immunity and mediates inflammatory responses through activation of Caspase-1. Several studies have shown that NLRP3 inflammasome activation contributes to the onset and development of AF. Ecological dysregulation of the gut microbiota has been associated with the development of AF, and some evidence suggests that gut microbiota components, functional byproducts, or metabolites may induce or exacerbate the development of AF by directly or indirectly modulating the NLRP3 inflammasome. In this review, we report on the interconnection of NLRP3 inflammasomes and gut microbiota and whether this association is related to the onset and persistence of AF. We discuss the potential value of pharmacological and dietary induction in the management of AF in the context of the association between the NLRP3 inflammasome and gut microbiota. It is hoped that this review will lead to new therapeutic targets for the future management of AF.
In clinical practice, atrial fibrillation (AF) is a relatively common arrhythmia (1), often caused by multiple risk factors, and is linked to an increased risk of mortality and morbidity associated with stroke and heart failure (2, 3). In 2010, the number of people suffering from AF was calculated to be about 33.5 million worldwide, and the incidence and prevalence have been on the rise since the 1990s, posing a great threat to human health (4). AF can be classified as paroxysmal, persistent, or permanent. The majority of patients initially experience brief, self-terminating episodes, but with the appropriate triggers and substrates, the disease will advance and become more persistent (5). The mechanics behind AF are complex and yet not fully understood. The main mechanisms include electrical remodeling and calcium remodeling of atrial myocytes, DNA repair and mitochondrial dysfunction, and inflammasome activation, et al (6). There is growing evidence that uncontrolled inflammatory signaling cause structural remodeling and electrical conduction damage in AF (7).
The NOD-like receptor protein 3(NLRP3) inflammasomes is an intracellular multiprotein complex that plays an important role in the body’s intrinsic immune response. Various foreign pathogens or intracellularly generated danger signals can be recognized by the NLRP3 inflammasome, which mediates cellular pyroptosis and causes subsequent inflammatory cell recruitment and inflammatory cascade responses that are essential for maintaining host defense functions (8). NLRP3 inflammasome-mediated inflammatory response is closely related to intestinal barrier integrity, intestinal microbial composition and intestinal metabolites (9).
The gut microbiota affects metabolism and immune regulation, and microbiota-derived molecules, whether produced by microorganisms or transformed, are major players in the dialogue with immune cells (10). Under normal conditions, the gut microbiota is a symbiosis of the organism and participates in the immune function of the intestinal internal environment and does not cause harmful immune responses. When the gut microbiota is disturbed and the intestinal barrier is malfunctioning, bacterial components and metabolites circulate throughout the body with the bloodstream, causing chronic inflammatory reactions and cascading effects.
According to the available evidence, NLRP3 inflammasomes activation is associated with the development and progression of AF, and the possible mechanisms involved are leading to increased RyR 2 protein expression and RyR 2-mediated synaptic SR Ca2+ release events as well as inducing myocardial fibrosis, and contributing to the maintenance of substrates in AF (11, 12). According to recent studies, AF is exacerbated by gut microbial imbalances, and possible causes include targeting AF substrates or inhibiting risk factors that promote the development of AF substrates (13).
The activation of NLRP3 inflammasome and the metabolic disturbance of gut microbiota both affect the process of AF, and whether there is a causal relationship between these three factors needs to be further investigated. Using a fecal microbiota transplantation (FMT) rat model, a study demonstrated that high AF susceptibility in older rats can be transmitted to younger hosts via FMT, possibly associated with a significant increase in circulating lipopolysaccharide (LPS) and glucose levels leading to upregulation of NLRP3 inflammasomes expression (14), which demonstrated that the interaction between the NLRP3 inflammasomes and the gut microbiota on the development of AF. In this review, we report the interconnection of NLRP3 inflammasomes and gut microbiota, as well as evidence linking NLRP3 inflammasomes to AF, and attempt to identify new possible pathogenic pathways from the perspective of gut microbiota-derived metabolites and whether this association is relevant to the occurrence and persistence of AF. We discuss the potential value from pharmacological and dietary induction in AF management in the context of the association between NLRP3 inflammasomes and gut microbiota. This study may provide references for new therapeutic targets of AF in the future.
Inflammasomes were discovered 20 years ago as a protein complex that plays an important role in the activation of interleukin-1β (IL-1β), a key substance in inflammation, and inflammasomes are also considered to be an important component of innate immunity (15). Innate immune responses are primarily based on pattern recognition, and infectious agents share multiple molecules, called pathogen-associated molecular patterns(PAMPs) or damage-/hazard-associated molecular patterns (DAMPs), which have similar chemical properties. A small subset of pattern recognition receptors (PRRs) that recognize multiple pathogens (16, 17). Nucleotide-binding oligomerization domain NOD-like receptors (NLRs) are PRRs that recognize multiple pathogens and risk-associated products (18) Inflammasomes are multiprotein complexes assembled from PRRs following the detection of pathogenic microbes and danger signals in the host cell cytoplasm (18). To date, the best studied and validated inflammasome types are NLRP3 inflammasomes, which recognize a variety of stimuli, especially DAMPs, and are implicated in the pathogenesis of aseptic inflammatory diseases such as arthritis, atherosclerosis, et al (19–21).
The NLRP3 inflammasome consists of a sensor (NLRP3), an adapter (ASC), and an effector(caspase-1) (22). Upon pathogen stimulation, NLRP3 is capable of forming oligomers that will recruit CARD-containing adapter protein apoptosis-associated spot-like proteins into the inflammasome via per-PYD-PYD or CARD-CARD homotypic structural domain interactions (23). ASC acts as a bridge that connects the NLRP3 sensor to the effector caspase-1 to assemble into a complete inflammasome, with activated caspase-1 cleaving immature forms of the pro-inflammatory cytokines IL-1β and interleukin-18(IL-18) to form mature forms (24). In addition, caspase-1 can also process gasdermin D(GSDMD), releasing its cleaved N-terminal structural domain (N-GSDMD) and forming a plasma membrane pore, which leads to proinflammatory necrotic cell death, also called pyroptosis, if the load on the plasma membrane pore is high enough (25). The opening of NT-GSDMD pores also promotes the release of cytokines and other inflammatory mediators (26) (Figure 1).
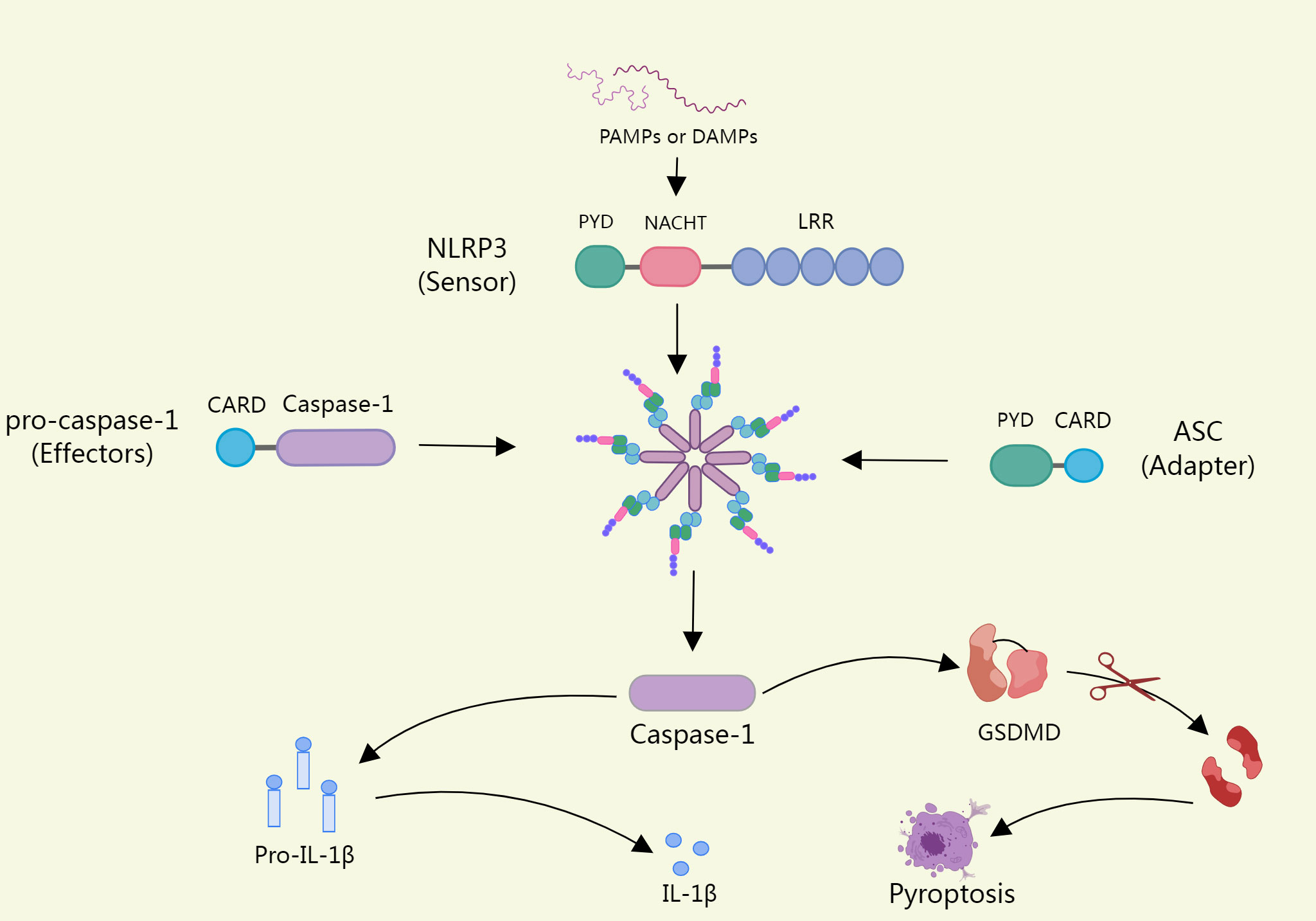
Figure 1 Upon pathogen stimulation, NLRP3 recruits ASCS into inflammasomes. ASC acts as a bridge linking the NLRP3 sensor to the effector Caspase-1, which assembles into intact inflammasomes, and activated caspase-1 cleaves the immature forms of Proinflammatory cytokine il-1β and Il-18, form into a mature form. In addition, Caspase-1 can also process GSDMD (Gasdermin D), release its lytic n-terminal domain (n-GSDMD), and form plasma membrane pores, which can lead to pyroptosis if a load of plasma membrane pores is high enough.
Two signaling processes, priming and activation, are necessary for the NLRP3 inflammasomes to become active (27).The activation signal of the NLRP3 inflammasome is relatively unique in that most PRRs only provide a little amount of specificity for one or two closely related PAMPs or DAMPs, whereas NLRP3 can be activated by a variety of extraneous stimuli, such as viral RNA, microbial toxins and bacterial surface components, uric acid crystals, ATP, β-amyloid peptides, glycolytic products, and many other PAMPs (22).
In the typical pathway, several PAMPs or DAMPs promote NLRP3 inflammasome formation by inducing numerous molecular and cellular processes such K+ efflux, Cl- efflux, mitochondrial dysfunction, reactive oxygen species (ROS) release, synthesis of mitochondrial DNA, and lysosomal disruption (16). In this process, most of the stimuli are activated by triggering intracellular K+ efflux, which in turn activates NLRP3 inflammasomes (28, 29).
LPS has also been shown to activate NLRP3 inflammasomes via an atypical pathway, and oxidized phospholipids in LPS activate caspase-4/5 (caspase -11 in mice), which cleave GSDMD and promote pore formation by inducing N-GSDMD insertion into the plasma membrane. The GSDMD pore allows for K+ efflux and subsequent activation of the NLRP3 inflammasome, which activates caspase-1 (30, 31). Another alternative inflammatory pathway differs from the above two approaches in that in response to LPS, human monocytes secrete IL-1B independently of classical inflammasome stimulation and instead propagate inflammatory signals through the TLR4-TRIF-RIPK1-FADDCASP8 signaling pathway upstream of NLRP3 (32) (Figure 2).
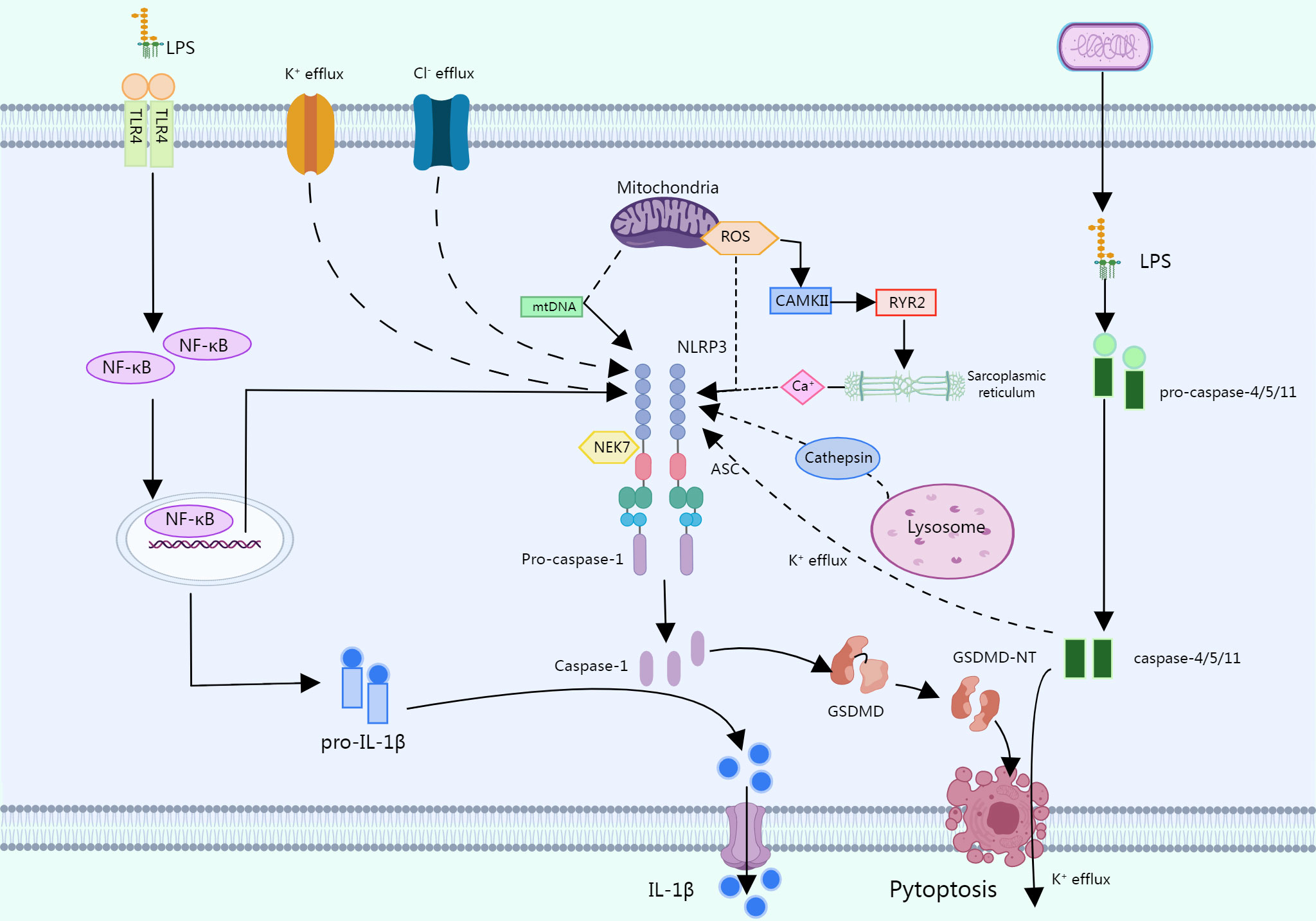
Figure 2 In a typical pathway, multiple PAMPs or DAMPs promote the assembly of the NLRP3 inflammasome by causing multiple molecular and cellular events such as K+ efflux, CI- efflux, mitochondrial dysfunction, etc. LPS can activate the NLRP3 through an atypical pathway, oxidized phospholipids in LPS activate caspase-4/5 (caspase-11 in mice), and these caspases cleave GSDMD, promoting pore formation by inducing N-GSDMD insertion into the plasma membrane. The GSDMD pore allows K+ efflux followed by activation of the NLRP3 inflammasome, which activates caspase-1. In response to LPS, human monocytes secrete IL-1b independent of classical inflammasome stimulation but instead propagate inflammatory signals through the TLR 4-TRIF-RIPK 1-FADDCASP 8 signaling pathway upstream of NLRP3.
The human gut microbiota consists of trillions of microbial cells and thousands of bacterial species (33). A healthy gut microbiota is characterized by stability, abundance, and diversity. Firmicutes and Bacteroidetes make up 90% of the total gut microbiota. The state of the gut microbiota is considered healthy when its ratio (F/B) is low (34).
The gut microbiota is not only involved in the digestion and absorption of food in the body, but also regulates the immune system. There is a bidirectional interaction between the host innate immune system and the gut microbiota, whereby the innate immune system, upon sensing information about the metabolic state of the gut microbiota, signals the host to generate the appropriate cascade of responses, and may also modulate the composition and function of the microbiota. Inflammasome pathways are important mediators of innate immunity and can be activated by various gut microbiota and their metabolites (35). Such interactions are critical for maintaining tissue homeostasis, and perturbations between the two have emerged as key drivers of various chronic disease states (e.g., metabolic syndrome, inflammatory bowel disease, atherosclerosis, cancer, etc.) (36).
Gut microbiota derivatives and metabolites, including bacterial components, functional gut microbiota byproducts, and gut microbiota metabolites. The integrity of the intestinal epithelium prevents pathogenic invasion in the systemic circulation, thus preventing immune and inflammatory diseases (37). Lipopolysaccharide (LPS) is a major component of the outer wall of Gram-negative bacteria. It can be shed and released into the extracellular space when bacteria are disrupted or pass through outer membrane vesicles (38). In some cases, when the intestinal barrier is breached, LPS enters the human circulation through the gut and causes activation of inflammatory pathways elsewhere in the body (39). Bile acids (BAs) are synthesized by the liver and excreted with the bile into the intestine, where they are part of the digestive juices, play an important role in fat metabolism, and are typical functional by-products of the gut microbiota. The gut microbiota can promote the absorption of dietary fats and fat-soluble vitamins by influencing the type and amount of BAs in the host (40). In addition, there are many gut microbiota metabolites, typically short chain fatty acids (SCFAs), trimethylamine (TMA), and indoxyl sulfates, which affect many important metabolic pathways (Figure 3).
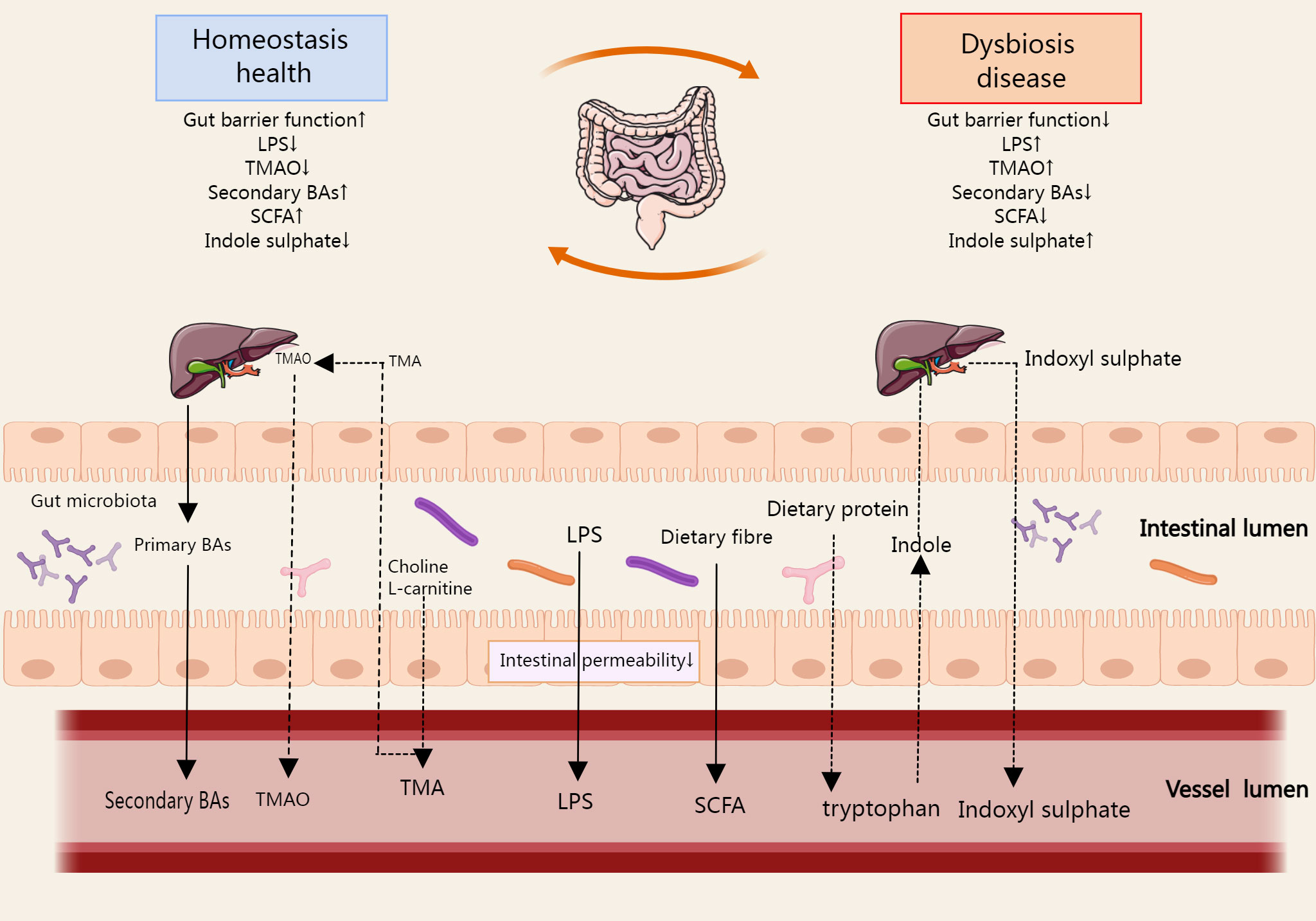
Figure 3 The metabolites produced during the metabolic process of gut microbial ginseng can be absorbed in the host gut, affecting the immune cell function of the gut and circulating from the gut into the host body. SCFA, TMA, Bas, and indoxyl sulfate are the typical metabolites, which affect many important metabolic pathways. In some cases, LPS enters the systemic circulation via the gut when the gut barrier function is reduced. The arrows in the figure refer to the direction of metabolism of gut microbiota derivatives and metabolites in the gut, blood circulation, and liver.
The onset and maintenance of AF are intimately correlated with the activation of the NLRP3 inflammasome (Table 1). Activation of NLRP3 Inflammasomes in atrial myocytes is one of the important mechanisms in the pathogenesis and progression of AF (11). Clinically, inflammatory indicators including IL-1 and IL-18 are linked to the development of AF and can serve as predictors of the recurrence of AF after ablation (7, 41, 42, 44, 45). Yao et al. found that NLRP3 inflammasome activity was enhanced in atrial myocytes (AMs) from patients with AF and dogs with rapid atrial pacing and that AM-restricted activation of NLRP3 in mice promoted ectopic discharge and AF-sustained substrates, thereby inducing AF (11). Enhanced activation of NLRP3-inflammasome in AMs increases caspase-1 cleavage and caspase-1-mediated apoptosis, releases inflammatory cytokines, recruits macrophages and other immune cells, and induces myocardial fibrosis and promotes the formation of AF maintenance substrates (11). NLRP3 inflammasome is hyperactivated in left atrial cells of patients with postoperative atrial fibrillation (POAF) (43), showing that the NLRP3 inflammasome may have a role in the development of AF.Metabolic disorders and cardiovascular diseases, such as obesity, diabetes, hypertension, and intestinal ecological disorders, can activate NLRP3 inflammasomes and other inflammatory cytokines in atrial cardiomyocytes, leading to atrial lesions and promoting AF (46–49). In addition, oxidative stress-induced calcium release is associated with the development of AF and activation of the NLRP3 inflammasome. Elevated levels of atrial reactive nitrogen (RNS) and reactive oxygen species (ROS) closely influence the development of AF (50, 51). Calcium ions are key molecular regulators of NLRP3 inflammasome assembly and activation (52, 53), increased intracellular ROS levels mediate the oxidation and activation of calmodulin-dependent kinase II (CaMKII), which in turn activates Ryanodine Receptor 2 (RYR2), leading to Ca2+ release from the sarcoplasmic reticulum (SR), thereby triggering NLRP3 inflammasome activation (54). Epicardial adipose tissue (EAT) adjacent to myocardium without fascial border (55). In the atria, EAT is associated with the incidence and severity of AF through the release of adipokines and inflammatory mediators leading to paracrine effects (44, 56–59). Related studies showed that EAT-mediated IL-1β secretion and expression were significantly higher in POAF patients than in the no-POAF group, but differences in NLRP3 inflammasome expression between groups were not confirmed in the experiments (44).
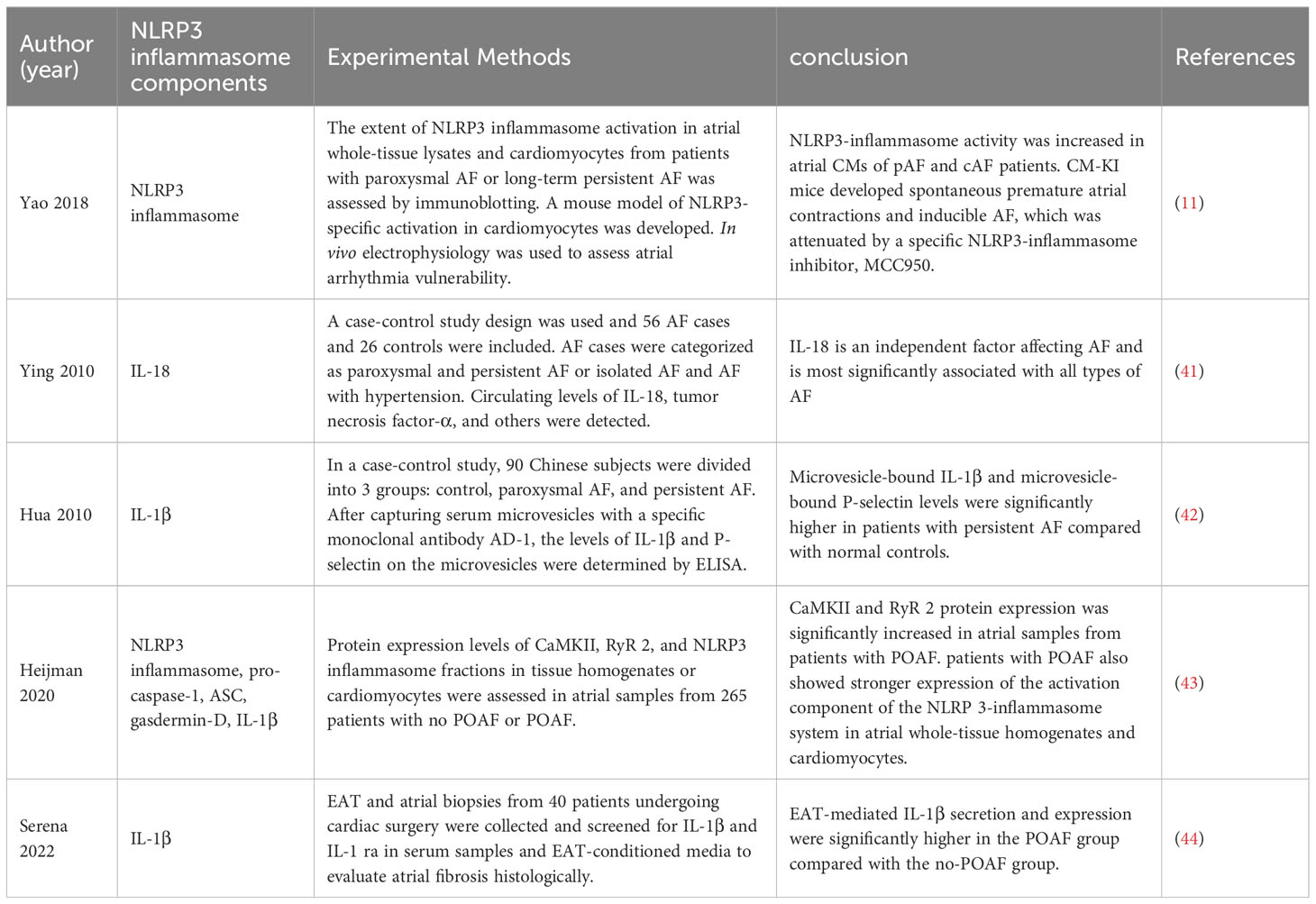
Table 1 Correlation studies of AF and NLRP3 inflammasome components.
The relationship between gut microbiota and AF is unclear, and recent preclinical and observational cohort studies suggest that imbalances in the composition of the gut microbiota are a factor in AF. Imbalances in the gut microbiota may contribute to obesity, hypertension, and type 2 diabetes, all of which are risk factors for the development of AF. In a small case-control study comparing 50 patients with AF to 50 matched controls, patients with AF had a disturbed microbial composition of the gut microbiota and dysregulated metabolic activity (60). Another retrospective clinical study analyzed a group of 6,763 randomly selected individuals to investigate the relationship between AF prevalence and gut microbiota. Although the gut microbiota categories did not enable differentiating between patients with prevalent or episodic AF and those without AF, the results showed a tendency to identify various genera and species. The bacterial composition shifted toward a spectrum similar to that of the hypertension and heart failure microbiomes, highlighting their common underlying pathophysiology (61). In addition, a number of derivatives and metabolite changes resulting from gut microbiota dysbiosis have also been shown to be associated with the development and exacerbation of AF (Table 2). Although unhealthy lifestyle, obesity, and other AF risk factors also contribute to gut microbiota dysbiosis, and the magnitude of the association between the two is influenced by many confounding factors, it may provide new insights into the mechanisms of AF and future therapeutic targets.
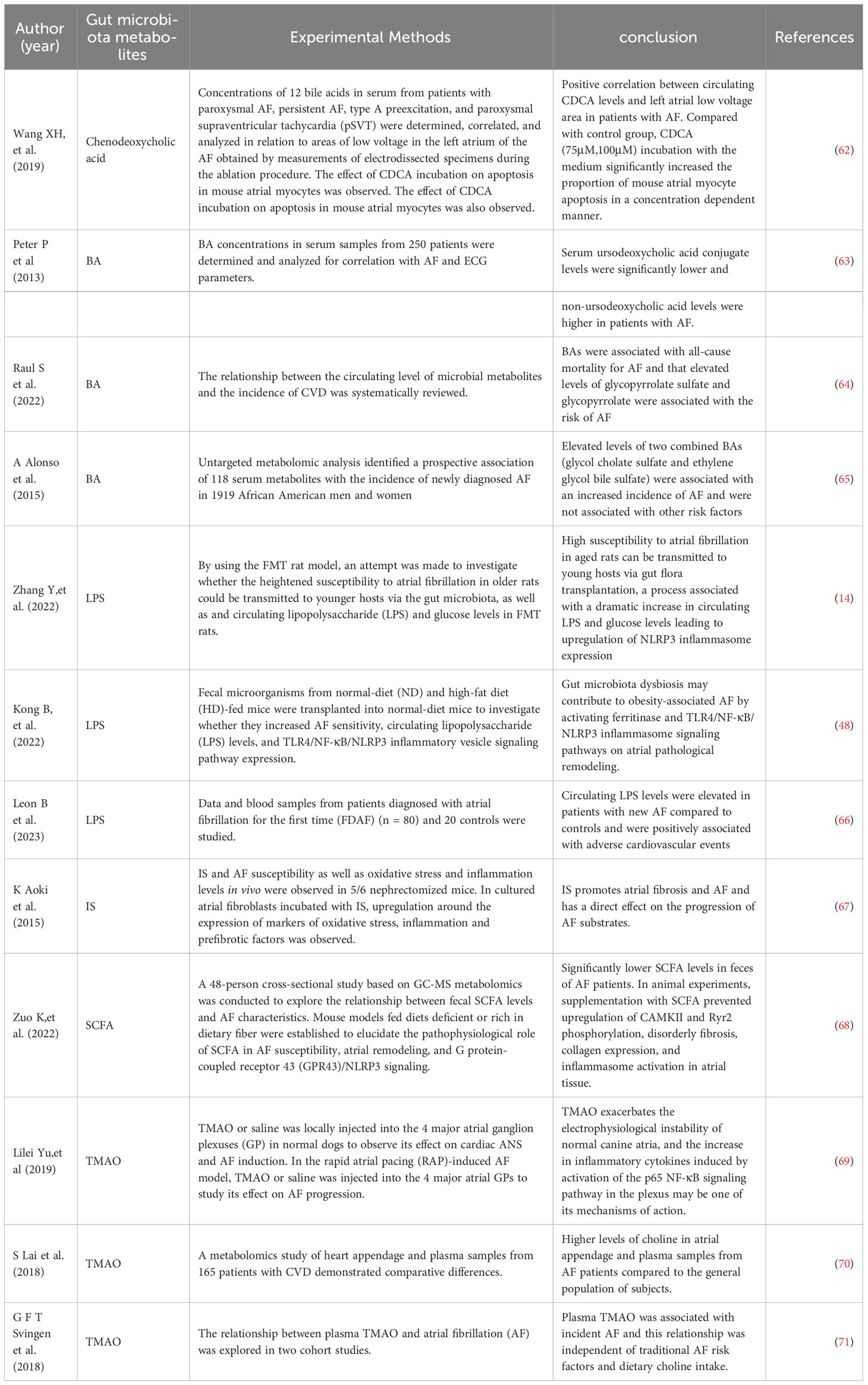
Table 2 Correlation studies of AF and gut microbiota derivatives and metabolites.
The association between the onset and progression of AF and the NLRP3 inflammasome is direct. The activation of NLRP3 inflammasomes can induce or exacerbate AF. Gut microbiota metabolites and derivatives differentially modulate the NLRP3 inflammasome (Table 3). And this section will explore the links that exist between gut microbiota derivatives or metabolites and NLRP3 inflammasomes and AF in the context of both direct and indirect evidence in this regard (Figure 4).
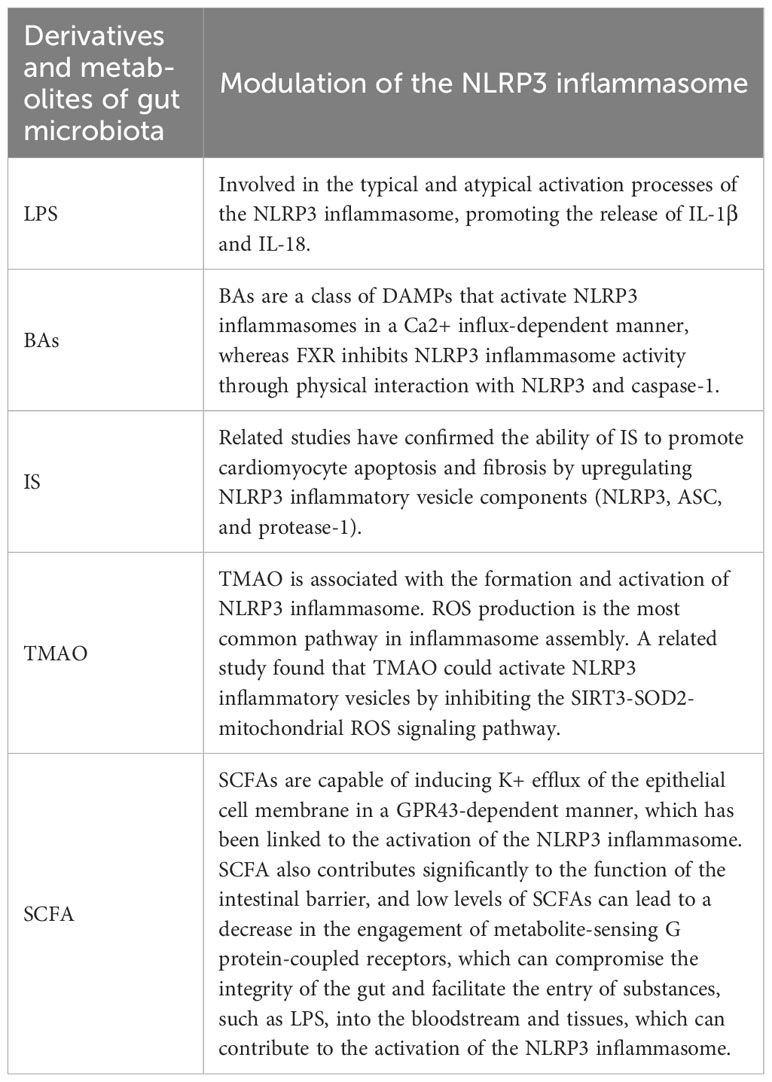
Table 3 Modulation of the NLRP3 inflammasome by gut microbial derivatives and metabolites.
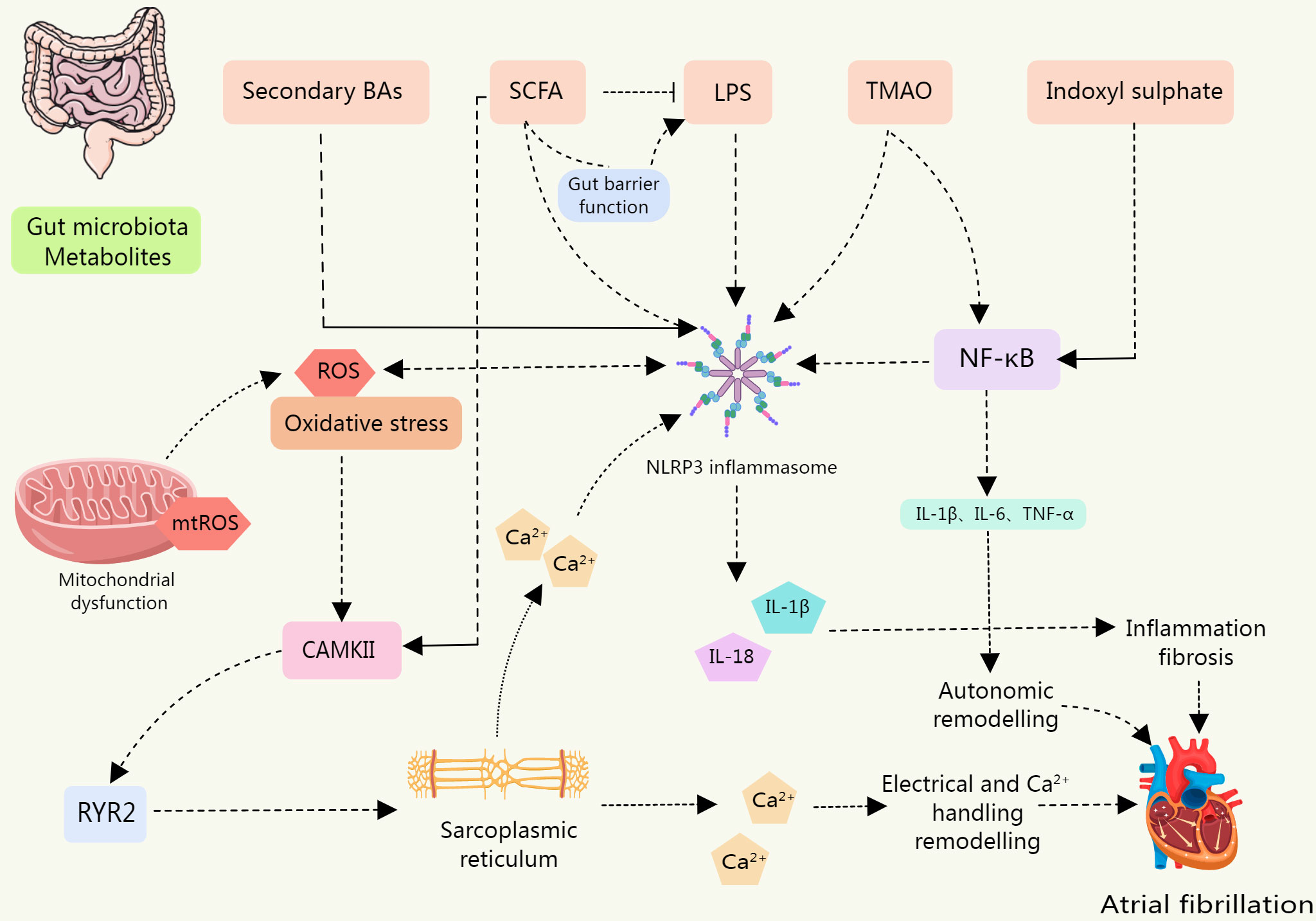
Figure 4 The levels of NLRP3 inflammasome, IL-1β, and IL-18 were upregulated in IS-treated cardiomyocytes, leading to cardiomyocyte apoptosis; BAs are a class of DAMPs that activate the NLRP3 inflammasome in a calcium influx dependent manner; LPS is involved in the typical and atypical activation of NLRP3 inflammasome. The sharp increase in circulating LPS level is associated with the up-regulation of NLRP3 inflammasome expression in rats, which increases their susceptibility to AF. SCFA is a potential contributor to the pathogenesis of AF. SCFAs promote the improvement of intestinal barrier function. Low SCFA levels lead to decreased intestinal integrity and promote the entry of substances such as LPS into blood and tissues. SCFA supplementation can prevent the up-regulation of phosphorylation of CAMK II and RyR2, and prevent disordered fibrosis, collagen expression, and NLRP3 inflammasome activation in atrial tissue. TMAO may be involved in the activation of the p65 NF-κB signaling pathway by activating NLRP3. Metabolites of gut microbiota can induce inflammation, myocardial fibrosis, autonomic remodeling, electrical remodeling, and other pathological processes of AF by directly or indirectly activating NLRP3 inflammasome.
LPS, a chemical component unique to the outer wall layer of Gram-negative bacteria, can induce inflammatory expression through activation of the receptor Toll-like receptor 4 (TLR4), eliciting a cascade response of immune stimulation and toxic pathophysiological activity in the body (72). LPS is involved in the typical and atypical activation processes of the NLRP3 inflammasome, promoting the release of IL-1β and IL-18, which are the main pathways of inflammation (73). Increased intestinal permeability contributes to the translocation of LPS on the intestinal epithelium, which leads to a moderate increase in blood LPS levels and causes systemic inflammation, defined as metabolic endotoxemia (74). LPS-induced metabolic endotoxemia is associated with obesity and increased cardiovascular risk and insulin resistance (75).
Recent evidence suggests that LPS is associated with the pathogenesis of AF. Animal experiments have shown that LPS increases the expression levels of inflammatory cytokines and L-type calcium channel proteins, shortens the effective atrial expiration period (ERP), and thus promotes the development of AF (11, 76). Zhang et al. demonstrated by using a FMT rat model that the high susceptibility of aged rats to AF can be transmitted to younger hosts, a process associated with a dramatic increase in circulating LPS and glucose levels leading to an upregulation of NLRP3 inflammasome expression (14). Kong et al. found that transplantation of fecal microorganisms derived from high-fat diet-fed mice into normal diet mice resulted in significantly increased susceptibility to AF, significantly higher circulating LPS levels, enhanced expression of intracardial ferritin, and enhanced activation of the TLR4/NF-κB/NLRP3 inflammasome signaling pathway relative to controls. Significant improvement in atrial fibrosis and reduction in susceptibility to AF induced by obesity-related gut ecological dysregulation through inhibition of ferritin excess or NLRP3 inflammasome signaling pathways (48).
LPS does not appear to directly cause new-onset AF, and in a study in which 652 healthy men (all without a history of AF) were infused with LPS and underwent continuous cardiac monitoring to assess rhythm, experimental LPS excitation resulted in a significant increase in acute inflammation-related indices but did not increase the propensity for new-onset AF in young, low-risk individuals. A similar conclusion was obtained in an animal experiment in which an experimental autoimmune myocarditis model was established and AF was induced by atrial electrical stimulation, and AF was induced in the chronic phase of myocarditis rats, whereas AF did not occur in the acute phase of myocarditis or LPS-infused rats (77). It may suggests that acute inflammation per se does not increase the incidence of AF induction, and that atrial structural remodeling due to inflammation and hemodynamic effects is necessary to induce AF. A study showed that circulating LPS levels were elevated in patients with new AF compared to controls and were positively associated with adverse cardiovascular events, suggesting that improving the intestinal barrier and reducing endotoxemia may be a potential target for treating cardiovascular diseases(CVD) and preventing complications (66).
An essential part of bile, BAs is crucial for the metabolism of glucolipids and the release of stored energy (78). BAs are produced in the liver and kept in the gallbladder. After eating, BAs are secreted into the intestine and metabolized by gut microorganisms. A total of 5% of BAs are expelled in feces, while 95% of BAs are reabsorbed in the ileum and transported back to the liver through enterohepatic circulation (79). Through gut flora transformation, BAs regulate multiple metabolic pathways in the host through nuclear farnesoid X receptor (FXR) and G protein-coupled membrane receptor (GPCR)-mediated signaling. Also, by triggering the innate immune response, BAs may have a direct or indirect impact on the composition of the gut’s microbiota (80). Primary BA (e.g. goose deoxycholic acid) is formed by combining with amino acids (e.g. taurine or glycine) to form bile salts, which are then further secreted into the intestine. The primary BA is further converted by the gut microbiota to form secondary BA (e.g. ursodeoxycholic acid). Dysbiosis of the gut microbiota regulates the BA ratio, decreasing the concentration of secondary BA and increasing the concentration of primary BA (13).
BA is a metabolite that circulates and binds to its receptor expressed in multiple tissues, cells, and organs throughout the body. BAs serve as crucial mediators of intestinal microecology and NLRP3 inflammasome activation (81). There is growing evidence that activation of the NLRP3 inflammasome by BAs is a major feature of systemic chronic low-grade inflammation. BAs are a class of DAMPs that activate NLRP3 inflammasomes in a Ca2+ influx-dependent manner, whereas FXR inhibits NLRP3 inflammasome activity through physical interaction with NLRP3 and caspase-1 (82). It has been found that BAs have different regulatory effects on NLRP3 in different inflammatory states. Under non-inflammatory conditions, supplementation with BAs activated the NLRP3 inflammasome in THP-1 differentiated macrophages and promoted inflammation. However, in LPS-induced inflammatory macrophages, BAs inhibited NLRP3 inflammasomes and attenuated inflammation (83).
A Study has shown that serum levels of ursodeoxycholic acid are significantly lower and non-ursodeoxycholic acid levels are higher in patients with AF (63). Another study measured serum concentrations of 12 bile acids separately in patients with different types of AF and found that circulating levels of goose deoxycholic acid (CDCA) were elevated in AF patients. CDCA caused a dose-dependent increase in atrial myocyte apoptosis in mice, suggesting that CDCA may play an important role in the structural remodeling process of AF (62). A systematic review of gut microbial-derived metabolites showed that BAs were associated with all-cause mortality for AF and that elevated levels of glycopyrrolate sulfate and glycopyrrolate were associated with the risk of AF (64). A prospective cohort study based on 1919 African Americans showed that increased levels of two combined BAs (ethylene glycol cholate sulfate and ethylene glycol cholate sulfate) were associated with increased incidence of AF and were independent of other risk factors (65).
The function of BA in immune system regulation has been validated. The regulatory effects of BA on the NLRP3 inflammasome differ across various states (81). While existing studies have demonstrated a link between AF and BA metabolism to a certain extent, and have characterized serum BA levels in AF patients, the specific mechanisms underlying how BA metabolism affects AF remain less well-studied. No research has explored the association between AF and BAs from inflammatory signaling pathways, indicating a potential area for future investigation.
Indoxyl sulfate (IS), a specific product of the gut microbiota metabolizing dietary tryptophan (from protein foods), is one of the most common uremic toxins. As dietary tryptophan reaches the colon, it is transformed into indole by microbial action and absorbed into the bloodstream. From there, the indole is further metabolized by the liver to produce IS and finally excreted by the kidneys (84). CKD is associated with a higher incidence of AF, but the mechanism is unclear (85). An animal study showed that IS increases the occurrence of pulmonary venous and atrial arrhythmias through oxidative stress, which may be associated with the occurrence of AF in CKD patients (86). Another animal experiment showed that mice with higher levels of IS, in vivo oxidative stress and inflammation after 5/6 nephrectomy were more prone to induce AF, while nephrectomy-induced changes were attenuated after AST-120 (an adsorbent for uremic toxins) reduced IS circulating levels (67). There is no direct evidence that IS affects AF through activation of the NLRP3 inflammasome, but there is correlative evidence confirming the ability of IS to upregulate NLRP3 inflammasome components (NLRP3, ASC, and procaspase-1), which contributes to cardiomyocyte apoptosis and fibrosis. In vitro treatment of mouse cardiomyocytes with IS resulted in upregulation of NLRP3 inflammasome, IL-1β and IL-18 levels in IS-treated cardiomyocytes through activation of the NF-κB signaling pathway, leading to apoptosis (87). To avoid AF brought on by renal insufficiency, IS may be a prospective therapeutic target and a risk factor for AF in renal failure.
The gut microbiota is engaged in the fermentation of dietary fiber and glucose to create SCFA, such as acetate, butyrate, and propionate, which are vital for preserving the in vivo homeostasis of the gut microbiota and host immunity. SCFAs can directly activate G-coupled receptors and inhibit histone deacetylases, which can repair gut barrier dysfunction and affect various physiological processes (88, 89). Related studies have shown that fiber can promote the release of SCFAs through intestinal microbial fermentation, that SCFAs can induce epithelial cell membrane K+ efflux in a GPR43-dependent manner, that this mechanism is associated with NLRP3 activation, and that IL-1β release following inflammasome activation contributes to intestinal internal environmental stability (90). SCFAs might have a role in the pathophysiology of AF. SCFAs contribute to intestinal barrier function, and low SCFA levels lead to decreased metabolite sensing G protein-coupled receptor engagement, which compromises intestinal integrity and promotes the entry of substances such as LPS into blood and tissues (91). A metabolomics-based cross-sectional study showed significantly lower fecal SCFA levels in AF patients (68). Animal experiments have shown that dietary fiber deficiency in mice increases susceptibility to AF during pacing, and supplementation with SCFAs may have a protective effect. Supplementation with SCFAs prevents upregulation of CAMKII and RyR2 phosphorylation and prevents disordered fibrosis, collagen expression and NLRP3 inflammasome activation in atrial tissue (68).
Choline, L-carnitine, betaine, and other choline-containing substances are the major dietary precursors of trimethylamine oxide (TMAO), which is a significant gut microbe-dependent metabolite (92). The gut microbiota is a key factor in the production of TMAO and can metabolize nutrient precursors from the diet into TMA (93, 94). TMA is absorbed through the intestine and transported through the circulatory system to the liver, where it is processed by hepatic flavin monooxygenase (FMO) to form TMAO (95, 96). In mice, the addition of TMAO, carnitine or choline to the diet alters the microbial composition of the cecum (93). In metabolic diseases, with increased dietary choline and L-carnitine intake, the microbiota is altered, resulting in elevated plasma TMAO levels (97). TMAO is associated with the formation and activation of NLRP3 inflammasomes and may be an important initiating mechanism for turning on the endothelial inflammatory response leading to endothelial dysfunction (98). A related study found that TMAO can activate NLRP3 inflammasome and promote vascular inflammation by inhibiting SIRT3-SOD2-mitochondrial ROS signaling pathway (99). ROS generation is the most common pathway in inflammasome assembly (100). In contrast, the SIRT3-SOD2 linkage pathway deacetylates mitochondrial proteins and limits the accumulation of mitochondrial ROS (101).
Elevated TMAO concentrations may be associated with diet, changes in the composition of the gut microbiota, intestinal dysbiosis, or impaired gut-blood barrier, and can increase the risk of cardiovascular disease, and metabolic syndrome (102, 103). These are risk factors that promote AF and can increase susceptibility to AF. By locally injecting TMAO or saline into the four major atrial ganglion plexuses (GPs) of normal dogs or dogs in an AF-induced model, Yu et al. found that TMAO increased atrial electrophysiological instability in normal dogs and exacerbated acute electrical remodeling in the AF model by promoting autonomic remodeling. These alterations may be associated with the activation of the p65 NF-κB signaling pathway and increased inflammatory cytokines in GPs (69). Cold exposure is an important risk factor for AF (104). Cold exposure increased the susceptibility of rats to AF and also led to a decrease in the abundance of A. muciniphila in the rat intestine, which increased the level of TMAO by modulating changes in microbial enzymes. Similarly, human participants’ plasma TMAO levels increased over time as the temperature dropped. Elevated TMAO enhanced infiltration of M1 macrophages in the atria, increased the expression of Casp1-p20 and cleaved GSDMD, induced apoptosis, and ultimately led to structural remodeling of the atria. However, mice with conditional deletion of caspase1 exhibit resistance to cold-related AF (105). These are components associated with the NLRP3 inflammasome, but NLRP3 inflammasome expression was not further investigated in this study. Direct mechanistic studies related to TMAO, AF, and NLRP3 inflammasomes are almost nonexistent, and this is a new area worth exploring. In addition, a metabolomic study of atrial appendage samples and plasma samples from 165 patients with cardiovascular disease (CVD) revealed higher levels of choline in atrial appendage and plasma samples from AF patients compared to the general population of subjects (70). The results of two cohort studies showed that plasma TMAO was positively associated with long-term AF events, a prospective relationship independent of traditional AF risk factors and potential confounders includes dietary intake of TMAO precursors (e.g., choline and betaine) (71).
Targeting inflammatory signaling has become an emerging option for AF management. At this stage, NLRP 3 inhibitors, caspase-1 inhibitors, and IL-1β antagonists have been developed successively (106). Regulation of NLRP3 inflammasome itself or its activation mechanism can inhibit the release of inflammatory factors and apoptosis, thereby alleviating myocardial fibrosis. Inhibition of the NLRP3 inflammatory pathway also affects gut Microbiota (107).
The diaryl sulfonylurea compound MCC950 is a specific inhibitor of NLRP3 and is generally believed to act on the assembly portion of the NLRP3 inflammasome. K+ efflux and the interaction of NEK7 with NLRP3 are considered to be potential targets of MCC950 (108). In the FMT rat model, significant elevation of LPS and glucose levels led to upregulation of NLRP3 inflammasome expression, which promoted the development of AF, and inhibition of NLRP3 inflammasome with MCC950 led to reduced susceptibility to AF (14).
Canakinumab is a monoclonal antibody that blocks IL-1β-mediated inflammatory pathways (109). A randomized controlled study of patients with persistent AF showed that anti-inflammatory treatment with canakinumab after electrical cardioversion did not reduce AF recurrence (110), but promising trends were seen in this experiment. Canakinumab reduced major cardiac events in patients with atherosclerosis in the CANTOS trial (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study) (111).
Colchicine is a microtubule-destroying drug that was first used in the treatment of gout and has a variety of anti-inflammatory effects. The anti-inflammatory mechanism of colchicine is unclear, but there is evidence that it reduces inflammasome-mediated IL-1β and IL-18 production by inhibiting the assembly and activation of the NLRP3 inflammasome (112). ACC/AHA guidelines suggest that colchicine may be considered for the prevention of POAF (Class IIb evidence level B) (113). Some current clinical evidence evaluates the effect of colchicine on cardiovascular diseases such as pericarditis, postoperative and post-ablation AF, and coronary artery disease (114–116). Colchicine has been tested in several clinical trials for the treatment of POAF (117–119). However, the designs of these studies differed in terms of a drug loading dose, AF treatment modality, time to treatment initiation (pre/postoperative), and duration of follow-up. Therefore, additional trials are needed to demonstrate the potential value of colchicine in the treatment of different forms of AF.
Short-and long-term changes in gut microbiota are closely related to diet. Diets rich in animal fats and saturated fats may alter the intestinal flora by increasing LPS, increasing TMAO, and decreasing SCFA (120) Altered intestinal permeability to LPS may be the trigger of low-grade systemic inflammation. Cani et al. found that a high-fat diet increased the proportion of LPS-containing microbiota in the mouse gut and dysregulated inflammatory tone, triggering weight gain and insulin resistance (74). High-fiber diets are associated with increased SCFA production in the gut, and when dietary fiber is in short supply, not only is SCFA production reduced, but it also results in the gut microbiota using less favorable substrates, such as amino acids and host mucins, for energy (88, 121, 122). In addition, the type of diet can affect microbial-derived TMA and may influence TMAO levels. Compared to vegans/vegetarians, omnivorous human subjects consuming L-carnitine produced more TMAO (123).
The Mediterranean diet is plant-based, rich in fiber and omega-3 fatty acids, and low in animal protein and saturated fat. Ghosh et al. analyzed the gut microbiota before and after a 12-month MedDiet intervention (NU-AGE diet) for elderly subjects in 612 non-frail or prematurely aged subjects from five European countries (UK, France, Netherlands, Italy, and Poland) and found that adherence to the Mediterranean diet modulated microbiota changes associated with increased production of short/branched-chain fatty acids and decreased production of secondary BAs, p-cresol, ethanol and carbon dioxide (124). A study investigating the dietary habits of AF patients found that AF patients were less likely to report adherence to a Mediterranean diet, a lower intake of plant foods such as nuts, vegetables, and fruits, and a preference for white meat over red meat (125), but the sample size included in that trial was small and retrospective and larger clinical studies are needed to confirm the extent to which high-quality dietary patterns such as the Mediterranean diet affect the onset of AF.
However, there is still a lack of studies based on dietary habits, gut microbiota, and the development of AF, and whether dietary habits can affect AF by influencing changes in the gut microbiota and its metabolites remains a hot topic for further research.
There is no clear evidence whether targeted modulation of gut microecology, including administration of probiotics, administration of prebiotics, or gut flora transplantation, can influence AF progression. Probiotics mainly include Lactobacillus spp. and Bifidobacterium spp. and have the functions of enhancing the barrier function of the gastrointestinal tract, inhibiting the growth of pathogens, and suppressing their harmful toxins (126). Probiotics can mitigate AF-related risk factors (obesity, abnormal lipid metabolism, inflammation, etc.) (127).
Prebiotics are not digested and absorbed by the host, but can selectively promote the metabolism and proliferation of beneficial bacteria in the body, thus improving host health. An animal study found that resveratrol attenuated TMAO-induced atherosclerosis by reducing TMAO levels and through gut microbiota remodeling (128). Oligogalactose increases SCFA and reduces high-fat diet-induced LPS production (129).
FMT is an intervention to restore a patient’s intestinal flora by transferring a specially prepared stool sample from a healthy donor to a recipient (130). Reduced atrial NLRP3 inflammasome activity and decreased atrial fibrosis in aged rats by transferring intestinal flora from juvenile to aged rats (14). In another study, the transplantation of cecum contents from normotensive rats into spontaneously hypertensive rats lowered blood pressure, while normotensive rats had increased blood pressure after FMT in spontaneously hypertensive rats (131). FMT may be a potential pathway and research hotspot for the treatment of CVD in the future. The exact effect of FMT on the AF matrix and its mechanism still need more investigation.
Activation of the NLRP3 inflammasome and dysregulation of intestinal microecology is a potential target factor that may provide new therapeutic avenues for AF. The interactions between NLRP3 inflammasome and intestinal microecological metabolic processes have been gradually elucidated, both of which affect the developmental process of AF, and whether there is a synergistic or causal relationship needs the further investigation. Gut microbial dysbiosis may promote increased AF susceptibility or AF substrates by influencing their derivatives and metabolites to modulate oxidative stress responses, regulate metabolism, and upregulate NLRP3 inflammasome expression, leading to cardiac inflammatory responses, myocardial fibrosis, or metabolic disturbances. In this review, less high-quality direct evidence demonstrated a relationship between the gut microbiota-AF-NLRP3 inflammasome axis. Only SCFA and LPS have been shown to be directly associated with the NLRP3 inflammasome and AF. TMAO, BAs and IS although all shown to be associated with NLRP3 inflammasome activation and AF, respectively, have not yet been found to be directly associated with all three. This review collates existing studies on gut microbiota derivatives and metabolites and immune response and AF occurrence, with a view to providing a reference for in-depth research in this area.
More valuable researches should inspire the development of new theoretical and drug targets in AF. This article may provide insights into the use of pharmacological inhibition of NLRP3 inflammasome activation, dietary modification, and re-establishment of gut microbiota for the clinical management of AF and its comorbidities. However, most studies used single-factor intervention analysis in this manuscript, which cannot better explain the relationship between the three. Therefore, it is urgent to explore the specific regulatory mechanism of NLRP3 inflammasome and intestinal microecology of AF patients through more comprehensive researches.
YX: Writing – original draft, Writing – review & editing. LY: Writing – review & editing. XL: Writing – review & editing. ZX: Writing – review & editing. XW: Writing – review & editing. HG: Writing – review & editing. YC: Writing – review & editing. XM: Writing – original draft, Writing – review & editing. JL: Writing – original draft, Writing – review & editing. JZ: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was financially supported by the CACMS Innovation Fund (Grant No. CI2021A00915) and the National Natural Science Foundation of China (Grant No. 81573817).
The authors would like to thank Xiyuan Hospital of the Chinese Academy of Traditional Chinese Medicine and the National Clinical Research Center of Cardiology of Traditional Chinese Medicine for supporting this work. Thank the reviewers for allowing us to improve the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sagris M, Vardas EP, Theofilis P, Antonopoulos AS, Oikonomou E, Tousoulis D. Atrial fibrillation: pathogenesis, predisposing factors, and genetics. Int J Mol Sci (2021) 23(1):6. doi: 10.3390/ijms23010006
2. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke (1991) 22:983–8. doi: 10.1161/01.str.22.8.983
3. Wang TJ, Larson MG, Levy D, Vasan RS, Leip EP, Wolf PA, et al. Temporal relations of atrial fibrillation and congestive heart failure and their joint influence on mortality: the Framingham Heart Study. Circulation (2003) 107:2920–5. doi: 10.1161/01.Cir.0000072767.89944.6e
4. Dai H, Zhang Q, Much AA, Maor E, Segev A, Beinart R, et al. Global, regional, and national prevalence, incidence, mortality, and risk factors for atrial fibrillation, 1990-2017: results from the Global Burden of Disease Study 2017. Eur Heart J Qual Care Clin Outcomes (2021) 7:574–82. doi: 10.1093/ehjqcco/qcaa061
5. Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res (2014) 114:1483–99. doi: 10.1161/circresaha.114.302226
6. Brundel B, Ai X, Hills MT, Kuipers MF, Lip GYH, de Groot NMS, et al. Atrial fibrillation. Nat Rev Dis Primers (2022) 8:21. doi: 10.1038/s41572-022-00347-9
7. Li N, Brundel B. Inflammasomes and proteostasis novel molecular mechanisms associated with atrial fibrillation. Circ Res (2020) 127:73–90. doi: 10.1161/circresaha.119.316364
8. Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
9. Man SM. Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat Rev Gastroenterol Hepatol (2018) 15:721–37. doi: 10.1038/s41575-018-0054-1
10. Lavelle A, Sokol H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol (2020) 17:223–37. doi: 10.1038/s41575-019-0258-z
11. Yao C, Veleva T, Scott L Jr., Cao S, Li L, Chen G, et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation (2018) 138:2227–42. doi: 10.1161/circulationaha.118.035202
12. Yarmohammadi F, Hayes AW, Karimi G. Possible protective effect of resolvin D1 on inflammation in atrial fibrillation: involvement of ER stress mediated the NLRP3 inflammasome pathway. Naunyn Schmiedebergs Arch Pharmacol (2021) 394:1613–9. doi: 10.1007/s00210-021-02115-0
13. Gawałko M, Agbaedeng TA, Saljic A, Müller DN, Wilck N, Schnabel R, et al. Gut microbiota, dysbiosis and atrial fibrillation. Arrhythmogenic mechanisms and potential clinical implications. Cardiovasc Res (2022) 118:2415–27. doi: 10.1093/cvr/cvab292
14. Zhang Y, Zhang S, Li B, Luo Y, Gong Y, Jin X, et al. Gut microbiota dysbiosis promotes age-related atrial fibrillation by lipopolysaccharide and glucose-induced activation of NLRP3-inflammasome. Cardiovasc Res (2022) 118:785–97. doi: 10.1093/cvr/cvab114
15. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi: 10.1016/s1097-2765(02)00599-3
16. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci (2019) 20:3328. doi: 10.3390/ijms20133328
17. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22:240–73. doi: 10.1128/cmr.00046-08
18. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
19. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
20. Takahashi M. NLRP3 inflammasome as a key driver of vascular disease. Cardiovasc Res (2022) 118:372–85. doi: 10.1093/cvr/cvab010
21. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol (2021) 18:2114–27. doi: 10.1038/s41423-021-00740-6
22. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
23. Coll RC, Schroder K, Pelegrín P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci (2022) 43:653–68. doi: 10.1016/j.tips.2022.04.003
24. Christgen S, Place DE, Kanneganti TD. Toward targeting inflammasomes: insights into their regulation and activation. Cell Res (2020) 30:315–27. doi: 10.1038/s41422-020-0295-8
25. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J (2016) 35:1766–78. doi: 10.15252/embj.201694696
26. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi: 10.1038/nri3452
27. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
28. Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and Nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem (1994) 269:15195–203. doi: 10.1016/S0021-9258(17)36591-2
29. Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J (1995) 14:1607–14. doi: 10.1002/j.1460-2075.1995.tb07149.x
30. Elliott EI, Miller AN, Banoth B, Iyer SS, Stotland A, Weiss JP, et al. Cutting edge: mitochondrial assembly of the NLRP3 inflammasome complex is initiated at priming. J Immunol (2018) 200:3047–52. doi: 10.4049/jimmunol.1701723
31. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov (2018) 17:688. doi: 10.1038/nrd.2018.149
32. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity (2016) 44:833–46. doi: 10.1016/j.immuni.2016.01.012
33. Gentile CL, Weir TL. The gut microbiota at the intersection of diet and human health. Sci (New York N.Y.) (2018) 362:776–80. doi: 10.1126/science.aau5812
34. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA (2010) 107:14691–6. doi: 10.1073/pnas.1005963107
35. Jimenez-Duran G, Triantafilou M. Metabolic regulators of enigmatic inflammasomes in autoimmune diseases and crosstalk with innate immune receptors. Immunology (2021) 163:348–62. doi: 10.1111/imm.13326
36. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature (2016) 535:65–74. doi: 10.1038/nature18847
37. Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell (2016) 167:1339–1353.e21. doi: 10.1016/j.cell.2016.10.043
38. Qian XH, Song XX, Liu XL, Chen SD, Tang HD. Inflammatory pathways in Alzheimer's disease mediated by gut microbiota. Ageing Res Rev (2021) 68:101317. doi: 10.1016/j.arr.2021.101317
39. Tang WHW, Li DY, Hazen SL. Dietary metabolism, the gut microbiome, and heart failure. Nat Rev Cardiol (2019) 16:137–54. doi: 10.1038/s41569-018-0108-7
40. Xiang D, Yang J, Liu L, Yu H, Gong X, Liu D. The regulation of tissue-specific farnesoid X receptor on genes and diseases involved in bile acid homeostasis. BioMed Pharmacother (2023) 168:115606. doi: 10.1016/j.biopha.2023.115606
41. Luan Y, Guo Y, Li S, Yu B, Zhu S, Li S, et al. Interleukin-18 among atrial fibrillation patients in the absence of structural heart disease. Europace (2010) 12:1713–8. doi: 10.1093/europace/euq321
42. Wang H, Yan HM, Tang MX, Wang ZH, Zhong M, Zhang Y, et al. Increased serum levels of microvesicles in nonvalvular atrial fibrillation determinated by ELISA using a specific monoclonal antibody AD-1. Clin Chim Acta (2010) 411:1700–4. doi: 10.1016/j.cca.2010.07.005
43. Heijman J, Muna AP, Veleva T, Molina CE, Sutanto H, Tekook M, et al. Atrial myocyte NLRP3/caMKII nexus forms a substrate for postoperative atrial fibrillation. Circ Res (2020) 127:1036–55. doi: 10.1161/circresaha.120.316710
44. Cabaro S, Conte M, Moschetta D, Petraglia L, Valerio V, Romano S, et al. Epicardial adipose tissue-derived IL-1β Triggers postoperative atrial fibrillation. Front Cell Dev Biol (2022) 10:893729. doi: 10.3389/fcell.2022.893729
45. Weymann A, Popov AF, Sabashnikov A, Ali-Hasan-Al-Saegh S, Ryazanov M, Tse G, et al. Baseline and postoperative levels of C-reactive protein and interleukins as inflammatory predictors of atrial fibrillation following cardiac surgery: a systematic review and meta-analysis. Kardiologia polska (2018) 76:440–51. doi: 10.5603/KP.a2017.0242
46. Scott L Jr., Fender AC, Saljic A, Li L, Chen X, Wang X, et al. NLRP3 inflammasome is a key driver of obesity-induced atrial arrhythmias. Cardiovasc Res (2021) 117:1746–59. doi: 10.1093/cvr/cvab024
47. Wu X, Liu Y, Tu D, Liu X, Niu S, Suo Y, et al. Role of NLRP3-inflammasome/caspase-1/galectin-3 pathway on atrial remodeling in diabetic rabbits. J Cardiovasc Transl Res (2020) 13:731–40. doi: 10.1007/s12265-020-09965-8
48. Kong B, Fu H, Xiao Z, Zhou Y, Shuai W, Huang H. Gut microbiota dysbiosis induced by a high-fat diet increases susceptibility to atrial fibrillation. Can J Cardiol (2022) 38:1962–75. doi: 10.1016/j.cjca.2022.08.231
49. Qiu H, Ji C, Liu W, Wu Y, Lu Z, Lin Q, et al. Chronic kidney disease increases atrial fibrillation inducibility: involvement of inflammation, atrial fibrosis, and connexins. Front Physiol (2018) 9:1726. doi: 10.3389/fphys.2018.01726
50. Dudley SC Jr., Hoch NE, McCann LA, Honeycutt C, Diamandopoulos L, Fukai T, et al. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation (2005) 112:1266–73. doi: 10.1161/circulationaha.105.538108
51. Kim YM, Guzik TJ, Zhang YH, Zhang MH, Kattach H, Ratnatunga C, et al. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ Res (2005) 97:629–36. doi: 10.1161/01.Res.0000183735.09871.61
52. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature (2012) 492:123–7. doi: 10.1038/nature11588
53. Ajoolabady A, Nattel S, Lip GYH, Ren J. Inflammasome signaling in atrial fibrillation: JACC state-of-the-art review. J Am Coll Cardiol (2022) 79:2349–66. doi: 10.1016/j.jacc.2022.03.379
54. Yang X, An N, Zhong C, Guan M, Jiang Y, Li X, et al. Enhanced cardiomyocyte reactive oxygen species signaling promotes ibrutinib-induced atrial fibrillation. Redox Biol (2020) 30:101432. doi: 10.1016/j.redox.2020.101432
55. Iozzo P. Myocardial, perivascular, and epicardial fat. Diabetes Care (2011) 34 Suppl 2:S371–9. doi: 10.2337/dc11-s250
56. Abe I, Teshima Y, Kondo H, Kaku H, Kira S, Ikebe Y, et al. Association of fibrotic remodeling and cytokines/chemokines content in epicardial adipose tissue with atrial myocardial fibrosis in patients with atrial fibrillation. Heart Rhythm (2018) 15:1717–27. doi: 10.1016/j.hrthm.2018.06.025
57. Sacks HS, Fain JN, Cheema P, Bahouth SW, Garrett E, Wolf RY, et al. Depot-specific overexpression of proinflammatory, redox, endothelial cell, and angiogenic genes in epicardial fat adjacent to severe stable coronary atherosclerosis. Metab Syndrome Related Disord (2011) 9:433–9. doi: 10.1089/met.2011.0024
58. van der Heijden CAJ, Verheule S, Olsthoorn JR, Mihl C, Poulina L, van Kuijk SMJ, et al. Postoperative atrial fibrillation and atrial epicardial fat: Is there a link? Int J Cardiol Heart Vasc (2022) 39:100976. doi: 10.1016/j.ijcha.2022.100976
59. Chahine Y, Macheret F, Ordovas K, Kim J, Boyle PM, Akoum N. MRI-quantified left atrial epicardial adipose tissue predicts atrial fibrillation recurrence following catheter ablation. Front Cardiovasc Med (2022) 9:1045742. doi: 10.3389/fcvm.2022.1045742
60. Zuo K, Li J, Li K, Hu C, Gao Y, Chen M, et al. Disordered gut microbiota and alterations in metabolic patterns are associated with atrial fibrillation. GigaScience 8 (2019) 8:giz058. doi: 10.1093/gigascience/giz058
61. Palmu J, Börschel CS, Ortega-Alonso A, Markó L, Inouye M, Jousilahti P, et al. Gut microbiome and atrial fibrillation-results from a large population-based study. EBioMedicine (2023) 91:104583. doi: 10.1016/j.ebiom.2023.104583
62. Wang XH, Li Z, Zang MH, Yao TB, Mao JL, Pu J. Circulating primary bile acid is correlated with structural remodeling in atrial fibrillation. J Interv Card Electrophysiol (2020) 57:371–7. doi: 10.1007/s10840-019-00540-z
63. Rainer PP, Primessnig U, Harenkamp S, Doleschal B, Wallner M, Fauler G, et al. Bile acids induce arrhythmias in human atrial myocardium–implications for altered serum bile acid composition in patients with atrial fibrillation. Heart (British Cardiac Society) (2013) 99:1685–92. doi: 10.1136/heartjnl-2013-304163
64. Sanchez-Gimenez R, Ahmed-Khodja W, Molina Y, Peiró OM, Bonet G, Carrasquer A, et al. Gut microbiota-derived metabolites and cardiovascular disease risk: A systematic review of prospective cohort studies. Nutrients (2022) 14:2654. doi: 10.3390/nu14132654
65. Alonso A, Yu B, Qureshi WT, Grams ME, Selvin E, Soliman EZ, et al. Metabolomics and incidence of atrial fibrillation in African Americans: the atherosclerosis risk in communities (ARIC) study. PloS One (2015) 10:e0142610. doi: 10.1371/journal.pone.0142610
66. Blöbaum L, Witkowski M, Wegner M, Lammel S, Schencke PA, Jakobs K, et al. Intestinal barrier dysfunction and microbial translocation in patients with first-diagnosed atrial fibrillation. Biomedicines (2023) 11:176. doi: 10.3390/biomedicines11010176
67. Aoki K, Teshima Y, Kondo H, Saito S, Fukui A, Fukunaga N, et al. Role of indoxyl sulfate as a predisposing factor for atrial fibrillation in renal dysfunction. J Am Heart Assoc (2015) 4:e002023. doi: 10.1161/jaha.115.002023
68. Zuo K, Fang C, Liu Z, Fu Y, Liu Y, Liu L, et al. Commensal microbe-derived SCFA alleviates atrial fibrillation via GPR43/NLRP3 signaling. Int J Biol Sci (2022) 18:4219–32. doi: 10.7150/ijbs.70644
69. Yu L, Meng G, Huang B, Zhou X, Stavrakis S, Wang M, et al. A potential relationship between gut microbes and atrial fibrillation: Trimethylamine N-oxide, a gut microbe-derived metabolite, facilitates the progression of atrial fibrillation. Int J Cardiol (2018) 255:92–8. doi: 10.1016/j.ijcard.2017.11.071
70. Lai S, Hua X, Gao R, Zeng L, Song J, Liu J, et al. Combinational biomarkers for atrial fibrillation derived from atrial appendage and plasma metabolomics analysis. Sci Rep (2018) 8:16930. doi: 10.1038/s41598-018-34930-6
71. Svingen GFT, Zuo H, Ueland PM, Seifert R, Løland KH, Pedersen ER, et al. Increased plasma trimethylamine-N-oxide is associated with incident atrial fibrillation. Int J Cardiol (2018) 267:100–6. doi: 10.1016/j.ijcard.2018.04.128
72. Honda T, Inagawa H. Utility of in vitro cellular models of low-dose lipopolysaccharide in elucidating the mechanisms of anti-inflammatory and wound-healing-promoting effects of lipopolysaccharide administration in vivo. Int J Mol Sci 24 (2023) 24:14387. doi: 10.3390/ijms241814387
73. Moretti J, Blander JM. Increasing complexity of NLRP3 inflammasome regulation. J Leukocyte Biol (2021) 109:561–71. doi: 10.1002/jlb.3mr0520-104rr
74. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes (2007) 56:1761–72. doi: 10.2337/db06-1491
75. Manco M, Putignani L, Bottazzo GF. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocrine Rev (2010) 31:817–44. doi: 10.1210/er.2009-0030
76. Okazaki R, Iwasaki YK, Miyauchi Y, Hirayama Y, Kobayashi Y, Katoh T, et al. lipopolysaccharide induces atrial arrhythmogenesis via down-regulation of L-type Ca2+ channel genes in rats. Int Heart J (2009) 50:353–63. doi: 10.1536/ihj.50.353
77. Hoyano M, Ito M, Kimura S, Tanaka K, Okamura K, Komura S, et al. Inducibility of atrial fibrillation depends not on inflammation but on atrial structural remodeling in rat experimental autoimmune myocarditis. Cardiovasc Pathol Off J Soc Cardiovasc Pathol (2010) 19:e149–57. doi: 10.1016/j.carpath.2009.07.002
78. Xiang J, Zhang Z, Xie H, Zhang C, Bai Y, Cao H, et al. Effect of different bile acids on the intestine through enterohepatic circulation based on FXR. Gut Microbes (2021) 13:1949095. doi: 10.1080/19490976.2021.1949095
79. Gonzalez FJ. Nuclear receptor control of enterohepatic circulation. Compr Physiol (2012) 2:2811–28. doi: 10.1002/cphy.c120007
80. Sah DK, Arjunan A, Park SY, Jung YD. Bile acids and microbes in metabolic disease. World J Gastroenterol (2022) 28:6846–66. doi: 10.3748/wjg.v28.i48.6846
81. Guan B, Tong J, Hao H, Yang Z, Chen K, Xu H, et al. Bile acid coordinates microbiota homeostasis and systemic immunometabolism in cardiometabolic diseases. Acta Pharm Sinica. B (2022) 12:2129–49. doi: 10.1016/j.apsb.2021.12.011
82. Hao H, Cao L, Jiang C, Che Y, Zhang S, Takahashi S, et al. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell Metab (2017) 25:856–867.e5. doi: 10.1016/j.cmet.2017.03.007
83. Liao C, Wang D, Qin S, Zhang Y, Chen J, Xu R, et al. Inflammatory-dependent bidirectional effect of bile acids on NLRP3 inflammasome and its role in ameliorating CPT-11-induced colitis. Front Pharmacol (2022) 13:677738. doi: 10.3389/fphar.2022.677738
84. Leong SC, Sirich TL. Indoxyl sulfate-review of toxicity and therapeutic strategies. Toxins (2016) 8:358. doi: 10.3390/toxins8120358
85. Chin LH, Hsu YJ, Hsu SC, Chen YH, Chang YL, Huang SM, et al. The regulation of NLRP3 inflammasome expression during the development of cardiac contractile dysfunction in chronic kidney disease. Oncotarget (2017) 8:113303–17. doi: 10.18632/oncotarget.22964
86. Chen WT, Chen YC, Hsieh MH, Huang SY, Kao YH, Chen YA, et al. The uremic toxin indoxyl sulfate increases pulmonary vein and atrial arrhythmogenesis. J Cardiovasc Electrophysiol (2015) 26:203–10. doi: 10.1111/jce.12554
87. Yamaguchi K, Yisireyili M, Goto S, Cheng XW, Nakayama T, Matsushita T, et al. Indoxyl sulfate activates NLRP3 inflammasome to induce cardiac contractile dysfunction accompanied by myocardial fibrosis and hypertrophy. Cardiovasc Toxicol (2022) 22:365–77. doi: 10.1007/s12012-021-09718-2
88. Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell (2016) 165:1332–45. doi: 10.1016/j.cell.2016.05.041
89. Peng L, Li ZR, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr (2009) 139:1619–25. doi: 10.3945/jn.109.104638
90. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun (2015) 6:6734. doi: 10.1038/ncomms7734
91. Zhu Y, Shui X, Liang Z, Huang Z, Qi Y, He Y, et al. Gut microbiota metabolites as integral mediators in cardiovascular diseases (Review). Int J Mol Med (2020) 46:936–48. doi: 10.3892/ijmm.2020.4674
92. Liu J, Zhao M, Zhou J, Liu C, Zheng L, Yin Y. Simultaneous targeted analysis of trimethylamine-N-oxide, choline, betaine, and carnitine by high performance liquid chromatography tandem mass spectrometry. J Chromatography. B Analytical Technol Biomed Life Sci (2016) 1035:42–8. doi: 10.1016/j.jchromb.2016.09.026
93. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med (2013) 19:576–85. doi: 10.1038/nm.3145
94. al-Waiz M, Mikov M, Mitchell SC. and R L Smith, The exogenous origin of trimethylamine in the mouse. Metabolism (1992) 41:135–6. doi: 10.1016/0026-0495(92)90140-6
95. Lang DH, Yeung CK, Peter RM, Ibarra C, Gasser R, Itagaki K, et al. Isoform specificity of trimethylamine N-oxygenation by human flavin-containing monooxygenase (FMO) and P450 enzymes: selective catalysis by FMO3. Biochem Pharmacol (1998) 56:1005–12. doi: 10.1016/s0006-2952(98)00218-4
96. Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. New Engl J Med (2013) 368:1575–84. doi: 10.1056/NEJMoa1109400
97. Agus A, Clément K, Sokol H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut (2021) 70:1174–82. doi: 10.1136/gutjnl-2020-323071
98. Boini KM, Hussain T, Li PL, Koka S. Trimethylamine-N-oxide instigates NLRP3 inflammasome activation and endothelial dysfunction. Cell Physiol Biochem Int J Exp Cell Physiology Biochemistry Pharmacol (2017) 44:152–62. doi: 10.1159/000484623
99. Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc (2017) 6:e006347. doi: 10.1161/jaha.117.006347
100. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11:136–40. doi: 10.1038/ni.1831
101. Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell (2010) 40:893–904. doi: 10.1016/j.molcel.2010.12.013
102. Al-Rubaye H, Perfetti G, Kaski JC. The role of microbiota in cardiovascular risk: focus on trimethylamine oxide. Curr Problems Cardiol (2019) 44:182–96. doi: 10.1016/j.cpcardiol.2018.06.005
103. Gatarek P, Kaluzna-Czaplinska J. Trimethylamine N-oxide (TMAO) in human health. EXCLI J (2021) 20:301–19. doi: 10.17179/excli2020-3239
104. Fustinoni O, Saposnik G, Esnaola y Rojas MM, Lakkis SG, Sposato LA. Higher frequency of atrial fibrillation linked to colder seasons and air temperature on the day of ischemic stroke onset. J Stroke Cerebrovascular Dis Off J Natl Stroke Assoc (2013) 22:476–81. doi: 10.1016/j.jstrokecerebrovasdis.2013.03.009
105. Luo Y, Zhang Y, Han X, Yuan Y, Zhou Y, Gao Y, et al. Akkermansia muciniphila prevents cold-related atrial fibrillation in rats by modulation of TMAO induced cardiac pyroptosis. EBioMedicine (2022) 82:104087. doi: 10.1016/j.ebiom.2022.104087
106. Wang Y, Liu X, Shi H, Yu Y, Yu Y, Li M, et al. NLRP3 inflammasome, an immune-inflammatory target in pathogenesis and treatment of cardiovascular diseases. Clin Transl Med (2020) 10:91–106. doi: 10.1002/ctm2.13
107. Xu L, Zhang C, He D, Jiang N, Bai Y, Xin Y. Rapamycin and MCC950 modified gut microbiota in experimental autoimmune encephalomyelitis mouse by brain gut axis. Life Sci (2020) 253:117747. doi: 10.1016/j.lfs.2020.117747
108. Li H, Guan Y, Liang B, Ding P, Hou X, Wei W, et al. Therapeutic potential of MCC950, a specific inhibitor of NLRP3 inflammasome. Eur J Pharmacol (2022) 928:175091. doi: 10.1016/j.ejphar.2022.175091
110. Krisai P, Blum S, Schnabel RB, Sticherling C, Kühne M, von Felten S, et al. Canakinumab after electrical cardioversion in patients with persistent atrial fibrillation: A pilot randomized trial. Circulation. Arrhythmia Electrophysiology (2020) 13:e008197. doi: 10.1161/circep.119.008197
111. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
112. Pappritz K, Lin J, El-Shafeey M, Fechner H, Kühl U, Alogna A, et al. Colchicine prevents disease progression in viral myocarditis via modulating the NLRP3 inflammasome in the cardiosplenic axis. ESC Heart Fail (2022) 9:925–41. doi: 10.1002/ehf2.13845
113. January CT, Wann LS, Alpert JS, Calkins H, Cigarroa JE, Cleveland JC Jr, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation (2014) 130:e199–267. doi: 10.1161/cir.0000000000000041
114. Imazio M, Brucato A, Ferrazzi P, Pullara A, Adler Y, Barosi A, et al. Colchicine for prevention of postpericardiotomy syndrome and postoperative atrial fibrillation: the COPPS-2 randomized clinical trial. Jama (2014) 312:1016–23. doi: 10.1001/jama.2014.11026
115. Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, et al. Colchicine in patients with chronic coronary disease. New Engl J Med (2020) 383:1838–47. doi: 10.1056/NEJMoa2021372
116. Imazio M, Brucato A, Cemin R, Ferrua S, Maggiolini S, Beqaraj F, et al. A randomized trial of colchicine for acute pericarditis. New Engl J Med (2013) 369:1522–8. doi: 10.1056/NEJMoa1208536
117. Zarpelon CS, Netto MC, Jorge JC, Fabris CC, Desengrini D, Jardim Mda S, et al. Colchicine to reduce atrial fibrillation in the postoperative period of myocardial revascularization. Arq Bras Cardiol (2016) 107:4–9. doi: 10.5935/abc.20160082
118. Tabbalat RA, Alhaddad I, Hammoudeh A, Khader YS, Khalaf HA, Obaidat M, et al. Effect of low-dose colchiciNe on the inciDence of atrial fibrillation in open heart surgery patients: END-AF low dose trial. J Int Med Res (2020) 48:300060520939832. doi: 10.1177/0300060520939832
119. Deftereos S, Giannopoulos G, Efremidis M, Kossyvakis C, Katsivas A, Panagopoulou V, et al. Colchicine for prevention of atrial fibrillation recurrence after pulmonary vein isolation: mid-term efficacy and effect on quality of life. Heart Rhythm (2014) 11:620–8. doi: 10.1016/j.hrthm.2014.02.002
120. Singh RK, Chang HW, Yan D, Lee KM, Ucmak D, Wong K, et al. Influence of diet on the gut microbiome and implications for human health. J Transl Med (2017) 15:73. doi: 10.1186/s12967-017-1175-y
121. Claesson MJ, Jeffery IB, Conde S, Power SE, O'Connor EM, Cusack S, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature (2012) 488:178–84. doi: 10.1038/nature11319
122. Makki K, Deehan EC, Walter J, Bäckhed F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe (2018) 23:705–15. doi: 10.1016/j.chom.2018.05.012
123. Koeth RA, Lam-Galvez BR, Kirsop J, Wang Z, Levison BS, Gu X, et al. l-Carnitine in omnivorous diets induces an atherogenic gut microbial pathway in humans. J Clin Invest (2019) 129:373–87. doi: 10.1172/jci94601
124. Ghosh TS, Rampelli S, Jeffery IB, Santoro A, Neto M, Capri M, et al. Mediterranean diet intervention alters the gut microbiome in older people reducing frailty and improving health status: the NU-AGE 1-year dietary intervention across five European countries. Gut (2020) 69:1218–28. doi: 10.1136/gutjnl-2019-319654
125. Neumann FA, Jagemann B, Makarova N, Börschel CS, Aarabi G, Gutmann F, et al. Mediterranean diet and atrial fibrillation: lessons learned from the AFHRI case-control study. Nutrients (2022) 14:3615. doi: 10.3390/nu14173615
126. Oniszczuk A, Oniszczuk T, Gancarz M, Szymańska J. Role of gut microbiota, probiotics and prebiotics in the cardiovascular diseases. Molecules (2021) 26:1172. doi: 10.3390/molecules26041172
127. Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, et al. Salt-responsive gut commensal modulates T(H)17 axis and disease. Nature (2017) 551:585–9. doi: 10.1038/nature24628
128. Chen ML, Yi L, Zhang Y, Zhou X, Ran L, Yang J, et al. Resveratrol attenuates trimethylamine-N-oxide (TMAO)-induced atherosclerosis by regulating TMAO synthesis and bile acid metabolism via remodeling of the gut microbiota. mBio (2016) 7:e02210–15. doi: 10.1128/mBio.02210-15
129. Chen Q, Liu M, Zhang P, Fan S, Huang J, Yu S, et al. Fucoidan and galactooligosaccharides ameliorate high-fat diet-induced dyslipidemia in rats by modulating the gut microbiota and bile acid metabolism. Nutrition (2019) 65:50–9. doi: 10.1016/j.nut.2019.03.001
130. Wang JW, Kuo CH, Kuo FC, Wang YK, Hsu WH, Yu FJ, et al. Fecal microbiota transplantation: Review and update. J Formosan Med Assoc = Taiwan yi zhi (2019) 118(Suppl 1):S23–s31. doi: 10.1016/j.jfma.2018.08.011
Keywords: atrial fibrillation, NLRP3 inflammasome, gut microbiota, mechanism, treatment
Citation: Xing Y, Yan L, Li X, Xu Z, Wu X, Gao H, Chen Y, Ma X, Liu J and Zhang J (2023) The relationship between atrial fibrillation and NLRP3 inflammasome: a gut microbiota perspective. Front. Immunol. 14:1273524. doi: 10.3389/fimmu.2023.1273524
Received: 06 August 2023; Accepted: 06 November 2023;
Published: 21 November 2023.
Edited by:
Gabriel Mbalaviele, Washington University in St. Louis, United StatesReviewed by:
Xiqing Bian, Macau University of Science and Technology, Macao SAR, ChinaCopyright © 2023 Xing, Yan, Li, Xu, Wu, Gao, Chen, Ma, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingchun Zhang, emhhbmdqaW5nY2h1bjI3NkAxMjYuY29t; Jiangang Liu, bGl1amlhbmdhbmcyMDAyQHNpbmEuY29t; Xiaojuan Ma, YWJjX214akBhbGl5dW4uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.