
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 12 December 2023
Sec. Viral Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1272166
 Julia K. Büttner1
Julia K. Büttner1 Sara Becker2Annette Fink1
Sara Becker2Annette Fink1 Melanie M. Brinkmann3,4Rafaela Holtappels1
Melanie M. Brinkmann3,4Rafaela Holtappels1 Matthias J. Reddehase1†
Matthias J. Reddehase1† Niels A. Lemmermann1,2*†
Niels A. Lemmermann1,2*†CD8 T cells are important antiviral effectors in the adaptive immune response to cytomegaloviruses (CMV). Naïve CD8 T cells can be primed by professional antigen-presenting cells (pAPCs) alternatively by “direct antigen presentation” or “antigen cross-presentation”. In the case of direct antigen presentation, viral proteins are expressed in infected pAPCs and enter the classical MHC class-I (MHC-I) pathway of antigen processing and presentation of antigenic peptides. In the alternative pathway of antigen cross-presentation, viral antigenic material derived from infected cells of principally any cell type is taken up by uninfected pAPCs and eventually also fed into the MHC class-I pathway. A fundamental difference, which can be used to distinguish between these two mechanisms, is the fact that viral immune evasion proteins that interfere with the cell surface trafficking of peptide-loaded MHC-I (pMHC-I) complexes are absent in cross-presenting uninfected pAPCs. Murine cytomegalovirus (mCMV) models designed to disrupt either of the two presentation pathways revealed that both are possible in principle and can substitute each other. Overall, however, the majority of evidence has led to current opinion favoring cross-presentation as the canonical pathway. To study priming in the normal host genetically competent in both antigen presentation pathways, we took the novel approach of enhancing or inhibiting direct antigen presentation by using recombinant viruses lacking or overexpressing a key mCMV immune evasion protein. Against any prediction, the strongest CD8 T-cell response was elicited under the condition of intermediate direct antigen presentation, as it exists for wild-type virus, whereas the extremes of enhanced or inhibited direct antigen presentation resulted in an identical and weaker response. Our findings are explained by direct antigen presentation combined with a negative feedback regulation exerted by the newly primed antiviral effector CD8 T cells. This insight sheds a completely new light on the acquisition of viral immune evasion genes during virus-host co-evolution.
Cytomegaloviruses (CMVs) belong to the β-subfamily of the herpesviruses [reviewed in (1)]. The medical relevance of human CMV (hCMV) is based on its pathogenicity and the resulting multiple organ CMV disease in the absence of immune protection. Risk is associated with congenital infection of the fetus and infection of immunocompromised patients with genetic or acquired immunodeficiencies. Patients with hematopoietic malignancies who undergo hematoablative therapy with subsequent hematopoietic cell transplantation (HCT) are at risk of developing symptomatic manifestations in the period before full reconstitution. In the case of allogeneic HCT, the risk is further enhanced by immunosuppressive therapy aimed at preventing graft-versus-host disease (GvHD). Likewise, CMV disease poses a threat to recipients of allogeneic solid organ transplantation (SOT) immunosuppressed for preventing graft rejection [reviewed in (2, 3)].
The mild and mostly unnoticed infection in the immunocompetent host reflects a largely reduced viral pathogenicity due to the control of viral replication by antiviral effector mechanisms of innate and adaptive immunity. Particularly during transient immunodeficiency in HCT patients, efficient reconstitution of antiviral CD8 T cells is associated with positive prognosis in both clinical CMV infection (4) and in experimental models [(5), reviewed in (6)]. Accordingly, the adoptive transfer of antiviral CD8 T cells is a promising immunotherapeutic approach to prevent CMV pneumonia and other organ manifestations of CMV infection in HCT recipients (3, 7–11).
Due to the strict host-specific replication of CMVs, hCMV cannot be studied in animal models (12, 13). As a versatile model system for natural host-virus pairs, the infection of mice with murine CMV (mCMV) has been established by many groups for investigating basic principles of viral pathogenesis and antiviral immune control (14–24). These principles are shared between different pairs of CMVs and their respective hosts, as coevolution has led to biological convergences in host-virus adaptation [reviewed in (9)].
For both hCMV and mCMV, CD8 T cells have been identified as major effectors in preventing viral pathology during acute infection (25, 26). In addition, the importance of CD8 T cells in the long-term surveillance of latent mCMV infection (27) is suggested by the expansion of certain viral epitope-specific populations of activated KLRG1high CD62Llow CD8 T cells over time, a phenomenon termed “memory inflation (MI)” [for reviews, see (28–31)], and by an enhanced viral transcriptional activity during latency in the absence of such “inflating” CD8 T cells (32). Surprisingly, given the importance of CD8 T cells in the immune control of CMV and the interest in how MI is induced and maintained, the mechanisms underlying CMV-specific priming of naïve CD8 T cells are still not fully understood and remain controversial.
Viral antigenic peptides are presented, bound to MHC class-I (MHC-I) molecules as pMHC-I complexes, by professional antigen-presenting cells (pAPCs) to naïve CD8 T cells by two different mechanisms, direct presentation and cross-presentation. In the case of direct presentation, endogenous viral proteins are processed in infected pAPCs, whereas in the case of cross-presentation uninfected pAPCs take up exogenous antigens and introduce them into the MHC-I pathway of antigen processing and presentation (33). As a potential source of direct antigen presentation, mCMV can infect pAPCs, such as dendritic cells (DCs) (34, 35) and CD169+ macrophages (36).
However, all CMVs code for proteins that manipulate the MHC-I pathway of antigen presentation and are known as viral regulators of antigen presentation (vRAP) [(35), for more recent reviews see (19, 37)]. For mCMV, three vRAPs have been described: the positive regulator m04/gp34 and the negative regulators m06/gp48 and m152/gp40. Recent work has shown that m04 and m06, which belong to the same protein family (37), compete for pMHC-I cargo and annihilate each other in their function (38). For this reason, m152 remains as the functionally relevant immunoevasin of mCMV (38) that traps pMHC-I complexes in a cis-Golgi compartment (39–42), thereby reducing their number available for interaction with CD8 T-cell receptors (TCRs) (43). Notably, recent work has shown that a reduction of the number of cell surface pMHC-I molecules raises the avidity threshold required for TCRs of antiviral CD8 T cells to recognize infected cells and protect against infection [reviewed in (44)]. As a consequence, the recognition of infected cells by virus-specific CD8 T cells is impaired by the action of m152 in vitro and in vivo (35, 45, 46). Besides downmodulating pMHC-I, m152 has also been shown to interfere with cell surface expression of RAE-1 family ligands of the activating NK-cell receptor NKG2D (47–49), thereby preventing NK-cell activation (50, 51). Furthermore, m152 targets STING to reduce induction of type I antiviral interferons (52). This multi-functionality indicates a key role for m152 in subverting the antiviral defense.
Since vRAPs have been shown to be functional in pAPCs after hCMV and mCMV infection (34, 35, 53–55), it has been hypothesized that cross-presentation is the major pathway of antigen presentation leading to effective CD8 T-cell priming by counteracting viral immune evasion (56–58). Consistent with this, MHC-I-negative fibroblasts infected with a spread-deficient mCMV, which prevents direct presentation in the first round and precludes further rounds of infection, have been shown to activate mCMV-specific CD8 T cells after immunization of WT mice (59). Furthermore, Batf3-/- mice, which lack cross-presenting CD8α+ and CD107+ DCs, showed impaired priming (60). All these results clearly indicate that, in principle, cross-presentation can occur during mCMV infection.
On the other hand, there is evidence that priming by cross-presentation does not exclude a role for direct presentation. While Batf3-/- mice completely lack CD8+ DCs, which could contribute also to direct priming, infection of CD11c-Rac mice, which are selectively defective in the uptake of exogenous antigens by DCs, showed an mCMV-specific priming comparable to that in WT mice (57). In analogy, mCMV-specific priming was not impaired in mice treated with the TLR9 agonist CpG to prevent cross-presentation (61). In addition, there is evidence that the downregulation of pMHC-I is less efficient in DCs and macrophages than it is in fibroblasts or endothelial cells (62–64). Overall, it remained controversial whether direct or cross-presentation is the canonical pathway for the induction of CD8 T-cell responses to CMV.
Previous approaches are characterized by blocking either direct antigen presentation or cross-presentation, and thus were, by concept, unable to decide which pathway is taken with preference in normal mice. Here we present a novel approach in which priming of CD8 T cells is studied under conditions of enhanced or reduced immune evasion, compared to infection with WT virus, by using recombinant viruses in which the key immune evasion protein m152 is overexpressed or deleted, respectively. If priming is achieved by direct antigen presentation, the CD8 T-cell response is expected to be reduced after enhanced immune evasion and increased after reduced immune evasion. Surprisingly, our data on CD8 T-cell priming in a regional lymph node (RLN) did not reveal such a difference. Rather, up- or down-modulation of immune evasion gene expression led to the same response magnitude in the net effect. It thus appeared more than logical to conclude that priming is not by direct antigen presentation, thereby providing indirect evidence and an argument for priming by cross-presentation. However, this tempting conclusion ignores the fact that the modulation of direct antigen presentation has an influence on the recognition of infected cells by the newly primed CD8 T cells. In a negative feedback loop, a high number of CD8 T cells is efficiently primed by enhanced direct antigen presentation limiting viral replication and thus the number of APCs available for further rounds of T-cell stimulation. In contrast, a low number of CD8 T cells generated initially after reduced direct antigen presentation inefficiently limits viral replication and thus leads to a higher number of infected APCs driving further rounds of antigen presentation. From this “immune evasion paradox”, we conclude that direct antigen presentation is the canonical pathway of mCMV-specific CD8 T-cell priming within RLNs.
P815 (No. TIB-64, haplotype H-2d) and EL4 (No. TIB-39, haplotype H-2b) cells were obtained from the American Tissue Culture Collection (ATCC) and cultivated in RPMI supplemented with 5% fetal calf serum (FCS) and antibiotics, or in DMEM with 10% FCS and antibiotics, respectively. Primary murine embryo fibroblasts (MEF) were cultivated in MEM supplemented with 10% FCS and antibiotics. A CD8 T-lymphocyte line (CTLL) specific for the immunodominant viral epitope IE1 (YPHFMPTNL) (65) was generated from spleen-derived memory CD8 T cells of latently infected BALB/c mice by four rounds of restimulation with synthetic peptide (66).
Virus derived from BAC plasmid pSM3fr (67) was used as “wild-type” (WT) virus, mCMV-WT. Recombinant viruses mCMV-Δm152 (40), mCMV-Δm157 (68), and mCMV-m152StopΔm157 (52) have been described previously.
BALB/c, C57BL/6, and C57BL/6-Unc93b13d/3d [briefly Unc93b13d/3d (69)] mice were bred and housed under specified-pathogen-free (SPF) conditions in the Translational Animal Research Center (TARC) at the University Medical Center of the Johannes Gutenberg-University Mainz, Germany, and at the central animal facility of HZI Braunschweig, Germany.
Recombinant plasmids were constructed according to established procedures, and enzyme reactions were performed as recommended by the manufacturers. Throughout, the fidelity of PCR-based cloning steps was verified by sequencing (GATC, Freiburg, Germany).
Mutagenesis of full-length mCMV BAC plasmid pSM3fr was performed in DH10B by using the two-step replacement method as described (67, 70), resulting in the BAC plasmid pSM3fr_m152.IE+E. For this, the shuttle plasmid pST76K_ie2_m152 was used to integrate the open reading frame (ORF) m152 for ectopic expression under the control of the ie2 promoter. In the first step, the intermediate plasmid pST76K_ie1/3-ie2 was generated by subcloning a PmlI-cleaved 5,557bp fragment of pUCAMB (71), containing nucleotides 181,415 to 186,972 (GenBank accession no. NC_004065) of the mCMV immediate-early (IE) region into the SmaI site of the shuttle plasmid pST76-KSR (70). In a subsequent step, a 1,452bp PCR fragment, encompassing the ie2 promoter and ORF m152, was introduced into the HpaI cleaved vector to generate pST76K_ie2_m152. The fragment was generated by a touchdown PCR with primer pair m152-HpaI-fwd GAAGTTAAC184,240CATATAAAAGCTGTCCCCCATGCCATTCGA184,269-211,468TCAGACGCGGGCTACTCCCGAAAGAGTAAC211,439 and m152-HpaI-rev GGAGTTAAC210,056TGACTAATAAGTTATCTTTATTGTACAAGTGTTGTGTGTTATCCCTGAGCCCATTCCCAG210,115 (HpaI restriction sites are indicated in bold letters) using ProofStart Taq DNA polymerase (catalog no. 202205; QIAGEN, Hilden, Germany) and cycler conditions as follows: an initial step for 5 min at 95°C was followed by 18 cycles for 30 s at 94°C, 120 s with temperatures decreasing by 1°C per cycle starting from 62°C, and 90 s at 68°C each, followed by 12 cycles for 30 s at 94°C, 120 s at 45°C, and 90 s at 68°C.
Reconstitution and purification of a high-titer virus stock of mCMV-m152.IE+E was performed as described (72).
For in vitro assays, MEF were infected with the indicated viruses at a multiplicity of infection (MOI) of 4 with centrifugal enhancement of infectivity (72–74). Intraplantar infection of 8-to-10-week-old mice was performed in the left hind footpad with 1x105 PFU of the respective virus.
For log-linear in vivo virus growth curves, BALB/c mice were immunocompromised by hematoablative treatment with a single 6.5 Gy dose of total-body γ-irradiation and were infected with the respective virus. Quantification of viral genome load in lungs and spleen was performed on days 2, 4, 6, 8, and 10 by qPCR as described previously (72).
Depletion of NK cells or of CD8 T cells was performed 24 h prior to infection by i.v. injection of 25µl rabbit antiserum directed against asialo-GM1 (catalog no. 986-100001; Wako Chemicals, Osaka, Japan) or of 1mg purified antibody directed against CD8 (clone YTS169.4), respectively (75).
To determine viral genome load in lungs and spleen, DNA of infected mice was isolated from the respective tissues with the DNeasy blood & tissue kit (catalog no. 69504; QIAGEN) according to the manufacturer’s instructions. Viral and cellular genomes were quantitated in absolute numbers by M55-specific and pthrp-specific qPCRs normalized to a log10-titration of standard plasmid pDrive_gB_PTHrP_Tdy (72, 76).
Viral transcripts were quantitated from total RNA extracted from infected MEF or from lymph nodes (75), and 500 ng RNA was used as template for RT-qPCR. Absolute quantification of E1 or m152 transcripts using in vitro transcripts as standard has been described previously (77). Spliced E1 transcripts (78, 79) were chosen as a surrogate for viral replication that otherwise would be confounded by inoculum viral DNA. It is important to recall that a PFU of mCMV equals 500 copies of viral genomic DNA (73), so that intraplantar infection with 1x105 PFU corresponds to 5x107 copies, which critically confounds the quantitation of de novo viral DNA replication in the RLN, in particular at early times. E1 (M112-113) expression is essential for viral DNA replication (80) and the quantity of E1 transcripts correlates with the number of infected cells.
Custom peptide synthesis to a purity of > 80% was performed by JPT Peptide Technologies (Berlin, Germany). Synthetic peptides representing antigenic peptides in mouse haplotype H-2b were M38 (SSPPMFRVP), M45 (HGIRNASFI), M57 (SCLEFWQRV), M122/IE3 (RALEYKNL), m139 (TVYGFCLL), and m141 (VIDAFSRL) (81). Those for mouse haplotype H-2d were m04 (YPGSLYRRF), m18 (SGPSRGRII), M45 (VGPALGRGL), M83 (YPSKEPFNF), M84 (AYAGLFTPL), M105 (TYWPVVSDI), m123/IE1 (YPHFMPTNL), m145 (CYYASRTKL), and m164 (AGPPRYSRI) (8). The synthetic peptides were used for exogenous loading of target cells in the ELISpot assay.
An interferon gamma (IFNγ) enzyme-linked immunospot (ELISpot) assay was performed for quantification of IFNγ-secreting CD8 T cells after sensitization by peptide-loaded stimulator cells. Frequencies of mCMV-specific CD8 T cells were determined by incubation of graded numbers of immunomagnetically-purified CD8 T cells, derived from the RLN, which is the popliteal lymph node in the case of intraplantar infection, with stimulator cells (P815 or EL4, as it applied) that were exogenously loaded with synthetic peptides at a saturating concentration of 10-7M (75). IE1 epitope presentation after endogenous antigen processing in infected BALB/c MEF was determined using short-term IE1-CTLL (82) as responder cells. Spots were counted automatically based on standardized criteria using Immunospot S4 Pro Analyzer (CTL, Shaker Heights, OH, USA) and CTL-Immunospot software V5.1.36. Frequencies of IFNγ-secreting cells and the corresponding 95% confidence intervals were calculated by intercept-free linear regression using Mathematica, version 8.0.4.
For a selective arrest of viral gene expression in the IE phase, MEFs were infected in the presence of 50µg/ml cycloheximide (CHX) to block protein synthesis reversibly. At 3 h after infection, the culture medium was replaced by fresh medium containing 5 µg/µl actinomycin D (ActD) as described previously (83).
IE1-CTLL (5x105 cells) were co-cultivated with 1x105 mCMV-infected, IE phase-arrested MEFs (BALB/c, haplotype H-2d) for 5h at 37°C in the presence of brefeldin A (BD GolgiPlug, final concentration 1:1,000; catalog no. 555029; BD Biosciences). Thereafter, the CTLL cells were fixed, permeabilized with BD Cytofix/Cytoperm (catalog no. 554722, BD Biosciences) and stained with FITC-conjugated anti-mouse IFNy antibody (clone XMG1.2, catalog no. 554411, BD Biosciences) for cytofluorometric (CFM) analysis performed with flow cytometer Cytomics FC500 and CXP analysis software (Beckman Coulter).
An mCMV ORF library of expression plasmids spanning the entire mCMV genome (81) was used for ORF-specific stimulation of ex vivo isolated CD8 T cells with transfected SV-40 fibroblasts, followed by CFM detection of intracellular IFNγ.
The expression of mCMV proteins was detected by Western blot analysis as described (40). In brief, lysates of infected MEF were prepared and 30µg of total protein amount was subjected to separation by 12.5% SDS-PAGE, followed by blotting onto polyvinylidene difluoride membrane and protein labeling with the respective antibodies. The following antibodies were used: m152 (M3D10, monoclonal antibody, kindly provided by E. Kremmer, Helmholtz Zentrum München, Munich, Germany), IE1 (Croma 101, monoclonal antibody, kindly provided by S. Jonjic, University of Rijeka, Rijeka, Croatia), and IE2 (αIE2-N, polyclonal antibody, rabbit, Peptide Specialty Laboratories, Heidelberg, Germany). Detection of antibody binding was visualized by chemiluminescence using the ECLplus Western blotting detection system (catalog no. RPN2132; Amersham Bioscience, Little Chalfont, United Kingdom) and Lumi-Film (catalog no. 11666657001, Roche Applied Science, Mannheim, Germany).
To evaluate statistical significance of differences between two independent sets of data, the unpaired t-test with Welch’s correction of unequal variances was used. Differences are considered statistically significant for P values <0.05 and highly significant for P values <0.001. In cohort analyses of viral epitope-specific CD8 T cells by the ELISpot assay, differences are considered statistically significant if 95% confidence intervals do not overlap.
Virus doubling times (vDT) and their 95% confidence intervals (CI) were calculated from the slopes of log-linear regression lines determined by linear regression analysis (84–86). Calculations were performed with Graph Pad Prism 10 (Graph Pad Software, San Diego, CA). It should be noted that vDT values vary between different organs but are a constant for each organ independent of the viral replication parameter tested, that is, identical for viral genomic DNA copy numbers measured by qPCR, infectious virus expressed as PFU, or numbers of infected tissue cells determined by immunohistological detection of viral proteins (85).
Conclusions on the mechanism of CD8 T-cell priming are usually based on measuring the magnitude of the primary immune response. This includes the tacit assumption that antigen presentation requirements are identical for sensitization of naïve CD8 T cells by antigen presentation in the immunological synapse (87, 88), which is the initial priming event that requires pAPCs, and for subsequent clonal expansion. Accordingly, the terms of “priming” and “primary response” are usually used as synonyms, not least because the initial priming event is difficult to access. Thus, while a response indicates a successful initial priming, one must keep in mind that one actually looks at the combined result of priming and subsequent clonal expansion.
To provide evidence for or against either of the two pathways of antigen presentation, we studied here the mCMV-specific CD8 T-cell response in an RLN draining a site of local infection, specifically in the popliteal lymph node after intraplantar infection. The RLN is the anatomical structure where direct priming is said to occur in the peripheral interfollicular region (89–91). Our work builds on a previous study (75) in which we have shown that mCMV rapidly reaches the RLN, infects cells in the peri-subcapsular sinus region, and generates virus-specific effector CD8 T cells as early as by day 3 after virus exposure. Notably, this finding is in perfect accordance with the more recent study by Reynoso and colleagues (92) showing that lymph node conduits rapidly transport virions to infect pAPCs in the RLN paracortex followed by rapid direct T-cell priming within the T-cell zone.
In the specific case of CMV infections, however, viral interference with the MHC-I pathway of direct antigen presentation was expected to prevent or at least severely inhibit CD8 T-cell priming. To test this prediction, we analyzed the mCMV-specific CD8 T-cell response in Unc93b13d/3d mice. This strain on C57BL/6 genetic background is known to lack endosomal TLR3, 7, and 9 signaling and is impaired in exogenous antigen processing, resulting in a blockade of cross-presentation (69, 93–96). Accordingly, CD8 T-cell priming in these mice ought to largely depend on direct presentation. Previous reports on mCMV infection in this mutant mouse strain revealed an impaired cytokine production but comparable frequencies of M45-specific hepatic CD8 T cells (69, 97). Herein, we compared the CD8 T-cell response in C57BL/6 WT and Unc93b13d/3d mice not just for the M45 epitope but for a panel of known mCMV peptides presented in the H2b haplotype (Figure 1).
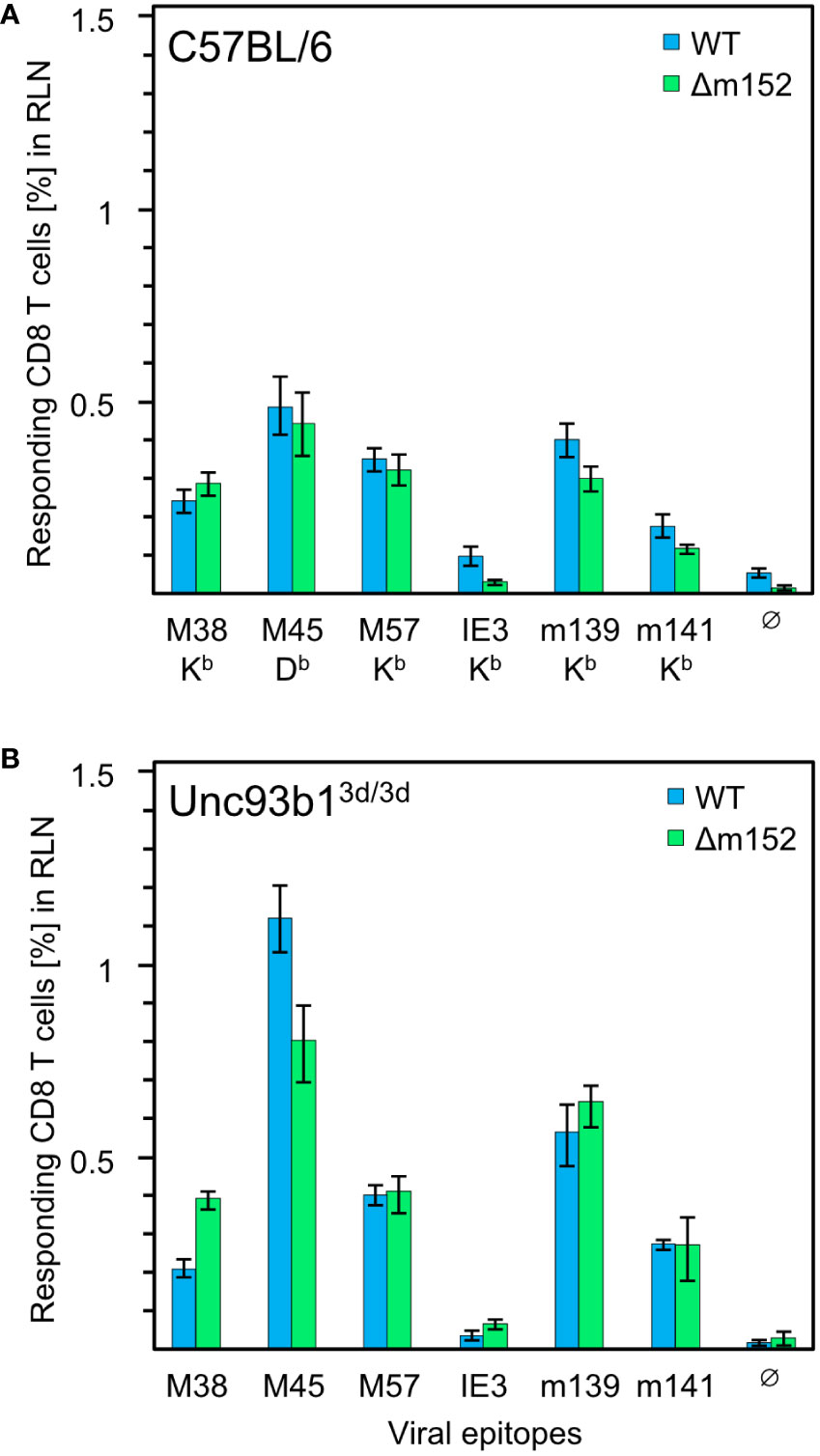
Figure 1 CD8 T-cell response induced by mCMV in presence and absence of the cross-presentation pathway. (A) CD8 T-cell response in C57BL/6 mice (n=5 per group/virus, tested as cohorts). (B) CD8 T-cell response in Unc93b13d/3d mice (n=4 per group/virus, tested as cohorts) that are deficient in antigen cross-presentation. CD8 T cells were isolated from the draining regional lymph node (RLN), that is, the popliteal lymph node, on day 7 after intraplantar infection with 1x105 PFU each of either mCMV-WT (WT) or mCMV-Δm152 (Δm152), and used as effector cells in an IFNγ-based ELISpot assay. EL4 cells exogenously loaded with synthetic peptides at a saturating concentration of 10−7 M were used as APCs. The panel of tested antigenic peptides and the corresponding peptide-presenting MHC-I molecules are indicated. Bars represent cohort average most probable numbers (MPN) of responding cells determined by intercept-free linear regression analysis, and error bars represent the corresponding 95% confidence intervals. Test groups are considered significantly different if the 95% confidence intervals do not overlap. Ø, no peptide added.
Overall, no qualitative differences in the immunodominance patterns were found. To our surprise, the magnitude of the CD8 T-cell response to some of the peptides tested, most markedly for M45, was even slightly higher in Unc93b13d/3d mice, but certainly not lower. This finding is consistent with data reported for C57BL/6 mice in which cross-presentation was suppressed by CpG pretreatment (61). In summary, the magnitude of the primary CD8 T-cell response is surprisingly not at all inhibited by viral interference with direct antigen presentation, which is known to be effective at the cellular level following infection with mCMV-WT in several cell types tested, including pAPCs, and in mice of haplotypes H-2b and H-2d (35, 98).
Given the remarkable finding that a CD8 T-cell response occurs despite genetic prevention of cross-presentation and despite viral interference with direct antigen presentation, we wondered whether the magnitude of the response would at least benefit from improved direct antigen presentation. For this, we compared infection with mCMV-WT and the immune evasion gene deletion mutant mCMV-Δm152 both under conditions with an open (Figure 1A) or closed (Figure 1B) antigen cross-presentation pathway in C57BL/6 and Unc93b13d/3d mice, respectively. The result was most striking: for the entire panel of viral epitopes tested, improved direct antigen presentation failed to improve the response, regardless of whether the antigen cross-presentation pathway was accessible or not. For C57BL/6 mice, earlier work by the group of A.B. Hill (98, 99) has already shown that an enhancement of direct antigen presentation by deletion of the viral key immune evasion gene m152 has little impact on the magnitude of the CD8 T-cell response, having suggested that priming may rather depend on antigen cross-presentation. Based on the data in Unc93b13d/3d mice, however, this explanation has now become obsolete, and we are faced with the riddle that the CD8 T-cell response in mice with the genetic background of C57BL/6 is resilient and buffered in that it neither depends on antigen cross-presentation nor does it appear to be notably influenced by enhanced direct antigen presentation.
Selectively in mice with the genetic background of C57BL/6, the viral protein m157 restricts viral replication by serving as an activatory ligand of Ly49H+ NK cells (100, 101). It has been shown that the activation of Ly49H+ NK cells by m157 suppresses the mCMV-specific CD8 T-cell response, most likely by reducing the number of infected pAPCs available for direct antigen presentation (102). Thus, Ly49H+ NK cells are a relevant player that certainly contributes to the magnitude of the CD8 T-cell response in C57BL/6 mice.
For clarification, we investigated the impact of NK cells on the CD8 T-cell response in the draining RLN measured on day 7 after intraplantar infection of C57BL/6 mice, and correlated the response magnitude with viral replication at this site on day 3 (Figure 2). In the presence of NK cells and CD8 T cells, mCMV-WT and mCMV-Δm152 induced similar CD8 T-cell responses (Figure 2A1), consistent with the preceding, independent experiment (Figure 1A). Notably, transcription of viral gene E1, which serves as a surrogate for viral replication, was identical for both viruses and on a low level (Figure 2A2). In contrast, pan-NK cell depletion prior to infection led to an increase in the response magnitude for the immunodominant viral peptides M45, M57, and m139, but only when direct antigen presentation was enhanced by deletion of m152 (Figure 2B1). As it was predicted, depletion of NK cells led to a greatly enhanced viral replication in the RLN (Figure 2B2) compared to the control group with no NK-cell depletion (Figure 2A2). Deletion of m152 resulted in a slight, but statistically significant, reduction in viral replication compared to WT virus infection (Figure 2B2). This reduction was caused by recently primed CD8 T cells, as the difference was abolished by the depletion of CD8 T cells (Figure 2C).
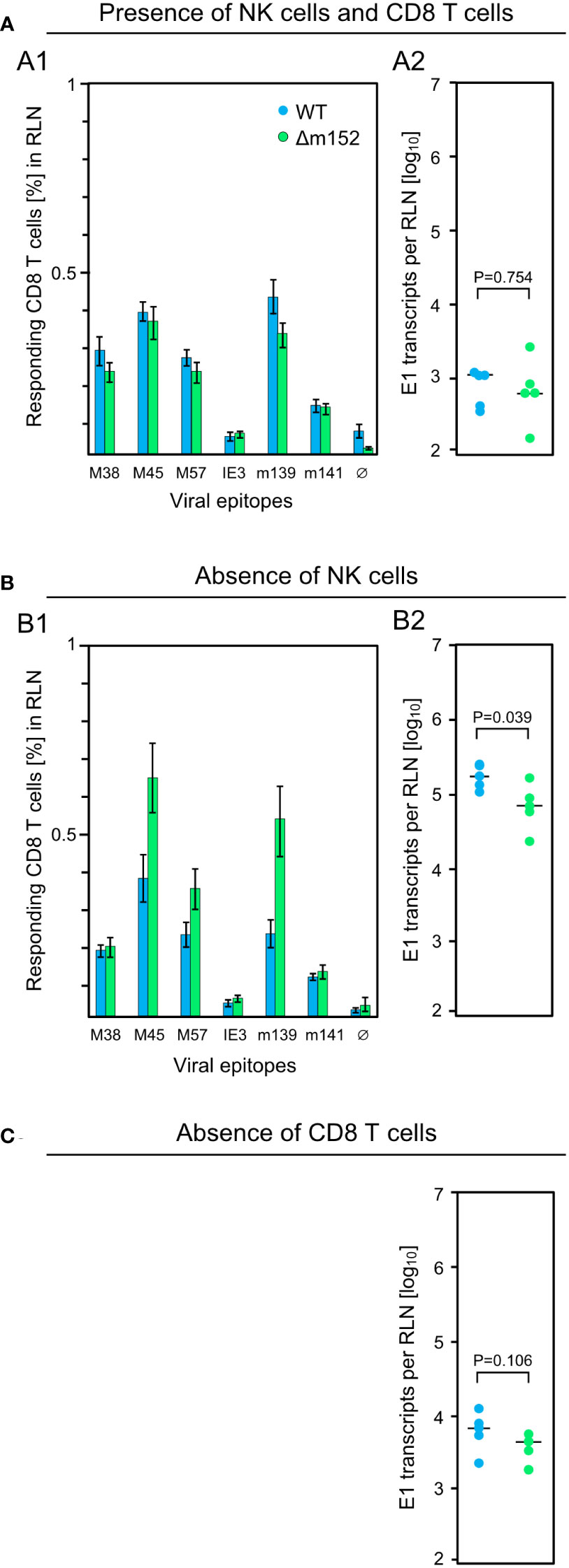
Figure 2 Influence of lymphocyte subsets on the CD8 T-cell response and the intranodal viral replication in C57BL/6 mice. Intraplantar infection of immunocompetent C57BL/6 mice (n=5 per group) was performed with 1x105 PFU each of either mCMV-WT (WT) or mCMV-Δm152 (Δm152). (A) No depletion of lymphocyte subsets. (B) Depletion of NK cells one day before infection. (C) Depletion of CD8 T cells one day before infection. The CD8 T-cell response in the RLN on day 7 post-infection was assessed by an IFNγ-based ELISpot assay (A1, B1), as explained in greater detail in the legend of Figure 1. As a surrogate for viral replication in the presence of otherwise confounding viral inoculum DNA, spliced E1 transcripts present in the RLN were quantitated by RT-qPCR on day 3 post-infection (A2, B2, C). Symbols represent individual mice and horizontal bars indicate median values. P values were calculated based on the log-transformed data with Welch´s unpaired t-test (two-sided). Differences are considered significant for P < 0.05.
Upon first impression, it may be irritating that not all viral epitopes show the same response pattern. Epitope hierarchy, however, is a general observation and is explained by differences in many consecutive steps in the MHC-I pathway of antigen processing and presentation. Key variables, which are even used in epitope prediction algorithms, include the efficacy of peptide-generating proteasomal cleavage and peptide binding affinity to the presenting MHC-I molecule. Cell surface density of pMHC-I complexes determines the cooperative TCR binding avidity and thus the intensity and duration of TCR signaling. This in turn defines the proliferation rate of CD8 T cells for clonal expansion and thus, finally, the response magnitude. As a consequence, differences that do not reach statistical significance for subdominant viral epitopes can reach statistical significance for dominant viral epitopes. For this reason, we always tested a panel of epitopes, and conclusions must be drawn from the overall picture rather than from single epitopes. Our data did not reveal a critical influence of the type of the presenting MHC-I molecule, which are Kb and Db in C57BL/6 mice.
These findings were essentially reproduced in an independent experiment using the alternative approach of testing the influence of m152 on the response magnitude in infected C57BL/6 mice in the absence specifically of Ly49H+ NK cell activation via Ly49H-m157 ligation, instead of pan-NK cell depletion (Figure 3). For this, we compared response magnitude and viral replication in the RLN after infection with mCMV-Δm157, expressing m152, and mCMV-m152StopΔm157, lacking m152 expression, both in the absence of Ly49H+ NK cell activation. Notably, deletion of m152 increased the overall CD8 T-cell response (Figure 3A1), more or less paralleling the findings after pan-NK cell depletion (Figure 2B). Under these conditions, too, early recognition of infected cells is indicated by a reduction in viral replication in the RLN after infection with mCMV-m152StopΔm157 (Figure 3A2). Again, this antiviral control in the absence of Ly49H+ NK cell activation is mediated by recently primed CD8 T cells, as depletion of CD8 T cells led to an increase in viral replication in the RLN (Figure 3B compared to Figure 3A2).
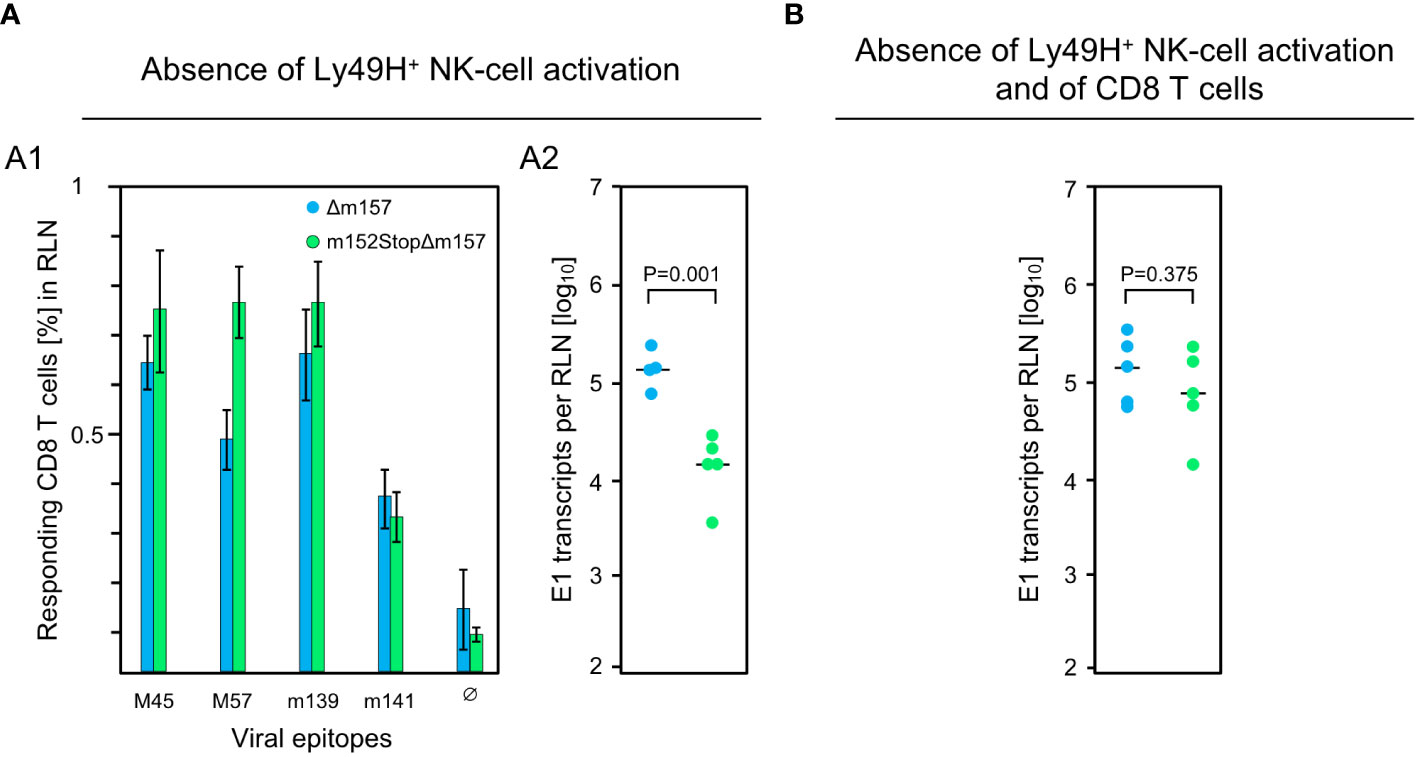
Figure 3 Impact of Ly49H+ NK cells on CD8 T-cell response and intranodal viral replication in C57BL/6 mice. (A) Absence of Ly49H+ NK-cell activation. (B) Absence of Ly49H+ NK-cell activation and of CD8 T cells. Intraplantar infection of immunocompetent C57BL/6 mice (n=5 per group) was performed with 1x105 PFU each of either mCMV-Δm157 (Δm157) or mCMV-m152StopΔm157 (m152StopΔm157). (A1) The CD8 T-cell response in the RLN on day 7 post-infection was assessed by an IFNγ-based ELISpot assay, as explained in greater detail in the legend of Figure 1. (A2, B) Spliced E1 transcripts present in the RLN on day 3 post-infection were quantitated by RT-qPCR. Depletion of CD8 T cells in (B) was performed on the day before infection. Symbols represent data from individual mice and horizontal bars indicate median values. P values were calculated based on the log-transformed data with Welch´s unpaired t-test (two sided). Differences are considered significant for P < 0.05.
The results of the two approaches are consistent in having demonstrated that early viral replication in the RLN of C57BL/6 mice is mainly controlled by Ly49H+ NK cells. While an enhanced direct antigen presentation did not improve the overall CD8 T-cell response magnitude in the presence of the NK-cell response (Figure 2A1), depletion or missing activation of NK cells disclosed an improvement (Figures 2B1, 3A1). Importantly, a high CD8 T-cell response corresponded to low intranodal viral replication, and a low response corresponded to high viral replication (Figures 2B, 3A). This clearly argues against antigen cross-presentation for which the opposite should apply: high virus replication and thus high amounts of antigenic proteins available for uptake by uninfected pAPCs ought to correspond to a high CD8 T-cell response and, accordingly, low virus replication ought to correspond to a low CD8 T-cell response. In summary, although masked by the strong activity of Ly49H+ NK cells, CD8 T cells in C57BL/6 mice are primed primarily by direct antigen presentation.
Since the mCMV-specific CD8 T-cell response in C57BL/6 mice is masked by the strong response of Ly49H+ NK cells, we decided to switch to the analysis of the mCMV-specific CD8 T-cell response in BALB/c mice, which do not express Ly49H and accordingly lack this functionally dominant subset of NK cells. Based on the evidence that the CD8 T-cell response in C57BL/6 mice is driven by direct antigen presentation (see above), and assuming that this is also the case in BALB/c mice, we reasoned that the best evidence for direct antigen presentation would be to show that enhanced and inhibited direct antigen presentation correspond to a high and low CD8 T-cell response, respectively. Increased direct antigen presentation compared to infection with the WT virus is achieved by deletion of the immune evasion gene m152 in virus mCMV-Δm152. For a more strongly inhibited direct antigen presentation compared to infection with the WT virus, we constructed a “super-evasion” virus mCMV-m152.IE+E as a new study tool.
In mCMV-WT infection, m152 is expressed quite early in the Early (E) phase of the viral replication cycle (24, 39). Antigens expressed even earlier, that is, in the Immediate-Early (IE) phase, may profit from a head start advantage of presentation before immune evasion can operate. After infection with mCMV-m152.IE+E, m152 is expressed from the IE phase onward.
Ectopic expression in the IE phase was achieved by insertion mutagenesis placing ORF m152 under the control of the ie2 enhancer-promoter, thereby disrupting the ie2 gene. Expression as an E phase protein occurred from its authentic position in the mCMV genome (Figure 4A). For verifying immune evasion operating already in the IE phase, viral gene expression in BALB/c-derived fibroblasts was metabolically arrested in the IE phase (Figure 4B1). This led to an enhanced synthesis of IE proteins IE1 and IE2, and absence of the m152 protein, after infection with either mCMV-WT or the deletion mutant mCMV-Δm152, whereas all three glycosylation isoforms of m152 (40) were strongly expressed and IE2 was absent after infection with mCMV-m152.IE+E (Figure 4B2, ). Presentation of the antigenic peptide IE1 (YPHFMPTNL, presented by Ld) was tested functionally with an IE1 epitope-specific CTLL (IE1-CTLL) used as responder cells in an IFNγ-based ELISpot assay with IE phase-arrested infected cells as stimulator cells (Figure 4B3) as well as by intracellular cytofluorometric staining of IFNγ in IE1-CTLL cells sensitized by IE phase-arrested infected cells (Figure 4B4). In both assays, IE1 peptide was presented without significant difference after infection with either mCMV-WT or the mCMV-Δm152 deletion mutant, since m152 is not expressed in the IE phase anyway. In contrast, presentation was blocked in IE phase-arrested cells after infection with mCMV-m152.IE+E (Figures 4B3, B4).
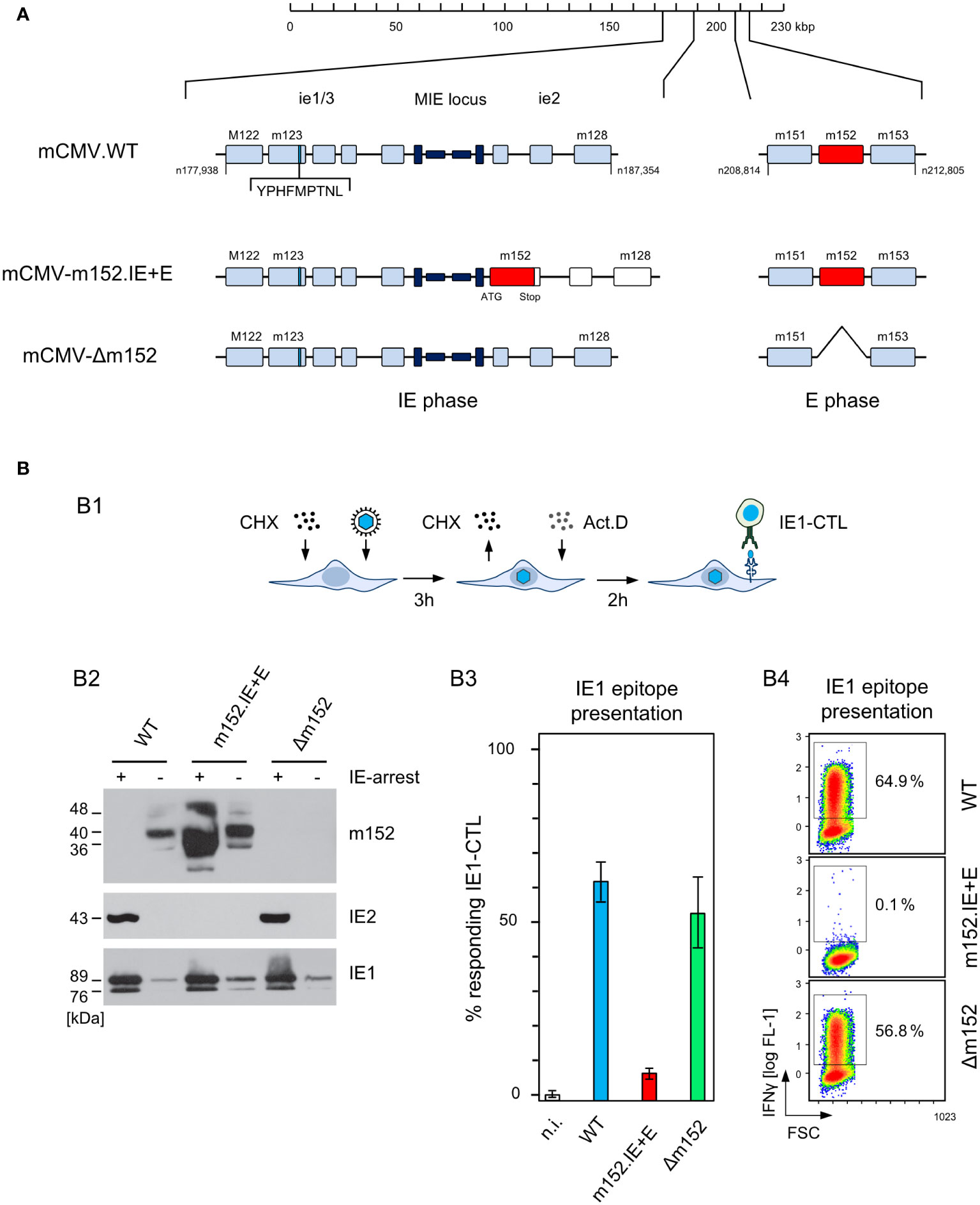
Figure 4 Construction and in vitro characterization of recombinant virus mCMV-m152.IE+E. (A) Maps, illustrating the mutagenesis design. A map of the mCMV genome is shown at the top of the sketch. The genomic region that includes the parental E phase gene m152 is shown expanded on the right-hand side. The “Major Immediately Early (MIE)” locus, consisting of the MIE promotor-enhancer-enhancer-promotor (dark blue) flanked by transcription units ie1/3 and ie2, is shown expanded on the left-hand side. Exons are indicated by boxes (light blue). The coding sequence of the IE1 peptide located in exon 4 of the ie1/3 transcription unit is marked. Recombinant virus mCMV-m152.IE+E, expressing m152 in both the IE phase and the E phase, was generated by introducing the ORF m152 by two-step BAC mutagenesis into the MIE locus under the control of the ie2 enhancer-promoter element, thereby disrupting the ie2 gene. (B) Immune evasion under selective IE phase conditions. (B1) Experimental protocol for arresting infection in the IE phase. BALB/c-derived mouse embryonal fibroblasts (MEF) were infected in the presence of cycloheximide (CHX) that was replaced at 3 h post-infection with actinomycin D (ActD). The thus IE phase-arrested MEF were harvested for analyses at 5 h post-infection. (B2) Western blot analysis of m152 (40kDa and additional glycosylation isoforms), IE2 (43kDa) and IE1 (89/76kDa) protein expression in IE phase-arrested (+) or untreated (–) MEF infected with the indicated viruses. (B3) IFNγ-based ELISpot analysis quantifying cells of an IE1-CTLL that were sensitized by IE phase-arrested BALB/c-derived MEF infected with the indicated viruses. Bars represent most probable numbers (MPN) of responding cells determined by intercept-free linear regression analysis, and error bars represent the corresponding 95% confidence intervals. Test groups are considered significantly different if the confidence intervals do not overlap. (n.i.) not infected. (B4) Intracellular IFNγ-staining of IE1-CTLL cells at 5 h after co-cultivation with IE phase-arrested MEF infected with the indicated viruses. Two-dimensional color-coded density plots (with red and blue representing highest and lowest density, respectively) show intracellular IFNγ expression (ordinate; fluorescence intensity) versus the forward scatter (abscissa; FSC, linear scale of channels) with 50,000 cells analyzed. The percentages of IFNγ+ cells present in the demarcated gates are indicated.
Since infected cells in vivo are not normally arrested in the IE phase, we studied the kinetics of m152 transcription in untreated cells infected with either mCMV-WT, expressing m152 only in the E phase and onward, or mCMV-m152.IE+E, expressing m152 additionally already in the IE phase (Figure 5A). The transcription data were then correlated with presentation of the antigenic IE1 peptide detected by sensitization of IE1-CTLL cells (Figure 5B). In accordance with the concept of constructing mCMV-m152.IE+E, m152 was expressed faster and inhibited antigen presentation earlier compared to infection with mCMV-WT. Specifically, at both times chosen, the IE1 peptide was not detectably presented by cells infected with mCMV-m152.IE+E, whereas it was always presented after infection with mCMV-Δm152. Under conditions of “physiological” immune evasion gene expression, as it applies to infection with mCMV-WT, the IE1 peptide was presented early in the time course but almost absent later (Figure 5B). We thus conclude that direct antigen presentation is largely inhibited at early and later times in cells infected with mCMV-m152.IE+E, whereas it is not inhibited at any time after infection with mCMV-Δm152, and only at later times after infection with mCMV-WT.
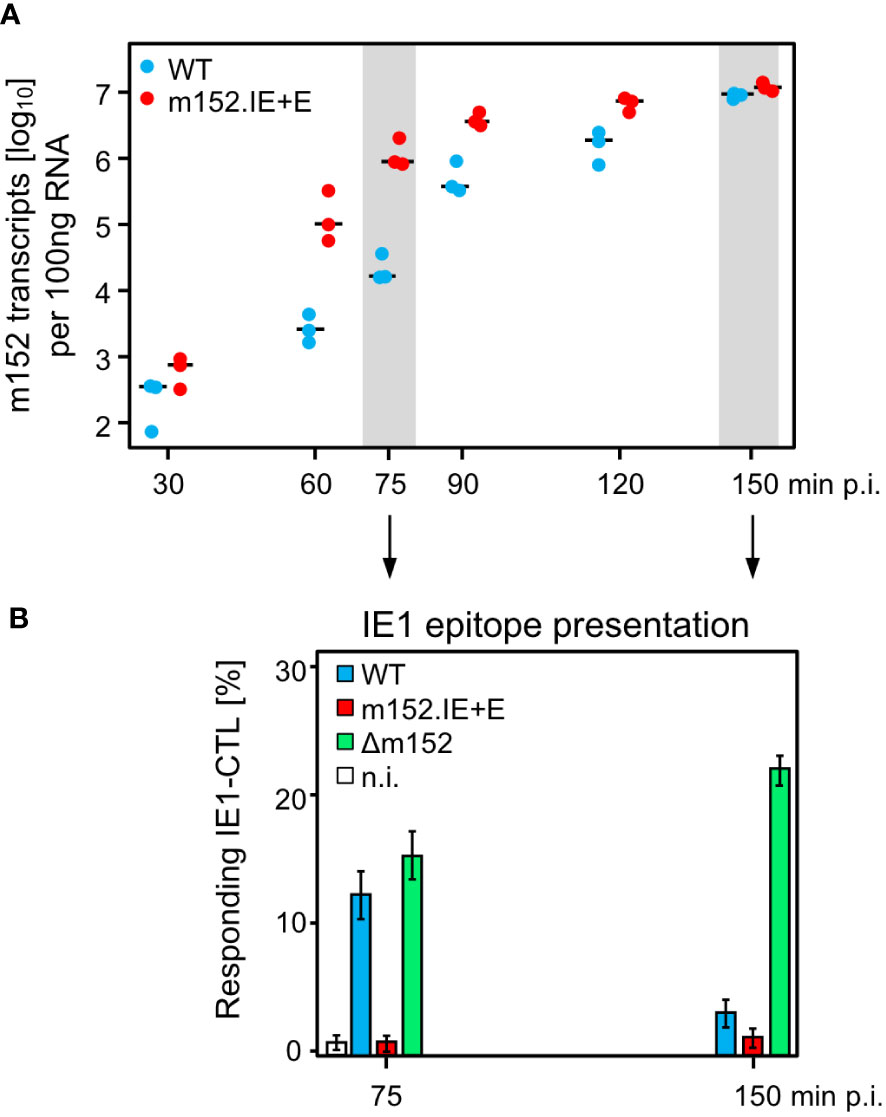
Figure 5 mCMV-m152.IE+E infection inhibits IE1 peptide presentation from the start of viral gene expression. (A) Quantitation of m152 transcripts in the time-course. BALB/c-derived MEF were infected with the indicated viruses and m152 transcripts were quantitated by RT-qPCR, normalized to 100 ng of total RNA, at the indicated times post-infection. Symbols represent biological replicates (n=3). Median values are indicated by horizontal bars. (B) IE1-peptide presentation at selected times. IE1-peptide presentation was detected at the indicated times based on sensitization of IE1-CTLL cells in an IFNγ-based ELISpot assay by BALB/c-derived MEF infected with the viruses indicated. To avoid ongoing transcription in the stimulator cells during the ELISpot assay time, the transcription inhibitor ActD was added at the indicated times of cell harvest. Bars represent the percentage of responding cells, and error bars indicate the 95% confidence intervals determined by intercept-free linear regression analysis. Test groups are considered significantly different if the 95% confidence intervals do not overlap. (n.i.) no infection.
BALB/c mice are competent in both antigen presentation pathways and therefore theoretically have the choice between direct antigen presentation and antigen cross-presentation. This raised the question of which pathway is used as the canonical pathway for priming of naïve CD8 T cells and for subsequent clonal expansion or whether the pathways can replace each other in case of need. Assuming that direct antigen presentation is the preferred mode of priming, one expects a response magnitude in a rank order defined by the quantity of pMHC-I complexes on the surface of infected APCs and thus reciprocal to the strength of immune evasion. Specifically, the response should be strongest after infection with mCMV-Δm152, intermediate after infection with mCMV-WT, and lowest after infection with mCMV-m152.IE+E.
The results of the experiment (Figure 6A) did not match this prediction. For all antigenic peptides tested, the magnitude of the CD8 T-cell response was essentially the same for the extremes of lowest and highest direct antigen presentation after infection with mCMV-m152.IE+E and mCMV-Δm152, respectively (Figure 6A). It seemed to be an obvious conclusion that direct antigen presentation is not the mode of priming. However, the results for mCMV-WT do not fit this interpretation, as the CD8 T-cell response to this virus was higher compared to the two immune evasion virus mutants for the epitope panel tested (Figure 6A, Supplementary Figure 1) and was broader in terms of the epitope specificity repertoire determined by using a viral genome-wide ORF expression library (Supplementary Figure 1). So, if direct presentation plays no role at all, as the two antipodal mutants suggest, why is the response after infection with mCMV-WT the best, with immune evasion and direct antigen presentation being in between?
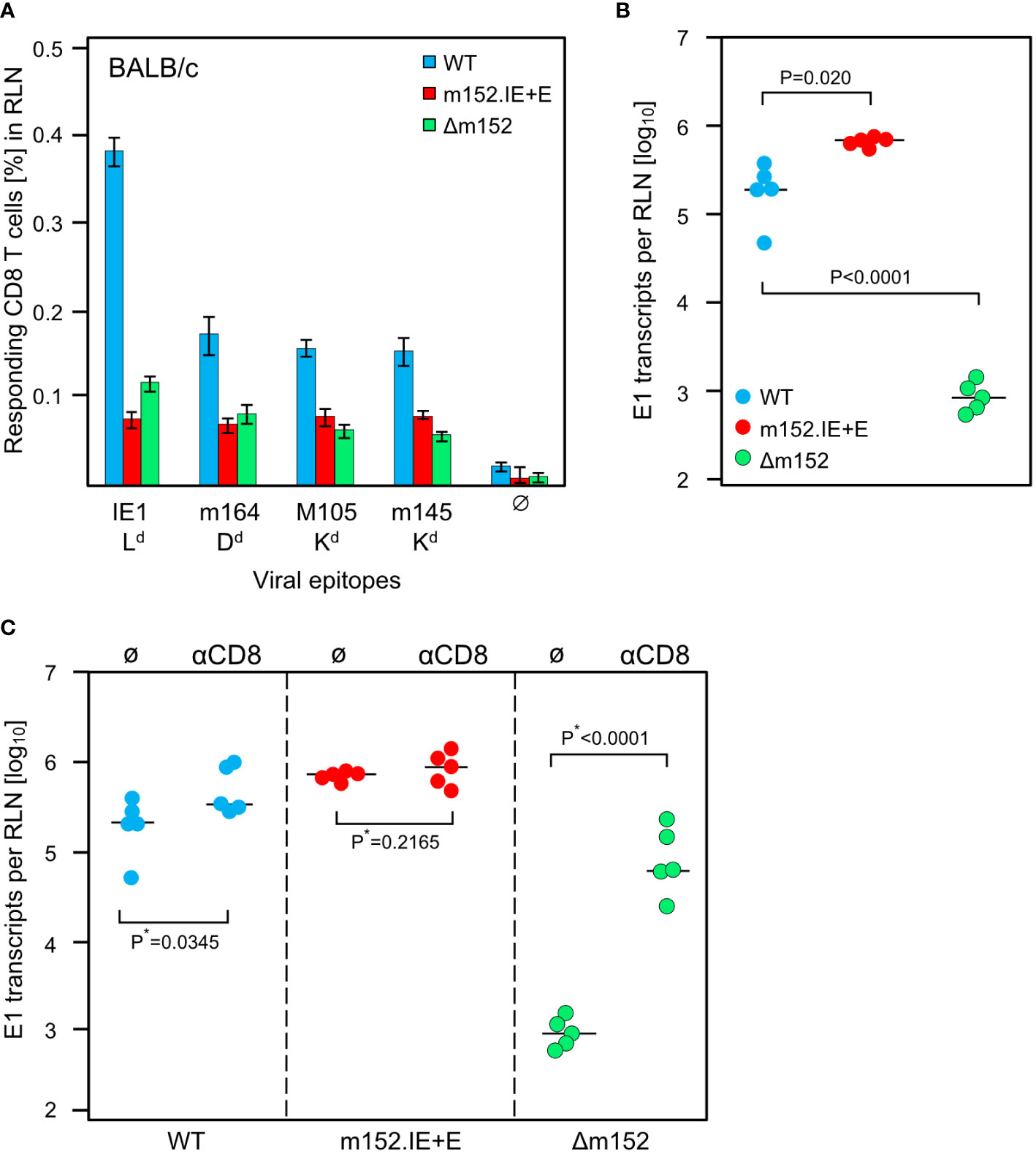
Figure 6 Impact of the strength of viral immune evasion on CD8 T-cell response magnitude and control of intranodal viral replication. Intraplantar infection of BALB/c mice (n=5 per group) was performed with 1x105 PFU each of either mCMV-WT (WT) or mCMV-m152.IE+E (m152.IE+E) or mCMV-Δm152 (Δm152). (A) CD8 T-cell response in the draining RLN on day 7 post-infection. Responding CD8 T cells were quantitated in an IFNγ-based ELISpot assay, with P815 cells used as APCs that were exogenously-loaded with the indicated synthetic peptides. Bars represent cohort average frequencies of epitope-specific CD8 T cells determined by intercept-free linear regression analysis, error bars indicate the 95% confidence intervals. Test groups are considered significantly different if the confidence intervals do not overlap. Ø, no peptide added. (B) Viral replication in the RLN on day 3 post-infection, determined by quantitating spliced E1 transcripts by RT-qPCR. Dots represent data for individual mice. Median values are indicated by horizontal bars. (C) Intranodal viral replication in mice depleted of CD8 T cells on the day before intraplantar infection (αCD8) or in mice left undepleted under otherwise identical conditions (Ø). Note that data for undepleted mice are the same as in (B) and are shown again to facilitate the comparison with results from mice depleted of CD8 T cells. P*-values are calculated from log-transformed data using Welch´s unpaired t-test (*one-sided).
Based on our experience with the C57BL/6 model (Figures 2, 3) and our published previous work in the BALB/c model (75), we quantitated replication of the three viruses in the draining RLN, the site where priming and clonal expansion take place, on day 3 after infection (Figure 6B). As an important control, replication differences caused by genetically-determined viral replicative fitness were excluded by showing identical replication of the three viruses in immune-deficient mice (Supplementary Figure 2). Therefore, replication differences in the RLN must reflect differences in immune control. Notably, unlike the sizes of the CD8 T-cell responses, the intranodal viral replication fulfilled the logic, that is, highest replication corresponds to strongest immune evasion of mCMV-m152.IE+E, lowest replication corresponds to weakest immune evasion of mCMV-Δm152, and intermediate replication corresponds to intermediate immune evasion of mCMV-WT (Figure 6B).
A possible contribution of recently primed virus-specific CD8 T cells to the control of intranodal virus replication was tested by CD8 T-cell depletion prior to infection (Figure 6C). It may be instructive that our previous work has already shown the presence of antiviral effector CD8 T cells in the RLN after 3 days of infection (75). In accordance with almost missing antigen presentation on cells infected with mCMV-m152.IE+E, intranodal viral replication was not detectably controlled by CD8 T cells, whereas, in accordance with optimal antigen presentation on cells infected with mCMV-Δm152, intranodal viral replication was almost prevented when CD8 T cells were present. Again, results for mCMV-WT were in between.
In summary, up to this point, the diametrically different levels of intranodal viral replication of mCMV-m152.IE+E and mCMV-Δm152 in BALB/c mice reflect missing and strong antiviral control, respectively, by the just generated effector CD8 T cells. Surprisingly, the result is almost the same low level of CD8 T-cell response, while the intermediate level of immune evasion by mCMV-WT results in the best response, generating high numbers of antiviral CD8 T cells exported from the RLN for controlling viral replication at distant sites of viral pathogenesis.
Before arriving at a conclusion, it was important to consider the possibility that direct antigen presentation and antigen cross-presentation may not be mutually exclusive. An alternative explanation for the comparable magnitude of response following high and low direct antigen presentation could be that the response to mCMV-Δm152 is actually driven by high direct antigen presentation, whereas the response to the super-evasion virus mCMV-m152.IE+E may be due to antigen cross-presentation being used as an alternative pathway, aided by large amounts of antigenic material derived from many dying cells. This tempting idea, though, was refuted by comparing the response to the two viruses in the cross-presentation deficient Unc93b13d/3d mice, revealing no notable difference for the panel of viral epitopes tested (Figure 7A).
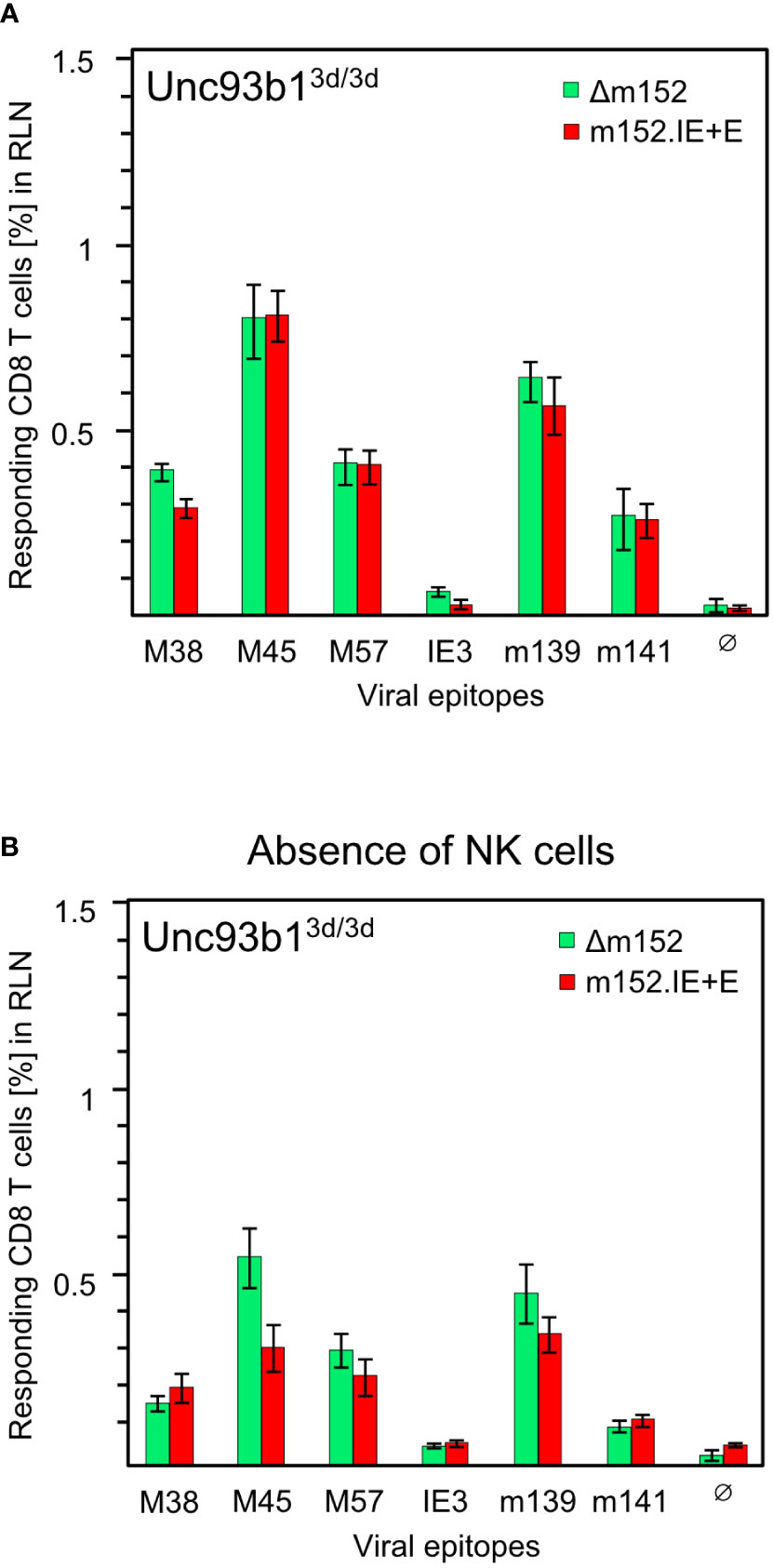
Figure 7 CD8 T-cell responses in absence of antigen cross-presentation. Equivalence of responses after high and low direct antigen presentation in presence (A) and in absence (B) of NK cells. CD8 T-cell responses were measured in Unc93b13d/3d mice (n=4 per experimental group) that are deficient in antigen cross-presentation. CD8 T cells were isolated from the draining regional lymph node (RLN), that is, the popliteal lymph node, on day 7 after intraplantar infection with 1x105 PFU each of either mCMV-Δm152 (Δm152) or mCMV-m152.IE+E (m152.IE+E), and were used as effector cells in an IFNγ-based ELISpot assay. (A) No depletion of NK cells. Data are from the experiment of Figure 1B, where only the comparison between mCMV-WT and mCMV-Δm152 is shown. (B) Depletion of pan-NK cells one day before infection. For further details, see the Legend to Figure 1. Note that (A, B) represent independent experiments, so that response magnitudes can be compared reliably only between the two viruses in either experiment.
As m152 also impacts on the NK-cell response by downmodulating ligands of the activatory NK-cell receptor NKG2D (47–51), high and low NK-cell responses to mCMV-Δm152 and mCMV-m152.IE+E, respectively, could indirectly affect the CD8 T-cell responses to these two viruses differentially. However, except for a minor difference in the case of viral epitope M45, the magnitude of the CD8 T-cell responses to the two extremes of immune evasion remained comparable after pan-NK cell depletion under conditions of absent antigen-cross presentation in Unc93b13d/3d mice (Figure 7B). As a consequence, all our results in both C57BL/6 and BALB/c mice must be explained on the basis of direct antigen presentation.
CMVs are often discussed as being masters in evading innate and adaptive immune control (19, 58, 103), whereas host counter-measures to ensure immune surveillance of CMV were rarely considered (104). Numerous reports published over decades specifically dealt with viral proteins, so-called immunoevasins, which subvert the MHC-I pathway of direct antigen presentation to CD8 T cells [for reviews, see (105–109)]. This may have left the medical research community with the false impression that CMVs are not controlled by CD8 T cells.
This view, however, conflicts with the undisputable fact that acute CMV infections are rapidly and tightly controlled by the immune system, with CD8 T cells being identified as the main antiviral effector cells that terminate productive acute infection and surveil latent infection for preventing virus reactivation (5, 7, 32, 77, 110). Accordingly, CMVs do not harm the immunocompetent host but cause severe and often lethal, tissue-destructing organ infection in the immunocompromised host. This fundamental observation applies to humans as well as to the mouse model [reviewed in (2, 3, 6, 9)].
CMVs are host-species specific, and different CMV species share homologous genes as well as genes with analogous function, but also possess “private genes” that have evolved for adaptation to the respective host in eons of virus-host co-evolution. So, the evolutionary acquisition and maintenance of a gene involved in limiting direct viral antigen presentation to host CD8 T cells must be expected to have a benefit for both virus and host. One hypothesis is that limiting direct antigen presentation to host CD8 T cells dampens the immune response to avoid virus clearance and to allow the virus to reach cellular niches for surviving in a state of latency. Our data now support a completely new view on the role of viral immune evasion.
It was our original aim to identify the canonical pathway of viral antigen presentation to CD8 T cells, that is, to decide between direct antigen presentation and cross-presentation. Previous studies in mouse models of CMV infection have shown that mice can mount a virus-specific CD8 T-cell response as a “plan B” by either pathway when the respective other pathway is closed genetically or experimentally (57, 59–61, 111). Specifically, as also shown here, Unc93b13d/3d mice genetically deficient in cross-presentation can perfectly develop a CD8 T-cell response by direct antigen presentation. On the other hand, mice immunized with infected MHC-I-deficient cells can develop a CD8 T-cell response by cross-presentation (59). All these reports are undoubtedly correct. However, it remained open to question which pathway is used as “plan A” when both pathways are principally accessible.
To tackle the problem, we used the novel approach of keeping the immunogenetics of the host constant and, instead, to genetically modify the virus in its immune evasion potential. This was done by enhancing and reducing direct antigen presentation relative to mCMV-WT by infection with mCMV-Δm152 and the newly constructed recombinant virus mCMV-m152.IE+E, respectively. If priming of naïve virus-specific CD8 T cells and subsequent clonal expansion are by direct antigen presentation, the magnitude of the CD8 T-cell response should have been high in case of infection with mCMV-Δm152, with which direct antigen presentation is not inhibited, and low in case of infection with mCMV-m152.IE+E, with which direct antigen presentation is strongly inhibited. The result was amazing in that the CD8 T-cell response was almost identical for the two extremes of particularly high and low direct antigen presentation, suggesting that direct antigen presentation is not the mechanism of priming. What prevented us from drawing this rash conclusion was the non-fitting finding that intermediate immune evasion, and thus intermediate inhibition of direct antigen presentation, by infection with mCMV-WT led to the best CD8 T-cell response.
Our current results concerning the response magnitude after infection with mCMV-Δm152 compared to mCMV-WT reproduce our previously published finding of a reduced response after infection with an immune evasion gene deletion virus despite enhanced direct antigen presentation (75). We called this phenomenon the “immune evasion paradox” and proposed as the mechanism a reduction of the amount of antigen available for cross-presentation due to a “negative feedback control” of intranodal viral replication effected by the just recently generated CD8 effector T cells (75). Our new data obtained with the super-evasion virus mCMV-m152.IE+E contradict a mechanism that involves antigen cross-presentation, because enhanced supply with antigenic material for cross-presentation by a high level of virus production did not improve the CD8 T-cell response. Furthermore, a contribution of antigen cross-presentation is now ruled out by analogous results obtained with Unc93b13d/3d mice genetically lacking the antigen cross-presentation pathway.
An aspect that requires explanation is the puzzling result that a CD8 T-cell response also occurs under conditions in which antigen cross-presentation is genetically excluded and direct antigen presentation is largely reduced, as is the case when Unc93b13d/3d mice are infected with the mCMV-m152.IE+E super-evasion virus. As we have recently reviewed, a tiny number of pMHC-I complexes reaching the cell surface despite interference by immune evasion proteins can suffice for recognition by high-avidity CD8 T cells (44). In addition, IFNγ is known to counteract immune evasion in the MHC-I pathway of direct antigen presentation (112, 113) by promoting MHC class-I synthesis (114) and the proteasomal processing of antigenic proteins (115). Of note, CMV immune evasion is less efficient in murine (62) and human (64) macrophages that can serve as pAPCs for direct antigen presentation.
These principles apply to BALB/c mice and C57BL/6 mice with the difference that virus replication in the RLN is restricted selectively in C57BL/6 mice by Ly49H+ NK cells and that even in absence of NK-cell activity the negative feedback control of viral replication by the primed CD8 T cells is less pronounced in C57BL/6 mice (Figures 2, 3) compared to BALB/c mice (Figure 6C). This difference is most obvious in the case of enhanced direct antigen presentation after infection with the immune evasion gene deletion mutant.
The negative feedback is not just a hypothesis based on our functional data, but has a structural correlate in the observation by intravital microscopy that CD8 T cells primed in the peripheral interfollicular T-cell zone of the RLN migrate back to a cortical region just underneath the subcapsular sinus, where they attack infected cells (91). The sketch in Figure 8 summarizes the results and illustrates the proposed mechanisms. In essence, in a “negative feedback loop”, the level of direct antigen presentation during the initial priming event determines the number of primed CD8 T cells that then restrict the numbers of infected pAPCs available for driving subsequent clonal expansion.
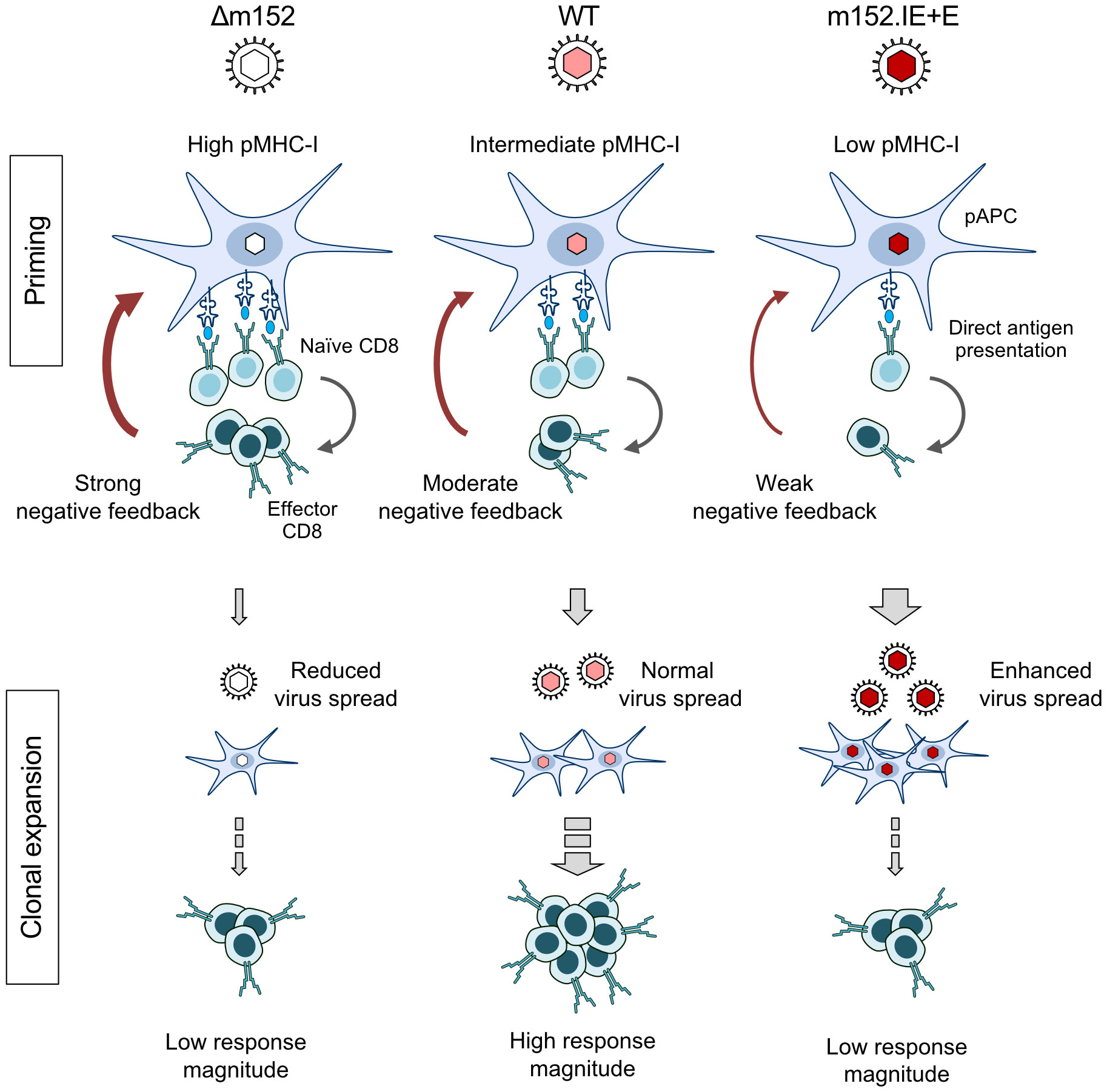
Figure 8 Graphical abstract. Immunoevasin-dependent CD8 T-cell response magnitude regulated by negative feedback on direct antigen presenting cells by recently primed effector CD8 T cells. (pAPC), professional antigen-presenting cells; (pMHC-I), MHC-I molecules presenting antigenic peptides. Symbols on CD8 T cells represent viral epitope-specific T-cell receptors. Growing red-color intensity symbolizes increasingly enhanced expression of immune evasion molecule m152, and thus increasing strength of immune evasion.
While we have now identified direct antigen presentation as the canonical pathway for mounting a primary CD8 T-cell response in the “immunocompetent mouse model” of CMV infection in both a genetically-resistant and a genetically-susceptible mouse strain, the findings may have an even more important bearing for our understanding of viral interference with the MHC-I pathway of direct antigen presentation. We find it utmost intriguing that mCMV-WT, the virus naturally selected during virus-host co-evolution, induced the best CD8 T-cell response, whereas both prevention as well as enhancement of m152-mediated immune evasion diminished the response. It appears that optimal calibration of the strength of viral interference with the MHC-I pathway of antigen presentation serves to still allow sufficient priming of naïve CD8 T cells to initiate a response but also to moderate the “negative feedback” that otherwise would inhibit clonal expansion of the primed CD8 T cells. So, unexpectedly, viral interference with direct antigen presentation is beneficial for mounting a protective CD8 T-cell response. This sheds a completely new light on the physiological role of viral immune evasion.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The animal study was approved by the ethics committee of the “Landesuntersuchungsamt Rheinland-Pfalz” according to German federal law §8 Abs. 1 TierSchG (animal protection law), permission number 177-07/G09-1-004. The study was conducted in accordance with the local legislation and institutional requirements.
JB: Data curation, Formal analysis, Investigation, Writing – review & editing, Validation, Visualization. AF: Formal analysis, Writing – review & editing, Investigation. SB: Formal analysis, Investigation, Writing – review & editing. MB: Writing – review & editing, Conceptualization, Resources. RH: Formal analysis, Writing – review & editing, Data curation, Funding acquisition, Supervision, Validation. MR: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft. NL: Data curation, Formal analysis, Funding acquisition, Project administration, Supervision, Conceptualization, Validation, Visualization, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Deutsche Forschungsgemeinschaft (DFG), Clinical Research Group KFO 183 (MR, and NL), SFB 900 (MB; Project No. 158989968), SFB 1292 (SB, RH, MR, NL; Project No. 318346496). MB is supported by the SMART BIOTECS alliance between the Technische Universität Braunschweig and the Leibniz Universität Hannover, an initiative supported by the Ministry of Science and Culture (MWK) of Lower Saxony, Germany, and the Helmholtz Association (W2/W3-090). NL is a member of the DFG-funded Cluster of Excellence ImmunoSensation -EXC2151- at the University Bonn.
The authors thank Angélique Renzaho and Kirsten Freitag for expert technical assistance and Daria Lohschelders for her help during manuscript preparation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1272166/full#supplementary-material
Supplementary Figure 1 | Magnitude and specificity repertoire of the acute CD8 T-cell response response in BALB/c mice. Frequencies of CD8 T cells responding to infection with either mCMV-WT (WT, two upper panels) or mCMV-m152.IE+E (m152.IE+E, two lower panels) were determined by intracellular IFNγ-staining, using as stimulator cells either an mCMV genome-wide ORF library of transfectants (left panels) or P815 cells exogenously-loaded with the indicated synthetic antigenic peptides at the saturating concentration of 10-7 M (right panels). Responder cells were CD8 T cells isolated from the spleen on day 7 after intraplantar infection with 1x105 PFU each of either of the two viruses. Note that the comparison of ORF library data for mCMV-WT and mCMV-ΔvRAP, which is equivalent to mCMV-Δm152, has been published previously (116), and supports the conclusions.
Supplementary Figure 2 | Viral replicative fitness in host organs. Immunocompromised BALB/c mice (6.5 Gy of total body γ-irradiation) were infected with 1x105 PFU of mCMV-WT (WT), mCMV-m152.IE+E (m152IE+E) or mCMV-Δm152 (Δm152). Viral replicative fitness was assessed by the viral doubling times (vDT), measured by M55/gB-specific qPCR in total DNA extracted from the organs indicated. Symbols represent individual mice. vDT values and their 95% confidence intervals were calculated from log-linear regression lines with ordinate intercept, including all data collected over the entire time course. Dashed curves border the respective 95% confidence areas.
1. Davison AJ, Holton M, Aidan D, Dargan DJ, Gatherer D, Hayward GS. Comparative genomics of primate cytomegaloviruses. In: Reddehase MJ, editor. Cytomegaloviruses: from molecular pathogenesis to intervention. Norfolk: Caister academic press (2013). p. 1–22.
2. Boppana SB, Britt WJ. Synopsis of clinical aspects of human cytomegalovirus disease. In: Reddehase MJ, editor. Cytomegaloviruses: from molecular pathogenesis to intervention. Norfolk: Caister academic press (2013). p. 1–25.
3. Griffiths P, Reeves M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat Rev Microbiol (2021) 19:759–73. doi: 10.1038/s41579-021-00582-z
4. Reusser P, Riddell SR, Meyers JD, Greenberg PD. Cytotoxic T-lymphocyte response to cytomegalovirus after human allogeneic bone marrow transplantation: pattern of recovery and correlation with cytomegalovirus infection and disease. Blood (1991) 78:1373–80. doi: 10.1182/blood.V78.5.1373.1373
5. Reddehase MJ, Weiland F, Münch K, Jonjic S, Lüske A, Koszinowski UH. Interstitial murine cytomegalovirus pneumonia after irradiation: characterization of cells that limit viral replication during established infection of the lungs. J Virol (1985) 55:264–73. doi: 10.1128/JVI.55.2.264-273.1985
6. Reddehase MJ. Mutual Interference between cytomegalovirus and reconstitution of protective immunity after hematopoietic cell transplantation. Front Immunol (2016) 7:294. doi: 10.3389/fimmu.2016.00294
7. Steffens HP, Kurz S, Holtappels R, Reddehase MJ. Preemptive CD8 T-cell immunotherapy of acute cytomegalovirus infection prevents lethal disease, limits the burden of latent viral genomes, and reduces the risk of virus recurrence. J Virol (1998) 72:1797–804. doi: 10.1128/jvi.72.3.1797-1804.1998
8. Ebert S, Podlech J, Gillert-Marien D, Gergely KM, Büttner JK, Fink A, et al. Parameters determining the efficacy of adoptive CD8 T-cell therapy of cytomegalovirus infection. Med Microbiol Immunol (2012) 201:527–39. doi: 10.1007/s00430-012-0258-x
9. Reddehase MJ, Lemmermann NAW. Mouse model of cytomegalovirus disease and immunotherapy in the immunocompromised host: predictions for medical translation that survived the “Test of Time”. Viruses (2018) 10:693. doi: 10.3390/v10120693
10. Neill L, Peggs K. Cell therapy for cytomegalovirus infection. Expert Opin Biol Ther (2021) 21:649–59. doi: 10.1080/14712598.2021.1857720
11. Holtappels R, Becker S, Hamdan S, Freitag K, Podlech J, Lemmermann NA, et al. Immunotherapy of cytomegalovirus infection by low-dose adoptive transfer of antiviral CD8 T cells relies on substantial post-transfer expansion of central memory cells but not effector-memory cells. PloS Pathog (2023) 19:e1011643. doi: 10.1371/journal.ppat.1011643
12. Adler B, Sattler C, Adler H. Herpesviruses and their host cells: a successful liaison. Trends Microbiol (2017) 25:229–41. doi: 10.1016/j.tim.2016.11.009
13. Azab W, Dayaram A, Greenwood AD, Osterrieder N. How host specific are herpesviruses? Lessons from herpesviruses infecting wild and endangered mammals. Annu Rev Virol (2018) 5:53–68. doi: 10.1146/annurev-virology-092917-043227
14. Krmpotic A, Bubic I, Polic B, Lucin P, Jonjic S. Pathogenesis of murine cytomegalovirus infection. Microbes Infection (2003) 5:1263–77. doi: 10.1016/j.micinf.2003.09.007
15. Benedict CA, Crozat K, Degli-Esposti MA, Dalod M. Host genetic models in cytomegalovirus immunology. In: Reddehase MJ, editor. Cytomegaloviruses: from molecular pathogenesis to intervention. Norfolk: Caister academic press (2013). p. 258–84.
16. Holtappels R, Ebert S, Podlech J, Fink A, Böhm V, Lemmermann NAW, et al. Murine model for cytoimmunotherapy of CMV disease after haematopoietic cell transplantation. In: Reddehase MJ, editor. ytomegaloviruses: from molecular pathogenesis to intervention. Norfolk: Caister academic press (2013). p. 352–79.
17. Biron CA, Tarrio ML. Immunoregulatory cytokine networks: 60 years of learning from murine cytomegalovirus. Med Microbiol Immunol (2015) 204:345–54. doi: 10.1007/s00430-015-0412-3
18. Brizić I, Lisnić B, Brune W, Hengel H, Jonjić S. Cytomegalovirus infection: mouse model. Curr Protoc Immunol (2018) 122:e51. doi: 10.1002/cpim.51
19. Berry R, Watson GM, Jonjic S, Degli-Esposti MA, Rossjohn J. Modulation of innate and adaptive immunity by cytomegaloviruses. Nat Rev Immunol (2020) 20:113–27. doi: 10.1038/s41577-019-0225-5
20. Fisher MA, Lloyd ML. A review of murine cytomegalovirus as a model for human cytomegalovirus disease-Do mice lie? Int J Mol Sci (2020) 22:214. doi: 10.3390/ijms22010214
21. Bonavita CM, Cardin RD. Don’t go breaking my heart: MCMV as a model for HCMV-associated cardiovascular diseases. Pathogens (2021) 10:619. doi: 10.3390/pathogens10050619
22. Brizić I, Lisnić B, Krstanović F, Brune W, Hengel H, Jonjić S. Mouse models for cytomegalovirus infections in newborns and adults. Curr Protoc (2022) 2:e537. doi: 10.1002/cpz1.537
23. Bruce K, Ma J, Lawler C, Xie W, Stevenson PG, Farrell HE. Recent advancements in understanding primary cytomegalovirus infection in a mouse model. Viruses (2022) 14:1934. doi: 10.3390/v14091934
24. Lodha M, Muchsin I, Jürges C, Juranic Lisnic V, L’Hernault A, Rutkowski AJ, et al. Decoding murine cytomegalovirus. PloS Pathog (2023) 19:e1010992. doi: 10.1371/journal.ppat.1010992
25. Sissons JGP, Wills MR. How understanding immunology contributes to managing CMV disease in immunosuppressed patients: now and in future. Med Microbiol Immunol (2015) 204:307–16. doi: 10.1007/s00430-015-0415-0
26. Holtappels R, Podlech J, Freitag K, Lemmermann NA, Reddehase MJ. Memory CD8 T cells protect against cytomegalovirus disease by formation of nodular inflammatory foci preventing intra-tissue virus spread. Viruses (2022) 14:1145. doi: 10.3390/v14061145
27. Reddehase MJ, Lemmermann NAW. Cellular reservoirs of latent cytomegaloviruses. Med Microbiol Immunol (2019) 208:391–403. doi: 10.1007/s00430-019-00592-y
28. Seckert CK, Griessl M, Büttner JK, Scheller S, Simon CO, Kropp KA, et al. Viral latency drives “memory inflation”: a unifying hypothesis linking two hallmarks of cytomegalovirus infection. Med Microbiol Immunol (2012) 201:551–66. doi: 10.1007/s00430-012-0273-y
29. Klenerman P, Oxenius A. T cell responses to cytomegalovirus. Nat Rev Immunol (2016) 16:367–77. doi: 10.1038/nri.2016.38
30. Cicin-Sain L. Cytomegalovirus memory inflation and immune protection. Med Microbiol Immunol (2019) 208:339–47. doi: 10.1007/s00430-019-00607-8
31. Welten SPM, Baumann NS, Oxenius A. Fuel and brake of memory T cell inflation. Med Microbiol Immunol (2019) 208:329–38. doi: 10.1007/s00430-019-00587-9
32. Simon CO, Holtappels R, Tervo H-M, Böhm V, Däubner T, Oehrlein-Karpi SA, et al. CD8 T cells control cytomegalovirus latency by epitope-specific sensing of transcriptional reactivation. J Virol (2006) 80:10436–56. doi: 10.1128/JVI.01248-06
33. Heath WR, Carbone FR. Cross-presentation in viral immunity and self-tolerance. Nat Rev Immunol (2001) 1:126–34. doi: 10.1038/35100512
34. Andrews DM, Andoniou CE, Granucci F, Ricciardi-Castagnoli P, Degli-Esposti MA. Infection of dendritic cells by murine cytomegalovirus induces functional paralysis. Nat Immunol (2001) 2:1077–84. doi: 10.1038/ni724
35. Holtappels R, Gillert-Marien D, Thomas D, Podlech J, Deegen P, Herter S, et al. Cytomegalovirus encodes a positive regulator of antigen presentation. J Virol (2006) 80:7613–24. doi: 10.1128/JVI.00723-06
36. Hsu KM, Pratt JR, Akers WJ, AChilefu SI, Yokoyama WM. Murine cytomegalovirus displays selective infection of cells within hours after systemic administration. J Gen Virol (2009) 90:33–43. doi: 10.1099/vir.0.006668-0
37. Becker S, Fink A, Podlech J, Reddehase MJ, Lemmermann NA. Host-adapted gene families involved in murine cytomegalovirus immune evasion. Viruses (2022) 14:128. doi: 10.3390/v14010128
38. Becker S, Fink A, Podlech J, Giese I, Schmiedeke JK, Bukur T, et al. Positive role of the MHC class-I antigen presentation regulator m04/gp34 of murine cytomegalovirus in antiviral protection by CD8 T cells. Front Cell Infect Microbiol (2020) 10:454. doi: 10.3389/fcimb.2020.00454
39. Ziegler H, Thale R, Lucin P, Muranyi W, Flohr T, Hengel H, et al. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity (1997) 6:57–66. doi: 10.1016/s1074-7613(00)80242-3
40. Fink A, Renzaho A, Reddehase MJ, Lemmermann NAW. The p36 isoform of murine cytomegalovirus m152 protein suffices for mediating innate and adaptive immune evasion. Viruses (2013) 5:3171–91. doi: 10.3390/v5123171
41. Janßen L, Ramnarayan VR, Aboelmagd M, Iliopoulou M, Hein Z, Majoul I, et al. The murine cytomegalovirus immunoevasin gp40 binds MHC class I molecules to retain them in the early secretory pathway. J Cell Sci (2016) 129:219–27. doi: 10.1242/jcs.175620
42. Ramnarayan VR, Hein Z, Janßen L, Lis N, Ghanwat S, Springer S. Cytomegalovirus gp40/m152 uses TMED10 as ER anchor to retain MHC class I. Cell Rep (2018) 23:3068–77. doi: 10.1016/j.celrep.2018.05.017
43. Lemmermann NAW, Gergely K, Böhm V, Deegen P, Däubner T, Reddehase MJ. Immune evasion proteins of murine cytomegalovirus preferentially affect cell surface display of recently generated peptide presentation complexes. J Virol (2010) 84:1221–36. doi: 10.1128/JVI.02087-09
44. Hamdan S, Reddehase MJ, Holtappels R. Cytomegalovirus immune evasion sets the functional avidity threshold for protection by CD8 T cells. Med Microbiol Immunol (2023) 212:153–63. doi: 10.1007/s00430-022-00733-w
45. Krmpotic A, Messerle M, Crnkovic-Mertens I, Polic B, Jonjic S, Koszinowski UH. The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J Exp Med (1999) 190:1285–96. doi: 10.1084/jem.190.9.1285
46. Holtappels R, Podlech J, Pahl-Seibert M-F, Jülch M, Thomas D, Simon CO, et al. Cytomegalovirus misleads its host by priming of CD8 T cells specific for an epitope not presented in infected tissues. J Exp Med (2004) 199:131–6. doi: 10.1084/jem.20031582
47. Arapovic J, Lenac T, Antulov R, Polic B, Ruzsics Z, Carayannopoulos LN, et al. Differential susceptibility of RAE-1 isoforms to mouse cytomegalovirus. J Virol (2009) 83:8198–207. doi: 10.1128/JVI.02549-08
48. Zhi L, Mans J, Paskow MJ, Brown PH, Schuck P, Jonjić S, et al. Direct interaction of the mouse cytomegalovirus m152/gp40 immunoevasin with RAE-1 isoforms. Biochemistry (2010) 49:2443–53. doi: 10.1021/bi902130j
49. Lis N, Hein Z, Ghanwat SS, Ramnarayan VR, Chambers BJ, Springer S. The murine cytomegalovirus immunoevasin gp40/m152 inhibits NKG2D receptor RAE-1γ by intracellular retention and cell surface masking. J Cell Sci (2021) 134:jcs257428. doi: 10.1242/jcs.257428
50. Krmpotić A, Busch DH, Bubić I, Gebhardt F, Hengel H, Hasan M, et al. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat Immunol (2002) 3:529–35. doi: 10.1038/ni799
51. Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, et al. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med (2003) 197:1245–53. doi: 10.1084/jem.20021973
52. Stempel M, Chan B, Juranić Lisnić V, Krmpotić A, Hartung J, Paludan SR, et al. The herpesviral antagonist m152 reveals differential activation of STING-dependent IRF and NF-κB signaling and STING’s dual role during MCMV infection. EMBO J (2019) 38:e100983. doi: 10.15252/embj.2018100983
53. Raftery MJ, Schwab M, Eibert SM, Samstag Y, Walczak H, Schönrich G. Targeting the function of mature dendritic cells by human cytomegalovirus: a multilayered viral defense strategy. Immunity (2001) 15:997–1009. doi: 10.1016/s1074-7613(01)00239-4
54. LoPiccolo DM, Gold MC, Kavanagh DG, Wagner M, Koszinowski UH, Hill AB. Effective inhibition of K(b)- and D(b)-restricted antigen presentation in primary macrophages by murine cytomegalovirus. J Virol (2003) 77:301–8. doi: 10.1128/jvi.77.1.301-308.2003
55. Mathys S, Schroeder T, Ellwart J, Koszinowski UH, Messerle M, Just U. Dendritic cells under influence of mouse cytomegalovirus have a physiologic dual role: to initiate and to restrict T cell activation. J Infect Dis (2003) 187:988–99. doi: 10.1086/368094
56. Arrode G, Davrinche C. Dendritic cells and HCMV cross-presentation. Curr Top Microbiol Immunol (2003) 276:277–94. doi: 10.1007/978-3-662-06508-2_13
57. Nopora K, Bernhard CA, Ried C, Castello AA, Murphy KM, Marconi P, et al. MHC class I cross-presentation by dendritic cells counteracts viral immune evasion. Front Immunol (2012) 3:348. doi: 10.3389/fimmu.2012.00348
58. Brinkmann MM, Dağ F, Hengel H, Messerle M, Kalinke U, Čičin-Šain L. Cytomegalovirus immune evasion of myeloid lineage cells. Med Microbiol Immunol (2015) 204:367–82. doi: 10.1007/s00430-015-0403-4
59. Snyder CM, Davis-Poynter N, Farrell HE, Allan JE, Bonnett EL, Doom CM, et al. Cross-presentation of a spread-defective MCMV is sufficient to prime the majority of virus-specific CD8+ T cells. PloS One (2010) 5:e9681. doi: 10.1371/journal.pone.0009681
60. Torti N, Walton SM, Murphy KM, Oxenius A. Batf3 transcription factor-dependent DC subsets in murine CMV infection: Differential impact on T-cell priming and memory inflation. Eur J Immunol (2011) 41:2612–8. doi: 10.1002/eji.201041075
61. Ong ML, Wikstrom ME, Fleming P, Estcourt MJ, Hertzog PJ, Hill GR, et al. CpG pretreatment enhances antiviral T-cell immunity against cytomegalovirus. Blood (2013) 122:55–60. doi: 10.1182/blood-2012-12-471227
62. Hengel H, Reusch U, Geginat G, Holtappels R, Ruppert T, Hellebrand E, et al. Macrophages escape inhibition of major histocompatibility complex class I-dependent antigen presentation by cytomegalovirus. J Virol (2000) 74:7861–8. doi: 10.1128/jvi.74.17.7861-7868.2000
63. Magri G, Muntasell A, Romo N, Sáez-Borderías A, Pende D, Geraghty DE, et al. NKp46 and DNAM-1 NK-cell receptors drive the response to human cytomegalovirus-infected myeloid dendritic cells overcoming viral immune evasion strategies. Blood (2011) 117:848–56. doi: 10.1182/blood-2010-08-301374
64. Frascaroli G, Lecher C, Varani S, Setz C, van der Merwe J, Brune W, et al. Human macrophages escape inhibition of major histocompatibility complex-dependent antigen presentation by cytomegalovirus and drive proliferation and activation of memory CD4+ and CD8+ T cells. Front Immunol (2018) 9:1129. doi: 10.3389/fimmu.2018.01129
65. Reddehase MJ, Rothbard JB, Koszinowski UH. A pentapeptide as minimal antigenic determinant for MHC class I-restricted T lymphocytes. Nature (1989) 337:651–3. doi: 10.1038/337651a0
66. Holtappels R, Thomas D, Podlech J, Reddehase MJ. Two antigenic peptides from genes m123 and m164 of murine cytomegalovirus quantitatively dominate CD8 T-cell memory in the H-2d haplotype. J Virol (2002) 76:151–64. doi: 10.1128/jvi.76.1.151-164.2002
67. Wagner M, Jonjic S, Koszinowski UH, Messerle M. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J Virol (1999) 73:7056–60. doi: 10.1128/JVI.73.8.7056-7060.1999
68. Chan B, Gonçalves Magalhães V, Lemmermann NAW, Juranić Lisnić V, Stempel M, Bussey KA, et al. The murine cytomegalovirus M35 protein antagonizes type I IFN induction downstream of pattern recognition receptors by targeting NF-κB mediated transcription. PloS Pathog (2017) 13:e1006382. doi: 10.1371/journal.ppat.1006382
69. Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol (2006) 7:156–64. doi: 10.1038/ni1297
70. Borst E-M, Pósfai G, Pogoda F, Messerle M. Mutagenesis of herpesvirus BACs by allele replacement. Methods Mol Biol (2004) 256:269–79. doi: 10.1385/1-59259-753-X:269
71. Grzimek NK, Dreis D, Schmalz S, Reddehase MJ. Random, asynchronous, and asymmetric transcriptional activity of enhancer-flanking major immediate-early genes ie1/3 and ie2 during murine cytomegalovirus latency in the lungs. J Virol (2001) 75:2692–705. doi: 10.1128/JVI.75.6.2692-2705.2001
72. Lemmermann NAW, Podlech J, Seckert CK, Kropp KA, Grzimek NKA, Reddehase MJ, et al. CD8 T-cell immunotherapy of cytomegalovirus disease in the murine model. In: Kabelitz D, Kaufmann S, editors. Methods in microbiology: immunology of infection. London: Academic Press (2010). p. 369–420. doi: 10.1016/S0580-9517(10)37016-4
73. Kurz S, Steffens HP, Mayer A, Harris JR, Reddehase MJ. Latency versus persistence or intermittent recurrences: evidence for a latent state of murine cytomegalovirus in the lungs. J Virol (1997) 71:2980–7. doi: 10.1128/JVI.71.4.2980-2987.1997
74. Podlech J, Holtappels R, Grzimek NKA, Reddehase MJ. Animal models: murine cytomegalovirus. In: Kabelitz D, Kaufmann S, editors. Methods in microbiology: immunology of infection. London: Academic Press (2002). p. 493–525.
75. Böhm V, Simon CO, Podlech J, Seckert CK, Gendig D, Deegen P, et al. The immune evasion paradox: immunoevasins of murine cytomegalovirus enhance priming of CD8 T cells by preventing negative feedback regulation. J Virol (2008) 82:11637–50. doi: 10.1128/JVI.01510-08
76. Simon CO, Seckert CK, Dreis D, Reddehase MJ, Grzimek NKA. Role for tumor necrosis factor alpha in murine cytomegalovirus transcriptional reactivation in latently infected lungs. J Virol (2005) 79:326–40. doi: 10.1128/JVI.79.1.326-340.2005
77. Griessl M, Renzaho A, Freitag K, Seckert CK, Reddehase MJ, Lemmermann NAW. Stochastic episodes of latent cytomegalovirus transcription drive CD8 T-cell “memory inflation” and avoid Immune evasion. Front Immunol (2021) 12:668885. doi: 10.3389/fimmu.2021.668885
78. Bühler B, Keil GM, Weiland F, Koszinowski UH. Characterization of the murine cytomegalovirus early transcription unit e1 that is induced by immediate-early proteins. J Virol (1990) 64:1907–19. doi: 10.1128/JVI.64.5.1907-1919.1990
79. Ciocco-Schmitt GM, Karabekian Z, Godfrey EW, Stenberg RM, Campbell AE, Kerry JA. Identification and characterization of novel murine cytomegalovirus M112-113 (e1) gene products. Virology (2002) 294:199–208. doi: 10.1006/viro.2001.1311
80. Perez KJ, Martínez FP, Cosme-Cruz R, Perez-Crespo NM, Tang Q. A short cis-acting motif in the M112-113 promoter region is essential for IE3 to activate M112-113 gene expression and is important for murine cytomegalovirus replication. J Virol (2013) 87:2639–47. doi: 10.1128/JVI.03171-12
81. Munks MW, Gold MC, Zajac AL, Doom CM, Morello CS, Spector DH, et al. Genome-wide analysis reveals a highly diverse CD8 T cell response to murine cytomegalovirus. J Immunol (2006) 176:3760–6. doi: 10.4049/jimmunol.176.6.3760
82. Pahl-Seibert M-F, Juelch M, Podlech J, Thomas D, Deegen P, Reddehase MJ, et al. Highly protective in vivo function of cytomegalovirus IE1 epitope-specific memory CD8 T cells purified by T-cell receptor-based cell sorting. J Virol (2005) 79:5400–13. doi: 10.1128/JVI.79.9.5400-5413.2005
83. Reddehase MJ, Keil GM, Koszinowski UH. The cytolytic T lymphocyte response to the murine cytomegalovirus. II. Detection of virus replication stage-specific antigens by separate populations of in vivo active cytolytic T lymphocyte precursors. Eur J Immunol (1984) 14:56–61. doi: 10.1002/eji.1830140111
84. Wilhelmi V, Simon CO, Podlech J, Böhm V, Däubner T, Emde S, et al. Transactivation of cellular genes involved in nucleotide metabolism by the regulatory IE1 protein of murine cytomegalovirus is not critical for viral replicative fitness in quiescent cells and host tissues. J Virol (2008) 82:9900–16. doi: 10.1128/JVI.00928-08
85. Kropp KA, Simon CO, Fink A, Renzaho A, Kühnapfel B, Podlech J, et al. Synergism between the components of the bipartite major immediate-early transcriptional enhancer of murine cytomegalovirus does not accelerate virus replication in cell culture and host tissues. J Gen Virol (2009) 90:2395–401. doi: 10.1099/vir.0.012245-0
86. Lemmermann NAW, Krmpotic A, Podlech J, Brizic I, Prager A, Adler H, et al. Non-redundant and redundant roles of cytomegalovirus gH/gL complexes in host organ entry and intra-tissue spread. PloS Pathog (2015) 11:e1004640. doi: 10.1371/journal.ppat.1004640
87. Bromley SK, Iaboni A, Davis SJ, Whitty A, Green JM, Shaw AS, et al. The immunological synapse and CD28-CD80 interactions. Nat Immunol (2001) 2:1159–66. doi: 10.1038/ni737
88. Lanzavecchia A, Sallusto F. Antigen decoding by T lymphocytes: from synapses to fate determination. Nat Immunol (2001) 2:487–92. doi: 10.1038/88678
89. Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat Immunol (2002) 3:265–71. doi: 10.1038/ni762
90. Bousso P, Robey E. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat Immunol (2003) 4:579–85. doi: 10.1038/ni928
91. Hickman HD, Takeda K, Skon CN, Murray FR, Hensley SE, Loomis J, et al. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol (2008) 9:155–65. doi: 10.1038/ni1557
92. Reynoso GV, Weisberg AS, Shannon JP, McManus DT, Shores L, Americo JL, et al. Lymph node conduits transport virions for rapid T cell activation. Nat Immunol (2019) 20:602–12. doi: 10.1038/s41590-019-0342-0
93. Janssen E, Tabeta K, Barnes MJ, Rutschmann S, McBride S, Bahjat KS, et al. Efficient T cell activation via a Toll-Interleukin 1 Receptor-independent pathway. Immunity (2006) 24:787–99. doi: 10.1016/j.immuni.2006.03.024
94. Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim Y-M. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol (2007) 177:265–75. doi: 10.1083/jcb.200612056
95. Kim Y-M, Brinkmann MM, Paquet M-E, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature (2008) 452:234–8. doi: 10.1038/nature06726
96. Maschalidi S, Nunes-Hasler P, Nascimento CR, Sallent I, Lannoy V, Garfa-Traore M, et al. UNC93B1 interacts with the calcium sensor STIM1 for efficient antigen cross-presentation in dendritic cells. Nat Commun (2017) 8:1640. doi: 10.1038/s41467-017-01601-5
97. Crane MJ, Gaddi PJ, Salazar-Mather TP. UNC93B1 mediates innate inflammation and antiviral defense in the liver during acute murine cytomegalovirus infection. PloS One (2012) 7:e39161. doi: 10.1371/journal.pone.0039161
98. Gold MC, Munks MW, Wagner M, Koszinowski UH, Hill AB, Fling SP. The murine cytomegalovirus immunomodulatory gene m152 prevents recognition of infected cells by M45-specific CTL but does not alter the immunodominance of the M45-specific CD8 T cell response in vivo. J Immunol (2002) 169:359–65. doi: 10.4049/jimmunol.169.1.359
99. Gold MC, Munks MW, Wagner M, McMahon CW, Kelly A, Kavanagh DG, et al. Murine cytomegalovirus interference with antigen presentation has little effect on the size or the effector memory phenotype of the CD8 T cell response. J Immunol (2004) 172:6944–53. doi: 10.4049/jimmunol.172.11.6944
100. Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science (2002) 296:1323–6. doi: 10.1126/science.1070884
101. Smith HRC, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci U.S.A. (2002) 99:8826–31. doi: 10.1073/pnas.092258599
102. Mitrović M, Arapović J, Jordan S, Fodil-Cornu N, Ebert S, Vidal SM, et al. The NK cell response to mouse cytomegalovirus infection affects the level and kinetics of the early CD8(+) T-cell response. J Virol (2012) 86:2165–75. doi: 10.1128/JVI.06042-11
103. Mancini M, Vidal SM. Mechanisms of natural killer cell evasion through viral adaptation. Annu Rev Immunol (2020) 38:511–39. doi: 10.1146/annurev-immunol-082619-124440
104. Reddehase MJ. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat Rev Immunol (2002) 2:831–44. doi: 10.1038/nri932
105. Wiertz E, Hill A, Tortorella D, Ploegh H. Cytomegaloviruses use multiple mechanisms to elude the host immune response. Immunol Lett (1997) 57:213–6. doi: 10.1016/s0165-2478(97)00073-4
106. Hengel H, Brune W, Koszinowski UH. Immune evasion by cytomegalovirus–survival strategies of a highly adapted opportunist. Trends Microbiol (1998) 6:190–7. doi: 10.1016/s0966-842x(98)01255-4
107. Alcami A, Koszinowski UH. Viral mechanisms of immune evasion. Trends Microbiol (2000) 8:410–8. doi: 10.1016/s0966-842x(00)01830-8
108. Yewdell JW, Hill AB. Viral interference with antigen presentation. Nat Immunol (2002) 3:1019–25. doi: 10.1038/ni1102-1019
109. Powers C, DeFilippis V, Malouli D, Früh K. Cytomegalovirus immune evasion. Curr Top Microbiol Immunol (2008) 325:333–59. doi: 10.1007/978-3-540-77349-8_19
110. Podlech J, Holtappels R, Wirtz N, Steffens HP, Reddehase MJ. Reconstitution of CD8 T cells is essential for the prevention of multiple-organ cytomegalovirus histopathology after bone marrow transplantation. J Gen Virol (1998) 79:2099–104. doi: 10.1099/0022-1317-79-9-2099
111. Busche A, Jirmo AC, Welten SPM, Zischke J, Noack J, Constabel H, et al. Priming of CD8+ T cells against cytomegalovirus-encoded antigens is dominated by cross-presentation. J Immunol (2013) 190:2767–77. doi: 10.4049/jimmunol.1200966
112. Hengel H, Lucin P, Jonjić S, Ruppert T, Koszinowski UH. Restoration of cytomegalovirus antigen presentation by gamma interferon combats viral escape. J Virol (1994) 68:289–97. doi: 10.1128/JVI.68.1.289-297.1994
113. Fink A, Lemmermann NAW, Gillert-Marien D, Thomas D, Freitag K, Böhm V, et al. Antigen presentation under the influence of “immune evasion” proteins and its modulation by interferon-gamma: implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med Microbiol Immunol (2012) 201:513–25. doi: 10.1007/s00430-012-0256-z
114. Zhou F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol (2009) 28:239–60. doi: 10.1080/08830180902978120
115. Kloetzel PM. Antigen processing by the proteasome. Nat Rev Mol Cell Biol (2001) 2:179–87. doi: 10.1038/35056572
Keywords: CD8 T cell response, m152/gp40, antigen presentation, antigen cross-presentation, antigen presenting cell (APC), murine cytomegalovirus (mCMV)
Citation: Büttner JK, Becker S, Fink A, Brinkmann MM, Holtappels R, Reddehase MJ and Lemmermann NA (2023) Direct antigen presentation is the canonical pathway of cytomegalovirus CD8 T-cell priming regulated by balanced immune evasion ensuring a strong antiviral response. Front. Immunol. 14:1272166. doi: 10.3389/fimmu.2023.1272166
Received: 04 August 2023; Accepted: 30 November 2023;
Published: 12 December 2023.
Edited by:
Stipan Jonjic, University of Rijeka, CroatiaReviewed by:
Anne Halenius, University of Freiburg Medical Center, GermanyCopyright © 2023 Büttner, Becker, Fink, Brinkmann, Holtappels, Reddehase and Lemmermann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Niels A. Lemmermann, bGVtbWVybWFubkB1bmktYm9ubi5kZQ==
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.