 Iris Lodewijk
Iris Lodewijk Marta Dueñas
Marta Dueñas Jesus M. Paramio
Jesus M. Paramio Carolina Rubio
Carolina Rubio
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 03 October 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1272681
This article is part of the Research Topic Advances of Novel Approaches to Enhance Therapeutic Efficacy and Safety in Human Solid Cold Tumor View all 12 articles
Targeted therapies are the state of the art in oncology today, and every year new Tumor-associated antigens (TAAs) are developed for preclinical research and clinical trials, but few of them really change the therapeutic scenario. Difficulties, either to find antigens that are solely expressed in tumors or the generation of good binders to these antigens, represent a major bottleneck. Specialized cellular mechanisms, such as differential splicing and glycosylation processes, are a good source of neo-antigen expression. Changes in these processes generate surface proteins that, instead of showing decreased or increased antigen expression driven by enhanced mRNA processing, are aberrant in nature and therefore more specific targets to elicit a precise anti-tumor therapy. Here, we present promising TAAs demonstrated to be potential targets for cancer monitoring, targeted therapy and the generation of new immunotherapy tools, such as recombinant antibodies and chimeric antigen receptor (CAR) T cell (CAR-T) or Chimeric Antigen Receptor-Engineered Natural Killer (CAR-NK) for specific tumor killing, in a wide variety of tumor types. Specifically, this review is a detailed update on TAAs CD44v6, STn and O-GD2, describing their origin as well as their current and potential use as disease biomarker and therapeutic target in a diversity of tumor types.
Immunotherapy, cell therapies, and vaccines are areas of active research and development aimed at harnessing the body’s immune system to fight diseases including cancer and infectious diseases. Antigens play a crucial role in these therapies as they elicit an immune response which leads to the activation and targeting of immune cells against specific disease-associated targets.
Tumor-associated antigens (TAAs) are structures expressed by tumor cells that are recognized by the immune system as foreign (1). These antigens can be used as targets for cancer immunotherapies such as adoptive cell transfer (ACT) or cancer vaccines (2, 3). With the growth of technological development and new research techniques every year new TAAs are being developed in order to reach preclinical research and clinical trials, but few of them actually change the therapeutic landscape. Difficulties either to find antigens that are solely expressed in tumors or the generation of good antibodies or specific molecules that recognize these TAAs represent a major bottleneck.
Basic research in specialized cellular machineries such as post-translational modifications (PTMs) and differential splicing is a good source of neo-antigen expression (4–6). Post-translational modifications (PTMs), including phosphorylation, methylation, glycosylation, sialylation and others, introduce structural chemical changes to existing proteins. Even though these proteins are self-proteins, the attachment of an additional glycan or sialyl-group transforms them into foreign antigens. PTMs occurring on tumor cells can therefore give rise to TAAs. Regarding differential splicing, the generation of TAAs can arise from an alternative splicing process during gene expression by which particular exons of a gene are either included or excluded from the final mRNA (7).
Changes in these processes generate surface proteins that, instead of increasing or decreasing protein antigens, as happens with EGFR or other surface proteins driven by increased mRNA processing, produce surface molecules that are aberrant in nature and therefore more specific as targets to elicit a specific anti-tumor therapy. Overall, ongoing research and advancements in understanding the immune system, genomics, and protein engineering are continually expanding the range of potential antigens that can be targeted for immunotherapy, cell therapies, and vaccine development.
It is worth noting that the development and approval of new antigens for immunotherapy, cell therapies, and vaccines involve a complex and rigorous process of preclinical and clinical trials to assess safety and efficacy. Regulatory agencies such as the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) play a critical role in evaluating and approving these therapies and vaccines (8).
In this review, we introduce a selection of promising TAAs that have shown potential as targets for cancer monitoring, targeted therapy, and the development of new immunotherapy tools. These tools include recombinant antibodies and CAR-T or CAR-NK cells, which can be utilized for precise tumor eradication in various types of tumors. This comprehensive review focuses on the latest information about three specific TAAs: CD44v6, STn, and O-GD2 the first is a variant generated from differential splicing of the well-known CD44 surface marker and the other two are very good examples of cancer associated antigens generated by aberrant glycosylation patterns such as sialylation and O-glycosylation. The review covers their origins, as well as their current and potential applications as disease biomarkers and therapeutic targets across a diverse range of tumor types.
The role of the CD44 adhesion protein family in neoplastic transformation and invasive potential of carcinomas has been widely considered (9). CD44 represents a complex transmembrane glycoprotein encoded by a single gene on the short arm of chromosome 11 (Figure 1, upper). In humans, the CD44 encoding gene consists of 20 exons that give rise to a wide variety of multifunctional glycoproteins due to alternative splicing processes. The standard form, known as CD44s glycopeptide, represents the smallest and most abundant CD44 structure, comprised of exons 1-5 and 16-20. The constant region of the variable isoforms (CD44v) is represented by the same exons, while alternative splicing of the remaining exons (exons 6-15 or CD44v1-10) gives rise to the different CD44v isoforms. It is of note that isoform CD44v1 has not been detected in humans due to the presence of a stop codon in exon 6 (also known as exon v1) (10). The different isoforms of CD44 generated by alternative splicing can act as TAAs and have been implicated in tumor progression and immune evasion. Some CD44 isoforms, such as CD44v6 and CD44v9, have been found to be frequently expressed in many types of cancer, including pancreatic (11, 12), colon (13, 14), and prostate cancer (15, 16). These isoforms are associated with increased invasiveness, metastasis, and resistance to apoptosis.
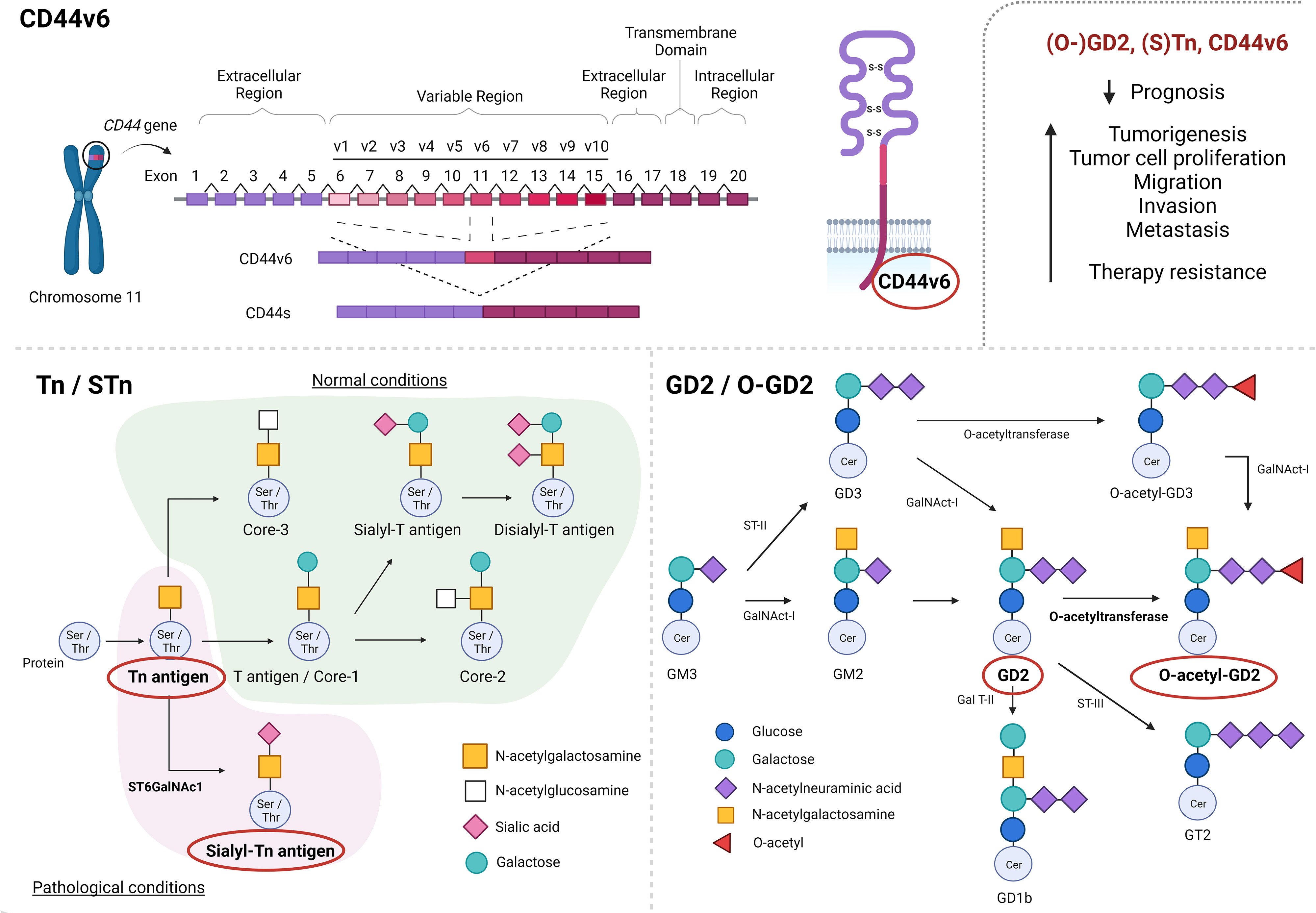
Figure 1 Schematic representation of CD44-alternative splicing, giving rise to CD44 standard form (CD44s) and CD44 isoforms (CD44v1-10), as well as alternative glycosylation processes, generating O-glycans sialyl-Tn (STn) and O-acetyl-GD2 (O-GD2). CD44v6, (S)Tn and (O-)GD2 have been described to negatively impact prognosis as well as to promote tumorigenesis, tumor progression and therapy resistance. Created with BioRender.com.
Alterations of CD44 glycoprotein expression have been shown to play an important role in the progression of various malignancies (17). Specifically, overexpression of the CD44v6 variant has been found in the majority of squamous cell carcinomas and a variety of adenocarcinomas, but has not frequently been observed in non-epithelial tumors (18). In this way, CD44v6 has been described to negatively impact the prognosis of patients with multiple myeloma (19), colorectal cancer (CRC) (20, 21), osteosarcoma (22), esophageal carcinoma (22), gastric cancer (23) as well as head and neck squamous cell carcinoma (HNSCC) (24, 25) patients, among others. Moreover, altered CD44v6 expression has been associated with tumor development, migration, invasion and metastatic potential in a broad variety of tumor types, such as HNSCC (24), oral cancer (26), laryngeal carcinoma (27), esophageal squamous cell carcinoma, gastric cancer (28–30), pancreatic cancer (11, 31), liver cancer (32, 33), CRC (34), lung cancer (35, 36), breast carcinoma (37) and gynecologic malignancies (38) such as ovarian (39, 40) and prostate cancer (41), among others (Figure 1).
Circulating tumor cells (CTCs) highly expressing CD44v6 were found in blood samples from metastatic CRC patients (34, 42). Nicolazzo et al. (2020) (43) described treatment failure in those patients with CD44v6-positive CTCs undergoing first-line chemotherapy and proposed that the presence of those cells might be a valuable predictive biomarker of therapy resistance. Indeed, many studies have associated CD44v6 expression with chemoresistance. For example, Gaggianesi et al. (2022) (44) demonstrated that tumor microenvironmental cytokines promote CD44v6 expression in CRC stem cells, conferring resistance to standard anti-tumor therapeutic options, whereas Wang et al. (2019) (45) found that the downregulation of CD44v6 augments chemosensitivity of CRC cells in vitro. CD44v6 has been described to enhance CRC therapy resistance by a variety of mechanisms, including activation of the PI3K/Akt/mTOR pathway leading to increased multidrug resistance, up-regulation of the MAPK/ERK pathway stimulating autophagosome formation, and blocking of the Fas-FasL interaction preventing cell apoptosis. Pereira et al. (2020) (46) found similar results, showing that CD44v6+ gastric cancer cells were more resistant to chemotherapy treatment compared to the CD44v6 cells. Even though the exact mechanism in gastric cancer remains unknown, they proposed that therapy resistance might be the result of concomitant CD44v6 expression and either STAT3 or P38 activation, depending on the cellular context. Wang et al. (2021) (45) proposed the involvement of CD44v6 expression in cisplatin resistance in ovarian cancer cells due to the interaction of CD44v6 with P-glycoprotein (P-gp) and, therefore, the acquisition of multidrug resistance. Sagawa et al. (2016) (47) found a correlation between CD44v6 expression and chemoradiotherapy resistance in nasopharyngeal carcinoma, and prostate cancer chemosensitivity was associated with CD44v6 by Ni et al. (2020) (41). Both studies indicated the activation of the PI3K/Akt/mTOR pathway as the major player in the acquisition of therapy resistance. Specifically, in the case of prostate cancer, the interaction between CD44v6 and hyaluronic acid, a component of the extracellular matrix of epithelial and connective tissues, could augment multidrug resistance through up-regulation of the PI3K/Akt/mTOR pathway.
Even though some studies hypothesized the potential role of the majority of CD44 isoforms, such as CD44v3-5 and CD44v8-10, in tumorigenesis and cancer progression, none has been as extensively described as CD44v6 (48). The extensive description of all these different isoforms and their potential implication in tumor development and/or disease progression goes beyond the scope of this review. Beyond alternative splicing process giving rise to different CD44 isoforms, extensive posttranslational modifications, such as N- and O-glycosylation processes as well as the addition of glycosaminoglycans add another layer of complexity to the CD44 transmembrane glycoprotein structure and function (49).
Altered glycosylation processes are known to be widely involved in tumorigenesis and cancer progression (50). In early cancer stages, the normal synthesis of glycans present in normal epithelial tissues is often impaired due to altered expression of glycosyltransferases, which causes the biosynthesis of truncated carbohydrate structures (such as Tn and STn) which can be observed in a variety of tumor types (49). In the same way, altered ganglioside expression (like GD2 and O-GD2 expression) due to modified glycosyl- and sialyltransferase expression has been found in different cancers such as melanomas and neuroblastomas (51). The role of Tn/STn and GD2/O-GD2 expression in tumorigenesis and cancer progression will be discussed below (Figure 1, lower).
Truncated O-glycan Tn (GalNAcα1-Ser/Thr) and its sialylated variant sialyl-Tn (STn) (Neu5Acα2,6GalNAcα1-Ser/Thr) represent two modifications observed in a variety of proteins which give rise to two tumor-associated antigens that are not expressed in normal cells. Even though O-glycan Tn formation does occur in normal tissue, the Tn glycopeptide represents an immature structure which is normally elongated into other structures by additional glycosyltransferases. Furthermore, altered expression of ST6GalNAc1 glycosyltransferase modifies the Tn glycopeptide by the addition of a sialyl-acid, resulting in STn glycopeptide formation (Figure 1). Therefore, the presence of both O-glycan Tn and STn are described to be restricted to pathological conditions, including tumorigenesis (49).
The structural simplicity and biological complexity of the Tn antigen have been extensively reviewed by Ju et al. (2011) (52). Even though the exact role of the Tn antigen in tumor development and progression is only starting to be unraveled, the presence of the Tn antigen on tumor cells was observed as early as 1969 (53). The first correlation between the Tn antigen and cancer was made by Springer et al., who described expression of the Tn antigen in 90% of breast carcinomas. Subsequently, several studies have reported altered levels of the Tn antigen in many tumor types, such as lung, gastric, CRC, bladder, cervical, prostate and ovarian tumors (54–56). Additionally, altered expression of the Tn antigen has been associated with tumor progression, migratory capacity and metastasis as well as poor prognosis in a wide variety of cancer types, including lung cancer (57), pancreatic cancer (58), CRC (59, 60), breast carcinomas and cervical cancer (59, 61).
Similar to the Tn antigen, the presence of the STn antigen has been reported in several tumor types, such as lung cancer, gastric cancer, pancreatic cancer, CRC, breast carcinomas, cervical cancer, prostate and ovarian cancer (62–64). Nevertheless, STn expression in tumors has been described to be rather heterogeneous, with STn-positive cells ranging from 5% to 100% independent of tumor type origin (64). A growing body of evidence also suggests that the role of STn in tumorigenesis might be cancer type-specific and/or organ-specific as STn can be carried by different glycoproteins. Increased expression of STn was associated with cell proliferation and metastasis in gastric, breast and pancreatic cancer (65). In prostate cancer, however, Munkley et al. (2015) (66) described that STn expression is likely to promote cancer cell dissemination and invasion, and this expression is up-regulated in primary prostate carcinoma. Concordantly, Davidson et al. (2000) (67, 68) reported higher levels of STn-positive ovarian cancer cells at the invasive site of primary tumors than in metastatic lesions, and Ferreira et al. (2013) (69) reported increased migratory and invasive capacity of bladder cancer cells upon STn expression. The current hypothesis behind these observations suggests that the STn antigen inhibits tumor formation by reduction of cell-cell and cell-matrix interactions, which facilitates tumor cell spreading. Even though the STn antigen does not seem to provide cell adhesive properties needed for the formation of metastatic lesions, these specific adhesive characteristics are essential for extravasation and invasive capacity. Accordingly, a transient role for STn in cancer progression has been proposed.
Recently, STn expression has also been associated with tumor-microenvironment interactions. Interestingly, a protective role for glycan STn with regard to tumor cell recognition and degradation has been described (70). Either by receptor masking or the inhibition of cytolytic activity, STn has been reported to play an essential role in immunosuppression (71, 72). In bladder cancer, STn expression has been suggested to induce a tolerogenic phenotype in innate and adaptive immune cells. However, extensive studies in a variety of cancer types will be needed to further unravel the role of STn in cancer progression, invasion and metastasis (73).
The STn antigen can be detected in serum when considerable tumor size is reached, either due to O-glycoprotein secretion from tumors or by its expression on CTCs. Therefore, the presence of the STn antigen in serum is thought to be correlated with advanced cancer and, thus, poor prognosis. Indeed, Carvalho et al. (2020) (74) recently reported an association between STn expression and advanced bladder cancer stage and grade. However, the correlation between the STn antigen and prognosis seems to be rather ambiguous and cancer type-specific (64). STn expression was associated with poor prognosis in ovarian cancer (75, 76), but has not been associated with overall survival in either lung (77) or cervical cancer (68). Additionally, contradictory findings have been published for several tumor types, such as breast cancer, esophageal cancer, gastric cancer and CRC, reflecting the potential cancer subtype-specific role of the STn antigen in tumorigenesis and cancer progression (64).
As mentioned above, STn can be carried by different glycoproteins and glycosylation processes add another layer of complexity to the CD44 glycoprotein. With these observations in mind, CD44 has indeed been reported as carrier protein for STn in gastric and colon cancer. Campos et al. (2015) (78) described the presence of glycan STn on CD44 glycoprotein which leads to altered CD44 molecular features such as molecular weight and antibody recognition in gastric cancer. Mereiter et al. (2019) (79) also reported activation of the receptor tyrosine kinase RON due to increased colocalization of CD44v6 with this receptor in the presence of STn, leading to enhanced hyaluronan binding capacity. Additionally, improved CD44v9 detection by the expression of immature O-glycan structures, such as STn, has recently been proposed by Moreira et al. (2020) (80). Finally, Singh et al. (2001) (81) showed the presence of the STn antigen on CD44 splice variants in CRC, further emphasizing the abovementioned interaction between STn and CD44. Taken together, it might be clear that not only STn or CD44 (v) expression, but also their combination might be very useful with regard to biomarker detection as well as therapeutic targeting.
Even though the exact biological mechanisms remain to be elucidated, altered activity of glycosyl- and sialyltransferases seems to be mainly responsible for the modification of ganglioside expression in tumorigenesis (51). For example, increased N-acetylgalactosaminyltransferase I (GM2/GD2 synthase) expression has been found to provoke enhanced GD2 ganglioside levels in melanoma and neuroblastomas (82). Additionally, even though high expression levels of GD2 have been associated with reduced apoptosis as well as enhanced tumor cell proliferation, adhesion, angiogenesis, migration and invasion capacity in a variety of tumor types such as breast cancer (83, 84), bladder cancer (85), lung cancer (86), osteosarcoma (87), Ewing sarcoma (88), retinoblastoma (89) and brain tumors (90), some expression of this ganglioside has been also observed in some normal tissue in healthy adults, like the central nervous system and peripheral nerves (90), Even so, this ganglioside has been used as a biomarker of cancer in serum samples (91) associated with advanced disease and poor prognosis in neuroblastoma by (92), and (85) in high-grade bladder cancer compared to low-grade disease (84). Also, a correlation between GD2 expression and malignant phenotypes of lung cancer has been described by Yoshida et al. (2002) (86) and Esaki et al. (2020) (93).
Even though GD2 expression might be a valuable prognostic marker and biomarker, its expression on healthy cells complicates its potential as therapeutic target (detailed below). Therefore, interest has currently focused on O-acetyl-GD2 (O-GD2), which is formed by the addition of an O-acetyl ester to the GD2 backbone by 9 (7)-O-Acetyl transferase (Figure 1, lower right). Fleurence et al. (2017) (51) and Cavdarli et al. (2019) (91) have extensively modeled the complex biosynthetic processes giving rise to the different members of the O-acetylated ganglioside family. Interestingly, O-GD2 has been found to be coexpressed with GD2 on tumor cells. Indeed, the presence of O-GD2 in GD2 positive tumors, such as lung carcinoma, melanoma, osteocarcoma, brain tumors and neuroblastoma, has been confirmed by various studies (94, 95), whereas no expression of O-GD2 was observed in either peripheral nerves or a large variety of other healthy tissues (96).
Even though these findings strongly support O-GD2’s potential as valuable biomarker and therapeutic target, its regulatory mechanisms of expression as well as its role in tumorigenesis remain largely unknown due to the complexity of studying this antigen within the extensive biosynthesis network of the whole ganglioside family. Fleurence et al. (2017) (51) suggested that O-GD2 expression in a cell type depends on the balance and activity of at least 4 different enzymes involved in the biosynthetic model of the ganglioside family.
Due to its expression pattern, CD44v6 has been considered an attractive target for antibody-based cancer therapy (17). Thus far, a variety of recombinant antibodies and antibody-drug conjugates have been evaluated in different phase I clinical trials, mainly for their use in HNSCC treatment (96–104). In addition, CD44v6-directed CAR-T cell therapies have shown promising results in preclinical studies, which has led to a phase I/IIa trial in AML and MM patients to study efficacy, safety and feasibility of CD44v6-directed CAR-T cell therapy (105). Even though CAR-T cell therapy for hematological cancers has been very successful, its use for the treatment of solid cancers remains very challenging. Therefore, a variety of studies has evaluated the potential of CD44v6-directed CAR-T cell therapy in solid CD44v6-expressing tumors (105–108). Recently, CD44v6-directed CAR-cytokine induced killer cells (CIK) have demonstrated anti-tumor activity in preclinical studies among different cancer types such as high grade soft tissue sarcomas where the anti-sarcoma activity of CD44v6-CAR-CIK bipotential killers was confirmed in a STS xenograft model in which killing activity was significantly higher compared with unmodified CIK, especially at low effector/target (E/T) ratios: 98% vs 82% (E/T = 10:1) and 68% vs 26% (1:4), (p<0.0001) (109) (Figure 2). An extensive overview on the development and potential of the abovementioned immunotherapy strategies can be found below.
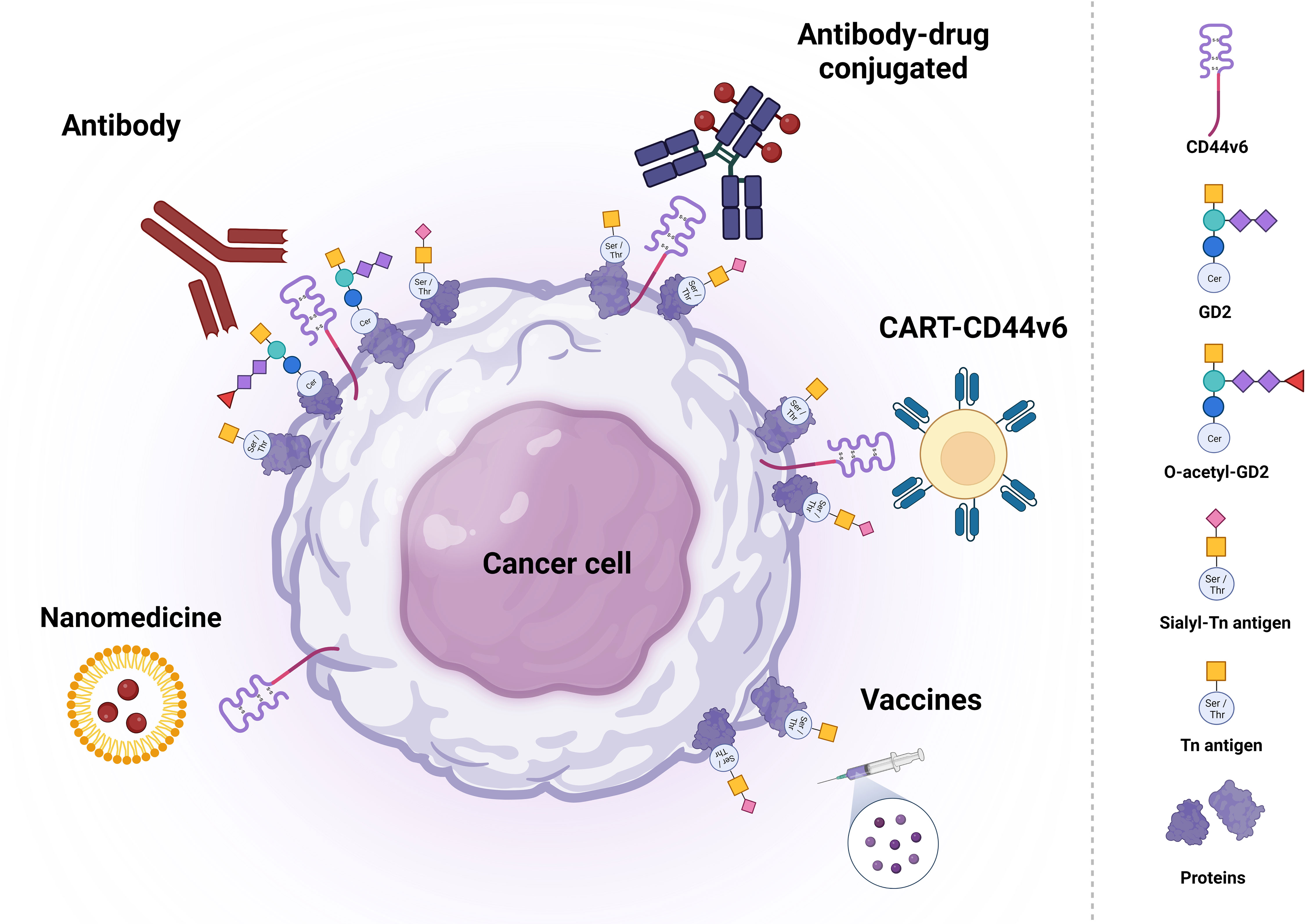
Figure 2 Schematic representation of potential therapeutic strategies based on CD44v6, (S)Tn and (O-)GD2 targeting. Each type of therapy is grouped with different antigens. Created with BioRender.com.
The first studies into the potential of anti-CD44v6 antibody-based immunotherapy were performed almost 30 years ago, when anti-CD44v6 antibodies VFF18 and U36 were characterized as candidates for immunotherapy in squamous cell carcinoma (97–99).
Heider et al. (1996) (97) showed essential therapeutic features for radiolabeled the anti-CD44v6 VFF18 antibody in an in vivo model of human epidermoid squamous cell carcinoma. Additionally, a first clinical study was performed by Bree et al. (1995) (98), who demonstrated high and selective tumor uptake for the radiolabeled 99mTc-labeled anti-CD44v6 U36 antibody in HNSCC patients. In agreement with the previous finding, Van Hal et al. (1996) (99) demonstrated that the CD44v6 antigen targeted by anti-U36 is an appropriate target for the treatment of head-and-neck carcinoma by the use of radiolabeled anti-CD44v6 U36 antibody. The biodistribution studies in vitro, after which a first phase I clinical trial with 186RE-labeled chimeric antibody U36 among patients with HNSCC showed excellent tumor targeting (100). Even though stable disease as well as reduction in tumor size were observed and the antibody was found to be well-tolerated, myelotoxicity represented a dose-limiting factor. Preclinical characterization of two other radiolabeled anti-CD44v6 antibodies, 111In- and 177Lu-labeled U36 antibodies, using HNSCC in vitro and in vivo models has also been described as showing a tumor uptake of 72 h.p.i which makes it interesting for therapies (110, 111) cancer detection tool (Table 1).
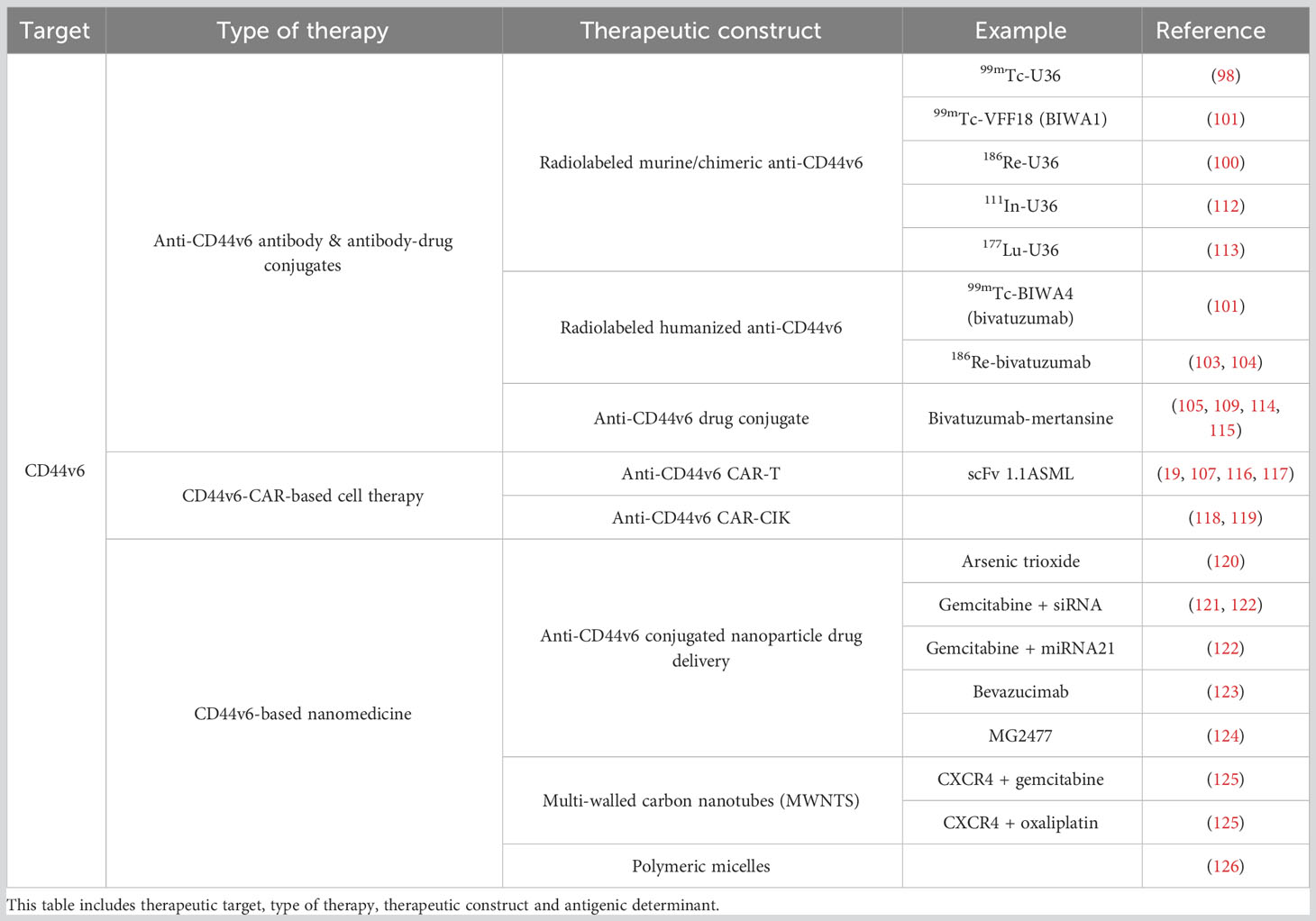
Table 1 Overview of the CD44v6 directed therapies described in this study.
Another phase I clinical study reported high tumor uptake and well-tolerated administration of the murine 99mTc-labeled anti-CD44v6 antibody BIWA1 (initially called VFF18) in patients with HNSCC (101) showing a tumor intake with a mean value of 14.2+/-8.4% of the injected dose/kg tumor tissue and a mean tumor:blood ratio of 2.0+/-1.4 at 40 h after injection with a mean biological half-life in blood (34.5+/-6.1 h). With the aim of clinical application, a humanized 99mTc-labeled anti-CD44v6 antibody BIWA4 antibody, known as bivatuzumab, was then developed, and a phase I clinical trial with HNSCC patients showed selective and high tumor uptake by radioimmunoscintigraphy showing targeting of primary tumors and lymph node metastases in 8 of 10 and 1 of 5 patients, respectively. The highest tumor uptake and tumor to nontumor ratios was observed for the 50-mg dose group with a tumor intake of 26.2 +/- 3.1% of bivatuzumab, without evidence of safety concerns (102). At the same time, phase I clinical studies were performed with HNSCC patients and patients with recurrent or metastatic HNSCC receiving humanized 186Re-labeled bivatuzumab, which showed anti-tumor activity and high tumor to non-tumor targeting ratio (103, 104). Several parallel phase I clinical trials in patients with HNSSC, esophageal carcinoma and metastatic breast cancer were also started with antibody-drug conjugate Bivatuzumab mertansine (105, 109, 114, 115), but the clinical development of this drug was discontinued after a lethal outcome due to toxic epidermal necrolysis in one of the trials. Nevertheless, in 2012, Gurtner et al. (127) reported improved local tumor control at acceptable toxicity levels in an in vivo model using lower doses of Bivatuzumab mertansine (the maytansine derivative DM1 as cytotoxic agent) in combination with fractioned irradiation (Table 1).
Other therapeutic strategies based on anti-CD44v6 antibody constructions, which have recently generated interest, include the use of chimeric antigen receptor (CAR) T cell approaches. In 1996, Hekele et al. (110) reported tumor growth suppression in a rat pancreatic adenocarcinoma in vivo model when receiving genetically manipulated cytotoxic T lymphocytes (CTLs), expressing the scFv of rat-specific anti-CD44v6 antibody 1.1ASML. The same 1.1ASML antibody was used for the construction of a bispecific F(ab9)2 antibody conjugate (BAC) which recognizes both the CD44v6 tumor cell moiety and the complement receptor CR3 on macrophages in order to redirect and induce efficient macrophage-mediated tumor cytotoxicity. However, in vitro and in vivo models of the abovementioned pancreatic carcinoma showed lower anti-tumor activity for the BAC compared to the naked anti-CD44v6 1.1ASML antibody (111). So far, promising results regarding CAR-T cell mediated anti-tumor activity have been described for several cancer types such as leukemia (19), multiple myeloma (19, 116), HNSCC (117), lung adenocarcinoma (107) and ovarian cancer (107). In in vivo models of acute myeloid leukemia (AML) and multiple myeloma (MM), CD44v6 CAR-T cells were found to provide significant anti-tumor activity without affecting either CD44v6-expressing keratinocytes or hematopoietic stem cells (19). A phase I/IIa clinical trial has subsequently been performed to study safety, efficacy and feasibility of CD44v6 CAR-T cell immunotherapy in AML and MM patients mediating a potent antitumor effects against primary AML and MM while sparing normal hematopoietic stem cells and CD44v6-expressing keratinocytes (105) mediating a potent antitumor effects against primary AML and MM while sparing normal hematopoietic stem cells and CD44v6-expressing keratinocytes (105) mediating a potent antitumor effects against primary AML and MM while sparing normal hematopoietic stem cells and CD44v6-expressing keratinocytes (105) mediating a potent antitumor effects against primary AML and MM while sparing normal hematopoietic stem cells and CD44v6-expressing keratinocytes (105). Additionally, Porcellini et al. (2020) (107) showed CD44v6 CAR-T cell infiltration and proliferation at the tumor site and tumor growth inhibition, leading to enhanced overall survival in xenograft mouse models of ovarian and lung adenocarcinoma with more than 30% of CD44v6-CAR T treated mice still alive after 2 months of observation (106, 117). T cells expressing CD44v6 CAR have been shown to be effective against lung and ovarian adenocarcinomas in mice (107), urothelial carcinoma (106) and pancreatic adenocarcinoma (108). Recently, studies have also focused on optimization of the CAR structure in order to improve CD44v6 CAR-T cell functionality (105) and generating not just CAR-T but CAR-NK cells, which in comparison to CAR-T are proposed to show less cytokine release syndrome associated problems (105) and generating not just CAR-T but CAR-NK cells, which in comparison to CAR-T are proposed to show less cytokine release syndrome associated problems (105) and generating not just CAR-T but CAR-NK cells, which in comparison to CAR-T are proposed to show less cytokine release syndrome associated problemsRaferty et al. (108) demonstrated the efficacy of CD44v6-CAR-NKs in triple negative breast cancer model demonstrating cytotoxic function in both 2D and 3D models of triple-negative breast cancer (108). Nearby to CAR-T and CAR-NK cells, CD44v6-directed CAR-cytokine induced killer cells (CIK) have demonstrated anti-tumor activity in in vitro and in vivo models of high-grade soft tissue sarcomas such as undifferentiated pleomorphic sarcoma, liposarcoma, fibrosarcoma, leiomyosarcoma and gastrointestinal stromal tumor, among others (118). Moreover, a recent preclinical study proposed the use of CAR-CIK in leukemia patients due to intrinsic anti-tumor activity as well as enhanced safety (119).
Finally, CD44v6-targeting nanomedicine has been explored as a promising tool for cancer therapy. Qian et al. (2013) described the use of anti-CD44v6 conjugated nanoparticles carrying the anti-cancer agent Arsenic trioxide in a pancreatic cancer mouse model and its successful accumulation at the tumor site, followed by tumor growth control (120). The same animal model was used for the evaluation of nanoparticle-mediated co-delivery of a chemotherapeutic agent and target genes, either Gemcitabine and small-interfering RNA or Gemcitabine and microRNA-21 (121, 122). Both strategies confirmed CD44v6-directed tumor cell targeting and they were found to provide efficient inhibitory effects on tumor growth and metastasis. Kennedy et al. (2018) (128) reported the specificity and stability of anti-CD44v6 Fab-conjugated poly(lactic-co-glycolic acid (PLGA) nanoparticles in vivo in a gastric cancer model, emphasizing its potential as a carrier for cancer therapy in solid tumors, and Baião et al. (2020) (123) described the application of anti-CD44v6 PLGA-PEGylated nanoparticles for bevacizumab delivery in a colorectal cancer model demonstrating intracellular levels of bevacizumab significantly higher in cells incubated with v6 Fab-PLGA-PEG NPs and these nanoparticles resulted in a significant decrease in the intracellular VEGF compared to untargeted nanoparticles and free bevacizumab. In addition, the use of anti-CD44v6 conjugated PEGylated, organic-modified silica (ORMOSIL) nanoparticles carrying the anti-cancer agent 3N-cyclopropylmethyl-7-phenyl-pyrrolo-quinolinone (MG2477) has been shown to provide significantly increased cytotoxic activity towards CD44v6-expressing cells demonstrating that nanoparticles conjugated with a smaller amount of targeting agent being more effective than the ones conjugated with a larger amount of antibody, an effect that will probably be dependent of the affinity of the antigen-antibody interaction (123) demonstrating that nanoparticles conjugated with a smaller amount of targeting agent being more effective than the ones conjugated with a larger amount of antibody, an effect that will probably be dependent of the affinity of the antigen-antibody interaction (123) demonstrating that nanoparticles conjugated with a smaller amount of targeting agent being more effective than the ones conjugated with a larger amount of antibody, an effect that will probably be dependent of the affinity of the antigen-antibody interaction (123) demonstrating that nanoparticles conjugated with a smaller amount of targeting agent being more effective than the ones conjugated with a larger amount of antibody, an effect that will probably be dependent of the affinity of the antigen-antibody interaction (123). Finally, multi-walled carbon nanotubes (MWNTS) and polymeric micelles have been considered as vectors for drug delivery in cancer therapy. Recently, Yin and Qian (2021) (125) showed enhanced anti-tumor activity after treatment with anti-CD44v6 MWNTS, carrying CXCR4 and either gemcitabine or oxaliplatin, in in vitro and in vivo models of ovarian cancer, whereas Andrade et al. (2021) (126) described the efficacy of anti-CD44v6 polymeric micelles as an anticancer agent carrier (Table 1).
Although the above experimental evidences require further proof, the targeted nanoparticle development present clinical potential and provide a launching point for future improvements and therapeutic and/or diagnostic opportunities.
Based on the restricted expression of O-glycans Tn and STn among human carcinomas, these antigens represent an excellent feature for targeted therapy. As for CD44v6, several recombinant antibodies and antibody-drug conjugates have been evaluated in a variety of phase I/II clinical studies for their anti-tumor efficacy in different tumor types (129–133). Additionally, STn-based CAR-T cell therapy has shown promising results in preclinical studies and, subsequently, reached phase I clinical trials for the treatment of solid tumors (134–136). Nevertheless, the best characterized STn-targeting therapy remains a STn-based vaccine, which has been extensively evaluated in phase III clinical trials (51) (Figure 2). An extensive overview on the development and potential of the abovementioned immunotherapy strategies can be found below.
Many Tn-recognizing antibodies have been generated over the last 35 years. The Tn antigen seems to represent an easy antigenic determinant due to its simple chemical structure (GalNAcα1-Ser/Thr). However, even though Trabbic et al. (2018) (137) published the recognition of a single Tn residue by the anti-Tn Kt-IgM-8 antibody, a single Tn determinant has often been found not be enough for anti-Tn antibody function (138). Accordingly, either the presence of a peptide backbone or multiple consecutive Tn residues has been found to be essential for a wide variety of anti-Tn antibodies. Reis et al. (1998) (89) reported that the PMH1 antibody requires an additional MUC2 apomucin peptide chain, whereas the Tn-MUC1 complex was found to represent the antigenic determinant for several anti-Tn antibodies, including SM3 (139), PankoMab (140) and 5E5 (141). Furthermore, multiple anti-Tn antibodies, such as MLS128 (142), 83D4 (143), KM3413 (144) and Ca3638 (145), have been described to require at least two consecutive Tn residues. Finally, the importance of the Tn backbone composition has been described for some antibodies. For example, Mazal et al. (2013) (146) demonstrated specific recognition of a tri-Serine Tn backbone by the 15G9 antibody (Table 2).
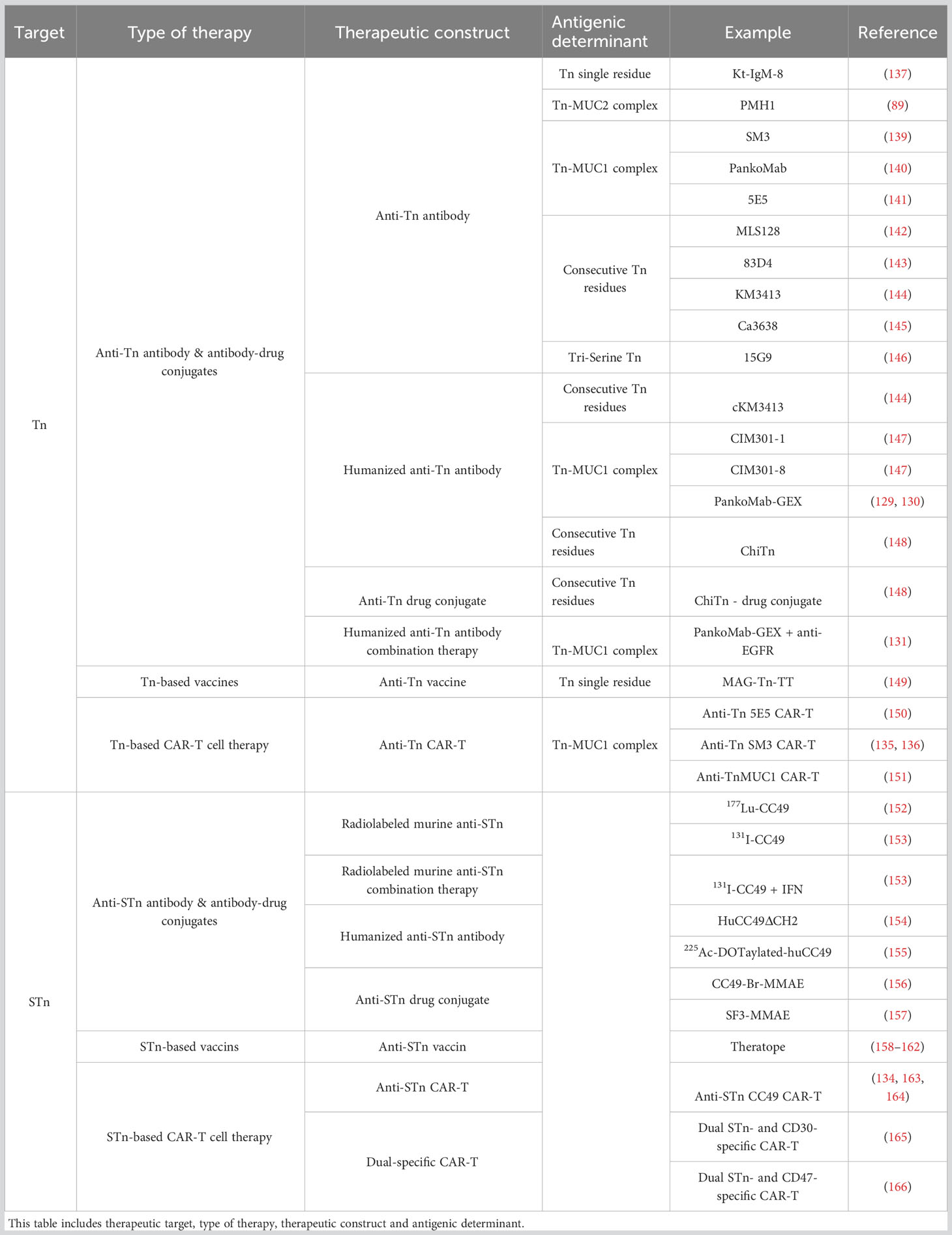
Table 2 Overview of the (S)Tn directed therapies described in this study.
Some of the abovementioned mouse anti-Tn antibodies have been converted into chimeric or humanized antibodies for therapeutic use in the clinic. The mouse-human chimeric antibody, cKM3413, was found to induce ADCC and direct killing activity, which was accompanied by increased survival in vivo (144). Additionally, the humanized 5E5 antibodies CIM301-1 and CIM301-8 were reported to enhance NK cell activation and cytotoxicity in vitro (147), and humanized PankoMab-GEX was described to be safe, well-tolerated and exhibit promising anti-tumor activity in advanced disease in ovarian cancers, in a phase I clinical trial (129, 130). In 60 evaluable patients with ovarian cancer the clinical benefit included one complete response in a patient treated 483 d and confirmed disease stabilization in 19 patients lasting a median (range) of 23 (10–102) weeks (126). Additionally, the combination of PankoMab-GEX with an anti-EGFR antibody was evaluated in a phase I clinical study and showed anti-tumor activity in lung cancer and CRC patients (131). From this clinical trial were 2 and 4 RECIST partial responses in the first and second part of the study, all in CRC patients. There were 2 responses in each subgroup and the duration of best response was 7.2 months. The PFS for NSCLC was 5.3 months and 2 heavily pretreated patients achieved a prolonged control of disease of 10.6 and 9.4 months. Sedlik et al. (147) has used another mouse-human chimeric antibody, ChiTn, derived from the mouse anti-83D4 antibody, as an ADC for cancer treatment. The ChiTn antibody selectively accumulated in the solid tumor, but not in healthy tissue. When conjugated to saporin (SAP) or to auristatin F, the Chi-Tn ADC exhibited effective cytotoxicity to Tn-positive tumor cells in vitro and conjugated to MMAF also induced a delay of tumor growth in vivo, validating for the first time the use of an anti-Tn antibody as an effective ADC (148).
The anti-STn antibody CC49 has shown promising. Radiolabeled 177Lu-CC49 was initially demonstrated to provide significant anti-tumor activity in vivo (152), which led to its evaluation in phase I and phase I/II radioimmunotherapy clinical trials (132, 133). Here, the 177Lu-CC49 antibody was shown to be well-tolerated and anti-tumor effects were confirmed in patients with chemoresistant ovarian cancer. A differently radiolabeled CC49 antibody, 131I-CC49, was tested in patients with hormone-resistant metastatic prostate cancer. Phase II clinical studies demonstrated enhanced tumor uptake and anti-tumor activity for this antibody in the presence of interferon (IFN) which promoted increased tumor antigen expression and localization compared to the use of the 131I-CC49 antibody alone (153). However, the generation of human anti-murine antibody responses and bone marrow suppression represented two major adverse side-effects, which limited the therapeutic use of these radiolabeled anti-CC49 antibodies. A solution was reported by Rogers et al. (2005) (154) who presented the development of a humanized CC49 antibody, huCC49ΔCH2, and demonstrated less bone marrow toxicity as well as prolonged median survival in vivo, probably due to the acceptance of higher radiation doses without limiting side effects and off-side toxicity. In 2021, Minnix et al. (155) described the use of another humanized CC49 antibody construction, 225Ac-labeled DOTaylated-huCC49, in an in vivo murine model of ovarian cancer, which showed reduced tumor growth and increased survival without considerable off-side toxicity. Minnix et al. (2020) (156) also proposed an alternative therapeutic strategy using antibody-drug conjugates. The same abovementioned murine model was used to evaluate treatment with the anti-STn antibody-drug conjugate, CC49-Br-monomethyl auristatin E (MMAE, developed from CC49 antibody and monomethyl auristatin E). Reduced tumor growth as well as increased survival were found in mice receiving CC49-Br-MMAE treatment. Finally, another anti-STn antibody-drug conjugate, SF3-MMAE, was reported to be well tolerated and inhibit tumor growth in murine breast and colon cancer models (157) (Table 2).
Another therapeutic strategy is the use of Tn- and STn-based vaccines. In a phase I clinical trial among breast cancer patients with high-risk of relapse, Rosenbaum et al. (2020) (149) evaluated the efficiency of the multiple antigenic glycopeptide-Tn-tetanus toxoid-derived TT830-844 (MAG-Tn-TT) vaccine. This study revealed high levels of Tn-specific antibodies which were found to induce complement dependent cytotoxicity (CDC) and subsequent tumor cell death in all vaccinated patients. It is of note that carbohydrates can induce immune tolerance towards the tumor, which complicates the clinical success of glycan-based cancer vaccines. The development of efficient Tn-based vaccines, such as the MAG-Tn-TT vaccine, might therefore be a complicated process rather than standard therapeutic strategy (167–170).
The same holds for the STn antigen. The generation of an efficient STn-based anti-tumor immunotherapy is challenging due to its poor immunogenicity (71, 171). However, surprisingly, the best characterized STn-targeting therapy is a STn-based vaccine called Theratope. As delayed tumor growth by efficient antibody responses was observed in in vivo models receiving Theratope (158, 159), anti-STn immune responses and increased survival were initially also observed in phase I and II clinical studies with Theratope-treated pancreatic, colorectal, breast and ovarian cancer patients (160–162). Nevertheless, phase III clinical trials with metastatic breast cancer patients did not show this overall beneficial anti-tumor activity of Theratope, either alone or in combination treatment with endocrine therapy (172). An extensive review on Theratope´s composition and all clinical studies performed with this STn-based vaccine was published by Julien et al. (2012) (51).
The construction of anti-STn CAR-T cells and their anti-tumor activity in vitro was first described by Hombach et al. (163) in 1997, after which therapeutic benefit of anti-STn CC49-based CAR-T cells was proposed based on in vivo models of colon and endometrial carcinoma by McGuinness et al. (164) in 1999. Based on these preliminary findings, several anti-STn CAR-T cell engineering strategies have been evaluated. For example, dual-specific CAR-T cells targeting a second additional antigen, such as CD30 or CD47, have been developed to potentiate the anti-tumor activity of anti-STn CAR-T cells (165, 166). Indeed, Shu et al. (2021) (166) reported delayed tumor growth in low STn expressing tumors when treated with the dual STn- and CD47-specific CAR-T cells, but not after therapy with the single anti-STn CAR-T cells in an in vivo ovarian cancer model. It is of note that single anti-STn CAR-T cell therapy was found to be sufficient to obtain the same result in high STn expressing tumors. Finally, the first phase I clinical trials with anti-STn CC49-based CAR-T cells as immunotherapy for solid tumors were published by Hege et al. (134) in 2017. Even though these anti-STn engineered T cells were found to be relatively safe, no considerable clinical efficacy was observed in metastatic CRC patients. Here, rapid clearance of those anti-STn CAR-T cells was proposed as one of the limitations to be overcome for significant therapeutic benefit (Table 2).
CAR-T cell strategies targeting Tn or STn have been extensively studied as well. Posey et al. (2016) (150) used the scFv of the abovementioned anti-Tn 5E5 antibody for the development of anti-Tn CAR-T cells, which subsequently showed anti-tumor activity in murine models of pancreatic cancer and leukemia. Anti-Tn CAR-T cells with alternative Tn recognition domains have also been developed and showed therapeutic benefit in in vivo models of head and neck cancer (173) and breast and gastric carcinoma (174). In this last case, as an alternative to the use of αβ T cells a subtype of γδ T cells were used, and comparison of both CAR-Ts demonstrated a better cytotoxic effect with the γδ CAR-T cells both in vitro and in vivo. Additionally, anti-Tn SM3 antibody specificity has been used for the generation of multiple CAR-T constructs, which have been tested in a variety of clinical studies (135, 136). Significant clinical efficacy without evidence of adverse side-effects and safety concerns has been observed in phase I trials including patients with metastatic seminal vesicle cancer (135) and lung cancer (136). Finally, therapeutic efficacy as well as tolerability and safety of anti-TnMUC1 CAR-T cells are being evaluated in a phase I clinical trial among patients with TnMUC1+ multiple myeloma and solid tumors such as lung cancer, breast cancer, pancreatic cancer and ovarian cancer, by Gutierrez et al. (2021) (151). Preliminary efficacy assessed by RECIST v1.1 at Day +28 demonstrate stable disease in all patients that followed lymph depletion chemotherapy.
GD2 might be the most relevant glycan in the clinic. Two different antibodies, namely anti-GD2 antibodies 3F8 and 14.18, have been the basis for the generation of clinically successful anti-GD2 therapy (Figure 2).
After a variety of phase I clinical studies indicating clinical safety and effectiveness without significant toxicity, either after single or repeated ch14.18 antibody administration (175, 176), a phase II clinical trial showed that the use of ch14.18 combined with granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin-2 (IL2) compared to the ch14.18 alone in metastatic neuroblastoma patients was associated with a significantly improved outcome as compared with standard therapy in patients with high-risk neuroblastoma (177). This observation was emphasized by Simon et al. (2004) (178), who demonstrated that the administration of ch14.18 alone did not improve progression-free survival in stage 4 neuroblastoma patients. In 2010, Yu et al. (179) reported a significant increase in event-free survival and overall survival for high-risk neuroblastoma patients treated with a combination of anti-GD2 mAb ch14.18 with IL2 and GM-CSF compared to patients who received standard treatment in a phase III clinical study. Based on these results, Dinutuximab (anti-GD2 ch14.18 antibody) in combination with IL2 and GM-CSF was approved by the United States Food and Drug Administration (FDA) in 2015 for the treatment of high-risk neuroblastoma patients (180). The mechanism of action behind Dinutuximab is based on GD2 binding and subsequent induction of antibody-dependent cell-mediated cytotoxicity (ADCC) as well as CDC, after which recruitment of granulocytes and NK cells finally provoke tumor cell death. In 2017, Dinutuximab β (ch14.18/CHO) was approved by the European Commission for the treatment of high-risk neuroblastoma patients in Europe, whereas Naxitamab (a humanized mAb Hu3F8) in combination with GM-CSF was approved by the FDA in 2020 for the treatment of relapsed high-risk neuroblastoma patients or those who show refractory disease in the bone or bone marrow (181). Currently, several clinical trials are ongoing which will evaluate the potential of Dinutuximab and other hu14.18- and hu3F8-based anti-GD2 antibodies as therapeutic strategies for neuroblastoma and other cancer types including lung cancer, melanoma and osteosarcoma (138, 182). Additionally, alternative therapeutic constructs of anti-GD2 antibodies are being extensively studied, such as radiolabeled antibodies, antibodies modified for drug delivery, GD2-based vaccines, GD2-specific chimeric antigen receptor (CAR) T cells and bispecific antibodies, among others (138, 182). Accordingly, the number of clinical trials evaluating anti-GD2 antibodies for cancer immunotherapy is rapidly growing. An extensive review on approved anti-GD2 antibody-based cancer treatments as well as the many ongoing clinical trials was recently published by Nazha et al. (2020) (182) and Berois et al. (2022) (138).
However, a major bottleneck to the clinical application of anti-GD2-based immunotherapy is the presence of significant adverse side-effects such as neuropathic pain, due to GD2 expression among peripheral nerves (96). In addition to further studies into strategies that circumvent or diminish those significant side effects, alternative approaches for anti-GD2 cancer immunotherapy are being evaluated. Accordingly, the O-GD2 antigen has shown great potential as therapeutic target for cancer immunotherapy due to its absence on healthy tissues, supposing safer therapeutic options and improved treatment tolerance (96). Decades ago Cerato et al. (1997) (183) developed a mouse 8B6-antibody specific for O-GD2 (Table 3). Over time, this antibody has been proven to be as efficient as anti-GD2 antibodies regarding the induction of ADCC and CDC (95, 96, 184). Additionally, Cochonneau et al. (2013) (185) proposed a role for the anti-O-GD2 8B6 antibody in tumor cell death by the induction of cell cycle arrest and apoptosis. Even though the exact mechanism behind this observation remains to be elucidated, the apoptosis inducing activity of this O-GD2 antibody might clinically be of major importance, especially in the treatment of tumors that are able to protect themselves from immunological cytotoxicity. For clinical purposes, an alternative mouse/human chimeric version of the 8B6 antibody (c.8B6), maintaining antigen binding affinity and specificity characteristics, was developed by Terme et al. (2014) (184) (Table 3). The c.8B6 O-GD2 antibody was found to be as effective as the ch14.18 GD2 antibody, but did not provoke any significant adverse side effects in an in vivo model, emphasizing the potentially major clinical benefit of O-GD2-based therapeutic approaches. Nevertheless, whereas many clinical trials with anti-GD2-based antibody therapies are ongoing, clinical trials with an anti-O-GD2 c.8B6 antibody are still awaited.
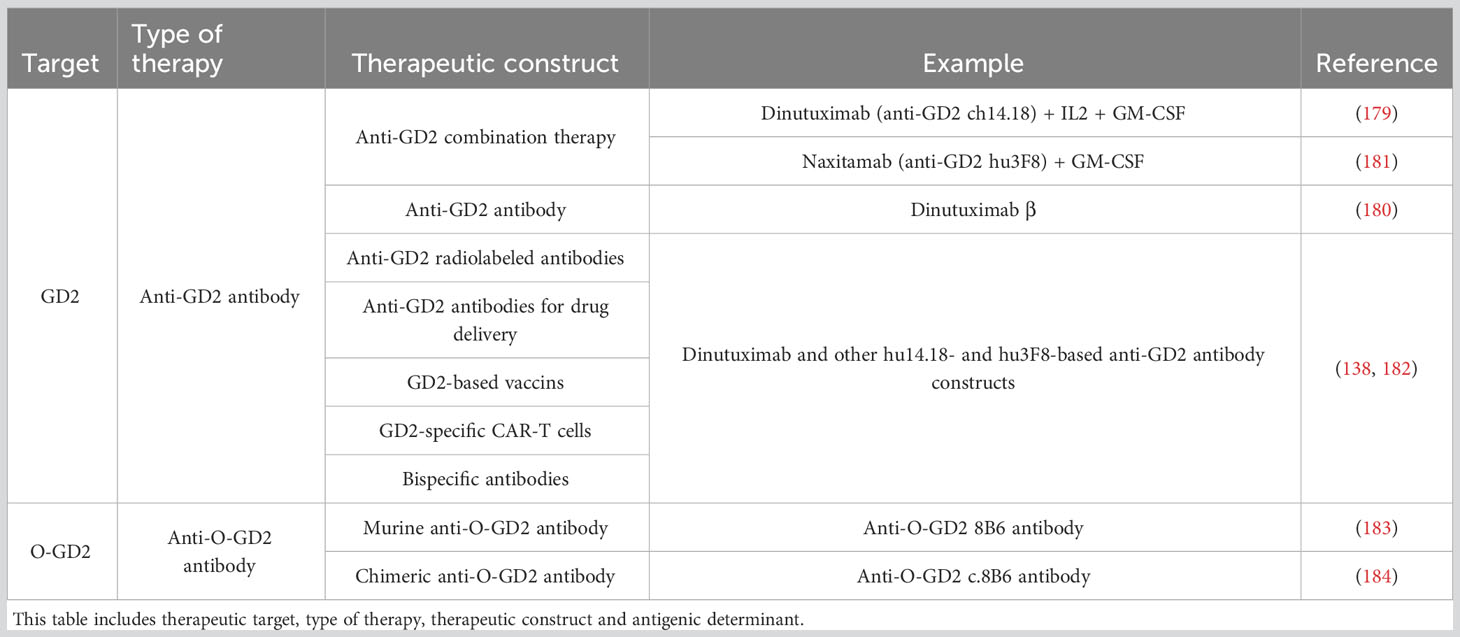
Table 3 Overview of the (O-)GD2-directed therapies described in this study.
Taken together, the reviewed antigens represent potential biomarkers and attractive therapeutic targets for personalized medicine in cancer treatment.
The three mentioned TAAs could serve as biomarkers in liquid biopsy and provide valuable information about the presence, progression, and characteristics of tumors. Liquid biopsy involves the analysis of various biomarkers, including circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), and proteins, obtained from a patient’s blood or other bodily fluids (Lodewijk et al., 2018). TAAs can be detected and measured in these samples and offer insights into the underlying cancer biology. Even though some studies have reported the presence of CD44v6, STn and GD2 antigens in serum, no extensive evaluations of their potential as valuable diagnostic, prognostic and/or predictive biomarkers in liquid biopsy have been described. It is of note that the presence of promising biomarker and therapeutic target O-GD2 in serum has not been reported. However, this might be explained by both the recent interest and limited approaches to study the regulatory mechanisms of O-GD2 expression, and further studies into the expression of this antigen are needed to examine the presence of this glycan in serum. Importantly, CD44v6 expression has only been detected on CTCs, whereas STn glycans have been found to be present either on CTCs or as secreted products from tumors. Contrarily, GD2 expression has not been observed on CTCs and is found in the serum as secreted products either from the tumor or exosomes. This observation emphasizes the importance of evaluation of different components of liquid biopsies in order to characterize the presence of newly described antigens in liquid biopsies. Moreover, liquid biopsies not only include serum, but various biological fluids, such as pleural liquid, cerebrospinal fluid, saliva and urine. Therefore, dependent on the cancer type, it might be important to examine CD44v6, STn and O-GD2 expression in other fluids additional to serum. For example, as expression of all those antigens has been observed in lung cancer, the evaluation of pleural liquid might be of great interest. It’s important to note that the choice of TAAs as biomarkers in liquid biopsy depends on the specific type of cancer and the unique genetic alterations associated with it. Different cancers have distinct TAAs, and ongoing research continues to identify and validate novel TAAs for liquid biopsy applications. The utilization of TAAs as biomarkers in liquid biopsy holds promise for non-invasive cancer detection, monitoring, and personalized treatment decisions.
In conclusion, the identification and targeting of tumor-associated antigens have opened up exciting new avenues in cancer treatment. Immunotherapies based on TAAs, such as immune checkpoint inhibitors, cancer vaccines, and CAR-T-cell therapy, have shown great promise in improving patient outcomes. However, it is important to note that the field of TAA-based therapies is still evolving, and further research is needed to optimize treatment strategies, overcome resistance mechanisms, and broaden the application to a wider range of cancer types. Nonetheless, the progress made so far suggests the tremendous potential for the future of cancer treatment.
CR: Conceptualization, Supervision, Writing – review & editing, Data curation, Validation, Visualization, Funding acquisition, Investigation, Project administration. IL: Conceptualization, Data curation, Investigation, Resources, Writing – original draft. MD: Conceptualization, Data curation, Funding acquisition, Investigation, Supervision, Validation, Visualization, Writing – original draft. JP: Conceptualization, Data curation, Funding acquisition, Investigation, Supervision, Validation, Visualization, Writing – review & editing.
This study was cofounded by Instituto de Salud Carlos III (ISCIII) through the Project PI20/00813 to MD and co-funded by the European Union; European Regional Development Fund (FEDER) Grants from Science and Innovation (SAF2015-66015-R, and PID2019-110758RB-I00 to JMP), Instituto de Salud Carlos III (CIBERONC no. CB16/12/00228 to JMP; DTS20/00043; DTS22/00002 to MD), Project "FINANCED BY NEXTGENERATIONEU FUNDS, WHICH FINANCE THE ACTIONS OF THE RECOVERY AND RESILIENCE MECHANISM (MRR)". Funding entity: CARLOS III HEALTH INSTITUTE (ISCIII). ISCIII project code: AC22/00015 - Title: "Circulating tumor microenvironment components as predictors of response to immunotherapy in urothelial cancer" - PI: Marta Dueñas Porto. Project also funded by the Scientific Foundation of the Spanish Association Against Cancer (FCAECC) with project ID TRNSC213883DUEN, Transcan-3 JTC2022 and Fundación Eugenio Rodríguez Pascual (FERP-2022-79 to CR). IL is supported by a predoctoral fellowship from AECC (Spanish Ass. against Cancer), Predoctoral AECC 2019 grant number PRDMA19024LODE.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Zhang JY, Looi KS, Tan EM. Identification of tumor-associated antigens (TAAs) as diagnostic and predictive biomarkers in cancer. Methods Mol Biol (2009) 520:1. doi: 10.1007/978-1-60327-811-9_1
2. Wang RF, Rosenberg SA. Human tumor antigens for cancer vaccine development. Immunol Rev (1999) 170:85–100. doi: 10.1111/j.1600-065X.1999.tb01331.x
3. Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother (2005) 54(3):187–207. doi: 10.1007/s00262-004-0560-6
4. Matsushima S, Ajiro M, Iida K, Chamoto K, Honjo T, Hagiwara M. Chemical induction of splice-neoantigens attenuates tumor growth in a preclinical model of colorectal cancer. Sci Transl Med (2022) 14(673):eabn6056. doi: 10.1126/scitranslmed.abn6056
5. Sun R, Kim AMJ, Lim SO. Glycosylation of immune receptors in cancer. Cells (2021) 10(5):1100. doi: 10.3390/cells10051100
6. Liu Z, Ren Y, Weng S, Xu H, Li L, Han X. A new trend in cancer treatment: the combination of epigenetics and immunotherapy. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.809761
7. Srivastava AK, Guadagnin G, Cappello P, Novelli F. Post-translational modifications in tumor-associated antigens as a platform for novel immuno-oncology therapies. Cancers (Basel) (2022) 15(1):138. doi: 10.3390/cancers15010138
8. Kudrin A. Cancer vaccines: what do we need to measure in clinical trials? Hum Vaccin Immunother (2014) 10(11):3236–40. doi: 10.4161/hv.27586
9. Harjunpää H, Asens ML, Guenther C, Fagerholm SC. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front Immunol (2019) 10(MAY):1078. doi: 10.3389/fimmu.2019.01078
10. Spiegelberg D, Nilvebrant J. CD44v6-targeted imaging of head and neck squamous cell carcinoma: antibody-based approaches. Contrast Media Mol Imaging (2017) 2017:2709547. doi: 10.1155/2017/2709547
11. Li Z, Chen K, Jiang P, Zhang X, Li X, Li Z. CD44v/CD44s expression patterns are associated with the survival of pancreatic carcinoma patients. Diagn Pathol (2014) 9(1):79. doi: 10.1186/1746-1596-9-79
12. Zhou G, Chiu D, Qin D, Niu L, Cai J, He L, et al. Detection and clinical significance of cd44v6 and integrin-β1 in pancreatic cancer patients using a triplex real-time RT-PCR assay. Appl Biochem Biotechnol (2012) 167(8):2257–68. doi: 10.1007/s12010-012-9752-2
13. Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell (2014) 14(3):342–56. doi: 10.1016/j.stem.2014.01.009
14. Wielenga VJ, Heider KH, Offerhaus GJ, Adolf GR, van den Berg FM, Punta H, et al. Expression of CD44 variant proteins in human colorectal cancer is related to tumor progression. Cancer Res (1993) 53:4754–6.
15. Ghatak S, Hascall VC, Markwald RR, Misra S. Stromal hyaluronan interaction with epithelial CD44 variants promotes prostate cancer invasiveness by augmenting expression and function of hepatocyte growth factor and androgen receptor. J Biol Chem (2010) 285(26):19821. doi: 10.1074/jbc.M110.104273
16. Ni J, Cozzi PJ, Hao JL, Beretov J, Chang L, Duan W, et al. CD44 variant 6 is associated with prostate cancer metastasis and chemo-/radioresistance. Prostate (2014) 74(6):602–17. doi: 10.1002/pros.22775
17. Kuncová J, Urban M, Mandys V. Expression of CD44s and CD44v6 in transitional cell carcinomas of the urinary bladder: comparison with tumour grade, proliferative activity and p53 immunoreactivity of tumour cells. APMIS (2007) 115(11):1194–205. doi: 10.1111/j.1600-0643.2007.00602.x
18. Heider KH, Kuthan H, Stehle G, Munzert G. CD44v6: A target for antibody-based cancer therapy. Cancer Immunology Immunother (2004) 53(7):567–79. doi: 10.1007/s00262-003-0494-4
19. Casucci M, Di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood (2013) 122(20):3461–72. doi: 10.1182/blood-2013-04-493361
20. Zhao LH, Lin QL, Wei J, Huai YL, Wang KJ, Yan HY. CD44v6 expression in patients with stage II or stage III sporadic colorectal cancer is superior to CD44 expression for predicting progression. Int J Clin Exp Pathol (2015) 8(1):692.
21. Garouniatis A, Zizi-Sermpetzoglou A, Rizos S, Kostakis A, Nikiteas N, Papavassiliou AG. FAK, CD44v6, c-Met and EGFR in colorectal cancer parameters: tumour progression, metastasis, patient survival and receptor crosstalk. Int J Colorectal Dis (2013) 28(1):9–18. doi: 10.1007/s00384-012-1520-9
22. Shiozaki M, Ishiguro H, Kuwabara Y, Kimura M, Mitsui A, Naganawa Y, et al. Expression of CD44v6 is an independent prognostic factor for poor survival in patients with esophageal squamous cell carcinoma. Oncol Lett (2011) 2(3):429. doi: 10.3892/ol.2011.264
23. Almeida GM, Pereira C, Park JH, Lemos C, Campelos S, Gullo I, et al. CD44v6 high membranous expression is a predictive marker of therapy response in gastric cancer patients. Biomedicines (2021) 9(9):1249. doi: 10.3390/biomedicines9091249
24. Kawano T, Nakamura Y, Yanoma S, Kubota A, Furukawa M, Miyagi Y, et al. Expression of E-cadherin, and CD44s and CD44v6 and its association with prognosis in head and neck cancer. Auris Nasus Larynx (2004) 31(1):35–41. doi: 10.1016/j.anl.2003.09.005
25. Xu YP, Zhao XQ, Sommer K, Moubayed P. Correlation of matrix metalloproteinase-2, -9, tissue inhibitor-1 of matrix metalloproteinase and CD44 variant 6 in head and neck cancer metastasis. J Zhejiang Univ Sci (2003) 4(4):491–501. doi: 10.1631/jzus.2003.0491
26. Kashyap T, Pramanik KK, Nath N, Mishra P, Singh AK, Nagini S, et al. Crosstalk between Raf-MEK-ERK and PI3K-Akt-GSK3β signaling networks promotes chemoresistance, invasion/migration and stemness via expression of CD44 variants (v4 and v6) in oral cancer. Oral Oncol (2018) 86:234–43. doi: 10.1016/j.oraloncology.2018.09.028
27. Liu B, Kong W, Gong S, Yang C, Wang G, Zhu L. Relationship between the expression of CD44v6 and development, progress, invasion and metastasis of laryngeal carcinoma. J Huazhong Univ Sci Technolog Med Sci (2005) 25(3):351–3. doi: 10.1007/BF02828165
28. Sun Y, Yu X, Li M, Zou Z. Expression of CD44v6 and lymphatic vessel density in early gastric cancer tissues and their clinical significance. Pak J Med Sci (2019) 35(2):549. doi: 10.12669/pjms.35.2.464
29. Lourenço BN, Springer NL, Ferreira D, Oliveira C, Granja PL, Fischbach C. CD44v6 increases gastric cancer Malignant phenotype by modulating adipose stromal cell-mediated ECM remodeling. Integr Biol (Camb) (2018) 10(3):145. doi: 10.1039/c7ib00179g
30. Liang S, Li HL, Han GY, Cui JH. CD44V6 regulates gastric carcinoma occurrence and development through up-regulating VEGF expression. Eur Rev Med Pharmacol Sci (2017) 21(22):5121–8. doi: 10.26355/eurrev_201711_13828
31. Xie Z, Gao Y, Ho C, Li L, Jin C, Wang X, et al. Exosome-delivered CD44v6/C1QBP complex drives pancreatic cancer liver metastasis by promoting fibrotic liver microenvironment. Gut (2022) 71(3):568–79. doi: 10.1136/gutjnl-2020-323014
32. Wrana F, Dötzer K, Prüfer M, Werner J, Mayer B. High dual expression of the biomarkers CD44v6/α2β1 and CD44v6/PD-L1 indicate early recurrence after colorectal hepatic metastasectomy. Cancers (Basel) (2022) 14(8):1939. doi: 10.3390/cancers14081939
33. Jha RK, Ma Q, Chen S, Sha H, Ding S. Relationship of fibronectin and CD44v6 expression with invasive growth and metastasis of liver cancer. Cancer Invest (2009) 27(3):324–8. doi: 10.1080/07357900802375753
34. De Angelis ML, Francescangeli F, Nicolazzo C, Xhelili E, La Torre F, Colace L, et al. An orthotopic patient-derived xenograft (PDX) model allows the analysis of metastasis-associated features in colorectal cancer. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.869485
35. Yang L, Yang J, Jacobson B, Gilbertsen A, Smith K, Higgins LA, et al. SFPQ Promotes Lung Cancer Malignancy via Regulation of CD44 v6 Expression. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.862250
36. Eren B, Sar M, Oz B, Oner Dincbas FH. MMP-2, TIMP-2 and CD44v6 expression in non-small-cell lung carcinomas †. Ann Acad Med Singap (2008) 37(1):32–9.
37. Rustamadji P, Wiyarta E, Bethania KA. CD44 variant exon 6 isoform expression as a potential predictor of lymph node metastasis in invasive breast carcinoma of no special type. Int J Breast Cancer (2021) 2021:1586367. doi: 10.1155/2021/1586367
38. Soon CH, Jae YS, Jae KL, Nak WL, Sun HK, Bom WY, et al. Significance of CD44v6 expression in gynecologic Malignancies. J Obstetrics Gynaecology Res (2006) 32(4):379–86. doi: 10.1111/j.1447-0756.2006.00422.x.
39. Wang Y, Yang X, Xian SHU, Zhang LI, Cheng Y. CD44v6 may influence ovarian cancer cell invasion and migration by regulating the NF-κB pathway. Oncol Lett (2019) 18(1):298. doi: 10.3892/ol.2019.10306
40. Wang Y, Yang X, Yuan M, Xian S, Zhang L, Yang D, et al. Promotion of ovarian cancer cell invasion, migration and colony formation by the miR-21/Wnt/CD44v6 pathway. Oncol Rep (2019) 42(1):91. doi: 10.3892/or.2019.7153
41. Ni J, Cheung BB, Beretov J, Duan W, Bucci J, Malouf D, et al. CD44 variant 6 is associated with prostate cancer growth and chemo-/radiotherapy response in vivo. Exp Cell Res (2020) 388(2):111850. doi: 10.1016/j.yexcr.2020.111850
42. Belthier G, Homayed Z, Grillet F, Duperray C, Vendrell J, Krol I, et al. CD44v6 defines a new population of circulating tumor cells not expressing epCAM. Cancers (Basel) (2021) 13(19):4966. doi: 10.3390/cancers13194966
43. Nicolazzo C, Loreni F, Caponnetto S, Magri V, Vestri AR, Zamarchi R, et al. Baseline CD44v6-positive circulating tumor cells to predict first-line treatment failure in patients with metastatic colorectal cancer. Oncotarget (2020) 11(45):4115–22. doi: 10.18632/oncotarget.27794
44. Gaggianesi M, Mangiapane LR, Modica C, Pantina VD, Porcelli G, Di Franco S, et al. Dual inhibition of myc transcription and PI3K activity effectively targets colorectal cancer stem cells. Cancers (Basel) (2022) 14(3):673. doi: 10.3390/cancers14030673
45. Wang Y, Chen G, Dai F, Zhang L, Yuan M, Yang D, et al. miR-21 induces chemoresistance in ovarian cancer cells via mediating the expression and interaction of CD44v6 and P-gp. Onco Targets Ther (2021) 14:325–36. doi: 10.2147/OTT.S286639
46. Pereira C, Ferreira D, Mendes N, Granja PL, Almeida GM, Oliveira C. Expression of CD44v6-containing isoforms influences cisplatin response in gastric cancer cells. Cancers (Basel) (2020) 12(4):858. doi: 10.3390/cancers12040858
47. Sagawa K, Uwa N, Daimon T, Sakagami M, Tsujimura T. Expression of CD44 variant isoforms, CD44v3 and CD44v6, are associated with prognosis in nasopharyngeal carcinoma. J Laryngol Otol (2016) 130(9):843–9. doi: 10.1017/S0022215116008525
48. Chen C, Zhao S, Karnad A, Freeman JW. The biology and role of CD44 in cancer progression: therapeutic implications. J Hematol Oncol (2018) 11(1):1–23. doi: 10.1186/s13045-018-0605-5
49. Radhakrishnan P, Dabelsteen S, Madsen FB, Francavilla C, Kopp KL, Steentoft C, et al. Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc Natl Acad Sci USA (2014) 111(39):E4066–75. doi: 10.1073/pnas.1406619111
50. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer (2015) 15(9):540–55. doi: 10.1038/nrc3982
51. Fleurence J, Fougeray S, Bahri M, Cochonneau D, Clémenceau B, Paris F, et al. Targeting O-acetyl-GD2 ganglioside for cancer immunotherapy. J Immunol Res (2017) 2017:5604891. doi: 10.1155/2017/5604891
52. Ju T, Otto VI, Cummings RD. The tn antigen—Structural simplicity and biological complexity. Angew Chem Int Ed Engl (2011) 50(8):1770. doi: 10.1002/anie.201002313
53. Prokop O, Uhlenbruck G. N-acetyl-D-galactosamine in tumor cell membranes: demonstration by means of Helix agglutinins. Med Welt (1969) 46:2515–9
54. Dombek GE, Ore AS, Cheng J, Matsumoto Y, Glickman JN, Fleishman A, et al. Immunohistochemical analysis of Tn antigen expression in colorectal adenocarcinoma and precursor lesions. BMC Cancer (2022) 22(1):1281. doi: 10.1186/s12885-022-10376-y
55. Springer GF. Immunoreactive T and Tn epitopes in cancer diagnosis, prognosis, and immunotherapy. J Mol Med (Berl) (1997) 75(8):594–602. doi: 10.1007/s001090050144
56. Springer GF. T and Tn, general carcinoma autoantigens. Science (1984) 224(4654):1198–206. doi: 10.1126/science.6729450
57. Laack E, Nikbakht H, Peters A, Kugler C, Jasiewicz Y, Edler L, et al. Lectin histochemistry of resected adenocarcinoma of the lung: helix pomatia agglutinin binding is an independent prognostic factor. Am J Pathol (2002) 160(3):1001–8. doi: 10.1016/S0002-9440(10)64921-8
58. Hofmann BT, Schlüter L, Lange P, Mercanoglu B, Ewald F, Fölster A, et al. COSMC knockdown mediated aberrant O-glycosylation promotes oncogenic properties in pancreatic cancer. Mol Cancer (2015) 14(1):109. doi: 10.1186/s12943-015-0386-1
59. Dong X, Jiang Y, Liu J, Liu Z, Gao T, An G, et al. T-synthase deficiency enhances oncogenic features in human colorectal cancer cells via activation of epithelial-mesenchymal transition. BioMed Res Int (2018) 2018:9532389. doi: 10.1155/2018/9532389
60. Konno A, Hoshino Y, Terashima S, Motoki R, Kawaguchi T. Carbohydrate expression profile of colorectal cancer cells is relevant to metastatic pattern and prognosis. Clin Exp Metastasis (2002) 19(1):61–70. doi: 10.1023/A:1013879702702
61. Numa F, Tsunaga N, Michioka T, Nawata S, Ogata H, Kato H. Tissue expression of Sialyl Tn antigen in gynecologic tumors. J Obstet Gynaecol (Tokyo 1995) (1995) 21(4):385–9. doi: 10.1111/j.1447-0756.1995.tb01027.x
62. Munkley J. The role of sialyl-tn in cancer. Int J Mol Sci (2016) 17(3):275. doi: 10.3390/ijms17030275
63. Fu C, Zhao H, Wang Y, Cai H, Xiao Y, Zeng Y, et al. Tumor-associated antigens: Tn antigen, sTn antigen, and T antigen. HLA (2016) 88(6):275–86. doi: 10.1111/tan.12900
64. Julien S, Videira PA, Delannoy P. Sialyl-tn in cancer: (how) did we miss the target? Biomolecules (2012) 2(4):435–66. doi: 10.3390/biom2040435
65. Thomas D, Sagar S, Caffrey T, Grandgenett PM, Radhakrishnan P. Truncated O-glycans promote epithelial-to-mesenchymal transition and stemness properties of pancreatic cancer cells. J Cell Mol Med (2019) 23(10):6885–96. doi: 10.1111/jcmm.14572
66. Munkley J. The role of sialyl-tn in cancer. Int J Mol Sci (2016) 17(3):275. doi: 10.3390/ijms17030275
67. Davidson B, Berner A, Nesland JM, Risberg B, Kristensen GB, Tropé CG, et al. Carbohydrate antigen expression in primary tumors, metastatic lesions, and serous effusions from patients diagnosed with epithelial ovarian carcinoma: evidence of up-regulated Tn and Sialyl Tn antigen expression in effusions. Hum Pathol (2000) 31(9):1081–7. doi: 10.1053/hupa.2000.9776
68. Davidson B, Gotlieb WH, Ben-Baruch G, Kopolovic J, Goldberg I, Nesland JM, et al. Expression of carbohydrate antigens in advanced-stage ovarian carcinomas and their metastases-A clinicopathologic study. Gynecol Oncol (2000) 77(1):35–43. doi: 10.1006/gyno.1999.5708
69. Alexandre Ferreira J, Videira PA, Lima L, Pereira S, Silva M, Carrascal M, et al. Overexpression of tumour-associated carbohydrate antigen sialyl-Tn in advanced bladder tumours. Mol Oncol (2013) 7(3):719–31. doi: 10.1016/j.molonc.2013.03.001
70. Bhatia R, Gautam SK, Cannon A, Thompson C, Hall BR, Aithal A, et al. Cancer-associated mucins: role in immune modulation and metastasis. Cancer Metastasis Rev (2019) 38(1-2):223–36. doi: 10.1007/s10555-018-09775-0
71. Monti P, Leone BE, Zerbi A, Balzano G, Cainarca S, Sordi V, et al. Tumor-derived MUC1 mucins interact with differentiating monocytes and induce IL-10highIL-12low regulatory dendritic cell. J Immunol (2004) 172(12):7341–9. doi: 10.4049/jimmunol.172.12.7341
72. Ohta M, Ishida A, Toda M, Akita K, Inoue M, Yamashita K, et al. Immunomodulation of monocyte-derived dendritic cells through ligation of tumor-produced mucins to Siglec-9. 2010. Biochem Biophys Res Commun (2010) 402(4):663–9. doi: 10.1016/j.bbrc.2010.10.079
73. Carrascal MA, Severino PF, Guadalupe Cabral M, Silva M, Ferreira JA, Calais F, et al. Sialyl Tn-expressing bladder cancer cells induce a tolerogenic phenotype in innate and adaptive immune cells. Mol Oncol (2014) 8(3):753–65. doi: 10.1016/j.molonc.2014.02.008
74. Quintela A, Videira P, Barata PC, Sonpavde G, Carvalho S, Oliveira MI, et al. Phenotypic analysis of urothelial exfoliated cells in bladder cancer via microfluidic immunoassays: sialyl-tn as a novel biomarker in liquid biopsies. Front Oncol (2020) 10:1774. doi: 10.3389/fonc.2020.01774
75. Kobayashi H, Terao T, Kawashima Y. Serum sialyl Tn as an independent predictor of poor prognosis in patients with epithelial ovarian cancer. J Clin Oncol (2016) 10(1):95–101. doi: 10.1200/JCO.1992.10.1.95
76. Kudoh K, Kikuchi Y, Kita T, Tode T, Takano M, Hirata J, et al. Preoperative determination of several serum tumor markers in patients with primary epithelial ovarian carcinoma. Gynecol Obstet Invest (1999) 47(1):52–7. doi: 10.1159/000010062
77. D’Amico TA, Aloia TA, Moore MBH, Herndon JE, Brooks KR, Lau CL, et al. Molecular biologic substaging of stage I lung cancer according to gender and histology. Ann Thorac Surg (2000) 69(3):882–6. doi: 10.1016/S0003-4975(99)01522-2
78. Campos D, Freitas D, Gomes J, Magalhães A, Steentoft C, Gomes C, et al. Probing the O-glycoproteome of gastric cancer cell lines for biomarker discovery. Mol Cell Proteomics (2015) 14(6):1616–29. doi: 10.1074/mcp.M114.046862
79. Mereiter S, Martins ÁM, Gomes C, Balmaña M, Macedo JA, Polom K, et al. O-glycan truncation enhances cancer-related functions of CD44 in gastric cancer. FEBS Lett (2019) 593(13):1675–89. doi: 10.1002/1873-3468.13432
80. Moreira IB, Pinto F, Gomes C, Campos D, Reis CA. Impact of truncated O-glycans in gastric-cancer-associated CD44v9 detection. Cells (2020) 9(2):264. doi: 10.3390/cells9020264
81. Singh R, Campbell BJ, Yu LG, Fernig DG, Milton JD, Goodlad RA, et al. Cell surface-expressed Thomsen-Friedenreich antigen in colon cancer is predominantly carried on high molecular weight splice variants of CD44. Glycobiology (2001) 11(7):587–92. doi: 10.1093/glycob/11.7.587
82. Ruan S, Lloyd K. Glycosylation pathways in the biosynthesis of gangliosides in melanoma and neuroblastoma cells: relative glycosyltransferase levels determine ganglioside patterns. Cancer Res (1992) 52(20):5725–31.
83. Mansoori M, Mirzaei A, Rad IA, Mahmodlou R, Mansouri F, Zanjani LS, et al. Upregulation of ganglioside GD2 synthase (GD2S), as a new putative cancer stem cell marker in breast carcinomas. Med J Islam Repub Iran (2021) 35(1):1–9. doi: 10.47176/mjiri.35.148
84. Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest (2012) 122(6):2066. doi: 10.1172/JCI59735
85. Vantaku V, Donepudi SR, Ambati CR, Jin F, Putluri V, Nguyen K, et al. Expression of ganglioside GD2, reprogram the lipid metabolism and EMT phenotype in bladder cancer. Oncotarget (2017) 8(56):95620. doi: 10.18632/oncotarget.21038
86. Yoshida S, Kawaguchi H, Sato S, Ueda R, Furukawa K. An Anti-GD2 Monoclonal Antibody Enhances Apoptotic Effects of Anti-cancer Drugs against Small Cell Lung Cancer Cells via JNK (c-Jun Terminal Kinase) Activation. Jpn J Cancer Res (2002) 93(7):816. doi: 10.1111/j.1349-7006.2002.tb01324.x
87. Shibuya H, Hamamura K, Hotta H, Matsumoto Y, Nishida Y, Hattori H, et al. Enhancement of Malignant properties of human osteosarcoma cells with disialyl gangliosides GD2/GD3. Cancer Sci (2012) 103(9):1656–64. doi: 10.1111/j.1349-7006.2012.02344.x
88. Bailey K, Cost C, Davis I, Glade-Bender J, Grohar P, Houghton P, et al. Emerging novel agents for patients with advanced Ewing sarcoma: a report from the Children’s Oncology Group (COG) New Agents for Ewing Sarcoma Task Force [version 1; peer review: 3 approved]. F100Res (2019) 8(F1000 Faculty Rev):493. doi: 10.12688/f1000research.18139.1
89. Reis CA, Sørensen T, Mandel U, David L, Mirgorodskaya E, Roepstorff P, et al. Development and characterization of an antibody directed to an @-N-acetyl-D-galactosamine glycosylated MUC2 peptide. Glycoconj J (1998) 15:51–62. doi: 10.1023/A:1006939432665
90. Lammie GA, Cheung NKV, Gerald W, Rosenblum M, Cordon-Cardo C. Ganglioside GD2 expression in the human nervous system and in neuroblastomas - An immunohistochemical study. Int J Oncol (1993) 3(5):909–15. doi: 10.3892/ijo.3.5.909
91. Cavdarli S, Groux-Degroote S, Delannoy P. Gangliosides: the double-edge sword of neuro-ectodermal derived tumors. Biomolecules (2019) 9(8):311. doi: 10.3390/biom9080311
92. Valentino L, Moss T, Olson E, Wang HJ, Elashoff R, Ladisch S. Shed tumor gangliosides and progression of human neuroblastoma. Blood (1990) 75(7):1564–7. doi: 10.1182/blood.V75.7.1564.1564
93. Esaki N, Ohkawa Y, Hashimoto N, Tsuda Y, Ohmi Y, Bhuiyan RH, et al. ASC amino acid transporter 2, defined by enzyme-mediated activation of radical sources, enhances Malignancy of GD2-positive small-cell lung cancer. Cancer Sci (2018) 109(1):141. doi: 10.1111/cas.13448
94. Ye JN, Cheung NV. A novel O-acetylated ganglioside detected by anti-GD2 monoclonal antibodies. Int J Cancer (1992) 50(2):197–201. doi: 10.1002/ijc.2910500207
95. Fleurence J, Cochonneau D, Fougeray S, Oliver L, Geraldo F, Terme M, et al. Targeting and killing glioblastoma with monoclonal antibody to O-acetyl GD2 ganglioside. Oncotarget (2016) 7(27):41172. doi: 10.18632/oncotarget.9226
96. Alvarez-Rueda N, Desselle A, Cochonneau D, Chaumette T, Clemenceau B, Leprieur S, et al. A monoclonal antibody to O-acetyl-GD2 ganglioside and not to GD2 shows potent anti-tumor activity without peripheral nervous system cross-reactivity. PloS One (2011) 6(9):e25220. doi: 10.1371/journal.pone.0025220
97. Heider KH, Sproll M, Susani S, Patzelt E, Beaumier P, Ostermann E, et al. Characterization of a high-affinity monoclonal antibody specific for CD44v6 as candidate for immunotherapy of squamous cell carcinomas. Cancer Immunol Immunother (1996) 43(4):245–53. doi: 10.1007/s002620050329
98. de Bree R, Roos J, Quak J, den Hollander W, Snow G, van Dongen GV. Radioimmunoscintigraphy and biodistribution of technetium-99m-labeled monoclonal antibody U36 in patients with head and neck cancer. Clin Cancer Res (1995) 1(6):591–8.
99. Van Hal NL, Ams Van Dongen G, Rood-knippels EM, Van Der Valk P, Snow GB, Brakenhoff RH. Monoclonal antibody U36, a suitable candidate for clinical a CD44 isoform immunotherapy of squamous-cell carcinoma, recognizes. Int J Cancer (1996) 68:520–7. doi: 10.1002/(SICI)1097-0215(19961115)68:4<520::AID-IJC19>3.0.CO;2-8
100. Colnot DR, Quak JJ, Roos JC, van Lingen A, Wilhelm AJ, van Kamp GJ, et al. Phase I therapy study of 186Re-labeled chimeric monoclonal antibody U36 in patients with squamous cell carcinoma of the head and neck. J Nucl Med (2000) 41(12):1999–2010.
101. Stroomer JWG, Roos JC, Sproll M, Quak JJ, Heider KH, Wilhelm BJ, et al. Safety and biodistribution of 99mtechnetium-labeled anti-CD44v6 monoclonal antibody BIWA 1 in head and neck cancer patients. Clin Cancer Res (2000) 6(8):3046–55.
102. Colnot DR, Roos JC, De Bree R, Wilhelm AJ, Alain AJ, Gertraud K, et al. Safety, biodistribution, pharmacokinetics, and immunogenicity of 99m Tc-labeled humanized monoclonal antibody BIWA 4 (bivatuzumab) in patients with squamous cell carcinoma of the head and neck. Cancer Immunol Immunother (2003) 52:576–82. doi: 10.1007/s00262-003-0396-5
103. Börjesson PKE, Postema EJ, Roos JC, Colnot DR, Marres HAM, van Schie MH, et al. Phase I therapy study with 186Re-labeled humanized monoclonal antibody BIWA 4 (Bivatuzumab) in patients with head and neck squamous cell carcinoma. Clin Cancer Res (2003) 9(10 II):343–54.
104. Postema EJ, Börjesson PKE, Buijs WCAM, Roos JC, Marres HAM, Boerman OC, et al. Dosimetric analysis of radioimmunotherapy with 186Re-labeled bivatuzumab in patients with head and neck cancer. J Nucl Med (2003) 44(10):1690–9.
105. Tijink BM, Buter J, De Bree R, Giaccone G, Lang MS, Staab A, et al. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin Cancer Res (2006) 12(20 Pt 1):6064–72. doi: 10.1158/1078-0432.CCR-06-0910
106. Grunewald CM, Haist C, König C, Petzsch P, Bister A, Nößner E, et al. Epigenetic priming of bladder cancer cells with decitabine increases cytotoxicity of human EGFR and CD44v6 CAR engineered T-cells. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.782448
107. Porcellini S, Asperti C, Corna S, Cicoria E, Valtolina V, Stornaiuolo A, et al. CAR T cells redirected to CD44v6 control tumor growth in lung and ovary adenocarcinoma bearing mice. Front Immunol (2020) 11:99. doi: 10.3389/fimmu.2020.00099
108. Greco B, Malacarne V, DeGirardi F, Scotti GM, Manfredi F, Angelino E, et al. Disrupting N-glycan expression on tumor cells boosts chimeric antigen receptor T cell efficacy against solid Malignancies. Sci Transl Med (2022) 14(628):eabg3072. doi: 10.1126/scitranslmed.abg3072
109. Sauter A, Kloft C, Gronau S, Bogeschdorfer F, Erhardt T, Golze W, et al. Pharmacokinetics, immunogenicity and safety of bivatuzumab mertansine, a novel CD44v6-targeting immunoconjugate, in patients with squamous cell carcinoma of the head and neck. Int J Oncol (2007) 30(4):927–35. doi: 10.3892/ijo.30.4.927
110. Hekele A, Dall P, Moritz D, Wels’ W, Groner B, Herrlich P, et al. Growth retardation of tumors by adoptive transfer of cytotoxic T lymphocytes reprogrammed by Cd44v6-specific SCFV:C-Chimera. Int J Cancer (1996) 68:232–8. doi: 10.1002/(SICI)1097-0215(19961009)68:2<232::AID-IJC16>3.0.CO;2-C
111. Somasundaram C, Arch R, Matzku S, Zöller M. Development of a bispecifie F(ab’)2 conjugate against the complement receptor CR3 of macrophages and a variant CD44 antigen of rat pancreatic adenocarcinoma for redirecting macrophage-mediated tumor cytotoxicity. Cancer Immunol Immunother (1996) 42(6):343–50. doi: 10.1007/s002620050292
112. Sandström K, Nestor M, Ekberg T, Engström M, Anniko M, Lundqvist H. Targeting CD44v6 expressed in head and neck squamous cell carcinoma: preclinical characterization of an 111In-labeled monoclonal antibody. Tumor Biol (2008) 29(3):137–44. doi: 10.1159/000143399
113. Nestor M, Andersson K, Lundqvist H. Characterization of 111In and 177Lu-labeled antibodies binding to CD44v6 using a novel automated radioimmunoassay. J Mol Recognition (2008) 21(3):179–83. doi: 10.1002/jmr.883
114. Rupp U, Schoendorf-Holland E, Eichbaum M, Schuetz F, Lauschner I, Schmidt P, et al. Safety and pharmacokinetics of bivatuzumab mertansine in patients with CD44v6-positive metastatic breast cancer: Final results of a phase I study. Anticancer Drugs (2007) 18(4):477–85. doi: 10.1097/CAD.0b013e32801403f4
115. Riechelmann H, Sauter A, Golze W, Hanft G, Schroen C, Hoermann K, et al. Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Oral Oncol (2008) 44(9):823–9. doi: 10.1016/j.oraloncology.2007.10.009
116. Ding L, Hu Y, Huang H. Novel progresses of chimeric antigen receptor (CAR) T cell therapy in multiple myeloma. Stem Cell Investig (2021) 8(2):1–10. doi: 10.21037/sci-2020-029
117. Haist C, Schulte E, Bartels N, Bister A, Poschinski Z, Ibach TC, et al. CD44v6-targeted CAR T-cells specifically eliminate CD44 isoform 6 expressing head/neck squamous cell carcinoma cells. Oral Oncol (2021) 116:105259. doi: 10.1016/j.oraloncology.2021.105259
118. Leuci V, Casucci GM, Grignani G, Rotolo R, Rossotti U, Vigna E, et al. CD44v6 as innovative sarcoma target for CAR-redirected CIK cells. Oncoimmunology (2018) 7(5):e1423167. doi: 10.1080/2162402X.2017.1423167
119. Circosta P, Donini C, Gallo S, Giraudo L, Gammaitoni L, Rotolo R, et al. Full chimaeric CAR.CIK from patients engrafted after allogeneic haematopoietic cell transplant: Feasibility, anti-leukaemic potential and alloreactivity across major human leukocyte antigen barriers. Br J Haematol (2023) 200(1):64–9. doi: 10.1111/bjh.18469
120. Qian C, Wang Y, Chen Y, Zeng L, Zhang Q, Shuai X, et al. Suppression of pancreatic tumor growth by targeted arsenic delivery with anti-CD44v6 single chain antibody conjugated nanoparticles. Biomaterials (2013) 34(26):6175–84. doi: 10.1016/j.biomaterials.2013.04.056
121. Li J, Chen Y, Zeng L, Lian G, Chen S, Li Y, et al. A nanoparticle carrier for co-delivery of gemcitabine and small interfering rna in pancreatic cancer therapy. J BioMed Nanotechnol (2016) 12(8):1654–66. doi: 10.1166/jbn.2016.2269
122. Li Y, Chen Y, Li J, Zhang Z, Huang C, Lian G, et al. Co-delivery of microRNA-21 antisense oligonucleotides and gemcitabine using nanomedicine for pancreatic cancer therapy. Cancer Sci (2017) 108(7):1493. doi: 10.1111/cas.13267
123. Baião A, Sousa F, Oliveira AV, Oliveira C, Sarmento B. Effective intracellular delivery of bevacizumab: Via PEGylated polymeric nanoparticles targeting the CD44v6 receptor in colon cancer cells. Biomater Sci (2020) 8(13):3720–9. doi: 10.1039/D0BM00556H
124. Morillas-Becerril L, Peta E, Gabrielli L, Russo V, Lubian E, Nodari L, et al. Multifunctional, CD44v6-targeted ORMOSIL nanoparticles enhance drugs toxicity in cancer cells. Nanomaterials (2020) 10(2):298. doi: 10.3390/nano10020298
125. Yin W, Qian SM. CD44v6-O-MWNTS-loaded gemcitabine and CXCR4 siRNA improves the anti-tumor effectiveness of ovarian cancer. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.687322
126. Andrade F, Rafael D, Vilar-Hernández M, Montero S, Martínez-Trucharte F, Seras-Franzoso J, et al. Polymeric micelles targeted against CD44v6 receptor increase niclosamide efficacy against colorectal cancer stem cells and reduce circulating tumor cells in vivo. J Controlled Release (2021) 331:198–212. doi: 10.1016/j.jconrel.2021.01.022
127. Gurtner K, Hessel F, Eicheler W, Dörfler A, Zips D, Heider KH, et al. Combined treatment of the immunoconjugate bivatuzumab mertansine and fractionated irradiation improves local tumour control in vivo. Radiotherapy Oncol (2012) 102:444–9. doi: 10.1016/j.radonc.2011.10.013
128. Kennedy PJ, Sousa F, Ferreira D, Pereira C, Nestor M, Oliveira C, et al. Fab-conjugated PLGA nanoparticles effectively target cancer cells expressing human CD44v6. Acta Biomater (2018) 81:208–18. doi: 10.1016/j.actbio.2018.09.043
129. Fiedler W, Dedosso S, Cresta S, Weidmann J, Tessari A, Salzberg M, et al. A phase I study of PankoMab-GEX, a humanised glyco-optimised monoclonal antibody to a novel tumour-specific MUC1 glycopeptide epitope in patients with advanced carcinomas. Eur J Cancer (2016) 63:55–63. doi: 10.1016/j.ejca.2016.05.003
130. Ledermann J, Sehouli J, Zurawski B, Raspagliesi F, De Giorgi U, Banerjee S, et al. A double-blind, placebo-controlled, randomized, phase 2 study to evaluate the efficacy and safety of switch maintenance therapy with the anti-TA-MUC1 antibody PankoMab-GEX after chemotherapy in patients with recurrent epithelial ovarian carcinoma. Ann Oncol (2017) 28:v626. doi: 10.1093/annonc/mdx440.035
131. Garralda E, del Conte G, Macchini M, Matos I, Klinghammer KF, Gadea OSS, et al. Activity results of the GATTO study, a phase Ib study combining the anti-TA-MUC1 antibody gatipotuzumab with the anti-EGFR tomuzotuximab or panitumumab in patients with refractory solid tumors. Journal of Clinical Oncology (2021) 39(15_suppl):2522–2. doi: 10.1200/JCO.2021.39.15_suppl.2522
132. Meredith RF, Alvarez RD, Partridge EE, Khazaeli MB, Lin CY, Macey DJ, et al. Intraperitoneal radioimmunochemotherapy of ovarian cancer: A phase I study. Cancer Biother Radiopharm (2001) 16(4):305–15. doi: 10.1089/108497801753131381
133. Alvarez RD, Partridge EE, Khazaeli MB, Plott G, Austin M, Kilgore L, et al. Intraperitoneal radioimmunotherapy of ovarian cancer with177Lu-CC49: A phase I/II study. Gynecol Oncol (1997) 65(1):94–101. doi: 10.1006/gyno.1996.4577
134. Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer (2017) 5(1):22. doi: 10.1186/s40425-017-0222-9
135. You F, Jiang L, Zhang B, Lu Q, Zhou Q, Liao X, et al. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified Anti-MUC1 chimeric antigen receptor transduced T cells. Sci China Life Sci (2016) 59(4):386–97. doi: 10.1007/s11427-016-5024-7
136. Lin Y, Chen S, Zhong S, An H, Yin H, McGowan E. Phase I clinical trial of PD-1 knockout anti-MUC1 CAR-T cells in the treatment of patients with non-small cell lung cancer. Ann Oncol (2019) 30:xi12. doi: 10.1093/annonc/mdz448
137. Trabbic KR, Kleski KA, Shi M, Bourgault JP, Prendergast JM, Dransfield DT, et al. Production of a mouse monoclonal IgM antibody that targets the carbohydrate Thomsen-nouveau cancer antigen resulting in in vivo and in vitro tumor killing. Cancer Immunology Immunother (2018) 67(9):1437–47. doi: 10.1007/s00262-018-2206-0
138. Berois N, Pittini A, Osinaga E. Targeting tumor glycans for cancer therapy: successes, limitations, and perspectives. Cancers (2022) 14(3):645. doi: 10.3390/cancers14030645
139. Möller H, Serttas N, Paulsen H, Burchell JM, Taylor-Papadimitriou J, Meyer B. NMR-based determination of the binding epitope and conformational analysis of MUC-1 glycopeptides and peptides bound to the breast cancer-selective monoclonal antibody SM3. Eur J Biochem (2002) 269(5):1444–55. doi: 10.1046/j.1432-1033.2002.02787.x
140. Danielczyk A, Stahn R, Faulstich D, Löffler A, Märten A, Karsten U, et al. PankoMab: A potent new generation anti-tumour MUC1 antibody. Cancer Immunology Immunother (2006) 55(11):1337–47. doi: 10.1007/s00262-006-0135-9
141. Tarp MA, Sørensen AL, Mandel U, Paulsen H, Burchell J, Taylor-Papadimitriou J, et al. Identification of a novel cancer-specific immunodominant glycopeptide epitope in the MUC1 tandem repeat. Glycobiology (2007) 17(2):197–209. doi: 10.1093/glycob/cwl061
142. Numata Y, Nakada H, Fukui S, Kitagawa H, Ozaki K, Inoue M, et al. A monoclonal antibody directed to Tn antigen. Biochem Biophys Res Commun (1990) 170(3):981–5. doi: 10.1016/0006-291X(90)90488-9
143. Osinaga E, Bay S, Tello D, Babino A, Pritsch O, Assemat K, et al. Analysis of the fine specificity of Tn-binding proteins using synthetic glycopeptide epitopes and a biosensor based on surface plasmon resonance spectroscopy. FEBS Lett (2000) 469(1):24–8. doi: 10.1016/S0014-5793(00)01248-5
144. Ando H, Matsushita T, Wakitani M, Sato T, Kodama-Nishida S, Shibata K, et al. Mouse-human chimeric anti-tn igG1 induced anti-tumor activity against jurkat cells in vitro and in vivo. Biol Pharm Bull (2008) 31(9):1739–44. doi: 10.1248/bpb.31.1739
145. Matsumoto Y, Kudelka MR, Hanes MS, Lehoux S, Dutta S, Jones MB, et al. Identification of Tn antigen O-GalNAc-expressing glycoproteins in human carcinomas using novel anti-Tn recombinant antibodies. Glycobiology (2020) 30(5):282. doi: 10.1093/glycob/cwaa044
146. Mazal D, Lo-Man R, Bay S, Pritsch O, Dériaud E, Ganneau C, et al. Monoclonal antibodies toward different Tn-amino acid backbones display distinct recognition patterns on human cancer cells. Implications for effective immuno-targeting of cancer. Cancer Immunology Immunother (2013) 62(6):1107–22. doi: 10.1007/s00262-013-1425-7
147. Gong Y, Wolterink RGJK, Gulaia V, Cloosen S, Ehlers FAI, Wieten L, et al. Defucosylation of tumor-specific humanized anti-muc1 monoclonal antibody enhances nk cell-mediated anti-tumor cell cytotoxicity. Cancers (Basel) (2021) 13(11):2579. doi: 10.3390/cancers13112579
148. Sedlik C, Heitzmann A, Viel S, Ait Sarkouh R, Batisse C, Schmidt F, et al. Effective antitumor therapy based on a novel antibody-drug conjugate targeting the Tn carbohydrate antigen. Oncoimmunology (2016) 5(7):e1171434. doi: 10.1080/2162402X.2016.1171434
149. Rosenbaum P, Artaud C, Bay S, Ganneau C, Campone M, Delaloge S, et al. The fully synthetic glycopeptide MAG-Tn3 therapeutic vaccine induces tumor-specific cytotoxic antibodies in breast cancer patients. Cancer Immunology Immunother (2020) 69(5):703–16. doi: 10.1007/s00262-020-02503-0
150. Posey AD, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T cells targeting the cancer-associated tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity (2016) 44(6):1444. doi: 10.1016/j.immuni.2016.05.014
151. Gutierrez R, Shah PD, Hamid O, Garfall AL, Posey A, Bishop MR, et al. Phase I experience with first in class TnMUC1 targeted chimeric antigen receptor T-cells in patients with advanced TnMUC1 positive solid tumors. Journal of Clinical Oncology (2021) 39(15_suppl):e14513–3. doi: 10.1200/JCO.2021.39.15_suppl.e14513
152. Meredith RF, Partridge EE, Alvarez RD, Khazaeli MB, Plott G, Russell CD, et al. Intraperitoneal radioimmunotherapy of ovarian cancer with lutetium-177-CC49. J Nucl Med (1996) 37(9):1491–6.
153. Meredith RF, Khazaeli MB, Macey DJ, Grizzle WE, Mayo M, Schlom J, et al. Phase II study of interferon-enhanced 131I-labeled high affinity CC49 monoclonal antibody therapy in patients with metastatic prostate cancer. Clin Cancer Res (1999) 5(10 Suppl):3254s–8s.
154. Rogers BE, Roberson PL, Shen S, Khazaeli MB, Carpenter M, Yokoyama S, et al. Intraperitoneal radioimmunotherapy with a humanized anti-TAG-72 (CC49) antibody with a deleted CH2 region. Cancer Biother Radiopharm (2005) 20(5):502–13. doi: 10.1089/cbr.2005.20.502
155. Minnix M, Li L, Yazaki PJ, Miller AD, Chea J, Poku E, et al. TAG-72–targeted α-radionuclide therapy of ovarian cancer using 225Ac-labeled DOTAylated-huCC49 antibody. J Nucl Med (2021) 62(1):55. doi: 10.2967/jnumed.120.243394
156. Minnix M, Li L, Yazaki P, Chea J, Poku E, Colcher D, et al. Improved targeting of an anti-TAG-72 antibody drug conjugate for the treatment of ovarian cancer. Cancer Med (2020) 9(13):4756. doi: 10.1002/cam4.3078
157. Prendergast JM, Galvao da Silva AP, Eavarone DA, Ghaderi D, Zhang M, Brady D, et al. Novel anti-Sialyl-Tn monoclonal antibodies and antibody-drug conjugates demonstrate tumor specificity and anti-tumor activity. MAbs (2017) 9(4):615. doi: 10.1080/19420862.2017.1290752
158. Longenecker BM, Reddish M, Koganty R, Maclean GD. Immune responses of mice and human breast cancer patients following immunization with synthetic sialyl-tn conjugated to KLH plus detox adjuvant. Ann N Y Acad Sci (1993) 690(1):276–91. doi: 10.1111/j.1749-6632.1993.tb44016.x
159. Fung PYS, Madej M, Koganty RR, Longenecker BM. Active specific immunotherapy of a murine mammary adenocarcinoma using a synthetic tumor-associated glycoconjugate. Cancer Res (1990) 50:4308–14.
160. MacLean GD, Reddish MA, Koganty RR, Longenecker BM. Antibodies against mucin-associated sialyl-Tn epitopes correlate with survival of metastatic adenocarcinoma patients undergoing active specific immunotherapy with synthetic STn vaccine. J Immunother (1996) 19(1):59–68. doi: 10.1097/00002371-199601000-00007
161. Holmberg LA, Sandmaier BM. Vaccination with Theratope® (STn-KLH) as treatment for breast cancer. Expert Rev Vaccines (2014) 3(6):655–63. doi: 10.1586/14760584.3.6.655
162. MacLean GD, Miles DW, Rubens RD, Reddish MA, Longenecker BM. Enhancing the effect of THERATOPE STn-KLH cancer vaccine in patients with metastatic breast cancer by pretreatment with low-dose intravenous cyclophosphamide. J Immunother Emphasis Tumor Immunol (1996) 19(4):309–16. doi: 10.1097/00002371-199607000-00006
163. Hombach A, Heuser C, Sircar R, Tillmann T, Diehl V, Kruis W, et al. T cell targeting of TAG72+ tumor cells by a chimeric receptor with antibody-like specificity for a carbohydrate epitope. Gastroenterology (1997) 113(4):1163–70. doi: 10.1053/gast.1997.v113.pm9322511
164. McGuinness RP, Ge Y, Patel SD, Kashmiri SVS, Lee HS, Hand PH, et al. Anti-tumor activity of human T cells expressing the CC49-ζ chimeric immune receptor. Hum Gene Ther (1999) 10(2):165–73. doi: 10.1089/10430349950018968
165. Hombach AA, Rappl G, Abken H. Blocking CD30 on T cells by a dual specific CAR for CD30 and colon cancer antigens improves the CAR T cell response against CD30– tumors. Mol Ther (2019) 27(10):1825. doi: 10.1016/j.ymthe.2019.06.007
166. Shu R, Evtimov VJ, Hammett MV, Nguyen NYN, Zhuang J, Hudson PJ, et al. Engineered CAR-T cells targeting TAG-72 and CD47 in ovarian cancer. Mol Ther Oncolytics (2021) 20:325. doi: 10.1016/j.omto.2021.01.002
167. Slovin SF, Ragupathi G, Musselli C, Olkiewicz K, Verbel D, Kuduk SD, et al. Fully synthetic carbohydrate-based vaccines in biochemically relapsed prostate cancer: Clinical trial results with α-N-acetylgalactosamine-O- serine/threonine conjugate vaccine. J Clin Oncol (2003) 21(23):4292–8. doi: 10.1200/JCO.2003.04.112
168. Kuduk SD, Schwarz JB, Chen XT, Glunz PW, Sames D, Ragupathi G, et al. Synthetic and immunological studies on clustered modes of mucin-related Tn and TF O-linked antigens: The preparation of a glycopeptide-based vaccine for clinical trials against prostate cancer. J Am Chem Soc (1998) 120(48):12474–85. doi: 10.1021/ja9825128
169. Lo-Man R, Vichier-Guerre S, Bay S, Leclerc C. Anti-tumor immunity provided by a synthetic multiple antigenic glycopeptide displaying a tri-tn glycotope. J Immunol (2001) 166(4):2849–54. doi: 10.4049/jimmunol.166.4.2849
170. Laubreton D, Bay S, Sedlik C, Artaud C, Ganneau C, Dériaud E, et al. The fully synthetic MAG-Tn3 therapeutic vaccine containing the tetanus toxoid-derived TT830-844 universal epitope provides anti-tumor immunity. Cancer Immunology Immunother (2016) 65(3):315. doi: 10.1007/s00262-016-1802-0
171. Lakshminarayanan V, Thompson P, Wolfert MA, Buskas T, Bradley JM, Pathangey LB, et al. Immune recognition of tumor-associated mucin MUC1 is achieved by a fully synthetic aberrantly glycosylated MUC1 tripartite vaccine. Proc Natl Acad Sci USA (2012) 109(1):261–6. doi: 10.1073/pnas.1115166109
172. Miles D, Roché H, Martin M, Perren TJ, Cameron DA, Glaspy J, et al. Phase III multicenter clinical trial of the sialyl-TN (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist (2011) 16(8):1092. doi: 10.1634/theoncologist.2010-0307
173. Mei Z, Zhang K, Lam AKY, Huang J, Qiu F, Qiao B, et al. MUC1 as a target for CAR-T therapy in head and neck squamous cell carinoma. Cancer Med (2020) 9(2):640. doi: 10.1002/cam4.2733
174. Zhai X, You F, Xiang S, Jiang L, Chen D, Li Y, et al. MUC1-Tn-targeting chimeric antigen receptor-modified Vγ9Vδ2 T cells with enhanced antigen-specific anti-tumor activity. Am J Cancer Res (2021) 11(1):79.
175. Handgretinger R, Anderson K, Lang P, Dopfer R, Klingebiel T, Schrappe M, et al. A phase I study of human/mouse chimeric antiganglioside GD2 antibody ch14.18 in patients with neuroblastoma. Eur J Cancer (1995) 31A(2):261–7. doi: 10.1016/0959-8049(94)00413-y
176. Yu AL, Uttenreuther-Fischer MM, Huang CS, Tsui CC, Gillies SD, Reisfeld RA, et al. Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J Clin Oncol (1998) 16(6):2169–80. doi: 10.1200/JCO.1998.16.6.2169
177. Yu A, Batova A, Alvarado C, Rao V, Castleberry R. Usefulness of a chimeric anti-GD2 (ch14. 18) and GM-CSF for refractory neuroblastoma: a POG phase II study. Proc Am Soc Clin Oncol (1997) 27(1):85–91.
178. Simon T, Hero B, Faldum A, Handgretinger R, Schrappe M, Niethammer D, et al. Consolidation treatment with chimeric anti-GD2-Antibody ch14.18 in children older than 1 year with metastatic neuroblastoma. J Clin Oncol (2004) 22(17):3549–57. doi: 10.1200/JCO.2004.08.143
179. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med (2010) 363(14):1324. doi: 10.1056/NEJMoa0911123
180. Dhillon S. Dinutuximab: First global approval. Drugs (2015) 75(8):923–7. doi: 10.1007/s40265-015-0399-5
181. Markham A. Naxitamab: first approval. Drugs (2021) 81(2):291–6. doi: 10.1007/s40265-021-01467-4
182. Nazha B, Inal C, Owonikoko TK. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front Oncol (2020) 10:1000. doi: 10.3389/fonc.2020.01000
183. Cerato E, Birkle S, Portoukalian J, Mezazigh A, Chatal JF, Aubry J. Variable region gene segments of nine monoclonal antibodies specific to disialogangliosides (GD2, GD3) and their O-acetylated derivatives. Hybridoma (1997) 16(4):307–16. doi: 10.1089/hyb.1997.16.307
184. Terme M, Dorvillius M, Cochonneau D, Chaumette T, Xiao W, Diccianni MB, et al. Chimeric Antibody c.8B6 to O-Acetyl-GD2 Mediates the Same Efficient Anti-Neuroblastoma Effects as Therapeutic ch14.18 Antibody to GD2 without Antibody Induced Allodynia. PloS One (2014) 9(2):e87210. doi: 10.1371/journal.pone.0087210
Keywords: CD44v6, STn, O-GD2, Cancer, Targeted Therapy, O-glycans, Tn, GD2
Citation: Lodewijk I, Dueñas M, Paramio JM and Rubio C (2023) CD44v6, STn & O-GD2: promising tumor associated antigens paving the way for new targeted cancer therapies. Front. Immunol. 14:1272681. doi: 10.3389/fimmu.2023.1272681
Received: 04 August 2023; Accepted: 14 September 2023;
Published: 03 October 2023.
Edited by:
Yanqing Liu, Columbia University, United StatesReviewed by:
Xue Jiang, Duke University, United StatesCopyright © 2023 Lodewijk, Dueñas, Paramio and Rubio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolina Rubio, Q2Fyb2xpbmEuUnViaW9AZXh0ZXJub3MuY2llbWF0LmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.