 Fang Zhao
Fang Zhao- 1Department of Rheumatology of The First Hospital and Institute of Innovation and Applied Research in Chinese Medicine, Hunan University of Chinese Medicine, Changsha, Hunan, China
- 2Xiangya School of Pharmaceutical Sciences, Central South University, Changsha, Hunan, China
- 3Institute (College) of Integrative Medicine, Dalian Medical University, Dalian, Liaoning, China
- 4Department of Neurosurgery, The Second Xiangya Hospital, Central South University, Changsha, Hunan, China
- 5State Key Laboratory of Dampness Syndrome of Chinese Medicine, The 2nd Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, China
Angiogenesis plays a key role in the pathological process of inflammation and invasion of the synovium, and primarily drives the progression of rheumatoid arthritis (RA). Recent studies have demonstrated that the Notch signaling may represent a new therapeutic target of RA. Although the Notch signaling has been implicated in the M1 polarization of macrophages and the differentiation of lymphocytes, little is known about its role in angiogenesis in RA. In this review, we discourse the unique roles of stromal cells and adipokines in the angiogenic progression of RA, and investigate how epigenetic regulation of the Notch signaling influences angiogenesis in RA. We also discuss the interaction of the Notch-HIF signaling in RA’s angiogenesis and the potential strategies targeting the Notch signaling to improve the treatment outcomes of RA. Taken together, we further suggest new insights into future research regarding the challenges in the therapeutic strategies of RA.
1 Introduction
Rheumatoid arthritis (RA) is a prototypic autoimmune disease characterized by progressive inflammatory synovitis. Severe RA is accompanied by clinical sequelae and has a 50% risk of cardiovascular mortality. In global epidemiology, rheumatoid arthritis has a very high global age-standardized prevalence (7.4% of the total population) and incidence (8.2% of the total population) (1). In the pathogenesis of RA, inflammation is the background response that drives resident interstitial synovial cells to acquire a pro-angiogenic phenotype, resulting in dramatic changes in synovial structure and function (2). Angiogenesis plays an early and key role in the process of synovitis. In the inflammatory environment, vessels firstly undergo a period of high inflammation, followed by a period of intense increase in blood vessel growth. Newly formed blood vessels provide oxygen and nutrition for proliferative synovitis cells, further promoting the progression of synovitis and promoting joint invasion and destruction. Angiogenesis is essential for supplying nutrients and oxygen to the proliferative joints of patients with RA, relying on an extremely complex network system consisting of highly interactive and mutually promoted extracellular matrix and stromal cells (3).
Angiogenesis is the process of the formation of new capillaries that develop from preexisting vessels. Angiogenesis is critical for many physiological processes, including embryonic organ development, tissue repair, remodeling, and wound repair (4), and it initiates when vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) bind to several cognate receptors on endothelial cells (ECs). Activated ECs induce the disassembly of tight junctions, leading to an increase in vascular permeability. Subsequently, matrix metalloproteinases (MMPs) degrade the vascular basement membrane, accelerating endothelial migration, proliferation, and the stability of new tubes. Interconnected tubules accelerate the construction of vessel networks, which are regulated by Ang1, a pro-angiogenic factor, to potentiate pericyte recruitment and facilitate blood flow (5).
Notch signaling is a highly conserved intercellular signaling mechanism that directs cell fate specification and proliferation and controls cell differentiation and processes in both embryonic and adult tissues (6). Notch signaling is a pivotal regulator of vessel formation and is involved in EC-pericyte interactions and the coupling of osteogenesis and angiogenesis (7). Notch signaling mainly consists of four parts: ligands, receptors, DNA-binding proteins, and downstream gene transcription (8). The epigenetic regulation of Notch signaling determines the specificity of Notch target genes in different cell types (9). As for the effect of the epigenetic regulation of Notch signaling in angiogenesis, it is worth further investigation.
Many human diseases, including rheumatoid arthritis, are associated with aberrant Notch signaling (10). Several studies have shown that the Notch signaling pathway is critical for hypoxia-induced proliferation and angiogenesis (11). Under hypoxic conditions, the interaction between Notch-1 and hypoxia-inducible factor (HIF)-1α regulates invasion and angiogenesis in the progression of inflammatory arthritis (12). Notably, no review article has been published to date from the perspective of Notch signaling regulation of angiogenesis in RA.
Therefore, in this review, we have summarized the unique roles of stromal cells and adipokines and their influence on the progression of RA angiogenesis. A comprehensive description of Notch signaling and its epigenetic regulation is provided. We describe the role of the epigenetic regulation of Notch signaling in stromal cells and adipokines during angiogenesis. Then, we further elucidate the interaction of HIF-Notch signaling in angiogenesis. Finally, we discuss the potential strategies to target Notch signaling for the treatment of RA. Based on our discussion, we suggest new insights for future research regarding the challenges of developing novel treatment strategies for RA.
2 Angiogenesis in rheumatoid arthritis
Angiogenesis in rheumatoid arthritis is an intricate process that depends on an extremely complex network system involving the extracellular matrix and stromal cells, by which innate immune cells, adaptive immune cells, vascular endothelial cells, and adipokines can turn on the switch for vessel formation to enhance cartilage erosion and pannus formation (Figure 1).
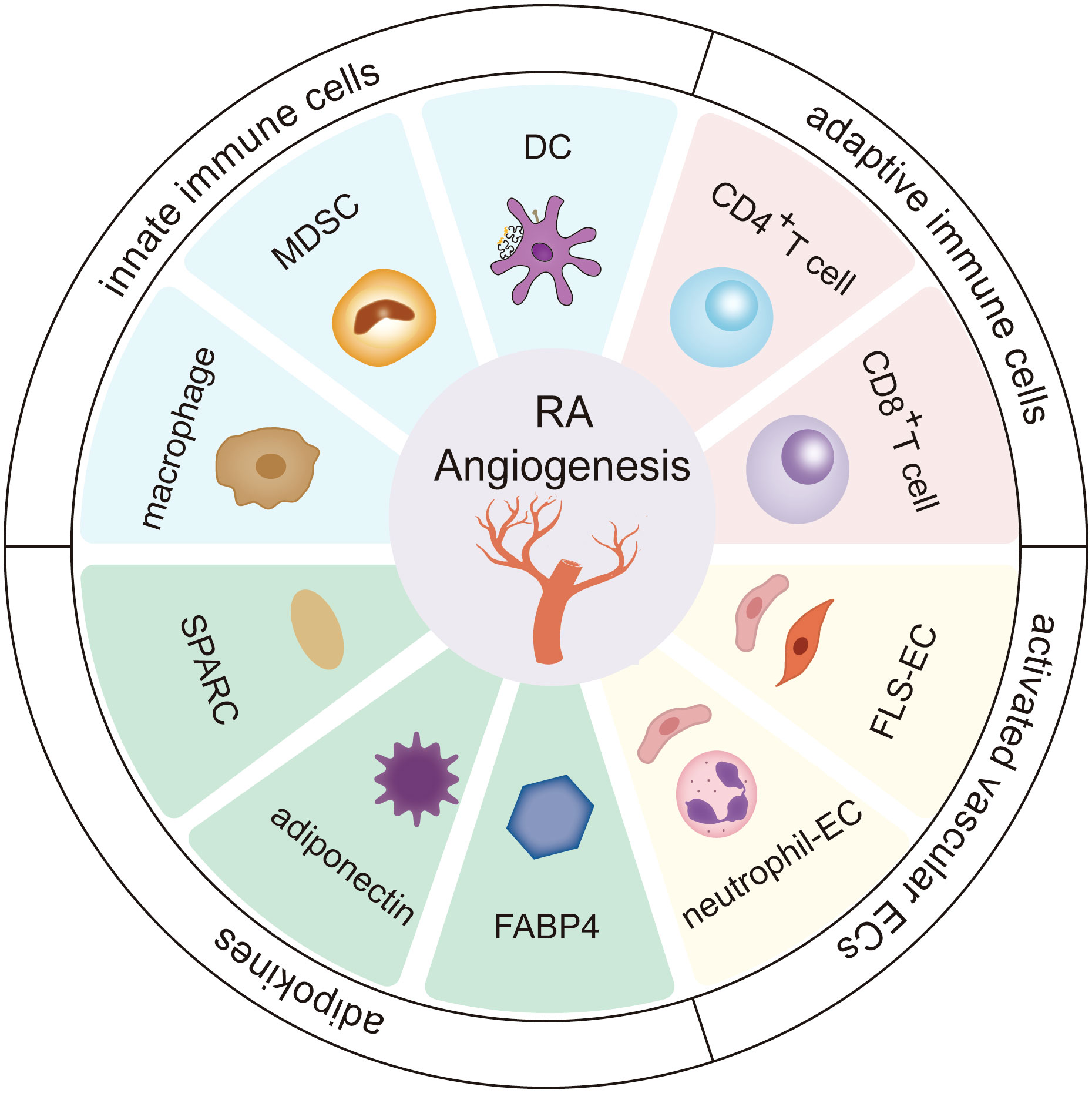
Figure 1 The role of stromal cells and adipokines in RA angiogenesis. Innate immune cells (including dendritic cells, cells-Myeloid-derived suppressor cells, and macrophages), adaptive immune cells (including CD4+ T cells and CD8+ T cells), vascular endothelial cells, and adipokines (including fatty acid-binding protein 4 (FABP4), adiponectin, secreted protein acidic and rich in cysteine (SPARC)) greatly influence the progression of angiogenesis in the RA microenvironment. The cross-talk interactions of stromal cells and adipokines form a continuous cycle, further deteriorating RA.
2.1 Innate immune cells-dendritic cells
Dendritic cells (DCs), which connect the innate and adaptive immune systems, are heterogeneous cell populations essential for immunity and maintaining immune tolerance due to their specialized antigen-presenting capabilities (13). DCs also represent potential therapeutic targets for angiogenesis and lymphangiogenesis in chronic and tumor-associated inflammation (14).
The process involves the following actions: (I) DCs secrete and release angiogenic growth factors, including vascular endothelial growth factor-A (VEGF-A), FGF2, and endothelin-1 (ET-1). These factors have the properties of promoting angiogenesis by activating transcription factors, including CREB, HIF-1α, and STAT3 (15). (II) DCs release angiogenic mediators directly or indirectly by expressing pro-angiogenic chemokines. The upregulated CXCL8, CCL2, and CCL21 chemokines can directly induce angiogenesis by binding to ECs (16, 17). Whereas, the actions of CXCL1, CXCL2, CXCL3, CXCL5, and CXCL7 are indirect, occurring via recruitment of NK cells, neutrophils, monocytes, and macrophages, which in turn promote angiogenesis by producing pro-angiogenic transmitter substances that induce endothelial cell aggregation (18–20). (III) DCs may contribute to the activation of vasculogenic events through transdifferentiation into ECs upon exposure to VEGF (14).
Recent studies have found that the precursors of conventional dendritic cells (pre-cDCs), monocyte-derived DCs (mo-DCs), plasmacytoid DCs (pDCs), and Langerhans cells (LCs), which belong to the most studied specialized subtypes, are major players in the chemotaxis and migration of DCs during inflammation and immunity (21). Studies have indicated that an increased level of pre-DCs in the peripheral blood of RA patients with relatively poor prognoses is a key indicator of treatment resistance (22). Mo-DCs induce deleterious Th17 cell responses and participate in the chronic inflammatory microenvironment and bone loss in RA (23). ERK1/2 modulated mo-DCs may also contribute to regulating angiogenesis (24). pDCs are a major link between anomalous immunity and angiogenesis in rosacea (25). LCs, fibroblasts, and macrophages have been demonstrated to provide the basis for ECs behavior in the angiogenic niche during murine skin repair (26). The LCs density was positively correlated with disease activity in patients (27). Moreover, DCs maturation affects the synovial microenvironment in RA (13, 17, 28). Collectively, these results suggest that DCs are the primary initiators of inflammation-associated angiogenesis and that different DCs subtypes are essential for RA development.
2.2 Innate immune cells-myeloid-derived suppressor cells
Hematopoietic stem cells (HSCs) differentiate into granulocyte-monocyte progenitor cells (GMPs) via common myeloid progenitor cells (CMPs). Disruption of the differentiation of myeloid ancestors in chronic inflammatory conditions (autoimmune diseases, chronic infections, and cancer) contributes to the development of immature myeloid cells, termed myeloid-derived suppressor cells (MDSCs), which belong to a heterogeneous cell population (29). MDSCs represent a state of pathological activation of neutrophils and monocytes with substantial immunosuppressive activity (30). Dilated MDSCs exert both pro- and anti-inflammatory effects in animal models (31). The confirmed pro-inflammatory effects of MDSCs include the following: (1) MDSCs promote the response of Th17 cells by IL-1β signaling, potentiating the inflammatory response (32). (2) MDSCs also differentiate into osteoclasts via interaction with Th17 cells and upregulation of nuclear factor κβ ligand-RANK signaling, enhancing bone erosion and synovitis (33). (3) MDSCs suppress the immune-suppressing Treg cell response. MDSCs exhibit the following anti-inflammatory effects: (1) MDSCs have an inhibitory effect on DCs maturation. (2) MDSCs depress the proliferation of autoreactive T cells and reduce Th17 cell responses. (3) MDSCs may potentially reduce macrophage numbers, leading to a reduction in pro-inflammatory factors secreted by macrophages, namely TNF-α and GM-CSF (31). (4) MDSCs potentiate Tregs with anti-inflammatory effects (Figure 2).
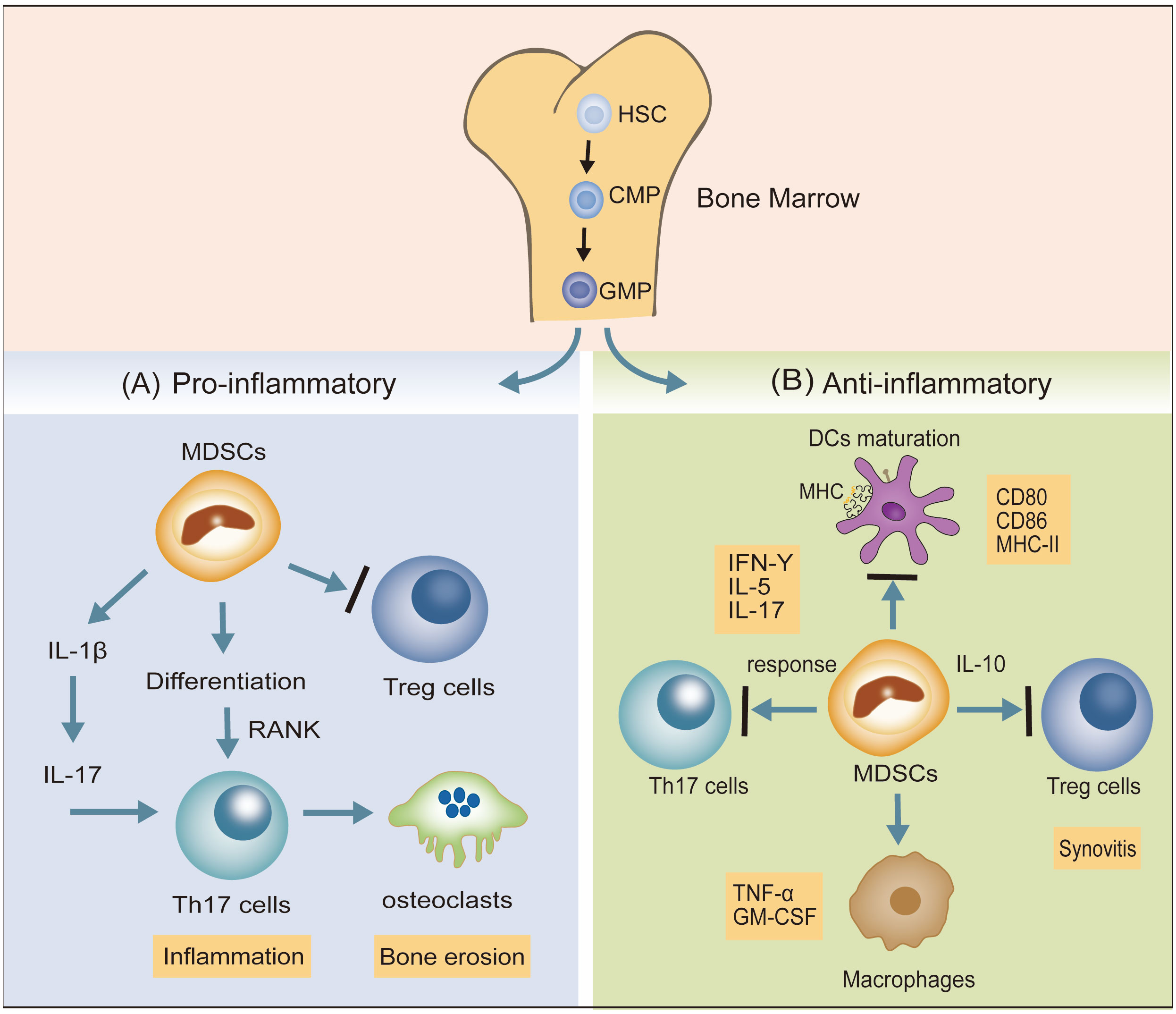
Figure 2 The dual inflammatory actions (pro- and anti-) of MDSCs in RA. (A) MDSCs promote the response of Th17 cells via IL-1β signaling and also differentiate into osteoclasts by interacting with Th17 cells and upregulating nuclear factor κβ ligand-RANK signaling, potentiating the inflammatory response. MDSCs may suppress Treg cell responses. (B) MDSCs inhibit the expression of DCs maturation biomarkers (CD80, CD86, MHC-II), suppress Th17 cell responses, and reduce the pro-inflammatory factors (TGF-α, GM-CSF) secreted by macrophages. MDSCs also potentiate Treg cells to depress the progress of synovitis.
MDSCs contribute to angiogenesis and tumor growth by secreting cytokines and enzyme, including vascular endothelial growth factor (VEGF), bFGF and MMP9 (34). It has been corroborated that CXCR2, a pivotal chemokine in MDSC recruitment, is expressed by MDSCs and is involved in endometriotic growth and angiogenesis (35). A2B receptor stimulation induces VEGF release, driving immune suppression by promoting the expansion of MDSCs, thereby accelerating tumor angiogenesis and worsening the impaired immune system (36, 37). In the course of bone defect reconstruction and its associated angiogenesis, MDSCs can augment the proliferation of ECs and increase vascular thickness and capillary density (38, 39). The role of MDSCs in RA-induced angiogenesis and their putative inflammatory-regulating mechanisms require further investigation.
2.3 Innate immune cells-macrophages
Macrophages are essential components of innate immunity and exhibit significant heterogeneity and polarization (40). Under the stimulation of various pathological factors, macrophages polarize to various phenotypes (mainly M1 and M2) depending on the state of and changes in the environment, and thereafter perform different functions with varying effects (41). M1 macrophages suppress sprouting angiogenesis, whereas M2 macrophages potentiate the formation of the vascular network and prolong dysregulated innate immunity by secreting a series of pro-angiogenic growth factors, angiogenic CXC chemokines, and angiogenesis-associated factors (42, 43). These induction factors augment the migration and proliferation of ECs, further activating the angiogenic switch and promoting angiogenesis (19). A recent study indicated that miRNAs play well-established roles in the interactions between M2 macrophages and ECs. M2 macrophages activate the upregulation of PCAT6 by miR-4723-5p and modulate the expression of VEGFR2, facilitating the angiogenesis of triple-negative breast cancer (44). M2 macrophage-derived exosomal miR-155-5p and miR-221-5p promote the angiogenic ability of endothelial cells (45). M2 macrophage-derived exosomes from adipose-derived stem cells trigger an exosomal pro-angiogenic effect on mouse ischemic hindlimbs by upregulating exosomal miRNA-21 delivery (46). Furthermore, M2 macrophages can deliver miR-501-3p through exosomes to elevate tube formation and repress the apoptosis of ECs through the TGF-β signaling pathway during the progression of pancreatic ductal adenocarcinoma (47). miR-199b-3p is responsible for M2 macrophage-mediated angiogenesis and immunomodulation (48). Accumulating evidence suggests that macrophages stimulated by CCL21 directly exert angiogenic effects by elevating the production of VEGF, IL-8, and Ang1 and also indirectly induce Th17 cell polarization to activate vascularization via IL-17 production in RA joints (49). IL-18 has been demonstrated to play a role in triggering macrophage M2 polarization by amplifying the expression of CD163, contributing to angiogenesis in RA (50). These findings emphasize the importance of macrophage polarization in promoting pathologic angiogenesis through cell–cell interactions with the endothelium and regulation of chemokines and cytokines in RA (Figure 3).
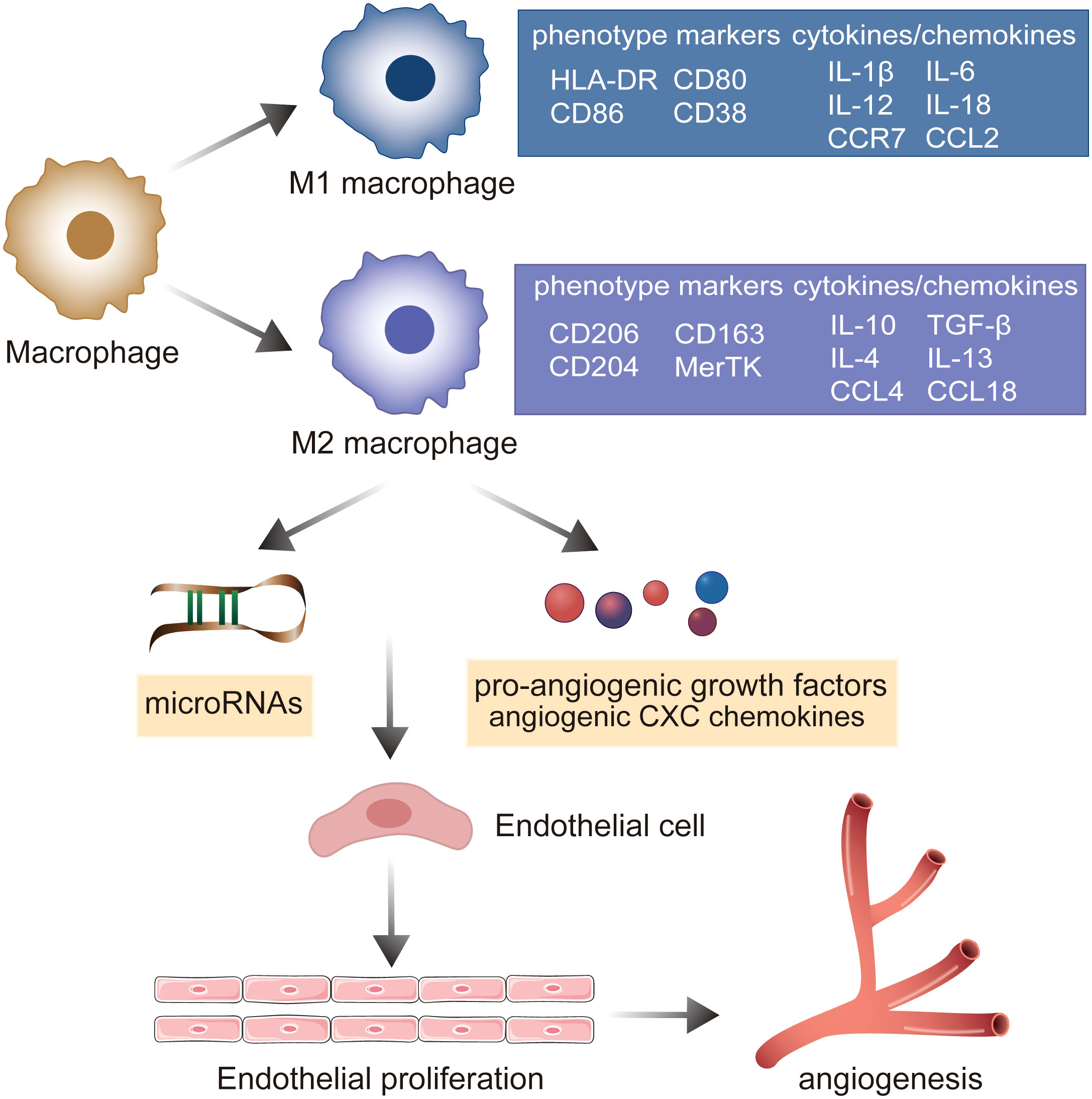
Figure 3 The role of macrophage polarization in the evolution of angiogenesis in RA. Under the stimulation of various pathological factors, macrophages polarize into pro-inflammatory M1 macrophages and M2 macrophages involved in the resolution of inflammation. M2 macrophages mediate microRNAs, pro-angiogenic growth factors, and angiogenic CXC chemokines to establish the cell–cell interaction with endothelium, promoting the ECs’ proliferation and potentiating the formation of the vascular network.
2.4 Adaptive immune cells
The angiogenic response in RA is orchestrated by CD4+ T cell subsets, especially Th17 and regulatory T (Treg) cells (51). Th17 cells, as classical proinflammatory mediators, induce the production of immune inflammatory cells and cytokines, which favor the induction of autoimmune-derived chronic inflammation and joint destruction (52). Stimulated by PlGF, an angiogenic factor homologous to VEGF, the differentiation of Th17 cells has been demonstrated to be significant in linking CD4+ T cells and endothelial cells. CD4+ T cell-derived PlGF promotes the production of IL-17 and favors the activation of ECs in RA (53). Moreover, the Th17/IL-17 related-signaling pathway has been shown to potentiate cell proliferation and foster the expression of pro-angiogenic molecules in osteoblasts (54). Tregs inhibit the activity of Th17 and other effector T cells to maintain their self-tolerance and immunosuppressive state (55). Treg cells that secrete anti-inflammatory cytokines suppress the proliferation of fibroblast-like synoviocytes (FLSs) and inflammatory angiogenesis in RA joints (56). Therefore, the crosstalk between CD4+ T cells and ECs, and the regulatory Treg/Th17 cell balance are essential for the progression of RA-induced angiogenesis.
In addition to CD4+ T cells, peripheral blood CD8+T cells from patients with active and in-remission RA exhibit a potently activated status and proinflammatory potential (57). Plasma VEGF levels and VEGF expression in CD8+T cells positively correlate with the levels of inflammatory factors in patients with RA (58). Studies have shown that CD8+T cells modulate pathological angiogenesis by promoting inflammation and chemokine recruitment in ocular neovascular diseases (59), and in type-2 diabetes, CD8+T cells’ plasticity regulates revascularization, ECs function, and angiogenesis (60). However, the role of CD8+T cells in RA angiogenesis remains poorly understood.
2.5 Activated ECs
The vascular endothelium is responsible for the dynamic maintenance of vascular tone, angiogenesis, permeability, and hemostasis and provides an anti-inflammatory, antioxidant, and antithrombotic interface for the body (61, 62). Central to the cardiovascular complications of rheumatoid arthritis are EC activation and endothelial dysfunction (63). Sustained and exaggerated activation of ECs promotes vascular endothelial dysfunction in the synovial joints and arteries of patients with RA, which can destroy inter-endothelial cell gaps and endothelial integrity. In addition, the release of vasoactive substances from the endothelium, including vascular endothelial growth factor (VEGF), nitric oxide (NO), and MMPs, further initiates endothelial proliferation, migration, and tube formation, leading to pathological angiogenesis (62, 64–66).
Neutrophils play an important role in every phase of RA, from disease initiation to chronic inflammation (67). Emerging evidence suggests that neutrophils are implicated in rheumatic disease-associated vascular inflammation via the regulation of endothelial barriers at the blood–vessel interface (68). NETosis, a modulated mechanism of neutrophil cell death, can be used to evaluate RA activity and the development of atherosclerosis in RA (69, 70). Supernatants of neutrophils cultured with serum from patients with RA, who had been treated with infliximab or tocilizumab, suppressed endothelial dysfunction by inhibiting prothrombotic mediators, pro-inflammatory factors, and adhesion molecules (69). Furthermore, tumor-infiltrating neutrophils contain a series of intracellular VEGF molecules that can be rapidly released upon stimulation to activate the angiogenic switch (71). Neutrophil extracellular traps, secreted by dying neutrophils, trigger nuclear factor κB-dependent endothelial angiogenesis in pulmonary hypertension (72). Neutrophils have been suggested to provide a functional link between inflammatory angiogenesis and RA.
In addition to neutrophil-EC interactions in RA models, FLSs also play a vital role. Under pathological conditions, angiogenesis and FLSs hyperproliferation synergistically trigger panel formation of pannus (73). Synovial layer FLSs polarize the interstitial macrophages and secrete pro-inflammatory mediators and pro-angiogenic factors that cause vessel sprouting (74). Furthermore, the aberrant proliferation of FLSs disrupts the dynamic balance between pro- and anti-inflammatory factors, promoting the proliferation and migration of ECs to constitute new blood vessels and amplify synovial inflammation in the RA process and joint dysfunction (75). FLSs can express a surface biomarker, namely fibroblast-activated protein-α (FAP-α), to initiate local angiogenesis by recruiting and co-interaction with abnormal cells (76). Additionally, the vasculature potently promotes the pro-inflammatory phenotype of synovial layer FLSs in the context of RA joint inflammation (77). The presence of vessels provides relative positional information for the inflammatory FLSs and enables them to respond to the maximum extent of their inflammatory potential. Inflammatory FLSs, trafficked from the intimal lining of the synovial membrane to the proximal vessel, express high levels of the pro-inflammatory marker Thy1 (CD90), which plays a large role in cartilage destruction (74). It has been confirmed that the angiogenic cytokines PlGF and VEGF secreted from synoviocytes can represent both disease activity and synovitis severity as well as the corresponding therapeutic responses to biologics in RA patients (78). Thus, the crosstalk between FLSs and ECs supports the notion that angiogenesis is essential for synovial inflammation in joints with RA (Figure 4).
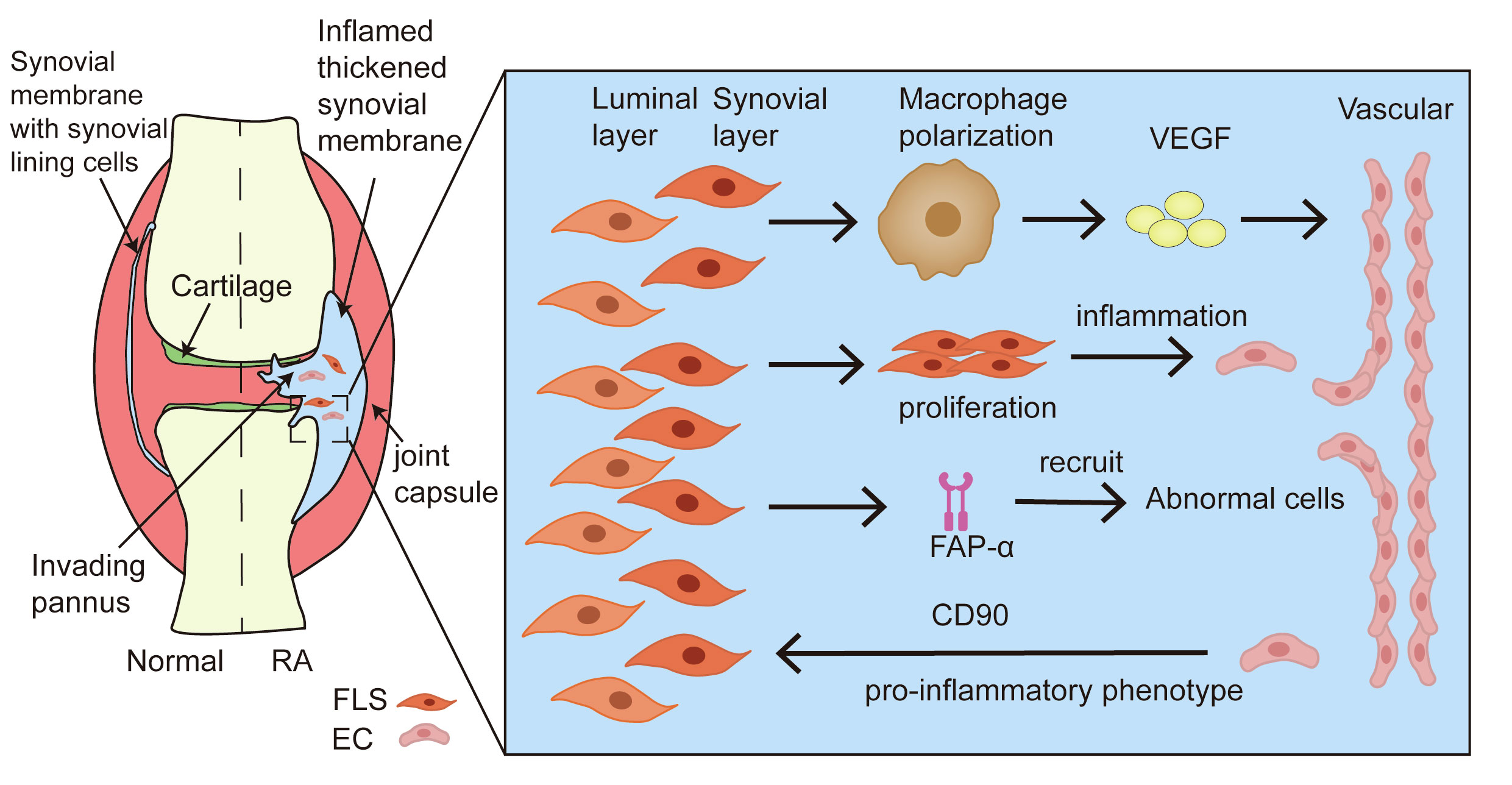
Figure 4 Crosstalk between FLSs and ECs in angiogenesis of RA. During the formation of pannus in the synovium tissue of RA, FLSs promote resident macrophage polarization, accelerating the secretion of pro-inflammatory mediators and pro-angiogenic factors. The aberrant proliferation of FLSs amplifies the ECs response through the activation of inflammation. FAP-α recruits and co-interacts with abnormal cells to induce local angiogenesis. In addition, ECs provide the pro-inflammatory phenotype of FLS to foster the development of synovitis.
2.6 Adipokines
Adipose tissue is an endocrine organ that releases a series of highly biologically active factors called adipokines, fatty acid-binding protein (FABP) 4, adiponectin, and secreted protein acidic and rich in cysteine (SPARC) (79). In the affected joints of RA patients, adipokines are implicated in both innate and adaptive immunity, increase the binding of rheumatoid arthritis synovial fibroblasts (RASFs) to EC, and are involved in the inflammatory process and cartilage reconstitution (80, 81). Recent evidence supports the link between adipokines and angiogenesis in type 2 diabetes mellitus and breast cancer (82, 83). However, the role of adipokines in angiogenesis in RA joints remains unclear.
2.6.1 FABP4
FABP4 is a lipid chaperone protein belonging to the cytoplasmic FABP multigene family that regulates lipid responses and free fatty acid levels in cells (84). Several studies have demonstrated that the enhanced expression of FABP4 is correlated with RA, insulin resistance, type 2 diabetes mellitus, and cardiovascular disease (85, 86). FABP4 is expressed in the synovial lining layer by macrophages and CD8+ T cells, contributing to the inflammatory infiltration of the affected RA joint. FABP4 inhibitors can reduce relative inflammatory effector functions by inhibiting the secretion of IL-10 (86, 87). Furthermore, FABP4 has been found to influence de novo nucleotide synthesis in EC proliferation, tumor angiogenesis and growth, and asthma (88, 89). In RA patients and AIA rats, FABP4 is present in the intrasynovial adipose tissue and vessels, and it is regarded as a pivotal protein in pathologic angiogenesis and an increased risk of atherosclerotic changes (86, 90). Recent studies have shown that FABP4 is secreted by synovial M1-polarized macrophages and could be regulated by the mTORC1 pathway. BMS309403 (a FABP4 inhibitor) treatment degrades synovitis, angiogenesis, and cartilage degradation in vivo and in vitro (91).
2.6.2 Adiponectin
Adiponectin is present in macrophages and synoviocytes and modulates proinflammatory cytokine secretion during the pathophysiology of RA (92, 93). Adiponectin can exacerbate B cell proliferation and differentiation by activating the PI3K/Akt1/STAT3 axis (94). Adiponectin can also stimulate the generation of RA FLSs T follicular helper cells by regulating the secretion of the soluble factor IL-6 (95). Adiponectin triggers the expression of osteopontin, which promotes osteoclast precursor migration in synovial fibroblasts and causes osteoclastogenesis and bone erosion in collagen-induced arthritis (CIA) mice (96). However, local AdipoR1 inhibition effectively ameliorated joint inflammation and bone destruction in vivo (97). In addition, elevated levels of circulating adiponectin are positively correlated not only with disease activity and synovial thickening degree in developing RA but also with higher all-cause mortality and cardiovascular mortality in RA (98–100). Some studies have shown that adiponectin can foster the production of VEGF, vascular cell adhesion molecules-1, and MMPs, including MMP-1 and MMP-13, causing RA joint inflammation and connective tissue vascularization (92). Adiponectin induces VEGF-dependent angiogenesis in synovial fibroblasts in rheumatoid arthritis via MEK/ERK signaling and miRNA-106a-5p (101). Adiponectin, secreted by synovial cells, activates cell migration, invasion, and angiogenesis, thereby promoting the pannus of arthritic joints (102). This result adds support to the notion that adiponectin plays a critical role in angiogenic activity in inflamed joints in RA.
2.6.3 SPARC
SPARC is a nonstructural matricellular protein of the extracellular matrix (ECM) involved in cell-cell and cell-ECM adhesive interactions. SPARC affects immune system functions, including DCs migration, MDSCs proliferation, and functional differentiation (103, 104). Because of these properties, SPARC has been demonstrated to be associated with RA susceptibility in the Chinese Han population (105). A growing number of studies have suggested that the downregulation of SPARC is likely to be a major precipitating event in the pathogenesis of rheumatoid arthritis (103). SPARC is expressed in the synovium and synovial fluid of the affected joints of patients with RA and the CIA model. SPARC-based nanomedicines have strong potential for rheumatoid arthritis therapy by inhibiting the secretion of pro-inflammatory cytokines (106). In addition, SPARC amplified M2-polarized macrophages to activate the proliferation, migration, and angiogenesis of cholangiocarcinoma cells by modulating PI3K/AKT signaling (107). Overexpression of SPARC is capable of disrupting the integrity of vascular endothelial cell layers, affecting matrix composition and cell adhesion, thus facilitating the invasion and metastasis of tumors (106, 108, 109). SPARC knockdown reduces the expression levels of inflammatory factors and VEGFR, downregulating ECs apoptosis, and angiogenesis in diabetic retinopathy (110). SPARC is a key factor in angiogenesis and endothelial barrier function which is well established; however, the underlying mechanism of RA-induced angiogenesis has not yet been defined.
3 Notch signaling modulates angiogenesis in RA
3.1 Notch signaling
3.1.1 The canonical Notch signaling pathway
The Notch signaling pathway is ubiquitous in all mammalian species and participates in cell differentiation, survival, proliferation, and cell fate decisions during development and morphogenesis (111). Notch signaling comprises four parts: ligands (Delta-like 1,3,4 or Jagged-1, 2), receptors (Notch 1–4), DNA-binding proteins, and downstream target genes. Notch receptors consist of three domains: an extracellular domain (NECD), a transmembrane domain (NTM), and an intracellular domain (NICD), which are partially similar to Notch ligands (112). The structure of extracellular domains in Notch ligands and receptors contains multiple EGF-like repeats. In signal-receiving cells, Notch precursors from the endoplasmic reticulum are glycosylated at the epidermal growth factor (EGF)-like repeat domain and translocated into the Golgi apparatus. Next, mature Notch receptors are cleaved by S1 and transported to the cell membrane. Through constitutive endocytosis, ubiquitin ligases promote Notch receptor ubiquitination and degradation via the proteasome. The remainder binds to Notch ligands on the cell membrane to transmit signals (8). The canonical Notch signaling pathway includes the following steps: (1) Mature Notch receptors bind to Notch ligands, exposing the S2 site for cleavage and initiating the Notch signal. (2) The product of S2 cleavage is further cleaved in the NTM at the S3 site, and NICD as the principal effector form of Notch signaling is released. (3) NICD is transferred from the cytoplasm to the nucleus, triggering the transcription of downstream Notch target genes (113) (Figure 5).
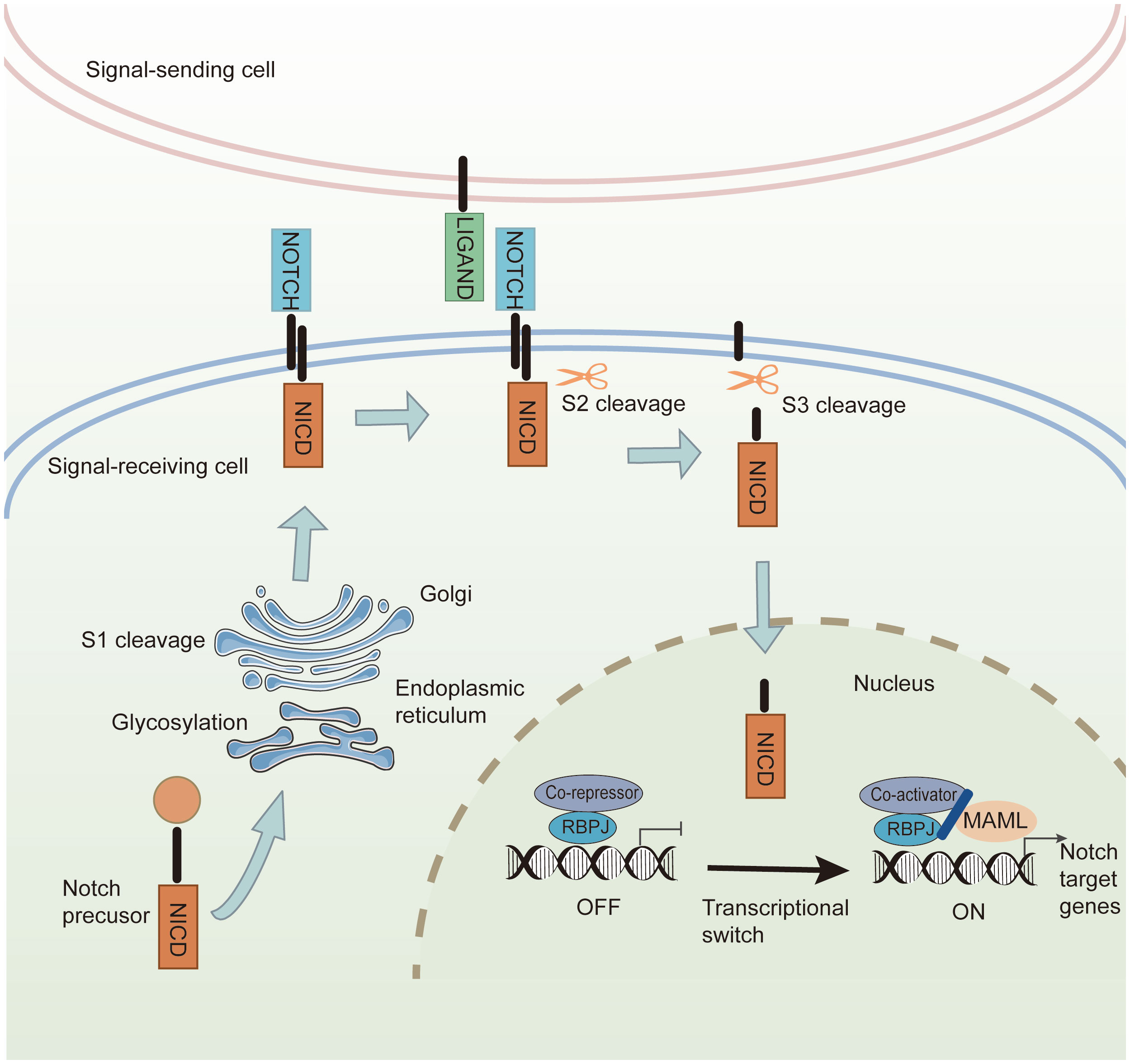
Figure 5 The canonical Notch signaling pathway. In the signal-receiving cell, Notch precursors are glycosylated by the endoplasmic reticulum and undergo S1 cleavage in the Golgi. The mature Notch receptors are transported into the cell membrane. Canonical Notch ligands bind to the Notch receptors, and Notch signaling induces cleavage at the S2 site. Then S3-cleavage is catalyzed by γ-secretase, causing the release of NICD. The NICD is transferred into the nucleus, where it interacts with transcriptional regulators and the coactivator to promote the transcription of Notch downstream target genes.
3.1.2 Notch signaling as a target controlling angiogenesis
Endothelial VEGF and DLL-4/Notch signaling constitute a feedback loop in angiogenesis (114–116). VEGF signaling induces the expression of the Notch ligand delta-like ligand 4 (DLL4), which activates Notch signal transduction in adjacent ECs. Notch signaling enhances VEGFR1 expression and inhibits the expression and function of VEGFR2 in neighboring cells. Thus, the level of VEGFR2 in targeted EC is higher than that in adjacent EC, which in turn increases the relative sensitivity to VEGF and endothelial proliferation (117). In contrast, DLL-4/Notch signaling is involved in a feedback loop with VEGF and confines the response of ECs, preventing excessive angiogenesis. This was confirmed by a subsequent study, which demonstrated that the levels of VEGF-A immunostaining and transcripts were significantly increased, and the expression of ESM1, a tip cell marker and VEGF-regulated gene product, was also upregulated in mice with EC-specific inactivation of the Delta-like 4 (Dll4) gene. This study further demonstrated that mice with loss of one or both VEGF-A alleles in ECs exhibited decreased levels of retinal vessel growth, branching, and EC area in the frontal region and in the central plexus. EC density and sprouting in proximity to the avascular region expressing VEGF-A were restored by the administration of an inhibitor of Notch signaling (115). Further studies are required to unravel the role of Notch signaling as an additional target to control angiogenesis.
3.1.3 Notch signaling involvement in angiogenesis of RA
There is a link between Notch signaling and RA pathogenesis. Stromal cells and adipokines have been implicated in multiple studies on RA, with roles in various pathways, including inflammatory arthritis and angiogenesis. An early study found that inhibition of Notch signaling by γ-secretase inhibitors (GSI) effectively blocked Th1 and Th17 cell responses and ameliorated CIA-induced arthritis (118). A follow-up study found that GSI reduced the severity of inflammatory arthritis by regulating the expansion and function of Tregs in both CIA and collagen antibody-induced arthritis (CAIA) mice (119). In TNF-Tg mice, thapsigargin, a Notch inhibitor, suppresses M1 macrophage formation, fosters the M2 macrophage phenotype, and alleviates joint lesions (120). Notch signaling plays a vital role in controlling osteoclast differentiation and bone-resorbing activity either directly acting on osteoclast precursors, or indirectly acting on cells of the osteoblast lineage and cells of the immune system (121). In RA-affected joints, Notch1 is overexpressed and activated in FLS, Th17 cells, and M1 macrophages, accelerating the production of pro-inflammatory cytokines TNF-α, IL-6, and IL-17, leading to inflammation, bone destruction, and joint bone loss (122).
Recent studies have suggested that endothelium-derived Notch signaling modulates synovial fibroblast identity. Notch signaling increases mural cell and CD90 (THY1)+ sublining fibroblast differentiation and promotes the development of inflammatory arthritis (123). In synovial explant cultures, Notch signaling modulates VEGF/Ang2-induced angiogenesis, EC invasion, and pro-inflammatory cytokine secretion (124). Administration of GSI suppresses the proliferation of RA FLSs and secretion of inflammatory cytokines as well as angiogenesis, suggesting the importance and versatility of Notch signaling in angiogenesis in RA (125).
The role of FABP4 in angiogenesis has been well described. Further research underscored that Dll4-Notch signaling modulates FABP4 mRNA and protein expression in human endothelial microvascular ECs. Notch signaling, a repressor of angiogenesis, regulates fatty acid transport across the endothelium by enhancing FABP4 expression, affecting endothelial metabolic and vascular remodeling in the adult heart (126). Chromatin immunoprecipitation showed that NICD associates with FOXO1-binding sites and RBPJκ-binding motifs in the FABP4 promoter, promoting gene transcription (126). Endothelial-specific genetic ablation of Rbp-jκ elevated cardiac blood vessel density and impaired heart function, causing heart hypertrophy and failure. FABP4 upregulation following stimulation of VEGF-A/VEGFR2 and/or the Notch pathway suggests that FABP4 expression depends on Notch-DLL4 signaling (127). FABP4 secreted by M1-polarized macrophages established an essential role in synovitis, angiogenesis, and cartilage degradation in RA (91). FABP4 regulated by Notch signaling may be involved in RA angiogenesis.
In a new report of RNA-sequence analysis, SPARC expression was found to be negatively correlated with Notch signaling in 144 adenoid cystic carcinomas. Exogenous SPARC reverses Notch-induced osteoclast differentiation, bone metastasis, and bone resorption (128). SPARC-overexpressing neuroblastoma cells inhibit the expression of Notch signaling and VEGFR2 phosphorylation and suppress tumor-induced angiogenesis by limiting the formation of endothelial capillary-like tubules, the proliferation of ECs, and the generation of pro-angiogenic factors, including VEGF, FGF, PDGF, and MMP-9 (129). These findings suggest that SPARC may associate with Notch signaling and play a key role in angiogenesis.
3.2 Epigenetic regulation of Notch signaling
Epigenetic regulation causes heritable changes in gene expression without DNA sequence changes and includes DNA methylation, histone modifications, non-coding RNAs (ncRNAs), chromatin remodeling, and RNA modifications (130, 131). Notch signaling is mainly regulated by histone modifications and non-coding RNA in regulatory regions (132).
3.2.1 The regulation of Notch signaling during angiogenesis by PTM in RA
Recent evidence indicates that post-translational modifications (PTMs) in histones are covalent modifications that play a pivotal role in controlling the dynamic fine-tuning of Notch activity (133). PTMs modulate the Notch pathway function or determine the final output of Notch signaling by regulating protein-protein interactions and establishing new binding sites. Notch ligands, receptors, and transcriptional complexes can be modified by PTMs (132). Several PTMs, including glycosylation, ubiquitination and SUMOylation, may participate in regulating Notch signaling during RA angiogenesis.
Glycosylation occurs predominantly in angiogenic regulators that modulate migration and proliferation of ECs. Notch regulation is influenced by glycosylation status, which modulates DLL4 and Jagged1 ligand responsiveness (134). According to the different connection modes of glycoprotein side chain and protein amino acid group, glycosylation is divided into N-linked and O-linked glycosylation. In the various modification types of O-linked glycosylation, O-GlcNAc glycosylation has been studied well in recent years due to the lack of fixed characteristic sequence and the complexity of structure (135). O-GlcNAc glycosylation has been demonstrated to play a critical role in activating Notch signaling, promoting Treg cell differentiation, and inhibiting T cell infiltration in autoimmune hepatitis (136, 137). O-GlcNAc glycosylation amplifies DLL4-mediated Notch signaling, which has been implicated in retinal angiogenesis and pathological vascularization (138). A higher expression of O-GlcNAcylation was exhibited in human RA synovial tissues and RASFs. Treatment of Thiamet G, an inhibitor of O-GlcNAcase, inhibited the IL-1β-induced IL-6 and IL-8 production in human RASFs in vitro (139). Furthermore, dynamic changes in O-GlcNAcylation are required for osteoclastogenesis. TNF-α fosters the occurrence of O-GlcNAcylation to promote osteoclastogenesis in inflammatory arthritis. Targeted pharmaceutical or genetic inhibition or knockdown O-GlcNAcylation could ameliorate bone loss in experimental arthritis (140). The role of glycosylation of Notch signaling in RA angiogenesis is worth further exploration.
Ubiquitylation of the Notch receptor is essential for NICD degradation and Notch incorporation into the intraluminal vesicles of maturing endosomes (141). Ubiquitin-specific peptidase 10 (USP10), a deubiquitinating enzyme, interacts with NICD1 to slow the ubiquitin-dependent turnover of the activated Notch-1 receptor, thereby eliminating the ubiquitination of NICD1 and stimulating Notch signaling in ECs (142). A previous study showed that B-cell chronic lymphocytic leukemia/lymphoma6-associated zinc finger protein (BAZF) undergoes polyubiquitination in a CUL3-based E3 ligase complex, accelerating the degradation of CBF1 and abridging vascular plexus formation by mediating VEGFR and Notch cross-signaling (143). The alteration of ubiquitin proteasomal pathway causes dysregulated cellular homeostasis, progressively leading to RA. For example, polyubiquitination of TRAF2, TRAF6, RIP-1 and NEMO activated TAK1 complex, which in turn is associate with IKK activation, ultimately leading to chronic joint damage in a patient of RA (144). Methotrexate, used for the treatment of RA, inhibits the K48-linked polyubiquitination of Numb and promotes Notch-1 ubiquitination, thus restricting the expression of Notch-1 (145). Therefore, ubiquitination of Notch signaling may be related to RA angiogenesis.
Studies have shown that NICD1 is modified by SUMOylation upon stress stimulation. SUMOylation of NICD1 can amplify nuclear localization and suppress the expression of Notch target genes (146). SUMOylation of NICD1 occurs in the RBPJ-associated molecular domain to modulate Notch target gene expression at unique lysine residue sites (K1774, K1780, K1781, and K1782) in several species, including mice, rats, and humans. Stress-induced SUMOylation of NICD1 increases nuclear localization and transcriptional activity but does not influence the formation of transcriptional complexes, further regulating NICD1 nuclear translocation and stability (146, 147). Given the effect of SUMOylation on angiogenesis, deSUMOylated VEGFR2 was tested in pathological angiogenesis. SUMO-specific protease 6, a member of the sentrin-specific protease (SENP) family (SENP1, 2, 3, 5, 6, and 7), is responsible for the depolymerization of SUMO chains, deSUMOylating VEGFR2, enhancing VEGFR2 transport to the surface, and triggering the VEGF signaling cascade in AGEs to induce EC angiogenesis (148). SUMOylated VEGFR2 at the lysine K1270 site accumulates in the Golgi apparatus of ECs, reducing its surface expression and distribution and leading to retarded progression of retinal vascular angiogenesis (149). Angiogenesis is regulated by the SUMOylation of Notch-activated Dll4. SENP1-mediated Notch-1 deSUMOylation limits the cleavage of N1ICD and triggers subsequent co-transcriptional activity in the nucleus, resulting in the activation of VEGFR signaling and endothelial angiogenesis. Endothelial SENP1 deletion significantly abrogates retinal vascularization by increasing the endothelial Notch-1 response (146). Recent research showed that the expression of SUMO-1 and SUMO-2 were elevated in FLSs of RA patients. SUMO-1 proteins are mostly localized in the nuclei of synovial sublining cells (150). The role of SUMOylation of Notch signaling in RA angiogenesis is unknown.
3.2.2 The regulation of Notch signaling during angiogenesis by lncRNAs/miRNAs in RA
Long nc-RNAs (lncRNAs) and short ncRNAs (such as microRNAs [miRNAs]) are a class of ncRNAs that regulate gene expression by interacting with DNA and RNA. A recent study revealed that Linc00514, a long nc-RNA, is highly expressed in clinical tissues and breast cancer cell lines. Overexpression of Linc00514 amplified M2 macrophage polarization via the Jagged1-mediated Notch signaling pathway to regulate breast cancer tumorigenicity (151). Additionally, lncRNA BREA2 disrupts WWP2-mediated ubiquitination of Notch-1 signaling to drive breast cancer metastasis (152). LINC01123 also activates Notch signaling to accelerate lung adenocarcinoma malignancy (153). In contrast, Notch signaling regulates the downstream target lncRNA LUNAR1, further triggering oncogenic activity in colorectal cancer and T-cell acute lymphoblastic leukemia (154, 155). LncRNAs involved in interactions with the Notch pathway have been linked to a variety of malignancies and tumor microenvironments (156).
The dysfunction of lncRNAs has been shown to be associated with angiogenesis through the modulation of Notch signaling. For instance, Meg3 is a tumor suppressor lncRNA that represses the expression of Notch and its target genes (Hes-1 and Hey-1) during angiogenesis after ischemic stroke (157). lncRNA Xist has been reported to be geared toward X-inactive specific transcripts and regulates the expression of Notch-1 in chronic compressive spinal cord injury-induced angiogenesis by binding to miR-32-5p (158). MALAT1 is decreased in the peripheral blood of RA patients and regulates changes in the Notch signaling pathway (159). It has been reported that lncRNA H19 inhibitors inhibit the proliferation and increase the apoptosis of synovial cells in RA by suppressing Notch signaling (160).
MiRNAs are a short functional endogenous class of RNA molecules that are 21–25 nucleotides long and are involved in most physiological and pathological events. Many studies have focused on the negative correlation between miRNAs and Notch signaling in cancer (161). However, miR-124 is a tumor-suppressive miRNA that is silenced by tumor-specific methylation during pancreatic cancer metastasis. MiR-124 recruits and activates macrophages and is transferred to a tumor-supporting M2-like phenotype by directly downregulating Notch signaling in cancer cells (162). Similarly, miR-34b-5p functions as a tumor suppressor in retinoblastoma by dysregulating the Notch signaling pathway (163). MiR-525-5p inhibits the activation of Notch and Wnt signaling pathways to reduce oxaliplatin resistance in breast cancer (164). In hepatic fibrosis, upregulation of miR-148a-5p inhibits the expression of the Notch2 and the Notch signaling pathway in human umbilical cord mesenchymal stem cells (165).
Many miRNAs interact with Notch and play pivotal roles in angiogenesis. For instance, miR-24-3p directly abrogates the expression of Notch-1 and DLL1, thereby reducing EC survival, proliferation, and angiogenic capacity in the limb muscles (166). MiR-652-3p can also limit the Notch ligand Jagged-1 to exert various effects on angiogenesis and immune cell regulation (167). MiR-223-3p has been reported to be a downstream molecule of Notch signaling that modulates the proliferation, differentiation, migration, and sprouting of ECs (168). Notch activation affects miR-223 transcription and enhances cytokine production by macrophages in patients with RA (169).
4 Notch signaling mediates hypoxia-induced angiogenesis in RA
Hypoxia is a key driving force inducing HIF stabilization. HIF-1α is a subunit of a heterodimeric transcription factor, and its expression is modulated by oxygen availability (170). HIF-1α is rapidly upregulated in hypoxia and degraded in normoxia. Increased HIF-1α is highly involved in the synovial tissues of patients with RA and is recognized as a crucial element in the perpetuation of angiogenesis and joint destruction in inflammatory arthritis (12). The interaction between HIF-1α and Notch signaling was elucidated in the hypoxic cellular response of cancer and RA synovial fibroblast cells (171). Activated N- and C-terminal regions of HIF-1α are involved in the hypoxia-dependent activation of Notch signaling. The HIF-1α-containing γ-secretase complex directly increases the cleavage of Notch ICD (172). The activated Notch signaling further potentiated the interaction of HIF-1α with the NICD and the CSL transcription factor, thereby increasing Notch signaling (173). Protein–protein interaction studies verified the crosstalk between HIF-1α-Notch signaling. Notch inhibitors limited the activation of HIF-1α (174, 175). Notch signaling inhibits hydroxylation of HIF-1α and recruitment of HIF-1α to corresponding sites, thereby enhancing the stability of HIF-1α (173). Recent studies revealed that under normoxia, factors inhibiting HIF hydroxylate NICD1 cause the recruitment of NICD1-deubiquitinating enzymes, consequently influencing non-degradative ubiquitin chains (176). HIF-1α-regulated miR-1275 exerts tumorigenic effects on cancer stem cell properties by modulating Notch signaling (177).
Several studies have shown that Notch signaling modulates hypoxia-induced RA angiogenesis both in vitro and in vivo (12). In RA synovial cultures, Notch-1 siRNA or antisense oligonucleotides suppress abnormal proliferation, the pro-inflammatory phenotype of FLSs, and hypoxia-induced angiogenesis (12). Notch signaling also regulates the hypoxia-induced invasion of RA-inflamed joints by influencing the VEGF/angiopoietin-2 pathway (10). The crosstalk between HIF-1α and Notch signaling has been shown to trigger the activation of synovial fibroblasts in RA. Silencing HIF-1α with small interfering RNA abridged the expression of N1ICD exposed to hypoxia in RA synovial fibroblast cells (RASFCs). Similarly, Notch-1 small interfering RNA abrogated VEGF, MMP2, and MMP9 expression in hypoxia-induced RASFCs, thus inhibiting their invasive capacity and pannus formation. However, N1ICD overexpression had the opposite effect, potentiating these processes (171).
HIF-Notch signaling has been shown to mediate angiogenesis by regulating the epigenetic modification factors in various diseases. HIF-Notch signaling has been shown to mediate angiogenesis in various diseases. Epigenetic modification factors have provided significant evidence of this.For instance, the MiR-497∼195 cluster, belonging to the miR-15 family, negatively regulated osteogenesis and angiogenesis through sustaining endothelial Notch and HIF-1α activity in murine long bones (178). In the TRIM family, TRIM28 is the most constitutively expressed transcription factor. The latest research on biological function in the cardiovascular system suggests that TRIM28 plays a pivotal role in controlling the VEGFR-Notch signaling circuit in sprouting angiogenesis by mediating HIF-1α and RBPJκ (179). In addition, HIF-1α-Notch-VEGF signaling has been reported to contribute to angiogenesis in temporomandibular joint osteoarthritis, missed abortion, and diabetic retinopathy (180–182).
5 Angiogenesis regulated by Notch signaling in RA: new insights into therapeutic strategies
Based on the various mechanisms mediated by Notch signaling, a suitable anti-RA approach has been developed, and promising novel strategies targeting the Notch pathway are currently under investigation (Table 1).
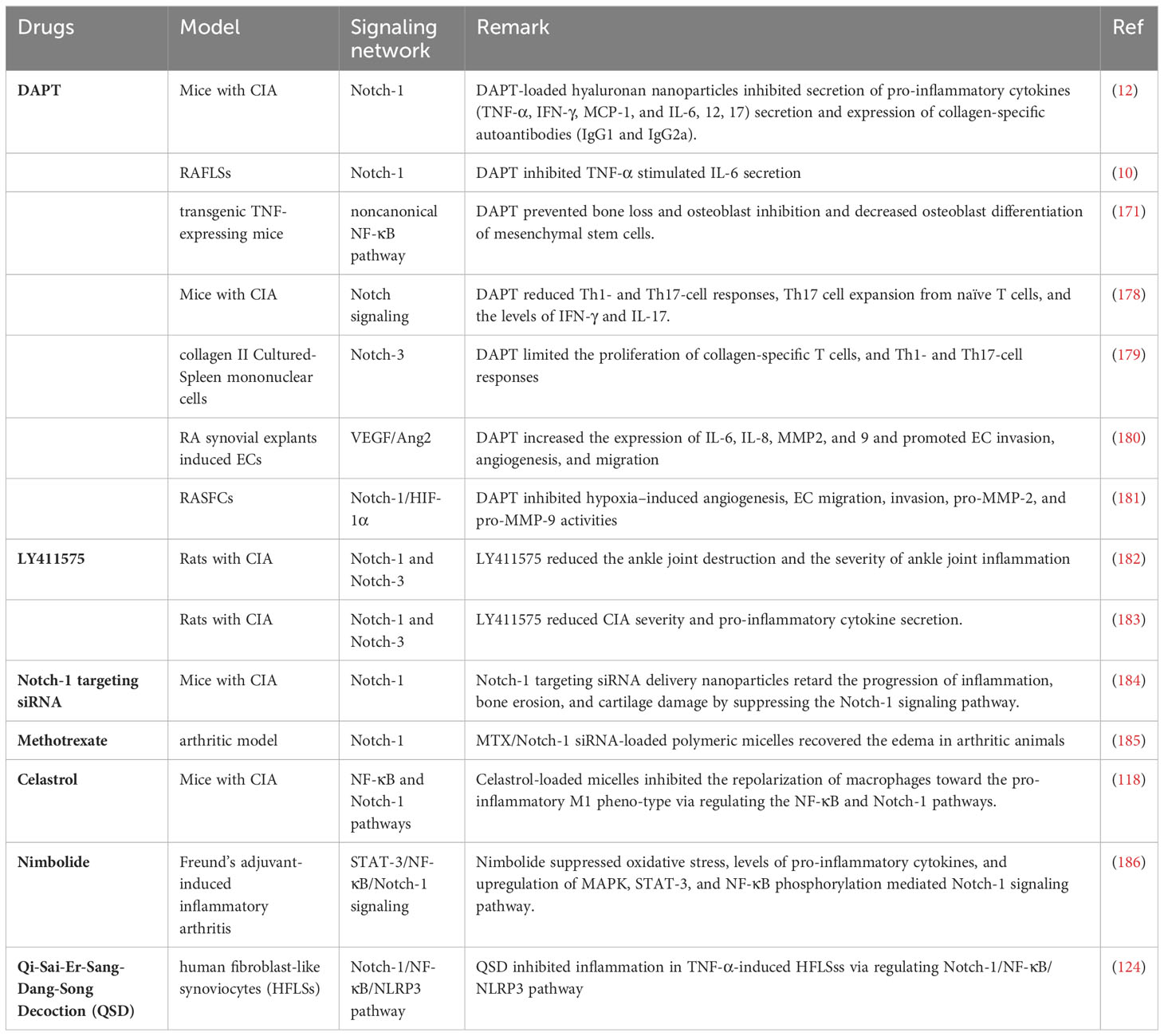
Table 1 Drugs targeting the Notch signaling assessed in RA.
The biological effects of Notch signaling are largely dependent on γ-secretase activity. Numerous studies have demonstrated that DAPT, an oral pharmacological inhibitor of active γ-secretase, attenuates arthritic symptoms and joint damage in CIA-treated and transgenic TNF-expressing mice (183–185). DAPT suppresses T cell proliferation and mediates Th1- and Th17-cell responses in RA both in vivo and in vitro (118, 186). The regulatory effect of Notch signaling on angiogenesis has been well established in several studies. DAPT inhibits EC invasion, migration, and hypoxia-induced angiogenesis in RASFCs (12, 124). LY411575 is another-secretase inhibitor that suppresses the expression of N1ICD and N3ICD, which can improve ankle joint destruction and reduce the production of pro-inflammatory cytokines (171, 187). Similarly, Notch-1 transfected by siRNA inhibited hypoxia-induced RASF invasion, angiogenesis and epithelial-mesenchymal transition in vitro, whereas overexpression of Notch-1 promoted these processes (188). Notch-1 targeting siRNA delivery nanoparticles inhibited inflammation, bone erosion, and cartilage damage in a CIA model, indicating that targeting Notch signaling could potentially prevent or delay the onset of RA in inflamed joints.
Methotrexate is a first- or second-line disease-modifying anti-rheumatic drug used to treat RA effectively. Nanomedicine-based MTX and Notch 1 siRNA delivery have been demonstrated to suppress paw thickness and disease progression by downregulating Notch-1 expression in an arthritic model (189). Some Chinese herbs and natural compounds have provided new sources for the development and treatment of RA. Celastrol is an active component of Tripterygium wilfordii. Celastrol-loaded polymeric micelles abrogate the repolarization of macrophages, swelling of joints, synovial inflammation, cartilage degradation, and bone erosion by modulating NF-κB and Notch signaling (190). Nimbolide, a natural chemical constituent of Azadirachta indica, suppresses oxidative stress and inflammation by inhibiting STAT-3/NF-κB/Notch-1 signaling in both IL-1β stimulated synovial fibroblasts and a Freund’s adjuvant-induced inflammatory arthritis model (191). Furthermore, in the Tibetan Hospital of the Tibet Autonomous Region, a classical herbal formula called Qi-Sai-Er-Sang-Dang-Song Decoction (QSD) is a standard medical prescription for treating RA. QSD alleviates inflammation in TNF-α-induced HFLSss by depressing the Notch-1/NF-κB/NLRP3 pathway (192).
6 Conclusions and future directions
Notch signaling is an important and complex player in many aspects of angiogenesis. The modulation of Notch signaling in the angiogenic processes of RA relies on different mechanisms involving stromal cells and adipokines. Although the available data suggest that DCs, MDSCs, M2 macrophages, CD4+ and CD8+ T cells, and, in part, adipokines can activate ECs and promote ECs proliferation and migration, further studies are necessary to verify the effects of Notch signaling regulated by glycosylation, ubiquitination, SUMOylation, and lncRNAs/miRNAs on RA angiogenesis. Notch signaling is an important factor in hypoxia-induced angiogenesis in RA, and targeting Notch signaling may represent an efficient therapy for angiogenesis in RA.
Author contributions
FZ: Writing – original draft, Conceptualization, Project administration; KA, LL and XC: Writing – review & editing, Conceptualization, Project administration; ZZ and JH: Writing – review & editing, Project administration; YH and HH: Writing – review & editing, Formal Analysis, Methodology.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by grants from the National Natural Science Foundation of China General Program (82274506), Science and Technology Innovation Program of Hunan Province (2020SK1020, 2021RC4035), Open-Competing Disciple Construction Project of Hunan University of Chinese Medicine (HNUCM) (22JBZ003), Hunan Furong Distinguished Scholar Program (XJT[2020]58), Hunan 121 Training Project for Innovative Talents (XRSH[2019]192), the Chinese Academy of Engineering Academician Liang Liu’s Workstation Project of HNUCM (KH[2021]4-21YS001) and World First-class Discipline Incubation Project of HNUCM (XJF[2022]57).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Finckh A, Gilbert B, Hodkinson B, Bae SC, Thomas R, Deane KD, et al. Global epidemiology of rheumatoid arthritis. Nat Rev Rheumatol (2022) 18:591–602. doi: 10.1038/s41584-022-00827-y
2. Elshabrawy HA, Chen Z, Volin MV, Ravella S, Virupannavar S, Shahrara S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis (2015) 18:433–48. doi: 10.1007/s10456-015-9477-2
3. Marrelli A, Cipriani P, Liakouli V, Carubbi F, Perricone C, Perricone R, et al. Angiogenesis in rheumatoid arthritis: a disease specific process or a common response to chronic inflammation? Autoimmun Rev (2011) 10:595–8. doi: 10.1016/j.autrev.2011.04.020
4. Annex BH, Cooke JP. New directions in therapeutic angiogenesis and arteriogenesis in peripheral arterial disease. Circ Res (2021) 128:1944–57. doi: 10.1161/CIRCRESAHA.121.318266
5. Kargozar S, Baino F, Hamzehlou S, Hamblin MR, Mozafari M. Nanotechnology for angiogenesis: opportunities and challenges. Chem Soc Rev (2020) 49:5008–57. doi: 10.1039/C8CS01021H
6. MacGrogan D, Munch J, de la Pompa JL. Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat Rev Cardiol (2018) 15:685–704. doi: 10.1038/s41569-018-0100-2
7. Luo Z, Shang X, Zhang H, Wang G, Massey PA, Barton SR, et al. Notch signaling in osteogenesis, osteoclastogenesis, and angiogenesis. Am J Pathol (2019) 189:1495–500. doi: 10.1016/j.ajpath.2019.05.005
8. Zhou B, Lin W, Long Y, Yang Y, Zhang H, Wu K, et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct Target Ther (2022) 7:95. doi: 10.1038/s41392-022-00934-y
9. Borggrefe T, Oswald F. The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci (2009) 66:1631–46. doi: 10.1007/s00018-009-8668-7
10. Keewan E, Naser SA. The role of notch signaling in macrophages during inflammation and infection: implication in rheumatoid arthritis? Cells-Basel (2020) 9. doi: 10.3390/cells9010111
11. Zhang J, Xiong Q, Yang L, Xue Y, Ke M, Li Z. Cytochrome P450 2J2 inhibits the proliferation and angiogenesis of retinal vascular endothelial cells by regulating the Notch signaling pathway in a hypoxia-induced retinopathy model. Bioengineered (2021) 12:10878–90. doi: 10.1080/21655979.2021.1994722
12. Gao W, Sweeney C, Connolly M, Kennedy A, Ng CT, McCormick J, et al. Notch-1 mediates hypoxia-induced angiogenesis in rheumatoid arthritis. Arthritis Rheum (2012) 64:2104–13. doi: 10.1002/art.34397
13. Canavan M, Marzaioli V, McGarry T, Bhargava V, Nagpal S, Veale DJ, et al. Rheumatoid arthritis synovial microenvironment induces metabolic and functional adaptations in dendritic cells. Clin Exp Immunol (2020) 202:226–38. doi: 10.1111/cei.13479
14. Bosisio D, Ronca R, Salvi V, Presta M, Sozzani S. Dendritic cells in inflammatory angiogenesis and lymphangiogenesis. Curr Opin Immunol (2018) 53:180–6. doi: 10.1016/j.coi.2018.05.011
15. Del PA, Salvi V, Soriani A, Laffranchi M, Sozio F, Bosisio D, et al. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell Mol Immunol (2023) 20:432–47.
16. Korbecki J, Grochans S, Gutowska I, Barczak K, Baranowska-Bosiacka I. CC chemokines in a tumor: A review of pro-cancer and anti-cancer properties of receptors CCR5, CCR6, CCR7, CCR8, CCR9, and CCR10 ligands. Int J Mol Sci (2020) 21(20):7619. doi: 10.3390/ijms21207619
17. Brandum EP, Jorgensen AS, Rosenkilde MM, Hjorto GM. Dendritic cells and CCR7 expression: an important factor for autoimmune diseases, chronic inflammation, and cancer. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22158340
18. Huang J, Zhou Q. Identification of the relationship between hub genes and immune cell infiltration in vascular endothelial cells of proliferative diabetic retinopathy using bioinformatics methods. Dis Markers (2022) 2022:7231046. doi: 10.1155/2022/7231046
19. Lee WS, Yang H, Chon HJ, Kim C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp Mol Med (2020) 52:1475–85. doi: 10.1038/s12276-020-00500-y
20. Sun K, Li YY, Jin J. A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Signal Transduct Target Ther (2021) 6:79. doi: 10.1038/s41392-020-00455-6
21. Liu J, Zhang X, Cheng Y, Cao X. Dendritic cell migration in inflammation and immunity. Cell Mol Immunol (2021) 18:2461–71. doi: 10.1038/s41423-021-00726-4
22. Yamada S, Nagafuchi Y, Wang M, Ota M, Hatano H, Takeshima Y, et al. Immunomics analysis of rheumatoid arthritis identified precursor dendritic cells as a key cell subset of treatment resistance. Ann Rheum Dis (2023) 82:809–19. doi: 10.1101/2022.12.21.22283652
23. Coutant F. Shaping of monocyte-derived dendritic cell development and function by environmental factors in rheumatoid arthritis. Int J Mol Sci (2021) 22. doi: 10.3390/ijms222413670
24. Berger S, Dyugovskaya L, Polyakov A, Lavie L. Short-term fibronectin treatment induces endothelial-like and angiogenic properties in monocyte-derived immature dendritic cells: involvement of intracellular VEGF and MAPK regulation. Eur J Cell Biol (2012) 91:640–53. doi: 10.1016/j.ejcb.2012.02.003
25. Mylonas A, Hawerkamp HC, Wang Y, Chen J, Messina F, Demaria O, et al. Type I IFNs link skin-associated dysbiotic commensal bacteria to pathogenic inflammation and angiogenesis in rosacea. JCI Insight (2023) 8. doi: 10.1172/jci.insight.151846
26. Wasko R, Bridges K, Pannone R, Sidhu I, Xing Y, Naik S, et al. Langerhans cells are essential components of the angiogenic niche during murine skin repair. Dev Cell (2022) 57:2699–713. doi: 10.1016/j.devcel.2022.11.012
27. Bitirgen G, Kucuk A, Ergun MC, Satirtav G, Malik RA. Corneal nerve loss and increased Langerhans cells are associated with disease severity in patients with rheumatoid arthritis. Eye (Lond) (2023) 37:2950–5. doi: 10.1038/s41433-023-02447-6
28. Marzaioli V, Canavan M, Floudas A, Flynn K, Mullan R, Veale DJ, et al. CD209/CD14(+) dendritic cells characterization in rheumatoid and psoriatic arthritis patients: activation, synovial infiltration, and therapeutic targeting. Front Immunol (2021) 12:722349. doi: 10.3389/fimmu.2021.722349
29. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
30. Grover A, Sanseviero E, Timosenko E, Gabrilovich DI. Myeloid-derived suppressor cells: A propitious road to clinic. Cancer Discovery (2021) 11:2693–706. doi: 10.1158/2159-8290.CD-21-0764
31. Ling Z, Yang C, Tan J, Dou C, Chen Y. Beyond immunosuppressive effects: dual roles of myeloid-derived suppressor cells in bone-related diseases. Cell Mol Life Sci (2021) 78:7161–83. doi: 10.1007/s00018-021-03966-9
32. Chen S, Guo C, Wang R, Feng Z, Liu Z, Wu L, et al. Monocytic MDSCs skew Th17 cells toward a pro-osteoclastogenic phenotype and potentiate bone erosion in rheumatoid arthritis. Rheumatol (Oxford) (2021) 60:2409–20. doi: 10.1093/rheumatology/keaa625
33. Navashenaq JG, Shabgah AG, Hedayati-Moghadam M, Ariaee N, Mohammadi H, Hemmatzadeh M, et al. The role of myeloid-derived suppressor cells in rheumatoid arthritis: An update. Life Sci (2021) 269:119083. doi: 10.1016/j.lfs.2021.119083
34. Yin Z, Li C, Wang J, Xue L. Myeloid-derived suppressor cells: Roles in the tumor microenvironment and tumor radiotherapy. Int J Cancer (2019) 144:933–46. doi: 10.1002/ijc.31744
35. Zhang T, Zhou J, Man G, Leung KT, Liang B, Xiao B, et al. MDSCs drive the process of endometriosis by enhancing angiogenesis and are a new potential therapeutic target. Eur J Immunol (2018) 48:1059–73. doi: 10.1002/eji.201747417
36. Rahma OE, Hodi FS. The intersection between tumor angiogenesis and immune suppression. Clin Cancer Res (2019) 25:5449–57. doi: 10.1158/1078-0432.CCR-18-1543
37. Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget (2015) 6:27478–89. doi: 10.18632/oncotarget.4393
38. Wang W, Zuo R, Long H, Wang Y, Zhang Y, Sun C, et al. Advances in the Masquelet technique: Myeloid-derived suppressor cells promote angiogenesis in PMMA-induced membranes. Acta Biomater (2020) 108:223–36. doi: 10.1016/j.actbio.2020.03.010
39. Li Y, Qiao K, Zhang X, Liu H, Zhang H, Li Z, et al. Targeting myeloid-derived suppressor cells to attenuate vasculogenic mimicry and synergistically enhance the anti-tumor effect of PD-1 inhibitor. iScience (2021) 24:103392. doi: 10.1016/j.isci.2021.103392
40. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22136995
41. Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage polarization and its role in liver disease. Front Immunol (2021) 12:803037. doi: 10.3389/fimmu.2021.803037
42. Qiu S, Xie L, Lu C, Gu C, Xia Y, Lv J, et al. Gastric cancer-derived exosomal miR-519a-3p promotes liver metastasis by inducing intrahepatic M2-like macrophage-mediated angiogenesis. J Exp Clin Cancer Res (2022) 41:296. doi: 10.1186/s13046-022-02499-8
43. Ikeda T, Nakamura K, Kida T, Oku H. Possible roles of anti-type II collagen antibody and innate immunity in the development and progression of diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol (2022) 260:387–403. doi: 10.1007/s00417-021-05342-6
44. Dong F, Ruan S, Wang J, Xia Y, Le K, Xiao X, et al. M2 macrophage-induced lncRNA PCAT6 facilitates tumorigenesis and angiogenesis of triple-negative breast cancer through modulation of VEGFR2. Cell Death Dis (2020) 11:728. doi: 10.1038/s41419-020-02926-8
45. Yang Y, Guo Z, Chen W, Wang X, Cao M, Han X, et al. M2 macrophage-derived exosomes promote angiogenesis and growth of pancreatic ductal adenocarcinoma by targeting E2F2. Mol Ther (2021) 29:1226–38. doi: 10.1016/j.ymthe.2020.11.024
46. Zhu D, Johnson TK, Wang Y, Thomas M, Huynh K, Yang Q, et al. Macrophage M2 polarization induced by exosomes from adipose-derived stem cells contributes to the exosomal proangiogenic effect on mouse ischemic hindlimb. Stem Cell Res Ther (2020) 11:162. doi: 10.1186/s13287-020-01669-9
47. Yin Z, Ma T, Huang B, Lin L, Zhou Y, Yan J, et al. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-beta signaling pathway. J Exp Clin Cancer Res (2019) 38:310. doi: 10.1186/s13046-019-1313-x
48. Xu H, Zhu Y, Hsiao AW, Xu J, Tong W, Chang L, et al. Bioactive glass-elicited stem cell-derived extracellular vesicles regulate M2 macrophage polarization and angiogenesis to improve tendon regeneration and functional recovery. Biomaterials (2023) 294:121998. doi: 10.1016/j.biomaterials.2023.121998
49. Van Raemdonck K, Umar S, Shahrara S. The pathogenic importance of CCL21 and CCR7 in rheumatoid arthritis. Cytokine Growth Factor Rev (2020) 55:86–93. doi: 10.1016/j.cytogfr.2020.05.007
50. Kobori T, Hamasaki S, Kitaura A, Yamazaki Y, Nishinaka T, Niwa A, et al. Interleukin-18 amplifies macrophage polarization and morphological alteration, leading to excessive angiogenesis. Front Immunol (2018) 9:334. doi: 10.3389/fimmu.2018.00334
51. Shang ZZ, Qin DY, Li QM, Zha XQ, Pan LH, Peng DY, et al. Dendrobium huoshanense stem polysaccharide ameliorates rheumatoid arthritis in mice via inhibition of inflammatory signaling pathways. Carbohydr Polym (2021) 258:117657. doi: 10.1016/j.carbpol.2021.117657
52. Paradowska-Gorycka A, Wajda A, Romanowska-Prochnicka K, Walczuk E, Kuca-Warnawin E, Kmiolek T, et al. Th17/treg-related transcriptional factor expression and cytokine profile in patients with rheumatoid arthritis. Front Immunol (2020) 11:572858. doi: 10.3389/fimmu.2020.572858
53. Yoo SA, Kim M, Kang MC, Kong JS, Kim KM, Lee S, et al. Placental growth factor regulates the generation of T(H)17 cells to link angiogenesis with autoimmunity. Nat Immunol (2019) 20:1348–59. doi: 10.1038/s41590-019-0456-4
54. Liu S, Li Y, Xia L, Shen H, Lu J. IL-35 prevent bone loss through promotion of bone formation and angiogenesis in rheumatoid arthritis. Clin Exp Rheumatol (2019) 37:820–5.
55. Jin S, Sun S, Ling H, Ma J, Zhang X, Xie Z, et al. Protectin DX restores Treg/T(h)17 cell balance in rheumatoid arthritis by inhibiting NLRP3 inflammasome via miR-20a. Cell Death Dis (2021) 12:280. doi: 10.1038/s41419-021-03562-6
56. Xin PL, Jie LF, Cheng Q, Bin DY, Dan CW. Pathogenesis and function of interleukin-35 in rheumatoid arthritis. Front Pharmacol (2021) 12:655114. doi: 10.3389/fphar.2021.655114
57. Carvalheiro H, Duarte C, Silva-Cardoso S, Da SJ, Souto-Carneiro MM. CD8+ T cell profiles in patients with rheumatoid arthritis and their relationship to disease activity. Arthritis Rheumatol (2015) 67:363–71. doi: 10.1002/art.38941
58. Wang Y, Song L, Sun J, Sui Y, Li D, Li G, et al. Expression of Galectin-9 and correlation with disease activity and vascular endothelial growth factor in rheumatoid arthritis. Clin Exp Rheumatol (2020) 38:654–61.
59. Deliyanti D, Figgett WA, Gebhardt T, Trapani JA, Mackay F, Wilkinson-Berka JL. CD8(+) T cells promote pathological angiogenesis in ocular neovascular disease. Arterioscler Thromb Vasc Biol (2023) 43:522–36. doi: 10.1161/ATVBAHA.122.318079
60. Liang C, Yang KY, Chan VW, Li X, Fung T, Wu Y, et al. CD8(+) T-cell plasticity regulates vascular regeneration in type-2 diabetes. Theranostics (2020) 10:4217–32. doi: 10.7150/thno.40663
61. Ungvari Z, Tarantini S, Kiss T, Wren JD, Giles CB, Griffin CT, et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol (2018) 15:555–65. doi: 10.1038/s41569-018-0030-z
62. Xu S, Ilyas I, Little PJ, Li H, Kamato D, Zheng X, et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev (2021) 73:924–67. doi: 10.1124/pharmrev.120.000096
63. Blum A, Adawi M. Rheumatoid arthritis (RA) and cardiovascular disease. Autoimmun Rev (2019) 18:679–90. doi: 10.1016/j.autrev.2019.05.005
64. England BR, Thiele GM, Anderson DR, Mikuls TR. Increased cardiovascular risk in rheumatoid arthritis: mechanisms and implications. BMJ (2018) 361:k1036. doi: 10.1136/bmj.k1036
65. Luczak A, Malecki R, Kulus M, Madej M, Szahidewicz-Krupska E, Doroszko A. Cardiovascular risk and endothelial dysfunction in primary sjogren syndrome is related to the disease activity. Nutrients (2021) 13. doi: 10.3390/nu13062072
66. Akhmedov A, Crucet M, Simic B, Kraler S, Bonetti NR, Ospelt C, et al. TNFalpha induces endothelial dysfunction in rheumatoid arthritis via LOX-1 and arginase 2: reversal by monoclonal TNFalpha antibodies. Cardiovasc Res (2022) 118:254–66. doi: 10.1093/cvr/cvab005
67. Wright HL, Lyon M, Chapman EA, Moots RJ, Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol (2020) 11:584116. doi: 10.3389/fimmu.2020.584116
68. Wang L, Luqmani R, Udalova IA. The role of neutrophils in rheumatic disease-associated vascular inflammation. Nat Rev Rheumatol (2022) 18:158–70. doi: 10.1038/s41584-021-00738-4
69. Perez-Sanchez C, Ruiz-Limon P, Aguirre MA, Jimenez-Gomez Y, Arias-de LRI, Abalos-Aguilera MC, et al. Diagnostic potential of NETosis-derived products for disease activity, atherosclerosis and therapeutic effectiveness in Rheumatoid Arthritis patients. J Autoimmun (2017) 82:31–40. doi: 10.1016/j.jaut.2017.04.007
70. Yu S, Liu J, Yan N. Endothelial dysfunction induced by extracellular neutrophil traps plays important role in the occurrence and treatment of extracellular neutrophil traps-related disease. Int J Mol Sci (2022) 23. doi: 10.3390/ijms23105626
71. Liang W, Ferrara N. The complex role of neutrophils in tumor angiogenesis and metastasis. Cancer Immunol Res (2016) 4:83–91. doi: 10.1158/2326-6066.CIR-15-0313
72. Aldabbous L, Abdul-Salam V, McKinnon T, Duluc L, Pepke-Zaba J, Southwood M, et al. Neutrophil extracellular traps promote angiogenesis: evidence from vascular pathology in pulmonary hypertension. Arterioscler Thromb Vasc Biol (2016) 36:2078–87. doi: 10.1161/ATVBAHA.116.307634
73. Zisman D, Safieh M, Simanovich E, Feld J, Kinarty A, Zisman L, et al. Tocilizumab (TCZ) decreases angiogenesis in rheumatoid arthritis through its regulatory effect on miR-146a-5p and EMMPRIN/CD147. Front Immunol (2021) 12:739592. doi: 10.3389/fimmu.2021.739592
74. Tuckermann J, Adams RH. The endothelium-bone axis in development, homeostasis and bone and joint disease. Nat Rev Rheumatol (2021) 17:608–20. doi: 10.1038/s41584-021-00682-3
75. Deng R, Bu Y, Li F, Wu H, Wang Y, Wei W. The interplay between fibroblast-like synovial and vascular endothelial cells leads to angiogenesis via the sphingosine-1-phosphate-induced RhoA-F-Actin and Ras-Erk1/2 pathways and the intervention of geniposide. Phytother Res (2021) 35:5305–17. doi: 10.1002/ptr.7211
76. Wang Z, Wang J, Lan T, Zhang L, Yan Z, Zhang N, et al. Role and mechanism of fibroblast-activated protein-alpha expression on the surface of fibroblast-like synoviocytes in rheumatoid arthritis. Front Immunol (2023) 14:1135384. doi: 10.3389/fimmu.2023.1135384
77. Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature (2019) 570:246–51. doi: 10.1038/s41586-019-1263-7
78. Kim JW, Kong JS, Lee S, Yoo SA, Koh JH, Jin J, et al. Angiogenic cytokines can reflect the synovitis severity and treatment response to biologics in rheumatoid arthritis. Exp Mol Med (2020) 52:843–53. doi: 10.1038/s12276-020-0443-8
79. Recinella L, Orlando G, Ferrante C, Chiavaroli A, Brunetti L, Leone S. Adipokines: new potential therapeutic target for obesity and metabolic, rheumatic, and cardiovascular diseases. Front Physiol (2020) 11:578966. doi: 10.3389/fphys.2020.578966
80. Neumann E, Hasseli R, Ohl S, Lange U, Frommer KW, Muller-Ladner U. Adipokines and autoimmunity in inflammatory arthritis. Cells-Basel (2021) 10. doi: 10.3390/cells10020216
81. Hasseli R, Frommer KW, Schwarz M, Hulser ML, Schreiyack C, Arnold M, et al. Adipokines and inflammation alter the interaction between rheumatoid arthritis synovial fibroblasts and endothelial cells. Front Immunol (2020) 11:925. doi: 10.3389/fimmu.2020.00925
82. Lelis DF, Freitas DF, MaChado AS, Crespo TS, Santos S. Angiotensin-(1-7), adipokines and inflammation. Metabolism (2019) 95:36–45. doi: 10.1016/j.metabol.2019.03.006
83. Barone I, Giordano C, Bonofiglio D, Ando S, Catalano S. The weight of obesity in breast cancer progression and metastasis: Clinical and molecular perspectives. Semin Cancer Biol (2020) 60:274–84. doi: 10.1016/j.semcancer.2019.09.001
84. Mukherjee A, Chiang CY, Daifotis HA, Nieman KM, Fahrmann JF, Lastra RR, et al. Adipocyte-induced FABP4 expression in ovarian cancer cells promotes metastasis and mediates carboplatin resistance. Cancer Res (2020) 80:1748–61. doi: 10.1158/0008-5472.CAN-19-1999
85. Chen S, Du J, Zhao W, Cao R, Wang N, Li J, et al. Elevated expression of FABP4 is associated with disease activity in rheumatoid arthritis patients. biomark Med (2020) 14:1405–13. doi: 10.2217/bmm-2020-0284
86. Andres CL, Kuklova M, Hulejova H, Vernerova Z, Pesakova V, Pecha O, et al. The level of fatty acid-binding protein 4, a novel adipokine, is increased in rheumatoid arthritis and correlates with serum cholesterol levels. Cytokine (2013) 64:441–7. doi: 10.1016/j.cyto.2013.05.001
87. Kraus FV, Keck S, Klika KD, Graf J, Carvalho RA, Lorenz HM, et al. Reduction of proinflammatory effector functions through remodeling of fatty acid metabolism in CD8+ T cells from rheumatoid arthritis patients. Arthritis Rheumatol (2023) 75:1098–109. doi: 10.1101/2022.09.22.22280236
88. Harjes U, Bridges E, Gharpure KM, Roxanis I, Sheldon H, Miranda F, et al. Antiangiogenic and tumour inhibitory effects of downregulating tumour endothelial FABP4. Oncogene (2017) 36:912–21. doi: 10.1038/onc.2016.256
89. Ghelfi E, Yu CW, Elmasri H, Terwelp M, Lee CG, Bhandari V, et al. Fatty acid binding protein 4 regulates VEGF-induced airway angiogenesis and inflammation in a transgenic mouse model: implications for asthma. Am J Pathol (2013) 182:1425–33. doi: 10.1016/j.ajpath.2012.12.009
90. Xie Y, Feng SL, Mai CT, Zheng YF, Wang H, Liu ZQ, et al. Suppression of up-regulated LXRalpha by silybin ameliorates experimental rheumatoid arthritis and abnormal lipid metabolism. Phytomedicine (2021) 80:153339. doi: 10.1016/j.phymed.2020.153339
91. Guo D, Lin C, Lu Y, Guan H, Qi W, Zhang H, et al. FABP4 secreted by M1-polarized macrophages promotes synovitis and angiogenesis to exacerbate rheumatoid arthritis. Bone Res (2022) 10:45. doi: 10.1038/s41413-022-00211-2
92. Szumilas K, Szumilas P, Sluczanowska-Glabowska S, Zgutka K, Pawlik A. Role of adiponectin in the pathogenesis of rheumatoid arthritis. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21218265
93. Laczna M, Kopytko P, Tkacz M, Zgutka K, Czerewaty M, Tarnowski M, et al. Adiponectin is a component of the inflammatory cascade in rheumatoid arthritis. J Clin Med (2022) 11.
94. Che N, Sun X, Gu L, Wang X, Shi J, Sun Y, et al. Adiponectin enhances B-cell proliferation and differentiation via activation of akt1/STAT3 and exacerbates collagen-induced arthritis. Front Immunol (2021) 12:626310. doi: 10.3389/fimmu.2021.626310
95. Liu R, Zhao P, Zhang Q, Che N, Xu L, Qian J, et al. Adiponectin promotes fibroblast-like synoviocytes producing IL-6 to enhance T follicular helper cells response in rheumatoid arthritis. Clin Exp Rheumatol (2020) 38:11–8.
96. Qian J, Xu L, Sun X, Wang Y, Xuan W, Zhang Q, et al. Adiponectin aggravates bone erosion by promoting osteopontin production in synovial tissue of rheumatoid arthritis. Arthritis Res Ther (2018) 20:26. doi: 10.1186/s13075-018-1526-y
97. Wang Y, Liu R, Zhao P, Zhang Q, Huang Y, Wang L, et al. Blockade of adiponectin receptor 1 signaling inhibits synovial inflammation and alleviates joint damage in collagen-induced arthritis. Clin Rheumatol (2022) 41:255–64. doi: 10.1007/s10067-021-05846-w
98. Lei Y, Li X, Gao Z, Liu Y, Zhang B, Xia L, et al. Association between adiponectin and clinical manifestations in rheumatoid arthritis. J Interferon Cytokine Res (2020) 40:501–8. doi: 10.1089/jir.2020.0080
99. Baker JF, England BR, George MD, Wysham K, Johnson T, Kunkel G, et al. Elevations in adipocytokines and mortality in rheumatoid arthritis. Rheumatol (Oxford) (2022) 61:4924–34. doi: 10.1093/rheumatology/keac191
100. Federico LE, Johnson TM, England BR, Wysham KD, George MD, Sauer B, et al. Circulating adipokines and associations with incident cardiovascular disease in rheumatoid arthritis. Arthritis Care Res (Hoboken) (2023) 75:768–77. doi: 10.1002/acr.24885
101. Huang CC, Law YY, Liu SC, Hu SL, Lin JA, Chen CJ, et al. Adiponectin promotes VEGF expression in rheumatoid arthritis synovial fibroblasts and induces endothelial progenitor cell angiogenesis by inhibiting miR-106a-5p. Cells-Basel (2021) 10. doi: 10.3390/cells10102627
102. Kim KS, Lee YA, Ji HI, Song R, Kim JY, Lee SH, et al. Increased expression of endocan in arthritic synovial tissues: effects of adiponectin on the expression of endocan in fibroblast-like synoviocytes. Mol Med Rep (2015) 11:2695–702. doi: 10.3892/mmr.2014.3057
103. Sangaletti S, Botti L, Gulino A, Lecis D, Bassani B, Portararo P, et al. SPARC regulation of PMN clearance protects from pristane-induced lupus and rheumatoid arthritis. iScience (2021) 24:102510. doi: 10.1016/j.isci.2021.102510
104. Sangaletti S, Talarico G, Chiodoni C, Cappetti B, Botti L, Portararo P, et al. SPARC is a new myeloid-derived suppressor cell marker licensing suppressive activities. Front Immunol (2019) 10:1369. doi: 10.3389/fimmu.2019.01369
105. Yang XL, Hu ZD, Wu Q, Liu X, Liu QJ, Zhang YC, et al. Association of polymorphisms in SPARC and NLRP2 genes with rheumatoid arthritis in a Chinese Han population. Mod Rheumatol (2015) 25:67–71. doi: 10.3109/14397595.2014.903595
106. Liu L, Hu F, Wang H, Wu X, Eltahan AS, Stanford S, et al. Secreted protein acidic and rich in cysteine mediated biomimetic delivery of methotrexate by albumin-based nanomedicines for rheumatoid arthritis therapy. ACS Nano (2019) 13:5036–48. doi: 10.1021/acsnano.9b01710
107. Deng SK, Jin Y, Jin Y, Wang JF. SPARC induces M2 polarization of macrophages to promote proliferation, migration, and angiogenesis of cholangiocarcinoma cells. Neoplasma (2022) 69:1101–7. doi: 10.4149/neo_2022_220324N333
108. Jing Y, Jin Y, Wang Y, Chen S, Zhang X, Song Y, et al. SPARC promotes the proliferation and metastasis of oral squamous cell carcinoma by PI3K/AKT/PDGFB/PDGFRbeta axis. J Cell Physiol (2019) 234:15581–93. doi: 10.1002/jcp.28205
109. Alkabie S, Basivireddy J, Zhou L, Roskams J, Rieckmann P, Quandt JA. SPARC expression by cerebral microvascular endothelial cells in vitro and its influence on blood-brain barrier properties. J Neuroinflamm (2016) 13:225. doi: 10.1186/s12974-016-0657-9
110. Luo L, Sun X, Tang M, Wu J, Qian T, Chen S, et al. Secreted protein acidic and rich in cysteine mediates the development and progression of diabetic retinopathy. Front Endocrinol (Lausanne) (2022) 13:869519. doi: 10.3389/fendo.2022.869519
111. Moldovan GE, Miele L, Fazleabas AT. Notch signaling in reproduction. Trends Endocrinol Metab (2021) 32:1044–57. doi: 10.1016/j.tem.2021.08.002
112. Krishna BM, Jana S, Singhal J, Horne D, Awasthi S, Salgia R, et al. Notch signaling in breast cancer: From pathway analysis to therapy. Cancer Lett (2019) 461:123–31. doi: 10.1016/j.canlet.2019.07.012
113. Meurette O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell (2018) 34:536–48. doi: 10.1016/j.ccell.2018.07.009
114. Spuul P, Daubon T, Pitter B, Alonso F, Fremaux I, Kramer I, et al. VEGF-A/notch-induced podosomes proteolyse basement membrane collagen-IV during retinal sprouting angiogenesis. Cell Rep (2016) 17:484–500. doi: 10.1016/j.celrep.2016.09.016
115. Pitulescu ME, Schmidt I, Giaimo BD, Antoine T, Berkenfeld F, Ferrante F, et al. Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nat Cell Biol (2017) 19:915–27. doi: 10.1038/ncb3555
116. Huang Z, Huang S, Song T, Yin Y, Tan C. Placental angiogenesis in mammals: A review of the regulatory effects of signaling pathways and functional nutrients. Adv Nutr (2021) 12:2415–34. doi: 10.1093/advances/nmab070
117. Hasan SS, Fischer A. Notch signaling in the vasculature: angiogenesis and angiocrine functions. Cold Spring Harb Perspect Med (2023) 13. doi: 10.1101/cshperspect.a041166
118. Jiao Z, Wang W, Hua S, Liu M, Wang H, Wang X, et al. Blockade of Notch signaling ameliorates murine collagen-induced arthritis via suppressing Th1 and Th17 cell responses. Am J Pathol (2014) 184:1085–93. doi: 10.1016/j.ajpath.2013.12.010
119. Choi BY, Choi Y, Park JS, Kang LJ, Baek SH, Park JS, et al. Inhibition of Notch1 induces population and suppressive activity of regulatory T cell in inflammatory arthritis. Theranostics (2018) 8:4795–804. doi: 10.7150/thno.26093
120. Wang Y, Han CC, Cui D, Li Y, Ma Y, Wei W. Is macrophage polarization important in rheumatoid arthritis? Int Immunopharmacol (2017) 50:345–52. doi: 10.1016/j.intimp.2017.07.019
121. Yu J, Canalis E. Notch and the regulation of osteoclast differentiation and function. Bone (2020) 138:115474. doi: 10.1016/j.bone.2020.115474
122. Tian Y, Xu Y, Fu Q, Chang M, Wang Y, Shang X, et al. Notch inhibits chondrogenic differentiation of mesenchymal progenitor cells by targeting Twist1. Mol Cell Endocrinol (2015) 403:30–8. doi: 10.1016/j.mce.2015.01.015
123. Wei K, Korsunsky I, Marshall JL, Gao A, Watts G, Major T, et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature (2020) 582:259–64. doi: 10.1038/s41586-020-2222-z
124. Gao W, Sweeney C, Walsh C, Rooney P, McCormick J, Veale DJ, et al. Notch signalling pathways mediate synovial angiogenesis in response to vascular endothelial growth factor and angiopoietin 2. Ann Rheum Dis (2013) 72:1080–8. doi: 10.1136/annrheumdis-2012-201978
125. Sucur A, Filipovic M, Flegar D, Kelava T, Sisl D, Lukac N, et al. Notch receptors and ligands in inflammatory arthritis - a systematic review. Immunol Lett (2020) 223:106–14. doi: 10.1016/j.imlet.2020.04.010
126. Harjes U, Bridges E, McIntyre A, Fielding BA, Harris AL. Fatty acid-binding protein 4, a point of convergence for angiogenic and metabolic signaling pathways in endothelial cells. J Biol Chem (2014) 289:23168–76. doi: 10.1074/jbc.M114.576512
127. Jabs M, Rose AJ, Lehmann LH, Taylor J, Moll I, Sijmonsma TP, et al. Inhibition of endothelial notch signaling impairs fatty acid transport and leads to metabolic and vascular remodeling of the adult heart. Circulation (2018) 137:2592–608. doi: 10.1161/CIRCULATIONAHA.117.029733
128. Zhang Y, Liu X, Zhu L, Zhou Z, Cui Y, Zhou CX, et al. Notch activation promotes bone metastasis via SPARC inhibition in adenoid cystic carcinoma. Oral Dis (2023). doi: 10.1111/odi.14573
129. Gorantla B, Bhoopathi P, Chetty C, Gogineni VR, Sailaja GS, Gondi CS, et al. Notch signaling regulates tumor-induced angiogenesis in SPARC-overexpressed neuroblastoma. Angiogenesis (2013) 16:85–100. doi: 10.1007/s10456-012-9301-1
130. Chi Z, Lu Y, Yang Y, Li B, Lu P. Transcriptional and epigenetic regulation of PD-1 expression. Cell Mol Life Sci (2021) 78:3239–46. doi: 10.1007/s00018-020-03737-y
131. Esteve-Puig R, Bueno-Costa A, Esteller M. Writers, readers and erasers of RNA modifications in cancer. Cancer Lett (2020) 474:127–37. doi: 10.1016/j.canlet.2020.01.021
132. Condorelli AG, El HM, Zambruno G, Nystrom A, Candi E, Castiglia D. Notch-ing up knowledge on molecular mechanisms of skin fibrosis: focus on the multifaceted Notch signalling pathway. J BioMed Sci (2021) 28:36. doi: 10.1186/s12929-021-00732-8
133. Antfolk D, Antila C, Kemppainen K, Landor SK, Sahlgren C. Decoding the PTM-switchboard of notch. Biochim Biophys Acta Mol Cell Res (2019) 1866:118507. doi: 10.1016/j.bbamcr.2019.07.002
134. Bousseau S, Vergori L, Soleti R, Lenaers G, Martinez MC, Andriantsitohaina R. Glycosylation as new pharmacological strategies for diseases associated with excessive angiogenesis. Pharmacol Ther (2018) 191:92–122. doi: 10.1016/j.pharmthera.2018.06.003
135. Zhang L, Ten HK. O-Linked glycosylation in Drosophila melanogaster. Curr Opin Struct Biol (2019) 56:139–45. doi: 10.1016/j.sbi.2019.01.014
136. Hao X, Li Y, Wang J, Ma J, Zhao S, Ye X, et al. Deficient O-glcNAc glycosylation impairs regulatory T cell differentiation and notch signaling in autoimmune hepatitis. Front Immunol (2018) 9:2089. doi: 10.3389/fimmu.2018.02089
137. Chen J, Dong X, Cheng X, Zhu Q, Zhang J, Li Q, et al. Ogt controls neural stem/progenitor cell pool and adult neurogenesis through modulating Notch signaling. Cell Rep (2021) 34:108905. doi: 10.1016/j.celrep.2021.108905
138. Sawaguchi S, Varshney S, Ogawa M, Sakaidani Y, Yagi H, Takeshita K, et al. O-GlcNAc on NOTCH1 EGF repeats regulates ligand-induced Notch signaling and vascular development in mammals. Elife (2017) 6. doi: 10.7554/eLife.24419
139. Umar S, Singh AK, Chourasia M, Rasmussen SM, Ruth JH, Ahmed S. Penta-o-galloyl-beta-d-Glucose (PGG) inhibits inflammation in human rheumatoid arthritis synovial fibroblasts and rat adjuvant-induced arthritis model. Front Immunol (2022) 13:928436. doi: 10.3389/fimmu.2022.928436
140. Li YN, Chen CW, Trinh-Minh T, Zhu H, Matei AE, Gyorfi AH, et al. Dynamic changes in O-GlcNAcylation regulate osteoclast differentiation and bone loss via nucleoporin 153. Bone Res (2022) 10:51. doi: 10.1038/s41413-022-00218-9
141. Schnute B, Shimizu H, Lyga M, Baron M, Klein T. Ubiquitylation is required for the incorporation of the Notch receptor into intraluminal vesicles to prevent prolonged and ligand-independent activation of the pathway. BMC Biol (2022) 20:65. doi: 10.1186/s12915-022-01245-y
142. Lim R, Sugino T, Nolte H, Andrade J, Zimmermann B, Shi C, et al. Deubiquitinase USP10 regulates Notch signaling in the endothelium. Science (2019) 364:188–93. doi: 10.1126/science.aat0778
143. Ohnuki H, Inoue H, Takemori N, Nakayama H, Sakaue T, Fukuda S, et al. BAZF, a novel component of cullin3-based E3 ligase complex, mediates VEGFR and Notch cross-signaling in angiogenesis. Blood (2012) 119:2688–98. doi: 10.1182/blood-2011-03-345306
144. Behl T, Chadha S, Sachdeva M, Kumar A, Hafeez A, Mehta V, et al. Ubiquitination in rheumatoid arthritis. Life Sci (2020) 261:118459. doi: 10.1016/j.lfs.2020.118459
145. Wu Q, Chen X, Qiao C, Cao X, Du Q, Yuan Y, et al. Methotrexate and Triptolide regulate Notch signaling pathway by targeting the Nedd4-Numb axis. Int Immunopharmacol (2023) 114:109595. doi: 10.1016/j.intimp.2022.109595
146. Zhu X, Ding S, Qiu C, Shi Y, Song L, Wang Y, et al. SUMOylation negatively regulates angiogenesis by targeting endothelial NOTCH signaling. Circ Res (2017) 121:636–49. doi: 10.1161/CIRCRESAHA.117.310696
147. Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling–are we there yet? Nat Rev Drug Discovery (2014) 13:357–78. doi: 10.1038/nrd4252
148. He Q, Chen Z, Li J, Liu J, Zuo Z, Lin B, et al. SENP6-mediated deSUMOylation of VEGFR2 enhances its cell membrane transport in angiogenesis. Int J Mol Sci (2023) 24. doi: 10.3390/ijms24032544
149. Zhou HJ, Xu Z, Wang Z, Zhang H, Zhuang ZW, Simons M, et al. SUMOylation of VEGFR2 regulates its intracellular trafficking and pathological angiogenesis. Nat Commun (2018) 9:3303. doi: 10.1038/s41467-018-05812-2
150. Dehnavi S, Sadeghi M, Johnston TP, Barreto G, Shohan M, Sahebkar A. The role of protein SUMOylation in rheumatoid arthritis. J Autoimmun (2019) 102:1–7. doi: 10.1016/j.jaut.2019.05.006
151. Tao S, Chen Q, Lin C, Dong H. Linc00514 promotes breast cancer metastasis and M2 polarization of tumor-associated macrophages via Jagged1-mediated notch signaling pathway. J Exp Clin Cancer Res (2020) 39:191. doi: 10.1186/s13046-020-01676-x
152. Zhang Z, Lu YX, Liu F, Sang L, Shi C, Xie S, et al. lncRNA BREA2 promotes metastasis by disrupting the WWP2-mediated ubiquitination of Notch1. Proc Natl Acad Sci U.S.A. (2023) 120:e2088273176. doi: 10.1073/pnas.2206694120
153. Zhang M, Han Y, Zheng Y, Zhang Y, Zhao X, Gao Z, et al. ZEB1-activated LINC01123 accelerates the Malignancy in lung adenocarcinoma through NOTCH signaling pathway. Cell Death Dis (2020) 11:981. doi: 10.1038/s41419-020-03166-6
154. Trimarchi T, Bilal E, Ntziachristos P, Fabbri G, Dalla-Favera R, Tsirigos A, et al. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell (2014) 158:593–606. doi: 10.1016/j.cell.2014.05.049
155. Zhang Z, Li G, Qiu H, Yang J, Bu X, Zhu S, et al. The novel notch-induced long noncoding RNA LUNAR1 determines the proliferation and prognosis of colorectal cancer. Sci Rep (2019) 9:19915. doi: 10.1038/s41598-019-56536-2
156. Dang Q, Liu Z, Liu Y, Wang W, Yuan W, Sun Z, et al. LncRNA profiles from Notch signaling: Implications for clinical management and tumor microenvironment of colorectal cancer. Front Immunol (2022) 13:953405. doi: 10.3389/fimmu.2022.953405
157. Liu J, Li Q, Zhang KS, Hu B, Niu X, Zhou SM, et al. Downregulation of the long non-coding RNA meg3 promotes angiogenesis after ischemic brain injury by activating notch signaling. Mol Neurobiol (2017) 54:8179–90. doi: 10.1007/s12035-016-0270-z
158. Cheng X, Xu J, Yu Z, Xu J, Long H. LncRNA xist contributes to endogenous neurological repair after chronic compressive spinal cord injury by promoting angiogenesis through the miR-32-5p/notch-1 axis. Front Cell Dev Biol (2020) 8:744. doi: 10.3389/fcell.2020.00744
159. Wan L, Liu J, Huang C, Zhao L, Jiang H, Liu L, et al. [Decreased long-chain non-coding RNA MALAT1 expression and increased hsa-miR155-3p expression involved in Notch signaling pathway regulation in rheumatoid arthritis patients]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2020) 36:535–41.
160. Zhi LQ, Zhong Q, Ma JB, Xiao L, Yao SX, Wang X. LncRNA H19 inhibitor represses synovial cell proliferation and apoptosis in rats with rheumatoid arthritis via Notch signaling pathway. Eur Rev Med Pharmacol Sci (2020) 24:4088–94. doi: 10.26355/eurrev_202008_22456
161. Ghafouri-Fard S, Glassy MC, Abak A, Hussen BM, Niazi V, Taheri M. The interaction between miRNAs/lncRNAs and Notch pathway in human disorders. BioMed Pharmacother (2021) 138:111496. doi: 10.1016/j.biopha.2021.111496
162. Geng Y, Fan J, Chen L, Zhang C, Qu C, Qian L, et al. A notch-dependent inflammatory feedback circuit between macrophages and cancer cells regulates pancreatic cancer metastasis. Cancer Res (2021) 81:64–76. doi: 10.1158/0008-5472.CAN-20-0256
163. Zhang S, Cui Z. MicroRNA-34b-5p inhibits proliferation, stemness, migration and invasion of retinoblastoma cells via Notch signaling. Exp Ther Med (2021) 21:255. doi: 10.3892/etm.2021.9686
164. Yao Y, Li X, Cheng L, Wu X, Wu B. Circular RNA FAT atypical cadherin 1 (circFAT1)/microRNA-525-5p/spindle and kinetochore-associated complex subunit 1 (SKA1) axis regulates oxaliplatin resistance in breast cancer by activating the notch and Wnt signaling pathway. Bioengineered (2021) 12:4032–43. doi: 10.1080/21655979.2021.1951929
165. Zhou Q, Rong C, Gu T, Li H, Wu L, Zhuansun X, et al. Mesenchymal stem cells improve liver fibrosis and protect hepatocytes by promoting microRNA-148a-5p-mediated inhibition of Notch signaling pathway. Stem Cell Res Ther (2022) 13:354. doi: 10.1186/s13287-022-03030-8
166. Marchetti M, Meloni M, Anwar M, Zen A, Sala-Newby G, Slater S, et al. MicroRNA-24-3p targets notch and other vascular morphogens to regulate post-ischemic microvascular responses in limb muscles. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21051733
167. Stevens MT, Saunders BM. Targets and regulation of microRNA-652-3p in homoeostasis and disease. J Mol Med (Berl) (2021) 99:755–69. doi: 10.1007/s00109-021-02060-8
168. Wang R, Yang Z, Liang L, Feng X, Che B, Zhang X, et al. Notch activation suppresses endothelial cell migration and sprouting via miR-223-3p targeting Fbxw7. In Vitro Cell Dev Biol Anim (2022) 58:124–35. doi: 10.1007/s11626-022-00649-y
169. Ogando J, Tardaguila M, Diaz-Alderete A, Usategui A, Miranda-Ramos V, Martinez-Herrera DJ, et al. Notch-regulated miR-223 targets the aryl hydrocarbon receptor pathway and increases cytokine production in macrophages from rheumatoid arthritis patients. Sci Rep (2016) 6:20223. doi: 10.1038/srep20223
170. Tao J, Miao R, Liu G, Qiu X, Yang B, Tan X, et al. Spatiotemporal correlation between HIF-1alpha and bone regeneration. FASEB J (2022) 36:e22520. doi: 10.1096/fj.202200329RR
171. Chen J, Cheng W, Li J, Wang Y, Chen J, Shen X, et al. Notch-1 and notch-3 mediate hypoxia-induced activation of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheumatol (2021) 73:1810–9. doi: 10.1002/art.41748
172. O'Brien KA, Murray AJ, Simonson TS. Notch signaling and cross-talk in hypoxia: A candidate pathway for high-altitude adaptation. Life (Basel) (2022) 12. doi: 10.3390/life12030437
173. Kim S, Lee M, Choi YK. The role of a neurovascular signaling pathway involving hypoxia-inducible factor and notch in the function of the central nervous system. Biomol Ther (Seoul) (2020) 28:45–57. doi: 10.4062/biomolther.2019.119
174. Fearon U, Canavan M, Biniecka M, Veale DJ. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat Rev Rheumatol (2016) 12:385–97. doi: 10.1038/nrrheum.2016.69
175. Gao W, McCormick J, Connolly M, Balogh E, Veale DJ, Fearon U. Hypoxia and STAT3 signalling interactions regulate pro-inflammatory pathways in rheumatoid arthritis. Ann Rheum Dis (2015) 74:1275–83. doi: 10.1136/annrheumdis-2013-204105
176. Ferrante F, Giaimo BD, Friedrich T, Sugino T, Mertens D, Kugler S, et al. Hydroxylation of the NOTCH1 intracellular domain regulates Notch signaling dynamics. Cell Death Dis (2022) 13:600. doi: 10.1038/s41419-022-05052-9
177. Jiang N, Zou C, Zhu Y, Luo Y, Chen L, Lei Y, et al. HIF-1a-regulated miR-1275 maintains stem cell-like phenotypes and promotes the progression of LUAD by simultaneously activating Wnt/beta-catenin and Notch signaling. Theranostics (2020) 10:2553–70. doi: 10.7150/thno.41120
178. Yang M, Li CJ, Sun X, Guo Q, Xiao Y, Su T, et al. MiR-497 approximately 195 cluster regulates angiogenesis during coupling with osteogenesis by maintaining endothelial Notch and HIF-1alpha activity. Nat Commun (2017) 8:16003. doi: 10.1038/ncomms16003
179. Wang Y, Singh AR, Zhao Y, Du T, Huang Y, Wan X, et al. TRIM28 regulates sprouting angiogenesis through VEGFR-DLL4-Notch signaling circuit. FASEB J (2020) 34:14710–24. doi: 10.1096/fj.202000186RRR
180. Chen Y, Zhao B, Zhu Y, Zhao H, Ma C. HIF-1-VEGF-Notch mediates angiogenesis in temporomandibular joint osteoarthritis. Am J Transl Res (2019) 11:2969–82.
181. Fang Y, Yu S, Ma Y, Sun P, Ma D, Ji C, et al. Association of Dll4/notch and HIF-1a -VEGF signaling in the angiogenesis of missed abortion. PloS One (2013) 8:e70667. doi: 10.1371/journal.pone.0070667
182. Ai X, Yu P, Luo L, Sun J, Tao H, Wang X, et al. Berberis dictyophylla F. inhibits angiogenesis and apoptosis of diabetic retinopathy via suppressing HIF-1alpha/VEGF/DLL-4/Notch-1 pathway. J Ethnopharmacol (2022) 296:115453. doi: 10.1016/j.jep.2022.115453
183. Heo R, Park JS, Jang HJ, Kim SH, Shin JM, Suh YD, et al. Hyaluronan nanoparticles bearing gamma-secretase inhibitor: in vivo therapeutic effects on rheumatoid arthritis. J Control Release (2014) 192:295–300. doi: 10.1016/j.jconrel.2014.07.057
184. Zhang H, Hilton MJ, Anolik JH, Welle SL, Zhao C, Yao Z, et al. NOTCH inhibits osteoblast formation in inflammatory arthritis via noncanonical NF-kappaB. J Clin Invest (2014) 124:3200–14. doi: 10.1172/JCI68901
185. Jiao Z, Wang W, Ma J, Wang S, Su Z, Xu H. Notch signaling mediates TNF-alpha-induced IL-6 production in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Clin Dev Immunol (2012) 2012:350209. doi: 10.1155/2012/350209
186. Jiao Z, Wang W, Xu H, Wang S, Guo M, Chen Y, et al. Engagement of activated Notch signalling in collagen II-specific T helper type 1 (Th1)- and Th17-type expansion involving Notch3 and Delta-like1. Clin Exp Immunol (2011) 164:66–71. doi: 10.1111/j.1365-2249.2010.04310.x
187. Chen J, Li J, Chen J, Cheng W, Lin J, Ke L, et al. Treatment of collagen-induced arthritis rat model by using Notch signalling inhibitor. J Orthop Translat (2021) 28:100–7. doi: 10.1016/j.jot.2021.01.003
188. Kim MJ, Park JS, Lee SJ, Jang J, Park JS, Back SH, et al. Notch1 targeting siRNA delivery nanoparticles for rheumatoid arthritis therapy. J Control Release (2015) 216:140–8. doi: 10.1016/j.jconrel.2015.08.025
189. Zhao G, Zhang H. Notch-1 siRNA and Methotrexate towards a Multifunctional Approach in Rhematoid Arthritis Management: a Nanomedicine Approach. Pharm Res (2018) 35:123. doi: 10.1007/s11095-018-2401-x
190. An L, Li Z, Shi L, Wang L, Wang Y, Jin L, et al. Inflammation-targeted celastrol nanodrug attenuates collagen-induced arthritis through NF-kappaB and notch1 pathways. Nano Lett (2020) 20:7728–36. doi: 10.1021/acs.nanolett.0c03279
191. Anchi P, Swamy V, Godugu C. Nimbolide exerts protective effects in complete Freund's adjuvant induced inflammatory arthritis via abrogation of STAT-3/NF-kappaB/Notch-1 signaling. Life Sci (2021) 266:118911. doi: 10.1016/j.lfs.2020.118911
192. Su J, Tao Y, Liu J, Sun J, Zeng Y, Meng X, et al. Tibetan medicine Qi-Sai-Er-Sang-Dang-Song Decoction inhibits TNF-alpha-induced rheumatoid arthritis in human fibroblast-like synoviocytes via regulating NOTCH1/NF-kappaB/NLRP3 pathway. J Ethnopharmacol (2023) 310:116402. doi: 10.1016/j.jep.2023.116402
Keywords: rheumatoid arthritis, angiogenesis, Notch signaling, adipokines, epigenetic regulation
Citation: Zhao F, He Y, Zhao Z, He J, Huang H, Ai K, Liu L and Cai X (2023) The Notch signaling-regulated angiogenesis in rheumatoid arthritis: pathogenic mechanisms and therapeutic potentials. Front. Immunol. 14:1272133. doi: 10.3389/fimmu.2023.1272133
Received: 03 August 2023; Accepted: 16 October 2023;
Published: 26 October 2023.
Edited by:
Long Li, Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital, ChinaReviewed by:
Neng-Yu Lin, National Taiwan University, TaiwanDavid Baker, Leiden University Medical Center (LUMC), Netherlands
Copyright © 2023 Zhao, He, Zhao, He, Huang, Ai, Liu and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelong Ai, YWlrZWxvbmdAY3N1LmVkdS5jbg==; Liang Liu, bGxpdUBnenVjbS5lZHUuY24=; Xiong Cai, Y2FpeGlvbmdAaG51Y20uZWR1LmNu